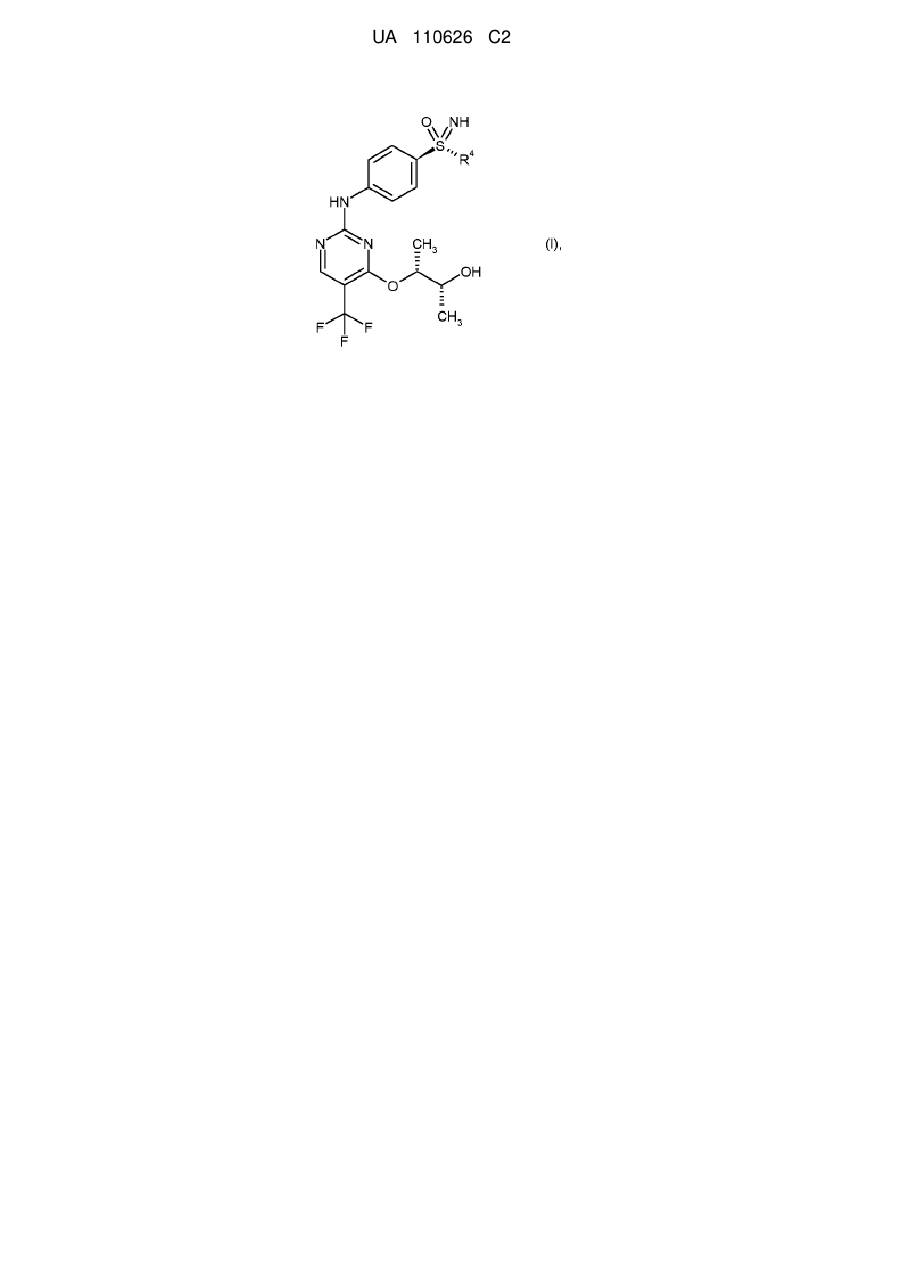
Спосіб одержання пан-інгібіторів циклінзалежної кінази формули (i), а також проміжні продукти цього процесу одержання
Номер патенту: 110626
Опубліковано: 25.01.2016
Автори: Крюгер Йоахім, Грес Йорг, Ловіс Каі, Хассфельд Йорма




Формула / Реферат
1. Спосіб одержання сполук загальної формули (І)
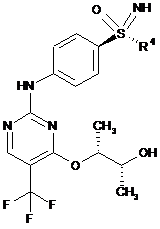 , (I)
, (I)
в якій
R4 означає С1-С6-алкільну групу або С3-С7-циклоалкільне кільце,
в якому здійснюють принаймні одну із наведених далі стадій:
І.а) алкілування 4-нітротіофенолу в присутності карбонату калію в N-метилпіролідиноні (NMP) з одержанням нітрофенілсульфіду формули (I-1)
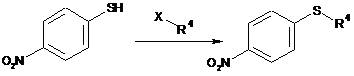 , I-1
, I-1
в якій
X означає Вr, Сl, І, O-SO2-CH3 або О-SО2-(4-метилфеніл),
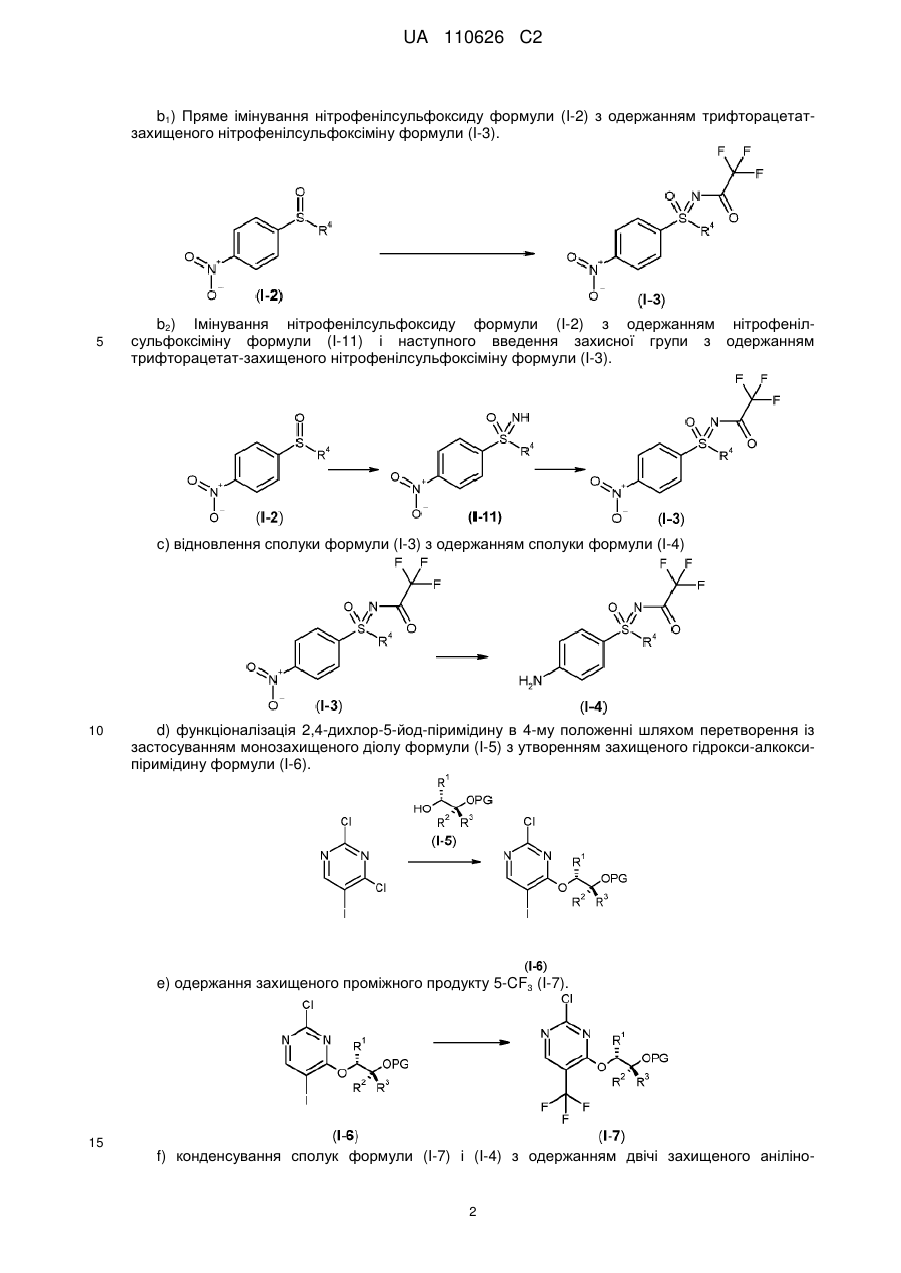
І.b) окиснювальне амінування нітрофенілсульфіду формули (I-1) з одержанням трифторацетатзахищеного нітрофенілсульфіліміну формули (I-10)
 ,
,
I.с) окиснення трифторацетатзахищеного нітрофенілсульфіліміну формули (I-10) з одержанням трифторацетатзахищеного нітрофенілсульфоксіміну формули (I-3) і наступне відщеплення захисних груп з одержанням нітрофенілсульфоксіміну формули (I-11)
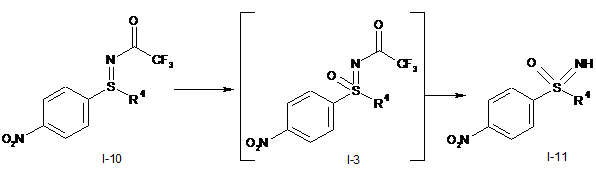 ,
,
l.d) розщеплення рацемату нітрофенілсульфоксіміну формули (I-11) за допомогою (+)-ди-O-п-толуоїл-D-винної кислоти
 ,
,
причому R-енантіомер нітрофенілсульфоксіміну формули (I-11-R) потім вивільняють із солей і знову вводять трифторацетатну захисну групу з утворенням R-енантіомера трифторацетатзахищеного нітрофенілсульфоксіміну формули (I-3-R),
І.e) гідрування трифторацетатзахищених нітрофенілсульфоксімінів формули (I-3-R) з одержанням трифторацетатзахищених аніліносульфоксімінів формули (I-4-R) із застосуванням легованого залізом паладієвого каталізатора
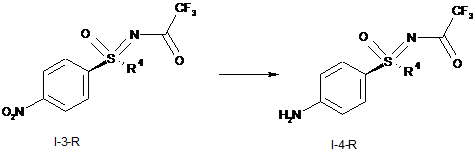 ,
,
I.f) одержання (2R,3R)-3-(бензилокси)бутан-2-олу (I-5-А) у двостадійному процесі з утворенням (4R,5R)-4,5-диметил-2-феніл-1,3-діоксолану (I-12-А), причому першу стадію проводять із застосуванням піримідин-п-толуолсульфонату в толуолі, а потім проводять відновлення гідриду діізобутилалюмінію в толуолі
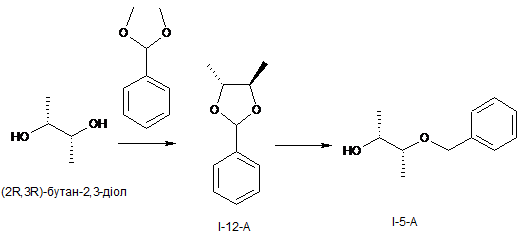 ,
,
I.g). конденсування сполуки (І-5-А) з 2,4-дихлор-5-трифторметилпіримідином з одержанням 4-{[(2R,3R)-3-(бензилокси)бутан-2-іл]окси}-2-хлор-5-(трифторметил)-піримідину (І-7-А) із застосуванням літієвих основ в етерних розчиниках
 ,
,
I.h) одержання солей бензолсульфонової кислоти двічі захищених анілінопіримідинів формули (I-8-R-BSA) шляхом здійснення каталізованого бензолсульфоновою кислотою конденсування сполук (1-7-А) і (I-4-R)
 ,
,
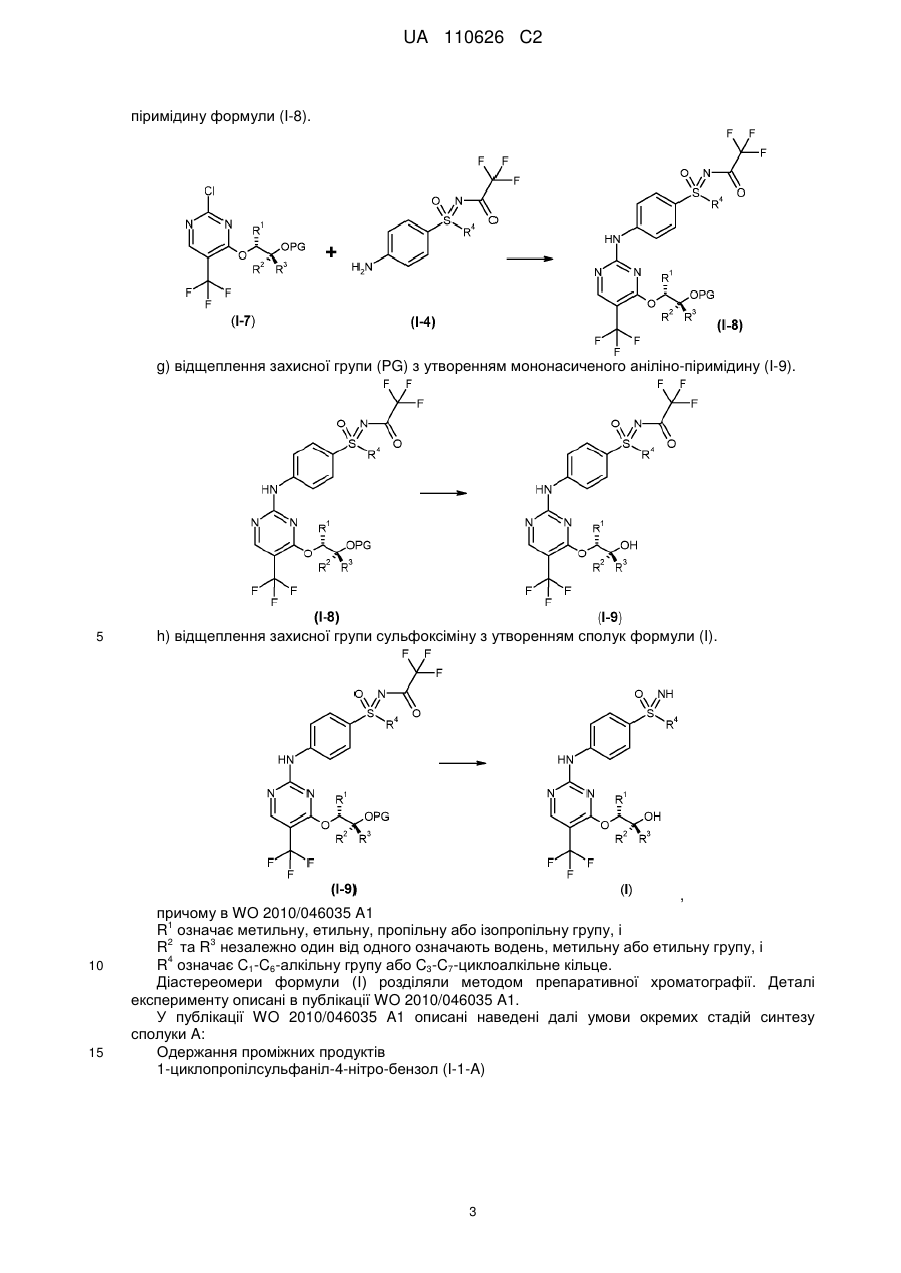
І.і) відщеплення захисних груп у солях бензолсульфонової кислоти двічі захищених анілінопіримідинів формули (I-8-R-BSA) шляхом гідрування із застосуванням паладію на активованому вугіллі і водню в метанолі, а також шляхом обробки карбонатом калію в метанолі з одержанням сполук формули (І)
 .
.
2. Спосіб за пунктом 1, причому на стадії І.b) 1,3-дибром-5,5-диметилгідантоїн застосовують як окиснювальний засіб, а трифторацетамід застосовують як реагент.
3. Спосіб за пунктом 1, причому на стадії І.с) проводять окиснення із застосуванням пероксомоносульфату калію (Охоnе®).
4. Спосіб за пунктом 1, причому на стадії I.d) проводять кристалізацію нітрофенілсульфоксіміну формули (I-11) із застосуванням (+)-ди-О-п-толуоїл-О-винної кислоти в ацетонітрилі або пропіонітрилі.
5. Спосіб за пунктом 1, причому на стадії I.g) як літієву основу застосовують гексаметилдисилазид літію, а як етерний розчинник застосовують тетрагідрофуран.
6. Сіль формули (I-11-R-D-Tol-Tart) нітрофенілсульфоксімінів формули (I-11-R) з (+)-ди-О-п-толуоїл-О-винною кислотою
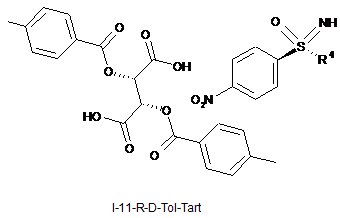 ,
,
причому R4 означає С1-С6-алкільну групу або С3-С7-циклоалкільне кільце.
7. Сіль за пунктом 6, що є сіллю формули (I-11-A-R-D-Tol-Tart)
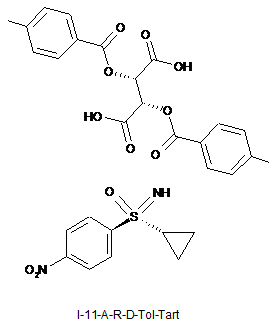 .
.
8. Проміжний продукт формул (I-8-R-BSA)
 ,
,
причому R4 означає С1-С6-алкільну групу або С3-С7-циклоалкільне кільце.
9. Проміжний продукт за пунктом 8, що є проміжним продуктом формули (I-8-A-R-BSA)
 .
.
Текст
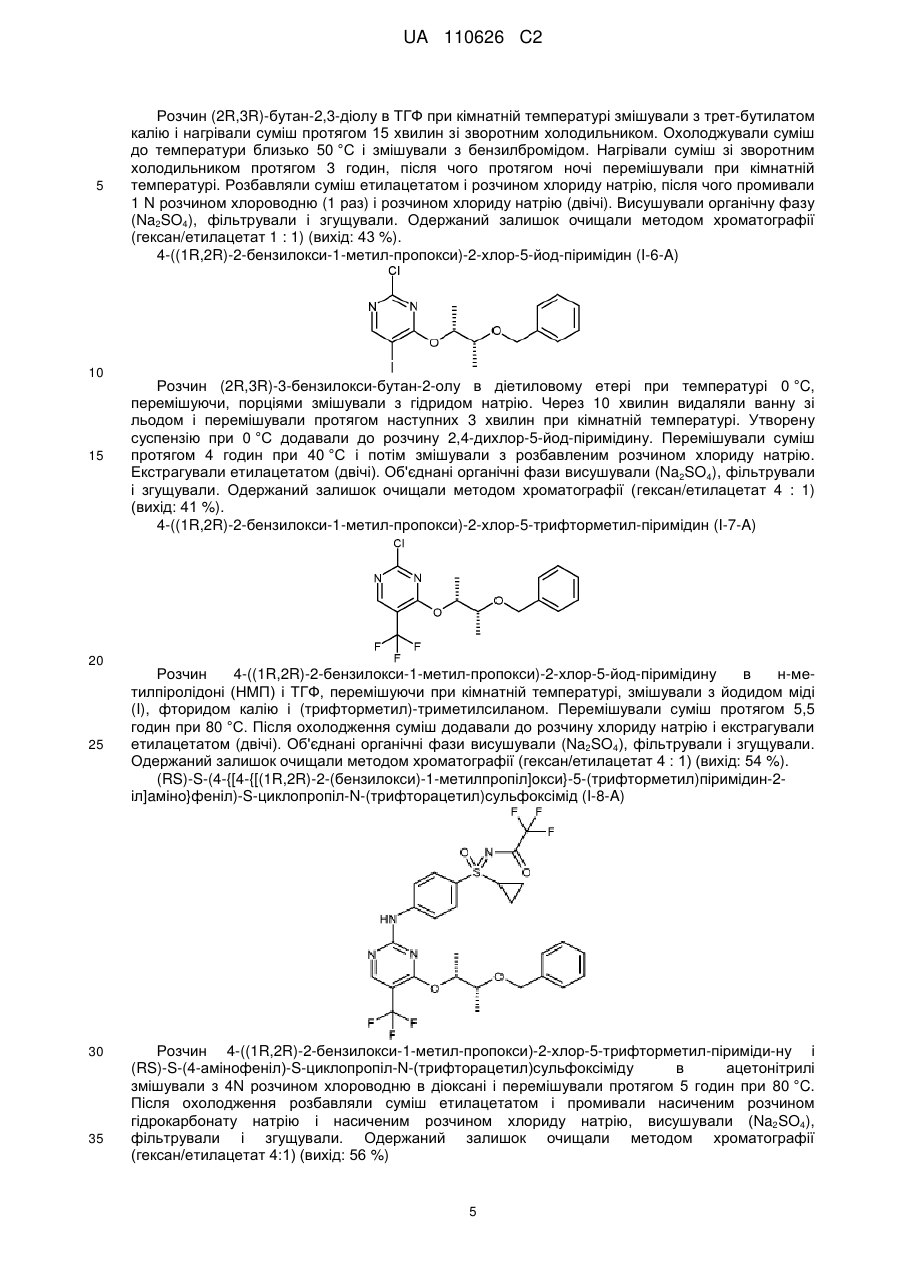
Реферат: Винахід стосується нового способу одержання пан-інгібіторів циклінзалежної кінази формули (І), а також проміжних продуктів цього процесу одержання. NH O S R 4 HN N N CH3 OH O F F F CH3 (I) UA 110626 C2 (12) UA 110626 C2 UA 110626 C2 5 10 15 20 25 30 Винахід стосується нового способу одержання пан-інгібіторів циклін-залежної кінази формули (I), а також проміжних продуктів цього процесу одержання. Новий спосіб стосується сполук формули (I), зокрема сполуки (2R,3R)-3-{[2-{[4-(Sциклопропілсульфонімідоїл)феніл]аміно}-5-(трифторметил)піримідин-4-іл]oкси}бутан-2-ол (сполука A), протипухлинна активність яких зумовлена механізмом цитотоксичної дії. Було розроблено спосіб одержання сполуки загальної формули (I), в якій 4 R означає C1-C6-алкільну групу або C3-C7-циклоалкільне кільце, який є придатним до масштабування (scale-up) і позбавлений недоліків способів одержання з рівня техніки. Цей спосіб є особливо придатним для одержання сполуки A Сполука А В основу заявки було покладено такі визначення: C1-C6-алкіл C1-C6-алкільною групою в кожному випадку є нерозгалужений або розгалужений алкільний залишок, такий як, наприклад, метильний, етильний, пропільний, ізопропільний, бутильний, ізобутильний, втор-бутильний, трет-бутильний, пентильний, ізопентильний або гексильний залишок. C3-C7-циклоалкіл C3-C7-циклоалкільним кільцем є циклопропільне, циклобутильне, циклопентильне, циклогексильне або циклогептильне кільце. Сполуки загальної формули (I), зокрема також сполука A, і спосіб їх одержання описані в публікації WO 2010/046035 A1, причому їх розкриття відображає найближчий рівень техніки. Спосіб згідно з публікацією WO 2010/046035A 1 є 10-стадійним конвергентним процесом із загальним виходом для найдовшої послідовності близько 7 %. Спосіб згідно з WO 2010/046035 A1 включає принаймні одну із описаних далі стадій: a) Окиснення нітрофенілсульфіду формули (I-1) з одержанням нітрофеніл-сульфоксиду формули (I-2). 1 UA 110626 C2 b1) Пряме імінування нітрофенілсульфоксиду формули (I-2) з одержанням трифторацетатзахищеного нітрофенілсульфоксіміну формули (I-3). 5 b2) Імінування нітрофенілсульфоксиду формули (I-2) з одержанням нітрофенілсульфоксіміну формули (I-11) і наступного введення захисної групи з одержанням трифторацетат-захищеного нітрофенілсульфоксіміну формули (I-3). с) відновлення сполуки формули (I-3) з одержанням сполуки формули (I-4) 10 d) функціоналізація 2,4-дихлор-5-йод-піримідину в 4-му положенні шляхом перетворення із застосуванням монозахищеного діолу формули (I-5) з утворенням захищеного гідрокси-алкоксипіримідину формули (I-6). e) одержання захищеного проміжного продукту 5-CF3 (I-7). 15 f) конденсування сполук формули (I-7) і (I-4) з одержанням двічі захищеного аніліно 2 UA 110626 C2 піримідину формули (I-8). g) відщеплення захисної групи (PG) з утворенням мононасиченого аніліно-піримідину (I-9). 