Фармацевтична композиція та спосіб її отримання
Номер патенту: 110882
Опубліковано: 25.02.2016
Автори: Лі Син Хьон, Лім Хюн Те, Кхім Те Сун, Схін Че Соо, Кхім Тон Йон, Сун Чун Хо







Формула / Реферат
1. Фармацевтична композиція, яка містить: гранулу, яка містить 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид як активний інгредієнт та поверхнево-активну речовину, яка являє собою щонайменше один тип матеріалу, вибраний із групи, що включає рицинову олію, лецитин (яєчний) та жирно кислотний макроголгліцерид.
2. Фармацевтична композиція за п. 1, де фармацевтична композиція складена у гранули, порошки, пігулки, капсули або рідку суспензію, при цьому містить активний інгредієнт у кількості від 20 мг до 1000 мг.
3. Фармацевтична композиція за п. 1, де активний інгредієнт має середній розмір частинок приблизно 50 мкм або менше.
4. Фармацевтична композиція за п. 1, де рицинова олія вибрана з групи, що включає поліетиленгліколю гліцерингідростеарат, кремофор RH 40 або кремофор RH 60.
5. Фармацевтична композиція за п. 1, де жирнокислотний макроголгліцерид являє собою Gelucire 44/14.
6. Фармацевтична композиція за п. 1, де фармацевтична композиція додатково містить наповнювач, який являє собою щонайменше один тип матеріалу, вибраний із групи, що включає целюлозу, її похідне, у тому числі мікрокристалічну целюлозу, гідроксипропілцелюлозу, гідроксіетилцелюлозу, гідроксипропілметилцелюлозу, гідрат лактози, ангідрат лактози, дикальційфосфат, легкий кремнієвий ангідрид, крохмаль, маніт, полівінілпіролідон, поліетиленгліколь та еудрагіт.
7. Фармацевтична композиція за п. 6, де наповнювач застосований у кількості від 0,01 до 60 масових часток відносно 100 масових часток активного інгредієнта.
8. Фармацевтична композиція за п. 1, яка додатково містить розпушувач, засіб, що обумовлює плинність, або їх суміш.
9. Фармацевтична композиція за п. 1, де поверхнево-активна речовина включена у концентрації від 0,1 масової частки до 30 масових часток відносно загальної ваги композиції.
10. Фармацевтична композиція за п. 8, де розпушувач являє собою щонайменше один тип матеріалу, вибраний із групи, що включає поперечно-зшитий полівінілпіролідон, кросповідон, натрію кроскармелозу, натрію крохмальгліколат, крохмаль та альгінат.
11. Фармацевтична композиція за п. 8, де співвідношення розпушувача до поверхнево-активної речовини складає 0,66 або більше.
12. Спосіб отримання фармацевтичної композиції, яка містить 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид, при цьому спосіб включає етап вологого/сухого гранулювання із застосуванням поверхнево-активної речовини, органічного розчинника або співрозчинника (водного/органічного розчинника) як солюбілізувальний засіб активного інгредієнта фармацевтичної композиції,
де поверхнево-активна речовина являє собою щонайменше один тип матеріалу, вибраний із групи, що включає рицинову олію, лецитин (яєчний) та жирнокислотний макроголгліцерид;
органічний розчинник вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон; та
співрозчинник містить воду та органічний розчинник, вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон.
13. Спосіб за п. 12, де рицинова олія вибрана з групи, що включає поліетиленгліколю гліцерингідростеарат, кремофор RH 40 або кремофор RH 60.
14. Спосіб за п. 12, де жирнокислотний макроголгліцерид являє собою Gelucire 44/14.
15. Спосіб за п. 12, що додатково включає етап отримання гранулюючої рідини із застосуванням поверхнево-активної речовини із органічним розчинником або співрозчинником.
16. Спосіб за п. 15, де застосовувана гранулююча рідина має розчинну здатність 0,2 мг/мл або більше.
17. Спосіб отримання фармацевтичної композиції, при цьому спосіб включає етапи:
отримання порошкоподібної суміші із застосуванням 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду та щонайменше одного типу фармацевтично прийнятного наповнювача;
отримання вологих гранул шляхом об'єднання порошкоподібної суміші з поверхнево-активною речовиною, органічним розчинником або співрозчинником (водним/органічним розчинником) як гранулюючу рідину;
отримання гранул шляхом висушування вологих гранул; та
здійснення способу добору,
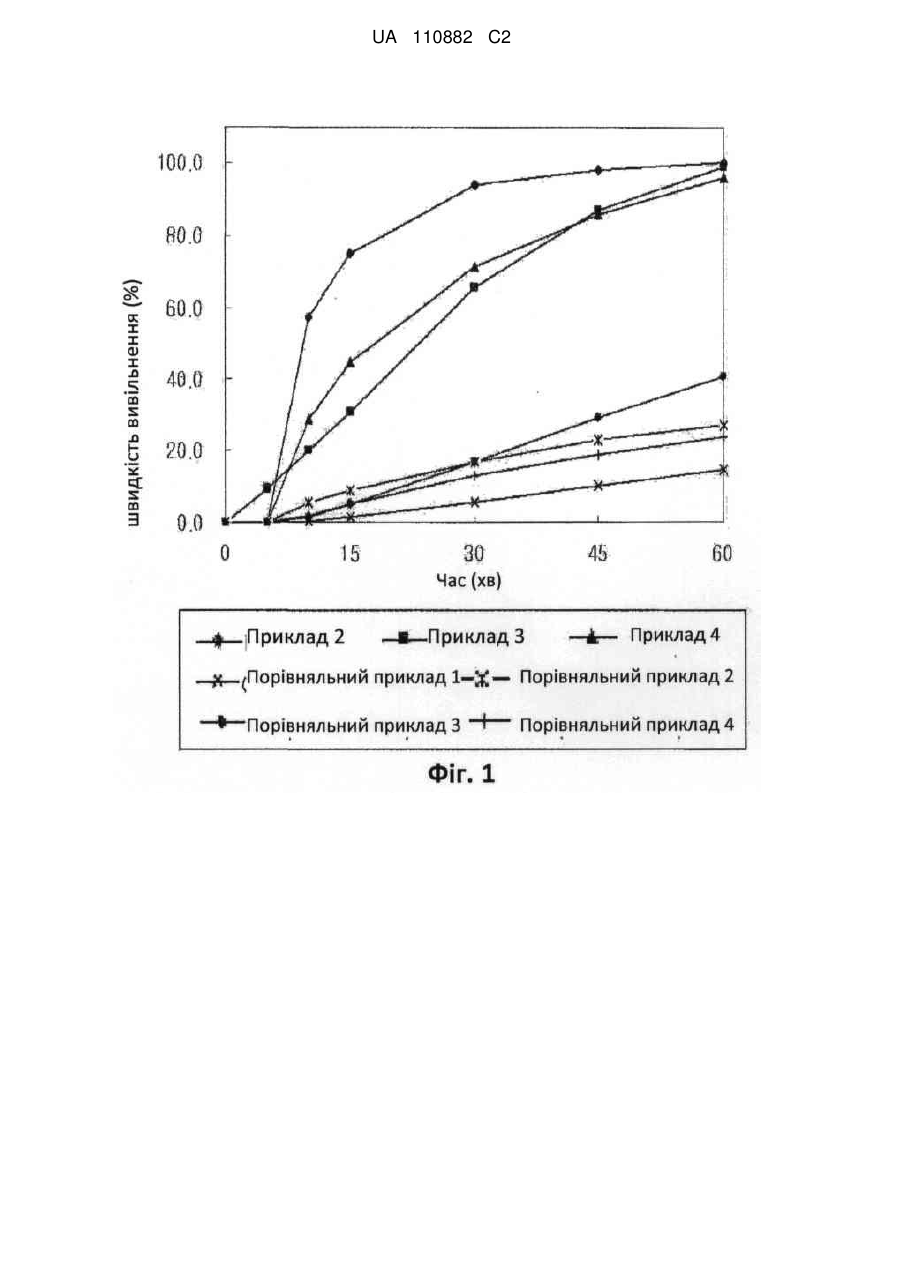
де поверхнево-активна речовина являє собою щонайменше один тип матеріалу, вибраний із групи, що включає рицинову олію, лецитин (яєчний) та жирнокислотний макроголгліцерид;
органічний розчинник вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон; та
співрозчинник містить воду та органічний розчинник, вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон.
18. Спосіб за п. 17, де рицинова олія вибрана з групи, що включає поліетиленгліколю гліцерингідростеарат, кремофор RH 40 або кремофор RH 60.
19. Спосіб за п. 17, де жирнокислотний макроголгліцерид являє собою Gelucire 44/14.
