Макроліди, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі, спосіб лікування бактеріальної інфекції з використанням таких сполук
Номер патенту: 77210
Опубліковано: 15.11.2006
Автори: Дам'яні Федеріка, Тібаско Єссіка, Б'єнтінезі Іларіа, Лучуро Серджо, Шенфельд Вольфґанґ, Пайо Альфредо, Павловіч Дражен, Марсіч Наташа, Мутак Шт'ьєпан, Думіч Мільєнко, Куалья Анна, Лазаревскі Горяна, Ераковіч Весна, Чірако Мануела, Стімач Владо, Аліґоджич Сулейман, Марусіч-Істук Зоріца, Бердік Андреа, Джерек Марко, Андреотті Даніеле, Бйонді Стефано, Хутінеч Антун








Формула / Реферат
1. Макроліди формули (І)
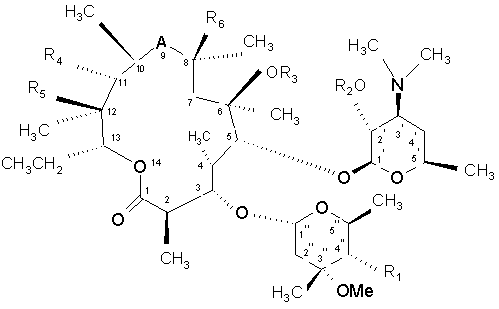 , (І)
, (І)
у якій
А - бівалентний радикал, вибраний з: -С(O)-, -C(O)NH-, -NHC(O)-, -N(СН3)-СН2-, -СН2-N(СН3)- або -С(NОСН2OСН2СН2OСН3)-;
R1 - OC(O)(CH2)nXR7;
R2 - гідроген або група, яка захищає гідроксил;
R3 - гідроген, С1-4алкіл або С3-6алкеніл, як варіант, заміщений 9-10-членним конденсованим біциклічним гетероарилом;
R4 - гідроксил або С3-6алкенілоксил, як варіант, заміщений 9-10-членним конденсованим біциклічним гетероарилом;
R5 - гідроксил, або
R4 і R5 разом з проміжними атомами утворюють циклічну групу структури:
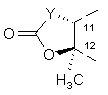 ,
,
де Y є бівалентним радикалом, вибраним з: -СН2-, -CH(CN)-, -O-, -N(R8)- і -CH(SR8)-;
R6 - гідроген або фтор;
R7 - гетероциклічна група структури:
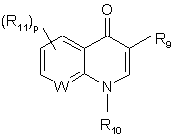
або
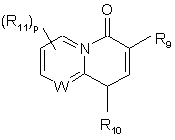 ,
,
R8 - гідроген або С1-4алкіл, заміщений групою, вибраною з:
як варіант, заміщеного фенілу,
як варіант, заміщеного 5- або 6-членного гетероарилу,
як варіант, заміщеного 9-10-членного конденсованого біциклічного гетероарилу;
R9 - гідроген, C(O)OR12, C(O)NHR12 або C(O)CH2NO2;
R10 - гідроген, С1-4алкіл, С3-7циклоалкіл, як варіант, заміщений фенілом або бензилом;
R11 - галоген, С1-4алкіл, С1-4тіоалкіл, С1-4алкоксил, NH2, NH(С1-4алкіл) або N(C1-4алкіл)2;
R12 - гідроген або С1-4алкіл;
R13 - гідроген, С1-4алкіл, С3-7циклоалкіл, як варіант, заміщений фенілом або бензилом, ацетил або бензоїл;
Х - -U(CH2)mZ,- або Х є групою, вибраною з:
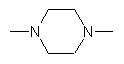
або
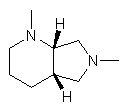 ,
,
U і Z незалежно є двовалентним радикалом, вибраним з:
-N(R13)-, -O-, -S(O)q-, -N(R13)C(O)-, -C(O)N(R13)-, -N[С(O)R13]-,
W - атом карбону або нітрогену;
n - 0 або ціле число від 1 до 5;
m – ціле число від 2 до 8;
p - 0, 1 або 2;
q дорівнює 0, 1 або 2;
і їх фармацевтично прийнятні солі і сольвати.
2. Сполука за п. 1, яка відрізняється тим, що Х є NH(CH2)2-3NH.
3. Сполука за п. 1 або п. 2, яка відрізняється тим, що R7 є гетероциклічною групою структури:
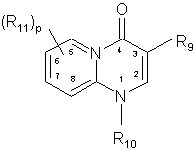 ,
,
де R9, R10, R11 і p мають значення, визначені у п. 1.
4. Сполука, вибрана з групи, яку складають:
4”-O[3-[[2-((3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]-етил]аміно]пропіоніл]-6-O-метил-8а-аза-8а-гомоеритроміцин А;
11,12-карбонат-4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]етил]аміно]пропіоніл]-11,12-дидезоксіазитроміцин;
11,12-карбонат-4”-O-[3-[[2-[(3-карбокси-1-циклопропіл-1,4-дигідро-6-фтор-4-оксо-7-хінолініл)аміно]етил]аміно]пропіоніл]-11,12-дидезоксіазитроміцин;
11,12-карбонат-4”-O-[3-[[2-[(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-7-хінолініл)-аміно]етил]аміно]пропіоніл]-11,12-дидезоксіазитроміцин;
4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]-етил]аміно]пропіоніл]азитроміцин;
4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]-етил]аміно]пропіоніл]-11,12-дидезокси-11,12-(етиламінокарбонілокси)-6-O-метилеритроміцин А;
4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]-етил]аміно]пропіоніл]-6-O-метилеритроміцин А;
4”-O-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-4-оксо-7-хінолініл)аміно]етил]аміно]-пропіоніл]-6-O-метилеритроміцин А;
4”-O-[3-[[2-[(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-7-хінолініл)аміно]етил]-аміно]пропіоніл]-6-O-метилеритроміцин А;
4”-O-[3-[[2-[(3-карбокси-1-циклопропіл-4-оксо-1,4-дигідро-6-хінолініл)аміно]етил]-аміно]пропіоніл]-6-O-метилеритроміцин А;
(11S,11аR)-4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]етил]аміно]пропіоніл]-11-(карбоксиціанометил)-11-дезокси-6-О-метил-еритроміцин А;
(11S, 11aR)-11-(карбоксиціанометил)-4”-O-[3-[[2-[(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-7-хінолініл)аміно]етил]аміно]пропіоніл]-11-дезокси-6-O-метилеритроміцин А;
4”-O-[3-[4-(3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]-етил]аміно]пропіоніл]рокситроміцин;
4”-O-(3-(2-(3-карбокси-7-хлор-1-циклопропіл-4-оксо-1,4-дигідро-6-хінолініл)аміно)-етил)метиламіно)пропіоніл)-11,12-дидезокси-11,12-(етиламінокарбонілокси)-6-O-метилеритроміцин А;
4”-O-(3-[(2-[(3-карбоксиметил-7-хлор-1-циклопропіл-4-оксо-1,4-дигідро-6-хінолініл)-аміно]етил)аміно]пропіоніл)-6-О-метилеритроміцин А;
11,12-(амінокарбонілокси)-4”-О-{3-[(2-[(3-карбокси-1-циклопропіл-4-оксо-1,4-дигідро-6-хінолініл)аміно]етил)аміно]пропіоніл}-11,12-дидезокси-6-O-метилеритроміцинА;
4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]-етил]метиламіно]пропіоніл]азитроміцин;
11,12-карбонат-4”-O-[(3-[[2-[(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)аміно]етил]метиламіно]пропіоніл]-11,12-дидезоксіазитроміцин;
4”-O-(3-{[2-(7-хлор-1-циклопропіл-3-метоксикарбоніл-4-оксо-1,4-дигідрохінолін-6-іламіно)-етил]-аміно}-пропіоніл)азитроміцин;
4”-O-(3-{[2-(7-хлор-1-циклопропіл-3-метоксикарбоніл-4-оксо-1,4-дигідрохінолін-6-іламіно)етил]метиламіно}пропіоніл)азитроміцин;
4”-O-{3-[(2-[(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-7-хінолініл)аміно]етил)-метиламіно]пропіоніл}азитроміцин і
4”-O-[3-[[2-[(3-карбоксі-1-етил-1,4-дигідро-4-оксо-6-хінолініл)аміно]етил]аміно]-пропіоніл]-6-O-метилеритроміцин А.
5. Сполука за будь-яким з пп. 1-4, призначена для застосування в терапії.
6. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що її застосовують у приготуванні ліків, призначених для застосування в терапії системних або місцевих бактеріальних інфекцій організму людини або тварини.
7. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що її застосовують для лікування або профілактики системних або місцевих бактеріальних інфекцій організму людини або тварини.
8. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (II)
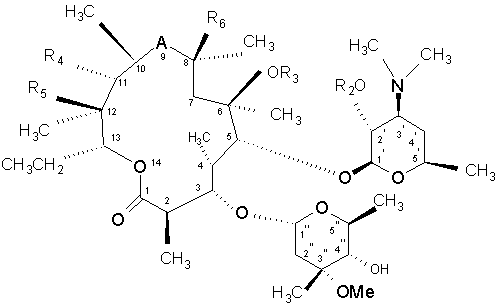 , (II)
, (II)
з придатним активованим похідним кислоти (III)
HOC(O)(CH2)nXR7, (ІІІ)
де n є цілим числом від 1 до 5, а Х і R7 мають значення, визначені в п. 1, з отриманням сполуки формули (І), де n є цілим від 1 до 5;
і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій:
і) видаляють захисну групу R2 і
іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати.
9. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (II), у якій 4”-гідроксил придатним чином активовано, зі сполукою формули XR7 (IV), де R7, m і Z мають значення, визначені в п.1, а Х є -U(CH2)mZ-, де U є групою, вибраною з -N(R13)-, -О- і -S-, з отриманням сполуки формули (I), де n=0, а U є групою, вибраною з -N(R13)-, -O- і -S-;
і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій:
і) видаляють захисну групу R2 і
іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати.
10. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (V)
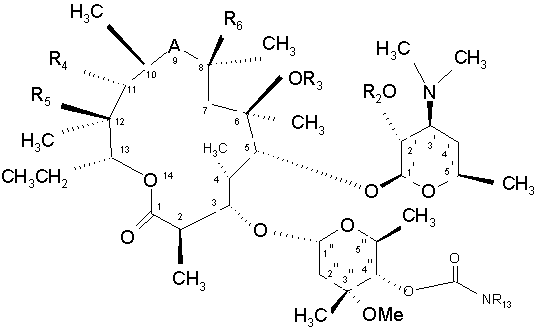 , (V)
, (V)
де R13 має значення, визначене в п. 1, з придатним активованим похідним карбонової кислоти
HOC(O)C(O)(CH2)mZR7 (VIIb),
де R7 і Z мають значення, визначені в п. 1, з отриманням сполуки формули (І), де n=0, a U є -N(R13)С(O)-;
і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій:
і) видаляють захисну групу R2 і
іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольват.
11. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (VII)
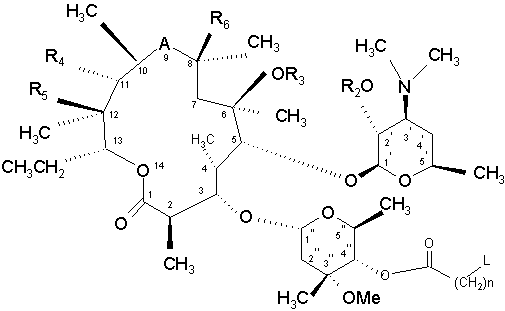 (VII)
(VII)
зі сполукою формули
XR7 (IV),
де R7 і Х мають значення, визначені в п. 1, де U є групою, вибраною з -N(R13)-, -О- і -S-, a L є придатною відщеплюваною групою, з одержанням сполуки формули (І), де n дорівнює 1-5, а U є групою, вибраною з -N(R13)-, -О- і -S-;
і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій:
і) видаляють захисну групу R2 і
іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати.
12. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (IX) 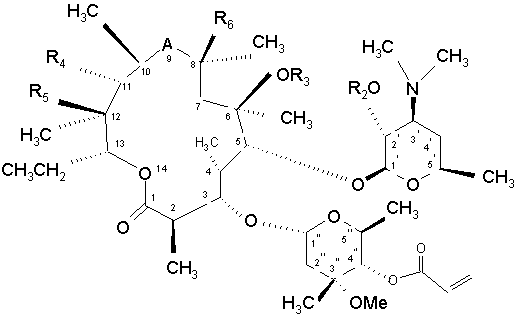 , (IX)
, (IX)
зі сполукою формули
XR7 (IV),
де R7 і Х мають значення, визначені в п. 1, де U є групою, вибраною з -N(R13)-, -О- і -S-, з отриманням сполуки формули (І), де n=2, а U є групою, вибраною з -N(R13)-, -O- і -S-,
і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій:
і) видаляють захисну групу R2 і
іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати.
13. Фармацевтична композиція, що містить сполуку за будь-яким з пп. 1-4 у суміші з одним або більше фармацевтично прийнятними носіями або ексципієнтами.
14. Спосіб лікування бактеріальної інфекції в організмі людини або організмі будь-якої тварини, у якому вводять ефективну кількість сполуки за будь-яким з пп. 1-4.
Текст
1. Макроліди формули (І) 3 77210 4 R13 - гідроген, С1-4алкіл, С3-7циклоалкіл, як варіант, 4”-O-[3-[[2-[(3-карбокси-1-циклопропіл-1,4-дигідрозаміщений фенілом або бензилом, ацетил або 4-оксо-7-хінолініл)аміно]етил]-аміно]пропіоніл]-6-Oбензоїл; метилеритроміцин А; Х - -U(CH2)mZ,- або Х є групою, вибраною з: 4”-O-[3-[[2-[(3-карбокси-1-циклопропіл-4-оксо-1,4дигідро-6-хінолініл)аміно]етил]-аміно]пропіоніл]-6O-метилеритроміцин А; N N (11S,11аR)-4”-O-[3-[[2-[(3-карбокси-7-хлор-1циклопропіл-1,4-дигідро-4-оксо-6або хінолініл)аміно]етил]аміно]пропіоніл]-11(карбоксиціанометил)-11-дезокси-6-О-метилH N еритроміцин А; (11S, 11aR)-11-(карбоксиціанометил)-4”-O-[3-[[2N [(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-7хінолініл)аміно]етил]аміно]пропіоніл]-11-дезокси-6H , O-метилеритроміцин А; U і Z незалежно є двовалентним радикалом, виб4”-O-[3-[4-(3-карбокси-7-хлор-1-циклопропіл-1,4раним з: дигідро-4-оксо-6-хінолініл)аміно]-N(R13)-, -O-, -S(O)q-, -N(R13)C(O)-, -C(O)N(R13)-, етил]аміно]пропіоніл]рокситроміцин; N[С(O)R13]-, 4”-O-(3-(2-(3-карбокси-7-хлор-1-циклопропіл-4W - атом карбону або нітрогену; оксо-1,4-дигідро-6-хінолініл)аміно)n - 0 або ціле число від 1 до 5; етил)метиламіно)пропіоніл)-11,12-дидезоксиm – ціле число від 2 до 8; 11,12-(етиламінокарбонілокси)-6-Op - 0, 1 або 2; метилеритроміцин А; q дорівнює 0, 1 або 2; 4”-O-(3-[(2-[(3-карбоксиметил-7-хлор-1і їх фармацевтично прийнятні солі і сольвати. циклопропіл-4-оксо-1,4-дигідро-6-хінолініл)2. Сполука за п. 1, яка відрізняється тим, що Х є аміно]етил)аміно]пропіоніл)-6-О-метилеритроміцин NH(CH2)2-3NH. А; 3. Сполука за п. 1 або п. 2, яка відрізняється тим, 11,12-(амінокарбонілокси)-4”-О-{3-[(2-[(3-карбоксищо R7 є гетероциклічною групою структури: 1-циклопропіл-4-оксо-1,4-дигідро-6O (R11)p хінолініл)аміно]етил)аміно]пропіоніл}-11,12R9 дидезокси-6-O-метилеритроміцинА; N 4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4дигідро-4-оксо-6-хінолініл)аміно]N етил]метиламіно]пропіоніл]азитроміцин; 11,12-карбонат-4”-O-[(3-[[2-[(3-карбокси-1R10 , циклопропіл-1,4-дигідро-4-оксо-6де R9, R10, R11 і p мають значення, визначені у п. 1. хінолініл)аміно]етил]метиламіно]пропіоніл]-11,124. Сполука, вибрана з групи, яку складають: дидезоксіазитроміцин; 4”-O[3-[[2-((3-карбокси-7-хлор-1-циклопропіл-1,44”-O-(3-{[2-(7-хлор-1-циклопропіл-3дигідро-4-оксо-6-хінолініл)аміно]метоксикарбоніл-4-оксо-1,4-дигідрохінолін-6етил]аміно]пропіоніл]-6-O-метил-8а-аза-8аіламіно)-етил]-аміно}-пропіоніл)азитроміцин; гомоеритроміцин А; 4”-O-(3-{[2-(7-хлор-1-циклопропіл-311,12-карбонат-4”-O-[3-[[2-[(3-карбокси-7-хлор-1метоксикарбоніл-4-оксо-1,4-дигідрохінолін-6циклопропіл-1,4-дигідро-4-оксо-6іламіно)етил]метиламіно}пропіоніл)азитроміцин; хінолініл)аміно]етил]аміно]пропіоніл]-11,124”-O-{3-[(2-[(3-карбокси-1-циклопропіл-1,4-дигідродидезоксіазитроміцин; 4-оксо-7-хінолініл)аміно]етил)11,12-карбонат-4”-O-[3-[[2-[(3-карбокси-1метиламіно]пропіоніл}азитроміцин і циклопропіл-1,4-дигідро-6-фтор-4-оксо-74”-O-[3-[[2-[(3-карбоксі-1-етил-1,4-дигідро-4-оксо-6хінолініл)аміно]етил]аміно]пропіоніл]-11,12хінолініл)аміно]етил]аміно]-пропіоніл]-6-Oдидезоксіазитроміцин; метилеритроміцин А. 11,12-карбонат-4”-O-[3-[[2-[(3-карбокси-15. Сполука за будь-яким з пп. 1-4, призначена для циклопропіл-1,4-дигідро-4-оксо-7-хінолініл)застосування в терапії. аміно]етил]аміно]пропіоніл]-11,126. Сполука за будь-яким з пп. 1-4, яка відрізнядидезоксіазитроміцин; ється тим, що її застосовують у приготуванні ліків, 4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,4призначених для застосування в терапії системних дигідро-4-оксо-6-хінолініл)аміно]або місцевих бактеріальних інфекцій організму етил]аміно]пропіоніл]азитроміцин; людини або тварини. 4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,47. Сполука за будь-яким з пп. 1-4, яка відрізнядигідро-4-оксо-6-хінолініл)аміно]ється тим, що її застосовують для лікування або етил]аміно]пропіоніл]-11,12-дидезокси-11,12профілактики системних або місцевих бактеріаль(етиламінокарбонілокси)-6-O-метилеритроміцин А; них інфекцій організму людини або тварини. 4”-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл-1,48. Спосіб отримання сполуки за п. 1, у якому здійсдигідро-4-оксо-6-хінолініл)аміно]нюють реакцію сполуки формули (II) етил]аміно]пропіоніл]-6-O-метилеритроміцин А; 4”-O-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-4-оксо7-хінолініл)аміно]етил]аміно]-пропіоніл]-6-Oметилеритроміцин А; 5 4 6 7 8 1 3 2 5 H3C CH3 A R4 9 8 10 R5 7 12 CH3CH2 O 14 3' 2' O 2 CH3 (II) 2'' 3'' 9 10 R5 7 12 H3C CH3CH2 O 14 2' 5 3 2 O 3' 1' 4 1 CH3 N CH3 6 H3 C 13 R2O O O O 5'' 1'' 2'' CH3 H3C 3'' 4' 5' O CH3 CH3 (V) O 4'' O OMe NR13 , де R13 має значення, визначене в п. 1, з придатним активованим похідним карбонової кислоти HOC(O)C(O)(CH2)mZR7 (VIIb), де R7 і Z мають значення, визначені в п. 1, з отриманням сполуки формули (І), де n=0, a U є N(R13)С(O)-; і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій: і) видаляють захисну групу R2 і іі) перетворюють отриману сполукуформули (І) у її фармацевтично прийнятну сіль і сольват. Винахід стосується нових напівсинтетичних макролідів, які володіють антибактеріальною активністю. Зокрема, винахід стосується 14- або 15членних макролідів, заміщених у 4"-положенні, CH3 N 2' 5 3' 4' 5' 1' 4 O 3 1 H3C OR3 11 O 14 R2O CH3 6 H3 C OH CH3 8 7 13 H3C OR3 11 CH3CH2 4'' R6 A CH3 8 12 , з придатним активованим похідним кислоти (III) HOC(O)(CH2)nXR7, (ІІІ) де n є цілим числом від 1 до 5, а Х і R7 мають значення, визначені в п. 1, з отриманням сполуки формули (І), де n є цілим від 1 до 5; і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій: і) видаляють захисну групу R2 і іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати. 9. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (II), у якій 4”гідроксил придатним чином активовано, зі сполукою формули XR7 (IV), де R7, m і Z мають значення, визначені в п.1, а Х є -U(CH2)mZ-, де U є групою, вибраною з -N(R13)-, -О- і -S-, з отриманням сполуки формули (I), де n=0, а U є групою, вибраною з -N(R13)-, -O- і -S-; і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій: і) видаляють захисну групу R2 і іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати. 10. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (V) H3C 10 R5 2 O O O OMe H3C R4 9 H3C 5'' 1'' CH3 4' O CH3 O O O R6 A R4 5' 1' 4 H3C N 5 3 1 CH3 R2O CH3 6 H3 C 13 H3C OR3 11 H3C 77210 6 11. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (VII) R6 5'' 1'' 2'' CH3 3'' (VII) O 4'' L O (CH2)n OMe H3C CH3 O CH3 зі сполукою формули XR7 (IV), де R7 і Х мають значення, визначені в п. 1, де U є групою, вибраною з -N(R13)-, -О- і -S-, a L є придатною відщеплюваною групою, з одержанням сполуки формули (І), де n дорівнює 1-5, а U є групою, вибраною з -N(R13)-, -О- і -S-; і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій: і) видаляють захисну групу R2 і іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати. 12. Спосіб отримання сполуки за п. 1, у якому здійснюють реакцію сполуки формули (IX) H3C R6 CH3 A R4 9 10 R5 8 7 12 H3C O 1 14 R2O 2' 5 O 3 O 3' O O 2'' O CH3 5'' 1'' CH3 3'' 4' 5' 1' 4 2 CH3 N CH3 6 H3 C 13 CH3CH2 H3C OR3 11 CH3 (IX) O 4'' O OMe HC , зі сполукою формули XR7 (IV), де R7 і Х мають значення, визначені в п. 1, де U є групою, вибраною з -N(R13)-, -О- і -S-, з отриманням сполуки формули (І), де n=2, а U є групою, вибраною з -N(R13)-, -O- і -S-, і після цього, якщо потрібно, проводять з отриманою сполукою одну або більше таких операцій: і) видаляють захисну групу R2 і іі) перетворюють отриману сполуку формули (І) у її фармацевтично прийнятну сіль і сольвати. 13. Фармацевтична композиція, що містить сполуку за будь-яким з пп. 1-4 у суміші з одним або більше фармацевтично прийнятними носіями або ексципієнтами. 14. Спосіб лікування бактеріальної інфекції в організмі людини або організмі будь-якої тварини, у якому вводять ефективну кількість сполуки за будь-яким з пп. 1-4. 