Сірководневі похідні нестероїдних протизапальних лікарських засобів
Номер патенту: 104274
Опубліковано: 27.01.2014
Автори: Уоллейс Джон Л., Календо Джузеппе, Чіріно Джузеппе, Сантагада Вінченцо





Формула / Реферат
1. Сполука загальної формули:
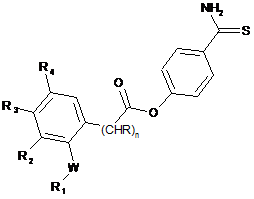 ,
,
де
W є простим зв'язком, N або О;
R є воднем або метилом;
R1 є воднем; ацетилом або фенілом, необов'язково заміщеним 2-3 замісниками, вибраними з галогену або метилу;
R2 є воднем;
R3 є воднем; С1-6алкілом; фенілом або бензоїлом;
R4 є воднем, галогеном або метилом;
R2 і R3 разом утворюють ароматичне вуглеводневе кільце, заміщене метоксигрупою;
n=0 або 1.
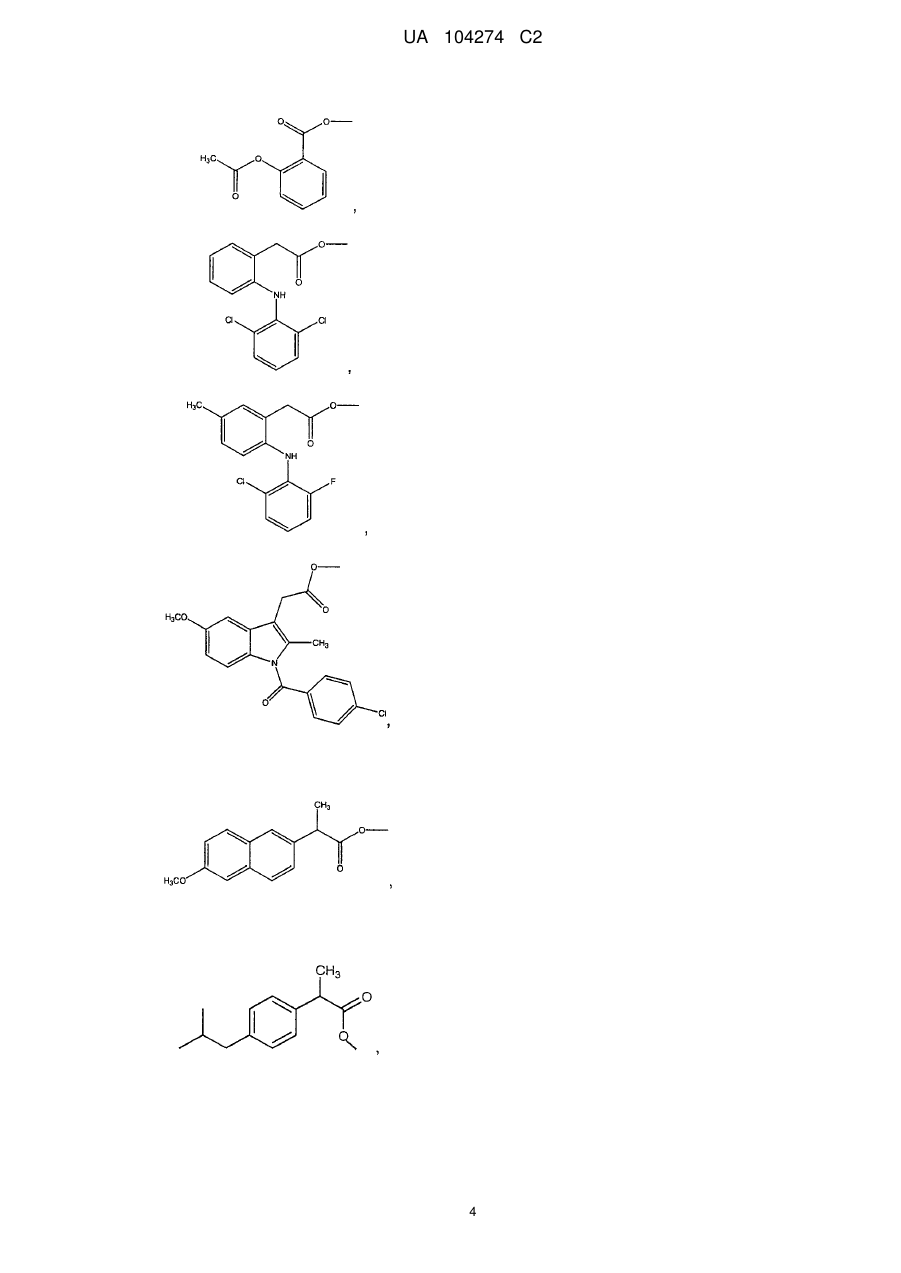
2. Сполука за п. 1, яка являє собою 4-тіокарбамоїлфеніловий складний ефір 2-(6-метоксинафталін-2-іл)пропіонової кислоти.
3. Сполука за п. 1, яка являє собою 4-тіокарбамоїлфеніловий складний ефір 2-ацетоксибензойної кислоти.
4. Сполука за п. 1, яка являє собою 4-тіокарбамоїлфеніловий складний ефір [2-(2,6-дихлорфеніламіно)феніл]оцтової кислоти.
5. Сполука за п. 1, яка являє собою 4-тіокарбамоїлфеніловий складний ефір [2-(2-хлор-6-фторфеніламіно)-5-метилфеніл]оцтової кислоти.
6. Сполука за п. 1, яка являє собою 4-тіокарбамоїлфеніл 2-(4-ізобутилфеніл)пропіонат.
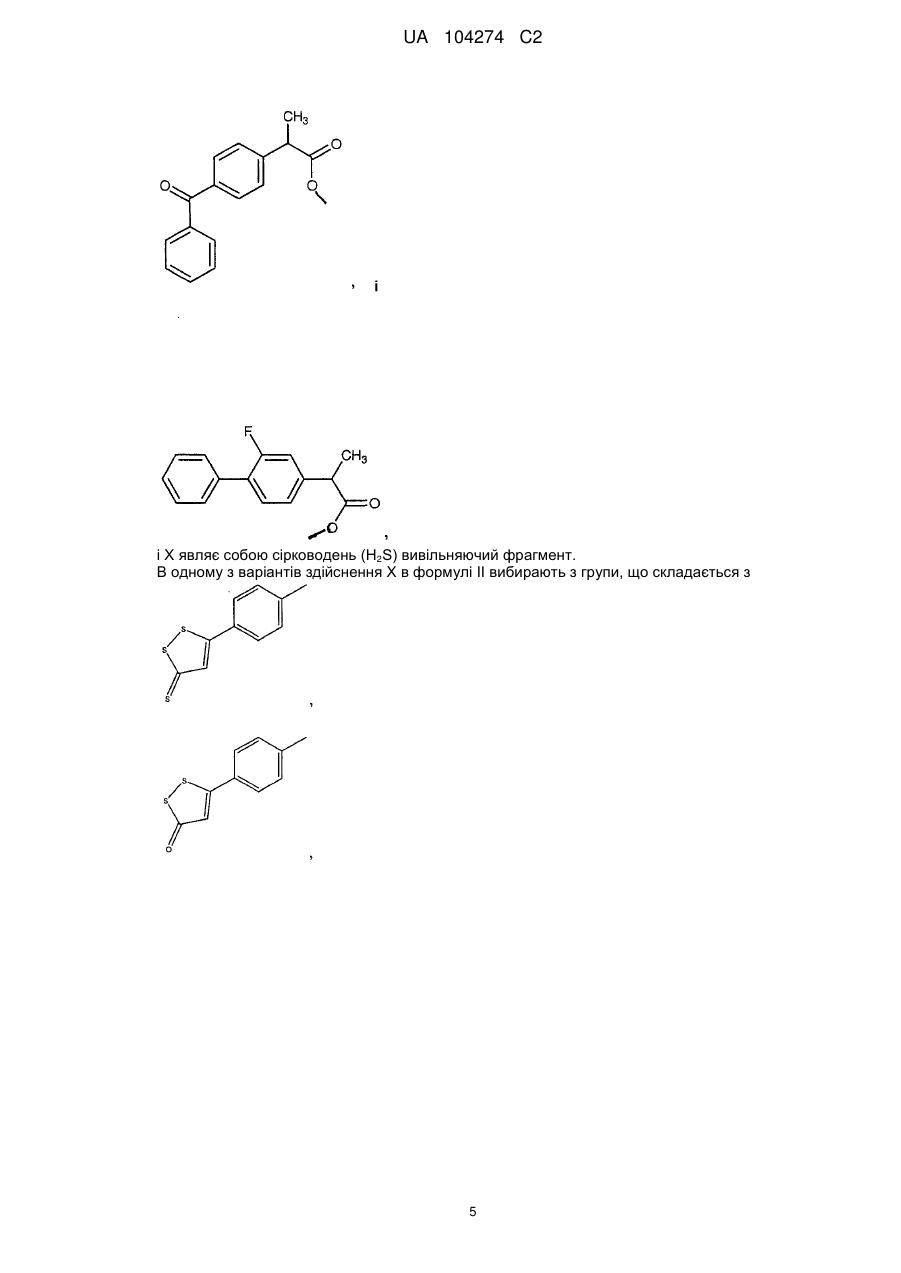
7. Сполука за п. 1, яка являє собою 4-тіокарбамоїлфеніл 2-(4-оксофеніл)фенілпропіонат.
8. Сполука за п. 1, яка являє собою 4-тіокарбамоїлфеніл 2-(3-фтор-4-біфеніліл)пропіонат.
9. Фармацевтична композиція, яка містить сполуку за будь-яким з пп. 1-8 і фармацевтично прийнятний ексципієнт або носій.
10. Застосування сполуки за будь-яким з пп. 1-8 для лікування запалення у пацієнта, який потребує такого лікування, що включає введення пацієнту полегшуючої запалення кількості сполуки.
11. Застосування сполуки за будь-яким з пп. 1-8 для лікування болю у пацієнта, який потребує такого лікування, що включає введення пацієнту полегшуючої біль кількості сполуки.
12. Застосування сполуки за будь-яким з пп. 1-8 для лікування гарячкового стану у пацієнта, який потребує такого лікування, що включає введення пацієнту полегшуючої гарячковий стан кількості сполуки.
