Спосіб отримання похідних ізоксазолу







Формула / Реферат
Способ получения производных изоксазола общей формулы
где А - группа общей формулы
где R1- водород, отличающийся тем, что соединение общей формулы
где В- группа общей формулы
или
где R1 - имеет указанные значения, подвергают циклизации путем обработки избытком реактива дегидратации, такого как хлористый тионил, в инертном растворителе.
Текст
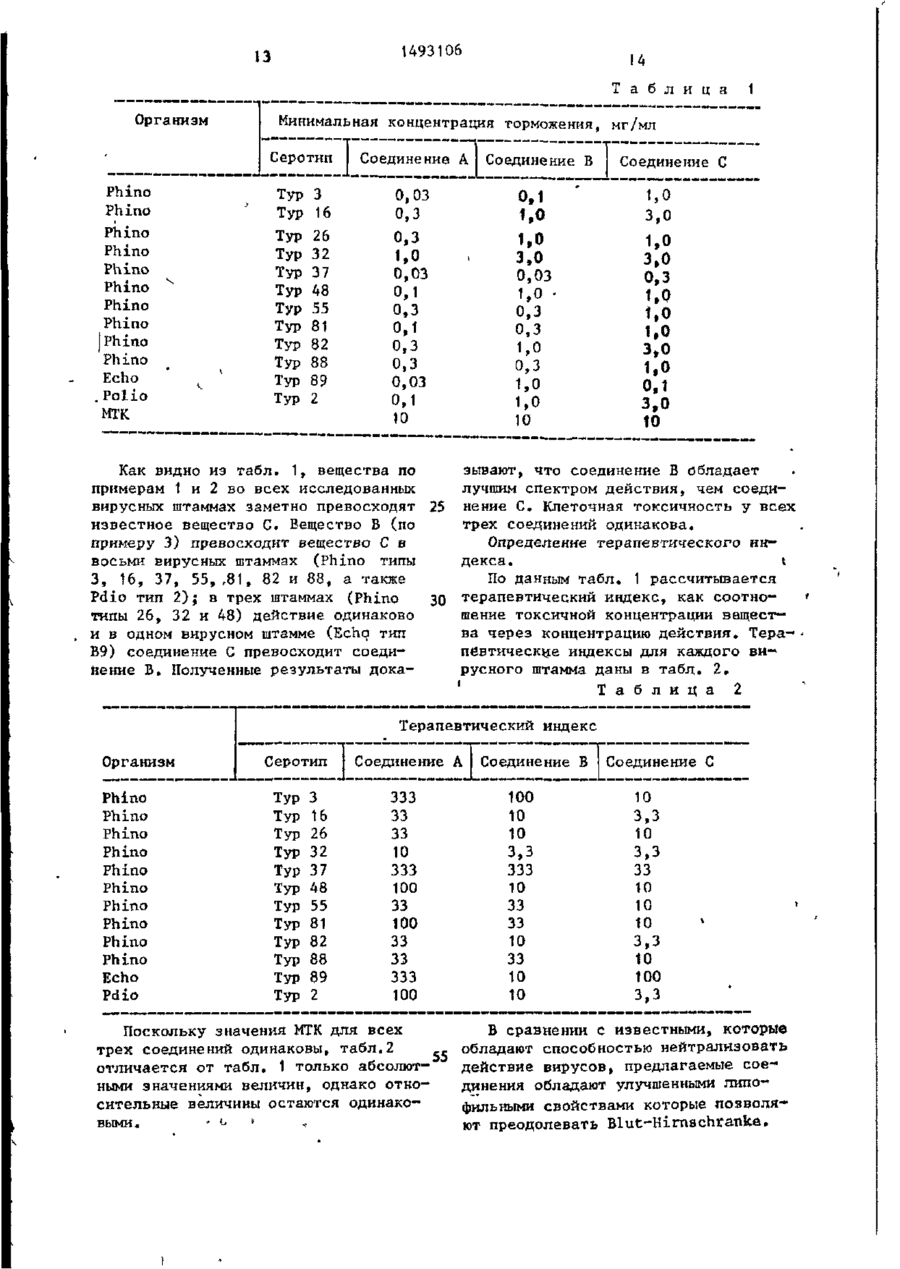
Изобретение касается гетероциклических веществ, в частности получения производных изоксазола общей Изобретение относится к способу получения новых производных изоксазола общей формулы . НС 3 где А - группа общей формулы о или N. где R, - водород, обладающих антивирусной активностью. \ ф-лы I: А-0(СН 2 ) 7 -С=СН-С(СН 3 )=К-0, где А - группа ф-лы II или III: -C=CH-CH-CK-S (II); -C=CH-S-CHK-CH (III) при К - - O N - C C H ^ - O , которые обладают антивирусной активностью, что может быть использовано в медицине. Цель - создание более активных веществ указанного класса. Синтез ведут циклизацией соединений ф-лы: IV • или V: H0-(CH l )^-NH-C(0)-X 1 -0-(CH l ) 7 -C=CH-C(CH3)=N-0 (IV); Н -C(0)-X 2 -0-(CH 2 b-C=CH-C(CH 3 )=N-0 (V), где X, - J T H L и Х д помощью агента дегидратации и хлористого тионила в среде инертного растворителя. Новые соединения при аыти,вирусной активности обладают лучшими в сравнении с известными липофильными свойствами, которые позволяют преодолевать Blut-Hirnschranke. 2 табл. Цель изобретения - создание новых соединений изоксазольного ряда, обладающих улучшенной антивирусной активностью. . ' П р и м е р 1. 5-[7-[5-(4,5-Дигидро-2-оксазолил)-2-тиенил ^-оксигептилЗ-3-метилизоксазол. 0,60 г (1,64 ммоль) амида N-(2-оксиэтил)-5-[7-(3-метил-5-изоксазо-, лил)-гептилокси ]-2-тиофенкарбоновой кислоты вводят при 0°С в 2,5 мл хлористого тионила. Смесь перемешивают і в течение 15 мин при 0°С, после чего в вакууме удаляют избыточное коли 'В 1493106 чество хлористого тионила. Остаток • раствором соляной кислоты, водную распределяют между насыщенным растфазу еще три раза экстрагируют хловором кислого углекислого натрия и ристым метиленом, объединенные оргаэтиловым эфиром уксусной кислоты. 5анические фазы сушат над сернокислым тем еще дважды производят экстрагинатрием и упаривают. рование этиловым эфиром уксусной кисНеочищенный продукт (около 45 г) лоты. Объединенные органические растпорциями перегоняют в трубке с шароворы сушат над сернокислым натрием вым расширеиием(температура воздушпри добавлении активированного угля, 10 ной бани 80°С, 0,2 мбар). а затем" упаривают. Выход 26,9%, окрашенное в желтоНеочищенный продукт (0,52 г окраватый цвет маслообразное вещество. r 1 шенного в желтоватый цвет кристаллиH-NMR (CDC13) J , РРт: 5,81 (з; ческого вещества) очищают с помощью 1Н; Isox - Н 4 ) ; 3,51 (t, J=6,6 