Антибактеріальні 3,4-дигідро-1н-[1,8]нафтиридинони, заміщені циклопента[c]піролом
Номер патенту: 111210
Опубліковано: 11.04.2016
Автори: Коул Аніл, Жільмон Жером Еміль Жорж, Арну Ерік П'єр Александр, Мотт Магалі Мадлен Сімон, Лансуа Давід Франсіс Ален, Балеманс Уенді Міа Альберт








Формула / Реферат
1. Сполука формули (І)
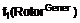 , (I)
, (I)
де
А являє собою -СºС- або
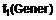 ,
,
зв'язок ![]() являє собою одинарний зв'язок або подвійний зв'язок,
являє собою одинарний зв'язок або подвійний зв'язок,
X являє собою вуглець або азот, і, якщо X являє собою азот, то зв'язок ![]() являє собою одинарний зв'язок;
являє собою одинарний зв'язок;
Ζ1 являє собою СН або N;
R1 являє собою водень, С1-4алкіл або галоген;
R2 являє собою водень, С1-4алкіл або галоген;
R3 являє собою водень, С1-6алкіл, гідроксил або галоген;
R4 являє собою водень; галоген; С1-6алкіл; С2-6алкеніл; С2-6алкініл; С1-6алкілокси; С1-4алкілоксикарбоніл; амінокарбоніл; моно- або ді(С1-4алкіл)-амiнокарбоніл; арил; арилокси; арилкарбоніл; арилсульфоніл; гетероарил; С1-6алкіл, заміщений ціаногрупою; С1-6алкіл, заміщений арилом або арилоксигрупою; або С1-6алкіл, заміщений гетероарилом;
арил є фенілом; фенілом, заміщеним одним, двома або трьома замісниками, кожний з яких окремо вибирається з галогену, гідроксилу, С1-4алкілу, С1-4алкокси, полігалогенС1-4алкілу, полігалогенС1-4алкокси, ціано-, нітро- та аміногрупи;
гетероарил являє собою фураніл, тіофеніл, піроліл, піразоліл, імідазоліл, ізоксазоліл, тіазоліл, триазоліл, тетразоліл, ізотіазоліл, тіадіазоліл, оксадіазоліл, піридиніл, піридазиніл, піримідиніл, піразиніл, бензо[1,3]діоксоліл, бензофураніл, бензотіазоліл, індоліл, 2,3-дигідро-1Н-індоліл, тетрагідротіофеніл або хінолініл, де кожний гетероарил може бути заміщений одним або двома замісниками, кожний з яких окремо вибирається з галогену, ціаногрупи, С1-4алкілу, С1-4алкокси, С1-4алкоксикарбонілу або фенілу;
або її фармацевтично прийнятна сіль приєднання кислоти.
2. Сполука за п. 1, де:
Ζ1 являє собою СН;
R1 являє собою водень або С1-4алкіл;
R2 являє собою водень або С1-4алкіл.
3. Сполука за п. 1 або п. 2, де
А являє собою -СºС- або
,
зв'язок ![]() являє собою одинарний зв'язок або подвійний зв'язок,
являє собою одинарний зв'язок або подвійний зв'язок,
X являє собою вуглець або азот, і, якщо X являє собою азот, то зв'язок ![]() являє собою одинарний зв'язок;
являє собою одинарний зв'язок;
R1 є воднем;
R2 є воднем;
R3 є воднем, гідроксилом або галогеном;
R4 є воднем; галогеном; C1-6алкілом; C1-6алкілокси; C1-4алкілоксикарбонілом;
амінокарбонілом; моно- або ді(C1-4алкіл)-амінокарбонілом; арилом; арилокси; арилсульфонілом; гетероарилом; C1-6алкілом, заміщеним ціаногрупою; C1-6алкілом, заміщеним арилом або арилоксигрупою; або C1-6алкілом, заміщеним гетероарилом;
арил є фенілом; фенілом, заміщеним одним замісником, вибраним з галогену, C1-4алкілу, C1-4алкілокси- та ціаногрупи;
гетероарил є фуранілом, тіофенілом, піразолілом, ізоксазолілом, тіазолілом, триазолілом, тетразолілом, тіадіазолілом, піридинiлом або піримідинілом, де кожний гетероарил може бути заміщений одним замісником, вибраним з галогену, ціаногрупи, C1-4алкілу, C1-4алкокси або C1-4алкілкарбонілу;
або її фармацевтично прийнятна сіль приєднання кислоти.
4. Сполука за будь-яким з пп. 1-3, де R1 є воднем і R2 є воднем.
5. Сполука за будь-яким з пп. 1-4, де R3 являє собою водень.
6. Сполука за будь-яким з пп. 1-5, де R4 є арилом.
7. Сполука за будь-яким з пп. 1-5, де R4 є гетероарилом.
8. Сполука за будь-яким з пп. 1-5, де R4 є C1-6алкілом, заміщеним арилом.
9. Сполука за будь-яким з пп. 1-8, де X являє собою азот, а зв'язок ![]() являє собою одинарний зв'язок.
являє собою одинарний зв'язок.
10. Сполука за будь-яким з пп. 1-9, де
А являє собою
.
11. Фармацевтична композиція, яка містить фармацевтично прийнятний носій і терапевтично ефективну кількість сполуки за будь-яким з пп. 1-8.
12. Спосіб одержання фармацевтичної композиції за п. 11, де терапевтично ефективну кількість сполуки за будь-яким з пп. 1-10 ретельно перемішують з фармацевтично прийнятним носієм.
13. Сполука формули (І) за будь-яким з пп. 1-10 для застосування як лікарського засобу.
14. Сполука формули (І) за будь-яким з пп. 1-10 для застосування у лікуванні бактеріальних інфекцій.
15. Сполука за п. 14, де бактеріальна інфекція викликана бактерією, яка експресує фермент FabI.
16. Спосіб одержання сполук формули (І) за п. 1:
(і) реакцією проміжної сполуки формули (II) із проміжною сполукою формули (III)
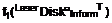 ;
;
(іі) у випадку сполук формули (І), в яких А являє собою -C(R2)=C(R1)-, реакцією проміжної сполуки формули (V) із проміжною сполукою формули (VI)
 ,
,
де Ха1 являє собою придатну відхідну групу, а інші змінні - як визначено у п. 1;
або за бажанням сполуку формули (І) перетворюють у фармацевтично прийнятну сіль приєднання кислоти, або навпаки, сіль приєднання кислоти сполуки формули (І) перетворюють у форму вільної основи, використовуючи луг.
Текст
Реферат: Даний винахід стосується нових сполук формули (І), що інгібують активність ферменту FabI, які внаслідок цього є корисними у лікуванні бактеріальних інфекцій. Він також стосується фармацевтичних композицій, які містять ці сполуки, та хімічних способів одержання таких сполук. R4 O N X C O Z1 A N R3 N H (I) UA 111210 C2 (12) UA 111210 C2 UA 111210 C2 5 10 15 20 25 30 Даний винахід стосується нових сполук формули (I), що інгібують активність ферменту FabI, які внаслідок цього є корисними у лікуванні бактеріальних інфекцій. Він також стосується фармацевтичних композицій, які містять ці сполуки, та хімічних способів одержання таких сполук. Сполуки даного винаходу є антибактеріальними сполуками, які інгібують білок FabI, NADHзалежний фермент еноїл-ацилтранспортуючий білок-редуктазу (ACP), у шляху біосинтезу жирних кислот. Синтаза жирних кислот (FAS) бере участь у загальному шляху біосинтезу насичених жирних кислот усіх організмів, але структурна організація FAS серед них істотно відрізняється. Відмітними характеристиками FAS хребетних і дріжджів є те, що усі види ферментативної активності записані на одному або двох поліпептидних ланцюгах, і те, що білок, який переносить ацил (ACP), присутній у вигляді комплексу. На відміну від цього, у бактеріальній FAS кожна стадія синтезу каталізується певним монофункціональним ферментом, а ACP являє собою окремий білок. Внаслідок цього існує можливість селективного інгібування бактеріальної FAS шляхом блокування однієї із стадій синтезу, використовуючи інгібуючий агент. NADH-залежна еноїл-ACP редуктаза (FabI) бере участь в останній стадії чотирьохстадійної реакції, включеної у кожний цикл бактеріального біосинтезу жирних кислот. Так, фермент FabI є біосинтетичним ферментом у загальному шляху синтезу бактеріального біосинтезу жирних кислот. Було показано, що фермент FabI являє собою найважливішу ціль у основних патогенів, таких як E. Coli (Heath et al. J. Biol. Chem. 1995, 270, 26538; Bergler et al. Eur. J. Biochem. 2000, 275, 4654). Отже сполуки, що інгібують FabI, можуть бути корисними в якості антибактеріальних засобів. Сполуки, що характеризуються інгібуючою дією щодо ферменту FabI, розкриті у публікаціях WO-01/26652, WO-01/26654 і WO-01/27103. Заміщені сполуки нафтиридинону, що характеризуються інгібуючою дією щодо FabI, розкриті у публікаціях WO-03/088897, WO2007/043835 і WO-2008/098374. Міжнародна патентна заявка WO 2007/053131 розкриває різні сполуки для можливого застосування в якості інгібіторів FabI. Міжнародна патентна заявка WO 2011/061214 також розкриває різні сполуки для можливого застосування в якості інгібіторів FabI. Однак жоден із цих документів не розкриває конденсований біциклічний фрагмент, безпосередньо приєднаний до карбонільного фрагмента, що перебуває у положенні α до алкену. Даний винахід стосується сполуки формули (I) R4 X O N C Z1 O A (I) NH N R3 , де R1 35 40 45 50 R2 A являє собою –C≡C– або ; зв'язок являє собою одинарний або подвійний зв'язок, X являє собою вуглець або азот, і якщо X являє собою азот, то зв'язок являє собою одинарний зв'язок; Z1 являє собою CH або N; 1 R є воднем, C1-4алкілом або галогеном; 2 R є воднем, C1-4алкілом або галогеном; 3 R є воднем, C1-6алкілом, гідроксилом або галогеном; 4 R є воднем; галогеном; C1-6алкілом; C2-6алкенілом; C2-6алкінілом; C1-6алкілоксі; C14алкілоксикарбонілом; амінокарбонілом; моно- або ди(C1-4алкіл)-амінокарбонілом; арилом; арилоксі; арилкарбонілом; арилсульфонілом; гетероарилом; C 1-6алкілом, заміщеним ціаногрупою; C1-6алкілом, заміщеним арилом або арилоксігрупою; або C 1-6алкілом, заміщеним гетероарилом; арил є фенілом; фенілом, заміщеним одним, двома або трьома замісниками, кожний з них окремо вибирається з галогену, гідроксилу, C 1-4алкілу, C1-4алкокси, полігалогенC1-4алкілу, полігалогенC1-4алкокси, ціано-, нітро- та аміногрупи; гетероарил є фуранілом, тіофенілом, піролілом, піразолілом, імідазолілом, ізоксазолілом, тіазолілом, триазолілом, тетразолілом, ізотіазолілом, тіадіазолілом, оксадіазолілом, 1 UA 111210 C2 5 10 15 20 25 30 35 40 45 50 55 60 піридинілом, піридазинілом, піримідинілом, піразинілом, бензо[1,3]діоксолілом, бензофуранілом, бензотіазолілом, індолілом, 2,3-дигідро-1H-індолілом, тетрагідротіофенілом або хінолінілом, де кожний гетероарил може бути заміщений одним або двома замісниками, кожний з них окремо вибирається з галогену, ціаногрупи, C1-4алкілу, C1-4алкокси, C1-4алкілкарбонілу або фенілу; або її фармацевтично прийнятної солі приєднання кислоти. Як використано у попередніх визначеннях: - термін "галоген" є загальним для атомів фтору, хлору, брому та йоду; - C1-4алкіл позначає вуглеводневі радикали із прямим і розгалуженим ланцюгом, що містять від 1 до 4 атомів вуглецю, такі як, наприклад, метил, етил, пропіл, бутил, 1-метилетил, 2метилпропіл тощо; - C1-6алкіл включає C1-4алкіл і його більш високі гомологи, що містять 5 або 6 атомів вуглецю, такі як, наприклад, 2-метилбутил, пентил, гексил тощо; - полігалогенC1-4алкіл визначений як полігалоген-заміщений C1-4алкіл (як визначено тут і вище), заміщений 2-6 атомами галогену, такий як дифторметил, трифторметил, трифторетил тощо. Як використовується в описі, щоразу, коли використовується термін "сполука формули (I)”, передбачається, що він також включає фармацевтичні солі приєднання, які здатні утворювати сполуки формули (I), і сольвати, які можуть утворювати сполуки формули (I) або фармацевтично прийнятні солі приєднання кислоти сполук формули (I). Визначення "сполуки формули (I)” по суті включає всі стереоізомери сполуки формули (I) або у вигляді чистого стереоізомера, або у вигляді суміші двох або більше стереоізомерів. Енантіомери являють собою стереоізомери, які є неспівпадаючими один з одним дзеркальними зображеннями. Суміш 1:1 пари енантіомерів являє собою рацемат або рацемічну суміш. Діастереомери (або діастереоізомери) є стереоізомерами, які не є енантіомерами, тобто вони не співвідносяться як дзеркальні зображення. Якщо сполука містить дизаміщену циклоалкільну групу, замісники можуть перебувати у цис- або транс-конфігурації. Тому, даний винахід включає енантіомери, діастереомери, рацемати, цис-ізомери, транс-ізомери та їх суміші. Абсолютна конфігурація визначається згідно із системою Кана-Інгольда-Прелога. Конфігурація асиметричного атома визначається за допомогою або R, або S. Виділені сполуки, абсолютна конфігурація яких не відома, можуть бути позначені за допомогою (+) або (-), залежно від напрямку, у якому вони обертають плоскополяризоване світло. Якщо вказаний певний стереоізомер, це означає, що зазначений стереоізомер практично вільний від інших ізомерів, тобто зв'язаний з менше ніж 50 %, переважно менше ніж 20 %, більш переважно менше ніж 10 %, ще більш переважно менше ніж 5 %, зокрема менше ніж 2 % і найбільш переважно менше ніж 1 % інших ізомерів. Таким чином, якщо сполука формули (I) визначена як (R), то це означає, що сполука практично не містить (S) ізомеру; якщо сполука формули (I) зазначена, наприклад, як E, то це означає, що сполука практично не містить Z ізомеру; якщо сполука формули (I) зазначена, наприклад, як цис, це означає, що сполука практично не містить транс-ізомеру. Терміни "стереоізомери" або "стереохімічно ізомерні форми" раніше і далі у даному документі застосовуються взаємозамінно. Фахівці у даній галузі техніки можуть легко визначити абсолютну стереохімічну конфігурацію сполук формули (I) та використовуваних при їх одержанні проміжних сполук, застосовуючи добре відомі способи, такі як, наприклад, рентгенівська дифракція. Деякі сполуки формули (I) можуть також існувати в їх таутомерній формі. Такі форми, хоча вони явно і не зазначені у наведеній вище формулі, повинні бути включені в об'єм даного винаходу. Крім того, деякі сполуки формули (I) і використовувані при їх одержанні проміжні сполуки можуть проявляти поліморфізм. Слід розуміти, що даний винахід охоплює будь-які поліморфні форми, які мають корисні у терапевтичному лікуванні описаних вище станів властивості. Фармацевтично прийнятні солі приєднання кислот, що згадуються зараз і раніше, включають нетоксичні форми солей приєднання кислот, які мають терапевтичну дію, що можуть бути утворені сполукою формули (I). Фармацевтично прийнятні солі приєднання кислоти можна легко одержати шляхом обробки основної форми такою відповідною кислотою. Відповідні кислоти включають, наприклад, неорганічні кислоти, такі як галогенводневі кислоти, наприклад соляна або бромистоводнева кислота, сірчану, азотну, фосфорну та подібні кислоти; або органічні кислоти, такі як, наприклад, оцтову, пропіонoву, гліколеву, молочну, піровиноградну, щавлеву (тобто етандіову), малонову, бурштинову (тобто бутандіову), малеїнову, фумарову, яблучну, винну, лимонну, метансульфонову, етансульфонову, бензолсульфонову, п 2 UA 111210 C2 5 10 15 20 25 30 35 40 45 толуолсульфонову, цикламову, саліцилову, п-аміносаліцилову, памову та подібні кислоти. І навпаки, зазначені сольові форми можна перетворити шляхом обробки відповідною основою на вільну основну форму. Сполуки формули (I) можуть існувати як у несольватованій, так і в сольватованій формах. Термін "сольват" використовується у даному контексті для позначення асоціації молекул, що включає сполуку даного винаходу та одну або декілька молекул фармацевтично прийнятного розчинника, наприклад, води або етанолу. Термін "гідрат" використовується, якщо розчинником є вода. Термін "FabI" відомий у даному рівні техніки та стосується бактеріального ферменту, який, імовірно, діє еноїл-ацилтранспортуючий білок-редуктаза (ACP), на останній стадії чотирьох реакцій, що бере участь у кожному циклі бактеріального біосинтезу жирних кислот. Передбачається, що цей фермент широко розповсюджений у бактеріях. Сполуки формули (I), які можна назвати, включають ті, у яких: (i) Z1 являє собою CH, і, отже, сполука формули I являє собою наступне: R4 O (I) O X N C A NH N R3 , де 1 2 (ii) якщо R або R являють собою галоген, то вони переважно є F або Cl; 1 (iii) R являє собою водень або C1-4алкіл; та/або 2 (iv) R являє собою водень або C1-4алкіл. Переважні сполуки формули (I) включають ті, у яких A являє собою подвійний зв'язок (а не потрійний зв'язок), тобто переважно, щоб: R1 R2 A являв собою . Становлять інтерес такі сполуки формули (I), які є сполуками формули (I), де застосовується одне або декілька з наступних обмежень: 1 2 a) R і R являють собою водень; або 3 b) R являє собою водень; або 3 c) R являють собою водень, галоген або гідроксил; або 4 d) R являє собою водень або галоген; або 4 e) R являє собою арил; або 4 f) R являє собою C1-6алкіл; або 4 g) R являє собою арилоксі або арилсульфоніл; або 4 h) R являє собою C1-6алкіл, заміщений арилом; або 4 i) R являє собою гетероарил; або 4 j) R являє собою C1-6алкіл, заміщений гетероарилом; або k) гетероарил являє собою фураніл, тіофеніл, піразоліл, ізоксазоліл, тіазоліл, триазоліл, тетразоліл, тіадіазоліл, піридиніл або піримідиніл; або l) X являє собою вуглець; або m) X являє собою азот, і зв'язок являє собою одинарний зв'язок. Першою групою сполук є сполуки формули (I) R4 O (I) O X N C A NH N R3 , де R1 R2 A являє собою –C≡C– або ; зв'язок являє собою одинарний зв'язок або подвійний зв'язок, X являє собою вуглець або азот, і якщо X являє собою азот, то зв'язок одинарний зв'язок; 3 являє собою UA 111210 C2 1 5 10 15 R є воднем; 2 R є воднем; 3 R є воднем, гідроксилом або галогеном; 4 R є воднем, галогеном; C1-6алкілом; C1-6алкілоксі; C1-4алкілоксикарбонілом; амінокарбонілом; моно- або ди(C1-4алкіл)-амінокарбонілом; арилом; арилоксі; арилсульфонілом; гетероарилом; C1-6алкілом, заміщеним ціаногрупою; C1-6алкілом, заміщеним арилом; або C16алкілом, заміщеним гетероарилом; арил є фенілом; фенілом, заміщеним одним замісником, вибраним з галогену, C 1-4алкілу, C14алкілоксі та ціаногрупи; гетероарил є фуранілом, тіофенілом, піразолілом, ізоксазолілом, тіазолілом, триазолілом, тетразолілом, тіадіазолілом, піридинілом або піримідинілом; де кожний гетероарил може бути заміщений одним замісником, вибраним з галогену, ціаногрупи, C1-4алкілу, C1-4алкілоксі або C1-4алкілкарбонілу; або їх фармацевтично прийнятна сіль приєднання кислоти. Другою групою сполук формули (I) є такі сполуки формули (I), де A являє собою –C≡C-. Третьою групою сполук формули (I) є такі сполуки формули (I), де R1 R2 A являє собою . Переважні сполуки формули (I) включають ті, у яких кільце, що містить Х, являє собою одне з наступного: 20 25 30 35 40 тобто подвійні цикли, які містять цис-конфігурацію у точці сполучення кілець (трансконфігурація викличе напруження в кільці), що можуть бути рацемічним або окремим енантіомерами. Як пояснюється тут і далі, якщо абсолютна стереохімія одного енантіомера не відома/не була відома, хіральні вуглеці у точці сполучення кілець можуть бути позначені жирними або штриховими лініями (а не у вигляді клинів). Більш переважні сполуки формули (I) включають ті, у яких конденсоване біциклічне кільце, що містить Х, являє собою одне з наступного: де у вищезгаданих конденсованих подвійних циклах сполуки можуть бути рацемічним або окремим енантіомером (якщо відповідна симетрія відсутня, і існування енантіомерів можливе), як описано тут і вище. Переважно у сполуках формули (I): 3 4 (i) Присутній щонайменше один замісник R або R , який не являє собою водень; 3 4 3 (ii) Один з R і R (наприклад, R ) являє собою водень, гідроксил або галоген (наприклад, 3 4 4 фтор), а інший з R і R (наприклад, R ) являє собою замісник, відмінний від водню; 3 (iii) R являє собою водень, гідроксил або галоген (наприклад, фтор) і більш переважно 3 являє собою водень (тобто R фактично не присутній); 4 4 (iv) R являє собою замісник, відмінний від водню (тобто присутній замісник R , який не являє собою водень); 4 (v) R являє собою замісник, відмінний від водню, який приєднаний до X, при цьому будь-які із зазначених вище можуть бути взяті разом або у комбінації. Наприклад, у комбінації можуть бути взяті (iii), (iv) та/або (v), щоб забезпечити особливо переважні сполуки формули (I) нижче: 4 UA 111210 C2 4 5 10 15 20 25 30 35 40 45 50 в яких R являє собою замісник, відмінний від водню. Особливо переважні замісники, які 4 можуть бути представлені R (тут та в інших частинах документа), включають: (i) необов'язково заміщений арил; (ii) необов'язково заміщений гетероарил; (iii) C1-6алкіл, заміщений арилом або гетероарилом (причому останні дві арильні та гетероарильна групи у свою чергу необов'язково заміщені, як визначено у даному документі); (iv) арилокси (у якого арильний фрагмент необов'язково заміщений, як визначено у даному документі); (v) арилсульфоніл (у якого арильний фрагмент необов'язково заміщений, як визначено у даному документі); (vi) C1-6алкіл, який є незаміщеним (наприклад, етил, метил, ізопропіл); (vii) ди(C1-4алкіл)амінокарбоніл (наприклад, -C(O)N(CH3)2); (viii) амінокарбоніл (-C(O)NH2); (ix) C1-4алкілоксикарбоніл (наприклад, -C(O)O-CH2CH3); (x) галоген (наприклад, фтор); (xi) C2-6алкініл (наприклад, -CC); (xii) C1-6алкокси (наприклад, -OCH3). 4 Особливо переважно, щоб група R містила ароматичний фрагмент, а отже, наведені вище (i), (ii), (iii), (iv) і (v) є особливо переважними). 4 У випадку, якщо R являє собою наведений вище (i), то арильна група переважно є фенілом, причому зазначена група може бути незаміщеною або заміщеною одним або двома (наприклад, одним) замісниками, вибраними з C1-4алкілоксі, галогену, C1-4алкілу або ціаногрупи (наприклад, OCH3, хлор, фтор, метил або ціаногрупа). 4 У випадку, якщо R являє собою наведений вище (ii), то гетероарильна група є моноциклічним 5- або 6-членним кільцем, що містить від одного до чотирьох гетероатомів, наприклад, тієніл (наприклад, 2- або 3-тієніл), піридил (наприклад, 4-піридил або 3-піридил), піразоліл (наприклад, 5-піразоліл, 4-піразоліл або 1-піразоліл), фураніл (наприклад, 2- або 3фураніл), тіазоліл (наприклад, 2-тіазоліл), ізоксазоліл (наприклад, 4-ізоксазоліл), піроліл (наприклад, 1-піроліл), триазоліл (наприклад, 1,2,3-триазол-1-іл, 1,2,3-триазол-2-іл або 1,2,4триазол-2-іл), тіадіазоліл (наприклад, 1,3,4-тіадіазол-2-іл), піримідиніл (наприклад, 5піримідиніл), тетразоліл (наприклад, 1,2,3,4-тетразол-2-іл, 1,2,3,4-тетразол-1-іл), імідазоліл (наприклад, 2-імідазоліл). Такі гетероарильні групи можуть бути незаміщені або заміщені одним або двома (наприклад, двома або, переважно, одним) замісниками, вибраними з галогену, ціаногрупи, C1-4алкілу (наприклад, C1-2алкіл), C1-4алкілоксі (наприклад, C1-2алкілоксі) та C14алкілкарбонілу (наприклад, C 1-2алкілкарбоніл), наприклад, -OCH3, метил, галоген (наприклад, хлор), ціаногрупа та -C(O)-CH3. 4 У випадку, якщо R являє собою наведений вище (iii), то C1-6алкільна група є метилом, тобто -CH3, заміщеним арилом (наприклад, фенілом, таким як незаміщений феніл) або гетероарилом (наприклад, 5- або 6-членною моноциклічною гетероарильною групою, що містить один або два (наприклад, один) гетероатоми, утворюючи таким чином, наприклад, тієнільну групу, таку як 2тієнільна група; і така гетероарильна група переважно є незаміщеною). 4 У випадку, якщо R являє собою наведені вище (iv) або (v), арил є переважно незаміщеним 4 фенілом, а отже група R є - O-фенілом або -S(O)2-фенілом. 4 Більш переважно група R являє собою наведені вище (i) або (ii), тобто арил або 4 гетероарил. Ще більш переважно група R являє собою наведений вище (i), особливо незаміщений феніл. Найбільш переважні сполуки формули (I) включають ті, у яких конденсований біциклічний фрагмент, що містить X, являє собою: 4 у яких R є таким, як визначено у даному документі. Можуть бути корисними такі сполуки, які 4 4 безпосередньо поруч із подвійним зв'язком містять або N(R ) фрагмент, або C(R ) фрагмент. Це 5 UA 111210 C2 5 10 15 20 25 відбувається тому, що форма атома азоту (наприклад, будучи за своєю природою більш 4 плоскою у порівнянні з фрагментом CR , який не перебуває безпосередньо поруч із подвійним зв'язком), або присутність подвійного зв'язку у кільці, що містить X, може допомогти орієнтувати 4 R групу (якщо вона присутня) таким чином, що сполука у цілому (наприклад, з огляду орієнтації 4 замісника R ) проявляє кращі/поліпшені властивості зв'язування щодо бактеріального ферменту FabI. Отже, ці сполуки даного винаходу можуть бути корисними у тому розумінні, що присутність подвійного зв'язку може приводити до поліпшеного зв'язування з ферментом FabI або його інгібування. Отже, сполуки даного винаходу можуть бути корисними сполуками (наприклад, у порівнянні з відомими сполуками) завдяки цим властивостям, які можуть внаслідок цього приводити до кращої активності, ефективності тощо. Сполуки формули (I) зазвичай можуть бути отримані реакцією проміжної сполуки формули (II) із проміжною сполукою формули (III) у щонайменше одному інертному розчиннику і необов'язково у присутності щонайменше одного придатного зв'язуючого реагенту та/або придатної основи, причому зазначений спосіб додатково необов'язково включає перетворення сполуки формули (I) у її сіль приєднання та/або одержання її стереохімічно ізомерних форм. Може бути зручним активувати карбонову кислоту формули (III) шляхом додавання ефективної кількості прискорювача реакції. Необмежуючі приклади таких прискорювачів реакції включають карбонілдіімідазол, N, N'-дициклогексил-карбодіімід або 1-(3-диметиламінопропіл)-3етилкарбодіімід, гідроксибензотриазол, гексафторфосфат бензотриазолілокситрис(диметиламіно)-фосфонію, гексафторфосфат тетрапіролідинфосфонію, гексафторфосфат бромтрипіролідинфосфонію або їх функціональне похідне. Сполуки формули (I) також можуть бути отримані реакцією проміжної сполуки формули (II) із проміжною сполукою формули (IV), де Y являє собою гідроксил або галоген. Реакцію можна проводити в інертному розчиннику, такому як, наприклад, дихлорметан або диметилформамід, і необов'язково у присутності придатної основи, такої як, наприклад, діізопропілетиламін (DIPEA). 