Антивірусні нуклеозиди
Номер патенту: 91255
Опубліковано: 12.07.2010
Автори: Ванг Пейюань, Чун Буйонг-Квон, Сарма Кешаб, Кларк Джеремі

















Формула / Реферат
1. Сполука за формулою (І)
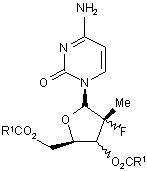 , (I)
, (I)
де:
R1 вибраний із групи, що складається із етилу, ізопропілу, ізобутилу, і n-пропілу;
або її гідрати, сольвати та солі приєднання кислот.
2. Сполука за п. 1, де R1 є етил або ізопропіл.
3. Сполука за п. 1 або 2, де R1 є ізопропіл, а сполукою є гідрохлорид або сульфат.
4. Сполука за будь-яким із пп. 1-3, де R1 є ізопропіл, а сполукою є гідрохлорид.
5. Медикамент, що містить сполуку за будь-яким із пп. 1-4.
6. Медикамент за п. 5, що призначений для лікування хвороби, опосередкованої вірусом гепатиту С (HCV).
7. Медикамент за п. 5 або 6, який відрізняється тим, що цей медикамент вводять пацієнту, що цього потребує, у терапевтично ефективній дозі.
8. Медикамент за п. 5 або 6, який відрізняється тим, що цей медикамент вводять пацієнту, що цього потребує, у дозі в інтервалі від 0,1 до 10 г на день.
9. Медикамент за п. 5 або 6, який відрізняється тим, що цей медикамент вводять, крім того, щонайменше з одним модулятором імунної системи і/або щонайменше з одним антивірусним засобом, який інгібує реплікацію HCV-вірусу.
10. Фармацевтична композиція, яка містить терапевтично ефективну кількість сполуки за будь-яким із пп. 1-4, змішану з щонайменше одним фармацевтично доступним носієм, розріджувачем або ексципієнтом.
11. Фармацевтична композиція за п. 10, що призначена для лікування хвороби, опосередкованої вірусом гепатиту С (HCV).
12. Фармацевтична композиція за п. 11, яка відрізняється тим, що терапевтично ефективна кількість сполуки складає від 0,1 до 10 г на день.
13. Фармацевтична композиція за будь-яким із пп. 10-12, яка, крім того, містить щонайменше один модулятор імунної системи і/або щонайменше один антивірусний засіб, який інгібує реплікацію HCV-вірусу.
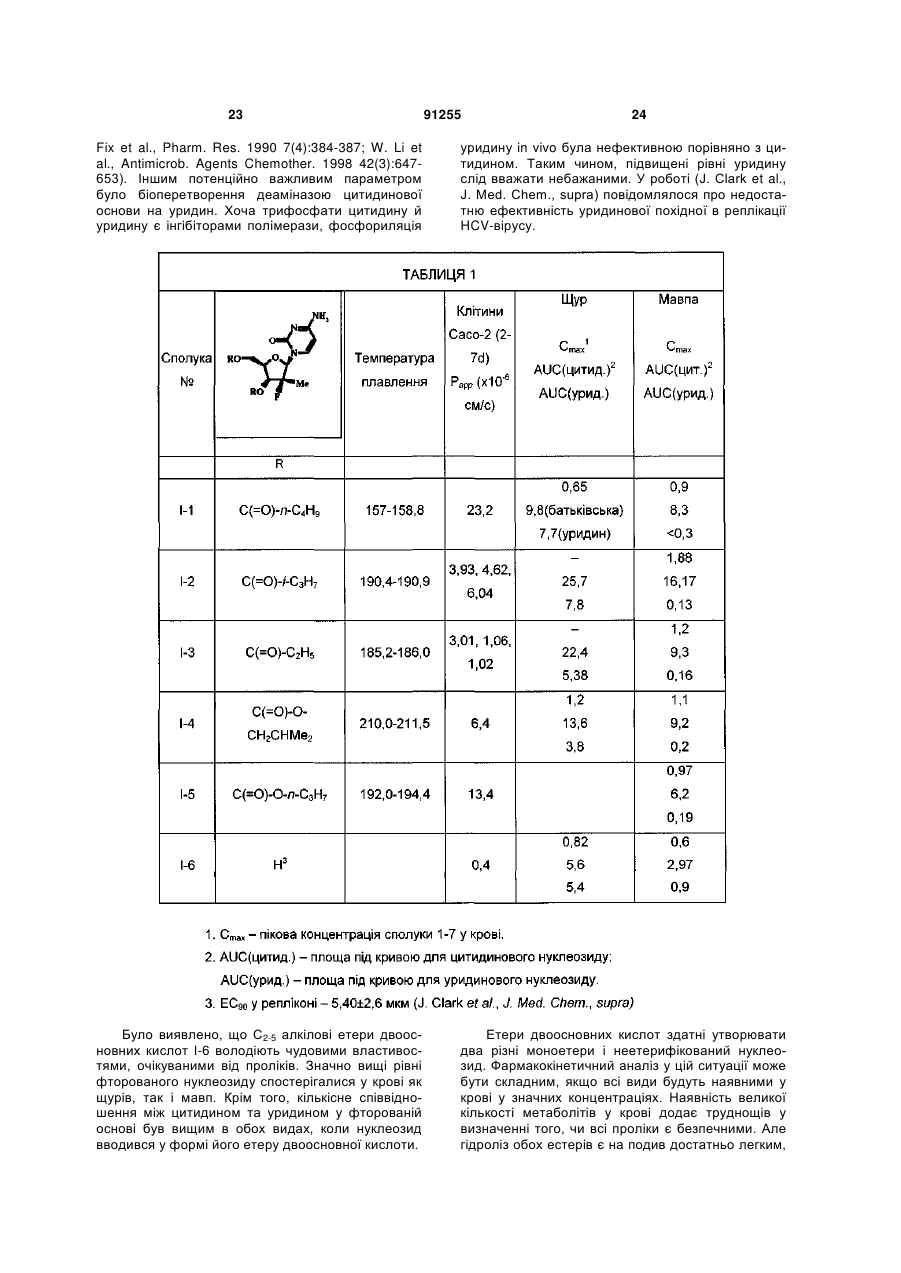
14. Спосіб одержання сполуки формули І, в якому здійснюють процес селективного О-ацилування нуклеозиду II, в результаті чого одержують О-ацилнуклеозид І в основних умовах реакції
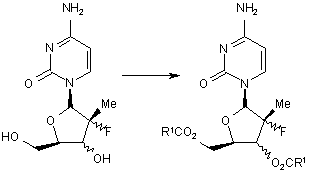 ,
,
(ІІ)
(І)
де R1 вибраний із групи, що складається із етилу, ізопропілу, ізобутилу, і n-пропілу, причому зазначений процес включає у себе такі стадії:
(і) розчинення II і DMAP у гетерогенній водній розчинювальній суміші і додавання водної основи для встановлення величини рН в інтервал від 7,5 до 12;
(іі) необов'язково додавання достатнього насиченого водного розчину NaCl для утворення двофазної реакційної суміші;
(ііі) додавання ацилувального засобу і додаткової основи, достатньої для підтримання величини рН в інтервалі приблизно від 7,5 до 12;
(iv) моніторингу реакції і безперервного додавання зазначеного ацилувального засобу і зазначеної основи, коли перетворення досягає достатнього рівня;
(v) необов'язково приведення в контакт О-ацилнуклеозиду з фармацевтично доступною кислотою для утворення солі приєднання кислоти О-ацилнуклеозиду.
Текст
1. Сполука за формулою (І) 2 (19) 1 3 91255 NH2 NH2 N N N O N O Me O HO F OH Me R1CO2 O F O2CR1 , (ІІ) (І) де R1 вибраний із групи, що складається із етилу, ізопропілу, ізобутилу, і n-пропілу, причому зазначений процес включає у себе такі стадії: (і) розчинення II і DMAP у гетерогенній водній розчинювальній суміші і додавання водної основи для Даним винаходом пропонуються ацильовані нуклеозиди, що є проліками інгібітору вірусу гепатиту С (HCV: Hepatitis С Virus) - РНК-залежної РНК-вірусної полімерази. Введені пероральним шляхом ці сполуки легко поглинаються зі шлунково-кишкового тракту та ефективно повертаються у стан первинного нуклеозиду в крові. Ці проліки є інгібіторами РНК-залежноої РНК-вірусної реплікації і можуть застосовуватися як інгібітори NS5Bполімерази HCV-вірусу, інгібітори HCV-реплікації і для лікування інфекційного гепатиту С у ссавців. Даний винахід стосується нуклеозидних проліків, що є інгібіторами реплікації HCV-вірусу. Зокрема, винаходом пропонується застосування ацильованих піримідинових нуклеозидних сполук, які забезпечують поліпшене поглинання ліків при введенні нуклеозиду пероральним шляхом. Вірус гепатиту С являє собою головну проблему охорони здоров'я і є лідером серед причин хронічної хвороби печінки в усьому світі (Воуеr, N. et al., J. Hepatol. 2000 32:98-112). Пацієнти, інфіковані вірусом HCV, є в групі ризику виникнення у них розвитку цирозу печінки і далі - гепатоклітинної карциноми. Отже, HCV-вірус є головним показанням для трансплантації печінки. Згідно з даними Всесвітньої організації охорони здоров'я сьогодні у світі нараховується 200 мільйонів HCV-інфікованих, а кожного року цим вірусом інфікується від 3 до 4 мільйонів людей. Приблизно 20% інфікованих спромагаються позбавитися HCV-вірусу, але у решти він залишається прихованим протягом усього їхнього життя. Від десяти до двадцяти відсотків хронічно інфікованих кінець кінцем наражаються на руйнівний цироз або рак печінки. Ця вірусна хвороба передається парентеральним шляхом через забруднену кров і кров'яні продукти, забруднені голки шприців, статевим шляхом або по вертикалі - від інфікованих матерів або матерів-носіїв інфекції народженим ними дітям. Сучасні методи лікування HCVінфекції, які є обмеженими імунотерапією рекомбінантним -інтерфероном, застосовуваним окремо або в комбінації з його нуклеозидним аналогом рибавірином, мають недостатньо задовільну кліні 4 встановлення величини рН в інтервал від 7,5 до 12; (іі) необов'язково додавання достатнього насиченого водного розчину NaCl для утворення двофазної реакційної суміші; (ііі) додавання ацилувального засобу і додаткової основи, достатньої для підтримання величини рН в інтервалі приблизно від 7,5 до 12; (iv) моніторингу реакції і безперервного додавання зазначеного ацилувального засобу і зазначеної основи, коли перетворення досягає достатнього рівня; (v) необов'язково приведення в контакт Оацилнуклеозиду з фармацевтично доступною кислотою для утворення солі приєднання кислоти Оацилнуклеозиду. чну ефективність через резистентність, що швидко проти цих ліків розвивається. Таким чином, існує нагальна потреба в поліпшених терапевтичних засобах, здатних ефективно долати хронічну HCVінфекцію. HCV-вірус був віднесений до сімейства Flaviviridae, до якого входять такі роди, як flaviviruses, pestiviruses і hepaciviruses, включаючи віруси гепатиту С (Rice, С. М., Flaviviridae: The viruses and their replication (Віруси та їх реплікація), in: Fields Virology, Editors: Fields, B. N., Knipe, D. M., and Howley, P. M., lippincott-Raven Publishers, Philadelphia, Pa., Chapter 30, 931-959, 1996). HCVвірус має оболонку і містить геном із одноланцюгової РНК, що має позитивний зміст і є завдовжки приблизно 9,4 т.п.н. Цей вірусний геном складається із нетрансльованої 5'-ділянки (UTR: untranslated region), довгої відкритої рамки зчитування (ORF: open reading frame), що кодує поліпротеїновий попередник із приблизно 3011 амінокислот, і коротку 3'-UТР-ділянку. Ділянка 5'-UTR є найбільш висококонсервативною частиною геному HCV і відіграє важливу роль в ініціації та регулюванні трансляції поліпротеїну. Генетичний аналіз HCV-вірусу дозволяє ідентифікувати шість головних генотипів, у котрих спостерігається більш ніж 30% розбіжність їхніх DNAпослідовностей. Кожний із цих генотипів містить низку більш близько споріднених підтипів, розбіжності нуклеотидних послідовностей у котрих складають 20-25% (Simmonds, P. 2004 J. Gen. Virol. 85:3173-88). Було виділено більше 30 таких підтипів. У Сполучених Штатах приблизно 70% HCVінфікованих мають інфекцію типів 1а та 1b. В Азії домінуючим є тип 1b (X. Forns and J. Bukh, Clinics in liver Disease 1999 3:693-716; J. Bukh et al., Semin. Uv. Dis. 1995 15:41-63). Шкода, але інфекції 1 типу є більш стійкими до терапії, ніж 2-й і 3-й генотипи (N. N. Zein, Clin. Microbiol Rev., 2000 13:223-235). Генетична організація і поліпротеїновий процесінг неструктурної білкової частини рамки ORF вірусів pestivirus і hepacivirus є дуже схожими. Ці віруси з позитивно-ланцюговими РНК мають одну 5 велику ORF-рамку, що кодує всі вірусні протеїни, необхідні для реплікації вірусів. Ці протеїни експресуються як поліпротеїн, котрий піддається котрансляційному і посттрансляційному процесінгу як клітинними, так і вірус-кодованими протеїназами, з утворенням зрілих вірусних протеїнів. Вірусні протеїни, відповідальні за реплікацію вірусної геномної РНК, розташовані на ділянці карбоксильного кінця. Дві третини ORF-рамки звуться неструктурними (NS) протеїнами. Як у вірусів pestivirus, так і у вірусів hepacivirus, зрілі неструктурні (NS) протеїни в порядку послідовності від амінокінця кодувальної ділянки неструктурного протеїну до карбоксильного кінця ORF-рамки складаються із р7, NS2, NS3, NS4A, NS4B, NS5A і NS5B. Неструктурні протеїни вірусів pestivirus і hepacivirus мають загальні області послідовностей, для котрих є типовими специфічні білкові функції. Наприклад, протеїни NS3 вірусів обох вищезазначених груп мають мотиви амінокислотних послідовностей, характерні для серинпротеїназ і геліказ (Gorbalenya et al., Nature 1988 333:22; Bazan and Fletterick Virology 1989 171:637-639; Gorbalenya et al., Nucleic Add Res. 1989 17, 38893897). Подібним чином протеїни NS5B вірусів pestivirus і hepacivirus мають мотиви, характерні для РНК-спрямованих РНК-полімераз (Kооnіn, Е. V. and Dolja, V. V. Crit. Rev. Biochem. Molec. Biol. 1993 28:375-430). Фактичні ролі і функції NS-протеїнів вірусів pestivirus і hepacivirus у життєвому циклі цих вірусів є абсолютно аналогічними. В обох випадках серинпротеїназа протеїну NS3 є відповідальною за весь протеолітичний процесінг попередників поліпротеїнів вправо від її положення в ORF-рамці зчитування (Wiskerchen and Collett Virology 1991 184:341-350; Bartenschlager et al., J. Virol. 1993 67:3835-3844; Eckart et al., Biochem. Biophys. Res. Comm. 1993 192:399-406; Grakoui et al., J. Virol. 1993 67:2832-2843; Grakoui et al., Proc. Natl. Acad. Sd. USA 1993 90:10583-10587; Ilijikata et al., J. Virol 1993 67:4665-4675; Tome et al., J. Virol 1993 67:4017-4026). Протеїн NS4A в обох випадках діє як спільний з NS3 серинпротеазою кофактор (Bartenschlager et al., J. Virol 1994 68:5045-5055; Failla et al., J. Virol 1994 68: 3753-3760; Xu et al., J Virol 1997 71:53 12-5322). Протеїн NS3 обох вірусів також діє як геліказа (Kim et al., Biochem. Biophys. Res. Comm. 1995 215: 160-166; Jin and Peterson Arch. Biochem. Biophys. 1995, 323:47-53; Warrener and Collett J. Virol 1995 69:1720-1726). I нарешті, протеїни NS5B вірусів pestivirus і hepacivirus володіють передбаченою РНК-спрямованою РНКполімеразною активністю (Behrens et al., EMBO 1996 15:12-22; Lechmann et al., J. Virol. 1997 71:8416-8428; Yuan et al., Biochem. Biophys. Res. Comm. 1997 232:231-235; Hagedom, PCT WO 97/12033; Zhong et al., J. Virol 1998 72:9365-9369). Сьогодні медицина має у своєму розпорядженні доволі обмежену кількість схвалених терапевтичних засобів для лікування HCV-інфекції. Огляди відомих і нових терапевтичних методів і засобів лікування HCV-інфекції та інгібування HCV NS5B полімерази можна знайти в роботах (R. G. Gish, Sem. Liver. Dis., 1999 19:5; Di Besceglie, A M. 91255 6 and Bacon, B. R., Scientific American, October: 1999 80-85; G. Lake-Bakaar, Current and Future Therapy for Chronic Hepatitis С Virus liver Disease (Сучасна і майбутня терапія хронічних хвороб печінки, викликаних вірусом гепатиту С), Curr. Drug Targ. Infect. Dis. 2003 3(3):247-253; P. Hoffmann et al., Патенти останніх років, що стосуються експериментальної терапії інфекції вірусом гепатиту С (1999-2002), Exp. Opin. Ther. Patents 2003 13(11): 1707-1723; F. F. Poordad et al., Developments in Hepatitis С therapy during 2000-2002 (Розробки в галузі терапії гепатиту С за 2000-2002 роки), Exp. Opin. Emerging Drugs 2003 8(І):9-25; Μ. P. Walker et al., Promising Candidates for the treatment of chronic hepatitis С (Перспективні засоби-кандидати для лікування хронічного гепатиту С), Exp. Opin. Investig. Drugs 2003 12(8): 1269-1280; S.