Hz; хроматографии на колонке (1:35, си2Н; -CILjCl); 2,68 (t(.J = 6,6 Hz; 15 1 ликагель 60, размер зерен 0,0405ox ~ C H 2 - ) ; 2,23 (в; ЗН; I J O x 0,063; элюирующее средство: этиловый CH3-); 1,85 - 1,15 (m; 10H; -(CH2)y-). эфир уксусной кислоты и петролейный Метиловый эфир 5-Г7-(3-метил-5эфир в соотношении 3:1). -изоксазолил)-гептилокси J-2-тиофенВыход 0,29 г бесцветного кристал- 20 карбоновой кислоты. лического вещества (50,7% от теоре9,3 г (43,1 ммоль) 5-(7-хлоргептически рассчитанного значения). тил)-3-нетилизоксазола и 7,12 г Т.пл. = 69-70°С (из диизопропилового (47,4 ммоль) иодида натрия в течение эфира). 24 ч нагревают в 60 мл абсолютного Исходный материал получают описан- 25 ацетона при температуре кипения реакционной смеси с обратным холодильниным ниже способом. ком. Непосредственно после этого ре'Н-NMR: (CDC1 3 ) СГ, ррт: 7,27; акционную смесь охлаждают, выделив7,22; 6,18; 6,13 (АВ; 2Н; Th-H 3 und 3 шийся в осадок хлористый натрий отT h h H)) ; 5 7 9 - 4 5,79 ) - H 44 ) ; 30 фильтровывают, промывают небольшим 4,05 (t; J = 6,6 Hz; 2H; -OCH,-); количеством ацетона, после чего . 4,50-3,92 (A^B 7 ; 4H; -O-CH 2 -CH 2 -N-); фильтрат смешивают с 7,16 г 2,70 (t, J = 6,6 Hz; 2H; I S o x (45,3 ммоль) метилового эфира 5-окси- СИi"); 2,25 (s; 3H; IS o x - C H 3 ) ; -2-тиофенкарбоновой кислоты и 13,1 г 1,85-1,12 (m; ІОН; - ( C H 2 ) 5 - ) . (94,8 ммоль) углекислого калия. Смесь 5-(7~Хлоргептгоі)~3-метилизоксазол. -зс нагревают с обратным холодильником 21,0 г (0,216 моль) 3,5-дйметилпри температуре ее кипения в течение изоксазола растворяют в 200 мл абсо2 ч, охлаждают и затем упаривают. Ослютного тетрагидрофурана, раствор охтаток распределяют между водой и диэлаждают до -80°С, после чего при тиловьтм эфиром, после чего водную 40 указанной температуре к раствору в фазу дважды экстрагируют диэтиловым течение 40 мин прибавляют по каплям эфиром. Объединенные органические 160 мл раствора н-бутиллития (1,35 М растворы промывают небольшим колираствор в н-гексане, 0,216 моль") . За' чеством насыщенного раствора гидротем смесь дополнительно перемешивасульфита натрия, сушат над сернокис45 лым натрием при добавлении активироют в течение 15 мин при -75°С. Непосредственно после этого реакванного угля, а .з,атем упаривают. . ционную смесь прибавляют по каплям к Неочищенный продукт (11,9 г 82% раствору 53,5 г (0,217 моль) 1-иодот теоретически рассчитанного значе6-хлоргексана в 150 мл абсолютного ния) перекристаллизовывают из диизотетрагидрофурана таким образом, что- 50 пропилового эфира). бы температура не поднималась выше Выход 5,2 г окрашенного в светло—, -60° С. После завершения прибавления розовый цвет кристаллического вещестреакционную смесь дополнительно пева (35,5% от теоретически рассчитан-' ремешивают в течение 15 мин при -60°С, а затем температуре смеси да- 55 кого значения). Т. пл. = 55-56**С (из диизопропилового эфира). ют возможность подняться до комнатГ Н - NMR (CDClj) -2-тиофенкарбоновая кислота. 