2 1 Сполуки формули (I), в яких A являє собою -C(R )=C(R )-, також можуть бути отримані реакцією проміжної сполуки формули (V) із проміжною сполукою формули (VI), 30 35 40 де Xa1 являє собою придатну групу, що відходить, таку як придатна галогенідна група (наприклад, хлор-, йод- і особливо бром-), а інші позначення, як визначено тут і вище, за придатних умов реакції, наприклад, в умовах реакції сполучення у присутності металевого каталізатора (наприклад, реакція сполучення у присутності дорогоцінного металу, де дорогоцінний метал є, наприклад, паладієвою основною), зокрема в умовах реакції Хека з використанням переважно каталізатора на основі паладію, такого як ацетат паладію, тетракіс(трифенілфосфін)паладій(0), біс(трифенілфосфін)паладію(II) дихлорид, [1,1’біс(дифенілфосфіно)фероцен]паладію(II) дихлорид або подібні (бажано, щоб каталізатором був ацетат паладію), наприклад, необов'язково у присутності придатного розчинника (наприклад, ацетонітрилу або подібного йому), основи (наприклад, амінної основи, такої як N, Nдіізопропіламін або подібної їй) і ліганду (наприклад, трифенілфосфіну, три-O-толілфосфіну або 6 UA 111210 C2 5 10 15 20 25 30 35 40 45 подібного їм). Реакцію можна проводити у запаяній трубці та/або в мікрохвильовій печі. Вихідні матеріали та деякі проміжні сполуки є відомими сполуками, та їх можна придбати або одержати згідно традиційних процедур реакцій, загальновідомих у даному рівні техніки. Для сполук, у яких Z1 являє собою CH, проміжні сполуки (IV) і (VI) можуть бути отримані, як описано у даному документі, або згідно традиційних процедур реакцій, загальновідомих у даному рівні техніки. Це також може бути застосовним у випадку відповідних проміжних сполук, у яких Z1 являє собою N. Однак такі сполуки можуть бути також отримані у відповідності за наступною схемою: Умови: a) NBS, ACN, кип'ятіння зі зворотним холодильником, 3 год., 70 %; b) LiAlH4 1M в THF, THF, 5 °C до кімнатної температури, протягом ночі, 20 %; c) PBr3, DCM, кімнатна температура, протягом ночі, 90 %; f) диметил малонат, NaOMe в MeOH, MeOH, кімнатна температура, протягом ночі, 25 %; g) NaOH, MeOH, кип'ятіння зі зворотним холодильником, 4год, HCl, кип'ятіння зі зворотним холодильником, протягом ночі; h) DIEA, Pd(OAc)2, три-O-толілфосфін, ACN, DMF, мікрохвилі, 180 °C, 25 хв. Сполуки формули (I), отримані в описаних тут і вище способах, можуть бути синтезовані у формі рацемічних сумішей енантіомерів, які можна відокремити один від одного, дотримуючись відомих з рівня техніки процедур розділення. Ці сполуки формули (I), які одержують у рацемічній формі, можуть бути перетворені у відповідні форми діастереомерної солі шляхом реакції з придатною хіральною кислотою. Згадані форми діастереомерної солі потім розділяють, наприклад, за допомогою селективної або фракційної кристалізації, а енантіомери виділяють звідти за допомогою лугу. Альтернативний спосіб розділення енантіомерних форм сполук формули (I) включає рідинну хроматографію з використанням хіральної нерухомої фази. Зазначені чисті стереохімічні ізомерні форми також можна одержати з відповідних чистих стереохімічних ізомерних форм придатних вихідних матеріалів за умови, що реакція протікає стереоспецифічно. Якщо необхідний певний стереоізомер, бажано, щоб зазначена сполука була синтезована стереоспецифічними способами одержання. У цих способах переважно використовують енантіомерно чисті вихідні матеріали. Сполуки, описані у даному документі, є інгібіторами ферменту FabI, як проілюстровано прикладами нижче (включаючи фармакологічний приклад 1). З огляду на ці інгібуючі фермент FabI властивості, сполуки, описані у даному документі, корисні у лікуванні бактеріальних інфекцій. Наприклад, ці сполуки корисні у лікуванні бактеріальних інфекцій, таких як, наприклад, інфекції верхніх дихальних шляхів (наприклад, отит середнього вуха, бактеріальний трахеїт, гострий епіглотит, тиреоїдит), нижніх дихальних відділів (наприклад, емпієма, абсцес легень), кардіологічні (наприклад, інфекційний ендокардит), шлунково-кишкові (наприклад, секреторна діарея, абсцес селезінки, позачеревинний абсцес), ЦНС (наприклад, церебральний абсцес), ока (наприклад, блефарит, кон'юнктивіт, кератит, ендофтальміт, пресептальний та орбітальний целюліт, дакріоцистит), нирок і сечовивідних шляхів (наприклад, епідидиміт, внутрішньонирковий та перинефральний абсцес, синдром токсичного шоку), шкіри (наприклад, імпетиго, фолікуліт, шкірні абсцеси, целюліт, ранові інфекції, бактеріальний міозит), а також кісток та суглобів (наприклад, септичний артрит, остеомієліт). Крім того, сполуки можуть бути корисні у комбінації з відомими антибіотиками. Таким чином, даний винахід також стосується сполук формули (I) для застосування в якості лікарського засобу, особливо для застосування у лікуванні бактеріальних інфекцій, зокрема, бактеріальних інфекцій, викликаних бактеріями, що експресують фермент FabI. Відтак дані сполуки можуть застосовуватися у виробництві лікарського засобу для лікування бактеріальних інфекцій, зокрема, бактеріальних інфекцій, викликаних бактерією, що експресує фермент FabI. 7 UA 111210 C2 5 10 15 20 25 30 35 40 45 50 55 Крім того, даний винахід передбачає спосіб лікування бактеріальних інфекцій, який включає введення суб'єкту, що потребує цього, сполуки формули (I), яка інгібує фермент FabI. Суб'єкт, який потребує лікування, має бактеріальну інфекцію або зазнав впливу інфекційної бактерії, симптоми якої можуть бути полегшені шляхом уведення терапевтично ефективної кількості сполук даного винаходу. Наприклад, у суб'єкта, який потребує лікування, може бути інфекція, проти якої сполуки формули (I) можуть бути введені в якості лікування. В іншому прикладі суб'єкт, який потребує лікування, може мати відкриту рану або опік, для яких сполуки формули (I) можуть бути введені в якості лікування. Зазвичай суб'єкта лікують проти наявної у нього бактеріальної інфекції. У суб'єкта може бути бактеріальна інфекція, викликана Bacillus anthracis, Citrobacter sp., Escherichia coli, Francisella tularensis, Haemophilus influenza, Listeria monocytogenes, Moraxella catarrhalis, Mycobacterium tuberculosis, Neisseria meningitidis, Proteus mirabilis, Proteus vulgaris, Salmonella sp., Serratia sp., Shigella sp., Stenotrophomonas maltophilia, Staphylococcus aureus або Staphylococcus epidermidis. Переважно, суб'єкту проводять лікування (профілактичне або терапевтичне), направлене проти бактеріальної інфекції, викликаної бактерією, яка експресує фермент FabI. Термін "проводити лікування" і "лікування", як використовується у даному документі, стосується лікувального, паліативного та профілактичного лікування, включаючи реверсування, полегшення, уповільнення прогресу захворювання, розладу або стану, щодо застосовний такий термін, або одного або декількох симптомів такого захворювання, розладу або стану. "Терапевтично ефективна кількість" сполуки даного винаходу являє собою кількість, що при введенні суб'єкту, який потребує лікування, поліпшує прогноз для суб'єкта, наприклад, сповільнює виникнення та/або знижує у суб'єкта тяжкість одного або декількох асоційованих з бактеріальною інфекцією симптомів. Необхідна для введення суб'єкту кількість сполуки, що розкривається, буде залежати від конкретного захворювання, способу введення та характеристик суб'єкта, таких як загальний стан здоров'я, наявність інших захворювань, вік, стать, генотип, вага тіла та переносимість лікарських засобів. Фахівець у даній галузі зможе визначити необхідні дози залежно від цих та інших факторів. Сполуки можуть бути випробувані в одному з декількох біологічних досліджень для того, щоб визначити концентрацію сполуки, яка необхідна для заданої фармакологічної дії. Крім того, даний винахід передбачає фармацевтичні композиції, що містять щонайменше один фармацевтично прийнятний носій і терапевтично ефективну кількість сполуки формули (I). З метою одержання фармацевтичних композицій згідно даного винаходу ефективну кількість певної сполуки, в основній формі або у формі солі приєднання кислоти, в якості активного інгредієнта поєднують в однорідній суміші зі щонайменше одним фармацевтично прийнятним носієм, причому носій може приймати широку різноманітність форм залежно від форми препарату, бажаної для введення. Ці фармацевтичні композиції знаходяться переважно у стандартній лікарській формі, придатній, переважно, для перорального введення, ректального введення, введення через шкіру або парентеральної ін'єкції. Наприклад, при одержанні композицій у пероральній лікарській формі можна використовувати будь-який із звичайних рідких фармацевтичних носіїв, таких як, наприклад, вода, гліколі, олії, спирти тощо, у випадку пероральних рідких препаратів, таких як суспензії, сиропи, міцні настої та розчини; або тверді фармацевтичні носії, такі як крохмалі, цукри, каолін, змащувальні речовини, зв'язуючі речовини, розпушувачі тощо, у випадку порошків, пігулок, капсул і таблеток. Завдяки простоті їх введення, таблетки і капсули представляють найбільш зручну пероральну стандартну лікарську форму, і в такому випадку безумовно використовують тверді фармацевтичні носії. У випадку композицій для парентеральних ін'єкцій фармацевтичний носій в основному буде містити стерильну воду, хоча, з метою поліпшення розчинності активного інгредієнта можуть бути включені й інші компоненти. Розчини для ін'єкцій можуть бути отримані шляхом використання фармацевтичного носія, що містить фізіологічний розчин, глюкозу або їх суміш. Суспензії для ін'єкцій можуть бути отримані шляхом використання придатних рідких носіїв, суспендуючих агентів тощо. У композиціях, придатних для введення через шкіру, фармацевтичний носій може необов'язково містити засіб для збільшення проникнення та/або придатний змочувальний засіб, необов'язково об'єднані із придатними добавками будь-якої природи у малих кількостях, які не чинять значний шкідливий вплив на шкіру. Згадані добавки можуть бути вибрані для сприяння введенню активного інгредієнта в шкіру та/або для полегшення приготування бажаних композицій. Дані композиції для зовнішнього застосування можна вводити різними шляхами, наприклад, у вигляді трансдермального пластиру, точкового нанесення або мазі. Солі приєднання сполук формули 8 UA 111210 C2 5 10 15 20 25 30 35 40 45 50 55 60 (I) внаслідок їхньої підвищеної розчинності у воді у порівнянні з відповідною формою основи є більш придатними при одержанні водних композицій. Особливо зручним є складання фармацевтичних композицій даного винаходу в стандартній лікарській формі для простоти введення і рівномірності дозування. "Стандартна лікарська форма", як використовується у даному документі, стосується фізично дискретних одиниць, придатних у якості стандартних доз, при цьому кожна одиниця містить попередньо задану кількість активного інгредієнта, розраховану для одержання бажаного терапевтичного ефекту, разом з необхідним фармацевтичним носієм. Прикладами таких стандартних лікарських форм є таблетки (у тому числі ділимі таблетки або таблетки, покриті оболонкою), капсули, пігулки, пакети з порошкоподібним продуктом, пластинки, розчини для ін'єкцій або суспензії, чайні ложки з верхом, столові ложки з верхом тощо, а також їх відокремлені кратні кількості. У випадку перорального введення фармацевтичні композиції даного винаходу можуть приймати форму твердої лікарської форми, наприклад, таблеток (як жувальних, так і у формі для проковтування цілими), капсул або желатинових капсул, отриманих традиційними способами з використанням фармацевтично прийнятних наповнювачів і носіїв, таких як зв'язуючі агенти (наприклад, попередньо пептизований кукурудзяний крохмаль, полівінілпіролідон, гідроксипропілметилцелюлоза тощо), наповнювачі (наприклад, лактоза, мікрокристалічна целюлоза, фосфат кальцію тощо) змащувальні речовини (наприклад, стеарат магнію, тальк, діоксид кремнію тощо), розпушувачі (наприклад, картопляний крохмаль, натрію крохмальгліколят тощо), змочувальні агенти (наприклад, лаурилсульфат натрію) тощо. Такі таблетки також можуть мати покриття, отримане способами, відомими у даному рівні техніки. Рідкі препарати для перорального прийому можуть приймати форму, наприклад, розчинів, сиропів або суспензій, або можуть бути складені у вигляді сухого продукту для змішування перед вживанням з водою та/або іншим придатним рідким носієм. Такі рідкі препарати можуть бути отримані традиційними способами, необов'язково з іншими фармацевтично прийнятними добавками, такими як суспендуючі агенти (наприклад, сироп сорбітолу, метилцелюлоза, гідроксипропілметилцелюлоза або гідрогенізовані харчові жири), емульгатори (наприклад, лецитин або гуміарабік), неводні носії (наприклад, мигдальна олія, жирні естери або етиловий спирт), підсолоджувачі, ароматизатори, коригувальні агенти та консерванти (наприклад, метилабо пропіл-п-гідроксибензоати або сорбінова кислота). Фармацевтично прийнятні підсолоджувачі, придатні для фармацевтичних композицій даного винаходу, включають