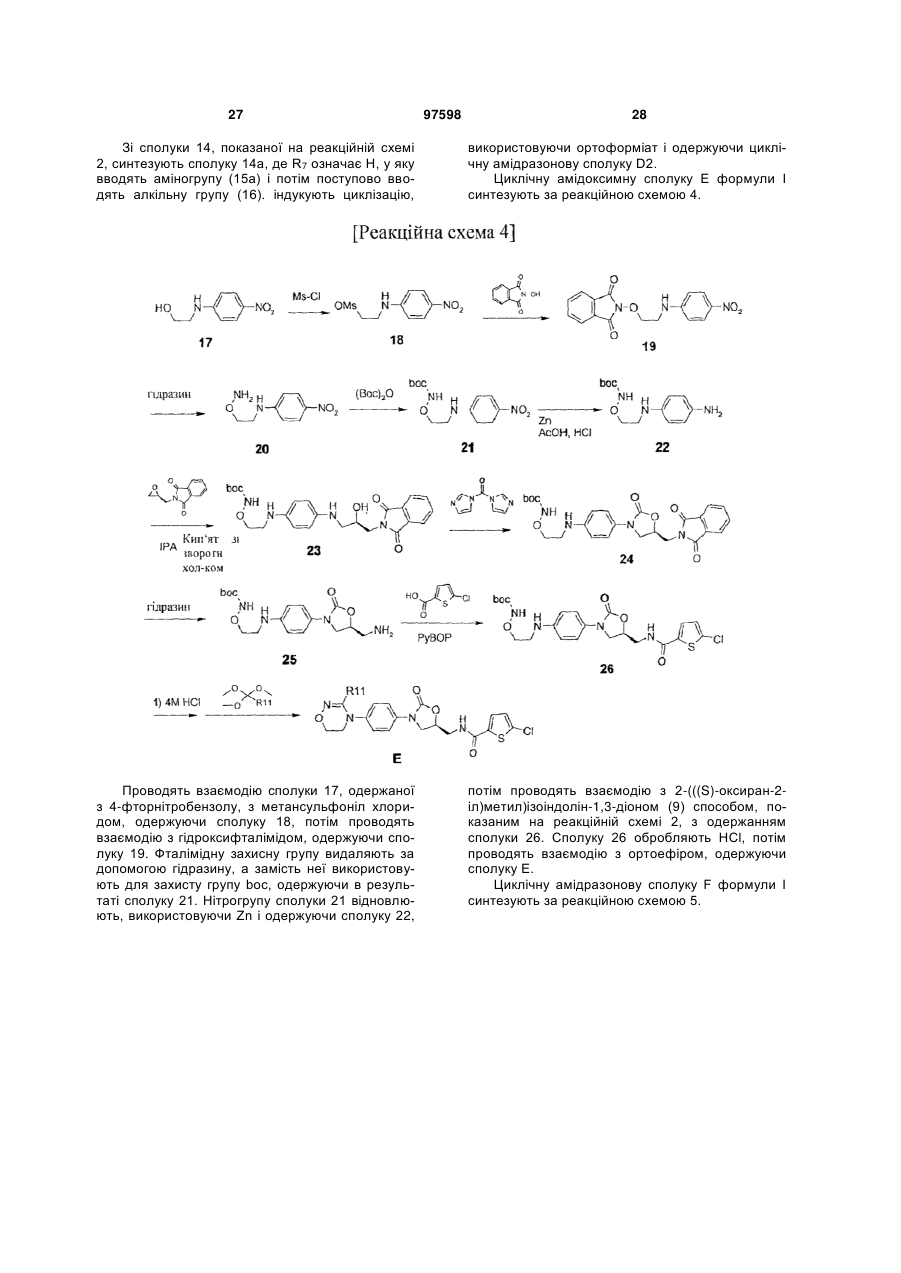
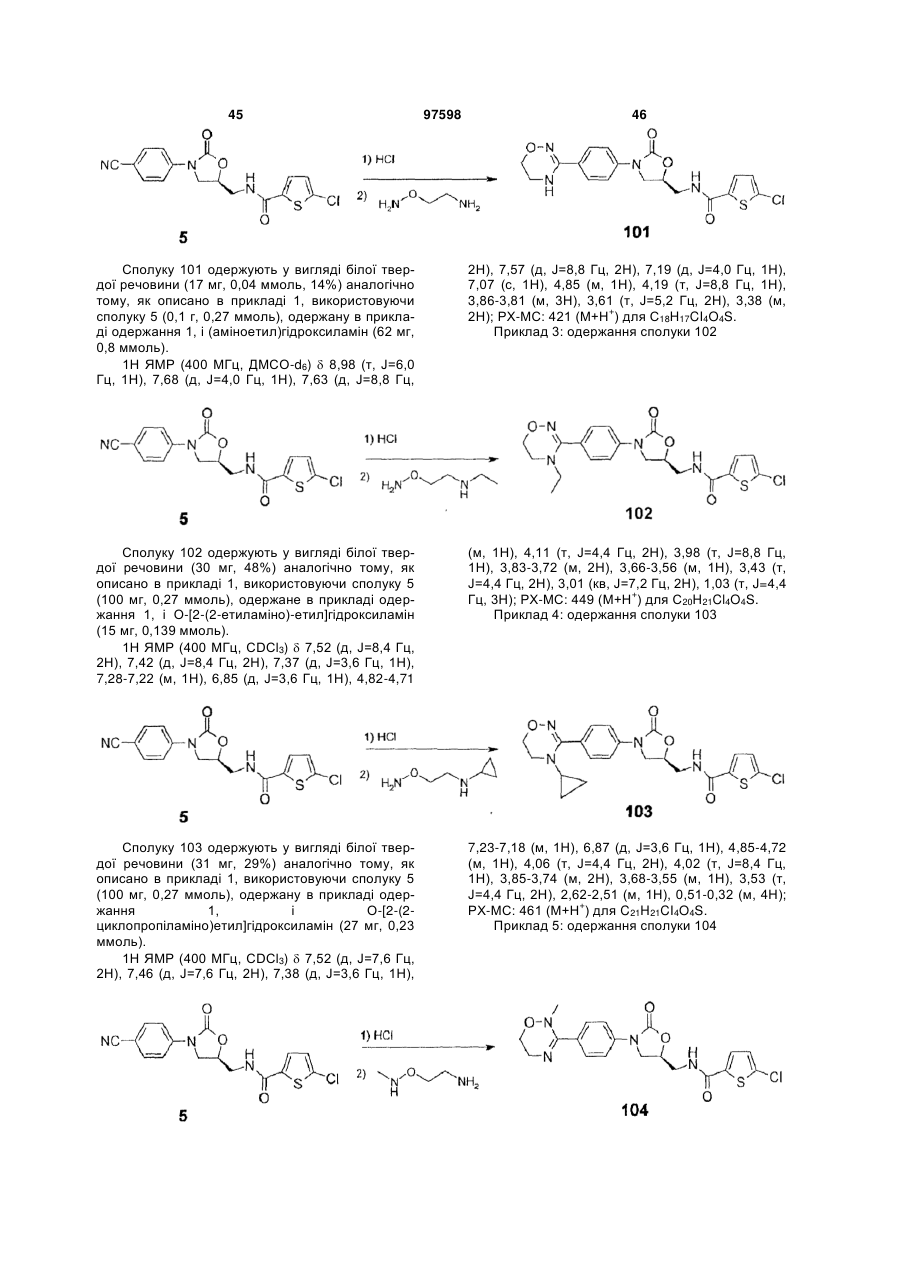
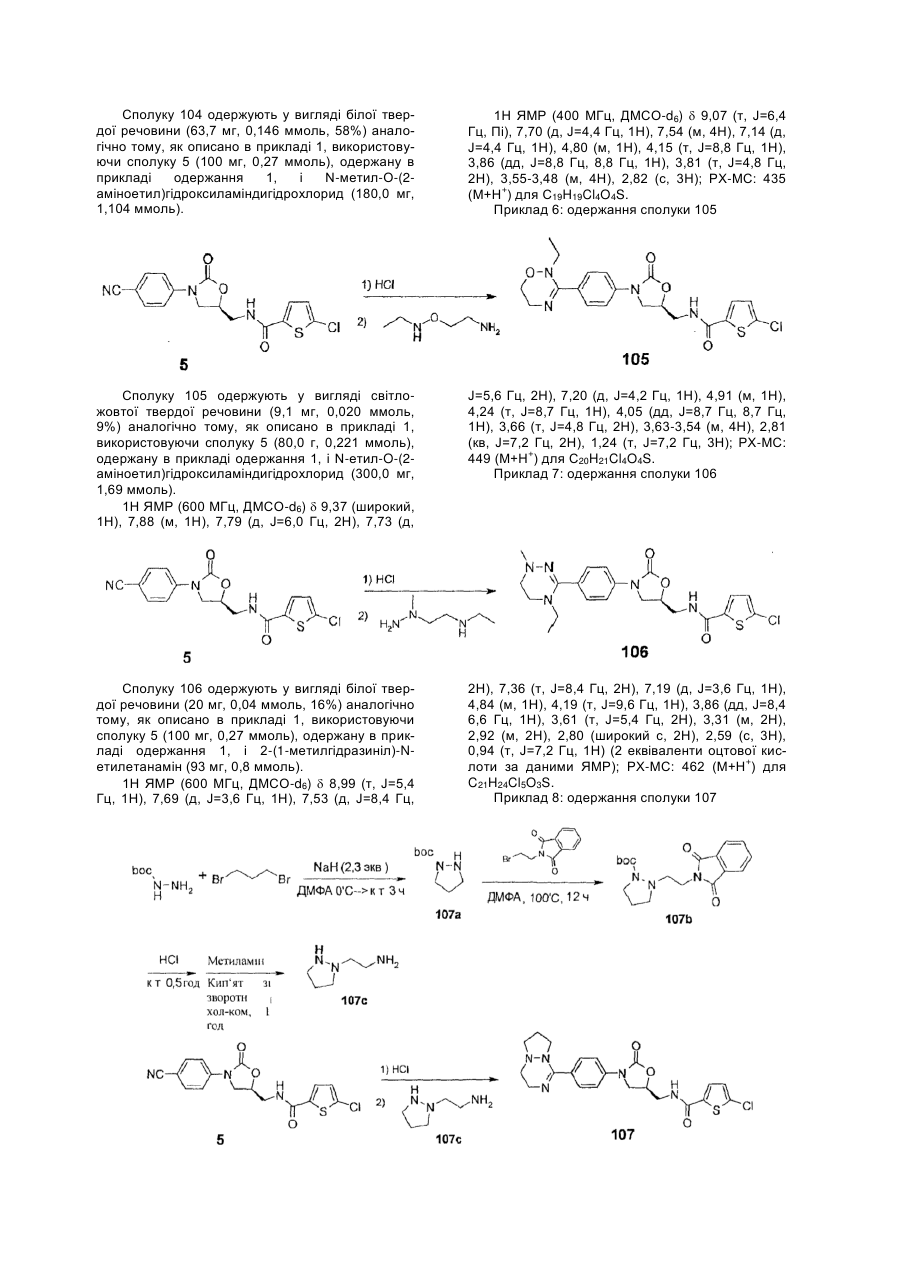
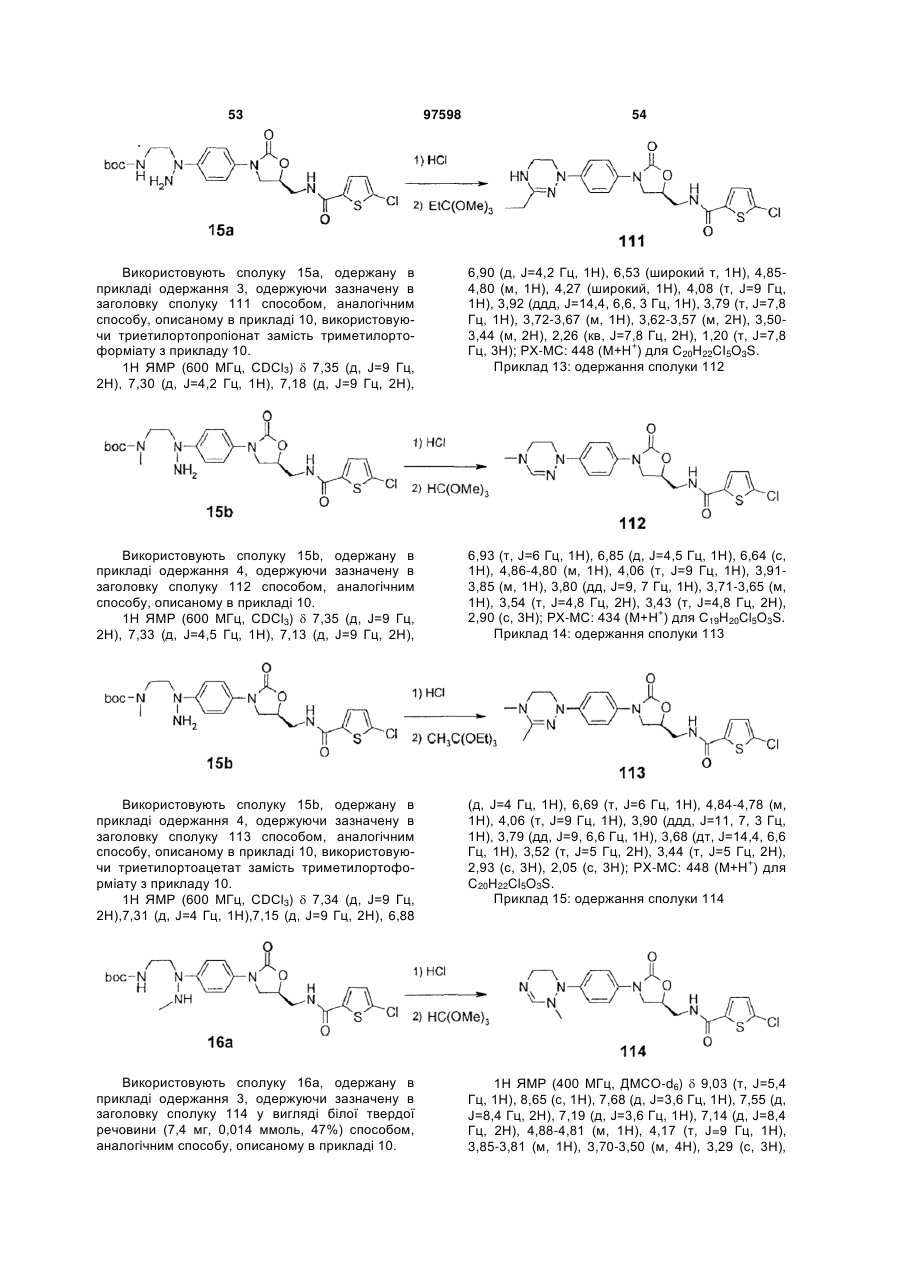
Інгібітори fxа з циклічним амідоксимом або циклічним амідразоном як р4 субодиниця, способи їх одержання і їх фармацевтичні композиції і похідні
Номер патенту: 97598
Опубліковано: 27.02.2012
Автори: Чає Санг Єун, Лі Хіанг Соок, Воо Сунг Хо, Чо Йоунг Лаг, Баєк Сунг Йоон, Кім Йєон Ок, Парк Дзу Хіун, Дзо Санг Хой, Сонг Хо Йоунг, Парк Тає Кіо, Кім Йонг Зу, Парк Хеє Сок, Лі Дає Йон





















Формула / Реферат
1. Оксазолідинонове похідне з циклічною амідоксимною або циклічною амідразоновою групою, представлене формулою 1, або його проліки, гідрат, сольват, ізомер або фармацевтично прийнятна сіль,
[Формула 1]
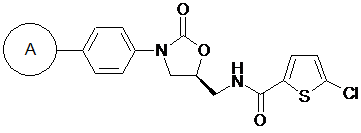 ,
,
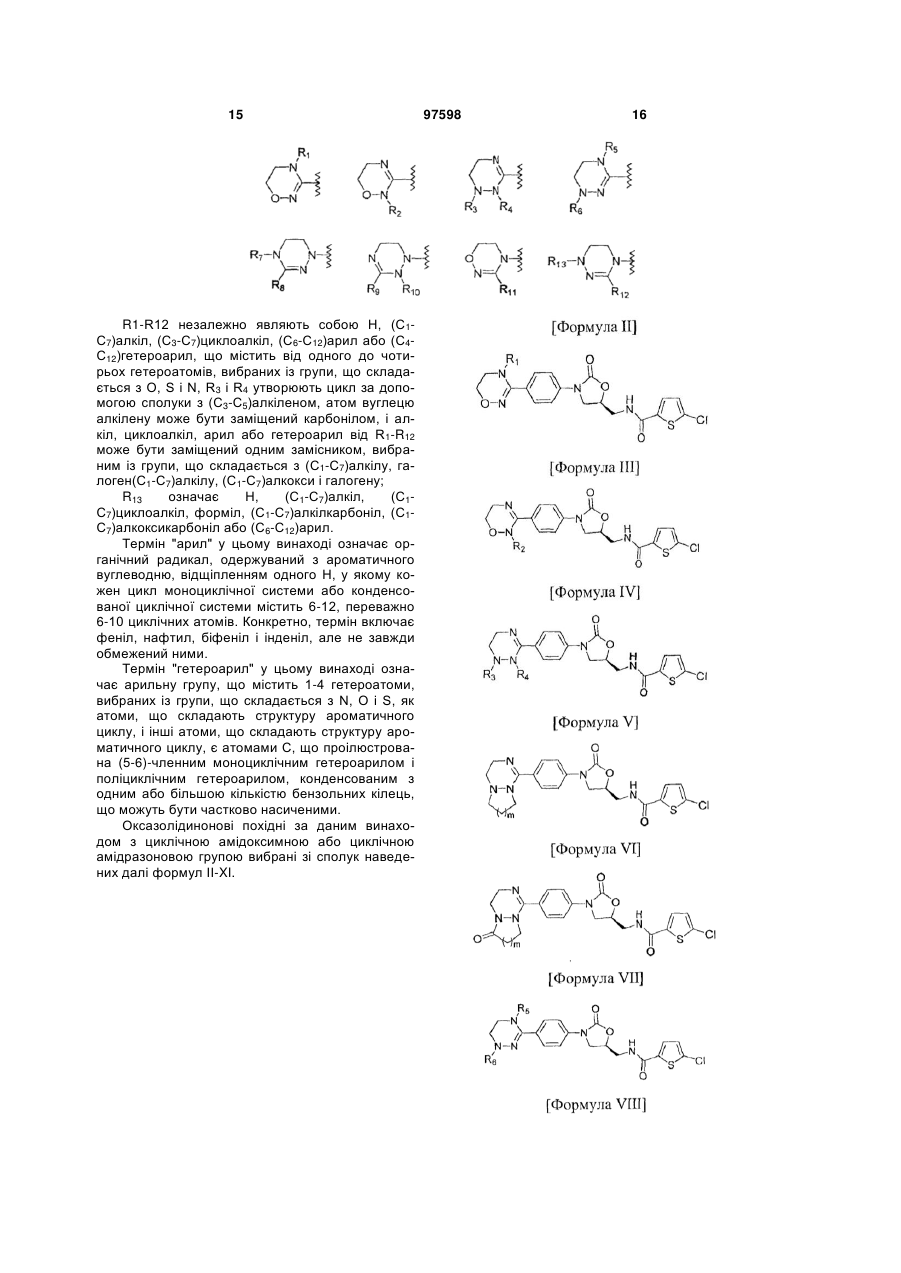
де цикл А означає залишок, вибраний із групи, що включає наступні структури:
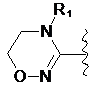 ,
, 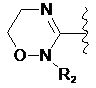 ,
, 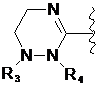 ,
, 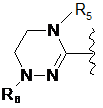 ,
,
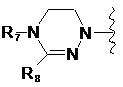 ,
, 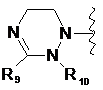 ,
, 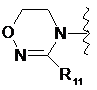 ,
, 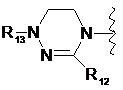 ,
,
де R1-R12 незалежно являють собою Н, (С1-С7)алкіл, (С3-С7)циклоалкіл, (С6-С12)арил або (С4-С12)гетероарил, що містить від одного до чотирьох гетероатомів, вибраних із групи, що складається з О, S і N, R3 і R4, утворюють цикл за допомогою (С3-С5)алкілену, атом вуглецю алкілену може бути заміщений карбонілом, і алкіл, циклоалкіл, арил або гетероарил у R1-R12 можуть бути заміщені будь-яким замісником, вибраним із групи, що складається з (С1-С7)алкілу, галоген(С1-С7)алкілу, (С1-С7)алкокси і галогену;
R13 означає Н, (С1-С7)алкіл, (С3-С7)циклоалкіл, форміл, (С1-С7)алкілкарбоніл, (С1-С7)алкоксикарбоніл або (С6-С12)арил.
