Макроциклічні інгібітори вірусу гепатиту с
Номер патенту: 95239
Опубліковано: 25.07.2011
Автори: Де Кок Герман Августінус, Антонов Дмітрій, Розенквіст Еса Анніка Крістіна, Канберг Піа Сесілія, Нілссон Карл Магнус, Самуельссон Бенгт Бертіл, Ліндстром Матс Стефан, Рабуассон П'єр Жан-Марі Бернар, Сіммен Кеннет Алан






























Формула / Реферат
1. Сполука, що має формулу
і її N-оксиди, солі і стереоізомери, у якій пунктирна лінія позначає можливий подвійний зв'язок між атомами С7 і С8:
R1 позначає водень або С1-6-алкіл;
R2 позначає водень або С1-6-алкіл; і
n = 3,4, 5 або 6.
2. Сполука за п. 1, яка відрізніться тим, що має формулу (Іа):
 .
.
3. Сполука за будь-яким із пп. 1, 2, яка відрізняється тим, що має формулу (Іb):
 .
.
4. Сполука за будь-яким із пп. 1-3, яка відрізняється тим, що n = 4 або 5.
5. Сполука за будь-яким із пп. 1-3, яка відрізняється тим, що R1 позначає водень або метил.
6. Сполука за будь-яким із пп. 1-3, яка відрізняється тим, що R2 позначає водень.
7. Сполука за будь-яким із пп. 1-3, яка відрізняється тим, що R2 позначає метил.
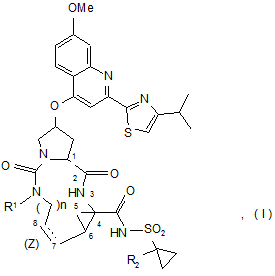
8. Сполука за будь-яким із пп. 1-3, яка відрізняється тим, що сполука вибрана з
 ,
,
 ,
,
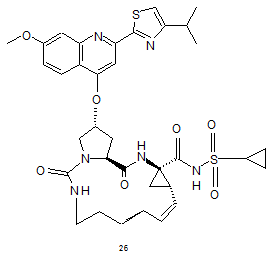 ,
,
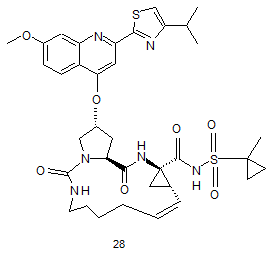 ,
,
 .
.
9. Сполука за будь-яким із пп. 1-8, яка відрізняється тим, що не є N-оксидом або сіллю.
10. Фармацевтична композиція, що містить носій та як активний інгредієнт противірусно ефективну кількість сполуки за будь-яким із пп. 1-9.
11. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає:
отримання сполуки формули (І), у якій зв'язок між С7 і С8 являє собою подвійний зв'язок, яка є сполукою формули (Іа), як визначено в п. 2, шляхом утворення подвійного зв'язку між С7 і С8, зокрема шляхом реакції обміну олефінів, із супутньою циклізацією з утворенням макроциклу, як показано в наступній реакційній схемі:
 .
.
12. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає:
перетворення сполуки формули (Іа) у сполуку формули (І), у якій зв'язок між С7 і С8 у макроциклі являє собою простий зв'язок, тобто сполуку формули (Іb), як визначено в п. 3, шляхом відновлення подвійного зв'язку С7-С8 у сполуці формули (Іа).
13. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає:
введення в реакцію циклопропілсульфонаміду (IV) із проміжною сполукою (III) через реакцію утворення аміду, як показано в наступній реакційній схемі:
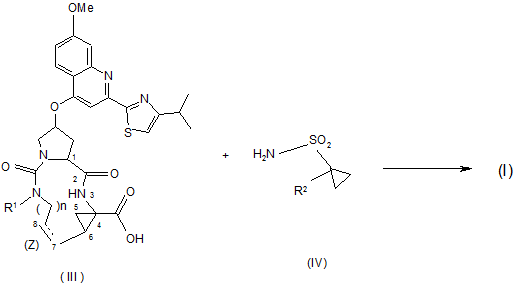 .
.
14. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає:
етерифікацію проміжної сполуки (V) хіноліном формули (VI), як показано в наступній реакційній схемі:
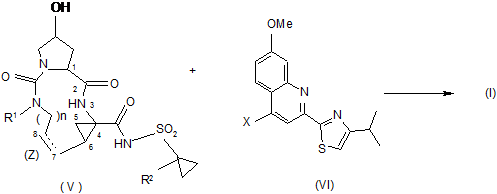 ,
,
у якій X в (VI) позначає гідрокси або группу, що видаляється; причому зазначена реакція, зокрема, є реакцією О-арилування, у якій X позначає группу, що видаляється, або реакцією Mitsunobu, у якій X позначає гідрокси.
15. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає:
отримання сполуки формули (І), у якій R1 позначає водень, причому зазначена сполука представлена (Іd), з відповідної захищеної по азоту проміжної сполуки (VII), у якій PG позначає захисну групу атома азоту:
 .
.
16. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає:
перетворення сполук формули (І) одна в одну реакцією перетворення функціональної групи.
17. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає:
отримання форми солі шляхом введення в реакцію вільної форми сполуки формули (І) з кислотою або основою.
Текст
1. Сполука, що має формулу 3 95239 шляхом реакції обміну олефінів, із супутньою циклізацією з утворенням макроциклу, як показано в наступній реакційній схемі: S O N 4 N OMe O O H N N O O O N O N S N O N H S N O , 16 R1 N O 2 S N 1 O N ( HN )n (Ia) O 3 5 4 8 SO2 HN 6 7 R2 O O H N N O O S N H O HN O , 9 S O N N O O O OMe H N N S O N H O NH ( II ) . 12. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає: перетворення сполуки формули (Іа) у сполуку формули (І), у якій зв'язок між С7 і С8 у макроциклі являє собою простий зв'язок, тобто сполуку формули (Іb), як визначено в п. 3, шляхом відновлення подвійного зв'язку С7-С8 у сполуці формули (Іа). 13. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає: введення в реакцію циклопропілсульфонаміду (IV) із проміжною сполукою (III) через реакцію утворення аміду, як показано в наступній реакційній схемі: O N N O , 26 S S O N N R1 N H O OH ( III ) S O 6 7 (IV) H N N O , 28 S 14. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає: етерифікацію проміжної сполуки (V) хіноліном формули (VI), як показано в наступній реакційній схемі: OH N OMe N N 1 + O O O 2 O O H N N O HN 5 4 (Z) O O O )n R2 O 3 8 O NH HN (I) O 2 N ( SO2 H2 N + 1 O N O R1 N ( HN )n 5 8 S N H O 33 . 9. Сполука за будь-яким із пп. 1-8, яка відрізняється тим, що не є N-оксидом або сіллю. 10. Фармацевтична композиція, що містить носій та як активний інгредієнт противірусно ефективну кількість сполуки за будь-яким із пп. 1-9. 11. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає: отримання сполуки формули (І), у якій зв'язок між С7 і С8 являє собою подвійний зв'язок, яка є сполукою формули (Іа), як визначено в п. 2, шляхом утворення подвійного зв'язку між С7 і С8, зокрема (Z) N O 3 4 6 SO2 HN (I) N X S 7 R2 (V) , у якій X в (VI) позначає гідрокси або группу, що видаляється; причому зазначена реакція, зокрема, є реакцією О-арилування, у якій X позначає группу, що видаляється, або реакцією Mitsunobu, у якій X позначає гідрокси. 15. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає: 1 отримання сполуки формули (І), у якій R позначає водень, причому зазначена сполука представлена (Іd), з відповідної захищеної по азоту проміжної сполуки (VII), у якій PG позначає захисну групу атома азоту: (VI) 5 95239 OMe N N N N O S N Видалення захисної групи HN )n N H 5 8 4 6 O 2 O 3 1 O O 2 (Z) S 1 O N PG ( HN SO2 N ( HN )n O 3 5 8 (Z) 7 4 6 HN SO2 7 R2 (VII ) 16. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає: перетворення сполук формули (І) одна в одну реакцією перетворення функціональної групи. 17. Спосіб отримання сполуки за будь-яким із пп. 1-9, що включає: отримання форми солі шляхом введення в реакцію вільної форми сполуки формули (І) з кислотою або основою. OMe O 6 R2 ( I-d ) . Даний винахід відноситься до макроциклічних сполук, що мають інгібуючу активність відносно реплікації вірусу гепатиту С (HCV). Він також відноситься до композицій, що включають ці сполуки у якості активних інгредієнтів, а також до способів отримання цих сполук і композицій. Вірус гепатиту С є провідною причиною хронічного захворювання печінки в усьому світі і на ньому сфокусовані значні медичні дослідження HCV є членом сімейства вірусів Flaviviridae з роду hepacivirus, і близько пов'язані з родом flavivirus, що включає безліч вірусів, що беруть участь у розвитку захворювань людини, таких як вірус Денге і вірус жовтої лихоманки, і до сімейства pestivirus тварин, що включає вірус вірусної діареї великої рогатої худоби (BVDV) HCV є вірусом, що містить РНК з позитивним одноланцюговим геномом, що містить приблизно 9 600 основ. Геном включає як 5', так і 3' нетрансльовані області, які встановлюють вторинні структури РНК, і центральну відкриту рамку зчитування, що кодує єдиний полі протеїн із приблизно 3010-3030 амінокислот. Цей поліпротеїн кодує десять генних продуктів, які походять від поліпротеїну-попередника в ході впорядкованої серії ко- і посттрансляційних ендопротеолітичних розщеплень, опосередковуваних як хазяйськими, так і вірусними протеазами. Вірусні структурні білки включають білок центрального нуклеокапсиду й два глікопротеїда оболонки Е1 і Е2. Неструктурні (НС) білки кодують деякі суттєві вірусні ферментативні функції (геліказа, полімераза, протеаза), а також білки з невідомою функцією. Реплікація вірусного генома опосередковується РНК-залежною РНК полімеразою, що кодується неструктурним білком 5b (NS5B). На додаток до полімерази, було показано, що функції вірусної гелікази й протеази, які обидві кодуються у біфункціональному білці NS3, є суттєвими для реплікації РНК HCV. На додаток до сироваткової протеази NS3, HCV також кодує металопротеїназу в області NS2. Після початкової гострої інфекції у більшості інфікованих людей розвивається хронічний гепатит, тому що HCV реплікується вибірково в гепатоцитах, але не є безпосередньо цитопатичним. Зокрема, відсутність сильної Т-лимфоцитної відповіді й висока схильність вірусу до мутацій, очевидно, забезпечують високу частоту хронічної інфекції. Хронічний гепатит може розвиватися до фіброзу печінки, що призводить до цирозу, термінальної стадії захворювання печінки, і НСС (гепа тоцелюлярний рак), що робить його провідною причиною трансплантацій печінки. Існує 6 головних генотипів HCV і більше 50 підтипів, які географічно розподілені по-різному HCV типу 1 є переважним генотипом у Європі й США. Широка генетична гетерогенність HCV має важливі діагностичні і клінічні прояви, можливо пояснюючи труднощі у розробці вакцин і відсутності реакції на терапію. Передача HCV може відбуватися через контакт із зараженими продуктами крові або кров'ю, наприклад, після переливання крові або внутрішньовенного використання лікарського засобу. Впровадження діагностичних тестів, що використовується у скринінгу крові, призвело до тенденції до зниження частоти посттрансфузійних випадків зараження HCV. Однак, приймаючи до уваги повільну прогресію до термінальної стадії захворювання печінки, існуючі інфекції будуть і надалі являти серйозну медичну й економічну проблему протягом багатьох десятиліть. Існуючі терапії HCV засновані на (пегільованому) альфа інтерфероні (IFN-α) у комбінації з рибавірином. Ця комбінована терапія призводить до пролонгованої вірусологічної відповіді у більше ніж 40 % пацієнтів, інфікованих генотипом 1 вірусу, і приблизно 80 %, інфікованих генотипами 2 і 3. Крім обмеженої ефективності по відношенню до HCV типу 1, ця комбінована терапія має значні побічні ефекти й погано переноситься багатьма пацієнтами. Головні побічні ефекти включають грипо-подібні симптоми, гематологічні аномалії й нейропсихіатричні симптоми. Отже, існує потреба у більш ефективних, зручних і краще переносимих методах лікування. Нещодавно два пептидоміметичні інгібітори HCV-протеази привернули увагу як клінічні кандидати, а саме, BILN-2061, розкритий в WO00/59929, і VX-950, розкритий в WO03/87092. Безліч подібних інгібіторів HCV-протеази було також розкрито в академічній і патентній літературі Уже стало очевидно, що пролонговане введення BILN-2061 або VX-950 призводить до відбору мутантів HCV, які є резистентними до відповідного лікарського засобу, так званих drug escape mutants. Ці drug escape mutants мають характерні мутації в геномі HCV-протеази, особливо D168V, D168A та/або A156S. Відповідно, пацієнтам, які не піддаються лікуванню, потрібні додаткові лікарські засоби і іншими властивостями резистентності, і комбіно 7 вана терапія з використанням багатьох лікарських засобів, імовірно, буде нормою в майбутньому, навіть для лікування першої лінії. Експерименти з лікарськими засобами проти HIV, і зокрема, з інгібіторами HIV-протеази, додатково підтвердили, що субоптимальні фармакокінетики й складні режими введення швидко призводять до ненавмисних порушень режиму лікування. Це у свою чергу означає, що мінімальна 24годинна концентрація (мінімальна плазмова концентрація) відповідних препаратів в HIV-режимі часто падає нижче порога ІС90 або ED90 більшу частину дня. Вважається, що мінімальний 24годинний рівень щонайменше ІС50, і більш реалістично, ІC90 або ED90, є суттєвим для вповільнення розвитку drug escape mutants. Досягнення необхідних фармакокінетик і метаболізму лікарського засобу для забезпечення таких мінімальних рівнів висуває суворі вимоги до розробки лікарського засобу. Виражена пептидоміметична природа інгібіторів HCV-протеази з рівня техніки із множинними пептидними зв'язками є фармакокінетичною перешкодою для ефективного режиму введення. Існує потреба в інгібіторах HCV, які могли б перебороти недоліки існуючої терапії HCV, такі як побічні ефекти, обмежена ефективність, прояв резистентності й порушення режиму. Даний винахід стосується інгібіторів HCV, які перевершують аналоги з рівня техніки по відношенню до одного або більше наступних фармакологічних властивостей, тобто, потенціалу, зниженої цитотоксичності, поліпшеної фармакокінетики, поліпшеного профілю резистентності, прийнятної дози і розміру таблетки. Даний винахід стосується інгібіторів реплікації HCV, які можуть бути представлені формулою (І): і їх N-оксидів, солей і стереоізомерів, у якій пунктирна лінія позначає можливий подвійний зв'язок між атомами С7 і С8, 1 R позначає водень або С1-6-алкіл, 2 R позначає водень або С1-6-алкіл, і n = 3, 4, 5 або 6 Даний винахід стосується двох підгруп інгібіторів реплікації HCV, які можуть бути представлені формулою (I-а) і (I-b) 95239 8 1 і їх N-оксидів, солей і стереоізомерів, у якій R , R і n мають визначені тут значення. Винахід також відноситься до способів отримання сполук формули (l), їх N-оксидів, солей приєднання, четверинних амінів, комплексів з металами і стереохімічно ізомерних форм, їх проміжних продуктів і застосування проміжних продуктів в отриманні сполук формули (І). Винахід відноситься до сполук формули (І) per se, їх N-оксидам, солям приєднання, четверинним амінам, комплексам з металами і стереохімічно ізомерним формам для застосування у якості лікарського засобу. Винахід також відноситься до фармацевтичних композицій, що включають носій і ефективну у противірусному відношенні кількість сполуки формули (I) як воно визначено тут. Фармацевтичні композиції можуть включати комбінації зазначених сполук із іншими анти-HCV засобами. Винахід також відноситься до зазначених фармацевтичних композицій для введення пацієнтові, що страждає на інфекцію HCV. Винахід також відноситься до застосування сполуки формули (І) або u N-оксиду, солі приєднання, четверинного аміну, комплексу з металом або стереохімічно ізомерних форм для отримання лікарського засобу для інгібування реплікації HCV. Або винахід відноситься до способу інгібування реплікації HCV у теплокровної тварини, що включає введення ефективної кількості сполуки формули (І) або її N-оксиду, солі приєднання, четверинного аміну, комплексу з металом або стереохімічно ізомерних форм. Раніше й надалі використовуються наступні визначення, якщо не зазначене інше. У рамках винаходу "С1-6-алкіл" як група або частина групи позначає насичений вуглеводневий радикал з прямим або розгалуженим ланцюгом, що має від 1 до 6 атомів вуглецю, такий як, наприклад, метил, етил, 1-пропіл, 2-пропіл, 1-бутил, 22 9 бутил, 2-метил-1-пропіл, 1-пентил, 2-пентил, 3пентил, 1-гексил, 2-гексил, 2-метил-1-бутил, 2метил-1-пентил, 2-етил-1-бутил, 3-метил-2-пентил і т. п. Серед С1-6-алкілів цікаві С1-4-алкіли. Щораз надалі термін "сполуки формули (І)" або "сполуки за винаходом" або подібні терміни включають сполуки формули (І), кожну й будь-яку з підгруп цих сполук, їх проліки, N-оксиди, солі приєднання, четверинні аміни, комплекси з металами й стереохімічно ізомерні форми. Один варіант здійснення включає сполуки формули (І) або будь-яку підгрупу сполук формули (І), визначених тут, а також N-оксиди, солі і можливі стереоізомерні форми. Інший варіант здійснення включає сполуки формули (І) або будь-яку підгрупу сполук формули (І), визначених тут, а також солі і можливі стереоізомерні форми. Сполуки формули (I) мають кілька хіральних центрів і існують як стереохімічно ізомерні форми. Термін "стереохімічно ізомерні форми" у рамках винаходу позначає всі можливі сполуки, що складаються з тих же самих атомів, що поєднані тією же самою послідовністю зв'язків, але мають різні просторові структури, які не є взаємозамінними, які можуть мати сполуки формули (І). Відносно випадків, де (R) або (S) використовується для визначення абсолютної конфігурації хірального атома в складі замісника, позначення роблять, беручи до уваги цілу сполуку, а не окремо замісник. Якщо не вказане або не позначене інше, хімічне позначення сполуки охоплює суміш усіх можливих стереохімічно ізомерних форм, які зазначена сполука може охоплювати. Зазначена суміш може містити всі діастереоізомери та/або енантіомери основної молекулярної структури зазначеної сполуки. Усі стереохімічно ізомерні форми сполук згідно даному винаходу, як у чистій формі, так і в суміші один з одним, входять у рамки даного винаходу. Чисті стереоізомерні форми сполук і проміжних продуктів, як вони зазначені тут, визначають як ізомери, що в основному не містять інших енантіомерних або діастереомерних форм однієї й тієї ж основної молекулярної структури зазначених сполук або проміжних продуктів. Зокрема, термін "стереохімічно чисті" відноситься до сполук або проміжних продуктів, що мають стереоізомерний надлишок від щонайменше 80 % (тобто мінімум 90 % одного ізомеру і максимум 10 % інших можливих ізомерів) до стереоізомерного надлишку 100 % (тобто 100 % одного ізомеру і відсутність іншого), зокрема, до сполук або проміжних продуктів, що мають стереоізомерний надлишок від 90 % до 100 %, більш переважно, що має стереоізомерний надлишок від 94 % до 100 %, і найбільше переважно, що мають стереоізомерний надлишок від 97 % до 100 %. Терміни "енантіомерно чистий" і "діастереомерно чистий" повинні розумітись у такий же спосіб, але відносяться до енантіомерного надлишку і діастереомерного надлишку, відповідно, розглядаємої суміші. Чисті стереоізомерні форми сполук і проміжних продуктів за винаходом можуть бути отримані з використанням відомих з рівня техніки процедур. 