10 0,98 г (2,90 ммоль) метилового эфира 5-С7-(3-метил-5-изоксазолил)гептнлокси]-2-тиофенкарбоновой кислоты нагревают в 8 мл этилового спирта идо мл воды с обратным холодильни4 ком < температуры кипения смеси, пос-(5 ле чего к полученному раствору в течение 10 мин прибавляют по каплям 0,18 г (3,19 ммоль) гидроокиси калия, растворенной в 6 мл воды и 4 мл этилового спирта. Непосредственно после 20 этого реакционную смесь дополнительно перемешивают в течение 2,5 ч с обратным холодильником при температуре ее кипения. После охлаждения большую часть 25 смеси отгоняют, остаток распределяют между водой и диэтиловым эфиром, а водную фазу затем подкисляют прибавлением 2 н. соляной кислоты до рН= 1,5. Затем применяют 80 мл последне- 30 го, объединенные органические растворы сушат над сернокислым натрием при ' добавлении активированного угля и сушат. Выход 0,90 г бесцветного кристал35 лического вещества (95,8% от теоретически рассчитанного значения). Т.пл. = 96-97 С (из диизопропиловрго эфира). *H-NMR (CDCX3) rf, ррт: 10,5 40 (S.Breit; 1H; - О Н ) ; 7,64; 7,59; 6,26; 6,21: (АВ; 2Н; Th - н 3 и Th - Н 4 ) ; 5,80 (s; Н; Isox" Н } ; 4,09 (t, J = =6,6 Hz; 2H; -ОСТЦ-); 2,69 (t, J = = 6,6 Hz; 2H; І 5 0 ) Г C H t - ) ; 2,26 (s; 45 3H; I s o x - C H 3 ) ; 1,95 - 1,30 (m; • 10H; - ( C H 2 ) S - ) . N- (2-0ксиэтил) амид-5- [ 7- ( 3-метил— -5-изок£азолил)-гептилокси ]-2-тиофенкарбоновой кислоты. 50 К 0,81 г (2,51 ммоль) 5-[7-(3-метил-5-изоксазолил)-гептилокси]-2-тиофенкарбоновой кислоты при перемешивании и охлаждении медленно прибавляют по каплям 2 мл хлористого тионила, в 55 результате чего образовывается прозрачный раствор. Затем реакционную смесь дополнительно перемешивают в течение 30 мин при комнатной темпера туре и непосредственно после это^о производят отгонку в вакууме избыточного количества хлористого тионила.• Остаток растворяют в 6 мл абсолютного хлористого метилена, после чего при 15 С к приготовленному раствору прибавляют по каплям раствор 0,34 г (5,51 ммоль) этаноламина в 5 мл абсолютного хлористого метилена. Реакционную смесь дополнительно перемешивают в течение 1 ч при комнатной температуре, несколько упаривают и затем распределяют между водой и этиловым эфиром уксусной кислоты. Водную фазу еще раз экстрагируют небольшим количеством этилового эфира уксусной кислоты, объединенные органические растворы промывают водой, сушат над сернокислым натрием при добавлении активированного угля и упаривают. Выход 0,81 г окрашенного в желтоватый цвет кристаллического вещества (88,2% от теоретически рассчитанного значения). Т. пл. = 107-1Ю°С (из ацетонитрила). r H-NMR (CDC13) cf, ррт: 7,25; 7,20; 6,17; 6 , U (АВ, 2Н, Th-H 3 и T h - H 4 ) , 6,73 ( t . b r e i t , 1H, N-H), 5,80 ( s , 1H, Isox" H 4)» 4 , 0 4 ( t , J=6,6 H, 2H, Th-OCH 2 -); 3,78 ( t , J=4,9 Hz; 2H; -ОЦ-ОН); 3,57 ( t , J=5,1 Hz; 2H, -N-CH 2 -); 2,69 ( t , J=6,6 Hz; 2H; CH2~), 2,25 ( s , 3_H, 1,85-1,12 (m, 10H, - ( C H 2 ) 5 - ) . П р и м е р 2." 