Спосіб одержання епоксиду (варіанти)
Номер патенту: 45968
Опубліковано: 15.05.2002
Автори: Мелігрес Петер Є., Упадхай Віна, Аскін Девід, Россен Кай, Волант Рольф П., Енг Кен К., Рейдер Пол Дж.

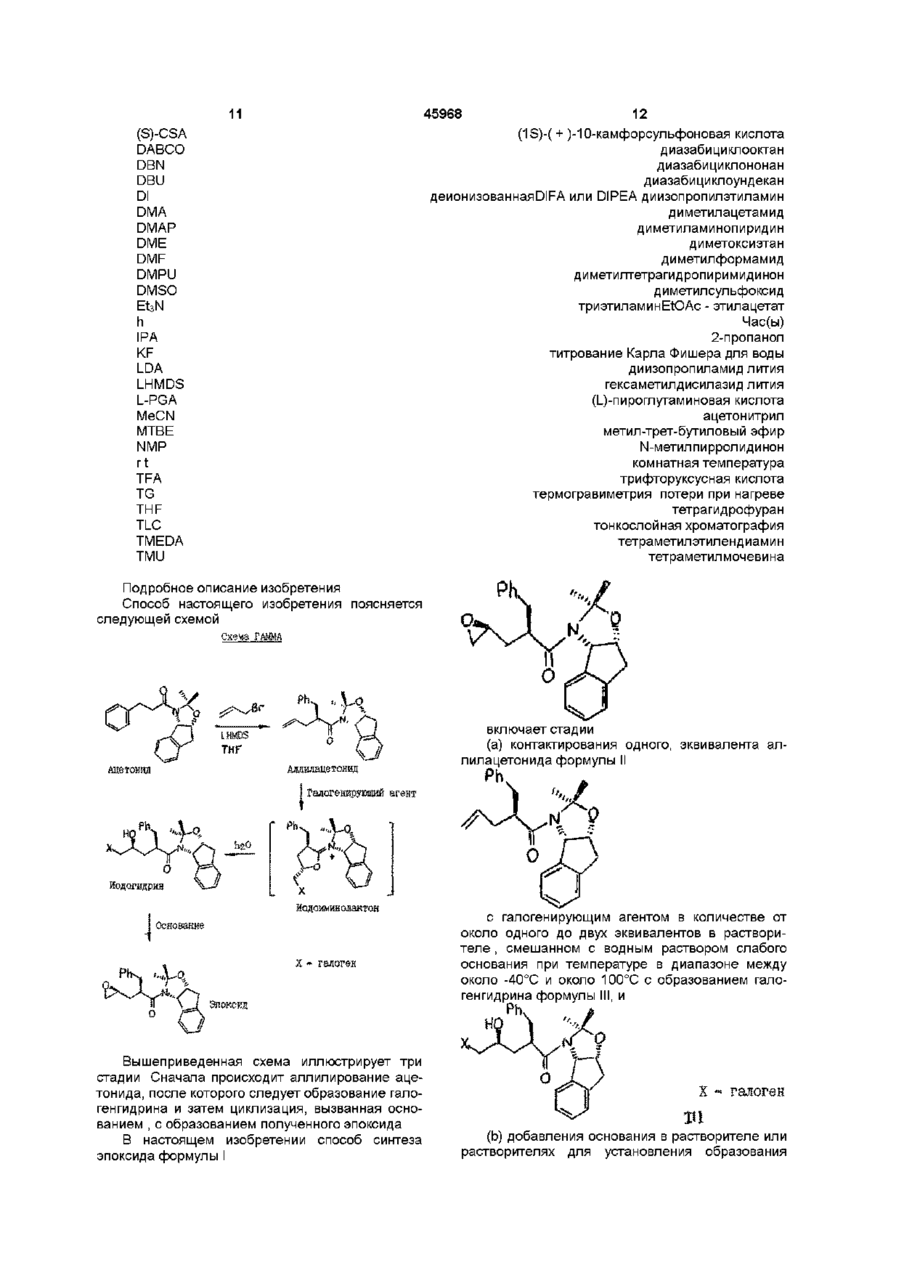
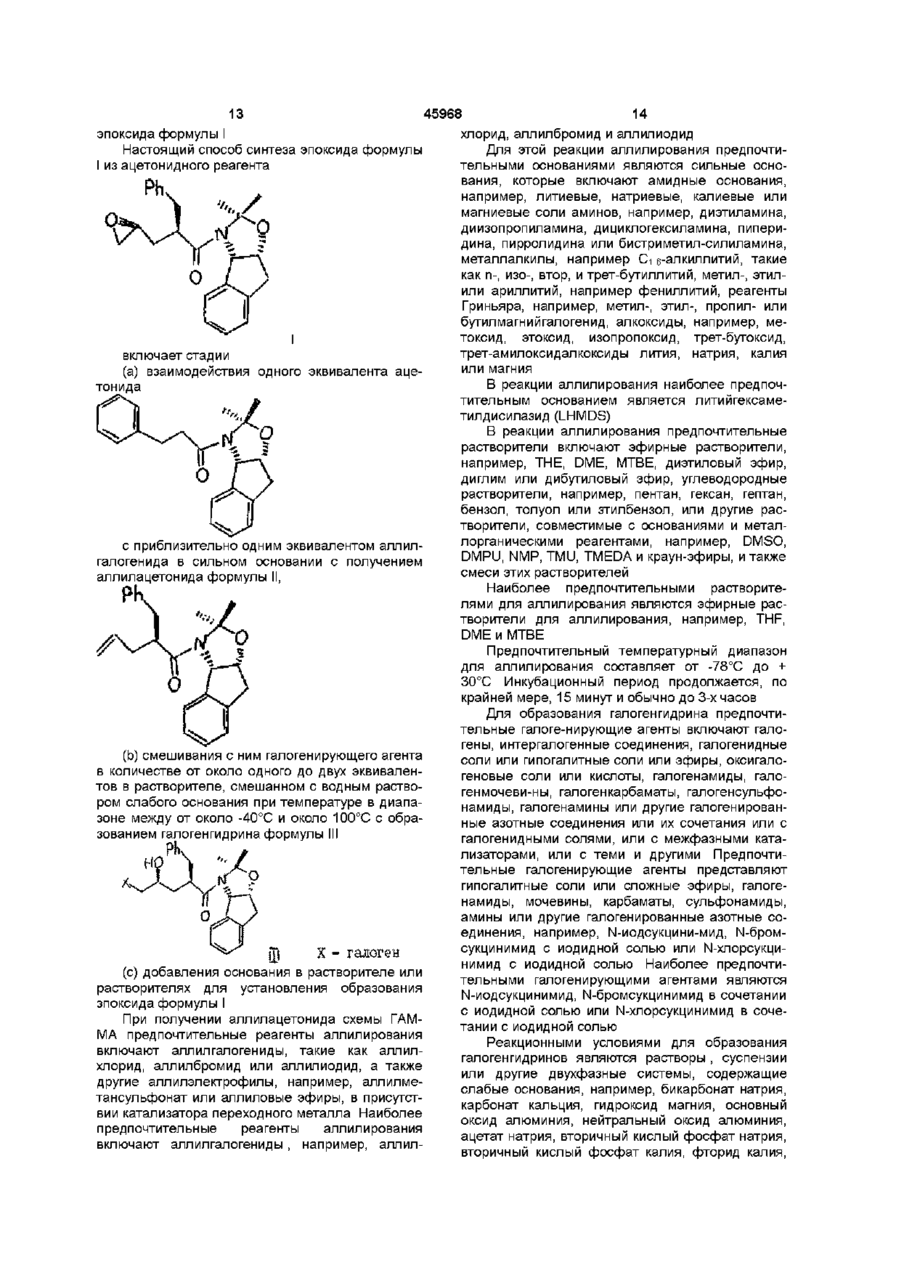
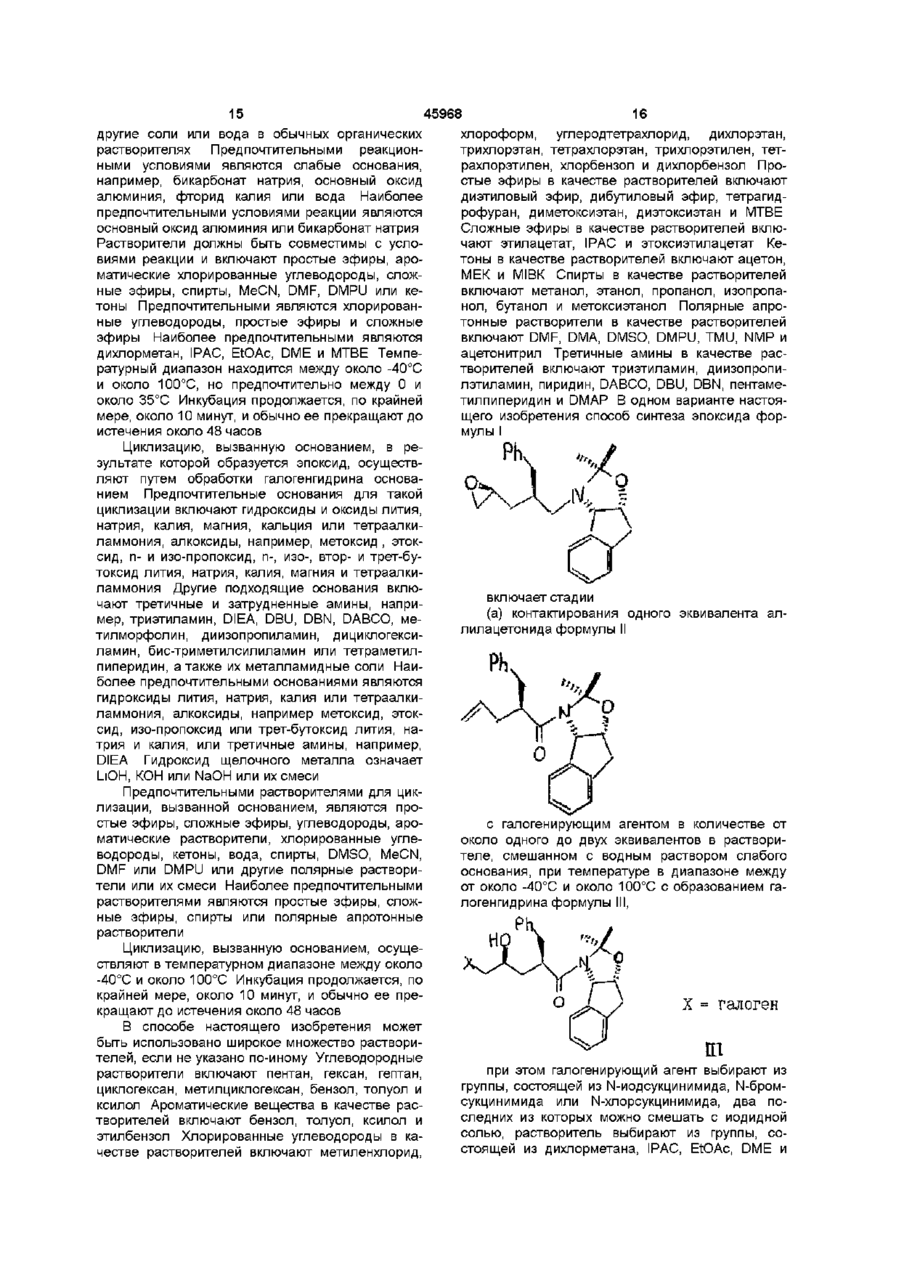
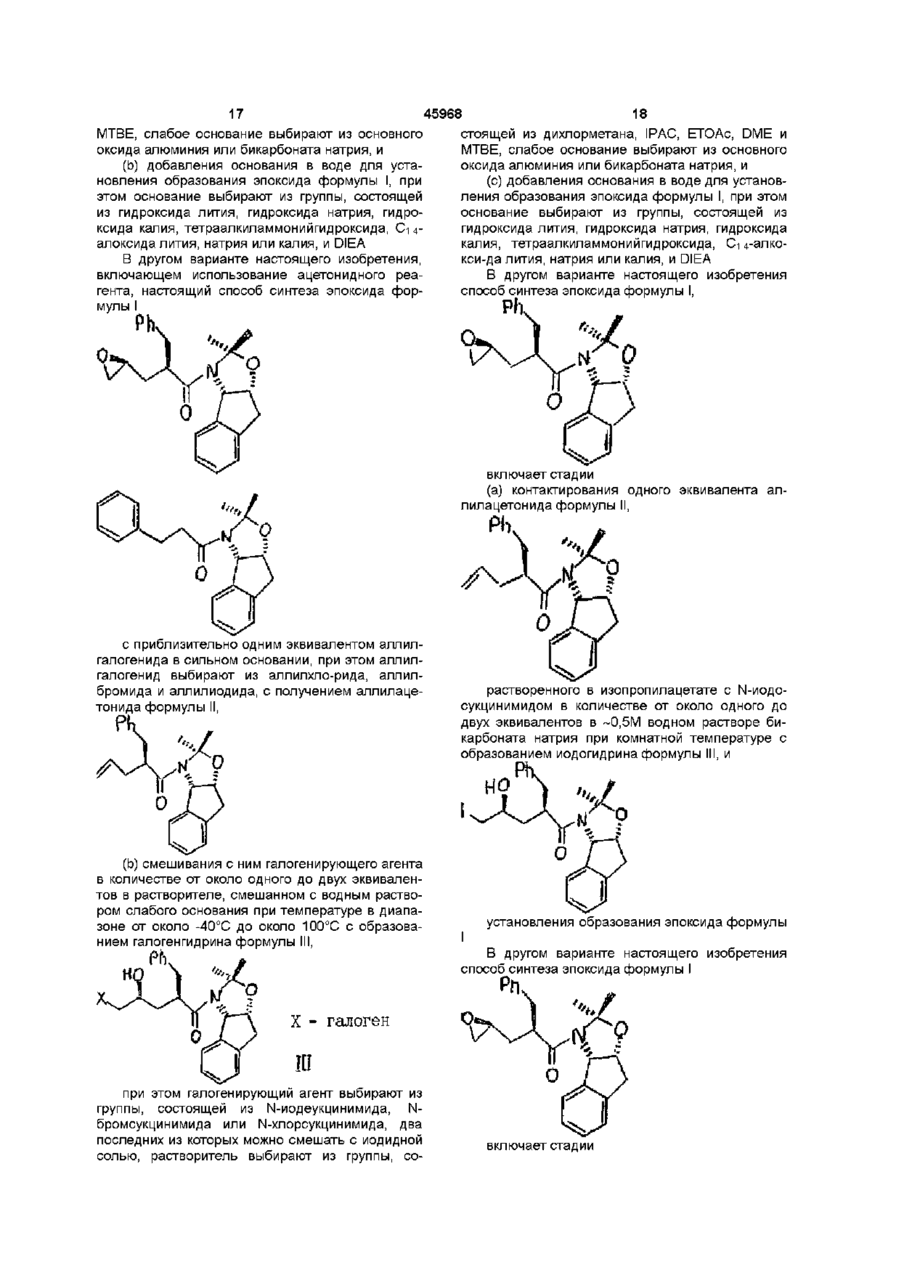






Формула / Реферат
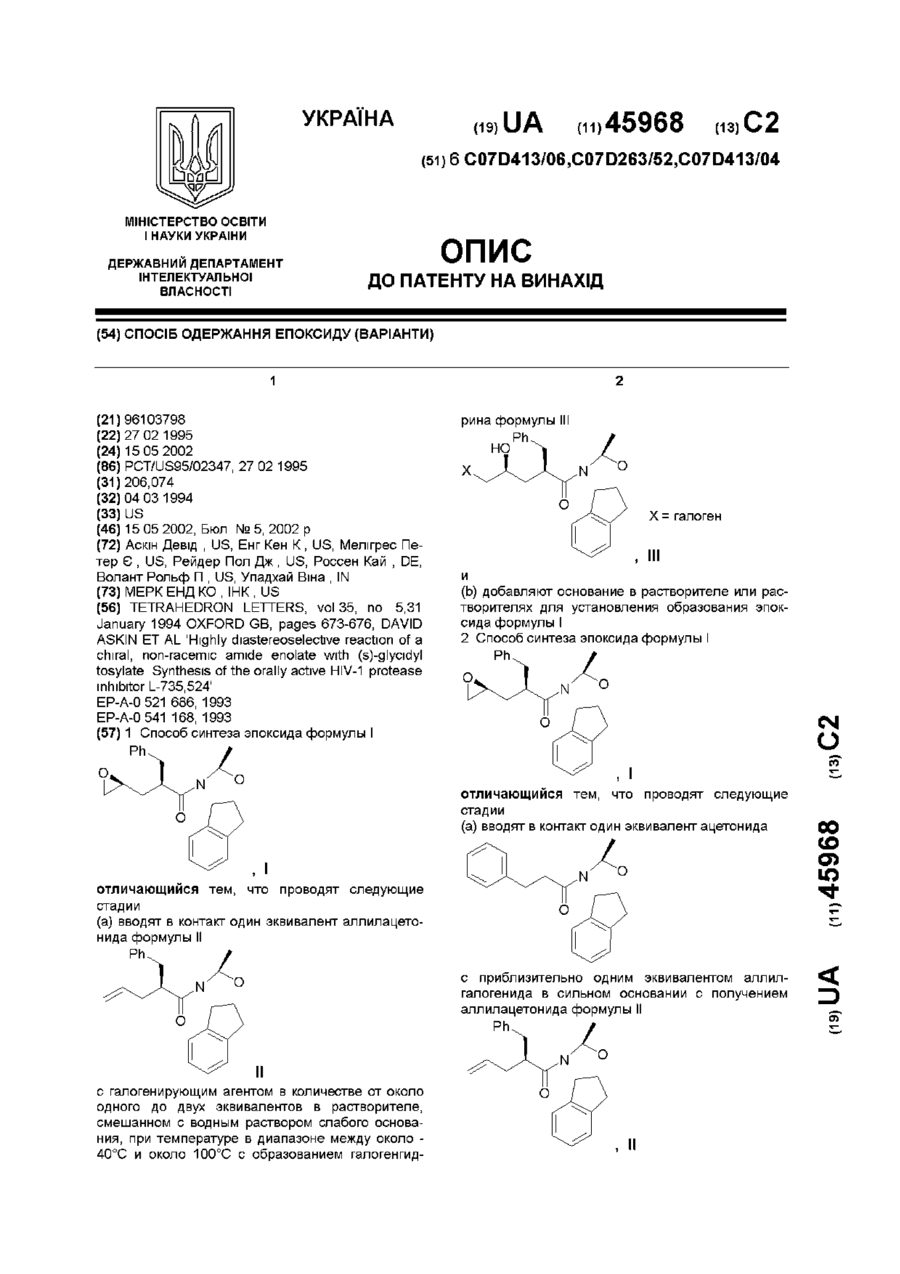