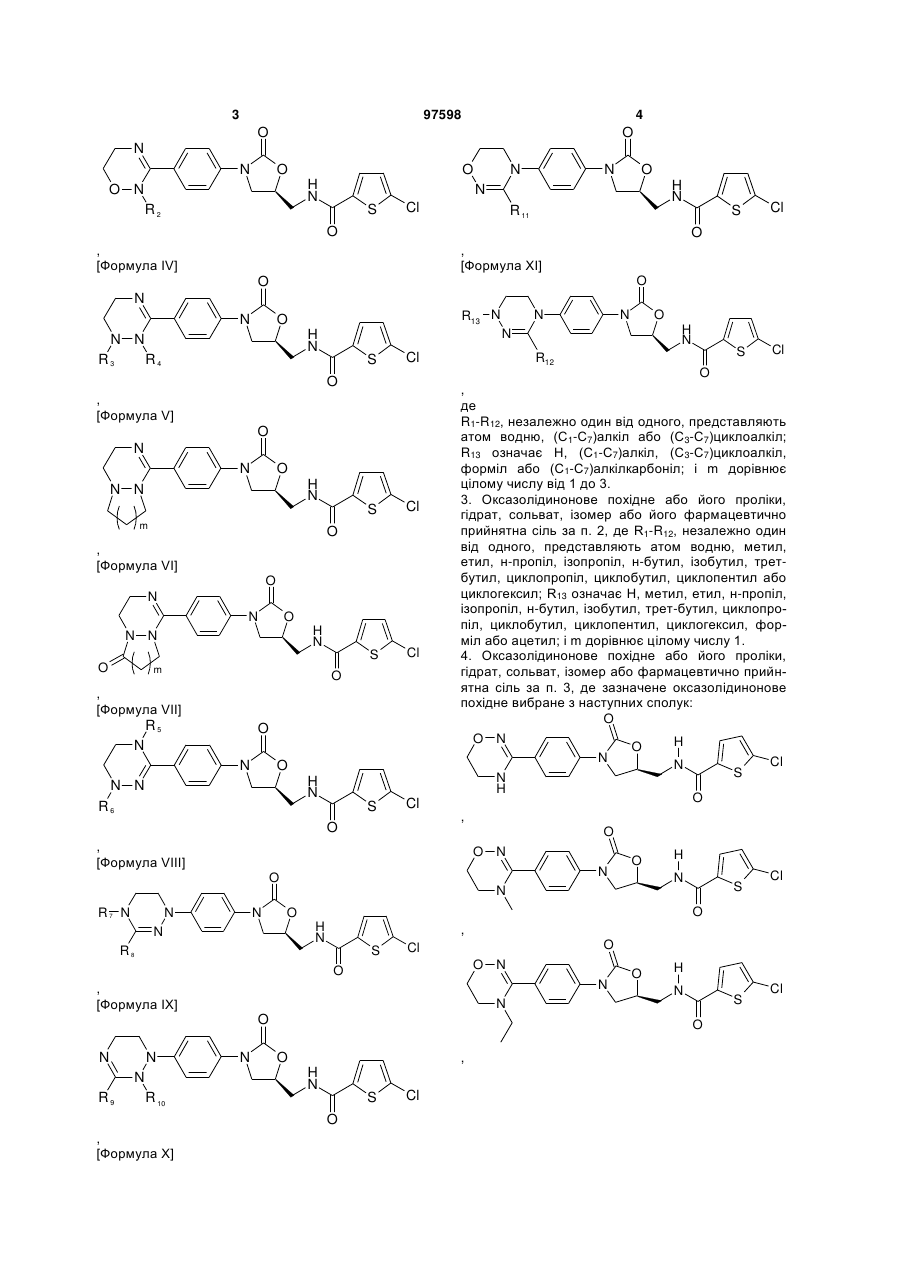
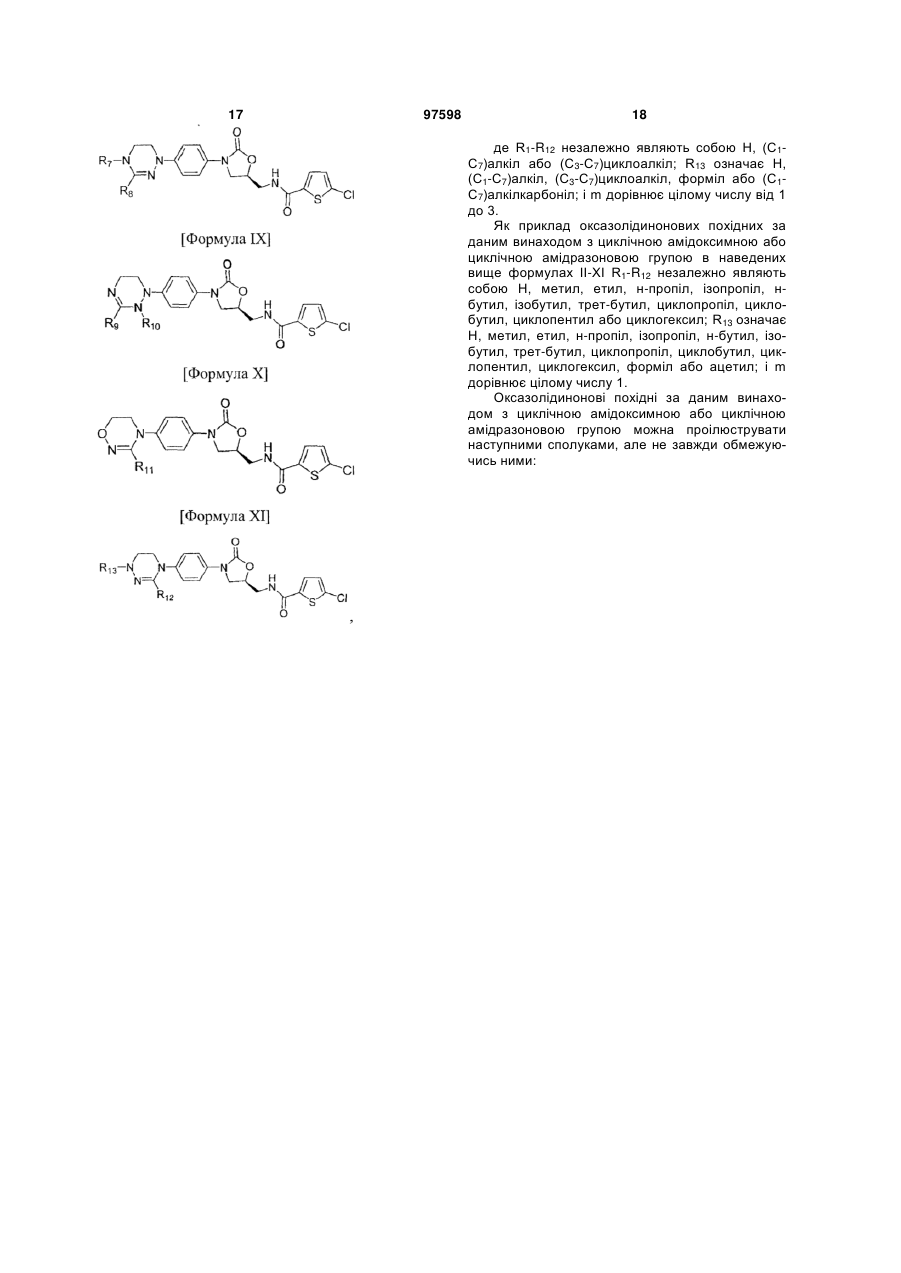
2. Оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або фармацевтично прийнятна сіль за п. 1, де оксазолідинонове похідне вибране з наступних формул ІІ-ХІ:
[Формула II]
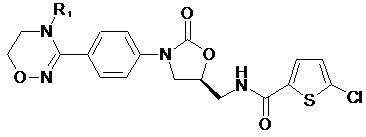 ,
,
[Формула III]
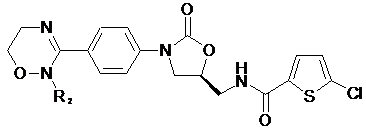 ,
,
[Формула IV]
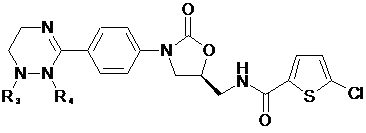 ,
,
[Формула V]
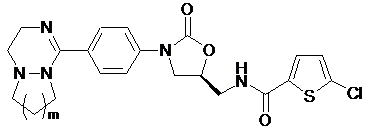 ,
,
[Формула VI]
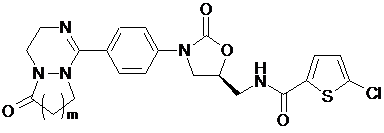 ,
,
[Формула VII]
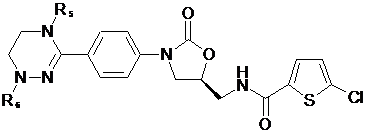 ,
,
[Формула VIII]
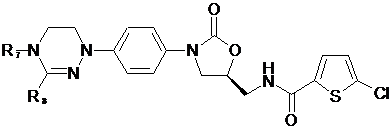 ,
,
[Формула IX]
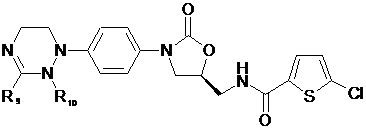 ,
,
[Формула X]
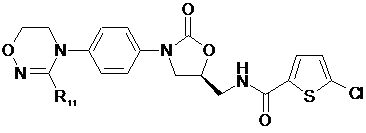 ,
,
[Формула XI]
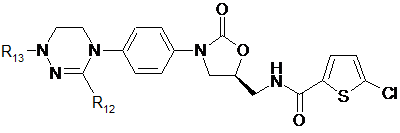 ,
,
де
R1-R12, незалежно один від одного, представляють атом водню, (С1-С7)алкіл або (С3-С7)циклоалкіл; R13 означає Н, (С1-С7)алкіл, (С3-С7)циклоалкіл, форміл або (С1-С7)алкілкарбоніл; і m дорівнює цілому числу від 1 до 3.
3. Оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або його фармацевтично прийнятна сіль за п. 2, де R1-R12, незалежно один від одного, представляють атом водню, метил, етил, н-пропіл, ізопропіл, н-бутил, ізобутил, трет-бутил, циклопропіл, циклобутил, циклопентил або циклогексил; R13 означає Н, метил, етил, н-пропіл, ізопропіл, н-бутил, ізобутил, трет-бутил, циклопропіл, циклобутил, циклопентил, циклогексил, форміл або ацетил; і m дорівнює цілому числу 1.
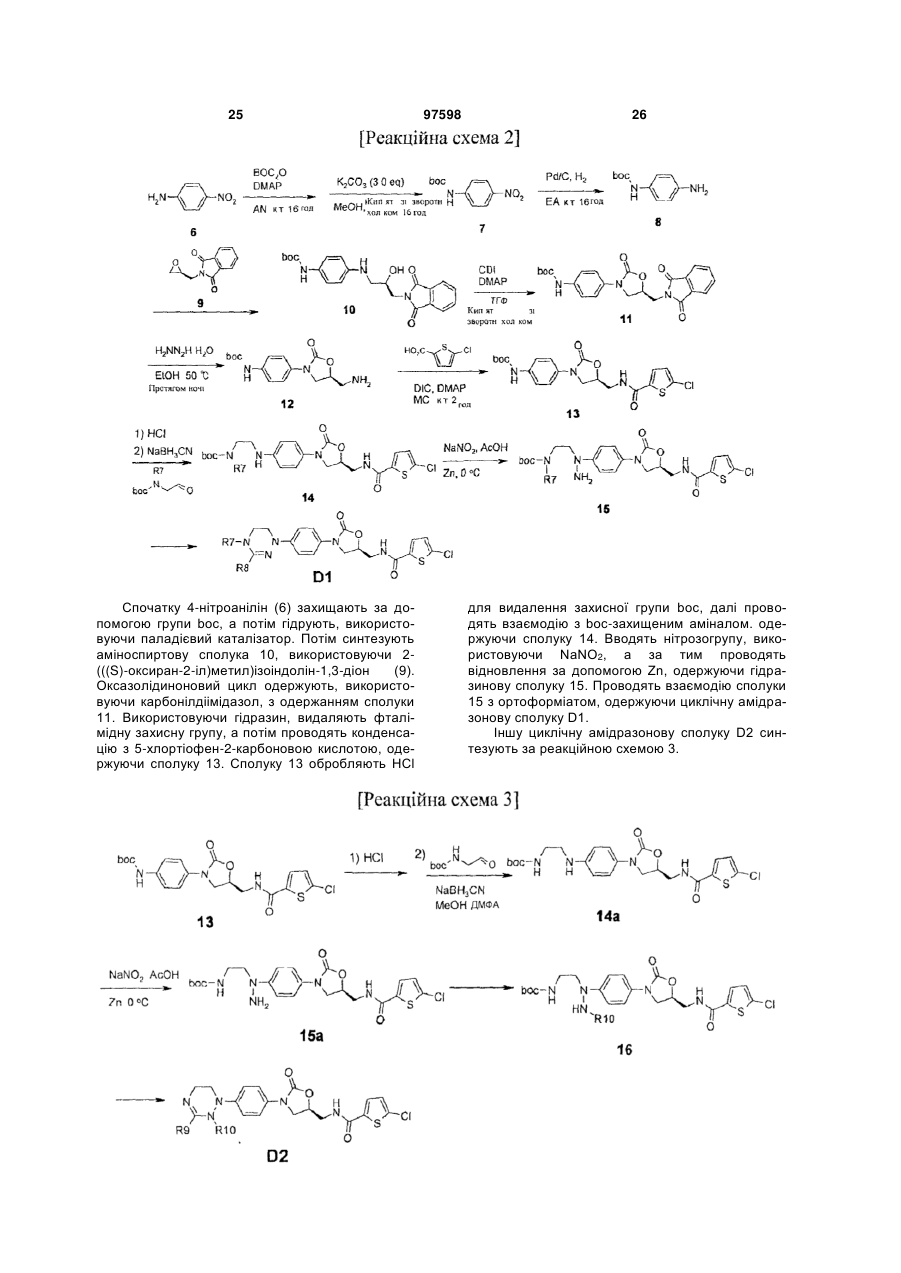
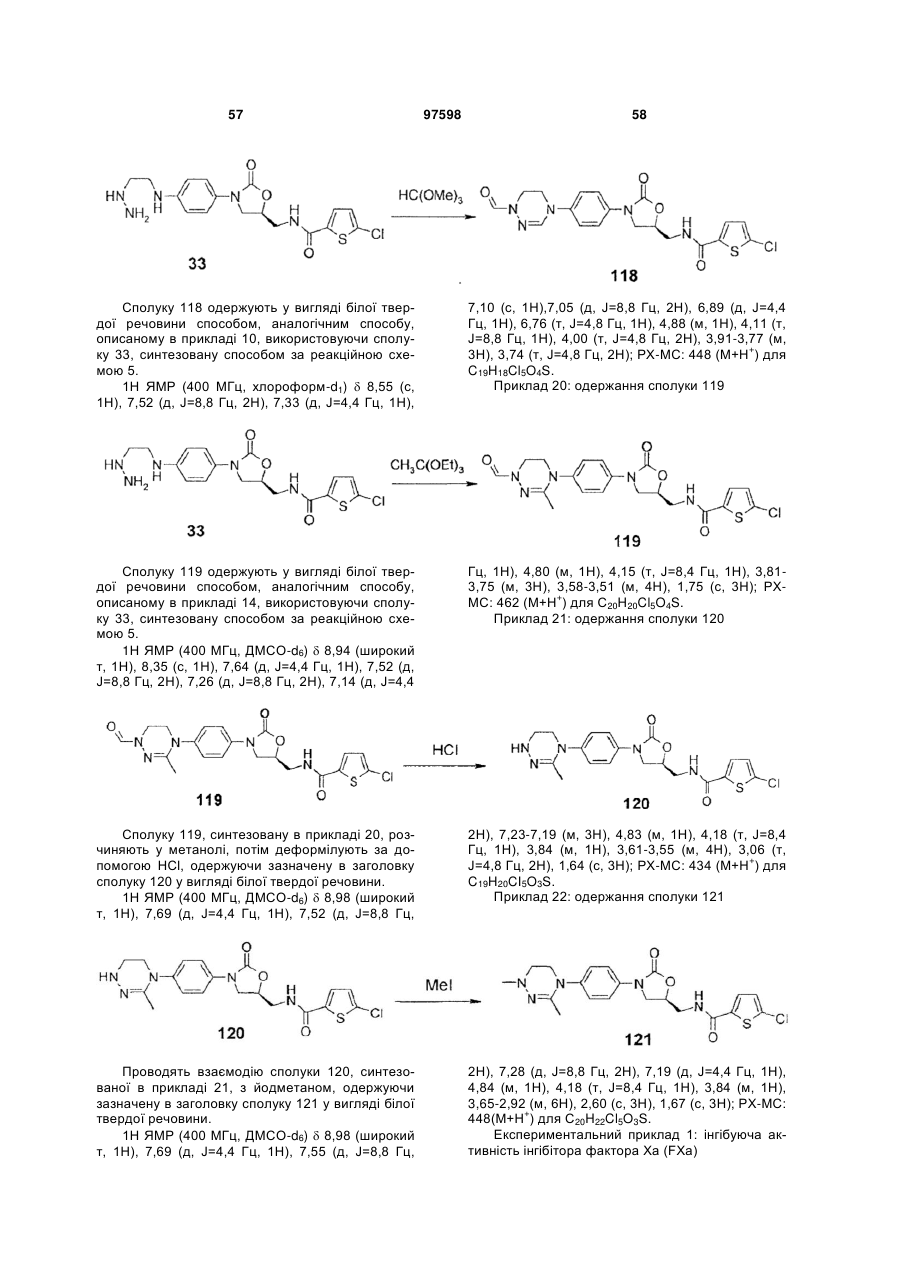
4. Оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або фармацевтично прийнятна сіль за п. 3, де зазначене оксазолідинонове похідне вибране з наступних сполук:
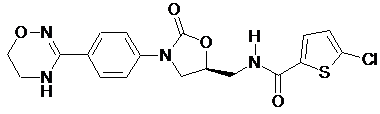 ,
,
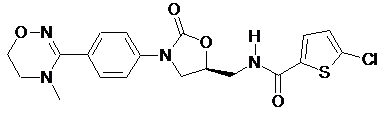 ,
,
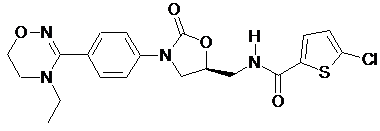 ,
,
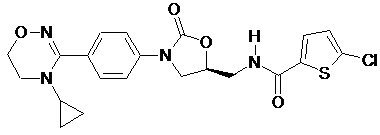 ,
,
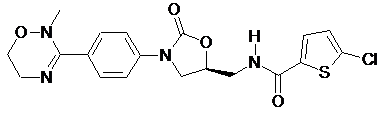 ,
,
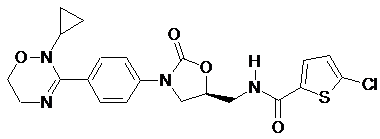 ,
,
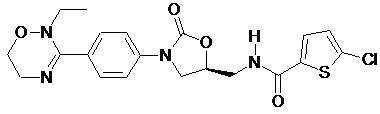 ,
,
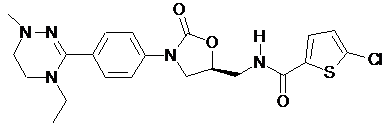 ,
,
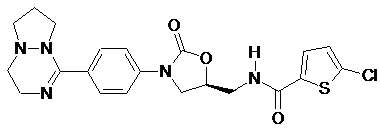 ,
,
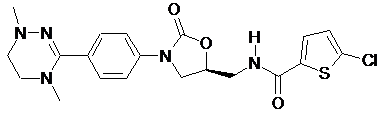 ,
,
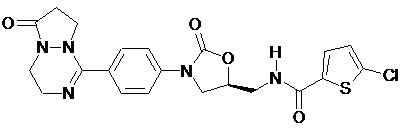 ,
,
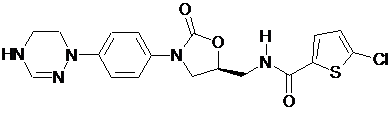 ,
,
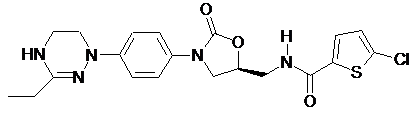 ,
,
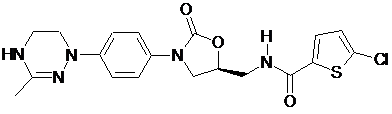 ,
,
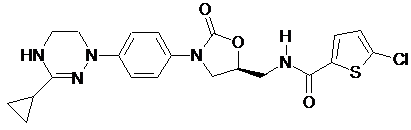 ,
,
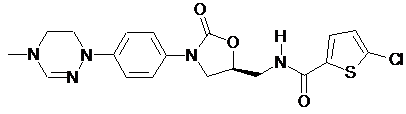 ,
,
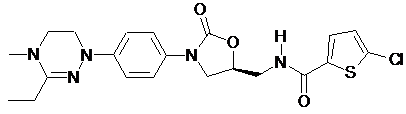 ,
,
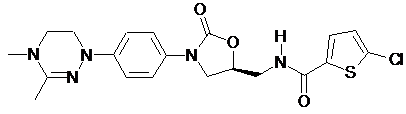 ,
,
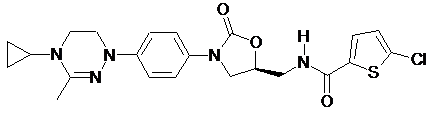 ,
,
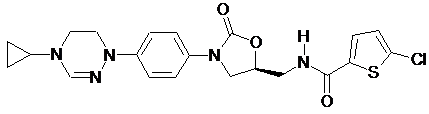 ,
,
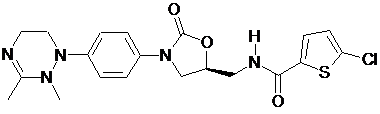 ,
,
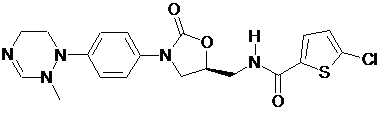 ,
,
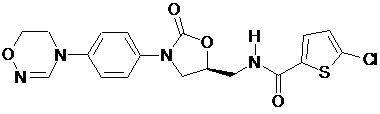 ,
,
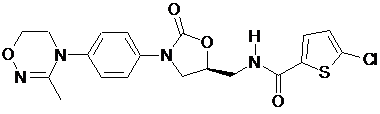 ,
,
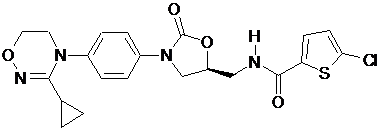 ,
,
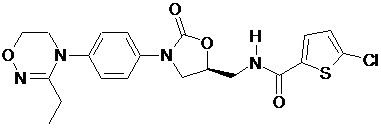 ,
,
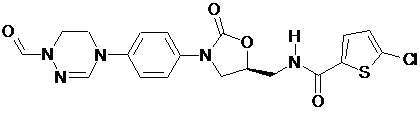 ,
,
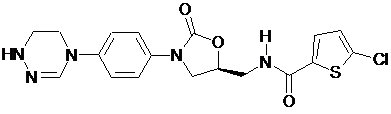 ,
,
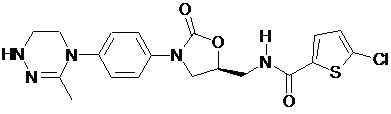 ,
,
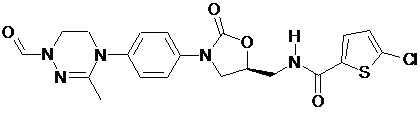 ,
,
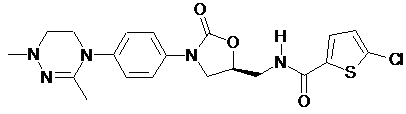 .
.
5. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або його фармацевтично прийнятну сіль за будь-яким з пп. 1-4.
6. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або його фармацевтично прийнятну сіль за будь-яким з пп. 1-4, для профілактики або лікування тромбозу, інфаркту міокарда, артеріосклерозу, запалення, удару, стенокардії, рестенозу, переміжної кульгавості, флеботромбозу, легеневої емболії, артеріального тромбозу, ішемії міокарда або тромбоемболії.
7. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його пролiки, гідрат, сольват, ізомер або фармацевтично прийнятну сіль за будь-яким з пп. 1-4 у комбінації з тромболітичним лікарським засобом, для профілактики або лікування захворювання коронарної артерії, захворювання церебральних артерій або захворювання периферичних артерій.
8. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або фармацевтично прийнятну сіль за будь-яким з пп. 1-4, для консервування крові, плазми або продуктів крові in vitro.
Текст
1. Оксазолідинонове похідне з циклічною амідоксимною або циклічною амідразоновою групою, представлене формулою 1, або його проліки, гідрат, сольват, ізомер або фармацевтично прийнятна сіль, [Формула 1] O 3 97598 4 O O N N O O H N O N N N O H N N S R2 Cl S R 11 O Cl O , [Формула IV] , [Формула XI] O O N N S R4 Cl O , [Формула V] O N N O H N N N S Cl S Cl S m Cl O , [Формула VI] O N N O H N N N O m O , [Формула VII] R5 N O N O H N N N R6 O , [Формула VIII] O R7 N N N O Cl O N N O H N S N O N Cl O , де R1-R12, незалежно один від одного, представляють атом водню, (С1-С7)алкіл або (С3-С7)циклоалкіл; R13 означає Н, (С1-С7)алкіл, (С3-С7)циклоалкіл, форміл або (С1-С7)алкілкарбоніл; і m дорівнює цілому числу від 1 до 3. 3. Оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або його фармацевтично прийнятна сіль за п. 2, де R1-R12, незалежно один від одного, представляють атом водню, метил, етил, н-пропіл, ізопропіл, н-бутил, ізобутил, третбутил, циклопропіл, циклобутил, циклопентил або циклогексил; R13 означає Н, метил, етил, н-пропіл, ізопропіл, н-бутил, ізобутил, трет-бутил, циклопропіл, циклобутил, циклопентил, циклогексил, форміл або ацетил; і m дорівнює цілому числу 1. 4. Оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або фармацевтично прийнятна сіль за п. 3, де зазначене оксазолідинонове похідне вибране з наступних сполук: O O N H O N Cl N S N H O , O O N H O N Cl N S N , [Формула IX] O O , H N S R 10 O , [Формула X] S R12 O R9 H N , S N O O H N R8 N N O N N N N H N N N R3 R13 N O Cl Cl 5 97598 6 O O O N O N H N S N Cl HN N H O N N S N Cl O O , O , O O N O N HN H N S N N O N N Cl O O HN O N N H S N Cl O HN , N O O N N N S Cl O , N O N O N H N H S Cl , O O N N , O N H N S N O N Cl O O N N Cl O N N , H O N S N Cl N N O O N H N S N , Cl O O N N , O N O H N S N Cl N N N O H N S N O Cl O , , O O O N N N O H N N S N O , S N O Cl , O N S O H N N N H N O Cl , O N S O N O N H O N N O Cl , O N S N , O N H Cl N N O H N S N O , Cl 7 O N N 97598 8 O H O N N S N Cl HN N O N H N S N Cl O O , , O O N N H O N N S N Cl O N H N S N Cl , , O O N N O O O HN H O N O N N S N Cl N N O H N S N Cl O O , , O O O N O N N H N S N Cl O O N H O N N S N Cl O , O O N H O N N S N Cl O , O O N N N O H N S N N O H N S N Cl O , O N Cl O , Галузь техніки Даний винахід стосується нових оксазолідинонових похідних з циклічною амідоксимною або циклічною амідразоновою групою, представлених формулою І, їх фармацевтично прийнятних солей, способів їх одержання і фармацевтичних композицій, що їх містять. . 5. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або його фармацевтично прийнятну сіль за будь-яким з пп. 1-4. 6. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або його фармацевтично прийнятну сіль за будь-яким з пп. 1-4, для профілактики або лікування тромбозу, інфаркту міокарда, артеріосклерозу, запалення, удару, стенокардії, рестенозу, переміжної кульгавості, флеботромбозу, легеневої емболії, артеріального тромбозу, ішемії міокарда або тромбоемболії. 7. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його пролiки, гідрат, сольват, ізомер або фармацевтично прийнятну сіль за будь-яким з пп. 1-4 у комбінації з тромболітичним лікарським засобом, для профілактики або лікування захворювання коронарної артерії, захворювання церебральних артерій або захворювання периферичних артерій. 8. Фармацевтична антикоагулянтна композиція, яка містить оксазолідинонове похідне або його проліки, гідрат, сольват, ізомер або фармацевтично прийнятну сіль за будь-яким з пп. 1-4, для консервування крові, плазми або продуктів крові in vitro. , 9 97598 10 де цикл А являє собою залишок, вибраний із групи, що включає наступні структури: Антитромботичний і антикоагулянтний ефект нових оксазолідинонових похідних за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою, представлених формулою І, приписують інгібуванню активної коагулюючої протеази, відомої як фактор Ха, або інших активних серинових протеаз, таких як фактор VIIa, фактор ІХа або тромбін. Рівень техніки Фактори згортання крові розподілені в плазмі, ого притому що різні типи факторів, від 1 фактора ого згортання до 13 фактора згортання, працюють по каскадному типу, забезпечуючи в результаті згортання крові. Механізм, по якому індивідуальні фактори згортання крові беруть участь у згортанні крові, показаний на Фіг. Як показано на Фігурі, згортання крові відбувається в результаті ряду складних реакцій. У цілому, інактивовані попередники активуються специфічними активними факторами згортання крові (зазначені індексом "а", доданим після номера фактора коагуляції). Потім активуються наступні фактори згортання крові. Більшість з цих активованих факторів згортання крові являють собою ферменти сімейства серинових протеаз. Вони прилипають до поверхні активованого тромбоцита на місці рани, поступово активують фактори згортання крові і, зрештою, продукують фібриновий згусток, приводячи до зупинки кровотечі. Тромбін є багатофункціональним чинником коагуляції, що залучений у кінцеві стадії коагуляційного каскаду. Протромбін, попередник тромбіну, активується комплексом протромбінази, що скла++ дається з фактора Va, фактора Ха, Са і фосфоліпідів (PL), одержуючи тромбін, що перетворює фібриноген у фібрин. Генеровані фібрини покривають агрегований тромбоцит, індукуючи згортання крові. На закінчення, фібрини зшиваються фактором ХІIIа, продукуючи стабільний фібриновий згусток. Для утворення комплексу протромбінази, фактор X активується у фактор Ха, що опосередковано, головним чином, комплексом Хази. Фактор ++ VIIla, фактор ІХа, Са і фосфоліпіди (PL), що генеруються по внутрішньому шляху, або фактор ++ VIIa, фактор тканини (TF) і Са , що генеруються по зовнішньому шляху, працюють як комплекс Хази. Тромбін також активує фактор V і фактор VIII. Якщо відбувається надвиробництво тромбіну, сама кровоносна судина може забитися. Щоб уник нути закупорювання, тромбін запускає дію по інгібуванню згортання крові. Тобто, тромбін зв'язується з тромбомодуліном, активуючи протеїн С. Активований протеїн С (АРС), утворює комплекс із протеїном S, інактивує фактор Va і фактор VIIla. Фактично, фактор Ха сам по собі є сериновою протеазою і включений у складний процес згортання крові. Фактор Ха, як істотний член комплексу протромбінази, діє як каталізатор для перетворення протромбіну в тромбін. Тромбін перетворить фібриноген у фібринові мономери, і генеровані в такий спосіб фібринові мономери включаються в утворення і стабілізацію тромбу. Таким чином, у результаті надмірного або невідповідного продукування тромбіну може відбуватися тромбоемболія. Отже, інгібування самого тромбіну або генерації тромбіну може приводити до зниження утворення фібрину, залученого в утворення тромбу, і попередженню тромбоемболії. Коротенько, інгібування фактора Ха призводить до інгібування утворення тромбіну, за допомогою чого можна попередити або послабити тромбоемболію. Сполука, представлена в даному винаході формулою І, і його фармацевтично прийнятна сіль можуть інгібувати фактор Ха, що, у кінцевому рахунку, відповідно до викладеної вище логіки, веде до попередження тромбоемболічних захворювань (МІ, нападу, РЕ і ін.). Серед сполук, відомих як інгібітори фактора Ха, типовими прикладами інгібіторів білка є антистазин (ATS) і кліщовий антикоагулянтний пептид (ТАР). ATS (що складається з 119 амінокислот) являє собою природний пептид, що виділяється з п'явок, який має значення Kі 0,05 нМ щодо фактора Ха. ТАР також являє собою пептид, що виділяється із кліщів, що складений з 60 амінокислот і має значення Ki 0,5 нМ щодо фактора Ха. Однак ці інгібітори мають обмежене клінічне застосування; тільки гепаринів або його сульфатовані полісахаридні аналоги мають клінічне застосування з деяким обмеженням. Як інгібітор згортання крові, зокрема, інгібітор фактора Ха, розроблена низькомолекулярна сполука, що описана в W095291S9. Тим часом, у WO9933800 описаний інгібітор фактора Ха, що має індольні фрагменти. Крім того, різні інгібітори фактора Ха розкриті й у процесі розробки. Наприклад, гетероциклічна сполука, що містить атом азоту (WO2004058743), імідазольні похідні (WO2004050636), піразольні похідні (WO2004056815), індол-2-карбоксамідні похідні 11 97598 12 (WO2003044014), оксибензамідні похідні (WO200205I831), гуанідин/амідинові похідні (WO2002046159), аміно-біциклічні піразинон/піридинонові похідні (WO2004002405) і ін. Щоб бути клінічно придатними як інгібітори FXa, ці молекули повинні мати високий антитромботичний ефект, високу