Сполука 2-алкіліндазолу для лікування певних розладів, пов’язаних з цнс, спосіб її одержання (варіанти) та фармацевтична композиція на її основі
Номер патенту: 98311
Опубліковано: 10.05.2012
Автори: Омбрато Розелла, Фурлотті Гвідо, Каццолла Нікола, Поленцані Лоренцо, Мауджері Катеріна, Алісі Марія Алессандра





Формула / Реферат
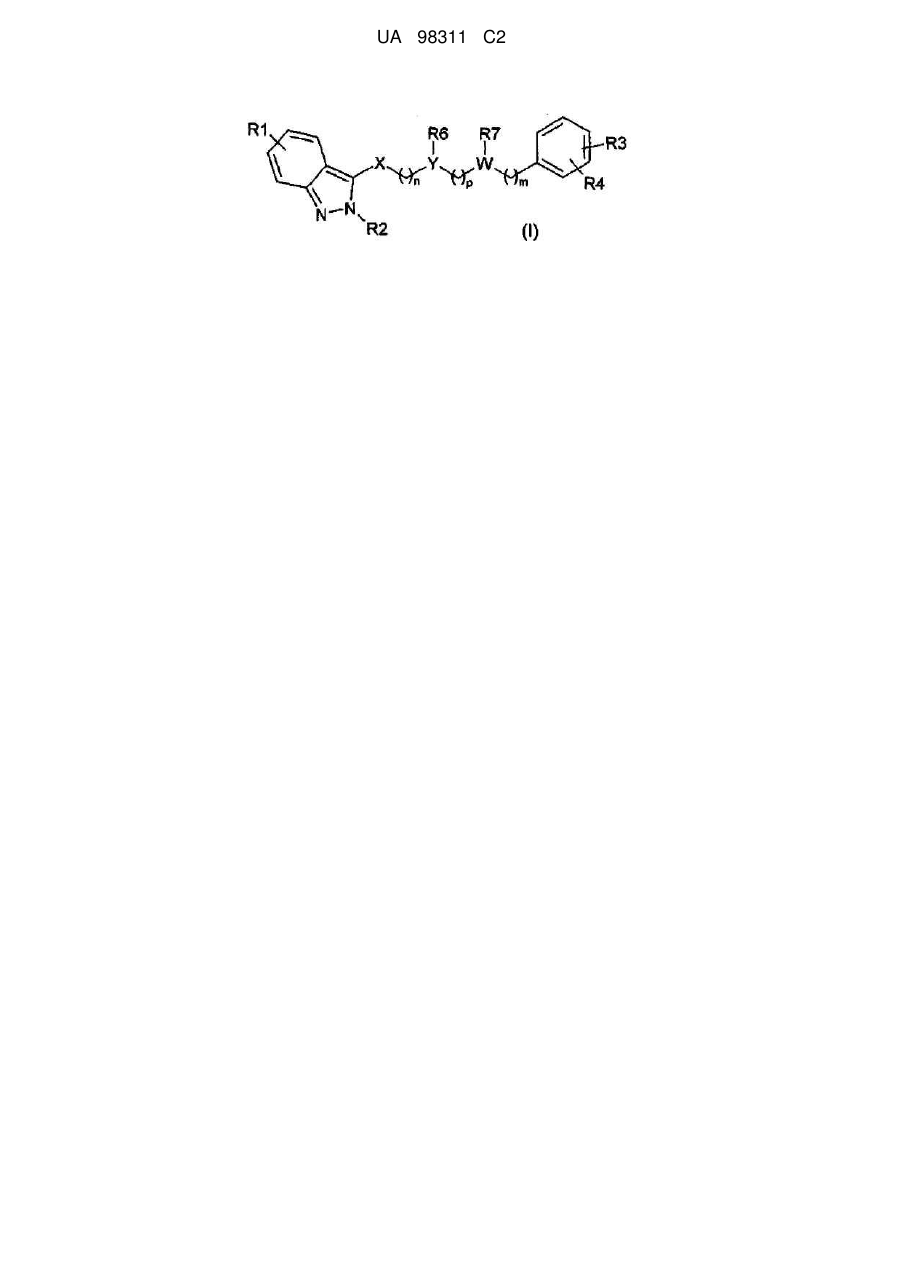
1. Сполука 2-алкіліндазолу загальної формули (I):
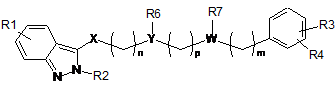 , (I)
, (I)
у якій
X являє собою -C(O)N(R5)-;
Y являє собою CH або N;
W являє собою CH або N;
за умови, що принаймні один з Y та W являє собою атом азоту;
n являє собою ціле число, вибране з 1, 2 та 3;
m являє собою ціле число, вибране з 0, 2 та 3;
p являє собою ціле число, вибране з 1 та 2;
R1 являє собою H, метил або метоксигрупу;
R2 являє собою метил, етил, ізопропіл або метоксіетил;
R3 являє собою H, 4-гідроксигрупу, 4-метоксигрупу, 3-хлор або 4-фтор;
R4 являє собою H або 2-фтор;
R5 являє собою H, метил, метоксигрупу або, разом з R6, утворює 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин;
R6, разом з R5 або R7, утворює 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин;
R7 являє собою H, етил, фенілетил, 4-фторфенілетил, 2,4-дифторфенілетил або, разом з R6, утворює 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин,
або її солі приєднання з фармацевтично прийнятною органічною або неорганічною кислотою.
2. Сполука за п. 1, яка відрізняється тим, що X являє собою -C(O)N(R5)-, Y являє собою CH; W являє собою N; n являє собою ціле число, вибране з 1 та 2; m являє собою 2; p являє собою ціле число, вибране з 1 та 2; R1 являє собою H або метоксигрупу; R2 являє собою метил або метоксіетил; R3 являє собою H, 4-гідроксигрупу, 4-метоксигрупу або 4-фтор; R4 являє собою H або 2-фтор; R5 являє собою H або, разом з R6, утворює 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R6, разом з R5 або R7, утворює 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R7 являє собою H, етил або, разом з R6, утворює 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин.
3. Спосіб одержання (i) сполуки 2-алкіліндазолу формули (I), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, p та m приймають значення, представлені вище у п. 1, або (ii) її солі з фармацевтично прийнятною органічною або неорганічною кислотою, де у способі:
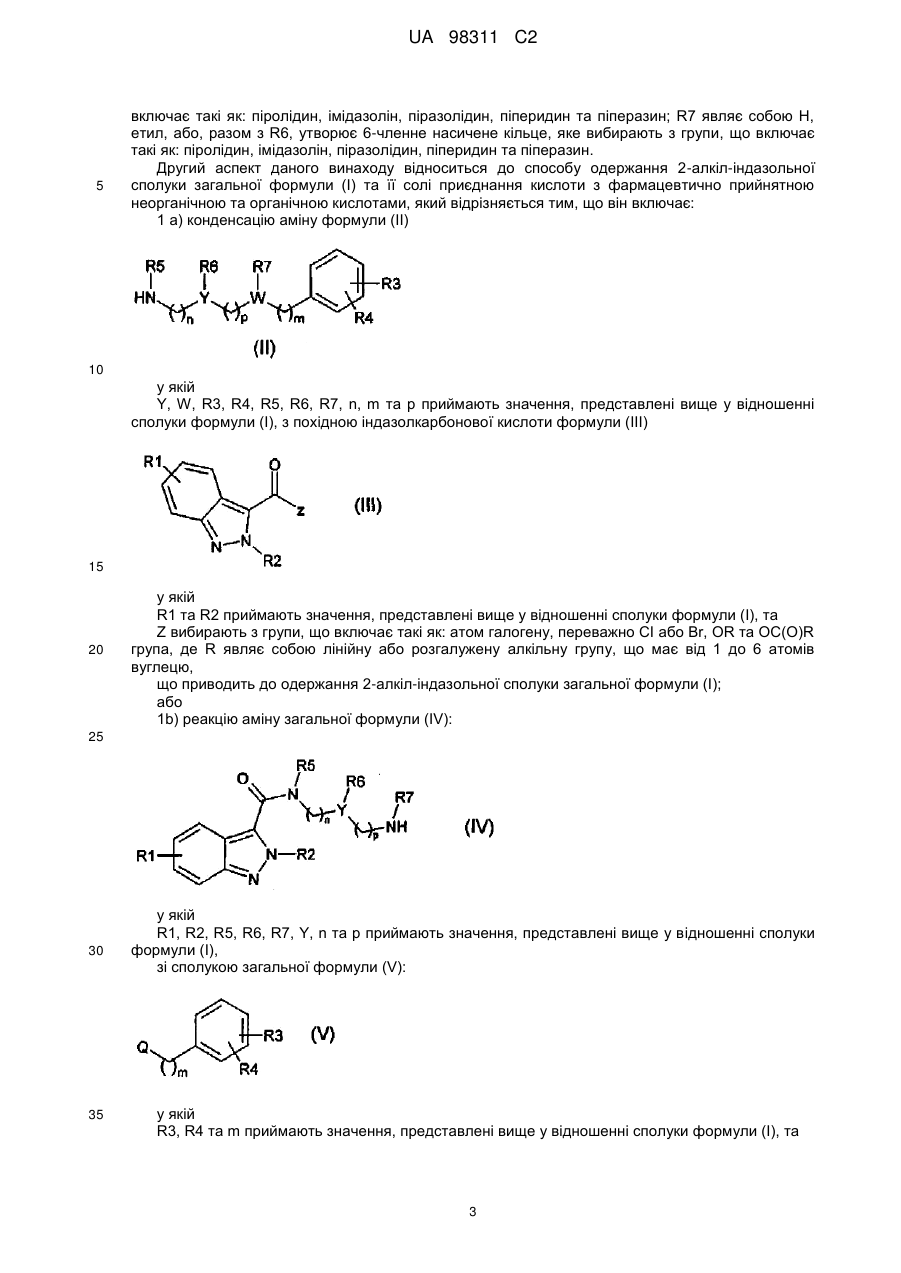
1a) проводять реакцію конденсації аміну формули (II)
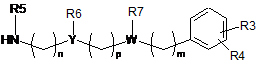 , (II)
, (II)
у якій
Y, W, R3, R4, R5, R6, R7, n, m та p приймають значення, представлені вище у п. 1,
з похідною індазолкарбонової кислоти формули (III)
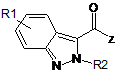 , (III)
, (III)
у якій
R1 та R2 приймають значення, представлені вище у п. 1, та
Z вибирають з групи, що включає такі як: атом галогену, переважно Cl або Br, OR та OC(O)R групу, де R являє собою лінійну або розгалужену алкільну групу, що має від 1 до 6 атомів вуглецю,
що приводить до одержання сполуки 2-алкіліндазолу загальної формули (I), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, p та m приймають значення, представлені вище у п. 1, та
2) необов’язково одержання солі приєднання кислоти сполуки 2-алкіліндазолу загальної формули (I), отриманої таким чином, з фармацевтично прийнятною органічною або неорганічною кислотою.
4. Спосіб одержання (i) сполуки 2-алкіліндазолу формули (I), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, p та m приймають значення, представлені вище у п. 1, або (ii) її солі з фармацевтично прийнятною органічною або неорганічною кислотою, де у способі:
1b) проводять реакцію аміну загальної формули (IV):
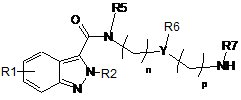 , (IV)
, (IV)
у якій
R1, R2, R5, R6, R7, Y, n та p приймають значення, представлені вище у п. 1,
зі сполукою загальної формули (V):
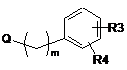 , (V)
, (V)
у якій
R3, R4 та m приймають значення, представлені вище у п. 1, та
Q являє собою відхідну групу, вибрану з групи, що включає такі як: атом галогену, переважно Cl або Br, мезилатну групу (MeSO3-) та тозилатну групу (п-MePhSO3-),
що приводить до одержання сполуки 2-алкіліндазолу загальної формули (I), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, p та m приймають значення, представлені вище у п. 1, та
2) необов’язково одержання солі приєднання кислоти сполуки 2-алкіліндазолу загальної формули (I), отриманої таким чином, з фармацевтично прийнятною органічною або неорганічною кислотою.
5. Спосіб одержання (i) сполуки 2-алкіліндазолу формули (I), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, p та m приймають значення, представлені вище у п. 1, або (ii) її солі з фармацевтично прийнятною органічною або неорганічною кислотою, де у способі:
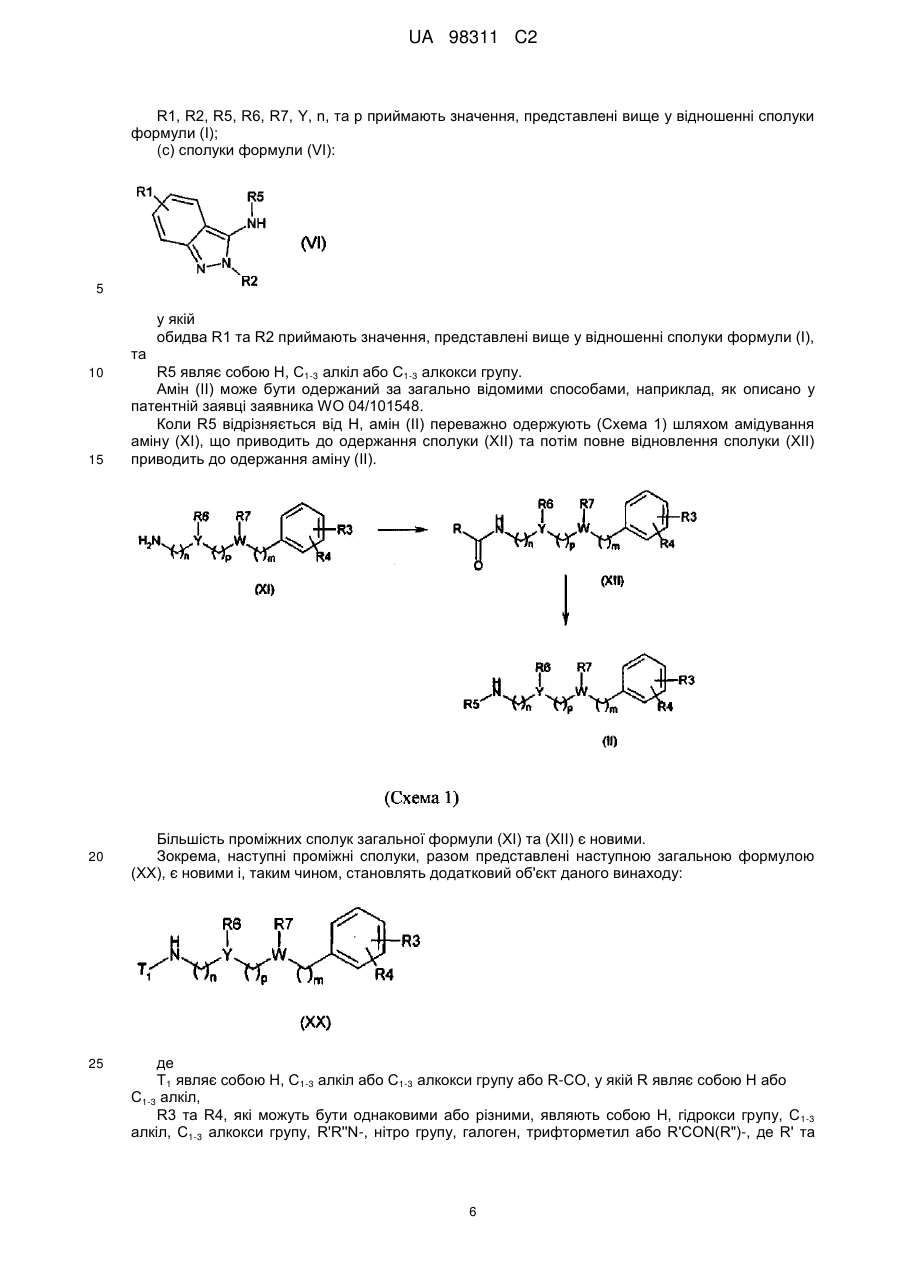
1c) проводять реакцію конденсації аміну загальної формули (VI):
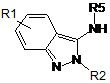 , (VI)
, (VI)
у якій
R1, R2 та R5 приймають значення, представлені вище у п. 1,
з похідною карбонової кислоти загальної формули (VII):
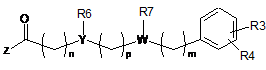 , (VII)
, (VII)
у якій
R3, R4, R6, R7, Y, W, n, m та p приймають значення, представлені вище у п. 1, та
Z приймає значення, представлені вище у відношенні сполуки формули (III),
що приводить до одержання сполуки 2-алкіліндазолу загальної формули (I), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, p та m приймають значення, представлені вище у п. 1; та
2) необов’язково одержання солі приєднання кислоти сполуки 2-алкіліндазолу загальної формули (I), отриманої таким чином, з фармацевтично прийнятною органічною або неорганічною кислотою.
6. Проміжна сполука формули (II):
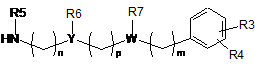 , (II)
, (II)
у якій
R3, R4 та n приймають значення, представлені в п. 1,
R5 являє собою C1-3алкіл,
R6 та R7 разом утворюють 5- або 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин,
Y являє собою CH,
W являє собою N,
p являє собою ціле число, вибране з 0 та 1,
m являє собою 2.
7. Проміжна сполука формули (IV):
, (IV)
у якій
R1, R2, R5, R6, R7, Y, n та p приймають значення, представлені вище у п. 1.
8. Проміжна сполука формули (VI):
, (VI)
у якій
R1 та R2 приймають значення, представлені в п. 1, та
R5 являє собою H, C1-3алкіл або C1-3алкоксигрупу.
9. Проміжна сполука формули (XX):
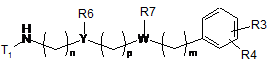 , (XX)
, (XX)
у якій
T1 являє собою H, C1-3алкіл або C1-3алкоксигрупу, або R-CO, у якій R являє собою H або C1-3алкіл;
R3, R4, n та p приймають значення, представлені в п. 1,
R6 та R7 разом утворюють 5- або 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин,
Y являє собою CH,
W являє собою N, та
m являє собою 2.
10. Проміжна сполука загальної формули (XXX):
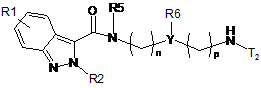 , (XXX)
, (XXX)
у якій
R1, R2, n та p приймають значення, представлені в п. 1,
T2 являє собою H, захисну групу (P), вибрану з групи, що включає такі як: 9-флуоренілметилкарбамат, трет-бутилкарбамат, алілкарбамат, N-бензил та N-бензиліден, або групу R-CO, у якій R являє собою C1-3алкіл,
R5 та R6 разом утворюють 5- або 6-членне насичене кільце, вибране з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин, та
Y являє собою CH.