щонайменше один інтенсивний підсолоджував, такий як аспартам, ацесульфам калію, цикламат натрію, алітам, підсолоджувач дигідрохалькон, монелін, стевіозид, сукралоза (4,1',6'-трихлор-4,1',6'-тридеоксигалактосахароза), або, переважно, сахарин, сахарин натрію або кальцію і необов'язково щонайменше один об'ємний підсолоджувач, тякий як сорбіт, маніт, фруктоза, сахароза, мальтоза, ізомальт, глюкоза, гідрогенізований сироп глюкози, ксиліт, карамель або мед. Інтенсивні підсолоджувачі зазвичай використовують у малих кількостях. Наприклад, у випадку сахарину натрію, згадана концентрація може змінюватися від приблизно 0,04 % до 0,1 % (вага/об'єм) кінцевої сполуки. Об'ємний підсолоджувач може бути ефективно використаний у більш високих концентраціях, що змінюються від приблизно 10 % до приблизно 35 %, переважно від приблизно 10 % до 15 % (вага/об'єм). Фармацевтично прийнятні ароматизатори, які можуть маскувати інгредієнти з гірким смаком у складах з низьким дозуванням, переважно є фруктовими ароматизаторами, такими як вишневий, малиновий, чорносмородиновий або полуничний ароматизатори. Комбінація двох ароматизаторів може дати дуже гарний результат. У складах з високим дозуванням можуть знадобитися більш сильні фармацевтично прийнятні ароматизатори, такі як карамельношоколадний, м'ятний прохолодний, Fantasy тощо. Кожний ароматизатор може бути присутнім у кінцевій композиції у концентрації, що змінюється від приблизно 0,05 % до 1 % (вага/об'єм). Переважно використовують комбінації згаданих сильних ароматизаторів. Бажано використовувати ароматизатор, який не зазнає будь-якої зміни або втрати смаку та/або кольору в умовах складання препарату. Композиції формули (I) можуть бути складені для парентерального введення шляхом ін'єкції, переважно внутрішньовенної, внутрішньом'язової або підшкірної ін'єкції, наприклад, болюсної ін'єкції або безперервного внутрішньовенного вливання. Сполуки для ін'єкції можуть бути оформлені як стандартна доза, наприклад, в ампулах або багатодозових контейнерах, включаючи доданий консервант. Вони можуть бути оформлені як суспензії, розчини або емульсії в масляних або водних носіях, і можуть містити агенти для складання препарату, такі як ізотонізуючі, суспендуючі, стабілізуючі та/або диспергуючі агенти. Альтернативно, активний інгредієнт може бути представлений у порошковій формі для змішування перед вживанням з придатним носієм, наприклад, стерильною, апірогенною водою. 9 UA 111210 C2 5 10 15 20 25 30 35 40 45 50 55 60 Сполуки формули (I) можуть бути складені у ректальні композиції, такі як супозиторії або утримуючі клізми, наприклад, що містять традиційні супозиторні основи, такі як масло какао та/або інші гліцериди. Фахівці в лікуванні бактеріальних захворювань, пов'язаних з інгібуванням ферменту FabI, можуть легко визначити терапевтично ефективну кількість сполуки формули (I) з результатів випробувань, представлених тут і далі. У більшості випадків передбачається, що терапевтично ефективна доза буде становити від приблизно 0,001 мг/кг до приблизно 50 мг/кг ваги тіла, більш переважно від приблизно 0,01 мг/кг до приблизно 10 мг/кг ваги тіла пацієнта, який проходить лікування. Може виявитися доцільним вводити терапевтично ефективну дозу у вигляді двох або більше субдоз із відповідними інтервалами протягом дня. Згадані субдози можуть бути складені у формах стандартної дози, причому кожна може містити від приблизно 0,1 мг до приблизно 1000 мг, зокрема, від приблизно 1 до приблизно 500 мг активного інгредієнта на форму стандартної дози. Точне дозування і частота введення залежать від конкретної використовуваної сполуки формули (I), конкретного стану, що підлягає лікуванню, тяжкості стану, що підлягає лікуванню, віку, ваги та загального фізичного стану певного пацієнта, а також від іншого медикаментозного лікування, яке може приймати пацієнт, як це добре відомо фахівцям у даній галузі. Крім того, згадана "терапевтично ефективна кількість" може бути зменшена або збільшена залежно від відповідної реакції пацієнта, що проходить лікування, та/або залежно від оцінки лікаря, що призначає сполуки даного винаходу. Діапазони ефективної добової кількості, наведені вище і далі, є лише рекомендаційними. Сполуки формули (I) можуть мати перевагу в тому, що вони можуть бути більш ефективними, менш токсичними, діючими більш тривалий час, більш сильними, давати менше побічних ефектів, легше всмоктуватися, та/або мати поліпшену фармакокінетичну криву (наприклад, мати більш високу біологічну доступність та/або більш низький кліренс), та/або мати корисні фармакологічні, фізичні або хімічні властивості у порівнянні із сполуками, відомими у даному рівні техніки, як при застосуванні у перерахованих вище випадках, так і в інших 4 випадках. Сполуки також можуть демонструвати такі переваги через наявність NR фрагмента 4 або CR фрагмента, який знаходиться безпосередньо поруч із подвійним зв'язком у кільці, що містить X. Наприклад, сполуки формули (I) можуть мати перевагу в тому, що вони мають гарну або поліпшену термодинамічну стабільність (наприклад, у порівнянні зі сполуками, відомими у рівні техніки; і, наприклад, як визначено за допомогою відомого способу та/або описаного тут способу). Сполуки формули (I) можуть також мати перевагу в тому, що вони мають широкий спектр дії у порівнянні з антибактеріальними засобами (наприклад, більш широкий спектр антибактеріальної дії у порівнянні зі сполуками, відомими у рівні техніки; і, наприклад, як визначено відомими випробуваннями та/або описаними тут випробуваннями). Сполуки формули (I) можуть також мати перевагу в тому, що вони мають гарні або поліпшені фармакокінетичні характеристики in vivo та біодоступність при пероральноми прийомі. Вони можуть також мати перевагу в тому, що вони мають гарну або поліпшену ефективність in vivo. Наприклад, сполуки даного винаходу можуть бути пристосовані для внутрішньовенного складу/дозування і, таким чином, можуть демонструвати поліпшену in vivo ефективність при внутрішньовенному введенні. Сполуки також можуть демонструвати такі переваги через 4 4 наявність NR фрагмента або CR фрагмента, який знаходиться безпосередньо поруч із подвійним зв'язком в кільці, що містить X. Експериментальна частина Абревіатури "DMF" визначається як N, N-диметилформамід, "DCM" або "CH2Cl2" визначається як дихлорметан, "MeOH" визначається як метанол, "EtOH" визначається як етанол, "MgSO4" визначається як сульфат магнію, та "THF" визначається як тетрагідрофуран, "AcOEt" або "EtOAc" визначається як етилацетат, "DIPEA" визначається як діізопропілетиламін, "EDCI" визначається як N'-(етилкарбонімідоїл)-N, N-диметил-1,3пропандіамін моногідрохлорид, "HOBT" визначається як 1-гідрокси-1H-бензотриазол, "DIPA" визначається як діізопропіламін, "K2CO3" визначається як карбонат калію, "TFA" визначається як трифтороцтова кислота, "NH4OH" визначається як гідроксид амонію, "NaHCO 3" визначається як гідрокарбонат натрію, "Et2O" визначається як діетиловий етер, "Na2SO4" визначається як сульфат натрію, "CH3CN" визначається як ацетонітрил, "NaOH" визначається як гідроксид натрію, "н-BuLi" визначається як н-бутиллітій, "i-PrOH" визначається як ізопропанол, "Pd(OAc)2" визначається як ацетат паладію, "DMA" визначається як диметилацетамід, "Et3N" визначається як триетиламін. 10 UA 111210 C2 Стереохімічне подання Сполуки формули (I) мають щонайменше два асиметричні атоми вуглецю, як показано нижче, де асиметричні атоми вуглецю ідентифікуються за позначенням *: 5 Через напруження у кільці в системі із двох кільцеподібних п'ятичленних кілець можна одержати тільки "цис" форми, а не "транс" форми. Сполуки формули (I), де система із двох кільцеподібних п'ятичленних кілець має ’цис’конфігурацію 10 Кожна з описаних вище "цис" сполук складається з рацемічної суміші двох енантіомерів, а виділені жирним зв'язки або штрихові зв'язки використовувалися для позначення цих відповідних стереохімічних конфігурацій. У випадку, коли така "цис" сполука розділяється на два окремі енантіомери, стереохімічна * * конфігурація окремого енантіомера позначається як R або S , що вказує на відносну * * стереохімію. Відповідно, окремий енантіомер, позначений як (R ,S ), може мати або абсолютну (R, S) конфігурацію, або (S, R) конфігурацію. Якщо абсолютна стереохімія конкретного хірального атома вуглецю в окремому енантіомері відома, виділені жирним і штрихові зв'язки були замінені на клинчасті зв'язки для позначення того, що сполука є окремим енантіомером, який має відому абсолютну стереохімію. A. Синтез проміжних сполук Приклад А.1 15 20 25 30 35 Розчин 6-бром-3,4-дигідро-1H-[1,8]нафтиридин-2-ону (1,0 г, 4,4 ммоль), трет-бутилакрилату (2,56 мл, 17,62 ммоль) і N, N-діізопропілетиламіну (1,46 мл, 8,81 ммоль) в ацетонітрилі (20 мл) і DMF (7 мл) перемішували і дегазували, використовуючи газ азот протягом 10 хвилин. Додавали три-o-толілфосфін (0,27 г, 0,88 ммоль) та ацетат паладію (II) (47 % у перерахуванні на Pd) (0,099 г, 0,44 моль) і отриману суміш обробляли у мікрохвильовій печі (1600 Вт, 180 °C, 35 хвилин). Реакційну суміш випарювали до сухого стану, розчиняли у суміші DCM/метанол (8/2) (50 мл), фільтрували через тонку подушку із целіту та промивали дихлорметаном. Органічний шар промивали водою, сушили (MgSO4), фільтрували і випарювали до сухого стану. Залишок розчиняли у холодному етанолі (10 мл) і перемішували при 5 °C протягом 5 хвилин, осад відфільтровували, промивали холодним етанолом (3 мл) і сушили під вакуумом з одержанням 950 мг проміжної сполуки (1). Проміжну сполуку (1) (4,1 г, 14,95 ммоль) розчиняли у суміші трифтороцтової кислоти (23,2 мл) в DCM (41 мл). Реакційний склад перемішували при кімнатній температурі протягом 30 хвилин. Реакційну суміш концентрували при зниженому тиску. Отриману тверду речовину розтирали з діетиловим етером, відфільтровували та сушили під вакуумом з одержанням 3,97 г проміжної сполуки (2). 11 UA 111210 C2 5 Проміжну сполуку (2) розтирали протягом ночі у суміші HCl у діоксані (4 М, 48 мл), тверду речовину відфільтровували, промивали діетиловим етером і сушили під вакуумом з одержанням 3,9 г проміжної сполуки (3). Приклад А.2 15 Розчин трет-бутилового етеру аліл-проп-2-ініл-карбамової кислоти (CAS 147528-20-9, 45 г, 0,23 моль), карбонілу кобальту (17,5 г, 46,1 ммоль) і 1,1,3,3-тетраметил-2-тіосечовини (36,6 г, 0,277 ммоль) у толуолі (1,8 л) перемішували і нагрівали при 70 °C протягом 5 годин в автоклаві під тиском CO (2-3 бар). Отриману суміш фільтрували через тонку подушку із целіту і випарювали до сухого стану. Залишок розчиняли в DCM і фільтрували через тонку подушку із целіту для одержання прозорого розчину. Його випарювали до сухого стану з одержанням 85,7 г неочищеного залишку. Його очищали за допомогою препаративної рідинної хроматографії на (силікагель 20-45µм, 1000 г, рухома фаза (градієнт DCM/AcOEt від 95/5 до 80/20). Очищені фракції збирали і випарювали розчинник з одержанням 36,5 г проміжної сполуки (4). 20 Суміш проміжної сполуки (4) (37,6 г, 0,168 моль) і 10 % паладію на вугіллі (7,5 г) в етилацетаті (750 мл) гідрогенізували при кімнатній температурі протягом 30 хвилин при тиску 3 бар у реакторі закритого типу. Отриману суміш фільтрували через тонку подушку із целіту та випарювали до сухого стану з одержанням 38,2 г проміжної сполуки (5). 10 25 30 35 40 н-BuLi 1,6M у гексані (64 мл, 0,102 моль) додали по краплях при -20 °C, в атмосфері N2, до розчину діізопропіламіну (14,3 мл, 0,102 моль) у сухому THF (140 мл), потім суміш перемішували при -20 °C протягом 20 хвилин. Потім додавали розчин проміжної сполуки (5) (19,1 г, 84,8 ммоль) у сухому THF (190 мл) при -78 °C і перемішували отриману суміш протягом 1 години при -78 °C. Додавали розчин N-феніл-трифторметану сульфоніміду (36,4 г, 0,102 моль) у сухому THF (110 мл) при -78 °C, потім суміші давали нагрітися до кімнатної температури і перемішували протягом ночі. Суміш випарювали до сухого стану. Залишок розчиняли у DCM, промивали водним розчином NaHCO3, сушили (MgSO4) і випарювали до сухого стану з одержанням 27,7 г проміжної сполуки (6). Розчин проміжної сполуки (6) (9,3 г, 26,0 ммоль) і фенілборної кислоти (3,81 г, 31,2 ммоль) у розчині 2 M карбонату калію (26 мл) і диметилового етеру етиленгліколю (93 мл) продували N2 протягом 10 хвилин, потім додавали тетракіс(трифенілфосфін)паладій (3,0 г, 2,6 ммоль). Реактор закритого типу нагрівали при 80 °C, використовуючи мікрохвильову піч із однією багаторежимною камерою системи CEM Mars з потужністю в діапазоні від 0 до 400 Вт протягом 30 хвилин. Отриманий розчин охолоджували до кімнатної температури, додавали воду й EtOAc, відділяли органічний шар, промивали водою, а потім сольовим розчином, сушили (MgSO 4) і випарювали до сухого стану. Очищення залишку виконували за допомогою флешхроматографії на силікагелі (330г, 15-40µм, гептан/EtOAc від 100/0 до 80/20). Очищені фракції збирали й випарювали до сухого стану з одержанням 4,3 г проміжної сполуки (7). 