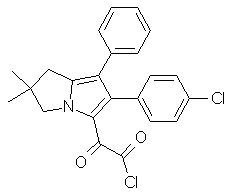
6-(4-хлорфеніл)-2,2-диметил-7-феніл-2,3-дигідро-1н-піролізин-5-ілоцтова кислота, спосіб її одержання (варіанти), фармацевтична композиція на її основі та проміжна сполука
Номер патенту: 72580
Опубліковано: 15.03.2005
Автори: Стрігель Ханс-Гюнтер, Меркле Філіпп, Каммермайєр Томас, Лауфер Штефан







Формула / Реферат
1. Спосіб одержання сполуки формули І
 , I
, I
який полягає в тому, що сполуку формули III
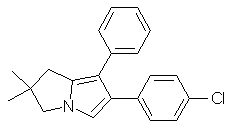 III
III
піддають взаємодії з оксалілхлоридом і одержаний продукт обробляють гідразином і гідроксидом лужного металу у водній фазі при температурі в діапазоні від 120 до 180°С в присутності аліфатичного одно- або двоатомного спирту з температурою кипіння принаймні 140°С, після завершення обробки шляхом додавання ефіру утворюють трифазну систему і підкисленням середньої фази одержують сполуку формули І.
2. Спосіб за п. 1, який полягає в тому, що як простий ефір використовують діетиловий ефір, метилтрет-бутиловий ефір або тетрагідрофуран.
3. Спосіб за п. 1 або 2, який полягає в тому, що обробку гідразином і гідроксидом лужного металу проводять у присутності етиленгліколю, монометилового ефіру етиленгліколю або діетиленгліколю.
4. Спосіб за п. 3, який полягає в тому, що обробку гідразином і гідроксидом лужного металу проводять у присутності діетиленгліколю.
5. Спосіб за будь-яким з пп. 1-4, який полягає в тому, що одержуваний взаємодією з оксалілхлоридом продукт спочатку обробляють гідразином, а потім гідроксидом лужного металу.
6. Спосіб за п. 5, який полягає в тому, що обробку гідроксидом лужного металу проводять при температурі в діапазоні від 120 до 180°С.
7. Спосіб за п. 6, який полягає в тому, що при обробці компоненти, які є леткими при температурі обробки, видаляють принаймні частково.
8. Спосіб за будь-яким з пп. 1-7, який полягає в тому, що середню фазу перед підкисленням змішують із сумішшю води з простим ефіром, що не змішується з водою.
9. Спосіб за п. 8, який полягає в тому, що цільовий продукт одержують з ефірної фази шляхом додавання до ефірної фази аліфатичного або циклоаліфатичного вуглеводню з температурою кипіння вище температури кипіння простого ефіру.
10. Спосіб за п. 8 або 9, який полягає в тому, що для одержання необхідного продукту простий ефір відганяють принаймні частково.
11. Спосіб за будь-яким з пп. 1-10, який полягає в тому, що для одержання сполуки формули III 2-бензил-4,4-диметил-1-піролін піддають взаємодії з ![]() галоген-4-хлорацетофеноном у полярному органічному розчиннику в присутності гідрокарбонату лужного металу і/або карбонату лужного металу у твердому вигляді.
галоген-4-хлорацетофеноном у полярному органічному розчиннику в присутності гідрокарбонату лужного металу і/або карбонату лужного металу у твердому вигляді.
12. Спосіб за п. 11, який полягає в тому, що як полярний органічний розчинник використовують метанол.
13. Спосіб за п. 11 або 12, який полягає в тому, що реакцію проводять у присутності твердого гідрокарбонату натрію.
14. Сполука формули II
 II.
II.
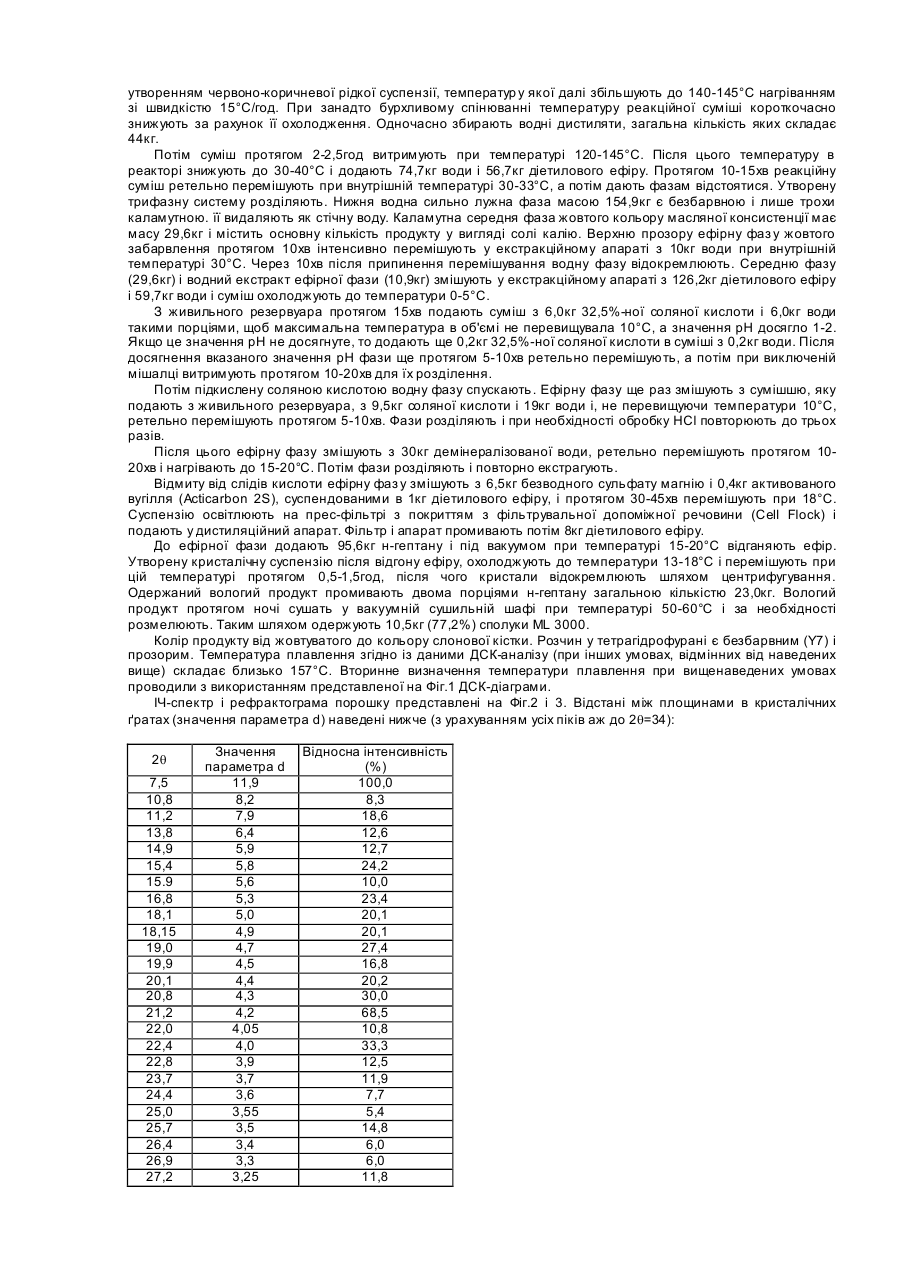
15. Сполука формули І
I
у кристалічному вигляді з єдиною ендотермою на ДСК-діаграмі, що знаходиться в діапазоні температур від 155 до 170°С.