-L. Tan et al., Hepatitis С Therapeutics: Current Status and Emerging Strategies (Терапія гепатиту С: сучасний стан і стратегії на майбутнє), Nature Rev. Drug Discov. 2002 1:867-881; R. De Francesco et al., Approaching a new era for Hepatitis С Virus therapy: inhibitors of the NS3-4A serine protease and the NS5B RNAdependant RNA Polymerase (Наближається нова ера в терапії гепатиту С: інгібітори NS3-4A серинпротеази і NS5B РНК-залежної РНК-полімерази), Antiviral Res. 2003 58:1-16; Q. M. Wang et al., Hepatitis С Virus encoded proteins: targets for antiviral therapy (Білки, кодовані вірусом гепатиту С: мішені для антивірусної терапії), Drugs of the Future 2000 25(9):933-8-944; J. A. Wu and Z. Hong, Targeting NS5B-Dependant RNA Polymerase for Anti-HCV Chemotherapy (NS5B РНК-залежна РНКполімераза - мішень для анти-HCV хіміотерапії) Cur. Drug Targ.-Inf. Dis. 2003 3:207-219). Сполуки, описані у цитованих вище оглядах, у даний час перебувають на різних стадіях процесів їх розробки. Комбінаційна терапія із застосуванням двох чи трьох засобів, націлених на одну і ту саму або на різні мішені, набула стану стандартного методу, який дозволяє уникнути розвитку стійких штамів вірусу або сповільнити його. Таким чином, сполуки, описані в цитованих вище оглядах, можуть застосовуватися в комбінаційній терапії разом зі сполуками згідно з даним винаходом, а ці огляди включені тут в усій їхній повноті шляхом посилання Рибавірин (амід 1а,1-((2R,3R,4S,5R)-3,4дигідрокси-5-гідроксиметилтетрагідрофуран-2-іл)1Н-[1,2,4]триазол-3-карбоновоі кислоти, VIRAZOLE ) є синтетичним аналогом антивірусного нуклеозиду з широким спектром застосування, який не індукує інтерферону Рибавірин володіє активністю in vitro проти декількох ДНК-1 РНКвірусів, включаючи Flavivindae (Gary L Davis, 7 Castroenterobgy 2000 118 8104-8114). При монотерапи рибавірин знижує сироваткові рівні амінотрансферази до нормальних у 40% пацієнтів, але не знижує сироваткові рівні HCV-PHK. Рибавірин виявляє також значну токсичність і, як відомо, викликає анемію Рибавірин є інгібітором інозинмонофосфатдегідрогенази. Рибавірин не отримав схвалення для монотерапи проти HCV, але затверджений як засіб комбінаційної терапії разом з інтерфероном -2а та інтерфероном -2b Вірамідин 1b є промедикаментом, що перетворюється на 1а в гепатоцитах Інтерферони (IFN) застосовувалися для лікування хронічного гепатиту впродовж майже десяти років Інтерферони являють собою глікопротеїни, які виробляються імунними клітинами у відповідь на вірусну інфекцію. Розрізняють два типи інтерферону до 1 типу входять кілька сннтерферонів та один -інтерферон, а до 2 типу належить інтерферон Інтерферони 1 типу виробляються, головним чином, інфікованими клітинами i захищають сусідні клітини від нової інфекції. Інтерферони інгібують реплікацію багатьох вірусів, включаючи HCV, а при їх використанні як єдиного засобу для лікування інфекційного гепатиту С вони пригнічують HCV-PHK у сироватці до настільки низьких рівнів, що вони не піддаються вимірюванням. Крім того, інтерферон нормалізує сироваткові рівні амінотрансферази. Проте, дія інтерферону є тимчасовою. При припиненні лікування у 70% пацієнтів наступає рецидив i лише у 10-15% підтримується вірусологічна відповідь з нормальними рівнями аланінтрансферази в сироватці (L-B Davis, supra). Одним із обмежень інтерферонової терапії у початковий період його застосування було швидке виведення цього білка із крові. Хімічна дериватизація інтерферону поліетиленгліколем (PEG) дала можливість виготовляти білки з суттєво поліпшеними фармакокінетичними властивостями. Продукт марки PEGASYS є кон'югатом інтерферону -2а з 40 кДа розгалуженим монометокси-PEG, а продукт марки PEG-INTRON є кон'югатом інтерферону -2b з 12 кДа монометокси-PEG. (В. A. Luxon et al., Clin. Therap. 2002 24(9): 13631383; A. Kozlowski and J. M. Harris, J. Control. Release, 2001 72:217-224). Інтерферон -2а та інтерферон -2b сьогодні допущені до застосування як засобів монотерапії в лікування вірусу HCV. Продукт марки ROFERONA (фірма Roche) є рекомбінантною формою інтерферону -2а. Продукт марки PEGASYS (Roche) є пегильованою (тобто модифікованою поліетиленгліколем) формою інтерферону -2а. Продукт марки INTRON-A (Schering Corporation) є рекомбінантною формою інтерферону -2b, a продукт марки PEG-INTRON (Schering Corporation) є пегильованою формою інтерферону -2b. Інші форми -інтерферону, а також -, -, - і -інтерферонів проходять сьогодні стадію клінічних розробок для лікування від HCV-інфекції. Такими матеріалами, що сьогодні розробляються, є, наприклад, продукт марки INFERGEN (інтерферон-альфакон-1) фірми InterMune, продукт марки 91255 8 OMNIFERON (натуральний інтерферон) фірми Viragen, продукт марки ALBUFERON фірми Human Genome Sciences, продукт марки REBIF (інтерферон -ia) фірми Ares-Serono, омегаІнтерферон фірми BioMedicine, пероральний альфа-Інтерферон фірми Amarillo Biosciences, а також -інтерферон, -інтерферон та інтерферон -ib фірми InterMune. Оптимальною комбінацією лікувальних засобів у терапії HCV-інфекції на сьогоднішній день вважається поєднання рибавірину з -інтерфероном. Поєднання рибавірину з PEG-IFN (див. нижче) дає пролонговану реакцію на вірус (SVR) у 54-56% пацієнтів. У 2 і 3 типів HCV SVR-реакція досягається у 80% пацієнтів (Walker, supra). Але ця комбінація дає також побічні ефекти, які становлять чималі клінічні проблеми. Так, наприклад, підшкірні ін'єкції -IFN супроводжуються депресією, грипоподібними симптомами та реакціями шкіри, а пролонгована дія рибавірину асоціюється з гемолітичною анемією. Серед інших макромолекулярних сполук, які зараз проходять передклінічну або клінічну стадію розробок для лікування вірусного гепатиту С, можна назвати: Інтерлейкін-10 фірми Schering-Plough, IP-SO1 фірми Intemeuron, Мерімебодіб (Merimebodib, VX-497) фірми Vertex, продукт марки HEPTAZYME фірми RPI, матеріал IDN-6556 фірми Idun Pharma., матеріал XTL-002 фірми XTL, засіб HCV/MFS9 фірми Chiron, продукт марки CIVACIR (імуноглобулін проти гепатиту С) фірми NABI, продукт марки ZADAXIN (тимозин -1) фірми SciClone, тимозин плюс пегильований інтерферон фірми SciClone, продукт марки CEPLENE ; терапевтична вакцина, спрямована на Е2, фірми Innogenetics, терапевтична вакцина фірми Intercell, терапевтична вакцина фірми Epimmune/Genencor, терапевтична вакцина фірми Меrіх, терапевтична вакцина Chron-VacC фірми Тrірер. Іншими вартими уваги макромолекулярними матеріалами є рибозими, мішенню котрих є РНК HCV-вірусу. Рибозими є короткими молекулами природного походження, які володіють ендонуклеазною активністю, що каталізує специфічне для даної послідовності розщеплення РНК. Альтернативним підходом є використання протизмістових олігонуклеотидів, що зв'язуються з РНК і стимулюють RN-азаН-опосередковане розщеплення. Були визначені численні перспективні молекулярні матеріали для їх розробки як терапевтичних засобів від HCV-вірусів. Серед них можна назвати, наприклад, NS2-NS3 аутопротеазу, N3-протеазу, N3-геліказу і NS5B-полімеразу. РНК-залежна РНКполімераза є абсолютно необхідною для реплікації одноголанцюгового РНК-геному з позитивним змістом і, у зв'язку з цим, притягає до себе увагу фахівців з медичної хімії. Нуклеозидні інгібітори NS5B полімерази можуть використовуватися як штучний субстрат, який приводить до завершення ланцюга, або як конкурентний інгібітор, що протидіє зв'язуванню нуклеотидів з полімеразою. Для того щоб виконувати функцію термінатора ланцюга, нуклеозидний аналог повинен поглинатися клітиною і перетворюва 9 91255 10 тися in vivo на трифосфат, щоб у такій формі конкурувати за сайт зв'язування нуклеотидів з полімеразою. Його перетворення на трифосфат зазвичай опосередковується клітинними кіназами, якими на потенційний нуклеозидий інгібітор полімерази накладаються додаткові структурні вимоги. Проте це обмежує можливість проводити пряму оцінку інгібіторної здатності нуклеозидів стосовно HCV реплікації всіма іншими методами, окрім аналізу на клітинах, який дозволяє здійснювати фосфориляцію in situ. У заявці WO 0190121, опублікованій 29 листопада 2001p., описана і розглянута на прикладах спрямована проти HCV-полімерази активність 1'алкіл- і 2'-алкіл-нуклеозидів, які мають показані вище формули 2 і 3. У заявці WO 01/92282 (автори J.-P. Sommadossi, P. Lacolla), опублікованій 6 грудня 2001p., описане і розглянуте на прикладах лікування викликаних вірусами Flaviviruses і Pestiviruses хвороб 1'-алкілі 2'-алкілнуклеозидами, які мають показані вище формули 2 і 3. У заявці WO 03/026675 (автор G. Gosselin), опублікованій 3 квітня 2003p., описані 4'-алкілнуклеозиди 4 для лікування хвороб, викликаних вірусами Flaviviruses i Pestiviruses. У заявці WO 2004003000 (автори J -P. Sommadossi et аl.), опублікованій 8 січня 2004p., описані 2'- і 3'-проліки 1'-, 2'-, 3'- і 4'-заміщених ( -D і -1-нуклеозидів). У заявці WO 2004/002422, опублікованій 8 січня 2004p., описаний 2'-С-метил-3'-Оваліновий естер-рибофуранзилцитидин для лікування хвороб, викликаних інфекціями Flaviviridae. Повідомлялося (Idenix) про клінічні випробування спорідненої сполуки NM283, яка вважалася валіновим естером за формулою 5 цитидинового аналога за формулою 2 (де В є цитозин). У заявці WO 2004/002999 (автори J.-P. Sommadossi et аl.,), опублікованій 8 січня 2004p., описана низка 2'- і 3'проліків 1', 2', 3' і 4'-розгалужених нуклеозидів для лікування хвороб, викликаних інфекціями flavivirus, включаючи HCV-вірус. У заявці WO2004/046331 (автори J.-Р Sommadossi et al.), опублікованій 3 червня 2004p., описані 2'-розгалужені нуклеозиди і мутація Flaviviridae. У заявці WO 03/026589 (автори G. Gosselin et al.), опублікованій 3 квітня 2003p., описані способи лікування вірусного гепатиту С за допомогою 4'-модифікованих нуклеозидів. У заявці WO 2005009418 (автори R. Storer et al.), опублікованій 3 лютого 2005p., описані пуринові нуклеозидні аналоги для лікування хвороб, викликаних вірусом Flaviviridae, включаючи HCV. Опубліковані також патентні заявки, в котрих описано застосування деяких нуклеозидних аналогів у лікуванні вірусного гепатиту С. У заявці WO 01/32153 (автор R. Storer), опублікованій 10 травня 2001p., описані похідні нуклеозидів для лікування вірусних хвороб. У заявці WO 01/60315 (автори Н. Ismaili et al.), опублікованій 23 серпня 2001p., описані способи лікування та профілактики нуклеозидними сполуками інфекційних хвороб, викликаних вірусом Flavivirus. У заявці WO 02/18404 (автори R. Devos et al.), опублікованій 7 березня 2002p., описані 4'-заміщені нуклеотиди для лікування HCV-вірусних хвороб. У заявці WO 01/79246 (автор K. A. Watanabe), опублікованій 25 жовтня 2001p., описані 2'- і 3'-гідроксиметилнуклеозидні сполуки для лікування вірусних хвороб. У заявці WO 02/32920, опублікованій 25 квітня 2002p., і в заявці WO 02/48 165, опублікованій 20 червня 2002ρ, (автори Stuyver et al.), описані нуклеозидні сполуки для лікування вірусних хвороб. У заявці WO 03/105770 (автори В. Bhat et al.), опублікованій 24 грудня 2003p., описана низка карбоциклічних нуклеозидних сполук, придатних для лікування HCV-інфекційних хвороб. У заявці WO 2004/007512, опублікованій 22 січня 2003р. (автори В. Bhat et al.), описані нуклеозидні сполуки, що інгібують РНК-залежну РНК-вірусну полімеразу. У цій публікації описані передусім 2'-метил2'-гідрокси-заміщені нуклеозиди. У заявці WO 2002/057425 (автори S. S. Carroll et al.), опублікованій 25 липня 2002p., описані нуклеозидні похідні, що є інгібіторами РНК-залежної вірусної полімерази, i способи лікування HCV-інфекційних хвороб. У заявці WO02/057287 (автори S. S. Carroll et al.), опублікованій 25 липня 2002p., описані споріднені 2 -метил- i 2 -метилрибозні сполуки, основою яких є необов'язково заміщений 7Н-пірол[2,3d]піримідиновий радикал за формулою 6. У цій же заявці описаний приклад 3p-метил-нуклеозиду. У роботі (S. S. Carroll et al., J ΒiοΙ Chem. 2003 278(14): 11979-11984) описане інгібування HCVвірусноі полімеразаи 2'-О-метилцитидином (6а). У 11 заявці WO 2004/009020 (автори D. В. Olsen et al.), опублікованій 29 січня 2004p., описана низка тюнуклеозидних сполук, що є інгібіторами РНКзалежнооі РНК-вірусної