12 UA 111210 C2 5 10 15 20 25 30 35 40 Трифтороцтову кислоту (44 мл) по краплях додавали до розчину проміжної сполуки (7) (14,5 г, 50,8 ммоль) в CH2Cl2 (44 мл). Отриманий розчин перемішували при кімнатній температурі протягом 30 хвилин, потім суміш охолоджували до 5 °C. Повільно додавали 3н NaOH до одержання лужного середовища, суміш двічі екстрагували, використовуючи CH2Cl2. Об'єднаний органічний шар промивали 3н NaОH, а потім водою, сушили над MgSO 4 і випарювали з одержанням 8,8 г рацемічної проміжної сполуки (8). Проміжну сполуку (8) очищали і розділяли за допомогою хіральної SFC на (CHIRALPAK ADH 5µм 250 × 20 мм). Рухома фаза (0,3 % ізопропіламін, 73 % CO2, 27 % iPrOH). Збирали чисті 20 фракції і видаляли розчинник з одержанням 3,9 г проміжної сполуки (10) (R*,S*) ([α] D = -53,19° 20 (589 нм, конц-я 0,3365 вага/об. %, DMF, 20 °C)) і 4 г проміжної сполуки (9) (S*,R*) ([α]D = +38,6° (589 нм, конц-я 0,285 вага/ об. %, DMF, 20 °C)). Проміжна сполука (9) 1 H ЯМР (400MГц, DMSO-d6) δ (м.д.) 7,43 (d, J=7,6 Гц, 2 H), 7,32 (t, J=7,6 Гц, 2 H), 7,20-7,26 (m, 1 H), 6,07 (d, J=2,0 Гц, 1 H), 3,30-3,39 (m, 1 H), 2,77-2,94 (m, 4 H), 2,66 (dd, J=3,0, 11,1 Гц, 1 H), 2,58 (dd, J=3,0, 11,1 Гц, 1 H), 2,46 (d, J=15,7 Гц, 1 H). Проміжна сполука (10) 1 H ЯМР (400MГц, DMSO-d6) δ (м.д.) 7,43 (d, J=7,6 Гц, 2 H), 7,32 (t, J=7,6 Гц, 2 H), 7,20-7,26 (m, 1 H), 6,07 (d, J=2,0 Гц, 1 H), 3,30-3,39 (m, 1 H), 2,77-2,94 (m, 4 H), 2,66 (dd, J=3,0, 11,1 Гц, 1 H), 2,58 (dd, J=3,0, 11,1 Гц, 1 H), 2,46 (d, J=15,7 Гц, 1 H). Приклад А.3 Розчин проміжної сполуки (6) (44,4 г, 111,82 ммоль) і 3-тіофенілборної кислоти (17,17 г, 134,19 ммоль) у розчині 2 M карбонату калію (112 мл) і диметилового етеру етиленгліколю (444 мл) у відкритій ємності продували N 2 протягом 10 хвилин, потім додавали тетракіс(трифенілфосфін)паладій (12,92 г, 223,65 ммоль). Розчин нагрівали при 78 °C, використовуючи мікрохвильову піч із однією багаторежимною камерою системи CEM MARS з потужністю у діапазоні від 0 до 400 Вт протягом 1 години. Суміш охолоджували до кімнатної температури, додавали воду й EtOAc. Отриману суміш фільтрували через подушку із целіту. Органічний шар відділяли, промивали водою, потім сольовим розчином, сушили над MgSO 4 і випарювали до сухого стану. Залишок очищали за допомогою препаративної рідинної хроматографії на (силікагель 20-45µм, 1000 г, рухома фаза (80 % гептану, 20 % AcOEt)). Очищені фракції збирали й концентрували з одержанням 16 г проміжної сполуки (11). Трифтороцтову кислоту (14,37 мл, 186,47 ммоль) додавали до розчину проміжної сполуки (11) (5,72 г, 18,65 ммоль) в CH2Cl2 (57 мл). Реакційну суміш перемішували при кімнатній температурі протягом 3 годин. K2CO3 (10 % водний розчин, 50 мл), а потім твердий K2CO3 додавали при 0 °C для підлуговування розчину. Органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану. Залишок очищали за допомогою препаративної рідинної хроматографії на (силікагель 20-45µм, 1000 г, рухома фаза (1 % NH4OH, 93 % DCM, 7 % MeOH)). Очищені фракції збирали й концентрували з одержанням 12 г проміжної сполуки (12). 13 UA 111210 C2 5 10 15 20 25 30 35 40 Проміжну сполуку (12) очищали і розділяли за допомогою хіральної SFC на (CHIRALPAK AD-H 5µм 250 × 20 мм). Рухома фаза (0,3 % ізопропіламін, 80 % CO2, 20 % метанол). Збирали 20 чисті фракції і видаляли розчинник з одержанням 5,8 г проміжної сполуки (14) (R*,S*) ([α] D = 20 12,4° (589 нм, конц-я 0,5 вага/об. %, DСM, 20 °C)) і 5,6 г проміжної сполуки (13) (S*,R*) ([α]D = +9,43° (589 нм, конц-я 0,35 вага/об. %, DСM, 20 °C)). Проміжна сполука (13) 1 H ЯМР (500MГц, DMSO-d6) δ (м.д.) 7,49 (dd, J=2,5, 5,0 Гц, 1 H), 7,31 (d, J=5,0 Гц, 1 H), 7,29 (d, J=2,5 Гц, 1 H), 5,88 (d, J=1,9 Гц, 1 H), 3,28-3,33 (br.s., 1 H), 2,75-2,87 (m, 4 H), 2,61 (dd, J=2,8, 10,7 Гц, 1 H), 2,54 (dd, J=3,3, 10,9 Гц, 1 H), 2,40-2,15(m, 2 H). Проміжна сполука (14) 1 H ЯМР (500MГц, DMSO-d6) δ (м.д.) 7,49 (dd, J=2,5, 5,0 Гц, 1 H), 7,31 (d, J=5,0 Гц, 1 H), 7,29 (d, J=2,5 Гц, 1 H), 5,88 (d, J=1,9 Гц, 1 H), 3,28-3,33 (br.s., 1 H), 2,75-2,87 (m, 4 H), 2,61 (dd, J=2,8, 10,7 Гц, 1 H), 2,54 (dd, J=3,3, 10,9 Гц, 1 H), 2,40-2,15(m, 2 H). Приклад А.4 Розчин проміжної сполуки (6) (108 г, 0,302 моль) і піридин-4-борної кислоти (49,5 г, 0,363 моль) в 2M водному карбонаті калію (302 мл, 0,604 моль) і диметиловому етері етиленгліколю (1,1 л) продували, використовуючи N2, протягом 5 хвилин, потім додавали тетракіс(трифенілфосфін)паладій (34,9 г, 0,030 моль), суміш нагрівали при 78 °C, використовуючи багаторежимну мікрохвильову піч (CEM Mars 5) з потужністю в діапазоні від 0 до 800 Вт протягом 1 години, охолоджували до кімнатної температури, додавали EtOAc, органічний шар відділяли, промивали водою, а потім сольовим розчином, сушили над MgSO4 і випарювали до сухого стану. Залишок очищали за допомогою препаративної рідинної хроматографії на (силікагель 15-40µм, 300 г, рухома фаза (0,1 % NH4OH, 97 % DCM, 3 % iPrOH). Збирали очищені фракції і видаляли розчинник з одержанням 47,6 г проміжної сполуки 15. Проміжну сполуку (16) очищали і виділяли за допомогою хроматографії на Chiralpak AD (20µм, 2000 г, 110 мм) з об'ємною швидкістю потоку 750 мл/хв. Рухома фаза являла собою 100 % метанол. Збирали чисті фракції і випарювали до сухого стану з одержанням 18,7 г 20 проміжної сполуки (18) (R*,S*) ([α]D = +55,75° (589 нм, конц-я 0,339 вага/об. %, DMF, 20 °C)) і 20 20,7 г проміжної сполуки (17) (S*,R*) ([α]D = -68,38° (589 нм, конц-я 0,253 вага/об. %, DMF, 20 °C)). Проміжна сполука (17) 1 H ЯМР (500 МГц, DMSO-d6) δ (м.д.) 8,52 (d, J=6,0 Гц, 2 H), 7,41 (d, J=6,0 Гц, 2 H), 6,50 (s, 1 H), 3,36-3,61 (m, 4 H), 2,81-3,02 (m, 3 H), 2,61-2,53 (m, 1 H), 1,36 (s, 9 H). Проміжна сполука (18) 1 H ЯМР (500 МГц, DMSO-d6) δ (м.д.) 8,52 (d, J=6,0 Гц, 2 H), 7,41 (d, J=6,0 Гц, 2 H), 6,50 (s, 1 H), 3,36-3,61 (m, 4 H), 2,81-3,02 (m, 3 H), 2,61-2,53 (m, 1 H), 1,36 (s, 9 H). Приклад А.5 14 UA 111210 C2 Проміжну сполуку (18) (24,8 г, 86,6 ммоль) додавали до HCl в діоксані (4 M, 108 мл) при 5 °C, потім суміш перемішували при кімнатній температурі протягом 90 хвилин. Отриманий осад відфільтровували, промивали діетиловим етером і сушили під вакуумом при 70 °C з одержанням 21,1 г проміжної сполуки (19). 5 10 15 20 25 30 35 40 Проміжну сполуку (20) одержували аналогічним способом, починаючи з проміжної сполуки (17). Приклад А-6. Реакцію проводили на 4-х партіях по 0,5 г кожна 6-бром-3,4-дигідро-1Н-[1,8]нафтиридин-2ону. Розчин 6-бром-3,4-дигідро-1H-[1,8]нафтиридин-2-ону (0,5 г, 2,20 ммоль), біс(пінаколато)дибору (0,67 г, 2,64 ммоль) та ацетату калію (0,648 г, 6,61 ммоль) в DMF (5 мл) і CH3CN (10 мл) перемішували і дегазували, використовуючи азот, протягом 10 хвилин. Додавали 1,1'-біс(дифенілфосфін)фероцендихлорпаладій(II) (0,161 г, 0,22 ммоль) і нагрівали при 120 °C, використовуючи мікрохвильову піч (Biotage initiator 60) з потужністю у діапазоні від 0 до 400 Вт, протягом 40 хвилин. Суміш випарювали до сухого стану, залишок розчиняли в DCM і воді, фільтрували через тонку подушку із целіту. Органічний шар фільтрату відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану. Залишок розчиняли в EtOH, відфільтровували та сушили з одержанням 0,36 г проміжної сполуки (21). Проміжну сполуку (21) (1,0 г, 3,65 ммоль), трет-бутилпропіонат (0,426 мл, 3,04 ммоль), оксид срібла(I) (1,06 г, 4,56 ммоль) і K2CO3 (0,84 г, 6,08 ммоль) у CH3CN (10 мл) і DMF (5мл) продували газом N2, потім додавали ацетат паладію(II) (47 % Pd) (0,034 г, 0,152 ммоль) і суміш нагрівали при 100 °C, використовуючи однорежимну мікрохвильову піч (Biotage initiator 60) з потужністю у діапазоні від 0 до 400 Вт протягом 20 хвилин. Додавали воду і EtOAc, суміш фільтрували через тонку подушку із целіту, органічний шар відділяли, промивали водою, а потім сольовим розчином, сушили (MgSO4) і випарювали до сухого стану. Отриманий залишок очищали за допомогою флеш-хроматографії на силікагелі (15-40µм, картридж 30 г, від CH2Cl2 до CH2Cl2/CH3OH/NH4OH: 98,5/1,5/0,1). Очищені фракції збирали і випарювали до сухого стану з одержанням 0,037 г проміжної сполуки (22). Проміжну сполуку (22) (0,053 г, 0,195 ммоль) розчиняли у розчині TFA/DCM (0,37 мл/0,5 мл). Реакційну суміш перемішували при кімнатній температурі протягом 30 хвилин. Реакційну суміш концентрували при зниженому тиску. Отриману тверду речовину розтирали з діетиловим етером, відфільтровували та сушили під вакуумом (80 °C) з одержанням 0,032 г проміжної сполуки (23). Приклад А.7 Умови мікрохвильової обробки: Biotage, 90 °C, 25 хвилин, повільно після 30 секунд попереднього перемішування. Розчин бромбензолу (0,228 мл, 2,64 ммоль), цис-2-третбутилокси-карбоніл-гексагідропіроло[3.4]піролу (0,6 г, 2,82 ммоль) і трет-бутоксиду натрію (0,624 15 UA 111210 C2 5 10 15 20 25 30 г, 6,5 ммоль) у толуолі (добре висушеному за допомогою молекулярних сит) (15 мл) перемішували та дегазували, використовуючи азот, протягом 10 хвилин. Додавали трис(дибензиліденацетон)дипаладій(0) (0,198 г, 0,216 ммоль) і 2-(ди-трет-бутилфосфіно)біфеніл (0,065 г, 0,216 ммоль) і отриману суміш повторно опромінювали, дотримуючись описаних вище умов мікрохвильової обробки. Додавали воду й етилацетат, а органічний шар відділяли і потім висушували (MgSO4), відфільтровували і концентрували. Очищення отриманого залишку проводили за допомогою флеш-хроматографії на силікагелі (15-40µм, 40 г, гептан/EtOAc 80/20). Очищені фракції збирали й концентрували з одержанням проміжної сполуки (24). TFA (4,54 мл, 58,95 ммоль) додавали до розчину проміжної сполуки (24) (1,7 г, 5,9 ммоль) в DCM (15 мл). Реакційну суміш перемішували при кімнатній температурі протягом 2 годин, додавали воду і DCM, K2CO3 (10 % водний розчин) додавали для підлуговування, а органічний шар відділяли, промивали водою, сушили (MgSO 4) і випарювали до сухого стану з одержанням проміжної сполуки (25) у вигляді масла. Наступні сполуки були отримані з використанням такої ж процедури, як і у прикладі A.7, де бромбензол був замінений на 2-бромтіофен, 2-броманізол, 2-бром-1-метилбензол, 2-бром-1хлорбензол, 3-бромпіридин, 2-бромтіазол, 4-бром-1-хлорбензол або 3-бром-1-хлорбензол, відповідно. Приклад А.8 Реакцію проводили в атмосфері N2. н-BuLi (1,6M у гексані) (3,33 мл, 5,33 ммоль) додавали по краплях при -20 °C до розчину DIPA (0,749 мл, 5,33 моль) в THF (8 мл), потім суміш перемішували при -20 °C протягом 20 хвилин. Потім додавали розчин проміжної сполуки (4) (1,0 г, 4,44 ммоль) в THF (10 мл) при -78 °C і перемішували отриману суміш протягом 30 хвилин при -78 °C. Додавали розчин N-феніл-трифторметану-метансульфоніміду (1,4 г, 4,88 моль) в THF (6 мл) при -78 °C, потім суміші дали нагрітися до кімнатної температури і перемішували протягом ночі. Суміш концентрували, а залишок очищали за допомогою флеш-хроматографії на силікагелі (40 г, 15-40 µм, гептан/EtOAc 70/30). Очищені фракції збирали і випарювали до сухого стану, одержуючи проміжну сполуку (34). Реакція в атмосфері азоту. Умови мікрохвильової обробки: Biotage initiator 60, 80 °C, 20 хвилин. Розчин проміжної сполуки (34) (0,42 г, 0,881 ммоль) і тіофен-2-борної кислоти (0,135 г, 1,06 ммоль) у K2CO3 (2 M, 0,88 мл) і диметиловому етері етиленгліколю (4 мл) продували N 2 16 UA 111210 C2 5 10 15 протягом 10 хвилин, потім додавали тетракіс(трифенілфосфін)паладій(0) (0,102 г, 0,088 ммоль). Суміш піддавали опромінювали піддавали впливу за описаних вище умов мікрохвильової обробки, охолоджували до кімнатної температури, додавали воду й EtOAc, органічний шар відділяли, промивали водою, а потім сольовим розчином, сушили (MgSO4) і випарювали до сухого стану. Очищення отриманого залишку проводили за допомогою флеш-хроматографії на силікагелі (10 г, 15-40µм, гептан 100 до гептан/EtOAc 80/20). Очищені фракції збирали і випарювали до сухого стану з одержанням проміжної сполуки (35). Суміш проміжної сполуки (35) (0,226 г, 0,776 ммоль) в TFA (0,7 мл) і DCM (4 мл) перемішували при кімнатній температурі протягом 1 години, потім реакційну суміш виливали в K2CO3 (10 % водний розчин) та екстрагували, використовуючи DCM. Органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану, одержуючи проміжну сполуку (36). Наступні сполуки були отримані з використанням такої ж процедури, як і у прикладі A.8b/A.8c, де тіофен-2-борна кислота була замінена на 2-метоксифеніл-борну кислоту або мурашину кислоту, відповідно. Приклад А.9 20 25 30 35 Умови мікрохвильової обробки: Biotage 60, 120 °C, 30 хвилин. Суміш цис-2-третбутилоксикарбоніл-гексагідропіроло[3.4]піролу (0,027 г, 0,13 ммоль), 2-бром-пропану (0,018 мл, 0,19 ммоль) і триетиламіну (0,088 мл, 0,64 моль) в DMF (0,2 мл) піддавали впливу випромінювання за описаних вище умов мікрохвильової обробки. Додавали воду і EtOAc, органічний шар відділяли, водний шар двічі екстрагували, використовуючи EtOAc, об'єднану органічну фазу промивали водою і сольовим розчином, сушили (MgSO 4) і випарювали до сухого стану, одержуючи проміжну сполуку (37). TFA (0,62 г, 8,02 ммоль) додавали до розчину проміжної сполуки (37) (0,204 г, 0,8 ммоль) в DCM (2 мл). Реакційну суміш перемішували при кімнатній температурі протягом 3 годин, додавали воду і DCM, 10 %-ний K2CO3 додавали для підлуговування, твердий NaCl додавали до насичення і відділяли органічний шар, промивали водою, сушили (MgSO 4) і випарювали до сухого стану з одержанням проміжної сполуки (38) у вигляді масла. Наступні сполуки були отримані з використанням такої ж процедури, як і у прикладі A.9, де 2-бромпропан був замінений на пропаргіл бромід, бензилсульфоніл хлорид або метансульфонат, відповідно. 