Текст
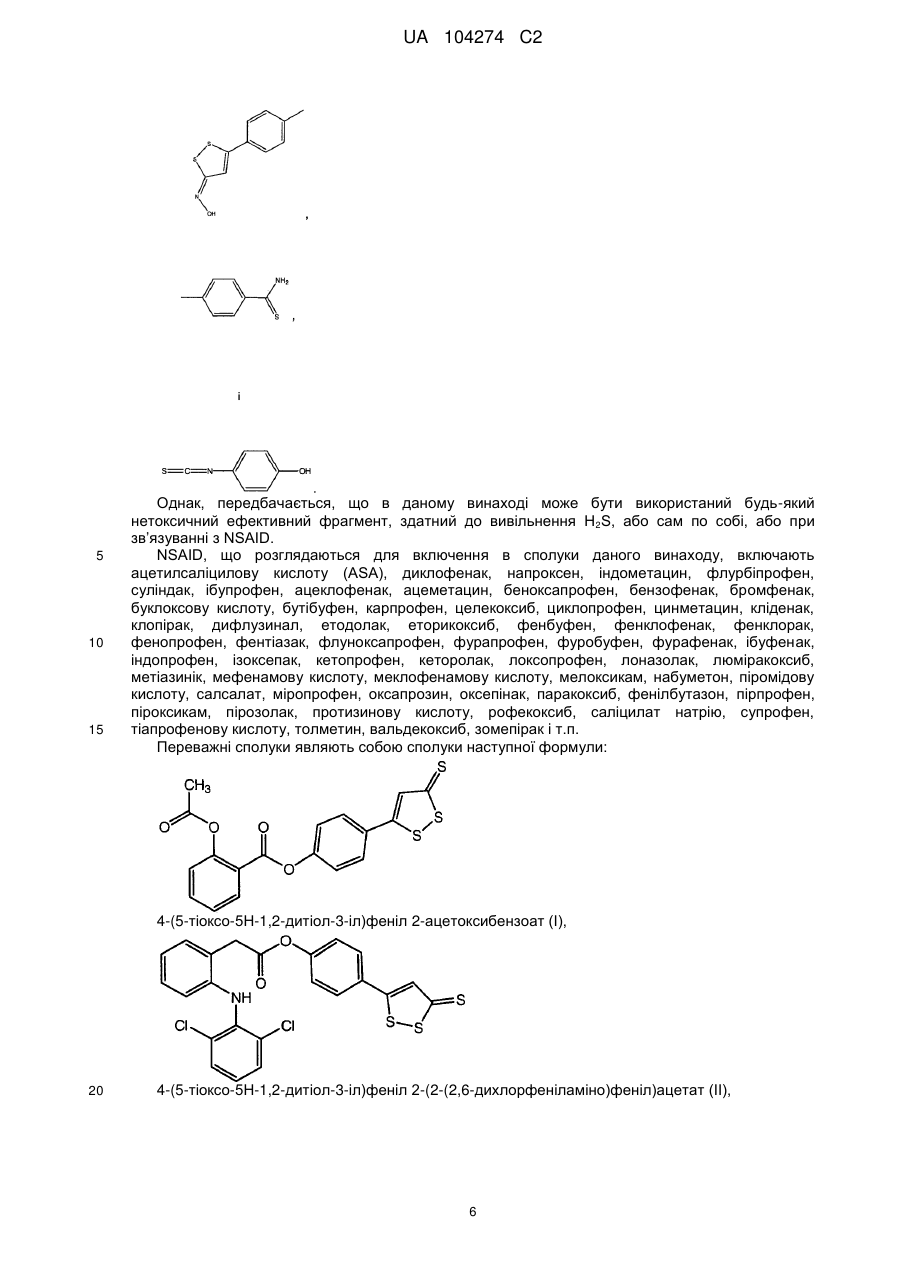
Реферат: Даний винахід стосується похідних нестероїдних протизапальних лікарських препаратів (NSAID), що мають поліпшені протизапальні властивості, придатні для лікування запалення, болю і гарячкового стану. Більш конкретно, отримані NSAID похідні з фрагментом, що вивільняє сірководень (H2S), для отримання нових протизапальних сполук, які мають ослаблені побічні дії. UA 104274 C2 5 10 15 20 25 30 35 40 45 50 55 Дана заявка подана як частково продовжена заявка PCT/CA2006/000484, подана 31 березня 2006, яка запитує пріоритет заявки PCT/CA2005/000819, поданої 27 травня 2005. Дана заявка додатково запитує пріоритет попередніх заявок на патент США №№ 60/807639, поданої 18 липня 2006 і 60/887188, поданої 30 січня 2007. Галузь техніки, до якої належить винахід Даний винахід стосується похідних нестероїдних протизапальних лікарських засобів (NSAID), що мають поліпшені протизапальні властивості, корисні при лікуванні запалення, болю і гарячкового стану. Більш конкретно, NSAID модифікують сірководень(H2S)-вивільняючим залишком з отриманням нових протизапальних сполук, що мають знижені побічні ефекти. Попередній рівень техніки Нестероїдні протизапальні лікарські засоби (NSAID) широко використовують для лікування різних станів, пов'язаних з болем, гарячкою і запаленням, включаючи остеоартрит, ревматоїдний артрит, подагру і анкілозувальний спондиліт. Їх також широко використовують для лікування гострого болю, пов'язаного з травмами і хірургічними процедурами (включаючи зубні процедури), і головного болю. В основному, вважається, що сприятлива дія NSAID може бути зумовлена їх здатністю пригнічувати синтез простагландинів за рахунок інгібування циклооксигенази-1 (СОХ-1) і циклооксигенази-2 (СОХ-2). Однак тривале застосування NSAID значною мірою обмежується їх здатністю викликати клінічно значущі ушкодження в шлунково-кишковому тракті (Wallace, J. L. Nonsteroidal antiinflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology. 1997; 112:1000-1016). Селективні інгібітори СОХ-2 розглядалися як поліпшення в порівнянні з традиційними NSAID, оскільки, як виявилося, вони в меншій мірі викликають ушкодження шлунково-кишкового тракту. Однак виник неспокій відносно серцево-судинної токсичності вказаних препаратів і, можливо, також традиційних NSAID (Grosser et al., Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006; 116: 4-15). Добре відомо, що NSAID стимулюють адгезію лейкоцитів і зменшення кровотоку в слізовій шлунку, і вказані дії являють собою важливий внесок в патогенез викликаного NSAID ушкодження шлунково-кишкового тракту (Wallace, 1997). Індукція адгезії лейкоцитів за рахунок неселективних і СОХ-2-селективних NSAID може також давати внесок в серцево-судинні ускладнення вказаних лікарських засобів. Нещодавно помітили, що сірководень (H2S) виявляє протизапальні і анальгезивні активності. H2S являє собою ендогенну речовину, що продукується в багатьох тканинах, і діє на багато функцій (Wang, Two's company, three's а crowd: can H 2S be the third endogenous gaseous transmitter? FASEB J 2002; 16: 1792-1798). Також було показано, що він є судинорозширювальним засобом і може пригнічувати адгезію лейкоцитів до ендотелію судин (Wang, 2002; Fiorucci et al., Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005; 129: 1210-1224). Додатково, Fiorucci et al. (2005) показали, що попередня обробка донором H 2S може зменшувати тяжкість індукованого NSAID ушкодження шлунково-кишкового тракту щура. Несподівано, автори даного винаходу показали в даний заявці, що протизапальна активність різних NSAID значною мірою поліпшується або при ковалентному зв’язуванні, або при утворенні солей NSAID з фрагментом H2S, що вивільняється. Додатково, було показано, що вказані похідні NSAID мають знижену побічну дію. Зокрема, автори показали, що NSAID похідні даного винаходу мають одну або декілька з наступних додаткових характеристик: (1) призводять до меншого ушкодження шлунково-кишкового тракту в порівнянні з традиційними NSAID; (2) прискорюють лікування виразок шлунку, що вже існували; і (3) в значно меншій мірі спричиняють підвищення системного кров'яного тиску в порівнянні з традиційними NSAID. Більше того, похідну NSAID даного винаходу знижують адгезію лейкоцитів до ендотелію судин, що може давати внесок в зниження побічної дії як на шлунково-кишковий тракт, так і на серцево-судинну систему. Суть винаходу В одному аспекті даного винаходу забезпечують похідні NSAID, причому вказані похідні включають H2S-вивільняючий фрагмент, який або ковалентно зв'язаний з NSAID або утворює сіль з NSAID. Несподівано, сполуки даного винаходу демонструють поліпшену протизапальну активність в моделі індукованого карагінаном набряку лапи щура в порівнянні з одним NSAID, одним H2S-вивільняючим фрагментом і комбінацією NSAID і H2S-вивільняючого фрагмента, що вводяться окремо, але одночасно. Більше того, похідні NSAID даного винаходу призводять до помірного короткочасного збільшення концентрацій H2S в плазмі. Без зв'язку з певною теорією, 1 UA 104274 C2 5 10 15 20 25 30 короткочасне збільшення концентрацій H2S в плазмі, яке все ще залишається в фізіологічному діапазоні, може давати внесок в їх поліпшену протизапальну активність. Несподівано, сполуки даного винаходу можуть також демонструвати поліпшену здатність по пригнічення активності циклооксигенази-2 (COX-2) і/або активності циклооксигенази-1 (COX-1) в порівнянні з їх відповідними немодифікованими NSAID аналогами. Така поліпшена здатність по пригнічення активності COX-2 і/або COX-1 може також давати внесок в поліпшену протизапальну активність. Більше того, сполуки даного винаходу, що мають поліпшену здатність з пригнічення активності COX-1, демонструють значне пригнічення вироблення тромбоксану В2 в тромбоцитах, що може давати внесок в знижену серцево-судинну токсичність. Додатково, сполуки даного винаходу в меншій мірі демонструють побічні дії в порівнянні з їх відповідними немодифікованими NSAID аналогами. Наприклад, деякі сполуки несподівано викликають значною мірою менше пошкоджень шлунку, ніж самі NSAID, незважаючи на те, що сполуки помітно пригнічують синтез шлункового простагландину. При тому, що збереження шлунку спостерігається при вказаних H2S-вивільняючих похідних NSAID, вказаний результат не досягається, якщо NSAID і H2S-вивільняючий фрагмент вводять щурам окремо, але одночасно. Без зв'язку з певною теорією, було показано, що сполуки даного винаходу зменшують адгезію лейкоцитів до ендотелію судин, що може давати внесок в їх безпеку для шлунку. Додатково, знижена адгезія лейкоцитів до ендотелію судин може знижувати побічну дію на серцевосудинну систему, яка часто спостерігається при тривалому застосуванні NSAID. Додатково, сполуки даного винаходу несподівано спричиняють менше збільшення систолічного кров'яного тиску при введенні щурам, що страждають на гіпертензію, в порівнянні з тим, що спостерігається при введенні звичайних NSAID. Знижена схильність до підвищення кров'яного тиску може знижувати побічні дії на серцево-судинну систему, які часто спостерігаються при тривалому застосуванні NSAID. Відповідно до даного винаходу забезпечують сполуки загальної формули: A-Y-X (Формула I), де А являє собою NSAID радикал, Y вибирають з групи, що складається з -С(О)O-, -С(О)NH, -С(О)OC(О)-, -С(О)NHCH2C(О)- або нуля, і Х являє собою фрагмент, здатний вивільнювати сірководень, або сам по собі, або при зв’язуванні з NSAID (що тут далі позначається як H 2Sвивільняючий фрагмент), або його фармацевтично прийнятну сіль, таким чином, що якщо Y являє собою нуль, похідне NSAID може являти собою сіль А і Х. У переважному варіанті здійснення винаходу Х в формулі I вибирають з групи, що складається з: 2 UA 104274 C2 5 10 Однак, передбачається, що в даному винаході може бути використаний будь-який нетоксичний ефективний фрагмент, здатний до вивільнення H2S, або сам по собі, або при зв’язуванні з NSAID. В одному з варіантів здійснення сполуки за винаходом мають наступну загальну формулу В-С(О)О-X (Формула II) де В-С(О)О- являє собою похідне NSAID, що має вільну карбоксильну групу, або карбоксизаміщеного NSAID, і X являє собою H2S-вивільняючий фрагмент, або його фармацевтично прийнятної солі. В одному з варіантів здійснення В-C(O)O- в формулі II вибирають з групи, що складається з: 3 UA 104274 C2 4 UA 104274 C2 і Х являє собою сірководень (H2S) вивільняючий фрагмент. В одному з варіантів здійснення Х в формулі II вибирають з групи, що складається з 5 UA 104274 C2 5 10 15 Однак, передбачається, що в даному винаході може бути використаний будь-який нетоксичний ефективний фрагмент, здатний до вивільнення H2S, або сам по собі, або при зв’язуванні з NSAID. NSAID, що розглядаються для включення в сполуки даного винаходу, включають ацетилсаліцилову кислоту (ASA), диклофенак, напроксен, індометацин, флурбіпрофен, суліндак, ібупрофен, ацеклофенак, ацеметацин, беноксапрофен, бензофенак, бромфенак, буклоксову кислоту, бутібуфен, карпрофен, целекоксиб, циклопрофен, цинметацин, кліденак, клопірак, дифлузинал, етодолак, еторикоксиб, фенбуфен, фенклофенак, фенклорак, фенопрофен, фентіазак, флуноксапрофен, фурапрофен, фуробуфен, фурафенак, ібуфенак, індопрофен, ізоксепак, кетопрофен, кеторолак, локсопрофен, лоназолак, люміракоксиб, метіазинік, мефенамову кислоту, меклофенамову кислоту, мелоксикам, набуметон, піромідову кислоту, салсалат, міропрофен, оксапрозин, оксепінак, паракоксиб, фенілбутазон, пірпрофен, піроксикам, пірозолак, протизинову кислоту, рофекоксиб, саліцилат натрію, супрофен, тіапрофенову кислоту, толметин, вальдекоксиб, зомепірак і т.