95239 10 Наприклад, енантіомери можуть бути відділені один від одного селективною кристалізацією їх діастереомерних солей з оптично активними кислотами або основами. Прикладами цього є винна кислота, дибензилвинна кислота, дитолуолвинна кислота й камфорсульфонова кислота. Альтернативно, енантіомери можуть бути відділені хроматографічними методами з використанням хіральних стаціонарних фаз Зазначені чисті стереохімічно ізомерні форми можуть також бути отримані з відповідних чистих стереохімічно ізомерних форм відповідних вихідних матеріалів, за умови, що реакція відбувається стереоспецифічно. Переважно, якщо бажаний певний стереоізомер, зазначену сполуку синтезують стереоспецифічними способами отримання. У цих способах переважно використовують енантіомерно чисті вихідні матеріали. Діастереомерні рацемати сполук формули (І) можуть бути отримані окремо звичайними способами. Відповідними способами фізичного поділу, які можуть використовуватись, є, наприклад, селективна кристалізація і хроматографія, наприклад хроматографія на колонках. Для деяких із сполук формули (I), їх N-оксидів, солей, сольватів, четверинних амінів або комплексів з металами і проміжних продуктів, що використовуються у їх отриманні, абсолютна стереохімічна конфігурація не була експериментально визначена. Фахівець може визначити абсолютну конфігурацію таких сполук, використовуючи відомі з рівня техніки способи, такі як, наприклад, дифракція рентгенівських променів. Даний винахід також включає всі ізотопи атомів, що зустрічаються на сполуках за винаходом Ізотопи включають атоми, що мають той же самий атомний номер, але різні масові числа. У якості загального прикладу і без обмеження, ізотопи водню включають тритій і дейтерій Ізотопи вуглецю включають С-13 і С-14. Термін "проліки" у цьому тексті позначає фармакологічно прийнятні похідні, такі як складні ефіри, аміди і фосфати, такі, що отриманий in vivo продукт біотрансформації цього похідного є активним лікарським засобом, як визначено у сполуких формули (І). У даний опис включена шляхом посилання робота Goodman and Gilman (The th Pharmacological Basis of Therapeutics, 8 ed, McGraw-Hill, Int Ed 1992, "Biotransformation of Drugs", р 13-15), що описує проліки загалом. Проліки переважно мають чудову розчинність у воді, збільшену біодоступність і легко метаболізуються в активні інгібітори in vivo. Проліки сполуки згідно з даним винаходом можуть бути отримані шляхом модифікації функціональних груп, присутніх у сполуці, таким чином, що модифікації відщеплюються, або звичайною маніпуляцією, або in vivo, з отриманням батьківської сполуки. Бажаними є фармацевтично прийнятні складноефірні проліки, які можуть гідролізуватись in vivo і є похідними сполук формули (І), що мають гідроксильну або карбоксильну групу. Гідролізуємий in vivo складний ефір являє собою складний ефір, що гідролізується в організмі людини або тварин з утворенням материнської кислоти або спирту. Прийнятні фармацевтично прийнятні складні ефі 11 ри для карбокси включають складні ефіри С1-6алкоксиметил, наприклад, метоксиметил, складні ефіри С1-6-алканоілоксиметила, наприклад, півалоілоксиметил, складні ефіри фталідилу, складні ефіри С3-8-циклоалкоксикарбонілокси-С1-6-алкіл, наприклад, циклогексилкарбоніл-оксиетил; складні ефіри 1,3-діоксолен-2-онілметила, наприклад, 5метил-1, 3-діоксолен-2-онілметил; і складні ефіри С1-6-алкоксикарбонілоксиетила, наприклад, 1етоксикарбоніл-оксиетил, які можна утворити по будь-якій карбоксильній групі в сполуках за винаходом. Гідролізуємий in vivo складний ефір сполуки формули (І), що містить гідроксильну групу, включає неорганічні складні ефіри, такі як фосфатні складні ефіри і α-ацилоксиалкілові ефіри і споріднені сполуки, що у результаті гідролізу складного ефіру in vivo розщеплюються, утворюючи материнську гідроксильну групу. Приклади αацилоксиалкілових ефірів включають ацетоксиметокси й 2,2-диметилпропіонілокси-метокси. Вибір гідролізуємих in vivo груп, що утворюють складний ефір, для гідрокси включає алканоіл, бензоїл, фенілацетил і заміщений бензоїл і фенілацетил, алкоксикарбоніл (приводячи до складних ефірів алкілкарбонату), діалкілкарбамоїл і N(діалкіламіноетил)-N-алкілкарбамоїл (приводячи до карбамінатів), діалкіламіноацетил і карбоксиацетил. Приклади замісників на бензоїлі включають морфоліно й піперазино, пов'язані з кільцевим атомом азоту через метиленову групу в положенні 3 або 4 бензоїльного кільця. Для терапевтичного застосування, солі сполук формули (І) являють собою солі, у яких протиіон є фармацевтично прийнятним. Однак, солі з кислотою і основою, які не є фармацевтично прийнятними, можуть також знайти застосування, наприклад, в отриманні або очищенні фармацевтично прийнятної сполуки. Усі солі, чи є вони фармацевтично прийнятними чи ні, включені в рамки даного винаходу. Фармацевтично прийнятні солі приєднання з кислотою і основою, як зазначено вище, включають терапевтично активні нетоксичні форми солі приєднання з кислотою і основою, які сполуки формули (І) можуть утворювати. Фармацевтично прийнятні солі приєднання з кислотою можуть бути отримані шляхом обробки форми основи відповідною кислотою. Прийнятні кислоти включають, наприклад, неорганічні кислоти, такі як галогеноводневі кислоти, наприклад, хлористоводнева або бромистоводнева кислота, сірчана, азотна, фосфорна і т. п. кислоти; або органічні кислоти, такі як, наприклад, оцтова, пропіонова, гідроксиоцтова, молочна, піровиноградна, щавлева (тобто, етандикислота), малонова, бурштинова (тобто, бутандикислота), малеїнова, фумарова, оксибурштинова (тобто, гідроксибутандикислота), винна, лимонна, метансульфонова, етансульфонова, бензолсульфонова, п-толуолсульфонова, цикламова, саліцилова, п-аміносаліцилова, памова і т. п. кислоти. Навпаки, зазначені форми солі можуть бути перетворені обробкою відповідною основою у форму вільної основи. 95239 12 Сполуки формули (І), що містять кислий протон, можуть також бути перетворені в їх форми солі приєднання з аміном або нетоксичним металом шляхом обробки відповідними органічними і неорганічними основами. Прийнятні форми солі з основою включають, наприклад, солі амонію, солі лужного і лужноземельного металу, наприклад, солі літію, натрію, калію, магнію, кальцію і т. п., солі з органічними основами, наприклад, солі бензатину, N-метил-D-глюкаміну, гідрабаміну, і солі з амінокислотами, такі як, наприклад, аргінін, лізин і т. п. Термін «сіль приєднання», як він використовується вище, також включає сольвати, які сполуки формули (І), а також їх солі можуть утворювати. Такі сольвати є, наприклад, гідратами, алкоголятами і т. п. Термін "четверинний амін", як він використовується вище, позначає солі четверинного амонію, які сполуки формули (І) можуть утворювати в результаті реакції між основним атомом азоту сполуки формули (I) і відповідним кватернізуючим агентом, таким як, наприклад, у разі потреби заміщений алкілгалогенід, арилгалогенід або арилалкілгалогенід, наприклад, метилйодид або бензилйодид. Можуть також використовуватись інші реагенти із групами, що легко видаляються, такі як алкілтрифторметансульфонати, алкілметансульфонати, і алкіл п-толуолсульфонати. Четверинний амін має позитивно заряджений азот. Фармацевтично прийнятні протиіони включають хлор, бром, йод, трифторацетат і ацетат. Обраний протиіон можна вводити, використовуючи іонообмінні смоли. Форми N-оксидів сполук за винаходом включають сполуки формули (І), у яких один або кілька атомів азоту окислені до так званого N-оксиду. Слід розуміти, що сполуки формули (І) можуть мати метал-зв'язувальні, хелатуючі, комплексоутворюючі властивості і тому можуть існувати як комплекси з металами або хелати з металами. Такі похідні сполук формули (І), що містять метал, входять у рамки даного винаходу. Деякі із сполук формули (І) можуть також існувати в їх таутомерній формі. Такі форми, хоча вони явно не позначені в зазначеній формулі, включені в рамки даного винаходу. Як згадано вище, сполуки формули (І) мають кілька центрів асиметрії. Щоб більш ефективно вказати кожний із цих центрів асиметрії використовують систему нумерації, як позначено в наступній структурній формулі. 13 95239 14 Центри асиметрії присутні в положеннях 1, 4 і 6 макроциклу, а також в атомі вуглецю 3' у пірролідиновому кільці. Кожний з цих центрів асиметрії може перебувати в R або S конфігурації. Стереохімія в положенні 1 переважно відповідає стереохімії конфігурації L-амінокислоти, тобто L-пірролідин-альфа-карбоновоі кислоти. Сполуки формули (І) включають циклопропільні групи, представлені у структурному фрагменті нижче: де С7 позначає вуглець у положенні 7, і атоми вуглецю в положенні 4 і 6 являють собою асиметричні атоми вуглецю циклопропанового кільця. Незважаючи на інші можливі центри асиметрії на інших сегментах сполук за винаходом, наявність цих двох центрів асиметрії означає, що сполуки можуть існувати як суміші діастереомерів, таких як діастереомери сполук формули (І), у яких вуглець у положенні 7 перебуває в конфігурації syn по відношенню до карбонілу, або syn по відношенню до аміду, як показано нижче. Один варіант здійснення відноситься до сполук формули (І), у якій вуглець у положенні 7 перебуває в конфігурації syn по відношенню до карбонілу. Інший варіант здійснення відноситься до сполук формули (І), у якій конфігурація при атомі вуглецю в положенні 4 є конфігурацією R. Специфічну підгрупу сполук формули (І) складають сполуки, у яких вуглець у положенні 7 перебуває в конфігурації syn по відношенню до карбонілу, і в яких конфігурація при атомі вуглецю в положенні 4 є конфігурацією R. Сполуки формули (І) також включають залишок пірролідин-альфа-карбонової кислоти. Переважними є сполуки формули (І), у яких замісник у положенні 1 (або 5') і замісник у положенні 3' перебувають у транс-конфігурації. Особливий інтерес являють сполуки формули (І), у яких положення 1 має конфігурацію, що відповідає L-пірролідинальфа-карбоновій кислоті, і замісник у положенні 3' перебуває в транс-конфігурації відносно положення 1. Переважно, сполуки формули (І) мають стереохімію, як позначено в структурі формули (Iс) нижче: Переважно, пунктирна лінія позначає подвійний зв'язок між атомами вуглецю 7 і 8 у сполуках формули (І), (I-с) або в будь-якій підгрупі сполук формули (І). Більш переважно, зазначений подвійний зв'язок між атомами вуглецю 7 і 8 перебуває в цис-конфігураціі. Варто розуміти, що зазначена вище підгрупа сполук формул (I-b), а також будь-яка інша підгрупа, визначена тут, також включає будь-які Nоксиди, солі приєднання, четверинні аміни, комплекси з металами і стереохімічно ізомерні форми таких сполук. Коли n = 2, група -СН2-, до якої відноситься "n", відповідає етандіілу у сполуках формули (І) або в будь-якій підгрупі сполук формули (І). Коли n = 3, група -СН2", до якої відноситься "n", відповідає пропандіілу у сполуках формули (І) або в будь-якій підгрупі сполук формули (I). Коли n = 4, група -СН2, до якої відноситься "n", відповідає бутандіілу у сполуках формули (І) або в будь-якій підгрупі сполук формули (І). Коли n = 5, група -СН2-, до якої відноситься "n", відповідає пентандіілу в сполуках формули (І) або в будь-якій підгрупі сполук формули (І). Коли n = 6, група -СН2-, до якої відноситься "n", відповідає гександіілу в сполуках формули (І) або в будь-якій підгрупі сполук формули (І). Окремі підгрупи сполук формули (I) складають сполуки, у яких n = 4 або 5. Переважні варіанти здійснення винаходи відносяться до сполук формули (І) або будь-якої з 1 підгруп сполук формули (І), у яких R позначає водень або метил. Варіанти здійснення винаходу відносяться до сполук формули (I) або будь-якої з підгруп сполук 2 формули (І), у яких R позначає водень або С1-4алкіл, тобто, метил, етил, пропіл, ізопропіл, бутил, трет-бутил або ізобутил. Підгрупу сполук за винаходом складають сполуки формули (І) або будь-яка з підгруп сполук 2 формули (І), у яких R позначає водень. Іншу підгрупу сполук за винаходом складають сполуки формули (І) або будь-яка з підгруп сполук 2 формули (І), у яких R позначає метил. Сполуки формули (І) складаються із трьох головних будівельних блоків, P1, Р2 і Р3, кожний з яких обмежений кривою синусоїдальною лінією. Будівельний блок Р1 додатково містить частину Р'. Карбонільна група, відзначена зірочкою, може бути частиною або будівельного блоку Р2, або будівельного блоку Р3. З'єднання будівельних блоків, Р1 з Р2, Р2 з Р3 і Р1 з Р', включає утворення амід 15 ної сполуки. З'єднання блоків Р1 і Р3 включає формування подвійного зв'язку. З'єднання будівельних блоків Р1, Ρ', Ρ2 і Р3 з отриманням сполук формули (І) може бути здійснене в будь-якій заданій послідовності. Одна з стадій включає циклізацію, за допомогою чого утворюється макроцикл. Процедури синтезу, описані далі, є застосовними також для рацематів, стереохімічно чистих проміжних продуктів або кінцевих продуктів, як будь-які стереоізомерні суміші. Рацемати або стереохімічні суміші можуть бути розділені на стереоізомерні форми на будь-якій стадії процедур синтезу. В одному варіанті здійснення проміжні продукти і кінцеві продукти мають стереохімію, визначену вище в сполуках формули (I-е). В одному варіанті здійснення сполуки (І) отримують утворенням спочатку амідних сполук і наступним утворенням подвійного зв'язку між Р3 і Ρ1 із супутньою циклізацією до макроциклу. У кращому варіанті здійснення сполуки (І), у яких зв'язок між С7 і С8 являє собою подвійний зв'язок, які є сполуками формули (I-а), як вона визначена вище, можуть бути отримані, як показано у наступній реакційній схемі: Формування макроциклу може бути виконане через реакцію обміну олефіну в присутності придатного металевого каталізатора, такого як, наприклад, каталізатор на основі рутенію, про який повідомляють Miller, S.J., Blackwell, H.E., Grubbs, R.H. J. Chem. Soc. 118, (1996), 9606-9614; 20 Kingsbury, J. S., Harrity, J. P. Α., Bonitatebus, P. J., Hoveyda, A. H., J.. Chem. Soc. 121, (1999), 791-799; і Huang et al., J. Chem. Soc. 121, (1999), 2674-2678; наприклад, каталізатор Hoveyda-Grubbs. Можуть використовуватись стабільні на повітрі каталізатори рутенію, такі як біс(трициклогексилфосфін)-3-феніл-1Н-інден-1іліден рутеній хлорид (NeoRlyst M1®) або 95239 16 біс(трициклогексилфосфін)-[(фенілтіо)метилен] рутеній (IV) дихлорид. Іншими каталізаторами, які можуть використовуватись, є насамперед Grubbs і каталізатори другого покоління, тобто, бензиліденбіс(трициклогексилфосфін)дихлоррутеній і (1,3-біс(2,4,6-триметилфеніл)-2імідазолідиніліден)дихлор(фенілметилен)(трициклогексилфосфін)рутеній, відповідно. Особливо цікавими є спочатку Hoveyda-Grubbs і каталізатори другого покоління, якими є дихлор(оізопропоксифенілметилен)(трициклогексилфосфін)-рутеній (II) і (2,4,6триметилфеніл)-1,3-біс-2-імідазолідиніліден), дихлор(о-ізопропоксифенілметилен)рутеній, відповідно. Також для цієї реакції можуть використовуватись інші каталізатори, що містять інші перехідні метали, такі як Мо. Реакції обміну можуть проводитися в придатному розчиннику, такому як, наприклад, прості ефіри, наприклад, ТГФ, діоксан; галогеновані вуглеводні, наприклад, дихлорметан, СНСl3, 1,2дихлоретан і т. п. У кращому варіанті здійснення, реакцію обміну проводять у толуолі. Ці реакції проводять при підвищених температурах в атмосфері азоту. Сполуки формули (І), у яких зв'язок між С7 і С8 у макроциклі являє собою простий зв'язок, тобто, сполуки формули (I-b), можуть бути отримані із сполук формули (I-а) шляхом відновлення подвійного зв'язку С7-С8 у сполуках формули (I-а). Це відновлення може бути здійснене каталітичним гідруванням воднем у присутності каталізатора на основі благородного металу, такого як, наприклад, Pt, Pd, Rh, Ru або нікель Ренея. Інтерес становить Rh на оксиді алюмінію. Реакцію гідрування переважно проводять у розчиннику, такому як, наприклад, спирт, такий як метанол, етанол, або простий ефір, такий як ТГФ, або їх суміші. Вода може також бути додана до цих розчинників або сумішей розчинника. Частина Р' може бути з'єднана з будівельним блоком Р1 на будь-якій стадії синтезу, тобто, до або після циклізації, або до чи після циклізації і відновлення, як пояснено вище. Р' може бути з'єднана з Р1 з утворенням амідної сполуки між обома групами. В одному варіанті здійснення групу Р' вводять на останній стадії синтезу сполук (І), як показано в наступній реакційній схемі, у якій G позначає групу: 17 У цій процедурі, циклопропілсульфонамід (IV) вводять у реакцію із проміжним продуктом (III) через реакцію утворення аміду, таку як будь-яка з процедур для формування амідної сполуки, описана далі. Зокрема, (111) може бути оброблена агентом сполучення, наприклад, Ν,Ν'карбонілдіімідазолом (CDI), EEDQ, IlDQ, EDCl