Спосіб одержання хіназолінових алкалоїдів
Номер патенту: 81287
Опубліковано: 25.12.2007
Автори: Моорманн Йоахім, Матуш Рудольф, Хоффманн Ханс-Райнер





Формула / Реферат
1. Спосіб одержання сполуки наступної формули (І)
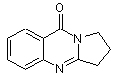 (I),
(I), 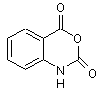 (II)
(II)
шляхом взаємодії сполуки формули (II) з 2-піролідоном, який відрізняється тим, що
використовують надлишок 2-піролідону, по відношенню до сполуки (II).
2. Спосіб за п. 1, який відрізняється тим, що в наступній стадії продукт реакції (І) виділяють безпосередньо з реакційної суміші шляхом кристалізації.
3. Спосіб за пп. 1 або 2, який відрізняється тим, що застосовують від 1,5 до 5 моль, переважно від 2 до 4 моль, особливо переважно від 2,5 до 3,5 моль 2-піролідону по відношенню до кількості сполуки (II).
4. Спосіб за будь-яким з попередніх пунктів, який відрізняється тим, що реакцію здійснюють при температурі від 50 до 200 °С.
5. Спосіб за п. 4, який відрізняється тим, що реакційну суміш спочатку нагрівають до температури від 70 до 130 °С, переважно до температури від 80 до 120 °С, із наступним нагріванням до температури від 140 до 200 °С, переважно від 150 до 190 °С.
6. Спосіб за п. 5, який відрізняється тим, що згадану початкову температуру підтримують протягом періоду від 0,5 до 2 годин, переважно від 1 до 1,5 годин і згадану наступну температуру підтримують протягом періоду від 1 до 8 годин, переважно протягом від 2 до 5 годин.
7. Спосіб за п. 6, який відрізняється тим, що в реакційну суміш після охолодження вносять затравочні кристали сполуки (І) і підтримують при кімнатній температурі, переважно принаймні при 25°С, протягом періоду від 24 годин до 7 днів, переважно протягом від 50 до 100 годин, залишають кристалізуватися.
8. Спосіб за п. 7, який відрізняється тим, що кристалізацію здійснюють при температурі від 30 до 70 °С, переважно при температурі від 40 до 60 °С.
9. Спосіб одержання сполуки наступної формули (III), що включає наступні стадії:
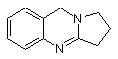 (III)
(III)
(A) одержання сполуки (І) згідно з способом за будь-яким з пунктів 1-8;
(B) реакцію відновлення з одержанням сполуки (III) у формі солі;
(C) виділення сполуки (III) з солі.
10. Спосіб за п. 9, який відрізняється тим, що продукт реакції, виділений шляхом кристалізації згідно з будь-яким з пунктів від 1 до 8, застосовують як сполуку (І).
11. Спосіб за пп. 9 або 10, який відрізняється тим, що реакцію відновлення (стадія В) здійснюють у присутності цинку і кислоти.
12. Спосіб за п. 11, який відрізняється тим, що сполуку (І), переважно у вигляді кристалізованої форми, спочатку розчиняють у льодяній оцтовій кислоті і потім додають цинк і хлорводневу кислоту.
13. Спосіб за п. 11, який відрізняється тим, що реакцію відновлення здійснюють в присутності водного розчину сірчаної кислоти і цинкового пилу.
14. Спосіб за будь-яким з пп. 9-13, який відрізняється тим, що в стадії (В) сполуку (III) виділяють шляхом кристалізації з реакційної суміші у вигляді солі.
15. Спосіб за будь-яким з пп. 9-14, який відрізняється тим, що в стадії (С) сполуку (III) виділяють з солі шляхом додавання основи, переважно шляхом додавання NaOH.
16. Спосіб за п. 15, який відрізняється тим, що стадію (С) здійснюють при нагріванні, і сполуку (III) виділяють з її солі у розплавленій формі.