полімерази. У публікації РСТ № WO 99/43691 від Університету Емори (Emory University) під назвою "2'Fluoronucleosides" описане застосування деяких 2'фторнуклеозидів у лікуванні HCV-інфекційних хвороб. У патенті США №6,348,587 від Університету Емори (Emory University) під назвою "2'Fluoronucleosides" описане сімейство 2'фторнуклеозидів, придатних для лікування гепатиту В, HCV- i ВІЛ-інфекцій та аномальної клітинної проліферації. При цьому розглядалися обидві конфігурації 2'-фторозамісника. У публікації [Eldrup et al., Oral Session V, Hepatitis C Virus, Flavivindae, 16th International Conference on Antiviral Research (Apr. 27, 2003p., Savannah, шт. Ga) (Усна сесія V, Вірус гепатиту С, Flavivindae, 16 Міжнародна конференція з антивірусних досліджень, 27 квітня, 2003p., Саванна, шт. Джорджія)] описаний взаємозв'язок між структурою та активністю 2'-модифікованих нуклеозидів для інгібування HCV. У публікації [Bhat et al., Oral Session V, Hepatitis C Virus, Flavivindae, 16th International Conference on Antiviral Research (Apr. 27, 2003p., Savannah, Ga.) (Усна сесія V, Вірус гепатиту С, Flavivindae, 16 Міжнародна конференція з антивірусних досліджень, 27 квітня, 2003p., Саванна, шт. Джорджія, p Α75)] описаний процес синтезу та фармакокінетичні властивості нуклеозидних аналогів, як потенційних інгібіторів реплікації РНК HCVвірусу. Автори цієї публікації повідомляли, що 2'модифіковані нуклеозиди демонстрували потужну інгібіторну активність у клітинних випробуваннях на репліконах. У публікації [Olsen et al., Oral Session V, Hepatitis С Virus, Flavivindae, 16th International Conference on Antiviral Research (Apr. 27, 2003p., Savannah, Ga.) (Усна сесія V, Вірус гепатиту С, Flavivindae; 16 Міжнародна конференція з антивірусних досліджень, 27 квітня, 2003p., Саванна, шт. Джорджія, p Α76) також описаний вплив 2'модифікованих нуклеозидів на реплікацію РНК HCV-вірусу. Ненуклеозидні алостеричні інгібітори ВІЛревертази довели свою терапевтичну ефективність при застосуванні їх як самих, так i в комбінаціях з нуклеозидними інгібіторами i з інгібіторами протеази. В літературі описано декілька класів ненуклеозидних HCV NS5B-інгібіторів, які сьогодні є на різних стадіях розробок - це, наприклад бензимідазоли (Н. Hashimoto et al., WO 01/47833, Η. Hashimoto et al., WO 03/000254, Ρ L Beauheu et al., WO 03/020240 A2; Ρ. L. Beauheu et al., US 6,448,281 Bl; Ρ. L. Beauheu et al., WO 03/007945 Al); індоли (Ρ. L. Beauheu et al., WO 03/0010141 A2); бензотіадіазини, наприклад за формулою 7, (D. Dhanak et al., WO 01/85172 Al; D. Dhanak et al., WO 03/037262 A2; K. J. Duffy et al., WO03/099801 Al, D.Chai et al., WO 2004052312, D.Chai et al., WO2004052313, D.Chai et al., WO02/098424, J. K. Pratt et al., WO 2004/041818 Al; J. K. Pratt et al., WO 2004/087577 Al); тіофени, наприклад за формулою 91255 12 8, (С. k. Chan et al., WO 02/100851); бензотіофени (D. С. Young i Τ. R. Bailey WO 00/18231); ркетопірувати (S. Attamura et al., US 6,492,423 Bl, A. Attamura et al., WO 00/06529); піримідини (С. Gardelh et al., WO 02/06246 Al); піримідиндioни (Τ. R. Bailey and D. С. Young WO 00/13708); триазини (Κ.-Η. Chung et al., WO 02/079187 Al); похідні роданіну (Τ. R. Bailey and D. С. Young WO 00/10573, J. С. Jean et al., WO 01/77091 A2); 2,4-дioксопірани (R. A. Love et al., EP 256628 A2); похідні фенілаланіну (Μ. Wang et al., J. Biol Chem. 2003 278:24892495). NS3-протеаза стала головною мішенню при розробці нового засобу анти-HCV-вірусної терапії. У заявці WO 98/22496 (автори Μ. R. Attwood et al.), опублікованій 28 травня 1998p., були описані інгібітори протеази з активним сайтом на механічній основі (М. R. Attwood et al., Antiviral Chemistry and Chemotherapy 1999 10:259-273; Μ. R. Attwood et al., Preparation and use of ammo acid derivatives as anti-viral agents (Виготовлення і застосування амінокислотих сполук в антивірусних засобах), German Patent Pub. DE 19914474). У заявці WO98/17679 (автори R. D. Tung et al.), опублікованій 30 квітня 1998p., описані пептидні інгібітори NS3-протеази на механічній основі. У заявці WO99/07734, опублікованій 18 лютого 1999p., i заявці WOOO/09543, опублікованій 9 серпня 1999, (автори Μ Llinas-Brunet et al.), описані пептидні інгібітори протеази. У заявці WOOO/59929, опублікованій 12 жовтня 2000p. (Υ. S. Tsantnzos et al.), описані макроциклічні трипептиди, котрі є потужними інгібіторами HCV NS3протеази. У низці споріднених патентів від Boehnnger-lngleheim описані споріднені інгібітори протеази i була створена база для ідентифікації трипептидної сполуки BILN 2061 (М. Llinas-Brunet et al., Biorg. Med. Chem. Lett. 2000 10(20):2267-70; J. Med Chem.2W4 47(26):6584-94, J. Med. Chem. 2004 47(7):1605-1608; Angew. Chem. Int. Ed. Eng. 2003 42(12): 1356-60). Інші трипептидні інгібітори були ідентифіковані фірмою Bristol-Myers Squibb та описані, поміж іншим, у заявці WO 03/099274, опублікованій 4 грудня 2003р., у заявці WO 2004/032827, опублікованій 22 квітня 22 2004p., у заявці WO 03/053349, опублікованій 3 липня 2003p., у заявці WO 2005/046712, опублікованій 26 травня 2005p., та в заявці WO 2005/051410, опублікованій 9 червня 2005р. У заявці WO 2004/072234, опублікованій 26 серпня 2004p., та в заявці WO 2004/093798, опублікованій 4 листопада 2004p., були описані трипептидні інгібітори протеази, ідентифіковані фірмою Enanta Pharmaceuticals. У заявці WO 2005/037214 13 (автори L. М. Blatt et аl.), опублікованій 28 квітня 2005, описані ще одні трипептидні похідні, що інгібують HCV NS3-протеазу. У заявці WO 2005/030796 (автори S. Venkatraman et аl.), опублікованій 7 квітня 2005p., описані макроциклічні інгібітори HCV NS3-серинпротеази. У заявці WO 2005/058821 (автори F. Velazquez et al.), опублікованій 30 червня 2005p., описані інгібітори HCV NS3/NS4a-серинпротеази. У заявці WO 02/48172 (автор Z. Zhu), опублікованій 20 червня 2002p., описані діарилпептиди, придатні служити інгібіторами NS3 протеази. У заявці WO 02/08187 та в заявці WO 02/08256 (автори A. Saksena et al.), опублікованих 31 січня 2002p., описані пептидні інгібітори HCV NS3-протеази. У заявці WO 02/08251 (автори М. Lim-Wilby et al.), опублікованій 31 січня 2002p., описані пептидні інгібітори NS3протеази. У патенті США №6,004,933 (автори L. W. Spruce et al.), опублікованому 21 грудня 1999p., описані гетероциклічні пептидні сполуки, що інгібують цистеїн-протеази, включаючи HCV ендопептидазу. Досліджувалися також інгібітори NS3-протеази на безсубстратній основі, такі як 2,4,6-тригідрокси3-нітробензамідні похідні (Sudo K. et al., BBRC 1997 238:643-647; Sudo K. et al., Antiviral Chemistry and Chemotherapy 1998 9:186), включаючи матеріали RD3-4082 і RD3-4078, де перший заміщувався на амідній частині 14-вуглецевим ланцюгом, а у другого піддавалася процесінгу парафеноксифенільна група. Матеріал SCH 68631, фенантренхінон, є інгібітором HCV-протеази (Chu M. et al., Tetrahedron Lett. 1996 37:7229-7232). інший матеріал тих самих авторів, SCH 351633, виділений із грибків РеnісіІІіum griseofulvum, був визнаний інгібітором протеазного ферменту (Chu Μ. et al., Bioorg. Med. Chem. Lett. 1999 9:1949-1952). Наномолярна потужність проти HCV NS3-протеази досягалася цими авторами у сконструйованих ними селективних інгібіторах на основі макромолекулярного егліну с. Матеріал „еглін с", виділений із п'явки, є потужним інгібітором декількох серинопротеаз і, зокрема, протеаз А і В S. griseus, -хімотрипсину, хімази і субтилізину (Qasim Μ. A. et al., Biochemistry 1997 36:1598-1607). Повідомлялося про похідні тіазолідину, які продемонстрували суттєве інгібування у випробуваннях методом HPLC-хроматографії з оберненою фазою на злитому білку NS3/4A з NS5A/5B субстратом (Sudo K. et al., Antiviral Research 1996 32:9-18); це були, зокрема, сполука RD-1-6250, яка мала злиту цинамоїлову частину, заміщену довгим алкіловим ланцюгом, а також сполуки RD4 6205 і RD4 6193. Тіазолідини і бензаніліди були ідентифіковані в роботах (N. Kakiuchi et al., in FEBS Let. 1998 421:217-220; N. Takeshita et al., Anal Biochem. 1997 247:242-246). Про здатність імідазолідинонів служити інгібіторами NS3-серинпротеази HCV-вірусу повідомлялося в заявці WO 02/08198 (заявник: фірма Schering Corporation), опублікованій 31 січня 2002p., і в заявці WO 02/48157 (заявник: фірма Bristol Myers Squibb), опублікованій 20 червня 2002p. У заявці WO 02/48116 (автори P. Glunz et 91255 14 al.), опублікованій 20 червня 2002p., описані примідинонові інгібітори NS3-протеази. Іншими ферментативними мішенями для засобів проти HCV-вірусу є внутрішня рибосомна ділянка проникнення вірусів (IRES: Internal Ribosomal Entry Site) і геліказа HCV-вірусу. Про інгібітори IRES-ділянки повідомлялося фірмами Immusol, Rigel Pharmaceuticals (R803) і Anadys (ANA 245 і ANA 246). Фірмою Vertex був описаний інгібітор HCV-гелікази. Комбінаційна терапія, здатна пригнічувати стійкі мутантні штами, стала вже добре усталеним методом антивірусної хіміотерапії. Описані тут нуклеозидні інгібітори можуть об'єднуватися з іншими нуклеозидними інгібіторами HCVполімерази, ненуклеозидними інгібіторами HCVполімерази та інгібіторами HCV-протеази. У зв'язку з появою та подальшою розробкою інших класів анти-HCV ліків, наприклад, інгібіторів ділянки проникнення вірусів, інгібіторів гелікази, інгібіторів IRES-ділянки, рибозимів і протизмістових олігонуклеотидів ці засоби також є чудовими кандидатами для комбінаційної терапії. Похідні інтерферону вже успішно комбінувалися з рибавірином та інтерферонами, а хімічно модифіковані інтерферони можуть ефективно використовуватися в комбінації з описаними тут нуклеозидами. Нуклеозидні похідні часто є потужними противірусними (наприклад, проти ВІЛ, HCV вірусу, вірусу простого герпесу, CMV вірусу) і протираковими хіміотерапевтичними засобами. Проте, на практиці їхня корисність часто обмежується двома факторами. По-перше, їхні низькі фармакокінетичні властивості часто стають обмеженням для поглинання нуклеозидів із травного тракту і для міжклітинної концентрації нуклеозидів, а по-друге, нижче оптимальних фізичні властивості обмежують вибір препаратів, які можуть використовуватися для підсилення постачання активного інгредієнта. А. Альбертом (A. Albert, Selective Toxidty, Chapman and Hall, London, 1951) був уведений термін "проліки", який означає сполуку, котра не володіє власною біологічною активністю, але може піддаватися метаболічному перетворенню на активну лікувальну речовину. Достатньо широкі огляди проліків нещодавно з'явилися в публікаціях (Р. Ettmayer et al., J. Med Chem. 2004 47(10):23932404; K. Beaumont et al., Curr. Drug Metab. 2003 4:461-485; H. Bundgaard, Design of Prodrugs: Bioreversible derivatives for various functional групи and хімічні entities in Design of Prodrugs (Розробка проліків: біооборотні похідні для різноманітних функціональних груп і хімічних субстанцій), Н. Bundgaard (ed) Elsevier Science Publishers, Amersterdam 1985; G. M. Pauletti et al., Adv. DrugDeliv. Rev. 1997 27:235-256;R. J. Jones and N. Bischofberger, Antiviral Res. 1995 27; 1-15 and С R. Wagner et al., Med. Res. Rev. 2000 20:417-45). У той час як каталіз метаболічного перетворення може здійснюватися специфічними ферментами, часто гідролазами, активна сполука може регенеруватися також шляхом неспецифічних хімічних процесів. 