стабільність в плазмі і печінці, що придатна селективністю відносно інших споріднених серинових протеаз (тромбін, трипсин, катепсин G і ін.), низькою токсичністю і задовільною біодоступністю. . Найбільш прогресивною сполукою, що містить оксазолідинон, аналогічно сполуці за даним винаГоловною тенденцією останніх досліджень ходом, є ривароксабан (формула А), що у даний FXa і інгібітору тромбіну є реалізація амідинових час знаходиться у фазі III клінічної оцінки. Деякі функцій. Передбачається, що амідинова функція, оксазолідинонові похідні, представлені формулою 189 так звана група Ρ1, зв'язує Asp , розташований 2, описані в WO 01/47919. Однак повідомляється, на дні кишені S1. І FXa, і тромбін розпізнають аргіщо деякі з цих сполук мають обмежену розчинніновий залишок у природному субстраті як сайт ність; конкретний приклад даної проблеми предР1. Амідинова група (у тому числі гуанідинові похіставляє ривароксабан. Розчинність ривароксабану дні), що заміщує гуанідин в аргініні, є високо гідстановить тільки 8 мг/л. У результаті слабкої розрофільною. Таким чином, інгібітори з амідиновою чинності може виникати множина практичних обфункцією звичайно не дуже добре абсорбуються, і межень, включаючи варіабельність і повільне рознавіть якщо вони абсорбуються, то дуже швидко кладання. Ці проблеми можна обійти, вводячи виводяться внаслідок властивої їм високої полярвисокорозчинний фрагмент. ності амідину (Drugs of the Future, 1999, 24(7), 771). Сам амідин характеризується сильною основністю (PKa: приблизно 12,5). Через формально позитивний заряд у фізіологічних умовах амідиновий інгібітор звичайно демонструє слабку абсорбцію. Отже, необхідно зробити зміни в напрямку менш основних альтернатив. Типовими прикладами таких речовин є піридинове похідне, амідразон, циклічний амін, алкіламінне похідне, амінобензизоксазол і ін. (US 6958356). Маються також фундаментально відмінні підходи до подолання цих проблем, що включають використання амідоксимів. Амідоксим легко синтезують, додаючи гідроксильну групу до амідинової структури, що являє собою проліки, ґрунтуючись на тому, що слабкий зв'язок N-O легко відновлюється in vivo до амідину. Цей підхід вигідно використовує той факт, що PKa амідоксиму помітно нижче (8-9), ніж відповідне значення для амідину. Іншим прикладом проліків такого ж типу є ксимелагатран. Ця тенденція поміУ WO 2004/83174 описане застосування піратна не тільки при дослідженні FXa-інгібітора, але зольних похідних, включаючи апіксабан. Деякі з також при дослідженні інгібітору тромбіну. Однак цих інгібіторів є циклічними амідиновими і сульфобільшість цих спроб виявляються не такими гарніламідиновими похідними, представленими форними, як очікувалося. Як спроба третього типу мулою С. вводять нейтральні групи Ρ1. На відміну від лік Однак не мас прецедентів, аналогічних даноіншого класу, FXa і інгібітори тромбіну мають тенму винаходу, що описують специфічне введення денцію демонструвати гарну ефективність, якщо циклічного амідоксиму або циклічного амідразону вони присутні в крові при високих концентраціях. в оксазолідиноновий каркас з метою одержання Крім того, дуже важлива концентрація в крові вільінгібіторів фактора Ха. Фактично, мало що відомо ного лікарського засобу, не зв'язаного з білками відносно обгрунтування присутності циклічного сироватки. У випадку інгібітору з нейтральною груамідоксиму або циклічного амідразону в конструкпою Р1 існує тенденція високого зв'язування білка, ції ліків. у результаті чого ефективність є меншою, ніж очікувалося. Для подолання цих проблем автори даного винаходу вводять відносно полярну групу в положення, відмінні від сайту Р1, при фіксованому положенні нейтральної групи на сайті P1. Паралельно з цією логікою включаються деякі інші важливі фактори для поліпшення фармацевтичного ефек 13 97598 14 ту, що наведено далі: 1) істотне підвищення рознаприклад, переміжної кульгавості. Крім того, окчинності у воді; 2) низьке зв'язування білку плазми; сазолідинонові похідні з циклічним амідоксимом якщо концентрація вільного лікарського засобу є або циклічним амідразоном за даним винаходом високою, ефективність у РТ-дослідженні також можуть служити як інгібітор відносно фактора VIIa, підвищується, навіть якщо FXa-єднальна афінність фактора ІХа і тромбіну, що являють собою факточастково знижується. ри коагуляції в каскаді згортання крові. Сайт, вибраний для введення полярної групи, Метою даного винаходу є забезпечення нових у даному винаході є підсайтом Р4 інгібітори, і логіоксазолідинонових похідних з циклічною амідокка вибору є наступною. Сайт S4 від FXa має Uсимною або циклічною амідразоновою групою, що подібний сайг зв'язування, оточений із трьох сторін демонструють властивість інгібування фактора Ха, Tyr99, Phe174 і Тrр215. Сайт зв'язування складеабо їх фармацевтично прийнятних солей. ний тільки з бічних ланцюгів ароматичних амінокиІншою метою даного винаходу є забезпечення слот, на відміну від тромбіну, оточеного Leu99, lle фармацевтичної композиції для протидії коагуля174 і Тrр215. Розходження використовують у консції, що містить як активний інгредієнт оксазолідитрукції ліків. нонові похідні з циклічною амідоксимною або цикS4 кишеня від FXa є придатною при взаємодії лічною амідразоновою групою, або їх фармацевтично прийнятні солі. з катіонним залишком, що звичайно називають "Також метою даного винаходу є забезпечення катіонною взаємодією". Фактично, деякі інгібітори фармацевтичної композиції, що містить як активконструюють і синтезують таким чином, щоб вони ний інгредієнт оксазолідинонові похідні з циклічмали позитивний заряд на сайтах Р4. У даному ною амідоксимною або циклічною амідразоновою винаході циклічний амідоксим або амідразон ввогрупою, або їх фармацевтично прийнятні солі, для дять у сайт Р4 для поліпшення розчинності у воді і лікування або профілактики тромбозу, інфаркту підвищення ефекту лікарського засобу по зниженміокарда, артеріосклерозу, запалення, удару, стеню зв'язування білку, як згадується вище. Причина нокардії, рестенозу, переміжної кульгавості, флевибору циклічної форми полягає в тому, що автоботромбозу, легеневої емболії, артеріального ри думають, що абсорбцію можна поліпшити, знитромбозу, ішемії або міокарда тромбоемболії. жуючи кількість NH зв'язків, що звичайно впливаКрім того, метою даного винаходу є забезпеють на абсорбцію. Відповідно до останніх чення фармацевтичної композиції для лікування досліджень, для ліків більш переважно мати менабо профілактичного лікування захворювання коше донорів водневих зв'язків (HBD), ніж акцепторів ронарної артерії, захворювання церебральних водневого зв'язку (НВА). Відповідно до правила артерій і захворювання периферичних артерій, що Ліпінськи, можна допустити наявність до 10 НВА, а характеристично піддаються спільному лікуванню кількість HBD обмежена тільки 5 (Adv. Drug оксазолідиноновими похідними з циклічною амідоDeliveiy Rev., 2001, 46, 3-26), і, зокрема, у випадку ксимною або циклічною амідразоновою групою, нових ліків середня кількість HBD становить прибабо їх фармацевтично прийнятними солями і тролизно 2, припускаючи, що число HBD обмежене мболітичним агентом. більш жорстко (J. Med. Chem. 2004, 47, 6338-48). Також, метою даного винаходу є забезпечення Амідоксимна або амідразонова група сама по собі застосування оксазолідинопових похідних з циклімає основний характер, що робить можливим чною амідоксимною або циклічною амідразоновою більш легке розділення-очищення-збереження у групою, або їх фармацевтично прийнятних солей вигляді солі і, як результат, очікується підвищена як антикоагулянту для in vitro консервації крові, розчинність у воді. або плазми продуктів крові. Коротенько, автори вводять амідоксим або Технічне рішення амідразон у сайт Р4. Для зниження числа HBD Даний винахід стосується нових оксазолідиноавтори використовують циклічну форму функції, нових похідних з циклічною амідоксимною або циодержуючи інгібітори, більш схожі на ліки. клічною амідразоновою групою, представлених Фактично, встановлено, що сполуки формули І формулою І, або їх фармацевтично прийнятних за даним винаходом мають зазначені вище пересолей, способів їх одержання й утримуючим їх ваги. Представлено розчинність у воді і рівень фармацевтичних композицій. зв'язування білку разом зі значеннями 2РТ і Ki. Розкриття винаходу Технічна задача Автори даного винаходу синтезували нові оксазолідинонові похідні з циклічною амідоксимною або циклічною амідразоновою групою, що має придатні властивості, які можна застосовувати для приготування фармацевтичних препаратів. Зокрема, оксазолідинонові похідні з циклічною амідоксимною або циклічною амідразоновою групою де, монструють FXa інгібуючу дію, так що їх можна застосовувати для лікування або профілактики де цикл А являє собою залишок, вибраний із тромбозу, інфаркту міокарда, артеріосклерозу, групи, що включає наступні структури; запалення, апоплексії, стенокардії, рецидивуючої стриктури після ангіопластики і тромбоемболії, 15 R1-R12 незалежно являють собою Η, (С1С7)алкіл, (С3-С7)циклоалкіл, (С6-С12)арил або (С4С12)гетероарил, що містить від одного до чотирьох гетероатомів, вибраних із групи, що складається з О, S і N, R3 і R4 утворюють цикл за допомогою сполуки з (С3-С5)алкіленом, атом вуглецю алкілену може бути заміщений карбонілом, і алкіл, циклоалкіл, арил або гетероарил від R1-R12 може бути заміщений одним замісником, вибраним із групи, що складається з (С1-С7)алкілу, галоген(С1-С7)алкілу, (С1-С7)алкокси і галогену; R13 означає Н, (С1-С7)алкіл, (С1С7)циклоалкіл, форміл, (С1-С7)алкілкарбоніл, (С1С7)алкоксикарбоніл або (С6-С12)арил. Термін "арил" у цьому винаході означає органічний радикал, одержуваний з ароматичного вуглеводню, відщіпленням одного Н, у якому кожен цикл моноциклічної системи або конденсованої циклічної системи містить 6-12, переважно 6-10 циклічних атомів. Конкретно, термін включає феніл, нафтил, біфеніл і інденіл, але не завжди обмежений ними. Термін "гетероарил" у цьому винаході означає арильну групу, що містить 1-4 гетероатоми, вибраних із групи, що складається з Ν, Ο і S, як атоми, що складають структуру ароматичного циклу, і інші атоми, що складають структуру ароматичного циклу, є атомами С, що проілюстрована (5-6)-членним моноциклічним гетероарилом і поліциклічним гетероарилом, конденсованим з одним або більшою кількістю бензольних кілець, що можуть бути частково насиченими. Оксазолідинонові похідні за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою вибрані зі сполук наведених далі формул II-XI. 97598 16 17 97598 18 де R1-R12 незалежно являють собою Η, (С1С7)алкіл або (С3-С7)циклоалкіл; R13 означає Н, (С1-С7)алкіл, (С3-С7)циклоалкіл, форміл або (С1С7)алкілкарбоніл; і m дорівнює цілому числу від 1 до 3. Як приклад оксазолідинонових похідних за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою в наведених вище формулах ІІ-ХІ R1-R12 незалежно являють собою Н, метил, етил, н-пропіл, ізопропіл, нбутил, ізобутил, трет-бутил, циклопропіл, циклобутил, циклопентил або циклогексил; R13 означає Н, метил, етил, н-пропіл, ізопропіл, н-бутил, ізобутил, трет-бутил, циклопропіл, циклобутил, циклопентил, циклогексил, форміл або ацетил; і m дорівнює цілому числу 1. Оксазолідинонові похідні за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою можна проілюструвати наступними сполуками, але не завжди обмежуючись ними: 19 97598 20 21 У цьому винаході, способи одержання оксазолідинонових похідних з циклічною амідоксимною або циклічною амідразоновою групою, представлених формулою І, проілюстровані за допомогою реакційних схем з 1 по 5. Однак наведений далі спосіб не може обмежувати способи одержання оксазолідинонових похідних за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою, представлених формулою І, і фахівцям у даній галузі добре відомо, що модифікація наведеного нижче способу дозволена і також включена в ознаки даного винаходу. Якщо зазначено іншого, замісники на реакційних схемах такі, як визначено для формули І. 97598 22 Як показано на реакційних схемах 1-5, спосіб одержання оксазолідинонових похідних з циклічною амідоксимною або циклічною амідразоновою групою, представлених формулою І, модифікують у відповідності зі структурою циклічної сполуки А у формулі І. Спочатку синтезують циклічні амідоксимні сполуки, такі як сполуки В1 і В2 на реакційній схемі 1 і сполука Ε на реакційній схемі 4. Також синтезують циклічні амідразонові сполуки, такі як сполуки С1 і С2 на реакційній схемі 1, сполука D1 на реакційній схемі 2 і сполука F на реакційній схемі 5. 