17. Спосіб за п. 16, який відрізняється тим, що сполуку (III), що присутня у вигляді розплавленої форми, охолоджують і після заморожування кристалізують з водного лужного розчину.
18. Спосіб за п. 9, який відрізняється тим, що сполуку (III) виділяють і відокремлюють від її солі у вигляді вільної основи в розплавленій формі.
19. Спосіб за п. 18, який відрізняється тим, що сполуку (III), що присутня у вигляді розплавленої форми, охолоджують і після заморожування кристалізують з водного лужного розчину.
20. Спосіб за п. 9, який відрізняється тим, що до сполуки (ІІІ) у формі солі додають основу, після чого сполуку (ІІІ) відокремлюють у рідкій формі.
Текст
1. Спосіб одержання сполуки наступної формули (І) O O O N N O H N (II) (I), шляхом взаємодії сполуки формули (II) з 2піролідоном, який відрізняється тим, що використовують надлишок 2-піролідону, по відношенню до сполуки (II). 2. Спосіб за п. 1, який відрізняється тим, що в наступній стадії продукт реакції (І) виділяють безпосередньо з реакційної суміші шляхом кристалізації. 3. Спосіб за пп. 1 або 2, який відрізняється тим, що застосовують від 1,5 до 5 моль, переважно від 2 до 4 моль, особливо переважно від 2,5 до 3,5 моль 2-піролідону по відношенню до кількості сполуки (II). 4. Спосіб за будь-яким з попередніх пунктів, який відрізняється тим, що реакцію здійснюють при температурі від 50 до 200 °С. 2 (19) 1 3 81287 4 13. Спосіб за п. 11, який відрізняється тим, що реакцію відновлення здійснюють в присутності водного розчину сірчаної кислоти і цинкового пилу. 14. Спосіб за будь-яким з пп. 9-13, який відрізняється тим, що в стадії (В) сполуку (III) виділяють шляхом кристалізації з реакційної суміші у вигляді солі. 15. Спосіб за будь-яким з пп. 9-14, який відрізняється тим, що в стадії (С) сполуку (III) виділяють з солі шляхом додавання основи, переважно шляхом додавання NaOH. 16. Спосіб за п. 15, який відрізняється тим, що стадію (С) здійснюють при нагріванні, і сполуку (III) виділяють з її солі у розплавленій формі. 17. Спосіб за п. 16, який відрізняється тим, що сполуку (III), що присутня у вигляді розплавленої форми, охолоджують і після заморожування кристалізують з водного лужного розчину. 18. Спосіб за п. 9, який відрізняється тим, що сполуку (III) виділяють і відокремлюють від її солі у вигляді вільної основи в розплавленій формі. 19. Спосіб за п. 18, який відрізняється тим, що сполуку (III), що присутня у вигляді розплавленої форми, охолоджують і після заморожування кристалізують з водного лужного розчину. 20. Спосіб за п. 9, який відрізняється тим, що до сполуки (ІІІ) у формі солі додають основу, після чого сполуку (ІІІ) відокремлюють у рідкій формі. Винахід стосується способів одержання хіназолінових алкалоїдів наступних формул (І) і (III). закриття циклу при нагріванні, феноксильна група замінюється бромом і потім, через наступне закриття циклу, одержується 2,3-триметилен-4хіназолон (= пегенон). Якщо пегенон необхідний як вихідна сполука для одержання деоксипеганіну, єдиним спосібом, який може бути взятим до уваги - окрім спосібу запропонованого SPATH та PLATZER згаданого вище - є, якщо взагалі, спосіб синтезу (іі) запропонований Morris і інш. Однак, цей спосіб є неекономічним через багатостадійність реакції. Деоксипеганін, переважно, одержують шляхом виділення з рути сірійської (Peganum harmala) або шляхом його синтезу. MORRIS, HANFORD та ADAMS (наведено вище, cтop.953) описують одержання деоксивазицину (= деоксипеганін) шляхом відновлення 2,3-(a-гідрокситриметилен)-4хіназолону або 2,3-(a-хлортриметилен)-4хіназолону, використовуючи льодяну оцтову кислоту і цинковий пил (реакція: відновлення Клемменсена). Далі екстрагують хлороформом, продукт виділяють за допомогою кристалізації з петролейного етеру. Невигідно, що вихідні сполуки (2,3-(a-гідрокситриметилен)-4-хіназолон або 2,3(a-хлортриметилен)-4-хіназолон) синтезуються виходячи з пеганіну (=вазицин). Згідно з SARGAZAKOV і інш. [Khim. Prir. Soedin. 