15 Фармацевтично доступними проліками називають сполуку, яка внаслідок метаболічного перетворення, наприклад гідролізу або окислювання, в хазяїні утворює сполуку згідно з даним винаходом. Таке біоперетворення повинно уникати утворення фрагментів з токсикологічним навантаженням. Типовими прикладами проліків є сполуки, які мають біологічно нестійкі захисні групи, зв'язані з функціональною частиною активної сполуки. При розробці пронуклеотидів застосовувалися методи алкілування, ацилування та інші методи ліпофільної модифікації гідроксильних груп у цукровій частині. Ці пронуклеотиди можуть піддаватися гідролізу або деалкілуванню in vivo, утворюючи в результаті активну сполуку. Факторами, що обмежують пероральну біодоступність ліків, часто є поглинання зі шлунковокишкового тракту та екскреція першого проходження стінками стравоходу і печінкою. Для оптимізації міжклітинного поглинання через шлунковокишковий тракт потребується величина D(7,4) більше нуля. Проте, оптимізація коефіцієнта розподілу не гарантує досягнення успіху. Проліки можуть потребувати уникнення активних транспортерів відтоку в ентероцитах. Внутрішньоклітинний метаболізм в ентероциті може приводити до пасивного транспорту або активного транспорту продуктів обміну речовин відтоковими насосами назад, у кишкову порожнину. Таким чином, проліки повинні бути стійкими до небажаних біоперетворень у крові до досягнення ними цільових клітин або рецепторів. Поряд з тим, що в деяких випадках потрібні проліки можна досить раціонально сконструювати на основі наявних у даній молекулі хімічних функціональних груп, хімічна модифікація активної сполуки приводить до створення цілком нової молекулярної субстанції, котра може мати небажані фізичні, хімічні та біологічні властивості, відсутні у батьківської сполуки. Обумовлені законом регуляторні вимоги стосовно продуктів обміну речовин можуть поставити складні проблеми, якщо численні шляхи метаболізму ведуть до великої кількості його продуктів. У результаті ідентифікація проліків залишається неясною і складною справою. Крім того, складними і коштовними виявляються спроби давати оцінку фармакокінетичним властивостям потенційним пролікам. Результати фармакокінетичних досліджень на тваринних моделях може бути важко екстраполювати на людину. Метою даного винаходу є створення нових сполук, способів і складів для лікування носія, інфікованого вірусом гепатиту С. Даний винахід спрямований на нові діацилові похідні 4-аміно-1-((2R,3R,4R,5R)-3-фтор-4гідрокси-5-гідроксиметил-3-метилтетрагідрофуран2-іл)-1Н-піримідин-2-ону (який має також назву (2R)2'-деокси-2-метил-2-фторцитидину). Даним винаходом пропонуються сполуки, котрі мають структуру, що відповідає формулі І: 91255 16 де: R1 вибраний із сукупності, що складається із С2-5 нерозгалуженого або розгалуженого алкілу, С2-5 нерозгалуженого або розгалуженого алкенілу, С2-5 нерозгалуженого або розгалуженого алкінілу, С2-5 нижчого галоїдалкілу, С3-6 циклоалкілу і С2-4 алкокси; а також їхні гідрати, сольвати та солі приєднання кислот. Сполуки згідно з даним винаходом є корисними в лікуванні розладу, опосередкованого HCV-вірусом. Винаходом пропонуються також способи лікування викликаних HCV-інфекцією хвороб за допомогою запропонованих винаходом сполук і фармацевтичних складів, які містять ці сполуки. В одному з варіантів здійснення даного винаходу пропонується сполука, що відповідає формулі І, де R1 є таким, як визначено вище. В іншому варіанті здійснення даного винаходу пропонується сполука, що відповідає формулі І, де R1 є етил, n-пропіл, ізопропіл, n-бутил або ізобутил. Передбачений також варіант здійснення винаходу, в якому запропонована сполука відповідає формулі І, де R1 є етил або ізопропіл. Можливим є також варіант здійснення винаходу, в якому запропонована сполука відповідає формулі І, де R1 є ізопропіл, а сполукою є гідрохлорид або сульфат. В одному з варіантів здійснення даного винаходу пропонується сполука, що відповідає формулі І, де R1 є ізопропіл, а сполукою є гідрохлорид. Передбачений також варіант здійснення винаходу, в якому запропонована сполука відповідає формулі І, де R1 є етокси, n-пропокси або ізопропокси. Згідно з ще одним варіантом здійснення винаходу пропонується сполука, що відповідає формулі І, для її застосування в терапії і, зокрема, для застосування в лікуванні хвороби, опосередкованої HCV-вірусом. У ще одному з варіантів здійснення даного винаходу пропонується застосування сполуки згідно з формулою І для виготовлення медикаменту, призначеного для лікування хвороби, опосередкованої HCV-вірусом. Сполука, що відповідає формулі І, може використовуватися, зокрема, у виготовленні медикаменту для введення його пацієнту, що цього потребує, у терапевтично ефективній дозі, котрою в кращому варіанті є доза в інтервалі від 0,1 до 10 г на день, у ще кращому варіанті - доза в інтервалі від 0,5 до 7 г на день, а в ще кращому варіанті доза від 1,0 г до 6,0 г на день. 17 Сполука, що відповідає формулі І, може, крім того, використовуватися для виготовлення медикаменту, який може містити також терапевтично ефективну кількість принаймні одного модулятора імунної системи, такого як інтерферон, хімічно дериватизований інтерферон, інтерлейкін, фактор некрозу пухлин або колонієстимулювальний фактор і/або щонайменше один антивірусний засіб, який інгібує реплікацію HCV-вірусу, тобто такий як інгібітор HCV-протеази, інший нуклеозидний інгібітор HCV-полімерази, ненуклеозидний інгібітор HCV-полімерази, та інгібітор HCV-гелікази, інгібітор HCV-примази або інгібітор злиття HCV-вірусу. Крім того, передбачений також варіант здійснення винаходу, в якому пропонується спосіб лікування хвороби, опосередкованої HCV-вірусом, де запропонований спосіб включає у себе введення пацієнту, що цього потребує, терапевтично ефективної дози сполуки згідно з формулою І відповідно до визначеного вище. Передбачений також варіант здійснення винаходу, в якому пропонується спосіб лікування хвороби, опосередкованої HCV-вірусом, де запропонований спосіб включає у себе введення пацієнту, що цього потребує, дози від 0,1 до 10 г на день сполуки згідно з формулою І відповідно до визначеного вище. В іншому варіанті здійснення винаходу зазначена доза лежить в інтервалі від 0,5 до 7 г на день, а в ще одному варіанті зазначена доза лежить в інтервалі від 1,0 до 6,0 г на день. В одному з варіантів здійснення даного винаходу пропонується спосіб лікування хвороби, опосередкованої HCV-вірусом, де запропонований спосіб включає у себе спільне введення пацієнту, що цього потребує, терапевтично ефективної дози сполуки згідно з формулою І відповідно до визначеного вище і терапевтично ефективної кількості принаймні одного модулятора імунної системи і/або щонайменше одного антивірусного засобу, який інгібує реплікацію HCV-вірусу. В іншому варіанті здійснення даного винаходу пропонується спосіб лікування хвороби, опосередкованої HCV-вірусом, де запропонований спосіб включає у себе спільне введення пацієнту, що цього потребує, терапевтично ефективної дози сполуки згідно з формулою І відповідно до визначеного вище і терапевтично ефективної кількості принаймні одного модулятора імунної системи, де зазначеним модулятором імунної системи є інтерферон, інтерлейкін, фактор некрозу пухлин або колонієстимулювальний фактор. В іншому варіанті здійснення даного винаходу пропонується спосіб лікування хвороби, опосередкованої HCV-вірусом, де запропонований спосіб включає у себе спільне введення пацієнту, що цього потребує, терапевтично ефективної дози сполуки згідно з формулою І відповідно до визначеного вище i терапевтично ефективної кількості принаймні одного модулятора імунної системи, де зазначеним модулятором імунної системи є інтерферон або хімічно дериватизований інтерферон. В іншому варіанті здійснення даного винаходу пропонується спосіб лікування хвороби, опосередкованої HCV-вірусом, де запропонований спосіб 91255 18 включає у себе спільне введення пацієнту, що цього потребує, терапевтично ефективної дози сполуки згідно з формулою І відповідно до визначеного вище i терапевтично ефективної кількості щонайменше однієї іншої антивірусної сполуки. Передбачений також варіант здійснення даного винаходу, в якому пропонується спосіб лікування хвороби, опосередкованої HCV-вірусом, де запропонований спосіб включає у себе спільне введення пацієнту, що цього потребує, терапевтично ефективної дози сполуки згідно з формулою І відповідно до визначеного вище i терапевтично ефективної кількості щонайменше однієї іншої антивірусної сполуки, де зазначеною сполукою є інгібітор HCV-протеази, інший нуклеозидний інгібітор HCV-полімерази, ненуклеозидний інгібітор HCV-полімерази, інгібітор HCV-гелікази, інгібітор HCV-примази або інгібітор злиття HCV-вірусу. В іншому варіанті здійснення даного винаходу пропонується фармацевтичний склад, який містить сполуку, що відповідає формулі І згідно з визначеним вище, змішану щонайменше з одним фармацевтично доступним носієм, розріджувачем або ексципієнтом. В одному з варіантів здійснення даного винаходу пропонується процес виготовлення сполуки згідно з формулою І відповідно до визначеного вище, де зазначений процес включає у себе стадії (i)-(v), нумерація яких відповідає п. 17, i які проілюстровані в розглянутих нижче прикладах. Запропонований процес включає у себе обробку сполуки І в основному водному розчині органічного середовища, яке може бути гомогенним або двофазним, ацилувальним засобом згідно з даним тут визначенням при наявності DMAP (диметиламінопіридину) та основи в кількості, достатній для підтримання рН розчину на рівні не нижче, ніж приблизно 7,5. Запропонований процес дозволяє проводити ацилування без супровідної реакції даної гетероциклічної основи. Використовуваний у даному описі термін "сполука" чи будь-яка інша "субстанція" може означати як одну, так i більше однієї субстанцій, наприклад, при згадуванні в тексті однієї сполуки, це може означати як одну, так i більше однієї сполук. Відповідно до цього, вирази "один", "один чи більше одного", "щонайменше один" i т.п. можуть використовуватися як взаємозамінні. Використовувані у даному описі терміни "необов'язковий" та "не обов'язково" означають, що подія або обставина, яку вони характеризують, може, але не обов'язково, мати місце, тобто що ця характеристика включає у себе i випадки, в котрих подія або обставина має місце, і випадки, в котрих вона не має місця. Наприклад, вираз "необов'язковий зв'язок" означає, що даний зв'язок може бути і може не бути наявним, і що дана характеристика передбачає можливість існування одинарного, подвійного або потрійного зв'язків. Використовуваний у даному описі термін "незалежно" означає, що певна змінна використовується в усіх випадках без урахування наявності або відсутності тої чи іншої змінної, що має таке саме або інше визначення в тій самій сполуці. Таким чином, у сполуці, в котрій змінний компонент R 19 згадується двічі і визначається як такий, що є "незалежно вуглець або азот", обидва R можуть бути вуглецями, обидва R можуть бути азотами, або один R може бути вуглець, а інший - азот. Використовуваний у даному описі термін "алкеніл" означає незаміщений вуглеводнений ланцюговий радикал, який складається із 2-10 атомів вуглецю, що мають один або два олефінові подвійні зв'язки, а в кращому варіанті - один олефіновий подвійний зв'язок. Так, запис "С2-10 алкеніл" означає алкеніл, який складається із 2-10 атомів вуглецю. Це може бути, наприклад, вініл, 1пропеніл, 2-пропеніл (аліл) або 2-бутеніл (кротил). Використовуваний у даному описі термін "алкіл" означає насичений, одновалентний вуглеводневий залишок у формі нерозгалуженого або розгалуженого ланцюга, що складається із 1-10 атомів вуглецю. Термін "нижчий алкіл" означає вуглеводневий залишок у формі прямолінійного або розгалуженого ланцюга, що складається із 1-6 атомів вуглецю. Запис "С1-10 алкіл" означає алкіл, що складається із 1-10 атомів вуглецю. Алкільними групами можуть бути, наприклад, групи нижчих алкілів, включаючи метил, етил, пропіл, i-пропіл, nбутил, i-бутил, t-бутил або пентил, ізопентил, неопентил, гексил, гептил та октил. Записи (ар)алкіл і (гетероарил)алкіл вказують на те, що дана алкільна група є необов'язково заміщеною, відповідно, арильною групою і гетероарильною групою. Використовуваний у даному описі термін "алкініл" означає вуглеводневий радикал у формі нерозгалуженого або розгалуженого ланцюга, що складається із 2-10 атомів вуглецю, а в кращому варіанті - із 2-5 атомів вуглецю, і має один потрійний зв'язок. Так, запис "С2-10 алкініл" означає алкініл, що складається із 2-10 атомів вуглецю. Це можуть бути, наприклад, етиніл, 1-пропініл, 2пропініл, 1-бутиніл, 2-бутиніл або 3-бутиніл. Використовуваний у даному описі термін "циклоалкіл" означає насичене карбоциклічне кільце, що містить від 3 до 8 атомів вуглецю, тобто таке, як циклопропіл, циклобутил, циклопентил, циклогексил, циклогептил або циклооктил. Так, запис "С3-7 циклоалкіл" означає