Текст
Реферат: Винахід стосується фармацевтичної композиції, яка містить гранулу, яка містить 4-метил-N-[3(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид як активний інгредієнт, та поверхнево-активну речовину, яка являє собою щонайменше один тип матеріалу, вибраний із групи, що включає рицинову олію, лецитин (яєчний) та жирнокислотний макроголгліцерид, та способу отримання фармацевтичної композиції. UA 110882 C2 (12) UA 110882 C2 UA 110882 C2 5 10 15 20 25 30 35 40 45 50 55 ГАЛУЗЬ ТЕХНІКИ Даний винахід відноситься до фармацевтичної композиції, яка включає 4-метил-[3-(4метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид, та до способу її отримання. РІВЕНЬ ТЕХНІКИ Іматинібу мезилат (глівек, загальнодоступний патентний документ Кореї № 1993-0021624), що зазвичай використовують у якості терапевтичного засобу проти хронічного мієлоїдного лейкозу, показав відмінну дію на стан пацієнтів з хронічним мієлоїдним лейкозом, але існує проблема, тому що останнім часом зустрічаються пацієнти, що мають стійкість до дії цього лікарського засобу. Зазвичай, вихідні матеріали для традиційних терапевтичних засобів проти хронічного мієлоїдного лейкозу мають надзвичайно низьку розчинність у воді та мають низьку швидкість вивільнення у шлунково-кишковий тракт через низьку розчинність. Таким чином, їх біопоглинальність є низькою. Також вони характеризуються сильними адгезивно-когезивними властивостями. Відповідно, при складанні у пігулки або капсули вони прилипають до змішувача, штампувального преса, прес-форми і т. п. Через це вони мають проблему низької придатності для обробки при складанні. Для того, щоб продемонструвати переважний терапевтичний ефект через зменшення недоліків терапевтичної сполуки, терапевтична сполука повинна бути у формі водного розчину та повинна неодмінно мати розчинність у воді. Відносно нерозчинна сполука демонструє недостатню та нерівномірну швидкість поглинання та, таким чином, має низький терапевтичний ефект. При цьому, нерозчинний лікарський засіб інколи демонструє недостатню швидкість поглинання. Для того, щоб ефективно збільшити розчинність надзвичайно нерозчинного лікарського засобу, виходячи із властивості хімічної структури лікарського засобу, можна застосовувати спосіб хімічної модифікації, такий як отримання більш розчинних похідних (наприклад, солі, естеру або розчинних проліків), або спосіб фізичної модифікації, такий як встановлення розміру частинок (з мікро розміром, з нано розміром), кристалічна модифікація, тверда дисперсія та ускладнення (включення)/розчинення. В патенті США № 5145684 розкривають типовий спосіб отримання активуючої композиції у формі наночастинок для нерозчинного лікарського засобу. Також, в зареєстрованих патентах США №№ 5518187 та 5862999 та патентах США №№ 5718388 та 5510118 розкривають спосіб отримання активуючої композиції у формі наночастинок. РОЗКРИТТЯ ТЕХНІЧНА ЗАДАЧА Декілька задач були порушені у розділі "Рівень техніки". Для вирішення даних задач виконали дослідження відносно похідного N-феніл-2-піримідин-аміну або його солі, що виявляє більш кращу ефективність ніж іматинібу мезилат. ВИРІШЕННЯ ЗАДАЧІ Як описано вище, у розділі "Рівень техніки", було порушено декілька задач відносно терапевтичних сполук проти лейкозу. Для того, щоб вирішити вищезгадані задачі винахідники даного винаходу дослідили похідне N-феніл-2-піримідин-аміну або його сіль, котрі демонструють більш високу ефективність лікарського засобу по відношенню до хронічного мієлоїдного лейкозу ніж іматинібу мезилат. Для вирішення задач винахідники шляхом повторюваних досліджень виявили, що 4-метилN-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)бензаміду гідрохлорид особливо придатний для пероральної фармацевтичної композиції з поліпшеною біологічною доступністю. Також вони виявили, що для цього матеріалу можливо збільшити розчинність та значно поліпшити швидкість вивільнення лікарського засобу при складанні цього матеріалу в гранули із застосуванням органічного розчинника або співрозчинника (водного/органічного розчинника) або співрозчинника, який містить поверхневоактивну речовину. Відповідно, об'єктом даного винаходу є забезпечення пероральної фармацевтичної композиції з поліпшеною біологічною доступністю та спосіб її отримання. В даному винаході 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид характеризується збільшеною розчинністю та значно поліпшеною швидкістю вивільнення лікарського засобу. Таким чином, можливо одержувати пероральну фармацевтичну композицію, яка включає даний матеріал, зі збільшеною біологічною доступністю та спосіб її отримання. Також, можливо поліпшувати розчинність нерозчинного лікарського засобу, та, таким чином, фармацевтична композиція, яка включає цей лікарський засіб, має стабільність та зменшення статичної електричної 1 UA 110882 C2 5 10 15 20 25 30 35 40 45 50 55 60 характеристики та адсорбуючу здатність, та є поліпшеною відносно швидкості вивільнення та біологічної доступності. ПЕРЕВАЖНІ ЕФЕКТИ ВИНАХОДУ Даний винахід забезпечує 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид, який характеризується кращою розчинністю та значно поліпшеною швидкістю розчинення лікарського засобу, фармацевтичну композицію, яка містить дану сполуку, що має поліпшену біологічну доступність, та спосіб їх отримання. Завдяки цьому розчинення нерозчинного лікарського засобу було поліпшено та фармацевтична композиція, яка містить дану сполуку, є стабільною, демонструє зменшені статичні електричні характеристики та характеристики поглинання, поліпшену швидкість розчинення та біологічну доступність. КОРОТКИЙ ОПИС ГРАФІЧНИХ МАТЕРІАЛІВ Вищезгадані та інші об'єкти, властивості та переваги даного винаходу будуть більш явними з наступного докладного опису у взаємозв'язку з доданими графічними матеріалами, на яких: на фіг. 1 показаний графік, що ілюструє швидкість вивільнення лікарського засобу (in vitro) у прикладах та порівняльних прикладах за даним винаходом; та на фіг. 2 показаний графік, що ілюструє PK-швидкість поглинання лікарського засобу в бігля в прикладах та порівняльних прикладах за даним винаходом; на фіг. 3 представлений графік, що ілюструє швидкість розчинення лікарського засобу, як виміряно in vitro, для прикладів за даним винаходом. СПОСІБ ЗДІЙСНЕННЯ ВИНАХОДУ Далі у даному документі переважний варіант здійснення даного винаходу буде описаний з посиланням на додані графічні матеріали. У подальшому описі та графічних матеріалах однакові номера позицій використані для позначення однакових або схожих компонентів, та отже повторення опису таких же або схожих компонентів буде опущено. Даний винахід відноситься до фармацевтичної композиції, яка включає похідне N-феніл-2піримідин-аміну або його сіль, переважно 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметилфеніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензамід або його фармацевтично прийнятну сіль, особливо переважно 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2іл-піримідин-2-іламіно)-бензаміду гідрохлорид, у якості терапевтичної сполуки та фармацевтично прийнятний наповнювач. Фармацевтична композиція може включати терапевтичну сполуку у кількості від 20 до 1000 мг. Також терапевтична сполука може бути включена у кількості від 40 ваг. % або більше та переважно 40~60 ваг. % відносно фармацевтичної композиції. Похідне N-феніл-2-піримідинаміну або його сіль переважно характеризуються середнім розміром частинок приблизно 50 мкм або менше та переважно приблизно 30 мкм або менше. Терапевтична сполука за даним винаходом може бути отримана у вигляді гранульованої суміші з наповнювачем, розпушувачем, зв'язувальним засобом, засобом, що обумовлює плинність і т. п. та може бути складена у форми для перорального застосування, такі як гранули, порошки, пігулки, тверді/м'які капсули (шляхом заповнення). У даному винаході з ціллю поліпшити розчинність переважно використовують органічний розчинник, співрозчинник (водний/органічний розчинник) або співрозчинник, який містить поверхнево-активну речовину. Поліпшення розчинності шляхом застосування такого матеріалу розглянуто у прикладах нижче у даному винаході. У даному описі вираз "терапевтична сполука" означає терапевтично або фармацевтично активну композицію, придатну зокрема для перорального застосування, та головним чином означає будь-яку сполуку, матеріал, лікарський засіб, лікарський препарат або активний інгредієнт, придатні для введення ссавцям (наприклад, людині). Така терапевтична сполука бере участь в інгібуванні