або бензотримазол-1-іл-окси-триспірролідинофосфонію гексафторфосфатом (комерційно доступним під маркою РуВОР®), у розчиннику, такому як ТГФ, з наступною реакцією з бажаним циклопропілсульфонамідом (IV) у присутності основи, наприклад, триалкіламіну, такого як триетиламін або діізопропілетиламін, або 1,8діазабіцикло-[5.4.0] ундец-7-ен (DBU), або діізопропілетиламін. Активація карбонової кислоти в (III), як описано у вищевказаних реакціях, може приводити до внутрішньої реакції циклізації до проміжного азалактону формули у якій R' і n мають визначені вище значення, і в якій стереогенні центри можуть мати стереохімічну конфігурацію, визначену вище, особливо як у (I-е). Проміжні продукти (ІІІ-а) можуть бути виділені з реакційної суміші з використанням звичайної методології, і виділений проміжний продукт (lll-а) вводять у реакцію з (IV), або реакційна суміш, що містить (ІІІ-а) може бути введена в реакцію далі з (IV) без виділення (lll-а). В одному варіанті здійснення, коли реакцію з агентом сполучення проводять у розчиннику, що не змішується з водою, реакційна суміш, що містить (III-а), може бути промита водою або слабким водним розчином основи, для видалення всіх водорозчинних побічних продуктів. У такий спосіб отриманий вимитий розчин може бути введений у реакцію з (IV) без додаткових стадій очищення. Виділення проміжних продуктів (III-а), з іншого боку, може забезпечити визначені переваги у тому, що виділений продукт, після додаткового очищення, може бути введений у реакцію з (IV), приводячи до меншої кількості побічних продуктів і сприяючи більш легкому проведенню реакції. Сполуки формули (I) можуть також бути отримані етерифікацією проміжної сполуки (V) з хіноліном формули (VI), як показано в наступній реакційній схемі: 95239 18 X у (VI) позначає гідроксил або группу, що видаляється, таку як галогенід, наприклад, бромід або хлорид, або арилсульфонільну групу, наприклад, мезилат, трифлат або тозилат і т. п. В одному варіанті здійснення реакція (V) з (VI) являє собою реакцію О-арилування, і X позначає группу, що видаляється. Ця реакція може бути здійснена Згідно процедур, описаним Ε. Μ. Smith et al. (J. Med. Chem. (1988), 31, 875-885). Зокрема, ця реакція проводиться в присутності основи, бажано, сильної основи, в інертному розчиннику реакції, наприклад, одному з розчинників, зазначених для утворення амідної сполуки. В одному варіанті здійснення вихідний матеріал (V) вводять у реакцію з хіноліном (VI) у присутності основи, що є досить сильною, щоб видділити водень від гідроксильної групи, наприклад, луги гідриду лужного металу, такого як LiH або гідрид натрію, або алкоголят лужного металу, такий як метилат або етилат натрію або калію, третбутилат калію, в інертному розчиннику реакції, такому як біполярний апротонний розчинник, наприклад, DMA, DMF і т. п. Отриманий алкоголят вводять у реакцію з арилуючим агентом (VII), у якому X позначає прийнятну группу, що видаляється, як зазначено вище Перетворення (V) в (І) з використанням реакції О-арилування не змінює стереохімічну конфігурацію при атомі вуглецю, що несе гідрокси або О-хінолін. Альтернативно, реакція (V) з (VI) може також бути здійснена через реакцію Mitsunobu (Mitsunobu, 1981, Synthesis, January, 1-28, Rano et al, Tetrahedron Lett, 1995,36,22, 3779-3792, Krchnak et al, Tetrahedron Lett, 1995,36,5, 6193-6196, Richter et al, Tetrahedron Lett, 1994, 35,27, 4705-4706). Ця реакція включає обробку проміжного продукту (V) хіноліном (VI), у якому X позначає гідроксил, у присутності трифенілфосфіну і активатора, такого як діалкілазокарбоксилат, наприклад, діетилазодикарбоксилат (DEAD), діізопропілазодикарбоксилат (DІAD) і т. п. Реакція Mitsunobu змінює стереохімічну конфігурацію при атомі вуглецю, що несе гідрокси або О-хінолін. 1 Сполуки формули (І), у яких R позначає водень, причому зазначені сполуки представлені формулою (I-d), можуть також бути отримані з відповідної захищеної азотом проміжної сполуки (VII), у якій PG позначає захисну групу для атома азоту. Прийнятні N-захисні групи описані далі. В одному варіанті здійснення PG в (VII) позначає бензил або заміщений бензил, особливо 4-метоксибензил. 19 Вихідні матеріали (VII) у зазначеній реакції можуть бути отримані Згідно процедур, описаних для отримання сполук формули (І), але з викорис1 танням проміжних продуктів, у яких група R позначає PG. Альтернативно, щоб отримати сполуки формули (І), спочатку формують амідний зв'язок між будівельними блоками Р2 і Р1 з наступним приєднанням будівельного блоку Р3 до групи Р2 у Р1-Р2 і наступним формуванням амідного зв'язку між групами Р3 і Р2 у Р2-Р1-Р3 із супутнім замиканням кільця. Знову ж таки, частина Р' може бути приєднана до будівельного блоку Р1 на будь-якій стадії синтезу сполук формули (І), наприклад, до або після з'єднання будівельних блоків Р2 і Р1; до або після з'єднання будівельних блоків Р3 і Р1; або до або після з'єднання будівельних блоків Р3 і Р2 і супутнього замикання кільця. Ще одним альтернативним методом синтезу є формування амідного зв'язку між будівельними блоками Р2 і Р3 з наступним приєднанням будівельного блоку Р1 до Р3 і останнім формуванням амідного зв'язку між Р1 і Р2 із супутнім замиканням кільця. Знову ж таки, частина Р' може бути приєднана до будівельного блоку Р1 на будь-якій стадії синтезу сполук формули (І), тобто, у даному випадку, до або після з'єднання будівельних блоків Р2 і Р3; до або після з'єднання будівельних блоків Р1 і Р3; до або після з'єднання будівельних блоків Р1 і Р2 із супутнім замиканням кільця. Будівельні блоки P1 і Р3 можуть бути з'єднані через формування подвійного зв'язку при атомах вуглецю 7 і 8, якщо бажано, з наступним відновленням подвійного зв'язку С7-С8. Сформований у такий спосіб блок Р1-Р3 може бути приєднаний до будівельного блоку Р2 і згодом циклізований, шляхом утворення амідних зв'язків. У бажаному варіанті здійснення будівельний блок Р1-Р3 не відновлюють і з'єднують як є з Р2 і циклізують, отримуючи сполуки (I-1). Будівельні блоки Ρ1 і Р3 у будь-якому з попередніх підходів можуть бути зв'язані шляхом утворення подвійних зв'язків, наприклад, реакцією обміну олефіну, описаної далі, або реакцією типу Wittig. Слід зазначити, що у сполуках формули (І) формування амідного зв'язку між блоками Р2 і Р3 може бути досягнуте у двох різних положеннях карбамідної групи. Перше формування амідной зв'язку включає введення в реакцію азоту пірролідинового кільця із суміжним активованим карбонілом (поміченим зірочкою), що є частиною будіве 95239 20 льного блоку Р3. Альтернативне друге формування амідного зв'язку включає реакцію активованого карбонілу, поміченого зірочкою, що є частиною 1 1 будівельного блоку Р2, із групою NHRR , у якій R. має значення, визначене для сполук формули (I) 1 або їхньої підгрупи, і в якій R може додатково бути захисною групою для атома азоту; і R позначає алкільну групу Р3. Активований помічений зірочкою карбоніл може бути введений реакцією пірролідину або аміну NHRR1 з фосгеном або похідним фосгену. Індивідуальні будівельні блоки можуть спочатку бути отримані і згодом з'єднані разом або альтернативно, попередники будівельних блоків можуть бути з'єднані разом і модифіковані на більш пізній стадії до молекул бажаної структури. Функціональні групи в кожному з будівельних блоків можуть бути захищені, щоб уникнути побічних реакцій. Формування амідних зв'язків може бути виконане з використанням стандартних процедур, таких як використовувані для сполучення з амінокислотами в синтезі пептидів. Останні включають дегідративне сполучення карбоксильної групи одного реагенту з аміногрупою іншого реагенту з утворенням з'єднувального амідного зв'язку. Формування амідного зв'язку може бути здійснене шляхом реакції вихідних матеріалів у присутності агента сполучення або шляхом перетворення карбоксильної функціональної групи в активну форму, таку як активний складний ефір, змішаний ангідрид або хлорангідрид чи бромангідрид карбонової кислоти. Загальні описи таких реакцій сполучення і реактивів, що там використовується, можуть бути знайдені у загальних підручниках з хімії пептидів, наприклад, М. Bodanszky, "Peptide Chemistry", 2nd rev. ed., Springer-Verlag, Берлін, Німеччина, (1993). Приклади реакцій сполучення з формуванням амідного зв'язку включають азидний спосіб, спосіб з використанням змішаного ангідриду карбонової кислоти (ізобутил хлорформіат), спосіб з використанням карбодііміду (дициклогексилкарбодііміду, діізопропілкарбодііміду або водорозчинного карбодііміду, такого як N-етил-N'-[(3диметиламіно)пропіл]карбодіімід), спосіб з використанням активного складного ефіру (пнітрофенілового складного ефіру, складного ефіру N-гідроксукциніміду), К-спосіб реагенту Woodward, спосіб з використанням 1,1-карбонілдіімідазолу (CDI або Ν,Ν-карбонілдіімідазолу), реактиви фосфору або окислювально-відновлювальні способи. Деякі із цих способів можуть бути посилені додаванням прийнятних каталізаторів, наприклад, у способі карбодііміду, додаванням 1гідроксибензотріазолу, DBU (1,8-діазабіцикло[5.4.0]ундец-7-ена) або 4-DMAP. Іншими агентами сполучення є (бензотріазол-1-ілокси)-трис(диметиламіно)фосфоній гексафторфосфат, окремо або в присутності 1-гідроксибензотріазола або 4-DMAP; або 2-(1H-бензотріазол-1-іл)N,N,N',N'-тетраметилуроній тетрафторборат, або О-(7-азабензотріазол-1-іл)-N,N,N',N'тетраметилуроній гексафторфосфат. Ці реакції сполучення можуть бути виконані або в розчині (рідка фаза) або у твердій фазі. 21 Бажане формування амідного зв'язку здійснюють, використовуючи N-етилоксикарбоніл-2етилокси-1,2-дигідрохінолін (EEDQ) або Nізобутилокси-карбоніл-^-ізобутилокси-1,2дигідрохінолін (IIDQ). На відміну від класичної ангідридної процедури, EEDQ і IIDQ не вимагають ані основи, ані низьких температур реакції. Як правило, процедура включає реакцію еквімолярньїх кількостей карбоксильних і аміно-компонентів в органічному розчиннику (може використовуватись широкий різновид розчинників). Тоді EEDQ або IIDQ додають у надлишку, і суміш перемішують при кімнатній температурі. Реакції сполучення переважно проводять в інертному розчиннику, такому як галогеновані вуглеводні, наприклад, дихлорметан, хлороформ, біполярні апротонні розчинники, такі як ацетонітрил, диметилформамід, диметилацетамід, диметилсульфоксид, НМРТ, прості ефіри, такі як тетрагідрофуран (ТГФ). У багатьох випадках реакції сполучення проводять у присутності прийнятної основи, такої як третинний амін, наприклад, триетиламін, діізопропілетиламін (DIPEA), N-метил-морфолин, Nметилпірролідин, 4-DMAP або 1,8діазабіцикло[5.4.0]ундец-7-ен (DBU). Температура реакції може складати від 0°С до 50°С, і час реакції може складати від 15 хвилин до 24 годин. Функціональні групи в будівельних блоках, які з'єднані разом, можуть бути захищені, щоб уникнути формування небажаних сполук. Прийнятні захисні групи, які можуть використовуватись, наведені, наприклад, в Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1999) and "The Peptides: Analysis, Synthesis, Biology", Vol. 9, Academic Press, New York (1987), що згадуються далі просто як Green. Карбоксильні групи можуть бути захищені як складний ефір, що може бути розщеплений з утворенням карбонової кислоти. Захисні групи, які можуть використовуватись, включають 1) складні алкілові ефіри, такі як метил, триметилсиліл і третбутил; 2) складні арил алкілові ефіри, такі як бензил і заміщений бензил; або 3) складні ефіри, які можуть бути розщеплені слабкою основою або м'якими відновлювачами, такі як складні ефіри трихлоретила і фенацила. Аміногрупи можуть бути захищені різними N-захисними групами, такими як: 1) ацильні групи, такі як форміл, трифторацетил, фталил і п-толуолсульфоніл; 2) ароматичні карбаматні групи, такі як бензилоксикарбоніл (Cbz або Z) і заміщені бензилоксикарбоніли, і 9-флуоренілметилоксикарбоніл (Fmoc); 3) аліфатичні карбаматні групи, такі як третбутилоксикарбоніл (Вос), етоксикарбоніл, діізопропілметокси-карбоніл і аллилоксикарбоніл; 4) циклічні алкілкарбаматні групи, такі як циклопентилоксикарбоніл і адамантилоксикарбоніл; 5) алкільні групи, такі як трифенілметил, бензил або заміщений бензил, такий як 4метоксибензил; 6) триалкілсиліл, такий як триметилсиліл або t.Bu диметилсиліл; і 95239 22 7) тіолмістячі групи, такі як фенілтіокарбоніл і дитіасукциноіл. Аміно-захисними групами, що представляють інтерес, є Вос і Fmoc. Переважно аміно-захисну групу розщеплюють до наступної стадії сполучення. Видалення N-захисних груп може бути зроблене з використанням відомих з рівня техніки процедур. Коли використовується Вос, здійснюють способи з використанням трифтороцтової кислоти, без домішок або в дихлорметані, або НСI у діоксані або в етилацетаті. Отриману сіль амонію потім нейтралізують або до сполучення, або in situ з основними розчинами, такими як водні буфери або третинні аміни в дихлорметані або ацетонітрилі або диметилформаміді. Коли використовується група Fmoc, у якості реактивів використовують піперидин або заміщений піперидин у диметилформаміді, але може використовуватись будь-який вторинний амін. Зняття захисту виконують при температурі від 0°С до кімнатної температури, звичайно приблизно 15-25°С або 20-22°С. Інші функціональні групи, які можуть брати участь у реакції сполучення будівельних блоків, можуть також бути захищені. Наприклад, гідроксильні групи можуть бути захищені як бензил або заміщені ефіри бензила, наприклад 4метоксибензиловий ефір, бензоїл або заміщені складні ефіри бензоїла, наприклад, складний ефір 4-нітробензоїла, або триалкілсилільними групами (наприклад, триметилсиліл або третбутилдиметилсиліл). Інші аміногрупи можуть бути захищені захисними групами, які можуть бути селективно відщеплені. Наприклад, коли у якості захисної групи для аміногрупи використовується Вос, то прийнятними є наступні захисні групи бічного ланцюга: птолуолсульфонільні (тозильні) групи можуть використовуватись для захисту інших аміногрупи, прості бензилові (Вn) ефіри можуть використовуватись для захисту гідроксильних груп, і складні ефіри бензила можуть використовуватись для захисту інших карбоксильних груп. Або коли Fmoc обраний для захисту α-аміногрупи, звичайно прийнятними є захисні групи на основі трет-бутила. Наприклад, Вос може використовуватись для інших аміногрупи, прості ефіри трет-бутила - для гідроксильних груп, і складні ефіри трет-бутила для інших карбоксильних груп. Будь-яка із захисних груп може бути видалена на будь-якій стадії процедури синтезу, але переважно, захисні групи будь-якої з функціональних груп, що не використовуються на стадіях реакції, видаляють після завершення будівництва макроциклу. Видалення захисних груп може бути здійснено будь-яким способом, що диктується вибором захисних груп, і ці способи є відомими фахівцеві. Проміжні продукти формули (II) можуть бути отримані реакцією проміжного продукту (VIII) з алкенаміном (IX) у присутності агента, що вводить карбоніл, як показано на наступній реакційній схемі. 23 95239 24 2 1 L позначає О-захисну групу PG або групу O-захисна група може бути будь-якою із груп, зазначених тут, і особливо являє собою бензоїл або заміщений бензоїл, такий як 4-нітробензоіл. Карбонілові (СО)-вводячі агенти включають фосген або похідні фосгену, такі як карбонілдіімідазол (CDi), і т. п. В одному варіанті здійснення (VIII) вводять у реакцію із (СО)-вводячим агентом у присутності прийнятної основи і розчинника, якими можуть бути основи і розчинники, що використовуються в реакціях утворення аміду, як описано вище. В окремому варіанті здійснення основою є гідрокарбонат, наприклад, NaHCO3 або третинний амін, такий як триетиламін і т. п., а розчинник являє собою простий ефір або галогенований вуглеводень, наприклад, ТГФ, СН2Сl2, СНСl2 і т. п. Після цього додають амін (IX), отримуючи, таким чином, проміжні продукти (XII) або (ХІІ-А), як у зазначеній схемі. Альтернативний шлях з використанням схожих умов реакції включає спочатку введення в реакцію (СО)-вводячого агенту з аміном (IX) і потім введення в реакцію у такий спосіб отриманого проміжного продукту з (VIII). 