5 - [ 7 - [ 5 - ( 4 , 5 - Д и - . гидро-2-оксазолил)-2-тиенилJ-оксигептилJ-3-метилизоксазол. •0,60 г (1,64 ммоль) Ы-(2-оксиэтил) амида 4-^-7-(3-мeтил-5-изoкcaзoлил)~ -reптилoкcиJ-2-тиoфeнкapбoнoвoй кислоты растворяют в 30 мл хлороформа, после чего приготовленный раствор смешивают при 0°С с 0,3 г (2,54 ммоль) хлористого тионила. Непосредственно после этого реакционную смесь перемешивают в течение 60 мин при 0°С, в результата чего получают сухой остаток. Остаток распределяют между насыщенным раствором кислого углекислого натрия и этиловым эфиром уксусной кислоты. Еще два раза производят экстрагирование этиловым эфиром уксусной кислоты, объединенные органические растворы сушат над сернокислым натрием при добавлении активированного угля и упаривают. Очистку неочищенного продукта с • помощью хроматографии на колонке осу 1493106 щєствлягат по аналогии с описанным в примере 1, Выход 0,24 г бесцветного кристаллического вещества (42% от теорети-» чески рассчитанного з н а ч е н и я ) . Т . п л . = 68-70 С (из диизопропилового эфира). П р и м е р 3. 5-У7-£2-(4,5-Дигидро-2-оксазолил)-4-тиенил J-оксигептшг^-З-метилиэоксазол. ^п 2,06 г ( 5 , 6 2 ммоль) Ы-(2-оксиэтил) амид-4-С7-(3-метил-5-изоксазолил)гептилоксиj-2-тиофенкарбоновой к и с лоты при 0°С вводят в 5 мл хлористого тионила. Смесь перемешивают в т е ч е 15ниє 10 мин при 0°С, после чего в в а кууме без нагревания удаляют избыточное количество хлористого тионила. Остаток распределяют между насыщенным раствором кислого углекислого 20 натрия и этиловым эфиром уксусной кислоты. Еще два раза производят э к страгирование этиловым эфиром уксусной кислоты, объединенные органические растворы сушат над сернокислым 25 натрием при добавлении активированного угля и упаривают. Неочищенное маслообразное вещество подвергают очистке с помощью хроматографии на колонке ( 1 : 4 0 , силикагель 60, размер зерен 30 от 0,040 до 0 , 0 6 3 ; элюнрующие средство: этиловый эфир уксусной кислоты и петролейный эфир в соотношении 3 : 1 ) . Выход 0 , 5 2 г окрашенного в желтоватый цвет кристаллического вещества ,_ (26,5% от теоретически рассчитанного з н а ч е н и я ) . Т. п л , = 67-68°С (из д и изопропилового э ф и р а ) . Исходный материал получают описанным ниже способом. 40 I H-NMR (CDC13) d\ р р т : 7,22 ( d , J=1,7 Hz; 1H, T h - H 3 ) , 6,37 (d, J=»1,7 Hz; 1H, T h - H 4 ) , 5,80 ( s , 1H, I 5 o x - H 4 ) , 4 , 5 0 - 3 , 9 2 ( A 2 B 2 , 4H, -O-CH^-CH 2 -N-), 3,93 ( t , J = 6 , 6 Hz; 2H, 4 5 -OCH 2 -), 2,70 ( t , J = 6 , 6 Hz: 2H, lSQK~ CH-2-), 2,25 ( s , 3H, I S 0 X - C H 3 ) , 1,851,20 (m, ЮН, - ( С Н г ) 5 - ) . Метиловый эфир 4—окси—2—тиофенкар— боновой кислоты. 