17 UA 111210 C2 Приклад А.10 5 10 15 20 25 30 35 40 Реакція в атмосфері N2. BuLi (1,6M у гексані) (4,8 мл, 7,70 ммоль) додавали по краплях при 78 °C до розчину тіазолу (0,5 мл, 7,05 моль) в Et 2O (5 мл), потім суміш перемішували протягом 30 хвилин. Додавали розчин проміжної сполуки (5) (1,44 г, 6,41 ммоль) в Et2O (7 мл), потім суміш перемішували і давали їй нагрітися до кімнатної температури протягом 2 годин. Додавали воду й EtOAc, органічний шар відділяли, промивали водою, потім сольовим розчином, сушили над MgSO4 і випарювали до сухого стану. Очищення отриманого залишку проводили за допомогою флеш-хроматографії на силікагелі (50 г, 15-40µм, від гептан/EtOAc 80/20 до гептан/EtOAc 50/50). Очищені фракції збирали і випарювали до сухого стану з одержанням проміжної сполуки (44). Суміш проміжної сполуки (44) (1,05 г, 3,38 ммоль) в HCl (37 % в H2O) (7 мл) у запаяній трубці нагрівали при 140 °C, використовуючи мікрохвильову піч (Biotage Initiator EXP 60) з потужністю у діапазоні від 0 до 400 Вт протягом 1 години. Реакційну суміш виливали в K 2CO3 (10 %-ний водний розчин), органічний шар відділяли, сушили (MgSO 4) і випарювали до сухого стану з одержанням 0,23 г залишку (1). Водний шар випарювали до сухого стану, тверду речовину суспендували в DCM і перемішували протягом 10 хвилин. Суспензію фільтрували і фільтрат випарювали до сухого стану з одержанням 0,29 г залишку (2). Залишки (1) і (2) об'єднували для очищення, яке проводили за допомогою флеш-хроматографії на силікагелі (15-40µм, 30 г, від CH2Cl2 до CH2Cl2/CH3OH/NH4OH: 90/10/1). Очищені фракції збирали і випарювали до сухого стану з одержанням 0,41 г проміжної сполуки (45). Приклад А.11 Трифторид діетиламіносірки (1,24 мл, 10,12 ммоль) по краплях додавали до розчину проміжної сполуки (5) (0,570 г, 2,53 ммоль) в DCM (6 мл), охолоджували на льодяній бані при 5 °C, суміш перемішували протягом 1 години при 5 °C, а потім протягом ночі при кімнатній температурі. Суміш охолоджували при 0 °C і додавали насичений NaHCO 3. Органічний шар екстрагували, використовуючи CH2Cl2, сушили (MgSO4), фільтрували і концентрували з одержанням проміжної сполуки (46). TFA (0,39 г, 5,12 ммоль) додавали до розчину проміжної сполуки (46) (0,146 г, 0,51 ммоль) в DCM (1,2 мл). Реакційну суміш перемішували при кімнатній температурі протягом 3 годин, додавали воду і DCM, K2CO3 (10 % водний розчин) додавали для підлуговування і відділяли органічний шар, промивали водою, сушили (MgSO 4) і випарювали до сухого стану з одержанням проміжної сполуки (47) у вигляді масла. Приклад А.12 Суміш проміжної сполуки (7) (0,3 г, 1,05 ммоль) і 10 %-ного сухого Pd/C (0,06 г) у МеОН (15 мл) гідрогенізували при кімнатній температурі та атмосферному тиску протягом 2 годин. Отриману суміш фільтрували через тонку подушку із целіту, промивали, використовуючи DCM, і фільтрат випарювали до сухого стану з одержанням проміжної сполуки (48). 18 UA 111210 C2 5 10 15 20 25 30 Суміш проміжної сполуки (48) (0,286 г, 0,995 ммоль) і TFA (0,9 мл) в DCM (6 мл) перемішували при кімнатній температурі протягом 30 хвилин, потім реакційну суміш виливали в K2CO3 (10 % водний розчин) та екстрагували, використовуючи DCM. Органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану, одержуючи проміжну сполуку (49). Приклад А.13 Реакція в атмосфері N2. Умови мікрохвильової обробки: Biotage initiator 60, 80 °C, 20 хвилин. Розчин проміжної сполуки (38) (0,45 г, 1,26 ммоль) і 2-хлорфенілборної кислоти (0,236 г, 1,51 ммоль) у K2CO3 (2 M, 1,26 мл) і диметиловому етері етиленгліколю (5 мл) продували N 2 протягом 10 хвилин, потім додавали тетракіс(трифенілфосфін)паладій(0) (0,146 г, 0,126 ммоль). Суміш піддавали впливу випромінювання за описаних вище умов обробки, охолоджували до кімнатної температури, додавали воду і DCM, органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану. Залишок очищали за допомогою препаративної рідинної хроматографії на (силікагелі 15µм, 150 × 30,0 мм). Рухома фаза (100 % DCM). Цільові фракції збирали, а розчинник випарювали з одержанням проміжної сполуки (50). Суміш проміжної сполуки (50) (0,3 г, 0,938 ммоль) і TFA (0,9 мл) в DCM (6 мл) перемішували при кімнатній температурі протягом 30 хвилин, потім реакційну суміш виливали в K 2CO3 (10 % водний розчин) та екстрагували, використовуючи DCM. Органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану, одержуючи проміжну сполуку (51). Наступні сполуки були отримані з використанням такої ж процедури, як і у прикладі A.13, де 2-хлорфенілборна кислота було замінено на 2-метилфенілборну кислоту, 1-метил-1H-піразол-5борний пінаколовий естер, фуран-2-борну кислоту, 2-фторфеніл-борну кислоту, фуран-3-борну кислоту, 2-ціанофенілборну кислоту, 5-диметилізоксазол-4-борну кислоту, піридин-3-борну кислоту, 1-метил-4-(4,4,5,5-тетраметил-1,3,2-діоксаборан-2-іл)-1H-піразол, бензилцинк бромід, 2-хлорпіридин-3-борну кислоту, пінаколат піримідил-5-борної кислоти, пінаколовий естер 1ВОС-піразол-4-борної кислоти, 5-метилфуран-2-борну кислоту або 4-метокси-3-піридинілборну кислоту, відповідно. 19 UA 111210 C2 Приклад А.14 5 10 15 20 Проміжну сполуку (34) (2,798 ммоль), паладій(II)ацетат (47 % Pd) (0,14 ммоль), K2CO3 (4,198 ммоль), триметилоцтову кислоту (0,84 ммоль) і трициклогексилфосфонію тетрафторборат (0,196 ммоль) продували N2 у запаяній трубці. Додавали тіазол (4,198 ммоль) і DMA (10 мл) і реакційну суміш нагрівали при 100 °C протягом ночі. Додавали воду і EtOAc, органічний шар відділяли, промивали водою і сольовим розчином, сушили над MgSO 4 і випарювали до сухого стану. Отриманий залишок очищали за допомогою флеш-хроматографії на силікагелі (картридж 30 г, 15-40 µм, від гептан/EtOAc 80/20 до гептан/EtOAc 60/40). Очищені фракції збирали і випарювали до сухого стану, одержуючи проміжну сполуку (67). Розчин проміжної сполуки (67) (0,24 г, 0,821 ммоль) в TFA (0,8 мл) і DCM (5 мл) перемішували при кімнатній температурі протягом 30 хвилин, потім реакційну суміш виливали в K2CO3 (10 % водний розчин) та екстрагували, використовуючи DCM. Органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану, одержуючи 0,1 г проміжної сполуки (68). Приклад А.15 25 Pd(OAc)2 (1,3 мг, 0,0056 ммоль) додавали до розчину проміжної сполуки (34) (0,1 г, 0,28 ммоль), 1,3-біс(дифенілфосфін)пропану (4,6 мг, 0,011 ммоль) та ацетату калію (0,041 г, 0,42 ммоль) в EtOH (0,25 мл) і THF (2 мл) в атмосфері азоту. Суміш перемішували в атмосфері монооксиду вуглецю під тиском 5 бар протягом 18 годин в автоклаві із нержавіючої сталі з одержанням проміжної сполуки (69). 30 Розчин проміжної сполуки (69) (0,2 г, 0,711 ммоль) в HCl (4М у діоксані) (2 мл) перемішували при кімнатній температурі протягом 30 хвилин, потім випарювали до сухого стану, одержуючи 0,13 г проміжної сполуки (70). Приклад А.16 Pd(OAc)2 (25 мг, 0,112 ммоль) додавали до розчину проміжної сполуки (34) (2,0 г, 5,6 ммоль), 1,3-біс(дифенілфосфін)пропану (92 мг, 0,22 ммоль) та ацетату калію (0,82 г, 8,4 ммоль) в EtOH (5 мл) і THF (40 мл) в атмосфері азоту, потім суміш перемішували в атмосфері 20 UA 111210 C2 5 10 15 20 25 30 35 40 монооксиду вуглецю під тиском 5 бар при 100 °C протягом 18 годин в автоклаві із нержавіючої сталі. Реакційну суміш виливали у воду і EtOAc, органічний шар відділяли, промивали водою, а потім сольовим розчином, сушили (MgSO4), фільтрували і випарювали до сухого стану. Очищення отриманого залишку проводили за допомогою флеш-хроматографії на силікагелі (1540µм, 40 г, від гептан/EtOAc 90/10 до гептан/EtOAc 70/30). Очищені фракції збирали і випарювали до сухого стану з одержанням 0,61 г проміжної сполуки (71). Суміш проміжної сполуки (71) (0,3 г, 1,18 ммоль), диметиламіну в THF (2 M,1,18 мл, 2,37 ммоль), EDCI (0,27 г, 1,42 ммоль), HOBt (0,19 г, 6,21 ммоль) і триетиламіну (0,25 мл, 1,78 ммоль) в DCM (3 мл) і THF (3 мл) перемішували протягом ночі при кімнатній температурі. Додавали воду і DCM, органічний шар відділяли, сушили (MgSO 4) і випарювали до сухого стану, одержуючи 0,37 г проміжної сполуки (72). Розчин проміжної сполуки (72) (0,37 г, 1,32 ммоль) в HCl (4М у діоксані) (4 мл) перемішували при кімнатній температурі протягом 30 хвилин, потім реакційну суміш виливали в K 2CO3 (10 % водний розчин) та екстрагували, використовуючи DCM. Органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану, одержуючи проміжну сполуку (73). Приклад А.17 Суміш проміжної сполуки (71) (0,3 г, 1,18 ммоль), 1,1,1,3,3,3-гексаметилдисилазану (0,23 г, 1,42 ммоль), EDCI (0,27 г, 1,42 ммоль), HOBt (0,19 г, 6,21 ммоль) і триетиламіну (0,25 мл, 1,78 ммоль) в DCM (3 мл) і THF (3 мл) перемішували протягом ночі при кімнатній температурі. Додавали воду і DCM, органічний шар відділяли, сушили (MgSO 4) і випарювали до сухого стану. Отриманий залишок очищали за допомогою флеш-хроматографії на силікагелі (15-40µм, 10 г, від CH2Cl2 до CH2Cl2/CH3OH/NH4OH: 94/6/0,1). Очищені фракції збирали і випарювали до сухого стану з одержанням 0,16 г проміжної сполуки (74). Розчин проміжної сполуки (74) (0,16 г, 0,634 ммоль) в HCl (4М у діоксані) (2 мл) перемішували при кімнатній температурі протягом 30 хвилин, потім випарювали до сухого стану, одержуючи 0,1 г проміжної сполуки (75). Приклад А.18 Розчин проміжної сполуки (34) (0,2 г, 0,56 ммоль), біс(пінаколато)дибору (0,171 г, 0,67 ммоль) та ацетату калію (0,165 г, 1,68 ммоль) у діоксані (2 мл) перемішували й дегазували, використовуючи N2, протягом 10 хвилин. Додавали 1,1'біс(дифенілфосфін)фероцендихлорпаладій (II) (0,041 г, 0,056 ммоль) і реакційну суміш нагрівали при 100 °C, використовуючи мікрохвильову піч (Biotage Initiator EXP 60) з потужністю в діапазоні від 0 до 400 Вт протягом 20 хвилин. Додавали воду і EtOAc, органічний шар відділяли, промивали водою, потім сольовим розчином, сушили над MgSO 4 і випарювали до сухого стану. Очищення отриманого залишку проводили за допомогою флеш-хроматографії на силікагелі (10 г, 15-40µм, від гептан/EtOAc 85/15 до гептан/EtOAc 70/30). Очищені фракції збирали і випарювали до сухого стану з одержанням проміжної сполуки (76). 