16. Сполука за п. 15 з наступними значимими піками в ІЧ-спектрі (хвильове число, см-1): 1706; 1600; 1536; 1487; 1463; 1450; 1441; 1413; 1395; 1383; 1369; 1293; 1219; 1177; 1099; 1013; 836; 765; 698.
17. Сполука за п. 15 або 16 із представленим в основному на фіг. 2 ІЧ-спектром.
18. Сполука за будь-яким з пп. 15-17 з наступними характерними значеннями параметра d (відстань між площинами в кристалічних решітках) на рентгенограмі: 11,9; 4,2; 4,0.
19. Сполука за п. 18 з наступними значеннями параметра d: 11,9; 8,2; 7,9; 6,4; 5,9; 5,8; 5,6; 5,3; 5,0; 4,9; 4,7; 4,5; 4,4; 4,3; 4,2; 4,05; 4,0; 3,9; 3,7; 3,6; 3,55; 3,5; 3,4; 3,3; 3,25; 3,2; 3,1; 2,95; 2,9; 2,85; 2,8; 2,75; 2,7; 2,65; 2,6.
20. Фармацевтичний засіб, який містить сполуку за будь-яким з пп. 15-19, необов'язково разом зі звичайними допоміжними речовинами.
21. Застосування сполук за будь-яким з пп. 15-19 для одержання фармацевтичного засобу, призначеного для лікування захворювань ревматичного характеру.
22. Спосіб одержання сполук за будь-яким з пп. 15-19, який полягає в тому, що до ефірного розчину сполуки формули І додають аліфатичний або циклоаліфатичний вуглеводень, температура кипіння якого вище температури кипіння простого ефіру, простий ефір за необхідності відганяють принаймні частково і таким шляхом одержують сполуку формули І.
Текст
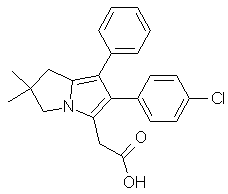
Даний винахід стосується способу одержання 6-(4-хлорфеніл)-2,2-диметил-7-феніл-2,3-Дигідро-1Hпіролізин-5-ілоцтової кислоти (ML 3000), а також нової поліморфної модифікації сполуки ML 3000, яка позначається далі як поліморфна модифікація А. ML 3000 представляє собою перспективний інгібітор циклооксигенази і 5-ліпоксигенази і тим самим придатний для лікування захворювань ревматичного характеру і для попередження захворювань, індукованих алергічними реакціями, як це описано, наприклад, у Drugs оf the Future, 20 (10), стор.1007-1009 (1095). У цій публікації також розглядається можливість одержання даної сполуки. Інші можливості одержання вищезгаданої сполуки описуються також у ЕР-А-397175, WO95/32970, WO95/32971, WO95/32972, Archiv der Pharmazie 312, стор.896-907 (1979) і 321, стор.159-162 (1988), J. Med. Chem 1994 (37), стор.1894-1897, Arch. Pharm. Med. Chem. 330, стор.307-312 (1997), При здійсненні всіх цих синтезів піролізиновий скелет структур ують за методом, проілюстрованим на наступній схемі: Реакцію проводять у метиленхлориді, етанолі або діетиловому ефірі. Бромистий водень, який утворюється в процесі реакції, уловлюють шляхом додавання водного розчину бікарбонату натрію. Введення залишку оцтової кислоти в положення 5 здійснюють потім за рахунок реакції з діазооцтовим ефіром або хлоридом ефіру щавлевої кислоти і наступного омилення або омилення і відновлення кетогрупи гідразином. У Arch. Pharm. 312, стор.896-907 (1979) розглянута наступна реакція: Цю реакцію проводять у бензолі, використовуваному як розчинник. Однак СОСОСІ-угр уповання в даному випадку переводять не в груп у оцтової кислоти, а піддають взаємодії з діетиламіном. У заявках WO95/32970, WO95/32971 і WO95/32972 описується введення залишку оцтової кислоти в сполуки, споріднені за їх структурою до сполуки ML 3000, здійснюване взаємодією цих піролізинових сполук з оксалілхлоридом або етилоксалілхлоридом і подальшим відновленням гідразином і гідроксидом калію (варіант Хуанга-Мінлона відновлення за Вольфом-Кіжнером). Більш докладні дані про проведення експериментів містяться лише в прикладі 5С в заявці WO95/32971. Відповідно до цієї методики піролізинову сполуку піддають взаємодії з оксалілхлоридом у тетрагідрофурані. До продукту реакції додають воду і гідрат гідразину, відганяють тетрагідрофуран, до залишку домішують діетиленгліколь і гідроксид калію і нагрівають до 140°С при одночасному видаленні води. Після цього реакційну суміш змішують з водою, підкислюють і розчиняють виділену карбонову кислоту в діетиловому ефірі. Продукт очищають шляхом перемішування ефірного розчину протягом деякого проміжку часу над сушильним агентом, таким як безводний сульфат натрію або сульфат магнію, залишають стояти, після чого насичений водою сульфат відфільтровують і наприкінці підведенням тепла випарюють ефір. Речовину, яка кристалізується з маточного розчину при його концентруванні, збирають і сушать. Для одержання сполуки ML 3000 у промисловому масштабі переважним є введення залишку оцтової кислоти з використанням оксалілхлориду. При цьому, однак, було встановлено, що при роботі відповідно до вищеописаної методики виділення й очищення сирого продукту, використовуваної в існуючи х у даний час способах, вихід продукту значно знижується, а на стадії очи щення і навіть у процесі сушіння знову утворюється цілий ряд продуктів розкладу, внаслідок чого виникає необхідність проведення додаткового дорогого очищення сполуки ML 3000, наприклад шляхом перекристалізації, з метою забезпечити її якість, необхідну для фармацевтичних продуктів. У вищевказаних способах одержання сполук, аналогічних за структурою до сполуки ML 3000, їх очищення і кристалізацію здійснюють нижчеописаними методами. Згідно із J. Med. Chem., 37, cтop.1894-1897 (1994) одержаний у результаті омилення етилового ефіру етанольно-лужний розчин натрієвої солі ML 3000 підкисляють фосфорною кислотою й екстрагують сумішшю з 3 частин діетилового ефіру і 1 частини метиленхлориду. Тверду речовину, яка утворюється після сушіння над сульфатом натрію, і після видалення суміші розчинників вторинно суспендують у дізопропіловому ефірі, відфільтровують і сушать. Що стосується одержання сполуки ML 3000, то можна послатися на Arch. Pharm. 321, стор.159-162 (1988) (більш докладно розглядається нижче). Згідно із Arch. Pharm. Med. Chem. 330, стор.307-312 (1997) гетероциклічні структурні аналоги сполуки ML 3000, які утворюються за методом Хуанга-Мінлону з попередників, що представляють собою ефіри 2оксооцтової кислоти, одержують шляхом концентрування елюатів, одержаних на заповненій силікагелем невеликій колонці з використанням діетилового ефіру. Згідно із Arch. Pharm. 321, стор.159-162 (1988) етилові ефіри деяких кислот, споріднених за їх структурою до сполуки ML 3000, піддають омиленню в етанольному розчині КОН, після омилення кислоти виділяють з водно-етанольного маточного розчину солей калію за допомогою 6%-ної фосфорної кислоти і розчиняють у діетиловому ефірі. Після зменшення об'єму кислоти адсорбують нейтральним оксидом алюмінію. Після елюювання ефіром нейтральних супровідних речовин карбонові кислоти десорбують з мінерального носія під дією водного розчину дигідрофосфату натрію і повторно розчиняють у діетиловому ефірі. Цей вторинний екстракт діетилового ефіру концентрують до утворення кристалів, кристали відокремлюють і після додавання пентану з метою осадження другої кристалічної фракції знову зменшують об'єм ефірного маточного розчину. Відповідно до дисертації Кіфера (Kiefer, Франкфурт, 1992) для одержання аналогічної піролізин-5ілоцтової кислоти відповідну 2-оксооцтову кислоту піддають перетворенню відповідно до відновного методу Хуанга-Мінлона. Перед виділенням у вільному вигляді піролізин-5-ілоцтової кислоти, що