ферментативної активності тирозинкінази зі збільшеною кількістю білка Bcr-Abl (онкобілка) та фосфорилюванні pCrkL, та придатна для отримання фармацевтичної композиції для цільової протиракової терапії хронічного мієлоїдного лейкозу та мієлопроліферативних порушень. Терапевтична сполука за даним винаходом являє собою похідне N-феніл-2-піримідин-аміну або його сіль, переважно являє собою 4-метил-N-[3-(4метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензамід або його фармацевтично прийнятну сіль та особливо переважно являє собою 4-метил-N-[3-(4метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид. В даному винаході 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид являє собою надзвичайно нерозчинний матеріал, який нерозчинний в органічному розчиннику, такому як етанол, метанол, або 2 UA 110882 C2 5 10 15 20 25 30 35 40 45 50 55 60 полярному розчиннику, такому як вода, та має низьку розчинність. Через низьку розчинність важко передати користь терапевтичної сполуки пацієнту (іншими словами, терапевтична сполука має низьку біопоглинальність). Відповідно, необхідно складати сполуку у форму з поліпшеною розчинністю та збільшеною біологічною доступністю. Даний винахід забезпечує фармацевтичну композицію зі збільшеною біологічною доступністю та спосіб її отримання. Гранули терапевтичної сполуки за даним винаходом можуть бути отримані з використанням фармацевтично прийнятного наповнювача. Більш того, при отриманні можна використовувати поверхнево-активну речовину, розпушувач або засіб, що обумовлює плинність, окремо або в комбінації. Сполука може бути складена у вигляді фізичної суміші, але з прикладів, наведених нижче, було виявлено, що сприятливою є гранульована форма. Наприклад, полівінілпіролідон, поліетиленгліколю гліцерингідростеарат або лецитин можуть бути піддані вологому гранулюванню з частинками 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду для отримання гранул. В даному винаході фармацевтично прийнятна поверхнево-активна речовина може бути щонайменше одним типом матеріалу, вибраним із групи, що включає поверхнево-активні речовини, такі як рицинова олія (наприклад, поліетиленгліколю гліцерингідростеарат, кремофор RH 40, кремофор RH 60), лецитин (яєчний), співполімер поліоксіетилену та поліоксипропілену, поліоксіетилен сорбітани або їх суміш, естер сорбіту та поліоксіетиленовий естер сорбіту та жирної кислоти (наприклад, полісорбат 80), лаурилсульфат натрію, жирнокислотний естер гліцерину, жирнокислотні макроголгліцериди (наприклад, Gelucire 44/14), але даний винахід не обмежується ними. Зокрема, придатним є поліетиленгліколю гліцерингідростеарат. Поверхнево-активну речовину можна переважно застосовувати у кількості від 1 до 50 масових часток та більш переважно від 5 до 20 масових часток по відношенню до 100 масових часток активного інгредієнта за даним винаходом. Також поверхнево-активну речовину можна включати у концентрації від 0,1 масової частки до 30 масових часток по відношенню до загальної ваги фармацевтичної композиції. У даному винаході наповнювач може бути щонайменше одним типом матеріалу, вибраним із групи, що включає целюлозу або її похідне (таку як мікрокристалічну целюлозу), гідроксипропілцелюлозу, гідроксіетилцелюлозу та гідроксипропілметилцелюлозу, гідрат та ангідрат лактози, дикальційфосфат, легкий кремнієвий ангідрид, крохмаль, маніт, полівінілпіролідон, поліетиленгліколь, еудрагіт, але даний винахід не обмежується ними. Наповнювач можна застосовувати у кількості від 0,01 до 60 масових часток та переважно від 1 до 50 масових часток по відношенню до 100 масових часток активного інгредієнта за даним винаходом. В даному винаході фармацевтично прийнятний розпушувач може бути щонайменше одним типом матеріалу, вибраним із групи, що включає поперечно-зшитий полівінілпіролідон або кросповідон, натрію кроскармелозу, натрію крохмальгліколат, крохмаль та альгінат, але даний винахід не обмежується ними. Розпушувач можна застосовувати у кількості від приблизно 1 до 40 ваг. % по відношенню до фармацевтичної композиції. В даному винаході фармацевтично прийнятний засіб, що обумовлює плинність, може бути щонайменше одним типом матеріалу, вибраним із групи, що включає стеарат магнію, тальк, легкий кремнієвий ангідрид, гліцерил бегенат, стеаринову кислоту та стеарилфумарат натрію. Засіб, що обумовлює плинність, можна застосовувати у кількості від 0 до 10 ваг. % та переважно від 0,5 до 2 ваг. % по відношенню до композиції. В прикладах за даним винаходом для отримання суміші можна обрати щонайменше один тип матеріалу з описаної вище групи. Зазвичай спосіб вологого гранулювання включає етап змішування, етап зв'язування з використанням рідини, етап висушування та етап розмірної обробки. При вологому способі, перший, етап змішування терапевтичної сполуки зі щонайменше одним фармацевтично прийнятним наповнювачем можна виконувати за допомогою придатного фармацевтичного обладнання. Наприклад, високошвидкісний змішувач, гранулятор із псевдозрідженим шаром, розпилювальну сушарку або ліофілізатор можна застосовувати для формування порошкоподібної суміші, але даний винахід не обмежується цим обладнанням для гранулювання. При вологому гранулюванні як рідкий розчинник, воду, так і полярний розчинник та органічні розчинники, такі як етанол, метанол, ізопропанол та ацетон, можна застосовувати окремо або в комбінації та органічний розчинник можна застосовувати окремо або в комбінації. Суміш, отриману вологим гранулюванням, можна висушувати за допомогою фармацевтичного обладнання для гранулювання або придатного обладнання для висушування. На етапі висушування гранули висушують до LOD приблизно 3 ваг. % або менше, наприклад, 2 ваг. % або менше. Після етапу висушування відповідний розмір гранул можна отримати за допомогою сита. Така гранульована суміш може бути складена у пігулки або 3 UA 110882 C2 5 10 15 20 25 30 35 40 45 50 55 60 складена у капсули шляхом наповнення капсули. Приклади капсули включають тверду желатинову капсулу та тверду HPMC капсулу. Розмір капсули варіює від № 00 до № 5, але даний винахід не обмежується ними. Терапевтична фармацевтична композиція може бути включена у капсулі у кількості від 20 мг до 1000 мг. Зокрема, капсула може включати терапевтичну фармацевтичну композицію у кількості від 20 мг до 800 мг. Наприклад, більш конкретно, капсула може включати терапевтичну фармацевтичну композицію у кількості 50 мг, 100 мг або 200 мг. Щодо кількості застосовуваних допоміжних речовин суміш терапевтичної сполуки та наповнювача може мати плинність та однорідність складу такі, щоб нею належним чином можна було заповнити капсулу. Для поліпшення швидкості розчинення терапевтичної сполуки та забезпечення високої швидкості вивільнення терапевтичної сполуки важливим є співвідношення розпушувача до солюбілізувального засобу (який є поверхнево-активною речовиною). Співвідношення розпушувач/солюбілізувальний засіб має бути 0,66 або більше (наприклад, кросповідон/поліетиленгліколю гліцерингідростеарат, 0,66~0,80). Також в даному винаході у переважній фармацевтичній композиції, отриманій способом сушіння розпилюванням, вміст наповнювача, поверхнево-активної речовини або масляного матеріалу може варіювати відповідно до властивості отримуваної композиції. Важливо коригувати кількості наповнювача, поверхнево-активної речовини, масляного матеріалу та активних матеріалів за даним винаходом для того, щоб коригувати концентрацію. У тих випадках, коли концентрація низька, ефективність препарату знижена та важко отримати дисперсну композицію. У тих випадках, коли концентрація висока, в'язкість підвищена та осад утворюється з дуже високою швидкістю під час диспергування. Таким чином, шийка може легко закорковуватися. Переважно у композиції наповнювач, за виключенням розчинника, характеризується концентрацією від 0,1 до 20 масових часток, поверхнево-активна речовина характеризується концентрацією від 0,1 до 50 масових часток, та масляний матеріал характеризується концентрацією від 0,1 до 5 масових часток. В даному винаході, наприклад, наповнювач може бути щонайменше одним типом матеріалу, вибраним із групи, що включає полівінілпіролідон, гідроксипропілцелюлозу, гідроксіетилцелюлозу, поліетиленгліколь та еудрагіт. Переважно застосовувати у кількості від 0,1 до 20 масових часток та більш переважно від 1 до 10 масових часток по відношенню до 100 масових часток 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-ілпіримідин-2-іламіно)-бензаміду гідрохлорид. В даному винаході поверхнево-активна