21 UA 111210 C2 5 10 15 20 25 30 35 40 45 Розчин проміжної сполуки (76) (0,45 г, 1,34 ммоль) і 2-бром-5-метил-1,3,4-тіадіазолу (0,288 г, 1,61 ммоль) у K2CO3 (2 M, 1,34 мл, 2,69 ммоль) і диметиловому етері етиленгліколю (5 мл) перемішували і дегазували за допомогою N2 протягом 10 хвилин. Додавали тетракіс(трифенілфосфін)паладій(0) (0,155 г, 0,134 ммоль) і реакційну суміш нагрівали при 150 °C, використовуючи однорежимну мікрохвильову піч (Biotage Initiator EXP 60) з потужністю в діапазоні від 0 до 400 Вт протягом 5 хвилин. Додавали воду і EtOAc, органічний шар відділяли, промивали сольовим розчином, сушили (MgSO4) і випарювали до сухого стану. Очищення отриманого залишку проводили за допомогою флеш-хроматографії на силікагелі (картридж 30 г, 15-40µм, від DCM до DCM/MeOH/NH4OH: 98/2/0,1). Очищені фракції збирали і випарювали до сухого стану з одержанням проміжної сполуки (77). Розчин проміжної сполуки (77) (0,14 г, 0,455 ммоль) в HCl (4М у діоксані) (2 мл) перемішували при кімнатній температурі протягом 30 хвилин, потім реакційну суміш виливали в10 %-ний водний K2CO3 та екстрагували, використовуючи DCM. Органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану, одержуючи 81 мг проміжної сполуки (78). Приклад А.19 BuLi (1,6M у гексані) (4,2 мл, 6,66 ммоль) додавали по краплях до розчину 1-метилімідазолу (0,53 мл, 6,66 моль) в THF (5 мл) в атмосфері азоту при -78 °C, потім отриману суміш перемішували при 0 °C протягом 1 години. Реакційну суміш охолоджували до -78 °C, додавали розчин проміжної сполуки (5) (1,0 г, 4,44 ммоль) в THF (10 мл). Реакційну суміш перемішували протягом 2 годин при -78 °C, потім давали охолонути до кімнатної температури і перемішували протягом ночі. Додавали воду і EtOAc, органічний шар відділяли, промивали водою і сольовим розчином, сушили над MgSO4 і випарювали до сухого стану. Отриманий залишок очищали за допомогою флеш-хроматографії на силікагелі (15-40µм, 30г, від CH2Cl2 до CH2Cl2/CH3OH/NH4OH: 95/5/0,1). Очищені фракції збирали і випарювали до сухого стану з одержанням 0,54 г проміжної сполуки (79). Суміш проміжної сполуки (79) (0,54 г, 1,76 ммоль) в HCl (37 % в H2O) (5 мл) у запаяній трубці нагрівали при 140 °C, використовуючи однорежимну мікрохвильову піч (Biotage Initiator EXP 60) з потужністю у діапазоні від 0 до 400 Вт протягом 1 години. Реакційну суміш випарювали до сухого стану з одержанням 0,47 г проміжної сполуки (80). Приклад А.20 Реакцію проводили в безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, петролейний етер/етилацетат 1/1, УФ/фосфомолібденова кислота). н-Бутиллітій, 2,5M у гексані (4,28 мл, 10,7 ммоль) по краплях додавали (5 хв.) до розчину діізопропіламіну (1,51 мл, 10,7 ммоль) в THF (16 мл) при -20 °C. Суміш перемішували 15 хвилин при -20 °C і потім охолоджували до -78 °C. Розчин проміжної сполуки (95) (2,00 г, 8,88 ммоль) в THF (20 мл) додавали (5 хв) при -78 °C. Суміш перемішували при -78 °C протягом 2 годин. Розчин 2-[N, N-біс(трифторметил-сульфоніл)аміно]піридину (3,50 г, 9,77 ммоль) в THF (12,5 мл) додавали (5 хвилин) при -78 °C. Суміші потім давали знову нагрітися до кімнатної температури і перемішували 17 годин. Суміш нагрівали при 50 °C 22 UA 111210 C2 5 10 15 20 протягом 4 годин. Реакційну суміш гасили додаванням насиченого водного хлориду амонію (100 мл) та екстрагували етилацетатом (3 × 100 мл). Об'єднані органічні шари сушили (сульфатом натрію), фільтрували і концентрували. До отриманого залишку (6,07 г) додавали дихлорметан (50 мл), потім суміш відфільтровували з одержанням 1,30 г білої твердої речовини. Фільтрат концентрували, а потім очищали за допомогою колонкової флеш-хроматографії на силікагелі (елюент: петролейний етер/EtOAc від 100/0 до 60/40). Фракції продукту збирали і випарювали розчинник з одержанням 1,02 г проміжної сполуки (81). Реакцію проводили в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (петролейний етер/етилацетат 8/2, УФ/фосфомолібденова кислота). 5-Ацетил-2-тієнілборну кислоту (0,057 г, 0,336 ммоль) і 2M водний карбонат калію (0,280 мл, 0,560 ммоль) додавали до розчину проміжної сполуки (81) (0,100 г, 0,280 ммоль) в 1,2-диметоксиетані (5 мл). Суміш продували аргоном і додавали тетракіс(трифенілфосфін)паладій(0) (0,032 г, 0,028 ммоль). Потім суміш нагрівали при 80 °C протягом ночі. Суміш охолоджували до кімнатної температури, додавали воду (10 мл) і EtOAc (10 мл). Органічний шар відділяли, промивали водою (10 мл), потім сольовим розчином (10 мл), сушили (сульфатом натрію), фільтрували і випарювали під вакуумом до сухого стану. Очищення залишку проводили за допомогою колонкової хроматографії на силікагелі (елюент: петролейний етер/етилацетат 8/2). Цільові фракції збирали і розчинник випарювали з одержанням 0,076 г проміжної сполуки (82). O H S с) Одержання (cis) NH проміжної сполуки (83) H 25 30 Реакцію проводили в безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (петролейний етер/етилацетат 9/1, УФ). HCl, 4М у діоксані (3,33 мл, 13,3 ммоль) додавали до розчину проміжної сполуки (82) (0,444 г, 1,33 ммоль) у діоксані (9 мл). Реакційну суміш перемішували при кімнатній температурі протягом 70 годин і потім концентрували до сухого стану, одержуючи 0,370 г проміжної сполуки (83). Наступні сполуки були отримані з використанням такої ж процедури, як і у прикладі A.20b/A.20c, де 5-ацетил-2-тієнілборну кислоту було замінено на 4-метилтіофен-2-борну кислоту, 2-хлортіофен-3-борну кислоту, 4-метил-3-тіофен-борну кислоту, 2-ацетил-3-тіофенборну кислоту, 5-ціанотіофен-2-борну кислоту, 5-хлортіофен-2-борну кислоту, пінаколовий естер 5-метилтіофен-2-борної кислоти, пінаколовий естер 3-метилтіофен-2-борної кислоти або пінаколовий естер 3-метокситіофен-2-борної кислоти, відповідно. Cl H NH (cis) H H S S H H H проміжна сполука (84) (cis) NH (cis) NH S проміжна сполука (85) проміжна сполука (86) 35 O NC H S NH H S (cis) NH (cis) H Cl S H проміжна сполука (87) H проміжна сполука (88) проміжна сполука (89) H H H S S S NH NH (cis) H проміжна сполука (90) (cis) NH H (cis) H проміжна сполука (91) Приклад А.21 23 NH O (cis) H проміжна сполука (92) UA 111210 C2 5 10 15 20 25 30 35 40 45 Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 9/1, фосфомолібденова кислота). Метиллітій 1,6M у діетиловому етері (3,29 мл, 5,26 ммоль) додавали до суспензії йодиду міді(I) (0,794 г, 4,17 ммоль) в THF (5,0 мл) при 0 °C. Через 1 годину через трубочку додавали розчин проміжної сполуки (81) (0,355 г, 0,993 ммоль) в THF (2,1 мл) при 0 °C, промиваючи за допомогою THF (2,1 мл). Реакційну суміш перемішували при кімнатній температурі протягом ночі. Суміш гасили за допомогою насиченого розчину NH 4Cl (14 мл) і сушили до сухого стану. Очищення залишку проводили за допомогою колонкової хроматографії на силікагелі (елюент: пентан/етилацетат 95/5). Фракції продукту збирали і випарювали розчинник з одержанням 0,180 г проміжної сполуки (93). Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 9/1, фосфомолібденова кислота). HCl (4М у діоксані) (2,02 г, 8,06 ммоль) додавали до розчину проміжної сполуки (93) (0,180 г, 0,806 ммоль) в 1,4-діоксані (4,3 мл), розчин перемішували при кімнатній температурі протягом 65 годин, потім концентрували до сухого стану, одержуючи 0,141 г проміжної сполуки (94) (110 %). Приклад А.22 Реакцію проводили в безводних умовах, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 50/50, проявник: УФ/фосфомолібденова кислота. Розчин проміжної сполуки (4) (6,93 г, 31,0 ммоль) в THF (180 мл) гідрогенізували при кімнатній температурі (атмосферному тиску) з паладієм на вуглеці, 10 ваг. % нанесення (1,65 г), у якості каталізатора протягом 15 годин. Каталізатор відфільтровували на clarcel, осад на фільтрі промивали дихлорметаном (50 мл) і об'єднані фільтрати концентрували при зниженому тиску до сухого стану. Очищення отриманого залишку (7,26 г) проводили за допомогою колонкової хроматографії на силікагелі (елюент: петролейний етер/етилацетат від 80/20 до 50/50). Фракції продукту збирали і випарювали розчинник з одержанням 6,70 г проміжної сполуки (95). Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 6/4, DCIP). Комплекс лантан трихлорид літій 0,6М у THF (3,70 мл, 2,22 ммоль) додавали до розчину проміжної сполуки (95) (0,500 г, 2,22 ммоль) в THF (15 мл). Суміш перемішували при кімнатній температурі протягом 1 години, потім охолоджували до 0 °C. Додавали по краплях розчин етилмагнію броміду 0,1М у THF (2,66 мл, 2,66 ммоль) і реакційній суміші давали нагрітися до кімнатної температури, перемішували протягом 18 годин. Реакційну суміш гасили додаванням насиченого водного хлориду амонію (50 мл) та екстрагували етилацетатом (3 × 50 мл). Об'єднані органічні шари сушили (Na2SO4), фільтрували і концентрували. Очищення отриманого залишку (0,635 г) проводили за допомогою колонкової хроматографії на силікагелі (елюент: петролейний етер/етилацетат від 9/1 до 7/3). Фракції продукту збирали і випарювали розчинник. Очищення отриманого залишку (0,410 г) проводили за допомогою колонкової хроматографії на силікагелі (елюент: петролейний етер/етилацетат 8/2). Фракції продукту збирали і випарювали розчинник з одержанням 0,235 г проміжної сполуки (96). 24 UA 111210 C2 5 10 15 20 25 30 35 40 45 Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням 1Н ЯМР. HCl у діоксані (4М, 2,30 мл, 9,20 ммоль) додавали до розчину проміжної сполуки (96) (0,235 г, 0,920 ммоль) у діоксані (2 мл). Реакційну суміш перемішували при 60 °C протягом 18 годин. Після охолодження до кімнатної температури осад відфільтровували на склоподібній фриті і промивали діетиловим етером (20 мл) з одержанням 0,126 г твердої речовини. Фільтрат випарювали до сухого стану з одержанням 0,077 г залишку. Тверду речовину й залишок поєднували і розчиняли у діоксані (2 мл). Додавали 4M HCl у діоксані (2,30 мл, 9,20 ммоль) і суміш перемішували при 60 °C протягом 24 годин, потім - при 100 °C протягом 72 годин. Реакційну суміш концентрували до сухого стану з одержанням 0,158 г проміжної сполуки (97). Приклад А.23 Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 1/1, фосфомолібденова кислота). Боргідрид натрію (0,893 г, 23,6 ммоль) додавали порціями протягом 30 хвилин до розчину проміжної сполуки (95) (2,66 г, 11,8 ммоль) у МеОН (60 мл) при 0 °C. Реакційну суміш перемішували протягом 1 години при 0 °C, а потім концентрували до сухого стану. Залишок розбавляли етилацетатом (200 мл) і промивали водою (100 мл), 1М водною соляною кислотою (100 мл) і сольовим розчином (100 мл). Органічний шар висушували (Na2SO4), фільтрували і концентрували з одержанням 2,27 г проміжної сполуки (98). Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 1/1, фосфомолібденова кислота). Метансульфоніл хлорид (0,930 мл, 11,9 ммоль) по краплях додавали до розчину проміжної сполуки (98) (2,27 г, 9,98 ммоль) і триетиламіну (4,17 мл, 29,9 ммоль) в DCM (50 мл) при 0 °C. Реакційну суміш перемішували протягом 1 години при кімнатній температурі, а потім концентрували до сухого стану. Залишок розбавляли етилацетатом (200 мл) і промивали водою (100 мл), сольовим розчином (100 мл), 1М водною соляною кислотою (100 мл) і знову сольовим розчином (100 мл). Органічний шар висушували (Na 2SO4), фільтрували і концентрували. Очищення отриманого залишку (2,52 г) проводили за допомогою колонкової хроматографії на силікагелі (елюент: петролейний етер/етилацетат від 8/2 до 5/5). Фракції продукту збирали і випарювали розчинник з одержанням 2,39 г проміжної сполуки (99). Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 8/2, нінгідрин/фосфомолібденова кислота). Проміжну сполуку (99) (0,300 г, 0,982 ммоль) розчиняли в DMF (3 мл) і суміш охолоджували до 0 °C. Додавали пірол (0,102 мл, 1,47 ммоль) і гідрид натрію, 60 %-у дисперсію у мінеральному маслі (0,0589 г, 1,47 ммоль) і реакційну суміш перемішували при кімнатній температурі 18 годин. Реакційну суміш розбавляли етилацетатом (50 мл) і промивали водою (2 × 50 мл), а потім сольовим розчином (3 × 50 мл). Органічний шар висушували (Na2SO4), фільтрували і концентрували. Очищення отриманого залишку (0,290 г) проводили за допомогою колонкової хроматографії на силікагелі (елюент: петролейний етер/етилацетат від 98/2 до 95/5, а потім до 90/10). Фракції продукту збирали і випарювали розчинник з одержанням 0,175 г проміжної сполуки (100). Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 6/4, фосфомолібденова кислота). 4М HCl у діоксані (1,58 мл, 6,33 ммоль) додавали до розчину проміжної сполуки (100) (0,175 г, 0,633 ммоль) у діоксані (3 мл). Реакційну суміш перемішували 25 UA 111210 C2 5 при 50 °C протягом 2 годин і концентрували до сухого стану, одержуючи 0,135 г проміжної сполуки (101). Наступні сполуки були отримані з використанням такої ж процедури, як і в прикладі A.23c/A.23d, де пірол був замінений на тетразол, піразол, 1,2,4-триазол, 1,2,3-триазол або фенол, відповідно. Приклад А.24 10 15 20 25 30 Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 8/2, нінгідрин/фосфомолібденова кислота). 25 % (ваг.) розчин метоксиду натрію в метанолі (0,449 мл, 1,96 ммоль) додавали до розчину проміжної сполуки (99) (0,300 г, 0,982 ммоль) у МеОН (4 мл). Реакційну суміш перемішували при кип'ятінні зі зворотним холодильником протягом 20 годин. Реакційну суміш концентрували до сухого стану. Залишок розбавляли етилацетатом (50 мл) і промивали водою (50 мл), а потім сольовим розчином (50 мл). Органічний шар висушували (Na2SO4), фільтрували і концентрували. Очищення отриманого залишку проводили за допомогою колонкової хроматографії на силікагелі (елюент: петролейний етер/етилацетат від 100/0 до 97/3, а потім до 1/1). Фракції продукту збирали і випарювали розчинник з одержанням 0,182 г проміжної сполуки (109). Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, елюент: петролейний етер/етилацетат 8/2, нінгідрин/фосфомолібденова кислота). 