присутня у реакційній суміші у вигляді солі калію, видаляють супровідні речовини (від нейтрального до лужного характеру) і домішки за допомогою попередньої екстракції водно-лужної фази, що містить продукт, за допомогою етилацетату. Лише після цього за допомогою 6н. НСl виділяють карбонову кислоту, яку розчиняють у діетиловому ефірі. Діетилефірні екстракти промивають водою, сушать і повністю видаляють розчинник до моменту утворення кристалічної твердої речовини, яку потім промивають холодним діетиловим ефіром. Одержані за відомими у даний час методами кристалічні порошкові зразки сполуки ML 3000 аналізували рентгенографічним методом у рефрактометрах для порошків і рефрактограм, спектри порошків, порівнювали між собою. Потім порошкові зразки досліджували методом диференціальної сканувальної калориметрії (ДСК) або методом термогравіметрії (ТГМ). Дані рефрактометричного аналізу порошків і ДСК свідчать про те, що після кристалізації з діетилового ефіру речовину одержують спочатку у ви гляді ефірного сольвату у формі голчастих кристалів. При кристалізації з етилацетату аналогічним шляхом одержують сольват з етилацетатом у вигляді ромбічних кристалів. При цьому було встановлено, що такі сольвати є нестабільними. Вони розкладаються у вакуумі і/або при підвищених температурах з неповною віддачею зв'язаного розчинника до власне кажучи аморфних речовин, які усе ще містять залишки розчинника й у яких після сушіння можна виявити підвищений вміст продуктів розкладання. За допомогою ДСК-методу в сольватів виявляють характерні для десольватації температурні переходи. Сира сполука ML 3000, яку за гідразиновим способом одержують у вигляді солі калію і яку потім виділяють з підкисленої мінеральною сіллю реакційної суміші, окрім важкорозчинних у воді солей калію містить також гідразин, побічні продукти і продукти розкладання (продукт декарбоксилювання, а також димер) у вигляді забруднюючих домішок. У зв'язку з цим виникає необхідність проведення додаткових операцій з очищення одержуваного продукту. Так, наприклад, для видалення домішок гідразину сирі кристалічні кислоти необхідно багатократно промивати розведеними мінеральними кислотами або екстрагувати їх розчин з метою мінімізувати вміст гідразину в чистому цільовому продукті. Ні один з опублікованих до даного часу способів не дозволяє одержувати матеріал, який мав би не обмежену якими-небудь факторами придатність для використання на людині. З урахуванням вищевикладеного в основу даного винаходу була покладена задача розробити спосіб одержання сполуки ML 3000, який би дозволяв одержувати цю сполуку з високим виходом і у вигляді чисти х кристалів, які мають певну форму. Несподівано було встановлено, що вказана задача вирішується завдяки тому, що відповідну піролізинову сполуку піддають взаємодії з оксалілхлоридом і гідразином, а реакційну суміш піддають спеціальній переробці. Крім того, було встановлено, що при такій переробці утворюється нова поліморфна модифікація сполуки ML 3000 (поліморфна модифікація А). Даний винахід відповідно до цього стосується способу одержання сполуки формули І (ML 3000) який полягає в тому, що сполуку формули III піддають взаємодії з оксалілхлоридом і одержаний продукт обробляють гідразином і гідроксидом лужного металу у водній фазі при підвищеній температурі, після завершення обробки шляхом додавання ефіру, шо не змішується або частково змішується з водою, утворюють трифазну систему і підкисленням середньої фази одержують сполуку формули І. На доданих до опису кресленнях показано: на Фіг.1 - ДСК-діаграма кристалічної поліморфної модифікації сполуки ML 3000 (поліморфна модифікація А), на Фіг.2 - ІЧ-спектр поліморфної модифікації А, на Фіг.3 - рентгенограма поліморфної модифікації А, на Фіг.4 - ДСК-діаграма етилацетатного сольвату сполуки ML 3000 (поліморфна модифікація С), на Фіг.5 - ІЧ-спектр поліморфної модифікації С, на Фіг.6 - рентгенограма поліморфної модифікації С, на Фіг.7 - ДСК-діаграма діетилового ефірату сполуки ML 3000 (поліморфна модифікація Е), на Фіг.8 - ІЧ-спектр поліморфної модифікації Ε і на Фіг.9 - рентгенограма поліморфної модифікації Е. Процес одержання 6-(4-хлорфеніл)-2,2-диметил-7-феніл-2,3-дигідро-1Н-піролізин-5-ілоцтової кислоти (ML 3000) виходячи зі сполуки формули IV відповідно до запропонованого у винаході способу можна проілюструвати на прикладі наступної реакційної схеми: Сполука формули IV є відомою. Ця сполука описана, наприклад, у Arch. Pharm. 321, стор.159-162 (1988). Ця сполука може бути одержана взаємодією бензилмагнійхлориду з 3,3-Диметил-4-хлорбутиронітрилом, як це описано в J. Med. Chem. 37, стор.1894-1987 (1994). Реакцію проводять в інертному розчиннику, такому як простий ефір або вуглеводень, наприклад у толуолі. У цьому випадку сполуку формули IV піддають взаємодії з w-бром-4-хлоацетофеноном V. Сполуку w-бром-4-хлоацетофенон і її одержання відомі й описані, наприклад, у Bull. Soc. Chim. Fr. 21, стор.69 (1989). Як правило, реакцію проводять у полярному органічному розчиннику. Придатними для цієї мети полярними органічними розчинниками є насамперед С 1-С4спирти, такі як метанол, етанол, ізопропанол або прості ефіри, такі як діетиловий ефір, ТГФ або діоксан. Компоненти реакції можна застосовувати в еквімолярних кількостях. Однак, як правило, w-бром-4-хлоацетофенон застосовують у надлишку, наприклад у кількості аж до 40мол. %. Для уловлювання бромистого водню, який виділяється в процесі реакції, працюють у присутності основи. Переважно застосовують неорганічну основу, насамперед гідрокарбонат лужного металу або карбонат лужного металу, при цьому особливо переважні відповідні сполуки натрію і калію. Неорганічну основу можна застосовувати у вигляді водного розчину. Однак особливо переважним виявилося застосування неорганічної основи у твердому вигляді. Останнє полегшує відділення неорганічних продуктів реакції і зменшує спектр утворюваних побічних продуктів. Неорганічну основу, як правило, застосовують у еквімолярній кількості щодо кількості бромистого водню, що виділяється. Доцільно, однак, використовувати неорганічну основу в надлишку, наприклад аж до 1,8 еквівалента. Крім того, виявилося доцільним проводити реакцію без доступу світла. Температуру реакції можна змінювати в широкому діапазоні. Переважно, однак, працювати в інтервалі від 0 до 50°С. Сполуку формули III можна одержувати звичайним образом, видаляючи утворювані солі і розчинник. Таким шляхом одержують сполуку формули III з виходом принаймні 40% і з чистотою принаймні 97%. Так, зокрема, вміст ізомеру з 4-хлофенільною групою в положенні 5 складає не більше порядку 1,5%, звичайно приблизно 1%. Потім сполуку формули, III піддають реакції з оксалілхлоридом. При цьому звичайно застосовують інертний розчинник, такий як простий ефір, зокрема діетиловий ефір, метил-mреm-бутиловий ефір, тетрагідрофураи або діоксан, вуглеводень, такий як толуол, або хлорований вуглеводень, такий як метиленхлорид. Переважно використовувати тетрагідрофуран. Як правило, температура реакції знаходиться в діапазоні від -20 до +30°С. Для підтримання цього температурного режиму екзотермічну реакцію контролюють