1 Якщо L являє собою PG , реакція (VIII) з (lХ) призводить до проміжних продуктів (ІІ-а). Вони можуть піддаватися реакції видалення захисної 1 групи, наприклад, якщо PG являє собою бензоїл або заміщений бензоїл, - реакції з гідроксидом лужного металу (LiOH, NaOH, КОН), особливо, 1 якщо PG являє собою 4-нітробензоіл, з LiOH, у водному середовищі, що включає воду й водорозчинні органічні розчинники, такі як алканол (метанол, етанол) і ТГФ. Отриманий спирт (тобто, проміжний продукт (ll-А), у якому L позначає водень), вводять у реакцію з проміжною сполукою (VI), як описано вище для реакції (V) з (VI), і ця реакція призводить до проміжних продуктів (II). Проміжні продукти формули (III) можуть бути отримані циклізацією спочатку проміжного складного ефіру (X) з утворенням макроциклічного складного ефіру (XI), що у свою чергу перетворюють у відповідну макроциклічну карбонову кислоту (III) у такий спосіб: L має значення, визначене вище, і PG позначає захисну групу карбоксильної групи, наприклад, одну із зазначених вище захисних груп карбоксильної групи, особливо С1-4-алкіловий або бензиловий складний ефір, наприклад, метиловий, етиловий або трет-бутиловий складний ефір. Група PG може бути видалена з використанням відомих способів, наприклад, складні метилові або етилові ефіри - обробкою гідроксидом лужного металу у водному середовищі, трет-бутилові складні ефіри слабкою кислотою, а бензилові складні ефіри сильною кислотою або каталітичним гідруванням. Якщо L являє собою радикал (а), ця послідовність реакцій призводить до проміжних продуктів (III). Вони можуть також бути отримані видаленням L, що є О-захисною групою і етерифікацією в такий спосіб отриманого спирту з проміжним продуктом (VI), як описано вище. Проміжні продукти формули (VII) можуть бути отримані циклізацією проміжного продукту (XII), у якому PG позначає захисну групу для атома азоту, як визначено вище, з отриманням проміжних продуктів (VII) з подвійним зв'язком у макроциклі (VIIA), які можуть бути відновлені до відповідних проміжних продуктів (VII) з простим зв'язком у цьому місці розташування у макроциклі (VII-b): L має значення, визначене вище. Якщо L позначає радикал (а), ця послідовність реакцій призводить до проміжних продуктів (Vll-a) або (VII-b). Вони можуть також бути отримані видаленням L, що є О-захисною групою, і етерифікацією в такий спосіб отриманого спирту із проміжним продуктом (VI), як описано вище. Сульфоніламідна група у зазначеній послідовності може бути складним ефі2 ром (тобто, групою -OPG , визначеною вище), який може бути видалений і конденсований із циклопропіламідом (IV) згідно процедур, зазначеним раніше. Циклопропілсульфонамідна група може бути введена на будь-якій стадії синтезу, або на останній стадії, як описано вище, або раніше, перед формуванням макроциклу, як показано на наступній схемі. 25 95239 26 вище, і реакція сполучення призводить до проміжного продукту (XV-a), від якого група PG може бути видалена з отриманням проміжного продукту (Хlllа), з використанням умов реакції, також зазначених вище: 2 L має значення, визначене вище, PG являє собою захисну групу карбоксильної групи, як ви1 значено вище, і L являє собою захисну групу ато1 ма азоту (PG, як визначено вище), або L позначає 1 групу (b), у якій R і n мають зна1 чення, визначені вище, або в якій R може також позначати захисну групу атома азоту (PG, як ви1 значено вище). Очевидно, коли R являє собою захисну групу атома азоту, він може бути видалений на бажаній стадії синтезу. Проміжні продукти (XIV), у яких L являє собою групу (b), відповідають проміжним продуктам (II) або (ll-а), і можуть бути додатково оброблені як визначено вище. З'єднання будівельних блоків Р1 і Р2 Будівельні блоки Р1 і Р2 з'єднують, використовуючи реакцію утворення аміду згідно процедур, описаним вище. Будівельний блок Р1 може мати 2 захисну групу карбоксильної групи PG (як у (XVIa)) або може вже бути пов'язаний із групою Р1' (як 2 у (XVI-b)). L позначає водень або групу L, як визначено вище. В одному варіанті здійснення, PG у цій реакції 3 являє собою групу Вос. Якщо додатково L позначає водень, вихідний матеріал являє собою ВосL-гідроксипролін. 2 1 Група L може бути О-захисною групою PG , 2 яку вводять на вихідний матеріал (XV), у якому L позначає водень і який може бути селективно розщеплений відносно груп PG. З'єднання будівельних блоків Р3 і Р2 Будівельні блоки Р3 і Р2 з'єднують, використовуючи реакцію утворення сечовини, Згідно процедур, описаним вище для сполучення (VII) з (IX). Загальна процедура представлена у наступній реакційній схемі, у якій L має значення, як визна2 чено вище, і L позначає групу -O-PG , або групу 1 25 У процедурі зазначеної схеми циклопропіламінокислоту (XVI-a) або (XVl-b) приєднують до кислотної функціональної групи будівельного блоку Р2, використовуючи реакцію утворення аміду, таку як стандартні умови пептидного сполучення, описані вище. Видалення захисної групи кислотної групи у (XIII) з використанням прийнятних умов для використовуємо! захисної групи, з наступним сполученням з циклопропілсульфонамідом (IV), як описано вище, з отриманням, знову таки, проміжного продукту (XIV). 1 В одному варіанті здійснення L позначає групу (b), і ці реакції включають приєднання Р1 до Р2Р3, що призводить до проміжних продуктів (X) або (II), зазначеним вище В іншому варіанті здійснення 1 L являє собою N-захисну групу PG, зазначену У (XVIII) R має значення, визначене вище, але може також позначати захисну групу атома азоту, що може бути видалена за допомогою засобу для видалення захисної групи атома азоту на 3 бажаній стадії синтезу. Якщо L в (XVIII) позначає 2 2 групу -OPG , група PG може бути видалена і отримана кислота може бути з'єднана із циклопропіламінокислотами (XVI-a) або (XVI-b), приводячи до проміжних продуктів (ХНІ) або (XIV), у яких L являє собою радикал (b). Будівельні блоки Р1, Р1', Р2 і Р3 для сполук формули (І) можуть бути отримані, виходячи з відомих проміжних продуктів. Багато таких синтезів описані далі більш докладно. Синтез будівельних блоків Р2 Будівельні блоки Р2 можуть бути отримані реакцією О-арилування, наприклад, Згідно процедур, описаним вище, як показано на схемі нижче, де L має значення, визначене вище, і особливо являє собою N-захисну групу PG, X має значення, визна2 чене вище, і L позначає гідрокси, групу -OPG , де 2 PG є захисною групою карбоксильної групи, такий як будь-яка із зазначених вище захисних груп кар 27 95239 28 4 боксильної групи; або L позначає групу Р1, таку як група (с) або (d), як визначено вище. Вихідний матеріал (XIX) вводять у реакцію з реагентом (VI), як описано вище для синтезу (Ι-d), виходячи з (V) і (VI). Аналогічно тому, як описано вище, ця реакція може бути проведена з утриманням (арилування з X, що є групою, що видаляється) або інверсією (реакція Mitsunobu) стереохімії при атомі вуглецю, що несе гідроксильну групу. В 4 арилуванні з X, що є групою, що видаляється, L може також позначати гідроксил, у реакції 4 2 Mitsunobu L є групою -OPG . 1 В одному варіанті здійснення група L позначає PG, що є Вос, а вихідний матеріал (VIII) є комерційно доступним Вос-1-гідроксипроліном, або будь-якоюй іншою його стереоізомерною формою. 4 2 Якщо L в (XX) позначає -OPG , то захисна 2 група карбоксильної групи PG може бути видалена згідно процедур, описаним вище, з отриманням гідроксипролінових похідних (XVII). В одному варі1 2 анті здійснення PG є групою Вос, a PG позначає нижчий алкіловий складний ефір, особливо метиловий або етиловий складний ефір. Гідроліз останнього складного ефіру до кислоти може бути здійснений у відповідності зі стандартними процедурами, наприклад, кислотним гідролізом із соляною кислотою в метанолі або етанолі, або гідроксидом металу, таким як гідроксид натрію або, переважно, гідроксид літію. Проміжні продукти (VI) можуть бути отримані відомими способами з використанням відомих вихідних матеріалів. Вони можуть бути отримані, як показано нижче: Ацилування по Фридель-Крафту 3метоксианіліну (ХХll), комерційно доступного або такого, що може бути отриманий відомими способами, з використанням агента ацилування, такого як ацетилхлорид і т. п., у присутності однієї або більше кислот Льюіса, таких як хлорид бору і хлорид алюмінію в розчиннику, такому як дихлорметан, забезпечує отримання (XXIII). Сполучення (ХХlll) з 4-ізопропіл-тіазол-2-карбоновою кислотою (XXIV), переважно в основних умовах, таких як у піридині, у присутності активатора для карбоксилатної групи, наприклад, РОСb, з наступним замиканням кільця і дегідратацією в основних умовах, таких як трет-бутоксид калію в трет-бутанолі, призводить до похідного хіноліну (VI-а). Останній може бути перетворений у (VI-b), у якому LG позначає группу, що видаляється, наприклад, реакцією (XII) з галогенуючим реагентом, наприклад фосфорилхлоридом і т. п., або з арилсульфонілхлоридом, наприклад, з тозилхлоридом. 2-карбокси-4-ізопропіл-тіазол (XXIV) синтезується відповідно відомим процедурам, зокрема, у такий спосіб: Етил тіооксамат (XXVI) вводять у реакцію з βбромкетон (XXVII), отримуючи складний ефір тіазолилу і карбонової кислоти (XXVII), який гідролизують у відповідну кислоту (XXIV). Складний етиловий ефір у цих проміжних продуктах може бути заміщений іншою захисною групою карбоксильної 2 групи PG , як визначено вище. Проміжний продукт (XXIII) може також бути отриманий, як описано у Brown et al. J. Med. Chem. 1989, 32, 807-826, або як зазначено у наступній схемі. 29 95239 30 хімічна дериватизація; або хіральна хроматографія на колонках. Похідне сульфаміду (XVI-b) може бути отримано, як зазначено у наступній реакційній схемі, 2 де R і PG мають значення, визначені вище. Вихідні матеріали етилацетилацетат і етоксиметилен малононітрил, які є комерційно доступними, вводять у реакцію в присутності придатної основи, такої як етилат натрію, і розчинника, такого як етанол і т. п. Ця реакція призводить до проміжного продукту (XXIX). Останній гідролизують, наприклад, за допомогою основи, такої як гідроксид лужного металу, наприклад, NaOH або LiOH, у придатному розчиннику, такому як етанол/вода, щоб отримати (XXX). Декарбоксилування проміжного продукту (XXX) у проміжний продукт (XXXI) виконують при підвищеній температурі, до припинення виділення газу, переважно у присутності основного розчинника, такого як хінолін. Метилування проміжного продукту (XXXI), зокрема, метилуючим реагентом, таким як МеІ, у присутності придатної основи (наприклад, K2СО3) у придатному розчиннику (такому як DMF і т. п.) дає (XXXII). Останній вводять у реакцію з реактивом Гриньяра, таким як MeMgBr, у присутності придатного розчинника (наприклад, ТГФ), з наступним гідролізом, наприклад, з використанням водяного розчину НСI, отримуючи проміжний продукт (XXIII). Синтез будівельних блоків Р1 Циклопропанамінокислота, що використовується в отриманні фрагмента Р1, є комерційно доступною або може бути отримана з використанням відомих процедур. Аміно-вініл-циклопропілетиловий складний ефір (XVI-a) може бути отриманий згідно процедури, описаній у WO00/09543, або як показано у на2 ступній схемі, де PG позначає захисну групу карбоксильної групи, як визначено вище: Обробка комерційно доступного або легко отримуваного іміну (ХХХlll) 1,4-дигалогенбутеном у присутності основи призводить до (XXXIV), що після гідролізу призводить до циклопропіл амінокислоти (XVI-a), що має аллильний замісник у конфігурації syn по відношенню до карбоксильної групи. Поділ енантіомерної суміші (XVI-a) призводить до (XVl-a-1). Поділ здійснюють, використовуючи відомі процедури, такі як ферментативний поділ, кристалізація з хіральною кислотою; або Реакція (XVI-c) із сульфонамідом (IV) являє собою процедуру утворення аміду, що може бути виконана згідно процедур, описаним вище. Ця реакція призводить до проміжних продуктів (XVId), від яких захисну групу аміногрупи видаляють стандартними методами, такими як описані вище. Це у свою чергу призводить до бажаним проміжним (XVI-b). Вихідні матеріали (XVI-c) можуть бути отримані із проміжних продуктів (XVI-a) введенням спочатку N-захисної групи PG і наступним вида2 ленням групи PG . Синтез будівельних блоків Р3 Будівельні блоки Р3 (IX) можуть бути отримані згідно методів, відомим з рівня техніки. Один із цих методів показаний на схемі нижче і у ньому як вихідні продукти використовують захищені аміни (XXXVI), особливо моноацильовані аміни, такі як трифторацетамід, або Вос-захищений амін. У цій схемі LG позначає N-захисну групу, як визначено вище, і зокрема, Вос або трифтораце1 тил; R і n мають значення, визначені вище, і при1 чому R може також бути додатковою захисною групою атома азоту, що може бути селективно відщеплена по відношенню до групи PG. LG являє собою видаляється группу, що, зазначену вище, 1 зокрема, LG позначає хлор або бром. Якщо R являє собою захисну групу атома азоту, вона може бути видалена за допомогою засобу зняття захисту з атома азоту на бажаній стадії синтезу. Моноацильовані аміни (XXXVI) обробляють сильною основою, такою як гідрид натрію, і потім вводять у реакцію з галоген-С3-8-алкенілом (XXXVII) з отриманням відповідного захищеного аміну (XXXVIII). Зняттям захисту з (XXXVIII) отримують (IX). Зняття захисту залежить від групи PG, таким чином, якщо PG являє собою Вос, видалення захисної групи може бути здійснене з використанням відносно слабкої кислоти, наприклад, трифтороцтової кислоти, або, якщо PG являє собою трифторацетил, видалення здійснюють з використанням основи, наприклад, гідроксида натрію. 1 Проміжні продукти (IX), у яких R позначає водень, може також бути отримані синтезом Габріеля алкеніламіна, що може бути виконаний обробкою фталіміду (XXXIX) основою, такою як 31 гідроксид калію, і (ХХХХ), з наступним гідролізом, з отриманням алкеніламіна (ІХ-а). У зазначеній схемі LG позначає галоген, n має значення, визначене вище. Сполуки формули (І) можуть бути перетворені у прийнятні N-оксидні форми згідно відомих процедур перетворення тривалентного азоту в його N-оксидну форму. Зазначену реакцію Ν-окислення можна виконувати, вводячи в реакцію вихідний матеріал формули (І) з придатним органічним або неорганічним перекисом. Прийнятні неорганічні перекиси, включають, наприклад, перекис водню, перекиси лужного металу або лужноземельного металу, наприклад, перекис натрію, перекис калію; прийнятні органічні перекиси можуть включати пероксикислоти, такі як, наприклад, бензолкарбопероксова кислота, або галоген-заміщена бензолкарбопероксова кислота, наприклад, 3хлорбензол-карбопероксова кислота, пероксоалканові кислоти, наприклад, пероксооцтова кислота, алкілгідропероксиди, наприклад, третбутилгідропероксид. Прийнятними розчинниками є, наприклад, вода, нижчі спирти, наприклад, етанол і т. п., вуглеводні, наприклад, толуол, кетони, наприклад, 2-бутанон, галогеновані вуглеводні, наприклад, дихлорметан, і суміші таких розчинників. Чисті стереохімічно ізомерні форми сполук формули (І) можуть бути отримані з використанням відомих процедур. Діастереоізомери можуть бути розділені фізичними способами, такими як селективна кристалізація, і методами хроматографії, наприклад, противоточним розподілом, рідинною хроматографією і т. п. Сполуки формули (І) можуть бути отримані як рацемічні суміші енантіомерів, які можуть бути відділені один від одного за допомогою відомих процедур поділу. Рацемічні сполуки формули (І), які є достатньо основними або кислими, можуть бути перетворені у відповідні діастереомерні сольові форми реакцією з прийнятною хіральною кислотою, відповідно, хіральною основою. Зазначені діастереомерні форми солі потім відокремлюють, наприклад, селективною або фракційною кристалізацією, і енантіомери вивільняють лугою або кислотою. Альтернативний спосіб поділу енантіомерних форм сполук формули (І) включає рідинну хроматографію, зокрема, рідинну хроматографію з використанням хіральної стаціонарної фази. Зазначені чисті стереохімічно ізомерні форми можуть також бути отримані з відповідних чистих стереохімічно ізомерних форм прийнятних вихідних матеріалів, за умови, що реакція проходить стереоспецифічно. Переважно, якщо бажаний певний стереоізомер, зазначена сполука може синтезуватися стереоспецифічними способами отримання У цих способах можуть переважно використовуватись енантіомерно чисті вихідні матеріали. 