5 10 15 h) відщеплення захисної групи сульфоксіміну з утворенням сполук формули (I). , причому в WO 2010/046035 A1 1 R означає метильну, етильну, пропільну або ізопропільну групу, і 2 3 R та R незалежно один від одного означають водень, метильну або етильну групу, і 4 R означає C1-C6-алкільну групу або C3-C7-циклоалкільне кільце. Діастереомери формули (I) розділяли методом препаративної хроматографії. Деталі експерименту описані в публікації WO 2010/046035 A1. У публікації WO 2010/046035 A1 описані наведені далі умови окремих стадій синтезу сполуки A: Одержання проміжних продуктів 1-циклопропілсульфаніл-4-нітро-бензол (I-1-A) 3 UA 110626 C2 5 10 15 20 25 30 Розчин циклопропантіолу в суміші тетрагідрофуран (ТГФ)/діетиловий етер порціями змішували з гідридом натрію і перемішували при кімнатній температурі. Потім порціями додавали 1-фтор-4-нітро-бензол. Суміш перемішували протягом 2 годин при 40 °C. Після охолодження суміш додавали у воду та екстрагували бензолом (тричі). Об'єднані органічні фази згущували і очищали залишок методом хроматографії (гексан/етилацетат 95 : 5) (вихід: 61 %). (RS)-1-циклопропансульфініл-4-нітро-бензол (I-2-A) Розчин 1-циклопропілсульфаніл-4-нітро-бензолу в ацетонітрилі змішували з хлоридом заліза (III) і перемішували при кімнатній температурі. Потім до суміші порціями додавали перйодну кислоту. Перемішували суміш протягом 30 хвилин, після чого, перемішуючи, виливали в охолоджений насичений розчин тіосульфату натрію. Екстрагували етилацетатом (двічі). Об'єднані органічні фази висушували (Na2SO4), фільтрували і згущували. Одержаний залишок очищали методом хроматографії (гексан/етилацетат 1 : 1) (вихід: 76 %). (RS)-S-циклопропіл-S-(4-нітрофеніл)-N-(трифторацетил)сульфоксімід (I-3-A) Суспензію (RS)-1-циклопропансульфініл-4-нітробензолу, трифторацетаміду, йодбензолдіацетату та оксиду магнію в дихлорметані в атмосфері аргону змішували з димером ацетату родію (II) і протягом ночі перемішували при кімнатній температурі. Одержану суміш фільтрували крізь целіт і згущували. Одержаний залишок очищали методом хроматографії (гексан/етилацетат 2 : 1) (вихід: 78 %). (RS)-S-(4-амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксімід (I-4-A) Розчин (RS)-S-циклопропіл-S-(4-нітрофеніл)-N-(трифторацетил)сульфоксіміду в етанолі і тетрагідрофурані (ТГФ) змішували з паладієм на вугіллі і піддавали гідруванню протягом однієї години під нормальним тиском при 25 °C. Знову змішували суміш із паладієм на вугіллі та гідрували протягом наступних 4,5 годин під нормальним тиском. Суміш фільтрували, фільтрат повторно змішували з паладієм на вугіллі, після чого гідрували протягом 45 хвилин. Суміш фільтрували і згущували (вихід: 93 %). (2R,3R)-3-бензилокси-бутан-2-ол (I-5-A) 4 UA 110626 C2 5 10 15 20 25 30 35 Розчин (2R,3R)-бутан-2,3-діолу в ТГФ при кімнатній температурі змішували з трет-бутилатом калію і нагрівали суміш протягом 15 хвилин зі зворотним холодильником. Охолоджували суміш до температури близько 50 °C і змішували з бензилбромідом. Нагрівали суміш зі зворотним холодильником протягом 3 годин, після чого протягом ночі перемішували при кімнатній температурі. Розбавляли суміш етилацетатом і розчином хлориду натрію, після чого промивали 1 N розчином хлороводню (1 раз) і розчином хлориду натрію (двічі). Висушували органічну фазу (Na2SO4), фільтрували і згущували. Одержаний залишок очищали методом хроматографії (гексан/етилацетат 1 : 1) (вихід: 43 %). 4-((1R,2R)-2-бензилокси-1-метил-пропокси)-2-хлор-5-йод-піримідин (I-6-A) Розчин (2R,3R)-3-бензилокси-бутан-2-олу в діетиловому етері при температурі 0 °C, перемішуючи, порціями змішували з гідридом натрію. Через 10 хвилин видаляли ванну зі льодом і перемішували протягом наступних 3 хвилин при кімнатній температурі. Утворену суспензію при 0 °C додавали до розчину 2,4-дихлор-5-йод-піримідину. Перемішували суміш протягом 4 годин при 40 °C і потім змішували з розбавленим розчином хлориду натрію. Екстрагували етилацетатом (двічі). Об'єднані органічні фази висушували (Na2SO4), фільтрували і згущували. Одержаний залишок очищали методом хроматографії (гексан/етилацетат 4 : 1) (вихід: 41 %). 4-((1R,2R)-2-бензилокси-1-метил-пропокси)-2-хлор-5-трифторметил-піримідин (I-7-A) Розчин 4-((1R,2R)-2-бензилокси-1-метил-пропокси)-2-хлор-5-йод-піримідину в н-метилпіролідоні (НМП) і ТГФ, перемішуючи при кімнатній температурі, змішували з йодидом міді (I), фторидом калію і (трифторметил)-триметилсиланом. Перемішували суміш протягом 5,5 годин при 80 °C. Після охолодження суміш додавали до розчину хлориду натрію і екстрагували етилацетатом (двічі). Об'єднані органічні фази висушували (Na2SO4), фільтрували і згущували. Одержаний залишок очищали методом хроматографії (гексан/етилацетат 4 : 1) (вихід: 54 %). (RS)-S-(4-{[4-{[(1R,2R)-2-(бензилокси)-1-метилпропіл]oкси}-5-(трифторметил)піримідин-2іл]aміно}феніл)-S-циклопропіл-N-(трифторацетил)сульфоксімід (I-8-A) Розчин 4-((1R,2R)-2-бензилокси-1-метил-пропокси)-2-хлор-5-трифторметил-піриміди-ну і (RS)-S-(4-амінофеніл)-S-циклопропіл-N-(трифторацетил)сульфоксіміду в ацетонітрилі змішували з 4N розчином хлороводню в діоксані і перемішували протягом 5 годин при 80 °C. Після охолодження розбавляли суміш етилацетатом і промивали насиченим розчином гідрокарбонату натрію і насиченим розчином хлориду натрію, висушували (Na2SO4), фільтрували і згущували. Одержаний залишок очищали методом хроматографії (гексан/етилацетат 4:1) (вихід: 56 %) 5 UA 110626 C2 (RS)-S-циклопропіл-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]oкси}-5(трифторметил)піримідин-2-іл]aміно}феніл)-N-(трифторацетил)сульфоксімід (I-9-A) 5 10 15 20 25 30 35 40 Розчин (RS)-S-(4-{[4-{[(1R,2R)-2-(бензилокси)-1-метилпропіл]oкси}-5(трифторметил)піримідин-2-іл]aміно}феніл)-S-циклопропіл-N-(трифторацетил)сульфоксіміду в етанолі змішували з паладієм на вугіллі (конц. 10 %) і гідрували під нормальним тиском при кімнатній температурі. Суміш фільтрували і згущували (вихід: 79 %). Одержання сполуки A Розчин (RS)-S-циклопропіл-S-(4-{[4-{[(1R,2R)-2-гідрокси-1-метилпропіл]oкси}-5-(трифторметил)піримідин-2-іл]aміно}феніл)-N-(трифторацетил)сульфоксіміду в 35 мл мета-нолу змішували з карбонатом калію і перемішували протягом 1,5 годин при кімнатній температурі. Розбавляли насиченим розчином хлориду натрію і екстрагували етилацетатом (тричі). Об'єднані органічні фази висушували (Na2SO4), фільтрували і згущували. Суміш діастереомерів розділяли шляхом препаративної високоефективної рідинної хроматографії (ВЕРХ) на чисті стереоізомери: Колонка: Chiralpak IA 5 мкм 250 30 мм Елюенти: гексан/етанол 8 : 2 Потік: 40,0 мл/хв. Детектор: UV 254 нм Температура: кімнатна температура Час утримання: 10,8-13,4 хв.; стереоізомер 1 13,6-18,5 хв.; стереоізомер 2 (сполука A) Такий спосіб одержання сполуки формули (I) згідно з публікацією WO 2010/046035 A1 є непридатним для виробничого процесу. Критичні аспекти описані далі: - Множину проміжних продуктів очищують методом хроматографії. У великих масштабах це потребує великих витрат часу і коштів. - Вихідний матеріал (I-1) одержують із циклопропілсульфіду, який не є комерційно доступним у великій кількості. - Для одержання сполуки (I-2) застосовували метод окиснення з утворенням рацематів. Тому стереоізомери наприкінці синтезу необхідно розділяти методом хроматографії. Оскільки розділення здійснюють лише наприкінці синтезу, загальний вихід послідовності реакцій синтезу значно