4 (1990), 506-507], гідрохлорид деоксипеганіну можна одержати шляхом циклоконденсації антранілової кислоти з 2піролідоном з одержанням гідрохлориду деоксивазицинону (= гідрохлорид пегенону) і наступного відновлення Клемменсена цього проміжного продукту з утворенням гідрохлориду деоксипеганіну. Реакцію циклоконденсації проводять в присутності трихлориду фосфору, з толуолом, що використовується як розчинник. Або після першої стадії (циклоконденсації), або після другої стадії (відновлення), необхідна багатократна екстракція хлороформом. Загалом, одержання 2кг гідрохлориду деоксипеганіну потребує приблизно 50л толуолу і 80л хлороформу. Спосіб синтезу описаний SARGAZAKOV і інш. є неприйнятним з декількох причин. Вихід Сполукою (III) є 1,2,3,9-тетрагідропіроло[2,1b]хіназолін. Ця сполука також відома під тривіальною назвою ідеоксипеганій і зустрічається в рослинах родини Zygophyllaceae. Завдяки його фармакологічншГвластивостям, деоксипеганін розглядається як член групи зворотноактивних інгібіторів холінестерази. Крім того, вона також діє як інгібітор моноаміноксидази. Деоксипеганін розглядається як медичний агент для терапевтичних цілей, наприклад, для лікування наркомани' і залежності від лікарських засобів (DE-A 199 06 978), для лікування деменції Альцгеймера (DE-A 199 06 975) або для лікування нікотинової залежності (DE-A 199 06 979). Сполука (І), відома під назвою пеген-(9)-он-(8), є важливим проміжним продуктом для синтезу деоксипеганіну і інших хіназолінових алкалоїдів цього типу. Синтез пеген-(9)-ону-(8) (коротко: jiereHOify можна провести згідно з SPATH та PLATZER [Вег. 6§ (1935), 2221-2224] шляхом взаємодії ангідриду ізатоїнової кислоти з еквімолярною кількістю піролідону. Потім, повинна проводитись високовакуумна перегонка при 140 160 °С для відокремлення побічних продуктів реакції. Цей спосіб є придатним тільки для одержання малих кількостей продукту (в межах граму), але не прийнятний для великотонажного виробництва. Згідно з MORRIS, HANFORD та ADAMS [J. Amer. Chem. Soc. 57 (1935), 951-954), пегенон (2,3триметилен-4-хіназолон) можна одержати (і) шляхом оксидування деоксивазицину за допомогою пероксиду водню. Деоксивазицин є ідентичним деоксипеганіну. Альтернативно, (іі), пегенон мона одержати шляхом багатостадійного синтезу, з g-феноксибутирилхлориду, що реагує з о-амінобензамідом у відповідний амід. Після 5 продукту становить приблизно тільки 50%, одержаний продукт (гідрохлорид деоксипеганіну) має ступінь чистоти 94-95%. Цей спосіб потребує великих кількостей органічних розчинників, особливо толуолу і хлороформу, також як і трихлориду фосфору. Це невигідно з точки зору екології, але також з точки зору безпечності і вартості. Крім того, цей спосіб потребує значних витрат часу і трудовитрат на проведення декількох стадій екстракції хлороформом для досягнення наведеного вище ступеня чистоти. З огляду на застосування деоксипеганіну як медичного агента, особливо невигідним є застосування хлорвуглеводнів. У вищезгаданих відомих способах синтезу, крім того існує недолік, що через високе співвідношення побічних продуктів реакції, відокремлення продуктів за допомогою способів кристалізації не можливо і може замість цього бути досягнуто тільки висовартісними