1. Способ синтеза эпоксида формулы I
отличающийся тем, что проводят следующие стадии:
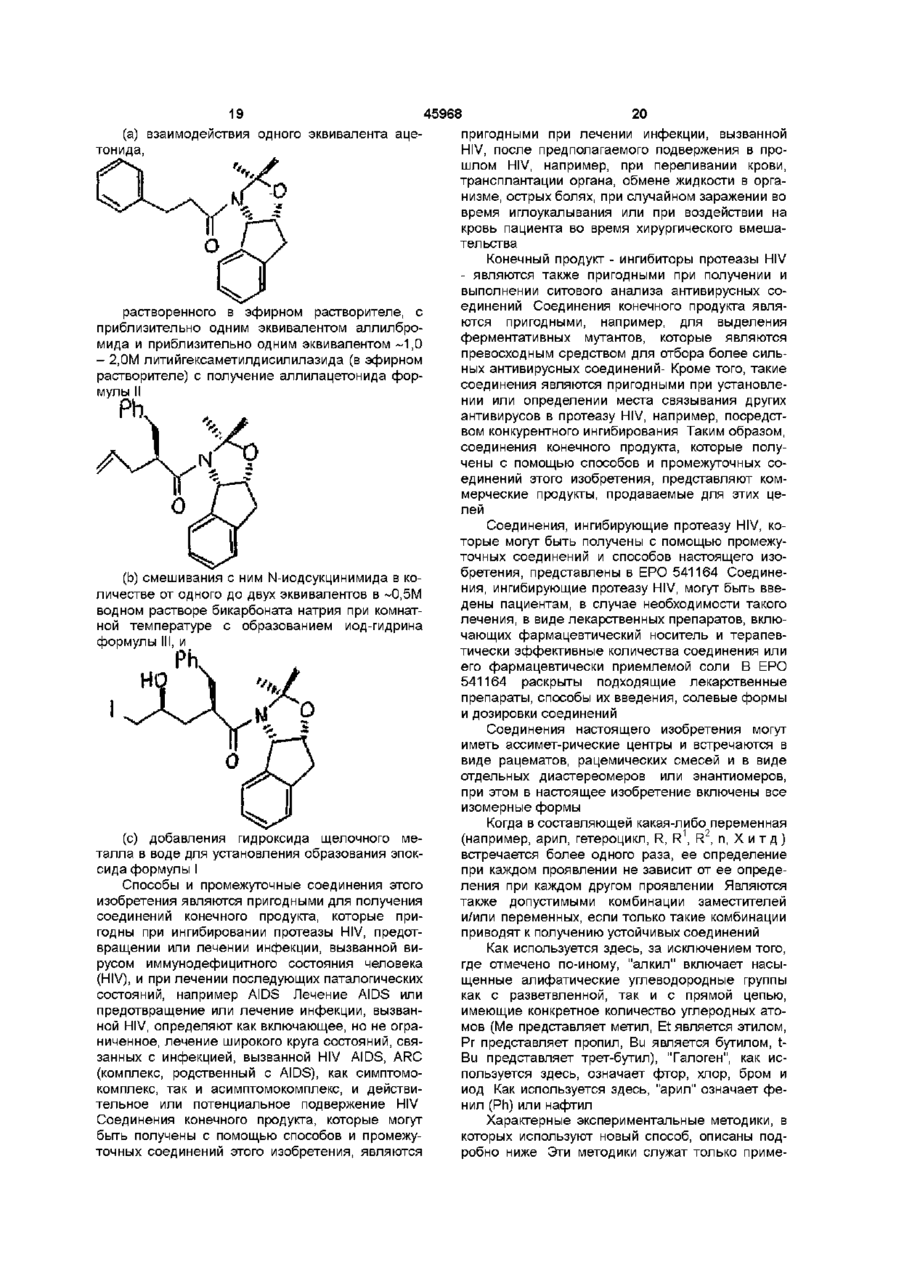
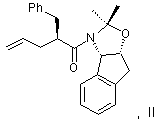
(а) вводят в контакт один эквивалент аллилацетонида формулы II
с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы ІІІ
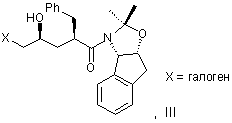
и
(b) добавляют основание в растворителе или растворителях для установления образования эпоксида формулы I.