зменшується. - При одержанні сполуки (I-3) використовують велику кількість димеру ацетату родію (II). Він є дорогою речовиною, і необхідно видаляти родій для уникнення забруднення активної речовини. Йодбензолдіацетат є непридатним для масштабування (scale-up), оскільки він не є комерційно доступним у великій кількості; він є також потенційно вибухонебезпечною речовиною. - Альтернативне одержання сполуки (I-3) з проміжним утворенням сполук (I-2) і (I-2/3) не може бути здійснене простим способом із причин, пов'язаних з необхідністю забезпечення безпеки технологічного процесу, оскільки в ньому застосовують токсичні і вибухонебезпечні речовини, такі як ацид натрію або О-мезитиленсульфоніл-гідроксиламін (MСГ). - Синтез сполуки (I-5-A) не є селективним, оскільки відбувається також подвійне алкилювання. Тому вихід становить лише 43 %. - Послідовність реакцій (I-6) - (I-7) не є конвергентною, оскільки трифторметильна група не введена вже в піримідиновий компонент. Вихід обох стадій є низьким. Оскільки процеси перетворення відбуваються з утворенням багатьох побічних компонентів, доводиться здійснювати дороге очищення методом хроматографії. - Проміжний продукт (I-8) утворюється у формі олії, яка може бути очищена лише методом 6 UA 110626 C2 5 10 15 хроматографії. Оброблення олії в технічному масштабі є дуже складним, а її стабільність при зберіганні порівняно з твердою речовиною є низькою. - На стадії (I-9) діастереомери розділяють методом препаративної хроматографії. Цей метод потребує великих витрат часу і коштів. Окрім цього, значно зменшується загальний вихід, оскільки розділення здійснюють лише на останній стадії синтезу. Ці аспекти мають бути поліпшені чи оптимізовані в рамках розширення синтезу до масштабу від кількасот грамів до кількох кілограмів. Тому задачею винаходу було розроблення способу одержання пан-інгібіторів циклінзалежної кинази загальної формули (I), зокрема сполуки A, позбавленого вищеописаних недоліків. I. Стадії відповідного винаходові способу одержання сполук загальної формули (I) Відповідний винаходові спосіб одержання відрізняється переважними стадіями, а також проміжними продуктами. Відповідний винаходові спосіб одержання сполук загальної формули (I) включає принаймні одну із описаних далі стадій: I.a). Алкілювання 4-нітротіофенолу в присутності розчину карбонату калію в Nметилпіролідиноні (NMП) з одержанням нітрофенілсульфіду формули (I-1) , 20 25 в якій X означає Br, Cl, I, O-SO2-CH3 або O-SO2-(4-метилфеніл). I.b). Окиснювальне амінування нітрофенілсульфіду формули трифторацетат-захищеного нітрофеніл-сульфіліміну формули (I-10) (I-1) з одержанням I.c) Окиснення трифторацетат-захищеного нітрофеніл-сульфіліміну формули (I-10) з одержанням трифторацетат-захищеного нітрофеніл-сульфоксіміну формули (I-3) і наступне видалення захисної групи з одержанням нітрофеніл-сульфоксіміну формули (I-11) I.d) Розщеплення рацемату нітрофеніл-сульфоксіміну формули (I-11) за допомогою (+)-ди-Oп-толуоїл-D-винної кислоти 7 UA 110626 C2 5 , причому R-енантіомер нітрофеніл-сульфоксіміну формули (I-11-R) потім вивільняють із солей і знову вводять трифторацетатну захисну групу з утворенням R-енантіомера трифторацетат-захищеного нітрофеніл-сульфокс-іміну формули (I-3-R). I.e) Гідрування трифторацетат-захищених нітрофеніл-сульфоксімінів формули (I-3-R) з одержанням трифторацетат-захищених аніліно-сульфоксімінів формули (I-4-R) із застосуванням легованого залізом паладієвого каталізатора 10 . I.f). Одержання (2R,3R)-3-(бензилокси)бутан-2-oлу (I-5-A) у двостадійному процесі з проміжним продуктом (4R,5R)-4,5-диметил-2-феніл-1,3-діоксоланом (I-12-A), причому першу стадію здійснюють із застосуванням розчину піридин-п-толуолсульфонату в толуолі, а потім здійснюють відновлення гідриду діізобутилалюмінію в толуолі, 15 I.g). Конденсування сполуки(I-5-A) з 2,4-дихлор-5-трифторметилпіримідином із одержанням 4-{[(2R,3R)-3-(бензилокси)бутан-2-іл]oкси}-2-хлор-5-(трифтор-метил)піримідину (I-7-A) із застосуванням літієвих основ в етерному розчиннику I.h) Одержання двічі захищених солей бензолсульфонової кислоти аніліно-піримідинів 8 UA 110626 C2 формули (I-8-R-BSA) шляхом здійснення каталізованого бензолсульфоновою кислотою конденсування сполуки (I-7-A) зі сполукою (I-4-R) 5 10 15 I.i) Відщеплення захисних груп у солях бензолсульфонової кислоти двічі захищених анілінопіримідинів формули (I-8-R-BSA) шляхом гідрування із застосуванням паладію на активованому вугіллі і водню в метанолі, а також шляхом обробки карбонату калію в метанолі з одержанням сполук формули (I) . Стадії одержання верхньої структурної ланки сполук згідно з формулою (I) I.a) Одержання нітрофеніл-сульфідів формули (I-1) Предмет винаходу стосується стадії алкілювання 4-нітрофенолу. Згідно з публікацією WO 2010/046035 A1 вихідний матеріал (I-1) одержували з циклопропілсульфіду. Ця речовина не є комерційно доступною у великій кількості. Тому аклілювання комерційно доступного 4-нітротіофенолу здійснювали із застосуванням агентів 4 алкілювання (X-R ) в присутності допоміжної основи, причому X означає Br, Cl, I, O-SO2-CH3 або O-SO2-(4-метилфеніл). Придатними до застосування основами є карбонати натрію, калію або цезію; переважним є карбонат калію. Придатними до застосування розчинниками є N,Nдиметилформамід. N-метилпіролідинон, диметилсульфоксид, N,N-диметилацетамід; 9 UA 110626 C2 переважним є N-метилпіролідинон. 5 10 15 20 25 30 35 40 45 Фіг. 1 Інші предмети винаходу стосуються окиснювального амінування нітрофеніл-сульфідів формули (I-1) з одержанням трифторацетат-захищених нітрофеніл-сульфілімінів формули (I-10) (фіг. 2) і наступного окиснення з одержанням нітрофеніл-сульфоксімінів формули (I-11) (фіг. 3). I.b) Одержання трифторацетат-захищених нітрофеніл-сульфілімінів формули (I-10) Фіг. 2 Рівень техніки для одержання сульфілімінів Метою було пряме амінування сульфідів з одержанням добре придатних для препаративного використання трифторацетат-захищених сульфілімінів із застосуванням простих вихідних матеріалів, таких як, наприклад, 2,2,2-трифторацетамід (CF3CONH2). У публікації Carreira et al. (Org. Lett. 1999, 1, 149-151) описане каталізоване міддю пряме амінування з одержанням трифторацетат-захищених сульфілімінів за допомогою літійованого гідроксиламіну трифтороцтової кислоти (ТФК), який, проте, спочатку необхідно одержувати двома стадіями, і який не є комерційно доступним. Це енантіоселективне перетворення може бути здійснене із застосуванням стехіометричної кількості нітридо-марганцевого комплексу (Helv. Chim. Acta. 2002, 3773-3783). Згідно з публікацією Bolm et al. (Tetrahedron Letters 2005), можливим є пряме імінування сульфідів без застосування металів. Пропонують використовувати п-нітрофенілсульфонамід (носиламід, Nos-NH2) та (діацетоксийод)бензол (PhI(OAc)2) і одержують носил-захищені сульфіліміни після нагрівання протягом 16 годин зі зворотним холодильником. Проте, ці сполуки є менш придатними до масштабування (scale-up), оскільки п-нітрофенілсульфонамідна захисна група дуже важко піддається видаленню, а (діацетоксийод)бензол не є комерційно доступним у великій кількості. Придатними до застосування окиснювальними засобами у відповідному винаходові перетворенні згідно з фіг. 2 є, зокрема, N-бромсукцинімід, йод, гіпобромід