23 Як показано на реакційній схемі 1, синтез циклічних амідоксимних сполук В1 і В2 формули І і циклічних амідразонових сполук С1 і С2 здійснюють, проводячи взаємодію 4-ціаноаніліну (1) і 2(((S)-оксиран-2-іл)метил)трет-бутилоксикарбонілу (2) з одержанням сполуки 3. Потім синтезують оксазолідинонову циклічна сполука 4, використовуючи 1,1-карбонілдіімідазол і DMAP, і реакційну суміш обробляють HCl для видалення захисної групи boc. Проводять конденсацію з 5хлортіофен-2-карбоновою кислотою з наступною обробкою HCl. На закінчення, проводять взаємодію реагенту з діамінною сполукою, одержуючи циклічні амідоксимні сполуки В1 і В2 формули І і циклічні амідразонові сполуки С1 і С2. 97598 24 У синтезі сполуки 3 можна одержати амінну сполуку, захищену фталімідом, використовуючи 2-(((S)-оксиран-2-іл)метил)ізоіндолін-1,3-діон (9) з реакційної схеми 2 замість 2-(((S)-оксиран-2іл)метил)трет-бутилоксикарбонілу (2), аналогічно способу за реакційною схемою 2. На реакційній схемі 1 описаний спосіб з використанням 2-(((S)оксиран-2-іл)метил)трет-бутилоксикарбонілу (2) і на реакційній схемі 2 описаний спосіб з використанням 2-(((S)-оксиран-2-іл)метил)ізоіндолін-1,3діону (9). Циклічні амідразонові сполуки D1 і D2 формули І можна розрізнити по розташуванню подвійного зв'язку, і їх синтезують за реакційною схемою 2 і реакційною схемою 3. 25 Спочатку 4-нітроанілін (6) захищають за допомогою групи boc, а потім гідрують, використовуючи паладієвий каталізатор. Потім синтезують аміноспиртову сполука 10, використовуючи 2(((S)-оксиран-2-іл)метил)ізоіндолін-1,3-діон (9). Оксазолідиноновий цикл одержують, використовуючи карбонілдіімідазол, з одержанням сполуки 11. Використовуючи гідразин, видаляють фталімідну захисну групу, а потім проводять конденсацію з 5-хлортіофен-2-карбоновою кислотою, одержуючи сполуку 13. Сполуку 13 обробляють HCl 97598 26 для видалення захисної групи boc, далі проводять взаємодію з boc-захищеним аміналом. одержуючи сполуку 14. Вводять нітрозогрупу, використовуючи NaNO2, а за тим проводять відновлення за допомогою Zn, одержуючи гідразинову сполуку 15. Проводять взаємодію сполуки 15 з ортоформіатом, одержуючи циклічну амідразонову сполуку D1. Іншу циклічну амідразонову сполуку D2 синтезують за реакційною схемою 3. 27 97598 28 Зі сполуки 14, показаної на реакційній схемі 2, синтезують сполуку 14а, де R7 означає Н, у яку вводять аміногрупу (15а) і потім поступово вводять алкільну групу (16). індукують циклізацію, використовуючи ортоформіат і одержуючи циклічну амідразонову сполуку D2. Циклічну амідоксимну сполуку Ε формули І синтезують за реакційною схемою 4. Проводять взаємодію сполуки 17, одержаної з 4-фторнітробензолу, з метансульфоніл хлоридом, одержуючи сполуку 18, потім проводять взаємодію з гідроксифталімідом, одержуючи сполуку 19. Фталімідну захисну групу видаляють за допомогою гідразину, а замість неї використовують для захисту групу boc, одержуючи в результаті сполуку 21. Нітрогрупу сполуки 21 відновлюють, використовуючи Zn і одержуючи сполуку 22, потім проводять взаємодію з 2-(((S)-оксиран-2іл)метил)ізоіндолін-1,3-діоном (9) способом, показаним на реакційній схемі 2, з одержанням сполуки 26. Сполуку 26 обробляють HCl, потім проводять взаємодію з ортоефіром, одержуючи сполуку Е. Циклічну амідразонову сполуку F формули І синтезують за реакційною схемою 5. 29 Вводять захист сполуки 17, одержаної з 4фторнітробензолу, за допомогою групи tbs (третбутилдиметилсиліл) і потім вводять захист амінної області за допомогою boc, одержуючи в результаті сполуку 27. Використовуючи паладієвий каталізатор, одержують амін, що піддають взаємодії з Cbz-Cl, одержуючи сполуку 28. Проводять взаємодію сполуки 28 із гліцидилбутилатом, одержуючи сполуку 29. Спиртову групу заміняють аміном з одержанням сполуки 30. Проводять взаємодію цієї сполуки з хлортіофенкарбоновою кислотою, одержуючи сполуку 31, потім проводять взаємодію з метансульфонілхлоридом. Реагент обробляють гідразином з одержанням сполуки 33. Проводять взаємодію сполуки 33 з ортоефіром, одержуючи сполуку F. Оксазолідинонові похідні за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою, представлені формулою І, можна застосовувати в медицині як фармацевтично активний інгредієнт, зокрема, для профілактики і лікування тромбозу, інфаркту міокарда, артеріосклерозу, запалення, апоплексії, стенокардії, рецидивуючої стриктури після ангіопластики, переміжної кульгавості, флеботромбозу, легеневої емболії, артеріального тромбозу, ішемії міокарда, нестабільної стенокардії на базі тромбозу і тромбоемболії, наприклад, кризу. Оксазолідинонові похідні за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою, представлені формулою І, або їх фармацевтично прийнятні солі можна використовувати для профілактики або лікування атеросклеротичного захворювання, у тому числі, захворювання коронарної артерії, захворювання церебральних артерій або захворювання периферичних артерій. Для лікування інфаркту міокарда оксазолідинонові похідні з циклічною амідоксимною або циклічною амідразоновою групою 97598 30 можна застосовувати разом із тромболітичним агентом (наприклад, альтеплазою, тенектеплазою і ін.). Зазначені сполуки також можна використовувати для профілактики реоклюзії після тромболізису, черезшкірної транслюмінальної коронарної ангіопластики (РТСА) і обхідного шунтування коронарної артерії. Оксазолідинонові похідні за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою, представлені формулою І, або їх фармацевтично прийнятні солі можна використовувати для профілактики післяоперативного повторного утворення тромбів. Їх також можна використовувати як антикоагулянт відносно штучного органу або гемодіалізу. Зазначену сполуку можна використовувати для промивання катетера і медичного допоміжного пристрою, застосовуваного in vivo. Крім того, їх також можна використовувати як антикоагулянтні композиції для ex vivo збереження крові, плазми і продуктів крові інших типів. Зазначені сполуки за даним винаходом також ефективні при лікуванні захворювання, зв'язаного зі згортанням крові, або захворювання, викликаного вторинним ураженням, наприклад, раку (у тому числі, метастатичного раку), запального захворювання, у тому числі, артриту і діабету. Оксазолідинонові похідні за даним винаходом з циклічною амідоксимною або циклічною амідразоновою групою, представлені формулою І, можна використовувати у вигляді фармацевтично прийнятних солей. Що стосується фармацевтично прийнятних солей, переважні адитивні солі кислот, одержувані з використанням фармацевтично прийнятної вільної кислоти. Вільну кислоту (неорганічну або органічну кислоту) можна використовувати, якщо вона є фармацевтично прийнятною. Приклади неорганічної вільної кислоти включають соляну кислоту, бромистоводневу 31 97598 32 кислоту, сірчану кислоту і фосфорну кислоту. Оксазолідинонові похідні за даним винахоПрикладами доступних органічних вільних кислот дом з циклічною амідоксимною або циклічною є лимонна кислота, оцтова кислота, молочна киамідразоновою групою, представлені формулою слота, малеїнова кислота, фумарова кислота, І, можна одержати як проліки, призначені для глюконова кислота, метансульфонова кислота, підвищення in vivo абсорбційної здатності або гліколева кислота, бурштинова кислота, 4розчинності, і можна застосовувати у вигляді гідтолуолсульфонова кислота, трифтороцтова кисрату або сольвату. Наприклад, як пояснюється лота, галактуронова кислота, ембонова кислота, нижче, приєднують групу, яку можна легко відоглютамінова кислота й аспарагінова кислота. кремити після in vivo абсорбції, або одержують Оксазолідинонові похідні за даним винаходом сполуку у вигляді солі, а саме у вигляді одного можуть включати гідрат солі, зокрема, якщо заабо більшої кількості гідратів або сольватів. Прозначена сіль є гігроскопічною, її переважно виколіки і гідрат або сольват солі також включені в ристовувати у вигляді кристалогідрату. ознаки даного винаходу. Для одержання ефекту від лікування можна визначити ефективну дозу оксазолідинонових похідних, представлених формулою І, їх гідратів, їх сольватів або їх фармацевтично прийнятних солей, приймаючи до уваги конкретні використовувані сполуки, спосіб введення, цільовий суб'єкт, цільове захворювання й ін., але переважною є доза 0,1-20 мг/кг (маси тіла) на день оксазолідинонової похідної сполуки, представленої формулою I. Денну дозу можна вводити один раз на день (одночасно) або кілька разів на день, поділивши денну ефективну дозу належним способом. Відповідно до препарату можна допустити пероральне введення, парентеральне введення (ін'єкцію) або локальне введення. Фармацевтичну композицію за даним винаходом для перорального введення можна приготувати, наприклад, у вигляді таблеток, порошків, сухих сиропів, жувальних таблеток, гранул, капсул, м'яких капсул, пігулок, напоїв, під'язикових препаратів і ін. Композицію за винаходом, приготовлену у вигляді таблеток, можна вводити суб'єкту будь-яким способом або шляхом, що доставляє ефективну дозу таблетки з біодоступністю, що може забезпечити пероральний шлях. Також можна визначити спосіб або шлях уведення відповідно до характеристик, стадій цільового захворювання й інших умов. Якщо композицію за винаходом формують у вигляді таблеток, то вона може додатково включати фармацевтично прийнятні наповнювачі. Вміст і характеристики наповнювача можна визначити по розчинності і хімічних властивостях обраної таблетки, шляху введення і виходячи зі стандартної фармацевтичної практики. Короткий опис фігур Зазначені вище й інші цілі, відмітні ознаки і переваги даного винаходу стануть ясні з наступного опису переважних варіантів, наданих у сполученні з прикладеними малюнками, на яких: на Фіг. наведена діаграма, що демонструє механізм згортання крові. Спосіб за винаходом Фактичні і переважні в даний час варіанти даного винаходу є ілюстративними, як показано в наступних прикладах. Однак зрозуміло, що фахівці в даній галузі під час обговорення даного розкритгя, можуть робити модифікації і виправлення в рамках духу й галузі даного винаходу. Приклад одержання 1: одержання сполуки 5 33 Одержання сполуки 3 4-Амінобензонітрил (1) (5 г, 42,30 ммоль) і 2(((S)-оксиран-2-іл)метил)трет-бутилоксикарбоніл (2) (8,79 г, 50,78 ммоль) додають до 2ізопропілового спирту (20 мл) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 12 годин. Реакційну суміш концентрують при зниженому тиску і потім відправляють на колонку, одержуючи зазначену в заголовку сполуку 3 у вигляді білої твердої речовини (7,30 г, 25,1 ммоль, 59%). 1Н ЯМР (400 МГц, хлороформ-d1) 7,41 (д, J=8,4 Гц, 4Н), 6,59 (д, J=8,4 Гц, 1Н), 4,95 (широкий с, 1Н), 4,80 (широкий с, 1Н), 3,97-3,93 (м, 1Н), 3,31-3,15 (м, 5Н), 1,46 (с, 9Н). Одержання сполуки 4 Одержану вище сполуку 3 (7,30 г, 25,05 ммоль), 1,1-карбонілдіімідазол (4,87 г, 30,06 ммоль) і диметиламінопіридин (1,53 г, 12,52 ммоль) поступово додають до тетрагідрофурану (70 мл) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 12 годин. Реакційну суміш концентрують при зниженому тиску, потім розчиняють у етилацетаті (300 мл). Після послідовного промивання 1н. розчином HCl (50 мл) і розчином бікарбонату натрію (50 мл) реакційну суміш сушать, над сульфатом натрію, а потім концентрують при зниженому тиску. Реакційну суміш промивають діетиловим ефіром (100 мл) з одержанням сполуки 4 у вигляді білої твердої речовини (6,60 г, 20,8 ммоль, 83%). 1Н ЯМР (400 МГц, хлороформ-d1) 7,67 (с, 4Н), 4,95 (широкий с, 1Н), 4,82-4,79 (м, 1Н), 4,07 (дд, J=8,8, 8,8 Гц, 1Н), 3,94 (дд, J=8,8, 6,8 Гц, 1Н), 3,56-3,54 (м, 2Н), 1,38 (с, 9Н). Одержання сполуки 5 97598 34 Одержану вище сполуку 4 (6 г, 18,90 ммоль) додають до етилацетату (10 мл), реакційну суміш додають до 4н. HCl, розчиненого в 1,4-діоксані (60 мл), з наступним перемішуванням при кімнатній температурі протягом однієї години. Одержану тверду речовину відфільтровують при зниженому тиску і потім послідовно промивають етилацетатом (20 мл) і діетиловим ефіром (30 мл). У результаті одержують гідрохлорид амінної сполуки з вилученими boc-групами у вигляді білої твердої речовини (4,60 г, 18,1 ммоль, 95,9%). 1Н ЯМР (400 МГц, хлороформ-d1) 8,36 (широкий с, 3Н), 7,90 (д, J=8,8 Гц, 2Н), 7,73 (д, J=8,8 Гц, 2Н), 5,03-4,96 (г, Η), 4,25 (дд, J=9,2, 9,2 Гц, 1H), 3,92 (дд, J=9,2, 6,4 Гц, 1Н), 3,28-3,25 (м, 2Н). Амінну сполуку (4,60 г, 18,13 ммоль), HOBt (2,75 г, 19,94 ммоль), EDC (4,17 г, 21,75 ммоль), 5-хлортіофен-2-карбонову кислоту (3,20 г, 19,04 ммоль) і триетиламін (5,70 мл, 39,88 ммоль) поступово додають до Ν,Ν'-диметилформаміду (50 мл) з наступним перемішуванням при кімнатній температурі протягом 12 годин. Реакційну суміш повільно додають до дистильованої води (800 мл) і одержану тверду речовину відфільтровують при зниженому тиску. Реакційну суміш промивають діетиловим ефіром (100 мл) з одержанням сполуки 5 у вигляді білої твердої речовини (5,70 г, 15,8 ммоль, 87%). 