50 50,0 г ( 0 , 3 4 7 моль) 4-окси-2-тиофенкарбоноврй кислоты и 58,3 г (0,694 моль) углекислого натрия н а г ревают в атмосфере азота в 900 мл а б солютного 2-бутанона до температуры с$ кипения, после чего к приготовленной смеси прибавляют по каплям в течение 20 мин 43,7 г (0,347 моль) диметилсульфата. Затем реакционную смесь д о 8 полнительно нагревают в течение 2,5 ч с обратным холодильником при'температуре кипения. Непосредственно после этого реакционную смесь упаривают в вакууме, остаток распределяют между насыщенным раствором углекислого натрия и диэтиловым эфиром, водную фазу еще пять раз экстрагируют диэтиловым эфиром, причем каждый раз применяют по 80 мл последнего. Объединенные о р ганические растворы сушат над серно-, кислым натрием при добавлении активированного угля, фильтруют и упаривают. Выход 49,6 окрашенного в желтоватый цвет кристаллического вещества (90% от теоретически рассчитанного з н а ч е н и я ) , Т. п л . = 84-85°С (из смеси диизопропилового эфира и петролейного эфира). ІН-NMR (CDC13) (Г , ррт: 7,22 ( d , J=1,7 Hz; 1Н, Th-Нз), 6,64 (d, J=* =1,7 Hz, 1H, T h - H 4 ) , 6,70 ( s , 1H, -OH), 3,88 ( s , 3H, -OCH3). 5-(7-Иодгептил)-3-метилизоксазол. 16,66 г (77,23 ммоль) 5-(7-хлоргептил)-3-метилизоксазола и 12,75 г (85,06 ммоль) йодистого натрия нагревают с обратным холодильником в 110 мл безводного ацетона при температуре кипения смеси. По данным гН-ЯМР~спектроскопии через 7 ч степень превышения составляет приблизительно 85%, а через 22 ч приблизительно 89%. Через 27 ч реакционную смесь упаривают и остаток распределяют между днхлорметаном и водой (при добавлении нескольких миллилитров 2н. соляной кислоты)• Водную фазу несколько раз экстрагируют дихлорметаном, причем суммарно применяют 250 мл последнего, и органическую фазу сушат над сернокислым натрием и упаривают. Выход 22,95 г окрашенной в коричневый цвет жидкости (96,7%) от теоретически рассчитанного значения), r H-NMR: (CDCI3) А ррт: 5,81 (s, Ш , I s o X - H 4 ) , 3,18 (t, J=6,6 Hz; 2H, - C H r ) , 2,69 (t, J=6,6 Hz); 2H, IS o x - C H ? - ) , 2,55 (в, ЗН, І 5 О Х - С Н 3 ) , 1,851,25 (m, 10H, -(CH^) 5 -). Метиловый эфир 4-[7-(3-метил-5-изоксазолил)-гептилокси J-2-тиофенкарбоновой кислоты. 6,42^-г (40,60 ммсль) метилового эфира 4-окси-2-тиофенкарбоновой кислоты и 11,88 г (38,67 ммоль) 5-(7 1493106 -иодгептил)-3-метилизоксазола в течение 8 ч нагревают с 5,34 г (ДО,60 ммоль) углекислого калия в 130 мл безводного ацетона при температуре кипения смеси с обратным холодильником. Реакционную смесь выдерживают в течение ночи, а затем упаривают. Остаток распределяют метсду 2 н. раствором гидроокиси натрия и 10 диэтилэвым эфиром, водную фазу еще несколько раз экстрагируют диэтиловым эфиром, причем суммарно применяют 150 мл последнего. Органическую фазу сушат над сернокислым натрием 15 при добавлении активированного угля и упаривают. Выход 12,56 г окрашенного в желтый цвет кристаллического вещества (95,3% от теоретически рассчитанного значе- 20 ния), Т. пл. = 58-60°С. r H-NMR (CDClj) гептилокси J-2-тиофенкарбоновая кислота. 