п. Переважні сполуки являють собою сполуки наступної формули: 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-ацетоксибензоат (I), 20 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетат (II), 6 UA 104274 C2 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл (III), 5 2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетат 4-(5-тіоксо-5Н-[1,2]дитіол-3-іл)феніловий метил-1Н-індол-3-іл]оцтової кислоти (IV), складний складний 4-(5-тіоксо-5Н-[1,2]дитіол-3-іл)феніловий іл)пропіонової кислоти (V), 10 ефір [1-(4-хлорбензоїл)-5-метокси-2 ефір 2-(6-метоксинафталін-2 4-(5-оксо-5Н-[1,2]дитіол-3-іл)феніловий складний ефір 2-ацетоксибензойної кислоти (VI), складний 4-(5-оксо-5Н-[1,2]дитіол-3-іл)феніловий дихлорфеніламіно)феніл]оцтової кислоти (VII), 7 ефір [2-(2,6 UA 104274 C2 4-(5-оксо-5Н-[1,2]дитіол-3-іл)феніловий метилфеніл]оцтової кислоти (VIII) 5 15 ефір [2-(2-хлор-6-фторфеніламіно)-5 4-(5-оксо-5Н-[1,2]дитіол-3-іл)феніловий складний ефір [1-(4-хлорбензоїл)-5-метокси-2-метил1Н-індол-3-іл]оцтової кислоти (IX), 4-(5-оксо-5Н-[1,2]дитіол-3-іл)феніловий іл)пропіонової кислоти (Х), 10 складний складний ефір 2-(6-метоксинафталін-2 4-(5-гідроксііміно-5Н-[1,2]дитіол-3-іл)феніловий складний ефір 2-ацетоксибензойної кислоти (XI), 4-(5-гідроксііміно-5Н-[1,2]дитіол-3-іл)феніловий дихлорфеніламіно)феніл]оцтової кислоти (ХII), 8 складний ефір [2-(2,6 UA 104274 C2 4-(5-гідроксііміно-5Н-[1,2]дитіол-3-іл)феніловий складний ефір [2-(2-хлор-6-фторфеніламіно)5-метилфеніл]оцтової кислоти (ХIII), 5 4-(5-гідроксііміно-5Н-[1,2]дитіол-3-іл)феніловий складний ефір [1-(4-хлорбензоїл)-5-метокси2-метил-1Н-індол-3-іл]оцтової кислоти (XIV), 4-(5-гідроксііміно-5Н-[1,2]дитіол-3-іл)феніловий іл)пропіонової кислоти (ХV), 10 15 складний ефір 2-(6-метоксинафталін-2 4-тіокарбамоїлфеніловий складний ефір 2-ацетоксибензойної кислоти (XVI), 4-тіокарбамоїлфеніловий складний ефір [2-(2,6-дихлорфеніламіно)феніл]оцтової кислоти (ХVII), 9 UA 104274 C2 4-тіокарбамоїлфеніловий складний метилфеніл]оцтової кислоти (ХVIII), 5 ефір [2-(2-хлор-6-фторфеніламіно)-5 4-тіокарбамоїлфеніловий складний ефір [1-(4-хлорбензоїл)-5-метокси-2-метил-1Н-індол-3іл]оцтової кислоти (XIX), 4-тіокарбамоїлфеніловий складний ефір 2-(6-метоксинафталін-2-іл)пропіонової кислоти (ХХ), 10 4-ізотіоціанатофеніл 2-ацетоксибензоат (XXI), 4-ізотіоціанатофеніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетат (ХXII), 10 UA 104274 C2 4-ізотіоціанатофеніл 2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетат (ХXIII), 4-(ізотіоціано)феніл 2-[1-(4-хлорбензоїл)-5-метокси-2-метиліндол-3-іл]ацетат (XXXIV), 5 4-ізотіоціанатофеніл 2-(2-метоксинафталін-6-іл)пропіонат (ХXV), 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-(4-ізобутилфеніл)пропіонат (ХXVI), 10 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-(3-бензоїлфеніл)пропіонат (XXVII), 11 UA 104274 C2 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-(2-фтор-4-біфеніліл)пропіонат (XXVIII), 4-тіокарбамоїлфеніл 2-(4-ізобутилфеніл)пропіонат (XXIX), 5 4-тіокарбамоїлфеніл 2-(4-оксофеніл)фенілпропіонат (XXX), 4-тіокарбамоїлфеніл 2-(2-фтор-4-біфеніліл)пропіонат (XXXI), 10 4-ізотіоціанатофеніл 2-(4-ізобутилфеніл)пропіонат (XXXII), 12 UA 104274 C2 4-(ізотіоціано)феніл 2-(4-оксофеніл)-фенілпропіонат (XXXIII) і 5 10 15 20 25 30 4-(ізотіоціано)феніл 2-(2-фтор-4-біфеніліл)пропіонат (XXXIV). Згаданий вище попередник NSAID (А) отримують згідно зі способами, відомими в th попередньому рівні техніки. Дивись, наприклад, The Merck Index, 13 Edition (2001), Merck & Co., Whitehouse Station, N.J., включений тут як посилання. Якщо доступні, можуть бути використані відповідні ізомери, включаючи оптичні ізомери. Фармацевтично прийнятні солі сполук даного винаходу, такі як, наприклад, солі лужних металів і лужно-земельних металів, нетоксичних амінів і амінокислот також являють собою частину даного винаходу. Переважні солі сполук даного винаходу являють собою солі аргініну і агматину. Також включені фармацевтично прийнятні солі приєднання кислоти. У переважному варіанті здійснення винаходу NSAID даного винаходу являють собою модифікований H2S-вивільняючий фрагмент 4-гідрокситіобензамід (що означається тут як TBZ). TBZ похідні рівним чином демонструють кращу загальну протизапальну активність і знижені побічні дії в порівнянні з похідними 5-п-гідроксифеніл-1,2-дитіол-3-тіону (ADT-OH). Несподівано, TBZ похідні генерують значною мірою більше H2S в порівнянні з ADT-OH похідними, що може давати внесок як в збільшення протизапальної активності, так і в зменшення побічної дії. Додатково, TBZ похідні зберігають здатність більш стабільно інгібувати СОХ-1/СОХ-2 в порівнянні з ADT-OH похідними. Фактично, багато TBZ похідних дійсно демонструють посилення інгібування СОХ-1 або інгібування СОХ-2, або обох. Більш того сполуки ХХ (TBZ похідне напроксену) показували значною мірою кращі результати в інгібуванні синтезу тромбоксану В2 в порівнянні з еквівалентним похідним ADT-OH, сполукою V (напроксен-ADT-OH), і сполука ХIХ (TBZ похідне індометацину) показувала значною мірою кращі результати в інгібуванні синтезу тромбоксану В2 в порівнянні з еквівалентним похідним ADT-OH, сполукою IV (ADT-OH похідне індометацину). Поліпшене інгібування тромбоксану В 2 може впливає на безпеку даних похідних відносно серцево-судинної системи. Сполуки даного винаходу можуть бути отримані, як показано на двох наступних схемах: Схема 1 Схема 1 нижче представлена при використанні як прикладу синтезу 4-(5-тіоксо-5Н-1,2,дитіол-3-іл)феніл 2-(2-(2,6-дихлорфеніламін)феніл)ацетату (Сполука II) 13 UA 104274 C2 5 10 15 20 25 30 35 NSAID, що мають вільну карбоксильну групу (або карбоксизаміщене NSAID), наприклад, диклофенак (1), спочатку розчиняли в диметилформаміді і додавали гідроксибензотриазол (HOBt) і 1,3-дициклогексилкарбодіімід (DCC). До вказаної суміші додавали фрагмент, що вивільняє сірководень, такий як 5-п-гідроксифеніл-1,2-дитіол-3-тіон (ADT-OH) (2) за умов, придатних для отримання сполук даного винаходу, таких як 4-(5-тіоксо-5H-1,2-дитіол-3-іл)феніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетат (3). Очевидно, що в даній схемі можна використовувати інші фрагменти, що вивільняють сірководень, такі як 4гідроксифенілізотіоціанат (що тут позначається як HPI). Схема 2 Схема 2 нижче представлена при використанні як прикладу синтезу 4тіокарбамоїлфенілового складного ефіру [2-(2,6-дихлорфеніламіно)феніл]оцтової кислоти (ХVII). На вказаній схемі використовують реагент Лавессона для приєднання сірковмісної групи до фрагмента, що вивільняє сірководень, після його ковалентного зв’язування з NSAID. NSAID, що має вільну карбоксильну групу (або карбоксизаміщене NSAID), наприклад, диклофенак (1), спочатку розчиняли в диметилформаміді і додавали гідроксибензотриазол (HOBt) і 1,3-дициклогексилкарбодіімід (DCC). До вказаної суміші додавали попередник, що вивільняє сірководень, такий як 4-гідроксибензамід, за умов, придатних для утворення попередника (наприклад, 4-карбамоїлфеніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетату (2)) сполуки даного винаходу, причому вказаний попередник не містить сірки. Додають відповідну сполуку, яка може додати сірковмісну групу, таку як реагент Лавессона, для отримання сполуки даного винаходу (наприклад, 4-тіокарбамоїлфенілового складного ефіру [2-(2,6дихлорфеніламіно)феніл]оцтової кислоти (3)). У додатковому аспекті даний винахід забезпечує фармацевтичну композицію сполуки даного винаходу і фармацевтично прийнятного ексципієнта або носія, особливо фармацевтичну композицію для застосування при лікуванні запального стану шлунково-кишкового (GI) тракту. Сполуки даного винаходу придатні для застосування для, не обмежуючись перерахованим, лікування запалення у пацієнта і для лікування інших захворювань, пов'язаних із запаленням, наприклад, як анальгетика при лікуванні болю і головного болю, або як жарознижуючого засобу для лікування гарячкового стану. Наприклад, сполуки даного винаходу корисні для лікування артриту, включаючи, але не обмежуючись перерахованим, ревматоїдний артрит, спондилоартропатії, подагричний артрит, остеоартрит, системний червоний вовчак і ювенільний артрит. Такі сполуки за винаходом придатні при лікуванні астми, бронхіту, спазмів при менструаціях, тендиніту, бурситу, шкіряних хвороб, таких як псоріаз, екзема, опіки і дерматит, і післяопераційних запалень, включаючи очну хірургію, таку як операції з видалення катаракти і 14 UA 104274 C2 5 10 15 20 25 30 35 40 45 50 55 рефракційну хірургію. Сполуки за винаходом також корисні для лікування шлунково-кишкових захворювань, таких як запальне захворювання кишечнику, хвороба Крона, гастрит, синдром роздратованого кишечнику і виразковий коліт, а також для запобігання або лікування раку, такого як колоректальний рак. Сполуки за винаходом придатні при лікуванні запалень при таких захворюваннях, як судинні захворювання, мігрені, вузликовий періартерит, тиреоїдит, апластична анемія, хвороба Ходжкіна, склеродома, ревматична атака, діабет I типу, захворювання нервово-м’язових з’єднань, включаючи злоякісну міастенію, захворювання білої речовини, включаючи множинний склероз, саркоїдоз, нефротичний синдром, синдром Бехчета, поліміозит, гінгівіт, нефрит, гіперчутливість, післятравматична пухлина, ішемія міокарда і т.