натрію, 1,3-дибром5,5-диметилгідантоїн, N-хлорсукцинімід і трихлорціанурова кислота в присутності основ, таких як карбонат цезію, трет-бутилат калію, трет-бутилат натрію, водний натрієвий луг, метанолат натрію, етанолат натрію, гідрид натрію (NaH), в розчинниках, таких як метанол, дихлорметан, тетрагідрофуран-вода, ацетонітрил, ацетонітрил-вода, тетрагідрофуран (ТГФ, пропіонітрил, метил-трет-бутиловий етер, 1,4-діоксан, хлорбензол. Переважним окиснювальним засобом є 1,3-дибром-5,5-диметилгідантоїн. Переважними комбінаціями розчинників із основами є комбінації ацетонітрил-карбонат цезію, 1,4-діоксан-гідрид натрію, дихлорметан-трет-бутилат калію, ацетонітрил-гідрид натрію, тетрагідрофуран-гідрид натрію або метил-трет-бутиловий етер-гідрид натрію. Бажана реакція повністю завершується вже при 20 °C за кілька годин без додавання каталізатора. Порівняно з відомими з літератури способами нове окиснювальне амінування, як зображено на фіг. 2, забезпечує наведені далі переваги: можна відмовитися від застосування дорогого і потенційно вибухонебезпечного (діацетоксийод)бензолу, а також від додавання солей металів; сульфоксид майже не утворюється, і реакція відбувається в м'яких умовах вже при 20 °C із застосуванням комерційно доступних компонентів і реагентів; 10 UA 110626 C2 5 трифторацетатна група дуже легко піддається гідролізу (наприклад, із застосуванням карбонату калію в метанолі) і тому має високу препаративну цінність. Не лише нітрофеніл-сульфіди формули (I-1) можуть бути піддані окиснювальному амінуванню згідно зі стадією I.b). Інші трифторацетат-захищені сульфіліміни також можуть бути одержані таким шляхом. У таблиці 1 наведені інші сульфіліміни, які можуть бути одержані таким чином. Таблиця 1 I Сульфілімін Речовина Вихід O CF3 N S 1 81 % O2 N O N S 2 CF3 CH3 88 % H3 C O N S 3 CF3 CH3 80 % MeO O N S 4 CF3 CH3 75 % Cl O N S 5 CF3 CH3 74 % O N S 6 CF3 CH3 I.c) Одержання нітрофеніл-сульфоксімінів формули (I-11) 11 71 % UA 110626 C2 Окиснення трифторацетат-захищеного нітрофеніл-сульфіліміну (I-10) з одержанням нітрофеніл-сульфоксіміну (I-11) здійснюють переважно із застосуванням пероксомоносульфату ® калію (Oxone ) як окиснювального засобу. 5 10 15 20 25 Фіг. 3 Бажана реакція окиснення протікає особливо швидко в лужному діапазоні pH. У цих умовах водночас відбувається відщеплення трифторацетатної групи, завдяки чому забезпечується можливість здійснення в разі необхідності наступної стадії відщеплення захисної групи однореакторним методом. Реакцію здійснюють особливо переважно в суміші метанолу з водою і додають ® тетраметиленсульфон (сульфолан) як солюбілізатор. Пероксомоносульфат калію (Oxone ) додають порціями і після додавання кожної порції встановлюють значення pH=10. I.d) Розщеплення рацематів нітрофеніл-сульфоксімінів формули (I-11). Інший предмет винаходу стосується розщеплення рацематів нітрофеніл-сульфоксімінів формули (I-11). Розщеплення рацематів здійснюють на описаній далі стадії. Фіг. 4 Неочікувано, наприклад для нітрофеніл-сульфоксімінів формули (I-11-A), було виявлено, що за допомогою (+)-ди-O-п-толуоїл-D-винної кислоти одержують співвідношення між енантіомерами у кристалізаті, яке становить 95:5. Як розчинники можуть бути застосовані ацетонітрил, пропіонітрил або толуол. Переважно застосовують ацетонітрил або пропіонітрил. Процес кристалізації може бути інтегрований у технологічний процес таким чином, що захисну трифторацетатну групу в (I-3) відщепляють із застосуванням розчину карбонату калію в метанолі, і необроблений нітрофеніл-сульфоксімін (I-11) із застосуванням (+)-ди-O-п-толуоїл-Dвинної кислоти перетворюють на (I-11-R-D-Tol-Tart.). Фіг. 5 12 UA 110626 C2 Толуоїл-D-винну кислоту видаляють із солі шляхом екстрагування із застосуванням основи, а введення захисної групи в оптично активний нітрофеніл-сульфоксімін формули (I-11-R) може бути здійснене однореакторним методом із застосуванням трифтороцтового ангідриду в присутності триетиламіну з одержанням сполуки (I-3-R). 5 10 15 20 25 30 Фіг. 6 I.e) Гідрування трифторацетат-захищених нітрофеніл-сульфоксімінів формули (I-3-R) з одержанням трифторацетат-захищених аніліно-сульфоксімінів формули (I-4-R) Інший предмет винаходу стосується гідрування трифторацетат-захищених нітрофенілсульфоксімінів (I-3-R) з одержанням трифторацетат-захищених аніліно-сульфоксімінів формули (I-4-R) в присутності легованого залізом паладієвого каталізатора. Відновлення нітрогрупи в сполуці (I-3-R) з одержанням відповідного аніліну (I-4-R) може бути ефективно здійснене шляхом гідрування іммобілізованих паладієвих каталізаторів. Переважними є леговані залізом паладієві каталізатори на вугіллі. Як розчинники можуть бути застосовані метанол, етанол, ізо-пропанол, тетрагідрофуран або оцтова кислота. Переважним є метанол. Фіг. 7 Стадії одержання нижньої структурної ланки сполук формули (I) I.f) Одержання (R,R)-диметилдіоксолану (I-12-A) і (R,R)-бензилбутандіолу (I-5-A) Інший предмет винаходу стосується одержання (4R,5R)-4,5-диметил-2-феніл-1,3-діоксолану (I-12-A) і (2R,3R)-3-(бензилокси)бутан-2-oлу (I-5-A) для нижньої структурної ланки сполук формули (I). Згідно з публікацією WO 2010/046035 A1 комерційно доступний (R,R)-бутан-2,3-діол із застосуванням бензилхлориду шляхом здійснення однієї стадії перетворюють на монобензильовану сполуку (I-5-A). Оскільки перетворення згідно з очікуваннями не дозволяє селективно одержати моно-сполуку, реакційну суміш необхідно очищати методом хроматографії, і тому вихід становить менше 50 %. Альтернативним є здійснення двостадійного процесу (Bioorg. Med. Chem. Lett. 2006, 16, 186190). 13 UA 110626 C2 5 10 15 20 25 30 35 Фіг. 8 Предметом винаходу є визначення експериментальних умов для повного перетворення, а також простого відокремлення і очищення, які придатні для здійснення в технічному масштабі. Проміжний продукт (4R,5R)-4,5-диметил-2-феніл-1,3-діоксолан (I-12-A) одержують відповідним чином шляхом перетворення бензальдегіду диметилацетату і надлишку (2R,3R)бутан-2,3-діолу в присутності піридин-п-толуолсульфонату в толуолі як розчиннику. Реакція тривала до повного завершення при 50 °C протягом 3 годин, причому метанол безперервно видаляли шляхом відгонки під зниженим тиском. У рамках водної обробки надлишок діолу видаляли шляхом екстрагування. Залишкову толуольну фазу можна було безпосередньо застосовувати на наступній стадії. Для наступного відновлення із застосуванням гідриду діізобутилaлюмінію (ДІБАЛ) застосовували 1,5 M розчин гідриду діізобутилалюмінію в толуолі при 55-60 °C. Для обробки його додавали до декагідрату сульфату натрію і видаляли розчинник відгонкою після фільтрування. Одержали хороший вихід сполуки (I-5-A) хорошої чистоти. Продукт без додаткового очищення можна було застосовувати на наступній стадії. I.g) Одержання 4-{[(2R,3R)-3-(бензилокси)бутан-2-іл]oкси}-2-хлор-5-(трифторметил)піримідину (I-7-A) Нуклеофільне монозаміщення атома хлору в комерційно доступному 2,4-дихлор-5трифторметилпіримідині здійснюють переважно в 2-му положенні. Фіг. 9 Неочікувано було винайдено, що шляхом варіювання умов заміщення може бути напрямлене в бажане 4-е положення. Було доведено, що літієві основи в етерних розчинниках при -30 °C дозволяють досягти високого ступеня перетворення, і в найкращому