швидкістю додавання оксалілхлориду і/або за рахунок охолодження реакційної суміші. Таким шляхом одержують сполуку формули II. Потім реакційну суміш піддають взаємодії з водою для гідролізу надлишку оксалілхлориду. При цьому несподівано було встановлено, що гідроліз сполуки формули II до відповідної карбонової кислоти не відбувається. Потім реакційну суміш обробляють реагентом, придатним для відновлення кетокарбонільної групи в положенні 5 з утворенням групи оцтової кислоти. Для цієї мети переважно застосування гідразину (відновлення за Вольфом-Кіжнером). Найбільш доцільним виявився варіант Хуанга-Мінтона, відповідно до якого реакцію з гідразином проводять у висококиплячому спирті в присутності гідроксиду лужного металу. При цьому доцільно до або після додавання висококиплячого спирту принаймні частково видаляти використовуваний у реакції розчинник. Після цього додають гідразин, насамперед гідрат гідразину, і підвищують температуру реакції приблизно до 70-80°С з метою відгону при необхідності залишків розчинника. Потім температуру реакції підвищують до 120-180°С, переважно до 130-160°С. Гідроксид лужного металу додають у твердому вигляді або у вигляді концентрованого водного лугу, переважно, однак, у твердому вигляді. Момент додавання гідроксиду не відіграє вирішальної ролі, і його доцільно додавати після видалення залишків розчинника, використовуваного для реакції з оксалілхлоридом. Переважно використовувати гідроксид калію. Як висококиплячий спирт застосовують насамперед одно- або двоатомний спирт із температурою кипіння принаймні 140°С. Придатними для цієї мети спиртами є етиленгліколь, монометиловий ефір етиленгліколю і т.д. і в першу чергу діетиленгліколь. Тривалість реакції, як правило, складає від 30 до 300хв. Компоненти реакційної суміші, які є леткими при температурі реакції і до яких в основному належать вода, гідразин і присутні ще за певних умов залишки використовуваного в реакції з оксалілхлоридом розчинника, доцільно видаляти, наприклад, шляхом дистиляції. Після завершення реакції реакційну суміш змішують з ефіром (ефірний розчинник) і з водою або з водою, яка містить електроліт (наприклад, яка містить NaCl). Переважно застосовують ефір, який змішується з водою в обмеженому ступені. Придатними для цієї мети ефірами є, наприклад, метил-mреm-бутиловий ефір, тетрагідрофуран і насамперед діетиловий ефір. У результаті додавання ефіру відбувається утворення трифазної системи. Верхня фаза представляє собою ефірну фазу, яка містить присутні в системі органічні домішки. Нижня фаза представляє собою сильно лужну водн у фазу, яка містить неорганічні компоненти. Середня фаза представляє собою масляну фазу, яка складається в основному із солі ML 3000 з використовуваним у реакції гідроксидом лужного металу, який є важкорозчинним в лужній водній фазі, що містить диетиленгліколь. Несподівано було встановлено, що середня фаза містить сіль ML 3000 з високим ступенем чистоти. Фази розділяють і середню фазу змішують із сумішшю води з лише простим ефіром, що обмежено змішується з водою, наприклад діетиловим ефіром або метил-mреm-бутиловим ефіром, і підкислюють до рН приблизно 1-2 неорганічною або органічною кислотою, такою як соляна кислота, сірчана кислота, фосфорна кислота, оцтова кислота, щавлева кислота або лимонна кислота. У цьому випадку сполука ML 3000 здатна розчинятися в ефірній фазі. За необхідності цю ефірну фазу можна піддавати звичайному очищенню на наступних стадіях екстракції з використанням відповідної кислоти або води. За необхідності можна також проводити наступне очищення шляхом обробки одержуваного продукту активованим вугіллям або іншими адсорбентами (наприклад бентонітом і т.д.). Кількість простого ефіру і води, які додають для утворення трифазної системи, не відіграє вирішальної ролі. Як правило, додають таку кількість простого ефіру і води, щоб забезпечити утворення і легке розділення фаз. Як правило, використовують від 5 до 10мас. частин води і від 3 до 20мас. частин простого ефіру в перерахунку на 1мас. частину вихідної сполуки. Сполуку ML 3000 можна виділяти з ефірної фази різними методами. Так, наприклад, простий ефір можна випарювати й одержувати сполуку ML 3000 шляхом кристалізації з етилацетату або ізопропанолу. При випарюванні ефіру відбувається кристалізація сольвату простого ефіру, у якому при використанні діетилового ефіру на 1 молекулу розчинника припадає 2 молекули ML 3000. При роботі з етилацетатом одержують відповідний сольват, який містить 1 молекулу етилацетату і 2 молекули ML 3000. Однак більш переважно додавати до ефірної фази принаймні вуглеводень з більш високою в порівнянні з простим ефіром температурою кипіння, при необхідності принаймні частково відганяти простий ефір, осаджену у твердому кристалічному вигляді сполуку ML 3000 відокремлювати від маточного розчину звичайним способом, наприклад шляхом фільтрації або центрифугування, і одержувати шляхом сушіння під невисоким вакуумом при дещо підвищеній температурі. Переважно використовують вуглеводень з температурою кипіння, що принаймні на 30°С, насамперед на 40°С, вище температури кипіння простого ефіру. Вуглеводень застосовують при необхідності після відгону простого ефіру в 2-15-кратному надлишку (за об'ємом). Використовуваний вуглеводень може представляти собою аліфатичний вуглеводень з розгалуженим або нерозгалуженим ланцюгом і переважно з 6-12 атомами вуглецю. Прикладами таких є н-гексан, н-гептан, ноктан, ізооктан, н-декан, циклогексан і циклогептан. Переважний н-гептан або суміш ізомерів гептану, а також циклогексан. Несподівано було встановлено також, що при обробці ефірного розчину сполуки ML 3000, наприклад вищезгаданої ефірної фази, вуглеводнем одержують нову, кристалічну модифікацію сполуки ML 3000, яка в основному не містить розчинника, а саме, поліморфну модифікацію А. Дана поліморфна модифікація А характеризується наявністю однієї єдиної ендотерми на ДСК-діаграмі (від 50 до 200°С), яка знаходиться в діапазоні температур від 155 до 170°С. ДСК-діаграма представлена на Фіг.1. Поліморфну модифікацію А можна одержати з аморфної форми сполуки ML 3000 або з її інших поліморфних модифікацій (поліморфних модифікацій С і Е, див. приклади 4 і 5) шляхом їх обробки вуглеводнем при підвищеній температурі, наприклад при температурі в діапазоні від 40 до 110°С. Доцільно проводити обробку вуглеводнем шляхом розмішування (дигерування). Для поліморфної модифікації А характерно, окрім того, наявність наступних значимих піків у ІЧ-спектрі (розтирання з КВr у співвідношенні 1:3, ІЧ-спектрометр перетворенням Фур'є типу Spektmm 2000 фірми Perkin Elmer; керування приладом здійснюють за допомогою програми Spektram 2.00; вимірювання проводять у дифузному відбитому світлі):хвильове число (см-1): 1706; 1601; 1536; 1487; 1463; 1450; 1441; 1413; 1395; 1383; 1369; 1293; 1219; 1177; 1099; 1013; 836; 765; 698. IЧ-спектр представлений на фіг.2. Рентгенограма (рефрактограма порошку) поліморфної модифікації А представлена на фіг.3. Поліморфна модифікація А має такі характерні значення параметра d: 11,9; 4,2; 4,0, (2q: 7,5; 21,2; 22,4). Поліморфна модифікація А