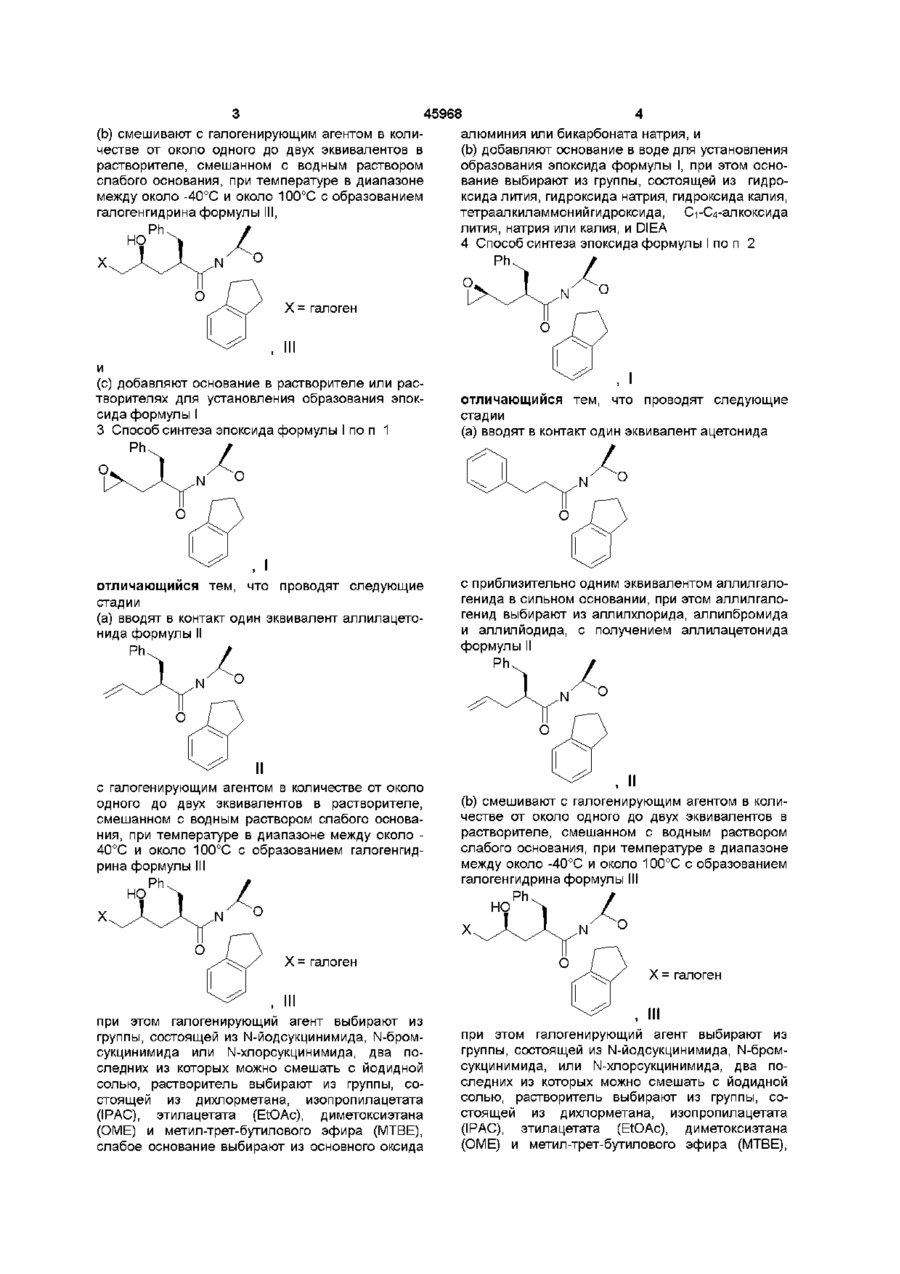
2. Способ синтеза эпоксида формулы I
отличающийся тем, что проводят следующие стадии:
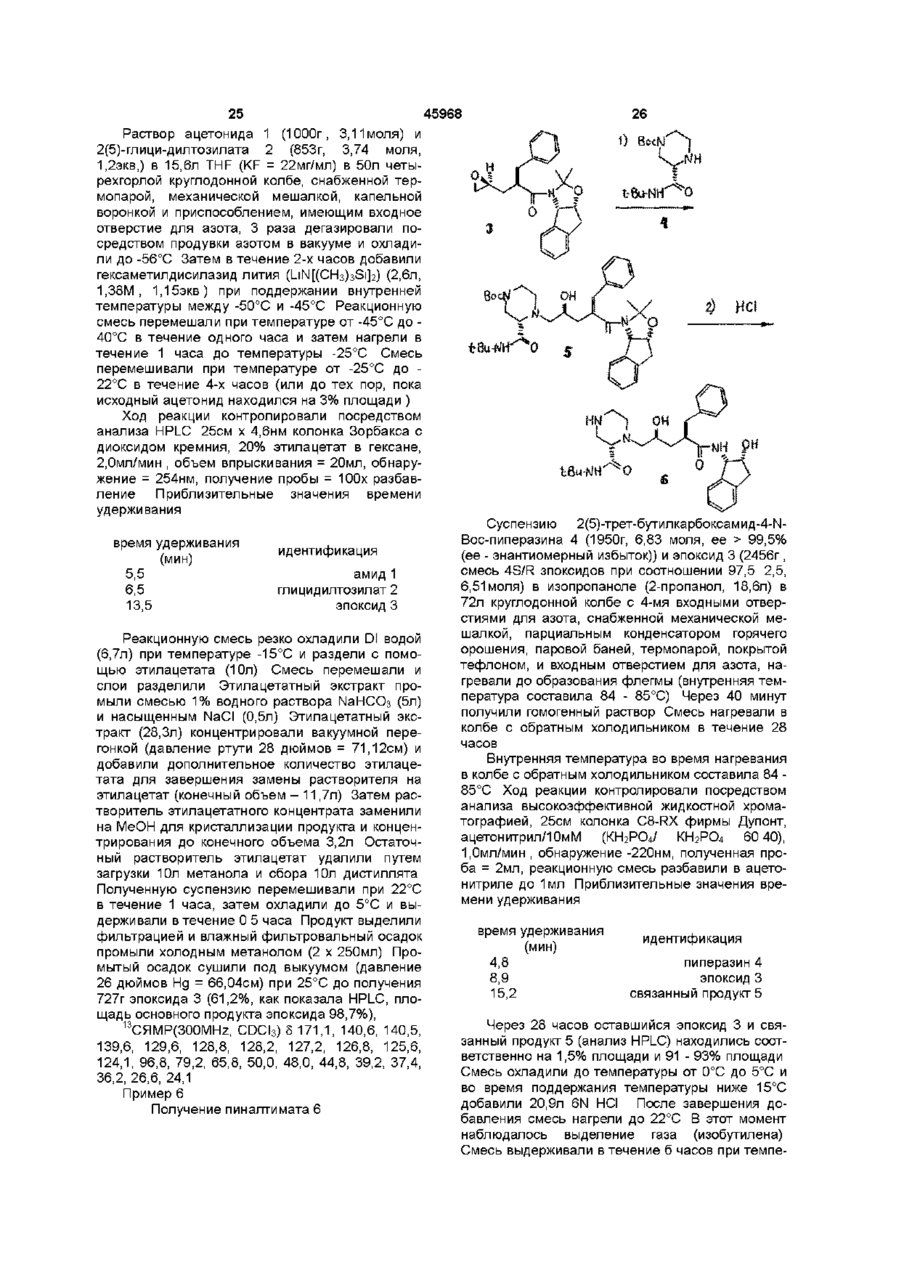
(а) вводят в контакт один эквивалент ацетонида
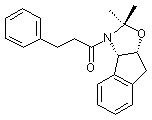
с приблизительно одним эквивалентом аллилгалогенида в сильном основании с получением аллилацетонида формулы II
(b) смешивают с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III,
и
(с) добавляют основание в растворителе или растворителях для установления образования эпоксида формулы I.
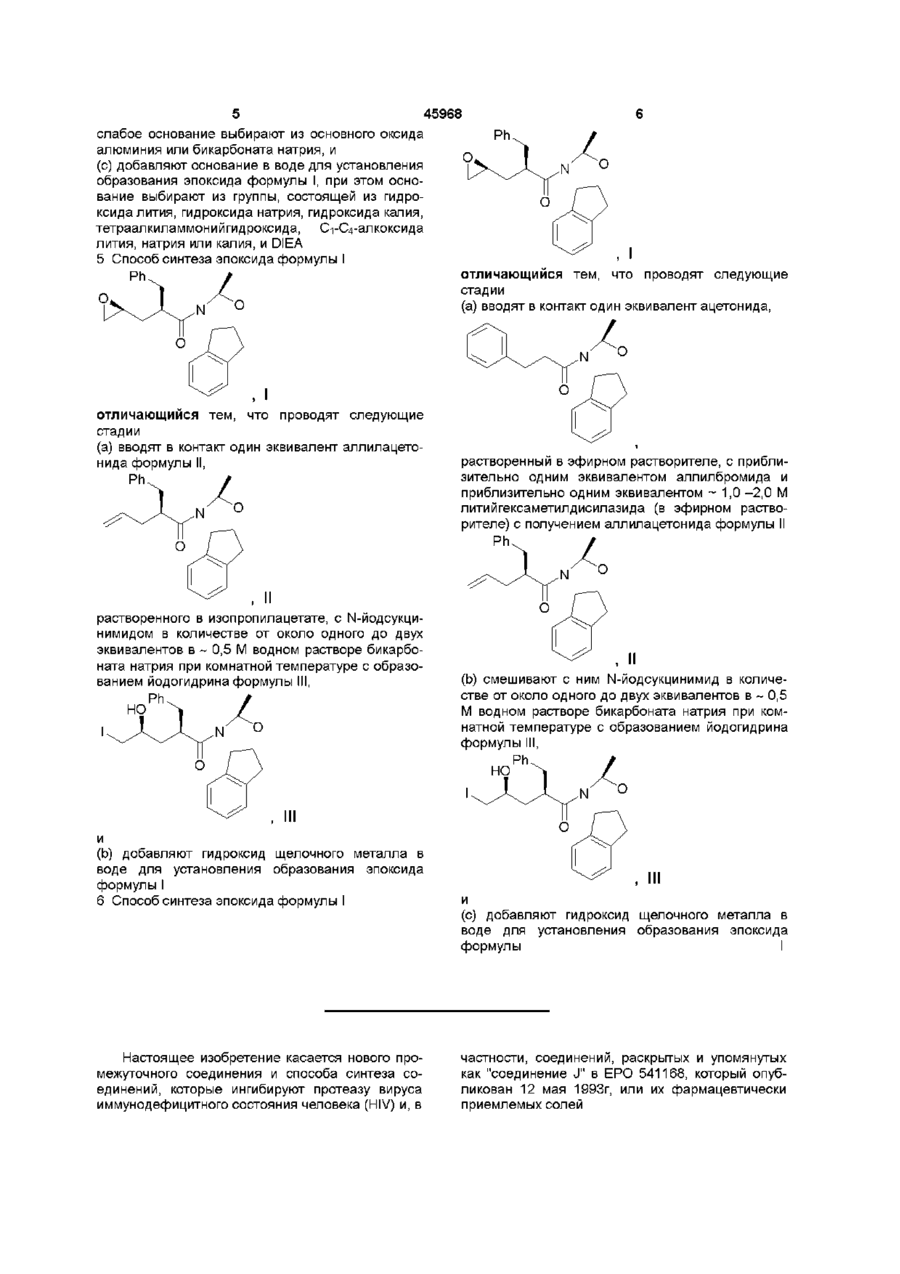
3. Способ синтеза эпоксида формулы I по п. 1
отличающийся тем, что проводят следующие стадии:
(а) вводят в контакт один эквивалент аллилацетонида формулы II
с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III
при этом галогенирующий агент выбирают из группы, состоящей из N-йодсукцинимида, N-бромсукцинимида или N-хлорсукцинимида, два последних из которых можно смешать с йодидной солью; растворитель выбирают из группы, состоящей из дихлорметана, изопропилацетата (ІРАС), этилацетата (EtOAc), диметоксиэтана (ОМЕ) и метил-трет-бутилового эфира (МТВЕ); слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и
(b) добавляют основание в воде для установления образования эпоксида формулы I, при этом основание выбирают из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия, тетраалкиламмонийгидроксида; С1-С4-алкоксида лития, натрия или калия; и DIEA.