3 способів їх одержання, композицій, що їх містять та їх застосування у медицині. Таким чином, згідно з винаходом, запропоновано сполуки загальної формули (І) 7 у якій А - бівалентний радикал, обраний серед: -С(О)-, -C(O)NH-, -NHC(O)-, -N(CH3)-CH2-, CH2-N(CH3)- або -C(NOCH2OCH2CH2OCH3)-; R1 - OC(O)(CH2)nXR7; R2 - гідроген або групу, що захищає гідроксил; R3 -гідроген, С1-4алкіл або С3-6алкеніл, як варіант, заміщений 9-10-членним конденсованим біциклічним гетероарилом; R4 - гідроксил або С3-6алкенілоксил, як варіант, заміщений 9-10-членним конденсованим біциклічним гетероарилом; R5 - гідроксил або R4 і R5 разом з проміжними атомами, утворюють циклічну групу структури: де Υ - бівалентний радикал, обраний серед: -СН2-, -CH(CN)-, -О-, -N(R8)- і -CH(SR8)-; R6 - гідроген або фтор; R7 - гетероциклічна група структури: або R8 - гідроген або С1-4алкіл, заміщений групою, обраною серед: як варіант, заміщеного фенілу, як варіант, заміщеного 5- або 6-членного гетероарилу, як варіант, заміщеного 9- або 10-членного конденсованого біциклічного гетероарилу; R9 - гідроген, C(O)OR12, C(O)NHR12 або C(O)CH2NO2; R10 - гідроген, С1-4алкіл, С3-7циклоалкіл, як варіант, заміщений фенілом або бензилом; R11 - галоген, С1-4алкіл, С1-4тіоалкіл, С1 77210 8 алкоксил, NH2, NН(С1-4алкіл) або N(С1-4алкіл)2; 4 R12 являє собою гідроген або С1-4алкіл; R13 являє собою гідроген, С1-4алкіл, С37циклоалкіл, як варіант, заміщений фенілом або бензилом, ацетилом або бензоїлом; X - -U(CH2)m-, або X - група, обрана серед: або U і Ζ незалежно є двовалентним радикалом, обраним серед: -N(R13)-, -О-, -S(O)q-, -N(R13)C(O)-, -C(O)N(R13)-, -N[C(O)R13]-, W - атом карбону або нітрогену; n - ціле, від 0 до 5; m - ціле, від 2 до 8; p - ціле, від 0 до 2; q - ціле, від 0 до 2; і їх фармацевтично прийнятних солей і сольватів. Придатні фармацевтично прийнятні солі сполук загальної формули (І) включають солі приєднання кислот, утворені з фармацевтично прийнятними органічними або неорганічними кислотами, наприклад гідрохлориди, гідроброміди, сульфати, алкіл- або арилсульфонати (наприклад метансульфонати або п-толуолсульфонати), фосфати, ацетати, цитрати, сукцинати, тартрати, фумарати і малеати. Сольвати можуть, наприклад, бути гідратами. У посиланнях на сполуку винаходу маються на увазі як сполуки формули (І), так і їх фармацевтично прийнятні солі приєднання кислот, а також фармацевтично прийнятні сольвати. Сполуки формули (І) і їх солі можуть утворювати сольвати (наприклад гідрати), і винахід включає всі такі сольвати. У загальній формулі (І), як її показано, суцільний клиноподібний зв'язок вказує, що цей зв'язок виступає над площиною паперу. Зв'язок, показаний штрихом, вказує, що цей зв'язок лежить за площиною паперу. Сполуки, у яких R2 є групою, що захищає гідроксил, ця група взагалі є проміжними сполуками для одержання інших сполук формули (І). Коли група OR2 є захищеною гідроксильною групою, бажано, щоб це був етер або ацилоксильна група. Прикладами особливо придатних етерних груп можуть бути групи, у яких R2 є триалкілсилілом (наприклад триметилсилілом). Коли група OR2 є ацилоксигрупою, то прикладами придатних груп R2 можуть бути ацетил або бензоїл. Коли R7 є гетероциклічной групою структури: 9 де W - карбон або нітроген, то зазначений гетероцикл з'єднаний у 7 або 6 положенні з Zгрупою, визначеною вище, або з одним з атомів нітрогену, що містяться структурах: або Коли R7 є гетероциклічною групою структури: то зазначений гетероцикл з'єднаний у 8 або 7 положенні з Z-групою, визначеною вище, або з одним з атомів нітрогену, що містяться структурах: або Коли R7 є гетероциклічной групою структури: то зазначений гетероцикл з'єднаний у 2 або 3 положенні з Z-групою, визначеною вище, або з одним з атомів нітрогену, що містяться у структурах: або 77210 10 Термін "С1-4алкіл", застосований тут стосовно групи або частини групи, стосується нерозгалуженої або розгалуженої алкільної групи, яка містить від 1 до 4 атомів карбону; прикладами таких груп є метил, етил, пропіл, ізопропіл, н-бутил, ізобутил, трет-бутил. Термін "С2-6алкенільна група", використаний тут стосовно групи або частини групи, стосується нерозгалуженої або розгалуженої алкенільної групи, яка містить від 2 до 6 атомів карбону; прикладами таких груп є 2-пропеніл, 1-пропеніл, ізопропеніл, 2-бутеніл, 2-пентеніл, 2-гексеніл тощо. Слід взяти до уваги, що в групах формули -О-С26алкеніл, подвійний зв'язок переважно не є суміжним з атомом оксигену. Термін " С3-7циклоалкільна група" стосується неароматичного моноциклічного карбоногідрогенового циклу з 3-7 атомами карбону, наприклад, циклопропілу, циклобутилу, циклопентилу, циклогексилу або циклогептилу. Термін "5- або 6-членний гетероарил", використаний тут стосовно групи або частини групи, стосується фуранілу, тіофенілу, імідазолілу, оксазолілу, тіазолілу, піразолілу, піридилу, піридинію, піридазинілу або піримідинілу. Термін "галоген" стосується атому фтору, хлору, брому або йоду. Термін "С1-4алкоксигрупа" може означати алкоксигрупу з нерозгалуженим або розгалуженим ланцюгом, наприклад метоксил, етоксил, пропоксил, проп-2-оксил, бутоксил, бут-2-оксил або метилпроп-2-оксил. Термін "9- - 10-членний конденсований біциклічний гетероарил", використаний тут стосовно групи або частини групи, стосується хінолінілу, ізохінолінілу, 1,2,3,4-тетрагідроізохінолінілу, бензофуранілу, бензімідазолілу, бензотіенілу, бензоксазолілу, 1,3-бензодіоксазолілу, індолілу, бензотіазолілу, фурилпіридину, оксазолопіридилу або бензотіофенілу. Терміни "як варіант, заміщений феніл", як варіант, заміщений 5- або 6-членний гетероарил" або, як варіант, "заміщений 9- 10-членний конденсований біциклічний гетероарил" стосуються групи, заміщеної 1-3 групами, обраними серед галогену, С1-4алкілу, С1-4алкоксилу, гідроксилу, нітрогрупи, ціаногрупи, аміногрупи, С14алкіламіногрупи або диС1-4алкіл-аміногрупи, фенілу або 5- або 6-членного гетероарилу. Бажаними сполуками формули (І) є ті, в яких R2 є гідрогеном. В одному з втілень R3 - гідроген або С1-4алкіл, наприклад метил, етил або пропіл. Коли А є -С(О)-, R6 переважно є гідрогеном, a R3 переважно є С1-4алкілом (наприклад, метилом), або R6 переважно є фтором, a R3 переважно є гідрогеном. Коли А позначає -С(О)-, R4 і R5 переважно є гідроксилом або R4 і R5 разом з проміжними атомами, утворюють циклічну групу структури: 11 де Υ переважно є -N(R8)-, a R8 є гідрогеном або С1-4алкілом, наприклад, метилом або етилом. Коли А є -NHC(O)-, -C(O)NH-, -N(CH3)-CH2-, H2N(CH3)- або -OCH2OCH2CH2OCH2, 41 або R5 є гідроксилом, або R4 і R5 разом з проміжними атомами, утворюють циклічну групу структури: то R6 переважно є гідрогеном, a R3 - гідрогеном, С1-4алкілом (наприклад, метилом) або 2пропенілом. Коли А є -NHC(O)-, -C(O)NH-, -N(CH3)-CH2-, CH2-N(CH3) або - C(NOCH2OCH2CH2OCH3)-, a R4 і R5 разом з проміжними атомами, утворюють циклічну групу структури: де Υ є бівалентним радикалом, обраним серед -СН2-, -CH(CN)-, -N(R8)-, то R6 переважно є гідрогеном, a R3 переважно є С1-4алкілом (наприклад, метилом) або 2-пропенілом. Коли n=0, U переважно обрано серед -N(R13)-, -О- і -S-. У сполуках формули (І) n переважно дорівнює 1 або 2. У цьому класі сполуки, в яких n дорівнює 2, є найкращими. Бажаний клас сполук формули (І) складають т, в яких X є NH(CH2)2-5NH або у цьому класі сполуки, в яких X є NH(CH2)2є найкращими. Інший бажаний клас сполук формули (І) складають ті, в яких X є N(R13)(CH2)23NH і R13 є С1-4алкілом, наприклад, метилом або ацетилом. У ще одному бажаному класі сполук формули (І) R7 є гетероциклілом формули: 3NH, 77210 12 У цьому класі найкращими є сполуки, в яких p дорівнює 0 або 1, R11 є галогеном (наприклад, хлором або фтором) у положенні 6 або 7, R9 є карбоксилом, і R10 є метилом, етилом або циклопропілом. Найкращий клас сполук винаходу складають ті, в яких R2 - гідроген, X - NH(CH2)2-3NH, R7 є гетероциклічною групою формули: де p дорівнює 0 або 1, R11 є галогеном (наприклад, хлором або фтором) у положенні 6 або 7, R9 є карбоксилом, і R10 - етил або циклопропіл. Інший бажаний клас сполук складають ті, в яких R9 - C(O)OR12 і R12 є С1-4алкілом, зокрема метилом. Ще один бажаний клас сполук складають ті, в яких R11 - С1-4алкоксил, наприклад, меток-сил в положенні 8. Коли R3 є С3-6алкенілом, заміщеним 9-10членним конденсованим біциклічним гетероарилом, 9-10-членний конденсований біциклічний гетероарил переважно є 2-хінолілом або 3хінолілом, R4 і R5 переважно є гідроксилом і R6 є гідрогеном. У цьому класі найкращими є сполуки, в яких R2 - гідроген, X є NH(CH2)2-3NH, R7 є гетероциклом формули: де p=1, R11 - галоген (наприклад хлор або фтор) у положенні 6 або 7, R9 є карбоксилом, a R10 - етил або циклопропіл. В одному з втілень Υ є бівалентним радикалом, обраним серед -СН2-, -CH(CN)-, -О- і N(Rg)-. Найбільш бажаними є такі сполуки винаходу: 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл1,4-дигідро-4-оксо-6-хінолініл(аміно]етил]аміно]пропіоніл]-8а-аза-8а-гомоеритроміцин А; 4"-O-[3-[[2-((3-карбокси-1-циклопропіл-1,4дигідро-6-фтор-4-оксо-7-хінолініл)аміно)етил]аміно]пропіоніл]-8а-аза-8а-гомоеритроміцин А; 4"-O-[3-[[2-((3-карбокси-7-хлор-1-циклопропіл1,4-дигідро-4-оксо-6-хінолініл(аміно(етил]аміно(пропіоніл]-6-O-метил-8а-аза-8агомоеритроміцин А; 4"-O-[3-[(2-[(3-карбокси-1-циклопропіл-1,4дигідро-6-фтор-4-оксо-7-хінолініл)аміно]етил]аміно]пропіоніл]-6-О-метил-8а-аза-8агомоеритроміцин А; 4"-O-[3-[[2-[(7-хлор-1-циклопропіл-1,4-дигідро3-метоксикарбоніл-4-оксо-6-хінолініл)аміно]етил]аміно]пропіоніл]-6-O-метил-8а-аза-8а 13 77210 14 гомоеритроміцин А; етил]аміно]пропіоніл]-рокситроміцин; 4"-O-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-44"-О-[3-[4-(3-карбокси-1-циклопропіл-1,4оксо-7-хінолініл)аміно]етил]аміно]-пропіоніл]-6-Одигідро-6-фтор-4-оксо-7-хінолініл)аміно]метил-8а-аза-8а-гомоеритроміцин А; етил]аміно]пропіоніл]-рокситроміцин; 4"-О-[3-[(2-[(3-карбокси-1-циклопропіл-1,44"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілдигідро-4-оксо-7-хінолініл)аміно]етил]1,4-дигідро-4-оксо-3-хінолініл)аміно]аміно]пропіоніл]-6-O-метил-8а-аза-8аетил]аміно]пропіоніл]-азитроміцин; гомоеритроміцин А; 11,12-карбонат-4"-О-[3-[[2-[(3-карбокси-14"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілциклопропіл-1,4-дигідро-6-фтор-4-оксо-71,4-дигідро-4-оксо-6-хінолініл)аміно]хінолініл)аміно]етил]аміно]пропіоніл]-11,12етил]аміно]пропіоніл]-6-О-етил-8а-аза-8адидезоксиазитроміцин; гомоеритроміцин А; 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл1,4-дигідро-4-оксо-3-хінолініл)аміно]1,4-дигідро-4-оксо-6-хінолініл)аміно]етил]аміно]пропіоніл]-6-О-метилазитроміцин; етил]аміно]пропіоніл]-6-О-пропіл-8а-аза-8а11,12-карбонат-4"-О-[3-[[2-[(3-карбокси-7-хлоргомоеритроміцин А; 1-цикпопропіл-1,4-дигідро-4-оксо-64"-O-[3-([2-((3-карбокси-7-хлор-1-циклопропілхінолініл)аміно]етил]аміно]пропіоніл]-11,121,4-дигідро-4-оксо-6-хінолініл(амінодидезоксиазитроміцин; (етил]аміно(пропіоніл]-6-О-метил-9а-аза-9а11,12-карбонат-4"-О-[3-[[2-[(3-карбокси-1гомоеритроміцин А; циклопропіл-1,4-дигідро-6-фтор-4-оксо-74"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілхінолініл)аміно]етил]аміно]пропіоніл]-11,121,4-дигідро-4-оксо-6-хінолініл)аміно]дидезоксиазитроміцин. етил]аміно]пропіоніл]-11,12-дидезокси-6-О-метилБажаними є також такі сполуки винаходу: 11,12-(метиламінокарбонілокси)-еритроміцин А; 4"-О-[3-[[2-((3-карбокси-7-хлор-1-циклопропіл11,12-(амінокарбонілокси)-4"-О-[3-[[2-[(31,4-дигідро-4-оксо-6-хінолініл)аміно)карбокси-7-хлор-1-цикпопропіл-4-оксо-1,4-дигідроетил]аміно]пропіоніл]-6-О-метил-8а-аза-8а6-хінолініл)аміно]етил]аміно]пропіоніл]-11,12гомоеритроміцин А; дидезокси-6-О-метил-еритроміцин А; 11,12-карбонат-4"-О-[3-[[2-[(3-карбокси-7-хлор11,12-(амінокарбонілокси)-4"-О-[3-[[2-[(31-циклопропіл-1,4-дигідро-4-оксо-6карбокси-1-цикпопропіл-1,4-дигідро-6-фтор-4-оксохінолініл)аміно]етил]аміно]пропіоніл]-11,127-хінолініл)аміно]етил]аміно]пропіоніл]-11,12дидезокси-азитроміцин; дидезокси-6-О-метил-еритроміцин А; 11,12-карбонат-4"-О-[3-[[2-[(3-карбокси-14"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілциклопропіл-1,4-дигідро-6-фтор-4-оксо-71,4-дигідро-4-оксо-6-хінолініл)аміно]хінолініл)аміно]етил]аміно]пропіоніл]-11,12етил]аміно]пропіоніл]-6-О-метил-еритроміцин А; дидезокси-азитроміцин; 4"-O-[3-[[2-[(3-карбокси-1-циклопропіл-6-фтор11,12-карбонат-4"-О-[3-[[2-[(3-карбокси-11,4-дигідро-4-оксо-7-хінолініл)циклопропіл-1,4-дигідро-4-оксо-7-хінолініл)аміно]етил]аміно]пропіоніл]-6-О-метиламіно]етил]-аміно]пропіоніл]-11,12-дидезоксиеритроміцин А; азитроміцин; 4"-О-[3-[[2-[(3-карбокси-1-циклопропіл-4-оксо4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл1,4-дигідро-6-хінолініл)аміно]етил]1,4-дигідро-4-оксо-6-хінолініл)аміно]аміно]пропіоніл]-6-O-метил-еритроміцин А; етил]аміно]пропіоніл]-азитроміцин; 4"-О-[3-[[2-[(3-карбокси-7-хлор-1,4-дигідро-14"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілетил-4-оксо-6-хінолініл)аміно]етил]аміно]1,4-дигідро-4-оксо-6-хінолініл)аміно]пропіоніл]-6-О-метил-еритроміцин А і етил]аміно]пропіоніл]-11,12-дидезокси-11,124"-О-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-6(етиламінокарбонілокси)-6-O-метил-еритроміцин фтор-4-оксо-7-хінолініл)аміно]етил]А; аміно]пропіоніл]-6-О-метил-еритроміцин А; 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл4"-О-[3-[[2-[(3-карбокси-1-циклопропіл-1,41,4-дигідро-4-оксо-6-хінолініл)аміно]дигідро-4-оксо-7-хінолініл)аміно]етил]етил]аміно]пропіоніл]-6-O-метилеритроміцин А; аміно]пропіоніл]-6-O-метил-еритроміцин А; 4"-О-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-4(11S,11а)-11-(карбоксиціанометил)-4"-О-[3-[[2оксо-7-хінолініл)аміно]етил]аміно]-пропіоніл]-6-О[(3-карбокси-1,4-дигідро-1-етил-4-оксо-7метилеритроміцин А; хінолініл)аміно]етил]аміно]пропіоніл]-11-дезокси-64"-O-[3-[[2-[(3-карбокси-1-циклопропіл-1,4О-метил-еритроміцин А; дигідро-4-оксо-7-хінолініл)аміно]етил](11S,11а)-11-(карбоксиціанометил)-4"-O-[3-[[2аміно]пропіоніл]-6-O-метилеритроміцин А; [(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-74"-O-[3-[[2-[(3-карбокси-1-циклопропіл-4-оксохінолініл)аміно]етил]аміно]пропіоніл]-11-дезокси-61,4-дигідро-6-хінолініл)аміно]етил]О-метил-еритроміцин А; аміно]пропіоніл]-6-О-метилеритроміцин А; (11S,11а)-4"-О-[3-[[2-[(3-карбокси-7-хлор-1(11S,11а)-4"-О-[3-[[2-[(3-карбокси-7-хлор-1циклопропіл-1,4-дигідро-4-оксо-6циклопропіл-1,4-дигідро-4-оксо-6хінолініл)аміно]етил]аміно]пропіоніл]-11хінолініл)аміно]етил]аміно]пропіоніл]-11(карбоксиціанометил)-11-дезокси-6-Ометил(карбоксиціанометил)-11-дезокси-6-О-метилеритроміцин А; еритроміцин А; 4"-О-[3-[4-(3-карбокси-7-хлор-1-циклопропіл(11S,11а)-11-(карбоксиціанометил)-4"-O-[3-[[21,4-дигідро-4-оксо-6-хінолініл)аміно][(3-карбокси-1-циклопропіл-1,4-дигідро-4-оксо-7 15 77210 16 хінолініл)аміно]етил]аміно]пропіоніл]-11-дезокси-6ництва терапевтичного засобу для лікування або О-метилеритроміцин А; профілактики системних або місцевих бактеріаль4"-О-[3-[4-(3-карбокси-7-хлор-1-циклопропілних інфекцій організму людини або тварини. 1,4-дигідро-4-оксо-6-хінолініл)аміно]Згідно з ще одним аспектом винаходу, запроетил]аміно]пропіоніл]-рокситроміцин; поновано спосіб лікування організму людини або 4"-O-(3-(2-(3-карбокси-7-хлор-1-циклопропіл-4тварини, що не є людиною, для боротьби з бактеоксо-1,4-дигідро-6-хінолініл)аміно)ріальними інфекціями, при якому в організм ввоетил)метиламіно)пропіоніл)-11,12-дидезоксидять ефективну кількість сполуки формули (І) або 11,12-(етиламінокарбонілокси)-6-Ойого фізіологічно прийнятної солі. метилеритроміцин А; Хоча для застосування в терапії, як варіант, 4"-O-{3-[(2-[(3-карбоксиметил-7-хлор-1можна вводити сполуку винаходу у вигляді хімічної циклопропіл-4-оксо-1,4-дигідро-6-хінолініл)речовини як такої, бажано використовувати активаміно]етил)аміно]пропіоніл}-6-O-метилеритроміцин ний інгредієнт у вигляді фармацевтичного препаА; рату. 11,12-(амінокарбонілокси)-4"-O-{3-[(2-[(3Сполуки винаходу можуть бути пристосовані карбокси-1-циклопропіл-4-оксо-1,4-дигідро-6для введення будь-яким зручним шляхом для вихінолініл)аміно]етил)аміно]пропіоніл}-11,12користання у медицині або ветеринарії, і винахід; дидезокси-6-О-метилеритроміцин А; отже, винахід включає фармацевтичні композиції, 4"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілщо містять сполуку винаходу і пристосовані для 1,4-дигідро-4-оксо-6-хінолініл)аміно]використання у медицині або ветеринарії. Такі етил]метиламіно]пропіоніл]-азитроміцин; композиції можуть бути використані у традиційний 11,12-карбонат-4"-O-(3-((2-((3-карбокси-1спосіб задопомогою одного або більше придатних циклопропіл-1,4-дигідро-4-оксо-6-хінолініл)носіїв або ексципієнтів. Композиції винаходу вклюаміно)етил)метиламіно)пропіоніл)-11,12чають композиції у формі, спеціально пристосовадидезокси-азитроміцин; ній для парентерального, перорального, буккаль4"-О-(3-{[2-(7-хлор-1-циклопропіл-3ного, ректального, локального, імплантантного, метоксикарбоніл-4-оксо-1,4-дигідрохінолін-6офтальмологічного, назального або генетоіламіно)етил]аміно}пропіоніл)-азитроміцин; уретального застосування. 4"-О-(3-{[2-(7-хлор-1-циклопропіл-3Сполуки винаходу можуть бути пристосовані метоксикарбоніл-4-оксо-1,4-дигідро-хінолін-6для застосування в медицині або ветеринарії іламіно)-етил]метиламіно}пропіоніл)-азитроміцин; шляхом ін'єкції (наприклад внутрішньовенної бо4"-О-{3-[(2-[(3-карбокси-1-циклопропіл-1,4люсной ін'єкції або інфузіі, або внутрішньом'язодигідро-4-оксо-7-хінолініл)аміно]етил)вим, підшкірним або інтратекальним шляхом) і метиламіно]пропіоніл}-азитроміцин; і можуть бути представлені як одиничні дозовані 4"-О-[3-[[2-[(3-карбокси-1-етил-1,4-дигідро-4форми, в ампулах, або в інших контейнерах для оксо-6-хінолініл)аміно]етил]аміно]-пропіоніл]-6-Оодиничної дози, або в багатодозових контейнерах метилеритроміцин А. з додаванням консерванту за необхідності. КомпоСполуки винаходу також демонструють широзиції для ін'єкції можуть мати форму суспензій, кий спектр антибактеріальної активності проти розчинів або емульсій у масляних або водних ноширокого ряду клінічних патогенних мікроорганізсіях, і можуть містити допоміжні засоби, напримів. Наприклад, при використанні стандартного клад, суспендуючі, стабілізуючі, солюбілізуючі тесту з серійними розведеннями мікротитраційнноі/або диспергуючі агенти. У іншому варіанті активго бульйону було виявлено, що сполуки винаходу ний інгредієнт може мати форму стерильного помають корисні рівні активності проти широкого рошку для розведення перед використанням приряду патогенних мікроорганізмів, включаючи штадатним носієм, наприклад стерильною ми Staphylococcus aureus, Streptopococcus безпірогенною водою. pneumoniae, Moraxella catarrhalis, Streptococcus Сполуки винаходу також можуть бути пристоpyogenes, Haemophilus Influenzae, Chlamydia совані для введення людині або тварині у формі, pneumoniae, Mycoplasma pneumoniae і Legionella придатній для перорального або буккального ввеpneumophila. Сполуки винаходу, таким чином, модення, наприклад, у вигляді розчинів, гелів, сирожуть бути використані для лікування багатьох запів, рідин або суспензій для полоскання рота, або хворювань, викликаних патогенними бактеріями у сухого порошку для розведення перед викорислюдини і тваринах. Сполуки винаходу можуть бути танням водою або іншим придатним носієм, як також активними проти стійких штамів, наприклад варіант, з коригентами і барвниками. Також моштамів, стійких до еритроміцину. Зокрема, сполуки жуть бути використані тверді композиції, напривинаходу можуть бути активними проти стійких до клад, таблетки, капсули, льодяники, пастилки, піеритроміцину штамів Streptococcus pneumoniae і гулки, болюси, порошки, пасти, гранули, кульки Streptococcus pyogenes. або премікс-препарати. Тверді і рідкі композиції Отже, відповідно до іншого аспекту винаходу, для перорального введення можуть бути приготозапропоновано сполуку формули (І) або її фізіоловлені згідно з методами, добре відомими фахівгічно прийнятну сіль для застосування в терапії цям. Такі композиції також можуть містити одного або профілактиці системних або місцевих бактеріабо більше фармацевтично прийнятних носіїв і альних інфекцій у людини або тварини. ексципієнтів у твердій або рідкій формі. Відповідно до подальшого аспекту винаходу, Сполуки винаходу також можна вводити перозапропоновано застосування сполуки формули (І) рально у ветеринарії у формі великих рідких доз або його фізіологічно прийнятної солі для виробліків, наприклад, у вигляді розчину, суспензії або 17 77210 18 дисперсії активного інгредієнта з фармацевтично прийнятним носієм або ексципієнтом. Сполуки винаходу можуть також бути приготовлені у вигляді суппозиторіїв, наприклад, таких, що містять основи для супозиторіїв, традиційні в медицині або ветеринари, або у вигляді песаріїв, наприклад, на традиційній основі. Сполуки винаходу можуть бути пристосовані для локального введення при застосуванні в медицині і ветеринарії, у формі мазей, кремів, гелів, з придатною активованою похідною карбонолосьйонів, шампунів, порошків (включаючи порошвої кислоти (III) ки, що розпорошуються), пессаріїв, тампонів, розз подальшим (якщо необхідно) видаленням чинів, що розпорошуються, просочень, аерозолів, групи R2, яка захищає гідроксил. крапель (наприклад очних крапель або крапель у Придатні активовані похідні карбоксильної ніс) або зрошень. групи включають відповідні ацилгалогеніди, зміАерозолі, що розпорошуються, зручно вводити шаний ангідрид або активований етер, наприклад, за допомогою упаковок, що