стадіями екстракції' або високовакуумною дистиляцією. Тому об'єкт згідно з винаходом, описаний тут, визначає способи синтезування для одержання хіназолінових алкалоїдів вищезазначених формул (І) і (III) які: - забезпечують вихід і ступені чистоти продукту, що принаймні відповідають таким відомим способам або які перевершують їх; - забезпечують одержання деоксипеганіну (III) для застосування при виробництві медикаментів; - у значній мірі здійснюються без застосування речовин, що є шкідливими для навколишнього середовища або для здоров'я, особливо органічних розчинників; - мають гарний баланс сировини; - в основному забезпечують тільки такі побічні продукти, які можна легко переробляти; - можуть бути здійснені з використанням рентабельних вихідних матеріалів; - в цілому забезпечують більш просте і більш рентабельне одержання вищезгаданих сполук в промисловому масштабі. Ці й інші об'єкти несподівано досягаються за допомогою способів згідно з незалежними пунктами формули винаходу 1, 9, 18 і 20, також як згідно з подальшими втіленнями, описаними в залежних пунктах. Згідно з пунктом 1, в способі згідно з винаходом, сполука формули (II) (= ангідрид ізатоїнової кислоти) реагує з 2-піролідоном (= піролідинон) з одержанням сполуки формули (І). В цій реакції використовується надлишок 2-піролідону, відносно кількості використовуваної сполуки (II). Це означає, що кількість використовуваного 81287 6 піролідону є більшою ніж еквімолярна кількість. Переважно використовується від 1,5 до 5мол, особливо від 2 до 4мол і найбільше переважно від 2,5 до 3,5 мол піролідону, кожне з цих значень, стосується кількості використовуваної сполуки (II). Несподіваним наслідком є те, що утворення шкідливих продуктів реакції знижується і що продукт (І) ранньої стадії може бути легко кристалізованим. Крім того, можливо одержувати високі виходи (70%), з ступенем чистоти приблизно 99% (ЯМР). Завдяки чудовій здатності до кристалізації, можливо обходитися без дорогих способів очищення, таких як високовакуумна перегонка. Тому, що очищення продукту може бути досягнуто простим способом, шляхом кристалізації, процес може також бути здійснений у промисловому масштабі без будь-яких проблем. Процес заснований на наступному рівнянні реакції, представлений за допомогою прикладу: Реакцію здійснюють при додаванні теплоти, переважно у діапазоні температур від 50 до 200°С. Переважним чином, спочатку температура знаходиться в діапазоні від 70 до 130°С і потім підтримується температура в діапазоні від 140 до 200°С; особливо перевага надається, спочатку температурі від 80 до 120°С і пізніше від 150 до 190°С. Вищезгадана початкова температура переважно підтримується протягом періоду від 0,5 до 2 годин, особливо від 1 до 1,5 годин після початку реакції Вищезгадана наступна температура переважно підтримується протягом періоду від 1 до 8 годин, особливо від 2 до 5 годин. Процес переважно здійснюють, шляхом забезпечення піролідону і подальшим додаванням до нього частинами сполуки (II). Особливо вигідно виділити продукт (І) безпосередньо з реакційної суміші шляхом кристалізації. Для кристалізації сполуки (І), реакційну суміш залишають охолоджуватися і вносять до неї затравочні кристали. Кристалізацію здійснюють при кімнатній температурі, переважно принаймні при 25°С, з перевагою від 30 до 70°С, найбільше переважно від 40 до 60°С, за допомогою чого хід кристалізації прискорюється. Процес кристалізації здійснюється приблизно від 24 годин до 7 днів, переважно приблизно від 50 годин до 100 годин. Одержані кристали містять піролідон. Представлений винахід