характеризується вузьким діапазоном розподілу частинок за розмірами, при цьому середні розміри частинок (відповідно до лазерних дифракційних спектрів, одержаним з використанням системи фірми Helios-Sypatec: сухий диспергатор RODOS; фокальна відстань 50мм) складають від 30 до 50мкм. Для сольватів же характерний широкий діапазон розподілу частинок за розмірами з високою часткою дрібнозернистої фракції з величиною частинок приблизно 10мкм і високою часткою грубозернистої фракції з величиною частинок приблизно 1мм. Середній розмір частинок складає від 100 до 150мкм. У порівнянні з відомими сольватами й аморфною формою сполуки ML 3000 поліморфна модифікація А має істотні переваги. Завдяки кристалічній структурі поліморфна модифікація А відрізняється стабільністю при сушінні і зберіганні. У неї не спостерігаються фазові перетворення і вторинне агрегування, які мають місце в аморфної форми. Крім того, чистота поліморфної модифікації А вище, оскільки при її одержанні забруднюючі домішки в кристалічну структур у не проникають. Для поліморфної модифікація А характерно щільне зчеплення кристалів з відносно невеликою поверхнею. На відміну від аморфної форми сполуки ML 3000 поверхневі явища, такі як електростатичний заряд, адгезія, адсорбція і т.п., виявляються в поліморфній модифікації А лише в незначному ступені. Крім того, кристалічна структура забезпечує високу хімічну стабільність, тоді як аморфна форма сполуки ML 3000 має велику поверхню і внаслідок цього піддана більш сильному окисному розкладанню. Сольвати не мають стабільності при зберіганні, оскільки вони здатні виділяти розчинник навіть при кімнатній температурі. Фазове перетворення, яке при цьому відбувається, в аморфну форму, яка не містить розчинника, протікає неоднозначно. При старінні сольватів каверни в кристалічній структурі залишаються. Внаслідок цього речовини в значній мірі схильні до окисної деструкції. Крім того, при старінні сольватів утворюється продукт із широким діапазоном розподілу частинок за розмірами, що негативно впливає на його реологічні властивості і його подальшу оброблюваність. За допомогою запропонованого у винаході способу виходячи зі сполуки формули III одержують сполуку ML 3000 з високим виходом, що складає принаймні 70%, що істотно ви ще в порівнянні з рівнем техніки. Так, зокрема, відповідно до прикладу 5С, наведеному в WO95/32971, аналогічну до сполуки ML 3000 сполуку одержують лише з виходом 29%. Несподіваним чином сполуку ML 3000 вдається одержувати у вигляді кристалів, які не містять розчинник, з високою чистотою. Так, наприклад, вміст самого цільового продукту, визначений шляхом титрування з використанням тетрабутиламонійхлориду, складає майже 100%. Вміст важких металів складає менше 10част./млн, а кількість золи складає 0%. Сумарна кількість ізомерів і похідних сполук ML 3000 складає менше 0,2% (згідно із даними РХВР), вміст залишків розчинника також менше 0,2% (згідно із даними газової хроматографії в паровій фазі). Запропонована у винаході сполука (поліморфна модифікація) є ефективним інгібітором циклооксигенази і/або ліпоксигенази. Ця сполука відрізняється вираженою знеболювальною дією і рівномірною інгібувальною дією на ферменти циклооксигеназу (ЦО) і ліпоксигеназу (ЛО) (IC50LO/IC 50CO~1). Завдяки цьому така сполука може застосовуватися при лікуванні захворювань, які супроводжуються змінами метаболізму арахідонової кислоти. При цьому насамперед слід назватихвороби ревматичного характеру і профілактику захворювань, які індукуються алергічними реакціями. Тим самим запропонована у винаході сполука представляє собою ефективний протизапальний засіб, знеболювальний засіб, жарознижувальний засіб, засіб, який перешкоджає звуженню бронхів і, крім того, придатний для профілактики тромбозів і запобігання анафілактичному і септичному шоку, а також для лікування дерматологічних захворювань, таких як псоріаз, кропивниця, гострі і хронічні ексантеми алергічного і неалергічного генезу. Крім того, ця сполука придатна для лікування гіперхолестеринемії. Запропоновану у винаході сполуку можна призначати або як таку у вигляді терапевтичної активної речовини, або в суміші з іншими терапевтичними активними речовинами. Однак, як правило, її призначають у вигляді фармацевтичного засобу, тобто у вигляді суміші активної речовини з фармацевтично прийнятними допоміжними речовинами, насамперед з носіями або розріджувачами і/або з добавками. Сполуку або засіб можна застосовувати ентерально, наприклад орально або ректально, або парентерально, наприклад як засіб для підшкірного, внутрішньовенного або внутрішньом'язового введення, при цьому переважна, однак, оральна лікарська форма. Тип фармацевтичного засобу і фармацевтичного носія або розріджувача залежить від передбаченої методики введення. Засоби для перорального застосування можуть бути представлені, наприклад, у вигляді таблеток або капсул і можуть містити звичайні ексципієнти, такі як в'яжучі (наприклад сироп, гуміарабік, желатин, сорбіт, трагакант або полівінілпіролідон), наповнювачі (наприклад лактозу, цукор, кукурудзяний крохмаль, фосфат кальцію, сорбіт або гліцин), ковзні речовини (наприклад стеарат магнію, тальк, поліетиленгліколь або діоксид кремнію), розпушувачі (наприклад крохмаль) або змочувачі (наприклад лаурилсульфат натрію). Рідкі препарати для перорального застосування можуть бути представлені у вигляді водних або масляних суспензій, розчинів, емульсій, сиропів, еліксирів або спреїв і т.п. або у вигляді сухи х порошків для відновлення водою або яким-небудь іншим придатним для цієї мети носієм. Рідкі препарати такого типу можуть містити звичайні добавки, наприклад суспендувальні засоби, смакові добавки, розріджувачі або емульгатори. При парентеральному призначенні можна застосовувати розчини або суспензії зі звичайними фармацевтичними носіями. Лікування запропонованою у винаході сполукою проводять шляхом введення пацієнту, переважно грудній дитині, насамперед людині, ефективної кількості сполуки, яка визначається, як правило, відповідно до фармацевтичної практики. Чи показане таке лікування пацієнту й у якій формі його слід проводити, залежить від кожного конкретного випадку і вимагає відповідної медичної оцінки (постановка діагнозу), що враховує наявність ознак, симптомів і/або функціональних порушень, так само як і ризику розвитку певних ознак, симптомів і/або функціональних порушень, а також з урахуванням інших факторів. Лікування, як правило, проводять шляхом призначення сполуки за винаходом у з розрахунку на однократний або багатократний прийом на день, при необхідності разом або по черзі з іншими активними речовинами або препаратами, які містять активні речовини, і тому добова доза для пацієнта складає від приблизно 0,1 до приблизно 1000мг, особливо переважно від 0,5 до приблизно 100мг/кг ваги тіла. Нижче винахід більш докладно пояснюється на прикладах, які не обмежують його обсяг. Наведені в даному винаході ДСК-діаграми і рентгенограми (рефрактограми порошків) одержували в такий спосіб. ДСК-аналіз проводили з використанням системи ТА 4000 фірми Mettler (вимірювальний елемент типу DSC-20; процесор ТИ; оброблення