речовина може бути щонайменше одним типом матеріалу, вибраним із групи, що включає рицинову олію (наприклад, поліетиленгліколю гліцерингідростеарат, кремофор RH 40, кремофор RH 60), лецитин (яєчний), співполімер поліоксіетилену та поліоксипропілену, поліоксіетилен сорбітани або їх суміш, естер сорбіту та поліоксіетиленовий естер сорбіту та жирної кислоти (наприклад, полісорбат 80), лаурилсульфат натрію, жирнокислотний естер гліцерину, жирнокислотні макроголгліцериди (наприклад, Gelucire 44/14). Поверхнево-активну речовину переважно застосовують у кількості від 1 до 60 масових часток та більш переважно від 5 до 50 масових часток по відношенню до 100 масових часток похідного N-феніл-2-піримідин-аміну або його солі. В даному винаході масляний матеріал може бути щонайменше одним типом матеріалу, вибраним із групи, що включає природні та тваринні масла (наприклад, соєву олію, риб'ячий жир і т. п.), тригліцериди жирних кислот, вуглеводні, поліетиленгліколь 660 гідроксистеарат та подібне. Переважно застосовують у кількості від 0,1 до 1 масової частки по відношенню до 100 масових часток похідного N-феніл-2-піримідин-аміну або його солі. Фармацевтичну композицію за даним винаходом можна одержувати способами отримання, такими як вологе гранулювання /сухе гранулювання /гранулювання з розтопу. Іншими словами, композицію за даним винаходом можна одержати способом отримання вологим гранулюванням, способом отримання сухим гранулюванням або способом отримання гранулюванням з розтопу (спосіб екструзії гарячого розтопу). При способі отримання вологим гранулюванням композицію отримують через суспендування, висушування та диспергування з рідкої суміші, яка включає поверхнево-активну речовину, наповнювач або їх суміш, масляний матеріал, активний матеріал (похідне N-феніл-2-піримідин-аміну або його сіль), етанол та чисту воду. При способі отримання сухим гранулюванням композицію можна отримувати шляхом змішування поверхнево-активної речовини, наповнювача або їх суміші, масляного матеріалу, активного матеріалу (похідного N-феніл-2-піримідин-аміну або його солі), додавання рідкої суміші етанолу та чистої води у кількості 2 % або більше по відношенню до суміші та гранулювання стисненням отриманої суміші за допомогою стиснення вальцями. При способі отримання гранулюванням з розтопу композицію отримують шляхом змішування похідної суміші з рідкою сумішшю етанолу та чистої води у кількості 2 % або більше по відношенню до суміші. 4 UA 110882 C2 5 10 15 20 25 30 35 Наприклад, для отримання рідкої суспензії поверхнево-активну речовину, наповнювач або їх суміш та масляний матеріал додають з рідкою сумішшю етанолу та чистої води з наступним перемішуванням та потім отриману суміш піддають розчиненню, суспендуванню або змочуванню та повільно додають до 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметилфеніл]-3-(4-піразин-2-іл- піримідин-2-іламіно)-бензаміду гідрохлориду. При отриманні фармацевтичної композиції рідку суспензію можна висушувати за допомогою способу висушування, зазвичай застосовуваного в галузі фармакології, такого як сушіння розпилюванням, сушіння у вакуумі, ліофільне сушіння, або можна висушувати термічним сушінням за допомогою тиску при стисненні вальцями або термічним сушінням при способі отримання гранулюванням з розтопу. Носій, який можна застосовувати на етапі висушування, являє собою фармацевтично прийнятний матеріал та може включати наповнювач (такий як мікрокристалічна целюлоза, дикальційфосфат, легкий кремнієвий ангідрид), розпушувач(такий як кросповідон або його похідне, повідони, натрію крохмальгліколат, кроскармелоза, низько заміщена гідроксил-целюлоза), засіб, що обумовлює плинність (такий як стеарат магнію, стеаринова кислота), та поверхнево-активну речовину (таку як лаурилсульфат натрію). Фармацевтична композиція, яка включає 4-метил-N-[3-(4-метилімідазол-1-іл)-5трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензамід або його фармацевтично прийнятну сіль, особливо, 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид, разом з наповнювачем або поверхнево-активною речовиною, масляним матеріалом або їх сумішшю може бути складена у різні форми. Вона може бути складена у твердий препарат, такий як гранули, капсули, пігулки, порошки, або може бути складена у препарат-суспензію. Переважно, вона може бути складена у пігулки або капсули. Наприклад, фармацевтичну композицію, гранульовану за допомогою способу гранулювання, яка включає 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-ілпіримідин-2-іламіно)-бензаміду гідрохлорид, можна безпосередньо змішувати з фармацевтично прийнятним носієм. Потім сумішшю можна заповнювати капсулу, можна стискати у пігулку за допомогою таблеткової машини або можна покривати оболонкою або покривати ентеросолюбільною оболонкою для зовнішнього вигляду, смаку, безпеки та зручності. У даному винаході фармацевтично прийнятний носій необхідно вибирати таким чином, щоб можна було зберегти стабільність терапевтичної сполуки. Як відзначається у таблиці 1 нижче, при зберіганні гомогенної суміші терапевтичної сполуки (API) та носія при 40 °C, при 75 % RH (6 тижнів) не спостерігали зміну кольору, зменшення вмісту терапевтичного лікарського засобу або вироблення супутніх домішок при усіх носіях, крім лактози. У випадку 4-метил-N-[3-(4метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду на стабільність впливає вологість. У випадку носія з високим коефіцієнтом вологоутримання за своєю природою, такого як лактоза, під час процесу змішування можуть траплятися деякі труднощі, такі як зміна кольору та зменшення вмісту. Таблиця 1 Умова зберігання 40 °C, при 75 % RH, 6 тижнів Співвідношення суміші композиції (API: наповнювач) Наповнювач Зміна кольору Вміст (%) Домішки (%) 1: 1 мікрокристалічна целюлоза 100,1 100,1 дикальційфосфат 99,6 кросповідон 99,8 лактоза S-Mg зміна кольору 94,2 99,7 0 40 45 Далі у даному документі даний винахід буде описаний більш докладно з посиланням на приклади та порівняльні приклади. Проте, приклади та порівняльні приклади є тільки ілюстративними та не обмежують даний винахід. Приклади Приклад 1 Розчинність перевіряли відносно 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметилфеніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду (A) та іматинібу мезилату (B). 5 UA 110882 C2 5 10 15 20 25 30 35 40 45 50 55 Приклад 1-1, 1-2: у стакан на 100 мл вносили рицинову олію 60 (18 г) та до неї додавали воду (22,5 мл) та етанол (9 мл) з наступним перемішуванням протягом 1 години. Після того як матеріали розчинялися, до отриманої суміші додавали A та B кожний у кількості 0,5 г з наступним перемішуванням достатнім чином протягом приблизно 2 годин. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого продукту. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-5, 1-6: у стакан на 100 мл вносили рицинову олію 60 (18 г) та до неї додавали воду (22,5 мл) та ацетон (9 мл) з наступним перемішуванням протягом 1 години. Після того як матеріали розчинялися, до отриманої суміші додавали A та B кожний у кількості 0,5 г з наступним перемішуванням достатнім чином протягом приблизно 2 годин. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого продукту. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-9, 1-10: у стакан на 100 мл вносили рицинову олію 60 (18 г) та до неї додавали воду (22,5 мл) та IPA (ізопропіловий спирт, 9 мл) з наступним перемішуванням протягом 1 години. Після того як матеріали розчинялися, до отриманої суміші додавали A та B кожний у кількості 0,5 г з наступним перемішуванням достатнім чином протягом приблизно 2 годин. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого продукту. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-3, 1-4: у стакан на 100 мл вносили воду (22,5 мл) та етанол (9 мл) з наступним перемішуванням протягом 10 хвилин. До отриманої суміші додавали A та B кожний у кількості 0,5 г з наступним перемішуванням достатнім чином протягом приблизно 2 годин. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого продукту. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-7, 1-8: у стакан на 100 мл вносили воду (22,5 мл) та ацетон (9 мл) з наступним перемішуванням протягом 10 хвилин. До отриманої суміші додавали A та B кожний у кількості 0,5 г з наступним перемішуванням достатнім чином протягом приблизно 2 годин. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого продукту. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-11, 1-12: у стакан на 100 мл вносили воду (22,5 мл) та IPA (ізопропіловий спирт, 9 мл) з наступним перемішуванням протягом 10 хвилин. До отриманої суміші додавали A та B кожний у кількості 0,5 г з наступним перемішуванням достатнім чином протягом приблизно 2 годин. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого продукту. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-13, 1-14: у стакан на 100 мл вносили воду (31,5 мл) та до неї додавали A та B кожний у кількості 0,5 г з наступним перемішуванням достатнім чином протягом приблизно 2 годин. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого продукту. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-15, 1-16: у стакан на 100 мл вносили A та B кожний у кількості 0,5 г. Потім до них додавали етанол (5 мл кожний раз) до розчинення суміші. Іншими словами, етанол додавали до них до загальної кількості 31,5 мл. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого розчину. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. Приклад 1-17, 1-18: у стакан на 100 мл вносили A та B кожний у кількості 0,5 г. Потім до них додавали ацетон (5 мл кожний раз) до розчинення суміші. Іншими словами, ацетон додавали до них до загальної кількості 31,5 мл. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого розчину. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. 6 UA 110882 C2 5 10 15 20 25 30 35 Приклад 1-19, 1-20: у стакан на 100 мл вносили A та B кожний у кількості 0,5 г. Потім до них додавали IPA (ізопропіловий спирт, 5 мл кожний раз) до розчинення суміші. Іншими словами, IPA додавали до них до загальної кількості 31,5 мл. За допомогою 0,45 мкм GHP-фільтра та шприца фільтрували 5 мл отриманого розчину. Потім збирали 1 мл продукту та вносили у колбу на 100 мл та до нього додавали розчинювальну рідину (DMSO:MeOH=3:7) до позначки. Дану рідину використовували у якості контрольного розчину. 1) Аналіз на розчинність 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду (A) Приготування стандартного розчину: еталонний зразок (106,8 мг) A точно вимірювали та вносили в колбу на 50 мл та до нього додавали DMSO до позначки. 5 мл даної рідини збирали та вносили в колбу на 50 мл та додавали до неї розчинник (диметилсульфоксид: метанол = 30: 70) до позначки. 10 мл даної рідини вносили ще раз у колбу на 100 мл та до неї додавали розчинник до позначки. Дану рідину готували в концентраціях 5, 10, 15 та 20 ppm (частин на мільйон) та аналізували її градуйовану криву. За допомогою лінійного рівняння градуйованої кривої вираховували кількість розчинення дослідного зразка. - Умова аналізу колонка: Capsellpak MG II, C18(4,6 ×150 мм, 5 мкм) або подібна до неї колонка; температура колонки: 35 °C рухлива фаза: амоній-ацетатний буферний розчин (pH 4,0): метанол = 25: 75; детектор: UV 270 нм; швидкість потоку: 1,0 мл/хв.; об'єм проби, що вводиться: 10 мкл; спосіб отримання буферного розчину: вносили оцтову кислоту (100) (1,2 мл) та ацетат амонію (0,25 г) та до неї додавали воду з отриманням загального об'єму 1 л. За допомогою 0,2 моля/л соляної кислоти та 0,2 моля/л гідроксиду натрію pH доводили до 4,0. 2) Аналіз на розчинність іматинібу мезилату (B) Приготування стандартного розчину: еталонний зразок (20 мг) A точно вимірювали та вносили в колбу на 100 мл до позначки. 5 мл даної рідини збирали та вносили в колбу на 50 мл та до неї вносили розчинник (метанол: 0,1M соляна кислота=6:4) до позначки. Дану рідину готували в концентраціях 5, 10, 15 та 20 ppm та аналізували її градуйовану криву. За допомогою лінійного рівняння градуйованої кривої вираховували кількість розчинення дослідного зразка іматинібу. - Умова аналізу Утворюючий іонні пари реактив: натрію 1-октаносульфонат моногідрат (7,5 г) розчиняли у 800 мл води. Отриманий розчин підкислювали 10 % o-фосфорною кислотою до pH 2,5 та потім розводили до 1000 мл. Рухлива фаза A: утворюючий іонні пари реактив + метанол (420+580). Рухлива фаза B: утворюючий іонні пари реактив + метанол (40+960). Час (хв.) 0 15 25 30 Рухлива фаза A (%) 100 100 30 100 Рухлива фаза В (%) 0 0 70 0 40 45 Колонка: Symmetry C18, 5 мкм (Waters), довжина 150 мм, внутрішній діаметр 3,9 мм або подібна до неї колонка. Температура колонки: 25 °C. Детектор: UV 268 нм. Швидкість потоку: 1,2 мл/хв. Об'єм проби, що вводиться: 10 мкл. Час виконання: 30 хвилин. 7 UA 110882 C2 Таблиця 2 Приклад органічний розчинник Athanol (мл) ацетон (мл) IPA (мл) чиста вода (мл) поверхнево-активна речовина, поліетиленгліколю гліцерингідростеарат (г) A (г) розчинність (приблизно мг/мл) Приклад органічний розчинник Athanol (мл) Ацетон (мл) IPА (мл) чиста вода (мл) поверхнево-активна речовина, поліетиленгліколю гліцерингідростеарат (г) B (г) розчинність (приблизно мг/мл) 5 1-1 9 1-5 1-9 1-3 9 9 1-11 1-13 0 1-15 31,5 9 9 22,5 22,5 22,5 18 18 18 22,5 0 0,5 0,5 0,5 0,89 0,88 0,85 1-2 9 1-6 1-7 1 10 0,5 0,5 0,5 11,22 10,60 12,20 1-19 31,5 22,5 0 9 22,5 0 31,5 0 0 0 0 0 31,5 0 0 0,5 0,06 0,5 0,22 0,5 0,21 0,5 0,00 0,5 4,26 0,5 0,29 0,5 0,44 1-4 1-8 1-12 1-14 0 1-16 31,5 1-18 1-20 9 9 22,5 22,5 22,5 18 18 18 1-17 9 22,5 0 22,5 0 31,5 9 22,5 0 31,5 0 0,5 0,5 0,5 0,5 15,8 15,8 15,8 15,8 або або або або більше більше більше більше 0 0 0 0 31,5 0 0 0,5 4,21 0,5 0,16 0,5 0,33 Як відзначається в таблиці 2 вище, виявили, що 4-метил-N-[3-(4-метилімідазол-1-іл)-5трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид (A) більш нерозчинний у різних типах водного/органічного розчинника, ніж іматинібу мезилат (B). Приклад 2 Згідно з композицією, наведеною в таблиці 3 нижче, була складена фармацевтична композиція за даним винаходом, яка містить 4-метил-N-[3-(4-метилімідазол-1-іл)-5трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид. 10 Таблиця 3 Вага на капсулу Співвідношення (мг) (%) Інгредієнт грануляція 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид мікрокристалічна целюлоза гідрат дикальційфосфату легкий кремнієвий ангідрид кросповідон поліетиленгліколю гліцерингідростеарат після змішування кросповідон стеарат магнію Загалом 8 213,6 47,4 50 70 11 30 60 11,1 15,5 2,4 6,7 13,3 10 6 450,6 2,2 1,3 100 UA 110882 C2 5 10 15 20 За допомогою високошвидкісного змішувача 4-метил-N-[3-(4-метилімідазол-1-іл)-5трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид, мікрокристалічну целюлозу, гідрат дикальційфосфату, легкий кремнієвий ангідрид та кросповідон змішували з утворенням порошкоподібної суміші. Крім того в рідкій суміші етанол: вода, розчиняли поліетиленгліколю гліцерингідростеарат для отримання рідини для гранулювання. Рідину змішували з отриманою порошкоподібною сумішшю у високошвидкісному змішувачі з тим, щоб піддати порошкоподібну суміш вологій грануляції. Потім порошкоподібну суміш висушували за відповідних умов протягом необхідного часу за допомогою сушильної шафи та згодом складали у гранули. Висушені гранули відсіювали у відношенні оптимального розміру гранул за допомогою просіювання через сито та потім крім того додавали кросповідон та стеарат магнію для забезпечення готової гранульованої суміші. Даною гранульованою сумішшю заповнювали тверду капсулу. Приклад 3 Таким же чином, як описано в прикладі 2 композицію за даним винаходом таблетують за допомогою етапів змішування, гранулювання, сушіння, змащування, виготовлення пігулок та нанесення оболонки. Приклад 4 Згідно з композицією, наведеною у таблиці 4 нижче, складали фармацевтичну композицію за даним винаходом, з якої виключили поверхнево-активну речовину, застосовувану у прикладі 2. Таблиця 4 Вага на Співвідношення капсулу (мг) (%) Інгредієнт грануляція із застосуванням співрозчинника 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметилфеніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид мікрокристалічна целюлоза гідрат дикальційфосфату легкий кремнієвий ангідрид кросповідон після змішування кросповідон стеарат магнію Загалом 25 30 213,6 54,7 50 70 11 30 12,8 17,9 2,8 7,7 10 6 390,6 2,6 1,5 100 Таким же чином, як описано в прикладі 2 композицію за даним винаходом складають у вигляді гранульованої суміші та заповнюють нею капсулу, з якої виключили поліетиленгліколю гліцерингідростеарат у якості поверхнево-активної речовини. Порівняльний приклад 1 Згідно з тією ж композицією, як і композиція у прикладі 4, наведеною у таблиці 5 нижче, інгредієнти не гранулювали, а фізично змішували для отримання порошкоподібної суміші. Потім порівняно аналізували роль поверхнево-активної речовини та швидкість вивільнення відповідно до способів отримання. Згідно з тією ж композицією, як і композиція у таблиці 4, інгредієнти просто фізично змішували та отриманим порошком заповнювали капсулу. 