знаходяться під підтіоетер. Реакцію переважно здійснюють у придатвищеним тиском, з використанням придатного ному апротонному розчиннику, наприклад, галогепропеланта, наприклад дихлордифторметану, ногідрокарбоні (наприклад ДХМ) або Ν,Ν-ДМФ, як трихлорфторметану, дихлортетрафторетану, діокваріант, в присутності третинної органічної основи, сиду карбону або іншого придатного газу. наприклад, диметиламінопіридину або триетилаДля локального введення інгаляцією сполуки міну, або в присутності неорганічної основи (навинаходу можна вводити за допомогою небулайприклад гідриду натрію) при температурі від 0° до зера. 120°С. Фармацевтичні композиції для локального Сполуки формули (І), де n=0, a U - група, обвведення можуть містити інші активні інгредієнти, рана серед -N(R13)-, -О- і -S(O)q-, де q=0, можуть наприклад, кортикостероїди або протигрибкові бути отримані через взаємодію сполуки формули засоби, якщо це вважається доцільним. (II), у якій 4"-гідроксил є належним чином активоКомпозиції можуть містити від 0,01 до 99% акваний, з сполукою формули XR7 (IV), з подальшим тивного матеріалу. Для місцевого введення, на(якщо необхідно) видаленням групи R2, яка захиприклад, композиція має взагалі містити від 0,01 щає гідроксил. Придатні активовані похідні 4"до 10%, бажано 0,01-1% активного матеріалу. гідроксигрупи включають, наприклад, карбоніліміДля системного введення добова доза для лідазол. Реакцію переважно здійснюють у придаткування дорослої людини, лежить у межах від 2 до ному апротонному розчиннику, наприклад, галоге100мг/кг ваги тіла, бажано 5-60мг/кг ваги тіла, і її ногідрокарбоні (наприклад ДХМ) або Ν,Ν-ДМФ, як можна вводити в 1-4 рази на добу, наприклад, заваріант, в присутності третинної основи, наприлежно від шляху введення і стану пацієнта. Коли клад, диметиламінопіридину або триетиламіну, композиція містить одиничні дозування, кожна при температурі від 0° до 120°С. одиниця повинна має містити від 200мг до 1г актиСполуки формули (І), де n=0, a U - вного інгредієнта. Тривалість лікування має визнаN(R13)C(O)-, можуть бути отримані взаємодією чатися радше рівнем реакції, а не умовною кількіссполуки формули (V), тю днів. Сполуки загальної формули (І) і їх солі можуть бути отримані згідно з загальними методиками, наведеними далі. У подальшому описі, групи R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, A, X, Y, U, Z, W, n, m, p і q мають значення, визначені для сполук формули (І), якщо не визначено інше. Групи XR7 і ZR7 позначено XR7 і ZR7, згідно з визначеннями для формули (І), або як групи, що можна перетворити у XR7 і ZR7 відповідно. Перетворення певної групи в групу XR7 або ZR7 звичайно має місце, якщо при проведенні реакцій, описаних нижче, використовується захисна група. Повний опис методик захисту таких груп, а також способів обробки отриманих захищених похідних можна знайти, наприклад у [T.W. Greene and P.G.M Wuts in Protective Groups in Organic Synthesis 2nd ed, John Wiley & Son, Inc 1991]. Сполуки формули (І), де n - ціле число від 1 до 5, можуть бути отримані взаємодією 4" гідроксилу формули (II), з сполукою формули HOC(O)C(O)(CH2)bZR7(VI), з подальшим (якщо необхідно) видаленням групи R2, яка захищає гідроксил. Реакцію переважно здійснюють у придатному апротонному розчиннику, такому як галогенгідрокарбон (наприклад, ДХМ) або Ν,Ν-ДМФ, як варіант, в присутності третинної основи, наприклад, диметиламінопіридину або триетиламіну при температурі від 0° до 120°С. Сполуки формули (V) можуть бути отримані обробкою сполуки формули (II), у якій 4" гідроксил 19 77210 20 є належним чином активований, з аміном NR13(Vlla). Придатні активовані похідні 4" гідроксигрупи включають, наприклад, карбонілімідазол. Сполуки формули (І), де n=0, a U є C(O)N(R13)-, можуть бути отримані взаємодією 4" гідроксилу формули (II) з придатним активованим похідним карбонової кислоти HOC(O)N(R13)(CH2)mZR7(Vllb), з подальшим (якщо необхідно) видаленням групи R2, яка захищає гідроксил. У бажаному втіленні винаходу сполуки формули (І), де n дорівнює 1-5, a U - група, обрана серед -N(R13)-, -О-, -S-, можуть бути отримані чез сполукою формули XR7 (IV). Реакцію здійсрез взаємодію сполуки формули (VII), нюють у розчиннику, наприклад, ДМСО, Ν,Ν-ДМФ, 1-метилпіролидоні, галогенгідрокарбоні (наприклад ДХМ), етері (наприклад, ТГФ, диметоксиетані), ацетонітрилі або етилацетаті або спирті (наприклад метанолі, ізопропанолі) тощо, в присутності основи з подальшим, забажання, видаленням групи R2, яка захищає гідроксил. Сполуки формули (І) можуть бути перетворені в інші сполуки формули (І). Отже, сполуки формули (І), де U - -S(O)q і q дорівнює 1 або 2, можуть бути отримані окислюванням відповідної сполуки формули (І), де q=0. Окислювання краще здійснювати, використовуючи перкис-. лоти, наприклад пероксибензойну кислоту, з подальшою обробкою де n - ціле, від 1 до 5, a L - придатна відщепфосфіном, наприклад, трифенілфосфіном. Реаклювальна група, цію звичайно здійснюють в органічному розчинниз XR7 (IV), де U - група, обрана серед -N(R13)-, ку, наприклад, метиленхлориді. -О-, -S-. Реакцію переважно проводять у розчинниСполуки формули (II), де А - -C(O)NH- або ку, наприклад, галогенгідрокарбоні (наприклад, NHC(O)-, R4 або R5 є гідроксилом, R3 - гідроген, і ДХМ), етері (наприклад, ТГФ, диметоксиетані), R6 - гідроген, є відомими сполуками або можуть ацетонітрилі або етилацетаті тощо), ДМСО, Ν,Νбути отримані способами, аналогічними відомим ДМФ, 1-метилпіролидоні в присутності основи з фахівцям, наприклад, як це описано у ЕР 507595 і подальшим, за бажання, видаленням групи R2, яка ЕР 503932. захищає гідроксил. Прикладами основ, що можуть Сполуки формули (II), де А - -C(O)NH- або бути використані, є органічна основа, наприклад, NHC(O)-, R4 або R5 є гідроксилом, і R2 - С1-4алкіл диізопропілетиламін, триетиламін, 1,8або С3-6алкеніл, як варіант, заміщений 9-10діазабіцикло[5.4.0]ундец-7-ен, або неорганічна членним конденсованим біциклічним гетероариоснова, наприклад, гідроксид калію, гідроксид целом, і R6 - гідроген, є відомими сполуками або мозію, гідроксид тетраалкіламонію, гідрид натрію, жуть бути отримані способами, аналогічними відогідрид калію тощо. Придатні відщеплювальні групи мим фахівцям, наприклад, як це описано у WO включають галоген (наприклад хлор, бром або 9951616 і WO 0063223. йод) або сульфонілоксигрупу (наприклад тозилокСполуки формули (II), де А - -C(O)NH- або сил або метансульфонілоксил). NHC(O)-, -N(CH3)-CH2-, -CH2-N(CH3)-, R4 і R5 разом Сполуки формули (VII) можуть бути отримані з проміжними атомами, утворюють циклічну групу шляхом взаємодії сполуки формули (II), де R2 структури: група, що захищає гідроксил, з придатною активованою похідною карбонової кислоти HOC(O)(CH2)nL (VIII), де L - придатна відщеплювальна група, визначена вище. Придатні активовані похідні карбоксильної групи є подібними визначеним вище для карбонової кислоти (III). Реакцію здійснюють при умовах, визначених для реакції сполуки формули (II) з карбоновою кислотою (III). У іншому втіленні винаходу сполуки формули (І), де n=2, a U - група, обрана серед -N(R13)-, -О- і R3 - С1-4алкіл або С3-6алкеніл, як варіант, замі-S-, можуть бути отримані реакцією Міхаеліса спощений 9-10-членним конденсованим біциклічним луки формули (IX), де R2 - група, що захищає гідгетероарилом, і R6 позначає гідроген, є відомими роксил, сполуками або можуть бути отримані способами, аналогічними відомим фахівцям, наприклад, як це описано у USP №6262030. Сполуки формули (II), де А - -С(О)-, R4 або R5 є гідроксилом, або R4 і R5 разом з проміжними 21 атомами утворюють циклічну групу структури: 77210 22 R6- гідроген, і R3 є С1-4алкілом, можуть бути отримані декарбоксилюванням сполуки формули (X), де R14 - група, що захищає гідроксил, з подальшим (якщо необхідно) видаленням захисної групи R2 або R14. де Υ - бівалентний радикал, обраний серед -Оі -N(R8)-, і R3 - С1-4алкіл або С3-6алкеніл, як варіант, заміщений 9-10-членним конденсованим біциклічним гетероарилом, є відомими сполуками або можуть бути отримані способами, аналогічними відомим фахівцям, наприклад, як це описано у ЕР 307177, ЕР 248279, WO 0078773, WO 9742204. Сполуки формули (II), де А - -N(CH3)-CH2-, CH2-N(CH3)-, R4 або R5 є гідроксилом, або R4 і R5 разом з проміжними атомами утворюють циклічну групу структури: Декарбоксилювання можна проводити присутності літієвої солі, наприклад, хлориду літію, переважно в органічному розчиннику, наприклад, ДМСО. Сполуки формули (II), де А - С(О)-, R4 і R5 разом з проміжними атомами утворюють циклічну групу структури: і R6 - гідроген, є відомими сполуками або можуть бути отримані способами, аналогічними відомим фахівцям, наприклад, як це описано у [ЕР 508699 і J.Chem. Res.Synop (1988, p.152-153), USP 6262030]. Сполуки формули (II), де А С(NОСН2ОСН2СН2ОСН3)-, R4 або R5 є гідроксилом, або R4 і R5 разом з проміжними атомами утворюють циклічну групу структури: і R3 є С1-4алкілом, можуть бути отримані циклізацією хлорпохідної (XI) і R6 - гідроген, є відомими сполуками або можуть бути отримані способами, аналогічними відомим фахівцям, наприклад, як це описано у ЕР 284203. Сполуки формули (II), де А - С(О)-, a R4 і R5 разом з проміжними атомами утворюють циклічну групу структури: ціанідом калію. Цю реакцію зручно проводити в присутності розчинника, наприклад, Ν,Ν-ДМФ. Сполуки формули (X) можуть бути отримані реакцією з'єднання сполуки формули (XII) 23 77210 24 або у присутності сильної основи, наприклад, 1,8діазабіцикло[5.4.0]ундец-7-ену. Цю реакцію зручно здійснювати в органічному розчиннику, наприклад, ацетонітрилі, Ν,Ν-ДМФ тощо. Сполуки формули (XII) можуть бути отримані взаємодією сполук формули (XIII) можуть бути отримані взаємодією XR7 (IV), де X має значення, визначене вище, з R15OC(O)(CH2)n (XV), де R15 - група, що захищає карбоксил, a L - придатна відщеплювальна група, з подальшим видаленням R15. Сполуки формули (IV), де X - -U(CH2)m, У якій Ζ - -N(R13)-, -О-, або -S-, або X є групою, обраною серед: або с ацилхлоридом формули СІСОСН2СООМе (XIV) у присутності третинної основи, наприклад, піридину, диметиламінопіридину або триетиламіну при температурі від 0° до 30°С. Сполуки формули (XIII) можуть бути отримані через взаємодію сполуки формули (II), де R14 група, що захищає гідроксил, R4 і R5, разом із проміжними атомами, утворюють циклічну групу структури: с сильною основою, наприклад, 1,8 діазабіцикло[5.4.0]ундец-7-еном. Сполуки формули (XI) можуть бути отримані через взаємодію сполук формули (XIII) з придатною активованою похідною кислоти НОСОСН2СІ (XV). Наприклад, етерифікація може бути проведена через взаємодію з ангідридом (СІСН2СО)2О (XVI) у придатному апротонному розчиннику, наприклад, галогенгідрокарбоні (наприклад ДХМ) або Ν,Ν-ДМФ в присутності третинної основи, наприклад, піридину, диметиламінопіридину або триетиламіну при температурі від 0о до 120°С. Сполуки формули (III), де X - -U(CH2)m(Ri3b a U є -N(R13) -О- або -S-, або X є групою, обрану серед: можуть бути отримані взаємодією сполуки формули R7L (XVI), де L - придатна відщеплювальна група, наприклад, хлор, фтор або бром, з сполукою формули -U(CH2)m (XVII), у якій Ζ - -N(R13)-, -Оабо -S-, або з піперазином, або з октагідро-1Нпіроло[3,4-b]піридином. Придатні реагенти для захисту гідроксигрупи описано у роботі [T.W. Greene and P.G.M Wuts у Protective Groups in Organic Synthesis 2nd ed, John Wiley & Son, Inc 1991], включеній сюди посиланням. Приклади придатних реагентів для захисту гідроксигрупи включають оцтовий ангідрид, бензойний ангідрид або триалкілсилілхлорид в апротонному розчиннику. Прикладами апротонних розчинників є ДХМ, Ν,Ν-ДМФ, ДМСО, ТГФ тощо. Придатні групи R15, що захищають карбоксил, включають трет-бутилоксил, аліл або бензилоксил. Солі приєднання кислот сполук загальної формули (І) можуть бути отримані різними способами, відомими фахівцям. Наприклад, водний розчин кислоти, такий як соляна кислота, може бути доданий до водної суспензії сполуки формули (І), і отримана в результаті суміш випарена до сухості (ліофілізована) з одержанням солі приєднання кислоти у вигляді твердої речовини. У іншому варіанті сполука формули (І) може бути розчинена в придатному розчиннику, наприклад спирті, наприклад, ізопропанолі, і кислота може бути додана в тім же розчиннику або іншому придатному розчиннику. Отримана сіль приєднання кислоти може потім бути осаджена безпосередньо або через 25 77210 26 додання менш полярного розчинника, наприклад, EtOAc -етилацетат, діізопропі-ловий етер або гексан, і виділена фільтГАТУ гексафторфосфат О-(7рацією. азабензотриазол-1-іл)-N,N,N',N'Для кращого розуміння винаходу, з метою йотетраметилуронію, го ілюстрації далі наведено приклади. МеОН - метанол, У Препаратах і Прикладах, якщо не зазначене іРrOН - ізопропанол; інше: TEA - триетиламін, Спектри протонного магнітного резонансу (1НАсОН - оцтова кислота, ЯМР) були зняті на Bruker Avance DRX 300 і DRX ТГФ - тетрагідрофуран, 500 або Varian 500МГц у розчинах у хлороформі dАс2О - оцтовий ангідрид, 1, якщо не зазначено інше. Хімічні зсуви наведено ТФК - трифтороцтова кислота, петрол - петролейний етер з точкою кипіння у м.д. з зсувом униз ( ) відносно Me4Si, використа40-60°С. ного як внутрішній стандарт, і віднесені до синглеРозчини були висушені над безводним сультів (s), дублетів (d), дублетів (dd), триплетів (t), фатом натрію або карбонатом калію. квартетів (q) або мультиплетів (m). Проміжна сполука 1: Спектри карбонового магнітного резонансу 13 6-[(2-аміно-етил)аміно]-1-бензил-1,4-дигідро-4( С-ЯМР) були зняті на Bruker Avance DPX 75 і оксо-хінолін-3-карбонова кислота DRX 125 МГц у розчинах у хлороформі d-1, якщо До розчину 1-бензил-6-хлор-1,4-дигідро-4не зазначено інше. Хімічні зсуви наведено в м.д. з оксохінолін-3-карбонової кислоти (300мг) у 1зрушенням униз ( ) відносно Me4Si, використаного метил-2-піролидиноне (6мл) додавали етилендіаяк внутрішній стандарт. мін (170мг) в атмосфері аргону. Суміш нагрівали Мас-спектри отримані на системі Hewlett при 120°С протягом 18год. і потім при 145°С проPackard 1100 MSD, обладнаній бінарним насосом тягом 4год. Реакційну суміш охолоджували і вили(Agilent Technologies), що працює на позитивній вали у воду (12мл). Величину рН доводили оцтоіонізації в електрострумі. вою кислотою до 7,0-7,5. Реакційну суміш Дані LC/MS були отримані з використанням екстрагували ДХМ (5 30мл). Органічну фазу висистеми HP 1100 LC (Agilent Technologies), обладсушували над Na2SO4, фільтрували і концентрувананої Sedex Evaporative Light Scattering Detector ли під зниженим тиском. Сирий продукт очищали model 75 (Sedere), з'єднаним з Platform LCZ Mass на силікагелевій SPE-колонці (елюент: від ДХМ Spectometer (Micromass), що працює на позитивній 100% до ДXM/MeOH/NH4OH 85/13/2) з одержаніонізації в електрострумі. Умови хроматографічноням сполуки, зазначеної в заголовку (80мг). го аналізу: колонка Waters XTerra MS C18 13 С-ЯМР (75МГц, ДМСО-d6) : 176,1; 165,4; (4,6 30мм, 2,5мкм); швидкість потоку 0,8мл/хв; 149,0; 138,1; 134,2; 133,5; 131,9; 129,8; 129,5; рухлива фаза: водний розчин NH4OAc (10мМ, рН 129,2; 127,1; 126,4; 118,9; 58,3; 42,8; 42,2. 6,8) (А) і ацетонітрил (В). Проміжна сполука 2: Очищення LC здійснювали з стат7-[(2-аміно-етил)аміно]-1-бензил-1,4-дигідро-4препаративною системою Waters 600, обладнаною оксохінолін-3-карбонова кислота бінарною насосною системою і детектором JascoДо розчину 7-хлор-1-бензил-1,4-дигідро-4UV. Умови хроматографического аналізу: колонка оксохінолін-3-карбонової кислоти (1,0г) у 1-метилSupelcosil ABZ+Plus (10см 21,2мм, 5мкм); швид2-піролидиноні (10мл) додавали етилендіамін кість потоку 8мл/хв; рухлива фаза: водний розчин (0,95г). Суміш нагрівали при 130°С протягом NH4OAc (10мМ, рН 6,8) (А) і ацетонітрил (В). 10год. Реакційну суміш охолоджували і виливали в Колонкову хроматографію проводили на силіДХМ (20мл). Отриманий осад диспергували в Мекагелі 60 (сито 230-400 ASTM - Merck AG ОН і фільтрували з одержанням сполуки, зазначеDarmstaadt, Germany). Моніторинг TLC здійснюваної в заголовку (560мг). ли з використанням Merck 60 F254 як TLC1 Н-ЯМР (300МГц, ДМСО-d6) : 8,95 (s, 1H); пластини. 8,05 (d, 1H); 7,36 (m, 5H); 6,94 (d, 1H); 6,83 (m, 1H); Відмивання смоли здійснювали на Extract6,56 (s, 1H); 5,68 (s, 4H); 3,09 (m, 2H). clean Tube від Alltech. 13 С-ЯМР (75МГц, ДМСО-d6) : 176,3; 166,5; Очищення сирого продукту здійснювали з 153,6; 148,6; 142,3; 135,5; 128,7; 127,8; 126,8; SCX-картриджем від Varian. 126,7; 114,5; 94,9; 56,3; 45,8. У тексті використані наступні абревіатури: Проміжна сполука 3: розсіл - для насиченого водного розчину хло7-[(2-аміноетил)аміно]-1-етил-1,4-дигідро-4риду натрію, оксо-хінолін-3-карбонова кислота ДБУ -1,8-діазабіцикло[5.4.0]ундец-7-ен, До розчину 7-хлор-1-етил-4-оксо-1,4ДХЕ - 1,2-дихлоретан, дигідрохінолін-3-карбонової кислоти (1,7г) у 1ДХМ - дихлорметан, метил-2-піролидиноні (20мл) додавали етилендіаΝΜΠ - 1-метил-2-піролидинон, мін (2,1г). Суміш нагрівали при 130°С протягом ДІПЕА для Ν,Ν-діізопропілетиламіну, 10год. Реакційну суміш охолоджували і виливали в ДМАП - 4-диметиламінопіридин, ДХМ (20мл). Отриманий осад диспергували в МеДМФ - Ν,Ν-диметилформамід, ОН і фільтрували з одержанням сполуки, зазначеДМСО - диметилсульфоксид, ної в заголовку (830мг). ДМП - гідрохлорид 1-(3-диметиламінопропіл)MS; m/z (ES): 276 [МН]+. 3-етилкарбодиіміду, 1 Н ЯМР (500МГц, ДМСО-d6) : 8,80 (s, 1H), 8,03 Et2O - діетилетер, 27 77210 28 (d, 1H), 7,17 (m, 1H), 6,95 (dd, 1H), 6,65 (s, 1H), 4,46 ковий розчин охолоджували при кімнатній темпе(q, 2H), 3,28 (q, 2H), 2,84 (q, 2H), 1,40 (t, 3Н). ратурі і перемішували протягом ночі. Осад фільт13 рували, промивали водою і сушили при 110°С проC ЯМР (125МГц, ДМСО-d6) : 176,1, 166,8, тягом 1год. з одержанням суміші, що містить 153,7, 147,8, 141,8, 126,9, 115,0, 106,5, 95,9, 48,5, сполуки 7 і 8, зазначені в заголовку (14,18г). За 44,6, 39,9, 14,2. необхідності гідрохлориди перетворювали у відпоПроміжна сполука 4: відні вільні основи перед використанням з засто7-[(2-аміноетил)аміно]-1-циклопропіл-1,4суванням стандартних умов. дигідро-4-оксо-хінолін-3-карбонова кислота Проміжна сполука 7 До розчину 7-хлор-1-циклопропіл-1,4-дигідро1 4-оксохінолін-3-карбонової кислоти (2,0г) у 1Н-ЯМР (300МГц, CF3COOD) : 8,94 (s, 1H), метил-2-піролидиноні (20мл) додавали етилендіа8,40 (s, 1H), 7,40 (s, 1H), 3,85 (m, 1H), 3,76 (m, 2H), мін (2,3г). Суміш нагрівали при 130°С протягом 5,45 (m, 2H), 1,42 (m, 2H), 1,77 (m, 2H). 10год. Реакційну суміш охолоджували і виливали в Проміжна сполука 9: ДХМ (20мл). Отриманий осад диспергували в Ме7-[(2-амінобутил)аміно]-1-циклопропіл-1,4ОН і фільтрували з одержанням сполуки, зазначедигідро-4-оксо-хінолін-3-карбонова кислота ної в заголовку (910мг). До розчину 7-хлор-1-циклопропіл-1,4-дигідро1 4-оксохінолін-3-карбонової кислоти (1,0г) у 1Н-ЯМР (500МГц, ДМСО-d6) : 8,41 (s, 1H), метил-2-піролидиноні (10мл) додавали бутиленді7,89 (d, 1H), 7,15 (m, 1H), 6,88 (s, 1H), 6,84 (dd, 1H), амін (1,7г). Суміш нагрівали при 130°С протягом 6,56 (s, 1H), 3,56 (m, 1H), 3,16 (m, 2H), 2,75 (t, 2H), 16год. Реакційну суміш охолоджували і виливали в 1,17 (m, 2H), 1,01 (m, 2H). 