циклоалкіл, що складається із 3-7 атомів вуглецю в карбоциклічному кільці. Використовуваний у даному описі термін "галоїдалкіл" означає алкільну групу у формі нерозгалуженого або розгалуженого ланцюга відповідно до визначеного вище, де 1, 2, 3 чи більше атомів водню є заміщеними галогеном. Так, запис "С1-3 галоїдалкіл" означає галоїдалкіл, який складається із 1-3 атомів вуглецю і 1-8 галогенових замісників. Таким чином, це можуть бути, наприклад, 1фторметил, 1-хлорметил, 1-бромметил, 1йодометил, трифторметил, трихлорметил, трибромметил, трийодометил, 1-фторетил, 1хлоретил, 1-брометил, 1-йодетил, 2-фторетил, 2хлоретил, 2-брометил, 2-йодетил, 2,2-дихлоретил, 3-бромпропіл, 2,2,2-трифторетил тощо. Використовуваний у даному описі термін "алкокси" означає -О-алкільну групу, в котрій алкіл відповідає визначенню, даному вище. Так, групами алкокси можуть бути, наприклад, метокси, етокси, n-пропілокси, i-пропілокси, n-бутилокси, i 91255 20 бутилокси, t-бутилокси. Запис "нижчий алкокси" означає алкоксигрупу з групою "нижчого алкілу" згідно з визначеним вище. Запис "С1-10 алкокси" означає -О-алкіл, у котрому алкілом є С1-10 алкіл. Використовуваний у даному описі термін "діацилова" похідна означає дериватизовану нуклеозидну сполуку згідно з даним тут визначенням, у котрій групи 3'- і 5'-гідрокси є із естерів -OC(=O)R1 і OC(=O)R2, де R1 і R2 є такими, як визначено в п. 1 Формули винаходу. Використовуваний у даному описі термін "ацилувальний засіб" може означати або ангідрид, галоїдангідрид, хлоркарбонілалкоксид (наприклад, етилхлорформіат) або активовану похідну Nзахищеної альфа-амінокислоти. При цьому термін "ангідрид" стосується сполук, які мають загальну структуру, виражену формулою R1C(O)-O-C(O)R1, де R1 є таким, як визначено в п. 1 Формули винаходу, а термін "галоїдангідрид" стосується сполук, які мають загальну структуру, виражену формулою R1C(O)X, де X є галоген. Використовуваний тут термін "ацилімідазол" стосується сполук, які мають загальну структуру, виражену формулою R1C(O)X, де X є N-імідазоліл. Термін "активована похідна" тої чи іншої сполуки означає тимчасову хімічно активну форму первинної сполуки, яка робить цю сполуку активною у бажаній хімічній реакції, у котрій ця первинна сполука має лише помірну хімічну активність або зовсім не є хімічно активною. При цьому активація досягається шляхом утворення похідної або хімічного групування в молекулі з більшим умістом вільної енергії, ніж у первинній сполуці, що робить активовану форму більш здатною до хімічної взаємодії з іншим реагентом. У контексті даного винаходу особливо важливою є активація карбоксильної групи, у зв'язку з чим нижче більш докладно розглядаються відповідні засоби та групування що активують карбоксильну групу. Особливо важливими для цілей даного винаходу є ангідриди та хлориди карбонової кислоти. Використовуваний у даному описі вираз "гетерогенна водна розчинювальна суміш" означає суміш води та органічного спільного з нею розчинника, які утворюють двофазну або гетерогенну суміш. Така гетерогенна водна розчинювальна суміш може створюватися шляхом змішування води та спільного розчинника, який має обмежену розчинність у воді, або шляхом регулювання іонної сили водного компонента і, таким чином, обмеження розчинності спільного розчинника у водяній фазі. Використовуваний у даному описі вираз "гідроксид лужного металу" означає сполуку за загальною формулою МОН, де Μ є літій, натрій, калій або цезій, вираз "бікарбонат лужного металу" означає групу МНСО3, де Μ є натрій або калій, а вираз "карбонат лужного металу" означає групу М2СО3, де Μ є натрій або калій. Для фахівця в даній галузі цілком зрозуміло, що для підтримання потрібного рівня рН можуть використовуватися інші основи, які також охоплюються об'ємом даного винаходу. В описі даної заявки використовуються такі скорочення: ацетил (Ас), оцтова кислота (НОАс), 21 1-Ν-гідроксибензотриазол (HOBt), атмосфери (атм.), рідинна хроматографія високого тиску (HPLC), метил (Me), трет-бутоксикарбоніл (Вос), ацетонітрил (MeCN), ди-трет-бутилпірокарбонат або boc-ангідрид (ВОСаО), 1-(3диметиламінопропіл)-3-етилкарбодііміду гідрохлорид (EDCI), бензил (Вn), бутил (Вu), метанол (МеОН), бензилоксикарбоніл (cbz або Z), температура плавлення (т. пл.), карбоніл-діімідазол (CDI), MeSO2(мезил або Ms), 1,4діазабіцикло[2.2.2]октан (DABCO), мас-спектр (ms), метил-t-бутиловий етер (МТВЕ), 1,5діазабіцикло[4.3.0]нон-5-ен (DBN), 1,8діазабіцикло[5.4.0]ундец-7-ен (DBU), Nметилморфолін (NMM), N-метилпіролідон (NMP), 1,2-дихлоретан (DCE), Ν,Ν'дициклогексилкарбодіімід (DCC), піридиндихромат (PDC), дихлорметан (DCM), пропіл (Рr), фунт/кв.дюйм (psi), діізопропілетиламін (DIPEA, основа Ханіга (Hunig)), піридин (руг), кімнатна температура (rt або RT), Ν,Ν-диметилацетамід (DMA), трет-бутилдиметилсиліл або f-BuMe2Si, (TBDMS), 4-Ν,Ν-диметиламінопіридин (DMAP), триетиламін (Et3N або TEA), Ν,Ν-диметилформамід (DMF), диметилсульфоксид (DMSO), трифтороцтова кислота (TEA), тонкошарова хроматографія (TLC), етилацетат (EtOAc), тетрагідрофуран (THF), діетиловий етер (Et2O), триметилсиліл або Me3Si (TMS), етил (Et), р-толуолсульфокислоти моногідрат (TsOH or pTsOH), 4-Me-C6H4SO2- або тозил (Ts), ізопропіл (i-Рr), N-ypeтан-N-карбоксіангідрид (UNCA), етанол (EtOH). Використовувана з алкільним компонентом символіка згідно зі стандартною номенклатурою, включаючи префікси n (нормальний), і (ізо), втор (вторинний), трет (третинний) і нео, має свої звичайні значення (J. Rigaudy and D. P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford). У Табл. 1 наведені типові приклади сполук, що охоплюються об'ємом даного винаходу. Ці сполуки та поданий нижче опис процесів їх виготовлення дозволяють фахівцеві в даній галузі більш детально ознайомитися з перевагами та особливостями даного винаходу і полегшити здійснення його на практиці. їх слід розглядати не як обмеження об'єму даного винаходу, а лише як його репрезентативну ілюстрацію. У загальному випадку номенклатура, використовувана в даній заявці, ґрунтується на комп'ютеризованій системі AUTONOM v.4.0 інституту "Beilstein Institute" для створення систематичних номенклатур IUPAC (Міжнародного союзу з теоретичної і прикладної хімії). У разі розбіжності між зображеною тут структурою і даною їй назвою, більшої ваги слід надавати зображеній структурі. Крім того, якщо стереохімія структури або її частини не позначена, наприклад, жирними або переривчастими лініями, то цю структуру або її частину слід вважати такою, що охоплює всі її стереоізомери. У цих кільцевих системах використовується така система нумерації: 91255 22 Сполуки та їх виготовлення Про інгібіторну активність (2R)-2'-деокси-2'фтор-2'-С-метилцитидину щодо HCV-полімерази повідомлялося в публікаціях (J. L. Clark et al., 1 Med. Chem. 2005 48(17) :5504-8; J. Clark U.S. Publication No. 2005/0009737), включених тут в усій їхній повноті шляхом посилання. У клінічній практиці бажано спочатку вводити високі дози І-6 для того, щоб швидко інгібувати HCV-полімеразу і тим самим знизити вірусні рівні в умовах, в яких стає мінімальною можливість вірусу мутувати і селектувати в стійкі штами. Але досягти достатньо високих рівнів з батьківським нуклеозидом може бути достатньо важко. Поліпшити фармакокінетичні та фізичні властивості сполуки і завдяки цьому оптимізувати її біодоступність обіцяє стратегія проліків. У публікації (U.S. Publication 2005/0009737) пропонуються загальні підходи до застосування проліків на основі 2'-деокси-2'-фтор-2'-метилових нуклеозидів. Проте поряд з підступною простотою проліків-кандидатів в уяві, на практиці ідентифікувати сполуки з підходящими фізикохімічними і фармакодинамічними властивостями, перетвореннями in vivo і профілем безпеки є складною, багатосторонньою справою, що потребує значного експериментування. При цьому на шляху виявлення та ідентифікації підходящих проліків для перорального постачання встають перешкоди у вигляді вимушеного пошуку компромісних рішень між підтримуванням достатньої розчинності у воді, ліпофільності і хімічної стабільності з одного боку і забезпеченням швидкого й ефективного вивільнення активного інгредієнта після введення препарату з іншого боку. Крім того, необхідно мінімізувати виведення проліків, зумовлене неестеразним метаболізмом і транспортом (K. Beaumont et al., Curr. DrugMetab. 2003 4(6):461485). Інгібування та індукція цитохромних Р450ферментів також може продукувати негативні міжмедикаментозні взаємодії, що є небажаними. Було встановлено, що нижчі алкілові діестери сполуки І6, подані в Табл. 1, значно поліпшують біодоступність цієї сполуки. Фармакокінетичні властивості потенційних проліків оцінювали в дослідах як на щурах, так і на мавпах, у намаганні мінімізувати внутрішньовидову варіантність і генетичні поліморфізми, котрі можуть призводити до внутрішньовидових артефактів. У тій мірі, в якій ферментативні трансформації є відповідальними за гідроліз естерних зв'язків, специфічна афінність до проліків може залежати від специфічної структури естерази і/або пептидази, котрі можуть каталізувати цю трансформацію. Активність естерази щура часто виявлялася значно вищою за активність людської естерази (J. A. 23 91255 24 Fix et al., Pharm. Res. 1990 7(4):384-387; W. Li et al., Antimicrob. Agents Chemother. 1998 42(3):647653). Іншим потенційно важливим параметром було біоперетворення деаміназою цитидинової основи на уридин. Хоча трифосфати цитидину й уридину є інгібіторами полімерази, фосфориляція уридину in vivo була нефективною порівняно з цитидином. Таким чином, підвищені рівні уридину слід вважати небажаними. У роботі (J. Clark et al., J. Med. Сhеm., supra) повідомлялося про недостатню ефективність уридинової похідної в реплікації HCV-вірусу. Було виявлено, що С2-5 алкілові етери двоосновних кислот І-6 володіють чудовими властивостями, очікуваними від проліків. Значно вищі рівні фторованого нуклеозиду спостерігалися у крові як щурів, так і мавп. Крім того, кількісне співвідношення між цитидином та уридином у фторованій основі був вищим в обох видах, коли нуклеозид вводився у формі його етеру двоосновної кислоти. Етери двоосновних кислот здатні утворювати два різні моноетери і неетерифікований нуклеозид. Фармакокінетичний аналіз у цій ситуації може бути складним, якщо всі види будуть наявними у крові у значних концентраціях. Наявність великої кількості метаболітів у крові додає труднощів у визначенні того, чи всі проліки є безпечними. Але гідроліз обох естерів є на подив достатньо легким, 25 91255 і єдиним метаболітом, який спостерігався у крові у значних кількостях, окрім батьківського нуклеозиду, був 3'-моноетер, який швидко перетворювався на сполуку I-6. У цілях подальшої оцінки властивостей імовірних проліків та можливості їх застосування для лікування людей досліджували їх транспортування через Сасо-2-клітини. Клітини Сасо-2 зазвичай використовуються для оцінки очікуваних поглинання і проникності молекул (G. Gaviraghi et al., in Pharmacokinetic optimization in drug research. Biological, Pharmacokinetic and Computational Strategies. B. Testa et al., eds. Wiley Interscience VCH, Zurich 2001 pp. 3-14). За результатами цих 26 досліджень проникність Сасо-2-клітин була доступною для С2.5 алкілових етерів двоосновних кислот І-6. Окрім ефективного біоперетворення in vivo проліки для перорального введення повинні також володіти прийнятними для готування ліків фізикохімічними властивостями і забезпечувати поглинання із кишки у відділ, де може відбуватися бажане біоперетворення. Особливо важливими з цього погляду є розчинність у воді, коефіцієнт розподілу і стабільність у шлункових та інтестинальних рідинах. Величини цих параметрів наведені в Табл. 2. Таблиця 2 I-1 І-2 І-3 І-4 I-5 І-6 KO/W1 3,39 2,33 1,27 3,00 2,20 -0,94 log D2 ND7 1,67 0,9 ND ND ND Розчинність3 0,016 0,186 3,78 0,0139 0,13 76 SGF4 Т1/2(год.) 37°C 18,6 25,1 9 >711 >10,411 >1211 SIF5 ТГ1/2(год.) 37°C >8,411 866 72 >1511 >1111 >1211 1. Розрахований коефіцієнт розподілу октанол/вода 2. Визначений шляхом вимірювань експериментальний коефіцієнт розподілу при рН7,4 3. Розчинність у водному середовищі, воді