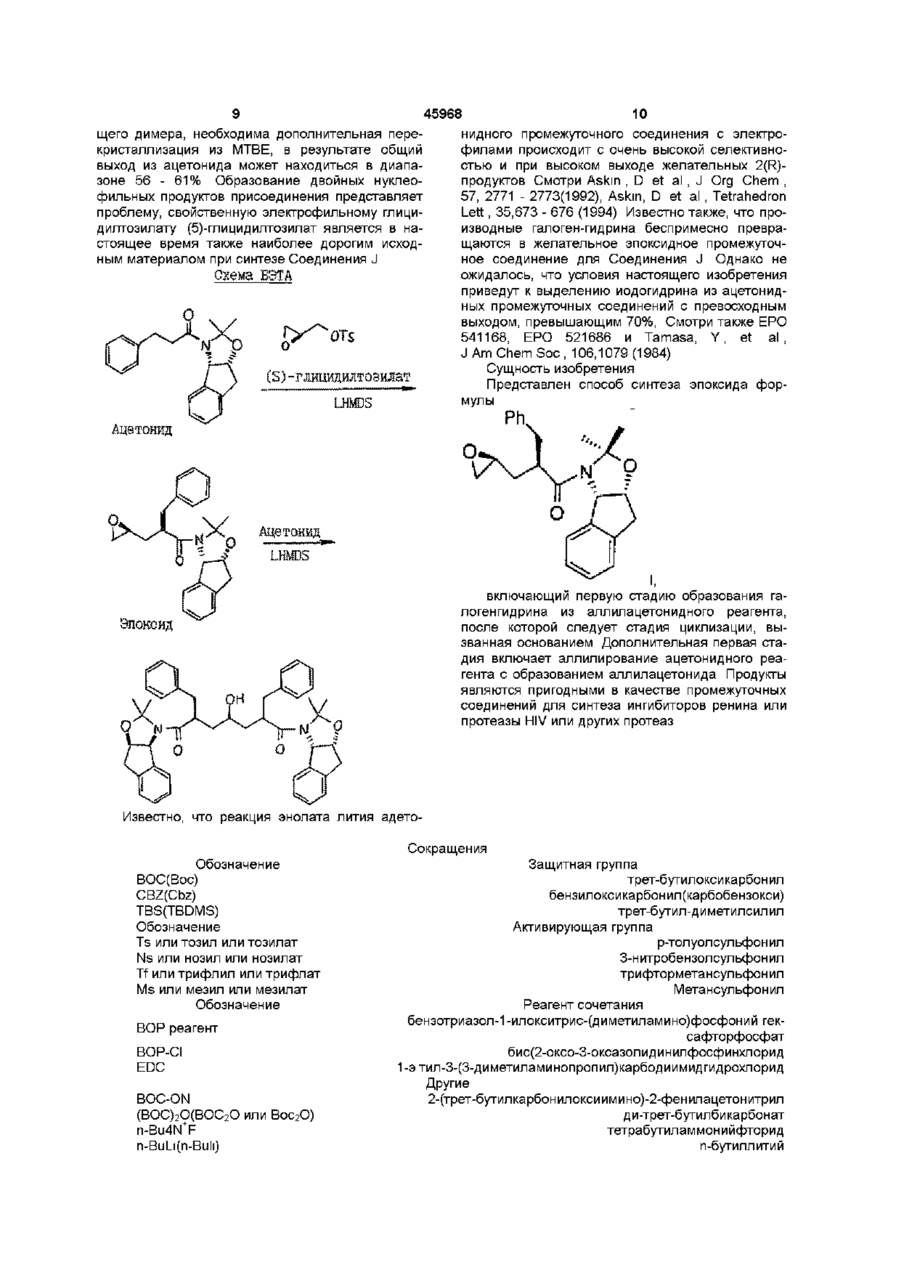
4. Способ синтеза эпоксида формулы I по п. 2
отличающийся тем, что проводят следующие стадии:
(а) вводят в контакт один эквивалент ацетонида
с приблизительно одним эквивалентом аллилгалогенида в сильном основании, при этом аллилгалогенид выбирают из аллилхлорида, аллилбромида и аллилйодида, с получением аллилацетонида формулы II
(b) смешивают с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III
при этом галогенирующий агент выбирают из группы, состоящей из N-йодсукцинимида, N-бромсукцинимида, или N-хлорсукцинимида, два последних из которых можно смешать с йодидной солью; растворитель выбирают из группы, состоящей из дихлорметана, изопропилацетата (IPAC), этилацетата (EtOAc), диметоксиэтана (ОМЕ) и метил-трет-бутилового эфира (МТВЕ); слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и
(с) добавляют основание в воде для установления образования эпоксида формулы I, при этом основание выбирают из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия, тетраалкиламмонийгидроксида; С1-С4-алкоксида лития, натрия или калия; и DIEA.
5. Способ синтеза эпоксида формулы I
отличающийся тем, что проводят следующие стадии:
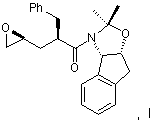
(а) вводят в контакт один эквивалент аллилацетонида формулы II,
растворенного в изопропилацетате, с N-йодсукцинимидом в количестве от около одного до двух эквивалентов в ~ 0,5 М водном растворе бикарбоната натрия при комнатной температуре с образованием йодогидрина формулы III,
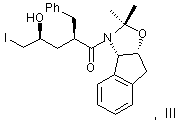
и
(b) добавляют гидроксид щелочного металла в воде для установления образования эпоксида формулы I.
6. Способ синтеза эпоксида формулы I
отличающийся тем, что проводят следующие стадии:
(а) вводят в контакт один эквивалент ацетонида,
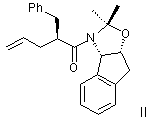
растворенный в эфирном растворителе, с приблизительно одним эквивалентом аллилбромида и приблизительно одним эквивалентом ~ 1,0 –2,0 М литийгексаметилдисилазида (в эфирном растворителе) с получением аллилацетонида формулы II
(b) смешивают с ним N-йодсукцинимид в количестве от около одного до двух эквивалентов в ~ 0,5 М водном растворе бикарбоната натрия при комнатной температуре с образованием йодогидрина формулы III,
и
(с) добавляют гидроксид щелочного металла в воде для установления образования эпоксида формулы I.
Текст
1 Способ синтеза эпоксида формулы I Ph. рина формулы Ph НО О X = галоген (Ь) добавляют основание в растворителе или растворителях для установления образования эпоксида формулы I 2 Способ синтеза эпоксида формулы I Ph. О отличающийся тем, что проводят следующие стадии (а) вводят в контакт один эквивалент ацетонида О отличающийся тем, что проводят следующие стадии (а) вводят в контакт один эквивалент аллилацетонида формулы II Ph. О с приблизительно одним эквивалентом аллилгалогенида в сильном основании с получением аллилацетонида формулы II Ph. О с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около 40°С и около 100°С с образованием галогенгид 00 (О ю 45968 (b) смешивают с галогенирующим агентом в колиалюминия или бикарбоната натрия, и честве от около одного до двух эквивалентов в (Ь) добавляют основание в воде для установления растворителе, смешанном с водным раствором образования эпоксида формулы I, при этом оснослабого основания, при температуре в диапазоне вание выбирают из группы, состоящей из гидромежду около -40°С и около 100°С с образованием ксида лития, гидроксида натрия, гидроксида калия, галогенгидрина формулы III, тетраалкиламмонийгидроксида, СгС4-алкоксида лития, натрия или калия, и DIEA Ph. 4 Способ синтеза эпоксида формулы І по п 2 НО • ^ Ph. -о X = галоген (с) добавляют основание в растворителе или растворителях для установления образования эпоксида формулы I 3 Способ синтеза эпоксида формулы І по п 1 Ph. отличающийся тем, что проводят следующие стадии (а) вводят в контакт один эквивалент ацетонида О отличающийся тем, что проводят следующие стадии (а) вводят в контакт один эквивалент аллилацетонида формулы II Ph. О с приблизительно одним эквивалентом аллилгалогенида в сильном основании, при этом аллилгалогенид выбирают из аллилхлорида, аллилбромида и аллилйодида, с получением аллилацетонида формулы II Ph. О с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около 40°С и около 100°С с образованием галогенгидрина формулы III Ph. НО • ^ -о X = галоген при этом галогенирующий агент выбирают из группы, состоящей из N-йодсукцинимида, N-бромсукцинимида или N-хлорсукцинимида, два последних из которых можно смешать с йодидной солью, растворитель выбирают из группы, состоящей из дихлорметана, изопропилацетата (IPAC), этилацетата (ЕЮ Ас), диметоксиэтана (ОМЕ) и метил-трет-бутил о во го эфира (МТВЕ), слабое основание выбирают из основного оксида (Ь) смешивают с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III Ph. НО • ^ -о X = галоген при этом галогенирующий агент выбирают из группы, состоящей из N-йодсукцинимида, N-бромсукцинимида, или N-хлорсукцинимида, два последних из которых можно смешать с йодидной солью, растворитель выбирают из группы, состоящей из дихлорметана, изопропилацетата (IPAC), этилацетата (ЕЮ Ас), диметоксиэтана (ОМЕ) и метил-трет-бутил о во го эфира (МТВЕ), 45968 слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и (с) добавляют основание в воде для установления образования эпоксида формулы I, при этом основание выбирают из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия, тетраалкиламмонийгидроксида, СгС4-алкоксида лития, натрия или калия, и DIEA 5 Способ синтеза эпоксида формулы I Ph. отличающийся тем, что проводят следующие стадии (а) вводят в контакт один эквивалент ацетонида, О отличающийся тем, что проводят следующие стадии (а) вводят в контакт один эквивалент аллилацетонида формулы II, Ph. О растворенный в эфирном растворителе, с приблизительно одним эквивалентом аллилбромида и приблизительно одним эквивалентом - 1 , 0 -2,0 М литийгексаметилдисилазида (в эфирном растворителе) с получением аллилацетонида формулы II Ph. О растворенного в изопропилацетате, с N-йодсукцинимидом в количестве от около одного до двух эквивалентов в ~ 0,5 М водном растворе бикарбоната натрия при комнатной температуре с образованием йодогидрина формулы III, Ph. НО -о (Ь) смешивают с ним N-йодсукцинимид в количестве от около одного до двух эквивалентов в ~ 0,5 М водном растворе бикарбоната натрия при комнатной температуре с образованием йодогидрина формулы III, Ph. НО -о (Ь) добавляют гидроксид щелочного металла в воде для установления образования эпоксида формулы I 6 Способ синтеза эпоксида формулы I Настоящее изобретение касается нового промежуточного соединения и способа синтеза соединений, которые ингибируют протеазу вируса иммунодефицитного состояния человека (HIV) и, в (с) добавляют гидроксид щелочного металла в воде для установления образования эпоксида формулы I частности, соединении, раскрытых и упомянутых как "соединение J" в ЕРО 541168, который опубликован 12 мая 1993г, или их фармацевтически приемлемых солей 8 45968 ЕРО 541168, который опубликован 12 мая 1993 г Ранее синтез Соединения J и родственных соединений осуществляли посредством 12-стадийного процесса, в котором использовали гидрои он ксизащищенный дигидро-5(5)-гидроксиметил-3(2Н)фуранон, который алкилировали, и который включалзамену спиртовой отщепляемой группы на алкилированный фуранон с пиперидиновой составляющей Затем связанный продукт гидролизовали для разрыва фуранонового кольца на Соединение J оксикислотную составляющую и, в конечном счеЭти соединения имеют значение при предотте, кислоту связывали в2(Н)-гидрокси-1(5)вращении инфекции, вызванной HIV, лечении инаминоиндан Эта методика описана в ЕРО541168 фекции, вызванной HIV, и лечении синдрома приЧрезвычайная продолжительность этого процесса обретенного иммунодефицита (СПИДа) (AIDS) (12стадий) приводит к большим трудозатратам, и он требует применения многих дорогостоящих Более конкретно, настоящий способ включает реагентов и дорогостоящего исходного матеполучение эпоксидного промежуточного соединериала Способ, для реализации которого требуния для получения указанного выше соединения ется меньшее количество стадий реакции и/или "J", являющегося ингибитором протеазы HIV Споболее эффективных реагентов, обеспечит эконособ относится к образованию иодогидрина из алмическую выгоду и снижение продолжительности лилацетонида, при этом сначала образуется пропроцесса межуточное соединение иодоиминолактон Циклизация иодогидрина, вызванная основанием, приИодлактамизация олефинового третичного водит затем к получению эпоксидного промежуамида А происходит, как известно, с последуюточного соединения Образование иодогидрина щим гидролизом заряженного промежуточного происходит при высокой диастереоселективности, соединения иодоиминолактама В, при этом в капри этом в этом процессе наблюдается реальное честве единственного выделенного продукта поотсутствие гидролиза амидной связи лучают иодолактон С (Схема АЛЬФА) Смотри Ретровирус, обозначенный как вирус иммукоTamaru, Y et al , J Am Chern Soc , 106, 1079 -1085 дефицитного состояния человека (HIV), представ(1984), Trost, B M e t a l , eds Comprehensive Orляет причинный фактор комплекса заболеваний, ganic Synthesis, Selectivity, Strategy, который включает прогрессирующую деструкцию & Efficiency in Modern Organic Chemistry, Volиммунной системы (синдром приобретенного имume 4, Pergamon Press, New York, 1991, p 398 мунодефицита AIDS) и дегенерацию центральной 421 Известно, что в этом процессе происходит и периферической нервной системы Этот вирус очень эффективное перемещение хиральности из ранее был известен как LAV , HTLV-III или ARV 2-положения в 4-положение, при этом получают Общей особенностью репликации ретровируса 2,4-синтетические продукты (представленные соявляется экстенсивная посттрансляционная обраответствующей оксикислотой D) с высокой диаботка предшественника полипротеинов посредстстереоселективностью вом вирусно-кодированной протеазы для генераСхема АЛЬФА ции зрелых