13 ДХМ (20мл). Отриманий осад диспергували в МеС-ЯМР (125МГц, ДМСО-d6) : 176,1, 166,5, ОН і фільтрували з одержанням сполуки, зазначе153,6, 147,1, 143,7, 126,7, 114,3, 106,1, 44,5, 39,7, ної в заголовку (450мг). 35,3, 7,4. MS; (ES) m/z: 316,4 [МН]+. Проміжні сполуки 5 і 6: Проміжна сполука 10: 6-[(2-аміноетил)аміно]-7-хлор-1,4-дигідро-16-[(2-аміноетил)аміно]-3-карбоксихінолін-7етил-4-оксохінолін-3-карбонова кислота (5) і хлор-1-циклопропіл-1,4-дигідро-4-оксо-метиловий 7-[(2-аміноетил)аміно]-1,4-дигідро-1-етил-6етер фтор-4-оксо-хінолін-3-карбонова кислота (6) Суспензію проміжної сполуки 7 (120мг) у роз7-хлор-1,4-дигідро-1-етил-6-фтор-4чині НСІ у МеОН (3%, 30мл) диспергували за дооксохінолін-3-карбонову кислоту (250мг) розчиняпомогою ультразвуку в ультразвуковій водяній ли в Ν,Ν-диметилацетаміді (5мл) і додавали етиванні при 60°С протягом 3год. І потім при кімнатній лендіамін (223мг). Реакційну суміш перемішували температурі протягом 48год. Розчинник випарюпротягом 10год. при 100°С, потім залишали на ніч вали під зниженим тиском і сирий продукт очищапри кімнатній температурі і знову нагрівали при ли флеш-хроматографією (елюент: Ме100°С протягом 7год. Додавали воду (5мл) і отриОН/ДХМ/NН4ОН 9/5/0,5) з одержанням сполуки, маний осад фільтрували, промивали водою (5мл) і зазначеної в заголовку (80мг). сушили з одержанням суміші, що містить сполуки, 1 Н-ЯМР (300МГц, ДМСО-d6) : 8,37 (s, 1H), зазначені в заголовку (120мг). + 8,04 (s, 1H), 7,36 (s, 1H), 5,77 (t, 1H), 3,37 (s, 3Н, ОMS; m/z (ES): 310,2 [MH] (Проміжна сполука Ме), 3,64 (m, 1H), 3,20 (q, 2Н), 2,85 (t, 2H), 1,23 (m, 5), 294,2 [MH]+ (Проміжна сполука 6). 2H), 1,08 (m, 2H). Проміжні сполуки 7 і 8: Проміжні сполуки 11 і 12: Гідрохлорид 6-[(2-аміноетил)аміно]-7-хлор-16-[(3-третбутоксикарбоніламінопропіл)аміно]-7циклопропіл-1,4-дигідро-4-оксо-хінолін-3хлор-1-циклопропіл-1,4-дигідро-4-оксохінолін-3карбонової кислоти (7) і карбонова кислота (11) і Гідрохлорид 7-[(2-аміноетил)аміно]-17-[(3-третбутоксикарбоніламінопропіл)аміно]-1циклопропіл-1,4-дигідро-6-фтор-4-оксо-хінолін-3цикпопропіл-1,4-дигідро-6-фтор-4-оксохінолін-3карбонової кислоти (8) карбонова кислота (12) 7-хлор-1-циклопропіл-1,4-дигідро-6-фтор-4У герметичній тубі 1-N-(трет-бутоксикарбоніл)оксохінолін-3-карбоновукислоту (56,3г) і етиленді3-пропандіамін (5мл) додавали до розчину 7-хлорамін (36г) розчиняли в Ν,Ν-диметилацетаміді 1-циклопропіл-1,4-дигідро-6-фтор-4-оксохінолін-3(650мл) при 100°С і перемішували протягом карбонової кислоти (3г) у ДМСО (6мл). Реакційну 8,5год. при 115°С. У реакційну суміш, охолоджену суміш перемішували при 110°С протягом 15год. до кімнатної температури, додавали воду (700мл). Залишали реакційну суміш охолоджуватися при Реакційну суміш перемішували при кімнатній тем50°С і виливали в лід, додавали оцтову кислоту до пературі протягом 2год., охолоджували при 0-5°С і досягнення рН=5. Осад, що з'явився, відокремлюперемішували протягом 1год. Отриманий осад вали фільтрацією і промивали етером з одержанфільтрували, промивали холодною водою, холодням суміші 11 і 12 у вигляді твердої жовтої речоним EtOH, і сушили при 110°С під зниженим тисвини (4,4г). Суміш очищали флеш-хроматографією ком протягом 1год. Сирий продукт обробляли НСІ (елюент: від EtOAc/циклогексан/АсОН 80/15/5 до (6%-й водний розчин), нагріваючи протягом 1год. у ЕЮАс/АсОН 90/10) з одержанням сполуки 11. заприсутності деревного вугілля. Після фільтрації значеної в заголовку (801мг) і сполуки 12. зазнарозчин охолоджували до 35-40°С і одержували ченої в заголовку (780мг). перший осад. Цей осад фільтрували, промивали Проміжна сполука 13: водою і сушили при 110°С протягом 1год. Сполуку 6-[(3-амінопропіл)аміно]-7-хлор-1-циклопропіл7, зазначену в заголовку (6,4г), одержували у ви1,4-дигідро-4-оксохінолін-3-карбонова кислота гляді гідрохлориду. Після першого осадження мат 29 77210 30 Розчин проміжної сполуки 11 (100мг) у 10% відокремлювали фільтрацією і промивали етером ТФК/ДХМ (1мл) перемішували при кімнатній темз одержанням 40:60 суміші сполук, зазначених у пературі протягом 2год. Розчинник частково випазаголовку (4,4г). рювали під зниженим тиском. Залишок розбавляПроміжні сполуки 17 і 18: ли ДХМ (2мл) і концентрували під зниженим Амід 6-[(2тиском п'ять разів. Остаточне випарювання розтретбутоксикарбоніламіноетил)аміно]-7-хлор-1чинника дало сполуку, зазначену в заголовку циклопропіл-1,4-дигідро-4-оксохінолін-3(103мг). карбонової кислоти (17) і MS; m/z (ES): 336 [МН]+. Амід 7-[(21 третбутоксикарбоніламіноетил)аміно]-1Н-ЯМР (500МГц, ДМСО) 8,56 (s, 1Н), 8,22 (s, циклопропіл-1,4-дигідро-6-фтор-4-оксохінолін-31Н), 7,35 (s, 1Н), 6,54 (bt, 1Н), 3,82 (m, 1Н), 2,69 (t, карбонової кислоти (18) 2Н), 2,49 (m, 2Н), 1,71 (m, 2Н), 1,28 (m, 2Н), 1,16 До розчину 40:60 суміші проміжних сполук 15 і (m, 2Н). 16 (1г) у безводному ДХМ (20мл) і безводному ТГФ Проміжна сполука 14: (30мл) при -5°С додавали N-метилморфолін 7-[(3-амінопропіл)аміно]-1-циклопропіл-1,4(0,56мл), а потім краплями додавали ізобутилхлодигідро-6-фтор-4-оксохінолін-3-карбонова кислота рформат (0,64мл). Реакційну суміш перемішували Розчин проміжної сполуки 12 (100мг) у 10% протягом 1год. при кімнатній температурі і додаТФК/ДХМ (1мл) перемішували при кімнатній темвали NH4OH (33%, 20мл). Реакційну суміш конценпературі протягом 2год. Розчинник частково випатрували під зниженим тиском і додавали насичерювали під зниженим тиском. Залишок розбавляний водний розчин NH4CI (20мл). Отриману суміш ли ДХМ (2мл) і концентрували під зниженим тиском п'ять разів. Остаточне випарювання розекстрагували ДХМ (3 20мл), об'єднану органічну чинника дало сполуку, зазначену в заголовку фазу висушували над Na2SO4, фільтрували і кон(103,4мг). центрували під зниженим тиском. Сирий продукт MS; m/z (ES): 320 [МН]+. очищали флеш-хроматографією (елюент 1 ДХМ/МеОН від 99:1 до 95:5) з одержанням 40:60 Н-ЯМР (500МГц, ДМСО) : 8,56 (s, 1H), 7,78 суміші сполук, зазначених у заголовку (528мг). (d, 1H), 7,31 (bm, 1H), 7,15 (d, 1H), 3,72 (m, 1 Η), Проміжні сполуки 19 і 20: 2,70 (t, 2H), 2,50 (m, 2H), 1,74 (m, 2H), 1,29 (m, 2H), сіль трифтороцтової кислоти аміду 6-[(21,15 (m, 2H). аміноетил)аміно]-7-хлор-1-циклопропіл-1,4Проміжні сполуки 13 і 14 (суміш) дигідро-4-оксо-хінолін-3-карбонової кислоти (19) і 6-[(3-амінопропіл)аміно]-7-хлор-1-циклопропілсіль трифтороцтової кислоти аміду 7-[(21,4-дигідро-4-оксохінолін-3-карбонова кислота і аміноетил)аміно]-1-цикпопропіл-1,4-дигідро-67-[(3-амінопропіл)аміно]-1-циклопропіл-1,4фтор-4-оксо-хінолін-3-карбонової кислоти (20) дигідро-6-фтор-4-оксохінолін-3-карбонова кислота 40:60 суміш проміжних сполук 17 і 18 (528мг) До розчину 7-хлор-1-циклопропіл-1,4-дигідророзчиняли в розчині ТФК (30мл, 10% у ДХМ) і реа6-фтор-4-оксо-хінолін-3-карбонової кислоти кційну суміш перемішували при кімнатній темпера(300мг) у ДМСО (1мл) додавали N-(треттурі протягом ночі. Реакційну суміш концентрували бутоксикарбоніл)-3-пропандіамін (0,37мл), і суміш під зниженим тиском з одержанням 40:60 суміші перемішували при 110°протягом 3год. Реакційну сполук, зазначених ν заголовку (544мг). суміш розбавляли ДХМ (3мл), промивали водою Аналіз HPLC/MS (рухлива фаза: А/В від 95/5 (3 50мл), сушили над Na2SO4, фільтрували і кондо 10/90 за 10хв, 10/90 протягом 5хв, масовий інцентрували під зниженим тиском. Сирий продукт тервал 100-800а.е.м.): час утримання: 6,19хв.(321 розчиняли в НСООН (5мл) і перемішували при [МН]+) і 5,63, (305 [МН]+). кімнатній температурі протягом 12год. Розчинник Проміжна сполука 21: видаляли in vacuo, Сирий продукт промивали кіль2'-О-ацетил-8а-аза-8а-гомоеритроміцин А ка разів ДХМ (3 50мл) і Et2O (1 50мл) і щоразу До розчину 8а-аза-8а-гомоеритроміцину А розчинник випарювали під зниженим тиском. (2,0г) у ДХМ (100мл) додавали NaНСО3 (900мг) і Отриманий сирий продукт очищали на SCXАс2О (0,280мл). Отриманий розчин перемішували картриджі (елюент : ДХМ, потім МеОН і NH3 (0,2N протягом ночі при кімнатній температурі. До цієї у МеОН)) з одержанням суміші сполук, зазначених суміші додавали розсіл (100мл) і воду (50мл). Віу заголовку (180мг). докремлювали органічний шар, промивали розсоПроміжні сполуки 15 і 16: лом (50мл), сушили над Na2SO4, фільтрували і 6-[(2-третбутоксикарбоніламіноетил)аміно]-7випарювали під зниженим тиском з одержанням хлор-1-циклопропіл-1,4-дигідро-4-оксохінолін-3сполуки, зазначеної в заголовку (2,1г). карбонова кислота (15) і MS; m/z(ES): 791,1 [МН]+. 7-[(2-третбутоксикарбоніламіноетил)аміно]-1Проміжна сполука 22: циклопропіл-1,4-дигідро-6-фтор-4-оксохінолін-32'-О-ацетил-4"-О-пропеноїл-8а-аза-8акарбонова кислота (16) гомоеритроміцин А У герметичній тубі N-(третПроміжну сполуку 21 (290мг) розчиняли в тобутоксикарбоніл)етилендіамін (5мл) додавали до луолі (10мл) і розчинник випарювали. Цю операрозчину 7-хлор-1-циклопропіл-6-фтор-4-оксо-1,4цію повторювали двічі. Залишок знову розчиняли в дигідрохінолін-3-карбонової кислоти (3г) у ДМСО толуолі (15мл) і розчин перемішували під аргоном. (6мл). Реакційну суміш перемішували при 110°С Додавали TEA (0,470мл), а потім 3протягом 15год. Реакційній суміші давали охолохлорпропіонілхлорид (0,101мл) трьома порціями нути при 50°С і виливали в лід, додавали оцтову за 10хв. Через 15год. додавали TEA (0,235мл) і 3кислоту до досягнення рН=5. Осад, що з'явився, 31 77210 32 хлорпропіонілхлорид (0,050мл). Ще через 3год. отриману суспензію перемішували протягом 15год. додавали TEA (0,170мл) і 3-хлорпропіонілхлорид при кімнатній температурі. Ацетон випарювали під (0,025мл). Через 1год. додавали насичений водзниженим тиском, додавали ДХМ (100мл) і Н2О ний розчин NaHCO3 (30мл) і водну фазу екстрагу(100мл). Величину рН отриманого розчину доводили до 9,5 доданням NaOH (2M). Відокремлювавали толуолом (3 20мл). Органічну фазу висушули органічний шар і промивали розсолом (50мл), вали над Na2SO4, фільтрували і концентрували під сушили над К2СО3, фільтрували і фільтрат концезниженим тиском з одержанням сполуки, зазначентрували під зниженим тиском. Залишок очищали ної в заголовку (261мг). флеш-хроматографією (елюент: етил-ацетат/нПроміжна сполука 23: гексан/диетиламін 6/3/0,2) з одержанням сполук 27 4"-O-пропеноїл-8а-аза-8а-гомоеритроміцин А (0,835г) і 28(1,38г), зазначених в заголовку. Проміжну сполуку 22 (225мг) розчиняли в МеMS; m/z (ES): 777,5 [МН]+. ОН (30мл) і перемішували протягом ночі. РозчинПроміжна сполука 29: ник випарювали під зниженим тиском з одержан2'-O0-ацетил-6-O-етил-8а-аза-8аням сполуки, зазначеної в заголовку (221мг). гомоеритроміцин А MS; m/z (ES): 803,7 [МН]+. До розчину проміжної сполуки 27 (735мг) у Проміжна сполука 24: ДХМ (20мл) додавали NaHCO3 (358мг) і оцтовий 2'-O-ацетил-6-O-метил-8а-аза-8аангідрид (0,098мл) з перемішуванням при кімнатгомоеритроміцин А ній температурі протягом 20год. Додавали насичеДо розчину 6-О-метил 8а-аза-8аний розчин NaHCO3 (20мл), водний шар промивагомоеритроміцину А (1,62г) у ДХМ (100мл), додавали NaHCO3 (713мг), і Ас2О (0,230мл) і перемішули ДХМ (2 10мл), органічний шар промивали вали протягом ночі. До цієї суміші додавали розсіл розсолом (20мл), сушили над К2СО3, фільтрували і (100мл) і воду (50мл). Відокремлювали органічний фільтрат концентрували під зниженим тиском з шар, промивали розсолом (50мл), сушили над одержанням сполуки, зазначеної в заголовку Na2SO4, фільтрували і випарювали під зниженим (661мг). тиском з одержанням сполуки, зазначеної в загоMS; m/z(ES): 819,4 [МН]+. ловку (1,67г). Проміжна сполука 30: MS; m/z (ES): 805,1 [МН]+. 2'-О-ацетил-6-О-етил-4"-О-пропеноїл-8а-азаПроміжна сполука 25: 8а-гомоеритроміцин А 2'-О-ацетил-4"-О-пропеноїл-6-О-метил-8а-азаДо розчину проміжної сполуки 29 (149мг) у су8а-гомоеритроміцин А хому толуолі (5мл) в атмосфері нітрогену додаваДо розчину проміжної сполуки 24 (1,80г) у сули TEA (0,229мл) при кімнатній температурі, потім хому толуолі (30мл) в атмосфері аргону одною трьома порціями протягом 3год. додавали 3порцією додавали TEA (2,90мл). До реакційної хлорпропіонілхлорид (0,05мл). Додавали насичесуміші протягом декількох хв., додавали 3ний розчин NaHCO3 (5мл), водний шар промивали хлорпропіонілхлорид (0,63мл). Реакційну суміш толуолом (2 5мл), органічний шар промивали розперемішували протягом 2год. До реакційного розсолом (10мл), сушили над К2СО3, фільтрували, і чину додавали ще два еквіваленти TEA і один екфільтрат концентрували під зниженим тиском з вівалент 3-хлорпропіонілхлориду і перемішували одержанням сполуки, зазначеної в заголовку протягом ще 2год. Реакцію гасили доданням наси(139мг). ченого NaНСО3 (60мл). Шари розділяли і водний MS m/z (ES) 873,4 [МН]+. шар екстрагували толуолом (3 30мл). Об'єднані Проміжні сполуки 31 і 32: толуолові екстракти промивали розсолом (20мл), 6-О-(2-пропеніл)-8а-аза-8а-гомоеритроміцин А сушили над К2СО3, фільтрували і концентрували (31) і під зниженим тиском з одержанням сполуки, за6-O-(2-пропеніл)-9а-аза-9а-гомоеритроміцин А значеної в заголовку (1,8г). (32) MS; m/z (ES): 859 [МН]+. До суспензії 9(Е)- і 9(Z)-оксимів 6-O-2Проміжна сполука 26: пропеніл-еритроміцину А (4,25г) в ацетоні/воді 1/1 4"-О-пропеноїл-6-О-метил-8а-аза-8а(250мл), охолодженої до 0°С, додавали NaHCO3 гомоеритроміцин А (6,34г). Потім краплями додавали розчин pПроміжну сполуку 25 (1,8г) розчиняли в МеОН толуолсульфонілхлориду (7,19г) в ацетоні (55мл) (100мл) і розчин перемішували при кімнатній темза 30хв. і отриману суспензію перемішували пропературі протягом 24год. і потім при 60°С протягом тягом 15год., дозволяючи нагрітися до кімнатної 2год. Розчинник видаляли під зниженим тиском і температури. Ацетон випарювали під зниженим залишок очищали флеш-хроматографією (елюент тиском і додавали ДХМ (100мл) і воду (100мл). МеОН/ДХМ/NН4ОН 5/90/0,5) з одержанням сполуВеличину рН розчину доводили до 9,5 доданням ки, зазначеної в заголовку (1,54г). NaOH (2M). Органічний шар промивали розсолом MS; m/z (ES): 817,5 [МН]+. (50мл), сушили над К2СО3, фільтрували і випарюПроміжні сполуки 27 і 28: вали під зниженим тиском. Залишок очищали 6-О-етил-8а-аза-8а-гомоеритроміцин А (27) і флеш-хроматографією (елюент: EtOAc/H6-O-етил-9а-аза-9а-гомоеритроміцин А (28) гексан/Et2NH 6/3/0,2) з одержанням сполуки 31. До суспензії 9(Е)- і 9(Z)-оксимів 6-Озазначеної в заголовку (2,36г) і сполуки 32, зазнаетилеритроміцину А (3,76г) в ацетоні і воді (230мл, ченої в заголовку (0,97г). суміш 1/1) при 0°С додавали NaHCO3 (5,69г). КраMS; m/z (ES): 789,4 [МН]+. плями протягом 30хв. додавали розчин рПроміжна сполука 33: толуолсульфонілхлориду (6,46г) в ацетоні (55мл) і 6-О-пропіл-8а-аза-8а-гомоеритроміцин А 33 77210 34 Розчин проміжної сполуки 31 (2,3г) у МеОН розчинник концентрували під зниженим тиском з (40мл) підкислювали до рН 5-5,5 доданням НСІ одержанням сполуки, зазначеної в заголовку (1М). Потім додавали 10% Pd/C (0,82г) і суміш гід(0,94г). рогенували при 20бар протягом 20год. МеОН виMS; m/z (ES): 862 [МН]+. парювали під зниженим тиском, потім додавали Проміжна сполука 38: ДХМ (50мл) і воду (50мл). Величину рН доводили 11-O-[3-(3-хіноліл)-2-пропеніл] -8а-аза-8адо 9,5 з використанням NaOH (1M). Водну фазу гомоеритроміцин А екстрагували ДХМ (10мл) і органічний шар промиПроміжну сполуку 37 (0,9г) і цисвали розсолом (50мл), сушили над К2СО3, фільтпропенілхіноліл-трет-бутилкарбонат (0,630г) розрували і випарювали під зниженим тиском з одерчиняли в толуолі (18мл), потім розчинник видаляжанням сполуки, зазначеної в заголовку (1,84г). ли під зниженим тиском. Цю операцію повторюваMS;m/z(ES):791,4[MH]+. ли двічі, потім залишок розчиняли в толуолі (25мл) Проміжна сполука 34: і приблизно 7мл розчинника видаляли in vacuo. 2'-O-ацетил-6-O-пропіл-8а-аза-8аЗалишковий розчин переносили у тригорлу круггомоеритроміцин А лодонну колбу і дезоксигенували аргоном. ДодаДо розчину проміжної сполуки 33 (0,27г) у ДХМ вали трис(дибензиліденацетон)дипалладій (0) (10мл) при кімнатній температурі додавали (40мг) і 1,4-біс(дифенілфосфін)бутан (37мг) і суміш NaHCO3 (131мг) і оцтовий ангідрид (0,039мл). нагрівали при 80сС протягом 1год. Розчинник виОтриману суспензію перемішували при кімнатній даляли під зниженим тиском. Залишок розчиняли температурі протягом 20год. Додавали насичений в МеОН (200мл), додавали мурашину кислоту, розчин NaHCO3 (10мл), і водний шар екстрагували доводячи рН до 5 і розчин перемішували протягом ночі. Суміш випарювали під зниженим тиском з ДХМ (2 10мл). Органічну фазу промивали розсоодержанням сирого продукту, який очищали лом (10мл), сушили над К2СО3, фільтрували, і вифлеш-хроматографією (елюент: парювали під зниженим тиском з одержанням споСНСІ3/МеОН/МН4ОН 6/1/0,1) з одержанням сполулуки, зазначеної в заголовку (0,25г). ки, зазначеної в заголовку (260мг). MS; m/z (ES): 833,4 [МН]+. MS;m/z(ES):917[MH]+. Проміжна сполука 35: Проміжна сполука 39: 2'-O-ацетил-4"-O-пропеноїл-6-O-пропіл-8а-аза2'-О-ацетил-11-О-[3-(3-хіноліл)-2-пропеніл]-8а8а-гомоеритроміцин А аза-8а-гомоеритроміцин А До розчину проміжної сполуки 34 (149мг) у суДо розчину проміжної сполуки 38 (0,30г) при хому толуолі (5мл) при кімнатній температурі в кімнатній температурі додавали NaHCO3 (900мг) і атмосфері нітрогену додавали TEA (0,229мл), поАс2О (0,280мл). Отриманий розчин перемішували тім трьома порціями щогодини додавали 3протягом ночі. До цієї суміші додавали розсіл хлорпропіонілхлорид (0,052мл) і перемішували (100мл) і воду (50мл). Відокремлювали органічний протягом 2год. Додавали насичений розчин шар, промивали розсолом (50мл), сушили, фільтNaHCO3 (5мл) і водну фазу промивали толуолом рували і випарювали під зниженим тиском з одер(2 5мл). Органічний розчин промивали розсолом жанням сполуки, зазначеної в заголовку (0,30г). (10мл), сушили над К2СО3, фільтрували, і фільтрат MS; m/z (ES): 958 [МН]+. випарювали під зниженим тиском з одержанням Проміжна сполука 40: сполуки, зазначеної в заголовку (139мг). 4"-О-пропеноїл-11-О-[3-(3-хіноліл)-2-пропеніл]MS; m/z (ES): 887,5 [МН]+. 8а-аза-8а-гомоеритроміцин А Проміжна сполука 36: До розчину проміжної сполуки 39 у сухому то2'-О-ацетил-11,12-карбонат-11,12-дидезоксилуолі (5мл) додавали TEA (0,26мл) і 34"-О-(1Н-1-імідазолілкарбоніл)-6-О-метил-8а-азахлорпропіонілхлорид (0,070мл). Реакційну суміш 8а-гомоеритроміцин А перемішували протягом 0,5год. потім гасили доРозчин проміжної сполуки 24 (2,0г) у сухому данням насиченого водного розчину бікарбонату ДМФ (20мл) охолоджували до 0°С у воднонатрію (15мл). Водну фазу екстрагували толуолом крижаній ванні в атмосфері аргону. Потім одною (10мл). Об'єднані толуолові екстракти промивали порцією додавали 1,1'-карбонілдіімідазол (3,20г), а розсолом (20мл), сушили над карбонатом калію, потім порціями додавали Na (60%-а масляна суфільтрували і концентрували під зниженим тиском. спензія, 0,47г). Отриману суміш перемішували в Сирий продукт розчиняли в МеОН (60мл) і переатмосфері аргону при 0°С протягом 1год. Реакцію мішували при кімнатній температурі протягом гасили доданням краплями холодної води (100мл), 24год. Розчинник випарювали з одержанням спопід час чого осаджувався твердий матеріал. Тверлуки, зазначеної в заголовку (0,25г). ду речовину фільтрували і сушили з одержанням MS; m/z (ES): 971 [МН]+. сполуки, зазначеної в заголовку (2,02г). + Проміжна сполука 41: MS; m/z (ES): 925,9 [МН] . 