п. Сполуки також корисні для лікування очних захворювань, таких як ретиніт, ретинопатія, увеїт, очна світлобоязнь і гостра травма тканин ока. Сполуки також придатні для лікування легеневих запалень, таких як пов'язані з вірусними інфекціями, і кістозного фіброзу. Сполуки також корисні для лікування певних порушень центральної нервової системи, таких як кортикальне недоумство, включаючи хворобу Альцгеймера. Сполуки за винаходом придатні як протизапальні засоби, такі як для лікування артриту, з додатковою перевагою, що перебуває в значно менш шкідливій побічній дії. Вказані сполуки також корисні для лікування алергічного риніту, синдрому ускладненого дихання, синдрому ендотоксичного шоку, атеросклерозу і ушкодження центральної нервової системи внаслідок удару, ішемії і травм. Сполуки також корисні при лікуванні болю, такого як, але не обмежуючись перерахованим, післяопераційний біль, зубний біль, м’язовий біль і біль внаслідок ракового захворювання. Крім корисності для лікування людини, вказані сполуки також придатні для лікування ссавців, включаючи коней, собак, кішок, щурів, мишей, овець, свиней і т.д. Залежно від конкретного стану або захворювання, що піддається лікуванню, пацієнту можуть бути введені сполуки даного винаходу в будь-якій придатній терапевтично ефективній і безпечній дозі, як може бути легко визначено фахівцем в даній галузі. Вказані сполуки найбільш бажано вводити в дозах в діапазоні від приблизно 1 до приблизно 2000 мг на день, у вигляді однієї дози або розділених доз, хоча неминуче будуть спостерігатися варіації залежно від маси і стану пацієнта, що піддається лікуванню, і конкретного вибраного шляху введення. Зрозуміло, що дози будуть залежати від конкретного NSAID, що використовується для отримання сполук даного винаходу. Однак найбільш бажане дозування, що лежить в діапазоні від приблизно 0,1 до приблизно 100 мг/кг, переважно, в діапазоні приблизно від 5 до 90 мг/кг, і, більш переважно, в діапазоні приблизно від 5 до 50 мг. Проте можуть спостерігатися варіації залежно від маси і стану пацієнтів, що піддаються лікуванню, і їх індивідуальної реакції на вказаний лікарський засіб, а також від типу вибраного фармацевтичного препарату і періоду часу і інтервалу, в ході якого проводять таке введення. У деяких випадках дози нижче межі вказаного діапазону можуть виявитися більше ніж адекватними, тоді як в інших випадках можуть застосовуватися ще більші дози, що не викликають ніякої шкідливої побічної дії, за умови, що такі великі дози спочатку розділяють на декілька невеликих доз для введення протягом дня. Сполуки даного винаходу можна вводити в формі будь-якого фармацевтичного препарату, природа якого буде залежати від шляху введення. Фармацевтичні композиції можуть бути отримані звичайними способами, при використанні сумісних фармацевтично прийнятних ексципієнтів або носіїв. Приклади таких композицій включають капсули, таблетки, черезшкірні пластирі, льодяники, пастилки, спреї, сиропи, порошки, гранули, гелі, еліксири, супозиторії і т.п., препарати розчинів для негайного прийому, препарати для ін'єкцій, ректальні, назальні, очні, вагінальні препарати і т.д. Переважний шлях введення являє собою пероральний і ректальний шлях введення. Для перорального введення можуть застосовуватися таблетки, що містять різні ексципієнти, такі як мікрокристалічна целюлоза, цитрат натрію, карбонат кальцію, дикальційфосфат і гліцин, спільно з різними дезінтегрантами, такими як крохмаль (переважно, кукурудзяний, картопляний крохмаль або тапіокі), альгінова кислота і певні складні силікати, разом із зв'язувальними для гранулювання, такими як полівінілпіролідон, сахароза, желатин і гуміарабік. Додатково, в цілях таблетування, можуть бути використані лубриканти, такі як стеарат магнію, лаурилсульфат натрію і тальк. Тверді композиції схожого типу можна також застосовувати як наповнювачі в желатинових капсулах; переважні матеріали, пов'язані з вказаним застосуванням, також включають лактозу або молочний цукор, а також поліетиленгліколі з високою молекулярною масою. Якщо для перорального введення бажана водна суспензія і/або еліксир, активні інгредієнти можна комбінувати разом з підсолоджувальними або смаковими агентами, фарбувальною речовиною і, при бажанні, емульгуючими і/або суспендувальними агентами, разом з такими розріджувачами як вода, етанол, пропіленгліколь, гліцерин і їх різні комбінації. 15 UA 104274 C2 5 10 15 20 25 30 35 40 45 50 55 Дозована форма може бути призначена для негайного вивільнення, контрольованого вивільнення, тривалого вивільнення, відстроченого вивільнення або цільового відстроченого вивільнення. Визначення вказаних термінів відомі фахівцям в даній галузі. Крім того, на профіль вивільнення дозованої форми може впливати склад полімерної суміші, склад матриксу з покриттям, склад з множини частинок, склад з множини частинок з покриттям, склад на основі іонообмінної смоли, осмотичний склад або склад біоруйнованих полімерів. Поза зв'язком з будь-якою теорією, вважається, що на вивільнення може бути наданий вплив за рахунок сприятливої дифузії, розчинення, ерозії, іонного обміну, осмосу або їх комбінації. У випадку парентерального введення можна використовувати розчин активної сполуки або в кунжутній або в арахісовій олії або у водному розчині пропіленгліколю. Водний розчин повинен бути відповідним чином забуферений (переважно, рН більше 8), і, при необхідності, рідкий розріджувач спочатку приводять в ізотонічний стан. Водні розчини придатні для внутрішньовенного введення. Отримання всіх таких розчинів при стерильних умовах легко здійснювати звичайними фармацевтичними технологічними способами, добре відомими фахівцям в даній галузі. Наступні приклади далі описують і дозволяють звичайному фахівцеві в даній галузі застосовувати і використовувати винахід. Однак потрібно розуміти, що дані варіанти здійснення представлені з метою ілюстрації винаходу і не повинні розглядатися як такі, що обмежують обсяг вимог винаходу, який визначений формулою винаходу. Короткий опис малюнків На фігурі 1 показана оцінка ушкодження шлунку, виміряна у щурів, що піддаються лікуванню носієм, диклофенаком і двом похідним диклофенаку даного винаходу, сполукою II і сполукою XVII. На фігурі 2 показана кількість шлункового простагландину Е2 (PGE2), продукована у щурів, що піддаються лікуванню носієм, диклофенаком, сполукою II і сполукою XVII. На фігурі 3 показана оцінка ушкодження шлунку, виміряна у щурів, що піддаються лікуванню носієм, напроксеном, і двома похідними напроксену за даним винаходом, сполукою V і сполукою XX. На фігурі 4 показана кількість тромбоксану В2, що синтезується в крові щурів, які згадуються на фігурі 3. На фігурі 5 показана загальна довжина виразок тонкого кишечника у щурів, що піддаються лікуванню носієм, диклофенаком і сполукою II. На фігурі 6 показаний процент гематокриту у щурів до і після лікування носієм, диклофенаком і сполукою II. На фігурі 7 показана кількість ексудативного PGE 2, продукованого в підшкірній кишені щурів, при використанні аналізу повітряної кишені у щурів, що піддаються лікуванню носієм, диклофенаком, сполукою II і сполукою XVII. На фігурі 8 показана кількість тромбоксану B2 (TXB2) цільної крові у щурів, представлених на фігурі 7. На фігурі 9 показане інгібування збільшення об'єму лапи у щурів, що піддаються лікуванню носієм, диклофенаком і сполукою II. На фігурі 10 показане інгібування збільшення об'єму лапи у щурів, що піддаються лікуванню носієм, диклофенаком і сполукою XVII. На фігурі 11 показана кількість ексудативного PGE2, продукованого в підшкірній кишені щурів, при використанні аналізу повітряної кишені у щурів, що піддаються лікуванню носієм, напроксеном, сполукою V і сполукою XX. На фігурі 12 показаний синтез тромбоксану (нг/мл) в крові людини (in vitro) як функція від концентрації індометацину, сполуки IV і сполуки XIX. 2 На фігурі 13 показана площа поверхні