вирусных белков, необходимых для скопления вирусов и их функционирования Ингибирование такой обработки приводит к предотвращению получения обычного инфекционного EfeO вируса Kohl, N Е et al , Proc Nat'1 Acacl Sci , 85, 4686(1988) показали, что генетическая инактивация протеазы HIV приводит к получению недоразвитых неинфекционных вирусных частиц Эти результаты показывают, что ингиби-рование проА теазы HIV представляет жизнеспособный способ лечения AIDS и предотвращения или лечения инфекции, вызванной HIV Нуклеотидная последовательность HIV показывает присутствие рої гена в одной открытой рамке для чтения [Ratner, L et al, Nature, 313, 277, (1985)] Гомология аминокислотной последовательности обеспечивает доказательство того, что рої последовательность кодирует ревертирующую транскриптазу, эндонуклеазу и протеазу HIV [Ton, H et al , EMBO J , 4, 1267 (1985), Power, M D et al , Science, 231, 1567 (1986), Pearl, L H et al , Nature, 329, 351 (1987)1 Соединения конечного продукта, включая соединение J, которые могут быть получены из новых промежуточных соединений и способа этого изобретения, представляют ингибиторы протеазы HIV, и раскрыты в С £> В другом существующем способе ацетонид взаимодействует с (Э)-глицидилтозилатом в присутствии сильного основания LHMDS с образованием эпоксида (смотри схему БЭТА) Поскольку как исходный материал (З)-глицидилтозилат, так и продукт представляют эпоксиды, анион ацетонида взаимодействует также с полученным эпоксидом, поэтому, кроме полученного эпоксида с выходом 71%, образуется около 20% двойных побочных продуктов присоединения После кристаллизации из МеОН для обеспечения эпоксида, не содержа щего димера, необходима дополнительная перекристаллизация из МТВЕ, в результате общий выход из ацетонида может находиться в диапазоне 56 - 6 1 % Образование двойных нуклеофильных продуктов присоединения представляет проблему, свойственную электрофильному глицидилтозилату (5)-глицидилтозилат является в настоящее время также наиболее дорогим исходным материалом при синтезе Соединения J Схема БЭТА (Е)-глицидилтоэилат шы» LHMDS 45968 10 нидного промежуточного соединения с электрофилами происходит с очень высокой селективностью и при высоком выходе желательных 2(R)продуктов Смотри Askm , D et al , J Org Chem , 57, 2771 - 2773(1992), Askm, D et al , Tetrahedron Lett, 35,673 - 676 (1994) Известно также, что производные галоген-гидрина беспримесно превращаются в желательное эпоксидное промежуточное соединение для Соединения J Однако не ожидалось, что условия настоящего изобретения приведут к выделению иодогидрина из ацетонидных промежуточных соединений с превосходным выходом, превышающим 70%, Смотри также ЕРО 541168, ЕРО 521686 и Tamasa, Y, et al , J Am Chem Soc , 106,1079 (1984) Сущность изобретения Представлен способ синтеза эпоксида формулы Ph. Ацетонид х4 Ц О ы Ацетонмд LHMDS включающий первую стадию образования галогенгидрина из аллилацетонидного реагента, после которой следует стадия циклизации, вызванная основанием Дополнительная первая стадия включает аллилирование ацетонидного реагента с образованием аллилацетонида Продукты являются пригодными в качестве промежуточных соединений для синтеза ингибиторов ренина или протеазы HIV или других протеаз Эпоксид Известно, что реакция энолата лития адетоСокращения Обозначение ВОС(Вос) CBZ(Cbz) TBS(TBDMS) Обозначение Ts или тозил или тозилат Ns или нозил или нозилат Tf или трифлил или трифлат Ms или мезил или мезилат Обозначение ВОР реагент BOP-CI EDC BOC-ON (ВОС)2О(ВОС2О или Вос2О) n-Bu4N+F n-BuLi(n-Buh) Защитная группа трет-бутилоксикарбонил бензилоксикарбонил(карбобензокси) трет-бутил-диметилсилил Активирующая группа р-толуолсульфонил 3-нитробензолсульфонил трифторметансульфонил Метансульфонил Реагент сочетания бензотриазол-1-илокситрис-(диметиламино)фосфоний гексафторфосфат бис(2-оксо-3-оксазолидинилфосфинхлорид 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид Другие 2-(трет-бутилкарбонилоксиимино)-2-фенилацетонитрил ди-трет-бутилбикарбонат тетрабутиламмонийфторид п-бутиллитий 11 45968 12 (1S)-( + )-10-камфорсульфоновая кислота диазабициклооктан диазабициклононан диазабициклоундекан деионизованнаяОІРА или DIPEA диизопропилэтиламин диметилацетамид диметиламинопиридин диметоксиэтан диметилформамид диметилтетрагидропиримидинон д и мети л сульфоксид триэтиламинЕЮАс - этилацетат Час(ы) 2-пропанол титрование Карла Фишера для воды диизопропиламид лития гексаметилдисилазид лития (І_)-пироглутаминовая кислота ацетонитрил метил-трет-бутиловый эфир N-метилпирролидинон комнатная температура три фтору ксусная кислота термогравиметрия потери при нагреве тетрагидрофуран тонкослойная хроматография тетраметилэтилендиамин тетраметил мочевина (S)-CSA DABCO DBN DBU Dl DMA DMAP DME DMF DMPU DMSO Et3N h I PA KF LDA LHMDS L-PGA MeCN MTBE NMP rt TFA TG THF TLC TMEDA TMU Подробное описание изобретения Способ настоящего изобретения поясняется следующей схемой РІ Схема включает стадии (а) контактирования одного, эквивалента аллилацетонида формулы II Ph. Иодогидрин йодоишнояастон Основание X - галоген Эпокскд Вышеприведенная схема иллюстрирует три стадии Сначала происходит аллилирование ацетонида, после которого следует образование галогенгидрина и затем циклизация, вызванная основанием , с образованием полученного эпоксида В настоящем изобретении способ синтеза эпоксида формулы I с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе , смешанном с водным раствором слабого основания при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III, и а Ph. "'0 X - галоген Ш (Ь) добавления основания в растворителе или растворителях для установления образования 13 45968 эпоксида формулы I Настоящий способ синтеза эпоксида формулы Iиз ацетонидного реагента включает стадии (а) взаимодействия одного эквивалента ацетонида с приблизительно одним эквивалентом аллилгалогенида в сильном основании с получением аллилацетонида формулы II, (Ь) смешивания с ним галоген и рую ще го агента в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания при температуре в диапазоне между от около -40°С и около 100°С с образованием галогенгидрина формулы III Pk Ij X - галоген (с) добавления основания в растворителе или растворителях для установления образования зпоксида формулы I При получении аллилацетонида схемы ГАММА предпочтительные реагенты аллилирования включают аллилгалогениды, такие как аллилхлорид, аллилбромид или аллилиодид, а также другие аллилэлектрофилы, например, аллилметансульфонат или аллиловые эфиры, в присутствии катализатора переходного металла Наиболее предпочтительные реагенты аллилирования включают аллилгалогениды , например, алл ил 14 хлорид, аллилбромид и аллилиодид Для этой реакции аллилирования предпочтительными основаниями являются сильные основания, которые включают амидные основания, например, литиевые, натриевые, калиевые или магниевые соли аминов, например, диэтиламина, диизопропиламина, дициклогексиламина, пиперидина, пирролидина или бистриметил-силиламина, металлалкилы, например Сі є-алкиллитий, такие как п-, изо-, втор, и трет-бутиллитий, метил-, этилили ариллитий, например фениллитий, реагенты Гриньяра, например, метил-, этил-, пропил- или бутилмагнийгалогенид, алкоксиды, например, метоксид, этоксид, изопропоксид, трет-бутоксид, трет-амилоксидалкоксиды лития, натрия, калия или магния В реакции аллилирования наиболее предпочтительным основанием является литийгексаметилдисилазид (LHMDS) В реакции аллилирования предпочтительные растворители включают эфирные растворители, например, THE, DME, МТВЕ, диэтиловый эфир, диглим или дибутиловый эфир, углеводородные растворители, например, пентан, гексан, гептан, бензол, толуол или зтилбензол, или другие растворители, совместимые с основаниями и металлорганическими реагентами, например, DMSO, DMPU, NMP, TMU, TMEDA и краун-эфиры, и также смеси этих растворителей Наиболее предпочтительными растворителями для аллилирования являются эфирные растворители для аллилирования, например, THF, DME и МТВЕ Предпочтительный температурный диапазон для аллилирования составляет от -78°С до + 30°С Инкубационный период продолжается, по крайней мере, 15 минут и обычно до 3-х часов Для образования галогенгидрина предпочтительные галоге-нирующие агенты включают галогены, интергалогенные соединения, галогенидные соли или гипогалитные соли или эфиры, оксигалогеновые соли или кислоты, галогенамиды, галогенмочеви-ны, галогенкарбаматы, гал о ген сул ьфонамиды, галогенамины или другие галогенированные азотные соединения или их сочетания или с галогенидными солями, или с межфазными катализаторами, или с теми и другими Предпочтительные галогенирующие агенты представляют гипогалитные соли или сложные эфиры, галогенамиды, мочевины, карбаматы, сульфонамиды, амины или другие галогенированные азотные соединения, например, N-иодсукцини-мид, N-бромсукцинимид с иодидной солью или N-хлорсукцинимид с иодидной солью Наиболее предпочтительными галогенирующими агентами являются N-иодсукцинимид, N-бромсукцинимид в сочетании с иодидной солью или N-хлорсукцинимид в сочетании с иодидной солью Реакционными условиями для образования галогенгидринов являются растворы , суспензии или другие двухфазные системы, содержащие слабые основания, например, бикарбонат натрия, карбонат кальция, гидроксид магния, основный оксид алюминия, нейтральный оксид алюминия, ацетат натрия, вторичный кислый фосфат натрия, вторичный кислый фосфат калия, фторид калия, 15 45968 16 другие соли или вода в обычных органических хлороформ, углеродтетрахлорид, дихлорэтан, растворителях Предпочтительными реакционтрихлорэтан, тетрахлорэтан, трихлорэтилен, тетными условиями являются слабые основания, рахлорэтилен, хлорбензол и дихлорбензол Пронапример, бикарбонат натрия, основный оксид стые эфиры в качестве растворителей включают алюминия, фторид калия или вода Наиболее диэтиловый эфир, дибутиловый эфир, тетрагидпредпочтительными условиями реакции являются рофуран, диметоксиэтан, диэтоксиэтан и МТВЕ основный оксид алюминия или бикарбонат натрия Сложные