11,34 г (33,61 ммоль) метилового эфира 4-£7-(3-метил-5-иэоксазолил)-гептилокси]-2-тиофенкарбоновой кислоты нагревают с обратным холодильником в 95 мл этилового спирта и 45 мл воды до температуры кипения, после чего к приготовленному раствору прибавляют по каплям 2,18 г (38,9 ммоль) гидроокиси калия, растворенного в 70 мл воды и 46 мл этилового спирта. После нагревания в течение 3 ч при температуре кипения с обратным холодильником реакционную смесь охлаждают, упаривают, остаток распределяют между водой н диэтиловым эфиром, а водную фазу после подкисления 2 н. соляной кислотой до рН=1 еще несколько раз экстрагируют диэтиловым эфиром. Объединенные органические растворы сушат над сернокислым натрием при добавлении активированного угля и упаривают. Выход 9,28 г окрашенного в желтый цвет кристаллического вещества (85,6% от теоретически рассчитанного значения) . Т. пл. = 110-113°С. 30 35 40 45 50 55 О Неочищенный продукт непосредственно может быть применен на последующей стадии или может быть перекристаллизован из дшгзочропилового эфира, в результате чего образуется бесцветное кристаллическое вещество. ІН-NMR (CDC13)
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of isoxazole derivatives
Назва патенту російською?????? ????????? ??????????? ??????????
МПК / Мітки
МПК: C07D 413/04, C07D 261/08, C07D 413/12, A61K 31/42, A61P 31/12, C07D 333/00, C07D 413/14
Мітки: ізоксазолу, отримання, похідних, спосіб
Код посилання
<a href="https://ua.patents.su/8-5548-sposib-otrimannya-pokhidnikh-izoksazolu.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання похідних ізоксазолу</a>
Спосіб отримання похідних бензаміду, або їх кислотно-адитивних солей, або оптичних ізомерів

Номер патенту: 4762
Опубліковано: 28.12.1994
Автори: Геста Леннарт Фрорвалл, Ян Ола Густав Лундстрем, Свен Ове Егрен, Стен Інгвар Ремсбі
МПК: A61K 31/40, A61K 31/60, A61P 1/08, C07D 207/09, A61P 25/18
Мітки: бензаміду, ізомерів, спосіб, солей, оптичних, похідних, кислотно-адітивних, отримання
Формула / Реферат:
Способ получения производных бензамида общей формулы (I)где R1 и R2 одинаковы или различны и означают водород, галоид, низший алкил; R3 - низший алкил или бензил, незамещенный или замещенный фтором;А1 и А2 - оба водород или каждый в отдельности водород или низший алкил,или их кислотно-аддитивных солей, или оптических изомеров, отличающийся тем, что соединение общей формулы (II) где R1, R2 и R3 имеют...