випадку співвідношення між 4-ізомером і 2-ізомером становить 1,2:1. Як розчинники можуть бути застосовані, наприклад, тетрагідрофуран, 1,2-диметоксіетан, 1,4- діоксан, метил-третбутиловий етер, діізопропіловий етер, н-дибутиловий етер, 2-метил-тетрагідрофуран або циклопентил-метиловий етер. Переважним є тетрагідрофуран. Як основи можуть бути застосовані, наприклад, гексаметилдисилазид літію, н-бутиллітій, діізопропіламід літію або літій-2,2,6,6-тетраметилпіперидин. Переважним є гексаметилдисилазид літію. Діапазон температур становить від -78 °C до +20 °C. На основі цього було розроблено відповідну винаходові стадію способу, в якому застосовують гексаметилдисилазид літію в тетрагідрофурані при -30 °C, що дозволяє одержати бажаний ізомер (I-7-A) після хроматографії з виходом до 46 % чистотою понад 95 Fl%. Конденсування верхньої та нижньої структурних ланок і одержання сполук згідно з формулою (I) I.h) Одержання аніліно-піримідинів формули (I-8-R-BSA) Для об'єднання обох компонентів (I-7-A) і (I-4-R) в сполуку (I-8-R) здійснюють реакцію 14 UA 110626 C2 5 10 15 20 25 конденсування. Це перетворення здійснюють у кислому середовищі. Придатними до застосування кислотами є, наприклад, хлороводень, п-толуолсульфонова кислота, бензолсульфонова кислота, метансульфонова кислота. Переважною є бензолсульфонова кислота. Вільні основи (I-8-R) в звичайному випадку перебувають у формі олії, що ускладнює їх очищення, а також зберігання. Неочікувано було винайдено, що при застосуванні бензолсульфонової кислоти утворювані солі бензолсульфонової кислоти (I-8-R-BSA) викристалізовуються з реакційної суміші. Солі (I-8-R-BSA) можуть бути очищені шляхом кристалізації; вони є стабільними при зберіганні. Фіг. 10 Альтернативно можна застосовувати толуолсульфонову кислоту або метансульфонову кислоту. I.i) Одержання сполук формули (I) На обох останніх стадіях здійснюють відщеплення захисних груп (фіг. 11). Фіг. 11 У результаті гідрування під нормальним тиском протягом кількох годин із застосуванням паладію на вугіллі та водню в метанолі одержують проміжний продукт формули (I-9-R-BSA). Проміжний продукт формули (I-9-R-BSA) може бути безпосередньо перетворений на кінцевий продукт. Відщеплення групи може бути завершене за допомогою карбонату калію, і кристалізація на останній стадії відбувається з етилового естеру оцтової кислоти/н-гептану. II. Проміжні продукти Іншими предметами винаходу є наведені далі проміжні продукти. II. a) Трифторацетат-захищені нітрофеніл-сульфіліміни формули (I-10), зокрема (I-10-A) II. b) Нітрофеніл-сульфоксіміни формули (I-11-R), зокрема (I- 11-A) і (1-11-A-R) 15 UA 110626 C2 II. c) (R)-енантіомери трифторацетат-захищених нітрофеніл-сульфоксімінів формули (I-3-R), зокрема (I-3-A-R) 5 II. d) Солі (I-11-R-D-Tol-Tart.) нітрофеніл-сульфоксімінів формули (I-11-R) з (+)-ди-O-птолуоїл-D-винної кислоти, зокрема (I-11-A-R-D-Tol-Tart.) I.e) Аніліно-піримідини формули (I-8-R-BSA), зокрема (I-8-A-R-BSA)) 10 15 , причому 4 R в кожному випадку означає C1-C6-алкільну групу або C3-C7-циклоалкільне кільце. I-A. Одержання сполуки A Одержання верхньої структурної ланки сполуки А I-A.a) Одержання циклопропіл-нітрофеніл-сульфіду (I-1-A) На першій стадії послідовності реакцій здійснюють алкілювання 4-нітротіофенолу із застосуванням бромциклопропану в присутності карбонату калію. Бажане перетворення здійснюють у N-метилпіролідиноні (NMP) протягом 8-10 годин при переважній температурі 135 °C. 16 UA 110626 C2 5 10 15 20 25 30 Фіг. 12 Відокремлення (I-1-A) здійснювали шляхом виливання реакційної суміші на льодяну воду; відокремлювали необроблений кристалізат хорошої чистоти, що становить в типовому випадку 89-93 Fl% при виході 82-87 %. I-A.b) Одержання трифторацетат-захищеного циклопропіл-нітрофеніл-сульфіл-іміну (I-10-A) Фіг. 13 На другій стадії здійснюють окиснювальне амінування з одержанням трифторацетатзахищеного циклопропіл-нітрофеніл-сульфіліміну (I-10-A). У рамках широкого скринінгу досліджували перетворення циклопропіл-нітрофеніл-сульфіду (I-1-A) на трифторацетат-захищений циклопропіл-нітрофеніл-сульфілімін (I-10-A). Окиснювальними засобами, які є придатними до застосування у відповідному винаходові перетворенні згідно з фіг. 13, є N-бромсукцинімід, 1,3-дибром-5,5-диметилгідантоїн у присутності таких основ, как трет-бутилат калію, гідрид натрію, в розчинниках, таких як дихлорметан, тетрагідрофуран або ацетонітрил. Бажану реакцію здійснюють в діапазоні температур 0-50 °C, причому переважна температура становить 20 °C. Як окиснювальні засоби досліджували 1,3-дибром-5,5-диметилгідантоїн, N-хлорсукцинімід і трихлорціанурову кислоту в присутності основ, таких як трет-бутилат калію, трет-бутилат натрію, водний натрієвий луг. гідроксид натрію, метанолат натрію, гідрид натрію, в таких розчинниках, як метанол, дихлорметан, тетрагідрофуран-вода, ацетонітрил, ацетонітрил-вода, тетрагідрофуран, пропіонітрил, метил-трет-бутиловий етер, діоксан, хлорбензол. Фіг. 14 В процесі перетворення спочатку гідрид натрію закладали в тетрагідрофуран і по краплях додавали циклопропіл-нітрофеніл-сульфід (I-1-A) із трифторацетамідом. Охолоджуючи, додавали розчин 1,3-дибром-5,5-диметилгідантоїну в тетрагідрофурані і перемішували при кімнатній температурі. Суміш піддавали відновленню (із застосуванням сульфіту натрію) і викристалізовували продукт із суміші діізопропіловий етер/н-гептан. Одержали продукт (I-10-A) із хорошим виходом і чистотою. I-A.c) Одержання 1-(циклопропілсульфонімідоїл)-4-нітробензолу (I-11-A) Окиснення трифторацетат-захищеного нітрофеніл-сульфіліміну ((I-10-A)) з одержанням 1(циклопропілсульфонімідоїл)-4-нітробензолу (I-11-A) здійснюють переважно із застосуванням ® пероксомоносульфату калію (Oxone ) як окиснювального засобу. 17 UA 110626 C2 5 10 15 20 25 Фіг. 15 Реакцію здійснювали в суміші метанолу з водою і додавали тетраметиленсульфон ® (сульфолан) як солюбілізатор. Пероксомоносульфат калію (Oxone ) додавали порціями і після додавання кожної порції встановлювали значення pH=10. Через 5 годин відсоток перетворення на бажаний рацемічний 1-(циклопропілсульфонімідоїл)-4-нітробензол (I-11-A) становив уже 99 %. Продукт піддавали водній обробці (сульфіт натрію), після чого викристалізовували із органічної фази (метилeнхлорид) після висушування над сульфатом магнію з н-гептану. I-A.d) Розщеплення рацемату 1-(циклопропілсульфонімідоїл)-4-нітробензолу (I-11-A) Для розщеплення рацемату здійснювали описану далі стадію: Фіг. 16 Неочікувано було винайдено, що при застосуванні розчину (+)-ди-O-п-толуоїл-D-винної кислоти в ацетонітрилі одержують співвідношення між енантіомерами в кристалізаті принаймні 95:5. Вихід становив 40-45 %. Альтернативно ацетонітрилу може бути використаний також пропіонітрил. Шляхом перекристалізації з ацетонітрилу або пропіонітрилу можна додатково поліпшити оптичну чистоту. Інтегрувати процес кристалізації в технологічний процес можна шляхом відщеплення захисної трифторацетатної групи в (I-3-A) із застосуванням карбонату калію в метанолі і перетворення необробленого нітрофеніл-сульфоксіміну (I-11-A) із застосуванням (+)-ди-O-птолуоїл-D-винної кислоти в ацетонітрилі на (I-11-A-D-Tart.). Оптично активний нітрофеніл-сульфоксімін вивільняють шляхом лужного екстрагування, після чого однореакторним методом вводять захисні групи із застосуванням трифтороцтового ангідриду в присутності триетиламіну, одержуючи сполуку (I-3-A-R). 