9 UA 110882 C2 Таблиця 5 Порівняльний приклад 1 СпіввідСпіввідВага Вага ношення ношення (мг) (мг) (%) (%) 4-метил-N-[3-(4-метилімідазол1-іл)-5-трифторметил-феніл]-3213,6 (4-піразин-2-іл-піримідин-2іламіно)-бензаміду гідрохлорид мікрокристалічна целюлоза 50 гідрат дикальційфосфату 70 легкий кремнієвий ангідрид 11 поліетиленгліколю гліцерингідростеарат кросповідон 40 стеарат магнію 6 Загалом (мг) 390,6 5 10 15 20 Порівняльний приклад 4 Порівняльний Порівняльни приклад 2 й приклад 3 54,7 213,6 47,4 12,8 17,9 2,8 50 70 11 8,9 1,3 100,0 Співвідношення (%) 213,6 213,6 78,1 60 21,9 273,6 100,0 13,3 40 6 450,6 Вага (мг) 11,1 15,5 2,4 60 Вага (мг) 10,2 1,5 100,0 213,6 Порівняльний приклад 2 Згідно з тією ж композицією, як і композиція у прикладі 2, поліетиленгліколю гліцерингідростеарат фізично домішували та отриманою сумішшю заповнювали капсулу. Порівняльний приклад 3 213,6 мг 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-ілпіримідин-2-іламіно)-бензаміду гідрохлориду складали у форму капсули шляхом заповнення порожньої капсули. Порівняльний приклад 4 213,6 мг 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-ілпіримідин-2-іламіно)-бензаміду гідрохлориду та поліетиленгліколю гліцерингідростеарат готували у фізичну суміш та заповнювали нею капсулу. Тестовий приклад 1. Профіль розчинності Капсули з прикладу 2 ~ 4 та порівняльного прикладу 1 ~ 4 перевіряли згідно зі способом 2 (спосіб із застосуванням лопатевої мішалки) з числа способів тестування на вивільнення за Корейською фармакопеєю, у котрому температуру вивільнення доводять до 37 °C шляхом занурення. Тест на вивільнення здійснювали за допомогою середовища розчинення (pH 1,2) у кількості 900 мл (доповненого SLS 1 %) за швидкості мішалки 100 об./хв. Кількість розчиненого лікарського матеріалу вимірювали за допомогою HPLC. Швидкість вивільнення (%) 4-метил-N[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)бензаміду гідрохлориду показана та відзначена на фіг. 1 та в таблиці 6 з проміжками часу. Таблиця 6 Приклад 2 Приклад 3 Приклад 4 Порівн прикл 1 Порівн прикл 2 Порівн прикл 3 Порівн прикл 4 25 30 5 хв. 0,0 9,5 0,1 0,0 0,0 0,0 0,0 10 хв. 57,3 20,1 28,6 0,4 5,7 1,4 2,1 15 хв. 75,2 31,0 44,9 1,6 9,1 5,2 5,3 30 хв. 94,1 65,6 71,4 5,8 16,9 16,8 13,0 45 хв. 98,1 86,9 85,9 10,4 22,8 29,3 18,9 60 хв. 100,0 99,1 95,9 14,9 26,9 40,5 23,9 Як показано на фіг. 1 та в таблиці 6, коли капсули з прикладу 2 та прикладу 4 порівнювали одна з одною, виявляли, що додавання неіонної поверхнево-активної речовини підвищувало швидкість вивільнення. Також, коли капсули з прикладу 2 та порівняльного прикладу 4 порівнювали одну з одною, виявляли, що спосіб грануляції значно підвищував швидкість вивільнення. Фізичні суміші з порівняльних прикладів 1, 2 та 4 показували низьку швидкість розчинення у порівнянні з капсулою з порівняльного прикладу 3. 10 UA 110882 C2 5 10 Приклад 5. Отримання фармацевтичної композиції шляхом способу сушіння розпилюванням Розчиняли 20 г поліетиленгліколю гліцерингідростеарату та 27 г полівінілпіролідону у 1870 мл рідкої суміші етанолу та чистої води (1: 2,5). Отриманий продукт перемішували за допомогою механічної мішалки поки 100 г 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду додавали до нього та суспендували. Отриманий продукт перемішували протягом 3 годин за швидкості обертання 300 об./хв або більше та гомогенізували при тиску від 10000 до 15000 за допомогою мікрофлюїдайзера. Потім за допомогою розпилювальної сушарки отримували фармацевтичну композицію. Спосіб сушіння розпилюванням здійснювали з температурою повітря на вході 100 ~ 120 °C та температурою повітря на виході від 70 до 90 °C. Згідно з інгредієнтами та вмістом, наведеними у таблиці 7 нижче, композицію отримували тим же способом, що описаний у прикладі 5. Рідка суміш етанолу та чистої води була присутня у кількості 10 мл відносно 1 г сукупності усіх використаних інгредієнтів. Таблиця 7 4-метил-N-[3-(4-метилімідазол-1-іл)5-трифторметил-феніл]-3-(4-піразин2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид полівінілпіролідон Gelucire 44/14 cremophor RH 40 лецитин поліетиленгліколю гліцерингідростеарат Загалом (г) 6 100 7 100 50 20 Приклад 9 10 11 100 100 100 70 5 20 195 8 100 10 12 100 20 13 100 14 100 5 5 10 30 2 50 10 10 50 50 30 30 15 40 5 10 5 50 20 30 202 170 200 170 175 120 160 160 15 20 25 30 Приклад 15. Отримання фармацевтичної композиції шляхом способу гранулювання із псевдозрідженим шаром Розчиняли 30 г поліетиленгліколю гліцерингідростеарату у 234 мл рідкої суміші етанолу та чистої води. Отриманий продукт перемішували за допомогою механічної мішалки поки 100 г 4метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)бензаміду гідрохлориду додавали до нього та суспендували. Отриманий продукт перемішували протягом 3 годин за швидкості обертання 300 об./хв або більше та гомогенізували. Потім належним чином змішували носії, такі як мікрокристалічна целюлоза (20 г), кросповідон (15 г), легкий кремнієвий ангідрид (5 г) та дикальційфосфат (25 г). Суміш вносили у гранулятор із псевдозрідженим шаром та псевдозріджували поки рідку суспензію висушували розпиленням для того, щоб отримати фармацевтичну композицію гранулярного типу Згідно з інгредієнтами та вмістом, наведеними у таблиці 8 нижче, гранули, що містили фармацевтичну композицію, отримували таким же способом, який описаний у прикладі 15. Рідка суміш етанолу та чистої води була присутня у кількості 1,2 мл відносно 1 г сукупності усіх використаних інгредієнтів. 11 UA 110882 C2 Таблиця 8 4-метил-N-[3-(4-метилімідазол-1-іл)5-трифторметил-феніл]-3-(4-піразин2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид Gelucire 44/14 полівінілпіролідон гідроксипропілцелюлоза поліетиленгліколь 6000 поліетиленгліколю гліцерингідростеарат лецитин гідроксипропіл-бета-циклодекстрин мікрокристалічна целюлоза кросповідон натрію крохмальгліколат легкий кремнієвий ангідрид дикальційфосфат Загалом (г) 5 10 15 20 25 30 35 16 100 17 100 18 100 Приклад 19 20 100 100 21 100 22 100 23 100 20 2 5 15 5 15 10 10 25 30 25 25 20 5,5 20 200,5 5,5 25 222,5 10 30 5 5 15 10 15 30 30 10 5 25 15 20 25 20 15 5 30 210,0 5 15 200,0 6 20 221,0 5 20 190,0 5 20 30 15 20 20 20 6 5 15 20 181,0 205,0 Приклад 24. Отримання капсули 50 г фармацевтичної композиції з прикладу 14 змішували з мікрокристалічною целюлозою 4 %, кросповідоном 6 %, дикальційфосфатом 6 % та стеаратом магнію 1,5 % для отримання капсули, що містить 100 мг 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду. Приклад 25. Отримання капсули 50 г фармацевтичної композиції з прикладу 13 змішували з мікрокристалічною целюлозою 4 %, кросповідоном 4,2 %, дикальційфосфатом 6 % та стеаратом магнію 1,5 % для отримання капсули, що містить 100 мг 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду. Приклад 26. Отримання капсули 100 г гранул, які містять фармацевтичну композицію з прикладу 15, змішували з кросповідоном 5 % та стеаратом магнію 1,5 % для отримання капсули, що містить 100 мг 4метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)бензаміду гідрохлориду. Приклад 27. Отримання капсули 100 г гранул, які містять фармацевтичну композицію з прикладу 21, змішували з кросповідоном 5 % та стеаратом магнію 1,5 % для отримання капсули, що містить 100 мг 4метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин -2-іл-піримідин-2іламіно)-бензаміду гідрохлориду. Приклад 28. Отримання капсули 50 г фармацевтичної композиції з прикладу 10 змішували з мікрокристалічною целюлозою 4 %, кросповідоном 4,2 %, дикальційфосфатом 6 % та стеаратом магнію 1,5 % для отримання капсули, що містить 100 мг 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду. Тестовий приклад 2. Тест на розчинність Для того, щоб визначити поліпшення розчинності фармацевтичної композиції, яка включає 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2іламіно)-бензаміду гідрохлорид або його похідне, здійснювали тест на вивільнення лікарського засобу із застосуванням капсули, отриманої шляхом змішування фармацевтичної композиції з прикладу 5 з мікрокристалічною целюлозою (20 %), натрію крохмальгліколатом (6 %), легким кремнієвим ангідридом (1 %) та стеаратом магнію (1,5 %), капсули, отриманої шляхом змішування гранул, що містять фармацевтичну композицію з кожного з прикладу 16 та прикладу 21, із засобом, що обумовлює плинність, та заповнення капсули сумішшю та капсули з кожного з прикладів 24-28. 