Спосіб отримання похідних бензаміду, або їх солей, або рацемічних сумішей, або стерєоізомерів

Номер патенту: 4753
Опубліковано: 28.12.1994
Автори: Свен Ове Егрен, Геста Леннарт Фрорвалл
МПК: A61K 9/48, C07C 65/00, C07C 67/00, A61P 25/18, C07C 51/347, C07C 51/00, C07D 207/09, A61K 31/40, A61K 9/20
Мітки: стерєоізомерів, спосіб, сумішей, отримання, похідних, рацемічних, бензаміду, солей
Формула / Реферат:
Способ получения производных бензамида общей формулыгде R1 - алкил с 1-3 С;R2 и R3 - одинаковые или различные и означают водород, хлор или бром,или их солей, или рацемических смесей, или сте-реоизмеров, отличающийся тем, что соединение общей формулыгде R1, R2 и R3 имеют вышеуказанные значения, подвергают взаимодействию с амином общей формулыв среде органического растворителя и целевой...
Спосіб отримання похідних (5 е)-13,14,18,18,19, 19-гексадегідро-3-окса-6а-карбапростагладіна-12 чи їх солей

Номер патенту: 4797
Опубліковано: 28.12.1994
Автори: Хельмут Форбрюген, Міхаель-Харольд Таун, Мартін Хаберей, Вернер Скубалла, Бернд Радюхель, Екехард Шіллінгер, Клаус-Штеффен Штюрцєбехер
МПК: C07D 309/12, C07C 59/00, C07C 67/00, A61K 31/557, C07C 405/00, C07C 51/347, A61P 9/12, C07C 51/00
Мітки: 19-гексадегідро-3-окса-6а-карбапростагладіна-12, е)-13,14,18,18,19, спосіб, солей, отримання, похідних
Формула / Реферат:
Способ получения производных (5Е)-13, 14, 18, 18, 19, 19 - гексадегидро-З-окса-6а-карбапростагландина-12 общей формулы Ігде R - метильная или этильная группа, или их солей с трис-(оксиметил)аминометаном, отличающийся тем, что сосдинение общей формулыгде R- указано выше,ТГП- тетрагидропиранильный остаток, подвергают этерификации бромуксусной кислотой в присутствии гидрида натрия и после этого отщепляют...
Спосіб отримання похідних імідазола або його фармацевтично прийнятих солей

Номер патенту: 2697
Опубліковано: 26.12.1994
Автори: Джон Джонес Вітотес Данкіе, Дейвід Джон Керіні
Мітки: похідних, отримання, імідазола, спосіб, прийнятих, солей, фармацевтично
Текст:
...5 Гц). пластинках для ТСХ. Масс-спектр 325. ЯМР (200 мГц, Е. 1~(4~Аминобензил)~5-гидроксиме3 э 8,00-6,80 (м, 8Н), тил-2-(2*-метоксиэтил~-4-«зшоримидазол. 5,15 ( с , 2Н>, 4,45 ( с , 2Н), 3,60 ( т , Соединение СИНТеЗИРУЮТ П Примеру О 2Н, 5 Г ц ) , 3,15 ( с , ЗН), 2,75 54, Е из 5-тидроксимет'ил—2~(2-меток~ ( т , 2Н, 5 Г ц ) 0 П р и м е р ы сиэгил)— 1 — (4—нитроб ензил) — 4—хлорими— 57—71„ Соединения, синтезированные по дазола ( 2 , 2 г, 6,75...
Спосіб отримання похідних стероідів

Номер патенту: 4789
Опубліковано: 28.12.1994
Автори: Жермен Костерусс, Жан Жорж Тетш, Роже Дераєдт, Даніель Філібер
МПК: A61P 3/08, C07J 41/00, A61P 43/00, A61P 9/10, C07J 1/00, A61K 31/58, C07J 63/00, C07J 43/00, A61K 31/565, C07J 7/00, A61P 25/20, C07J 51/00, A61P 5/38, A61K 31/57, A61P 9/12, C07J 21/00, C07J 71/00, A61P 37/04
Мітки: похідних, стероідів, отримання, спосіб
Формула / Реферат:
Способ получения производных общей фор-где R1- фенил, замещенный ди - (С1-С4) - алкиламиногруппой, возможно окисленной по азоту, ди - (С1-С4) - алкиламино - (С1-С4) - алкилгруппой, возможно окисленной по азоту, пирролидинильной группой, ди-(С1-С4)-алкиламино-(С1-С4)-алкилтиогруппой, ди- (С1-С4)-алкиламино-(С1-С4)-алкилоксигруппой, триметилсилильной группой, или R1-N-(С1-С4)-алкилдигидроиндолил, пиридинил; R2- метил, этил;...