2'-O-ацетил-6-O-метил-9а-аза-9аПроміжна сполука 37: гомоеритроміцин А 2'-О-ацетил-4"-О-триметилсиліл-8а-аза-8аДо розчину 6-О-метил-9а-аза-9агомоеритроміцин А гомоеритроміцину А (4,0г) у ДХМ (60мл) і ацетоні Проміжну сполуку 21 (1,0г) розчиняли в ДХМ (10мл) додавали NaHCO3 (2,2г) і Ас2О (1,5мл). (10мл) і охолоджували до 0-5°С, потім додавали Реакційну суміш перемішували протягом 1год. піридин (0,31мл) і триметилсилілхлорид (0,33мл) і потім додавали ДХМ (75мл) і воду (75мл). Органіотриману суміш перемішували протягом 2год. Речну фазу відокремлювали, промивали розсолом акційну суміш промивали водним розчином (50мл), сушили над Na2SO4, фільтрували і випаNa2PO4 (30мл), водою (30мл) і розсолом (30мл), і 35 77210 36 рювали під зниженим тиском з одержанням сполутягом 20год. Розчин концентрували під зниженим ки, зазначеної в заголовку (4,02г). тиском і додавали ДХМ (50мл) і воду (50мл). ВеПроміжна сполука 42: личину рН отриманої суміші доводили до 9,5 до2'-O-ацетил-4"-O-пропеніл-6-O-метил-9а-азаданням NaOH (1M). Водну фазу промивали ДХМ 9а-гомоеритроміцин А (30мл), і органічні розчини поєднували і промивали До розчину проміжної сполуки 41 (3,0г) у сухорозсолом (50мл), сушили над К2СО3 і випарювали му толуолі (30мл), додавали TEA (2,07мл) в атмопід зниженим тиском з одержанням сполуки, засфері аргону. Реакційну суміш охолоджували до значеної в заголовку (0,97г). 15°С і краплями додавали 3-хлорпропіонілхлорид MS;m/z(ES):791,4[MH]+. (0,71мл). Водяну баню видаляли і реакційну суміш Проміжна сполука 47: перемішували протягом 2год., потім додавали TEA 2'-О-ацетил-6-О-пропіл-9а-аза-9а(1мл) і 3-хлорпропіонілхлорид (0,48мл) і реакційгомоеритроміцин А ний розчин перемішували протягом додаткових До розчину проміжної сполуки 46 (0,97мг) у 2год. Додавали насичений водний розчин NaHCO3 ДХМ (50мл) при кімнатній температурі додавали (60мл) і EtOAc (30мл) і екстрагували водну фазу NaHCO3 (0,46г) і Ас2О (0,139мл). Отриману суспензію перемішували при кімнатній температурі EtOAc (2 30мл). Об'єднані органічні екстракти супротягом 20год. Додавали насичений водний розшили над К2СО3, фільтрували і концентрували під зниженим тиском з одержанням сполуки, зазначечин NaHCO3 (50мл) і екстрагували ДХМ (2 20мл). ної в заголовку (3,17г). Об'єднаний органічний розчин промивали розсоMS; m/z (ES): 859 [МН]+. лом (40мл), сушили над . К2СО3, фільтрували і Проміжна сполука 43: випарювали під зниженим тиском з одержанням 4"-О-пропеноїл 6-О-метил-9а-аза-9асполуки, зазначеної в заголовку (0,95г). гомоеритроміцин А MS; m/z (ES): 833,4 [МН]+. Проміжну сполуку 42 (2,9г) розчиняли в МеОН Проміжна сполука 48: (100мл), і розчин перемішували при кімнатній тем2'-О-ацетил-4"-О-пропеноїл-6-О-пропіл-8а-азапературі протягом 24год., і потім при 60°С протя8а-гомоеритроміцин А гом 2год. Розчинник випарювали і залишок очиДо розчину проміжної сполуки 47 (0,95г) у сущали флеш-хроматографією (елюент: хому толуолі (25мл) при кімнатній температурі в MeOH/ДХМ/NH4OH 5/90/0,5) з одержанням сполуатмосфері нітрогену додавали триетиламін ки, зазначеної в заголовку (1,8г). (0,958мл), потім трьома порціями щогодини додаMS;m/z(ES):817[MH]+. вали 3-хлорпропіонілхлорид (0,218мл), і потім пеПроміжна сполука 44: ремішували протягом 2год. Додавали насичений 2'-О-ацетил-6-О-етил-9а-аза-9аводний розчин NaHCO3 (25мл) і водну фазу прогомоеритроміцин А мивали толуолом (2 25мл). Органічну фазу проДо розчину проміжної сполуки 28 (1,33г) у ДХМ мивали розсолом (50мл), сушили над К2СО3, філь(30мл) при кімнатній температурі додавали трували, і випарювали під зниженим тиском з NaHCO3 (650мг) і Ас2О (0,178мл). Отриману суодержанням сполуки, зазначеної в заголовку спензію перемішували при кімнатній температурі (0,92г). протягом 20год. Додавали насичений водний розMS; m/z (ES): 887,4 [МН]+. чин NaHCO3 (20мл), водну фазу екстрагували ДХМ Проміжна сполука 49: (2 20мл), і об'єднаний органічний розчин проми2'-О-ацетил-11,12-карбонат-11,12-дидезоксивали розсолом (20мл), сушили над К2СО3, фільт4"-О-пропеноїл-азитроміцин рували і випарювали під зниженим тиском з одерРозчин проміжної сполуки 2'-О-ацетил-11,12жанням сполуки, зазначеної в заголовку (1,29г). карбонат-11,12-дидезоксиазитроміцину (10,9г) у MS; m/z (ES): 819,4 [ΜΗ]*. толуолі (300мл) перемішували при кімнатній темПроміжна сполука 45: пературі в атмосфері аргону. До цього розчину 2'-O-ацетил-4"-O-пропеноїл-6-O-етил-8а-азадвома порціями за 10хв. додавали TEA (12,66мл) і 8а-гомоеритроміцинА 3-хлорпропіонілхлорид (1,94мл). Через 20хв. розДо розчину проміжної сполуки 44 (0,21г) у сучин розбавляли насиченим водним розчином хому толуолі (5мл) при кімнатній температурі в NaHCO3 (300мл) і екстрагували толуолом атмосфері нітрогену додавали TEA (0,321мл). По(4 80мл). Зібрану органічну фазу висушували, тім трьома порціями за 3год. додавали 3фільтрували і концентрували під зниженим тиском хлорпропіонілхлорид (0,073мл) і потім перемішуз одержанням сполуки, зазначеної в заголовку вали протягом 2год. Додавали насичений розчин (11,0г). NaHCO3 (5мл) і водну фазу промивали толуолом MS; m/z (ES): 872 [МН]+. (2 10мл). Органічний розчин промивали розсолом Проміжна сполука 50: (10мл), сушили над К2СО3, фільтрували, і випарю11,12-карбонат-11,12-дидезокси-4"-Oвали під зниженим тиском з одержанням сполуки, пропеноїл азитроміцин зазначеної в заголовку (0,20г). Розчин проміжної сполуки 49 (11,0г) у МеОН + MS; m/z (ES): 873,4 [МН] . (200мл) перемішували при кімнатній температурі Проміжна сполука 46: протягом 48год. Розчинник випарювали під зниже6-О-пропіл-9а-аза-9а-гомоеритроміцин А ним тиском з одержанням сполуки, зазначеної в До розчину проміжної сполуки 32 (1,27г) у Мезаголовку (9,81г). ОН (40мл) додавали водний розчин НСІ (1М)до MS; m/z (ES): 829,1 [МН]+. 1 одержання рН=5-5,5. До розчину додавали 10% Н-ЯМР (500МГц,) : 6,45 (d, 1H), 6,17 (dd, 1H), Pd/C (0,45г) і суміш гідрогенували при 15бар про5,87 (d, 1H), 5,11 (d, 1H), 4,88 (dd, 1H), 4,77 (d, 1Н), 37 77210 38 4,53 (d, 1Н), 4,47-4,40 (m, 3Н), 3,72 (m, 1Н), 3,60 (d, 2'-О-ацетил-11,12-(амінокарбонілокси)-11,121Н), 3,33 (s, 3Н), 3,25 (dd, 1H), . 2,87-2,85 (m, 2Н), дидезокси-6-О-метил-еритроміцин А 2,58 (m, 1Н), 2,44-2,38 (m, 2Н), 2,32 (s, 6Н), 2,21 (s, До розчину 11,12-(амінокарбонілокси)-11,123Н), 2,06 (m, 1H), 2,00 (m, 1Н), 1,92 (m, 1H), 1,84 дидезокси-6-О-метилеритроміцину А у ДХМ (50мл) (m, 1H), 170-1,56 (m, 4H), 1,45 (s, 3Н), 1,40 (dd, 1H), при кімнатній температурі додавали NaHCO3 1,29 (s, 3Н), 1,25 (m, 1Н), 1,22 (d, 3Н), 1,18 (d, 6H), (478мг). До цього розчину додавали Ас2О 1,12 (s, 3Н), 108-1,06 (2d, 6H), 0,93 (m, 6H). (0,153мл) і перемішували протягом ночі. До цієї Проміжна сполука 51: суміші додавали розсіл (50мл) і воду (20мл). Відо4"-O-пропеноїл-азитроміцин кремлювали органічний шар, промивали розсолом До розчину проміжної сполуки 50 (1,3г) в аце(20мл), сушили, фільтрували і випарювали під тонітрилі (50мл) при кімнатній температурі додазниженим тиском з одержанням сполуки, зазначевали насичений водний розчин карбонату калію ної в заголовку (1,2г). (30мл). Отриману суміш нагрівали до 70°С протяMS; m/z(ES): 816,2 [МН]+. гом 8год. Суміш розбавляли водою (100мл), екстПроміжна сполука 56: 2'-О-ацетил-11,12-(амінокарбонілокси)-11,12рагували EtOAc (4 30мл), зібрану органічну фазу дидезокси-6-О-метил-4"-О-пропеноїл-еритроміцин висушували, фільтрували і концентрували під А зниженим тиском. Сирий продукт очищали флешПроміжну сполуку 55 розчиняли в толуолі хроматографією (елюент: ДXM/MeOH/NH3 (50мл) і розчинник випарювали. Це повторювали 90/9/0,5) з одержанням сполуки, зазначеної в задвічі. Після цього залишок знову розчиняли в тоголовку (530мг). луолі (45мл) і перемішували під аргоном. До цього MS; m/z (ES): 804 [MH]+. розчину додавали TEA (1,8мл) і 3Проміжна сполука 52: хпорпропіонілхлорид (0,40мл) (трьома порціями за 2'-O-ацетил-4"-O20хв). Через 20хв. додавали насичений водний імідазолілкарбонілазитроміцин розчин NaHCO3 (50мл). Водний розчин екстрагуДо розчину 2'-О-ацетилазитроміцину (0,8г) у сухому толуолі (30мл) в атмосфері аргону додававали толуолом (3 50мл), об'єднаний органічний ли К2СО3 (420м) і 1,1'-карбонілдиімідазол (0,170г). розчин сушили над К2СО3 і розчинник видаляли Реакційну суміш перемішували при кімнатній темпід зниженим тиском з одержанням сполуки, запературі протягом 20год. і при 40°С протягом 5год. значеної в заголовку (1,04г). Додавали насичений розчин NaHCO3 (30мл) і водMS; m/z(ES): 870,1 [МН]+. Проміжна сполука 57: ну фазу промивали толуолом (2 30мл). Об'єдна11,12-(амінокарбонілокси)-11,12-дидезокси-6ний органічний розчин висушували над К2СО3 і O-метил-4"-O-пропеноїл-еритроміцин А концентрували під зниженим тиском з одержанням Розчин проміжної сполуки 56 у МеОН (30мл) сполуки, зазначеної в заголовку (0,82г). перемішували при кімнатній температурі протягом MS; m/z (ES): 885 [МН]+. ночі. Розчинник концентрували під зниженим тисПроміжна сполука 53 ком з одержанням сполуки, зазначеної в заголовку 2'-О-ацетил-6-О-метил-азитроміцин (1,2г). До розчину 6-О-метил-азитроміцину (205мг) у MS; m/z(ES): 828,1 [MH]+. ДХМ (10мл) додавали NaHCO3 (112,8мг) і Ас2О 1 (0,030мл). Суміш перемішували при кімнатній темН-ЯМР (500МГц) : 6,44 (d, 1H), 6,13 (dd, 1H), пературі протягом 20год., потім додавали розсіл 5,89 (d, 1H), 5,11 (dd, 1H), 4,98 (d, 1H), 4,75 (d, 1H), (20мл). Розчин екстрагували ДХМ і органічну фазу 4,60 (d, 1H), 4,36 (m, 1H), 3,80 (d, 1H), 3,73 (m, 1H), висушували. Розчинник концентрували під зниже3,70 (s, 1H), 3,62 (d, 1H), 3,34 (m, 1Η), 3,32 (s, ЗН), ним тиском з одержанням сполуки, зазначеної в 3,22 (m, 1Н), 2,95 (s, 3Н). 13 заголовку (220мг). С-ЯМР (125МГц) : 218,4; 176,9; 166,2; 158,8; MS; m/z (ES): 805 [МН]+. 132,0; 128,4; 102,5; 96,2; 84,3; 80,7; 79,1; 78,8; 77,8; Проміжна сполука 54: 76,1; 73,2, 71,4; 68,2; 65,83; 63,6; 58,2; 58,2; 50,6; 4"-О-пропеноїп-6-О-метил-азитроміцин 49,9; 45,6; 45,5; 41,1; 40,7; 40,0; 39,9; 37,8; 35,5; Розчин проміжної сполуки 53 (215мг) у ДХМ 22,4,21,9; 21,4; 20,1; 18,6; 18,5; 16,1; 14,1; 13,6; (10мл) перемішували в атмосфері аргону протягом 10,8; 9,5. 10хв. Двома порціями додавали TEA (0,222мл) і 3Проміжна сполука 58: хлорпропіонілхлорид (0,051мл). Через 15хв. знову 2'-О-ацетил-11,12-дидезокси-6-О-метил-11,12додавали TEA (0,111мл) і 3-хлор-пропіонілхлорид (метиламінокарбонілокси)-еритроміцин А (0,026мл). Через 12год. додавали TEA (0,111мл) і 11,12-дидезокси-6-О-метил-11,123-хлорпропіонілхлорид (0,026мл). Реакційну суміш (метиламінокарбонілокси)-еритроміцин А (0,87г) перемішували при кімнатній температурі протягом розчиняли в ДХМ (20мл) і ацетоні (3мл). Додавали 2год. Додавали насичений водний розчин NaHCO3 твердий NaHCO3 (0,6г) і Ас2О (0,6мл) і реакційну (20мл). Шари розділяли і органічну фазу висушусуміш перемішували протягом 1год., потім додавали. Розчинник концентрували під зниженим тисвали ДХМ (50мл) і воду (50мл). Органічну фазу ком і залишок (240мг) розчиняли в МеОН (20мл) і відокремлювали, промивали розсолом (20мл), перемішували при кімнатній температурі протягом сушили над К2СО3, фільтрували і концентрували 14год. Розчинник концентрували під зниженим під зниженим тиском з одержанням сполуки, затиском з одержанням сполуки, зазначеної в загозначеної в заголовку (0,875г). ловку (220мг). MS; m/z (ES): 829,2 [МН]+. MS; m/z(ES): 817,6 [МН]+. Проміжна сполука 59: Проміжна сполука 55: 11,12-дидезокси-6-О-метил-11,12 39 77210 40 (метиламінокарбонілокси)-4"-O-пропеноїлMS; m/z (ES): 843,2 [МН]+. еритроміцин А Проміжна сполука 64: До розчину проміжної сполуки 58 (0,85г) у су11,12-дидезокси-11,12хому толуолі (10мл) в атмосфері аргону додавали (етиламінокарбонілокси)-6-О-метил-4"-ОTEA (0,85мл) і 3-хлорпропіонілхлорид (0,196мл, пропеноїл-еритроміцин А двома порціями за 20хв) і реакційну суміш переДо розчину проміжної сполуки 63 (1,45г) у сумішували протягом 1год. Потім додавали насичехому толуолі (17мл) в атмосфері аргону додавали ний розчин NaHCO3 (30мл) і суміш екстрагували TEA (1,42мл) і 3-хлорпропіонілхлорид (0,49мл, трьома порціями за 20хв). Реакційну суміш перетолуолом (2 60мл). Об'єднаний органічний розчин мішували протягом 1год., потім додавали насичепромивали розсолом (20мл), сушили над К2СО3, ний розчин NaHCO3 (50мл) і водну фазу екстрагуфільтрували і концентрували під зниженим тиском. Залишок розчиняли в МеОН (70мл) і нагрівали при вали толуолом (2 70мл). Об'єднаний органічний 60° протягом 4год., потім залишали стояти при розчин промивали розсолом (20мл), сушили над кімнатній температурі протягом 24год. Розчинник К2СО3, фільтрували і концентрували під зниженим концентрували під зниженим тиском з одержанням тиском. Залишок розчиняли в МеОН (100мл) і насполуки, зазначеної в заголовку (620мг). грівали при 60°С протягом 4год., і потім при кімнаMS; m/z (ES): 841 [MH]+. тній температурі протягом 24год. Розчинник випа1 рювали і сирий продукт очищали флешН-ЯМР (300МГц) : 6,44 (d, 1H), 6,13 (dd, 1H), хроматографією (елюент: MeOH/ДXM/NH4OH 5,90 (d, 1H), 4,97 (m, 2H), 4,75 (d, 1H), 4,60 (d, 1H), 5/90/0,5) з одержанням сполуки, зазначеної в за4,36 (m, 1H), 3,77 (m, 3Н), 3,64 (d, 1H), 3,55 (s, 1H). головку (1,4г). Проміжна сполука 60: MS; m/z (ES): 855 [МН]+. 2'-О,4"-О-діацетил-11 -дезокси-10,11 1 Н-ЯМР (500МГц) : 6,44 (d, 1H), 6,13 (dd, 1H), дидегідро-12-О-(1Н-1-імідазолкарбоніл)-6-Ометилеритроміцин А 5,89 (d, 1H), 4,99 (m, 2H), 4,75 (d, 1H), 4,60 (d, 1H), Розчин 2'-О,4"-О-діацетил-6-О4,38 (m, 1H), 3,77-3,55 (m, 6H), 3,32 (s, ЗН), 3,20 метилеритроміцину А (4,0г) розчиняли в ДМФ (m, 1H), 3,06 (m, 1 Η), 3,05 (s, 3Н), 2,95 (m, 1H), (20мл) при 0°С в атмосфері аргону і додавали Ν,Ν2,61-2,52 (m, 2H), 2,43 (d, 1H), 2,31 (s, 6H), 1,98карбонілдіімідазол (1,6г). Потім за 30хв. порціями 1,89 (m, 2H), 1,73 (d, 1H), 1,68-1,64 (m, 2H), 1,55додавали Na (1,2м, 50%-а суспензія в олії). Реак1,49 (m, 1H), 1,39 (s, 3Н), (d, 1H),1,38 (s, 3Н), 1,25ційну суміш перемішували протягом 4год., потім 1,12 (m), 1,02 (d, 3Н), 0,84 (t, 3Н). 13 додавали Н2О (60мл). Осад фільтрували, промиС-ЯМР (125МГц) : 216,1; 176,3; 165,8; 157,2; вали водою і сушили з одержанням сполуки, за131,5; 128,0; 102,2; 96,0; 82,6, 79,8; 78,7; 77,6; 76,1; значеної в заголовку (4,1г). 75,9; 73,7, 71,1; 67,9; 65,3; 63,2; 60,0; 50,5; 49,5; Проміжна сполука 61: 45,6; 45,1; 40,3; 38,9; 38,8; 38,7; 38,6; 35,2; 28,8; 2'-О,4"-О-діацетил-11,12-дидезокси-11,1221,9, 21,7; 21,0; 20,1; 18,9,18,6,18,3; 15,9; 14,2; (етиламінокарбонілокси)-6-0-метил-еритроміцин А 14,1; 12,7; 10,3; 9,1. Проміжну сполуку 60 (3,5г) розчиняли в CH3CN Проміжна сполука 65: (35мл) і Н2О (4мл) при кімнатній температурі і реа2'-О-ацетил-11,12-дидезокси-11,12-[(N-(4кційний розчин охолоджували до 0°С у воднофенілбутил)аміно)карбонілокси]-6-О-метилкрижаній ванні в атмосфері аргону. Додавали еритроміцин А EtNH2 (1,7г) і реакційну суміш перемішували проДо розчину 11,12-дидезокси-11,12-[(N-(4тягом 20год. Додавали воду (50мл) і осад фільтруфенілбутил)аміно)карбонілокси]-6-О-метилвали, промивали водою і сушили з одержанням еритроміцину А (0,550г) у ДХМ (17мл) додавали сполуки, зазначеної в заголовку (2,6г). оцтовий ангідрид (0,101мл) і NaHCO3 (0,140г). СуПроміжна сполука 62: міш перемішували при кімнатній температурі про11,12-дидезокси-11,12тягом 24год., потім додавали насичений розчин (етиламінокарбонілокси)-6-О-метил-еритроміцин А NaHCO3 (25мл). Органічну фазу відокремлювали, Проміжну сполуку 61 (2,5г) розчиняли в МеОН промивали розсолом (15мл) і водою (15мл), суши(140мл), додавали насичений водний розчин ли і випарювали з одержанням сполуки, зазначеК2СО3 (10мл), і реакційну суміш перемішували при ної в заголовку (0,530г). 75°С протягом 4год. і при кімнатній температурі MS; m/z (ES): 904 [MH]+. протягом ночі. Зменшували об'єм розчину вдвічі Проміжна сполука 66: під зниженим тиском і до залишку додавали EtOAc 2'-О-ацетил-11,12-дидезокси-11,12-[(N-(4(100мл) і воду (50мл). Відокремлювали органічний фенілбутил)аміно)карбонілокси]-6-O-метил-4"-Ошар, сушили і випарювали з одержанням сполуки, пропеноїл-еритроміцин А зазначеної в заголовку (2,4г). До розчину проміжної сполуки 65 (0,500г) у суПроміжна сполука 63: хому толуолі (8мл) в атмосфері аргону додавали 2'-О-ацетил-11,12-дидезокси-11,12двома порціями TEA (0,440мл) і 3(етиламінокарбонілокси)-6-О-метил-еритроміцин А хлорпропіонілхлорид (0,116мл). Реакційну суміш Проміжну сполуку 62 (1,5г) розчиняли в ДХМ перемішували при кімнатній температурі протягом (40мл) і ацетоні (5мл). Додавали твердий NaHCO3 0,5год. Додавали насичений розчин NaHCO3 (1,2г) і Ас2О (0,8мл) і суміш перемішували протя(25мл), і водну фазу екстрагували толуолом гом 1год. Потім додавали ДХМ (60мл) і воду (2 10мл). Об'єднані органічні екстракти промивали (60мл) і органічну фазу відокремлювали, промиварозсолом (25мл), сушили над К2СО3, фільтрували і ли розсолом (20мл), сушили і випарювали з одерконцентрували під зниженим тиском з одержанням жанням сполуки, зазначеної в заголовку (1,5г). сполуки, зазначеної в заголовку (0,450г). 41 77210 42 Проміжна сполука 67: ацетоні (6мл). Реакційну суміш потім перемішува11,12-дидезокси-11,12-[(N-(4ли при кімнатній температурі протягом 1год., фільфенілбутил)аміно)карбонілокси]-6-O-метил-4"-Oтрували, і фільтрат концентрували під зниженим пропеноїл-еритроміцин А тиском. Сирий продукт розчиняли в EtOAc (10мл), Проміжну сполуку 66 (0,450г) перемішували в додавали воду (10мл), і водну фазу екстрагували МеОН (110мл) при кімнатній температурі протягом EtOAc (3 10мл). Об'єднані органічні шари проми48год. Випарювання під зниженим тиском дало вали насиченим водним розчином Na2S2O5 (10мл), сполуку, зазначену в заголовку (0,420г). сушили, фільтрували -і концентрували під знижеMS; m/z (ES): 959 [МН]+. ним тиском з одержанням сполуки, зазначеної в Проміжна сполука 68: заголовку (546мг). 1 2'-О-ацетил-6-О-метил-4"-О-пропеноїлН-ЯМР (500МГц) : 5,00 (d, 1H), 4,81 (bt, 1H), еритроміцин А 4,74 (d, 1H), 4,66 (d, 1H), 4,34 (m, 1H), 3,76 (m, 1Н), До розчину 2'-O-ацетил-6-O-метил3,71 (d+d, 2Н), 3,37 (s, 3Н), 2,40 (d, 1Н), 2,10 (s, еритроміцину А (1,1г) у ДХМ (20мл) при 0°С дода3Н), 1,67 (m, 2H), 1,32 (m, 1H), 1,24 (d,3H), 1,13 вали піридин (1,7мл) і акрилоїлхлорид (1,1мл). (d,3H). Через 2год. додавали піридин (1,7мл) і акрилоїлхПроміжна сполука 71: лорид (1,1мл). Реакційну суміш гасили насиченим 6-О-пропіл-еритроміцин А розчином NH4СІ (10мл) і екстрагували ДХМ До розчину 6-O-(2-пропеніл)-еритроміцину А (3 20мл). Органічну фазу промивали насиченим (2,5г) у МеОН (60мл) додавали Pd/C (10%, 0,85г) і розчином NaHCO3 (10мл), водою (10мл), сушили суміш гідрогенували при кімнатній температурі при над Na2SO4, фільтрували і випарювали під зниже15бар протягом 20год. Каталізатор відфільтровуним тиском. Сирий продукт очищали флешвали і промивали МеОН, і розчинник випарювали хроматографією (ДXM/MeOH/NH3 95/5/0,5) з одерпід зниженим тиском. Додавали ДХМ (50мл) і воду жанням сполуки, зазначеної в заголовку (470мг). (50мл) і рН доводили до 9,5 доданням NaOH (1M). Проміжна сполука 69: Органічну фазу висушували над К2СО3, фільтру6-O-метил-4"-O-пропеноїл-еритроміцин А вали і випарювали під зниженим тиском з одерПроміжну сполуку 68 (1,82м, ммоль) розчиняжанням сполуки, зазначеної в заголовку (2,27г). ли в МеОН (100мл) і перемішували при 60°С проMS; m/z (ES): 776,6 [MH]+. тягом 4год., і потім при кімнатній температурі проПроміжна сполука 72: тягом 16год. Розчинник випарювали під зниженим 2'-О-ацетил-6-О-пропіл-еритроміцин А тиском і сирий продукт очищали флешДо розчину проміжної сполуки 71 (1,0г) у ДХМ хроматографіею (елюент: MeOH/ДXM/NH4OH (30мл) при кімнатній температурі додавали 5/90/0) з одержанням сполуки, зазначеної в загоNaHCO3 (0,487г) і Ас2О (0,134мл). Розчин переміловку (1,4г). шували протягом 24год., потім додавали воду + MS; m/z (ES): 802 [МН] . (20мл), рН доводили до 9,5 доданням NaOH (1M), 1 Н-ЯМР (500МГц) : 6,44 (d, 1H), 6,13 (dd, 1H), органічний шар висушували над К2СО3, фільтру5,89 (d, 1H), 5,07 (d, 1H), 5,00 (d, 1H), 4,75 (d, 1H), вали і випарювали під зниженим тиском з одер4,60 (d, 1H), 4,38 (m, 1H), 3,97 (s, 1H), 3,80-3,73 (m, жанням сполуки, зазначеної в заголовку (1,0г). 