эфиры в качестве растворителей вклюРастворители должны быть совместимы с услочают этилацетат, ІРАС и это кс и этил ацетат Кевиями реакции и включают простые эфиры, аротоны в качестве растворителей включают ацетон, матические хлорированные углеводороды, сложМЕК и МІВК Спирты в качестве растворителей ные эфиры, спирты, MeCN, DMF, DMPU или кевключают метанол, этанол, пропанол, изопропатоны Предпочтительными являются хлорированнол, бутанол и метоксиэтанол Полярные апроные углеводороды, простые эфиры и сложные тонные растворители в качестве растворителей эфиры Наиболее предпочтительными являются включают DMF, DMA, DMSO, DMPU, TMU, NMP и дихлорметан, IPAC, ЕЮАс, DME и МТВЕ Темпеацетонитрил Третичные амины в качестве расратурный диапазон находится между около -40°С творителей включают триэтиламин, диизопропии около 100°С, но предпочтительно между 0 и лэтиламин, пиридин, DABCO, DBU, DBN, пентамеоколо 35°С Инкубация продолжается, по крайней тилпиперидин и DMAP В одном варианте настоямере, около 10 минут, и обычно ее прекращают до щего изобретения способ синтеза эпоксида фористечения около 48 часов мулы I Циклизацию, вызванную основанием, в результате которой образуется эпоксид, осуществляют путем обработки галогенгидрина основанием Предпочтительные основания для такой циклизации включают гидроксиды и оксиды лития, натрия, калия, магния, кальция или тетраалкиламмония, алкоксиды, например, метоксид , этоксид, п- и изо-пропоксид, п-, изо-, втор- и трет-бутоксид лития, натрия, калия, магния и тетраалкиламмония Другие подходящие основания включают третичные и затрудненные амины, например, триэтиламин, DIEA, DBU, DBN, DABCO, метил морфолин, диизопропиламин, дициклогексиламин, бис-триметилсилиламин или тетраметилпиперидин, а также их металламидные соли Наиболее предпочтительными основаниями являются гидроксиды лития, натрия, калия или тетраалкиламмония, алкоксиды, например метоксид, этоксид, изо-пропоксид или трет-бутоксид лития, натрия и калия, или третичные амины, например, DIEA Гидроксид щелочного металла означает LiOH, КОН или NaOH или их смеси Предпочтительными растворителями для циклизации, вызванной основанием, являются простые эфиры, сложные эфиры, углеводороды, ароматические растворители, хлорированные углеводороды, кетоны, вода, спирты, DMSO, MeCN, DMF или DMPU или другие полярные растворители или их смеси Наиболее предпочтительными растворителями являются простые эфиры, сложные эфиры, спирты или полярные апротонные растворители Циклизацию, вызванную основанием, осуществляют в температурном диапазоне между около -40°С и около 100°С Инкубация продолжается, по крайней мере, около 10 минут, и обычно ее прекращают до истечения около 48 часов В способе настоящего изобретения может быть использовано широкое множество растворителей, если не указано по-иному Углеводородные растворители включают пентан, гексан, гептан, циклогексан, метилциклогексан, бензол, толуол и ксилол Ароматические вещества в качестве растворителей включают бензол, толуол, ксилол и этилбензол Хлорированные углеводороды в качестве растворителей включают метиленхлорид, Рк включает стадии (а) контактирования одного эквивалента аллилацетонида формулы II с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между от около -40°С и около 100°С с образованием галогенгидрина формулы III, X = галоген m при этом галогенирующий агент выбирают из группы, состоящей из N-иодсукцинимида, N-бромсукцинимида или N-хлорсукцинимида, два последних из которых можно смешать с иодидной солью, растворитель выбирают из группы, состоящей из дихлорметана, IPAC, EtOAc, DME и 17 45968 18 МТВЕ, слабое основание выбирают из основного стоящей из дихлорметана, IPAC, ETOAc, DME и оксида алюминия или бикарбоната натрия, и МТВЕ, слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и (Ь) добавления основания в воде для установления образования эпоксида формулы I, при (с) добавления основания в воде для установэтом основание выбирают из группы, состоящей ления образования эпоксида формулы I, при этом из гидроксида лития, гидроксида натрия, гидрооснование выбирают из группы, состоящей из ксида калия, тетраалкиламмонийгидроксида, Сі 4гидроксида лития, гидроксида натрия, гидроксида алоксида лития, натрия или калия, и DIEA калия, тетраалкиламмонийгидроксида, Сі 4-алкокси-да лития, натрия или калия, и DIEA В другом варианте настоящего изобретения, включающем использование ацетонидного реаВ другом варианте настоящего изобретения гента, настоящий способ синтеза эпоксида форспособ синтеза эпоксида формулы I, мулы РІХ включает стадии (а) контактирования одного эквивалента аллилацетонида формулы II, с приблизительно одним эквивалентом аллилгалогенида в сильном основании, при этом аллилгалогенид выбирают из аллилхло-рида, аллилбромида и аллилиодида, с получением аллилацетонида формулы II, (Ь) смешивания с ним галоген и рую ще го агента в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания при температуре в диапазоне от около -40°С до около 100°С с образованием галогенгидрина формулы III, растворенного в изопропилацетате с N-иодосукцинимидом в количестве от около одного до двух эквивалентов в ~0,5М водном растворе бикарбоната натрия при комнатной температуре с образованием иодогидрина формулы III, и I установления образования эпоксида формулы В другом варианте настоящего изобретения способ синтеза эпоксида формулы I р X = галоген ш при этом галогенирующий агент выбирают из группы, состоящей из N-иодеукцинимида, Nбромсукцинимида или N-хлорсукцинимида, два последних из которых можно смешать с иодидной солью, растворитель выбирают из группы, со включает стадии 19 45968 20 (а) взаимодействия одного эквивалента ацепригодными при лечении инфекции, вызванной HIV, после предполагаемого подвержения в протонида, шлом HIV, например, при переливании крови, трансплантации органа, обмене жидкости в организме, острых болях, при случайном заражении во время иглоукалывания или при воздействии на кровь пациента во время хирургического вмешательства Конечный продукт - ингибиторы протеазы HIV - являются также пригодными при получении и выполнении ситового анализа антивирусных соединений Соединения конечного продукта являрастворенного в эфирном растворителе, с ются пригодными, например, для выделения приблизительно одним эквивалентом аллилброферментативных мутантов, которые являются мида и приблизительно одним эквивалентом -1,0 превосходным средством для отбора более силь- 2,ОМ литийгексаметилдисилилазида (в эфирном ных антивирусных соединений- Кроме того, такие растворителе) с получение аллилацетонида форсоединения являются пригодными при установлемулы II нии или определении места связывания других антивирусов в протеазу HIV, например, посредством конкурентного ингибирования Таким образом, соединения конечного продукта, которые получены с помощью способов и промежуточных соединений этого изобретения, представляют коммерческие продукты, продаваемые для этих целей Ph. (b) смешивания с ним N-иодсукцинимида в количестве от одного до двух эквивалентов в ~0,5М водном растворе бикарбоната натрия при комнатной температуре с образованием иод-гидрина формулы III, и Н РК (с) добавления гидроксида щелочного металла в воде для установления образования эпоксида формулы I Способы и промежуточные соединения этого изобретения являются пригодными для получения соединений конечного продукта, которые пригодны при ингибировании протеазы HIV, предотвращении или лечении инфекции, вызванной вирусом иммунодефицитного состояния человека (HIV), и при лечении последующих паталогических состояний, например AIDS Лечение AIDS или предотвращение или лечение инфекции, вызванной HIV, определяют как включающее, но не ограниченное, лечение широкого круга состояний, связанных с инфекцией, вызванной HIV AIDS, ARC (комплекс, родственный с AIDS), как симптомокомплекс, так и асимптомокомплекс, и действительное или потенциальное подвержение HIV Соединения конечного продукта, которые могут быть получены с помощью способов и промежуточных соединений этого изобретения, являются Соединения, ингибирующие протеазу HIV, которые могут быть получены с помощью промежуточных соединений и способов настоящего изобретения, представлены в ЕРО 541164 Соединения, ингибирующие протеазу HIV, могут быть введены пациентам, в случае необходимости такого лечения, в виде лекарственных препаратов, включающих фармацевтический носитель и терапевтически эффективные количества соединения или его фармацевтически приемлемой соли В ЕРО 541164 раскрыты подходящие лекарственные препараты, способы их введения, солевые формы и дозировки соединений Соединения настоящего изобретения могут иметь ассимет-рические центры и встречаются в виде рацематов, рацемических смесей и в виде отдельных диастереомеров или энантиомеров, при этом в настоящее изобретение включены все изомерные формы Когда в составляющей какая-либо переменная (например, арил, гетероцикл, R, R1, R2, п, X и т д ) встречается более одного раза, ее определение при каждом проявлении не зависит от ее определения при каждом другом проявлении Являются также допустимыми комбинации заместителей и/или переменных, если только такие комбинации приводят к получению устойчивых соединений Как используется здесь, за исключением того, где отмечено по-иному, "алкил" включает насыщенные алифатические углеводородные группы как с разветвленной, так и с прямой цепью, имеющие конкретное количество углеродных атомов (Me представляет метил, Et является этилом, Рг представляет пропил, Ви является бутилом, tBu представляет трет-бутил), "Галоген", как используется здесь, означает фтор, хлор, бром и иод Как используется здесь, "арил" означает фенил (Рп) или нафтил Характерные экспериментальные методики, в которых используют новый способ, описаны подробно ниже Эти методики служат только приме 22 21 45968 ром и не являются ограничениями для нового способа этого изобретения Основание Пример 1 Превращение ацетонида в аллилацетонид LHMDS THF Эпоксид АЦЄТОШД Ацетонид Аллилбромид Литийгексаметилдисилилазид (LHMDS) 1,0М BTHF Тетрагидрофуран (THF) 32,1г 12,70г 105мл 200мл В 200мл THF в 100мл трехгорлой колбе, снабженной капельной воронкой, растворили ацетонид и дегазировали путембарботирования в азоте в течение 20 минут Смесь охладили до 25°С и через шприц добавили аллилбромид В капельную воронку под давлением азота через канюлю перенесли LHMDS LHMDS медленно, в течение 20 минут, прокапали в реакционную смесь, перемешиваемую магнитной мешалкой В то время, когда температура охлаждающей ванны была равна -30°С, внутренняя температура достигла -14°С Смесь выдержали в течение 30 минут при температуре от-20°С до-15°С Добавили воду (100мл) и IPAC (100мл) и температуру подняли до 5°С Нижнюю водную фазу выбросили за ненадобностью, и органическую фазу промыли 100мл 0.2HCI в 3% водном растворе NaCI, 30мл рассола и 30мл 0,5М бикарбоната натрия Органическую фазу выпарили (55°С, ЮОторр) до образования масла, добавили 40мл IPAC, и смесь опять выпарили до образования масла В этот момент сырой аллилацетонид можно было непосредственно подать на следующую стадию или очистить кристаллизацией из гексана-ІРАС ЗО 1 или метилциклогексана-ІРАС 30 1 до получения аллилацетонида в виде белого кристаллического твердого вещества с выходом 87% Данные 13С ЯМР для основного ротамера аллилацетонида (62,5MHz) 171,0 129,6 126,6 96,8 8.0 140,4 128,6 125,6 78,9 65,6 36.1 140,2 128,2 124,0 47,5 26,6 Пример 2 Превращение аллилацетонида в иодогидрин и циклизация до эпоксида с помощью NIS V ЙОДОГЙДРШ Аллилацетонид (сырой из вышеприведенного получения) N-иодсукцинимид (NIS) Водный раствор бикарбоната натрия (0,5М) Изопропилацетат (IPAC) -0,1 моля 29,24г 350мл 300мл Сырой аллилацетонид растворили в IPAC и перемешали с водным раствором бикарбоната натрия и NIS в течение 17 часов Добавили водный раствор бисульфата натрия (38 - 40%) и отделили верхнюю органическую фазу Органическую фазу промыли 300мл воды и 2x100мл рассола В этот момент раствор сырого иодогидрина в IPAC можно было непосредственно перенести на следующую стадию или раствор можно было выпарить и кристаллизовать из метил циклогексана IPAC до получения иодогидрина в виде бледно-желтого кристаллического твердого вещества, 13С ЯМР, вращение при точке плавления Иодогидрин (раствор сырого иодогидрина в IPAC из вышеприведенного получения) Моногидрат гидроксида лития Вода -0,1 моль 50г 200мл Данные С ЯМР для основного ротамера иодогидрина (62,5MHz) 172,2 129,5 126,8 79,1 40,6 24,3 140 ,6 140 ,4 139 ,3 128 ,8 128 ,2 127 ,2 125 ,7 124 ,0 96, 9 68, 7 65, 8 43, 7 39. 0 іі 36.2 26. 5 16,3 част на миллион Пример 3 Превращение аллилацетонида в иодогидрин и циклизация до эпоксида с помощью NCS/Nal Иодогидрин в IPAC смешали с гидроксидом лития в воде в течение 3-х часов при 25 - 30°С Верхнюю органическую фазу промыли 200мл воды и 200мл рассола и сушили над ~2г сульфата магния Раствор IPAC отфильтровали и выпарили (50 - 60°С, ЮОторр) до ~50мл, когда начал кристаллизоваться эпоксид Смесь охладили до 25°С В Течение 30 минут и 10мл порциями в течение 30 минут при перемешивании добавили 75мл метил циклогексана Смесь выдержали в течение 1 часа, и кристаллы отфильтровали и промыли 2х20мл метилцикло 23 45968 24 гексана и сушили до получения 24,Юг (64%) эпокванны из смеси льда и воды После добавления сида в виде белого кристаллического твердого смесь выдерживали при температуре от 18°С до вещества со степенью чистоты 99,9А%, опреде20°С в течение 30 минут и контролировали поленной HPLC, Маточный раствор и промывки высредством анализа HPLC (высокоэффективной парили до масла и растворили в 40мл IPAC Расжидкостной хроматографией) на исчезновение (-)твор обработали Юг углерода Дарко G60 в течецис-1-аминоиндан-2-ола ние 2-х часов при 25°С и фильтровали через проХод реакции контролировали посредством кладку из Solka Floe Фильтрат выпарили до ~20мл анализа высокоэффективной жидкостной хромаи добавили 40мл метилциклогексана Кристаллитографией (HPLC) 25см колонка C8-RX фирмы ческий эпоксид отфильтровали и промыли 2x10мл Дупонт, ацетонитрил/ЮмМ (КН2РО4/К2НРО4) метилциклогексана до получения еще 4,96г (13%) 60 40, 1,0мл/мин, объем впрыскивания = 20мл, эпоксида со степенью чистоты 96,2А%, опредеобнаружение = 200нм, получение пробы при 500 х ленной HPLC Превращение иодогидрина в эпокразбавлении Приблизительные величины вресид можно также-осуществлять путем добавления мени удерживания 1,7М калий-трет-бутоксида в THF (0,70мл, 1,2ммоля) или 5М гидроксида калия в метаноле время удерживания (мин) идентификация (0,24мл, 1,2ммоля) или DIEA (155мг, 1,2ммоля) к цис-аминоин6,3 раствору иодогидрина (505мг, 1,0ммоль) в IPAC (2 данол - Змл), последующей промывки 2х2мл воды и кристаллизации из метилциклогексана-ІРАС Реакционную смесь обработали пиридиний-ртолуолсульфонатом (241 г, 0,96 моля, 0,16экв) и перемешивали в течение 10 минут (рН смеси поАллилацетонид 26,15г сле разбавления 1мл пробы равным объемом воN-хлорсукцинамид (MCS) 22,7г ды находился между 4,3 - 4,6) Затем добавили 2Иодид натрия 25,5г метоксипропен (1,27л, 13,24моля, 2,2экв) и реВодный раствор бикарбоната на350мл акционную смесь нагрели до 38 - 40°С в течение трия (0,5М) 2-х часов Реакционную смесь охладили до 20°С и Изопропилацетат (IPAC) 300мл разделили этилацетатом (12л) и 5% водным раствором ІЧаНСОз (Юл) Смесь перемешали и слои NCS и Nal смешали вместе в 200мл воды в разделили Этилацетатный экстракт промыли 5% течение 20 минут Когда смесь стала темно-ководным раствором ІЧаНСОз (Юл) и водой (4л) ричневой, тогда тотчас же отделили темное тверЭтилацетатный экстракт сушили посредством атдое вещество Твердое вещество растворили и мосферной перегонки и растворитель изменили при дополнительной выдержке цвет поблекнул и на циклогексан (общий объем - 30л) В конце пестал светло-желтым В IPAC растворили сырой регонки и концентрированны раствор горячего аллилацетонид и перемешали с водным раствоциклогексана медленно охладили до 25°С для ром бикарбоната натрия и полученным выше кристаллизации продукта Затем полученную суссветло-желтым раствором в течение 17 часов пензию охладили до 10°С и выдерживали в течеДобавили водный раствор бисульфита натрия (38 ние 1 часа Продукт выделили фильтрацией и - 40%) и отделили верхнюю органическую фазу влажный фильтровальный осадок промыли хоОрганическую фазу промыли 300мл воды и лодным (Ю°С) циклогексаном (2х800мл) Промы2хЮ0мл рассола В этот момент раствор сырого тый осадок сушили под вакуумом (давление ртути иодогидрина в IPAC можно было непосредственно 26 дюймов = 66,04см) при 40°С до получения перенести на следующую стадию или раствор 1,65кг ацетонида 1 (86,4%, площадь определенможно было выпарить и кристаллизовать из меная HPLC, 98%), 1ЯМР (300,13MHz, CDCI3, более тилцикло-гексана-ІРАС до получения иодогидрина важный ротамер) 57,36 - 7,14 (m, 9H), 5,03 (d, J = в виде бледно-желтого кристаллического твердого 4,4, 1Н), 4,66 (т, 1Н) 3,15 (т, 2Н), 3,06 (широк, s, вещества 2Н), 2,97 (т, 2Н), 1,62 (s, ЗН), 1,37 (s, ЗН), 13ЯМР Пример 4 (75,5MHz, CDCI3, более важный ротамер) 5с Раствор (-)-цис-1 -аминоиндан-2-ола 168,8, 140,9, 140,8, 140,6, 128,6, 128,5, 128,4, 5,93моля) в 127,1, 126,3, 125,8, 124,1, 96,5, 78,6, 65,9, 38,4, 36,2, 31,9, 26,5, 24,1 нщ 17,8л сухого THF (тетрагидрофурана) (KF = 55мг/мл) (KF означает титрование Карла Фишера для воды) и триэтиламин (868мл, 6,22моля) в 50л круглодонной колбе, снабженной термопарой, механической мешалкой, приспособлением, имеющим входное отверстие для азота, и барботером, охладили до 15°С Затем в течение 75 минут добавили 3-фенил-пропионилхлорид (ЮООг, 5,93моля) при установлении внутренней температуры между 14 - 24°С с помощью охлаждающей Аналит вычислено для C21H23NO2 С, 78,47, Н.7,21, N,4,36 Найдено С, 78,65, Н 7,24, N, 4,40 Пример 5 Получение эпоксида 3 с использованием тозилата Основание 25 45968 Раствор ацетонида 1 (ЮООг, 3,11моля) и 2(5)-глици-дилтозилата 2 (853г, 3,74 моля, 1,2экв,) в 15,6л THF (KF = 22мг/мл) в 50л четырехгорлой круглодонной колбе, снабженной термопарой, механической мешалкой, капельной воронкой и приспособлением, имеющим входное отверстие для азота, 3 раза дегазировали посредством продувки азотом в вакууме и охладили до -56°С Затем в течение 2-х часов добавили гексаметилдисилазид лития (LiNffCHsbSib) (2,6л, 1,38М , 1,15экв ) при поддержании внутренней температуры между -50°С и -45°С Реакционную смесь перемешали при температуре от -45°С до 40°С в течение одного часа и затем нагрели в течение 1 часа до температуры -25°С Смесь перемешивали при температуре от -25°С до 22°С в течение 4-х часов (или до тех пор, пока исходный ацетонид находился на 3% площади ) 26 Ход реакции контролировали посредством анализа HPLC 25см х 4,6нм колонка Зорбакса с диоксидом кремния, 20% этилацетат в гексане, 2,0мл/мин , объем впрыскивания = 20мл, обнаружение = 254нм, получение пробы = ЮОх разбавление Приблизительные значения времени удерживания время удерживания (мин) 5,5 6,5 13,5 идентификация амид 1 глицидилтозилат 2 эпоксид 3 Реакционную смесь резко охладили DI водой (6,7л) при температуре -15°С и раздели с помощью этилацетата (Юл) Смесь перемешали и слои разделили Этилацетатный экстракт промыли смесью 1% водного раствора ЫаНСОз (5л) и насыщенным NaCI (0,5л) Этилацетатный экстракт (28,3л) концентрировали вакуумной перегонкой (давление ртути 28 дюймов = 71,12см) и добавили дополнительное количество этилацетата для завершения замены растворителя на этилацетат (конечный объем - 11,7л) Затем растворитель этилацетатного концентрата заменили на МеОН для кристаллизации продукта и концентрирования до конечного объема 3,2л Остаточный растворитель этилацетат удалили путем загрузки Юл метанола и сбора Юл дистиллята Полученную суспензию перемешивали при 22°С в течение 1 часа, затем охладили до 5°С и выдерживали в течение 0 5 часа Продукт выделили фильтрацией и влажный фильтровальный осадок промыли холодным метанолом (2 х 250мл) Промытый осадок сушили под выкуумом (давление 26 дюймов Нд = 66,04см) при 25°С до получения 727г эпоксида 3 (61,2%, как показала HPLC, площадь