в мм виразок шлунку у щурів на наступний день після лікування протягом одного тижня носієм, диклофенаком, сполукою XVII і сполукою XX. На фігурі 14 показане підвищення систолічного кров'яного тиску (мм ртутного стовпа) у щурів, що піддаються лікуванню носієм, диклофенаком, сполукою II, напроксеном і сполукою XX. На фігурі 15 показана концентрація сірководню в плазмі при пероральному лікуванні щурів 50 мкмоль/кг сполукою II. На фігурі 16 показана кількість сірководню, що отримується зі сполуки II і сполуки XVII при інкубації в буфері і гомогенаті печінки. Детальний опис винаходу Отримання сполук 16 UA 104274 C2 5 10 15 20 25 30 35 40 45 Проводили тонкошарову хроматографію на пластинках Macherey-Nagel силікагелю 50 з флуоресцентним індикатором, і пластинки виявляли УФ світлом (254 нм). Для колонкової хроматографії використовували Kieselgel 60. Всі реагенти для синтезу придбали від AldrichSigma Chemical Company і використовували без очищення. Розчинники були аналітичної міри чистоти або вище і були використані в тому вигляді, як отримувались. Роторний випарник Buchi R-114 використовували для видалення розчинників у вакуумі. Структури сполук підтверджували 1 13 спектроскопічно протонним Н-ЯМР і С-ЯМР. Спектри реєстрували при використанні приладу Varian Mercury Plus 400. Хімічні зсуви співвідносили з Me 4Si як внутрішнімстандартом. Масаспектри синтезованих продуктів отримували на мас-спектрометрі Applied Biosystem API 2000. Точку плавлення вимірювали на апараті Buchi В-540. Чистоту кінцевої сполуки визначали при використанні зворотно-фазовою ВЕРХ. Колонку приєднували до інжектору Rheodyne model 7725, ВЕРХ системи Waters 600, настроюваного детектору абсорбції Waters 486, встановленого на 215 або 235 нм, і самописця Waters 746. Синтезовані сполуки показали задовільні результати елементного аналізу; якщо аналізи представлені виключно символами елементів, то результати лежать в діапазоні ±0,4% від теоретичних величин. Приклад 1 Синтез 4-(5-тіоксо-5Н-[1,2]дитіол-3-іл)фенілового складного ефіру [2-(2,6дихлорфеніламіно)феніл]оцтової кислоти (Сполука II) Схема 1 Синтез 5-п-гідроксифеніл-1,2-дитіол-3-тіону (2; ADT-OH) Анезол (31 г, 0,21 моль) і сірку (44,8 г, 1,40 моль) нагрівали в N,N-диметилформаміді (250 мл) протягом 8 годин; після видалення розчинника залишок практично повністю був розчинний в толуолі. Спроба екстрагувати розчин в толуолі 2н-водним гідроксидом натрію призвела до оранжевого твердого осаду (8,5 г, т. пл. вище 300 °C). Вказаний продукт розчиняли в киплячій воді і, після додавання соляної кислоти, отримували 2 у вигляді оранжевого осаду (6,2 г, вихід 13%) т.пл. 188-189 °C. 1 Н-ЯМР (ДМСО-d6) δ: 6,86 (д, 2H), 7,68 (с, 1H), 7,75 (д, 2H), 10,51 (с, -ОН); MS (ESI), m/z 225(M-). Синтез 4-(5-тіоксо-5Н-[1,2]дитіол-3-іл)фенілового складного ефіру [2-(2,6дихлорфеніламіно)феніл]оцтової кислоти (3) До розчину 1 (диклофенак, 890 мг, 3,0 ммоль) в 50 мл N,N-диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (445 мг, 3,3 ммоль) і DCC (дициклогексилкарбодіімід) (680 мг, 3,3 ммоль). До реакційної суміші додавали 5-пгідроксифеніл-1,2-дитіол-3-тіон (2; 678 г, 3 ммоль) і перемішували протягом 1 год при 0 °C і 3 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску і Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, сушили над безводним MgSO4, фільтрували і випарювали розчинник. Неочищений продукт 3 завантажували на відкриту колонку з силікагелем і елюювали CH2Cl2/MeOH (9/1), з отриманням 4-(5-тіоксо-5Н-[1,2]дитіол-3-іл)фенілового складного ефіру [2(2,6-дихлорфеніламіно)феніл]оцтової кислоти (1,1 г, 74% вихід). 1 Н-ЯМР (ДМСО-d6): δ 4,12 (с, 2H), 6,21 (д, 1H), 6,87 (т, 1H), 7,14 (т, 1H), 7,19 (д, 1H), 7,22 (т, 1H), 7,34 (д, 2H), 7,54 (д, 2H), 7,80 (с, 1H), 7,97 (д, 2H); 13 С-ЯМР (ДМСО-d6): δ 37,4, 116,1, 121,0, 122,3, 123,5, 123,7, 127,0, 128,7, 129,3, 129,8, 132,0, 132,2, 136,4, 137,7, 143,8, 154,2, 170,3, 173,3, 213,2. + MS (EI), м/е 504 (М ) т.пл.: 83-86 °C Приклад 2 17 UA 104274 C2 Синтез 4-тіокарбамоїлфенілового складного ефіру [2-(2,6-дихлорфеніламіно)феніл]оцтової кислоти (Сполука XVII) 5 10 15 20 25 30 Схема 2 Синтез 4-карбамоїлфеніл 2-[2-(2,6-дихлорфеніламіно)феніл]ацетату (5) До розчину 1 (диклофенак, 890 мг, 3,0 ммоль) в 50 мл N,N-диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (445 мг, 3,3 ммоль) і DCC (680 мг, 3,3 ммоль). До реакційної суміші додавали 4-гідроксибензамід (4, 616, мг, 4,5 ммоль) і перемішували протягом 1 год при 0 °C і 3 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску і отриманий таким чином маслянистий залишок розчиняли в хлороформі; органічний шар промивали розсолом, сушили над безводним MgSO 4, фільтрували і випарювали розчинник. Неочищений продукт 5 завантажували на відкриту колонку з силікагелем і елюювали CH2Cl2/MeOH (9/1) з отриманням 4-карбамоїлфеніл [2-(2,6дихлорфеніламіно)феніл]ацетату (5) (212 мг, 17% вихід). Синтез 4-тіокарбамоїлфенілового складного ефіру [2-(2,6-дихлорфеніламіно)феніл]оцтової кислоти (6) 4-карбамоїлфеніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетат (5, 480 мг, 1,14 ммоль) і реагент Лавессона (460 мг, 1,14 ммоль) розчиняли в 20 мл безводного бензолу. Реакційну суміш нагрівали до 50 °C і перемішували протягом 6 год. Розчинник видаляли при зниженому тиску; неочищений залишок очищували на колонці з силікагелем (дихлорметан/метиловий спирт 9,5/0,5) з отриманням чистої сполуки 6 (446 мг, 91% вихід). 1 Н-ЯМР (CDCl3): δ 4,07 (с, 2Н), 6,59 (д, 1H), 6,67 (с, 1H), 6,98 (т, 1H), 7,14 (т, 1Н), 7,19 (д, 1H), 7,28 (т, 1H), 7,33 (д, 2Н), 7,63 (с, 1H), 7,97 (д, 2H); 13 С-ЯМР (ДМСО-d6): δ 38,8, 118,8, 121,8, 122,6, 123,7, 124,4, 128,7, 129,1, 129,6, 131,2, 137,2, 137,8, 142,9, 153,5, 170,5, 193,2, 201,7. + MS (EI), м/е 431 (М ) т.пл.: 170-172 °C Приклад 3 Синтез 4-(5-тіоксо-5Н-[1,2]дитіол-3-іл)фенілового складного ефіру [2-(2-хлор-6фторфеніламіно)феніл]оцтової кислоти (Сполука III) Схема 1 Синтез метилфеніл)ацетату (3) 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл-2-(2-(2-хлор-6-фторфеніламіно)-5 18 UA 104274 C2 5 10 15 20 25 30 35 40 До розчину 1 (люміракоксиб, 600 мг, 2,03 ммоль) в 40 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (301 мг, 2,23 ммоль) і DCC (459 мг, 2,23 ммоль). До реакційної суміші додавали 5-п-гідроксифеніл-1,2-дитіол-3-тіон (2, 504 мг, 2,23 ммоль) і перемішували протягом 1 год при 0 °C і 3 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, сушили над безводним MgSO 4, фільтрували і випарювали розчинник. Неочищений продукт 3 завантажували на відкриту колонку з силікагелем і елюювали CH 2Cl2 з отриманням 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл-2-(2-(2-хлор-6-фторфеніламіно)5метилфеніл)ацетату (3) (299 мг, 37% вихід). 1 Н-ЯМР (ДМСО): δ 2,32 (с, 3Н), 4,02 (с, 2H), 6,41 (с, 1H), 6,71 (д, 1H), 6,93 (т, 1H), 6,95 (д, 2H), 7,14 (д, 1H), 7,19 (д, 2H), 7,39 (с, 1H), 7,66 (д, 2H); 13 С-ЯМР (ДМСО): δ 20,8, 38,7, 115,2, 119,2, 122,5, 123,2, 124,0, 126,1, 127,2, 129,3, 130,3, 131,7, 132,2, 133,6, 136,4, 140,3, 153,7, 154,4, 156,8, 170,3, 171,6,215,7. + MS (EI), м/е 503 (М ) т.пл.: 131-133 °C Приклад 4 Синтез 4-тіокарбамоїлфеніл 2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетату (Сполука XVIII) Схема 2 Синтез 4-карбамоїлфеніл 2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетату (5) До розчину 1 (люміракоксиб, 223 мг, 0,75 ммоль) в 15 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (111 мг, 0,825 ммоль) і DCC (170 мг, 0,825 ммоль). До реакційної суміші додавали 4-гідроксибензамід (4, 154 мг, 1,125 ммоль) і перемішували протягом 1 год при 0 °C і 3 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в хлороформі; органічний шар промивали розсолом, сушили над безводним MgSO4, фільтрували і випарювали розчинник. Неочищений продукт 5 завантажували на відкриту колонку з силікагелем і елюювали CH 2Cl2/MeOH (9/1) з отриманням 4-карбамоїлфеніл-2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетату (5) (111 мг, 35% вихід). Синтез 4-тіокарбамоїлфеніл 2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетату (6) Розчиняли 4-карбамоїлфеніл 2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетат, 5 (110 мг, 0,27 ммоль) і реагент Лавессона (109 мг, 0,27 ммоль) в 15 мл безводного бензолу. Реакційну суміш нагрівали до 60 °C і перемішували протягом 3 год. Розчинник видаляли при зниженому тиску; неочищений залишок очищували на колонці з силікагелем (дихлорметан/метиловий спирт 9,5/0,5) з отриманням чистої сполуки 6 (59 мг, 51% вихід). 1 Н-ЯМР (CDCl3): δ 2,32 (с, 3Н), 4,01 (с, 2H), 6,46 (с, 1H), 6,70 (д, 1H), 6,92 (т, 1H), 7,01 (д, 2H), 7,11 (д, 2H), 7,19 (д, 1H), 7,62 (з, NH), 7,84 (д, 2H); 13 С-ЯМР (ДМСО-d6): δ 20,8, 30,7, 115,1, 119,2, 122,0, 122,3, 124,1, 124,9, 126,1, 128,2, 129,2, 132,3, 134,8, 138,6, 140,9, 153,7, 154,6, 156,2, 170,4, 201,7. 19 UA 104274 C2 + MS (EI), м/е 429 (М ) т.