11. Фармацевтична композиція, що містить ефективну дозу щонайменше однієї сполуки 2-алкіліндазолу формули (I), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, p та m приймають значення, зазначені у п. 1, або її солі з фармацевтично прийнятною органічною або неорганічною кислотою, та щонайменше один фармацевтично прийнятний наповнювач.
12. Фармацевтична композиція за п. 11, яка відрізняється тим, що містить таку кількість сполуки 2-алкіліндазолу формули (I), або її фармацевтично прийнятної солі приєднання кислоти, щоб забезпечити рівень введення від 0,0001 до 100 мг/кг/день.
13. Фармацевтична композиція за п. 12, яка відрізняється тим, що містить таку кількість сполуки 2-алкіліндазолу формули (I), або її фармацевтично прийнятної солі приєднання кислоти, щоб забезпечити рівень введення від 0,001 до 50 мг/кг/день.
14. Фармацевтична композиція за п. 13, яка відрізняється тим, що містить таку кількість сполуки 2-алкіліндазолу формули (I), або її фармацевтично прийнятної солі приєднання кислоти, щоб забезпечити рівень введення від 0,01 до 10 мг/кг/день.
15. Застосування фармацевтичної композиції за будь-яким з пп. 11-14 для лікування патологічного стану, який вибирають з групи, що включає такі як: розлади сну, шизофренія, неспокій, розлади гладких м’язів як шлунково-кишкової системи, так і серцево-судинної системи.
Текст
Реферат: Сполука 2-алкіліндазолу та її фармацевтично прийнятні солі приєднання кислоти, спосіб та проміжні сполуки для їх одержання, фармацевтична композиція, що містить їх, та застосування останньої. Сполука 2-алкіліндазолу має наступну загальну формулу (І), у якій R1, R2, R3, R4, R6, R7, X, Y, W, n, р та m приймають значення, представлені у описі. R6 X n N N R2 R7 Y R1 W p R3 m R4 UA 98311 C2 (12) UA 98311 C2 UA 98311 C2 5 10 15 20 25 30 35 Даний винахід відноситься до 2-алкіл-індазольної сполуки, до способу та проміжних сполук для її одержання, до фармацевтичної композиції, що містить її, та до застосування останньої. Зокрема, даний винахід відноситься до 2-алкіл-індазольної сполуки, що має селективну спорідненість до 5-НТ2 рецептору або до інших серотонінових рецепторів, таких як 5-НТ1, 5-НТ3, 5-НТ4, 5-HT5, 5-НТ6 та 5-НТ7 рецептори. У відношенні різних підтипів 5-НТ2 рецептору, сполуки даного винаходу мають селективну спорідненість до 5-НТ2А рецептору або до 5-НТ2B рецептору та в основному показують переважну спорідненість до 5-НТ2А рецептору у порівнянні з 5-НТ2С рецептором. Навіть більш особливо, даний винахід відноситься до сполуки, яка може бути використана у лікуванні деяких патологій, які залучають 5-НТ2A рецептор, наприклад, таких як деякі розлади центральної нервової системи, такі як розлади сну, шизофренія та неспокій, а також розлади гладких м'язів або шлунково-кишкової системи або серцево-судинної системи. Серотоніновий 2А рецептор (5-НТ2А) являє собою рецептор, сполучений із білком G, присутній у багатьох видах та широко розповсюджений у організмі людини. Спосіб передачі сигналу, що є результатом активації рецептору, повністю не зрозумілий. Відомо, що активація цього рецептору, через білок G, сполучений із ним, приводить до активації різних ферментів, таких як фосфоліпаза С (з наступним гідролізом фосфатидилінозитол дифосфату та виробленням інозитол трифосфату) або фосфоліпаза А2 (яка приводить до вивільнення 2+ арахідонової кислоти). Крім того, рецепторна відповідь приводить до потоку іонів кальцію (Са ) у клітину та до активації функціональних білків, таких як протеїнкіназа С. Даний рецептор присутній у центральній нервовій системі, у клітинах судинних та шлунково-кишкових гладких м'язів та у тромбоцитах. Наразі, була знайдена 2-алкіл-індазоламідна сполука, що має переважну спорідненість до 5НТ2А рецептору. Ця біологічна активність помітно відрізняється від активності відомих 2-алкіл-індазольних сполук, описаних як пестициди (арил або етер-арил 2-алкіл-індазоламіди, WO 94/05642), як інсектициди (піридинові сполуки 2-метил-індазоламіду, ЕР-А-0 726 266) або як антагоністи CRF1 рецептору (7-арил або 7-етер-арил-2-алкіл-індазоламід, US 2004/0 110 815). Ця біологічна активність також відрізняється від активності 1-алкіл-індазольних сполук із знеболюючою активністю (WO 04/101548). Більш того, сполуки даного винаходу мають структуру, яка особливо відрізняється від відомих сполук з переважною спорідненістю до 5-НТ2А рецептору та яка досягла клінічних фаз розробки, наприклад, EMD-281014 (7-{4-[2-(4-фтор-феніл)-етил]-піперазин-1-карбоніл}-1Н-індол3-карбонітрилу гідрохлорид) у фазі І для безсоння та MDL-100907 ((R)-(+)-(2,3-диметоксифеніл)-{1-[2-(4-фтор-феніл)-етил]-піперидин-4-іл}-метанол) у фазі II для безсоння. Перший аспект даного винаходу відноситься до 2-алкіл-індазольної сполуки загальної формули (І): 40 45 50 у якій X являє собою -C(O)N(R5)- або -N(R5)C(O)-; Y являє собою СН або N; W являє собою СН або N; за умови, що принаймні один з Y та W являє собою атом азоту; n являє собою ціле число, що вибирають з 0, 1, 2 та 3; m являє собою ціле число, що вибирають з 0, 1, 2 та 3; р являє собою ціле число, що вибирають з 0, 1 та 2; R1 являє собою Н, С1-3 алкіл, С1-3 алкокси групу, С1-3 алкіл-СО-, СООН, R'R''NCO-, R'R''NSO2, R'SO2-, галоген, R'R''N-або R'CON(R")-; R2 являє собою лінійний або розгалужений С 1-3 алкіл, С1-3 алкіл-С1-3 алкокси групу, арил-С1-3 алкіл, де арильна група може бути заміщена за допомогою 1 або 2 замісників, які вибирають з таких як: галоген, С1-3 алкіл та C1-3 алкокси група; 1 UA 98311 C2 5 10 15 20 25 30 35 40 45 50 55 60 R3 та R4, які можуть бути однаковими або різними, являють собою Н, гідрокси групу, С 1-3 алкіл, С1-3 алкокси групу, R'R''N-, нітро групу, галоген, трифторметил або R'CON(R")-; R5 являє собою Н, С1-3 алкіл, С1-3 алкокси групу або, разом з R6, утворює 5- або 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R6, разом з R5 або R7, утворює 5- або 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R7 являє собою Н, лінійний або розгалужений С1-6 алкіл, арил-С1-3 алкіл, де арильна група може бути заміщена за допомогою 1 або 2 замісників, які вибирають з таких як: галоген, С 1-3 алкіл, гідрокси група, С1-3 алкокси група, нітро група, трифторметил та R'R'N-, або R7, разом з R6, утворює 5- або 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R' та R", які можуть бути однаковими або різними, являють собою Н або С 1-3 алкіл; та її солей приєднання з фармацевтично прийнятною органічною та неорганічною кислотами. Типові приклади фармацевтично прийнятних кислот включають такі як: щавлева кислота, малеїнова кислота, метансульфонова кислота, паратолуолсульфонова кислота, бурштинова кислота, лимонна кислота, винна кислота, молочна кислота, соляна кислота, фосфорна кислота та сірчана кислота. Краще, коли X являє собою -C(O)N(R5)-, Y являє собою СН або N; W являє собою СН або N; за умови, що принаймні один з Y та W являє собою атом азоту; n являє собою ціле число, що вибирають зі, 2 та 3; m являє собою ціле число, що вибирають з 0, 2 та 3; р являє собою ціле число, що вибирають з 1 та 2; R1 являє собою Н, метил або метокси групу; R2 являє собою метил, етил, ізопропіл або метоксиетил; R3 являє собою Н, 4-гідрокси групу, 4-метокси групу, 3-хлор або 4-фтор; R4 являє собою Н або 2-фтор; R5 являє собою Н, метил, метокси групу або, разом з R6, утворює 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R6, разом з R5 або R7, утворює 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R7 являє собою Н, етил, фенілетил, 4-фторфенілетил, 2,4-дифторфенілетил або, разом з R6, утворює 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин. Краще, коли X являє собою -N(R5)C(O)-, Y являє собою СН; W являє собою N; n являє собою ціле число, що вибирають з 0 та 1; m являє собою 2; р являє собою 2; R1 являє собою Н, метил або метокси групу; R2 являє собою метил, етил, ізопропіл або метоксиетил; R3 являє собою Н, 4-гідрокси групу, 4-метокси групу, 3-хлор або 4-фтор; R4 являє собою Н або 2-фтор; R5 являє собою Н, метил, або метокси групу; R6, разом з R7, утворює 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R7, разом з R6, утворює 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин. Навіть ще краще, X являє собою -C(O)N(R5)-, Y являє собою СН; W являє собою N; n являє собою ціле число, що вибирають з 1 та 2; m являє собою 2; р являє собою ціле число, що вибирають з 1 та 2; R1 являє собою Н або метокси групу; R2 являє собою метил або метоксиетил; R3 являє собою Н, 4-гідрокси групу, 4-метокси групу або 4-фтор; R4 являє собою Н або 2-фтор; R5 являє собою Н або, разом з R6, утворює 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R6, разом з R5 або R7, утворює 6-членне насичене кільце, яке вибирають з групи, що 2 UA 98311 C2 5 включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин; R7 являє собою Н, етил, або, разом з R6, утворює 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин. Другий аспект даного винаходу відноситься до способу одержання 2-алкіл-індазольної сполуки загальної формули (І) та її солі приєднання кислоти з фармацевтично прийнятною неорганічною та органічною кислотами, який відрізняється тим, що він включає: 1 а) конденсацію аміну формули (II) 10 у якій Y, W, R3, R4, R5, R6, R7, n, m та р приймають значення, представлені вище у відношенні сполуки формули (І), з похідною індазолкарбонової кислоти формули (III) 15 20 у якій R1 та R2 приймають значення, представлені вище у відношенні сполуки формули (І), та Z вибирають з групи, що включає такі як: атом галогену, переважно СІ або Br, OR та OC(O)R група, де R являє собою лінійну або розгалужену алкільну групу, що має від 1 до 6 атомів вуглецю, що приводить до одержання 2-алкіл-індазольної сполуки загальної формули (І); або 1b) реакцію аміну загальної формули (IV): 25 30 35 у якій R1, R2, R5, R6, R7, Y, n та р приймають значення, представлені вище у відношенні сполуки формули (І), зі сполукою загальної формули (V): у якій R3, R4 та m приймають значення, представлені вище у відношенні сполуки формули (І), та 3 UA 98311 C2 5 10 15 20 25 30 35 40 45 Q являє собою відхідну групу, яку вибирають з групи, що включає такі як: атом галогену, переважно СІ або Вr, мезилатна група (МеSО3-) та тозилатна група (п-MePhSО3-), що приводить до одержання 2-алкіл-індазольної сполуки загальної формули (І); або 1с) конденсацію аміну загальної формули (VI): у якій R1, R2 та R5 приймають значення, представлені вище у відношенні сполуки формули (І), з похідною карбонової кислоти загальної формули (VII): у якій R3, R4, R6, R7, Y, W, n, m, та р приймають значення, представлені вище у відношенні сполуки формули (І), та Z приймає значення, представлені вище у відношенні сполуки формули (III), що приводить до одержання 2-алкіл-індазольної сполуки загальної формули (І); та 2) можливе утворення солі приєднання кислоти 2-алкіл-індазольної сполуки загальної формули (І), що таким чином одержують з фармацевтично прийнятною органічною або неорганічною кислотою. Стадія 1(а) може бути проведена за загально відомими технологіями. Наприклад, амін формули (II) вводять у реакцію з ацилгалогенідом формули (III), у якій Z, переважно, являє собою атом хлору, необов'язково у присутності прийнятної основи. Типові приклади прийнятних основ включають триетиламін та діізопропілетиламін. Переважно, зазначену реакцію проводять у присутності прийнятного розчинника при температурі від 0 до 140 °C, впродовж часу від 0,5 до 24 годин. Переважно, температура реакції коливається у інтервалі від 10 до 60 °C, тоді як час реакції становить від 0 до 12 годин. Як правило, використовуваний розчинник є апротонним, полярним або неполярним. Приклади прийнятних неполярних апротонних розчинників включають ароматичні вуглеводні, такі як толуол та ксилол. Приклади прийнятних полярних апротонних розчинників включають дихлорметан та N, N-диметилформамід. Переважно використовують неполярний апротонний розчинник. Сполука, отримана в стадії 1(а), також може бути очищена за загально відомими технологіями, такими як флеш хроматографія та кристалізація. Стадія 1(b) також може бути проведена за загально відомими технологіями. Наприклад, амін формули (IV) вводять у реакцію зі сполукою формули (V), у якій Q, переважно, являє собою атом брому або метансульфонільну групу, необов'язково у присутності прийнятної основи. Типові приклади прийнятних основ включають карбонат калію та карбонат натрію. Переважно, зазначену реакцію проводять у присутності прийнятного розчинника при температурі від кімнатної температури до 160 °C та впродовж часу від 1 до 48 годин. Переважно, температура реакції коливається у інтервалі від кімнатної температури до 100 °C, тоді як час реакції становить від 6 до 24 годин. Як правило, використовуваний розчинник є полярним, протонним або апротонним. Приклади прийнятних полярних протонних розчинників включають спирти, такі як етанол, в той час як приклад прийнятного полярного апротонного розчинника являє собою ацетон. 