результатів вимірювання здійснювали за. допомогою програми ТА-72). Швидкість нагрівання складала 5°С/хв, а в діапазоні температур плавлення дорівнювала 2°С/хв. Рефрактограми порошків одержували за допомогою рефрактометра для порошків типу STOE Powder Diffraction System фірми Stoe, Дармштадт, з використанням монохромного CuKal-випромінювання. Приклад 1 6-(4-хлофеніл)-2,2-диметил-7-феніл-2,3-дигідро-1H-піролізин У реактор об'ємом 250л після триразового вакуумування і продувки азотом послідовно завантажують 4,64кг (190,9моля) магнію і 18,8кг діетилового ефіру. Ефір нагрівають зі зворотним холодильником. При виключеній мішалці додають 0,03кг йоду і 0,5кг (4моля) бензилхлориду, при цьому одразу ж починається реакція між магнієм і галогенідом (знебарвлення і помутніння). При включеній мішалці в реактор протягом 2год з живильного резервуара подають розчин 23,5кг (185,6моля) бензилхлориду в 37,8кг діетилового ефіру, при цьому спостерігається бурхливе кипіння утвореної чорно-сірої суміші. Після закінчення додавання розчин Гриньяра ще протягом 2год кип'ятять зі зворотним холодильником. Потім при цій температурі з живильного резервуара протягом 1,5год подають розчин 17,7кг (134,6моля) дистильованого 4-хлор-3,3диметилбутиронітрилу в 48,5кг діетилового ефіру. Далі реакційну суміш протягом ще 2год нагрівають зі зворотним холодильником. Після цього із суспензії ясно-сірого кольору при нормальному тиску відганяють діетиловий ефір. Потім відбирають 54-59кг дистиляту (необхідний час складає 2год). При цьому реакційна суміш зберігає здатність до перемішування. До залишку додають 106,3кг толуолу. Температура всередині реактора складає 43°С. Потім аж до досягнення внутрішньої температури 85-90°С відганяють суміш ефір/толуол (приблизно 36-40кг). Залишок перетворюється в густу суспензію, яка ще зберігає здатність до перемішування, без кірки. Цю суспензію переносять у реактор, у який попередньо завантажують 76,7кг льоду 38,5кг 32%-ної соляної кислоти. При внесенні суспензії в цей реактор температура фаз підвищується з 0 до 23°С. Значення рН при цьому повинно знаходитися в інтервалі від 0,5 до 1,5 (рН=1,0). Після нагрівання реактора до температури всередині нього 40-45°С фази протягом 1,75-2год інтенсивно перемішують між собою. Потім для розділення фаз при цій же температурі і при виключеній мішалці витримують протягом 10-15хв. Потім водну фазу, яка містить продукт, відокремлюють (147кг). Водну фазу о холоджують у екстракційному апараті до температури в діапазоні від -8 до 0°С і потім підлуговують 33,2кг 24%-ного аміаку, при цьому швидкість подачі аміаку регулюють таким чином, щоб температура всередині апарата не перевищувала максимум 5°С. Значення рН складає 10,5-11. Піддану підлуговуванню водну фазу протягом 30-40хв при 10-25°С ретельно перемішують з 106,3кг діетилового ефіру і потім залишають стояти протягом 25-30хв для розділення фаз. Прозору водн у фазу жовтуватого кольору (170кг) відокремлюють і відкидають. Прозору ефірну фазу жовтувато-зеленого кольору повністю концентрують під вакуумом (0,7-0,8мбара) і в результаті одержують 95кг ефірного дистиляту (1,40год). Як залишок від перегонки одержують 20,6кг ясно-зеленого масла, яке містить 86,7% 2-бензил-4,4диметил-1-піроліну. 20,6кг цього залишку (86,7%-ного), відповідно 17,9кг (95,5моля) 2-бензил-4,4-диметил-1піроліну, 29,7кг (127,2моля, 1,33 еквівалента) w-брому-4-хлорацетофенону і 226,6кг метанолу попередньо завантажують у реактор об'ємом 500 л. Після додавання 12,7кг (151,2моля, 1,58 еквівалента) гідрокарбонату натрію реакційну суміш перемішують без доступ у світла при температурі від 17 до 24°С з утворенням суспензії бежевого кольору. Реакцію продовжують до залишкового вмісту піролінової сполуки в суміші менше 5%. Через 17год відбирають пробу, яку аналізують газовою хроматографією на вміст піролінової сполуки. У результаті проведеного аналізу одержують значення 2%. Після цього суспензію піддають центрифугуванню при внутрішній температурі від 18 до 22°С і одержану в результаті центрифугування тверду речовину промивають двома порціями метанолу загальною кількістю 14,4кг. Маса ще вологого продукту жовтуватого кольору складає 25,8кг. Усе ще вологий сирий продукт (25,8кг) суспендують у 150кг води, потім протягом 15хв нагрівають до температури в діапазоні від 50 до 60°С і впродовж 40хв перемішують при цій температурі. Охолоджену до 40°С суспензію (40хв) центрифугують і одержану в результаті центрифугування тверду кристалічну речовину ясно-жовтого кольору промивають двома порціями води загальною кількістю 27кг. Потім протягом 12-24год продукт сушать під вакуумом при 50-60°С. Таким шляхом одержують 18,6кг 6-(4-хлорфеніл)-2,2-диметил-7феніл-2,3-дигідро-1H-піролізину із вмістом золи 0,33% і вмістом ізомеру (5-(4-хлорфеніл)-2,2-диметил-7-феніл2,3-дигідро-1H-піролізину) 1,0%. Приклад 2 6-(4-хлорфеніл)-2,2-диметил-7-феніл-2,3-дигідро-1Н-піролізин-5-іл оцтова кислота (сполука ML 3000) У реактор об'ємом 250л після триразового вакуумування і продувки азотом попередньо завантажують 11,5кг (35,7ммоля) 6-(4-хлорфеніл)-2,2-диметил-7-феніл-2,3-дигідро-1Н-піролізину в 60кг тетрагідрофурану (ТГФ). При подачі азоту під тиском 0,5 бара розчин жовтого кольору охолоджують до 10-15°С. Потім в атмосфері азоту з живильного резервуара протягом 35хв порціями подають 6,8кг (54,7моля) оксалілхлориду таким чином, щоб вн утрішня температура не перевищувала 20°С. Після завершення подачі оксалілхлориду утворену темно-зелену рідку суспензію протягом 20-30хв перемішують при температурі від 18 до 25°С. У реактор об'ємом 500л пошарово завантажують 18кг льоду. Потім протягом 5хв у реактор порціями подають суспензію з температурою 25°С таким чином, щоб температура в об'ємі суміші не перевищувала 20°С. Реакційну суміш ще протягом 10-20хв перемішують при температурі від 25 до 35°С. Розчин, який усе ще зберігає зелений колір, при температурі від 25 до 35°С розбавляють 62,2кг діетиленгліколю. Потім з живильного резервуара при охолодженні протягом 10-15хв подають 14,9кг (298 молів) гідрату гідразину. Внутрішня температура при цьому підвищується максимум до 40-45°С. Шляхом ступеневого підвищення температури протягом 1,5год суспензію, яка набула бежевого забарвлення, нагрівають до температури 7075°С, відганяючи при цьому ТГФ. Кількість відігнаного ТГФ, який збирається аж до досягнення температури 75°С, складає 45,4кг. Реакційну суміш о холоджують до 50-55°С і впродовж 45хв пошарово змішують з 8-10 порціями гідроксиду калію (КОН), загальна кількість якого складає 26,4кг, при цьому внутрішня температура вже при промиванні першими 5кг КОН підвищується до 65-70°С, а спочатку густа суспензія забарвлюється в жовтий колір, стає більш рідкою, і протягом короткого проміжку часу спостерігається слабке кипіння з дефлегмацією. Потім цю суспензію нагрівають до 90°С зі швидкістю 15°С/год, при цьому починаючи з температури 85°С спостерігається легке спінювання, і суспензія загущується. Потім при швидкості нагрівання 2°С/год температуру підвищують до 102°С і одночасно через реакційну суміш при більш високій швидкості обертання мішалки через заглибну трубку