2H), 3,66 (d, 1H), 3,46 (s, 1H), 3,32 (s, 3Н), 3,21-3,18 MS; m/z(ES): 818,6 [MH]+. (m, 2H), 3,04 (s, 3 Η), 3,00 (m, 1Η), 2,92 (m, 1Н), Проміжна сполука 73: 2,56 (m, 2Н), 2,43 (d, 1Н), 2,31 (s, 6Н). 2'-O-ацетил-4"-O-пропеноїл-6-O13 пропілеритроміцин А С-ЯМР (75МГц) : 221,0; 175,7; 165,8; 131,5; Проміжну сполуку 72 (1,0г) розчиняли в сухому 128,0; 102,1; 96,0; 80,5, 78,8, 78,3; 78,0; 76,6; 74,3, толуолі (40мл) під N2, додавали TEA (0,513мл) і 372,7; 71,1; 69,1; 67,8; 65,3; 63,2:50,7; 49,5; 45,3; хлорпропіонілхлорид (0,116мл), і розчин перемі44,9; 40,3; 39,2; 38,8; 37,2; 35,2; 28,9; 21,7; 21,1; шували протягом 30хв. Через 1год. додавали ще 19,7; 18,3; 18,0; 15,9; 12,3; 10,6; 9,1. TEA (0,513мл) і через 2год. 3-хлорпропіонілхлорид Проміжна сполука 70 (0,116мл). Реакційну суміш гасили доданням наси2'-О-ацетил-4"-О-іодетаноїл-6-О-метилченого водного розчину NaHCO3 (30мл), органічеритроміцинА ний шар промивали розсолом, сушили над К2СО3, До розчину 2'-О-ацетил-6-О-метилфільтрували і концентрували під зниженим тиском еритроміцину А (600мг) у безводному ДХМ (40мл) з одержанням сполуки, зазначеної в заголовку додавали піридин (0,18мл) і хлорацетилхлорид (1,01г). (0,06мл) і реакційну суміш перемішували при кімПроміжна сполука 74: натній температурі протягом 15хв. Додавали ще 4"-О-пропеніл-6-О-пропіл-еритроміцинА піридину (0,18мл) і хлорацетилхлориду (0,06мл). Проміжну сполуку 73 (1,01г) розчиняли в МеЧерез 15хв. втретє додавали піридин (0,18мл) і ОН (50мл) і перемішували при кімнатній темперахлорацетилхлорид (0,06мл), після чого через ще турі протягом 20год. Розчинник видаляли під зни30хв. знову додавали піридин (0,18мл) і хлорацеженим тиском і додавали ДХМ (50мл) і воду тилхлорид (0,06мл). Реакційну суміш перемішува(30мл). Величину рН доводили до 9,5 доданням ли протягом 1год., потім додавали розсіл (20мл). NaOH (1Μ) і органічну фазу висушували над Водну фазу екстрагували ДХМ (3 20мл) і об'єднані К2СО3, фільтрували і випарювали під зниженим органічні шари промивали насиченим водним розтиском з одержанням сполуки, зазначеної в загочином NaHCO3 (20мл). Органічну фазу висушуваловку (0,92г). ли, фільтрували і концентрували під зниженим MS; m/z (ES): 830,6 [МН]+. тиском. До сирого продукту, розчиненого в ацетоні Проміжна сполука 75: (4мл), додавали розчин йодиду натрію (570мг) в 43 77210 44 11,12-карбонат-2',4"-О-діацетил-11,12чин гідроксиду літію (1М, 4мл) і реакційну суміш дидезокси-6-О-метил-еритроміцин А перемішували при кімнатній температурі протягом До розчину 2',4"-О-діацетил-6-О-метил48год. Розчин НСІ (10% про/про) повільно додаваеритроміцину А (200г) у безводному ДХМ (1,6л) ли до досягнення рН=8 і розчинник частково випапри 0°С в атмосфері нітрогену додавали піридин рювали під зниженим тиском. Розчин розбавляли (117мл) і розчин трифосгену (71,2г) у ДХМ водою (20мл) і екстрагували ДХМ (8 20мл). Зібра(400мл). Отриману суміш перемішували при 0°С ну органічну фазу висушували над Na2SO4, фільтпротягом 30хв. і потім при кімнатній температурі рували і концентрували під зниженим тиском. Сипротягом 15год. Суміш потім розбавляли водою рий продукт очищали флеш-хроматографією (750мл) і екстрагували ДХМ (2 500мл). Органічну (елюент: ДХМ/МеОН 85/15) з одержанням сполуки, зазначеної в заголовку (150мг). фазу промивали водою (3 300мл), сушили, фільтMS; m/z (ES): 797 [МН]+. рували і випарювали під зниженим тиском з одерTLC: МеОН/ДХМ 10/90 (Rf=0,23). жанням сполуки, зазначеної в заголовку (200г). Проміжна сполука 80: MS; m/z (ES): 858 [МН]+. (11S,11а)-2'-О-ацетил-11Проміжна сполука 76: (карбоксиціанометил)-11-дезокси-6-О-метил11-дезокси-2',4"-О-діацетил-10,11-дидегідро-6еритроміцин А О-метил-еритроміцин А До розчину проміжної сполуки 79 (150мг) у До розчину проміжної сполуки 75 (50,5г) у 2/1 безводному ДХМ (20мл) додавали NaHCO3 (48мг), суміші безводний толуол/EtOAc (675мл) при кімнаа потім оцтовий ангідрид (0,020мл). Реакційну сутній температурі додавали ДБУ (9,24мл). Отримаміш перемішували при кімнатній температурі прону суміш нагрівали до 85°С протягом 8год. Додали тягом ночі. Відфільтровували NaHCO3, органічну ще ДБУ (4,4мл), і реакційну суміш перемішували фазу промивали водою (3 20мл), об'єднані органіпри кімнатній температурі протягом 5год. Суміш розбавляли розсолом (250мл), екстрагували чні фази висушували, фільтрували і концентрували під зниженим тиском з одержанням сполуки, EtOAc (2 350мл), сушили і фільтрували. Випарюзазначеної в заголовку (0,145г). вання розчинника під зниженим тиском і кристаліMS; m/z (ES): 839 [МН]+. зація з суміші ацетон/вода дали сполуку, зазначеTLC: МеОН/ДХМ 5/95 (Rf=0,24). ну в заголовку (46г). Проміжна сполука 81: MS; m/z (ES): 814 [MH]+. (11S,17а)-2'-О-ацетил-4"-О-пропеноїл-11Проміжна сполука 77: (карбоксиціанометил)-11-дезокси-6-О-метил12-хлоретаноїл-11-дезокси-2',4"-О-діацетилеритроміцин А 10,11-дидегідро-6-О-метил-еритроміцин А До розчину проміжної сполуки 80 (90мг) у безДо розчину проміжної сполуки 76 (20г) у безводному ДХМ (10мл), охолодженому до 0°С, додаводному ДХМ (340мл) при 0°С додавали піридин вали піридин (0,174мл), і потім пропеноїлхлорид (6мл) і хлороцтовий ангідрид (8,4г) і дозволяли (0,174мл). Реакційну суміш нагрівали до кімнатної реакційній суміші нагрітися до кімнатної температемператури і перемішували протягом 24год., потури. Через 16год. реакційну суміш промивали водою (300мл), органічну фазу спочатку промиватім промивали водою (3 10мл). Потім органічну ли насиченим водним розчином NH4CI (150мл) і фазу спочатку промивали насиченим водним розпотім розсолом (150мл). Об'єднану водну фазу чином NaHCO3 (2 10мл), і потім розсолом екстрагували ДХМ (2 300мл). Органічну фазу ви(2 10мл). Об'єднану водну фазу екстрагували сушували, фільтрували і випарювали під знижеДХМ (2 10мл) і об'єднану органічну фазу висушуним тиском. Сирий продукт осаджували з суміші вали, фільтрували і концентрували під зниженим ацетон/вода з одержанням сполуки, зазначеної в тиском. Сирий матеріал фільтрували через силіказаголовку (20,4г). гель (елюент: ДХМ/МеОН 95/5) з одержанням спо+ MS; m/z (ES): 890 [МН] . луки, зазначеної в заголовку (90мг). Проміжна сполука 78: MS; m/z (ES): 893 [МН]+. (11S,11а)-11-(карбоксиціанометил)-11TLC: МеОН/ДХМ 5/95 (Rf=0,42). дезокси-2',4"-О-діацетил-6-О-метил-еритроміцин А Проміжна сполука 82: Проміжну сполуку 77 (27,4г) розчиняли в без2'-О-ацетил-рокситроміцин водному ДМФ (275мл) і додавали ціанід калію (5г). До розчину рокситроміцину (500мг) і NaHCO3 Суміш перемішували при кімнатній температурі (150мг) у безводному ДХМ (50мл) додавали оцтопротягом 4,5год., гасили насиченим водним розвий ангідрид (0,062мл). Реакційну суміш перемічином NaHCO3 (250мл) і екстрагували ДХМ шували при кімнатній температурі протягом ночі, (3 300мл). Органічну фазу висушували над фільтрували і фільтрат промивали водою Na2SO4, фільтрували і концентрували під зниже(3 20мл). Об'єднану водну фазу екстрагували ним тиском. Залишок розчиняли в ацетоні (100мл) ДХМ (3 20мл) і об'єднану органічну фазу висушуі сполуку осаджували доданням води (250мл). Фівали, фільтрували і концентрували під зниженим льтрація дала сполуку, зазначену в заголовку тиском з одержанням сполуки, зазначеної в заго(25,8г). ловку (510мг). + MS; m/z (ES): 881 [МН] . MS; m/z (ES): 879 [МН]+. Проміжна сполука 79: TLC: МеОН/ДХМ 5/95 (Rf=0,28). (11S,11а)-11-(карбоксиціанометил)-11Проміжна сполука 83: дезокси-6-О-метил-еритроміцин А 2'-О-ацетил-4"-О-пропеноїл-рокситроміцин До розчину проміжної сполуки 78 (300мг) у До розчину проміжної сполуки 82 (50мг) у безбезводному МеОН (40мл) додавали водний роз 45 77210 46 водному толуолі (10мл) додавали триетиламін (1979), 16(7), 1353-60] у 1-метил-2-піролидиноне (0,016мл) і 3-хлорпропіонілхлорид (0,008мл). Реа(15мл) додавали етилендіамін (1,9г). Суміш нагрікційну суміш перемішували при кімнатній темперавали при 130°С протягом 24год. Реакційну суміш турі протягом ночі. Додавали воду (3мл) і реакційохолоджували і виливали в ДХМ (20мл). Отриманий осад диспергували в МеОН і фільтрували з ну суміш екстрагували EtOAc (3 10мл). Об'єднану одержанням сполуки, зазначеної в заголовку органічну фазу висушували над Nа2SО4, фільтру(500мг). вали і концентрували під зниженим тиском. Сирий MS (ES) m/z: [MH]+ 262,3. матеріал фільтрували через силікагель (елюент: Проміжна сполука 88: ДХМ/МеОН 95/5) з одержанням сполуки, зазначе7-[(2-аміноетил)аміно]-1-циклопропіл-1,4ної в заголовку (49мг). дигідро-4-оксо-6-фтор-8-метокси-3MS; m/z (ES): 933 [МН]+. хінолінкарбонова кислота TLC: МеОН/ДХМ 5/95 (Rf=0,36). До розчину 1-циклопропіл-1,4-дигідро-4-оксоПроміжна сполука 84: 6,7-дифтор-8-метокси-3-хінолін-карбонової кисло6-(2-аміноетиламіно)-7-хлор-1-циклопропіл-1Нти (1,0г) у Ν,Ν-диметилацетаміді (10мл) додавали хінолін-4-он і N-BOC-етилендіамін (0,82мг). Суміш нагрівали при 7-(2-аміноетиламіно)-1-циклопропіл-6-фтор120°С протягом 12год. Додавали трифторцтову 1Н-хінолін-4-он кислоту (20мл) і реакційну суміш перемішували Розчин 40:60 суміші проміжних сполук 15 і 16 при кімнатній температурі протягом 24год. Роз(147мг) у водному НСІ (2N, 5мл) нагрівали при чинник випарювали під зниженим тиском. Сирий 100°С протягом 4 днів у герметичній тубі. Реакційпродукт розчиняли у воді (5мл) і доводили рН за ну суміш концентрували під зниженим тиском. Сидопомогою 20%-го NaOH до 7. Отриманий осад рий продукт очищали препаративною TLC (елюдиспергували в МеОН і фільтрували з одержанням ент: ДХМ/МеОН/ІМНЦОН 90/10/1,5) з одержанням сполуки, зазначеної в заголовку (1,01г). сполук, зазначених ν заголовку (23мг, 70/30 суміш MS; m/z (ES): 336,0 [МН]+. CI/F-похідних). 1 Приклад 1 Н-ЯМР (500МГц) для 6-(2-аміноетиламіно)4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл7-хлор-1-циклопропіл-1Н-хінолін-4-ону: 8,05 (s, 1,4-дигідро-4-оксо-6-хінолініл) амі1Н), 7,85 (d, 1H), 7,35 (s, 1H), 6,10 (d, 1H), 3,50-2,70 но]етил]аміно]пропіоніл]-8а-аза-8а(m, 5H), 1,30-0,98 (m, 4H). 1 гомоеритроміцин А Н-ЯМР (500МГц) для 7-(2-аміноетиламіно)До розчину проміжної сполуки 23 (20мг) у 1-циклопропіл-6-фтор-1Н-хінолін-4-ону: 7,81 (d, CH3CN (0,4мл) додавали проміжну сполуку 7 1Н), 7,65 (d, 1Н), 7,00 (d, 1Н), 6,03 (d, 1Н), 3,50-2,70 (16мг). Отриману суміш нагрівали при 70°С протя(m, 5Н), 1,30-0,98 (m, 4Н). гом 24год. Розчинник концентрували під зниженим Проміжна сполука 85: тиском і залишок очищали на силікагелевій SPEМетиловий етер 7-(2-аміно-етиламіно)-1колонці з використанням системи градієнта розциклопропіл-6-фтор-4-оксо-1,4-дигідрохінолін-3чинника ДХМ/МеОН/ІМН4ОН з одержанням сполукарбонової кислоти ки, зазначеної в заголовку (11мг). Суспензію проміжної сполуки 8 (2,0м, MS; m/z (ES): 1124,5 [MH]+. 6,55ммоль) у 3% H2SO4 у МеОН (170мл) перемі1 Н-ЯМР (500МГц) : 8,73 (s, 1H), 8,05 (s, 1H), шували при 60°С протягом 48год., причому за цей 7,53 (s, 1H), 5,86 (br,S, 1H), 5,31 (t, 1 Η), 5,12 (d, час реакційна суміш стає розчином. Реакційний 1H), 4,91 (dd, 1H), 4,70 (d, 1H), 4,53 (d, 1H), 4,33 (m, розчин нейтралізували і розчинник випарювали. 2H), 4,19 (m, 1H), 3,83 (m, 1H), 3,76-3,70 (m, 1H), Сирий продукт очищали флеш-хроматографією на 3,68-3,64 (m, 1H), 3,58-3,55 (m, 2H), 3,49 (s, 1H), силікагелі з використанням MeOH-CH2Cl23,39 (m, 1H), 3,29 (s, 3Н). NH4OH=9-5-0,5 і одержували сполуку, зазначену в Приклад 2 заголовку (720мг). 4"-О-[3-[[2-[(3-карбокси-1-циклопропіл-1,4Проміжна сполука 86: дигідро-6-фтор-4-оксо-77-[(2-амінобутил)аміно]-1-метил-1,4-дигідро-4хінолініл)аміно]етил]аміно]пропіоніл]-8а-аза-8аоксо-3-хінолінкарбонова кислота гомоеритроміцин А До розчину 7-хлор-1-метил-4-оксо-1,4-дигідроДо розчину проміжної сполуки 23 (20мг) у Ме3-хінолінкарбонової кислоти [1,0г, отриманої згідно ОН (0,4мл) додавали проміжну сполуку 8 (36мг). Н. Agui and Т. Nakagome, J. Heterocycl. Chem, Отриману суміш нагрівали при 70°С в ультразву(1979), 16(7), 1353-60] у 1-метил-2-піролидиноні ковій ванні протягом 60год. Суміш фільтрували і (10мл) додавали 1,4-діамінобутан (1,84г). Суміш фільтрат випарювали під зниженим тиском. Залинагрівали при 130°С протягом 17год. Реакційну шок очищали на силікагелевій SPE-колонці (елюсуміш охолоджували і виливали в ДХМ (20мл). ент: від ДХМ 100% до ДXM/MeOH/NH4OH 85/13/2) Отриманий осад диспергували в МеОН і фільтруз одержанням сполуки, зазначеної в заголовку вали з одержанням сполуки, зазначеної в заголов(13,6мг). ку (860мг). + + Ms (ES) m/z: 1108 [MH] . MS (ES) m/z: [MH] 290,3. Приклад 3 Проміжна сполука 87: 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-цикпопропіл7-[(2-аміноетил)аміно]-1-метил-4-оксо-1,41,4-дигідро-4-оксо-6дигідро-3-хінолінкарбонова кислота хінолініл)аміно]етил]аміно]пропіоніл]-6-О-метилДо розчину 7-хлор-1-метил-4-оксо-1,4-дигідро8а-аза-8а-гомоеритроміцин А 3-хінолінкарбонової кислоти [1,5г, отриманої згідно До розчину проміжної сполуки 25 (5г) у іРrОН Н. Agui and Т. Nakagome, J. Heterocycl. Chem, 47 77210 48 (40мл) додавали проміжну сполуку 7 (2,8г). Реакі перемішували при кімнатній температурі протяційну суміш перемішували при 80°С протягом гом 48год. Суспензію фільтрували і промивали 24год. Надлишок аміну відфільтровували з реакДХМ (10мл). Фільтрат концентрували під зниженим ційної суміші і додавали МеОН (100мл). Реакційну тиском з одержанням сполуки, зазначеної в загосуміш перемішували протягом 4 днів, потім фільтловку (150мг). + рували і випарювали під зниженим тиском з одерMS; m/z(ES): 1149,4 [МН] . жанням суміші сирого продукту (5,94г). Сирий проПриклад 7 дукт очищали флеш-хроматографією у системі 4"-О-[3-[4-(3-карбокси-1-циклопропіл-1,4градієнта розчинника ДХМ/МеОН/NН4ОН з одердигідро-4-оксо-7-хінолініл)-піперазиніл]пропіоніл]жанням сполуки, зазначеної в заголовку (1,385г). 6-O-метил-8а-аза-8а-гомоеритроміцин А MS; m/z(ES): 1139,8 [MH]+. Реакція проміжної сполуки 26 (10мг) і 11 циклопропіл-4-оксо-7-пиперазиніл-1,4Н-ЯМР (500МГц) : 8,73 (s, 1Н), 8,04 (s, 1Н), дигідрохінолін-3-карбонової кислоти (19мг), прове7,54 (s, 1H), 5,61 (d, 1H), 5,28 (t, 1H), 5,08 (d, 1Н), дена відповідно до методики, описаної для Прик4,94 (dd, 1H), 4,70 (d, 1H), 4,50 (d, 1H), 4,37 (m, 1H), ладу 6, давала сполуку, зазначену в заголовку 4,19 (m, 1H), 3,98 (d, 1H), 3,71-3,66 (m, 2Н), 3,58(4,2мг). 3,54 (m, 1Н), 3,50 (s, 1Н), 3,38 (m, 1Н), 3,31 (s, 3Н). 13 MS; m/z (ES): 1131,6 [MH]+. С-ЯМР (75МГц) : 177,5; 177,2; 174,3; 172,3; Приклад 8 167,3; 145,8; 143,1; 132,5; 127,6; 126,3; 118,0; 4"-O-[3-[4-(1-циклопропіл-1,4-дигідро-6-фтор-4107,6; 104,5; 102,6; 95,3; 80,1; 78,9; 78,8; 77,4; 74,2; оксо-7-хінолініл)-піперазиніл]пропіоніл]-6-О-метил72,9; 70,8; 70,3; 68,2; 65,4; 62,8; 51,8; 49,5; 47,5; 8а-аза-8а-гомоеритроміцин А 45,5; 44,4; 43,0; 42,8; 42,4; 42,3; 40,9; 40,4; 35,3; Реакція проміжної сполуки 26 (10мг) і 135,1; 34,7; 29,0; 23,9; 21,7; 21,6; 21,3; 21,2; 18,0; циклопропіл-5-фтор-4-оксо-7-пиперазиніл-1,416,1; 15,0; 11,2; 9,6; 9,3; 8,1. дигідро-4-хінолінона (18мг), проведена відповідно Приклад 4 до методики, описаної для Приклада 6, давала 4"-O-[3-[(2-[(3-карбокси-1-циклопропіл-1,4сполуку, зазначену в заголовку (5,1мг). дигідро-6-фтор-4-оксо-7MS; m/z (ES): 1105,6 [МН]+. хінолініл)аміно]етил]аміно]пропіоніл]-6-О-метилПриклад 9 8а-аза-8а-гомоеритроміцин А 4"-О-[3-[4-(3-карбокси-7-хлор-1-циклопропілДо розчину проміжної сполуки 26 (134мг) у і1,4-дигідро-4-оксо-6-хінолініл)-1РrOН (2,5мл) додавали проміжну сполуку 8 (100мг) пиперазиніл]пропіоніл]-6-O-метил-8а-аза-8аі струшували при 85°С протягом 14 днів. Реакційну гомоеритроміцин А суміш охолоджували до кімнатної температури і Реакція проміжної сполуки 26 (10мг) і 7-хлор-1фільтрували через силікагелевій картридж (2г), циклопропіл-4-оксо-6-пиперазиніл-1,4елюючи ДХМ (10мл). Розчинник випарювали з дигідрохінолон-3-карбонової кислоти (21мг), проодержанням 100мг сирого залишку, який очищали ведена відповідно до методики, описаної для Прина силікагелевій SPE-колонці в системі градієнта клада 6, давала сполуку, зазначену в заголовку розчинника flXM/MeOH/NH4OH з одержанням спо(4,8мг). луки, зазначеної. в заголовку (9,3мг). MS; m/z (ES): 1165,8 [MH]+. MS; m/z(ES): 1122,3 [МН]+. Приклад 10 Приклад 5 4"-О-[3-[(4-[(3-карбокси-1-циклопропіл-1,44"-О-[3-[[2-[(7-хлор-1-циклопропіл-1,4-дигідродигідро-6-фтор-4-оксо-73-метоксикарбоніл-4-оксо-6хінолініл)аміно]бутил]аміно]пропіоніл]-6-О-метилхінолініл)аміно]етил]аміно]пропіоніл]-6-О-метил8а-аза-8а-гомоеритроміцин А 8а-аза-8а-гомоеритроміцин А До розчину проміжної сполуки 26 (100мг) у іДо розчину проміжної сполуки 25 (100мг) у іРrOН (2мл) додавали 7-((4-амінобутил)аміно)-1РrOН (1мл) додавали проміжну сполуку 10 (60мг). циклопропіл-1,4-дигідро-6-фтор-4-оксохінолін-3Цю суміш нагрівали при 70°С протягом 18год. Розкарбонової кислоти (61мг) і струшували при 70°С чинник випарювали і залишок розчиняли в МеОН і протягом 2 днів. Розчинник випарювали і залишок перемішували при кімнатній температурі протягом очищали флеш-хроматографією у системі градієн24год. Розчинник випарювали і залишок очищали та розчинника ДХМ/МеОН/NН4ОН з одержанням на силікагелевій SPE-колонці (елюент: від ДХМ сполуки, зазначеної в заголовку (48мг). 100% до ДXM/MeOH/NH4OH 85/13/2) з одержанMS m/z (ES): 1150,9 [MH]+. ням сполуки, зазначеної в заголовку (62мг). + Приклад 11 MS; m/z (ES): 1152,6 [MH] . 