4 UA 98311 C2 5 10 15 20 25 Сполука, отримана у стадії 1(b), також може бути очищена за загально відомими технологіями, такими як флеш хроматографія та кристалізація. Стадія 1(с) також може бути проведена за загально відомими технологіями. Наприклад, амін формули (VI) вводять у реакцію з ацил-галогенідом формули (VII), у якій Z, переважно, являє собою атом хлору, необов'язково у присутності прийнятної основи. Типові приклади прийнятних основ включають триетиламін та діізопропілетиламін. Переважно, зазначену реакцію проводять у присутності прийнятного розчинника при температурі від 0 до 140 °C, впродовж часу від 0,5 до 24 годин. Переважно, температура реакції коливається у інтервалі від 10 до 60 °C, тоді як час реакції становить від 0 до 12 годин. Як правило, використовуваний розчинник є апротонним, полярним або неполярним. Приклади прийнятних неполярних апротонних розчинників включають ароматичні вуглеводні, такі як толуол та ксилол. Приклади прийнятних полярних апротонних розчинників включають дихлорметан та N, N-диметилформамід. Переважно використовують неполярний апротонний розчинник. Сполука, отримана у стадії 1(с), також може бути очищена за загально відомими технологіями, такими як флеш хроматографія та кристалізація. Утворення солі приєднання кислоти 2-алкіл-індазольної сполуки загальної формули (І) з фармацевтично прийнятною органічною або неорганічною кислотою також може бути проведене за загально відомими технологіями. Наприклад, воно може бути проведене шляхом розчинення сполуки формули (І) у прийнятному розчиннику та обробки розчину, отриманого таким чином, з органічним або водним розчином кислоти, що розглядається. Типові приклади прийнятних розчинників включають етанол, ізопропанол, етилацетат та діетиловий ефір. Соль, що утворюється, далі може бути відділена за загально відомими технологіями та, якщо прийнятно, очищена шляхом кристалізації. Деякі проміжні сполуки формули (II), (IV), та (VI) є новими, і, тому, вони складають додатковий аспект даного винаходу. Зокрема, наступні сполуки є новими: (а) сполуки формули (II): 30 35 40 у якій R3 та R4, які можуть бути однаковими або різними, являють собою Н, гідрокси групу, С1-3 алкіл, С1-3 алкокси групу, R'R''N-, нітро групу, галоген, трифторметил або R'CON(R")-, однак, за умови, що R3 та R4 обидва не являють собою атом Н, R5 являє собою С1-3 алкіл, R6 та R7 разом утворюють 5- або 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин, Y являє собою СН, W являє собою N, n являє собою ціле число, що вибирають зі, 2 та 3, р являє собою ціле число, що вибирають з 0 та 1, m являє собою 2, R' та R", які можуть бути однаковими або різними, являють собою Н або С 1-3 алкіл; (b) сполуки формули (IV): 45 у якій 5 UA 98311 C2 R1, R2, R5, R6, R7, Y, n, та р приймають значення, представлені вище у відношенні сполуки формули (І); (с) сполуки формули (VI): 5 у якій обидва R1 та R2 приймають значення, представлені вище у відношенні сполуки формули (І), та 10 15 20 25 R5 являє собою Н, С1-3 алкіл або С1-3 алкокси групу. Амін (II) може бути одержаний за загально відомими способами, наприклад, як описано у патентній заявці заявника WO 04/101548. Коли R5 відрізняється від Н, амін (II) переважно одержують (Схема 1) шляхом амідування аміну (XI), що приводить до одержання сполуки (XII) та потім повне відновлення сполуки (XII) приводить до одержання аміну (II). Більшість проміжних сполук загальної формули (XI) та (XII) є новими. Зокрема, наступні проміжні сполуки, разом представлені наступною загальною формулою (XX), є новими і, таким чином, становлять додатковий об'єкт даного винаходу: де Т1 являє собою Н, С1-3 алкіл або С1-3 алкокси групу або R-CO, у якій R являє собою Н або С1-3 алкіл, R3 та R4, які можуть бути однаковими або різними, являють собою Н, гідрокси групу, С 1-3 алкіл, С1-3 алкокси групу, R'R''N-, нітро групу, галоген, трифторметил або R'CON(R")-, де R' та 6 UA 98311 C2 5 10 15 20 25 R", які можуть бути однаковими або різними, являють собою Н або С 1-3 алкіл; однак, за умови, що R3 та R4 обидва не являють собою атом Н, R6 та R7, разом утворюють 5- або 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин, Y являє собою СН, W являє собою N, n являє собою ціле число, що вибирають зі, 2 та 3, р являє собою ціле число, що вибирають з 0, 1 та 2, та m являє собою 2. Індазолкарбонова кислота (III) може бути одержана за загально відомими способами. Наприклад шляхом алкілування складного ефіру індазол-3-карбонової кислоти (VIII) з алкілгалогенідом (IX) відповідно до Chem. Pharm. Bull. (1995) 43(11), 1912-1930, потім відділення 2алкільованої сполуки (X) від реакційної суміші шляхом хроматографії, потім піддавання 2алкільованої сполуки (X) основному гідролізу та, нарешті, перетворення зазначеної сполуки, отриманої таким чином, у бажану карбоксильну сполуку (III), як показано у наступній Схемі 2: Сполука (IV) може бути одержана, наприклад, з аміну (ХІІІ), у якому первинну амінну групу захищають за допомогою захисної групи Р (Схема 3), як описано, наприклад, Green T.W. та Wuts P.G.M., "Protective groups in organic synthesis." John Wiley & Sons Publ. (1991). Потім, сполуку (XIV), отриману таким чином, вводять у реакцію з сполукою (III). Далі, з аміду (XV), що утворюється, знімають захист відповідно до технологій, описаних Green T.W. та Wuts P.G.M., "Protective groups in organic synthesis." John Wiley & Sons Publ. (1991), утворюючи первинний амін (XVI), який ацилюють, що приводить до одержання аміду (XVII), потім відновлюють, як у процедурі, описаній у Схемі 1, що приводить до одержання сполуки (IV). 7 UA 98311 C2 5 10 15 20 Більшість проміжних сполук загальної формули (XV), (XVI) та (XVII) є новими. Зокрема, наступні проміжні сполуки, разом представлені наступною загальною формулою (XXX), є новими і, таким чином, становлять додатковий об'єкт даного винаходу: де R1 та R2 приймають значення, представлені вище у відношенні сполуки формули (І), Т2 являє собою Н, захисну групу (Р), яку вибирають з групи, що включає такі як: 9-флуореніл метилкарбамат, трет-бутилкарбамат, алілкарбамат, н-бензил та н-бензиліден, або група R-CO, у якій R являє собою С1-3 алкіл, R5 та R6, разом утворюють 5- або 6-членне насичене кільце, яке вибирають з групи, що включає такі як: піролідин, імідазолін, піразолідин, піперидин та піперазин, Y являє собою СН, n являє собою ціле число, що вибирають зі, 2 та 3, р являє собою ціле число, що вибирають з 0, 1 та 2, та m являє собою 2. Однак, відносно процедури (1с), амін (VI) може бути одержаний відповідно до загально відомих способів, наприклад, тих, що описані Fusco R., "The chemistry of heterocyclic compounds, Pyrazoles, Pyrazolines, Індазолез and condensed rings." (1967) Publ. Wiley N.Y., або Katritsky А. та Rees C.W., "Comprehensive heterocyclic chemistry." том 5 (1984) Publ. Pergamon Press. Сполука (VII) також може бути одержана за загально відомими способами, наприклад, тими, що описані у патентній заявці WO 04/101548. 8 UA 98311 C2 5 10 15 20 25 30 35 40 45 50 Інший аспект даного винаходу відноситься до фармацевтичної композиції, що включає ефективну дозу щонайменше однієї 2-алкіл-індазольної сполуки формули (І) або її солі з фармацевтично прийнятною органічною або неорганічною кислотою та щонайменше один фармацевтично прийнятний наповнювач. Переважно, фармацевтичну композицію даного винаходу одержують у прийнятних лікарських формах, що включають ефективну дозу щонайменше однієї 2-алкіл-індазольної сполуки формули (І) або її солі з фармацевтично прийнятною органічною або неорганічною кислотою та щонайменше один фармацевтично прийнятний наповнювач. Типові приклади патологічних станів, при яких можна б було мати користь з лікування за допомогою фармацевтичної композиції відповідно до даного винаходу, являють собою деякі розлади центральної нервової системи, такі як розлади сну, шизофренія та неспокій, а також розлади гладких м'язів як шлунково-кишкової системи, так і серцево-судинної системи. Тому, додатковий аспект даного винаходу відноситься до застосування описаної вище фармацевтичної композиції для лікування патологічного стану, який вибирають з групи, що включає такі як: розлади сну, шизофренія, неспокій, розлади гладких м'язів як шлунковокишкової системи, так і серцево-судинної системи. Приклади прийнятних лікарських форм включають таблетки, капсули, покриті таблетки, гранули, розчини та сиропи для перорального введення; лікарські пластири для трансдермального застосування; супозиторії для ректального введення та ін'єкційні стерильні розчини. Інші прийнятні лікарські форми являють собою форми з безперервним вивільненням та форми на основі ліпосом для перорального, ін'єкційного або трансдермального введення. Лікарські форми також можуть включати інші традиційні інгредієнти, такі як: консерванти, стабілізатори, поверхнево-активні речовини, буфери, солі для регулювання осмотичного тиску, емульгатори, підсолоджувачі, барвники, ароматизатори та подібні. Якщо є необхідність у спеціальних терапевтичних вимогах, фармацевтична композиція даного винаходу може містити інші фармакологічно активні інгредієнти, одночасне введення яких було б кращим. Кількість 2-алкіл-індазольної сполуки формули (І) або її фармацевтично прийнятної солі приєднання кислоти у фармацевтичній композиції даного винаходу може змінюватися у широких межах в залежності від відомих факторів, наприклад, таких як тип патології, серйозність хвороби, маса тіла пацієнта, лікарська форма, вибраний шлях введення, кількість введень на день та ефективність вибраної 2-алкіл-індазольної сполуки формули (І). Однак, оптимальна кількість може бути визначена спеціалістом, кваліфікованим у даній галузі техніки, легко та у плановому порядку. Як правило, кількість 2-алкіл-індазольної сполуки формули (І) або її фармацевтично прийнятної солі приєднання кислоти у фармацевтичній композиції даного винаходу буде такою, щоб забезпечити рівень введення від 0,0001 до 100 мг/кг/день. Переважно, рівень введення становить від 0,001 до 50 мг/кг/день, навіть більш переважно від 0,01 до 10 мг/кг/день. Лікарські форми фармацевтичної композиції даного винаходу можуть бути одержані за технологіями, які відомі хіміку-фармацевту, та включають змішування, гранулювання, пресування, розчинення, стерилізацію та подібне. Фармакологічні властивості сполук формули (І) відповідно до даного винаходу оцінювали за способами, описаними нижче, у розділах Тестування А, В та С. Коротко, спорідненість до щурячого 5-НТ2А рецептора демонструють стандартною методологією у: Leysen J.E., Niemegeers C.J., Van Nueten J.M., Laduron P.M. (1982) "[3Н]Кетансерін, a selective 3H-ligand for serotonin2 рецептор binding sites. Binding properties, brain distribution, and functional role." Molecular Pharmacology 21: 301-314. (Тестування А). Значення спорідненості деяких сполук формули (І) відповідно до даного винаходу до 5-НТ2А рецептору представлені у Таблиці 1, у якій спорідненість є більшою для вищого значення рKі. 9 UA 98311 C2 Таблиця 1 Сполука (№) 8 1 3 4 7 10 9 2 5 5 10 15 20 5-НТ2А(рKi) 7,80 7,49 7,32 7,32 7,04 6,58 6,20 6,09 5,80 Селективна спорідненість сполук формули (І) відповідно до даного винаходу до інших рецепторів або систем для транспортування та включення серотоніну також визначають, використовуючи стандартний метод (Тестування В). Ці тестування, що вимірюють відсоток інгібування, що викликають за допомогою сполук формули (І) відповідно до даного винаходу при концентрації 1 мкМ для рецепторів: 5-НТ1A, 5НТ1B, 5-НТ1D, 5-НТ2b, 5-НТ3, 5-НТ4, 5-НТ4(b), 5-НТ5(А), 5-НТ6 та 5-НТ7. Крім того, вимірюють відсоток інгібування, що викликають за допомогою сполук формули (І) відповідно до даного винаходу при 1 мкМ на систему переносу серотоніну та при 10 мкМ на клітинну систему для включення серотоніну. У всіх випадках, сполуки формули (І) відповідно до даного винаходу викликають відсоток інгібування, при концентрації, що використовують, менше ніж 50 %. Переважну спорідненість сполук формули (І) відповідно до даного винаходу до 5-НТ2A рецептору у порівнянні з 5-НТ2c рецептором також визначають, використовуючи стандартну методологію (Тестування В). У цих тестуваннях вимірюють відсоток інгібування, що викликають за допомогою сполук формули (І) відповідно до даного винаходу при шести різних концентраціях, з яких одержують рKі значення сполук у відношенні окремих рецепторів. Значення спорідненості деяких сполук формули (І) відповідно до даного винаходу до 5-НТ5A та 5-НТ2c рецепторів рекомбінантних клітин людини показані у Таблиці 2, у якій спорідненість є більшою для вищого значення рKі. Таблиця 2 Сполука (№) 8 1 3 25 30 35 40 (h)5-HT2a(pKi) 8,79 8,58 8,68 (h)5-HT2c(pKi) 7,27 7,05 6,83 Більше того, сполуки формули (І) відповідно до даного винаходу оцінюють in vivo у моделі "судоми голови" у мишей (Тестування С). Вона являє собою стандартне тестування для визначення будь-яких взаємодій з серотонінергічною системою, як описано у Sztanke K., Fidecka S., Kedzierska E., Karczmarzyk Z., Pihlaja K., Matosiuk D. (2005) "Antionociceptive activity of new imidazole carbonyl derivatives. Part 4. Synthesis and pharmacological activity of 8-aryl-3,4-dioxo-2H, 8H-6,7-dihydrimidazo[2,1c][1,2,4]triazine." Eur. J. Med. Chem. 40: 127-134, та у Corne S.J., Pickering R.W., Warner B.T. (1963) "Method for assessing the effects of drags on the central actions of 5hydroxytryptamine." Br. J. Pharmacol. Chemother. 20: 106-120. У цьому тестуванні, сполуки, що розглядають, викликали зниження у кількості "судом голови" у порівнянні з тваринами, яких лікують тільки за допомогою метилцелюлози (МТС), таким чином, показуючи здатність антагонізувати серотонінергічні ефекти, що викликають введенням 5-гідрокситриптофану (5-НТР). Значення інгібування, отримані у дослідженні "Судоми Голови" з досліджуваними сполуками, показані на Фігурі 1. Наступний опис призначений для додаткової ілюстрації даного винаходу, при цьому не обмежуючи його. Приклад 1 2-Метил-N-{[1-(2-фенілетил)піперидин-4-іл]метил}-2Н-індазол-3-карбоксаміду гідрохлорид гідрат (Сполука № 1) 10 UA 98311 C2 5 10 15 20 25 30 35 40 45 50 55 60 (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=R4=R5 = Н, n = 1, р = 2, m=2, Y=CH, W=N, R6+R7 =-СН2-СН2-) 1a) Метиловий складний ефір 2-метил-2Н-індазол-3-карбонової кислоти (Сполука X: R1 = Н, R2 = СН3) Карбонат калію (207,1 г; 1,498 моль) та, краплинним способом, метил йодид (31 мл; 0,50 моль) додають до суспензії метилового складного ефіру 1Н(2Н)-індазол-3-карбонової кислоти (74,0 г; 0,499 моль) у ацетоні (750 мл). Реакційну суміш нагрівають при зрошенні впродовж 4 годин, охолоджують та фільтрують. Розчинник видаляють випарюванням при зниженому тиску та залишок переносять двічі у етилацетат. Таким чином одержують приблизно 80 г сирої сполуки. Тверду речовину кристалізують двічі з суміші н-гексан/етилацетат, отримуючи метиловий складний ефір 2-метил-2Н-індазол-3карбонової кислоти (50,0 г). 1 Н-ЯМР(, DMSO-d6): 3,93 (s, 3Н); 4,17 (s, 3Н); 7,3-7,4 (m, 1Н); 7,5-7,6 (m, 1H); 7,7-7,8 (m, 1H); 8,0-8,1 (m, 1H). 1b) Хлорид 2-метил-2Н-індазол-3-карбонової кислоти (Сполука III; R1=H, R2=CH3, Z =СІ) Гідроліз метилового складного ефіру 2-метил-2Н-індазол-3-карбонової кислоти (1а) (50,0 г; 0,260 моль) проводять у воді (300 мл), що містить гідроксид натрію (20,9 г; 0,520 моль). Реакційну суміш кип'ятять при зрошенні впродовж 2 годин, охолоджують до кімнатної температури та підкислюють за допомогою 2N НСl до припинення осадження білої твердої речовини. Після фільтрування та висушування у печі у вакуумі, одержують 45,2 г 2-метил-2Ніндазол-3-карбонової кислоти, та її використовують без додаткового очищення у наступних реакціях. Тіоніл-хлорид (39 мл; 0,54 моль) додають до суспензії 2-метил-2Н-індазол-3-карбонової кислоти (45,2 г; 0,270 моль) у толуолі (350 мл) та реакційну суміш нагрівають при зрошенні впродовж 24 годин. Розчинник видаляють випарюванням при зниженому тиску. Залишок переносять двічі у толуол. Таким чином одержують 47,0 г хлориду 2-метил-2Н-індазол-3карбоновбї кислоти. 1 Н-ЯМР (,DMSO-d6): 3,97 (s, 3Н); 7,3-7,4 (m, 1H); 7,5-7,6 (m, 1H); 7,7-7,8 (m, 1H); 8,0-8,1 (m, 1H). 1c) 1-[1-(2-Фенілетил)-4-піперидиніл]метанамін (Сполука XI: R3 = R4 = Н, R6+R7 = -СН2-СН2-, n = 1, р = 2, m=2). Бензальдегід (28,0 г; 0,264 моль) додають, краплинним способом, до розчину 4амінометилпіперидину (30,2 г; 0,264 моль) у толуолі (100 мл). Отриманий таким чином розчин перемішують при кімнатній температурі. Через 3 години, розчинник видаляють випарюванням при зниженому тиску та залишок переносять двічі у толуолі. 