основного продукта эпоксида 98,7%), 13 СЯМР(300МНг, CDCI3) 5 171,1, 140,6, 140,5, 139,6, 129,6, 128,8, 128,2, 127,2, 126,8, 125,6, 124,1, 96,8, 79,2, 65,8, 50,0, 48,0, 44,8, 39,2, 37,4, 36,2, 26,6, 24,1 Пример 6 Получение пиналтимата 6 Суспензию 2(5)-трет-бутилкарбоксамид-4-г\ІВос-пиперазина 4 (1950г, 6,83 моля, ее > 99,5% (ее - энантиомерный избыток)) и эпоксид 3 (2456г, смесь 4S/R зпоксидов при соотношении 97,5 2,5, 6,51 моля) в изопропаноле (2-пропанол, 18,6л) в 72л круглодонной колбе с 4-мя входными отверстиями для азота, снабженной механической мешалкой, парциальным конденсатором горячего орошения, паровой баней, термопарой, покрытой тефлоном, и входным отверстием для азота, нагревали до образования флегмы (внутренняя температура составила 84 - 85°С) Через 40 минут получили гомогенный раствор Смесь нагревали в колбе с обратным холодильником в течение 28 часов Внутренняя температура во время нагревания в колбе с обратным холодильником составила 84 85°С Ход реакции контролировали посредством анализа высокоэффективной жидкостной хроматографией, 25см колонка C8-RX фирмы Дупонт, ацетонитрил/ЮмМ (КН2РО4/ КН2РО4 60 40), 1,0мл/мин , обнаружение -220нм, полученная проба = 2мл, реакционную смесь разбавили в ацетонитриле до 1мл Приблизительные значения времени удерживания время удерживания (мин) 4,8 8,9 15,2 идентификация пиперазин 4 эпоксид 3 связанный продукт 5 Через 28 часов оставшийся эпоксид 3 и связанный продукт 5 (анализ HPLC) находились соответственно на 1,5% площади и 91 - 93% площади Смесь охладили до температуры от 0°С до 5°С и во время поддержания температуры ниже 15°С добавили 20,9л 6N HCI После завершения добавления смесь нагрели до 22°С В этот момент наблюдалось выделение газа (изобутилена) Смесь выдерживали в течение б часов при темпе 27 45968 28 ратуре от 20°С до 22°С времени удерживания Ход реакции контролировали анализом HPLC те же самые условия, которые приведены выше Время удерживания Идентификация Приблизительные величины времени удержива(мин) ния 2,7 DMF 4,2 3-пиколилхлорид 4,8 Соединение J время удерживания идентификация 9,1 пиналтимат 6 (мин) цис-аминоинданол 7,0 пиналтимат 6 Смесь выдерживали при 68°С до тех пор, пока 11,9 остаток пиналтиматного соединения 6 находился связанный продукт 5 15,1 на поверхности < 0,3%,как следовало из анализа HPLC Смесь охладили до 0°С и медленно добавили 7,5л 50% NaOH для того, чтобы установить рН Смесь перемешали в течение 4-х часов при смеси равным 116, при поддержании темпера68°С, затем охладили до 25°С и разделили этилтуры во время добавления менее 25°С Смесь ацетатом (80л) и смесью, состоящей из 24л наразделили этилацетатом (40л) и водой (Зл) сыщенного водного раствора ЫаНСОз и дистиллиСмесь перемешали и слои разделили Органичерованной воды (14л) Смесь перемешали при скую фазу (60л) концентрировали под понижен55°С и слои разделили Этилацетатный слой три ным давлением (давление 29 дюймов Нд = раза промыли водой (20л) при 55°С Промытый 73,66см), растворитель заменили на DMF (K,Nэтилацетатный слой концентрировали при атмодиметилформа-мид) и концентрировали до конечсферном давлении до конечного объема котла ного объема 10,5л (KF = 1,8мг/мл) Анализ HPLC 30л В конце атмосферного концентрирования к показал, что выход 6 в этилацетате составил горячему раствору добавили воду (560мл), смесь 86,5% Пиналтиматное соединение 6 в DMF исохладили до 55°С и внесли затравку моногидрата пользовали непосредственно на следующей стаСоединения J Смесь охладили до 4°С и фильтродии без дополнительной очистки Для выделенвали для сбора продукта Продукт промыли хоного соединения 6 лодным этилацетатом (2хЗл) и сушили при 25°С и 13 атмосферном давлении до получения 2905г СЯМР(75 4MHz, CDCI3) 5 175,2, 170,5, 140,8, (70,7%) моногидрата соединения J в виде белого 140,5, 139,9, 129,1, 128,5, 127,9, 126,5, 125,2, твердого вещества 124,2, 73,0, 66,0, 64,8, 62,2, 57,5, 49,5, 47,9, 46,4,, 45,3, 39,6, 38,2, 28,9 Пример 7 Получение моногидрата Соединения J Пример 8 Пиразин-2-трет-бутилкарбоксамид 9 соон № 9Н м CONHt-Bu м« 2Пиразинкарбоновая кислота Оксалилхлорид трет-бутиламин (KF = 460мг/мл) ЕЮАс (KF = 56мг/мл) DMF 1-Пропанол Соединение J В раствор соединения 6 в DMF (10,5л, KF = 10мг/мл) с предыдущей стадии загрузили 8л высушенного на сите DMF (KF < 30мг/л) и смесь нагрели с помощью паровой бани под вакуумом 30 дюймов Нд (76,20см) для того, чтобы отогнать, главным образом, воду и/или остаточный изопропанол или этилацетат Конечный объем концентрата составил 13,5л (KF = 1,8мг/мл), и затем к раствору при 25°С добавили триэтиламин (28,6л, 20,5молей), после которого добавили 3-пиколилхло-ридгидрохлорид (96%, 1287г, 7,84моля) Полученную суспензию нагрели до 68°С Ход реакции контролировали посредством анализа HPLC при использовании тех же самых условий, что и на предыдущей стадии Приблизительные значения 3,35кг (27 мол ей) 3,46кг (27,2моля) 9,36л (89молей) 27л 120мл 30л Карбоновую кислоту 8 суспендировали в 27л ЕЮАс и 120мл DMF в 72л трехгорловои колбе при механическом перемешивании под азотом и суспензию охладили до 2°С Добавили оксалилхлорид, поддерживая температуру между 5 и 8°С Добавление завершили через 5 часов Во время экзотермического добавления выделились СО и СОг Образованный НСІ остался большей частью в растворе Присутствующий осадок представлял, по всей вероятности, хлорид пиразиновой кислоты Анализ образования хлорида кислоты осуществляли путем резкого охлаждения безводной пробы реакционного продукта трет-бутил-амином После завершения резкого охлаждения осталось
ДивитисяДодаткова інформація
МПК / Мітки
МПК: C07D 213/36, A61K 31/4418, C07D 263/52, C07D 413/06, A61K 31/44, A61P 31/12, A61K 31/4406, C07D 413/04, A61P 37/04
Мітки: варіанти, епоксиду, спосіб, одержання
Код посилання
<a href="https://ua.patents.su/17-45968-sposib-oderzhannya-epoksidu-varianti.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання епоксиду (варіанти)</a>
Спосіб асиметричного одержання флорфеніколу, спосіб регіовибіркового розкриття хірального епоксиду та спосіб одержання r, s- ізомеру флорфеніколу
Номер патенту: 43331
Опубліковано: 17.12.2001
Автори: Гуанг-Зонг Ву, Ванда Тормос I
МПК: C07C 231/00, C07C 233/18, C07D 303/00, C07D 263/14, C07B 53/00, C07C 233/25, C07C 315/00, C07C 317/32
Мітки: розкриття, регіовибіркового, хірального, епоксиду, спосіб, одержання, флорфеніколу, асиметричного, ізомеру
Формула / Реферат:
1. Способ асимметричного получения флорфеникола формулы (I)включающий получение оксазолина формулы (IV)обработку его агентом фторирования с последующим гидролизом кислотой, отличающийся тем, что получение оксазолина вышеприведенной формулы (IV) включает стадию региоизбирательного открытия хирального эпоксида формулы (II)путем последовательной обработки сильным основанием, кислотой Льюиса и...
Похідні аміно(тіо)ефірів, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі і спосіб її одержання

Номер патенту: 45952
Опубліковано: 15.05.2002
Автори: Грайнер Хартмут, Бартосцук Герд, Нобле Марк, Бруне Мішель, Цайе Жан-Жак, Бьоттхер Хеннінг, Бертелон Жан-Жак, Деван Ральф
МПК: C07D 311/58, C07D 311/64, C07C 255/54, C07D 333/20, C07C 217/16, C07D 213/38, C07D 405/12, C07C 217/20
Мітки: варіанти, похідні, аміно(тіо)ефірів, спосіб, основі, одержання, композиція, фармацевтична
Формула / Реферат:
1. Производные амино(тио)эфиров формулы I,где X представляет собой кислород, серу, или, если R0 и R1 вместе не являются алкиленовой цепью с 1-3 атомами, то СН2;Z представляет собой -(СН2)n1-(СНА)n2-(СН2)n3, причем n1 = 0, 1, 2 или 3; n2 = 0 или 1; n 3 = 0, 1, 2 или 3, при условии, что n1 + n2 + n3 < 4;R0 представляет собой водород или А;R1 представляет собой водород, А, ОА, фенокси, Ph, ОН, F, Cl, Br, CN,...
Спосіб одержання похідних 2,5-дихлорфенолу

Номер патенту: 26015
Опубліковано: 26.02.1999
Автори: Йозеф Драбек, Манфред Бьогер
МПК: C07C 43/225, C07C 41/00
Мітки: похідних, одержання, спосіб, 2,5-дихлорфенолу
Формула / Реферат:
Способ получения производных 2,5-дихлорфенола формулы (I):где R = NH2 или N=C=O, отличающийcя тем, что фенол формулы (II):в котором аминогруппу предварительно ацилируют, подвергают взаимодействию с гексафторпропеном и, в случае необходимости, полученное соединение формулы (I), где R означает NH2, обрабатывают фосгеном для получения соединения формулы I, где R означает N=C=O.
Спосіб одержання фінастериду, 17b-[n-трет-бутилкарбамоїл]-4-аза-5a-андрост-1-ен-3-он у поліморфній формі i та іi, способи їх одержання (варіанти)

Номер патенту: 41341
Опубліковано: 17.09.2001
Автори: Джеймс A. МакКаулі, Річард Дж. Варсолона, Ульф X. Доллінг
МПК: C07J 73/00, C07F 3/00, H04N 5/74, C07J 75/00
Мітки: варіанти, одержання, способи, фінастериду, 17b-[n-трет-бутилкарбамоїл]-4-аза-5a-андрост-1-ен-3-он, форми, спосіб, поліморфній
Формула / Реферат:
1. Способ получения финастерида формулы (2):отличающийся тем, что включает 1) контактирование сложного эфира формулы (1):где R представляет собой линейный или разветвленный (С1-С10)алкил, с т-бутиламиномагнийгалогенидом, где молярное соотношение т-бутиламиномагнийгалогенида и сложного эфира составляет, по меньшей мере, около 2:1, в инертном органическом растворителе в атмосфере инертного...
Спосіб одержання похідних пірідіна (його варіанти)

Номер патенту: 3637
Опубліковано: 27.12.1994
Автори: Стіг Айл Інгемар Карлссон, Педер Бєрнхарт Бернтссон, Ян Ернульф Гаардер, Бенгт Ріхард Люнг
Мітки: спосіб, пірідіна, його, одержання, варіанти, похідних
Текст:
...снижение кровяного давления на 20%. Соединения общей формулы ( I ) облаРезультаты сведены в табл.2. дают антигипертенсивным действием. Специфичность релаксации гладкой П р и м е р } . Ufk г 2-3-ДИХлор- 3S мышцы определяется следующим путем. бензальдегида, 3»2 г сложного этилоВ ванну подают изолированную ворот вого эфира З'аминокротоновой кислоную вену крыс и изолированную папилты, **,0 г 2-сложного метоксиэтилолярную сердечную мышцу тех же...
Наступний патент: Спосіб одержання етилового ефіру a-бромізовалеріанової кислоти
Випадковий патент: Газова електрична станція