і буфері рН 6,5, мг/мл 4. Стабільність у модельованій шлунковій рідині (SGF; рН 1,2) 5. Стабільність у модельованій інтестинальній рідині (SIF; рН 7,4) 6. Розчинність у SGF становить 13,3 мг/мл 7. ND: Не визначено 8. Середовищем є SIF, розчинність у SGF становить 13,6 мг/мл 9. Середовищем є SIF, розчинність у SGF становить 0,16 мг/мл 10. Вимірювання проводили у воді і буфері з рН 6 11. Була помічена незначна деградація упродовж даного часу 12. Визначено при рН 5,5 Коефіцієнт розподілу масло-вода PO/W (oilwater) (коефіцієнтом clogP є розрахований коефіцієнт PO/W) є властивістю, важливою для ліків перорального введення. Лікувальна субстанція повинна мати достатню розчинність у воді для її розчинення у препараті та рідинах шлунковокишкового тракту (Gl: gastrointestinal) і контактування з ендотеліальними клітинами у шлунку та кишечнику, а також розчинністю ліпідів, достатньою для проходження через двошарову ліпідну мембрану цих клітин і далі у кров. Оптимальні величини log Ρ сполуки для пероральної біодоступності лежать в інтервалі від 1 до 3 і демонструються сполуками згідно з даним винаходом. Символ PO/W означає коефіцієнт розподілу октанол/вода (octanol/water). Вираз clogP означає розрахований коефіцієнт PO/W. Комп'ютерна програма для обчислювання коефіцієнта PO/W є легко доступною для фахівців у галузі фармацевтики та медичної хімії. Використовуваний у даному описі термін "експериментальний коефіцієнт розподілу" означає експериментально визначений коефіцієнт розподілу між октанолом і буферизованим водним розчином. Розрахований коефіцієнт розподілу та експериментальний коефіцієнт розподілу у загальному випадку єподібними один одному, але останній залежить від величини рН водного розчину, у той час як PO/W від величини рН не залежить. Розчинність у воді призначених для перорального введення проліків повинна для оптимального фармацевтичного препарату бути не меншою 0,1 мг/мл, а період напіввиведення у модельованій шлунковій рідині і модельованій інтестинальній рідині повинен становити, відповідно, 1-2 год. і 2-4 год. для того, щоб забезпечувався достатній час проходження через шлунок і поглинання в кишечнику. Сполуки згідно з даним винаходом у кращому варіанті одержують в одностадійному процесі шляхом ацилування сполуки І-6 у водноорганічному розчиннику. Таким розчинником може бути або гомогенний водний розчин або двофазний розчин. Величину рН водно-органічного розчинника підтримують на рівні вище 7,5 шляхом додавання основи для нейтралізації кислоти, що виробляється внаслідок ацилування. Підходящою для цього основою може бути або гідроксид лужного чи лужноземельного металу або третинний амін. Реакцію проводять при наявності диметиламінопіридину (DMAP), відомого в даній галузі каталізатора процесів ацилування. Перевагою запропонованого даним винаходом процесу є те, що за його допомогою цільовий продукт можна одержу 27 вати без ацилування гетероциклічної основи. При цьому не потребується застосовувати захисні групи, а отже і виключити із процесу стадію захисту та зняття захисту. Запропонований процес докладно описаний нижче, у прикладах здійснення даного винаходу. Дозування і введення Сполуки згідно з даним винаходом можуть готуватися у різноманітних лікарських формах і з різноманітними носіями для перорального введення. Такими лікарськими формами можуть бути таблетки, таблетки з покриттям, тверді і м'які желатинові капсули, розчини, емульсії, сиропи або суспензії. Сполуки згідно з даним винаходом є ефективними при їх уведенні також у формі супозиторіїв та іншими способами. Найбільш вигідним способом їх уведення у загальному випадку є пероральний за підходящою денною схемою прийому, яка може регулюватися відповідно до тяжкості хвороби та реакції пацієнта на антивірусні препарати. Сполука або сполуки згідно з даним винаходом, а також їхні фармацевтично прйнятні солі, разом з одним чи більше звичайними ексципієнтами, носіями або розріджувачами можуть поміщатися у форми фармацевтичних складів і стандартних доз. У форми фармацевтичних складів і стандартних доз можуть входити звичайні інгредієнти у звичайних пропорціях з додатковими активними сполуками або без них, а стандартні лікарські форми можуть містити будь-яку підходящу ефективну кількість активного інгредієнта, яка відповідає заданому інтервалу денної дози. Фармацевтичні склади можуть мати тверді форми таблеток або заповнених капсул, напівтвердих препаратів, порошків, препаратів пролонгованої дії, рідкі форми суспензій, емульсій або заповнених капсул для перорального вживання, або форму супозиторіїв для ректального або вагінального введення. Типовий препарат при цьому містить приблизно від 5%(мас.) до 95%(мас.) активної сполуки або сполук. Використовувані тут терміни "препарат" і "лікарська форма" можуть означати як тверді, так і рідкі препарати з активною сполукою. При цьому для фахівця в даній галузі цілком зрозуміло, що активний інгредієнт може існувати у формах різних препаратів залежно від бажаної дози і фармакокінетичних параметрів. Використовуваний тут термін "ексципієнт" означає сполуку, яку використовують для виготовлення фармацевтичного складу і яка в загальному випадку є безпечною, нетоксичною, та ні біологічно ні іншим чином не є небажаною. Крім того, цей термін охоплює своїм значенням також ексципієнти, що є доступними для застосування у ветеринарії, а також у фармацевтиці для людей. Сполуки згідно з даним винаходом можуть уводитися відокремлено, але в загальному випадку призначаються для введення в суміші з одним чи більше підходящими фармацевтичними ексципієнтами, розріджувачами чи носіями, вибраними з урахуванням наміченого шляху введення і стандартної фармацевтичної практики. "Фармацевтично доступна сольова" форма активного інгредієнта також може надавати актив 91255 28 ному інгредієнту початкову бажану фармакокінетичну властивість, яка є відсутньою в його несольовій формі і навіть може позитивно впливати на фармакодинаміку активного інгредієнта в тому, що стосується його терапевтичної активності в організмі пацієнта. Вираз "фармацевтично доступна сіль" сполуки означає сіль, яка є фармацевтично доступною і володіє бажаною фармакологічною активністю батьківської сполуки. Такими солями можуть бути: (1) солі приєднання кислот, утворені з неорганічними кислотами, такими як соляна кислота, бромистоводнева кислота, сірчана кислота, азотна кислота, фосфорна кислота тощо; або з органічними кислотами, такими як гліколева кислота, піровиноградна кислота, молочна кислота, малонова кислота, яблучна кислота, малеїнова кислота, фумарова кислота, винна кислота, лимонна кислота, 3-(4-гідроксибензоїл)бензойна кислота, корична кислота, міндальна кислота, метансульфонова кислота, етансульфонова кислота, 1,2-етандисульфонова кислота, 2гідроксіетансульфонова кислота, бензолсульфонова кислота, 4-хлорбензолсульфонова кислота, 2-нафталінсульфонова кислота, 4толуолсульфокислота, камфоросульфонова кислота, лаурилсірчана кислота, глюконова кислота, глутамінова кислота, саліцилова кислота, муконова кислота і т. п. Цілком зрозуміло, що будь-яке згадування в даному описі фармацевтично доступних солей передбачає включення в нього також форми приєднання розчинника (сольвати) та кристалічні форми (поліморфи) згаданої солі приєднання кислоти відповідно до даних тут визначень. Твердими препаратами можуть бути, наприклад, порошки, таблетки, пілюлі, капсули, супозиторії та здатні диспергуватися гранули. Твердим носієм може бути одна чи більше речовин, які можуть бути також розріджувачами, коригентами смаку та запаху, соліюбілізаторами, мастилами, суспепндувальними агентами, сполучними, консервантами, дезінтеграторами таблетки, інкапсулювальним матеріалом тощо. У порошковому препараті носій, зазвичай, являє собою тонко диспергований твердий матеріал, що є сумішшю з тонко диспергованим активним компонентом. У таблетованому препараті активний компонент, як правило, є змішаним з носієм, який володіє необхідною зв'язуючою здатністю, у потрібних пропорціях і скомпактований у бажаних формі та розмірах. Підходящими носіями можуть бути, наприклад, карбонат магнію, стеарат магнію, тальк, цукор, лактоза, пектин, декстрин, крохмаль, желатин, трагакант, метилцелюлоза, натрійкарбоксиметилцелюлоза, низькоплавкий віск, масло какао і т. п. Препарати у твердих формах можуть містити, окрім активного компонента, барвники, коригенти смаку та запаху, стабілізатори, буфери, штучні та натуральні підсолоджувані, диспергатори, згущувачі, солюбілізатора тощо. Рідкі препарати, котрі також є підходящими для перорального введення, зазвичай мають форму емульсій, сиропів, еліксирів і водних суспензій. У первинному стані вони можуть мати також тверду форму, розраховану на перетворення її в рідку форму безпосередньо перед вживанням. Емульсії 29 можуть готуватися в розчинах і зокрема, наприклад, у водо-пропіленгліколевих розчинах, або ж можуть містити емульгатори, такі як лецитин, сорбітмоноолеат або гуміарабік. Водні суспензії можуть готуватися шляхом диспергування тонко диспергованого активного компонента у воді з в'язким матеріалом, таким як натуральна або синтетична смола, каучук, метилцелюлоза, натрійкарбоксиметилцелюлоза та інші добре відомі суспендувальні речовини. Сполуки згідно з даним винаходом можуть бути приготовані у препаратах для введення у формі супозиторіїв. Для цього низькоплавкий віск, наприклад, із суміші гліцеридів жирних кислот або масла какао спочатку розплавляють, і в ньому, наприклад, шляхом перемішування гомогенно розподіляють активний компонент. Після цього розплавлену гомогенну суміш виливають у відповідних розмірів форми та дають охолонути і затвердіти. Сполуки згідно з даним винаходом можуть бути приготовані у препаратах для вагінального введення. Такі препарати можуть мати форму вагінальних супозиторіїв, тампонів, кремів, гелей, паст, піни або спреїв, які окрім активного інгредієнта містять відповідні носії. Підходящі препарати, а також відповідні фармацевтичні носії, розріджувачі та ексципієнти описані в публікації (Remington: The Science and Practice of Pharmacy 1995, edited by E. W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania). Досвідчені науковці в галузі фармацевтики можуть створювати різноманітні варіанти розглянутих у даному описі препаратів відповідно до вибраних способів введення не виходячи при цьому за межі стабільності і не погіршуючи терапевтичної активності складів, запропонованих даним винаходом. Фахівці у даній галузі зможуть дуже легко модифікувати сполуки згідно з винаходом, наприклад, для надання їм більшої розчинності у воді або іншому носії шляхом незначних змін (наприклад, їхнього сольового складу). Можна буде також змінювати спосіб уведення і схему дозування тої чи іншої конкретної сполуки для регулювання її фармакокінетики і, таким чином, досягнення максимального корисного ефекту у пацієнтів. Використовуваний тут термін "терапевтично ефективна кількість" означає кількість, потрібну для придушення симптомів хвороби у пацієнта. Дозу регулюють відповідно до індивідуальних потреб у кожному конкретному випадку. Величина дози і режим дозування можуть варіювати в широких межах в залежності від цілого ряду факторів і, зокрема, від ступеню тяжкості хвороби, віку i загального стану здоров'я пацієнта, вживання пацієнтом інших медикаментів, вибраного способу i форми введення даної сполуки, від практичного досвіду лікаря-куратора i методик лікування, яким він віддає перевагу, тощо. Як при монотерапи, так i при комбінаційній терапії денна доза при пероральному введенні лежить в інтервалі приблизно від 0,1 до 10 г. Кращими є дози в інтервалі приблизно від 0,5 до 7,5 г на день, а ще кращими - від 1,5 до 6,0 г на день. У загальному випадку, курс лікуван 91255 30 ня сполуками згідно з винаходом починають великою "ударною дозою" для швидкого придушення або ліквідації вірусу, після чого дозу знижують до рівня, достатнього для запобігання рецидиву інфекції. Фахівці у галузі лікування зазначених тут хвороб зможуть без зайвого експериментування, i ґрунтуючись на власному досвіді, а також на інформації, поданій у даному описі, встановлювати