18 UA 110626 C2 5 10 15 20 25 Фіг. 17 Описаний в публікації WO 2010/046035 A1 метод одержання трифторацетат-захищеного нітрофеніл-сульфоксіміну (I-3-A) дозволяє отримати верхню структурну ланку в формі рацемату. I-A.e) Гідрування з одержанням трифторацетат-захищеного аніліно-сульфоксіміну (I-4-A-R) Фіг. 18 Перетворення нітрогрупи в сполуці (I-3-A-R) на відповідний анілін (I-4-A-R) здійснюють шляхом гідрування з іммобілізованими паладієвими каталізаторами. Особливо чистий продукт одержують при застосуванні легованих залізом паладієвих каталізаторів на вугіллі. Переважним розчинником є метанол. Трифторацетат-захищений аніліно-сульфоксімін (I-4-A-R) після кристалізації може бути відокремлений із виходом принаймні 88 %. Одержання нижньої структурної ланки сполуки A Синтез нижньої структурної ланки сполуки A здійснюють згідно з винаходом відповідно до I.g) та I.f). Конденсування верхньої та нижньої структурних ланок I-A.h) Одержання N-[(4-{[4-{[(2R,3R)-3-(бензилокси)бутан-2-іл]oкси}-5-(три6 фторметил)піримідин-2-іл]aміно}феніл)(циклопропіл)oксидо-лямбда -сульфа-нілiден]-2,2,2трифторацетаміду солі бензолсульфонової кислоти (I-8-A-R-BSA) На першій стадії здійснюють конденсування обох структурних ланок (I-7-A) і (I-4-A-R) з одержанням (I-8-A-R). Це перетворення здійснюють у кислому середовищі. Вільна основа (I-8-AR) перебуває в формі олії. Неочікувано було виявлено, що при застосуванні 1,4-діоксану як розчинника утворювана сіль бензолсульфонової кислоти (I-8-A-R-BSA) викристалізовується з реакційної суміші. Фіг. 19 Кристалізацію може бути завершена із застосуванням н-гептану; одержують бажану сполуку 19 UA 110626 C2 5 10 N-[(4-{[4-{[(2R,3R)-3-(бензилокси)бутан-2-іл]oкси}-5-(трифторметил)піримідин-2-іл]-aміно}6 феніл)(циклопропіл)oксидо-лямбда -сульфанілiден]-2,2,2-трифторацетаміду сіль бензолсульфонової кислоти з хорошим виходом. Сіль (I-8.A-R-BSA) є придатною до зберігання кристалічною речовиною; чистота відокремленої солі в типовому випадку становить близько 90 Fl%. I-A.i) Одержання сполуки A На обох останніх стадіях відокремлюють захисні групи (фіг. 20). Фіг. 20 Проміжний продукт I-9-A-R-BSA не відокремлювали, а безпосередньо перетворювали далі на кінцевий продукт. Відщеплення трифторацетатної групи доповнювали шляхом застосування кабонату калію в метанолі; викристалізовування кінцевого продукту відбувалося із суміші етиловий естер оцтової кислоти/гептан. 20 UA 110626 C2 Таблиця 2 7. Відповідність Поз. Найменування Структура Найменування згідно з номенклатурою IUPAC 1-(циклопропілсульфаніл)-4-нітробензол S Мол. маса 195,24 N-[(R)-циклопропіл(4нітрофеніл)oксидо6 лямбда -сульфанілiден]2,2,2-трифторацетамід 322,26 4 R I-1 нітрофенілсульфід O + N O I-1-A S циклопропілнітрофенілсульфід O2 N O S I-2 нітрофенілсульфоксид 4 R O + N O F I-3 трифторацетатзахищений нітрофенілсульфоксімін F F O N S O 4 R O + N O F I-3-R (R)-енантіомер трифторацетатзахищеного нітрофенілсульфоксіміну F F O N S O 4 R O + N O F I-3-A-R (R)-енантіомер трифторацетатзахищеного циклопропілнітрофенілсульфоксіміну F F O N O S O + N O 21 UA 110626 C2 Поз. Найменування Структура Найменування згідно з номенклатурою IUPAC F I-4 трифторацетатзахищений аніліносульфоксімін Мол. маса F F O N S O 4 R NH2 F I-4-R I-4-A-R (R)-енантіомер трифторацетатзахищеногоаніліносульфоксіміну (R)-енантіомер трифторацетатзахищеного циклопропіланіліносульфоксіміну F F O N S O 4 R NH2 F F F O S 292,28 180,25 4-{[(2R,3R)-3(бензилокси)бутан-2іл]oкси}-2-хлор-5(трифторметил)піримідин N N-[(R)-(4-амінофеніл)(циклопропіл)6 оксидо-лямбда сульфанілiден]-2,2,2трифторацетамід (2R,3R)-3(бензилокси)бутан-2-oл O 360,77 NH2 I-5-A O HO Cl I-6 захищений гідроксіалкоксипіримідин N N 1 R O OPG 2 R 3 R I Cl I-7 захищений CF3проміжний продукт N N 1 R O OPG 2 R F 3 R F F Cl I-7-A бензилзахищений CF3проміжний продукт N N O O F F F 22 UA 110626 C2 Поз. Найменування Структура Найменування згідно з номенклатурою IUPAC Мол. маса CF3 O N S I-8 двічі захищений аніліно-піримідин 4 R O HN N N OPG O CF3 CF3 O N S I-8RBSA сіль бензолсульфонової кислоти двічі захищеного аніліно-піримідину O SO3H 4 R HN N N O O CF3 CF3 O N S I-8A-RBSA сіль бензолсульфонової кислоти двічі захищенихого циклопропіл-анілінопіримідину O SO3H HN N N O O CF3 CF 3 N O S 4 R I-9 монозахищенийанілінопіримідин O HN N N OH O CF 3 O S I-9RBSA сіль бензолсульфонової кислоти моно- захищеного аніліно-піримідину CF 3 N 4 R HN N N OH O CF 3 23 O SO 3H N-[(4-{[4-{[(2R,3R)-3(бензилокси)бутан-2іл]oкси}-5-(трифторметил)-піримідин-2іл]aміно}феніл)(циклопропіл)oксидо6 лямбда сульфанілiден]-2,2,2трифторацетамід сіль бензолсульфонової кислоти (1:1) 774,76 UA 110626 C2 Поз. I-9-ARBSA Найменування сіль бензолсульфонової кислоти монозахищеного циклопропіланілінопіримідину Структура O Найменування згідно з номенклатурою IUPAC HN N SO3H N OH O CF 3 306,27 226,26 1-(R-циклопропілсульфон-імідоїл)-4нітробензол O 684,64 1-(циклопропілсульфонімідоїл)-4нітробензол S N-[циклопропіл(4-{[4{[(2R,3R)-3-гідроксибутан2-іл]oкси}5(трифторметил)піримідин2-іл]aміно}феніл)oксидо6 лямбда -сульфанілiден]2,2,2-трифторацетаміду сіль бензолсульфонової кислоти (1:1) N-[циклопропіл(44 нітрофеніл)-лямбда сульфанілiден]-2,2,2трифторацетамід CF 3 N Мол. маса 226,26 O I-10 трифторацетатзахищений нітрофенілсульфілімін N CF3 S 4 R O 2N I-10-A трифторацетатзахищений циклопропілнітрофенілсульфілімін O F N S F F O2 N NH O I-11 S нітрофенілсульфоксімін 4 R O2N O I-11-R R-енантіомер нітрофенілсульфоксіміну NH S O2 N O I-11-A циклопропілнітрофенілсульфоксімін NH S O2 N O I-11A-R R-енантіомер циклопропілнітрофенілсульфоксіміну 4 R NH S O2 N 24 UA 110626 C2 Поз. Найменування I-11-R-DTol-Tart. Структура сіль толуоїлD-винної кислоти Rенантіомера нітрофенілсульфоксіміну O Найменування згідно з номенклатурою IUPAC O O OH HO O O NH O S Мол. маса O 1-(R)-(циклопропілсульфон-імідоїл)-4нітробензол 4 R O2 N I-11-A-RD-TolTart. сіль толуоїлD-винної кислоти Rенантіомера циклопропілнітрофенілсульфоксіміну (2S,3S)-2,3-біс[(4метилбензоїл)oкси]сукцинова кислотa 1-(R-циклопропілсульфон-імідоїл)-4нітробензол (1:1) O HO NH O O O S 178,23 (2R,3R)-3-{[2-{[4-(Rциклопропілсульфонімідоїл)феніл]aміно}5-(трифторметил)піримідин-4іл]oкси}бутан-2-oл OH O 612,62 (4R,5R)-4,5-диметил2-феніл-1,3-діоксолан O O 430,45 O2 N O O I-12-A O NH S Сполука A HN Сполука A N N OH O F F F O NH S R Сполука формули (I) 4 HN N CH3 N OH O F F CH3 F 25 UA 110626 C2 Приклад Одержання 1-(циклопропілсульфаніл)-4-нітробензолу (I-1-A) 5 10 15 20 25 30 35 40 45 Розчин 80 г (0,51 моль) 4-нітротіофенолу (конц. 80 %) в 400 мл N-метилпіролідинону (NMP) протягом 30 хвилин додавали до суспензії 92,6 г (0,67 моль) карбонату калію в 400 мл NMP. При цьому температура становила 30 °C. До реакційної суміші додавали 93,6 г (0,77 моль) циклопропілброміду і перемішували суміш протягом 8 годин при 135-140 °C. Охолоджували суміш до 20 °C і змішували з 4,0 г активованого вугілля. Нагрівали до 65 °C, перемішували протягом однієї години, фільтрували і додатково промивали із застосуванням 80 мл NMP. Охолоджували суміш до 20 °C і протягом однієї години додавали до 3 л води. Фільтрували суміш і тричі промивали фільтраційний осад із застосуванням кожного разу 400 мл води. Потім перемішували з 800 мл 1 M водн. розчину соляної кислоти, знову фільтрували і тричі промивали із застосуванням кожного разу 400 мл води. Насамкінець висушували при 40 °C у вакуумі і одержали 86,6 г (86 %) цільової сполуки (I-1-A) чистотою 91,2 Fl.