53,4 г N-гексагідро-4піридинілметил-N-фенілметиліденамін одержують таким чином, та використовують у наступній реакції без додаткового очищення. N-Гексагідро-4-піридинілметил-N-фенілметиліденамін (53,4 г; 0,264 моль) розчиняють у абсолютному етанолі (500 мл), що містить безводний карбонат калію (73,0 г; 0,528 моль) та фенетил-бромід (39,5 мл; 0,290 моль). Реакційну суміш, отриману таким чином, кип'ятять при зрошенні впродовж 24 годин. Після охолодження до кімнатної температури, суспензію фільтрують та розчинник випарюють при зниженому тиску. Залишок, отриманий таким чином, суспендують у 3N НСl (200 мл) та перемішують при кімнатній температурі впродовж 2 годин. Кислу водну фазу промивають 4 рази етилацетатом та потім підлужують до рН приблизно 13 за допомогою 6N NaOH та екстрагують дихлоретаном. Органічну фазу, отриману таким чином, сушать над безводним Na2SO4 та розчинник видаляють випарюванням при зниженому тиску. Таким чином одержують 50,0 г 1-[1-(2-фенілетил)-4-піперидиніл]метанаміну. 1 Н-ЯМР (, CDCl3): 1,3-1,4 (m, 5H); 1,7-1,8 (m, 2H); 1,9-2,1 (m, 2Н); 2,5-2,6 (m, 4Н); 2,7-2,9 (m, 2Н); 2,9-3,1 (m, 2Н); 7,1-7,3 (m, 5Н). 1d) 2-Метил-N-{[1-(2-фенілетил)піперидин-4-іл]метил|-2Н-індазол-3-карбоксаміду гідрохлориду гідрат (Сполука I: R1=H, R2=CH3, X = -C(O)N(R5)-, R3=R4=R5=H, n=1, p=2, m=2, Y=CH, W=N, R6+R7 = -CH2-CH2-). 1-[1-(2-Фенілетил)-4-піперидиніл]метанамін (1c) (20,3 г; 0,0930 моль) додають до розчину хлориду 2-метил-2H-індазол-3-карбонової кислоти (1b) (18,0 г; 0,0930 моль) у толуолі (100 мл). Реакційну суміш перемішують при кімнатній температурі впродовж ночі та потім фільтрують. Тверду речовину переносять у 1N NaOH та екстрагують дихлорметаном (DCM). Органічні фази об'єднують та потім сушать над безводним Na2SO4, та розчинник видаляють випарюванням при зниженому тиску. 11 UA 98311 C2 5 Сиру сполуку, отриману таким чином, перетворюють у відповідний гідрохлорид шляхом розчинення у етилацетаті, обробки надлишком 5N хлористоводневого етанолу при кімнатній температурі впродовж 3 годин, видалення розчиннику випарюванням при зниженому тиску та кристалізації твердої речовини, що одержують з суміші етилацетат/абсолютний етанол. Одержують 7,2 г 2-метил-N-{[1-(2-фенілетил)піперидин-4-іл]метил}-2Н-індазол-3-карбоксаміду гідрохлориду гідрату. Тпл.= 130-140 °C Елементний аналіз для C23H31CIN4O2 Знайдено % Розраховано % С 64,16 64,10 Н 7,37 7,25 N 12,89 13,00 СІ 8,46 8,59 10 1 15 20 25 30 35 Н-ЯМР (, DMSO-d6): 1,59-2,10 (m, 5H); 2,80-3,40 (m, 10Н); 3,59 (d, J=12 Гц, 2Н); 4,31 (s, 3Н); 7,15-7,40 (m, 7Н); 7,69 (d, J=9 Гц, 1Н); 7,84 (d, J = 9Гц, 1H); 8,63 (t, J=6 Гц, 1Н); 10,50 J-10,70 (s широкий, 1Н). Приклад 2 N,2-Диметил-N-{[1-(2-фенілетил)піперидин-4-іл]метил}-2Н-індазол-3-карбоксаміду гідрохлориду напівгідрат (Сполука № 2) (Сполука І: R1=H, R2 = СН3, X = -C(O)N(R5)-, R3=R4 = Н, R5 = СН3, n = 1, р = 2, m = 2, Y = СН, W = N R6+R7 = -СН2-СН2-) 2а) N,2-Димeтил-N-{[1-(2-фeнiлeтил)пiпepидин-4-iл]мeтил}-2H-iндaзoл-3-кapбoкcaмiду гідрохлориду напівгідрат Після перемішуваная впродовж 1 години при кімнатній температурі, метил-йодид (0,83 мл; 0,013 моль) додають до суспензії 2-метил-N-{[1-(2-фенілетил)піперидин-4-іл]метил}-2Н-індазол3-карбоксаміду (1d) (5,0 г; 0,013 моль) та 60 % гідрид натрію (0,51 г; 0,013 моль) у тетрагідрофурані (THF) (50 мл). Суміш, отриману таким чином, перемішують при кімнатній температурі впродовж 48 годин. Потім додають дистильовану воду для видалення надлишку гідриду. Органічну фазу відділяють та концентрують випарюванням при зниженому тиску. Отриманий залишок очищують, використовуючи флеш хроматографію (елюенти CHCl3/CH3OH/NH3 у співвідношеннях 85/14/1) та перетворюють у гідрохлорид шляхом розчинення у етилацетаті, обробки надлишком 5N хлористоводневого етанолу при кімнатній температурі впродовж 3 годин та видалення розчиннику випарюванням при зниженому тиску. Сполуку, отриману таким чином, кристалізують з суміші етилацетат/абсолютний етанол. Таким чином одержують 5,2 г N,2-диметил-N-{[1-(2-феніл-етил)піперидин-4-іл]метил}-2Ніндазол-3-карбоксаміду гідрохлориду напівгідрату. Тпл. =206-207 °C Елементний аналіз рія C24H33CIN4O2 Знайдено % Розраховано % С 63,70 63,49 Н 7,44 7,55 1 40 45 50 N 12,26 12,34 СІ 7,82 7,81 Н-ЯМР(, DMSO-d6+D2O): 1,50-2,30 (m, 5Н); 2,80-3,80 (m, 14Н); 3,01 (s, 3Н); 4,15 (s, 3Н); 7,10-7,40 (m, 7Н); 7,61 (t, J = 8Гц, 1H); 7,69(d, J=9Гц, 1H). Приклад 3 N3-({1-[2-(4-Гідроксифеніл)етил]гексагідро-4-піридиніл}метил)-2-метил-2Н-3індазолкарбоксаміду гідрохлориду гідрат (Сполука № 3) (Сполука І: R1=H, R2 = СН3, X = -C(O)N(R5)-, R3=4-ОН, R4 = Н, R5 = Н, n = 1, р = 2, m=2, Y = СН, W=N, R6+R7 = -СН2-СН2-) 3а) 1-(2-(4-Гідроксифеніл)етил)-4-піперидиніл-метанамін (Сполука XI: R3=4-ОН, R4 = Н, R6+R7 = -СН2-СН2-, n = 1, р = 2, m=2) Одержання подібне до того, що описане для одержання (1с), виходячи з N-гексагідро-4піридинілметил-N-феніл-метиліденаміну (7,5 г, 0,037 моль) та 2-(4-гідроксифеніл)етил-броміду (7,5 г; 0,037 моль) [одержано як описано у Ada Chemica Scandinava (1967), 21 (1), 53-62]. Таким чином одержують 9,3 г1-(2-(4-гідроксифеніл)етил)-4-піперидиніл-метанаміну. 1 Н-ЯМР (, CDCl3 + D2O): 1,15-1,41 (m, 3Н); 1,74 (d, J=9 Гц, 2H); 1,9-2,1 (m, 2H); 2,4-2,6 (m, 4H); 2,65-2,75 (m, 2H); 3,01 (d, J=12Гц, 2H); 6,75 (d, J=9Гц, 2H); 7,00 (d, J=9Гц, 2Н). 3b) N3-({1-[2-(4-Гідроксифеніл)етил]гексагідро-4-піридиніл}метил)-2-метил-2Н-3індазолкарбоксаміду гідрохлориду гідрат 12 UA 98311 C2 5 10 1-(2-(4-Гідроксифеніл)етил)-4-піперидинілметанамін (3а) (5,6 г; 0,024 моль) додають невеликими частинами до розчину хлориду 2-метил-2Н-індазол-3-карбонової кислоти (1b) (4,7 г; 0,024 моль) у толуолі (100 мл). Суміш, отриману таким чином, перемішують впродовж 18 годин при кімнатній температурі. Потім її фільтрують. Залишок очищують декілька разів кристалізацією з суміші етилацетат/абсолютний етанол та перетворюють у гідрохлорид шляхом розчинення у етилацетаті, обробки надлишком 5N хлористоводневого етанолу при кімнатній температурі впродовж 3 годин, та видалення розчиннику випарюванням при зниженому тиску. Після кристалізації з суміші етилацетат/абсолютний етанол, одержують 5,0 г N3-({1-[2-(4гідроксифеніл)етил]гексагідро-4-піридиніл}метил)-2-метил-2Н-3-індазолкарбоксаміду гідрохлориду гідрату. Тпл.=236-238 °C Елементний аналіз [для C23H29CIN4O2 % Н2О Знайдено % Розраховано % С 62,43 62,43 Н 6,96 6,95 N 12,58 12,66 СІ 8,06 8,09 15 1 20 25 30 35 40 45 50 55 Н-ЯМР (, DMSO-d6): 1,5-2,1 (m, 5Н); 2,8-3,4 (m, 9Н); 3,57 (d, J=11 Гц, 2Н); 4,30 (s, 3Н); 6,73 (d, J=9Гц, 2Н); 7,06 (d, J = 9Гц, 2Н); 7,2-7,3 (m, 1Н); 7,3-7,4 (m, 1H); 7,68 (d, J=9 Гц, 1H); 7,84 (d, J=9 Гц, 1Н); 8,65 (t, J=6 Гц, 1Н); 9,2-9,5 (s широкий, 1Н); 10,4-10,7 (s широкий, 1Н). Приклад 4 N-({1-[2-(4-Гідроксифеніл)етил]піперидин-4-іл}метил)5-метокси-2-метил-2Н-індазол-3карбоксамід (Сполука № 4) (Сполука І; R1-5-СН3О, R2 = СН3, X = -C(O)N(R5)-, R3=4-ОН, R4 = Н, R5 = Н, n = 1, р = 2, m=2, Y4CH, W=N, R6+R7 =-СН2-СН2-) 4а) 5-Метокси-2-метил-2Н-індазол-3-карбонової кислоти метиловий складний ефір (Сполука X: R1=5-СН3О, R2 - СН3) Суміш 5 -метокси-індазол-3 -карбонової кислоти [отриманої відповідно до Gazzetta Chimica Italiana (1963) 93, 3-14] (11,8 г; 0,0610 моль), метанолу (200 мл) та сірчаної кислота (2 мл) перемішують при кімнатній температурі впродовж 4 годин. Суміш потім розбавляють за допомогою дистильованої води. Тверду речовину, що утворюється, відділяють фільтруванням, сушать у сушильній камері (9,6 г) та використовують без додаткового очищення у наступній реакції. Гідроксид калію (3,6 г; 0,064 моль) додають, невеликими частинами, до суспензії, що містить метиловий складний ефір 5-метокси-індазол-3-карбонової кислоти (9,6 г; 0,047 моль) та метилйодид (3,4 мл; 0,054 моль) у диметоксиетані (DME) (50 мл). Реакційну суміш нагрівають цри зрошенні впродовж 18 годин та потім охолоджують. Розчинник видаляють випарюванням при зниженому тиску. Тверду речовину переносять у толуол та промивають декілька разів водою та 6N NaOH. Розчинник далі випарюють при зниженому тиску, та отриманий залишок очищують флеш хроматографією (н-гексан/етилацетат = 7/3). Таким чином одержують 5,0 г 5-метокси-2метил-2Н-індазол-3-карбонової кислоти метилового складного ефіру. 1 Н-ЯМР(, CDCl3): 3,90 (s, 3Н); 4,03 (s, 3Н); 4,47 (s, 3Н); 7,0-7,1 (dd, J1=9,3 Гц, J2=2,3 Гц, 1H); 7,2-7,3 (d, J = 2,3 Гц, 1H); 7,6-7,7 (dd, J1 =9,3 Гц, J2=0,7 Гц, 1Н). 4b) Хлорангідрид 5-метокси-2-метил-2Н-індазол-3-карбонової кислоти (Сполука III: R1=5-СН3О, R2 - СН3; Z = СІ) Гідроксид натрію (1,0 г; 0,025 моль) додають до суспензії метилового складного ефіру 5метокси-2-метил-2Н-індазол-3-карбонової кислоти (одержано як описано у попередньому Прикладі 4а) (2,8 г; 0,013 моль) у дистильованій воді (40 мл). Суміш, отриману таким чином, залишають взаємодіяти при зрошенні впродовж 4 годин, охолоджують та підкислюють за допомогою 2N НСl до закінчення осадження 5-метокси-2-метил-2Н-індазол-3-карбонової кислоти, та після фільтрування її використовують без додаткового очищення у наступних реакціях. Тіоніл-хлорид (0,8 мл; 0,01 моль) додають до колби, що містить 5-метокси-2-метил-2Ніндазол-3-карбонову кислоту (1,1 г; 0,0050 моль) та толуол (30 мл). Реакційну суміш вводять у реакцію при зрошенні впродовж 5 годин та, після випарювання розчиннику при зниженому тиску, залишок переносять декілька разів у толуолі. Таким чином одержують 1,2 г хлорангідриду 5-метокси-2-метил-2Н-індазол-3-карбонової кислоти. 1 Н-ЯМР (, CDCl3): 3,90 (s, 3Н); 4,47 (s, 3Н); 7,0-7,1 (dd, J1 = 9,3 Гц, J2=2,3 Гц, 1Н); 7,2-7,3 (d, J=2,3 Гц, 1H); 7,6-7,7 (dd, J1=9,3 Гц, J2=0,7 Гц, 1H). 13 UA 98311 C2 5 10 15 20 25 30 35 40 45 50 55 4c) N-({1-[2-(4-Гідроксифеніл)етил]піперидин-4-іл}метил)-5-метокси-2-метил-2Н-індазол-3карбоксамід 1-(2-(4-Гідроксифеніл)етил)-4-піперидиніл-метанамін (3а) (1,25 г; 0,0050 моль) додають невеликими частинами до суспензії хлорангідриду 5-метокси-2-метил-2Н-індазол-3-карбонової кислотні (4b) (1,2 г; 0,0050 моль) у толуолі (30 мл). Суміш, отриману таким чином, залишаюсь взаємодіяти впродовж 4 годин при кімнатній температурі та нагрівають при зрошенні впродовж ще 4 годин. Потім суспензію охолоджують та фільтрують. Тверду речовину очищують кристалізацією з суміші н-гексан/етилацетат. Таким чином одержують 1,5 г N-({1-[2-(4-гідроксифеніл)етил]піперидин-4-іл}метил)-5метокси-2-метил-2H-індазол-3-карбоксаміду. Тпл. = 218-219°C (розклад) 1 Н-ЯМР (, DMSQ-d6+D2O): 1,21-1,3 (m, 2Н); 1,5-1,8 (m, 1H); 1,73 (d, J=12 Гц, 2H); 1,93 (t, J = 12 Гц, 2Н); 2,3-2,7 (m, 4Н); 2,93 (d, J=11 Гц, 2Н); 3,26 (d, J=6 Гц, 2Н); 3,82 (s, 3Н); 424 (s, 3Н); 6,66 + (d, J=9 Гц, 2Н); 6,9-7,1 (m, 4H); 7,59 (d, J=9 Гц, 1H); 8,40 (t, J = 6 Гц, 1H). MS показує 423 (MH ) основний пік. Приклад 5 N-({1-[2-(4-Mетоксифеніл)етил]піперидин-4-іл}метил)-2-метил-2Н-індазол-3-карбоксаміду гідрохлорид (Сполука № 5) (Сполука І: R1 = Н, R2 = СН3, X= -C(O)N(R5)-, R3=4-СН3О-, R4 = Н, R5 = Н, n = 1, р = 2, m=2, Y = СН, W=N, R6+R7 = -СН2-СН2-) 5а) 1-[2-(4-Метоксифеніл)етил]-4-піперидинілметанамін (Сполука XI: R3=4-СН3О, R4 = Н, R6+R7 = -СН2-CH2-, n=1, p=2, m=2) Зазначену сполуку удержують за способом, подібним тому, що описаний для одержання 1с), виходячи з N-гексагідро-4-піридинілметил-N-фенілметиліденаміну (12,9 г; 0,0631 моль) та 2-(4метоксифеніл)етил-броміду (15,0 г; 0,0698 моль) [одержано як описано у Acta Chemica Scandinava (1967), 21 (1), 53-62]. Одержують 5,0 г 1-[2-(4-метоксифеніл)етил]-4-піперидинілметанаміну. 1 H-ЯМР (, CDCl3): 1,1-1,4 (m, 3Н); 1,6-1,9 (m, 4H); 1,9-2,1 (m, 2H); 2,4-2,6 (m, 4Н); 2,7-2,8 (m, 2H); 2,9-3,1 (m, 2H); 3,77 (s, 3Н); 6,7-6,9 (m, 2H); 7,0-7,2 (m, 2H). 5b) N-({1-[2-(4-Метоксифеніл)етил]піперидин-4-іл}метил)-2-метил-2Н-індазол-3-карбоксаміду гідрохлорид 1-[2-(4-Метоксифеніл)етил]-4-піперидинілметанамін (5а) (6,8 г; 0,027 моль) додають невеликими частинами до суспензії хлорангідриду 2-метил-2Н-індазол-3-карбонової кислоти (1b) (5,3 г; 0,027 моль) у толуолі (200 мл). Суміш, отриману таким чином, перемішують впродовж 48 годин при кімнатній температурі та потім фільтрують. Тверду сполуку суспендують у 1N NaOH та екстрагують 3 рази дихлорметаном. Розчинник видаляють випарюванням при зниженому тиску та залишок кристалізують спочатку з суміші ізопропіловий ефір/ізопропанол та потім з суміші етилацетат/абсолютний етанол. Потім сполуку перетворюють у її гідрохлорид шляхом розчинення у етанолі, що супроводжують обробкою 5N хлористоводневим етанолом впродовж 3 годин при кімнатній температурі та потім видаленням розчиннику випарюванням при зниженому тиску. Після кристалізації з суміші етилацетат/абсолютний етанол, одержують 5,0 г N-({1-[2-(4метоксифеніл)етил]діперидин-4-іл}метил)-2-метил-2Н-індазол-3-карбоксаміду гідрохлориду. Тпл. = 80 °C (розклад) 1 Н-ЯМР (, DMSO-d6): 1,5-2,1 (m, 5Н); 2,8-3,5 (m, 8Н); 3,58 (d, J=12 Гц, 2Н); 3,73 (s, 3Н); 4,30 (s, 3H); 6,90 (d, J=9 Гц, 2Н); 7,1-7,4 (m, 4Н); 7,69 (d, J=9 Гц, 1Н); 7,84 (d, J=9Гц, 1Н); 8,67 (t, J=6 + Гц, 1Н); 10,7-11,0 (s широкий, 1Н). MS показує 407 (MH ) основний пік. Приклад 6 N3-2-Диметил-N3-[(1-(2-[4-(метилокси)феніл]етил}піперидин-4-іл)метил]-2Н-індазол-3карбоксаміду гідрохлориду гідрат (Сполука № 6) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=4-СН3О-, R4 = Н, R5 = СН3, n = 1, р = 2, m=2, Y=CH, W=N, R6+R7 = -СН2-СН2-) 6а) ({1-[2-(4-Метоксифеніл)етил]-4-піперидиніл}метил)формамід (Сполука XII: R3=4-СН3О, R4 = Н, R6+R7 = -СН2-СН2-, n = 1, р = 2, m=2, R=H) Мурашину кислоту (0,60 мл; 0,016 моль) додають краплинним способом, у атмосфері азоту, при температурі 0 °C, до колби, що містить оцтовий ангідрид (1,25 мл; 0,0112 моль). Суміш, отриману таким чином, нагрівають при зрошенні впродовж 2 годин та, після охолодженая до кімнатної температури, додають розчин 1-[2-(4-метоксифеніл)етил]-4-піперидинілметанаміну (5а) (1,24 г; 502 ммоль) у THF (10 мл). 14 UA 98311 C2 5 10 15 20 25 30 35 Суміш залишають взаємодіяти впродовж 18 годин. Суспензію потім концентрують випарюванням при зниженому тиску. Таким чином одержують 1,0 г ({1-[2-(4-метоксифеніл)етил]4-піперидиніл}метил)формаміду. GC/MS (m/z: 155 основний пік; 121; 110). 