пл.: 120-122 °C. Приклад 5 Синтез 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-ацетоксибензоату (Сполука I) 5 10 15 20 25 30 35 Схема 1 Синтез 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-ацетоксибензоату (3) До розчину 1 (ацетилсаліцилова кислота, 416 мг, 2,31 ммоль) в 40 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (343 мг, 2,54 ммоль) і DCC (523 мг, 2,54 ммоль). До реакційної суміші додавали 5-п-гідроксифеніл-1,2-дитіол-3-тіон (2, 574 мг, 2,54 ммоль) і перемішували протягом 1 год при 0 °C і 3 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, сушили над безводним MgSO 4, фільтрували і випарювали розчинник. Неочищений продукт завантажували на відкриту колонку з силікагелем і елюювали сумішшю етиловий ефір/петролейний ефір (1/1) з отриманням 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2ацетоксибензоату (3) (354 мг, 40% вихід). 1 Н-ЯМР (ДМСО-d6): δ 2,32 (с, 3Н), 7,20 (д, 1H), 7,33 (д, 2H), 7,40 (с, 1H), 7,41 (т, 1H), 7,67 (т, 1H), 7,73 (д, 2H), 8,21 (д, 1H). 13 С-ЯМР (ДМСО-d6): δ 21,3, 122,1, 123,4, 124,4, 126,6, 128,6, 129,7, 132,4, 135,4, 136,4, 151,6, 153,7, 162,6, 169,8, 171,9, 215,7. + MS (EI), м/е 389 (М ) т.пл.: 120-122 °C Приклад 6 Синтез 4-тіокарбамоїлфенілового складного ефіру 2-ацетоксибензойної кислоти (Сполука XVI) Схема 2 Синтез 4-карбамоїлфеніл 2-ацетоксибензоату (5) До розчину 1 (ацетилсаліцилова кислота, 500 мг, 2,77 ммоль) в 15 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (412 мг, 3,05 ммоль) і DCC (628 мг, 3,05 ммоль). До реакційної суміші додавали 4-гідроксибензамід (4, 418 мг, 3,05 ммоль) і перемішували протягом 1 год при 0 °C і 3 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в хлороформі; органічний шар промивали розсолом, сушили над безводним MgSO 4, фільтрували і випарювали розчинник. 20 UA 104274 C2 5 10 15 20 25 30 35 40 Неочищений продукт 5 завантажували на відкриту колонку з силікагелем і елюювали сумішшю CH2Cl2/MeOH (9/1), з отриманням 4-карбамоїлфеніл 2-ацетоксибензоату (5) (410 мг, 47% вихід). Синтез 4-тіокарбамоїлфеніл 2-(2-(2-хлор-6-фторфеніламіно)-5-метилфеніл)ацетату (6) Розчиняли 4-карбамоїлфеніл 2-ацетоксибензоат, 5 (410 мг, 1,37 ммоль) і реагент Лавессона (554 мг, 1,37 ммоль) в 35 мл безводного бензолу. Реакційну суміш нагрівали до 60 °C і перемішували протягом 3 год. Розчинник видаляли при зниженому тиску; неочищений залишок очищували на колонці з силікагелем (дихлорметан/метиловий спирт 9,5/0,5) з отриманням 470 мг неочищеної сполуки 6. Отриману сполуку очищували зворотно-фазовою ВЕРХ, що проводиться при використанні двох систем розчинників: А: 100% ацетонітрил в 0,1% ТФО (TFA), В: 100% H2O в 0,1% ТФО (лінійний градієнт від 10% А до 60% А через 35 хв, УФ-детектування при 254 нм, швидкість потоку 30 мл/хв) з отриманням чистої сполуки 6 (324 мг, 71% вихід). 1 Н-ЯМР (CDCl3): δ 2,30 (с, 3Н), 7,17 (д, 1H), 7,21 (д, 2Н), 7,40 (т, 1H), 7,66 (т, 1Н), 7,94 (д, 2H), 8,2 (д, 1H). 13 С-ЯМР (ДМСО-d6): δ 21,2, 121,9, 122,4, 124,3, 126,4, 128,7, 132,4, 135,1, 137,3, 151,5, 153,7, 162,7, 169,8, 201,8. + MS (EI), м/е 316 (М ) т.пл.: 154-156 °C Приклад 7 Синтез 4-(5-тіоксо-5H-[1,2]дитіол-3-іл)фенілового складного ефіру [1-(4-хлорбензоїл)-5метокси-2-метил-1-Н-індол-3-ілу]оцтової кислоти (Сполука IV) Схема 1 Синтез 4-[4-(5-тіоксо-5H-1,2-дитіол-3-іл)]феніл-2-[1-(4-хлорбензоїл)-5-метокси-2-метиліндол3-іл]ацетату (3) До розчину 1 (індометацин, 720 мг, 2,01 ммоль) в 30 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (301 мг, 2,21 ммоль) і DCC (456 мг, 2,21 ммоль). До реакційної суміші додавали 5-п-гідроксифеніл-1,2-дитіол-3-тіон (2, 500, мг, 2,21 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, 5% NaHCO3, 10% лимонною кислотою і потім сушили над безводним MgSO4, фільтрували і випарювали розчинник. Неочищений продукт завантажували на відкриту колонку з силікагелем і елюювали сумішшю дихлорметан/метиловий спирт (98/2) з отриманням 4-[4-(5-тіоксо-5H-1,2-дитіол-3-іл)]феніл-2-[1-(4-хлорбензоїл)-5-метокси-2-метиліндол-3-іл]ацетату (3) (257 мг, 23% вихід). 1 Н-ЯМР (CDCl3): δ 2,47 (с, 3Н), 3,84 (с, 3Н, ОСН3), 3,93 (с, 2H), 6,70 (д, 1H), 6,88 (д, 1H), 7,04 (с, 1H), 7,21 (д, 2H), 7,37 (с, 1H) 7,48 (д, 2Н), 7,65 (д, 2H), 7,67 (д, 2H) 13 С-ЯМР (ДМСО-d6): δ 13,6, 30,8, 56,0, 101,5, 111,6, 111,9, 115,3, 122,9, 128,4, 129,4, 129,6, 130,6, 131,1, 131,4, 133,9, 136,3, 136,6, 139,7, 153,8, 156,4, 167,5, 168,9, 170,4, 215,7. + MS (EI), м/е 567 (М ) т.пл.: 90-92 °C Приклад 8 Синтез 4-тіокарбамоїлфенілового складного ефіру [1-(4-хлорбензоїл)-5-метокси-2-метил-1Н-індол-3-ілу]оцтової кислоти (Сполука XIX) 21 UA 104274 C2 5 10 15 20 25 30 Схема 2 Синтез 4-карбамоїлфеніл-2-[1-(4-хлорбензоїл)-5-метокси-2-метиліндол-3-іл]ацетату (5) До розчину 1 (індометацин, 3 г, 8,38 ммоль) в 60 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (1,25 г, 9,22 ммоль) і DCC (1,9 г, 9,22 ммоль). До реакційної суміші додавали 4-гідроксибензамід (4, 1,72 г, 12,6 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, 5% NaHCO3, 10% лимонною кислотою і потім сушили над безводним MgSO4, фільтрували і випарювали розчинник. Неочищений продукт 5 завантажували на відкриту колонку з силікагелем і елюювали сумішшю CH2Cl2/MeOH (9,5/0,5) з отриманням 4-карбамоїлфеніл-2-[1(4-хлорбензоїл)-5-метокси-2-метиліндол-3-іл]ацетату (5) (479 мг, 12% вихід). Синтез 4-тіокарбамоїлфеніл-2-[1-(4-хлорбензоїл)-5-метокси-2-метиліндол-3-іл]ацетату (6) Розчиняли 4-карбамоїлфеніл-2-[1-(4-хлорбензоїл)-5-метокси-2-метиліндол-3-іл]ацетат, 5 (340 мг, 0,71 ммоль) і реагент Лавессона (287 мг, 0,71 ммоль) в 15 мл безводного бензолу. Реакційну суміш нагрівали до 60 °C і перемішували протягом 4 год. Розчинник видаляли при зниженому тиску; неочищений залишок очищували на колонці з силікагелем (дихлорметан/метиловий спирт 9,5/0,5) з отриманням 178 мг неочищеної сполуки 6. Отриману сполуку очищували зворотно-фазовою ВЕРХ, що проводиться при використанні двох систем розчинників: А: 100% ацетонітрил в 0,1% ТФО (TFA), В: 100% H 2O в 0,1% ТФО (лінійний градієнт від 10% А до 80% А через 30 хв, УФ-детектування при 254 нм, швидкість потоку 30 мл/хв) з отриманням чистої сполуки 6 (56 мг, 16% вихід). 1 Н-ЯМР (CDCl3): δ 2,45 (с, 3Н), 3,83 (с, 3Н, ОСН3), 3,91 (с, 2Н), 6,70 (д, 1H), 6,88 (д, 1H), 7,04 (с, 1H), 7,11 (д, 2H), 7,47 (д, 2H), 7,67 (д, 2H), 7,88 (д, 2H). 13 С-ЯМР (ДМСО-d6): δ 13,6, 30,8, 56,0, 101,5, 111,9, 112,0, 115,3, 121,7, 128,6, 129,4, 130,8, 131,2, 131,4, 134,0, 136,8, 137,1, 139,7, 156,2, 157,9, 167,6, 169,8, 201,8. + MS (EI), м/е 493 (М ) т.пл.: 224-226 °C Приклад 9 Синтез 4-(5-тіоксо-5Н-[1,2]дитіол-3-іл)фенілового складного ефіру 2-(6-метоксинафталін-2іл)пропіонової кислоти (Сполука V) 22 UA 104274 C2 5 10 15 20 25 30 35 40 45 Схема 1 Синтез 4-(5-тіоксо-5Н-1,2-дитіол-3-іл)феніл 2-(2-метоксинафталін-6-іл)пропіонату (3) До розчину 1 (напроксен, 595 мг, 2,58 ммоль) в 20 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (388 мг, 2,87 ммоль) і DCC (593 мг, 2,87 ммоль). До реакційної суміші додавали 5-п-гідроксифеніл-1,2-дитіол-3-тіон (2, 650, мг, 2,87 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, 5% NaHCO3, 10% лимонною кислотою і потім сушили над безводним MgSO4, фільтрували і випарювали розчинник. Неочищений продукт завантажували на відкриту колонку з силікагелем і елюювали дихлорметаном з отриманням 4-(5-тіоксо-5Н-1,2-дитіол-3іл)феніл 2-(2-метоксинафталін-6-іл) пропіонату (3) (406 мг, 36% вихід). 1 Н-ЯМР (ДМСО-d6): δ 1,59 (д, 3Н), 3,86 (с, 3Н, ОСН3), 4,24 (дд, 1H), 7,18 (д, 1Н), 7,22 (д, 2Н), 7,31 (с, 1Н), 7,50 (д, 1Н), 7,77 (с, 1H) 7,85 (д, 1H), 7,86 (с, 1H), 7,87 (д, 1H), 7,91 (д, 2H). 13 С-ЯМР (ДМСО-d6): δ 19,1, 45,2, 55,9, 106,5, 119,6, 123,5, 126,6, 126,9, 128,0, 129,2, 129,4, 129,5, 129,6, 129,9, 134,2, 135,6, 136,5, 154,2, 158,1, 173,2,216,2. + MS (EI), м/е 439 (М ) т.пл.: 111-113 °C Приклад 10 Синтез 4-тіокарбамоїлфенілового складного ефіру 2-(6-метоксинафталін-2-іл)пропіонової кислоти (Сполука XX) Схема 2 Синтез 4-карбамоїлфеніл 2-(2-метоксинафталін-6-іл)пропіонату (5) До розчину 1 (напроксен, 4 г, 17,4 ммоль) в 80 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (2,59 г, 19,14 ммоль) і DCC (2,59 г, 19,14 ммоль). До реакційної суміші додавали 4-гідроксибензамід (4, 3,58 г, 26,1 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, 5% NaHCO3, 10% лимонною кислотою і потім сушили над безводним MgSO 4, фільтрували і випарювали розчинник. Неочищений продукт 5 завантажували на відкриту колонку з силікагелем і елюювали сумішшю CH2Cl2/MeOH (9,5/0,5), з отриманням 4-карбамоїлфеніл-2-(2метоксинафталін-6-іл)пропіонату (5) (1,91 г, 32% вихід). Синтез 4-тіокарбамоїлфеніл 2-(2-метоксинафталін-6-іл)пропіонату (6) Розчиняли 4-карбамоїлфеніл-2-(2-метоксинафталін-6-ілу)пропіонат 5 (1,80 г, 4,34 ммоль) і реагент Лавессона (1,75 г, 4,34 ммоль) в 130 мл безводного бензолу. Реакційну суміш нагрівали до 60 °C і перемішували протягом 4 год. Розчинник видаляли при зниженому тиску; неочищений залишок очищували на колонці з силікагелем (дихлорметан/метиловий спирт 9,75/0,25) з отриманням 2,9 г неочищеної сполуки 6. Отриману сполуку очищували на відкритій колонці з силікагелем і елюювали сумішшю CH 2Cl2/MeOH (9,5/0,5), з отриманням чистої сполуки 6 (970 мг, 61% вихід). 