терапевтично ефективну кількість сполук згідно з даним винаходом для конкретних хвороб i пацієнтів. Терапевтична ефективність може встановлюватися за результатами тестів функціонування печінки i зокрема, наприклад, за рівнями білків, таких як сироваткові білки (наприклад, альбумін, фактори згортальної системи крові, лужна фосфатаза, амінотрансферази (наприклад, аланінтрансаміназа, аспартаттрансаміназа), 5'-нуклеозидаза, -глуамінілтранспептидаза тощо), синтезом білірубіну, синтезом холестерину i синтезом жовчної кислоти, за метаболічною функцією печінки i зокрема, наприклад, вуглеводному обміну, метаболізму амінокислот та аміаку. Інакше терапевтична ефективність може контролюватися шляхом вимірювання РНК HCV-вірусу. За результатами цих тестів можна встановлювати оптимальні дози. Запропонована активна сполука або м сіль може вводитися в комбінації з іншим антивірусним засобом, таким як рибавірин, інший нуклеозидний інгібітор HCV-полімерази, ненуклеозидний інгібітор HCV-полімерази, інгібітор HCV-протеази, інгібітор HCV-гелікази або інгібітор злиття HCV-вірусу. Коли активна сполука або її похідна чи сіль вводиться в комбінації з іншим антивірусним засобом, її активність може бути підвищеною порівняно з батьківською сполукою. При застосуванні комбінаційної терапії введення додаткових ліків може відбуватися одночасно або послідовно по відношенню до нуклеозидних похідних. Використовуваний тут термін "одночасне введення" може означати як введення лікувальних речовин в один i той самий момент часу, так i ведення їх в різні моменти часу. Введення двох чи більше лікувальних засобів в один i той самий момент часу може здійснюватися в одному препараті, що містить два чи більше активних інгредієнтів, або шляхом майже одночасного введення двох чи більше лікарських форм з одним активним засобом. Цілком зрозуміло, що під терміном "лікування" в даному описі маються на увазі також профілактика та лікування існуючих патологічних станів. Крім того, термін "лікування" стану, викликаного HCV-інфекцією означає як лікування або профілактику хвороби чи патологічного стану, асоційованих з HCV-інфекцією або опосередкованих нею, так і лікування їхніх клінічних симптомів. Приклад 1 (2R,3R,4R,5R)-5-(4-Аміно-2-оксо-2H-піримідин1-іл)-4-фтор-4-метил-3-пропіонілокситетрагідрофуран-2-ілметиловий естер пропіонової кислоти (І-3) 31 91255 32 До суспензії сполуки І-6 (30 г, 0,116 моль) , DMAP (1,4 г, 11,57 ммоль) у THF ( 300 мл) і води (150 мл) додали TEA (35,1 г, 0.347 моль), у результаті чого утворився світлий розчин (рН ~11). Приготовану таким чином реакційну суміш охолодили до 5-10° С і до цієї перемішуваної двофазної реакційної суміші по краплях додали пропіоновий ангідрид (30,1 г, 0,231 моль). За результатами моніторингу рН її величину підтримували в інтервалі рН 11-12 шляхом одночасного додавання розчину KOH. Контроль розвитку реакції вели за допомогою HPLC-хроматографічного аналізу і після додавання пропіонілхлориду реакційна суміш містила приблизно 52% етеру двоосновної кислоти, 30% моноетерів і 15% сировинного матеріалу. До цієї суміші в описаних вище умовах по краплях додали ще пропіонового ангідриду (45,2 г, 0,347 моль). Приготовану таким чином реакційну суміш залишили на ніч відстоюватися без перемішування. HPLC-хроматографічний аналіз органічної фази показав приблизно 96% етеру двоосновної кисло ти і приблизно 2% моноетеру. Фази суміші розділили, і водну фазу двічі екстрагували тетрагідрофураном (100 мл). Об'єднані органічні фази промили сольовим розчином. Органічну фазу профільтрували, піддали випарюванню, і залишок розчинили у воді (приблизно 500 мл), а потім розбавили ІРА (приблизно 100 мл). Оброблену таким чином суміш повільно, при перемішуванні охолодили до навколишньої температури. Осад, що утворився, відфільтрували, промили водою і гептаном, присушили при температурі приблизно 60°С у вакуумі протягом приблизно 60 год. і в результаті одержали 3,45 г (80,3%) сполуки І-3, яка за результатами HPLC-хроматографії мала чистоту 98,75%. Приклад 2 (2R,3R,4R,5R)-5-(4-Аміно-2-оксо-2Н-піримідин1-іл)-4-фтор-3-ізобутирилокси-4метилтетрагідрофуран-2-ілметиловий естер ізомасляної кислоти (І-2) До охолодженої на льоду суспензії сполуки I-6 (970 г, 3,74 моль) і DMAP (50 г, 0,412 моль) у THF (10 л) додали TEA (2,3 кг, 16,5 моль) і воду (7 л), у результаті чого утворився світлий розчин. До цієї суміші при перемішуванні, і підтримуючи її температуру на рівні приблизно 0°С, повільно додали ізобутирилхлорид (3 еквіваленти). Після цього додавали ще 1,2 екв., а потім 0,7 екв. Ізобутирилхлориду, аж поки HPLC-хроматографічний аналіз не показав практично повне завершення реакції (загальна кількість приблизно 1,95 кг). Приготовану таким чином реакційну суміш підкислили концентрованою НСІ до величини рН ~6,4, і органічну фазу промили етилацетатом EtOAc (2x10 л). Об'єднані екстракти промили водою (1x15 л). Органічну фазу профільтрували і концентрували у вакуумі. Залишок розчинили в ІРА (приблизно 20 кг), і до розчину додали гептан (14,2 кг). Після цього роз чин нагріли до температури приблизно 74-75°С, в результаті чого розчин став світлим, а потім приблизно 5 л видалили шляхом дистиляції. Далі розчин повільно охолодили до кімнатної температури. При температурі приблизно 42-43°С утворився преципітат. Охолодження розчину повільно продовжували до 5°С, а потім його перемішували протягом ночі. Утворену тверду речовину відфільтрували і фільтрат промили сумішшю ІРА/гептан (1:8) (13,4 кг), просушили у вакуумі при температурі 6070°С і в результаті отримали 1,295 кг (86,65%) сполуки І-2, яка за результатами HPLCхроматографії мала чистоту 99,45%. Приклад 3 (2R,3R,4R,5R)-5-(4-Аміно-2-оксо-2F-піримідин1-іл)-4-фтор-2-ізобутоксикарбонілоксиметил-4метилтетрагідрофуран-3-іловий естерізобутиловий естер вугільної кислоти (І-4) 33 Суспензію сполуки І-6 (700 мг), DMAP (33 мг) у THF (7 мл) розбавили сольовим розчином (7 мл). До цього розчину додавали розбавлений NaOH доти, аж поки не була досягнута величина рН ~11. Приготовану таким чином двофазну реакційну суміш охолодили в льодяній бані і до неї в умовах перемішування, і підтримуючи рН на рівні 11 добавками за потребою NaOH, по краплях добавили ізобутилхлорформіат (1,11 г). HPLC-аналіз показав, що більшість бікарбонату була забруднена ~15% монокарбонатів. До цього охолодженого на льоду розчину по краплях додали ще 1 екв. ізобутилхлорформіату. HPLC-аналіз показав майже повне перетворення первинного матеріалу. Отриману таким чином суміш залишили відстоюватися протягом ночі при кімнатній температурі. Після цього до неї додали EtOAc (приблизно 50 мл) і величину рН водної фази відрегулювали на рівень ~7,5 концентрованою НСІ. Фази розділили і органічну фазу промили водою (тричі) та піддали випарюванню до сухого стану, отримавши в результаті безколірну тверду речовину (приблизно 1,22 г). Цю тверду речовину розчинили в гарячому ацетоні, в результаті чого утворився світлий розчин, який повільно охолодили до кімнатної температури, у результаті чого утворилася тверда маса. Цю тверду речовину розбавили ІРА (приблизно 20 мл), внаслідок чого утворилася суспензія, яку профільтрували, а потім промили послідовно ІРА і гептаном та просушили, одержавши 0,85 г (68,5%) сполуки І-4, чистота якої за результатами HPLCхроматографії становила 97,5%. Приклад 4 Гідрохлорид(2R,3R,4R,5R)-5-(4-Аміно-2-оксо2Н-піримідин-1-іл)-4-фтор-4-метил-2пропоксикарбонілохуметилтетрагідрофуран-3іловий естер-пропілового естеру вугільної кислоти (І-5) До суспензії сполуки І-6 (700 мг, 2,70 ммоль), DMAP (33 мг, 0,27 ммоль) у THF (7 мл) і розбавленому сольовому розчині (7 мл) для встановлення її рН на ~11 додали розбавлений KOH. Утворену таким чином двофазну реакційну суміш охолодили до ~5°С і до неї по краплях при перемішуванні додали n-пропілхлорформіат (1,0 г). HPLC-аналіз показав утворення цільового продукту разом із ~20% монокарбонатів. До холодного розчину добавили ще дві кількості пропілхлорформіату (2x1 еквівалент) поки HPLC-аналіз не показав майже повне завершення реакції. Цю реакційну суміш розбавили EtOAc (30 мл) і величину рН водної фази відрегулювали на рівень ~6,5 концентрованою НСІ. Фази розділили й органічну фазу проми 91255 34 ли водою (тричі) та піддали випарюванню до сухого стану, отримавши в результаті безколірну тверду речовину (приблизно 1,1 г). Гарячий ІРА розчин підкислили 4 н. розчином НСІ (приблизно 1 мл) і піддали випарюванню до сухого стану. Утворену в результаті тверду речовину повторно розчинили в гарячому EtOH (приблизно 115 мл) і перемішували протягом ночі при кімнатній температурі. Утворилася тверда маса, яку відфільтрували, і залишок промили сумішшю МеОН/гептан (1:1). Тверду речовину, що залишилася, висушили у вакуумі при температурі приблизно 60°С, одержавши 0,325 г (25,8%) сполуки І-5, чистота якої за результатами HPLC-хроматографічного аналізу становила 97,5%. Приклад 5 Визначення фармакокінетичних параметрів у щурів У цьому експерименті використовували інтактних самців щурів IGS Wistar Han Crl:WI(GLx/BRI7Han)IGS BR (Hanover-Wistar) масою тіла 200-250 г. По кожному рівню дози експериментальної сполуки використовували групу із трьох щурів. Тваринам забезпечували нормальний доступ до їжі і води протягом усього експерименту. Випробувану речовину готували у формі водної суспензії, що містила Captex355EP, Capmul МСМ, EtOH і пропіленгліколь (30:20:20:30) з еквівалентом дози до 10 мг/кг сполуки І-6 і вводили перорально через шлунковий зонд. У щурів, котрі отримували випробувану речовину, відбирали зразки крові (0,3 мл) у 0,25, 0,5, 1, 3, 5 і 8 год. із яремної канюлі і в 24 год. шляхом серцевої пункції. До зразків добавляли оксалат калію і NaF і зберігали на льоду протягом всієї процедури відбирання зразків. Підготовані зразки одразу крутили на охолодженій центрифузі при температурі -4°С, й отримані зразки плазми зберігали до їх аналізу в морозильній камері при температурі -80° С Аліквоти плазми (0,05 мл) змішували з 0,1 мл ацетонітрилу. До них додавали внутрішній стандарт (0,05 мл у воді) і 0,02 мл контрольного розчинника. Крім того, готували набір калібрувальних стандартів шляхом змішування 0,05 мл аліквоти плазми від необроблених щурів (що не отримували випробуваної сполуки) з 0,1 мл ацетонітрилу, 0,02 мл аліквотами стандартного розчину у суміші метанол:вода (1:1) і 0,05 мл аліквотами внутрішнього стандарту у воді. Кожний зразок плазми і кожний калібрувальний стандарт піддавали ретельному вихровому перемішуванню, а потім центрифугували зі швидкістю 3000 об./хв. упродовж 5 хв. для осадження білка. Супернатант (по 100 мкл кожно 35 91255 го) після центрифугуваня переносили у 96лунковий планшет, в якому містилося 200 мкл водної рухомої фази для LC/MS/MS аналізу. Аналіз зразків Аналіз проліків здійснювали методом рідинної хроматографії високої розрізнювальної спроможності з подвійною мас-спектрометрією (HPLC/MS/MS). Для розділяння використовували колонку Thermo Aquasil C18 4,6x50 мм (5 мкМ). Іонізацію здійснювали методом розпорошування (ESI: electrospray ionization). Рухома фаза А містила 5 мМ ацетату амонію у воді з 0,1% мурашиної кислоти, а рухома фаза В містила МеОН з 0,1% мурашиної кислоти. Елюювання проводили з таким градієнтом при витраті 1 мл/хв.: Час 0 хв. 0,5 хв. 0,8 хв. 1,5 хв. 2,8 хв. 3,8 хв. 3,9 хв. 5 хв. А, % 95 95 85 85 10 10 95 95 В,% 5 5 15 15 90 90 5 5 Приклад 6 Визначення фармакокінетичних параметрів у дослідах на мавпах В експерименті використовували трьох самців мавп Cynomolgus з масою тіла 8-10 кг. Тваринам забезпечували нормальний доступ до їжі і води протягом усього експерименту. У час уведення ліків і в час негативної реакції реєстрували масу тіла тварин. Випробувану субстанцію готували у формі водної суспензії, яка містила гіпромелозу (2910, 50 цикл/с), Фармакопея США, полісорбат 80, NF, бензиловий спирт, NF (5,0, 4,0 і 9,0 мг/мл) і стерильну воду (в кількості, достатній для одержання 1,0 мл), з еквівалентом дози до 10 мг/кг сполуки І-6 і 0,5 мл/кг, і вводили пероральним способом через шлунковий зонд. У моменти часу 0, 0,083, 0,25, 0,5, 1, 3, 5, 8 і 24 год відбирали по одному зразку крові (0,5-1,0 мл). Обробку та аналіз зразків проводили так, як описано в експерименті з щурами. У кожної мавпи перед уведенням дози і в 0-8 год. брали 5 мл зразок сечі. Зразки сечі зберігали при температурі -80°С й аналізували методом LC/MS/MS. Стандартні криві будували за зразками плазми контрольної мавпи, що містили NaF та оксалат калію. Приклад 7 Методика досліджень на Сасо-клітинах Матеріали Порошки буфера Кребса-Хензеляйта (KrebsHenseleit), дигідрату хлориду кальцію і бікарбонату натрію були придбані у фірми Sigma (Сент-Луїс, шт. Міссурі). Сасо-2-клітини (пасаж ~100) були отримані від фірми Roche Basel. Високомодифіковане за способом Дульбеко середовище Ігла, 4-(2гідроксіетил)-1-піперазинетансульфонова кислота (HEPES), а бичача сироватка була отримана від фірми JRH Bioscience (Lenexa, шт. Канзас). Замінні амінокислоти мінімального підтримувального 36 середовища (MEM), L-глутамін, пеніцилін і стрептоміцин були отримані від фірми GIBCO Labs, Life Tech. LLC (Гранд-Айленд, шт. Нью-Йорк). Вставки клітинної культури Снепвела (Snapwell) (6,5 мм у 2 діаметрі, 1,12 см , розмір пор 0,4 мкм) були отримані від фірми Costar (Кембрідж, шт. Массачусетс). Клітинні культури Клітини вирощувалися у 75 см2 колбах і витримувалися при температурі 37°С і атмосфері із 5% СО2 і 95% повітря. Живильні середовища складалися із високомодифікованого за способом Дульбеко середовища Ігла, доповненого 5% бичачої сироватки, 25 мМ HEPES, 1% замінних амінокислот MEM, 1% L-глутаміну, 100 од./мл пеніциліну і 100 мкг/мл стрептоміцину. Культури кожного тижня ласувалися з індексом розведення 1-3. У дослідженнях проникності Сасо-2-клітини при 110-120 пасажів висівалися з густиною 400000 клітин/см2 на полікарбонатні фільтри Transwell у вставках Снепвела (Snapwell), після чого культивування клітин перед їх використанням тривало 7 днів. Буфер Кребса-Хензеляйта (Krebs-Henseleit) Бікарбонатний буфер Кребса-Хензеляйта, який містив 10 мМ глюкози і 2,5 мМ СаСІ2, регулювали на рівні рН 6,5 і 7,4 відповідно до інструкцій, вкладених у пакуванні. Порошкові солі розчиняли приблизно у 90% потрібного об'єму водою Millipore. До розчину додавали дигідрат хлориду кальцію і бікарбонат натрію, після чого регулювали його рН добавлянням 1 н. розчину НСІ або 1 н. розчину NaOH. Потім об'єм розчину доводили до кінцевого добавлянням води Millipore. Розчин стерилізували шляхом фільтрації через мембрану з 0,22 мікронними порами і до використання зберігали в холодильній шафі (~20°С). Готування клітин Диференційовані клітини отримувалися від центра постачання клітинних культур "Cell Culture Core Facility" і ставили їх на зрівноваження при 37°С в атмосфері 5% СО2 і 95% повітря. Вставки Снепвела (Snapwell), які мали моношари сасо-2клітин, промивали у зрівноваженому при 37°С буфері Кребса-Хензеляйта (Krebs-Henseleit) рН 7,4. Методика Клітинну вставку використовували як дифузійну камеру. Величини рН буфера КребсаХензеляйта (Krebs-Henseleit) у верхівковій і доннобічній камерах складали відповідно 6,5 і 7,4, а початкова концентрація субстрату на боці верхівки складала 100 мкМ. Клітини з випробуваною сполукою у верхівковій камері піддавалися попередньому інкубуванню впродовж приблизно 30 хвилин при температурі 37°С в атмосфері 5% СО2 і 95% повітря. Досліди починали, коли клітинні вставки з 100 мкМ сполуки у буфері Кребса-Хензеляйта (Krebs-Henseleit) з рН 6,5 переносили в новий планшент з попередньо зрівноваженим буфером у донно-бічній камері. Для аналізу відбирали зразки з боку донора у точці часу 0 хвилин і з обох боків донора і ресивера у точці часу 30 хвилин. Контроль після експерименту Для оцінки продуктивності дифузійної системи використовували барвник Люцифер-жовтий. Після останнього відбору зразків, що відповідали випро 37 буваним сполукам, до верхівкової камери додавали Люцифер-жовтий, щоб надати початкову концентрацію 100 мкМ. Через 60 хвилин інкубування із донної камери видаляли і піддавали аналізу 250 мкл її вмісту. Обчислення коефіцієнта проникності (Раpp). Рарр обчислювали за такою формулою: Papp V dC A C0 dt cm / sec У цій формулі V є об'єм (см3) розчину ресивера, А є площа поверхні (см2) вставки Снепвела (Snapwell), С0 є початкова концентрація (нМ), a dC/dt є зміна концентрації в камері ресивера в часі, тобто нахил (нМ/хв.) кривої концентрації в камері ресивера в часі. Концентрацію в кожній точці часу, в котрій проводили відбір зразка, коригували для врахування видаленої аліквоти або перенесення донорних вставок на нові планшети залежно від експерименту. Приклад 8 Фармацевтичні склади з досліджуваними сполуками для введення кількома способами готували так, як описано у Прикладі 8. Склад мокрого гранулярного препарату для перорального введення (А) Інгредієнт %(мас.) Усередині гранул Активний інгредієнт (І-2) 50,0 Маніт (Partech M200), Фармакопея 39,0 США Повідон K30, Фармакопея США 5,0 Натрійкроскармелоза, NF 4,0 Очищена вода, Фармакопея США Для грануляції Зовні гранул Кроскармелоза, Фармакопея США 2,0 Зазначені інгредієнти змішували, гранулювали і розподіляли у тверді желатинові капсули, що містили приблизно 500 мг активної сполуки. Склад для перорального введення (В) Інгредієнт %(мас.) Активний інгредієнт (I-2) 50,0 Маніт (Partech M200), Фармакопея США 30,0 Повідон K30, Фармакопея США 5,0 Натрійкроскармелоза, NF 9,5 Мікрокристалічна целюлоза 5,0 Стеарат магнію 0,5 Зазначені інгредієнти об'єднували і гранулювали, використовуючи як розчинник воду. Після цього препарат сушили і на таблетувальному стаКомп’ютерна верстка О. Гапоненко 91255 38 нку формували в таблетки, які містили приблизно 500 мг активної сполуки. Склад для перорального введення (С) Інгредієнт Активна сполука Карбоксиметилцелюлоза Метилпарабен Пропілпарабен Коригент смаку Барвники Дистильована вода Соляна кислота %(мас.) 6г 1г 0,15г 0,05 г 0,035 мл 0,5 мг Решта до 100 мл Скільки потрібно для встановлення рН 4 Зазначені інгредієнти змішували, утворюючи суспензію для перорального введення. Препарат у формі супозиторію (D) Інгредієнт Активний інгредієнт Поліетиленгліколь 1000 Поліетиленгліколь 4000 %(мас.) 1,0% 74,5% 24,5% Зазначені інгредієнти розплавляли разом, змішували на паровій бані і виливали у форми, які вміщали 2,5 г загальної маси. Ознаки даного винаходу розкриті в поданому вище описі та окреслені нижче у Формулі винаходу, виражені в їхніх специфічних формах або у визначенні засобу для виконання описаної функції, або способу чи процесу для досягнення описаного результату, відповідно до обставин, можуть окремо чи в будь-якій комбінації цих ознак використовуватися для здійснення даного винаходу в його різноманітних формах. Поданий вище детальний опис даного винаходу служить лише ілюстрацією та прикладом його здійснення, що дозволяють ясніше зрозуміти і повніше сприйняти його ідею та об'єм. Отже цілком зрозумілою є можливість внесення в наведені вище приклади різноманітних змін і модифікацій, які не виходять за межі об'єму, окресленого нижче у Формулі винаходу. Таким чином, зрозуміло також, що поданий вище опис має лише ілюстративну, а не обмежувальну ціль. У зв'язку з цим, об'єм даного винаходу визначається не поданим вище описом, а виключно Формулою винаходу разом з повним об'ємом еквівалентів, на які пункти Формули винаходу дають право. Усі цитовані в даній заявці патенти, патентні заявки і публікації включені цим в усій їхній повноті і для всіх цілей шляхом посилання в такій самій мірі, як би кожний із цих патентів, і кожна патентна заявка чи публікація були означені цим індивідуально. Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюAntiviral nucleosides
Автори англійськоюChun Byoung-Kwon, Clark Jeremy, Sarma Keshab, Wang Peiyuan
Назва патенту російськоюАнтивирусные нуклеозиды
Автори російськоюЧун Буйонг-Квон, Кларк Джереми, Сарма Кешаб, Ванг Пейюань
МПК / Мітки
МПК: A61K 31/7068, C07H 19/06, A61P 31/14
Мітки: антивірусні, нуклеозиди
Код посилання
<a href="https://ua.patents.su/19-91255-antivirusni-nukleozidi.html" target="_blank" rel="follow" title="База патентів України">Антивірусні нуклеозиди</a>
Заміщені дигідрохіназоліни, які мають антивірусні властивості

Номер патенту: 82519
Опубліковано: 25.04.2008
Автори: Зюссмаєр Франк, Ланг Дітер, Вунберг Тобіас, Хьюлетт Гай, Ціммерманн Хольгер, Гроссер Рольф, Ніколік Сузанне, Баумайстер Юдіт, Шое-Лооп Рудольф, Реєфшлєгер Юрген, Єске Маріо, Бетц Ульріх, Кельденіх Йорг, Нелль Петер, Хеннінгер Керстін, Лампе Томас
МПК: A61P 31/22, C07D 239/84
Мітки: дигідрохіназоліни, мають, антивірусні, властивості, заміщені
Формула / Реферат:
1. Сполука формули:
3-b-d-рибофуранозилтіазоло[4,5-d]піримідинові нуклеозиди та їх застосування

Номер патенту: 79764
Опубліковано: 25.07.2007
Автори: Руден Ерік Дж., Леннокс Джозеф Р., Аверетт Деврон Р., Веббер Стефен І.
МПК: A61P 37/02, A61K 31/7064, C07H 19/24
Мітки: застосування, 3-b-d-рибофуранозилтіазоло[4,5-d]піримідинові, нуклеозиди
Формула / Реферат:
(21) 20040604959 (57) 1. Сполуки, представлені Формулою І: , (I)де:R1 являє собою незалежно Н, -C(O)R3 або рацемічну, L- або D-амінокислотну групу -C(O)CHNH2R4, де R3 являє собою заміщений або незаміщений алкіл, а R4 являє собою Н або заміщений або незаміщений алкіл;R2 являє собою Н, OR5 або N(R6)2, де R5 являє собою незалежно Н...
Піперазинові проліки і заміщені піперидинові антивірусні засоби

Номер патенту: 88297
Опубліковано: 12.10.2009
Автори: Пек Шон К., Жанг Жонксінг, Саундарараджан Начімуту, Ліхі Девід Кеннет, Уеда Ясутсугу, Мінвелл Ніколас А., Кадоу Джон Ф., Сірар П'єр, Лєвеск Катя, Тораваль Домінік, Чен Чунг-Пін Х., Конноллі Тімоті П., Ванг Тао, Юнг Кап-Сун
МПК: A61P 31/18, A61K 31/437, C07D 471/04, C07F 9/09
Мітки: піперидинові, заміщені, антивірусні, піперазинові, проліки, засоби
Формула / Реферат:
1. Сполука за Формулою І, IдеX являє собою С або N, за умови, коли X має значення N, R1 не існує;W являє собою С або N, за умови, коли W має значення N, R2 не існує;V являє собою С;R1 являє собою водень, метокси або галоген; R2 являє собою водень;R3 являє собою метокси або гетероарил, кожен з яких може бути незалежно...
Нуклеозиди з біциклічною цукровою складовою, олігонуклеотид та антивірусна фармацевтична композиція

Номер патенту: 61997
Опубліковано: 15.12.2003
Автор: Ванг Гуанжі
МПК: C07H 19/10, C07G 3/00, C07H 21/00, C07H 19/06, C07H 19/20, C07H 19/04, C07H 19/16
Мітки: нуклеозиди, антивірусна, біциклічною, складовою, фармацевтична, цукровою, олігонуклеотид, композиція
Формула / Реферат:
1. Нуклеозиди з біциклічною цукровою складовою, які мають загальну формулу,де Z являє собою метилен,R1 є незалежно вибраним з групи, що містить аденін, цитозин, гуанін, гіпоксантин, урацил, тимін;R2, R3 є незалежно вибраними з групи, що містить Н, ОН, DMTO, TBDMSO, ВnО, ТНРО, АсО, BzO, OP(NiPr2)O(CH2)2CN, ОРО3Н, дифосфат, трифосфат; R2 та R3 разом можуть бути PhCHO2, TIPDSO2 або DTBSO2.2. Олігонуклеотид, що...
Нуклеозиди з модифікованими цукрами та олігонуклеотиди

Номер патенту: 45362
Опубліковано: 15.04.2002
Автори: Рамасамі Кандасамі, Ванг Гуанжі, Зайферт Уілфрід
МПК: C07H 21/00, C12N 15/09, A61K 31/7064, A61P 31/00, A61P 31/12, C07D 473/00, A61K 31/7072, A61K 31/7052, A61K 31/7076, A61P 35/00, A61K 31/7042, A61K 31/7068, A61K 48/00, A61K 31/70, A61K 31/708, A61K 31/7088, C07H 19/10, A61K 31/52, C07H 19/16, A61P 29/00, C07D 239/54, C07H 19/06
Мітки: модифікованими, нуклеозиди, олігонуклеотиди, цукрами
Формула / Реферат:
1. Нуклеозиды с модифицированными сахарами общей формулыгде В - нуклеозидное основание, такое, как тимин, любая арильная составляющая R'1, R'3, R'4 и R'5 представляет собой фенил, полицикл или гетероцикл;R2 - Н, X - S, О, СН2,и, если(I) R3 и R5 независимо выбраны из группы, состоящей из ОН, 2-цианоэтил-(N, N-диизопропил) фосфорамидитной или гидроксиблокирующей группы, то(A) Х представляет S,...
Попередній патент: Спосіб безперервного або періодичного добування металу або декількох металів зі шлаку, який містить зазначений метал або сполуку зазначеного металу
Наступний патент: Спосіб виробництва настою спиртового для напоїв
Випадковий патент: Напівпровідниковий фотоприймач з регульованою спектральною характеристикою світлоструму