1Н ЯМР (400 МГц, ДМСО-d6) 8,93 (т, 1=5,2 Гц, 1Н), 7,81 (д, J=9,2 Гц, 2Н), 7,69 (д, J=9,2 Гц, 2Н), 7,63 (д, J=4,0 Гц, 1Н), 7,15 (д, J=4,0 Гц, 1Н), 4,86-4,81 (м, 1Н), 4,17 (дд, J=9,2, 9,2 Гц, 1Н), 3,83 (дд, J=9,2, 5,2 Гц, 1Н), 3,57 (дд, J=5,2, 5,2 Гц, 2Н); + РХ-МС: 362 (М+Н ) для C16H12Cl3O3S. Приклад одержання 2: одержання сполуки 13 35 Одержання сполуки 7 4-Нітроанілін (20 г, 145 ммоль) розчиняють в ацетонітрилі (200 мл), до суміші додають ди-третбутилдикарбонат (63,2 г, 290 ммоль) і 4диметиламінопіридин (3,54 г, 29 ммоль) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 16 годин. Реакційний розчин охолоджують до кімнатної температури з наступним концентруванням при зниженому тиску, одержуючи коричневу тверду сполуку, що має дві групи boc (49 г, 145 ммоль, 100%). 1Н ЯМР (600 МГц, CDCl3) 8,25 (д, J=9 Гц, 2Н), 7,36 (д, J=9 Гц, 2Н), 1,45 (с, 18Н). Одержану сполуку (49 г, 145 ммоль) розчиняють у метанолі (200 мл), додають до розчину карбонат калію (60 г, 434 ммоль), потім кип'ятять зі зворотним холодильником при перемішуванні протягом 16 годин. Реакційний розчин охолоджують при кімнатній температурі, з наступним концентруванням при зниженому тиску. Проводять колонкову хроматографію (н-гексан/етилацетат, 6/1) реакційного розчину, одержуючи зазначену в заголовку сполуку 7 у вигляді світло-жовтої твердої речовини (17,6 г. 73,9 ммоль, 51%). 1Н ЯМР (600 МГц, CDCl3) 8,18 (д, J=9 Гц, 2Н), 7,53 (д, J=9 Гц, 2Н), 6,93 (широкий с, 1Н), 1,54 (с, 9Н). Одержання сполуки 8 Сполуку 7 (17,6 г, 73,9 ммоль) розчиняють у етилацетаті (200 мл), додають до розчину паладій на вугіллі (10% мас., 3,9 г), потім перемішують в атмосфері водню з балона. Через 16 годин реакційний розчин відфільтровують через селаіт з наступним концентруванням при зниженому тиску, одержуючи зазначену в заголовку сполуку 8 у вигляді світло-рожевої твердої речовини (15,4 г, 73,9 ммоль, 100%). 1Н ЯМР (600 МГц, CDCl3) 7,12 (широкий с, 2Н), 6,62 (д, J=9 Гц, 2Н), 6,31 (широкий с, 1Н), 3,53 (широкий с, 2Н), 1,50 (с, 9Н). Одержання сполуки 10 Сполуку 8 (13,5 г, 65,1 ммоль) розчиняють у 2-пропанолі (170 мл), додають до розчину (S)гліцидилфталімід (9) (14,6 г, 71,9 ммоль) з наступною взаємодією протягом 12 годин. Потім додатково додають (S)-гліцидилфталімід (9) (2,65 г, 13,0 ммоль) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 4 97598 36 годин. Реакційний розчин охолоджують при кімнатній температурі і потім концентрують при зниженому тиску. Виконують перекристалізацію з нгексану (500 мл), одержуючи зазначену в заголовку сполуку 10 у вигляді жовтої твердої речовини (26,8 г, 65,1 ммоль, 100%). 1Н ЯМР (600 МГц, CDCl3) 7,89-7,84 (м, 2Н), 7,78-7,73 (м, 2Н), 7,15 (широкий, 2Н), 6,63 (д, J=8 Гц, 2Н), 6,26 (широкий, 1Н), 4,18-4,12 (м, 1Н), 4,05 (широкий, 1Н), 3,94-3,86 (м, 2Н), 3,25 (дд, J=13, 4,5 Гц, 1Н), 3,15 (дд, J=13, 6,6 Гц, 1Н), 2,84 (д, J=4,8 Гц. 1Н), 1,50 (с, 9Н). Одержання сполуки 11 Сполуку 10 (26,8 г, 65,1 ммоль) розчиняють у тетрагідрофурані (200 мл), додають до розчину 1,1-карбонілдіімідазол (15,9 г, 98,1 ммоль) і 4диметиламінопіридин (1,59 г, 13,0 ммоль) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 16 годин. Реакційний розчин охолоджують і концентрують при зниженому тиску. Додають туди насичений розчин хлориду амонію (200 мл) з наступною екстракцією етилацетатом (250 мл 2). Зібраний органічний шар сушать над безводним сульфатом натрію, фільтрують, концентрують при зниженому тиску і потім відправляють на колонкову хроматографію (н-гексан/етилацетат/дихлорметан, 1/1/1), одержуючи зазначену в заголовку сполуку 11 у вигляді світло-жовтої твердої речовини (20,0 г, 45,7 ммоль, 70%). 1Н ЯМР (600 МГц, CDCl3) 7,91-7,87 (м, 2Н), 7,79-7,74 (м, 2Н), 7,43 (д, J=9 Гц, 2Н), 7,36 (д, J=9 Гц, 2Н), 6,48 (широкий с, 1Н), 5,00-4,95 (м, 1Н), 4,15 (дд, J=14, 7 Гц, 1Н), 4,11 (т, J=9 Гц, 1Н), 3,97 (дд, J=14, 6 Гц, 1Н), 3,89 (дд, J=9,6 Гц, 1Н), 1,52 (с, 9Н). Одержання сполуки 12 Сполуку 11 (15,3 г, 35,0 ммоль) розчиняють у етанолі (200 мл), додають до розчину гідразингідрат (3,40 мл, 70,0 ммоль) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 3 годин. Реакційний розчин охолоджують при кімнатній температурі. Одержану білу тверду речовину відфільтровують, і фільтрат концентрують при зниженому тиску. Додають дихлорметан (100 мл) і потім одержану тверду речовину видаляють фільтруванням. Фільтрат концентрують при зниженому тиску. Цей 37 97598 38 спосіб повторюють ще два рази, і потім реагент ммоль) з наступним перемішуванням при кімнатсушать, одержуючи зазначену в заголовку сполуній температурі протягом 2 годин. Реакційний ку 12 у вигляді білої твердої речовини (10,0 г, розчин концентрують при зниженому тиску з на32,5 ммоль, 93%). ступною перекристалізацією з розчину нгексан/діетиловий ефір (1/1, 200 мл), одержуючи 1Н ЯМР (600 МГц, CDCl3) 7,45 (д, J=9 Гц, зазначену в заголовку сполуку 13 у вигляді білої 2Н), 7,37 (д, J=9 Гц, 2Н), 6,65 (широкий с, 1Н), твердої речовини (5,0 г, 11,1 ммоль, 94%). 4,67-4,63 (м, 1Н),4,03 (т, J=9 Гц, 1Н), 3,82 (дд, J=9,7 Гц, 1Н), 3,09 (дд, J=14, 4 Гц, 1Н), 2,98 (дд, 1Н ЯМР (400 МГц, ДМСО-d6) 9,30 (с, 1Н), J=14, 6 Гц, 1Н), 1,52 (с, 9Н). 8,95 (т, J=6 Гц, 1Н), 7,67 (д, J=4 Гц, 1Н), 7,45-7,36 Одержання сполуки 13 (м, 4Н), 7,17 (д, J=4 Гц, 1Н), 4,82-4,74 (м, 1Н), Сполуку 12 (3,63 г, 11,8 ммоль) розчиняють у 4,11 (т, J=9 Гц, 1Н), 3,77 (дд, J=9, 6 Гц, 1Н), 3,57 хлороформі (50 мл), додають до розчину 5(т, J=5,6 Гц, 2Н), 1,45 (с, 9Н). хлортіофенкарбонову кислоту (2,30 г, 14,1 Приклад одержання 3: одержання сполуки ммоль) і додають 4-диметиламінопіридин (0,30 г, 16а 13,0 ммоль). Температуру знижують до 0°С. Додають Ν,Ν'-діїзопропілкарбодіімід (2,20 мл, 14,1 Одержання сполуки 14а Сполуку 13 (16,5 г, 36,5 ммоль) розчиняють у дихлорметані (150 мл), додають до розчину HCl (150 мл, 4М розчин 1,4-діоксану) з наступним перемішуванням при кімнатній температурі протягом 1 години. Реакційний розчин концентрують при зниженому тиску і сушать, одержуючи білу тверду сполуку (14,1 г, 36,3 ммоль, 99%). 1Н ЯМР (600 Мгц, ДМСО-d6) 9,07 (т, J=6 Гц, 1Н), 7,73 (д, J=3,6 Гц, 1Н), 7,63 (д, J=9 Гц, 2Н), 7,37 (д, J=9 Гц, 2Н), 7,20 (д, J=3,6 Гц, 1Н), 4,884,83 (м, 1Н), . 4,18 (т, J=9 Гц, 1Н), 3,87 (дд, J=9,6 Гц, 1Н), 3,61 (т, J=5,4 Гц, 2Н). Метанол (40 мл) і Ν,Ν-диметилформамід (15 мл) додають до одержаної вище сполуки (3,0 г, 7,73 ммоль), додають до суміші N-Boc-2аміноацетальдегід (1,48 г, 9,30 ммоль) і ціанборгідрид натрію (486 мг, 7,73 ммоль) з наступним перемішуванням при кімнатній температурі протягом 16 годин. Додають насичений розчин хлориду амонію (20 мл) і потім розчин концентрують при зниженому тиску. Ще додають туди насичений розчин хлориду амонію (50 мл) з наступною екстракцією етилацетатом (250 мл (2). Зібраний органічний шар сушать над безводним сульфатом натрію, фільтрують, концентрують при зниженому тиску і потім відправляють на колонкову хроматографію (н-гексан/етилацетат, 1/21/4), одержуючи зазначену в заголовку сполуку 14а у вигляді білої твердої речовини (2,76 г, 5,58 ммоль, 72%). 1H ЯМP (400 МГц, CDCl3) 7,30 (д, J=4 Гц, 1H), 7,18 (д, J=9 Гц, 2Н), 7,00 (т, J=6 Гц, 1Н), 6,80 (д, J=4 Гц, 1Н), 6,53 (д, J=9 Гц, 2Н), 4,84-4,74 (м, 2Н), 4,04 (широкий, 1Н), 3,98 (т, J=9 Гц, 1Н), 3,81 (ддд, J=14,4, 6, 3 Гц, 1Н), 3,74 (дд, J=9,6 Гц, 1Н), 3,66 (дт, J=14,8, 9 Гц, 1Н), 3,36-3,26 (м, 2Н), 3,18 (т, J=6 Гц, 2Н), 1,41 (с, 9Н). Одержання сполуки 15а Сполуку 14а (1,0 г, 2,0 ммоль) розчиняють в оцтовій кислоті (10 мл), повільно завантажують у розчин нітрит натрію (NaNO2) (170 мг, 2,46 ммоль), розчинений у дистильованій воді (2 мл), при 0°С з наступним перемішуванням протягом 30 хвилин. Потім завантажують туди цинкову амальгаму (650 мг цинку промивають 0,5% розчином ацетату ртуті(II) і потім промивають дистильованою водою, застосовують належним способом) з наступним перемішуванням при 0°С протягом 5 годин. Повільно додають насичений розчин карбонату натрію (50 мл) з наступною екстракцією етилацетатом (250 мл 2). Зібраний органічний шар сушать над безводним сульфатом натрію, фільтрують, концентрують при зниженому тиску і потім відправляють на колонкову хроматографію (н-гексан/етилацетат, 1/21/4), одержуючи зазначену в заголовку сполуку 15а у вигляді світло-жовтої твердої речовини (450 мг, 0,88 ммоль, 45%). 1Н ЯМР (600 МГц, CDCl3) 7,34 (д, J=3,6 Гц, 1Н), 7,32 (д, J=8,4 Гц, 2Н), 6,96 (д, J=8,4 Гц, 2Н), 6,90 (широкий, 1Н), 6,87 (д, J=3,6 Гц, 1Н), 4,94 (широкий с, 1Н), 4,87-4,81 (м, 1Н), 4,05 (т, J=9 Гц, 1Н), 3,90-3,83 (м, 1Н), 3,80 (дд, J=9, 6 Гц, 1Н), 3,75-3,67 (м, 1Н), 3,68 (широкий с, 2Н), 3,44 (широкий, 4Н), 1,41 (с, 9Н). Одержання сполуки 16а 39 97598 40 Сполуку 15а (150 мг, 0,29 ммоль) розчиняють ристовуючи оцтову кислоту, і повільно підвищуу метанолі (3 мл), додають до розчину формалін ють реакційну температуру до 50°С. Через 10 (0,10 мл, 37% мас. водяний розчин) з наступним годин реакційний розчин концентрують при зниперемішуванням при кімнатній температурі проженому тиску і розбавляють 2н. розчином HCl (30 тягом 1 години. Реакційний розчин концентрують мл) з наступною екстракцією дихлорметаном (25 при зниженому тиску і розбавляють дистильовамл 2). Зібраний органічний шар сушать над безною водою (15 мл) з наступною екстракцією дихводним сульфатом натрію, фільтрують, концентлорметаном (15 мл 2). Зібраний органічний шар рують при зниженому тиску і потім відправляють сушать над безводним сульфатом натрію, фільтна колонкову хроматографію (нрують і концентрують при зниженому тиску, одегексан/етилацетат, 1/11/3), одержуючи зазнаржуючи 110 мг світло-жовтої твердої речовини. чену в заголовку сполуку 16а у вигляді білої твеЦю тверду речовину розчиняють у метанолі (3 рдої речовини (15 мг, 0,029 ммоль). мл) і тетрагідрофурані (1 мл) і потім знижують Приклад одержання 4: одержання сполуки температуру до 0°С. Додають боргідрид натрію 15b (160 мг, 4,22 ммоль). Регулюють рН при 5, вико Одержання сполуки 14b Як пояснюється в синтезі сполуки 14а у прикладі одержання 3, сполуку 13 розчиняють у дихлорметані, додають до розчину HCl (4М 1,4діоксановий розчин) з наступним перемішуванням при кімнатній температурі протягом 1 години. Реакційний розчин концентрують при зниженому тиску і сушать, одержуючи білу тверду сполуку (500 мг, 1,29 ммоль). Додають метанол (10 мл) і Ν,Ν-диметилформамід (2 мл), додають N-Вос-Nметил-2-аміноацетальдегід (268 мг, 1,55 ммоль) і ціанборгідрид натрію (81 мг, 1,29 ммоль) з наступним перемішуванням при кімнатній температурі протягом 16 годин. Додають насичений розчин хлориду амонію (3 мл) і потім розчин концентрують при зниженому тиску. Ще додають насичений розчин хлориду амонію (30 мл) з наступною екстракцією етилацетатом (30 мл 2). Зібраний органічний шар сушать над безводним сульфатом натрію, фільтрують, концентрують при зниженому тиску і потім відправляють на колонкову хроматографію (н-гексан/етилацетат, 1/21/4), одержуючи зазначену в заголовку сполуку 14b у вигляді білої твердої речовини (544 мг, 1,07 ммоль, 83%). 1Н ЯМР (600 МГц, CDCl3) δ 7,31 (д, J=4 Гц, 1Н), 7,24 (д, J=9 Гц, 2H), 6,89 (д, J=4 Гц, 2Н), 6,65 (широкий, 1Н), 6,59 (д, J=9 Гц, 1Н), 4,85-4,79 (м, 1Н), 4,04 (т, J=9 Гц, 1Н), 3,90 (ддд, J=16,6, 7, 3 Гц, 1Н), 3,79 (дд, J=9, 6 Гц, 1Н), 3,74-3,67 (м, 1Н), 3,54-3,39 (м, 2Н), 3,26 (т, J=6 Гц, 2Н), 2,88 (с, 3Н), 1,46 (с, 9Н). Одержання сполуки 15b Сполуку 14b (405 мг, 0,80 ммоль) розчиняють в оцтовій кислоті (3 мл), повільно завантажують у розчин нітрит натрію (NaNO2) (66 мг, 0,96 ммоль), розчинений у дистильованій воді (0,5 мл), при 0°С з наступним перемішуванням протягом 30 хв. Потім завантажують туди цинк (130 мг, 1,99 ммоль) з наступним перемішуванням при 0°С протягом 3 годин. Повільно додають насичений розчин карбонату натрію (30 мл) з наступною екстракцією етилацетатом (30 мл 2). Зібраний органічний шар сушать над безводним сульфатом натрію, фільтрують, концентрують при зниженому тиску і потім відправляють на колонкову хроматографію (н-гексан/етилацетат, 1/21/4), одержуючи зазначену в заголовку сполуку 15b у вигляді світло-коричневої твердої речовини (81 мг, 0,15 ммоль, 19%). Приклад одержання 5: одержання сполуки 26 41 Одержання сполуки 18 4-фторнітробензол (5,2 г, 37 ммоль) розчиняють в ацетонітрилі (40 мл), додають до розчину 2-аміноетанол (5,2 г, 85 ммоль) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом ночі. Реакційний розчин охолоджують при кімнатній температурі, концентрують при зниженому тиску, розчиняють у етилацетаті, промивають послідовно водою, 1н. HCl і солоною водою. Реакційну суміш сушать над безводним сульфатом натрію, фільтрують і переганяють при зниженому тиску з одержанням сполуки 17. Сполуку 17 (15 г, 82,33 ммоль) і діїзопропілетиламін (27 мл, 164 ммоль) розчиняють у дихлормстані (150 мл), повільно завантажують у розчин метансульфонілхлорид (9,5 мл) при 0°С з наступним перемішуванням при кімнатній температурі протягом 2 годин. По завершенні взаємодії додають до реакційного розчину дихлорметан (800 мл). Реакційну суміш промивають розчином бікарбонату натрію (500 мл) і концентрують при зниженому тиску, одержуючи жовту тверду сполуку 18 (22 г, 82,00 ммоль, 99%). 