6b) 1-{1-[2-(4-Метоксифеніл)етил]-4-піперидиніл}-N-метилметанамін (Сполука II: R3=4-СН3О, R4=H, R5 = СН3, R6+R7 = -СН2-СН2-, n=1, р = 2, m = 2) Розчин боран-диметилсульфідного 2N комплексу у н-гексані (6 мл) додають краплинним способом до суспензії ({1-[2-(4-метоксифеніл)етил]-4-піперидиніл}метил)формаміду (1,0 г; 0,0041 моль) (6а) у 20 мл THF при 0 °C. Суміш нагрівають при зрошенні впродовж 5 годин та потім охолоджують до 0 °C. При цій температурі далі додають 2 мл метанолу та реакційну суміш енергійно перемішують впродовж 1 години. Далі, газоподібний НСl барботують у реакційну суміш до повного підкислення. Потім суміш нагрівають при зрошенні впродовж 1 години. Після її охолодження додають метанол (10 мл), та розчинник випарюють при зниженому тиску. Залишок переносять у воду та промиваюсь 3 рази етилацетатом. Водну фазу підлужують за допомогою 6N NaOH та екстрагують 3 рази дихлорметаном. Органічну фазу сушать над Na2SO4, фільтрують та випарюють при зниженому тиску. Таким чином одержують 0,6 г 1-{1-[2-(4-метоксифеніл)етил]-4-піперидиніл}-Nметилметанаміну. GC/MS (m/z: 127; 110; 96). 6с) N3-2-Диметил-N3-[(1-{2-[4-(метилокси)феніл]етил}піперидин-4-іл)метил]-2Н-індазол-3карбоксаміду гідрохлориду гідрат 1-{1-[2-(4-Метоксифеніл)етил]-4-піперидиніл}-N-метилметанамін (одержано як описано у попередньому Прикладі 6b) (1,4 г; 0,0052 моль) додають, невеликими частинами, до розчину хлорангідриду 2-метил-2Н-індазол-3-карбонової кислоти (1b) (1,1 г; 0,0061 моль) у толуолі (20 мл). Суміш, отриману таким чином, перемішують впродовж 18 годин при кімнатній температурі та потім фільтрують. Твердий залишок Переносять у 1N NaOH та екстрагують дихлорметаном. Об'єднані органічні фази сушать над Na2SO4 та розчинник видаляють випарюванням при зниженому тиску. Твердий залишок перетворюють у гідрохлорид шляхом розчинення у етанолі, додавання надлишку 5N хлористоводневого етанолу, перемішування впродовж 3 годин при кімнатній температурі та потім видалення розчиннику випарюванням при зниженому тиску. Після кристалізації з суміші етилацетат/абсолютний етанол, одержують 0,8 г N3-2-диметилN3-[(1-{2-[4-(метилокси)феніл] етил}піперидин-4-іл)метил]-2Н-індазол-3-карбоксаміду гідрохлориду гідрату. Тпл.=132,5-136,5 °С Елементний аналіз для C25H35ClN4O3 Знайдено % Розраховано % С 62,95 63,21 Н 7,65 7,43 1 40 45 50 55 N 11,61 11,79 СІ 7,59 7,76 Н-ЯМР(, DMSO-d6+D2O): 1,5-2,3 (m, 5Н); 2,7-3,7 (m, 13Н); 3,11 (s, 3Н); 3,73 (s, 3Н); 4,15 (s, 3Н); 4,90 (d, J = 8Гц, 2Н); 7,1-7,3 (m, 3Н); 7,33 (t, J=8 Гц, 1H); 7,59 (d, J=8 Гц, 1H); 7,69 (d, J = 9Гц, 1Н). Приклад 7 N-({1-[2-(4-Фторфеніл)етил]піперидин-4-іл}метил)-2-(2-метоксиетил)-2Н-індазол-3карбоксаміду гідрохлорид (Сполука № 7) (Сполука І: R1 = Н, R2 = СН2СН2ОСН3, X = -C(O)N(R5)-, R3=4-F, R4 = Н, R5 = Н, n = 1, р = 2, m=2, Y=CH, W=N, R6+R7 =-СН2-СН2-) 7а) 2-(2-Метоксиетил)-2Н-індазол-3 -етил-карбоксилат (Сполука III: R1 = H, R2 = СН3ОСН2СН2, Z = СН3СН2О) Карбонат калію (33,6 г; 0,243 моль) та, краплинним способом, 2-брометил-метиловий ефір (46 мл; 0,49 моль) додають до розчину етилового складного ефіру 1Н(2Н)-індазол-3-карбонової кислоти (15,4 г; 0,0811 моль) у абсолютному етанолі (200 мл). Реакційну суміш перемішують впродовж 48 годин. Розчинник потім видаляють випарюванням при зниженому тиску. Залишок переносять у хлороформ та промивають водою 3 рази. Органічну фазу сушать над Na2SО4, та, після фільтрування та випарювання розчиннику, одержують приблизно 10 г сирої сполуки. Її очищують флеш хроматографією, використовуючи н-гексан/етилацетат 7:3 суміш як елюент. Таким чином одержують 4,5 г 2-(2-метоксиетил)-2Ніндазол-3-етил карбоксилату. 15 UA 98311 C2 1 5 10 15 20 25 30 35 40 45 50 55 60 Н-ЯМР (, CDCl3): 1,50 (t, J = 6Гц, 3Н); 3,33 (s, 3Н); 3,91 (t, J = 6Гц, 2Н); 4,49 (q, J=6 Гц, 2Н); 5,13 (t, J = 6Гц, 2Н); 7,2-7,4 (m, 2H); 7,79 (d, J = 9 Гц, 1H); 8,02 (d, J=9 Гц, 1Н). 7b) 2-(2-Метоксиетил)-2Н-індазол-3-карбоніл-хлорид (Сполука III: R1 = Н, R2=CH3OCH2CH2, Z = СІ) Складний ефір 2-(2-метоксиетил)-2Н-індазол-3-етил-карбоксилату (одержано як описано у попередньому Прикладі 7а) (4,26 г; 0,0171 моль) гідролізують у водному розчині (30 мл) гідроксиду натрію (1,16 г; 0,0291 моль). Реакційну суміш нагрівають при зрошенні впродовж 2 годин, охолоджують до кімнатної температури та підкислюють за допомогою 6N НСl до закінчення осадження 2-(2-метоксиетил)-2Н-індазол-3-карбонової кислоти, яка, після відділення фільтруванням та висушування у печі у вакуумі, знаходиться у формі білої твердої речовини (3,6 г), яку використовують без додаткового очищення у наступній реакції. Тіоніл-хлорид (2,6 мл; 0,036 моль) додають до суспензії 2-(2-метоксиетил)-2Н-індазол-3карбонової кислоти (3,6 г; 0,016 моль) у толуолі (100 мл) та реакційну суміш нагрівають при зрошенні впродовж 24 годин. Розчинник видаляють випарюванням при зниженому тиску та залишок переносять двічі у н-гексан. Таким чином одержують 3,9 г 2-(2-метоксиетил)-2Ніндазол-3-карбоніл-хлориду. 1 Н-ЯМР(, CDCl3): 3,29 (s, 3Н); 3,84 (t, J = 6 Гц, 2Н); 5,11 (t, J = 6 Гц, 2Н); 7,1-7,4 (m, 2Н); 7,74 (d, J = 9 Гц, 1H); 7,99 (d, J = 9 Гц, 1H). 7c) 1-(2-Брометил)-4-фторбензол (Сполука V: R3-4-F, R4=H, m=2, Q=Br) 60,8 г 47 % бромоводневої кислоти (0,353 моль) повільно додають до розчину 2-(4фторфеніл)етанолу (20 г; 0,143 моль) у концентрованій сірчаній кислоті (8,5 мл; 0,16 моль). Суміш нагрівають при зрошенні впродовж 3 годин, потім охолоджують до кімнатної температури та додають 50 мл дихлорметану. Органічну фазу відділяють та промивають послідовно за допомогою 1N NaOH та водою та потім сушать над Na2SО4. Після фільтрування та випарювання розчиннику при зниженому тиску, одержують приблизно 13 г сирої сполуки, яку очищують флеш хроматографією (нгексан/етилацетат = 9/1). Таким чином одержують 10,6 г 1-(2-брометил)-4-фторбензолу. 1 Н-ЯМР (, CDCl3): 3,13 (t, J=9 Гц, 2Н); 3,54 (t, J=9 Гц, 2Н); 6,9-7,3 (m, 4H) 7d) 1-[2-(4-Фторфеніл)етил]-4-піперидину метанамін (Сполука XI: R3=4-F, R4=H, R6+R7 = -CH2-CH2-, n=1, p=2, m=2) N-гексагідро-4-тридинілметил-N-фенілметиліденамін (17,9 г; 0,0879 моль) розчиняють у абсолютному етанолі (100 мл), що містить безводний карбонат калію (24,3 г; 0,176 моль) та 1(2-брометил)-4-фторбензол (одержано як описано у попередньому Прикладі 7с) (18,0 г; 0,088 моль). Реакційну суміш нагрівають при зрошенні впродовж 24 годин. Після охолодження до кімнатної температури, суспензію фільтрують та розчинник випарюють при зниженому тиску. Отриманий залишок суспендують у 3N НСl (120 мл) та перемішують при кікнатній температурі впродовж 3 годин. Кислу водну фазу промивають 4 рази етилацетатом, потім підлужують до рН приблизно 13 за допомогою 6N NaOH та, нарешті, її екстрагують 3 рази етилацетатом. Органічну фазу, отриману таким чином, сушать над безводним Na2SO4 та, після фільтрування, розчинник видаляють випарюванням при зниженому тиску, що приводить до одержання 17,2 г 1-[2-(4фторфеніл)етил]-4-піперидину метанаміну. 1 Н-ЯМР (, CDCl3): 1,2-1,5 (m, 5H); 1,6-1,8 (m, 2H); 1,9-2,1 (m, 2H); 2,4-2,6 (m, 4Н); 2,7-2,8 (m, + 2H); 2,9-3,1 (m, 2Н); 6,9-7,2 (m, 4Н). MS показує 237 (МН ), 220 (основний пік). 7е) N-({1-[2-(4-Фторфеніл)етил]піперидин-4-іл}метил-2-(2-метоксиетил)-2Н-індазол-3карбоксаміду гідрохлорид 1-[2-(4-Фторфеніл)етил]-4-піперидину метанамін (одержано як описано у попередньому Прикладі 7d) (3,23 г; 0,0131 моль) та триетиламін (5,4 мл; 0,039 моль) додають до розчину 2-(2метоксиетил)-2Н-індазол-3-карбоніл-хлориду (одержано як описано у попередньому Прикладі 7b) (3,26 г; 0,0131 моль) у толуолі (50 мл). Реакційну суміш перемішують при зрошенні впродовж 5 годин та потім розчинник випарюють при зниженому тиску. Залишок переносять у етилацетат та промивають послідовно за допомогою 0,1N NaOH та водою. Органічну фазу сушать над безводним Na 2SO4 та, після фільтрування, розчинник видаляють випарюванням при зниженому тиску. Одержуюсь 4,7 г сирої сполуки, яку очищують флеш хроматографією (CHCl 3/CH3OH/NH3=98/2/0,2). Отриману сполуку (1,5 г) перетворюють у відповідний гідрохлорид шляхом розчинення у етилацетаті, обробки надлишком 5N хлористоводневого етанолу при кімнатній температурі впродовж 3 годин, видалення розчиннику випарюванням при зниженому тиску та нарешті кристалізацією з абсолютного етанолу. 16 UA 98311 C2 Таким чином одержують 0,6 г N-({1-[2-(4-фторфеніл)етил]піперидин-4-іл}метил)-2-(2метоксиетил)-2Н-індазол-3-карбоксаміду гідрохлориду. Тпл. = 193,0-194,0 °C Елементний аналіз для C25H31N4O2F. НСl 5 Знайдено % Розраховано % С 63,21 63,20 Н 6,79 6,79 N 11,69 11,80 СІ 7,50 7,46 1 10 15 20 25 Н-ЯМР (, DMSO-d6): 1,5-2,0 (m, 5Н), 2,8-3,7 (m, 10Н); 3,19 (s, 3Н); 3,80 (t, J=6 Гц, 2Н); 4,88 (t, J=6 Гц, 2Н); 7,1-7,4 (m, 6Н); 7,70 (d, J=9 Гц, 1Н); 7,82 (d, J=9 Гц, 1Н); 8,6-8,8 (m, 1H); 10,7-11,0 (s широкий, 1H). Приклад 8 N-({1-[2-(4-Фторфеніл)етил]піперидин-4-іл}метил)-2-метил-2Н-індазол-3-карбоксаміду гідрохлорид (Сполука № 8) (Сполука І: R1=H, R2 = СН3, X = -C(O)N(R5)-, R3=4-F, R4 = Н, R5 = Н, n = 1, р = 2, m = 2, Y = СН, W=N, R6+R7 = -СН2-СН2-) 8а) N-({1-[2-(4-Фторфеніл)етил]піперидин-4-іл}метил)-2-метил-2Н-індазол-3-карбоксаміду гідрохлорид Зазначену сполуку одержують як описано у Прикладі 7е), використовуючи 4,0 г хлорангідриду 2-метил-2Н-індазол-3-карбонової кислоти (одержано як описано у попередньому Прикладі 1b) (0,021 моль), толуол (100 мл), 1-[2-(4-фторфеніл)етил]-4-піперидин-метанамін (7d) (4,8 г; 0,021 моль) та триетиламін (8,6 мл; 0,062 моль). Одержують 10,7 г сирої сполуки, яку очищують флеш хроматографією (СНСl3/СН3ОН/NH3 - 97/3/0,3). Сполуку, отриману таким чином, (4,1 г) перетворюють у відповідний гідрохлорид шляхом розчинення у етанолі, обробки надлишком 5N хлористоводневого етанолу при кімнатній температурі впродовж 3 годин, видалення розчиннику випарюванням при зниженому тиску та нарешті кристалізацією з суміші абсолютний етанол/етилацетат. Таким чином одержують 4,3 г N-({1-[2-(4-фторфеніл)етил]піперидин-4-іл}метил)-2-метил-2Ніндазол-3-карбоксаміду гідрохлориду. Тпл. = 208,0-209,6 °C Елементний аналіз для C23H27N4OF. НСl 30 Знайдено % Розраховано % С 64,00 64,10 Н 6,54 6,55 1 35 40 45 50 N 12,89 13,00 СІ 8,34 8,23 Н-ЯМР (, DMSO-d6): 1,6-2,0 (m, 5H); 2,8-3,7 (m, 10Н); 4,32 (s, 3H); 7,1-7,4 (m, 6Н); 7,69 (d, J = 9Гц, 1H); 7,85 (d, J=9 Гц, 1H); 8,6-8,8 (m, 1H); 10,9-11,1 (s широкий, 1H). Приклад 9 2-(2,4-Дифторфеніл)-N-({1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4іл}метил)етанаміну гідрохлорид (Сполука № 9) (Сполука І: R1 = Н, R2 - СН3, X= -C(O)N(R5)-, R3=4-F, R4=2-F, R5+R6 = -СН2-СН2-, n=2, p=1, m=2, Y=CH, W=N, R7=H) 9а) 1-{1-[(2-Метил-2Н-індазол-3-іл)карбоніл]піперидин-4-іл]метанамін (Сполука XVI: R1= Н, R2 = СН3, R5+R6 = -СН2-СН2-, n = 2, р = 1) N-Гексагідро-4-піридинілметил-N-фенілметиліденамін (11,3 г; 0,0558 моль) та триетиламін (9,3 мл; 0,066 моль) додають до розчину хлорангідриду 2-метил-2Н-індазол-3-карбонової кислоти (одержано як описано у попередньому Прикладі 1b) (10,8 г; 0,0558 моль) у толуолі (150 мл). Суміш нагрівають при зрошенні впродовж 5 годин. Потім її охолоджують до кімнатної температури та фільтрують. Розчинник випарюють при зниженому тиску. Залишок перемішують при кімнатній температурі з 3N НСl (80 мл) впродовж 5 годин. Кислий розчин промивають 3 рази етилацетатом. Потім його підлужують за допомогою 12N NaOH. Лужну водну фазу екстрагують дихлорметаном 3 рази. Об'єднані органічні фази сушать над безводним Na2SО4. Далі їх фільтрують та розчинник випарюють при зниженому тиску. Таким чином одержують приблизно 16 г сирої сполуки, та очищують флеш хроматографією (CHCI 3/CH3OH/NH3=95/5/0,5). Одержують 13,4 г 1-{1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4-іл}метанаміну. GC/MS (m/z) показує 272, 159 (основний пік). 17 UA 98311 C2 5 10 15 20 25 30 35 9b) 2-[2,4-Дифторфеніл)етил метансульфонат (Сполука V: R3=2-F, R4=4-F, m=2, Q=CH3OSO2) Розчин (2,4-дифторфеніл)оцтової кислоти (15,0 г; 0,0871 моль) у етиловому ефірі (100 мл) повільно додають др суспензії алюмогідриду літію (6,58 г; 0,174 моль) у етиловому ефірі (100 мл). Суміш нагрівають при зрошенні впродовж 5 годин та потім доводять до кімнатної температури. Надлишок гідриду видаляють додаванням 1N HCl (150 мл). Кислу фазу відділяють та потім екстрагують 3 рази етиловим ефіром. Об'єднані органічні фази промивають двічі за допомогою 1N NaOH, сушать над Na2SO4 та, після фільтрування, розчинник випарюють при зниженому тиску. Таким чином одержують 6,4 г бажаної сполуки, та використовують без додаткового очищення у наступній реакції. Метансульфоніл-хлорид (3,3 мл; 0,043 моль) та триетиламін (5,9 мл; 0,043 моль) додають до розчину 2-(2,4-Дифторфеніл)етанолу (6,4 г; 0,0426 моль) у дихлорметані (100 мл). Зазначений розчин охолоджують впродовж 1 години при 0 °C та потім доводять до кімнатної температури впродовж 1 години. Реакційну суміш потім обробляють промиванням, послідовно, за допомогою 5N сірчаної кислоти (20 мл), води (50 мл), 5 % розчину NaHCO3 (50 мл) та води (50 мл). Органічну фазу потім сушать над Na2SO4, фільтрують та концентрують при зниженому тиску. Залишок переносять двічі у ССl4, таким чином отримуючи 6,42 г 2-(2,4-дифторфеніл)етилметансульфонату. 1 Н-ЯМР (, CDCl3): 2,92 (s, 3Н); 3,07 (t, J=6 Гц, 2Н); 4,40 (t, J=6 Гц, 2Н); 6,7-6,9 (m, 2H); 7,1-7,3 (m, 1Н). 9с) 2-(2,4-Дифторфеніл)-N({1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4іл}метил)етанаміну гідрохлорид K2СО3 (3,7 г; 0,027 моль) та 1-{1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4іл}метанамін (одержаний як описано у попередньому Прикладі 9а) (6,73 г; 0,0247 моль) додають до розчину 3-(2,4-дифторфеніл)етил-метансульфонату (9b) (6,42 г; 0,0271 моль) у абсолютному етанолі (150 мл). Суміш нагрівають при зрошенні впродовж 30 годин та потім розчинник випарюють при зниженому тиску. Залишок переносять у етилацетат та промивають 3 рази водою. Органічну фазу сушать над Na2SO4, фільтрують та концентрують при зниженому тиску. Таким чином одержують приблизно 15 г сирої сполуки, яку очищують флеш хроматографією (СНСl3/СН3ОН/NHз = 99/1/0,1). Сполуку, отриману таким чином, (2,5 г) перетворюють у її гідрохлорид шляхом розчинення у абсолютному етанолі та обробки надлишком 5N хлористоводневого етанолу впродовж 3 годин. Після випарювання розчиннику при зниженому тиску, твердий залишок кристалізують з суміші етанол/етилацетат, отримуючи 0,7 г 2-(2,4дифторфеніл)-N-({1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4-іл}метил)етанаміну гідрохлориду. Тпл. = 108,0-110,0 °C Елементний аналіз для C23H26N4OF2. НСl Знайдено % Розраховано % С 61,68 61,53 Н 5,97 6,06 N 12,46 12,48 СІ 8,00 7,90 40 1 45 50 55 Н-ЯМР (, DMSO-d6):1,0-2,2 (m, 5Н); 2,8-3,3 (m, 8Н); 3,4-4,9 (m, 2Н), 4,17 (s, 3Н); 7,0-7,5 (m, 5Н); 7,62 (d, J=9 Гц, 1H); 7,69 (d, J=9 Гц, 1H); 9,1-9,5 (s широкий, 2Н). Приклад 10 2-(4-Фторфеніл)-N-({1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4-іл}метил)етанаміну гідрохлорид (Сполука № 10) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=4-F, R4 = Н, R5+R6 = -СН2-СН2-, n = 2, р=1, m=2, Y=CH, W=N, R7=H) 10а) 2-(4-Фторфеніл)-N-({1-[2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4-іл}метил)етанаміну гідрохлорид Бажану сполуку одержують як описано у Прикладі 9с), використовуючи 4,10 г 1-(2брометил)-4-фторбензолу (одержаного як описано у попередньому Прикладі 7с) (0,0201 моль), 5,0 г 1-{1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4-іл}метан аміну (одержаного як описано у попередньому Прикладі 9а) (0,018 моль), 2,77 г K2СО3 (0,0201 моль) та 100 мл абсолютного етанолу. Таким чином одержують приблизно 7 г сирої сполуки, та очищують флеш хроматографією (СHСl3/СН3ОН/NH3 = 98/2/0,2). Отриману сполуку (3,5 г) перетворюють у її гідрохлорид шляхом розчинення у абсолютному етанолі та обробки надлишком 5N хлористоводневого етанолу 18 UA 98311 C2 5 впродовж 3 годин. Після випарювання розчиннику при зниженому тиску, твердий залишок кристалізують з суміші етанол/діізопропіловий ефір, отримуючи 2,5 г 2-(4-фторфеніл)-N-({1-[(2метил-2Н-індазол-3-іл)карбоніл]піперидин-4-іл}метил) етанаміну гідрохлориду. Тпл.