%. Необроблений матеріал можна було піддати додатковому очищенню. Для цього 90 г необробленого матеріалу розчиняли в 700 мл н-гептану, нагрівали до 65 °C, видаляли відгонкою близько 400 мл н-гептану і змішували із затравочними кристалами в процесі охолодження до 20 °C. Перемішували при 0-5 °C протягом однієї години, відфільтровували і промивали залишок із застосуванням 100 мл холодного н-гептану. Після висушування одержали 81 г цільової сполуки (I-1-A) чистотою 100 Fl.%. Результати аналізу методами ядерного магнітного резонансу (ЯМР) і мас-спектрометрії (МС): Luecking, Ulrich; Krueger, Martin; Jautelat, Rolf; Siemeister, Gerhard. Preparation of pyrimidinylamino-arylsulfoximines as cyclin dependent kinase (CDK) and/or vascular endothelial growth factor (VEGF) inhibitors: WO 2005037800, стор. 105. Метод високоефективної рідинної хроматографії (ВЕРХ) A: колонка Zorbax SB-Aq 150 3 мм, 3,5 M; градієнт: 0-20 хв. від 95 % водного буферного фосфатного розчину, pH=2,4/5 % ацетонітрилу до 20 % водного буферного фосфатного розчину, pH=2,4/80 % ацетонітрилу, потік: 0,5 мл/хв., детектування при довжині хвилі 210 нм, T = 45 °C; Час утримання сполуки (I-1-A) у методі A: 16,7 хв. 4 Одержання N-[циклопропіл(4-нітрофеніл)-лямбда -сульфанілiден]-2,2,2-трифтор-ацетаміду (I-10-A) Розчин 11,5 г (58,9 ммоль) 1-(циклопропілсульфаніл)-4-нітробензолу (I-1-A) і 10,0 г (88,4 ммоль) 2,2,2-трифторацетаміду в 46 мл тетрагідрофурану (ТГФ) при 0-5 °C протягом 30 хвилин додавали до суспензії 2,1 г (53 ммоль) гідриду натрію (конц. 60 % в мінеральному маслі) в 50 мл тетрагідрофурану. До реакційної суміші додавали розчин 25,3 г (88,4 ммоль) 1,3-дибром-5,5-диметил-гідантоїну в 86 мл тетрагідрофурану при 20-25 °C протягом 15 хвилин і перемішували протягом 14 годин. Для обробки змішували суміш із 70 мл 25 %-ного розчину сульфіту натрію і 140 мл толуолу. Тричі промивали органічну фазу із застосуванням кожного разу 140 мл води, згущували до близько 80 г у вакуумі, змішували з 40 мл н-гептану і продовжували перемішування протягом 1,5 годин при 20 °C. Фільтрували, двічі промивали із застосуванням кожного разу 25 мл н-гептану і висушували у вакуумі при 40 °C. Одержали 14,5 г цільової сполуки (I-10-A) чистотою 100 Fl%. Це відповідало виходу 80,8 %. Більший масштаб: Розчин 80,0 г (409,8 ммоль) 1-(циклопропілсульфаніл)-4-нітробензолу (I-1-A) і 69,5 г (614,6 ммоль) 2,2,2-трифторацетаміду в 320 мл ТГФ при 0-5 °C протягом 45 хвилин додавали до 26 UA 110626 C2 5 10 15 20 25 30 35 суспензії 14,8 г (368,8 ммоль) гідриду натрію (конц. 60 % в мінеральному маслі) в 360 мл ТГФ. Потім додавали розчин 175,7 г (614,6 ммоль) 1,3,-дибром-5,5-диметилгідантоїну в 550 мл ТГФ при 0-5 °C протягом 45 хвилин. Розморожували до 20 °C і відстоювали протягом 14 год. Для обробки додавали 560 мл 10 %-ного розчину лимонної кислоти і 1,1 л толуолу. Органічну фазу промивали 560 мл einer 25 %-ного розчину сульфіту натрію і тричі промивали із застосуванням кожного разу 640 мл води. Органічну фазу згущували у вакуумі до близько 750 г і змішували з 525 г н-гептану. Продовжували перемішування протягом однієї години при кімнатній температурі, відсмоктували і промивали із застосуванням 50 мл суміші толуол/гептан у співвідношенні 1:1. Після висушування у вакуумі одержали 90,7 г (вихід 72 %) цільової сполуки (I-10-A) чистотою 99,1 Fl%. Метод ВЕРХ A: час утримання для сполуки (I-10-A): 14,8 хв. + + МС (CI): [M+H] = 307, [M+NH4] = 324 1 H ЯМР (400 MГц, ДМСО-d6) δ млн.ч. 1,09 - 1,19 (м, 1 H) 1,21 - 1,38 (м, 3 H) 3,03 - 3,15 (м, 1 H) 8,13 - 8,23 (м, 2 H) 8,42 - 8,54 (м, 2 H). Одержання 1-(S-циклопропілсульфонімідоїл)-4-нітробензолу (I-11-A) До розчину 100,0 г (326,5 ммоль) (I-10-A) в 850 мл метанолу, 130 мл тетраметиленсульфону (сульфолану) і 590 мл води при 25 °C додавали 341,2 г (555,1 ммоль) пероксомоносульфату ® калію (Oxone ), розділеного на вісім порцій. Після додавання кожної порції встановлювали значення рН=10 за допомогою 47 %-ного водного розчину карбонату калію. В цілому було використано близько 350 мл розчину карбонату калію. Перетворення було завершене через годину при 25 °C. Додавали 960 мл дихлорметану і перемішували протягом однієї години при 20 °C. Відсмоктували рідину і двічі промивали залишок із застосуванням кожного разу 400 мл дихлорметану. Об'єднану органічну фазу промивали із застосуванням 400 мл водного розчину 10 %-ного сульфіту натрію і чотири рази із застосуванням кожного разу 1 л води. Після розділення фаз висушували над сульфатом магнію і згущували до близько 450 г. Додавали 100 мл н-гептану, згущували у вакуумі до близько 400 мл і продовжували перемішування протагом однієї години при 0-5 °C. Рідину відсмоктували і двічі промивали залишок із застосуванням кожного разу 100 мл холодного н-гептану. Насамкінець продукт висушували у вакуумі при 40 °C і одержали 68,5 г (92,8 %) цільової сполуки (I-11-A) чистотою 100 Fl%. Метод ВЕРХ A: час утримання для (I-11-A): 9,5 хв. + MS (CI): [M+H] = 227 1 H ЯМР (400 MГц, ДМСО-d6) δ млн.ч. 0,86 - 1,06 (м, 3 H) 1,15 (дт, J=9,96, 4,19 Гц, 1 H) 2,69 2,88 (м, 1 H) 4,65 (с, широкий, 1 H) 8,15 (д, J=8,80 Гц, 2 H) 8,41 (д, J=8,56 Гц, 2 H). Одержання (2S,3S)-2,3-біс[(4-метилбензоїл)oкси]бутанової дикислоти-N-[(R)-цикло-пропіл(46 нітрофеніл)oксидо-лямбда -сульфанілiден]-2,2,2-трифторацетаміду (1 : 1) (I-11-A-R-D-Tol-Tart.) До суспензії 64,9 г (286,8 ммоль) (I-11-A) в 1,3 л ацетонітрилу при 20 °C додавали 116,4 г 27 UA 110626 C2 5 10 15 (301,2 ммоль) ди-п-толуоїл-D-винної кислоти і перемішували протягом 16 годин при 20 °C. Відсмоктували рідину і двічі промивали залишок із застосуванням кожного разу 90 мл ацетонітрилу. Висушували у вакуумі при 40 °C до сухого стану і одержали 71,1 г (I-11-A-R-D-TolTart.). Це відповідало виходу 40,4 %. Метод ВЕРХ A: час утримання для сполуки (I-11-A-D-Tart): 9,6 хв. (36,2 %) і 14,7 хв. (63,8 %). Метод ВЕРХ B (L159-16EE): колонка Chiralpak IC (Firma DAICEL), довжина: 250 мм, внутрішній діаметр: 4,6 мм, розмір зерна: 5 мкм, градієнт: 1:1 н-гептан/i-пропанол, ізократний; потік: 1,0 мл/хв., детектування при довжині хвилі 252 нм, T = 35 °C Час утримання для R-енантіомера: 9,3 хв.; час утримання для S-енантіомера: 8,4 хв.; надлишок енантіомерів (ee): 99,6 %. + МС (ES+):[M+H] = 227; МС (ES-): [M-H] = 385 1 H ЯМР (400 MГц, ДМСО-d6) δ млн.ч. 0,91 - 1,06 (м, 3 H) 1,10 - 1,20 (м, 1 H) 2,41 (с, 6 H) 2,72 2,84 (м, 1 H) 4,64 (с, широкий, 1 H) 5,82 (с, 2 H) 7,40 (д, J=8,07 Гц, 4 H) 7,90 (д, J=8,07 Гц, 4 H) 8,09 - 8,20 (м, 2 H) 8,33 - 8,52 (м, 2 H) 13,85 (с, широкий, 2H). 