надалі стосується способів для одержання деоксипеганіну (сполука згідно з формулою III). Способи в основному включають три стадії: 7 (A) одержання сполуки (І) згідно з способом за будь-яким з пунктів 1 - 8; (B) реакцію відновлення, з утворенням сполуки (III) у формі солі; (C) виділення сполуки (III) з солі. Хід реакції відновлення (реакція Клемменсена) може бути визначений, за допомогою прикладу, наступним чином: Сполуку (III) одержують у формі солі; у випадку, представленому вище за допомогою прикладу, тетрахлорцинкатну сіль одержують, якщо реакцію відновлення здійснюють в присутності льодяної оцтової кислоти, цинку і хлорводневої кислоти. Особливо переважно, використовувана вихідна сполука (І) є продуктом реакції, виділеним шляхом кристалізації, цей продукт реакції може бути отриманий вищезгаданими описаними способами. Згідно з переважним втіленням винаходу, реакцію відновлення здійснюють, використовуючи льодяну оцтову кислоту, цинк (цинковий пил) і хлорводневу кислоту, переважним чином це здійснюють спочатку шляхом розчинення сполуки (І) у льодяній оцтовій кислоті і потім шляхом додавання цинку і хлорводневої кислоти. Згідно з подальшим переважним втіленням, реакцію відновлення здійснюють при додаванні цинку (у вигляді цинкового пилу) і сірчаної кислоти, без застосування льодяної оцтової кислоти. В результаті одержують гідросульфат сполуки (III): Реакцію переважно здійснюють при температурах приблизно від 50 до 90°С, особливо від 70 до 80°С; звичайно, реакція завершується через приблизно 2-3 години. Після завершення реакції, надлишок цинку видаляють відомими способами, наприклад фільтруванням через фільтрувальний папір. Сіль сполуки (III), одержана таким чином, може бути виділена простим і ефективним способом, шляхом кристалізації з реакційної суміші. Для цієї цілі в реакційну суміш вносять затравочні кристали. Коли кристалізація відбулася, матковий розчин що залишився, відділяють способами, відомими фахівцям даної галузі (наприклад фільтруванням). 81287 8 У вищезгаданій стадії (С) способів одержання сполуки формули (III) згідно з винаходом, сіль, одержану на стадії (В) потім перетворюють у вільну основу (III). Звичайно це здійснюють шляхом додавання основи, переважно NaOH до водного розчину солі. Згідно з особливо переважним втіленням, цю стадію здійснюють таким чином, що виділену основу одержують у вигляді розплавленої форми. Це досягають, виконуючи реакцію при температурі, що є вище ніж температура плавлення (86°С) вільної основи (III) (= деоксипеганіну), переважно при температурах у діапазоні від 90 до 100°С. Основа присутня у вигляді розплавленої форми може бути виділена простим способом, можливо після виморожування з реакційної суміші; прийнятні для цієї цілі способи відомі фахівцям цієї галузі. Згідно з подальшим особливо переважним втіленням, розплавлену основу (III) після того охолоджують і залишають для заморожування. Заморожений продукт розчиняють з одержанням насиченого розчину; переважно як розчинник використовують воду. З цього насиченого розчину, який підлуговують додаванням основи (переважно NaOH), кінцевий продукт деоксипеганіну може бути виділений у формі з високим ступенем чистоти за допомогою наступного процесу кристалізації. Таким чином винахід охоплює способи одержання деоксипеганіну (сполука формули (III)), що містять стадію, в якій згадану сполуку виділяють з реакційної суміші у вигляді рідкої форми. Більш особливо, винахід охоплює технологічні способи згаданого типу, що містять наступні стадії: - відновлення згаданої сполуки (І), до одержання сполуки (III), за допомогою чого