=198-199 °C Елементний аналіз для C23H27N4OF.НСl Знайдено % Розраховано % С 64,14 64,10 Н 6,54 6,55 N 13,00 13,00 СІ 8,24 8,23 1 10 15 20 25 30 35 40 45 50 Н-ЯМР (, DMSО-d6): 1,1-2,2 (m, 5Н); 2,7-3,3 (m, 8Н); 3,4-4,9 (m, 2Н), 4,17 (s, 3Н); 7,1-7,4 (m, 6Н); 7,61 (d, J=9 Гц, 1H); 7,68 (d, J=9 Гц, 1H); 9,0-9,4 (s широкий, 2Н). Приклад 11 N-Етил-2-(4-фторфеніл)-N-({1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4іл}метил)етанаміну гідрохлорид (Сполука № 11) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=4-F, R4 = Н, R5+R6 = -СН2-СН2-, n=2, p=1, m=2, Y=CH, W=N, R7=CH3CH2) 11а) N-Етил-2-(4-фторфеніл)-N-({1-[(2-метил-2Н-індазол-3-іл)карбоніл]піперидин-4іл}метил)етанаміну гідрохлорид K2СО3 (0,71 г; 0,0052 моль), йодид калію (10 мг; 0,062 ммоль) та етил-бромід (0,54 мл; 0,0072 моль) додають до розчину 2-(4-фторфеніл)-N-({1-[(2-метил-2Н-індазол-3іл)карбоніл]піперидин-4-іл}метил)етанаміну (10а) (1,7 г; 0,0043 моль) у абсолютному етанолі (20 мл). Суміш нагрівають при зрошенні впродовж 48 годин та потім розчинник випарюють при зниженому тиску. Залишок переносять у хлороформ та промивають водою двічі. Органічну фазу сушать над Na2SO4, фільтрують та концентрують при зниженому тиску. Отриману сиру сполуку (1,5 г) очищують флеш хроматографією (етилацетат/н-гексан/амоній = 96/4/0,2). Отриману сполуку (0,5 г) перетворюють у її гідрохлорид шляхом розчинення у абсолютному етанолі та обробки надлишком 5N хлористоводневого етанолу впродовж 3 годин. Після випарювання розчиннику при зниженому тиску, твердий залишок кристалізують з суміші етанол/етилацетат, отримуючи 0,15 г N-етил-2-(4-фторфеніл)-N-({1-[(2-метил-2Н-індазол-3-іл)карбоніл]-піперидин-4іл}метил)етанаміну гідрохлориду. 1 Н-ЯМР (, CDCl3): 0,6-2,4 (m, 5H); 1,44 (t, J=9 Гц, 3Н); 2,7-3,7 (m, 12H); 4,28 (s, 3Н); 6,9-7,4 (m, 6H); 7,46 (d, J=9 Гц, 1Н); 7,72 (d, J=9 Гц, 1Н); 12,0-12,7 (s широкий, 2Н). MS показує 423 + (MH ), 159 (основний пік). Приклад 12 N-({1-[2-(2-фторфеніл)етил]піперидин-4-іл}метил)-2-метил)-2Н-індазол-3-карбоксамід (Сполука № 12) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=2-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n=1, р=2, m=2, Y=CH, W=N) 12а) (1-[2-(2-фторфеніл)етил]-4-піперидин)метанамін (Сполука XI: R3=2-F, R4=H, R6+R7 =-СН2-СН2-, Y=CH, W=N, n=1, p=2, m=2) {1-[2-(2-Фторфеніл)етил]-4-піперидин}метанамін отримують відповідно до процедури, описаної у Прикладі 7d), використовуючи як реагенти N-гексагідро-4-піридинілметил-Nфенілметиліденамін (2,03 г; 10 ммоль), та 1-(2-брометил)-2-фторбензол (2,04 г; 10 ммоль). Одержують 1,42 г 1-[2-(2-фторфеніл)етил]-4-піперидин-метанаміну. 1 Н-ЯМР (, CDCl3): 1,0-1,4 (m, 2H), 1,5-2,1 (m, 5H), 2,4-3,0 (m, 10Н), 6,8-7,2 (m, 4H). 12b) N-({1-[2-(2-фторфеніл)етил]піперидин-4-іл}метил)-2-метил-2Н-індазол-3-карбоксамід Сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (880 мг; 4,5 ммоль) та 1-[2-(2фторфеніл)етил]-4-піперидин-метанамін (Приклад 12а) (1,06 г; 4,5 ммоль). Таким чином одержують 350 мг N-({1-[2-(2-фторфеніл)етил]піперидин-4-іл}метил)-2-(2метоксиетил)-2Н-індазол-3-карбоксаміду. Тпл. =128-130 °C Елементний аналіз ддя C23H27N4OF Знайдено % Розраховано % С 70,21 70,03 Н 6,79 6,90 19 N 14,39 14,20 UA 98311 C2 1 5 10 15 20 25 30 Н-ЯМР (DMSO-d6, 6 ppm): 1,15-1,35 (m, 2 Н), 1,50-1,83 (m, 3Н), 1,97 (t, J=10,82 Гц, 2Н), 2,422,57 (m, 2 Н), 2,77 (t, J=7,60 Гц, 2 Н), 2,94 (d, J=10,82 Гц, 2 Н), 3,25 (t, J=6,28 Гц, 2 Н), 4,28 (s, 3 Н), 7,06-7,38 (m, 6Н), 7,67 (d, J=8,77 Гц, 1Н), 7,77 (d, J=8,18 Гц, 1Н), 8,54 (t, J=5,70 Гц, 1Н). Приклад 13 2-етил-N-({1-[2-(4-фторфеніл)етил]піперидин-4-іл}метил)-2Н-індазол-3-карбоксаміду гідрохлорид (Сполука № 13) (Сполука І: R1 = Н, R2 = СН2СН3, X = -C(O)N(R5)-, R3=4-F, R4 = Н, R5 = Н, R6+R7 = -СН2СН2-, n=1, p=2, m=2, Y=CH, W=N) 13а) Метиловий складний ефір 2-етил-2Н-індазол-3-карбонової кислоти (Сполука X; R1 = Н, R2 = СН2СН3) Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 1а), використовуючи як реагенти метиловий складний ефір 1Н(2Н)-індазол-3-карбонової кислоти (7,4 г; 49,9 ммоль) та етил-бромід (3,73 мл; 50 ммоль). Одержують 4,5 г метилового складного ефіру 2-етил-2Н-індазол-3-карбонової кислоти. 1 Н-ЯМР (, DMSO-d6): 1,55 (t, J=7,3 Гц, 3Н), 4,09 (s, 3Н), 4,62 (q, J=7,3 Гц, 2 Н), 7,2-7,4 (m, 1H), 7,5-7,6 (m, 1), 7,7-7,8 (m, 1), 7,9-8,1 (m, 1H). 13b) 2-етил-2Н-індазол-3-карбоніл-хлорид (Сполука III: R1=H, R2=CH2CH3, Z=Cl) Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 1b), використовуючи як реагенти метиловий складний ефір 2-етил-2Н-індазол-3 -карбонової кислоти (Приклад 13а) (4,0 г; 19,6 ммоль). Таким чином одержують 3,5 г 2-етил-2Н-індазол-3-карбоніл-хлориду. 1 Н-ЯМР (, DMSO-d6): 1,51 (t, J=7,3 Гц, 3 Н), 4,59 (q, J=7,3 Гц, 2 H), 7,3-7,4 (m, 1H), 7,5-7,6 (m, 1), 7,7-7,8 (m, 1), 7,9-8,1 (m, 1H). 13c) N-({1-[2-(4-фторфеніл)етил]піперидин-4-іл}метил)-2-етил-2Н-індазол-3-карбоксаміду гідрохлорид Зазначену сполуку одержують як описано у Прикладі 7е), використовуючи 2-етил-2Ніндазол-3-карбоніл-хлорид (Приклад 13b) (1,05 г; 5,4 ммоль) та 1-[2-(4-фторфеніл)етил]-4піперидин-метанамін (Приклад 7d) (1,1г; 5,4 ммоль). Одержують 190 мг N-({1-[2-(4-фторфеніл)етил]піперидин-4-іл}метил)-2-етил-2Н-індазол-3карбоксаміду гідрохлориду. Тпл. = 182-186 °C Елементний аналіз для C24H29N4ОFHCl Знайдено % Розраховано % С 64,60 64,78 Н 6,54 6,80 N 12,69 12,59 СІ 8,04 7,97 35 Н-ЯМР (DMSO-d6, ppm): 1,46 (t, J=7,27 Гц, 3 Н), 1,51-1,75 (m, 2Н), 1,91 (br. s., 3Н), 2,663,84 (m, 10 Н), 4,68 (q, J=7,27 Гц, 2Н), 7,08-7,39 (m, 6 Н), 7,70 (d, J=8,59 Гц, 1Н), 7,80 (d, J = 8,26 Гц, 1Н), 8,65 (t, J=5,28 Гц, 1Н), 10,30 (br. s., 1Н). Приклад 14 N-(1-[2-(4-фторфеніл)етил]пiперидин-4-іл)-2-метил-2Н-індазол-3-карбоксаміду гідрохлорид (Сполука № 14) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=4-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n = 0, р=2, m=2, Y=CH, W=N) 14а) 1-[2-(4-фторфеніл)етил]піперидин-4-амін (Сполука XI: R3=4-F, R4=H, R6+R7 = -СН2-СН2-, Y=CH, W=N, n=0, р = 2, m=2) Бензальдегід (21,2 г; 0,2 моль) додають, краплинним способом, до розчину 4амінопіперидину (20 г; 0,2 моль) у толуолі (80 мл). Отриманий таким чином розчин перемішують при кімнатній температурі. Через 3 години розчинник видаляють випарюванням при зниженому тиску та залишок обробляють двічі толуолом. Таким чином одержують 31 г N(фенілметилен)піперидин-4-аміну, та використовують у наступних реакціях без додаткового очищення. Аліквоту цього продукту (1,88 г, 10 ммоль) використовують, відповідно до процедури, описаної у Прикладі 7d), разом з 1-(2-брометил)-4-фторбензолом (Приклад 7с) (2,03 г; 10 моль). Таким чином одержують 1,3 г 1-[2-(4-фторфеніл)етил]піперидин-4-аміну, та використовують у наступній реакції без додаткового очищення. 14b) Н-{1-[2-(4-фторфеніл)етил]піперидин-4-іл}-2-метил-2Н-індазол-3-карбоксаміду гідрохлорид 1 40 45 50 55 20 UA 98311 C2 5 Зазначену сполуку одержують як описано у Прикладі 7е), використовуючи як реагенти 2метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,15 г; 5,8 ммоль) та 1-[2-(4фторфеніл)етил]піперидин-4-амін (Приклад 14а) (1,1 г; 5,8 ммоль). Таким чином одержують 305 мг N-{1-[2-(4-фторфеніл)етил]піперидин-4-іл}-2-метил-2Ніндазол-3-карбоксаміду гідрохлориду. Тпл. =224-228 °C Елементний аналіз для C22H25N4OF. HCl Знайдено % Розраховано % 10 15 20 25 30 35 С 63,20 63,38 Н 6,44 6,29 Н-ЯМР (DMSO-d6, ppm): 2,09 (br. s., 4H), 2,72-3,83 (m, 8H), 4,09 (br. s., 1H), 4,29 (s, 3H), 7,08-7,26 (m, 3H), 7,32 (t, J=7,10 Гц, 3H), 7,68 (d, J=8,59 Гц, 1H), 7,75 (d, J=8,26 Гц, 1H); 8,78 (br. s., 1H), 10,63 (br. s., 1H). Приклад 15 N-(1-[2-(4-фторфеніл)етил]піролідин-3-іл)2-метил-2Н-індазол-3-карбоксамід (Сполука № 15) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5), R3=4-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n = 0, р=1, m=2, Y=CH, W-N) 15а) 1-[2-(4-фторфеніл)етил]піролідин-3-амін (Сполука XI: R3=4-F, R4=H, R6+R7 = -СН2-СН2-, Y-CH, W=N, n=0, p=1, m=2) Бензальдегід (21,2 г; 0,20 моль) додають, краплинним способом, до розчину 3амінопіролідину (17,2 г; 0,20 моль) у толуолі (80 мл). Отриманий таким чином розчин перемішують при кімнатній температурі. Через 3 години розчинник видаляють випарюванням при зниженому тиску та залишок обробляють двічі толуолом. Таким чином одержують 21 г N(фенілметилен)піролідин-3-аміну, та використовують у наступних реакціях без додаткового очищення. Аліквоту цього продукту (1,7 г, 10 ммоль) використовують, відповідно до процедури, описаної у Прикладі 7d), разом з 1-(2-брометил)-4-фторбензолом (Приклад 7с) (2,0 г; 10 ммоль). 1,4 г 1-[2-(4-фторфеніл)етил]піролідин-3-аміну одержують таким чином, та використовують у наступній реакції без додаткового очищення. 15b) N-{1-[2-(4-фторфеніл)етил]піролідин-3-іл}-2-метил-2Н-індазол-3-карбоксамід Зазначену сполуку одержують як описано у Прикладі 7е), використовуючи як реагенти 2метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,3 г; 6,7 ммоль) та 1-[2-(4фторфеніл)етил]піролідин-3-амін (Приклад 15а) (1,4 г; 6,7 ммоль). Таким чином одержують 235 мг N-{1-[2-(4-фторфеніл)етил]піролідин-3-іл}-2-метил-2Ніндазол-3-карбоксаміду. Тпл. =117-119 °C Елементний аналіз для C21H23N4ОF С 69,01 68,83 Н 6,44 6,33 50 N 15,29 15,29 Н-ЯМР (DMSO-d6, ppm): 1,83 (dddd, J=13,01, 7,67, 5,70, 5,48 Гц, 1 H), 2,11-2,26 (m, 1H), 2,53-2,82 (m, 7H), 2,92 (dd, J=9,35, 7,31 Гц, 1H), 4,27 (s, 3Н), 4,39-4,53 (m, 1H), 7,03-7,13 (m, 2H), 7,19 (ddd, J=8,33, 6,58, 0,88 Гц, 1H), 7,25-7,35 (m, 3H), 7,66 (ddd, J=8,50, 1,00, 0,80 Гц, 1H), 7,71 (ddd, J=8,33, 1,17, 1,02 Гц, 1H), 8,64 (d, J=7,02 Гц, 1H). Приклад 16 N-{1-[2-(4-фторфеніл)етил]піперидин-3-іл}-2-метил-2Н-індазол-3-карбоксамід (Сполука № 16) (Сполука І: R1 = Н, R2 = СН3, X =-C(O)N(R5)-, R3=4-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2СН2-, n=0, p=1, m=2, Y=CH, W=N) 16а) 1-[2-(4-фторфеніл)етил]піперидин-3-амін (Сполука XI: R3=4-F, R4 = Н, R6+R7 = -СН2-СН2-СН2-, Y = СН, W=N, n=0, р= 1, m = 2) Бензальдегід (21,2 г; 0,2 моль) додають, краплинним способом, до розчину 3амінопіперидину (20 г; 0,2 моль) у толуолі (80 мл). Отриманий таким чином розчин перемішують при кімнатній температурі. Через 3 години розчинник видаляють випарюванням при зниженому тиску та залишок обробляють двічі толуолом. Таким чином одержують 29 г N1 45 СІ 8,54 8,50 1 Знайдено % Розраховано % 40 N 13,29 13,44 21 UA 98311 C2 5 10 (фенілметилен)піперидин-3-аміну, та використовують у наступних реакціях без додаткового очищення. Аліквоту цього продукту (1,88 г, 10 ммоль) використовують, відповідно до процедури, описаної у Прикладі 7d), разом з 1-(2-брометил)-4-фторбензолом (Приклад 7с) (2,0 г; 10 ммоль). Таким чином одержують 1,5 г 1-[2-(4-фторфеніл)етил]піперидин-3-аміну. 1 Н-ЯМР ( ppm, CDCl3): 1,00-2,20 (m, 8 Н), 2,40-2,90 (m, 7 Н), 6,80-7,20 (m, 4 Н) 16b) N-1-[2-(4-фторфеніл)етил]піролідин-3-іл)-2-метил-2Н-індазол-3-карбоксамід Зазначену сполуку одержують як описано у Прикладі 7е), використовуючи як реагенти 2метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,13 г; 5,8 ммоль) та 1-[2-{4фторфеніл)етил]піперидин-3-амін (Приклад 16а) (1,3 г; 5,8 ммоль). Таким чином одержують 335 мг 1N-{1-[2-(4-фторфеніл)етил]піролідин-3-іл}-2-метил-2Ніндазол-3-карбоксаміду. Тпл. =194-197 °C Елементний аналіз для C22H25N4ОF 15 Знайдено % Розраховано % С 69,31 69,45 Н 6,64 6,62 N 14,79 14,73 Н-ЯМР (DMSO-d6, ppm): 1,38-1,64 (m, 2 Н), 1,66-1,92 (m, 2Н), 2,05-2,26 (m, 2Н), 2,53-2,62 (m, 2Н), 2,67-2,82 (m, 3Н), 2,95 (d, J=8,18 Гц, 1Н), 3,94-4,13 (m, 1Н), 4,28 (s, 3Н), 7,00-7,11 (m, 2Н), 7,15-7,38 (m, 4Н), 7,67 (d, J=8,48 Гц, 1Н), 7,72 (d, J=8,18 Гц, 1Н), 8,30 (d, J=8,30 Гц, 1H). Приклад 17 2-метил-N-[(1-{2-[4-(трифторметил)феніл]етил}піперидин-4-іл)метил]-2Н-індазол-3карбоксамід (Сполука № 17) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=4-CF3, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n=1, p=2, m=2, Y=CH, W=N) 17а) 1-(1-{2-[4-(трифторметил)феніл]етил}піперидин-4-іл)метанамін (Сполука XI: R3=4-CF3, R4=H, R6+R7 =-СН2-СН2-, Y=CH, W=N, n=1, р = 2, m = 2) 1-(1-{2-[4-(трифторметил)феніл]етил}піперидин-4-іл)метанамін отримують відповідно до процедури, описаної у Прикладі 7d), використовуючи як реагенти N-гексагідро-4піридинілметил-N-фенілметиліден-амін (2,36 г; 10 ммоль), та 1-(2-брометил)-4(трифторметил)бензол (2,5 г; 10 ммоль). Одержують 1,8 г 1-(1-{2-[4-(трифторметил)феніл]етил}піперидин-4-іл)метанаміну, та використовують у наступній реакції без додаткового очищення. 17b) 2-метил-N-[(1-{2-[4-(трифторметил)феніл]етил}піперидин-4-іл)метил]-2Н-індазол-3карбоксамід Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,28 г; 6,6 ммоль) та 1-(1-{2-[4-(трифторметил)феніл]етил}піперидин-4-іл)метанамін (Приклад 17а) (1,8 г; 6,6 ммоль). Таким чином одержують 415 мг 2-метил-N-[(1-{2-[4-(трифторметил)-феніл]етил}піперидин-4іл)метил]-2Н-індазол-3-карбоксаміду. Тпл. =174-176 °C Елементний аналіз для C24H27N4OF3 1 20 25 30 35 40 Знайдено % Розраховано % 45 50 С 64,71 64,85 Н 6,29 6,12 N 12,59 12,60 Н-ЯМР (DMSO-d6, ppm): 1,14-1,34 (m, 2Н), 1,52-1,81 (m, 3Н), 1,97 (t, J=10,82 Гц, 2Н), 2,54 (t, J=8,00 Гц, 2H), 2,83 (t, J=7,45 Гц, 2H), 2,95 (d, J = 11,11 Гц, 2Н), 3,25 (t, J =6,14 Гц, 2Н), 4,29 (s, 3Н), 7,16-7,25 (m, 1Н), 7,27-7,35 (m, 1Н), 7,46 (d, J = 8,18 Гц, 2Н), 7,62 (d, J = 8,18 Гц, 2Н), 7,67 (d, J = 8,77 Гц, 1 Н), 7,77 (d, J = 8,48 Гц, 1Н), 8,54 (t, J = 5,70 Гц, 1Н), Приклад 18 N-(1-[2-(2-фторфеніл)етил)піперидин-4-іл)-2-метил-2Н-індазол-3-карбоксамід (Сполука №18) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=2-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n = 0, р = 2, m=2, Y = СН, W-N) 18а) 1-[2-(2-фторфеніл)етил]піперидин-4-амін (Сполука XI: R3=2-F, R4 = Н, R6+R7 = -СН2-СН2-, Y = СН, W=N, n=0, р = 2, m=2) 1 22 UA 98311 C2 5 10 Зазначений продукт одержують, використовуючи процедуру, описану у Прикладі 7d), використовуючи як реагенти N-(фенілметилен)піперидин-4-амін (Приклад 14а) (1,8 г; 10 ммоль) та 1-(2-брометил)-2-фторбензол (2,5 г; 10 ммоль). Таким чином одержують 1,3 г 1-[2-(2-фторфеніл)етил]піперидин-4-амін, та використовують у наступній реакції без додаткового очищення. 