12 UA 110882 C2 5 У порівняльному прикладі 5 капсулу отримували шляхом змішування 4-метил-N-[3-(4метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду (67,5 %) з мікрокристалічною целюлозою (20 %), кросповідоном (10 %), легким кремнієвим ангідридом (1 %) та стеаратом магнію (1,5 %). Тест на розчинність здійснювали за умов відповідно до способу тесту на розчинність (спосіб 2) за Корейською фармакопеєю з використанням 900 мл середовища розчинення. Після тесту на розчинність протягом 45 хвилин, виконували HPLC-аналіз. Результати зазначені у таблиці 9. Таблиця 9 Порівняльний приклад 5 Приклад 5 Приклад 16 Приклад 21 Приклад 24 Приклад 25 Приклад 26 Приклад 27 Приклад 28 10 15 20 25 Швидкість вивільнення (%) 3,2 61 56 72 80 88 86 90 88 Як зазначено у таблиці вище, виявили, що швидкість вивільнення лікарського засобу фармацевтичної композиції за даним винаходом була значно поліпшена завдяки поліпшенню розчинності. Тестовий приклад 3. Тест на фармакокінетику Самців бігля (приблизно 10 кг) обирали, та зважували, та перевіряли на наявність будь-яких ран. Перед тим, як їм уводили тестовий матеріал, їх не годували протягом 16 годин. Кожна група включала 3 бігля. Застосовували капсулу з порівняльного прикладу 5, котру отримували шляхом змішування 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4-піразин-2іл-піримідин-2-іламіно)-бензаміду гідрохлориду з мікрокристалічною целюлозою, кросповідоном, легким кремнієвим ангідридом та стеаратом магнію та заповнення сумішшю капсули. Лікарські препарати з прикладу 24, прикладу 27, прикладу 28 та прикладу 2 вводили перорально та аналізували за допомогою LC-MS/MS. У результаті порівняльний приклад 5 показав площу під кривою концентрації плазми відносно кривої залежності від часу (AUC) 137,4 нг/мл у той час, як інші приклади показали AUC 922,0 нг/мл, 816,0 нг/мл та 1586,7 нг/мл. Іншими словами, у порівнянні з порівняльним прикладом 5 приклади показували вищий AUC у 5-10 разів або більше. Також виявили, що приклади показали у 4-18 разів вищу максимальну концентрацію в крові (Cmax) ніж у порівняльному прикладі 5. Результати відзначені на фіг. 2. Фармакокінетичні параметри зазначені у таблиці 10 нижче та площу під кривою концентрації плазми відносно кривої залежності від часу (AUC) вираховували за допомогою способу трапецій. 30 Таблиця 10 AUC12 (нг/мл) Сmах (нг/мл) 35 Порівняльний приклад 5 137,4 21,9 Приклад 24 1541,9 352,3 Приклад 27 802,2 119,0 Приклад 28 896,6 166,3 Приклад 2 1258,6 283,5 Тестовий приклад 4. Тест на стабільність Капсулу з прикладу 2 запаковували у PTP-упаковку та піддавали тесту на довгострокову стабільність (25±2 °C, 60±5 % RH) та тесту на високотемпературну стійкість (60±2 °C). Результати тесту на довгострокову стабільність (25±2 °C, 60±5 % RH) зазначені в таблиці 11 нижче, та результати тесту на високотемпературну стійкість (60±2 °C) зазначені в таблиці 12 нижче. Виходячи з результатів з таблиць 11 та 12 не спостерігали значної зміни у вмісті та зміни, пов'язаної з перебігом часу. 13 UA 110882 C2 Таблиця 11 Початковий 3 місяці 6 місяців 9 місяців 12 місяців 18 місяців 24 місяці 36 місяців Тестування вмісту 96,5 % 95,4 % 95,4 % 96,7 % 96,9 % 96,4 % 96,5 % 96,0 % Таблиця 12 Тестування вмісту 5 Початковий 98,4 % 1 тиждень 97,8 % 2 тижні 96,3 % 4 тижні 98,4 % Приклад 29 Згідно з наступною композицією, зазначеною у таблиці 13 нижче, фармацевтичну композицію без поверхнево-активної речовини складали як у прикладі 4. Таблиця 13 Компонент Гранулювання у співрозчиннику Гідрат 4-метил-N-[3-(4метилімідазол-1-іл)-5трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)бензаміду гідрохлориду 4-метил-N-[3-(4-метилімідазол-1іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)бензаміду дихлоргідрат 4-метил-N-[3-(4-метилімідазол1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іламіно)-бензаміду метансульфонат мікрокристалічна целюлоза гідрат дикальційфосфату легка безводна кремнієва кислота кросповідон після змішування кросповідон стеарат магнію 10 15 20 вага вага вага компокомпокомпо- співвідноспіввідноспіввіднонента в нента в нента в шення шення (%) шення (%) капсулі капсулі капсулі (%) (мг) (мг) (мг) Приклад 29 Приклад 30 Приклад 31 220,4 55,5 227,3 56,2 236,1 57,2 50 70 11 12,6 17,6 2,8 50 70 11 12,4 17,3 2,7 50 70 11 12,1 16,9 2,7 30 7,5 30 7,4 30 7,3 10 6 2,5 1,5 10 6 2,5 1,5 10 6 2,4 1,5 Дана композиція являє собою гранулярну суміш, котру отримували таким же способом, який описаний у прикладі 2, але з якої виключали поверхнево-активну речовину (поліетиленгліколю гліцерингідроксистеарат). Нею заповнювали капсулу. Приклад 30 Гранулярну суміш без поверхнево-активної речовини (поліетиленгліколю гліцерингідроксистеарату) отримували тим же способом, який описаний у прикладі 29. Нею заповнювали капсулу. Приклад 31 Гранулярну суміш без поверхнево-активної речовини (поліетиленгліколю гліцерингідроксистеарату) отримували тим же способом, який описаний у прикладі 29. Нею заповнювали капсулу. Тестовий приклад 5. Профіль розчинності Капсули, заповнені гранулярними сумішами, отриманими у прикладах 29-31, тестували за допомогою ванни при температурі розчинення 37 °C відповідно до способу 2 (спосіб із 14 UA 110882 C2 5 застосуванням лопатевої мішалки) з числа способів тестування розчинності за Корейською фармакопеєю. Використовували 900 мл буфера з pH 1,2 (доповненого SLS 1 %) у якості середовища розчинення та виконували тест на розчинність за швидкості мішалки 100 об./хв. Кількість розчиненого лікарського матеріалу вимірювали за допомогою HPLC. Швидкості розчинення (%) гідрату 4-метил-N-[3-(4-метилімідазол-1-іл)-5-трифторметил-феніл]-3-(4піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду, дигідрохлориду та метансульфонату у якості функції часу показані на фіг. 3 та у таблиці 14. Таблиця 14 Приклад 4 Приклад 29 Приклад 30 Приклад 31 10 15 5 хв. 0,1 0 0 0 10 хв. 28,6 6,8 1,4 2,7 15 хв. 44,9 23,7 3,4 6,3 30 хв. 71,4 59,3 8,2 11,4 45 хв. 85,9 77,1 11,5 15,6 60 хв. 95,9 86,0 14,0 18,2 Як показано у таблиці 14 та на фіг. 3, приклад 29 із застосуванням гідрату гідрохлориду у якості адитивної солі показав значно вищу швидкість розчинення у порівнянні з прикладами 30 та 31. Також, при порівнянні прикладу 4 та прикладу 29, виявили, що застосування гідрохлориду у якості адитивної солі приводить до більш відмінної швидкості розчинення. Хоча переважний варіант здійснення даного винаходу описували для цілей ілюстрації, фахівцям в даній галузі техніки буде зрозуміло, що різноманітні модифікування, додавання та заміни можливі без відхилення від обсягу та суті даного винаходу, який розкритий у доданій формулі винаходу. ФОРМУЛА ВИНАХОДУ 20 25 30 35 40 45 50 1. Фармацевтична композиція, яка містить: гранулу, яка містить 4-метил-N-[3-(4-метилімідазол1-іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид як активний інгредієнт та поверхнево-активну речовину, яка являє собою щонайменше один тип матеріалу, вибраний із групи, що включає рицинову олію, лецитин (яєчний) та жирно кислотний макроголгліцерид. 2. Фармацевтична композиція за п. 1, де фармацевтична композиція складена у гранули, порошки, пігулки, капсули або рідку суспензію, при цьому містить активний інгредієнт у кількості від 20 мг до 1000 мг. 3. Фармацевтична композиція за п. 1, де активний інгредієнт має середній розмір частинок приблизно 50 мкм або менше. 4. Фармацевтична композиція за п. 1, де рицинова олія вибрана з групи, що включає поліетиленгліколю гліцерингідростеарат, кремофор RH 40 або кремофор RH 60. 5. Фармацевтична композиція за п. 1, де жирнокислотний макроголгліцерид являє собою Gelucire 44/14. 6. Фармацевтична композиція за п. 1, де фармацевтична композиція додатково містить наповнювач, який являє собою щонайменше один тип матеріалу, вибраний із групи, що включає целюлозу, її похідне, у тому числі мікрокристалічну целюлозу, гідроксипропілцелюлозу, гідроксіетилцелюлозу, гідроксипропілметилцелюлозу, гідрат лактози, ангідрат лактози, дикальційфосфат, легкий кремнієвий ангідрид, крохмаль, маніт, полівінілпіролідон, поліетиленгліколь та еудрагіт. 7. Фармацевтична композиція за п. 6, де наповнювач застосований у кількості від 0,01 до 60 масових часток відносно 100 масових часток активного інгредієнта. 8. Фармацевтична композиція за п. 1, яка додатково містить розпушувач, засіб, що обумовлює плинність, або їх суміш. 9. Фармацевтична композиція за п. 1, де поверхнево-активна речовина включена у концентрації від 0,1 масової частки до 30 масових часток відносно загальної ваги композиції. 