1Н ЯМР (400 МГц, хлороформ-d1) 8,12 (д, J=9,2 Гц, 2Н), 6,60 (д, J=9,2 Гц, 2Н), 4,45 (т, J=5,6 Гц, 2Н), 3,63 (т, J=5,6 Гц, 2Н), 3,06 (с, 3Н); РХ-МС: + 183 (М+Н ) для C8H10N2O3S. Одержання сполуки 19 Сполуку 18 (22 г, 82,00 ммоль), гідроксифталімід (17,4 г, 107,04 ммоль) і триетиламін (17,3 мл, 123,5 ммоль) додають до ацетонітрилу (300 мл) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 6 годин. По завершенні взаємодії додають до реакційного розчину дихлорметан (1000 мл). Реакційну суміш промивають 0,5н. розчином HCl (500 мл) і насиченим розчином бікарбонату натрію (500 мл) і 97598 42 концентрують при зниженому тиску, одержуючи жовту тверду сполуку 19 (26 г, 79,44 ммоль, 97%). 1Н ЯМР (400 МГц, хлороформ-d1) 8,12 (д, J=9,2 Гц, 2Н), 7,88 (м, 2Н), 7,87 (м, 2Н), 6,65 (д, J=9,2 Гц, 2Н), 5,83 (широкий с, 1Н), 4,45 (т, J=4,8 + Гц, 2Н), 3,56 (м, 2Н); РХ-МС: 328 (М+Н ) для C16H13N3O5. Одержання сполуки 20 Сполуку 19 (79,44 ммоль) і гідразин (20 мл) додають до етанолу з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 2 годин. Реакційну суміш охолоджують при кімнатній температурі, фільтрують і концентрують при зниженому тиску з одержанням сполуки 20 (19 г, сира). Цю сполуку не очищають і використовують для наступного взаємодії. 1Н ЯМР (400 МГц, ДМСО-d6) 7,94 (д, J=9,6 Гц, 2Н), 7,34 (т, J=5,6 Гц, 1Н), 6,69 (д, J=9,5 Гц, 2Н), 3,68 (т, J=5,2 Гц, 2Н), 3,37 (м, 2Н); РХ-МС: + 198 (М+Н ) для C8H11N3O3. Одержання сполуки 21 Сполуку 20 (19 г) і карбонат натрію (21 г, 198 ммоль) розчиняють у діоксані (200 мл) і дистильованій воді (200 мл), повільно додають до розчину ди-трет-бутоксикарбоніл (26 г, 119 ммоль) з наступним перемішуванням при кімнатній температурі протягом 4 годин. По завершенні взаємодії реакційну суміш відфільтровують при зниженому тиску, промивають дистильованою водою (1000 мл) і сушать, одержуючи жовту тверду сполуку 21 (26 г, сира). 1Н ЯМР (400 МГц, хлороформ-d1) 8,10 (д, J=9,2 Гц, 2Н), 6,54 (д, J=9,2 Гц, 2Н), 5,50 (с, 1Н), 5,04 (с, 1Н), 3,91 (т, J=5,2 Гц, 2H), 3,45 (м, 2Н), + 1,43 (с, 9Н); РХ-МС: 298 (М+Н ) для C13H19N3O5. Одержання сполуки 22 Сполуку 21 (13 г, 40 ммоль), оцтову кислоту (132 мл) і концентровану HCl (10 мл, 300 ммоль) 43 97598 44 розчиняють у тетрагідрофурані (200 мл), додають Одержання сполуки 25 до розчину цинк (26 г, 400 ммоль) при 0°С з наСполуку 24 (1,6 г, 3,22 ммоль) і гідразин (1,6 ступним перемішуванням протягом 2 годин. Повімл, 32,20 ммоль) додають до етилового спирту льно додають 20% розчин аміаку (200 мл) при (30 мл) з наступним кип'ятінням зі зворотним хо0°С і потім додають диметиленхлорид (500 мл). лодильником при перемішуванні протягом 2 гоОрганічний шар відокремлюють, потім концентдин. Реакційну суміш охолоджують при кімнатній рують при зниженому тиску, одержуючи білу тветемпературі, фільтрують і концентрують при знирду сполуку 22 (6,5 г, 24,31 ммоль, 61%). женому тиску з одержанням сполуки 25 (1,3 г, неочищеної). Це сполуку не очищають і викорис1Н ЯМР (400 МГц, хлороформ-d1) 7,17 (с, товують для наступного взаємодії. 1Н), 6,6 (д, J=8,4 Гц, 2Н), 6,60 (д, J=8,4 Гц, 2Н), 4,03 (т, J=4,8 Гц, 2Н), 3,30 (т, J=4,8 Гц, 2Н), 1,47 1Н ЯМР (400 МГц, хлороформ-d1) 7,33 (д, + (с, 9Н); РХ-МС: 268 (М+Н ) для C13H21N3O3. J=9,2 Гц, 2Н), 6,66 (д, J=9,2 Гц, 2Н), 4,63 (м, 1H), Одержання сполуки 23 4,03 (м, 3Н), 3,77 (дд, J=6,4 2,0 Гц, 1Н), 3,34 (т, Сполуку 22 (6,5 г, 24,31 ммоль) і (S)J=5,2 Гц, 2Н), 3,03 (м, 2Н), 1,49 (с, 9Н); РХ-МС: + гліцидилфталімід (3,95 г, 19,45 ммоль) додають 367 (М+Н ) для C17H26N4O5. до ізопропілового спирту (100 мл), потім кип'ятять Одержання сполуки 26 зі зворотним холодильником при перемішуванні Сполуку 25 (1,3 г, 3,22 ммоль), 5-хлортіофенпротягом 6 годин. По завершенні взаємодії реак2-карбонову кислоту (0,68 г, 4,19 ммоль) і РуВОР ційну суміш концентрують при зниженому тиску з [(бензотриазол-1-ілокси)трипіролідинофосфоний наступною колонковою хроматографією, одергексафторфосфат] (2,5 г, 4,83 ммоль) додають до жуючи білу тверду сполуку 23 (9 г, неочищеної). Ν,Ν-диметилформаміду (20 мл), повільно додають до розчину діїзопропілетиламін (1,06 мл, 6,44 1Н ЯМР (400 МГц, хлороформ-d1) 7,85 (м, ммоль) при 0°С з наступним перемішуванням 2Н), 7,79 (м, 2Н), 6,33 (д, J=9,2 Гц, 2Н), 6,21 (д, протягом 1 години. По завершенні взаємодії доJ=9,2 Гц, 2Н), 4,07 (м, 3Н), 3,58-3,31 (м, 6Н), 1,46 + дають до реакційного розчину етилацетат (300 (с, 9Н); РХ-МС: 471 (М+Н ) для С24Н30N4Об. мл). Реакційну суміш двічі промивають дистильо Одержання сполуки 24 ваною водою (200 мл) і концентрують при знижеСполуку 23 (9 г, неочищенеу) і карбодііміданому тиску, одержуючи білу тверду сполуку 26 зол (3,5 г, 29,16 ммоль) додають до тетрагідро(1,6 г, 3,13 ммоль). фурану (150 мл) з наступним перемішуванням протягом 12 годин. По завершенні взаємодії реа1Н ЯМР (400 МГц, ДМСО-d6) 10,05 (с, 1Н), кційну суміш концентрують при зниженому тиску, 8,97 (т, J=5,6 Гц, 1Н), 7,70 (д, J=4,0 Гц, 1Н), 7,20 проводять колонкову хроматографію, одержуючи (м, 3Н), 6,60 (д, J=8,4 Гц, 2Н), 4,76 (м, 1Н), 4,07 (т, білу тверду сполуку 24 (1,6 г, 3,22 ммоль). J=8,8 Гц, 1Н), 3,83 (т, J=5,6 Гц, 2Н), 3,74 (м, 1Н), 3,57 (т, J=5,2 Гц, 2Н), 3,21 (т, J=5.6 Гц, 2Н), 1,40 1Н ЯМР (400 МГц, хлороформ-d1) 7,88 (м, + (с, 911); РХ-МС; 511 (М+Н ) для С22Н27Сl4О6. 2Н), 7,75 (м, 2Н), 7,28 (д, J=8,8 Гц, 2Н), 6,55 (д, Приклад 1: одержання сполуки 100 J=8,8 Гц, 2Н), 4,95 (м, 1Н), 4,17-3,82 (м, 6Н), 3,37 + (т, J=5,2 Гц, 2Н), 1,46 (с, 9Н); РХ-МС: 497 (М+Н ) для C25H28N4O7. Сполуку 5 (5,0 г, 13,8 ммоль), одержану в прикладі одержання 1, додають до безводного метанолу (200 мл), потім барботують газоподібний HCl при 0°С протягом 30 хв. Додають метанол (100 мл), потім знову барботують газоподібний HCl протягом більш 30 хв. з наступним перемішуванням при кімнатній температурі протягом 2 годин. Реакційну суміш концентрують при зниженому тиску для видалення НСІ, що залишається. Додають туди оцтову кислоту (120 мл) і 2(N-метиламіно)етилгідроксиламін гідрохлорид (3,5 г, 27,6 ммоль) з наступним кип'ятінням зі зворотним холодильником протягом ночі. Реакційний розчин концентрують при зниженому тиску, роз чиняють у етилацетаті і промивають насиченим розчином NаНСО3, потім розділяють суміш, використовуючи колонкову хроматографію й одержуючи зазначену в заголовку сполуку 100 у вигляді білої твердої речовини (1,85 г, 4,3 ммоль, 31%). 1Н ЯМР (400 МГц, CDCl3) 7,54 (д, J=9,0 Гц, 2Н), 7,44 (д, J=9,0 Гц, 2Н), 7,34 (д, J=4,2 Гц, 1Н), 6,89 (широкий т, 1Н), 4,80 (м, 1Н), 4,12 (т, J=4,8 Гц, 2Н), 4,04 (т, J=9,0 Гц. 1Н), 3,85-3,80 (м, 2Н), 3,69-3,64 (м, 1Н), 3,45 (т, J=4,8H7, 2H), 2,76 (с, + 3Н); РХ-МС; 435 (М+Н ) для C19H19Cl4O4S. Приклад 2: одержання сполуки 101 45 97598 46 Сполуку 101 одержують у вигляді білої твердої речовини (17 мг, 0,04 ммоль, 14%) аналогічно тому, як описано в прикладі 1, використовуючи сполуку 5 (0,1 г, 0,27 ммоль), одержану в прикладі одержання 1, і (аміноетил)гідроксиламін (62 мг, 0,8 ммоль). 1Н ЯМР (400 МГц, ДМСО-d6) 8,98 (т, J=6,0 Гц, 1Н), 7,68 (д, J=4,0 Гц, 1Н), 7,63 (д, J=8,8 Гц, 2Н), 7,57 (д, J=8,8 Гц, 2Н), 7,19 (д, J=4,0 Гц, 1Н), 7,07 (с, 1Н), 4,85 (м, 1Н), 4,19 (т, J=8,8 Гц, 1Н), 3,86-3,81 (м, 3Н), 3,61 (т, J=5,2 Гц, 2Н), 3,38 (м, + 2Н); РХ-МС: 421 (М+Н ) для C18H17CI4O4S. Приклад 3: одержання сполуки 102 Сполуку 102 одержують у вигляді білої твердої речовини (30 мг, 48%) аналогічно тому, як описано в прикладі 1, використовуючи сполуку 5 (100 мг, 0,27 ммоль), одержане в прикладі одержання 1, і О-[2-(2-етиламіно)-етил]гідроксиламін (15 мг, 0,139 ммоль). 1Н ЯМР (400 МГц, CDCl3) 7,52 (д, J=8,4 Гц, 2Н), 7,42 (д, J=8,4 Гц, 2Н), 7,37 (д, J=3,6 Гц, 1Н), 7,28-7,22 (м, 1Н), 6,85 (д, J=3,6 Гц, 1Н), 4,82-4,71 (м, 1Н), 4,11 (т, J=4,4 Гц, 2Н), 3,98 (т, J=8,8 Гц, 1Н), 3,83-3,72 (м, 2Н), 3,66-3,56 (м, 1Н), 3,43 (т, J=4,4 Гц, 2Н), 3,01 (кв, J=7,2 Гц, 2Н), 1,03 (т, J=4,4 + Гц, 3Н); РХ-МС: 449 (М+Н ) для C20H21Cl4O4S. Приклад 4: одержання сполуки 103 Сполуку 103 одержують у вигляді білої твердої речовини (31 мг, 29%) аналогічно тому, як описано в прикладі 1, використовуючи сполуку 5 (100 мг, 0,27 ммоль), одержану в прикладі одержання 1, і О-[2-(2циклопропіламіно)етил]гідроксиламін (27 мг, 0,23 ммоль). 1Н ЯМР (400 МГц, CDCl3) 7,52 (д, J=7,6 Гц, 2Н), 7,46 (д, J=7,6 Гц, 2Н), 7,38 (д, J=3,6 Гц, 1Н), 7,23-7,18 (м, 1Н), 6,87 (д, J=3,6 Гц, 1Н), 4,85-4,72 (м, 1Н), 4,06 (т, J=4,4 Гц, 2Н), 4,02 (т, J=8,4 Гц, 1Н), 3,85-3,74 (м, 2Н), 3,68-3,55 (м, 1Н), 3,53 (т, J=4,4 Гц, 2Н), 2,62-2,51 (м, 1Н), 0,51-0,32 (м, 4Н); + РХ-МС: 461 (М+H ) для C21H21CI4O4S. Приклад 5: одержання сполуки 104 Сполуку 104 одержують у вигляді білої твердої речовини (63,7 мг, 0,146 ммоль, 58%) аналогічно тому, як описано в прикладі 1, використовуючи сполуку 5 (100 мг, 0,27 ммоль), одержану в прикладі одержання 1, і N-метил-О-(2аміноетил)гідроксиламіндигідрохлорид (180,0 мг, 1,104 ммоль). 1Н ЯМР (400 МГц, ДМСО-d6) 9,07 (т, J=6,4 Гц, Пі), 7,70 (д, J=4,4 Гц, 1Н), 7,54 (м, 4Н), 7,14 (д, J=4,4 Гц, 1Н), 4,80 (м, 1Н), 4,15 (т, J=8,8 Гц, 1Н), 3,86 (дд, J=8,8 Гц, 8,8 Гц, 1Н), 3,81 (т, J=4,8 Гц, 2Н), 3,55-3,48 (м, 4Н), 2,82 (с, 3Н); РХ-МС: 435 + (М+Н ) для C19H19Cl4O4S. Приклад 6: одержання сполуки 105 Сполуку 105 одержують у вигляді світложовтої твердої речовини (9,1 мг, 0,020 ммоль, 9%) аналогічно тому, як описано в прикладі 1, використовуючи сполуку 5 (80,0 г, 0,221 ммоль), одержану в прикладі одержання 1, і N-етил-O-(2аміноетил)гідроксиламіндигідрохлорид (300,0 мг, 1,69 ммоль). 1Н ЯМР (600 МГц, ДМСО-d6) 9,37 (широкий, 1Н), 7,88 (м, 1Н), 7,79 (д, J=6,0 Гц, 2Н), 7,73 (д, J=5,6 Гц, 2Н), 7,20 (д, J=4,2 Гц, 1Н), 4,91 (м, 1Н), 4,24 (т, J=8,7 Гц, 1Н), 4,05 (дд, J=8,7 Гц, 8,7 Гц, 1Н), 3,66 (т, J=4,8 Гц, 2Н), 3,63-3,54 (м, 4Н), 2,81 (кв, J=7,2 Гц, 2Н), 1,24 (т, J=7,2 Гц, 3Н); РХ-МС: + 449 (М+Н ) для C20H21Cl4O4S. Приклад 7: одержання сполуки 106 Сполуку 106 одержують у вигляді білої твердої речовини (20 мг, 0,04 ммоль, 16%) аналогічно тому, як описано в прикладі 1, використовуючи сполуку 5 (100 мг, 0,27 ммоль), одержану в прикладі одержання 1, і 2-(1-метилгідразиніл)-Nетилетанамін (93 мг, 0,8 ммоль). 1Н ЯМР (600 МГц, ДМСО-d6) 8,99 (т, J=5,4 Гц, 1Н), 7,69 (д, J=3,6 Гц, 1Н), 7,53 (д, J=8,4 Гц, 2Н), 7,36 (т, J=8,4 Гц, 2Н), 7,19 (д, J=3,6 Гц, 1Н), 4,84 (м, 1Н), 4,19 (т, J=9,6 Гц, 1Н), 3,86 (дд, J=8,4 6,6 Гц, 1Н), 3,61 (т, J=5,4 Гц, 2Н), 3,31 (м, 2Н), 2,92 (м, 2Н), 2,80 (широкий с, 2Н), 2,59 (с, 3Н), 0,94 (т, J=7,2 Гц, 1Н) (2 еквіваленти оцтової кис+ лоти за даними ЯМР); РХ-МС: 462 (М+Н ) для C21H24Cl5O3S. Приклад 8: одержання сполуки 107 Na (3,3 г, 87,01 ммоль) додають до Ν,Νдиметилформаміду (30 мл) з наступним перемішуванням протягом 15 хв. Трет-бутилшабазит (5 г, 37,83 ммоль) розчиняють у Ν,Νдиметилформаміді (10 мл) і повільно додають до реакційної суміші при 0°С. Також додають туди дибромпропан (7,6 г, 37,83 ммоль) при 0°С з наступним перемішуванням при кімнатній температурі протягом 3 годин. Реакційну суміш охолоджують при 0°С і промивають дистильованою водою (50 мл). По завершенні взаємодії реакційну суміш розчиняють у етилацетаті (500 мл), потім три рази промивають розчином бікарбонату натрію (50 мл). Проводять колонкову хроматографію, одержуючи сполуку 107а у вигляді олії (2,4 г, 13,93 ммоль, 36,7%). 1Н ЯМР (400 МГц, хлороформ-d1) 3,85 (с, 1Н), 3,45 (т, J=7,2 Гц, 2Н), 3,04 (т, J=6,4 Гц, 2Н), + 2,03 (м, 2Н), 1,49 (с, 9Н); РХ-МС: 173 (М+Н ) для C8H16N2O2. Сполуку 107а (1,5 г, 8,71 ммоль), N-2бромфталімід (2,33 г, 8,71 ммоль) і карбонат калію (1,32 г, 9,58 ммоль) додають до Ν,Νдиметилформаміду (10 мл) з наступним перемішуванням при 100°С протягом 12 годин. Реакційну суміш розчиняють у етилацетаті (200 мл), потім промивають три рази розчином бікарбонату натрію (30 мл). Проводять колонкову хроматографію, одержуючи сполуку 107b у вигляді олії (1,7 г, 4,92 ммоль, 56,2%). 