18b) N-{1-[2-(2-фтopфeнiл)eтил]пiпepидин-4-iл}-2-мeтил-2H-iндaзoл-3-кapбoкcaмiд Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,14 г; 5,8 ммоль) та 1-[2-(2-фторфеніл)етил]піперидин-4-амін (Приклад 18а) (1,3 г; 5,8 ммоль). Таким чином одержують 188 мг N-{1-[2-(2-фторфеніл)етил]піперидин-4-іл}-2-метил-2Н-індазол-3карбоксаміду. Тпл. =164-166 °C Елементний аналіз для C22H25N4OF Знайдено % Розраховано % С 69,61 69,45 Н 6,59 6,62 N 14,59 14,73 15 Н-ЯМР (DMSO-d6, ppm): 1,64 (qd, J=11,55, 3,07 Гц, 2Н), 1,89 (d, J=11,98 Гц, 2Н), 2,13 (t, J=10,96 Гц, 2Н), 2,54 (t, J=8,48 Гц, 2Н), 2,79 (t, J=7,60 Гц, 2Н), 2,95 (d, J=11,69 Гц, 2Н), 3,73-3,98 (m, 1Н), 4,27 (s, 3Н), 7,07-7,39 (m, 6Н), 7,66 (dt, J = 8,70, 0,91 Гц, 1Н), 7,73 (dt, J=8,40, 0,91 Гц, 1Н), 8,48 (d, J=7,70 Гц, 1Н). Приклад 19 N-{2-[4-(4-фторфеніл)піперидин-1-іл]етил}-2-метил-2Н-індазол-3-карбоксамід (Сполука № 19) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5), R3=4-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n=2, р = 2, m=0, Y=N, W=CH) 19а) N-(2-аміноетил)4-(4-фторфеніл)піперидин (Сполука II: R5 = Н, R6+R7 = -СН2-СН2-, R3=4-F, R4 = Н, Y=N, W= СН, n = 2, р = 2, m = 0) K2О3 (1,61 г, 11,7 ммоль) додають невеликими частинами до розчину, що містить 4-(4фторфеніл)піперидину (1,05 г, 5,85 ммоль) у безводному ацетоні (25 мл). Потім розчин (2брометил)фталімід (164 г, 6,44 ммоль) у безводному ацетоні (25 мл) повільно додають до суміші при перемішуванні при кімнатній температурі. Наприкінці додавання (30 хвилин) суміш нагрівають до температури зрошення впродовж 24 годин. Наприкінці реакції, суміш охолоджують та фільтрують. Залишковий розчин потім розбавляють етилацетатом (50 мл) та екстрагують за допомогою 1,5N HCl(3 20 мл). Об'єднані кислі фази промивають етилацетатом (2 10 мл), потім сильно підлужують з 3N NaOH та екстрагують дихлорметаном (5 20 мл). Об'єднані органічні фази сушать над безводним Na2SO4 та випарюють при зниженому тиску. Отриманий залишок (607 мг) потім розчиняють у метанолі (11 мл) та обробляють гідразину моногідратом (2 мл, 41 ммоль) при 90 °C впродовж 2 годин. Реакційну суміш охолоджують до кімнатної температури, розбавляють дихлорметаном (50 мл) та екстрагують декілька разів з 3N NaOH (5 15 мл). Органічну фазу далі роблять безводною з Na 2SO4 та випарюють при зниженому тиску. Таким чином одержують 400 мг N-(2-аміноетил)-4-(4-фторфеніл)піперидину, та використовують у наступній реакції без додаткового очищення. 19b) N-{2-[4-(4-фторфеніл)піперидин-1-іл]етил}-2-метил-2Н-індазол-3-карбоксамід Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (240 мг; 1,22 моль) та N-(2-аміноетил)-4-(4-фторфеніл)піперидин (Приклад 19а) (400 мг). Таким чином одержують 190 мг N-{2-[4-(4-фторфеніл)піперидин-1-іл]етил}-2-метил-2Ніндазол-3-карбоксаміду. Тпл. =105-107 °C Елементний аналіз для C22H25FN4O 1 20 25 30 35 40 45 50 Знайдено % Розраховано % С 69,21 69,45 Н 6,62 6,62 23 N 14,99 14,73 UA 98311 C2 Н-ЯМР (DMSO-d6 ppm): 1,55-1,83 (m, 4Н), 2,05-2,19 (m, 2Н), 2,53-2,56 (m, 0Н), 2,59 (t, J=6,58 Гц, 2H), 3,06 (d, J=11,40 Гц; 2H), 3,42-3,58 (m, 2Н), 4,31 (s, 3Н), 7,11 (t, J=8,92 Гц, 2Н), 7,17-7,37 (m, 5Н), 7,68 (d, J = 8,48 Гц, 1Н), 7,92 (d, J=8,48 Гц, 1Н), 8,37 (t, J=5,41 Гц, 1Н). Приклад 20 N-(3-[4-(4-Фторфеніл)піперидин-1-іл]пропіл)-2-метил-2Н-індазол-3-карбоксамід (Сполука №20) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=4-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n = 3, р = 2, m=0, Y=N, W = СН) 20а) N-(3-амінопропіл)-4-(4-фторфеніл)піперидин (Сполука II: R5 = Н, R6+R7 = -СН2-СН2-, R3=4-F, R4 = Н, Y=N, W = СН, n = 3, р = 2, m = 0) Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 19а), використовуючи як реагенти 4-(4-фторфеніл)піперидин (1,10 г; 6,14 моль) та N-(3бромпропіл)фталімід (1,82 г, 6,78 ммоль). Таким чином одержують 1,6 г N-(3-амінопропіл)-4-(4-фторфеніл)піперидину, та використовують у наступній реакції без додаткового очищення. 20b) N-{3-[4-(4-фторфеніл)піперидин-1-іл]пропіл)-2-метил-2Н-індазол-3-карбоксамід Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (236 мг; 1,21 моль) та N-(3-амінопропіл)-4-(4-фторфеніл)піперидин (Приклад 20а) (550 мг). Таким чином одержують 120 мг N-{3-[4-(4-фторфеніл)піперидин-1-іл]пропіл}-2-метил-2Ніндазол-3-карбоксаміду. Тпл. =136-140 °C Елементний аналіз для C23H27FN4O 1 5 10 15 20 Знайдено % Розраховано % С 69,91 70,03 Н 6,72 6,90 N 14,29 14,20 25 Н-ЯМР(СDСl3, ррm): 1,26 (dq, J =12,9, 3,6 Гц, 2Н), 1,57 (bd, J=12,9 Гц, m, 2Н), 1,84 (q, J=5,7 Гц, 2Н), 1,94 (dt, J=11,7, 1,5 Гц, 2Н), 2,31 (tt, J=12,9, 4,4 Гц, 1Н), 2,58 (t, 1=5,7Гц, 2Н), 3,39 (bd, J=11,7 Гц, 2H), 3,70 (q, J=5,4 Гц, 2H), 4,46 (s, 3Н), 6,58 (dd, J=8,4, 5,4 Гц, 2H), 6,2 (t, J=8,7 Гц, 2H), 7,15-7,25 (m, 1H), 7,3-7,4 (m, 1H), 7,81 (t, J=8,7 Гц, 2H), 8,4 (bs, 1H). Приклад 21 N-(1-бензилпіперидин-4-іл)-2-метил-2Н-індазол-3-карбоксамід (Сполука №21) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3 = Н, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n=0, p=2, m=1, Y=CH, W=N) 21а) 1-бензилпіперидин-4-амін (Сполука XI; R3=H, R4=H, R6+R7 = -СН2-СН2-, Y=CH, W=N, n=0, р = 2, m=1) Зазначений продукт одержують, використовуючи процедуру, описану у Прикладі 7d), використовуючи як реагенти N-[фенілметилен]піперидин-4-амін (Приклад 14а) (1,88 г; 10 ммоль) та бензил-бромід (1,7 г; 10 моль). Таким чином одержують 1,7 г 1-бензилпіперидин-4-аміну, та використовують у наступній реакції без додаткового очищення. 21b) N-(1-бензилпіперидин-4-іл)-2-метил-2Н-індазол-3-карбоксамід Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,74 г; 8,9 ммоль) та 1-бензилпіперидин-4-амін (Приклад 21а) (1,7 г; 8,9 ммоль). Таким чином одержують 460 мг N-(1-бензилпіперидин-4-іл)-2-метил-2Н-індазол-3карбоксаміду. Тпл. =167-169 °C Елементний аналіз для C21H24N4O 1 30 35 40 45 Знайдено % Розраховано % С 72,21 72,39 Н 7,02 6,94 N 16,09 16,08 50 Н-ЯМР (DMSO-d6, ppm): 1,66 (dq, J=11,80, 2,90 Гц, 2Н), 1,82-1,94 (m, 2Н), 2,09 (t, J=10,67 Гц, 2Н), 2,83 (d, J= 11,69 Гц, 2Н), 3,49 (s, 2Н), 3,76-3,95 (m, 1H), 4,27 (s, 3Н), 7,19 (ddd, J=8,18, 6,87, 1,02 Гц, 1Н), 7,24-7,38 (m, 6Н), 7,66 (d, J=8,77 Гц, 1Н), 7,72 (d, J = 8,48 Гц, 1Н), 8,48 (d, J=7,89 Гц, 1Н). 1 24 UA 98311 C2 5 10 15 20 Приклад 22 N-{1-[3-(4-фторфеніл)пропіл]піперидин-4-іл}-2-метил-2Н-індазол-3-карбоксамід (Сполука № 22) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5), R3=4-F, R4 = Н, R5 = Н, R6+R7 = -СН2-СН2-, n = 0, р = 2, m = 3, Y = СН, W=N) 22а) 1-[3-(4-фторфеніл)пропіл]піперидин-4-амін (Сполука XI: R3=4-F, R4 = Н, R6+R7 = -СН2-СН2-, Y = СН, W=N, n=0, р = 2, m=3) Зазначений продукт одержують, використовуючи процедуру, описану у Прикладі 7d), використовуючи як реагенти N-(фенілметилен)піперидин-4-амін (Приклад 14а) (1,9 г; 10 моль) та 1-(3-бромпропіл)-4-фторбензол (2,2 г; 10 ммоль). Таким чином одержують 1,8 г 1-[3-(4-фторфеніл)пропіл]піперидин-4-аміну, та використовують у наступній реакції без додаткового очищення. 22b) N-{1-[3-(4-фторфеніл)пропіл]піперидин-4-іл}-2-метил-2Н-індазол-3-карбоксамід Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,49 г; 7,6 ммоль) та 1-[3-(4-фторфеніл)пропіл]піперидин-4-амін (Приклад 22а) (1,8 г; 7,6 ммоль). Таким чином одержують 165 мг N-{1-[3-(4-фторфеніл)пропіл]піперидин-4-іл}-2-метил-2Ніндазол-3-карбоксаміду. Тпл. =162-165 °C Елементний аналіз для C23H27N4OF Знайдено % Розраховано % С 69,91 70,03 Н 6,79 6,90 N 14,19 14,20 Н-ЯМР (DMSO-d6, ppm): 1,55-1,79 (m, 4Н), 1,88 (d, 2Н), 2,01 (t, J=10,82 Гц, 2H), 2,28 (t, J=7,16 Гц, 2H), 2,58 (t, J=7,60 Гц, 2H), 2,86 (d, J = 11,69 Гц, 2Н), 3,71-3,96 (m, 1Н), 4,27(s, 3Н), 7,09 (t, J=8,92 Гц, 2Н), 7,15-7,35 (m, 4 Н), 7,66 (d, J=8,48 Гц, 1Н), 7,73 (d, J=8,48 Гц, 1Н), 8,47 (d, J=7,60 Гц, 1Н). Приклад 23 N-({1-[2-(2,4-дифторфеніл)етил]піперидин-4-іл}метил)-2-метил-2Н-індазол-3-карбоксамід (Сполука № 23) (Сполука І: R1 = Н, R2 = СН3, X = -C(O)N(R5)-, R3=4-F, R4=2-F, R5 = Н, R6+R7 = -СН2СН2-, n=1, p=2, m=2, Y=CH, W=N) 23а) {1-[2-(2,4-дифторфеніл)етил]-4-піперидин)метанамін (Сполука XI: R3=4-F, R4=2-F, R6+R7 - -СН2-СН2-, Y = СН, W=N, n=1, р = 2, m =2) {1-[2-(2,4-дифторфеніл)етил]-4-піперидин}метанамін отримують відповідно до процедури, описаної у Прикладі 7d), використовуючи як реагенти N-гексагідро-4-піридинілметил-Nфенілметиліден-амін (2,0 г; 10 ммоль), та 1-(2-брометил)-2,4-дифторбензол (2,2 г; 10 моль). Одержують 1,9 г {1-[2-(2,4-дифторфеніл)етил]-4-піперидин}метанаміну, та використовують без додаткового очищення у наступній реакції. 23b) N-({1-[2-(2,4-дифторфеніл)етил]піперидин-4-іл}метил)-2-метил-2Н-індазол-3карбоксамід Зазначену сполуку одержують відповідно до процедури, описаної у Прикладі 7е), використовуючи як реагенти 2-метил-2Н-індазол-3-карбоніл-хлорид (Приклад 1b) (1,45 г; 7,4 ммоль) та {1-[2-(2,4-дифторфеніл)етил]-4-піперидин}метанамін (Приклад 23а) (1,9 г; 7,4 ммоль). Таким чином одержують 350 мг N-({1-[2-(2,4-дифторфеніл)етил]піперидин-4-іл}метил)-2метил-2Н-індазол-3-карбоксаміду. Тпл. =122-126 °C Елементний аналіз для C23H26N4OF2 1 25 30 35 40 45 Знайдено % Розраховано % 50 С 66,81 66,97 Н 6,39 6,35 N 13,39 13,58 Н-ЯМР (DMSO-d6, ppm): 1,23 (qd, J=11,79, 3,51 Гц, 2H), 1,48-1,80 (m, 3Н), 1,96 (t, J=10,96 Гц, 2Н), 2,47 (t, J=7,50 Гц, 2Н), 2,74 (t, J=7,45 Гц, 2Н), 2,93 (d, J=11,40 Гц, 2Н), 3,25 (t, J=6,28 Гц, 2Н), 4,29 (s, 3Н), 6,95-7,04 (m, 1Н), 7,09-7,25 (m, 2Н), 7,27-7,42 (m, 2Н), 7,67 (d, J=8,77 Гц, 1H), 7,77 (d, J=8,18 Гц, 1H), 8,54 (t, J=5,70 Гц, 1Н). Фармакологія 1 25 UA 98311 C2 5 10 15 20 25 30 35 40 45 50 55 60 Тестування А На зв'язування щурячого 5-НТ2А серотонінового рецептору впливають, використовуючи, як вихідний матеріал, препарат мембран з гомогенату кори головного мозку щура, що попередньо інкубують впродовж 15 хвилин при 37 °C та завчасно оброляють 100 нМ празозином та 100 нМ піриламіном для попередження зв'язування [3Н]-кетансеріну з 1 адренергічним та Н1 гістамінергічним рецепторами. Дослідження заміщення проводять, використовуючи 1 нМ [3Н]-кетансерін як радіоліганд. Цю концентрацію вибирають на основі досліджень насичення, використовуючи різні концентрації радіоліганду (від 0,04 нМ до 10 нМ), які дають можливість одержати Вmax 709,9 нМ та Kd 1,7 нМ. Неспецифічне зв'язування вимірюють у присутності 10 мкМ метисергіду. Зазначене дослідження проводять у Tris-HCl 50 мМ буфері (рН 7,4 при 37 °C), з інкубуванням впродовж 2 годин при 37 °C. Тестові сполуки розчиняють у DMSO, потім розбавляють у буфері (кінцева DMSO концентрація 0,01 %) та поміщають на 96-лункові планшети. Метисергід та/або кетансерін використовують як еталонні сполуки. Зв'язування ініціюють додаванням 200 мкл гомогенату (вміст білку 450 мкг/мл); після інкубування, мембрани фільтрують на скляних фільтрах (GF/B) (Unifilter, Packard), що обробляють з 0,3 % поліетиленіміном. Потім фільтри промивають буферним розчином та сушать у сушильній шафі впродовж 30 хвилин при 45 °C. Сцинтиляційну рідину додають до кожної лунки, та через 10 годин вимірюють радіоактивність впродовж 1 хвилини, використовуючи TopCount (Packard). Тестові сполуки досліджують у двох повторах при 8 концентраціях (від 10-12 до 10-5 М). ІС50 значення для кожної сполуки розраховують, використовуючи аналіз нелінійної регресії (GraphPad PRISM програмне забезпечення) та константи інгібування Kі визначають, використовуючи рівняння, описане Cheng Y. та Prussof W.H. (1973) "Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (150) of an enzyme reaction." Biochem. Pharmacol. 22: 3099-3108. Тестування В На зв'язування з серотоніновим 5-НТ1А рецептором впливають, використовуючи стандартні методи, описані у Martin G.R. та Humphrey P.P.A. (1994) "Receptor for 5-hydroxytryptamine: current perspectives on classification and nomenclature." Neuropharmacol. 33: 261-273 та у May J.A., McLaughlin M.A., Sharif N.A., Hellberg M.R. та Dean T.R. (2003) "Evaluation of the ocular hypotensive response of serotonin 5-HT1A and 5-НТ2 receptor ligands in conscious ocular hypertensive cynomolgus monkeys." J. Pharmacol. Exp. Ther. 306(1): 301-309. Рекомбінантні СНО клітини для експресії людського ферменту та метерголін як еталонну сполуку використовують як вихідний матеріал. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим 5-НТ1В рецептором впливають, використовуючи стандартні методи, описані у Hoyer D., Engel G. Та Kalkman НО. (1985) "Characterization of the 5-НТ1В 125 recognition site in rat brain: binding studies with (-)[ I]iodocyanopindolol." Eur. J.Pharmacol. 18: 112. Гомогенат з кори головного мозку щура Wistar використовують як вихідний матеріал, та серотонін як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим 5-НТ1D рецептором впливають, використовуючи стандартні методи, описані у: Heuring R.E. та Peroutka S.J. (1987) "Characterization of a novel 3Н-5hydroxytryptamine binding site subtype in bovine brain membranes." J. Neurosci. 7(3): 894-903. Гомогенат з бичачого мозкового хвостатого ядра використовують як вихідний матеріал, та серотонін як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим 5-НТ2А рецептором впливають, використовуючи стандартні методи, описані у Bonhaus D.W., Bach С., De Souza A., Salazar F.H., Matsuoka B.D., Zuppan P., Chan H.W., Eglen R.M. (1995) "The pharmacology and distribution of human 5-hydroxytryptamine2B (5-НТ2В) receptor gene products: comparison with 5-HT2A and5-HT2С receptors." Br. J. Pharmacol. 115(4): 622-628; та Saucier C, Albert P.R. (1997) "Identification of an endogenous 5hydroxytryptarnine2A receptor in NIH-3T3 cells: agonist-induced down-regulation involves decreases in receptor RNA and number." J. Neurochem. 68(5): 1998-2011. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ та потім при шести концентраціях для того, щоб знайти рKі значення окремих сполук. На зв'язування з серотоніновим 5-НТ2В рецептором впливають, використовуючи стандартну методологію, описану у Bonhaus D.W., Bach C, De-Souza A., Rich Salazar F.H., Matsuoka B.D., 26 UA 98311 C2 5 10 15 20 25 30 35 40 45 50 55 Zuppan P., Chan H.W. та Eglen R.M. (1995) "The pharmacology and distribution of human 5hydroxytryptamine2B (5-HT2B) receptor gene products: comparison with 5-HT2A and 5-HT2C receptors." Br. J. Pharmacol. 115: 622-628. CHO-K1 рекомбінантні клітини для експресії людського ферменту використовують як вихідний матеріал, та кетансерін як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим 5-НТ2С рецептором впливають, використовуючи стандартну методологію, описану у: Wolf W.A. and Schutz J.S. (1997) "The serotonin 5-HT2С receptor is a prominent serotonin receptor in basal ganglia: evidence from functional studies on serotonin-mediated phosphoinositide hydrolysis." J. Neurochem. 