95239 32 У наступному аспекті, даний винахід стосується фармацевтичної композиції, що містить терапевтично ефективну кількість сполуки формули (І), як вона визначена тут, або сполуки будь-якої з підгруп сполук формули (І), як визначено тут, і фармацевтично прийнятний носій. Терапевтично ефективна кількість у цьому контексті являє собою кількість, достатню для профілактичної дії проти, для стабілізації або зменшення вірусної інфекції, і зокрема HCV вірусної інфекції, в інфікованих пацієнтів або пацієнтів, для яких існує ризик інфікування. В іншому аспекті цей винахід відноситься до способу отримання фармацевтичної композиції, як вона визначена тут, що включає ретельне змішування фармацевтично прийнятного носія з терапевтично ефективною кількістю сполуки формули (І), як вона визначена тут, або сполуки будь-якої з підгруп сполук формули (І), як визначено тут. Тому, сполуки згідно даного винаходу або будь-яка їх підгрупа можуть бути складені в різні фармацевтичні форми для введення. У якості прийнятних композицій можуть бути названі всі композиції, що зазвичай використовуються для системного введення лікарських засобів. Для отримання фармацевтичних композицій за винаходом ефективна кількість визначеної сполуки, якщо є потреба, у формі солі або комплексу з металом, у якості активного інгредієнту комбінують у тісній суміші з фармацевтично прийнятним носієм, причому носій може мати різноманітні форми залежно від форми отримання, бажаної для введення. Ці фармацевтичні композиції переважно перебувають у вигляді стандартної лікарської форми, що підходить, зокрема, для перорального, ректального, трансшкірного введення або введення парентеральною ін'єкцією. Наприклад, при отриманні композицій у пероральній лікарській формі може використовуватись будь-яке звичайне фармацевтичне середовище, таке як, наприклад, вода, гліколі, масла, спирти і т. п., у випадку пероральних рідких препаратів, таких як суспензії, сиропи, еліксири, емульсії і розчини, або тверді носії, такі як крохмалі, цукор, каолін, лубриканти, в'яжучі, дезінтегратори і т. п. у випадку порошків, пігулок, капсул і таблеток Завдяки легкості введення, таблетки й капсули являють собою найбільш переважні пероральні стандартні лікарські форми, у яких очевидно використовуються тверді фармацевтичні носії. Для парентеральных композицій носій звичайно включає стерильну воду, щонайменше більшою частиною, хоча можуть бути включені інші інгредієнти, наприклад, для поліпшення розчинності. Наприклад, можуть бути отримані розчини для ін'єкцій, у яких носій включає сольовий розчин, розчин глюкози або суміш розчину глюкози й сольового розчину. Суспензії для ін'єкцій можуть також бути отримані, і в цих випадках можуть використовуватись прийнятні рідкі носії, суспендуючі агенти і т. п. Також у рамки винаходу включені тверді препаративні форми, які призначені для перетворення, незадовго до використання, у рідкі препаративні форми У композиціях, що прийнятні для трансшкірного введення, носій може включати засіб для посилення проникнення, та/або прийнятну змочувальну речовину, при потребі в комбінації з прийн 33 ятними добавками будь-якої природи у незначних пропорціях, причому ці добавки не спричиняють істотного шкідливого ефекту на шкіру. Сполуки згідно даного винаходу можуть також вводитися пероральною інгаляцією або вдуванням за допомогою способів і складів, що використовуються у даній області техніки для введення цим шляхом. Таким чином, загалом сполуки згідно даного винаходу можуть вводитися у легені у формі розчину, суспензії або сухого порошку, переважно, розчину. Будь-яка система, розроблена для доставки розчинів, суспензій або сухих порошків шляхом пероральної інгаляції або вдування, є прийнятною для введення сполук за винаходом. Таким чином, даний винахід також відноситься до фармацевтичної композиції, адаптованої для введення інгаляцією або вдуванням через рот, що включає сполуку формули (І) і фармацевтично прийнятний носій. Сполуки згідно даного винаходу можуть вводитися інгаляцією розчину у розпиляємих або аерозольних дозах. Особливо бажано складати зазначені фармацевтичні композиції у стандартній лікарській формі для простоти введення і однорідності дозування. Стандартна лікарська форма, як це поняття використовується тут, відноситься до фізично дискретних одиниць, що прийнятні як разові дози, причому кожна одиниця містить визначену кількість активного інгредієнту, обчислену таким чином, щоб спричинити бажаний терапевтичний ефект, у комбінації з необхідним фармацевтичним носієм. Прикладами таких стандартних лікарських форм є таблетки (включаючи таблетки без оболонки або таблетки, вкриті оболонкою), капсули, пігулки, супозиторії, пакетики з порошком, пластинки, розчини або суспензії для ін'єкцій і т. п., і їх упаковки з багатьма відділеннями. Сполуки формули (І) показують противірусні властивості Вірусні інфекції і пов'язані з ними захворювання, що піддаються лікуванню з використанням сполук і способів згідно даного винаходу, включає інфекції, викликані HCV і іншими патогенними флавівірусами, такі як жовта лихоманка, лихоманка Денге (типи 1-4), енцефаліт Сент-Луїс, японський енцефаліт, енцефаліт долини Мюррея, вірус Західного Нилу і вірус Kunjin. Захворювання, пов'язані з HCV, включають прогресивний фіброз печінки, запалення і некроз, що призводить до цирозу, термінальної стадії захворювання печінки і НСС, і для інших патогенних флавівірусів захворювання включають жовту лихоманку, лихоманку Денге, геморрагічну лихоманку і енцефаліт. Багато сполук за винаходом, крім того, активні відносно мутантних штамів HCV. Додатково, багато із сполук за винаходом показують сприятливий фармакокінетичний профіль і мають привабливі властивості по відношенню до біодоступності, включаючи прийнятний період напіврозпаду, AUC (область під кривою) і пікові значення і не показують несприятливих феноменів, таких як недостатньо швидкий початок дії і затримка у тканині. Противірусна активність in vitro проти HCV сполук формули (І) була протестована у клітинній системі реплікону HCV, заснованої на Lohmann et al., (1999) Science 285 110-113, з подальшими мо 95239 34 дифікаціями, описаними Krieger et al. (2001), Journal of Virology 75 4614-4624, що далі ілюструється секцією прикладів. Ця модель, хоча і не є повною моделлю інфекції для HCV, широко прийнята як найпотужніша і ефективна з доступних у цей час моделей автономної реплікації РНК HCV. Сполуки, що показують анти-HCV активність у цій клітинній моделі, розглядають у якості кандидатів на подальший розвиток відносно лікування інфекцій HCV у ссавців. Варто розуміти, що важливо відрізнити сполуки, які специфічно перешкоджають функціонуванню HCV, від сполук, які показують цитотоксичні або цитостатичні ефекти в моделі реплікону HCV, і, як наслідок, викликають зниження концентрації РНК HCV або зв'язаного ферменту-репортера. В області оцінки клітинної цитотоксичності відомі тести, засновані, наприклад, на активності митохондріальних ферментів, з використанням флуорогенних окислювально-відновних барвників, таких як діазорезорцин. Крім того, існують клітинні системи скринінгу для оцінки неселективного інгібування зв'язаної активності генарепортера, такі як люцифераза світляка. Прийнятні типи клітин можуть бути обладнані стабільною трансфекцією з геном репортера люциферази, експресія якого залежить від конститутивно активного генного промотору, і такі клітини можуть використовуватись у якості систем скринінгу для видалення неселективних інгібіторів. Внаслідок їхніх противірусних властивостей, особливо їх анти-HCV властивостей, сполуки формули (І) або будь-яка їх підгрупа, їх N-оксиди, солі приєднання, четверинні аміни, комплекси сполуки з металами і стереохімічно ізомерні форми є придатними для лікування людей, що мають вірусну інфекцію, особливо інфекцію HCV, і для профілактики цих інфекцій. Загалом, сполуки згідно даного винаходу можуть бути використані в лікуванні теплокровних тварин, інфікованих вірусами, особливо флавівірусами, таким як HCV. Сполуки згідно даного винаходу або будь-яка їх підгрупа можуть тому використовуватись у якості лікарських засобів. Зазначене використання у якості лікарських засобів або спосіб лікування включає системне введення інфікованим вірусом пацієнтам або пацієнтам, у яких можуть бути вірусні інфекції, кількості, ефективної для боротьби зі станами, пов'язаними з вірусною інфекцією, особливо інфекцією HCV. Даний винахід також відноситься до застосування сполук за винаходом або будь-якій їх підгрупи в отриманні лікарського засобу для лікування або профілактики вірусних інфекцій, особливо інфекцій HCV. Даний винахід, крім того, відноситься до способу лікування теплокровної тварини, інфікованої вірусом, або для якого існує ризик інфікування вірусом, особливо HCV, що включає введення противірусно ефективної кількості сполуки формули (І), як воно визначено тут, або сполуки будьякої з підгруп сполук формули (І), як визначено тут. Крім того, комбінація відомої анти-HCV сполуки, такої як, наприклад, інтерферон-α (IFN-α), пегільований інтерферон-α та/або рібавірин, і сполуки формули (I) може використовуватись у якості лі 35 карського засобу у комбінованій терапії. Термін "комбінована терапія" відноситься до продукту, що містить обов'язково (а) сполуку формули (І), і (b) у разі необхідності іншу анти-HCV сполуку, у якості комбінованого препарату для одночасного, роздільного або послідовного використання у лікуванні інфекцій HCV, зокрема, у лікуванні інфекцій HCV. Анти-HCV сполуки охоплюють засоби, обрані з інгібітору HCV полімерази, інгібітору HCV протеази, інгібітору іншої мішені у життєвому циклі HCV і імуномодулюючого засобу, противірусного засобу і їх комбінацій. Інгібітори HCV полімерази включають, але не обмежені ними, NM283 (валопіцитабін), R803, JTK109, JTK-003, HCV-371, HCV-086, HCV-796 і R1479. Інгібітори HCV протеаз (інгібітори NS2-NS3 і NS3-NS4A інгібітори) включають, але не обмежені ними, сполуки згідно WO02/18369 (див, наприклад, сторінку 273, рядки 9-22, і сторінка 274, рядок 4 сторінка 276, рядок 11), BILN-2061, VX-950, GS 9132 (АСН-806), SCH-503034 і SCH-6. Іншими засобами, які можуть використовуватись, є засоби, розкриті у WO98/17679, WO00/056331 (Vertex), WO98/22496 (Roche), WO99/07734, (Boehringer Ingelheim), WO2005/073216, WO2005/073195 (Medivir) і структурно подібні засоби. Інгібітори інших мішеней у життєвому циклі HCV, включаючи NS3 геліказу, інгібітори металопротеаз, інгібітори антисенсового олігонуклеотиду, такі як ISIS 14803, AVI-4065 і т. п., siPHK, такі як SIRPLEX-140-N і т. п., мала РНК-Шпилька (shRNA), що кодується вектором, ДНКзими, HCV-специфічні рібозими, такі як гептазим, RPI.13919 і т. п., інгібітори проникнення, такі як НереХ-С, HuMax-НерС і т. п., інгібітори альфа-глюкозидази, такі як целгозивир, UT-231B і т. п., KРЕ-02003002, і BrVN401. Імуномодулюючі засоби включають, але не обмежені ними, натуральні і рекомбінантні сполуки ізоформи інтерферону, включаючи α-інтерферон, β-інтерферон, -інтерферон, -інтерферон і т. п., такі як Intron A®, Roferon-A®, Canferon-F300®,0 Advaferon®, Infergen®, Humoferon®, Sumiferon MP®, Alfaferone®, IFN-beta®, Feron® і т. п., поліетиленгликоль дериватизовані (пегільовані) сполуки інтерферону, такі як ПЕГ інтерферон-α-2а (Pegasys®), ПЕГ інтерферон-α-2b (ΠΕΓ-Intron®), пегільований IFN-α-conl і т. п., склади довгої дії і дериватизати сполук інтерферону, такі як злитий з альбуміном інтерферон альбуферон і т. п., сполуки, що стимулюють синтез інтерферону в клітинах, такі як ресиквимод і т. п., інтерлейкіни, сполуки, які підсилюють розвиток типу 1 Т-хелперної відповіді, такі як SCV-07 і т. п., агоністи TOLL-подібного рецептора, такі як CpG-10101 (актилон), ізаторибін і т. п., тімозин α-1, ΑΝΑ-245, ΑΝΑ-246, гістамін дигідрохлорид, пропагерманий, тетрахлордекаоксид, ампліген, ІМР-321, KRN-7000, антитіла, такі як цивацир, XTL-6865 і т. п., і профілактичні і терапевтичні вакцини, такі як Inno Vac С, HCVElE2/MF59 і т. п. Інші противірусні агенти включають, але не обмежені ними, рібавірин, амантадин, вірамідин, нітазоксанід, тельбивудин, NOV-205, тарібавірин, інгібітори внутрішнього сайту посадки рибосоми, 95239 36 вірусні інгібітори широкого спектру, такі як інгібітори IMPDH (наприклад, сполуки згідно US5,807,876, US6,498,178, US6,344,465, US6,054,472, WO97/40028, WO98/40381, WO00/5633 1, і мікофенольні кислоти і їх похідні, і включаючи, але не обмежуючись ними, VX-950, меримеподіб (VX-497, VX-148, та/або VX-944), або комбінації будь-яких із зазначених засобів. Таким чином, для боротьби або лікування інфекцій HCV сполуки формули (І) можна вводити в комбінації з, наприклад, інтерфероном-α (IFN-α), пегільованим інтерфероном-α та/або рибавірином, а також з терапевтичними засобами, заснованими на антитілах, спрямованих до епітопів HCV, малої інтерферуючої РНК (siPHK), рибозимами, ДНКзимами, антисенсової РНК, антагоністами малої молекули, наприклад, протеази NS3, гелікази NS3 і полімерази NS5B. Відповідно, даний винахід відноситься до застосування сполуки формули (І) або будь-якої підгрупи цих сполук, як визначено вище, для отримання лікарського засобу, придатного для інгібування активності HCV у ссавця, інфікованого вірусами HCV, причому зазначений лікарський засіб використовується у комбінованій терапії, причому зазначена комбінована терапія переважно включає сполуки формули (І) і іншу інгібуючу HCV сполуку, наприклад, (пегільований) IFN-α та/або рибавірин. В іншому аспекті винахід відноситься до комбінацій сполуки формули (І), як визначено тут, і анти-HIV сполуки. Останнє переважно являє собою інгібітор HIV, що мають позитивний ефект на метаболізм та/або фармакокінетики лікарського засобу, що поліпшує біодоступність. Прикладом такого інгібітору HIV є ритонавір. Також даний винахід відноситься до комбінації, що включає (а) інгібітор HCV NS3/4a протеази формули (І) або його фармацевтично прийнятну сіль, і (b) ритонавір або його фармацевтично прийнятну сіль. Сполука ритонавір і його фармацевтично прийнятні солі і способи його отримання описані в WO94/14436. По відношенню до кращих лікарських форм ритонавіра див. US6,037,157, і документи, процитовані там US5,484,801, US08/402,690 і WO95/07696 і WO95/09614. Ритонавір має наступну формулу: У наступному варіанті здійснення, комбінація, що включає (а) інгібітор HCV NS3/4a протеази формули (І) або його фармацевтично прийнятну сіль, і (b) ритонавір або його фармацевтично прийнятну сіль, додатково включає додаткове анти-HCV сполуку, обрану із сполук, описаних тут. В одному варіанті здійснення даний винахід відноситься до способу отримання комбінації, як описано тут, що включає стадію комбінування інгібітору HCV NS3/4a протеази формули (І) або його 37 фармацевтично прийнятної солі і ритонавіра або його фармацевтично прийнятної солі. Альтернативний варіант здійснення винаходу відноситься до способу, у якому комбінація включає одне або більше додаткових засобів, як описано тут. Комбінації згідно даного винаходу можуть використовуватись у якості лікарських засобів. Зазначене використання у якості лікарських засобів або спосіб лікування включає системне введення HCV-інфікованим пацієнтам кількості, ефективної для боротьби зі станами, пов'язаними з HCV і іншими патогенними флаві- і пестивірусами. Отже, комбінації згідно даного винаходу можуть використовуватись для отримання лікарського засобу, придатного для лікування, профілактики або боротьби з інфекцією або захворюванням, пов'язаним з інфекцією HCV у ссавця, зокрема, для лікування станів, пов'язаних з HCV і іншими патогенними флаві- і пестивірусами. В одному варіанті здійснення даний винахід відноситься до фармацевтичної композиції, що включає комбінацію згідно будь-якого з варіантів здійснення, описаних тут, і фармацевтично прийнятний ексципієнт. Зокрема, даний винахід відноситься до фармацевтичної композиції, що включає (а) терапевтично ефективну кількість інгібітору HCV NS3/4a протеази формули (І) або його фармацевтично прийнятної солі, (b) терапевтично ефективну кількість ритонавіру або його фармацевтично прийнятної солі, і (с) фармацевтично прийнятний ексципієнт. У разі необхідності, фармацевтична композиція додатково містить додатковий засіб, обраний з інгібітору HCV полімерази, інгібітору HCV протеази, інгібітору іншої мішені в життєвому циклі HCV і імуномодулюючого засобу, противірусного агенту і їх комбінацій. Композиції можуть бути складені у прийнятні фармацевтичні лікарські форми, такі як лікарські форми, описані вище. Кожний з активних інгредієнтів може бути складений окремо, і склади можна вводити спільно або можна використовувати один склад, що містить обидва активних інгредієнта і якщо бажано, додаткові активні інгредієнти. У рамках винаходу термін "композиція" охоплює продукт, що включає зазначені інгредієнти, а також будь-який продукт, що походить, прямо чи побічно, від комбінації зазначених інгредієнтів. В одному варіанті здійснення комбінації за винаходом можуть також бути складені як комбінований препарат для одночасного, роздільного або послідовного використання у терапії HIV. У таких випадках сполуку загальної формули (І) або будьяку підгрупу цих сполук, складають у фармацевтичній композиції, що містить інші фармацевтично прийнятні ексципієнти, і ритонавір складають окремо у фармацевтичній композиції, що містить інші фармацевтично прийнятні ексципієнти. Бажано, ці дві окремі фармацевтичні композиції можуть бути частиною набору для одночасного, роздільного або послідовного використання. Таким чином, індивідуальні компоненти комбінації згідно даного винаходу можуть вводитися роздільно у різний час у ході терапії або одночасно у роздільних формах або у формі єдиної комбінації. Даний винахід повинне тому бути зрозуміле 95239 38 як таке, що охоплює всі такі режими одночасного або змінного лікування, і термін "введення" повинен інтерпретуватися відповідно. У бажаному варіанті здійснення, окремі лікарські форми вводять практично одночасно. В одному варіанті здійснення комбінація згідно даного винаходу містить кількість ритонавіра або його фармацевтично прийнятної солі, що є достатньою для клінічного поліпшення біодоступності інгібітору HCV NS3/4a протеази формули (І) відносно біодоступності, коли зазначений інгібітор HCV NS3/4a протеази формули (І) вводять індивідуально. В іншому варіанті здійснення комбінація згідно даного винаходу містить кількість ритонавіра або його фармацевтично прийнятної солі, що є достатньою, щоб збільшити щонайменше одну з фармакокінетичних змінних інгібітору HCV NS3/4a протеази формули (І), обраних з t½, Сmin, Сmax, CSS, AUC у 12 годин або AUC у 24 години, щодо зазначеної щонайменше однієї фармакокінетичної змінної, коли інгібітор HCV NS3/4a протеази формули (І) вводять індивідуально. Інший варіант здійснення відноситься до способу поліпшення біодоступності інгібітору HCV NS3/4a протеази, що включає введення людині комбінації як визначено тут, що включає терапевтично ефективну кількість кожного компонента зазначеної комбінації. У наступному варіанті здійснення винахід відноситься до застосування ритонавіра або його фармацевтично прийнятної солі, у