1Н ЯМР (400 МГц, хлороформ-d1) 7,89 (м, 2Н), 7,74 (м, 2Н), 3,94(т, J=4,4 Гц, 2Н), 3,90 (т, J=5,2 Гц, 2Н), 3,11 (т, J=6,0 Гц, 2Н), 3,06 (т, J=4,4 Гц, 2Н), 2,01 (м, 2Н), 1,48 (с, 9Н); РХ-МС: 346 + (М+Н ) для C18H23N3O4. Сполуку 107b (0,5 г, 1,45 ммоль) додають до 4М HCl (у діоксані) (5 мл) з наступним перемішуванням протягом 0,5 години. Реакційну суміш концентрують при зниженому тиску, додають до розчину метиловий спирт (5 мл) і метиламін (4 мл) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 1 години. Реакційну суміш концентрують при зниженому тиску з одержанням сполуки 107с, що більше не очищають і використовують для наступної взає+ модії. РХ-МС: 116 (М+Н ) для C5H13N3. Сполуку 5 (100 мг, 0,27 ммоль), одержану в прикладі одержання 1, додають до безводного метилового спирту (10 мл), потім барботують газоподібним HCl при 0°С протягом 30 хв. і перемішують при кімнатній температурі протягом 2 годин. Реакційну суміш концентрують при зниженому тиску для видалення HCl, що залишилася. Додають туди послідовно безводний метиловий спирт (10 мл) і сполуку 107с (300 мг, 0,8 ммоль) з наступним перемішуванням при кімнатній температурі протягом 12 годин і концентруванням при зниженому тиску. Потім проводять препаративну ТШХ, одержуючи зазначену в заголовку сполуку 107 у вигляді білої твердої речовини (60 мг, 0,13 ммоль, 48%). 1Н ЯМР (400 МГц, ДМСО-d6) 9,12 (т, J=5,6 Гц, 1Н), 7,77-7,64 (м, 5Н), 7,14 (д, J=4,4 Гц, 1Н), 4,84 (м, 1Н), 4,18 (т, J=8,8 Гц, 1Н), 3,90 (дд, J=8,8, 6,0 Гц, 1Н), 3,70 (т, J=6,8 Гц, 1Н), 3,65 (т, J=6,8 Гц, 1Н), 3,59-3,52 (м, 3Н), 3,25 (м, 1Н), 3,11 (м, 1Н), 2,95 (м, 1Н), 2,65 (м, 1Н), 2,55 (м, 1Н), 2,08 (м, + 2Н); РХ-МС: 460(М+Н ) для C21H22Cl5O3S. Приклад 9: одержання сполуки 108 Трет-бутилшабазит (трет-бутилкарбазат) (2,5 г, 18,91 ммоль). N-(брометил)фталімід (5,25 г, 20,80 ммоль) і карбонат калію (3,14 г, 22,70 ммоль) додають до Ν,Ν-диметилформаміду (30 мл) з наступним перемішуванням при 90°С протягом 12 годин. По завершенні взаємодії реакційну суміш розчиняють у етилацетаті (250 мл), потім три рази промивають розчином бікарбонату натрію (150 мл). Після концентрування при зни женому тиску проводять колонкову хроматографію, одержуючи білу тверду сполуку 108а (1 г, 3,2 ммоль, 19%). 1Н ЯМР (400 МГц, хлороформ-d1) 7,85 (м, 2Н), 7,73 (м, 2Н), 6,49 (с, 1Н), 4,12 (с, 1Н), 3,83 (т, J=3,6 Гц, 2Н), 3,05 (м, 2Н), 1,46 (с, 9Н); РХ-МС: + 306 (М+Н ) для C15H19N3O4. Сполуку 108а (0,9 г, 2,95 ммоль) і карбонат калію (1,03 г, 7,4 ммоль) розчиняють у Ν,Ν 51 97598 52 диметилформаміді (10 мл), додають до розчину що більше не очищають і використовують для 3-бромпропаноілхлорид (0,7 г, 3,6 ммоль), потім наступного взаємодії. + перемішують при 90°С протягом 5 годин. ДодаРХ-МС: 130 (М+Н ) для С3Н11N3О. ють етилацетат (100 мл), потім промивають розСполуку 108 одержують у вигляді білої тверчином бікарбонату натрію (30 мл). Після концентдої речовини (26 мг, 0,05 ммоль, 19%) аналогічно рування при зниженому тиску проводять тому, як описано в прикладі 1, використовуючи колонкову хроматографію, одержуючи сполуку сполуку 5 (0,1 г, 0,27 ммоль), одержану в прикла108b (у вигляді олії 230 мг, 0,64 ммоль, 22%). ді одержання 1, і сполуку 108с (103 мг, 0,64 ммоль). 1Н ЯМР (400 МГц, хлороформ-d1) 7,85 (м, 1Н ЯМР (600 МГц, ДМСО-d6) 9,09 (т, J=4,8 2Н), 7,73 (м, 2Н), 6,49 (с, 1Н), 4,12 (с, 1Н), 3,83 (т, J=3,6 Гц, 2Н), 3,05 (м, 2Н), 1,46 (с, 9Н); РХ-МС: Гц, 1Н), 7,79 (д, J=8,4 Гц, 2Н), 7,72-7,71 (м, 3Н), + 360 (М+Н ) для C18H21N3O5. 7,19 (д, J=4,2 Гц, 1Н), 4,89 (м, 1Н), 4,24 (т, J=9,0 Сполуку 108b (230 мг, 0,64 ммоль) додають Гц, 1Н), 4,12 (т, J=7,8 Гц, 2Н), 3,94-3,89 (м, 3Н), до 4М HCl (у діоксані) (2 мл) з наступним перемі3,62 (м, 2Н), 3,53 (т, J=4,8 Гц, 2Н), 2,80 (т, J=8,4 шуванням протягом 1 години. Реакційну суміш Гц, 2Н), 3,59-3,52 (м, 3Н), 3,25 (м, 1Н), 3,11 (м, концентрують при зниженому тиску, додають до 1Н), 2,95 (м, 1Н), 2,65 (м, 1Н), 2,55 (м, 1Н), 2,08 + розчину метиловий спирт (5 мл) і метиламін (2 (м, 2Н); РХ-МС: 474 (М+Н ) для C21H20CI5O4S. мл), потім перемішують протягом 1 години. РеакПриклад 10: одержання сполуки 109 ційну суміш концентрують при зниженому тиску з одержанням сполуки 108с (103 мг, 0,64 ммоль), Сполуку 15а (450 мг, 0,88 ммоль), одержану в прикладі одержання 3, розчиняють у дихлорметані (10 мл), додають HCl (4М 1,4-діоксановий розчин) (10 мл) з наступним перемішуванням при кімнатній температурі протягом 1 години. Реакційну суміш концентрують при зниженому тиску і сушать, одержуючи світло-жовту тверду сполуку (425 мг, 0,88 ммоль, 100%). Цю сполуку (392 мг, 0,81 ммоль) розчиняють в оцтовій кислоті (4 мл), додають триметилортоформіат (2 мл) з наступним кип'ятінням зі зворотним холодильником при перемішуванні. Через 10 годин після повного випарювання розчинника проводять колонкову хроматографію (дихлорметан/метанол (об./об.) 20/112/1), одержуючи зазначену в заголовку сполуку 109 у вигляді світло-жовтої твердої речовини (215 мг, 5,12 ммоль, 63%). 1Н ЯМР (400 МГц, CDCl3) 7,35 (д, J=9,2 Гц, 2Н), 7,33 (д, J=4,4 Гц, 1Н), 7,14 (д, J=9,2 Гц, 2Н), 7,01 (т, J=6,4 Гц, 1Н), 6,88 (с, 1Н), 6,85 (д, J=4,4 Гц, 1Н), 4,87-4,79 (м, 1Н), 4,06 (т, J=9 Гц, 1Н), 3,86 (ддд, J=14,4, 6, 3 Гц, 1Н), 3,81 (дд, J=9, 6,4 Гц, 1Н), 3,69 (дт, J=14,4, 6 Гц, 1Н), 3,62-3,58 (м, 2Н), + 3,55-3,51 (м, 2Н); РХ-МС: 420 (М+Н ) для C18H18Cl5O3S. Приклад 11: одержання сполуки 110 Використовують сполуку 15а, одержану в прикладі одержання 3, для одержання зазначеної в заголовку сполуки 110 способом, аналогічним способу, описаному в прикладі 10, використовуючи триетилереортоацетат замість триметилортоформіату з прикладу 10. 1Н ЯМР (600 МГц, CDCl3) 7,36 (д, J=9 Гц, 2Н), 7,30 (д, J=4 Гц, 1Н), 7,17 (д, J=9 Гц, 2Н), 6,90 (д, J=4,2 Гц, 1Н), 6,49 (широкий т, 1Η), 4,85-4,80 (м, 1Н), 4,28 (широкий, 1Н), 4,08 (т, J=9 Гц, 1Н), 3,95-3,90 (м, 1Н), 3,79 (т, J=7,8 Гц, 1Н), 3,72-3,67 (м, 1Н), 3,62-3,57 (м, 2Н), 3,49-3,44 (м, 2Н), 1,98 + (с, 3Н); РХ-МС: 434 (М+Н ) для С19Н20Сl5О3S. Приклад 12: одержання сполуки 111 53 97598 54 Використовують сполуку 15а, одержану в прикладі одержання 3, одержуючи зазначену в заголовку сполуку 111 способом, аналогічним способу, описаному в прикладі 10, використовуючи триетилортопропіонат замість триметилортоформіату з прикладу 10. 1Н ЯМР (600 МГц, CDCl3) 7,35 (д, J=9 Гц, 2Н), 7,30 (д, J=4,2 Гц, 1Н), 7,18 (д, J=9 Гц, 2Н), 6,90 (д, J=4,2 Гц, 1Н), 6,53 (широкий т, 1Η), 4,854,80 (м, 1Н), 4,27 (широкий, 1Н), 4,08 (т, J=9 Гц, 1Н), 3,92 (ддд, J=14,4, 6,6, 3 Гц, 1Н), 3,79 (т, J=7,8 Гц, 1Н), 3,72-3,67 (м, 1Н), 3,62-3,57 (м, 2Н), 3,503,44 (м, 2Н), 2,26 (кв, J=7,8 Гц, 2Н), 1,20 (т, J=7,8 + Гц, 3Н); РХ-МС: 448 (М+Н ) для C20H22CI5O3S. Приклад 13: одержання сполуки 112 Використовують сполуку 15b, одержану в прикладі одержання 4, одержуючи зазначену в заголовку сполуку 112 способом, аналогічним способу, описаному в прикладі 10. 1Н ЯМР (600 МГц, CDCl3) 7,35 (д, J=9 Гц, 2Н), 7,33 (д, J=4,5 Гц, 1Н), 7,13 (д, J=9 Гц, 2Н), 6,93 (т, J=6 Гц, 1Н), 6,85 (д, J=4,5 Гц, 1Н), 6,64 (с, 1Н), 4,86-4,80 (м, 1Н), 4,06 (т, J=9 Гц, 1Н), 3,913,85 (м, 1Н), 3,80 (дд, J=9, 7 Гц, 1Н), 3,71-3,65 (м, 1Н), 3,54 (т, J=4,8 Гц, 2Н), 3,43 (т, J=4,8 Гц, 2Н), + 2,90 (с, 3Н); РХ-МС: 434 (М+Н ) для C19H20Cl5O3S. Приклад 14: одержання сполуки 113 Використовують сполуку 15b, одержану в прикладі одержання 4, одержуючи зазначену в заголовку сполуку 113 способом, аналогічним способу, описаному в прикладі 10, використовуючи триетилортоацетат замість триметилортоформіату з прикладу 10. 1Н ЯМР (600 МГц, CDCl3) 7,34 (д, J=9 Гц, 2Н),7,31 (д, J=4 Гц, 1Н),7,15 (д, J=9 Гц, 2Н), 6,88 (д, J=4 Гц, 1Н), 6,69 (т, J=6 Гц, 1Н), 4,84-4,78 (м, 1Н), 4,06 (т, J=9 Гц, 1Н), 3,90 (ддд, J=11, 7, 3 Гц, 1Н), 3,79 (дд, J=9, 6,6 Гц, 1Н), 3,68 (дт, J=14,4, 6,6 Гц, 1Н), 3,52 (т, J=5 Гц, 2Н), 3,44 (т, J=5 Гц, 2Н), + 2,93 (с, 3Н), 2,05 (с, 3Н); РХ-МС: 448 (М+Н ) для C20H22Cl5O3S. Приклад 15: одержання сполуки 114 Використовують сполуку 16а, одержану в прикладі одержання 3, одержуючи зазначену в заголовку сполуку 114 у вигляді білої твердої речовини (7,4 мг, 0,014 ммоль, 47%) способом, аналогічним способу, описаному в прикладі 10. 1Н ЯМР (400 МГц, ДМСО-d6) 9,03 (т, J=5,4 Гц, 1Н), 8,65 (с, 1Н), 7,68 (д, J=3,6 Гц, 1Н), 7,55 (д, J=8,4 Гц, 2Н), 7,19 (д, J=3,6 Гц, 1Н), 7,14 (д, J=8,4 Гц, 2Н), 4,88-4,81 (м, 1Н), 4,17 (т, J=9 Гц, 1Н), 3,85-3,81 (м, 1Н), 3,70-3,50 (м, 4Н), 3,29 (с, 3Н), 55 97598 + 3,13-3,05 (м, 2Н); РХ-МС: 434 (М+Н ) для С19Н20Сl5О3S. 56 Приклад 16: одержання сполуки 115 Сполуку 26, одержану в прикладі одержання 5, додають до 4М HCl (2 мл) з наступним перемішуванням протягом однієї години. Реакційну суміш концентрують при зниженому тиску, додають триметилортоформіат (2 мл) і оцтову кислоту (4 мл) з наступним кип'ятінням зі зворотним холодильником при перемішуванні протягом 12 годин. Проводять колонкову хроматографію реакційної суміші, одержуючи зазначену в заголовку сполуку 115 у вигляді білої твердої речовини (11 мг, 0,03 ммоль, 8%). 1Н ЯМР (400 МГц, хлороформ-d1) 7,55 (с, 1Н), 7,50 (д, J=9,2 Гц, 2Н), 7,42 (широкий с, 1Н), 7,39 (д, J=4,0 Гц, 1Н), 7,03 (д, J=9,2 Гц, 2Н), 6,89 (д, J=4,0 Гц, 1Н), 4,86 (м, 1Н), 4,20 (т, J=4,8 Гц, 2Н), 4,12 (м, 1Н), 3,87 (м, 1Н), 3,83-3,74 (м, 4Н); + РХ-МС: 421 (М+Н ) для C18H17Cl4O4S. Приклад 17: одержання сполуки 116 Сполуку 116 одержують у вигляді білої твердої речовини способом, аналогічним способу, описаному в прикладі 14, використовуючи сполуку 26, одержану в прикладі одержання 5. 1Н ЯМР (400 МГц, ДМСО-d6) 8,93 (т, J=5,6 Гц, 1Н), 7,63 (д, J=4,2 Гц, 1Н), 7,50 (д, J=8,8 Гц, 2Н), 7,25 (д, J=8,8 Гц, 2Н), 7,14 (д, J=4,2 Гц, 1Н), 4,79 (м, 1Н), 4,13 (т, J=8,8 Гц, 1Н), 3,94 (т, J=4,6 Гц, 2Н), 3,79, 3,56-3,51 (м, 4Н), 1,56 (с, 3Н); РХ+ МС: 435 (М+Н ) для C19H19Cl4O4S. Приклад 18: одержання сполуки 117 Сполуку 26 (0,20 мг, 0,39 ммоль), одержану в прикладі одержання 5, циклопропанкарбонілхлорид (50 мг, 0,47 ммоль), піридин (61 мг, 0,78 ммоль) і 4-диметиламінопіридин (5 мг) додають до метиленхлориду (5 мл) з наступним перемішуванням протягом 2 годин. По завершенні взаємодії додають туди метиленхлорид (50 мл), потім двічі промивають дистильованою водою (10 мл). Після концентрування при зниженому тиску проводять колонкову хроматографію, одержуючи амідну сполука у вигляді білої твердої речовини (150 мг, 0,29 ммоль, 74%). 1Н ЯМР (400 МГц, ДМСО-d6) 9,90 (с, 1Н), 8,98 (т, J=6,0 Гц, 1Н), 7,69 (д, J=4,0 Гц, 1Н), 7,62 (д, J=8,8 Гц, 2Н), 7,42 (д, J=8,8 Гц, 2Н), 7,19 (д, J=4,0 Гц, 1Н), 4,84 (м, 1Н), 4,21 (т, J=8,8 Гц), 3,87 (дд, J=9,2, 6,4 Гц, 1Н), 3,78-3,74 (м, 4Н), 3,61 (т, J=5,6 Гц, 2Н), 1,22 (м, 1Н), 0,78 (м, 2Н), 0,59 (м, 2Н). Одержану вищу сполуку (0,15 г, 0,29 ммоль) додають до 4н. HCl (у діоксані) (2 мл) з наступним перемішуванням при кімнатній температурі протягом однієї години. Реакційну суміш концентрують при зниженому тиску. Додають толуол (5 мл) і оксихлорид фосфору (45 мг, 0,29 ммоль), потім кип'ятять зі зворотним холодильником при перемішуванні протягом 12 годин. Проводять колонкову хроматографію, одержуючи зазначену в заголовку сполуку 117 у вигляді білої твердої речовини (11 мг, 0,03 ммоль, 10%). 1Н ЯМР (400 МГц, хлороформ-d1) 7,54 (д, J=8,8 Гц, 2Н), 7,31 (д, J=4,4 Гц, 1Н), 7,25 (д, J=8,8 Гц, 2H), 6,90 (д, J=4,4 Гц, 1Н), 6,55 (т, J=4,8 Гц, 1Н), 4,88 (м, 1Н), 4,10 (т, J=4,8 Гц, 2Н), 3,89 (м, 2Н), 3,79 (м, 1Н), 3,67 (т, J=4,8 Гц, 2Н), 1,06 (м, + 1Н), 0,93 (м, 2Н), 0,58 (м, 2Н); РХ-МС: 461 (М+Н ) для C21H21Cl4O4S. Приклад 19: одержання сполуки 118 57 97598 58 Сполуку 118 одержують у вигляді білої твердої речовини способом, аналогічним способу, описаному в прикладі 10, використовуючи сполуку 33, синтезовану способом за реакційною схемою 5. 1Н ЯМР (400 МГц, хлороформ-d1) 8,55 (с, 1Н), 7,52 (д, J=8,8 Гц, 2H), 7,33 (д, J=4,4 Гц, 1Н), 7,10 (с, 1Н),7,05 (д, J=8,8 Гц, 2Н), 6,89 (д, J=4,4 Гц, 1Н), 6,76 (т, J=4,8 Гц, 1Н), 4,88 (м, 1Н), 4,11 (т, J=8,8 Гц, 1Н), 4,00 (т, J=4,8 Гц, 2Н), 3,91-3,77 (м, + 3Н), 3,74 (т, J=4,8 Гц, 2Н); РХ-МС: 448 (М+Н ) для C19H18Cl5O4S. Приклад 20: одержання сполуки 119 Сполуку 119 одержують у вигляді білої твердої речовини способом, аналогічним способу, описаному в прикладі 14, використовуючи сполуку 33, синтезовану способом за реакційною схемою 5. 1Н ЯМР (400 МГц, ДМСО-d6) 8,94 (широкий т, 1Н), 8,35 (с, 1Н), 7,64 (д, J=4,4 Гц, 1Н), 7,52 (д, J=8,8 Гц, 2Н), 7,26 (д, J=8,8 Гц, 2Н), 7,14 (д, J=4,4 Гц, 1Н), 4,80 (м, 1Н), 4,15 (т, J=8,4 Гц, 1Н), 3,813,75 (м, 3Н), 3,58-3,51 (м, 4Н), 1,75 (с, 3Н); РХ+ МС: 462 (М+Н ) для C20H20Cl5O4S. Приклад 21: одержання сполуки 120 Сполуку 119, синтезовану в прикладі 20, розчиняють у метанолі, потім деформілують за допомогою HCl, одержуючи зазначену в заголовку сполуку 120 у вигляді білої твердої речовини. 1H ЯМР (400 МГц, ДМСО-d6) 8,98 (широкий т, 1Н), 7,69 (д, J=4,4 Гц, 1Н), 7,52 (д, J=8,8 Гц, 2Н), 7,23-7,19 (м, 3Н), 4,83 (м, 1Н), 4,18 (т, J=8,4 Гц, 1Н), 3,84 (м, 1Н), 3,61-3,55 (м, 4Н), 3,06 (т, + J=4,8 Гц, 2Н), 1,64 (с, 3Н); РХ-МС: 434 (М+Н ) для C19H20CI5O3S. Приклад 22: одержання сполуки 121 Проводять взаємодію сполуки 120, синтезованої в прикладі 21, з йодметаном, одержуючи зазначену в заголовку сполуку 121 у вигляді білої твердої речовини. 