сполуку (III), одержують у формі солі; - додавання основи, за допомогою чого сполуку (III) виділяють з солі і відокремлюють у вигляді рідкої форми. Способи згідно з винаходом дозволяють просте й економічне виробництво згаданих сполук (І, III) у кількості і з ступенем чистоти, необхідних при виробництві медикаментів. Особливу перевагою є те, що можна обходитися без використання органічних розчинників. Способи згідно з винаходом у подальшому будуть проілюстровані прикладами. Приклад 1 (порівняльний приклад) Одержання пеген-9-(ону)-(8) (ідентичний з формулою (І)) шляхом взаємодії піролідону з еквімолярною кількістю ангідриду ізатоїнової кислоти згідно SPATH і PLATZER (згаданий вище, стор. 2221) і 2224. Два літри піролідону нагрівали з 4кг ангідриду ізатоїнової кислоти (молярне кількісне співвідношення 1:1,07) у резервуарі з хромистої сталі (ємність: 10л), при енергійному перемішуванні до температури 120°С до початку утворення газу. З наступним нагріванням протягом 10 хвилин до 160°С і потім протягом 30 хвилин до 190°С. З цієї реакційної суміші 558 г (= 3мол), все ще в гарячому рідкому стані, переносили в 1 9 літрову колбу (NS 29). Залишок, що залишився в сталевому резервуарі заморожували до склоподібної маси чорного кольору, що є не придатною для виділення пегенону. З реакційної суміші (558 г), перенесеної до колби, можна було переганяти за допомогою високовакуумної перегонки до 117г пегенону при нагріванні до 120°С з використанням перегінного холодильника; що відповідає 21% виходу. Після перекристалізації з етеру була визначена температура плавлення 111°С. Приклад 2 Синтез пегенону (Сполука згідно з формулою (І)) Один моль ангідриду ізатоїнової кислоти (формула (It)) піддавали взаємодії з 3 молями піролідону. В кінці взаємодії, піролідон поміщали в резервуар з хромистої сталі (30л), що нагрівається і нагрівали до приблизно 100°С, потім при безперервному перемішуванні додавали частинами ангідрид ізатоїнової кислоти. Приблизно через 1 годину, всю кількість ангідриду розчиняли в піролідоні. Після цього реакційну суміш нагрівали протягом 5 годин до температури від 155 до 160°С (протягом цього процесу, утворювалися СО2 і Н2О; дивіться вище) і нарешті нагрівали протягом короткого часу до температури від 170 до 180°С. Після охолодження до приблизно 50°С, в отриману таким чином масу, вносили кристали пегенону і залишали кристалізуватися протягом періоду приблизно від 50 до 100 годин при кімнатній температурі. Шляхом підвищення температури (принаймні 25°С), процес кристалізації може бути прискорений (тривалість приблизно тільки від 2 до 3 днів). "Матковий розчин" відділяли від кристалів шляхом фільтрування. В цілому, більше ніж 90% вихідних сполук, що використовується, були перетворені на пегенон. Вихід пегенону, що кристалізується був 40 %; частину пегенону, що залишилася, розчиняли в піролідоні. Кристали пегенону, що одержували все ще мають вміст 30мол.% піролідону, згідно з ЯМР; ступінь чистоти пегенону складає приблизно 99% згідно з ЯМР. У модифікації цього процесу, вихідні сполуки використовувалися у кількісному співвідношенні 1:2,5 (ангідрид ізатоїнової кислоти : піролідон). Частина пегенону, що кристалізується, могла таким чином бути збільшена до приблизно 55%, при ступені чистоти приблизно 99 %. Приклад 3 a) Відновлення Клемменсена сполуки (І) Кристали пегенону, одержані згідно з Прикладом 2 (2,5г; з вмістом 15ваг.