якості добавки для поліпшення щонайменше однієї з фармакокінетичних змінних інгібітору HCV NS3/4a протеази формули (І), обраних з t½, Сmin, Сmax, CSS, AUC у 12 годин або AUC у 24 години, за умови, що зазначене застосування не здійснюють в організмі тварин або людини. Термін "індивідуум" у рамках винаходу відноситься до тварини, переважно, ссавця, найбільше переважно, людині, що була об'єктом лікування, спостереження або експерименту. Біодоступність визначають у якості фракцію введеної дози, що досягла великого кола кровообігу t½ являє собою напівперіод або час, необхідний для зниження плазмової концентрації до половини и первісного значення CSS позначає концентрацію стаціонарного стану, тобто, концентрацію, при якій швидкість надходження лікарського засобу дорівнює швидкості видалення C min визначають як найнижчу (мінімальну) концентрацію, виміряну протягом інтервалу дозування С max являє собою найвищу (максимальну) концентрацію, виміряну протягом інтервалу дозування AUC визначають як область під кривої залежності "концентрація-час" для визначеного проміжку часу. Комбінації за винаходом можуть вводитися людям у діапазонах доз, визначених для кожного компонента, включеного у зазначені комбінації. Компоненти, включені у зазначені комбінації, можуть вводитися разом або окремо Інгібітори NS3/4a протеази формули (І) або будь-яка їх підгрупа і ритонавір або їх фармацевтично прийнятна сіль або складний ефір можуть мати рівні доз порядку від 0.02 до 5.0 грамів на добу. 39 Коли інгібітор HCV NS3/4a протеази формули (І) і ритонавір вводять у комбінації, вагове відношення інгібітору HCV NS3/4a протеази формули (І) до ритонавіру перебуває відповідно в діапазоні від приблизно 40:1 до приблизно 1:15, або від приблизно 30:1 до приблизно 1:15, або від приблизно 15:1 до приблизно 1:15, звичайно від приблизно 10:1 до приблизно 1:10, і більш звичайно від приблизно 8:1 до приблизно 1:8. Також можуть використовуватись вагові відношення інгібітору HCV NS3/4a протеази формули (І) до ритонавіру в межах від приблизно 6:1 до приблизно 1:6, або від приблизно 4:1 до приблизно 1:4, або від приблизно 3:1 до приблизно 1:3, або від приблизно 2:1 до приблизно 1:2, або від приблизно 1,5:1 до приблизно 1:1,5. В одному аспекті вагова кількість інгібітору HCV NS3/4a протеази формули (I) дорівнює або більше, ніж кількість ритонавіра, причому вагове відношення інгібітору HCV NS3/4a протеази формули (І) до ритонавіру перебуває відповідно в діапазоні від приблизно 1:1 до приблизно 15:1, звичайно від приблизно 1:1 до приблизно 10:1, і більш звичайно від приблизно 1:1 до приблизно 8:1. Також можуть використовуватись вагові відношення інгібітору HCV NS3/4a протеази формули (I) до ритонавіру в межах від приблизно 1:1 до приблизно 6:1, або від приблизно 1:1 до приблизно 5:1, або від приблизно 1:1 до приблизно 4:1, або від приблизно 3:2 до приблизно 3:1, або від приблизно 1:1 до приблизно 2:1 або від приблизно 1:1 до приблизно 1.5:1. Термін "терапевтично ефективна кількість" у рамках винаходу означає, що кількість активної сполуки або компонента або фармацевтичного засобу, що викликає біологічну або лікарську відповідь у тканині, системі, організмі тварини або людини, що вимагається у світлі даного винаходу з боку дослідника, ветеринара, лікаря або іншого клінічного фахівця, що включає полегшення симптомів захворювання, що лікується. Виходячи з того, що даний винахід відноситься до комбінацій, що включають два або більше засобів, "терапевтично ефективна кількість" являє собою кількість цих засобів, узятих разом так, щоб об'єднаний ефект викликав бажану біологічну або лікарську відповідь. Наприклад, терапевтично ефективна кількість композиції, що включає (а) сполуки формули (І) і (b) ритонавір, являє собою кількість сполуки формули (І) і кількість ритонавіра, що разом має комбінований ефект, що є терапевтично ефективним. У цілому противірусна ефективна добова кількість може складати від 0.01 мг/кг до 500 мг/кг маси тіла, більш переважно, від 0.1 мг/кг до 50 мг/кг маси тіла Може знадобитися введення·необхідної дози у вигляді однієї, двох, трьох, чотирьох або більше (під-)доз через прийнятні інтервали протягом дня. Зазначені (під-)дози можуть бути складені у якості стандартні лікарські форми, наприклад, що містять 1-1000 мг, і зокрема, 5-200 мг активного інгредієнта на стандартну лікарську форму. Точне дозування й частота введення залежать від конкретної використовуваної сполуки формули (I), конкретного стану, що піддається лікуванню, серйозності стану, що піддається лікуванню, віку, 95239 40 маси тіла, статі, ступеня порушення і загального фізичного стану конкретного пацієнта, а також іншого лікування, яке людина може отримувати, як відомо фахівцеві. Крім того, очевидно, що зазначена ефективна добова кількість може бути зменшена або збільшена залежно від реакції пацієнта та/або залежно від оцінки лікаря, що прописує сполуки згідно даного винаходу. Діапазони ефективних добових кількостей, згадані вище, є тільки тому приблизними. Згідно одному варіанту здійснення, інгібітор HCV NS3/4a протеази формули (I) і ритонавір можна спільно вводити кілька разів день, переважно, перорально, причому кількість сполук формули (І) на дозу становить від приблизно 1 до приблизно 2500 мг, і кількість ритонавіра на дозу становить від 1 до приблизно 2500 мг. В іншому варіанті здійснення кількості на дозу при спільному введенні один або два рази на добу складають від приблизно 50 до приблизно 1500 мг сполуки формули (І) і від приблизно 50 до приблизно 1500 мг ритонавіра. В іншому варіанті здійснення кількості на дозу при спільному введенні один або два рази на добу складають від приблизно 100 до приблизно 1000 мг сполуки формули (І) і від приблизно 100 до приблизно 800 мг ритонавіра. У ще одному варіанті здійснення кількості на дозу при спільному введенні один або два рази на добу складають від приблизно 150 до приблизно 800 мг сполуки формули (І) і від приблизно 100 до приблизно 600 мг ритонавіра. У ще одному варіанті здійснення кількості на дозу при спільному введенні один або два рази на добу складають від приблизно 200 до приблизно 600 мг сполуки формули (І) і від приблизно 100 до приблизно 400 мг ритонавіра. У ще одному варіанті здійснення кількості на дозу при спільному введенні один або два рази на добу складають від приблизно 200 до приблизно 600 мг сполуки формули (І) і від приблизно 20 до приблизно 300 мг ритонавіра. У ще одному варіанті здійснення кількості на дозу при спільному введенні один або два рази на добу складають від приблизно 100 до приблизно 400 мг сполуки формули (І) і від приблизно 40 до приблизно 100 мг ритонавіра. Приклади комбінацій сполука формули (I) (мг)/ритонавір (мг) при введенні один або два рази на добу включають 50/100, 100/100, 150/100, 200/100, 250/100, 300/100, 350/100, 400/100, 450/100, 50/133, 100/133, 150/133, 200/133, 250/133, 300/133, 50/150, 100/150, 150/150, 200/150, 250/150, 50/200, 100/200, 150/200, 200/200, 250/200, 300/200, 50/300, 80/300, 150/300, 200/300, 250/300, 300/300, 200/600, 400/600, 600/600, 800/600, 1000/600, 200/666, 400/666, 600/666, 800/666, 1000/666, 1200/666, 200/800, 400/800, 600/800, 800/800, 1000/800, 1200/800, 200/1200, 400/1200, 600/1200, 800/1200, 1000/1200 і 1200/1200. Інші приклади комбінацій сполука формули (І) (мг)/ритонавір (мг) при введенні один або два рази на добу включають 1200/400, 800/400, 600/400, 400/200, 600/200, 600/100, 500/100, 400/50, 300/50 і 200/50. В одному варіанті здійснення даний винахід відноситься до виробу, що включає композицію, ефективну для лікування інфекції HCV або для 41 інгібування протеази NS3 HCV; і пакувальний матеріал, що включає ярлик, на якому зазначено, що композиція може використовуватись для лікування інфекції вірусу гепатиту С; причому композиція включає сполуку формули (І) або будь-яку його підгрупу, або комбінацію, як описано тут. Інший варіант здійснення даного винаходу стосується набору або ємності, що включає сполуку формули (I) або будь-яку його підгрупу, або комбінацію згідно винаходу, що поєднує інгібітор HCV NS3/4a протеази формули (І) або його фармацевтично прийнятну сіль і ритонавір або його фармацевтично прийнятну сіль, у кількості, ефективній для використання у якості стандарту або реагенту у тесті або досліді на визначення здатності потенційних фармацевтичних препаратів інгібувати HCV NS3/4a протеазу, ріст HCV або і те і інше. Цей аспект винаходу може знайти застосування у програмах фармацевтичних досліджень. Сполуки і комбінації згідно даного винаходу можуть використовуватись у високопродуктивних тестах мішень-аналіт, як такі, що використовуються для вимірювання ефективності зазначеної комбінації у лікуванні HCV Приклади Наступні приклади призначені для ілюстрації, але не для обмеження даного винаходу. Приклад 1: Отримання N-[18-[2-(4ізопропілтіазол-2-іл)-7-метоксихінолін-4-іл-окси]2,15-діоксо-3,14,164,6 триазатрицикло[14.3.0.0 ]нонадец-7-ен-4карбоніл]-(циклопропіл)сульфаміду, з визначеною стереохімією, як зображено нижче у сполуці (9) Стадія А Розчин 2-(4-ізопропілтіазол-2-ил)-7метоксихінолін-4-ола (1, 3.6 г) у хлорангідриді фосфорної кислоти (20 мол) нагрівали при 100°С протягом 40 хвилин (реакцію перевіряли LC-MS). Потім реакційне середовище охолоджували до кімнатної температури, і надлишок хлорангідрида фосфорної кислоти випарювали. Залишкове масло розділяли між насиченим розчином NaHCO3 і екстрагували простим діетиловим ефіром (3 × 70 мол). Об'єднані органічні фракції промивали сольовим розчином, висушували над MgSO4, концентрували на роторному випарнику і пропускали через короткий фільтр із силікагелю (гексани), отримуючи 3.6г (62 %) бажаного продукту 2 у формі білого порошку. Стадія В 95239 42 До перемішуємого розчину Вос-4гідроксипроліна з визначеною стереохімією, як показано у формулі вище, (2.6 г, 112 ммоль) у диметилсульфоксиді (80 мл) додавали трет-бутоксид калію (З 8 г, 3 екв). Через приблизно 1 годину перемішування додавали 4-хлор-2-(4ізопропілтіазол-2-іл)-7-метоксихінолін (2, 3.6 г, 112 ммоль) і реакційну суміш перемішували при кімнатній температурі протягом ночі Потім реакційну суміш розбавляли водою (350 мл) і нейтралізували 1н НСl. Отриману суспензію екстрагували етилацетатом (3 × 100 мл), промивали сольовим розчином і висушували над MgSO4. Фільтрацією і концентрацією на роторному випарнику отримували після висушування протягом ночі в сильному вакуумі 3.6г (62 %) бажаного продукту 3. Чистота ВЕРХ >95 %, m/z = 514 (М+Н)+. Стадія С Кислоту 3 (3.6 г, 7 ммоль) змішували з ефіром 1-аміно-2-вініл-циклопропан-карбоновоі кислоти і етилгідрохлорида з визначеною стереохімією, як показано у формулі вище, (1.47 г, 7 6 ммоль), і потім розчиняли в DMF. Реакційну суміш змивали аргоном і остуджували у ванні з льодом, і D1PEA (1.5 мл) додавали у вигляді однієї порції. Потім реакційну суміш перемішували протягом 10-15 хвилин при 0°С, після чого додавали HATU (2.93 г, 7.7 ммоль) при 0°С у потоці аргону, у вигляді однієї порції. Через 40 хвилин при 0°С (реакцію перевіряли LC-MS) реакційну суміш концентрували на роторному випарнику (не до повної сухості), потім змішували з розчином насиченого NaHCO і екстрагували ЕtOАс (3 × 100 мл). Органічний шар промивали сольовим розчином, висушували над MgSO4 і концентрували на роторному випарнику. Очищенням хроматографією на колонках із силікагелем (DCM) і потім на силікагелі YMC (200 г, градієнт гексани/ЕА від 3:2 до 2:3) отримували 3.81 г (84 %) цільового продукту 4 у формі білого порошку. Стадія D Розчин 4 (3.81 г, 5.8 ммоль) в СН2Сl2 (30 мл) і трифтороцтової кислоті (30 мл) перемішували при кімнатній температурі приблизно протягом 1.5 годин. Потім розчинник випарювали й залишок розділяли між насиченим NaHCO3 (100 мл) і простим діетиловьім ефіром (3 × 100 мл). Шари простого 43 95239 44 діетилового ефіру поєднували, промивали сольовим розчином, висушували над MgSO4 і упарювали, отримуючи 3.13 г (98 3 %) цільового продукту 5: m/z 551 (М+Н)+. Стадія Ε NaНСО3 (10 г) додавали до розчину 5 (1.4 г, 2.5 ммоль) у тетрагідрофурані (50 мл). Потім при 0°С в атмосфері аргону додавали фосген (5 мл, 1.9 Μ у толуолі). Отриману суспензію перемішували протягом 40 хвилин при кімнатній температурі (контроль із LC-MS). Потім реакційну суміш відфільтровували і промивали ТГФ (2 × 30 мл). Фільтрат концентрували на роторному випарнику й повторно розчиняли в СН2Сl2 (50 мол). Додавали NaHCO3 (1. 0 г) і N-метилгепт-6-ениламін (1.5 г, 13 ммоль). Реакційну суміш перемішували при кімнатній температурі протягом ночі і потім відфільтровували. Очищенням хроматографією на силікагелі (ефір) отримували 1.42 г (84 %) цільового продукту 6 m/z 690 (М+Н)+. Стадія F Розчин 6 (1.42 г, 2 ммоль) у сухому дихлоретані (900 мол, 0.0023М розчин) барботували аргоном приблизно протягом 15 хв. Потім додавали каталізатор Hoveyda-Grubbs 1-ого покоління (120 мг, 12 мол %) і реакційну суміш нагрівали зі зворотним холодильником при перемішуванні з повільним потоком аргону протягом 16 годин. Реакційну суміш охолоджували до кімнатної температури і до суміші додавали паладієвий очисник МР-ТМТ (приблизно 200 мг). Через 2,5 години очисник видаляли фільтрацією і промивали 50 мол СН2Сl2. Отриманий розчин концентрували на роторному випарнику. Залишок очищали хроматографією на колонках із силікагелем YMC (100 г, EtOAc/гексан 1:1), отримуючи 806 мг (57 %) цільового продукту 7 m/z -662 (М+Н)+. Стадія G Гідроксид літію (300 мг) у воді (6 мол) додавали до розчину макроциклічного складного ефіру 7 (806 мг, 2.1 ммоль) у тетрагідрофурані (12 мол) і метанолі (6 мол). Через 1 год при 50°С об'єм зменшували наполовину упарюванням і додавали воду (30 мол). Підкисленням (рН = 2) з наступною екстракцією хлороформом отримували 760 мг цільового продукту 8 у формі білого порошку: m/z = 662 (М+Н)+. Стадія Η Розчин кислоти 8 (760 мг, 1.2 ммоль) і CDI (389 мг, 2.4 ммоль, 2 екв.) у сухому ТГФ (10 мл) нагрівали зі зворотним холодильником протягом 2 год. в атмосфері N2. У разі необхідності азалактонове похідне, якщо бажано, може бути виділено. Реакційну суміш охолоджували до кімнатної температури й додавали суміш сульфаміду, отриманого як описано в WO03/053349, (436 мг, 3.6 ммоль, 3 екв.) і DBU (0.5 мол, 3 ммоль) у сухому ТГФ (10 мл). Отриманий розчин нагрівали при 55°С протягом 18 год (перевірка LC-MS). Потім реакційну суміш охолоджували до кімнатної температури, розчинник випарювали, і залишок розділяли між EtOAc і водою (рН встановлювали рівним 3 0 за допомогою НСI). Сирий матеріал очищали хроматографією на колонках (ЕtOАс/петролейний ефір 1:1), отримуючи 380 мг цільового продукту, забрудненого 20 % вихідного сульфаміду (визначення ЯМР). Цей матеріал очищали хроматографією на попередньо заповнених колонках (градієнт від простого ефіру до суміші простий ефір/ТГФ, 3:1). Третім очищенням хроматографією на колонках (силікагель YMC 50 г, простий ефір, потім простий ефір-метанол 9:1) отримували 176 мг цільової сполуки у формі жовтуватого порошку, що далі очищали препаративної ВЕРХ, отримуючи 55 мг цільового продукту у формі жовтуватого порошку, m/z = 737, (М+Н)+, дані ЯМР (125 МГц, 13 CDCl3) С, δ 6.3, 6.5, 6.9, 22.0, 22.7, 22.8, 25.4, 26.7, 28.6, 29.1, 29.7, 31.3, 32.7, 34.7, 38.4, 45.3, 51.8, 55.8, 60.1, 77.0, 96.7, 107.5, 114.7, 116.5, 119.4, 123.2, 125.8, 136.4, 151.4, 153.1, 157.7, 160.2, 161.8 165.5, 168.2, 168.7, 178.2. Приклад 2: Синтез N-[17-(2-(4-ізопропілтіазол2-іл)-7-метоксихінолін-4-іл-окси)-13-метил-2,144,6 діоксо-3,13,15-триазатрицикло[13.3.0.0 ]октадец 45 7-ен-4-карбоніл](циклопропіл)сульфонаміда, з визначеною стереохімією, як зображено нижче у сполуці (16). ПРОЦЕДУРА А: Стадія А До перемішуємого розчину Вос-4гідроксипроліна з визначеною стереохімією, як показано у формулі вище, (2.59 г, 11.2 ммоль) у диметилсульфоксиді (80 мол) додавали третбутоксид калію (3.77 г, 33.6 ммоль). Після 1 години перемішування при кімнатній температурі додавали хлорид хіноліну 2 (3.57 г, 112 ммоль), і розчин перемішували при кімнатній температурі протягом ночі. Суміш розбавляли Н2О (350 мол), промивали ЕtOАс (100 мл) і підкислялася до рН приблизно 3 за допомогою 1М НСI. Отриману суспензію екстрагували ЕtOАс (3 × 100 мл), промивали сольовим розчином і висушували над MgSO4. Після розпарювання отримували сполуку 10 (3.60 г, 62 %). Стадія В Сполуку 10 (3.60 г, 7.02 ммоль) розчиняли в DMF (20 мл). Потім додавали ефір 1-аміно-2-вінілциклопропанкарбоновоі кислоти і етилгідрохлорида (1.47 г, 7.68 ммоль), і реакційну суміш змивали аргоном і охолоджували до 0°С. Додавали DIPEA (3.00 мол, 17 2 ммоль) і HATU (2.93 г, 7.66 ммоль), і реакційну суміш перемішували протягом 40 хвилин при 0°С. Розчин упарювали, змішували з насиченим NaHCO3 і екстрагували ЕtOАс (3 × 100 мл). Об'єднані органічні шари промивали сольовим розчином, висушували над MgSO4 і упарювали Флеш-хроматографією на колонці (гексани/ЕtOАс 3 2 -> гексани/ЕtOАс 2:3) отримували сполуку 11 (3.81 г, 84 %) у формі білого порошку. Стадія С Розчин 11 (3.81 г, 5.86 ммоль) у СН2Сl2 (30 мол) і ТФК (30 мл) перемішували при кімнатній 95239 46 температурі протягом 1.5 годин. Летучі компоненти випарювали. До отриманого масла додавали насичений