барботують азот. У результаті інтенсивного спінювання і додаткового газоутворення об'єм реакційної суміші збільшується вдвічі. За необхідності температуру реакційної суміші знижують шляхом її охолодження. При температурі 100-105°С піна починає опадати, що супроводжується утворенням червоно-коричневої рідкої суспензії, температур у якої далі збільшують до 140-145°С нагріванням зі швидкістю 15°С/год. При занадто бурхливому спінюванні температуру реакційної суміші короткочасно знижують за рахунок її охолодження. Одночасно збирають водні дистиляти, загальна кількість яких складає 44кг. Потім суміш протягом 2-2,5год витримують при температурі 120-145°С. Після цього температуру в реакторі знижують до 30-40°С і додають 74,7кг води і 56,7кг діетилового ефіру. Протягом 10-15хв реакційну суміш ретельно перемішують при внутрішній температурі 30-33°С, а потім дають фазам відстоятися. Утворену трифазну систему розділяють. Нижня водна сильно лужна фаза масою 154,9кг є безбарвною і лише трохи каламутною. її видаляють як стічну воду. Каламутна середня фаза жовтого кольору масляної консистенції має масу 29,6кг і містить основну кількість продукту у вигляді солі калію. Верхню прозору ефірну фаз у жовтого забарвлення протягом 10хв інтенсивно перемішують у екстракційному апараті з 10кг води при внутрішній температурі 30°С. Через 10хв після припинення перемішування водну фазу відокремлюють. Середню фазу (29,6кг) і водний екстракт ефірної фази (10,9кг) змішують у екстракційному апараті з 126,2кг діетилового ефіру і 59,7кг води і суміш охолоджують до температури 0-5°С. З живильного резервуара протягом 15хв подають суміш з 6,0кг 32,5%-ної соляної кислоти і 6,0кг води такими порціями, щоб максимальна температура в об'ємі не перевищувала 10°С, а значення рН досягло 1-2. Якщо це значення рН не досягнуте, то додають ще 0,2кг 32,5%-ної соляної кислоти в суміші з 0,2кг води. Після досягнення вказаного значення рН фази ще протягом 5-10хв ретельно перемішують, а потім при виключеній мішалці витримують протягом 10-20хв для їх розділення. Потім підкислену соляною кислотою водну фазу спускають. Ефірну фазу ще раз змішують з сумішшю, яку подають з живильного резервуара, з 9,5кг соляної кислоти і 19кг води і, не перевищуючи температури 10°С, ретельно перемішують протягом 5-10хв. Фази розділяють і при необхідності обробку НСl повторюють до трьох разів. Після цього ефірну фазу змішують з 30кг демінералізованої води, ретельно перемішують протягом 1020хв і нагрівають до 15-20°С. Потім фази розділяють і повторно екстрагують. Відмиту від слідів кислотиефірну фаз у змішують з 6,5кг безводного сульфату магнію і 0,4кг активованого вугілля (Acticarbon 2S), суспендованими в 1кг діетилового ефіру, і протягом 30-45хв перемішують при 18°С. Суспензію освітлюють на прес-фільтрі з покриттям з фільтрувальної допоміжної речовини (Cell Flock) і подають у дистиляційний апарат. Фільтр і апарат промивають потім 8кг діетилового ефіру. До ефірної фази додають 95,6кг н-гептану і під вакуумом при температурі 15-20°С відганяють ефір. Утворену кристалічну суспензію після відгону ефіру, охолоджують до температури 13-18°С і перемішують при цій температурі протягом 0,5-1,5год, після чого кристали відокремлюють шляхом центрифугування. Одержаний вологий продукт промивають двома порціями н-гептану загальною кількістю 23,0кг. Вологий продукт протягом ночі сушать у вакуумній сушильній шафі при температурі 50-60°С і за необхідності розмелюють. Таким шляхом одержують 10,5кг (77,2%) сполуки ML 3000. Колір продукту від жовтуватого до кольору слонової кістки. Розчин у тетрагідрофурані є безбарвним (Y7) і прозорим. Температура плавлення згідно із даними ДСК-аналізу (при інших умовах, відмінних від наведених вище) складає близько 157°С. Вторинне визначення температури плавлення при вищенаведених умовах проводили з використанням представленої на Фіг.1 ДСК-діаграми. ІЧ-спектр і рефрактограма порошку представлені на Фіг.2 і 3. Відстані між площинами в кристалічних ґратах (значення параметра d) наведені нижче (з урахуванням усіх піків аж до 2q=34): 2q 7,5 10,8 11,2 13,8 14,9 15,4 15.9 16,8 18,1 18,15 19,0 19,9 20,1 20,8 21,2 22,0 22,4 22,8 23,7 24,4 25,0 25,7 26,4 26,9 27,2 Значення параметра d 11,9 8,2 7,9 6,4 5,9 5,8 5,6 5,3 5,0 4,9 4,7 4,5 4,4 4,3 4,2 4,05 4,0 3,9 3,7 3,6 3,55 3,5 3,4 3,3 3,25 Відносна інтенсивність (%) 100,0 8,3 18,6 12,6 12,7 24,2 10,0 23,4 20,1 20,1 27,4 16,8 20,2 30,0 68,5 10,8 33,3 12,5 11,9 7,7 5,4 14,8 6,0 6,0 11,8 27,7 28,4 30,4 30,7 31,2 31,5 32,3 32,5 33,7 33,9 3,2 3,1 2,95 2,9 2,85 2,8 2,75 2,7 2,65 2,6 17,0 20,2 7,8 9,4 5,1 5,8 8,6 10,2 9,0 7,0 Вміст цільового продукту, визначений шляхом титрування з використанням тетрабутиламонійхлориду, складає близько 100,9%. Вміст важких металів:
ДивитисяДодаткова інформація
Назва патенту англійською6-(4-chlorophenyl)-2,2-dimethyl-7-phenyl-2,3-dihydro-1н-pyrolysine-5-yl acetic acid, a method for the preparation thereof (variants), a pharmaceutical composition based thereon and an intermediary compound
Назва патенту російською6-(4-хлорфенил)-2,2-диметил-7-фенил-2,3-дигидро-1н-пиролизин-5-илуксусная кислота, способ ее получения (варианты), фармацевтическая композиция на ее основе и промежуточное соединение
МПК / Мітки
МПК: A61P 11/08, A61P 7/02, C07D 487/04, A61P 3/06, A61P 29/00, A61P 35/04, A61P 17/04, A61P 37/08, A61P 43/00, A61P 17/00, A61P 17/06, A61P 19/02, A61K 31/407
Мітки: 6-(4-хлорфеніл)-2,2-диметил-7-феніл-2,3-дигідро-1н-піролізин-5-ілоцтова, фармацевтична, спосіб, одержання, основі, варіанти, проміжна, сполука, композиція, кислота
Код посилання
<a href="https://ua.patents.su/11-72580-6-4-khlorfenil-22-dimetil-7-fenil-23-digidro-1n-pirolizin-5-iloctova-kislota-sposib-oderzhannya-varianti-farmacevtichna-kompoziciya-na-osnovi-ta-promizhna-spoluka.html" target="_blank" rel="follow" title="База патентів України">6-(4-хлорфеніл)-2,2-диметил-7-феніл-2,3-дигідро-1н-піролізин-5-ілоцтова кислота, спосіб її одержання (варіанти), фармацевтична композиція на її основі та проміжна сполука</a>
[[[[[2-арил-2-гетероарилтіометил-1,3-діоксолан-4-іл]метокси]феніл]-1-піперазиніл]феніл]-2,4-дигідро-3н-1,2,4-триазалон, спосіб його одержання (варіанти), фармацевтична композиція, спосіб її одержання, проміжна

Номер патенту: 58486
Опубліковано: 15.08.2003
Автори: Ван Дер Ейкен Люк Альфонс Лео, Хендрікс Робер Жозеф Марія, Де Шаффуа Де Карселлес Дідьє Роберт Гай Габріель, Хірес Жан, Бакс Лео Якобус Жозеф
МПК: C07D 405/12, C07D 405/14, A61K 31/495, C07D 417/14, A61P 3/06, C07D 413/14, A61K 31/41, A61P 9/12
Мітки: 2-арил-2-гетероарилтіометил-1,3-діоксолан-4-іл]метокси]феніл]-1-піперазиніл]феніл]-2,4-дигідро-3н-1,2,4-триазалон, проміжна, варіанти, одержання, композиція, спосіб, фармацевтична
Формула / Реферат:
1. [[[[[2-арил-2-гетероарилтиометил-1,3-диоксолан-4-ил]метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-3Н-1,2,4-триазолон формулы (I) , (I)его N-окись, стереохимически изомерная форма или фармацевтически приемлемая кислотно-аддитивная соль, в которых А и В, взятые вместе, образуют двухвалентный радикал формулы:-N=CH- (a),-CH=N- (b),-СН2-СН2- (c),-СН=СН- ...