1 Н-ЯМР (ДМСО-d6): δ 1,59 (д, 3Н), 3,86 (с, 3Н, ОСН3), 4,24 (дд, 1H), 7,06 (д, 2Н), 7,18 (д, 1Н), 7,31 (с, 1Н), 7,50 (д, 1Н), 7,84 (с, 1H) 7,85 (д, 1H), 7,86 (с, 1H), 7,89 (д, 2H), 9,47 і 9,84 (с, 2H, NH2). 13 С-ЯМР (ДМСО-d6): δ 19,1, 45,2, 55,9, 106,5, 119,6, 121,6, 126,6, 126,9, 128,0, 129,4, 129,9, 134,2, 135,6, 137,8, 153,4, 158,1, 173,3, 199,7. + MS (EI), м/е 366 (М ) 23 UA 104274 C2 т.пл.: 196-198 °C Приклад 11 Синтез 4-тіокарбамоїлфеніл 2-(4-ізобутилфеніл)пропіонату (Сполука XXIX) 5 10 15 20 25 30 До розчину 1 (ібупрофен, 3,87 г, 18,8 ммоль) в 80 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (2,8 г, 20,7 ммоль) і DCC (4,27 г, 20,7 ммоль). До реакційної суміші додавали 4-гідроксибензамід (2, 3,9 г, 28 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, 5% NaHCO3, 10% лимонною кислотою і потім сушили над безводним MgSO4, фільтрували і випарювали розчинник. Неочищений продукт 3 завантажували на відкриту колонку з силікагелем і елюювали сумішшю CH 2Cl2/MeOH (9,5/0,5) з отриманням 4-карбамоїлфеніл-2-(4ізобутилфеніл)пропіонату (3) (2,48 г, 40% вихід). Синтез 4-тіокарбамоїлфеніл 2-(4-ізобутилфеніл)пропіонату (4) Розчиняли 4-карбамоїлфеніл 2-(4-ізобутилфеніл)пропіонат 3 (2,48 г, 7,62 ммоль) і реагент Лавессона (3,1 г, 7,62 ммоль) в 130 мл безводного бензолу. Реакційну суміш нагрівали до 60 °C і перемішували протягом 4 год. Розчинник видаляли при зниженому тиску. Неочищену сполуку очищували на відкритій колонці з силікагелем сумішшю CH 2Cl2/MeOH (9,5/0,5) з отриманням чистої сполуки 4 (1,45 г, 55% вихід). 1 Н-ЯМР (ДМСО-d6): δ 0,84 (д, 6H), 1,48 (д, 3H), 1,79-1,82 (м, 1H), 2,42 (д, 2H), 4,05 (дд, 1H), 7,05 (д, 2H), 7,15 (д, 2H), 7,28 (д, 2H) 7,88 (д, 2H), 9,49 і 9,87 (с, 2H, NH 2). 13 С-ЯМР (ДМСО-d6): δ 19,2, 22,9, 30,3, 44,9, 121,6, 127,9, 129,5, 130,0, 137,8, 138,0, 140,8, 153,3, 173,3, 199,6. + MS (EI), м/е 341 (М ) т.пл.: 121-123 °C Приклад 12 Синтез 4-тіокарбамоїлфеніл 2-(4-оксофеніл)фенілпропіонату (Сполука XXX) Синтез 4-карбамоїлфеніл 2-(4-оксофеніл)фенілпропіонату (3) 24 UA 104274 C2 5 10 15 20 25 30 35 40 45 До розчину 1 (кетопрофен, 3 г, 11,8 ммоль) в 80 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (1,76 г, 13 ммоль) і DCC (2,68 г, 13 ммоль). До реакційної суміші додавали 4-гідроксибензамід (2, 2,43 г, 17,7 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, 5% NaHCO3, 10% лимонною кислотою і потім сушили над безводним MgSO 4, фільтрували і випарювали розчинник. Неочищений продукт 3 завантажували на відкриту колонку з силікагелем і елюювали сумішшю CH2Cl2/MeOH (9,5/0,5) з отриманням 4-карбамоїлфеніл 2-(4оксофеніл)фенілпропіонату (3) (1,84 г, 42% вихід). Синтез 4-тіокарбамоїлфеніл 2-(4-оксофеніл)фенілпропіонату (4) Розчиняли 4-карбамоїлфеніл 2-(4-оксофеніл)фенілпропіонат (3) (1,84 г, 4,93 ммоль) і реагент Лавессона (2 г, 4,93 ммоль) в 100 мл безводного бензолу. Реакційну суміш нагрівали до 60 °C і перемішували протягом 4 год. Розчинник видаляли при зниженому тиску. Отриману сполуку очищували на відкритій колонці з силікагелем і елюювали сумішшю CH 2Cl2/MeOH (9,5/0,5) з отриманням чистої сполуки 4 (0,45 г, 23% вихід). 1 Н-ЯМР (ДМСО-d6): δ 1,53 (д, 3H), 4,25 (дд, 1H), 7,08 (д, 2H), 7,54-7,73 (м, 9H), 7,90 (д, 2H), 9,51 і 9,88 (с, 2H, NH2). 13 С-ЯМР (ДМСО-d6): δ 19,2, 44,9, 121,6, 129,3, 129,5, 129,8, 130,3, 132,6, 133,5, 137,6, 137,9, 138,1, 141,2, 153,3, 154,5, 156,1, 163,8, 172,9, 199,6. + MS (EI), м/е 390 (М ) т.пл.: 114-116 °C Приклад 13 Синтез 4-тіокарбамоїлфеніл 2-(3-фтор,4-феніл)фенілпропіонату (Сполука XXXI) Синтез 4-карбамоїлфеніл 2-(3-фтор,4-феніл)фенілпропіонату (3) До розчину 1 (флурбіпрофен, 2 г, 8,2 ммоль) в 80 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (1,22 г, 9,02 ммоль) і DCC (1,86 г, 9,02 ммоль). До реакційної суміші додавали 4-гідроксибензамід (2, 1,7 г, 12,2 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті; органічний шар промивали розсолом, 5% NaHCO3, 10% лимонною кислотою і потім сушили над безводним MgSO 4, фільтрували і випарювали розчинник. Неочищений продукт 3 завантажували на відкриту колонку з силікагелем і елюювали сумішшю CH2Cl2/MeOH (9,5/0,5) з отриманням 4-карбамоїлфеніл 2-(3фтор,4-феніл)фенілпропіонату (3) (1,09 г, 37% вихід). Синтез 4-тіокарбамоїлфеніл 2-(3-фтор,4-феніл)фенілпропіонату (4) Розчиняли 4-карбамоїлфеніл 2-(3-фтор,4-феніл)фенілпропіонат 3 (1,09 г, 3 ммоль) і реагент Лавессона (1,21 г, 3 ммоль) в 70 мл безводного бензолу. Реакційну суміш нагрівали до 60 °C і перемішували протягом 4 год. Розчинник видаляли при зниженому тиску. Отриману сполуку очищували на відкритій колонці з силікагелем і елюювали сумішшю CH 2Cl2/MeOH (9,5/0,5) з отриманням чистої сполуки 4 (0,35 г, 31% вихід). 1 Н-ЯМР (ДМСО-d6): δ 1,55 (д, 3Н), 4,21 (дд, 1H), 7,32-7,55 (м, 8H), 7,90 (д, 2H), 9,51 і 9,88 (с, 2H, NH2). 13 С-ЯМР (ДМСО-d6): δ 19,1, 44,7, 115,9, 116,2, 121,7, 124,8, 128,6, 129,3, 129,4, 129,5, 131,7, 135,8, 137,7, 142,6, 153,7, 158,3, 163,5, 173,1, 199,6. + MS (EI), м/е 380 (М ) 25 UA 104274 C2 т.пл.: 142-144 °C Приклад 14 Синтез 4-(ізотіоціано)феніл 2-(2-метоксинафталін-6-іл)пропіонату (Сполука XXV) 5 10 15 20 25 30 35 40 До розчину 1 (напроксен, 691 мг, 3 ммоль) в 20 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (446 мг, 3,3 ммоль) і DCC (619 мг, 3,3 ммоль). До реакційної суміші додавали 4-гідроксифенілізотіоціанат (2, 500 мг, 3,3 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті і видаляли осад. Випарювали розчинник і неочищений продукт завантажували на відкриту колонку з силікагелем і елюювали дихлорметаном з отриманням 4-(ізотіоціано)феніл 2-(2-метоксинафталін-6-іл)пропіонату (3) (230 мг, 21% вихід). 1 Н-ЯМР (ДМСО-d6): δ 1,57 (д, 3Н), 3,86 (с, 3Н, ОСН3), 4,20 (дд, 1H), 7,10 (д, 2Н), 7,15 (д, 1H), 7,29 (с, 1H), 7,43 (д, 2H), 7,48 (д, 1H), 7,78 (д, 1H), 7,80 (с, 1H), 7,83 (д, 1H). 13 С-ЯМР (ДМСО-d6): δ 19,1, 45,2, 55,9, 106,5, 119,6, 123,8, 126,6, 126,9, 128,0, 128,3, 129,2, 129,9, 134,2, 134,6, 135,7, 150,2, 158,1, 173,2, 215,1. + т.пл.: 66-68 °C; MS (EI), м/е 364 (М ) Приклад 15 Синтез 4-ізотіоціанатофеніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетату (Сполука XXII) 4-ізотіоціанатофеніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетат (3) До розчину 1 (диклофенак, 1717 мг, 5,8 ммоль) в 60 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол (862 мг, 6,38 ммоль) і DCC (1316 мг, 6,38 ммоль). До реакційної суміші додавали 4-гідроксифенілізотіоціанат (2, 965 мг, 6,38 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий такимчином маслянистий залишок розчиняли в етилацетаті і видаляли осад. Випарювали розчинник і неочищений продукт завантажували на відкриту колонку з силікагелем і елюювали сумішшю хлороформ/н-гексан в співвідношенні 9:1 з отриманням 4ізотіоціанатофеніл 2-(2-(2,6-дихлорфеніламіно)феніл)ацетату (3) (580 мг, 23% вихід). 1 Н-ЯМР (ДМСО-d6): δ 4,09 (с, 2H), 6,19 (д, 1H), 6,83 (т, 1H), 7,05 (т, 1H), 7,14 (шир.с, 1H, NH), 7,21 (д, 2H), 7,25 (д, 2H), 7,47-7,54 (м, 3H). 13 С-ЯМР (ДМСО-d6): δ 37,4, 116,1, 121,0, 122,7, 124,0, 127,1, 127,8, 128,3, 128,7, 129,8, 132,0, 132,2, 137,7, 144,0, 150,3, 170,5, 215,1. + т.пл.: 132-134 °C; MS (EI), м/е 364 (М ) Приклад 16 Синтез 4-ізотіоціанатофеніл 2-ацетоксибензоату (Сполука XXI) 4-ізотіоціанатофеніл 2-ацетоксибензоат (3) 26 UA 104274 C2 5 10 15 20 25 30 35 40 45 50 55 60 До розчину 1 (аспірин, 1200 мг, 6,67 ммоль) в 60 мл диметилформаміду додавали при перемішуванні при 0 °C протягом 1 год гідроксибензотриазол 992 мг, 7,34 ммоль) і DCC (1520 мг, 7,34 ммоль). До реакційної суміші додавали 4-гідроксифенілізотіоціанат (2, 1109 мг, 7,34 ммоль) і перемішували протягом 1 год при 0 °C і 2 год при кімнатній температурі. Після фільтрування фільтрат випарювали при зниженому тиску для видалення розчинника. Отриманий таким чином маслянистий залишок розчиняли в етилацетаті і видаляли осад. Випарювали розчинник і неочищений продукт завантажували на відкриту колонку з силікагелем і елюювали сумішшю хлороформ/н-гексан в співвідношенні 6:4, з отриманням 4ізотіоціанатофеніл 2-ацетоксибензоату (3) (150 мг, 7% вихід). 1 Н-ЯМР (CDCl3): δ 2,31 (с, 3Н), 7,17 (д, 1H), 7,19 (д, 2H), 7,29 (д, 2H), 7,38 (т, 1H), 7,66 (т, 1H), 8,20 (д, 1H). 13 С-ЯМР (CDCl3): δ 21,3, 122,2, 123,3, 124,4, 126,6, 127,2, 129,4, 132,4, 135,2, 149,3, 151,5, 163,0, 170,0, 215,1. + т.