NaHCO3 (100 мл), і рідкий розчин екстрагували простим діетиловим ефіром (3 × 100 мл). Органічні шари поєднували, промивали сольовим розчином, висушували над MgSO4 і упарювали, отримуючи сполуку 12 (3.13 г, 98 %). Стадія D До розчину сполуки 12 (1.41 г, 2 56 ммоль) у ТГФ (40 мол) додавали NaHCO3 (4 столових ложки) і фосген у толуолі (1.93 М, 4.0 мол, 7.7 ммоль). Суміш енергійно перемішували протягом 1 години при кімнатній температурі, після чого фільтрували й упарювали. Залишок розчиняли в СН2Сl2 (40 мл) і додавали NaHCO3 (3 столових ложки) і 5-енілметиламін гідрохлорид (770 мг, 5.15 ммоль). Після перемішування протягом ночі при кімнатній температурі реакційну суміш відфільтровували, додавали приблизно 3 г кремнезему, і рідкий розчин упарювали досуха Флеш-хроматографією на колонці (простий діетиловий ефір -> 3% метанолу в простому діетиловому ефірі) отримували сполуку 13 (1.57 г, 89 %) у формі жовтуватого порошку. Стадія Ε Сполуку 13 (1.53 г, 2.18 ммоль) розчиняли у 1, 2-дихлоретані (1.50 л) і розчин дегазовувували Ν2. Додавали каталізатор Hoveyda Grubbs 1-ого покоління (95 мг, 0.16 ммоль) і суміш нагрівали зі зворотним холодильником протягом ночі в атмосфері N2. Розчину дозволяли охолодитись до 60°С, після чого додавали 1 столову ложку поглинача МРТМТ. Реакційну суміш перемішували протягом 3 годин (охолоджуючи до кімнатної температури), відфільтровували і упарювали. Залишок розчиняли в СН2СІ2, додавали приблизно 3 г кремнезема і рідкий розчин упарювали досуха. Після флешхроматографії на колонці (простий діетиловий ефір -> 3% метанолу в простому діетиловому ефірі) отримували сполуку 14(1 09 г, 74 %) у формі білих призм. Стадія F 47 Сполуку 14 (1.00 г, 1.51 ммоль) розчиняли у ТНР/метанол/Н2О 2:1:1 (200 мл). Водний розчин LiOH (1 Μ, 15.1 мол, 15.1 ммоль) додавали по краплях при кімнатній температурі протягом 10 хвилин, і отриману реакційну суміш перемішували при кімнатній температурі протягом 20 годин. Розчин підкислювали до рН приблизно 1 за допомогою 1М НСI і концентрували, поки майже весь ТГФ і метанол не був видалений. Отриманий рідкий розчин екстрагували чотири рази СН2Сl2, і органічні фази поєднували, висушували (MgSO4) і упарювали, отримуючи сполуку 15 (960 мг, 100 %) у формі жовтуватого порошку. Стадія G Розчин сполуки 15 (960 мг, 1.51 ммоль) у ТГФ (75 мл) поміщали в атомсферу N2, після чого додавали карбонілдіімідазол (CDI) (510 мг, 3 14 ммоль). Суміш нагрівали зі зворотним холодильником в атмосфері N2 протягом 1 години і потім їй давали охолонути до кімнатної температури. У разі необхідності азалактонове похідне, якщо бажано, може бути виділено. Додавали циклопропілсульфонамін (550 мг, 4.54 ммоль) і DBU (530 мкл, 540 мг, 3.54 ммоль), і розчин перемішували в атмосфері N2 при 55°С протягом ночі. Розчинник випарювали і сирий залишок розчиняли в СН2СІ2. Додавали приблизно 3 г кремнезема, і рідкий розчин упарювали досуха Флеш-хроматографією на колонці (простий діетиловий ефір -> 5% метанолу в простому діетиловому ефірі) отримували сполуку 16 (960 мг, 86 %) у формі білого порошку. ПРОЦЕДУРА В: Альтернативний синтез кислоти 15 Стадія А Вос-захищений 4-пдроксипролін (10.2 г, 44.1 ммоль), HATU (18.7 г, 49.2 ммоль) і складний циклопропіловий ефір (9.31 г, 48.6 ммоль), обидва з визначеною стереохімією, як показано у формулах вище, розчиняли в DMF (120 мл) і охолоджували до 0°С. Додавали діізопропілетиламін (DlPEA) 95239 48 (30.0 мол, 172 ммоль). Розчину дозволяли нагрітися до кімнатної температури й перемішували протягом ночі. Додавали СН2Сl2 (~80 мл), і реакційну суміш промивали насиченим водним NaHCO3, лимонною кислотою, Н2О і сольовим розчином і потім висушували (MgSO4). Додавали приблизно 30 г кремнезему і рідкий розчин упарювали досуха. Очищенням флеш-хроматографією на колонці (простий діетиловий ефір -> 7% метанолу в простому діетиловому ефірі) отримували сполуку 17 (13.0 г, 80 %) у формі білого порошку. Стадія В Сполуку 17 (8.11 г, 22.0 ммоль), пнітробензойну кислоту (5.51 г, 33.0 ммоль) і Ph-tP (8.66 г, 33.0 ммоль) розчиняли в ТГФ (100 мол). Розчин охолоджували до 0°С, і додавали по краплях DIAD (6.50 мол, 33.0 ммоль). Реакційній суміші дозволяли нагрітися до кімнатної температури і перемішували протягом ночі. Додавали насичений водний розчин NaHCO3 (60 мл), і суміш екстрагували кілька разів СН2СІ2. Органічні фази поєднували і висушували (MgSO4), після чого додавали приблизно 40 г кремнезему і рідкий розчин упарювали досуха. Очищенням флеш-хроматографією на колонці (пентан/простий діетиловий ефір 2:1 -> пентан/простий діетиловий ефір 1:2 -> 2% метанолу у простому діетиловом ефірі) отримували Восзахищений проміжний продукт (9.50 г, 83 %) у формі білого порошку. Цей проміжний продукт (9.50 г, 18 4 ммоль) розчиняли в СН2Сl2 (66 мол), і розчин охолоджували до 0°С ТФК (33 мол) додавали по краплях, і суміш перемішували при кімнатній температурі протягом 2 годин. Летучі компоненти випарювали, додавали СН2Сl2 (100 мл) і додавали водний розчин Na2CO3 (0.50 Μ), до досягнення рН приблизно 8. Після поділу органічну фазу висушували (MgSO4) і упарювали, отримуючи сполуку 18 (7.68 г, 83 %) у формі жовтуватого порошку. Стадія С До розчину сполуки 18 (6.90 г, 16.5 ммоль) у ТГФ (240 мл) додавали NaHCO3 (15 столових ложок) і фосген у толуолі (1.93 М, 18.0 мл, 34.7 ммоль). Суміш енергійно перемішували протягом 1 години при кімнатній температурі, після чого її фільтрували і упарювали. Залишок розчиняли у СН2Сl2 (250 мл) і додавали NaHCO3 (15 столових ложок) і гекс-5-еніл-метиламін гідрохлорид (5.00 г, 33.4 ммоль). Після перемішування протягом ночі при кімнатній температурі реакційну суміш відфільтровували, додавали приблизно 17 г кремнезе 49 му, і рідкий розчин упарювали досуха. Флешхроматографією на колонці (простий діетиловий ефір -> 3% метанолу у простому діетиловому ефірі) отримували сполуку 19 (8.00 г, 87 %) у формі білого порошку. Стадія D Сполуку 19 (1.61 г, 2.90 ммоль) розчиняли у 1,2-дихлоретані (1.50 л), і розчин дегазовували з N2. Додавали каталізатор Hoveyda Grubbs 1-ого покоління (125 мг, 0.21 ммоль) і суміш нагрівали зі зворотним холодильником протягом ночі в атмосфері N2. Розчину дозволяли охолодитись до 60°С, після чого додавали 1 столову ложку поглинача МР-ТМТ. Реакційну суміш перемішували протягом 3 годин (охолоджуючи до кімнатної температури), відфільтровували і упарювали. Залишок розчиняли в СH2Сl2, додавали приблизно 3 г кремнезему і рідкий розчин упарювали досуха. Після флешхроматографії на колонці (простий діетиловий ефір -> 3% метанолу у простому діетиловому ефірі) отримували сполуку 20 (1.13 г, 74 %) у формі сіруватого порошку. Стадія Ε Сполуку 20 (200 мг, 0.38 ммоль) розчиняли в суміші ТГФ/метанол/вода (21.1, 20 мл) і охолоджували у ванні з льодом. Повільно додавали водний розчин LiOH (1 Μ, 1.9 мл, 1.9 ммоль). Суміш перемішували при 0°С протягом 4 годин і потім нейтралізували 1М НСI і екстрагували СH2Сl2. Органічний шар промивали насиченим водяним розчином NaHCO3, H2O і сольовим розчином. Після висушування (MgSO4) і розпарювання сирий залишок очищали флеш-хроматографією на колонці (2% метанолу в СН2СІ2 -> 4% метанолу в СH2Сl2), отримуючи сполуку 21 (115 мг, 80 %) у формі сіруватого порошку. Стадія F 95239 50 Сполуку 21 (100 мг, 0.26 ммоль), Ph3P (100 мг, 0.38 ммоль) і 2-(4-ізопропіл-тіазол-2-іл)-7метоксихінолін-4-ол (1) (117 мг, 0.39 ммоль) змішували у ТГФ (10-15 мл), і рідкий розчин охолоджували до 0°С у ванні з льодом. Додавали по краплях DIAD (77 мкл, 0.39 ммоль). Ванну з льодом тоді видаляли, і суміш перемішували при кімнатній температурі протягом 12 годин. Розчинник випарювали, і сирий продукт розчиняли у суміші ТНР/метанол/Н2О 2:1:1 (12 мл). Додавали водний розчин LiOH (1 Μ, 3.80 мл, 3.80 ммоль), і суміш перемішували при кімнатній температурі протягом ночі. Об'єм подвоювали додаванням води, і рідкий розчин екстрагували простим діетиловим ефіром. Водну фазу підкислювали до рН приблизно 1 за допомогою 1М НСI і екстрагували СН2Сl2. Фазу СН2Сl2 висушували (MgSO4) і упарювали. Бажаний продукт отримували флеш-хроматографією на колонці (2% метанолу у простому діетиловом ефірі до елюювання надлишку хіноліну 1, і потім 10% метанолу в СН2Сl2), отримуючи сполуку 15 (71 мг, 42 %). Приклад 3: Отримання {17-[2-(4ізопропілтіазол-2-іл)-7-метоксихінолін-4-ілокси]4,6 2,14-діоксо-3,13,15-триаза-трицикло[13.3.0.0 ]октадец-7-ен-4-карбоніл}-аміду циклопропансульфонової кислоти, з визначеною стереохімією, у якості показано в сполуці (26) нижче Стадія А: Отримання етилового ефіру 1-({1[гекс-5-еніл-(4-метоксибензил)-карбамоїл]-4-[2-(4ізопропілтіазол-2-іл)-7-метоксихінолін-4-ілокси]пірролідин-2-карбоніл}-аміно)-2вінілциклопропанкарбонової кислоти, з визначеною стереохімією, у якості показано у сполуці (22) нижче Сполуку 5, отриману у Прикладі 1, стадія D (1.97 г, 3.58 ммоль) змішували із приблизно 200 мг NaHCO3 (2 маленькі ложки) і ТГФ (20 мл). До реакційної суміші додавали 3 мол 1.9М фосгену в толуолі, і реакційну суміш перемішували приблизно протягом 1.5 години при кімнатній температурі. 51 Реакцію перевіряли LC-MS. Вихідний матеріал зник, і сформувався пік проміжного хлоркарбоніла (> 90 % згідно даним LC-MS). Реакційну суміш відфільтровували і концентрували в роторному випарнику Потім неї розчиняли в СН2Сl2 і гекс-5-еніл(п-метокси-бензил) амін (0.9 г) додавали разом з 100-150 мг (1 ложка) NaHCO3. Реакційну суміш перемішували при кімнатній температурі протягом ночі і потім фільтрували і очищали хроматографією на колонці із силікагелем (EtOAc/петролейний ефір), отримуючи чисту цільову сполуку 22 (1.42 г, 84 %). MS (Μ +)797. Стадія В: Отримання етилового ефіру 17-[2-(4ізопропілтіазол-2-іл)-7-метоксихінолін-4-ілокси]-13(4-метоксибензил)-2,14-діоксо-3,13,15-триаза4,6 трицикло[13.3.0.0 ]октадец-7-ен-4-карбонові кислоти, з визначеною стереохімією, як показано у сполуці (23) нижче Сполуку 22 (1.68 г, 2.1 ммоль), отриману на стадії А, розчиняли в сухому дихлоретані (дистильованому з використанням СаН, приблизно 800 мл), і барботували аргоном приблизно протягом 10 хв. Потім до розчину додавали каталізатор Hoveyda 1-ого покоління (88 мг, 7 мол %), і реакційну суміш нагрівали при 100°С при перемішуванні з повільним потоком аргону протягом 16 годин. Реакційну суміш охолоджували до кімнатної температури й додавали паладієвий поглинач МРТМТ (приблизно 100 мг), і суміш перемішували протягом 2.5 годин. Поглинач видаляли фільтрацією й промивали 100 мл СН2Сl2. Отриманий розчин концентрували на роторному випарнику і висушували в сильному вакуумі, отримуючи цільове сполуки (m/z 599, (М+Н)+), чистота ВЕРХ 79 % (діодна матриця), 95 % (ELSD) Вихід 63 %. Стадія С: Отримання 17-[2-(4-ізопропілтіазол2-іл)-метоксихінолін-4-ілокси]-13-(4метоксибензил)-2,14-діоксо-3,13,15-триаза4,6 трицикло[13.3.0.0 ]октадец-7-ен-4-карбонові кислоти, з визначеною стереохімією, як показано у сполуці (24) нижче 95239 52 Сполуку 23 (910 мг, 1.2 ммоль), отримана на стадії В, розчиняли в 30 мл ТГФ, 15 мл метанолу і 15 мл Н2О. До розчину додавали водний 1М розчин LiOH (10 мл). Реакційну суміш перемішували протягом 2 годин при 50°С, щоб дозволити усьому вихідному матеріалу ввійти в реакцію (перевірка LC-MS). Реакційну суміш підкислювали лимонною кислотою, екстрагували хлороформом (3 × 75 мл) і промивали сольовим розчином. Органічну фазу висушували з MgSO4. Сирий продукт очищали флеш-хроматографією (градієнт, починаючи із градієнту етилацетат/гексан 2:1), отримуючи цільову сполуку 10 (24) у формі твердої речовини жовтуватого кольору (864 мг). Чистота ВЕРХ 96 %. Стадія D: Отримання [17-[2-(4-ізопропілтіазол2-іл)-7-метоксихінолін-4-ілокси]-13-(4метоксибензил)-2,14-діоксо-3,13,15-триаза4,6 трицикло-[13.3.0.0 ]октадец-7-ен-4-карбоніл]аміду циклопропансульфонової кислоти, з визначеною стереохімією, як показано в сполуці (25) нижче Сполуки 24 (50 мг, 0.068 ммоль, 1 екв ) змішували з CDI (44 мг, 3 екв ) і розчиняли в ТГФ (1.0 мол) у мікрохвильовій ампулі на 2 мл. Ковпачок закривали і ампулу промивали аргоном. Активацію проводили в мікрохвильовій печі протягом 12 хвилин при 100°С (нормальний режим; без попереднього перемішування). Реакцію перевіряли LC-MS (100%-ве перетворення в активований проміжний + продукт - маса менше маси субстрату М -16). У разі необхідності, азалактонове похідне, якщо бажано, може бути виділено. Циклопропілсульфонамід (49 мг, 4 екв) змішували із ТГФ, і DBU (60 мкл, 4 екв ) додавали в окрему ампулу. Після повної активації суміш додавали шприцом у реакційну ампулу. Реакційну суміш нагрівали в мікрохвильовій печі при 100°С протягом 1 години. Реакційну суміш концентрували на роторному випарнику, змішували з ЕtOАс і водою і додавали 1 краплю 3М НСI. Водну фазу промивали EtOAc (3 × 15 мл). Об'єднані органічні фракції промивали сольовим розчином і висушували над MgSO4. Після відфільтровування осушувача і концентрації на роторному випарнику сирий продукт очищали хроматографією на колонці із силікагелем YMC (~70 мг, етилацетат, Rt = 0,7, вихідний сульфамід 0.6). Фракції, що містять бажаний продукт, поєднували і концентрували на роторному випарнику, отримуючи цільову сполуку 25 (83 мг, вихід 98 %) з 86%-ой чистотою. Сполуку використовували без додаткового очищення на наступній стадії. Стадія Ε: Отримання {17-[2-(4-ізопропілтіазол2-іл)-7-метоксихінолін-4-ілокси]-2,14-дюксо4,б 3,13,15-триаза-трицикло[13.3.0.0 ]октадец-7-ен-4 53 карбоніл}-аміду циклопропансульфонової кислоти з визначеною стереохімією, як показано в сполуці (26) нижче Сполуку 25 (65 мг, 0.077 ммоль) розчиняли в 4 мл DCM Додавали 2 мл ТФК і розчин перемішували протягом 20 хвилин. Реакційну суміш лили в ділильну воронку з NaHCO3 і СНСl3. Екстрагували СНСl3 (3 × 50 мл) і промивали NaHCO3 (2 × 50 мл) і сольовим розчином. Органічну фазу висушували з MgSO4 і концентрували на роторному випарнику. Сирий продукт очищали із флеш-хроматографією на силікагелі YMC (20 г, простий діетиловий ефір), отримуючи продукт 26 у формі твердої речовини білого кольору (25 мг, вихід 45 %). Приклад 4: Отримання [17-[2-(4-ізопропілтіазол-2-іл)-7-метоксихінолін-4-ілокси]-2,14-діоксо4,6 3,13,15-триаза-трицикло-[13.3.0.0 ]октадец-7-ен4-карбоніл]-аміду 1-метилциклопропансульфоновоі кислоти з визначеною стереохімією, як показано в сполуці (28) нижче 95239 54 циклопропілсульфонамід (отриманий, як описано в WO2004/043339), отримуючи цільову сполуку (27) (24.5 мг, 43 %) у формі твердої речовини білого кольору після очищення хроматографією на колонках із силікагелем (елюент - простий діетиловий ефір). Стадія В: Отримання [17-[2-(4-ізопропіл-тіазол2-іл)-7-метоксихінолін-4-ілокси]-2,14-діоксо4,6 3,13,15-триаза-трицикло-[13.3.0.0 ]октадец-7-ен4-карбоніл]-амід 1-метил-циклопропансульфонової кислоти з визначеною стереохімією, як описано в сполуці (28) нижче Сполуку 27 (20 мг, 0.023 ммоль) розчиняли в 3 мл метиленхлорида, додавали 1 мл ТФК і реакційну суміш перемішували при кімнатній температурі протягом 20 хв. ВЕРХ показала відсутність вихідного матеріалу. До реакційної суміші додавали розчин насиченого NaHCO3 (5 мл) і отриману суміш екстрагували СН2Сl2. Органічний екстракт промивали сольовим розчином, висушували над MgSO4 і концентрували в роторному випарнику. Отримане масло очищали хроматографією на колонках із силікагелем YMC (20 г, простий діетиловий ефір), отримуючи цільову сполуку (28) (14.7 мг, 85 %) у формі білого порошку. Чистота ВЕРХ > 95 %. Приклад 5: Отримання [18-[2-(4ізопропілтіазол-2-іл)-7-метоксіхінолін-4-ілокси]2,15-діоксо-3,14,16-триаза-трицикло4,6 [14.3.0.0 ]нонадец-7-ен-4-карбоніл]-амід 1-метилциклопропансульфонової кислоти з визначеною стереохімією, як показано в сполуці (33) нижче Стадія А: Отримання [17-[2-(4-ізопропіл-тіазол2-іл)-7-метоксіхінолін-4-ілокси]-13-(4метоксибензил)-2,14-діоксо-3,13,15-триаза4,6 трицикло-[13.3.0.0 ]октадец-7-ен-4-карбоніл]аміду 1-метил-циклопропансульфоновоі кислоти з визначеною стереохімією як показано в сполуці (27) нижче Стадія А: Отримання етилового ефіру 1-[[1[гепт-6-еніл-(4-метоксибензил)-карбамоїл]-4-[2-(4ізо-пропілтіазол-2-іл)-7-метоксихінолін-4-ілокси]пірролідин-2-карбоніл]-аміно]-2-вінілциклопропанкарбонової кислоти з визначеною стереохімією, як показано в сполуці (29) нижче Дотримувалися процедури, описаної на стадії D Прикладу 3, але замість циклопропілсульфонаміда використовували 1-метил 55 Цільову сполуку 29 отримували згідно процедури, описаної на стадії А Приклада 3, але використовуючи гепт-6-еніл-(п-метоксибензил)-амін замість гекс-5-еніл-(п-метоксибензил)-аміну. Стадія В: Отримання етилового ефіру 18-[2-(4ізопропілтіазол-2-іл)-7-метоксихінолін-4-ілокси]-14(4-метоксибензил)-2,15-діоксо-3,14,16-триаза4,6 трицикло[14.3.0.0 ]нонадец-7-ен-4-карбоновоі кислоти з визначеною стереохімією, як показано в сполуці (30) нижче 63 Сполуку, отриману на стадії А (29), обробляли згідно процедури, описаної на стадії В Приклада 3, отримуючи цільову сполуку (30). Стадія С: Отримання 18-[2-(4-ізопропілтіазол2-іл)-7-метоксихінолін-4-ілокси]-14-(4меткосибензил)-2,15-діоксо-3,14,16-триаза4,6 трицикло[14.3.0.0 ]нонадец-7-ен-4-карбонової кислоти з визначеною стереохімією, як показано в сполуці (31) нижче Сполуку, отриману на стадії В (30), обробляли згідно