1Н ЯМР (400 МГц, ДМСО-d6) 8,98 (широкий т, 1Н), 7,69 (д, J=4,4 Гц, 1Н), 7,55 (д, J=8,8 Гц, 2Н), 7,28 (д, J=8,8 Гц, 2Н), 7,19 (д, J=4,4 Гц, 1Н), 4,84 (м, 1Н), 4,18 (т, J=8,4 Гц, 1Н), 3,84 (м, 1Н), 3,65-2,92 (м, 6Н), 2,60 (с, 3Н), 1,67 (с, 3Н); РХ-МС: + 448(М+Н ) для C20H22Cl5O3S. Експериментальний приклад 1: інгібуюча активність інгібітора фактора Ха (FXa) 59 97598 60 1) Реагент і матеріал центрифугуванням при 2500g протягом 10 хв. при S-2765 (N-Z-D-Arg-Gly-Arg-pNA.2HCl), хромо4°С і зберігають при 70°С. генний субстрат, необхідний для визначення акb) Спосіб застосування spectramax: серійні тивності фактора Ха, одержують від Chromogenix. розведення розчину сполуки за даним винаходом Людський FXa одержують від Enzyme Research (5 мкл) змішують з цитратною плазмою (45 мкл), Laboratories. 96-ямковий мікропланшет одержупотім через 5 хв. додають STA-Neoplastine ють від Corning Life Sciences. (Diagnostica Stago, France) при 37°С. Безупинно 2) інгібуюча активність інгібітора FXa спостерігають поглинання при 340 нМ і визначаІнгібуючу активність оксазолідинонових похіють РТ як час (у секундах), коли поглинання при дних за даним винаходом з циклічною амідокси340 нМ досягає 0,1. Антикоагулянтну активність мною або циклічною амідразоновою групою, сполук визначають як концентрацію, необхідну представлених формулою І, відносно FXa визнадля збільшення в два рази часу згортання плазчають у такий спосіб. ми [2РТ (мкм)]. Антикоагулянтну активність споАктивність сполук щодо очищеного людськолук визначають як концентрацію, необхідну для го FXa визначають, використовуючи хромогенний збільшення в два рази часу згортання плазми субстрат S-2765 (Z-D-Arg-Gly-Arg-pNA.2HCl), у 96[2РТ (мкм)]. Людську плазму одержують з ямкових мікропланшетах при 37°С. ФерментатиDaejeon Red Cross Blood Center. Щурячу кров вну активність досліджують у 100 мМ Tris-HCl відбирають із сонної або артерії верхньої порожбуфері (р 7,8), що містить людський FXa (2,6 нМ), ньої вени під анестезією. Кров збирають у пласNaCl (150 мМ), PEG 8000 (0,1%), розведення дотикові пробірки, що містять 1/10 об'єму 3,8% цитсліджуваної сполуки (1% ДМСО) і S-2765 (300 рату натрію. Плазму одержують негайним мкМ). Реакцію ініціюють, додаючи субстрат, і бецентрифугуванням при 2500g протягом 10 хв. при зупинно протягом 5 хв. спостерігають поглинання 4°С і зберігають при -70°С. при 405 нМ, застосовуючи SpectraMax 190 2) Визначення антитромботичного ефекту з (Molecular Devices, USA). Розраховують констанвикористанням моделі артеріовенозного шунта ту інгібування (Ki) щодо людського FXa по рівнян(AV-шунт) у щурів. ню Ченга-Пруссофа (Ki=IC50/1+[S]/Km), де [S] Антитромботичний ефект оксазолідинонових означає концентрацію субстрату, і K m означає похідних за даним винаходом з циклічною амідоконстанту Міхаєліса-Ментен. Km визначають із ксимною або циклічною амідразоновою групою, графіка Лайнуівера-Берка. ІС50 означає кількість представлених формулою І, оцінюють, застосоінгібітора, необхідну для зниження вихідної швивуючи артеріовенозний (AV) шунт у щурів. Голодкості контролю на 50%. Значення ІС50 розраходним самцям щурів Sprague-Dawley масою 200вують, використовуючи програмне забезпечення 240 г і приблизно 7-тижневого віку проводять GraFit, версія 5.0.12 (Erithacus Software Ltd, UK). анестезію за допомогою внутрішньочеревинної Використовуване для розрахунку значення ін'єкції уретану (1,25 г/кг) або хлоралгідрату. ЩуKm становить 125 нМ, його одержують, змінюючи рам, що знаходиться під анестезією, проводять концентрацію субстрату при сталій концентрації артеріовенозне (AV) шунтування, як описано раферменту. ніше [Journal of Thrombosis and Haemostasis Експериментальний приклад 2: вплив на зго(2004) 3, 514] з незначною модифікацією. У ліву ртання крові загальну сонну артерію і праву яремну вену ввоЕфект оксазолідинонових похідних за даним дять канюлю з двома трубками довжиною 200 винаходом з циклічною амідоксимною або циклімм, заповненими фізіологічним розчином (РЕ-50, чною амідразоновою групою, представлених фоBecton Dickinson, USA). Поліетиленові трубки рмулою І, на згортання крові досліджують, визназ'єднують за допомогою силіконової трубки довчаючи протромбіновий час (РТ). жиною 8 мм (L/S13, MasterFlex, USA) із силіко1) Визначення РТ новою трубкою довжиною 50 мм (L/S16, a) Спосіб із застосуванням коагулометру: MasterFlex, USA), що містить бавовняну нитку протромбіновий час (РТ) визначають за допомодовжиною 75 мм. Сполуку або носій дають перогою Thrombotimer 4 - канального коагулометру рально за 60 хв. до відкриття шунта на 15 хв. (Behnk Elektronik, Germany). У дослідженнях виПотім бавовняну нитку видаляють і зважують. користовують цитратну плазму людини і щура. Значення ED50 розраховують, застосовуючи ліДля визначення РТ 100 мкл свіжорозмороженої нійний регресійний аналіз і використовуючи Excel плазми змішують з 3 мкл серійного розведення 2003 (Microsoft). досліджуваної сполуки або ДМСО. Після 5 хви3) Визначення часу кровотечі (ВТ), викорислинної інкубації при 37°С додають 200 мкл STAтовуючи модель кровотечі з хвоста щурів. Neoplastine (Diagnostica Stago, France) для початГолодним самцям щурів Sprague-Dawley маку утворення згустків. Антикоагулянтну активність сою 200-240 г і приблизно 7-тижневого віку просполук визначають як концентрацію, необхідну водять анестезію за допомогою внутрішньочередля збільшення в два рази часу згортання плазвинної ін'єкції пентобарбіталу натрію (60 мг/кг). ми [2РТ (мкм)]. Людську плазму одержують від Інгібітори FXa або носій дають перорально за 60 Daejeon Red Cross Blood Center. Щурячу кров хвилин до відсікання 2 мм кінчиків хвостів анесвідбирають із сонної артерії або верхньої порожтезованих щурів і вертикального занурення їх у ньої вени під анестезією. Кров збирають у пласфізіологічний розчин при 37°С. Визначають, що тикові пробірки, що містять 1/10 обсягу 3,8% цитчас до припинення безупинного плину крові скларату натрію. Плазму одержують негайним дає >30 сек. при максимальному часі спостере
ДивитисяДодаткова інформація
Назва патенту англійськоюFxa inhibitors with cyclic amidoxime or cyclic amidrazone group as subunit p4, processes for the preparation thereof, and pharmaceutical compositions and derivatives thereof
Автори англійськоюSong, Ho Young, Cho, Young Lah, Lee, Dae Yon, Park, Hee Sock, Baek, Sung Yoon, Chae, Sang Eun, Jo, Sang Hui, Kim, Yeon Ok, Lee, Hyang Sook, Park, Ju Hyun, Park, Tae Kyo, Woo, Sung Ho, Kim, Yong Zu
Назва патенту російськоюИнгибиторы fxа с циклическим амидоксимом или циклическим амидразоном как р4 субъединица, способы их получения и их фармацевтические композиции и производные
Автори російськоюСонг Хо Йоунг, Чо Йоунг Лаг, Ли Дае Йон, Парк Хее Сок, Баек Сунг Йоон, Чае Санг Еун, Дзо Санг Хой, Ким Йеон Ок, Ли Хианг Соок, Парк Дзу Хиун, Парк Тае Кио, Воо Сунг Хо, Ким Йонг Зу
МПК / Мітки
МПК: C07D 413/12
Мітки: амідоксимом, одержання, циклічним, похідні, амідразоном, інгібітори, субодиниця, композиції, способи, фармацевтичні
Код посилання
<a href="https://ua.patents.su/33-97598-ingibitori-fxa-z-ciklichnim-amidoksimom-abo-ciklichnim-amidrazonom-yak-r4-subodinicya-sposobi-kh-oderzhannya-i-kh-farmacevtichni-kompozici-i-pokhidni.html" target="_blank" rel="follow" title="База патентів України">Інгібітори fxа з циклічним амідоксимом або циклічним амідразоном як р4 субодиниця, способи їх одержання і їх фармацевтичні композиції і похідні</a>
Біциклічна сполука, способи її одержання, фармацевтичні композиції, що її містять,способи лікування та попередження різних захворювань, проміжні сполуки та способи їх одержання

Номер патенту: 72883
Опубліковано: 16.05.2005
Автори: Рамануджам Раджагопалан, Калчар Шіварамаййа, Лохрей Брадж Бхушан, Чакрабарті Ранджан, Баджі Ашок Чаннавеераппа, Лохрей Відіа Бхушан
МПК: A61K 9/10, A61K 9/14, A61P 9/10, A61P 3/10, A61P 17/06, A61P 13/12, C07D 413/06, A61K 9/08, A61K 9/48, A61P 3/06, A61P 25/28, C07D 265/36, A61P 3/04, A61K 31/538, A61K 9/20, C07D 279/00, A61P 19/10, A61K 31/5415
Мітки: проміжні, різних, лікування, захворювань, способи, містять,способи, композиції, фармацевтичні, попередження, біциклічна, сполуки, одержання, сполука
Формула / Реферат:
1. Сполука формули (І), (I)де групи R1, R2, R3, R4 і групи R5 і R6, коли вони приєднані до атома вуглецю, можуть бути однаковими або різними і означають водень, галоген, гідрокси або необов'язково заміщену групу, вибрану з алкілу, алкокси, фенілу, карбонової кислоти або сульфонової кислоти ; один або обидва замісники R5 і R6 можуть також означати оксогрупу,...
L-арабінодисахариди антрациклінів, способи їх одержання та фармацевтичні композиції, що містять ці речовини

Номер патенту: 69446
Опубліковано: 15.09.2004
Автори: Маджі Карло Альберто, Біджоні Маріо, Аніматі Фабіо, Береттоні Марко, Чіполлоне Амалія
МПК: A61P 35/00, C07H 15/252, A61K 31/704
Мітки: антрациклінів, фармацевтичні, способи, композиції, містять, одержання, l-арабінодисахариди, речовини
Формула / Реферат:
1. L-арабінодисахариди антрациклінів загальної формули (І) (І)та їх фармацевтично прийнятні солі,деR - ОН;R1 - Η або ОН;R2 - Η або ОН, або NH2.2. Сполуки формули (І) за п. 1, які входять у групу, яка складається з таких сполук:і) 7-O-[2,6-дидеокси-4-O-(2,3,6-тридеоксі-3-аміно--L-арабіногексопіранозил)--L-ліксогексопіранозил]-4-деметоксидоксорубіцинонгідрохлорид (R=R1=OH,...
Фармацевтичні композиції, що містять похідні 3-аміноазетидину, похідні і спосіб їхнього одержання

Номер патенту: 72319
Опубліковано: 15.02.2005
Автори: Боушард Херве, Філош Бруно, Хіттінгер Огюстін, Міерс Мішель, Ашард Даніель, Букерель Жан, Грізоні Серж
МПК: A61P 25/16, A61P 25/02, A61P 27/02, A61P 9/02, A61P 25/00, A61P 25/14, A61P 29/00, A61P 25/24, A61P 25/28, A61K 47/00, A61P 5/00, A61K 31/397, A61K 31/4709, A61P 1/08, A61P 11/06, A61P 1/14, A61K 31/4025, A61P 9/00, C07D 401/12, A61K 31/4427, C07D 403/12, C07D 403/08, C07D 205/00, C07D 401/06, A61P 25/22, A61P 35/00, A61P 37/00, C07D 409/12, A61K 31/506, A61P 23/00, A61P 1/00, A61P 3/04, A61P 25/18, A61P 25/08, A61P 11/00, A61K 31/4178, A61P 25/20, A61P 9/08
Мітки: одержання, містять, їхнього, фармацевтичні, спосіб, композиції, похідні, 3-аміноазетидину
Формула / Реферат:
1. Фармацевтична композиція, що містить як активний інгредієнт сполуку формули: , (І)у якійR1 означає радикал –NНСОR4 або -N(R5)-Y-R6,Υ означає CO або SO2,R2 і R3, однакові або різні, означають або ароматичний радикал, вибраний з фенілу, нафтилу і інденілу, що можуть бути незаміщеними або заміщеними одним або декількома замісниками:...
Похідні заміщених бензоконденсованим гетероариламідом тієнопіридинів, корисні як терапевтичні агенти, фармацевтичні композиції, що їх містять, і способи їх застосування

Номер патенту: 77303
Опубліковано: 15.11.2006
Автори: Палмер Сінтія Луїс, Хе Мінгїнг, Лоу Джіхонг, Кріппс Стефан Джеймс, Жоу Ру, Коллінз Майкл Раймонд, Ромінс Уільям Генрі III, Каніа Роберт Стівен, Діл Джудіт Гейл
МПК: A61K 31/444, A61P 9/00, A61K 31/4365, C07D 495/04, A61K 31/5377, A61P 15/08, A61P 9/10, A61P 35/00, A61P 43/00, A61P 1/18, A61P 27/02, A61P 17/06, A61K 31/506, A61P 17/00, A61P 13/08, A61P 17/12, A61P 29/00, A61P 11/00, A61P 25/00, A61P 3/10
Мітки: корисні, заміщених, фармацевтичні, застосування, способи, терапевтичні, містять, тієнопіридинів, гетероариламідом, агенти, бензоконденсованим, похідні, композиції
Формула / Реферат:
1. Сполука формули І: , Iв якій:Y означає -NH-, -O-, -S- або -СН2-;Z означає -O-, -S- або -N-;R14 означає С1-С6 алкіл, С1-С6 алкіламіно, С1-С6 алкілгідрокси, C3-C10 циклоалкільну, C3-C10 циклоалкіламіно, C1-C6 алкіл C3-C10 циклоалкільну або метилуреїдогрупу;R15 та R17 незалежно означають Н, галоген або С1-С6 алкільну групу,...
Похідні пурину, спосіб їх одержання і фармацевтичні композиції, що містять їх

Номер патенту: 68441
Опубліковано: 16.08.2004
Автор: Асслен Жан-Люк
МПК: A61K 31/52, C07D 473/16, A61K 31/522, C07D 473/26
Мітки: композиції, одержання, похідні, фармацевтичні, спосіб, пурину, містять
Формула / Реферат:
1. Сполука формули І:,в якійΖ означає двовалентний радикал -СН2-, -SO2-, -CO-, -COO-, -CONH- або -(CH2)2-NR3-;n означає ціле число 0 або 1;R1 вибирають з атома водню, радикалів арил, -СН2-арил, -SО2-арил, -СО-арил, гетероциклічний радикал, -СН2-гетероциклічний радикал, алкіл і -SО2-алкіл;R2 означає можливо заміщений...
Попередній патент: Спосіб виготовлення металопластикового або біпластикового виробу
Наступний патент: Спосіб і пристрій для регенерації етиленгліколю при одержанні поліетилентерефталату
Випадковий патент: Пристрій струмового захисту-2