Похідні 4-(амінометил)піперидинбензаміду, фармацевтична композиція на їх основі, спосіб їх одержання (варіанти), проміжна сполука та спосіб її одержання

Номер патенту: 71591
Опубліковано: 15.12.2004
Автори: Меулеманс Енн Луіз Габріель, Босманс Жан-Поль Рене Марі Андре, Гійсен Хенрикус Якобус Маріа, де Клейн Мішель Анна Жозеф
МПК: C07D 405/12, C07D 417/14, A61K 31/4523, A61K 31/453, A61P 1/14, A61K 31/454, A61P 1/10, C07D 405/14, C07D 513/04, A61P 1/08, A61K 31/4525, A61K 31/519, A61P 1/04
Мітки: 4-(амінометил)піперидинбензаміду, варіанти, спосіб, композиція, основі, похідні, одержання, проміжна, фармацевтична, сполука
Формула / Реферат:
1. Похідні 4-(амінометил)піперидинбензаміду формули (І)їх стереохімічно ізомерна форма, їх N-оксидна форма, їх проліки або їх фармацевтично прийнятна сіль приєднання кислоти та основи,у якій -R1-R2- означає бівалентний радикал формули-О-СН2-O- (а-1),-O-СН2-СН2- (а-2),-O-СН2-СН2-O- (а-3),-O-СН2-СН2-СН2- (а-4),-О-СН2-СН2-СН2-O- (а-5),-О-СН2-СН2-СН2-СН2- ...
Бензамід 3- або 4-заміщеного 4-(амінометил)піперидину, спосіб його одержання (варіанти), фармацевтична композиція на його основі, спосіб її одержання, проміжна сполука та спосіб її одержання

Номер патенту: 67745
Опубліковано: 15.07.2004
Автори: Босманс Жан-Поль Рене Марі Андре, Суркін Мішель, де Клеін Мішель Анна Жозеф
МПК: A61K 31/4427, A61K 31/443, C07D 211/42, A61K 31/453, A61P 43/00, A61K 31/502, C07D 405/12, A61K 31/445, A61K 31/454, A61K 31/501, A61P 1/10, A61K 31/4545, A61K 31/497, C07D 401/06, C07D 405/14, C07D 405/06, A61P 1/00, A61K 31/4525, C07D 211/44
Мітки: бензамід, спосіб, фармацевтична, 4-(амінометил)піперидину, одержання, основі, варіанти, 4-(заміщеного, сполука, композиція, проміжна
Формула / Реферат:
1. Сполука бензаміду 3- або 4-заміщеного 4-(амінометил)піперидину формули (I), (I)її стереоізомерні форми, N-оксиди або фармацевтично прийнятні солі з кислотами або лугами, деR1 і R2, взяті разом, утворюють бівалентний радикал формули-O-CH2-O- (a-1),-O-CH2-CH2- (a-2),-O-CH2-CH2-O- (a-3),-O-CH2-CH2-CH2-...
Похідні 4-діариламінопіперидину, їх використання, фармацевтична композиція на їх основі, спосіб лікування (варіанти), проміжна сполука

Номер патенту: 72287
Опубліковано: 15.02.2005
Автори: Волпоул Крістофер, Браун Вілл'ям
МПК: A61P 1/00, A61P 25/06, A61P 9/12, A61K 31/454, A61K 31/4525, A61P 25/36, A61P 29/00, C07D 409/06, A61P 25/02, A61P 1/12, A61P 25/04, A61P 25/22, A61K 31/4545, A61P 31/12, A61K 31/4535, C07D 401/06, C07D 211/58, A61P 13/02, A61P 25/24, A61P 35/00, A61P 25/32, A61P 37/02, C07D 405/06, A61K 31/4468, A61P 25/34, A61P 19/08, A61P 11/00, A61P 23/00
Мітки: 4-діариламінопіперидину, основі, проміжна, фармацевтична, лікування, варіанти, композиція, спосіб, похідні, сполука, використання
Формула / Реферат:
1. Сполука формули І, (І)деR1 вибрано з групи, яка складається з(і) фенілу;(іі) піридинілу;(ііі) тієнілу;(iv) фурилу
Похідні 1,2-анельованого хіноліну, спосіб їх одержання (варіанти), фармацевтична композиція, що їх містить, проміжна сполука та спосіб її одержання

Номер патенту: 71592
Опубліковано: 15.12.2004
Автори: Анжибо Патрік Рене, Бурдрез Ксав'єр Марк, Венет Марк Гастон
МПК: C07D 471/04, C07D 215/06, C07D 487/04, A61K 31/519, C07D 403/06, C07D 401/06, C07D 215/18, A61P 35/00, A61K 31/4709
Мітки: сполука, похідні, спосіб, одержання, хіноліну, проміжна, варіанти, 1,2-анельованого, фармацевтична, композиція, містить
Формула / Реферат:
1. Похідні 1,2-анельованого хіноліну формули (І)(І)або їх фармацевтично прийнятна кислотно-адитивна сіль чи стереохімічно ізомерна форма,у якій=Х1-Х2-Х3- означає тривалентний радикал формули =N-СR6=СR7- (х-1), =СR6-СR7=СR8- (х-6), =N-N=СR6- (х-2), =СR6-N=СR7- (х-7), ...
Попередній патент: Пристрій і спосіб для очищення дна водойми
Наступний патент: Спосіб хвильової діагностики нафтогазового покладу
Випадковий патент: Вихрострумовий накладний перетворювач для контролю феромагнітних матеріалів