% піролідону; це відповідає 1,83кг або 9,81мол чистого пегенону) були розчинені при приблизно 50°С в 6,1л (=100,4мол) льодяної оцтової кислоти і переносили у резервуар з хромистої сталі (50л). Потім маленькими порціями додавали 3,7кг (=56,4мол) цинкового пилу, протягом цього процесу температуру реакційної суміші підтримували при приблизно 60°С. Через приблизно 1 годину, маленькими порціями, 81287 10 протягом 2 годин при безперервному перемішуванні додавали 4х3л концентрованої НСІ (32%; відповідає 121,74мол). Після чого (через приблизно від 4 до 5 годин) надлишковий цинковий пил видаляли з реакційної суміші шляхом фільтрування через фільтрувальний папір. Після внесення затравочних кристалів, приблизно 66% деоксипеганінової солі можуть бути відділені кристалізацією. Матковий розчин, що залишився, концентрували шляхом випаровування, після чого 66% деоксипеганінової' солі знову кристалізували. В цілому, приблизно 90% деоксипеганінової солі можуть бути отримані у кристалічній формі. b) виділення деоксипеганінової' основи (сполуки (III)) Кристали (2,5кг), отримані таким чином, розчиняли в гарячій воді (приблизно 11л). Після того, додавали при перемішуванні NaOH (приблизно 8кг таблеток), при нагріванні суміші до температури від 95 до 100°С. Це надавало деоксипеганін у вигляді розплавленої форми і його відділяли з реакційної суміші. Після заморожування, продукт знову розчиняли в нагрітій воді з одержанням насиченого розчину, який підлуговували за допомогою NaOH. З цього насиченого лужного розчину, деоксипеганін виділяли шляхом кристалізації. Вихід деоксипеганіну, по відношенню до деоксипеганінової солі був більше ніж 90%, з ступенем чистоти 99,9% (ЯМР). Приклад 4 Відновлення Клемменсена сполуки (І) (Альтернативний спосіб) 175г кристалів пегенону (що містить 30мол.% піролідону; це відповідає 150г пегенону; дивіться Приклад 2) змішували з 750мл (20%) H2SO4 і нагрівали (приблизно 85°С) протягом приблизно 30 хвилин при перемішуванні. Після цього поступово додавали 260г цинкового пилу (тривалість: приблизно 2 години). Через приблизно 1 годину знову додавали H2SO4 (375мл, 40%). Через повних приблизно З годин, реакція завершувалася і надлишковий цинк відділяли шляхом фільтрування через фільтрувальну бумагу. Як описано в Прикладі 3 а), отриману деоксипеганінову сіль виділяли з реакційної суміші шляхом кристалізації. Виділення основи здійснювали згідно з способом, описаним в Прикладі Зb).
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for producing quinazoline alkaloids
Автори англійськоюMoormann Joachim, Hoffmann Hans-Rainer, Matusch Rudolf
Назва патенту російськоюСпособ получения хиназолиновых алкалоидов
Автори російськоюМоорманн Йоахим, Хоффманн Ханс-Райнер, Матуш Рудольф
МПК / Мітки
МПК: C07D 487/04
Мітки: хіназолінових, спосіб, одержання, алкалоїдів
Код посилання
<a href="https://ua.patents.su/5-81287-sposib-oderzhannya-khinazolinovikh-alkalodiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання хіназолінових алкалоїдів</a>
Спосіб одержання циталопраму (варіанти), s-циталопраму, проміжні кетони та спосіб одержання рацемічних сполук

Номер патенту: 72238
Опубліковано: 15.02.2005
Автори: Еллегор Петер, Петерсен Ханс, Рок Майкл Харольд
МПК: C07C 253/30, C07D 307/87, C07C 255/56
Мітки: сполук, проміжні, s-циталопраму, спосіб, кетони, одержання, рацемічних, циталопраму, варіанти
Формула / Реферат:
1. Спосіб одержання циталопраму, згідно з яким здійснюють реакцію сполуки формули IV, IVде R являє собою ацил, з 3-(N,N-диметиламіно)пропілмагнійгалогенідом, переважно з 3-(N,N-диметиламіно)пропілмагнійхлоридом, з одержанням циталопраму формули I, Iякий виділяють у вигляді основи або її фармацевтично прийнятної солі.2. Спосіб за п. 1, який відрізняється тим, що проміжну сполуку формули IV одержують...