69: 1449-1458. CHO-K1 рекомбінантні клітини для експресії людського ферменту використовують як вихідний матеріал, та SB242084 як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ та потім при шести концентраціях для того, щоб знайти рKі значення окремих сполук. На зв'язування з серотоніновим 5-НТ3 рецептором впливають, використовуючи стандартну методологію, описану у: Boess F.G., Steward L.J., Steele J.A., Liu D., Reid J., Glencorse T.A. та Martin I.L. (1997) "Analysis of the ligand binding site of the 5-HT3 receptor using site-directed mutagenesis: importance of glutamate 106." Neuropharmacology 36: 637-647 та у Millerk W.E., Fletcher P.W. та Teitler M. (1992) "Membrane-bound and solubilized brain 5-НТ3 receptor: improved radioligand binding assay using bovine area postrema or rat cortex and the radioligand [3H]GR65630, [3H]BRL43694, and [3H]LY278584." Synapse 11: 58-66. HEK293 рекомбінантні клітини для експресії людського ферменту використовують як вихідний матеріал, та MDL-72222 як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим 5-НТ4 рецептором впливають, використовуючи стандартну методологію, описану у: Grossman C.J., Kilpatrick G.J. та Bunce K.T. (1993) "Development of a radioligand binding assay for 5-HT4 receptors in guinea-pig and rat brain." Br. J. Pharmacol. 109: 618624. Гомогенат з мозкового стріатуму морської свинки використовують як вихідний матеріал, та RS-23597190 як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. Селективність тестових сполук до 5-НТ4(b) рецептору людських рекомбінантних клітин визначають, використовуючи процедуру, розроблену зі стандартної методології, описаної у Mialet J., Berque-Bestel І., Eftekhari P., Gastineau M., Giner M., Dahmoune Y., Donzeau-Gouge P., Hoebeke J., Langlois M., Sicsic S., Fischmeister R. та Lezoualc'h F. (2000) "Isolation of the serotoninergic 5-HT4(e) receptor from human heart and comparative analysis of its pharmacological profile in C6-glial and CHO cell lines." Br. J. Pharmacol. 129: 771-781. На зв'язування з 5-HT4(b) рецептором впливають, використовуючи препарат НЕК293 рекомбінантних клітин для людського 5-НТ4(b) рецептору як вихідний матеріал. У день експерименту, клітинну таблетку, що зберігали при -80 °C, відтаюють та тримають у льоді. Далі цю таблетку гомогенізують у буфері (25 мМ Tris-HCl pH 7,4; 0,1 мМ EDTA) та суспензію центрифугують при 30000 g впродовж 20 хвилин. Зазначену таблетку промивають три рази у промивному буфері та нарешті знову суспендують у інкубаційному буфері (25 мМ Tris-HCl pH 5 7,4; 0,5 мМ EDTA; 10 мМ MgSO4) при кінцевій концентрації приблизно 6 10 клітин/мл. Дослідження заміщення проводять, використовуючи 0,2 нМ [3H]-GR113808 як радіоліганд. Цю концентрацію вибирають на основі досліджень насичення, використовуючи різні концентрації радіоліганду (від 0,02 нМ до 2,5 нМ). Неспецифічне зв'язування вимірюють у присутності 10 мкМ рибосероду (Piboserod). Зазначене дослідження проводять у загальному об'ємі 250 мкл буферу 25 мМ Tris-HCl pH 7,4; 0,5 мМ EDTA; 10 мМ MgSO4 з інкубуванням впродовж 2 годин при 27 °C. Тестові сполуки розчиняють у DMSO та потім розбавляють у буфері (кінцева концентрація DMSO дорівнює 0,01 %) та поміщають на 96-лункові планшети. Рибосерод використовують як еталонну сполуку. Зв'язування ініціюють додаванням 200 мкл клітинного гомогенату (вміст білку 35-92 мкг/мл); після інкубування, мембрани фільтрують на скляних фільтрах (GF/B) (Unifilter, Packard), що обробляють з 0,1 % поліетиленіміном. Потім фільтри промивають буферним розчином та сушать у сушильній шафі впродовж 30 хвилин при 45 °C. Сцинтиляційну рідину додають до кожної лунки, та через 16 годин вимірюють радіоактивність впродовж 1 хвилини, використовуючи TopCount (Packard). Концентрацію білку визначають, використовуючи ВСА спосіб (Pierce) з BSA як стандартом. Тестові сполуки досліджують у двох повторах при 8 концентраціях (від 10-12 до 10-5 М), ІС50 значення розраховують для кожної сполуки, використовуючи аналіз нелінійної регресії (GraphPad PRISM програмне забезпечення) та константи інгібування Kі визначають, використовуючи рівняння, описане Cheng Y. та Prussof W.H. (1973) "Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (150) of an enzyme reaction." Biochem. Pharmacol. 22:3099-3108. 27 UA 98311 C2 5 10 15 20 25 30 35 40 45 На зв'язування з серотоніновим 5-HT5(A) рецептором впливають, використовуючи стандартну методологію, описану у: Rees S., den Daas 1., Foord S., Goodson S., Bull D., Kilpatriele G. та Lee M. (1994) "Cloning and characterization of the human 5-HT5A serotonin receptor." FEBS Lett. 355: 242-246. CHO-K1 рекомбінантні клітини для експресії людського ферменту використовують як вихідний матеріал, та метіотепін (methiothepin) як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим 5-НТ6 рецептором впливають, використовуючи стандартну методологію, описану у: Monsma F.J. Jr, Shen Y., Ward R.P., Hamblin M.W. and Sibley D.R. (1993) "Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drags." Моl. Pharmacol. 43: 320-327. HeLa рекомбінантні клітини для експресії людського ферменту використовують як вихідний матеріал, та метіотепін як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим 5-НТ7 рецептором впливають, використовуючи стандартну методологію, описану у: Roth B.L, Craigo S.C., Choudhary M.S., Uluer S., Monsma F.J. Jr, Shen Y., Meltzer H.Y. and Sibley D.R. (1994) "Binding of typical and atypical antipsychotic agents to 5hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors." J. Pharmacol. Exp. Ther. 268: 1403-1410 та у Shen Y., Monsma F.J. Jr, Metcalf M.A., Jose P.A., Hamblin M.W. and Sibley D.R. (1993) "Molecular cloning and expression of a 5-hydroxytryptamine 7 serotonin receptor subtype." J. Biol. Chem. 268: 18200-18204. CHO рекомбінантні клітини для експресії людського ферменту використовують як вихідний матеріал, та метіотепін як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На зв'язування з серотоніновим транспортером впливають, використовуючи стандартну методологію, описану у: Shearman L.P., McReynolds A.M., Zhou F.C., Meyer J.S. (1998) 125 "Relationship between [ I]RTI-55-labeled cocaine binding sites and the serotonin transporter in rat placenta." Am. J. Physiol. 275(6 Pt 1): C1621-1629 та у Wolf W.A. та Kuhn D.M. (1992) "Role of essential sulfhydryl groups in drug interactions at the neuronal 5-HT transporter. Differences between amphetamines and 5-HT uptake inhibitors." J. Biol. Chem. 267(29): 20820-20825. HEK293 рекомбінантні клітини для експресії людської транспортної системи використовують як вихідний матеріал, та флуоксепін як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 1 мкМ. На клітинне дослідження включення серотоніну впливають, використовуючи стандартну методологію, описану у: Gu Н., Wall S. та Rudnick G. (1994) "Stable expression of biogenic amine transporter reveals differences in inhibitor sensitivity, kinetics, and ion dependence." J. Biol. Chem. 269(10): 7124-7130. Використовують НЕК293 клітини, та флуоксепін як еталонну сполуку. Тестові сполуки досліджують у двох повторах при одній концентрації 10 мкМ. Тестування С Мишей CD-I чоловічої статі, що важать 25-30 г, використовують для дослідження судом голови у миші. Тварин обробляють внутрішньочеревинно тестовими сполуками (5 мг/кг), суспендованими у метилцелюлозі (МТС). Контрольних тварин обробляють тільки наповнювачем (МТС) тим же шляхом. Через півгодини після обробки тестовими сполуками, тваринам роблять внутрішньочеревинну ін'єкцію 5-гідрокситриптофану (5-НТР; 300 мг/кг), прекурсору серотоніну, для того, щоб викликати "судоми голови", характеристичне струшування голови, що викликають у тварин підвищенням у центральних рівнях серотоніну. Кількість судом голови, яка являє собою параметр для оцінки серотонінергічної відповіді, вимірюють у інтервали від 24 до 26 хвилин після введення 5-НТР. Як показано на Фіг. 1, сполуки даного винаходу викликають зменшення у кількості судом голови у порівнянні з тваринами, яких обробляють за допомогою тільки МТС, показуючи здатність антагонізування серотонінергічних ефектів, викликаних введенням 5-НТР. 50 ФОРМУЛА ВИНАХОДУ 1. Сполука 2-алкіліндазолу загальної формули (I): R6 X n N 55 N R7 Y R1 W p R3 m R4 R2 , (I) у якій 28
ДивитисяДодаткова інформація
Назва патенту англійською2-alkyl-indazole compound for the treatment of certain cns-related disorders, processes for the preparation thereof and pharmaceutical composition based thereon
Автори англійськоюAlisi, Maria, Alessandra, Cazzolla, Nicola, Furlotti, Guido, Maugeri, Caterina, Ombrato, Rosella, Polenzani, Lorenzo
Назва патенту російськоюСоединение 2-алкилиндазола для лечения определенных расстройств, связанных с цнс, способ его получения (варианты) и фармацевтическая композиция на его основе
Автори російськоюАлиси Мария Алессандра, Каццолла Никола, Фурлотти Гвидо, Мауджери Катерина, Омбрато Розелла, Поленцани Лоренцо
МПК / Мітки
МПК: A61P 25/00, A61K 31/435, C07D 401/12, C07D 401/06
Мітки: спосіб, композиція, варіанти, одержання, фармацевтична, 2-алкіліндазолу, певних, цнс, розладів, основі, лікування, пов'язаних, сполука
Код посилання
<a href="https://ua.patents.su/34-98311-spoluka-2-alkilindazolu-dlya-likuvannya-pevnikh-rozladiv-povyazanikh-z-cns-sposib-oderzhannya-varianti-ta-farmacevtichna-kompoziciya-na-osnovi.html" target="_blank" rel="follow" title="База патентів України">Сполука 2-алкіліндазолу для лікування певних розладів, пов’язаних з цнс, спосіб її одержання (варіанти) та фармацевтична композиція на її основі</a>
Гетероциклічна сполука, спосіб її одержання (варіанти), проміжні сполуки, фармацевтична композиція та спосіб лікування неврологічних захворювань або розладів (варіанти)

Номер патенту: 64006
Опубліковано: 16.02.2004
Автори: Палмер Майкл Джон, Уайтс Мартін Джеймс, Кемп Марк Ян, Сеннер Марк Аллен
МПК: A61P 27/16, A61P 25/00, A61P 37/00, C07D 413/04, A61P 21/00, C07D 471/14, C07D 401/04, A61P 25/28, A61K 31/454, C07D 413/14, A61P 19/00, A61K 31/4709, C07D 417/04, A61P 21/04, A61K 31/4545, A61K 31/4427, A61P 25/02, A61P 25/14, C07D 417/14, A61P 25/16, C07D 521/00, A61P 43/00, C07D 401/14, A61P 27/02
Мітки: фармацевтична, композиція, неврологічних, сполуки, одержання, проміжні, лікування, захворювань, гетероциклічна, варіанти, розладів, спосіб, сполука
Формула / Реферат:
1. Гетероциклічна похідна формули (І): , (I)або її фармацевтично прийнятна сіль, в якійA є нерозгалуженим C3-C5алкіленом, необов’язково, заміщеним C1-C6алкілом;X є O, S, NH або N(C1-C6алкіл);Y є О, S, NH або N(C1-C6алкіл);R є C-приєднаною 4-6-членною неароматичною гетероциклічною групою, що містить один атом азоту як гетероатом, згадана група, необов’язково, заміщена 1, 2 або 3 замісником(ами), кожний...
Водорозчинні проліки блокованих спиртів чи фенолів, фармацевтична композиція на їх основі, спосіб їх одержання, проміжна сполука (варіанти) та спосіб її одержання (варіанти)

Номер патенту: 73479
Опубліковано: 15.08.2005
Автори: Зігмунт Джан Дж., Сафаді Мухаммед С., Стелла Валентіно Дж., Георг Інгрід Гунда
МПК: C07F 9/655, C07F 9/6561, C07C 319/00, A61K 31/675, C07F 9/6515, A61K 31/7048, A61K 31/10, C07F 9/09, A61K 38/00, A61K 31/352, C07D 491/052, A61K 31/7042, C07K 7/64, A61K 31/661, A61K 31/355, C07D 311/72, C07H 17/04, A61K 31/437, A61P 25/00, A61K 31/665, A61P 35/00, C07C 323/12
Мітки: фармацевтична, композиція, основі, фенолів, проліки, спосіб, сполука, проміжна, спиртів, одержання, водорозчинні, блокованих, варіанти
Формула / Реферат:
1. Сполука відповідно до формули І:, IдеR-O- - залишок спиртвмісної або фенолвмісної фармацевтичної сполуки, крім таксолу і його похідних,R1 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота,R2 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота, іn являє...
Похідні дифенілацетидинону для лікування розладів ліпідного метаболізму, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі

Номер патенту: 78564
Опубліковано: 10.04.2007
Автори: Старке Інґемар, Шарет Туре, Дальстром Мікаель Ульф Йоган, Ліндквіст Анн-Марґрет, Лемурель Малін Аніта, Нордберґ Матс Петер
МПК: A61P 9/10, C07D 409/12, A61P 3/06, C07D 205/00
Мітки: метаболізму, лікування, спосіб, фармацевтична, ліпідного, композиція, варіанти, основі, похідні, одержання, дифенілацетидинону, розладів
Формула / Реферат:
1. Похідні дифенілацетидинону загальної формули,де:кільце А вибрано з групи: феніл або тієніл;Х вибрано з групи: -CR2R3-, -O-, -NRX- та -S(O)a-; де RX представляє гідроген або С1-6алкіл, а дорівнює 0-2;Y вибрано з групи: -CR4R5-, -O-, -NRZ- та -S(O)a-; де RZ представляє гідроген або С1-6алкіл, а дорівнює 0-2; причому є принаймні одна...
Похідні сульфонаміду для лікування захворювань, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі, проміжна сполука

Номер патенту: 82283
Опубліковано: 25.03.2008
Автори: Джеймс Кім, Лейн Шарлотт Еліс Луіз, Моузес Йан Брайан, Браун Алан Деніел, Томсон Ніколас Маррей
МПК: C07C 311/08
Мітки: захворювань, проміжна, сульфонаміду, сполука, одержання, основі, похідні, спосіб, лікування, фармацевтична, композиція, варіанти
Формула / Реферат:
1. Сполука загальної формули (1):де (CH2)n-C(=O)Q1 група розташована в мета- або пара-положенні, R1 і R2, незалежно, вибирають з Н і С1-С4 алкілу, n є 0, 1 або 2 і Q1 є групою, що вибирають з:і групи *-NR11-Q2-A, де р є 1 або 2, Q2 є С1-С4 алкілен, R11 є Н або С1-С4 алкіл і...
Фармацевтична композиція на основі тонкоподрібненого еплеренону (варіанти), спосіб її одержання та спосіб лікування розладів, опосередкованих альдостероном (варіанти)

Номер патенту: 74141
Опубліковано: 15.11.2005
Автори: Тхосар Шілпа С., Гокхал Раджів Д., Толверт Двайн С.
МПК: A61K 9/48, A61P 9/00, A61K 9/22, A61K 31/585
Мітки: розладів, альдостероном, одержання, варіанти, тонкоподрібненого, опосередкованих, лікування, еплеренону, спосіб, фармацевтична, композиція, основі
Формула / Реферат:
1. Фармацевтична композиція, яка містить тонкоподрібнений еплеренон, розміри щонайменше 90% частинок якого становлять від приблизно 25 мкм до приблизно 400 мкм, в кількості від приблизно 10 мг до приблизно 1000 мг і фармацевтично прийнятний носій.2. Фармацевтична композиція за п. 1, яка містить тонкоподрібнений еплеренон в кількості від приблизно 20 мг до приблизно 400 мг.3. Фармацевтична композиція за п. 1, яка містить...
Попередній патент: Композиції для знищення членистоногих у вигляді суспензійного концентрату та спосіб контролю членистоногого паразита
Наступний патент: Антагоністи прогестеронового рецептора
Випадковий патент: Спосіб кріплення гірничої виробки