4"-O-[3-[[3-[(3-карбокси-1-циклопропіл-6-фторПриклад 6 1,4-дигідро-4-оксо-74"-О-[3-[4-(3-карбокси-1-циклопропіл-1,4хінолініл)аміно]пропіл]аміно]пропіоніл]-6-О-метилдигідро-6-фтор-4-оксо-7-хінолініл)-18а-аза-8а-гомоеритроміцин А пиперазиніл]пропіоніл]-6-О-метил-8а-аза-8аДо розчину проміжної сполуки 25 (10мг) у ігомоеритроміцин А РrOН (3мл) додавали проміжну сполуку 14 (17мг) і Суміш проміжної сполуки 26 (100мг) і 1реакційну суміш нагрівали при 70°С протягом циклопропіл-1,4-дигідро-6-фтор-4-оксо-742год. Реакційну суміш фільтрували, промивали пиперазинілхінолін-3-карбонової кислоти (203мг) у тверду речовину і-РrOН, об'єднані фільтрати конМеОН (20мл) перемішували при 60°С протягом 2 центрували під зниженим тиском і перемішували в днів, потім при кімнатній температурі протягом 2 МеОН протягом 72год. Випарювання розчинника днів і потім при 60°С протягом 24год. Додавали давало сирий продукт, який очищали препаративполімерно-зв'язаний ізоціанат (600мг) і ДХМ (60мл) 49 77210 50 ною TLC (елюент: MeOH/NH4OH 97/3) з одержанночі, розчинник випарювали і залишок розчиняли в ням сполуки, зазначеної в заголовку (3,6мг). ДХМ (3мл). Додавали смолу з полімерно1 зв'язаного ізоціанату і суміш струшували при кімН-ЯМР (500МГц) : 8,73 (s, 1H), 7,93 (d, 1H), натній температурі протягом ночі. Смолу фільтру7,00 (bm, 1H), 6,91 (d, 1H), 5,60 (bd, 1H), 4,70 (d, вали і промивали ДХМ (3мл), МеОН (3мл) і ДХМ 1H), 4,40 (m, 1H), 4,20 (m, 1H), 3,50 (m, 1H), 3,40 (3мл). Зібрані органічні екстракти випарювали під (m, 2H), 2,94-2,90 (m, 4H), 2,65 (m, 2H), 1,95 (m, зниженим тиском. Залишок розчиняли в ДХМ (5мл) 2H), 1,39 (m, 2H), 1,16 (m, 2H). Приклад 12 і промивали водою (3 3мл). Органічну фазу вису4"-O-[3-[[3-[(3-карбокси-7-хлор-1-цикпопропілшували, фільтрували і концентрували під зниже1,4-дигідро-4-оксо-7ним тиском. Сирий матеріал очищали флешхінолініл)аміно]пропіл]аміно]пропіоніл]-6-O-метилхроматографією (елюент: ДХМ/МеОН 90/10) з 8а-аза-8а-гомоеритроміцин А одержанням сполуки, зазначеної в заголовку До розчину проміжної сполуки 25 (10мг) у і(18мг). РrOН (3мл) додавали проміжну сполуку 13 (20мг) і 1Н-ЯМР (500Мгц) (15,0 (bs, 1H), 8,71 (s, 1H), реакційну суміш нагрівали при 70°С протягом 8,12 (d, 1H), 5,57 (bd, 1H), 4,72 (d, 1H), 4,41 (q, 2Н), 42год. Реакційну суміш фільтрували, промивали 4,39 (m, 1Н), 4,19 (m, 1Н), 3,85 (m, 4Н), 2,83-2,55 тверду речовину і-РrOН, об'єднані фільтрати кон(m, 4Н), 2,64 (m, 4Н), 1,51 (t, 3Н). центрували під зниженим тиском і перемішували в Прикладів МеОН протягом 72год. Випарювання розчинника 4"-О-[3-[(2-[(1-бензил-3-карбокси-1,4-дигідро-4давало сирий продукт, який очищали препаративоксо-6-хінолініл)аміно]етил]аміно]-пропіоніл]-6-Оною TLC (елюент: MeOH/NH4OH 97/3) з одержанметил-8а-аза-8а-гомоеритроміцин А ням сполуки, зазначеної в заголовку (3,9мг). До розчину проміжної сполуки 26 (80мг) у і1 РrOН (1,5мл) додавали проміжну сполуку 1 (49мг). Н-ЯМР (500МГц) : 8,73 (s, 1H), 8,02 (s, 1H), Отриману суміш перемішували при 70°С протягом 7,52 (s, 1H), 5,86 (bs, 1H), 5,59 (bd, 1H), 4,70 (d, 18год. Додавали ДБУ (0,04мл) і реакційну суміш 1Η), 4,38 (m, 1Η), 3,55 (m, 1Η), 3,40 (m, 2H), 2,93 перемішували при 50°С протягом додаткових (m, 2H), 2,85 (m, 2H), 2,71-2,55 (m, 2H), 2,00 (m, 24год. Розчинник випарювали під зниженим тис2H), 1,40 (m, 2H), 1,16 (m, 2H). ком, і сирий продукт очищали флешПриклад 13 хроматографією (елюент: МеОН/ДХМ/NН4ОН 4"-О-[3-[(2-[(3-карбокси-1-циклопропіл-1,49/90/1,5) з одержанням сполуки, зазначеної в задигідро-4-оксо-7-хінолініл)аміно]головку (30мг). етил]аміно]пропіоніл]-6-O-метил-8а-аза-8аMS; m/z(ES): 1154,2 [МН]+. гомоеритроміцин А Приклад 17 До розчину проміжної сполуки 26 (100мг) у і4"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілРrOН (1мл) додавали проміжну сполуку 4 (105мг) і 1,4-дигідро-4-оксо-6струшували при 70°С протягом 22год. Розчинник хінолініл)аміно]етил]аміно]пропіоніл]-6-О-етил-8авипарювали і залишок розтирали з ДХМ (2мл). аза-8а-гомоеритроміцин А Отриману суспензію фільтрували і промивали До розчину проміжної сполуки 30 (139мг) у ДХМ (1мл). Фільтрат концентрували під зниженим МеОН (5мл) додавали проміжну сполуку 7 тиском. Сирий продукт осаджували з суміші (102,5мг) і перемішували при 60°С протягом 24год. EtOAc/н-гексан з одержанням сполуки, зазначеної Реакційну суспензію фільтрували, концентрували в заголовку (20мг). під зниженим тиском і залишок очищали флешMS; m/z(ES): 1105,0 [MH]+. хроматографією (елюент: ДXM/MeOH/NH4OH Приклад 14 9/1,3/0,2) з одержанням сполуки, зазначеної в за4"-O-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-4головку (37мг). оксо-7-хінолініл)аміно]етил]-аміно]пропіоніл]-6-ОMS; m/z(ES):1153,7 [МН]+. метил-8а-аза-8а-гомоеритроміцин А 13 С-ЯМР (500МГц) (5/м.д.): 177,5, 175,2, 172,3, Розчин проміжної сполуки 26 (90мг) і проміж167,4, 145,8, 143,1, 132,5, 127,6, 126,3, 118,0, ної сполуки 3 (90мг) у і-РrOН (1мл) нагрівали при 107,6,104,5,103,1, 97,4, 78,8, 78,7, 77,5, 74,1, 73,1, 70°С протягом 20год. Розчинник випарювали, за71,0, 70,9, 68,6, 65,4, 62,9, 49,5, 47,5, 45,4, 44,5, лишок розтирали з ДХМ (2мл). Отриману суспен42,8, 42,5, 41,9, 41,8, 40,7, 40,4, 35,8, 35,4, 34,5, зію фільтрували і промивали ДХМ (1мл). Матковий 23,2, 21,5, 21,4, 21,3, 17,5, 16,1, 15,4, 10,8, 10,5, розчин концентрували під зниженим тиском. Сирий 9,9, 8,1. продукт осаджували з суміші EtOAc/н-гексан з Приклад 18 одержанням сполуки, зазначеної в заголовку 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл(38мг). 1,4-дигідро-4-оксо-6MS; m/z(ES): 1093,3 [МН]+. хінолініл)аміно]етил]аміно]пропioніл]-6-О-пропілПриклад 15 8а-аза-8а-гомоеритроміцин А 4"-O-[3-[4-(3-карбокси-1-етил-6-фтор-1,4До розчину проміжної сполуки 48 (220мг) у дигідро-1,8]нафтиридиніл)-4-оксо-[1МеОН (5мл) додавали проміжну сполуку 7 (182мг) і пиперазиніл]пропіоніл]-6-О-метил-8а-аза-8аперемішували при 60°С протягом 24год. Реакційну гомоеритроміцин А суспензію фільтрували, випарювали метанол під До розчину проміжної сполуки 25 (20мг) у беззниженим тиском і залишок очищали флешводному МеОН (2мл) додавали 1-етил-6-фтор-4хроматографією (елюент: ДХМ/МеОН/NН4ОН оксо-7-пиперазин-1-іл-1,4-дигідро85/13/2) з одержанням сполуки, зазначеної в заго[1,8]нафтиридин-3-карбонову кислоту (22мг). Реаловку (23мг). кційну суміш перемішували при 40°С протягом 51 77210 52 MS; m/z(ES):1167,3 [МН]+. Приклад 22 13 С-ЯМР (500МГц) (5/м.д.): 177,5, 175,1, 172,2, 4"-О-[3-[[4-[(3-карбокси-1-цикпопропіл-1,4167,4, 145,8, 143,1, ,132,5, 127,7, 126,4, 118,0, дигідро-6-фтор-4-оксо-7107,6, 104,4, 102,9, 97,3, 78,9, 78,7, 77,4, 74,2, 73,0, хінолініл)аміно]бутил]аміно]пропіоніл]-6-О-метил71,1, 70,9, 68,5, 65,4, 62,9, 49,5, 47,5, 45,4, 44,5, 9а-аза-9а-гомоеритроміцин А 42,8, 42,5, 42,0, 40,7, 40,4, 35,8, 35,4, 34,6, 23,2, До розчину проміжної сполуки 43 (100мг) у і21,6, 21,5, 21,4, 21,3, 17,6, 16,2, 16,0, 10,8, 10,5, РrOН (2мл) додавали 7-((2-амінобутил)аміно)-19,9, 8,1. циклопропіл-1,4-дигідро-6-фтор-4-оксо-хінолін-3Приклад 19 карбонову кислоту (122мг) і струшували при 70°С 11,12-карбонат-4"-О-[[[2-[(3-карбокси-7-хлор-1протягом 24год. Розчинник концентрували під циклопропіл-1,4-дигідро-4-оксо-6зниженим тиском. Залишок очищали флешхінолініл)аміно]етил]аміно]карбоніл]-11,12хроматографією у системі градієнта розчинника дидезокси-6-О-метил-8а-аза-8а ДХМ/МеОН/NН4ОН з одержанням сполуки, зазна-гомоеритроміцин А ченої в заголовку (34мг). До розчину проміжної сполуки 36 (105мг) у MS; m/z (ES): 1150 [МН]+. ДМФ додавали проміжну сполуку 7 (73мг) і ДБУ Приклад 23 (0,2мл). Отриману суміш перемішували в атмос4"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілфері аргону при 60°С протягом 1год. Потім дода1,4-дигідро-4-оксо-6вали EtOAc (30мл) і насичений водний розчин хінолініл)аміно]етил]аміно]прогїюніл]-6-O-етил-9аNaHCO3 (30мл). Водну фазу промивали EtOAc аза-9а-гомоеритроміцинА До розчину проміжної сполуки 45 (200мг) у ме(2 15мл), і органічні розчини поєднували, сушили і танолі (5мл) додавали проміжну сполуку 7 (148мг) концентрували під зниженим тиском. Сирий залиі перемішували при 60°С протягом 24год. Реакційшок розчиняли в МеОН (50мл) і розчин перемішуну суспензію фільтрували, метанол випарювали вали протягом ночі. Розчинник випарювали і сирий під зниженим тиском і залишок очищали флешпродукт очищали флеш-хроматографією (елюент: хроматографією (елюент: ДХМ/МеОН/NН4ОН MeOH/ДXM/NH4OH 9/90/1,5) з одержанням сполу9/1,3/0,2) з одержанням сполуки, зазначеної в заки, зазначеної в заголовку (80мг). 1 головку (34мг). Н-ЯМР (500МГц) : 8,73 (s, 1H), 8,06 (s, 1H), MS; m/z (ES): 1153,0 [МН]+. 7,49 (s, 1H), 6,22 (d, 1H), 5,40 (t, 1H), 5,32 (t, 1Н), 13 С-ЯМР (500МГц) : 179,2, 177,1, 172,2, 167,4, 5,03 (d, 1H), 4,93 (dd, 1H), 4,60 (d, 1H), 4,41 (d, 1H), 145,9, 143,1, 132,5, 127,6, 126,3, 118,0, 107,6, 4,38 (d, 1H), 4,34 (m, 1H), 4,13 (m, 1H), 4,02 (m, 1Н), 104,5, 101,3, 96,1, 80,0, 79,0, 78,6, 77,7, 74,2, 73,8, 3,67-3,62 (m, 3Н), 3,57-3,55 (m, 2H), 3,47-3,43 (m, 72,8, 71,3, 67,3, 65,0, 63,3, 58,9, 50,8, 49,4, 47,6, 2H), 3,31 (s, 3H),3,18 (s, 3Н). 13 45,6, 44,5, 42,8, 40,6, 40,4, 39,8, 35,4, 35,2, 34,7, С-ЯМР (75МГц) : 177,5; 176,4; 170,4; 167,3; 34,3, 21,7, 21,3, 20,8, 20,7, 18,6, 18,3, 16,4, 16,1, 157,5; 153,3; 145,9; 142,9; 132,7; 127,5; 126,2; 15,9, 11,2, 9,7, 8,1. 118,1; 107,6; 103,9; 102,9; 96,1; 85,6; 82,2; 80,38; Приклад 24 79,7; 79,6; 75,4; 73,2; 70,7; 68,7; 65,3; 63,1; 51,1; 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл49,6; 45,8; 45,05; 42,6; 42,4; 42,0; 40,3; 39,8; 35,4; 1,4-дигідро-4-оксо-6-хінолініл)29,1; 22,8; 22,5; 21,9; 21,3; 21,1; 17,7; 14,6; 14,0; аміно]етил]аміно]пропіоніл]-6-O-пропіл-9а-аза-9а12,3; 11,0; 10,5; 8,1. гомоеритроміцин А Приклад 20 До розчину проміжної сполуки 48 (100мг) у ме4"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілтанолі (5мл) додавали проміжну сполуку 7 (73мг) і 4-оксо-1,4-дигідро-6перемішували при 60° протягом 24год. Реакційну хінолініл)аміно]етил]аміно]пропіоніл]-11-О-[3-(3суспензію фільтрували і розчинник випарювали під хіноліл)-2-пропеніл]-8а-аза-8а-гомоеритроміцин А зниженим тиском. Залишок очищали флешДо суспензії проміжної сполуки 40 (0,1г) у іхроматографією (елюент: ДXM/MeOH/NH4OH РrOН (4мл) додавали проміжну сполуку 7 (0,2г). 9/1,3/0,2) з одержанням сполуки, зазначеної в заОтриману суміш перемішували при 80°С протягом головку (10мг). 3 днів, потім розчинник випарювали і залишок MS; m/z (ES): 1167,3 [МН]+. очищали флеш-хроматографією (елюент: Приклад 25 СНСІ3/МеОН/NH4ОН 6/1/0,1) з одержанням сполу11,12-карбонат-4"-О-[3-[[2-[(3-карбокси-7-хлорки, зазначеної в заголовку (45мг). 1-циклопропіл-1,4-дигідро-4-оксо-6MS; m/z (ES): 1291,9 [МН]+. хінолініл)аміно]етил]аміно]пропіоніл]-11,12Приклад 21 дидезоксиазитроміцин 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-цикпопропілДо суспензії проміжної сполуки 50 (1,0г) у і1,4-дигідро-4-оксо-6-хінолініл)аміно]етил]аміно] РrOН (45мл) додавали проміжну сполуку 7 (2,0г). пропіоніл]-6-O-метил-9а-аза-9а-гомоеритроміцин А Цю суміш нагрівали при 70°С протягом 48год. РозДо розчину проміжної сполуки 43 (200мг) у ічинник випарювали і залишок очищали флешРrOН (2мл) додавали проміжну сполуку 7 (125мг) і хроматографією (елюент: СНСІ3/МеОН/NH3 струшували протягом 7год. при 70°С, потім при 6/1/0,1) з одержанням сполуки, зазначеної в заго80°С протягом ночі. Реакційну суміш фільтрували і ловку (120мг). фільтрат випарювали під зниженим тиском. ЗалиMS; m/z (ES): 1151 [MH]+. шок очищали флеш-хроматографією у системі 1 Н-ЯМР (500МГц) : (8,72s, 1H), 8,04 (s, 1H), градієнта розчинника ДXM/MeOH/NH4OH з одер7,52 (s, 1H), 5,10 (d, 1H), 4,87 (dd, 1H), 4,72 (d, 1Н), жанням сполуки, зазначеної в заголовку (48мг). 4,48 (d, 1Н), 4,44-4,37 (m, 3Н), 3,70 (m, 1Н), 3,58 (d, MS; m/z (ES): 1139,8 [MH]+. 53 77210 54 1Н), 3,31 (s, 3Н), 3,27 (m, 1H), 2,87-2,84 (m, 2H), карбонову кислоту (54мг) і ДБУ (0,2мл). Реакційну 2,67-2,55 (m, 3Н), 2,44-2,36 (m, 8H), 2,21 (s, 3Н), суміш перемішували при кімнатній температурі 2,07 (m, 1H), 2,00 (m, 1H), 1,92 (m, 1 Η), 1,87-1,79 протягом 24год. Потім додавали EtOAc (30мл) і (m, 2Η), 1,68-1,55 (m, 3Н), 1,45 (s, 3Н). Н2О (20мл). Величину рН реакційної суміші довоПриклад 26 дили до 9,5 доданням NaOH (1M). Шари розділя11,12-карбонат-4"-O-[3-[[2-[(3-карбокси-1ли, і водний шар промивали EtOAc (2 5мл). Об'єдциклопропіл-1,4-дигідро-6-фтор-4-оксо-7нані органічні шари концентрували під зниженим хінолініл)аміно]етил]аміно]пропіоніл]-11,12 дидезотиском. Залишок розчиняли в МеОН (20мл) і пексиазитроміцин ремішували при 60°С протягом 4год. і потім при До суспензії проміжної сполуки 50 (1,0г) у ікімнатній температурі протягом ночі. Розчинник РrOН (45мл) додавали проміжну сполуку 8 (2,0г). випарювали, і залишок очищали флешЦю суміш нагрівали при 70°С протягом 48год. Розхроматографією (елюент ДХМ/МеОН/NН4ОН чинник випарювали і залишок очищали флеш90/10/1,5) з одержанням сполуки, зазначеної в хроматографією (елюент: СНСI3/МеОН/NН3 заголовку (49мг). 6/1/0,1) з одержанням сполуки, зазначеної в загоMS; m/z (ES): 1109,54 [МН]+. 1 ловку (150мг). Н-ЯМР (500МГц) : 8,69 (s, 1H), 7,91 (d, 1H), + MS; m/z (ES): 1135 [MH] . 7,00 (d, 1H), 5,18 (d, 1H), 5,12 (brs, 1H), 4,96 (brs, Приклад 27 1H), 4,71 (dd, 1H), 4,55 (d, 1H), 4,53 (d, 1H), 4,34 (m, 11,12-карбонат-4"-O-[3-[[2-[(3-карбокси-11H), 4,24 (d, 1H), 3,76 (m, 1H), 3,66 (s, 1H), 3,60 (d, циклопропіл-1,4-дигідро-4-оксо-71H), 3,54 (m, 1H). 13 хінолініл)аміно]етил]-аміно]пропіоніл]-11,12С-ЯМР (125МГц) : 178,2; 176,3; 166,9; 156,0; дидезоксиазитроміцин 151,2; 147,9; 146,2; 141,9; 141,8; 139,7; 109,5; До суспензії проміжної сполуки 50 (200мг) у 109,3; 107,1; 101,6; 95,2; 94,1; 83,12; 78,5; 77,4; МеОН (2мл) додавали проміжну сполуку 4 (210мг). 73,6; 73,0; 72,9; 72,7; 70,4; 69,4; 67,0; 64,8; 62,8; Цю суміш нагрівали при 70°С протягом 20год. Роз62,1; 48,9; 44,7; 42,0; 41,6; 41,5; 39,9; 39,7; 35,6; чинник видаляли під зниженим тиском, потім до34,8; 34,4; 27,1; 29; 26,9; 26,2; 25,2; 21,4; 20,9; 20,6; давали ДХМ (10мл) і осад відфільтровували. Ор20,5; 17,2; 15,6; 14,1; 10,6; 8,7; 7,7; 6,7. ганічну фазу випарювали під зниженим тиском і Приклад 30 залишок очищали флеш-хроматографією (елюент: 4"-О-[[[2-[(3-карбокси-7-хлор-1-цикпопропілСНCI3/МеОН/NН3 6/1/0,1) з одержанням сполуки, 1,4-дигідро-4-оксо-6зазначеної в заголовку (55мг). хінолініл)аміно]етил]аміно]карбоніл]-азитроміцин MS ;m/z: (ES): 1117 [MH]+. До розчину проміжної сполуки 52 (150мг) у 1 Н-ЯМР (500МГц) ; 8,56 (s, 1H), 8,11 (d, 1H), ДМФ (2мл) додавали проміжну сполуку 7 (65мг) і 6,88 (s, 1H), 6,81 (d, 1H), 5,12 (d, 1H), 4,88 (dd, 1Н), ДБУ (0,4мл). Реакційну суміш перемішували при 4,73 (d, 1Н), 4,47-4,41 (m, 3Н), 3,68 (m, 1Н), 3,57 (d, кімнатній температурі протягом 24год., потім до1Н), 3,31 (s, 3Н), 3,29 (dd, 1H), 3,04 (m, 4Н), 2,87давали EtOAc (30мл) і Н2О (20мл). Величину рН 2,86 (m, 2Н), 2,68-2,63 (m, 3Н), 2,44-2,35 (m, 8Н), реакційної суміші доводили до 9,5 доданням NaOH 2,30 (s, 3Н), 2,06 (t, 1 Η), 1,99 (m, 1 Η), 1,93 (m, 1 (1M). Шари розділяли і водний шар екстрагували Η), 1,83 (m, 1 Η), 1,74 (br d, 1 Η), 1,66-1,57 (m, 3Н), EtOAc (2 15мл). Об'єднані органічні шари концен1,45 (s, 3Н). трували під зниженим тиском. Залишок розчиняли Приклад 28 в МеОН (20мл) і перемішували при 60°С протягом 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл4год., потім при кімнатній температурі протягом 1,4-дигідро-4-оксо-6ночі. Розчинник випарювали під зниженим тиском, хінолініл)аміно]етил]аміно]пропіоніл]-азитроміцин і залишок очищали флеш-хроматографією (елюДо суспензії проміжної сполуки 51 (70мг) в ент ДXM/MeOH/NH4OH 90/10/1,5) з одержанням ацетонітрилі (10мл) додавали проміжну сполука 7 сполуки, зазначеної в заголовку (60мг). (60мг) і ДБУ (0,05мл). Цю суміш нагрівали при MS; m/z (ES): 1097,1 [МН]+. 1 70°С протягом 18год. Розчинник випарювали, і Н-ЯМР (300МГц, CDCI3) : 8,72 (s, 1H), 8,05 (s, залишок очищали флеш-хроматографією (елюент: 1H), 7,47 (s, 1H), 5,39 (t, 1H), 5,25 (m, 1H), 5,18 (d, ДXM/MeOH/NH3 90/9/1,5) з одержанням сполуки, 1H), 4,70 (dd, 1H), 4,59 (d, 1H), 4,51 (d, 1H), 4,36 (m, зазначеної в заголовку (13мг). 1H), 4,25 (bs, 1H). + 13 MS; m/z (ES): 1125 [MH] . С-ЯМР (75МГц, CDCI3) : 178,8; 177,5; 167,3; 13 С-ЯМР (75МГц) : 178,5; 176,8; 171,7; 166,8; 157,7; 145,8; 142,9; 132,6; 127,5; 126,2; 118,2; 145,2; 142,5; 131,9; 127,0; 125,6; 117,5; 106,8; 107,6; 103,7; 102,4; 94,7; 83,3; 79,7; 77,9; 74,2; 73,7; 103,7; 101,8; 82,4; 78,2; 77,1; 76,9; 73,5; 73,0; 72,7; 73,6; 73,2; 70,9; 70,1; 67,8; 65,5; 63,3; 62,5; 49,5; 72,3; 70,3; 69,4; 67,3; 64,9; 62,4; 62,0; 48,9; 46,9; 45,2; 45,1; 42,2; 42,0; 40,4; 39,8; 36,3; 35,4; 35,1; 44,7; 43,8; 42,2; 41,9; 41,5; 39,8; 35,5; 34,8; 34,2; 29,5; 27,5; 26,7; 21,9; 21,6; 21,3; 21,2; 17,8; 16,2; 34,0; 28,4; 27,0; 26,1; 21,4; 21,3; 20,7; 20,6; 17,1; 14,7; 11,3; 9,2; 8,2; 7,4. 15,7; 13,8; 10,6; 8,3; 7,5; 6,5. Приклад 31 Приклад 29 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл4"-О-[[[4-[(3-карбокси-1-цикпопропіл-1,41,4-дигідро-4-оксо-3дигідро-6-фтор-4-оксо-7хінолініл)аміно]етил]аміно]пропіоніл]-6-О-метилхінолініл)аміно]бутил]аміно]карбоніл]азитроміцин азитроміцин До розчину проміжної сполуки 52 (130мг) у До розчину проміжної сполуки 54 (15мг) в ацеДМФ (1мл) додавали 7-((2-амінобутил)аміно)-1тонітрилі (0,6мл) додавали проміжну сполуку 7 циклопропіл-1,4-дигідро-6-фтор-4-оксохінолін-3(11,8мг) і ДБУ (0,010мл). Суміш перемішували 55 77210 56 протягом 12год. при 80°С. Розчинник випарювали Приклад 35 під зниженим тиском і залишок очищали на силіка4"-O-[3-[[2-[(3-карбокси-7-хлор-1 -циклопропілгелевій колонці (елюент: від ДХМ 100% до 1,4-дигідро-4-оксо-6-хінолініл)аміно]етил]-метилДХМ/МеОН/NН4ОН 85/13/2) з одержанням сполуаміно]пропіоніл]-11,12-дидезокси-6-О-метил-11,12ки, зазначеної в заголовку (2,2мг). (метиламінокарбонілокси)-еритроміцин А + MS; m/z (ES): 1138,7 [МН] . До розчину сполуки Прикладу 34 (150мг) у Приклад 32 СНСІ3 (3мл) додавали формальдегід (20мл) і му11,12-(амінокарбонілокси)-4"-О-[3-[[2-[(3рашину кислоту (18,2мл). Цю суміш нагрівали при карбокси-7-хлор-1-циклопропіл-4-оксо-1,4-дигідро60°С протягом 10год., потім додавали СНСІ3 6-хінолініл)аміно]етил]аміно]прогїюніл]-11,12(30мл) і насичений розчин NaHCO3 (20мл). Водну дидезокси-6-O-метил-еритроміцин А фазу промивали СНСІ3 (15мл) і об'єднані органічні До розчину проміжної сполуки 57 (20мг) в ацешари концентрували під зниженим тиском. Залитонітрилі (0,40мл) додавали проміжну сполуку 7 шок очищали флеш-хроматографією (елюент: (15мг). Цю суміш нагрівали при 70°С протягом ДXM/MeOH/NH4OH 90/10/1,5) з одержанням спо12год. Додавали ДБУ (0,030мл) і реакційну суміш луки, зазначеної в заголовку (76мг). перемішували при тій же температурі протягом MS; m/z (ES): 1176,8 [MH]+. 1 додаткових 3год. Розчинник випарювали і залишок Н ЯМР (500МГц, CDCI3) : 8,73 (s, 1H), 8,05 (s, очищали флеш-хроматографіею (елюент: від ДХМ 1H), 7,51 (s, 1H), 5,33 (t, 1H), 4,98 (d, 1H), 4,95 (dd, 100% до ДXM/MeOH/NH4OH 85/13/2) з одержан1H), 4,72 (d, 1H), 4,59 (d, 1H), 4,33 (m, 1H), 3,76 (d, ням сполуки, зазначеної в заголовку (5,8мг). 1H), 3,77-3,70 (m, 1H), 3,63 (d, 1H), 3,57 (m, 1Н), MS ;m/z (ES): 1148,8 [МН]+. 3,54 (s, 1Н), 3,35 (m, 2Н), 3,32 (s, 3Н), 3,27 (m, 1Н), 1 Н-ЯМР (500МГц) : 8,71 (s, 1H), 8,03 (s, 1H), 3,09 (s, 3Н), 3,03 (m, 1H), 3,01 (s, 3Н), 2,91 (m, 1H), 7,52 (s, 1H), 5,8 (s, 1H), 5,33 (bt, 1H), 5,10 (dd, 1H), 2,29 (S, 3H). 13 4,97 (d, 1H), 4,70 (d, 1H), 4,58 (d, 1H), 4,23-4,38 (m, C ЯМР (125МГц, CDCI3) : 215,7; 177,6; 1H), 3,79 (d, 1H), 3,71-3,74 (m, 1H), 3,67 (s, 1Н), 176,6; 171,8; 167,4; 145,8; 143,2; 132,5; 127,6; 3,61 (d, 1Н), 3,58 (d, 1Н), 3,43 (гл, 1Н), 3,31 (s, 3Н). 126,3; 118,0; 118,0; 107,6; 104,5; 101,9; 95,9; 82,8; Приклад 33 79,9; 78,6; 78,5; 76,0; 72,8; 71,0; 67,7; 65,2; 63,0; 11,12-(амінокарбонілокси)-4"-О-[3-[[2-[(362,1; 55,4; 52,8; 50,2; 49,5; 45,5; 45,2; 41,3; 40,7; карбокси-1-циклопропіл-1,4-дигідро-6-фтор-4-оксо38,9; 38,8; 35,4; 35,1; 32,7; 32,5; 21,9; 21,7; 21,1; 7-хінолініл)аміно]етил]аміно]пропіоніл]-11,1220,0; 18,5; 18,3; 15,9; 13,9; 13,7; 10,3; 9,2; 8,1. дидезокси-6-О-метил-еритроміцин А MS; m/z (ES): 855 [MH]+. До розчину проміжної сполуки 57 (20мг) в ацеПриклад 36 тонітрилі (0,40мл) додавали проміжну сполуку 8 4"-O-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл(15мг). Цю суміш нагрівали при 70° протягом 1,4-дигідро-4-оксо-612год. Додавали ДБУ (30мл) і перемішували при хінолініл)аміно]етил]аміно]пропіоніл]-11,12тій же температурі протягом додаткових 3год. Роздидезокси-11,12-(етиламінокарбонілокси)-6-Очинник випарювали під зниженим тиском і залишок метил-еритроміцин А очищали флеш-хроматографією (елюент: від ДХМ До суспензії проміжної сполуки 64 (100мг) у і100% до ДXM/MeOH/NH4OH 85/13/2) з одержанРrOН (2мл) додавали проміжну сполуку 7 (56мг) і ням сполуки, зазначеної в заголовку (4,0мг). ДБУ (0,02мл). Цю суміш нагрівали при 70°С протяMS; m/z (ES): 1132,4 [МН]+. гом 24год., потім розчинник випарювали і залишок Приклад 34 очищали флеш-хроматографією (елюент: 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілДXM/MeOH/NH4OH 90/10/1,5) з одержанням спо1,4-дигідро-4-оксо-6луки, зазначеної в заголовку (60мг). хінолініл)аміно]етил]аміно]пропіоніл]-11,12MS; m/z (ES): 1176,6 [MH]+. 