пл.: 84-86 °C; MS (EI), м/е 272 (М ) Приклад 17 Безпека сполук даного винаходу відносно шлунково-кишкового тракту Два похідних диклофенаку за даним винаходом, сполуки II і сполуки XVII оцінювали у відношенні їх безпеки для шлунково-кишкового тракту у щурів. Зокрема, вимірювали ушкодження шлунку, синтез шлункового PGE2, покриття виразками тонкого кишечнику і гематокрит. Самці щурів Wistar вагою 175-200 г голодували протягом 18 годин до перорального введення 1% карбоксиметилцелюлози (носій; 0,2 мл) самої по собі або з однією з наступних речовин, розчинених у вказаному носії: диклофенак (20 мг/кг), сполуки II (32 мг/кг), ADT-OH (12 мг/кг), диклофенак і ADT-OH, сполуки XVII (27,3 мг/кг) 4-гідрокситіобензамід (TBZ) (7,3 мг/кг), фрагмент, що вивільняє сірководень на сполуці XVII, або диклофенак і TBZ. Дози сполуки II і сполуки XVII являють собою дози, еквімолярні дозі 20 мг/кг диклофенаку. Схожим чином, дози ADT-OH і TBZ являють собою дози, еквімолярні дозам сполуки II і сполуки XVII, відповідно. У кожній групі було по 5 щурів. Через три години після введення сполук щурів, що досліджуються, умертвляли і сліпим способом вимірювали геморагічне ушкодження шлунку (в мм). «Результат ушкодження шлунку» отримували, підсумовуючи довжину всіх пошкоджень в шлунку. З посиланням на фігуру 1, ушкодження шлунку не спостерігалося в групах «носій», «сполука II» або «сполука XVII». Сполуки II і сполуки XVII призводили до значною мірою меншого пошкодженню шлунку в порівнянні з диклофенаком. Більш того, щадна відносно шлунку дія не спостерігалася при окремому введенні NSAID фрагмента (диклофенак) і H 2Sвивільняючого фрагмента сполуки II і сполуки XVII (ADT-OH і TBZ, відповідно), але тільки в один і той же час. Вказані спостереження були підтверджені при подальшому сліпому гістологічному аналізі. Зразки (100-200) тканини шлунку досліджували для вимірювання синтезу простагландину E2 (PGE2), як детально описано раніше (Wallace et al., Cyclooxygenase 1 contribute to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology 1998; 115: 101109, включено тут як посилання). Стисло, зразки тканини подрібнювали ножицями протягом 30 хвилин, потім поміщували в 1 мл буфери натрію фосфату (рН 7,4) і поміщували на водяну баню (37 °C), що струшується, на 20 хв. Безпосередньо після цього зразки центрифугували протягом 1 хв при 9000 g і надосадову рідину негайно заморожували при -80 °C для подальшого вимірювання концентрації PGE2 при використанні специфічного ELISA (Wallace et al., 1998). З посиланням на фігуру 2, можна побачити, що диклофенак (з або без супутнього введення ADT-OH або TBZ), сполуки II і сполуки XVII, всі, значно знижують рівень синтезу шлункового PGE2, що вказує на інгібування COX-1 і/або COX-2. ADT-OH і TBZ самі по собі не знижували синтез шлункового PGE2 в порівнянні з носієм. Таким чином, відсутність ушкодження шлунку у щурів, яким вводили сполуки II і сполуки XVII, як показано на фігурі 1, не пояснювалася змінами в здатності вказаних лікарських засобів пригнічувати синтез шлункового простагландину. Пригнічення синтезу шлункового PGE2 було практично повним при використанні вказаних лікарських препаратів в дозі, еквімолярній дозі використаного диклофенаку. На фігурі 3 видно, що два похідних напроксену даного винаходу (сполука V і XX) викликали значною мірою менше пошкоджень в порівнянні з самим напроксеном. Вказане дослідження проводили абсолютно таким же чином, як показано на фігурі 1. Напроксен, сполука V і сполука XX, кожну, вводили перорально в дозі 60 мкмоль/кг, і через 3 години сліпим методом оцінювали ушкодження шлунку. Ушкодження шлунку не піддавалося виявленню жодного з щурів, яким вводили сполуку V або сполуку XX. Кожна група складалася з 5 щурів. Вказані спостереження підтверджували при подальшому сліпому гістологічному аналізі. 27 UA 104274 C2 5 10 15 20 25 30 35 40 45 50 55 Інгібування COX-1 також вимірювали при використанні тих же щурів. Безпосередньо після збору ексудату з кишені у кожного щура з нижньої порожнистої вени відбирали 1 мл крові і поміщували в скляну пробірку і залишали скипатися протягом 45 хвилин, як описано раніше (Wallace et al. Gastroenterology 1998). Зразки потім центрифугували протягом 3 хв при 9000 g, надосадову рідину заморожували при -80 °C для подальшого вимірювання концентрацій тромбоксану B2 при використанні специфічного ELISA. Як показано на фігурі 4, напроксен, сполука V і сполука XX, всі, значно (*р
ДивитисяДодаткова інформація
Автори англійськоюWallace John L., Sparatore Anna, Santagada Vincenzo, Cirino Giuseppe
Автори російськоюУоллейс Джон Л., Чирино Джузеппе, Сантагада Винченцо, Календо Джузеппе
МПК / Мітки
МПК: A61P 29/00, C07D 339/00, C07C 331/00, C07C 327/00, A61K 31/385, C07D 209/28
Мітки: нестероїдних, протизапальних, засобів, сірководневі, похідні, лікарських
Код посилання
<a href="https://ua.patents.su/42-104274-sirkovodnevi-pokhidni-nesterodnikh-protizapalnikh-likarskikh-zasobiv.html" target="_blank" rel="follow" title="База патентів України">Сірководневі похідні нестероїдних протизапальних лікарських засобів</a>
Застосування композицій нестероїдних протизапальних засобів з кофеїном як препаратів з антиексудативною активністю

Номер патенту: 59396
Опубліковано: 10.05.2011
Автори: Сирова Ганна Олегівна, Бойко Євгеній Павлович, Шаповал Людмила Григоріївна, Наконечна Світлана Анатоліївна, Грабовецька Євгенія Романівна, Вакуленко Наталія Василівна
МПК: A61K 31/00
Мітки: антиексудативною, композицій, протизапальних, засобів, застосування, активністю, препаратів, кофеїном, нестероїдних
Формула / Реферат:
Застосування композицій нестероїдних протизапальних засобів з кофеїном як препаратів з антиексудативною активністю.
Спосіб підсилення анальгетичної дії периферичного генезу нестероїдних протизапальних і протиревматичних засобів, похідних оцтової та пропіонової кислот

Номер патенту: 56451
Опубліковано: 10.01.2011
Автори: Савельєва Олена Валеріївна, Петюніна Валентина Миколаївна, Бойко Євгеній Павлович, Сирова Ганна Олегівна, Бачинський Руслан Орестович
МПК: A61K 31/519
Мітки: нестероїдних, генезу, спосіб, підсилення, анальгетичної, периферичного, дії, засобів, оцтової, пропіонової, протиревматичних, протизапальних, кислот, похідних
Формула / Реферат:
Спосіб підсилення анальгетичної дії периферичного генезу лікарського засобу, що включає приєднання кофеїну, який відрізняється тим, що до нестероїдних протизапальних і протиревматичних засобів, похідних оцтової та пропіонової кислот, взятих із розрахунку на 1 кг ваги споживача, кофеїн приєднують в середньовіковій дозі.
Похідні хіноліну й ізохіноліну, заміщені в 5-положенні, спосіб їх одержання і їх застосування як протизапальних засобів
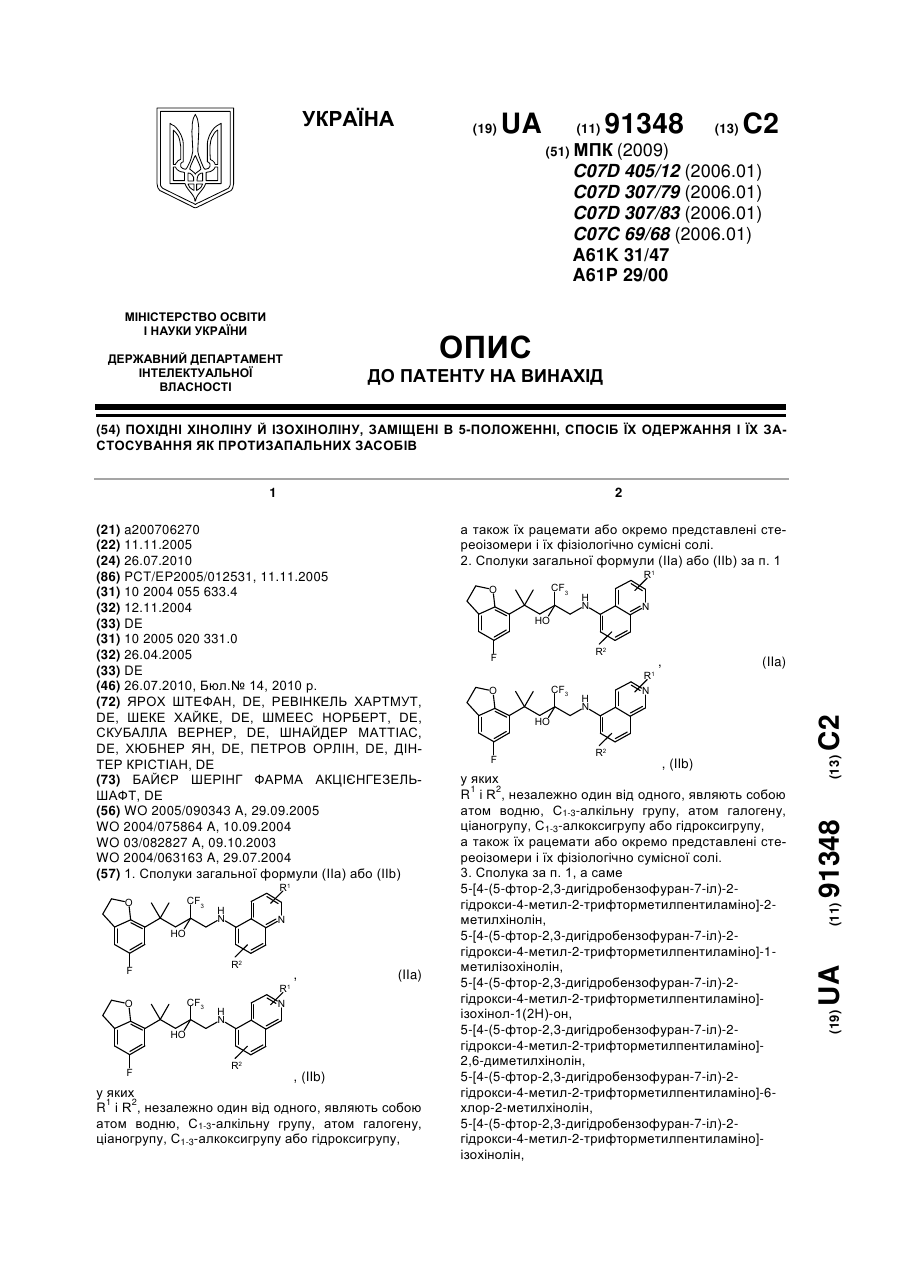
Номер патенту: 91348
Опубліковано: 26.07.2010
Автори: Шмеес Норберт, Ярох Штефан, Шеке Хайке, Хюбнер Ян, Петров Орлін, Скубалла Вернер, Дінтер Крістіан, Шнайдер Маттіас, Ревінкель Хартмут
МПК: C07D 405/12, C07D 307/83, C07C 69/68, A61P 29/00, A61K 31/47, C07D 307/79
Мітки: спосіб, заміщені, 5-положенні, протизапальних, хіноліну, застосування, похідні, ізохіноліну, одержання, засобів
Формула / Реферат:
1. Сполуки загальної формули (ІІа) або (ІІb), (IIa) , (IIb)у якихR1 і R2, незалежно один від одного, являють собою атом водню, С1-3-алкільну групу, атом галогену, ціаногрупу, С1-3-алкоксигрупу або гідроксигрупу, а також їх рацемати або окремо представлені стереоізомери і їх...
Похідні тетрагідронафталіну, спосіб їх одержання і їх застосування як протизапальних засобів

Номер патенту: 80644
Опубліковано: 10.10.2007
Автори: Шмеес Норберт, Бергер Маркус, Ревінкель Хартмут, Менгель Анне, Кроліківіч Конрад, Ярох Штефан, Скубалла Вернер, Шеке Хайке, Нгуєн Дуі, Бойрле Штефан
МПК: A61K 31/517, A61K 31/502, C07D 209/48, C07D 231/56, C07D 217/24, C07D 213/74, A61K 31/4406, A61K 31/4035, A61K 31/472, C07D 209/34, A61K 31/416, A61K 31/4709, C07D 239/74, A61K 31/404, C07D 215/38
Мітки: застосування, одержання, тетрагідронафталіну, засобів, спосіб, протизапальних, похідні
Формула / Реферат:
1. Сполуки загальної формули (І)(I),у якійR1 й R2, незалежно один від одного, представляють атом водню, гідроксигрупу, атом галогену, необов'язково заміщену С1-С10-алкільну групу, необов'язково заміщену С1-С10-алкоксигрупу, С1-С10-алкілтіогрупу, С1-С5-перфторалкільну групу, ціаногрупу, нітрогрупу, або R1 й R2 разом представляють групу, вибрану із груп...
N-ацильні похідні амінокислот, спосіб їх одержання, фармацевтична композиція і їх застосування як протиалергічних, протизапальних і гіполіпідемічних засобів

Номер патенту: 94586
Опубліковано: 25.05.2011
Автори: Кромова Татьяна Алєксандровна, Ковальова Віолєтта Лєонідовна, Нєбольсін Владімір Євгєньєвіч, Желтухіна Галіна Алєксандровна
МПК: C07D 233/64, A61P 37/08, A61P 9/00, A61K 31/405, C07D 209/20, A61P 3/06, A61K 31/4172, A61P 11/00
Мітки: композиція, застосування, похідні, фармацевтична, спосіб, засобів, гіполіпідемічних, одержання, амінокислот, протизапальних, протиалергічних, n-ацильні
Формула / Реферат:
1. N-ацильні похідні амінокислот загальної формули (І), (І) де n дорівнює 2 або 3; і R1 являє собою або , R2=Н, -СН3, -С2Н5, і їх...
Попередній патент: Мобільний комп’ютерний термінал “к.врт500″
Наступний патент: Конденсований амінопіридин як інгібітор hsp90
Випадковий патент: Спосіб прогнозування професійної надійності водіїв технологічного автотранспорту