6 Одержання N-[(R)-циклопропіл(4-нітрофеніл)oксидо-лямбда -сульфанілiден]-2,2,2трифторацетаміду (I-3-A-R) 40 Розчин 72,1 г (117,7 ммоль) сполуки (I-11-A-R-D-Tol-Tart.) у 720 мл дихлорметану протягом 60 хвилин перемішували з розчином 24,4 г карбонату калію в 350 мл води при 20 °C. Водну фазу екстрагували із застосуванням 360 мл дихлорметану, промивали об'єднану органічну фазу із застосуванням 720 мл води і висушували над сульфатом магнію. Фільтрували суміш і змішували фільтрат із 49 мл (353,1 ммоль) триетиламіну, а потім із 49,9 мл (353,1 ммоль) трифтороцтового ангідриду протягом 40 хвилин при 20-25 °C. Продовжували перемішування протягом 10 хвилин і потім додавали до 1,1 л насиченого розчину гідрокарбонату натрію. Після розділення фаз промивали із застосуванням 1,0 л води, висушували над сульфатом магнію і видаляли розчинник шляхом відгонки у вакуумі. Залишок викладали в 120 мл i-пропанолу і перемішували утворену суспензію протягом однієї години при 0-5 °C. Фільтрували і двічі промивали залишок із застосуванням кожного разу 30 мл холодного ізопропанолу. Висушували при 40 °C у вакуумі і одержали 27,3 г (75 %) сполуки (I-3-A-R.). Метод ВЕРХ A: час утримання для (I-3-A-R): 16,4 хв. (99,6 %). Метод ВЕРХ C (L159-10EE): колонка Chiralpak IC (компанія DAICEL), довжина: 250 мм, внутрішній діаметр: 4,6 мм, розмір зерна: 5 мкм, градієнт: 1:1 н-гептан/етанол, ізократний; потік: 1,0 мл/хв., детектування при довжині хвилі 240 нм, T = 35 °C Час утримання для R-енантіомера (I-3-A-R): 4,5 хв.; час утримання для S-енантіомера: 3,7 хв.; надлишок енантіомерів (ee): 100 %. + + MС (DCI): [M+H] = 323, [M+NH4] = 340 1 H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ млн.ч. 1,11 - 1,29 (м, 1 H) 1,36 - 1,52 (м, 2 H) 1,74 - 1,88 (м, 1 H) 2,69 - 2,89 (м, 1 H) 8,14 (д, J=8,80 Гц, 2 H) 8,47 (д, J=8,80 Гц, 2 H). 6 Одержання N-[(R)-(4-амінофеніл)(циклопропіл)oксидо-лямбда -сульфанілiден]-2,2,2трифторацетаміду (I-4-A-R): 45 Суспензію 40,0 г (124,1 ммоль) сполуки (I-3-A-R) і 10,0 г паладію на вугіллі (Pd/C: 5 % Pd, 1 % Fe, 55 % води) в 800 мл метанолу гідрували протягом семи годин під тиском 2,5 бар. Фільтрували крізь кізельгур і двічі додатково промивали із застосуванням кожного разу 200 мл метанолу. Фільтрат згущували у вакуумі, а потім додавали 800 мл води. Перемішували 20 25 30 35 28
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparing pan-cdk inhibitors of the formula (i), and intermediates in the preparation
Автори англійськоюKruger, Joachim, Gries, Jorg, Lovis, Kai, Hassfeld, Jorma
Автори російськоюКрюгер Йоахим, Грес Йорг, Ловис Каи, Хассфельд Йорма
МПК / Мітки
МПК: C07D 239/47
Мітки: одержання, кінази, також, процесу, цього, продукти, спосіб, пан-інгібіторів, проміжні, циклінзалежної, формули
Код посилання
<a href="https://ua.patents.su/39-110626-sposib-oderzhannya-pan-ingibitoriv-ciklinzalezhno-kinazi-formuli-i-a-takozh-promizhni-produkti-cogo-procesu-oderzhannya.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання пан-інгібіторів циклінзалежної кінази формули (i), а також проміжні продукти цього процесу одержання</a>
Спосіб одержання глюфозинату, проміжні продукти та спосіб одержання проміжних продуктів

Номер патенту: 66806
Опубліковано: 15.06.2004
Автор: Вілльмс Лотар
МПК: C07F 9/6571, C07F 9/32, C07F 9/30
Мітки: продуктів, глюфозинату, проміжні, проміжних, продукти, спосіб, одержання
Формула / Реферат:
1. Спосіб одержання сполук формули (І)у якій R* означає водень або алкіл із 1-4 атомами вуглецю, їхніх солей із кислотами або основами, який відрізняється тим, щоа) (стадія 1) тривалентну метилфосфорну сполуку формули (II) вводять у реакцію обміну з ненасиченим похідним формули (III) у присутності конденсуючого засобу або активатора і, при...
Індолінові похідні, а також їх фармацевтично прийнятні солі, що є антагоністами вазопресинових v1-рецепторів, спосіб їх одержання, проміжні продукти і фармацевтична композиція

Номер патенту: 27238
Опубліковано: 15.08.2000
Автори: Нісато Діно, Ваньон Жан, Тоннер Бернар, Серадей-Легаль Клодін, Плузан Клод
МПК: C07D 209/42, C07D 401/12, A61P 9/00, C07C 317/30, A61P 15/00, A61K 31/404, A61K 31/535, A61P 25/04, A61K 31/55, C07D 417/06, A61P 9/08, C07D 403/06, A61K 31/4427, A61P 1/04, A61P 9/10, A61K 31/40, A61K 31/445, C07D 401/06, A61K 31/403, A61P 25/18, A61P 9/12, A61P 13/02, A61K 31/495, C07C 311/08, A61K 31/54, A61K 31/425, A61P 7/02
Мітки: композиція, фармацевтично, одержання, антагоністами, також, вазопресинових, індолінові, солі, фармацевтична, продукти, прийнятні, похідні, v1-рецепторів, спосіб, проміжні
Текст:
...пл. ° С) (или их точкой кипения Т кип ) и/или их ЯМР спектром, снятым при 200 МГц в ДМСО и/или показателем вращения плоскости Бполяризации (альфаО), измеренной при 25 °С (если нет других указаний). Измеренное значение вращения плоскости поляризации зависит от количества остаточного растворителя, присутствующего в приготовленном продукте. За исключением особо указанных случаев обозначение "цисизомер" или "трансизомер" означает, что выделенное...
Біcпіперидини як протитромботичні агенти, спосіб їх одержання та проміжні продукти

Номер патенту: 65631
Опубліковано: 15.04.2004
Автори: Йю Крістоф, Жібуло Тьєррі, Анрі Маргеріт, Лезюр Бріжіт
МПК: C07D 417/14, A61P 43/00, A61K 31/4545, C07D 413/14, C07D 409/14, A61P 7/02, A61K 31/453, C07D 211/34, A61K 31/445, A61K 31/4465, C07D 401/14, A61K 31/443, A61K 31/4433, C07D 211/60, A61K 31/454, A61K 31/4535, A61K 31/4427, C07D 405/14
Мітки: продукти, агенти, протитромботичні, біcпіперидини, одержання, проміжні, спосіб
Формула / Реферат:
1. Сполуки формули:,Формула Ів якій:І) R1 або відбирають з- С1-С4-алкільної, С3-С12-моно- або біциклічної циклоалкільної, С2-С4-алкенільної або С2-С4-алкінільної груп, причому ці групи необов'язково заміщені групами, відібраними з галогенів і гідроксильної групи;- моно-, бі- або трициклічних С6-С14-арильних груп;- гетероарильних груп, відібраних з піридильної, тієнільної, фурильної,...
Спосіб одержання імідазо[1,2-c][2,3]бензодіазепінів та проміжні продукти при їх одержанні

Номер патенту: 79737
Опубліковано: 25.07.2007
Автори: Вінтер Ерік, Шнайдер Маттіас, Тільстам Ульф
МПК: C07D 487/12, C07D 487/04, C07D 235/02, A61K 31/5517, C07D 243/10, C07D 403/02, C07C 69/614, C07D 263/30
Мітки: імідазо[1,2-c][2,3]бензодіазепінів, одержанні, спосіб, проміжні, продукти, одержання
Формула / Реферат:
1. Спосіб одержання імідазо[1,2-с][2,3]бензодіазепінів загальної формули 1, (1)у якійR1 означає водень, С1-С6алкіл, нітрогрупу, галоген, ціаногрупу, С1-С4алкоксигрупу, -СF3, гідроксигрупу або С1-С6алканоїлоксигрупу,R2 і R3 мають ідентичні або різні значення і означають водень, галоген, С1-С6алкоксигрупу, гідроксигрупу, ціаногрупу,...
Тієнілазолілалкоксіетанаміни, спосіб їх одержання, проміжні продукти для їх одержання та фармацевтична композиція

Номер патенту: 58589
Опубліковано: 15.08.2003
Автори: АНДАЛУЗ-МАТАРО Блас, МЕРСЕ-ВІДАЛЬ Рамон, ФРІГОЛА КОНСТАНСА Хорді
МПК: A61K 31/381, A61K 31/4155
Мітки: проміжні, тієнілазолілалкоксіетанаміни, фармацевтична, одержання, композиція, продукти, спосіб
Формула / Реферат:
1. Похідна тієнілазолілалкоксіетанаміну загальної формули (І):, (І)деR1 - атом водню або атом галогену, або алкільний радикал з 1-4 атомами вуглецю;R2, R3 та R4, незалежно, - атом водню або алкільний радикал з 1-4 атомами вуглецю; таAz являє собою азотвмісне гетероциклічне ароматичне п'ятичленне N-метил-заміщене кільце, до складу якого входять від одного до трьох атомів азоту, загальної формули...
Попередній патент: Банка для харчового продукту, яка повторно закривається
Наступний патент: Покриття, що трансформується
Випадковий патент: Посуд для приготування їжі