13 дидезокси-6-О-метил-11,12С ЯМР (75МГц, CDCI3) : 216,1; 177,5; 176,3; (метиламінокарбонілокси)-еритроміцин А 172,1; 167,3; 157,2; 145,9; 143,1; 132,6; 127,6; До розчину проміжної сполуки 59 (100мг) у і126,3; 118,0; 107,6; 104,5; 102,0; 96,0; 82,6; 79,8; РrOН (2мл) додавали проміжну сполуку 7 (60мг) і 78,7; 77,7; 76,1; 72,7; 71,0; 67,8; 65,2, 63,1; 60,0; ДБУ (0,02мл). Цю суміш нагрівали при 70°С протя50,5; 49,5; 47,6; 45,6; 45,1; 44,4; 42,8; 40,4; 38,9; гом 36год., потім розчинник випарювали і залишок 38,8; 38,7; 38,6; 35,4; 35,1; 34,6; 29,5; 21,9; очищали флеш-хроматографією (елюент: 21,8,21,2; 20,1; 18,9; 18,3; 16,0; 14,2; 14,1; 12,5; ДХМ/МеОН/NН4ОН 90/10/1,5) з одержанням спо10,3; 9,2; 8,1. луки, зазначеної в заголовку (39мг). Приклад 37 MS; m/z (ES): 1162 [MH]+. 4"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропіл1 Н-ЯМР (500МГц) : 8,73 (s, 1H), 8,04 (s, 1H), 1,4-дигідро-4-оксо-67,54 (s, 1H), 5,27 (t, 1H), 4,96 (m, 2H), 4,70 (d, 1H), хінолініл)аміно]етил]аміно]пропіоніл]-11,124,56 (d, 1H), 4,34 (m, 1H), 3,75 (d, 1H), 3,72 (m, 1H), дидезокси-6-O-метил-11,12-[(Ν-(43,63 (d, 1H), 3,56 (m, 1H), 3,54 (s, 1Н). фенілбутил)аміно)карбонілокси]-еритроміцин А 13 С-ЯМР (75МГц) : 215,8; 177,5; 176,6; 172,2; До розчину проміжної сполуки 67 у і-РrOН 167,3, 157,7; 145,98; 143,1; 132,5, 127,6; 126,3; (10мл) і CH3CN (2мл) додавали проміжну сполуку 118,0; 107,6; 104,5; 102,1; 95,9; 82,8; 79,8; 78,7; 7 (0,5г). Цю суміш нагрівали при 80°С протягом 78,6; 77,6; 75,9; 72,7, 71,0; 67,8; 65,2; 63,0; 62,0; 24год., потім розчинник випарювали і залишок 50,2; 49,6; 47,6; 45,5; 45,2; 44,4; 42,8; 40,3; 38,9; очищали флеш-хроматографією (елюент: 38,8 (2С); 35,4; 35,1; 34,6; 32,5; 21,9; 21,8; 21,2; ДХМ/МеОН/NН4ОН 90/10/1,5) з одержанням спо20,0; 18,6; 18,3; 15,9; 13,9; 13,7; 10,3; 9,1; 8,1. луки, зазначеної в заголовку (1,0г). 57 77210 58 MS; m/z (ES): 1281 [МН]+. 4"-О-[3-[[2-[(3-карбокси-7-хлор1,4-дигідро-1Приклад 38 етил-4-оксо-64"-О-[3-[[2-[(3-карбокси-7-хлор-1-циклопропілхінолініл)аміно]етил]аміно]пропіоніл]-6-О1,4-дигідро-4-оксо-6метилеритроміцин А і хінолініл)аміно]етил]аміно]пропіоніл]-6-О-метил4"-О-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-6еритроміцинА фтор-4-оксо-7До суспензії проміжної сполуки 69 (500мг) в хінолініл)аміно]етил]аміно]пропіоніл]-6-Оацетонітрилі (10мл) додавали проміжну сполуку 7 метилеритроміцин А (300мг) і ДБУ (0,4мл). Цю суміш нагрівали при До розчину проміжної сполуки 69 (133мг) у і70°С протягом 18год. Розчинник випарювали під РrOН (2мл) додавали проміжну сполуку 5 і проміжзниженим тиском і залишок очищали флешну сполуку 6 (100мг, суміш 1/1). Реакційну суміш хроматографією (елюент ДXM/MeOH/NH4OH нагрівали при 80°С протягом 10 днів. Розчинник 90/10/1,5) з одержанням сполуки, зазначеної в випарювали і залишок очищали флешзаголовку (370мг). хроматографією (елюент: ДXM/MeOH/NH4OH MS; m/z (ES): 1124,7 [МН]+. 90/9/1,5) з одержанням сполук, зазначених у заго1 ловку у вигляді 1/1 суміші (140мг). Н-ЯМР (300МГц) : 8,73 (s, 1H), 8,04 (s, 1H), MS; m/z (ES): 112,1 [МН]+; 1096,1 [МН]+. 7,54 (s, 1H), 5,26 (t, 1H), 5,06 (dd, 1H), 4,99 (d, 1Η), Приклад 43 4,70 (d, 1Η), 4,57 (d, 1Η), 4,34 (m, 1Η). 13 4"-О-[3-[[3-[(3-карбокси-7-хлор-1-циклопропілС-ЯМР (75МГц) : 221,0; 177,5; 175,7; 172,2; 1,4-дигідро-4-оксо-6-хінолініл)167,284, 145,8; 143,1; 132,5, 127,6; 126,3; 118,0; аміно]пропіл]аміно]пропіоніл]-6-О107,7; 104,6; 102,2; 96,0; 80,4; 78,9; 78,8; 78,3; 78,2; метилеритроміцин А 76,6; 74,3; 72,7,71,1; 69,1; 67,8; 65,2; 63,0; 50,67. До розчину проміжної сполуки 68 (10мг) у іПриклад 39 РrOН (2,5мл) додавали проміжну сполуку 13 (20мг) 4"-O-[3-[[2-[(3-карбокси-1,4-дигідро-1-етил-4і реакційну суміш нагрівали при 70°С протягом оксо-7-хінолініл)аміно]етил]аміно]пропіоніл]-6-О32год. Реакційну суміш фільтрували, промивали метил-еритроміцин А тверду речовину і-РrOН, об'єднаний розчин концеРозчин проміжної сполуки 69 (100мг) і проміжнтрували під зниженим тиском і перемішували в ної сполуки 3 (103мг) у МеОН (1мл) струшували МеОН протягом 72год. Випарювання розчинника при 70°С протягом 16год. Розчинник випарювали і давало сирий продукт, який очищали препаративзалишок розтирали з ДХМ (2мл). Тверду речовину ною TLC (елюент: MeOH/NH4OH 97/3) з одержанфільтрували і промивали ДХМ (1мл). Матковий ням сполуки, зазначеної в заголовку (4,4мг). розчин концентрували під зниженим тиском. Сирий 1 Н-ЯМР (500МГц) : 8,73 (s, 1H), 8,02 (d, 1H), продукт осаджували з суміші EtOAc/н-гексан 1/1 з 7,53 (bs, 1H), 5,83 (bs, 1H), 4,70 (d, 1H), 4,36 (m, одержанням сполуки, зазначеної в заголовку 1H), 3,60-3,40 (m, 2H), 2,92 (m, 2H), 2,85 (m, 2H), (10мг). 2,65 (m, 2H), 2,00 (m, 2H), 1,39 (m, 2Н), 1,16 (m,2H). MS; m/z (ES): 1077,8 [МН]+. Приклад 44 Приклад 40 4"-O-[3-[[3-[(3-карбокси-1-циклопропіл-1,44"-О-[3-[[2-[(3-карбокси-1-циклопропіл-1,4дигідро-6-фтор-4-оксо-7-хінолініл)дигідро-4-оксо-7аміно]пропіл]аміно]пропіоніл]-6-Охінолініл)аміно]етил]аміно]пропіоніл]-6-О-метилметилеритроміцин А еритроміцин А До розчину проміжної сполуки 68 (7мг) у іРозчин проміжної сполуки 69 (100мг) і проміжРrOН (2мл) додавали проміжну сполуку 14 (14мг) і ної сполуки 4 (107мг) у МеОН (1мл) струшували реакційну суміш нагрівали при 70°С протягом при 70°С протягом 16год. Розчинник випарювали і 32год. Реакційну суміш фільтрували, тверду речозалишок розтирали з ДХМ (2мл). Тверду речовину вину промивали і-РrOН. Об'єднаний розчин концефільтрували і промивали ДХМ (1мл). Матковий нтрували під зниженим тиском і перемішували в розчин концентрували під зниженим тиском. Сирий МеОН протягом 72год. Випарювання розчинника продукт осаджували двічі з суміші EtOAc/н-гексан давало сирий продукт, який очищали препаратив1/1 з одержанням сполуки, зазначеної в заголовку ною TLC (елюент: MeOH/NH4OH 97/3) з одержан(15мг). ням сполуки, зазначеної в заголовку (3,6мг). MS; m/z (ES): 1090,8 [МН]+. 1 Приклад 41 Н-ЯМР (500МГц) : 8,73 (s, 1H), 7,94 (d, 1H), 4"-O-[3-[[2-[(3-карбокси-1-циклопропіл-1,47,02 (bs, 1H), 6,91 (d, 1H), 4,70 (d, 1H), 4,36 (m, 1Н), дигідро-6-фтор-4-оксо-73,50-3,30 (m, 4Н), 2,96-2,88 (m, 4Н), 2,70-2,55 (m, хінолініл)аміно]бутил]аміно]пропіоніл]-6-О-метил2Н), 1,39 (m, 2H), 1,16 (m, 2H). еритроміцин А Приклад 45 До розчину проміжної сполуки 69 (100мг) у і4"-О-[3-[[2-[(3-карбокси-1-циклопропіл-6-фторРrOН (2мл) додавали 7-((2-амінобутил)аміно)-11,4-дигідро-4-оксо-7-хінолініл)циклопропіл-1,4-дигідро-6-фтор-4-оксо-хінолін-3аміно]етил]аміно]пропіоніл]-6-О-метилеритроміцин карбонову кислоту (62мг). Цю суміш нагрівали при А 80°С протягом 72год., потім розчинник випарюваДо розчину проміжної сполуки 8 (50,4мг) у безли, і залишок очищали флеш-хроматографією водному ДМСО (1мл) додавали ДІПЕА (0,02мл) і (елюент: ДXM/MeOH/NH4OH 90/9/1,5) з одержанреакційну суміш перемішували при кімнатній темням сполуки, зазначеної в заголовку (141мг). пературі протягом 1год. Цю суміш додавали до MS; m/z (ES): 1136,1 [МН]+. розчину проміжної сполуки 68 (50мг) у ДМСО (1мл) Приклад 42 і реакційну суміш нагрівали при 80° протягом 59 77210 60 12год. Реакційну суміш охолоджували до кімнатної (3 3мл), об'єднану водну фазу екстрагували ДХМ температури і додавали ДХМ (10мл), і потім воду (3 3мл). Органічну фазу висушували над Na2SO4, (10мл). Органічну фазу промивали водою фільтрували і концентрували під зниженим тиском. (3 10мл), сушили, фільтрували і концентрували Сирий продукт очищали флеш-хроматографією під зниженим тиском. Сирий продукт розчиняли в (елюент: ДХМ/МеОН 90/10) з одержанням сполуки, с МеОН (0,5мл) і розчин нагрівали при 40 С протязазначеної в заголовку (12мг). 1 гом 12год. Розчинник випарювали під зниженим Н-ЯМР (500МГц) : 15,00 (bs, 1H), 8,71 (s, тиском, отриманий сирий продукт розчиняли в 1H), 8,12 (m, 1H), 4,72 (d, 1H), 4,41 (q, 2H), 4.35 (m, ДХМ (0,1мл). Додавали Et2O (0,5мл) і одержували 1Н), 3,85 (m, 4Н), 2,84-2,54 (m, 4Н), 2,64 (m, 4Н), осад. Тверду речовину фільтрували, і фільтрат 1,51 (t, 3Н). випарювали під зниженим тиском з одержанням Приклад 48 сполуки, зазначеної в заголовку (23мг). 4"-О-[3-[4-(3-карбокси-1-циклопропіл-6-фторHPLC/MS аналіз (рухлива фаза: А/В від 70/30 1,4-дигідро-4-оксо-7-хінолініл)-1до 45/55 за 10хв, від 45/55 до 10/90 за 5хв, 10/90 пиперазиніл]етаноїл]-6-О-метил-еритроміцин А протягом 5хв, масовий інтервал 150-1000а.е.м.): До розчину проміжної сполуки 70 (82мг) у час утримання :9,15хв. ДМСО (0,5мл) додавали попередньо приготовлеMS; m/z (ES): 1108 [MH]+. ний розчин ДІПЕА (0,054мл) і 1-циклопропіл-61 Н-ЯМР (500МГц) : 8,73 (s, 1Η), 7,97 (d, 1Η), фтор-4-оксо-7-пиперазин-1-іл-1,4-дигідрохінолін-37,00 (d, 1Η), 5,49 (bm, 1Η), 4,70 (d, 1Η), 4,35 (bm, карбонову кислоту (38мг) у ДМСО (1,3мл). Реак1H), 3,50 (m, 1H), 3,35 (m, 2H), 3,05 (m, 2H), 3,00 ційну суміш перемішували при кімнатній темпера(m, 2H), 2,68-2,50 (m, 2H), 1,38 (m, 2H), 1,20 турі протягом 6год. Додавали воду (10мл) і EtOAc (m,2H). (10мл) і водну фазу екстрагували EtOAc (3 10мл). Приклад 46 Органічну фазу висушували, фільтрували і конце4"-О-[3-[4-(3-карбокси-1-циклопропіл-6-фторнтрували під зниженим тиском. Сирий матеріал 1,4-дигідро-4-оксо-7-хінолініл)-1очищали флеш-хроматографією (елюент: пиперазиніл]пропіоніл]-6-О-метилеритроміцин А ДХМ/МеОН 90/10) і нагрівали в МеОН (12мл) при До розчину проміжної сполуки 68 (20мг) у Ме40°С протягом 16год. Випарювання розчинника під ОН (1мл) додавали ДІПЕА (0,021мл) і 1зниженим тиском давало сполуку, зазначена в циклопропіл-6-фтор-4-оксо-7-пиперазин-1-іл-1,4заголовку (37мг). 1 дигідро-хінолін-3-карбонову кислоту (40мг) і реакН-ЯМР (500МГц) ; 15,00 (bs, 1H), 8,76 (s, ційну суміш нагрівали при 40°С протягом 24год. 1H), 7,99 (d, 1H), 7,36 (d, 1H), 4,75 (d, 1H), 4,36 (m, Розчинник випарювали і залишок розчиняли в 1H), 3,55 (m, 1H), 3,45-3,25 (d+d, 2H), 3,41 (m, 4H), ДХМ (2мл). Додавали полімерно-зв'язаний ізоціа2,84 (m, 4H), 1,40-1,16 (m, 4H). нат (Aldrich, завантаження 2ммоль/м, 120мг) і суПриклад 49 міш струшували при кімнатній температурі протя4"-O-[2-[[2-[(3-карбокси-7-хлор-1-циклопропілгом ночі. Смолу фільтрували і промивали ДХМ 1,4-дигідро-4-оксо-6(2мл), МеОН (2мл) і ДХМ (2мл). Зібрані органічні хінолініл)аміно]етил]аміно]етаноїл]-6-Оекстракти випарювали під зниженим тиском. Заметилеритроміцин А лишок розчиняли в ДХМ (3мл) і промивали водою До розчину проміжної сполуки 70 (40мг) у (3 3мл), об'єднану водну фазу екстрагували ДХМ ДМСО (0,3мл) додавали попередньо приготовле(3 3мл). Органічну фазу висушували над Na2SO4, ний розчин ДІПЕА (0,036мл) і проміжну сполуку 7 фільтрували і концентрували під зниженим тиском. (23мг) у ДМСО (0,7мл). Реакційну суміш перемішуСирий матеріал очищали флеш-хроматографією вали при кімнатній температурі протягом 16год. (елюент: ДХМ/МеОН 90/10) з одержанням сполуки, Додавали воду (5мл) і EtOAc (10мл) і водну фазу зазначеної в заголовку (7,8мг). екстрагували EtOAc (3 10мл). Органічну фазу ви1Н-ЯМР (500МГц) : 15,00 (bs, 1Η), 8,79 (s, сушували, фільтрували і концентрували під зни1Η), 8,04 (d, 1Η), 7,35 (d, 1Η), 4,72 (d, 1Η), 4,35 (m, женим тиском. Сирий матеріал очищали флеш1H), 3,55 (m, 1H), 3,40-3,10 (m, 2H), 2,86-2,64 (m, хроматографією (елюент: ДХМ/МеОН 90/10) і на6H), 2,60 (m, 4H), 1,39 (m, 2H), 1,15 (m, 2Н). грівали в МеОН (12мл) при 50°С протягом 16год. Приклад 47 Випарювання розчинника під зниженим тиском 4"-О-[3-[4-(3-карбокси-1-етил-1,4-дигідро-6давало сполуку, зазначену в заголовку (11мг). 1 фтор-4-оксо-[1,8]нафтиридин)-1Н-ЯМР (500МГц) : 15,07 (bs, 1H), 8,74 (s, 1H), пиперазиніл]прогїюніл]-6-О-метилеритроміцин А 8,05 (s, 1H), 7,55 (s, 1H), 5,30 (t, 1H), 4,75 (d, 1Н), До розчину проміжної сполуки 68 (20мг) у без3,55 (m, 1Н), 3,41 (bm, 2H), 3,45-3,40 (m, 1H), 3,04водному МеОН (2мл) додавали 1-етил-6-фтор-43,00 (m, 2H), 1,39-1,1 (m, 4H). оксо-7-пиперазин-1-іл-1,4-дигідро[1,8]нафтиридинПриклад 50 3-карбонову кислоту (19мг). Реакційну суміш пе4"-О-[2-[[2-[(3-карбокси-1-циклопропіл-6-фторремішували при 40°С протягом ночі, розчинник 1,4-дигідро-4-оксо-7випарювали під зниженим тиском і залишок розчихінолініл)аміно]етил]аміно]етаноїл]-6-Оняли в ДХМ (3мл). Додавали полімерно-зв'язаний метилеритроміцин А ізоціанат (Aldrich, завантаження 2ммоль/м, 118мг) і До розчину проміжної сполуки 70 (46мг) у суміш струшували при кімнатній температурі проДМСО (0,3мл) додавали попередньо приготовлетягом ночі. Смолу фільтрували і промивали ДХМ ний розчин ДІПЕА (0,036мл) і проміжну сполуку 8 (3мл), МеОН (3мл) і ДХМ (3мл). Зібрані органічні (24мг) у ДМСО (0,7мл). Реакційну суміш перемішуекстракти випарювали під зниженим тиском. Завали при кімнатній температурі протягом 16год. лишок розчиняли в ДХМ (5мл) і промивали водою Додавали воду (5мл) і EtOAc (10мл) і водну фазу
ДивитисяДодаткова інформація
Назва патенту англійськоюMacrolids, process for their manufacture (variants), pharmaceutical composition, method for treatment of bacterial infections
Автори англійськоюPaio Alfredo
Назва патенту російськоюМакролиды, способ их получения (варианты), фармацевтическая композиция на их основе, способ лечения бактериальных инфекций с помощью этих соединений
Автори російськоюПайо Альфредо
МПК / Мітки
МПК: A61P 31/04, A61K 31/7048, C07H 17/08
Мітки: одержання, спосіб, композиція, варіанти, таких, макроліди, лікування, бактеріальної, сполук, інфекції, основі, використанням, фармацевтична
Код посилання
<a href="https://ua.patents.su/41-77210-makrolidi-sposib-kh-oderzhannya-varianti-farmacevtichna-kompoziciya-na-kh-osnovi-sposib-likuvannya-bakterialno-infekci-z-vikoristannyam-takikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Макроліди, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі, спосіб лікування бактеріальної інфекції з використанням таких сполук</a>
Похідні бензоксазиндіону, фармацевтична композиція на їх основі, спосіб посилення росту бактерій та спосіб лікування бактеріальної інфекції

Номер патенту: 45414
Опубліковано: 15.04.2002
Автори: РАЙСБРОДТ Рольф, ХАЙНІШ Лотар, Віттманн Стефен, МЮЛЛМАНН Юте
МПК: C07D 499/00, A61K 31/535, C07D 501/00, C07K 5/103, C07D 265/26, A61P 3/00, A61P 43/00, A61K 47/48, C07K 5/02, A61P 31/04, C07K 7/06, A61K 31/538, C07K 5/06
Мітки: бактеріальної, спосіб, основі, росту, бензоксазиндіону, похідні, інфекції, фармацевтична, лікування, композиція, бактерій, посилення
Формула / Реферат:
1. Похідні бензоксазиндіону формули Іде R1=Η, СО-С1-8-алкіл або СОО-С1-8-алкіл,R2=Η, С1-8-алкіл або галоген,і R3 вибраний з наступних груп з а) до f):a) -Z-CHR4-COR5, де Ζ - група формулиде:R6=Н, С1-8-алкіл або галоген,R7=Н, CO-С1-6-алкіл або СОО-С1-6-алкіл,n=ціле число від 1 до 10,m=1 або 2,R4 являє собою С1-8-алкіл; феніл; заміщений...
Похідні тієнопіридину, фармацевтична композиція з використанням таких сполук (варіанти), проміжна сполука

Номер патенту: 72881
Опубліковано: 16.05.2005
Автори: Соболов-Джейнс С'юзан Бет, Манчгоф Майкл Джон
МПК: A61P 35/00, A61K 31/444, A61K 31/4365, A61K 31/519, C07D 495/04, A61K 31/5377
Мітки: проміжна, сполука, фармацевтична, використанням, тієнопіридину, таких, варіанти, похідні, композиція, сполук
Формула / Реферат:
1. Тієнопіридин загальної формули (1) чи (2)або його фармацевтично прийнятні сіль або гідрат, в якихХ1 – CН, R1 – Н, C1–6 алкіл чи -C(O)(C1–6алкіл),R2 – C6-10арил чи 5–13-членний гетероцикл, причому вказаний R2 заміщено, як варіант, 1–5 замісниками R5,кожний R5 незалежно вибирають з галогену, ціаногрупи, нітрогрупи, трифлуорметоксилу, трифлуорметилу, азидогрупи, -C(O)R8, -C(O)OR8, -OC(O)R8, -OC(O)OR8,...
6-o-заміщені кетоліди з антибактеріальною активністю, спосіб їх одержання (варіанти), фармацевтична композиція та спосіб регулювання бактеріальної інфекції у ссавців
Номер патенту: 51730
Опубліковано: 16.12.2002
Автори: Платтнер Джекоб Дж., Чу Деніел Т., Ор Ят Сун, Ма Женкун, Кларк Річард Ф.
МПК: C07H 17/08, A61K 31/365, A61K 31/7048
Мітки: варіанти, композиція, бактеріальної, антибактеріальною, інфекції, регулювання, активністю, фармацевтична, одержання, кетоліди, 6-o-заміщені, ссавців, спосіб
Формула / Реферат:
1. Похідна еритроміцину, вибрана з групи, що складається з:, (II), (III) (IV)і, (V)або їх фармацевтично прийнятних солей чи складних ефірів, де Y і Z разом визначають групу X, причому X вибирають з групи, що складається з(1) =O(2) =N-ОН(3) =N-O-R1, де R1 вибраний з групи, що включає(a) незаміщений С1-C12-алкіл,(b) С1-C12-алкіл із замісником у вигляді арильної...
Похідні с-4″-заміщених макролідів, спосіб їх одержання, проміжні сполуки, фармацевтична композиція та спосіб лікування бактеріальної інфекції або протозойної інфекції

Номер патенту: 70298
Опубліковано: 15.10.2004
Автори: Бронк Браян Скотт, Ченг Хенгміао, Летавіц Майкл Ентоні, Канеко Такуші, Глейзер Едвард Алан, Янг Бінгвей Вера
МПК: A61K 31/7042, C07H 17/08, A61P 33/00, A61K 31/7052, A61P 31/04, A61K 31/7048, C07H 17/00
Мітки: сполуки, лікування, одержання, спосіб, композиція, макролідів, похідні, протозойної, фармацевтична, інфекції, проміжні, бактеріальної, с-4"-заміщених
Формула / Реферат:
1. Сполука формули:або її фармацевтично прийнятна сіль, в якій:X являє собою -CН(NR9R10)-, -C(О)-, -C(=NOR9)-, -CH2NR9- або -N(C1-C6алкіл)CH2-, де перша рисочка кожного значення групи X приєднана до C-10 вуглецю сполуки формули 1 і остання рисочка кожного значення групи X приєднана до C-8 вуглецю сполуки формули 1;R1 являє собою H, гідрокси або метокси;R2 являє собою гідрокси;R3 являє собою...
Похідні с-4″-заміщеного-9-деоксо-9а-а3а-9а-гомоеритроміцину а, спосіб їх одержання, проміжні сполуки, фармацевтична композиція та спосіб лікування бактеріальної інфекції або протозойної інфекції

Номер патенту: 67744
Опубліковано: 15.07.2004
Автори: Ченг Хенгміао, Бронк Браян Скотт, Летавіц Майкл Ентоні, Канеко Такуші, Глейзер Едвард Алан, Янг Бінгвей Вера
МПК: A61K 31/7052, C07H 17/08, A61P 33/02, A61K 31/7048, C07H 17/00, A61P 31/04
Мітки: бактеріальної, інфекції, композиція, фармацевтична, протозойної, одержання, спосіб, лікування, похідні, проміжні, с-4"-заміщеного-9-деоксо-9а-а3а-9а-гомоеритроміцину, сполуки
Формула / Реферат:
1. Сполука формули:або її фармацевтично прийнятна сіль, в якій:R1 являє собою H, гідрокси або метокси;R2 являє собою гідрокси;R3 являє собою C1-C10алкіл, C2-C10алкеніл, C2-C10алкініл, ціано, -CH2S(О)nR8, де n є цілим числом в інтервалі від 0 дo 2, -CH2OR8, -CH2N(COR9)R8, -CH2NR8R15, -(CH2)m(C6-C10арил) або -(CH2)m(5-10-членний гетероарил), де m є цілим числом в інтервалі від 0 дo 4, і де раніше загадані R3...
Наступний патент: Вміщений в раму поверхневий елемент-носій
Випадковий патент: Спосіб обробки титанового сплаву вт-6