10. Фармацевтична композиція за п. 8, де розпушувач являє собою щонайменше один тип матеріалу, вибраний із групи, що включає поперечно-зшитий полівінілпіролідон, кросповідон, натрію кроскармелозу, натрію крохмальгліколат, крохмаль та альгінат. 11. Фармацевтична композиція за п. 8, де співвідношення розпушувача до поверхнево-активної речовини складає 0,66 або більше. 12. Спосіб отримання фармацевтичної композиції, яка містить 4-метил-N-[3-(4-метилімідазол-1іл)-5-трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлорид, при 15 UA 110882 C2 5 10 15 20 25 30 цьому спосіб включає етап вологого/сухого гранулювання із застосуванням поверхнево-активної речовини, органічного розчинника або співрозчинника (водного/органічного розчинника) як солюбілізувальний засіб активного інгредієнта фармацевтичної композиції, де поверхнево-активна речовина являє собою щонайменше один тип матеріалу, вибраний із групи, що включає рицинову олію, лецитин (яєчний) та жирнокислотний макроголгліцерид; органічний розчинник вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон; та співрозчинник містить воду та органічний розчинник, вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон. 13. Спосіб за п. 12, де рицинова олія вибрана з групи, що включає поліетиленгліколю гліцерингідростеарат, кремофор RH 40 або кремофор RH 60. 14. Спосіб за п. 12, де жирнокислотний макроголгліцерид являє собою Gelucire 44/14. 15. Спосіб за п. 12, що додатково включає етап отримання гранулюючої рідини із застосуванням поверхнево-активної речовини із органічним розчинником або співрозчинником. 16. Спосіб за п. 15, де застосовувана гранулююча рідина має розчинну здатність 0,2 мг/мл або більше. 17. Спосіб отримання фармацевтичної композиції, при цьому спосіб включає етапи: отримання порошкоподібної суміші із застосуванням 4-метил-N-[3-(4-метилімідазол-1-іл)-5трифторметил-феніл]-3-(4-піразин-2-іл-піримідин-2-іламіно)-бензаміду гідрохлориду та щонайменше одного типу фармацевтично прийнятного наповнювача; отримання вологих гранул шляхом об'єднання порошкоподібної суміші з поверхнево-активною речовиною, органічним розчинником або співрозчинником (водним/органічним розчинником) як гранулюючу рідину; отримання гранул шляхом висушування вологих гранул; та здійснення способу добору, де поверхнево-активна речовина являє собою щонайменше один тип матеріалу, вибраний із групи, що включає рицинову олію, лецитин (яєчний) та жирнокислотний макроголгліцерид; органічний розчинник вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон; та співрозчинник містить воду та органічний розчинник, вибраний з групи, що включає етанол, метанол, ізопропанол та ацетон. 18. Спосіб за п. 17, де рицинова олія вибрана з групи, що включає поліетиленгліколю гліцерингідростеарат, кремофор RH 40 або кремофор RH 60. 19. Спосіб за п. 17, де жирнокислотний макроголгліцерид являє собою Gelucire 44/14. 16 UA 110882 C2 17 UA 110882 C2 Комп’ютерна верстка Л. Литвиненко Державна служба інтелектуальної власності України, вул. Василя Липківського, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут інтелектуальної власності”, вул. Глазунова, 1, м. Київ – 42, 01601 18
ДивитисяДодаткова інформація
Назва патенту англійськоюPharmaceutical composition and preparation method thereof
Автори англійськоюKim, Dong Yeon, Shin, Jae Soo, Lim, Hyun Tae, Kim, Dae Sung, Lee, Seung Hyoun, Sung, Jun Ho
Автори російськоюКхим Тон Йон, Схин Че Соо, Лим Хюн Те, Кхим Тэ Сун, Ли Син Хён, Сун Чун Хо
МПК / Мітки
МПК: A61K 31/505, A61K 9/20, A61K 31/685, A61P 35/00, A61K 9/16
Мітки: фармацевтична, композиція, спосіб, отримання
Код посилання
<a href="https://ua.patents.su/20-110882-farmacevtichna-kompoziciya-ta-sposib-otrimannya.html" target="_blank" rel="follow" title="База патентів України">Фармацевтична композиція та спосіб її отримання</a>
Похідні ізоіндоліну, фармацевтична композиція на їх основі, спосіб лікування хвороби альцгеймера, спосіб інгібування агрегації амілоїдних білків та спосіб виявлення амілоїдних відкладень

Номер патенту: 64842
Опубліковано: 15.03.2004
Автори: Уокер Лері Кресвелл, Оджеллі-Шафран Корінн Елізабет, Секкаб Аннетт Тереза, Лай Йінгджіє
МПК: A61K 31/4035, C07D 403/12, C07D 209/44, C07D 401/12, A61P 43/00, C07D 403/10, C07D 401/10
Мітки: хвороби, агрегації, білків, фармацевтична, виявлення, ізоіндоліну, основі, спосіб, відкладень, інгібування, композиція, похідні, амілоїдних, альцгеймера, лікування
Формула / Реферат:
1. Похідні ізоіндоліну формули Іабо їх фармацевтично прийнятні солі,де Χ - феніл або заміщений феніл,Υ - феніл, заміщений феніл, піридил або заміщений піридил,де заміщений феніл та заміщений піридил можуть мати 1-4 замісники, кожний з яких незалежно вибраний з -ОС1-С12алкілу, галогену, -С1-С6алкілу, фенілу, -C(O)NHR",...
19-норстероїди, галогеновані в положенні 17, спосіб їх отримання, фармацевтична композиція та лікарський засіб, проміжні продукти

Номер патенту: 66867
Опубліковано: 15.06.2004
Автори: Буалі Йасміна, Ван Де Вельд Патрік, НІК Франсуа, Може Жак
МПК: C07J 41/00, C07J 43/00, A61P 5/24, A61K 31/58, A61P 19/10, A61K 31/565
Мітки: композиція, 19-норстероїди, проміжні, лікарський, засіб, галогеновані, положенні, отримання, спосіб, фармацевтична, продукти
Формула / Реферат:
1. 19-норстероїди загальної формули (I):,в якій:R1 означає атом водню, радикал (СН2)m-Аr, (СО)-Аr, (СН2)m-Alk або (CO)-Alk, R2 означає вуглеводневий радикал, лінійний або розгалужений, насичений або ненасичений, що містить від 1 до 6 атомів вуглецю,Х означає атом галогену,Y означає простий зв'язок, O, NH, S, SO або SО2,Z...
Похідні піримідину, спосіб їх отримання, фармацевтична композиція на їх основі, спосіб її отримання, комбінація сполук та продукт для лікування віл

Номер патенту: 72458
Опубліковано: 15.03.2005
Автори: Де Корте Барт, Андрес Конраад Джозеф Людовійк Марсель, Янссен Пауль Адріаан Ян, Хеерес Ян, Кукла Міхаель Джозеф, Хо Чі Юнг, Янссен Марсель Аугуст Констант, де Йонге Марк Рене, ван Акен Коен Жеанн Альфонс, Людовісі Дональд Віл'ям, Койманс Люс'єн Маріа Хенрікус
МПК: C07D 239/42, C07D 239/48, C07D 239/50, A61K 31/506, C07D 239/46, C07D 401/12, C07D 239/47, C07D 409/12, A61P 31/12, A61K 31/505
Мітки: фармацевтична, комбінація, спосіб, отримання, сполук, похідні, продукт, основі, віл, піримідину, лікування, композиція
Формула / Реферат:
1. Похідна піримідину формули (I’):,її N-оксиди, фармацевтично прийнятні солі та стереохімічно ізомерні форми, де Q є атомом водню або -NR1R2, R1 та R2 кожний незалежно вибраний з атому водню, гідроксигрупи, С1-І2-алкілу, С1-І2-алкокси, С1-І2-алкілкарбоніл, С1-І2-алкоксикарбонілу, арилу, аміно, моно- або ді(С1-І2-алкіл)аміно, моно- або...
Похідні аденозину (варіанти), спосіб їх отримання, фармацевтична композиція та спосіб лікування пацієнта (варіанти)

Номер патенту: 64794
Опубліковано: 15.03.2004
Автори: Дайк Гайзел Джоун, Пеннелл Андрю Майкл Кеннет, Елдред Колін Дейвід, Песс Мартін, Казінс Річард Пітер Чарльз, Беис Дейвід Едмунд, Джадкінс Брайан Дейвід
МПК: A61K 31/70, A61P 9/00, C07H 19/16, A61P 25/00
Мітки: композиція, лікування, фармацевтична, варіанти, похідні, пацієнта, отримання, аденозину, спосіб
Формула / Реферат:
1. Похідні аденозину формули (Іb), Ibде Х - О або СН2;R2 - С1-3алкіл, С1-3алкоксил, галоген або гідроген;R3 - Н, феніл (як варіант, заміщений галогеном), 5-6-членний гетероарил, С1-6 алкоксил, С1-6алкілО(СН2)n, де n = 0-6, С3-7циклоалкіл, С1-6гідроксіалкіл, галоген або лінійний чи розгалужений С1-6алкіл, С1-6алкеніл або С1-6алкініл, що, як...
Сполуки 2,3-діарилпіразоло[1,5-в]піридазинів, спосіб їх отримання (варіанти) та фармацевтична композиція на їх основі

Номер патенту: 68358
Опубліковано: 16.08.2004
Автори: Бесвік Пол, Нейлор Елан, Кампбелл Ян, Мет'юз Нейл
МПК: C07D 237/00, C07D 487/04, C07D 231/00, A61K 31/50
Мітки: фармацевтична, 2,3-діарилпіразоло[1,5-в]піридазинів, сполуки, варіанти, спосіб, композиція, отримання, основі
Формула / Реферат:
1. Сполуки 2, 3-діарилпіразоло[1,5-В]піридазинів формули (I)(І)та їх фармацевтично прийнятні похідні, в яких:R0 – галоген, С1–6алкіл, С1–6алкоксил, С1–6алкоксил, заміщений одним чи більше атомами флуору, або О(СН2)nNR4R5, R1 та R2 незалежно один від одного вибрані з групи, яка складається з гідрогену, С1–6алкілу, С1–6алкілу, заміщеного одним чи...
Попередній патент: Ароматизуюча композиція для поліпшення тютюнового запаху повітря, яке видихається
Наступний патент: Спосіб отримання маточної суміші на основі вуглецевих нанонаповнювачів і надпластифікатора та її застосування в неорганічних отверджуваних системах
Випадковий патент: Система для гасіння вогню