процедури, описаної на стадії С Приклада 3, отримуючи цільову сполуку (31). Стадія D: Отримання [18-[2-(4-ізопропіл-тіазол2-іл)-7-метоксихінолін-4-ілокси]-14-(4метоксибензил)-2,15-діоксо-3,14,16-триаза4,6 трицикло[14.3.0.0 ]нонадец-7-ен-4-карбоніл]аміду 1-метил-циклопропансульфонової кислоти з визначеною стереохімією, як показано в сполуці (32) нижче 95239 56 64 Сполуку, отриману вище на стадії С (31), обробляли згідно процедури, описаної на стадії D Приклада 3, але використовуючи у якості основи замість DBU LiHMDS (4 екв , 1М розчин у ТГФ). Реакція була повною після закінчення 2 годин при кімнатній температурі. Цільову сполуку (32) отримували у формі твердої речовини білого кольору після очищення хроматографією на колонках із силікагелем і використовували на наступній стадії без додаткового очищення. Стадія Ε: Отримання [18-[2-(4-ізопропілтіазол2-іл)-7-метоксихінолін-4-ілокси]-2,15-діоксо4,6 3,14,16-триаза-трицикло-[14.3.0.0 ]нонадец-7-ен4-карбоніл]-амід 1-метил-циклопропансульфоновоі кислоти з визначеною стереохімією, як показано в сполуці (33) нижче Сполуку, отриману вище на стадії D (32) (100 мг), обробляли згідно процедури, описаної на стадії Ε Приклада 3, отримуючи цільову сполуку (33) (55 мг, 73 %) Приклад 6: Активність сполук формули (І) Тест з використанням репликонів Сполуки формули (I) були досліджені на активність в інгібуванні реплікації РНК HCV у клітинному тесті. Тест показав, що сполуки формули (І) показали активність проти репликонів HCV, функціональних у клітинній культурі. Клітинний тест був заснований на біцистронній конструкції експресії, як описано Lohmann et al. (1999) Science vol. 285 pp. 110-113, з модифікаціями, описаними Krieger et al. (2001) Journal of Virology 75: 4614-4624, у мультицільовіп стратегії скринига. В основному спосіб полягав у наступному. У тесті використовували стабільно трансфіковану лінію клітин Huh7 luc/neoR (далі Huh-Luc). Ця лінія клітин містить РНК, що кодує біцистронну експресіонну конструкцію, що включає області NS3-NS5B дикого типу HCV типу 1b, що транслюються із Внутрішнього Сайту Посадки Рибосоми (TRES) з вірусу енцефаломіокардиту (EMCV), яким передує репортерна ділянка (Ff-люцифераза), і 57 95239 R область селектуємого маркера (neo , неоміцин фофсфотрансфераза). Конструкція обмежена 5' і 3' NTR (нетрансльовані області) від HCV типу 1b. Безперервна культура клітин реплікону в присутR ності G418 (neo ) залежить від реплікації РНК HCV. Стабільно трансфіковані клітини реплікону, які експресують РНК HCV, що реплікується автономно і у великих кількостях, що кодує, серед іншого, люциферазу, використовуються для скринінгу противірусних сполук. Клітини реплікону висівали в 384-лункові планшети у присутності тестуємих і контрольних сполук, які додавали в різних концентраціях. Після інкубації протягом трьох днів вимірювали реплікацію HCV, тестуючи люциферазну активність (використовуючи стандартні субстрати і реактиви для люциферазного тесту і пристрій для візуалізації мікропланшетів Perkin Elmer ViewLux™ ultraHTS). Клітини реплікону в контрольних культурах показують високий рівень експресії люциферази при відсутності якого-небудь інгібітору. Інгібуючу активність сполуки на активність люциферази перевіряли на клітинах Huh-Luc, що дозволяє побудувати криву доза-відповідь для кожної тестуємої сполуки. Обчислювали значення EC50, які являють собою кількість сполуки, необхідну для 50 %-ого зниження рівня виявленої люциферазної активності, або більш точно, здатність генетично зв'язаного реплікону РНК HCV до реплікації. Тест інгібування Ціль цього тесту in vitro полягає у вимірі інгібування комплексів HCV NS3/4A протеази сполуками згідно даного винаходу. Цей тест показує, наскільки сполуки згідно даного винаходу були б ефекти 58 вні в інгібуванні протеолітичної активності HCV NS3/4A. Інгібування повнорозмірного ферменту NS3 протеази гепатиту С вимірювали по суті як описано в Poliakov, 2002 Prot Expression & Purification 25 363 371. Коротко, гідроліз депсипептидного субстрату, Ac-DED(Edans)EEAbu\|/[COO]ASK(Dabcyl)NH2 (AnaSpec, San Jose, США), вимірювали спектрофлуорометричним способом у присутності пептидного кофактора, KKGSWTVGRTVLSGK (Ake Engstrom, Department of Medical Biochemistry and Microbiology, Uppsala University, Швеція). [Landro, 1997 #Biochem 36 9340-9348]. Фермент (1 нМ) інкубували в 50 мМ HEPES, pH 7,5, 10 мМ DTT, 40% гліцерину, 0.1 % октил-D-глюкозиду з 25 мкМ кофактора NS4A і інгібітора при 30°С протягом 10 хвилин, після чого реакцію ініціювали додаванням 0.5 мкМ субстрату. Інгібітори розчиняли в диметилсульфоксиді, руйнували клітини ультразвуком протягом 30 секунд і перемішували у вортексі. Розчини між вимірами зберігали при -20°С. Кінцеву концентрацію диметилсульфоксида в тестуємому зразку встановлювали на рівні 3,3%. Ступінь гідролізу коректували для внутрішніх ефектів фільтра згідно відомих процедур. [Liu, 1999 Analytical Biochemistry 267 331-335]. Значення Кі оцінювали за допомогою аналізу нелінійної регресії (GraFit, Erithacus Software, Staines, MX, UK), використовуючи модель для конкурентного інгібування і фіксоване значення для Км (0.15 мкМ). Для всіх вимірів здійснювали мінімум два повтори. У наступній Таблиці 1 перераховані сполуки, які були отримані згідно будь-якого із зазначених прикладів. Активності протестованих сполук також показані. Таблиця 1 Номер приклада Приклад 1 Приклад 2 Приклад 3 Приклад 4 Приклад 5 Номер сполуки 9 16 26 28 33 Приклад 7: Здатність до проникнення сполук формули (І) У цьому прикладі вимірювали транспорт сполук формули (І) через клітини шлунково-кишкового тракту людини У тесті використовували відомі клітини Сасо-2 із числом пасажів від 40 до 60. Апікально (А) - базолатеральний (В) транспорт Кожна сполука була перевірена у 2-4 лунках Базолатеральні і апікальні лунки містили 1,5 мл і 0,4 мл транспортного буферу (ТВ), відповідно, і стандартна концентрація тестуємих речовин становила 10 мкм Крім того, всі тестуємі розчини і буфери містили 1% ДМСО До експерименту транспортні планшети були попередньо покриті культуральним середовищем, що містить 10%-у сироватку протягом 30 хвилин, щоб уникнути неспецифічного зв'язування з пластичним матеріалом Після 21-28 днів у культурі на фільтруючій підложці клітини були готові до експериментів на проникнення. ЕС50 (мкМ) Тест реплікону 7,0 × 10-3 5,5 × 10-2 6,4 × 10-3 2,3 × 10-3 3,6 × 10-3 Кі (нм) Ферментний тест 0,12 0,45 0,4 0,3 0,3 Транспортний планшет No. 1 складався з 3 рядів по 4 лунки кожний. Ряд 1 був позначений "Промивання", ряд 2 - "30 хвилин" і ряд 3 - "60 хвилин". Транспортний планшет No.2 складався з 3 рядів по 4 лунки, ряд 4 був позначений "90 хвилин", ряд 5 - "120 хвилин" і останній ряд не був позначений. Культуральне середовище з апікальних лунок видаляли, і вставки переносили в промивний ряд (No.1) у транспортному планшеті (планшет No.1) з 2 планшетів без вставок, які були вже отримані з 1,5 мл транспортного буфера (HBSS, 25 мМ HEPES, рН 7.4) у рядах 1-5. У скринінгу АВ ТВ у базолатеральній лунці також містив 1% бичачого сироваткового альбуміну. 0.5 мл транспортного буфера (HBSS, 25 мМ MES, рН 6.5) додавали до вставок і моношари клітин урівноважували в транспортній буферній системі протягом 30 хвилин при 37°С у шейкері Polymix. Після врівноважування з буферною сис 59 95239 темою значення трансепітеліального електричного опору (TEER) вимірювали у кожній лунці за допомогою пристрою chopstick EVOM. Значення TEER звичайно становили від 400 до 1000 Ω на лунку (залежно від використовуваного числа пасажів). Транспортний буфер (ТВ, рН 6.5) видаляли з апікального боку, і вставки переносили в ряд «30 хвилин» (No.2), а в апікальні (донорські) лунки додавали свіжі 425 мкл ТВ (рН 6.5), що включає тестуєму речовину. Планшети інкубували в шейкері Polymix при 37°С з низькою швидкістю коливань приблизно 150-300 об/хв. Після інкубації протягом 30 хвилин у ряді 2 вставки переносили в нові попередньо нагріті базолатеральні (реципієнтні) лунки кожні 30 хвилин; ряд З (60 хвилин), 4 (90 хвилин) і 5 (120 хвилин). 25 мкл проб відбирали з апікального розчину після закінчення ~2 хвилин і наприкінці експерименту. Ці проби являли собою донорські зразки від початку і кінця експерименту. 300 мкл відбирали від базолатеральних (реципієнтних) лунок у кожний розрахунковий момент часу і значення TEER вимірювали наприкінці експерименту. До всіх відібраних проб додавали ацетонітрил до кінцевої концентрації 50 % у пробах. Відібрані проби зберігали при -20°С до аналізу ВЕРХ або LC-MS. Базолатерально-апікальний транспорт Кожну сполуку перевіряли у 2-4 лунках. Базолатеральні і апікальні лунки містили 1,55 мл і 0,4 мл ТВ, відповідно, і стандартна концентрація тестуємих речовин становила 10 мкМ. Крім того, усі тестуємі розчини і буфери містили 1% ДМСО. До експерименту транспортні планшети були попередньо покриті культуральним середовищем, що містить 10%-у сироватку протягом 30 хвилин, щоб уникнути неспецифічного зв'язування із пластичним матеріалом. Після 21-28 днів у культурі на фільтруючій підложці клітини були готові до експериментів на проникнення. Культуральне середовище з апікальних лунок видаляли, і вставки переносили в промивний ряд (No. 1) у новому планшеті без вставок (транспортний планшет) Транспортний планшет містив 3 ряди з 4 лунок кожний. Ряд 1 був позначений як "промивання" і ряд 3 - як "експериментальний ряд". Транспортний планшет попередньо готували з використанням 1,5 мл ТВ (рн 7 4) у 60 промивному ряді No.1 із 1,55 мл ТВ (рн 7.4), що включає тестуєму речовину, в експериментальному ряді No.3 (донорський бік). 0.5 мл транспортного буфера (HBSS, 25 мМ MES, рН 6.5) додавали до вставок у ряді No 1 і моношари клітин урівноважували в транспортній буферній системі протягом 30 хвилин при 37°С у шейкері Polymix. Після врівноважування з буферною системою значення TEER вимірювали в кожній лунці за допомогою пристрою chopstick EVOM. Транспортний буфер (ТВ, рн 6.5) видаляли з апікального боку, і вставки переносили в ряд 3 і до вставок додавали 400 мкл свіжого ТВ, рн 6.5. Через 30 хвилин 250 мкл відбирали з апікальної (реципієнтної) лунки і заміняли свіжим транспортним буфером Після цього по 250 мкл зразка відбирали й заміняли свіжим транспортним буфером кожні 30 хвилин до кінця експерименту протягом 120 хвилин, і нарешті, значення TEER вимірювали наприкінці експерименту. 25 мкл проб відбирали від базолатерального (донорського) відділення після ~2 хвилин і наприкінці експерименту. Ці проби являли собою донорські проби від початку і кінця експерименту. До всіх зібраних проб додавали ацетонітрил до кінцевої концентрації 50 % у пробах Відібрані проби зберігали при -20°С до аналізу ВЕРХ або LC-MS Визначення кумулятивної абсорбованої фракції, FAcum залежно від часу. FAcum обчислювали по формулі. C A cum RI CDI де CRi, позначає концентрацію реципієнта наприкінці інтервалу і, і CDi - донорську концентрацію на початку інтервалу і. Повинна бути отримана лінійна залежність Визначення коефіцієнтів проникнення (Рарр, см/с) обчислювали по формулі. (k VR Papp ( A 60) -1 де k - швидкість транспорту (хв. ) визначена як крутизна, отримана лінійною регресією кумулятивної абсорбованої фракції (FAcum) залежно від часу (хв), VR позначає обсяг у реципієнтній камері 2 (мл), і А позначає область фільтра (см ). Таблиця 2 Еталонні сполуки Категорія абсорбції в організмі людини ПАСИВНИЙ ТРАНСПОРТ Низька (0-20%) Помірна (21-75%) Висока (76-100%) АКТИВНИЙ ТРАНСПОРТ Транспортна амінокислота активний відтік PGP-MDR1 Маркери Маннит Метотрексат Ацикловір Пропанолол Кофеїн % абсорбції в організмі людини 16 20 30 90 100 L-фенілаланін 100 Дигоксин 30 У наступній таблиці 3 показані результати -6 проникнення (Раpp), виражені в 10 см/с, для ре презентативної вибірки сполук згідно винаходу в тесті на проникнення, описаному вище.
ДивитисяДодаткова інформація
Назва патенту англійськоюMacrocyclic inhibitors of hepatitis c virus
Автори англійськоюDe Kock Herman Augustinus, Raboisson Pierre Jean-Marie Bernard, Simmen Kenneth Alan, Lindstroem Mats Stefan, Kahnberg Pia Cecilia, Antonov Dmitriy, Nilsson Karl Magnus, Samuelsson Bengt Bertil, Rosenquist Aasa Annica Kristina
Назва патенту російськоюМакроциклические ингибиторы вируса гепатита с
Автори російськоюДе Кок Герман Августинус, Рабуассон Пьер Жан-Мари Бернар, Симмен Кеннет Алан, Линдстром Матс Стефан, Канберг Пиа Сесилия, Антонов Дмитрий, Нилссон Карл Магнус, Самуэльссон Бенгт Бертил, Розенквист Эса Анника Кристина
МПК / Мітки
МПК: A61K 31/4709, C07K 5/06, C07K 5/078, A61P 31/14
Мітки: гепатиту, макроциклічні, інгібітори, вірусу
Код посилання
<a href="https://ua.patents.su/31-95239-makrociklichni-ingibitori-virusu-gepatitu-s.html" target="_blank" rel="follow" title="База патентів України">Макроциклічні інгібітори вірусу гепатиту с</a>
Макроциклічні інгібітори вірусу гепатиту с

Номер патенту: 93377
Опубліковано: 10.02.2011
Автори: Сальберг Свен Крістер, Белфраге Анна Карін Гертруд Ліннеа, Самуельссон Бенгт Бертіл, Ху Лілі, Велінг Хорст Юрген, Де Кок Герман Аугустінус, Рабуассон П'єр Жан-Марі Бернар, Нілссон Карл Магнус, Классон Бьорн Олоф, Ліндквіст Карін Шарлотта, Канберг Піа Сесілія, Ліндстром Матс Стефан, Розенквіст Еса Анніка Крістіна, Сіммен Кеннет Алан, Валльберг Ханс Крістіан
МПК: C07D 417/04, A61P 31/12, A61K 31/517, C07D 239/72
Мітки: вірусу, інгібітори, гепатиту, макроциклічні
Формула / Реферат:
1. Сполука формули І:або її N-оксиди, солі й стереоізомери, де А являє собою OR1, NHS(=O)рR2; де R1 являє собою водень, С1-С6алкіл, С0-С3алкіленкарбоцикліл, C0-С3алкіленгетероцикліл; R2 являє собою С1-С6алкіл, С0-С3алкіленкарбоцикліл, С0-С3алкіленгетероцикліл; р незалежно...
Макроциклічні карбонові кислоти і ацилсульфонаміди як інгібітори реплікації вірусу гепатиту с (вгс)

Номер патенту: 84579
Опубліковано: 10.11.2008
Автори: Кеннеді Ейпріл Лайн, Джіанг Ютонг, Кондроски Кевін Рональд, Блетт Лоренс М., Вуддард Бенджамін Т., Венгловскі Стівен Марк, Стенджел Пітер Джон, Джосі Джон Ентоні, Ендрюс Стівен Вейд, Маддуру Мачендер Р., Догерти Джордж Ендрю
МПК: C08H 1/00, A61K 38/00
Мітки: вірусу, гепатиту, кислоти, макроциклічні, інгібітори, карбонові, реплікації, вгс, ацилсульфонаміди
Формула / Реферат:
1. Сполука, що має формулу І, VIII або IX:, I, VIII , IXде:(a) R1 і R2 кожний незалежно являють собою Н, галоген, ціано, нітро, гідрокси, С1-6алкіл, С3-7циклоалкіл, C4-10алкілциклоалкіл, С2-6алкеніл,...
Макроциклічні пептиди, які є активними по відношенню до вірусу гепатиту с

Номер патенту: 78014
Опубліковано: 15.02.2007
Автори: Ллінас-Брунет Монтсе, Горус Вайда Дж.
МПК: C07K 5/083, A61K 31/7052, A61K 38/00, A61P 43/00, C07K 5/087, C07K 5/078, A61P 31/14, A61K 45/00, A61K 38/21
Мітки: гепатиту, активними, вірусу, макроциклічні, відношенню, пептиди
Формула / Реферат:
1. Сполука формули (І): , (I)у якій R1 означає гідрокси або NHSO2R1A, де R1A означає С1-С8алкіл, С3-С7циклоалкіл або С1-С6алкіл-С3-С7циклоалкіл, які всі необов'язково заміщені 1-3 замісниками, вибраними з ряду, який включає галоген, ціано, нітро, ОС1-С6алкіл, амідо, аміно або феніл, або R1А означає С6- або С10арил, необов'язково заміщений 1-3 замісниками,...
Гетероциклічні трипептиди як інгібітори вірусу гепатиту с

Номер патенту: 77757
Опубліковано: 15.01.2007
Автори: Бейлі Мюррей Д., Ллінас-Брунет Монтсе, Гіро Еліс
МПК: A61P 31/14, C07K 5/08, C12N 15/09, C07K 5/083, C07K 5/087, A61K 38/21, A61K 31/7056, A61K 38/00, C07D 417/14, A61P 31/00, A61P 1/16
Мітки: гепатиту, вірусу, гетероциклічні, інгібітори, трипептиди
Формула / Реферат:
1. Сполука формули (І): , (I)у якій R1 означає гідрокси або NHSO2R1A, де R1A означає С1-С8алкіл, С3-С7циклоалкіл або С1-С6алкіл-С3-С7циклоалкіл, які всі необов'язково заміщені 1-3 замісниками, вибраними з ряду, який включає галоген, ціано, нітро, ОС1-С6алкіл, амідо, аміно або феніл, або R1А означає С6- або С10арил, необов'язково заміщений 1-3 замісниками,...
Пептидні інгібітори вірусу гепатиту с, спосіб їх одержання та фармацевтична композиція

Номер патенту: 73480
Опубліковано: 15.08.2005
Автори: Пупар Марк-Андре, Тсантріцос Юла С., Гудро Наталі, Лінас-Брюне Монтсе, Кемерон Дейл, Ранкур Жан, Гіро Еліз, Бейлі Мьоррі Д.
МПК: A61P 31/12, A61K 38/55, C07K 1/10, A61K 45/00, C07K 5/107, C07K 5/10, A61P 31/14, A61K 31/13, C07K 7/06, C12N 15/09, A61K 31/7052, A61P 43/00, A61K 38/21, C07K 14/81, A61P 1/16, A61K 38/00
Мітки: вірусу, фармацевтична, спосіб, композиція, пептидні, інгібітори, одержання, гепатиту
Формула / Реферат:
1. Сполука формули (І), включаючи її рацемати, діастереоізомери і оптичні ізомери: (І),у якійа дорівнює 0 або 1,b дорівнює 0 або 1,Y означає Н або С1-С6алкіл,В означає Н, дансил або ацильне похідне формули R7-C(O)- ,R7 означає С1-С6алкіл або Het, який вибирають з групи, що містить: , , ,, , або,R6 означає С1-С6алкіл, заміщений карбоксилом, R5 означає...
Попередній патент: Імуногенна композиція
Наступний патент: Спосіб формування дифузно-відбиваючої інформації на поверхні світлопровідних елементів і пристрій, в якому реалізовано спосіб
Випадковий патент: Спосіб одержання каталізатора для окислення в газовій фазі з визначеним розподілом за розмірами частинок оксиду ванадію