4М HCl у діоксані (1,88 мл, 7,54 ммоль) додавали до розчину проміжної сполуки (109) (0,182 г, 0,754 ммоль) у діоксані (4 мл). Реакційну суміш перемішували при кімнатній температурі протягом 18 годин, а потім при 50 °C протягом 2 годин. Реакційну суміш концентрували до сухого стану з одержанням 0,139 г проміжної сполуки (110). Деякі із проміжних сполук, використовуваних в одержанні кінцевих сполук, можна, наприклад, придбати. В. Одержання кінцевих сполук Приклад В.1 Суміш проміжної сполуки (3) (9,4 г, 44,3 ммоль), проміжної сполуки (9) (8,5 г, 44,3 ммоль), 26 UA 111210 C2 5 10 15 20 25 30 35 40 45 гідроксибензотриазолу (6,0 г, 44,3 ммоль) і триетиламіну (15,4 мл, 0,111 ммоль) в CH 2Cl2 (160 мл) і THF (160 мл) перемішували протягом ночі при кімнатній температурі. Додавали воду (175 мл), осад відфільтровували, промивали водою/EtOH (50 мл). Тверду речовину суспендували в EtOH (50 мл) і перемішували 15 хвилин. Отриману суспензію відфільтровували і сушили під вакуумом при 70 °C з одержанням 7,3 г сполуки (14) у вигляді білого порошку (т.пл. = 266 °C), 20 ([α]D = -105,1° (589 нм, конц-я 0,1275 вага/об. %, CH2Cl2, 20 °C). 1 H ЯМР (500MГц, DMSO-d6) δ (м.д.) 10,64 (d, J=7,6 Гц, 1 H), 8,33 (dd, J=1,7, 9,6 Гц, 1 H), 8,04 (d, J=17,3 Гц, 1 H), 7,48 (d, J=7,6 Гц, 2 H), 7,31-7,42 (m, 3 H), 7,23-7,28 (m, 1 H), 6,99 (t, J=15,0 Гц, 1 H), 6,20 (d, J=6,0 Гц, 1 H), 3,38-4,04 (m, 5 H), 3,09-3,21 (m, 1 H), 2,85-3,04 (m, 3 H), 2,55-2,67 (m, 3 H). Приклад В.2 Суміш проміжної сполуки (14) (5,8 г, 30,32 ммоль), проміжної сполуки (3) (7,72 г, 30,32 ммоль), 1-гідроксибензотриазолу (4,92 г, 30,38 ммоль), EDCI (6,97 г, 30,38 ммоль) і триетиламіну (14,71 мл, 106,12 ммоль) в CH2Cl2 (100 мл) і THF (100 мл) перемішували протягом ночі при кімнатній температурі. Суміш виливали у воду. Осад відфільтровували, двічі промивали, використовуючи EtOH, і сушили під вакуумом при 65 °C. Цей осад перекристалізовували з EtOH, відфільтровували й сушили під вакуумом при 62 °C з одержанням 20 9,02 г сполуки (44) у вигляді білого порошку (т.пл. = 264 °C), ([α]D = +170,12° (589 нм, конц-я 0,2075 вага/об. %, CH2Cl2, 20 °C). 1H ЯМР (400MГц, DMSO-d6) δ (м.д.) 10,63 (d, J=4,5 Гц, 1 H), 8,32 (d, J=5,1 Гц, 1 H), 8,03 (d, J=10,6 Гц, 1 H), 7,52 (dd, J=2,8, 4,8 Гц, 1 H), 7,41 (br. s., 1 H), 7,36 (dd, J=4,8, 9,3 Гц, 2 H), 6,98 (dd, J=9,1, 15,7 Гц, 1 H), 6,01 (br. s., 1 H), 3,35-4,03 (m, 5 H), 2,94-3,21 (m, 2 H), 2,90 (q, J=7,9 Гц, 3 H), 2,52-2,62 (m, 2 H). Приклад В.3 Суміш проміжної сполуки (19) (21,1 г, 81,4 ммоль), проміжної сполуки (3) (17,3 г, 61,8 ммоль), 1-гідроксибензотриазолу (11,0 г, 81,4 ммоль), EDCI (15,6 г, 81,4 ммоль) і триетиламіну (47 мл, 0,339 ммоль) в CH2Cl2 (350 мл) і THF (350 мл) перемішували протягом ночі при кімнатній температурі. До суміші додавали воду. Осад відфільтровували, промивали водою/EtOH, а потім EtOH, і сушили під вакуумом при 70 °C з одержанням 12,7 г сполуки (40) у вигляді білого 20 порошку (т.пл. = 271 °C), ([α]D = +116,08° (589 нм, конц-я 0,2145 вага/об. %, CH2Cl2, 20 °C). 1 H ЯМР (400MГц, DMSO-d6) δ (м.д.) 10,63 (d, J=5,1 Гц, 1 H), 8,52 (d, J=5,6 Гц, 2 H), 8,33 (d, J=6,1 Гц, 1 H), 8,03 (d, J=13,6 Гц, 1 H), 7,41-7,46 (d, J=15,7 Гц, 2 H), 7,38 (d, J=4,0 Гц, 1 H), 6,98 (dd, J=11,6, 15,7 Гц, 1 H), 6,53 (d, J=8,1 Гц, 1 H), 3,37-4,04 (m, 5 H), 2,86-3,22 (m, 5 H), 2,58-2,70 (m, 2 H). Сполуку (41) отримували аналогічно шляхом взаємодії проміжної сполуки (20) із проміжною сполукою (3), дотримуючись такої ж процедури. 1 H ЯМР (500MГц, DMSO-d6) δ (м.д.) 10,63 (d, J=5,1 Гц, 1 H), 8,52 (d, J=5,6 Гц, 2 H), 8,33 (d, J=6,1 Гц, 1 H), 8,03 (d, J=13,6 Гц, 1 H), 7,41-7,46 (d, J=15,7 Гц, 2 H), 7,38 (d, J=4,0 Гц, 1 H), 6,98 (dd, J=11,6, 15,7 Гц, 1 H), 6,53 (d, J=8,1 Гц, 1 H), 3,37-4,04 (m, 5 H), 2,86-3,22 (m, 5 H), 2,58-2,70 (m, 2 H). 20 ([α]D = -115,85° (589 нм, конц-я 0,183 вага/об. %, CH2Cl2, 20 °C)). Приклад В.4 27 UA 111210 C2 5 10 15 20 25 30 35 40 45 Реакцію проводили в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (силікагель, CH2Cl2/метанол/триетиламін 95/5/0,1, УФ/фосфомолібденова кислота). 1-(3-Диметиламінопропіл)-3-етилкарбодіімід (.HCl) (1,70 г, 8,87 ммоль) додавали до суміші проміжної сполуки (3) (2,02 г, 7,39 ммоль), неочищеного цисгексагідро-циклопента[c]пірол-5(1H)ону (1,85 г, максимально 8,89 ммоль), 1гідроксибензотриазолу моногідрату (1,36 г, 8,87 ммоль) і N-етилдіізопропіламіну (6,32 мл, 36,9 ммоль) в DMF (75 мл). Суміш перемішували при кімнатній температурі протягом 18 годин. Суміш концентрували при зниженому тиску, розбавляли дихлорметаном (150 мл) і промивали насиченим водним NaHCO3 (100 мл). Водний шар знову екстрагували дихлорметаном (2 × 150 мл). Об'єднані органічні шари промивали сольовим розчином (400 мл), висушували (Na 2SO4), фільтрували і концентрували при зниженому тиску до сухого стану. Очищення отриманого залишку проводили за допомогою колонкової хроматографії на силікагелі (елюент: дихлорметан/метанол від 100/0 до 94/6). Фракції продукту збирали і випарювали розчинник. Лужні водні шари знову екстрагували дихлорметаном (3 × 300 мл). Об'єднані органічні шари промивали сольовим розчином (900 мл), сушили (Na2SO4), фільтрували і концентрували при зниженому тиску до сухого стану. Очищення отриманого залишку проводили за допомогою колонкової хроматографії на силікагелі (елюент: дихлорметан/метанол від 100/0 до 94/6). Фракції продукту збирали і випарювали розчинник. Цільові залишки об'єднували з одержанням 1,58 г проміжної сполуки (111). Реакцію проводили у безводних умовах в атмосфері аргону, спостерігаючи за її ходом з використанням тонкошарової хроматографії (петролейний етер/етилацетат 95/5, УФ/фосфомолібденова кислота). Комплекс лантан трихлорид літій хлорид 0,6М у THF (2,38 мл, 1,43 ммоль) додавали до суспензії проміжної сполуки (111) (0,464 г, 1,43 ммоль) в THF (18 мл). Суміш перемішували при кімнатній температурі протягом 1 години, потім охолоджували до 0 °C. По краплях додали розчин фенілмагній броміду, 0,1М у THF (3,57 мл, 3,57 ммоль). Реакційну суміш перемішували і дозволияли нагрітися знову до кімнатної температури протягом 3 днів. Додатково по краплях додали 1,0М розчин фенілмагній броміду в THF (2,85 мл, 2,85 ммоль, 2 еквівалента). Суміш перемішували при кімнатній температурі протягом 2 годин. Реакційну суміш гасили додаванням насиченого водного хлориду амонію (30 мл) та екстрагували етилацетатом (3 × 30 мл). Об'єднані органічні шари сушили (Na 2SO4), фільтрували і концентрували при зниженому тиску до сухого стану. Отриманий залишок очищали колонковою флешхроматографією на силікагелі (елюент: CH2Cl2/MeOH від 100/0 до 95/5). Розчинник із зібраних фракцій продукту випарювали. Залишок розтирали з діетиловим етером (2 × 3 мл) і потім сушили під вакуумом з одержанням 0,077 г сполуки (71). Приклад В.5 Суміш проміжної сполуки (23) (0,032 г, 0,097 ммоль), проміжної сполуки (8) (0,032 г, 0,145 ммоль), EDCI (0,022 г, 0,0116 ммоль), HOBT (0,016 г, 0,116 ммоль) і триетиламіну (0,049 мл, 0,349 ммоль) в DCM (1 мл) і THF (1 мл) перемішували протягом ночі при кімнатній температурі. Додавали воду, суміш екстрагували, використовуючи DCM, органічний шар відділяли, промивали водою, сушили (MgSO4) і випарювали до сухого стану. Залишок перекристалізовували з EtOH, тверду речовину відфільтровували, промивали EtOH і сушили (вакуум, 70 °C) з одержанням 0,015 г сполуки (73). У Таблиці F-1 перераховані сполуки, які були отримані згідно одного з наведених вище прикладів. 28
ДивитисяДодаткова інформація
Назва патенту англійськоюAntibacterial cyclopenta[c]pyrrole substituted 3,4-dihydro-1h-[1,8]naphthyridinones
Автори англійськоюGuillemont, Jerome, Emile, Georges, Lancois, David, Francis, Alain, Motte, Magali, Madeleine, Simone, Koul, Anil, Balemans, Wendy, Mia, Albert, Arnoult, Eric, Pierre, Alexandre
Назва патенту російськоюАнтибактериальные 3,4-дигидро-1н-[1,8]нафтиридиноны, замещенные циклопента[c]пирролом
Автори російськоюЖильмон Жером Эмиль Жорж, Лансуа Давид Франсис Ален, Мотт Магали Мадлен Симон, Коул Анил, Балеманс Уенди Миа Альберт, Арну Эрик Пьер Александр
МПК / Мітки
МПК: C07D 471/04, C07D 487/04, A61K 31/4375, A61P 31/04
Мітки: циклопента[c]піролом, 3,4-дигідро-1н-[1,8]нафтиридинони, заміщені, антибактеріальні
Код посилання
<a href="https://ua.patents.su/45-111210-antibakterialni-34-digidro-1n-18naftiridinoni-zamishheni-ciklopentacpirolom.html" target="_blank" rel="follow" title="База патентів України">Антибактеріальні 3,4-дигідро-1н-[1,8]нафтиридинони, заміщені циклопента[c]піролом</a>
Заміщені піролом 2-індолінони, фармацевтична композиція, спосіб модуляції каталітичної активності протеїнкінази та спосіб лікування або профілактики захворювання, пов’язаного з протеїнкіназою

Номер патенту: 73976
Опубліковано: 17.10.2005
Автори: Хоулі Майкл, Ліанг Конгксін, Лі Ксіаоюан, Шіразян Шахрзад, Сан Лі, Уей Чанг Чен, Войковскі Томас, Міллер Тодд, Танг Пенг Чо, Нематалла Асаад С.
МПК: A61P 35/00, A61K 31/506, A61P 29/00, A61K 31/55, A61P 19/02, C07D 401/12, C07D 403/14, A61P 43/00, A61P 3/10, A61K 31/404, A61P 17/06, A61K 31/4178, A61K 31/4439, A61K 31/496, C07D 403/12, A61K 31/5377, C07D 403/06, C07D 209/44, A61P 9/00, C07D 207/33, C07D 401/14, A61P 37/00, A61P 37/06, C07D 521/00
Мітки: лікування, 2-індолінони, пов'язаного, фармацевтична, заміщені, каталітичної, захворювання, піролом, протеїнкіназою, композиція, спосіб, модуляції, протеїнкінази, профілактики, активності
Формула / Реферат:
1. Сполука формули (I): , (І)у якій R1 вибраний з групи, яка включає водень, галоген, алкіл, циклоалкіл, арил, гетероарил, гетероаліциклічну групу, гідроксигрупу, алкоксигрупу, -C(O)R15, -NR13R14, -(CH2)rR16 та -C(O)NR8R9;R2 вибраний з групи, яка включає водень, галоген, алкіл, тригалометил, гідроксигрупу, алкоксигрупу, -NR13R14,...
1,3-дизаміщені похідні 6,7-дигідро-1н-циклопента[d]піримідин-2,4(3н, 5н)-діону, що проявляють спазмолітичну активність

Номер патенту: 40845
Опубліковано: 27.04.2009
Автори: Притула Тетяна Павлівна, Кононевич Юрій Миколайович, Демченко Анатолій Михайлович, Пупишева Олена Володимирівна, Мохорт Микола Антонович
МПК: A61K 31/517, C07D 239/00
Мітки: спазмолітичну, проявляють, похідні, 1,3-дизаміщені, 6,7-дигідро-1н-циклопента[d]піримідин-2,4(3н, активність, 5н)-діону
Формула / Реферат:
1,3-дизаміщені похідні 6,7-дигідро-1H-циклопента[d]піримідин-2,4(3H, 5H)-діону, загальної формули:,деR=(3-Cl, 4-Cl)-Ph, (4-Br)-Ph, CONHPh, CONHNH-SO2-(4-Me)Ph, CONH-(4-Cl)-Ph, CONH-(4-N-Ac)-Ph, CONH-(3-Cl, 4-OMe)-Ph, що проявляють спазмолітичну активність.
7-(1,3-бензотіазол-2-ілтіо)-3-метил-6,7-дигідро-1н-циклопента[d]піримідин-2,4(3н,5н)-діон, що проявляє аналгетичну активність

Номер патенту: 80383
Опубліковано: 27.05.2013
Автори: Кононевич Юрій Миколайович, Демченко Анатолій Михайлович, Шуть Дмитро Миколайович, Бухтіарова Тетяна Анатоліївна, Бершова Тетяна Анатоліївна, Бобкова Людмила Станіславівна, Ядловський Олег Євгенович
МПК: C07B 43/00, C07D 291/00
Мітки: активність, анальгетичну, 7-(1,3-бензотіазол-2-ілтіо)-3-метил-6,7-дигідро-1н-циклопента[d]піримідин-2,4(3н,5н)-діон, проявляє
Формула / Реферат:
7-(1,3-Бензотіазол-2-ілтіо)-3-метил-6,7-дигідро-1H-циклопента[d]піримідин-2,4(3H,5H)-діон формули:,що проявляє аналгетичну активність.
Заміщені 3-арил-10,11-дигідро-4,10-метанопіразоло[4,3-с][1,5] бензоксазоцин-4(1н)карбонові кислоти та спосіб їх одержання

Номер патенту: 95886
Опубліковано: 12.09.2011
Автори: Чебанов Валентин Анатолійович, Мурликіна Марина Володимирівна, Афанасіаді Людмила Михайлівна, Сахно Яна Ігорівна, Десенко Сергій Михайлович
МПК: C07D 498/08, C07D 231/54, C07D 498/18, C07D 267/00
Мітки: заміщені, спосіб, бензоксазоцин-4(1н)карбонові, 3-арил-10,11-дигідро-4,10-метанопіразоло[4,3-с][1,5, кислоти, одержання
Формула / Реферат:
1. Заміщені 3-арил-10,11-дигідро-4,10-метанопіразоло[4,3-с][1,5]бензоксазоцин-4(1H)карбонові кислоти загальної формули,де R1=C6H5, 4-CH3O-C6H4, 4-Br-C6H4, 4-Cl-C6H4, 4-C2H5-C6H4; R2=H, 6-CH3O, 8-Cl. 2. Спосіб одержання сполук загальної формули,де...
Заміщені дигідро-3-гало-1н-піразол-5-карбоксилати та способи їх одержання

Номер патенту: 81104
Опубліковано: 10.12.2007
Автори: Селбі Томас Пол, Фройденбергер Джон Герберт, Лам Джордж Філіп, Стівенсон Томас Мартін
МПК: C07D 401/04, A01N 43/56, C07D 231/22, C07D 231/06, C07D 231/16, C07D 231/08, C07D 231/14
Мітки: одержання, дигідро-3-гало-1н-піразол-5-карбоксилати, способи, заміщені
Формула / Реферат:
1. Сполука формули І , (I)де R1 являє собою галоген;кожна R2 являє собою незалежно C1-C4 алкіл, С2-С4 алкеніл, С2-С4 алкініл, С3-С6 циклоалкіл, C1-C4 галоалкіл, С2-С4 галоалкеніл, С2-С4 галоалкініл, С3-С6 галоциклоалкіл, галоген, CN, NO2, C1-C4 алкокси, С1-С4 галоалкокси, C1-C4 алкілтіо, C1-C4 алкілсульфініл, C1-C4 алкілсульфоніл, C1-C4 алкіламіно,...
Попередній патент: Спосіб модифікування метанвмісного газового потоку
Наступний патент: Гербіцидна композиція
Випадковий патент: Спосіб моделювання розлитого жовчного перитоніту