Спосіб виділення алкалоїдів ріжків з ріжків (варіанти)

Номер патенту: 81085
Опубліковано: 26.11.2007
Автори: Цвак Ладіслав, Родер Любомір, Голан Іржі
МПК: C07D 519/00
Мітки: спосіб, варіанти, ріжків, алкалоїдів, виділення
Формула / Реферат:
1. Спосіб виділення алкалоїду ріжків з ріжків, який включає екстракцію ріжків за допомогою суміші, що містить толуол і етанол, з одержанням первинного екстракту.2. Спосіб за п. 1, де суміш містить толуол і близько 5-30 % (об./об.) етанолу.3. Спосіб за п. 2, де суміш містить толуол і близько 10-20 % (об./об.) етанолу.4. Спосіб за п. 2, де екстракцію здійснюють при температурі близько 20-50° С.5. Спосіб за п. 4, де...
Спосіб одержання циталопраму та проміжні сполуки для його одержання

Номер патенту: 63034
Опубліковано: 15.01.2004
Автори: Сван Хенрік, Рок Майкл Харольд, Петерсен Ханс
МПК: A61K 31/343, C07B 61/00, A61P 25/24, A61P 29/00, C07D 307/81, C07D 307/87
Мітки: одержання, спосіб, циталопраму, сполуки, проміжні
Формула / Реферат:
1. Спосіб одержання циталопраму, під час якого здійснюють взаємодію сполуки формули IV:, IVу якій R являє собою галоген або групу СF3-(СF2)n-SO2-O-, де n являє собою ціле число в інтервалі 0-8, включно, із джерелом ціаніду в присутності паладієвого каталізатора і каталітичної кількості Сu+ або Zn2+, або з Zn(CN)2 у присутності паладієвого каталізатора, і виділяють відповідну 5-ціаносполуку, тобто циталопрам:, (I)у...
Тієнопіримідини, спосіб їх одержання, фармацевтична композиція та спосіб її одержання

Номер патенту: 66856
Опубліковано: 15.06.2004
Автори: Крістадлер Марія, Клюхен Франс-Вернер, Джонас Рохус, Шеллінг П'єр
МПК: A61P 9/00, C07D 495/04, A61K 31/519, A61P 43/00, A61P 15/10
Мітки: композиція, спосіб, одержання, тієнопіримідини, фармацевтична
Формула / Реферат:
1. Тієнопіримідини формули І, Iв якійR1, R2 кожний незалежно один від одного означає Н, А, ОА або Hal,R1 і R2 разом означають також алкілен з 3-5 С-атомами, -О-СН2-СН2-, -СН2-О-СН2-,-О-СН2-О- або -О-СН2-СН2-О-,Х означає R4, R5 або R6, одноразово заміщений R7,R4 означає прямий чи розгалужений алкілен з 1-10 С-атомами, де одна або...
Похідні циклопептидів, спосіб їх одержання, фармацевтична композиція, спосіб її одержання

Номер патенту: 55439
Опубліковано: 15.04.2003
Автори: ХЬОЛЬЦЕМАНН Гюнтер, ФІТТШЕН Клаус, ГОДМАН Сімон
МПК: C07K 7/56
Мітки: одержання, композиція, циклопептидів, фармацевтична, похідні, спосіб
Формула / Реферат:
1. Сполуки формули Іцикло(Arg-X-Asp-R1), (I)в якійХ являє собою Gly, Ala або NH-NH-CO,причому можуть братися до уваги також похідні вказаних амінокислот, і залишки амінокислот з'єднані один з одним через -аміно- і -карбоксигрупи по типу пептидного зв'язку,R1 являє собою залишок формули II(II),R2, R3, R4 кожний незалежно один від одного являє собою Н, А, Аr, R5-Ar, Het абоR5-Het,А...
Попередній патент: Вхідний регульований напрямний апарат відцентрового насоса
Наступний патент: Вкладиш підшипника ковзання і спосіб виготовлення вкладиша підшипника ковзання
Випадковий патент: Спосіб отримання хлорангідридів кислот