Сіль антагоніста ccr-2, спосіб модуляції активності рецептора хемокіну, спосіб лікування (варіанти) та фармацевтична композиція
Номер патенту: 81365
Опубліковано: 25.12.2007
Автори: Дженсен Марк, Ларсен Роберт, Сідлер Деніел Річард










Формула / Реферат
1. Сполука
 .
.
2. Спосіб модуляції активності рецептора хемокіну у ссавців, що включає введення ефективної кількості сполуки за п. 1.
3. Спосіб лікування запального та імунорегуляторного розладу або захворювання або поліпшення cтану при запальному та імунорегуляторному розладі або захворюванні, або зниження ризику запального та імунорегуляторного розладу або захворювання, що включає введення пацієнту ефективної кількості сполуки за п. 1.
4. Спосіб лікування ревматоїдного артриту або поліпшення стану при ревматоїдному артриті або зниження ризику ревматоїдного артриту, що включає введення пацієнту ефективної кількості сполуки за п. 1.
5. Фармацевтична композиція, що містить введений носій і сполуку за п. 1.
Текст
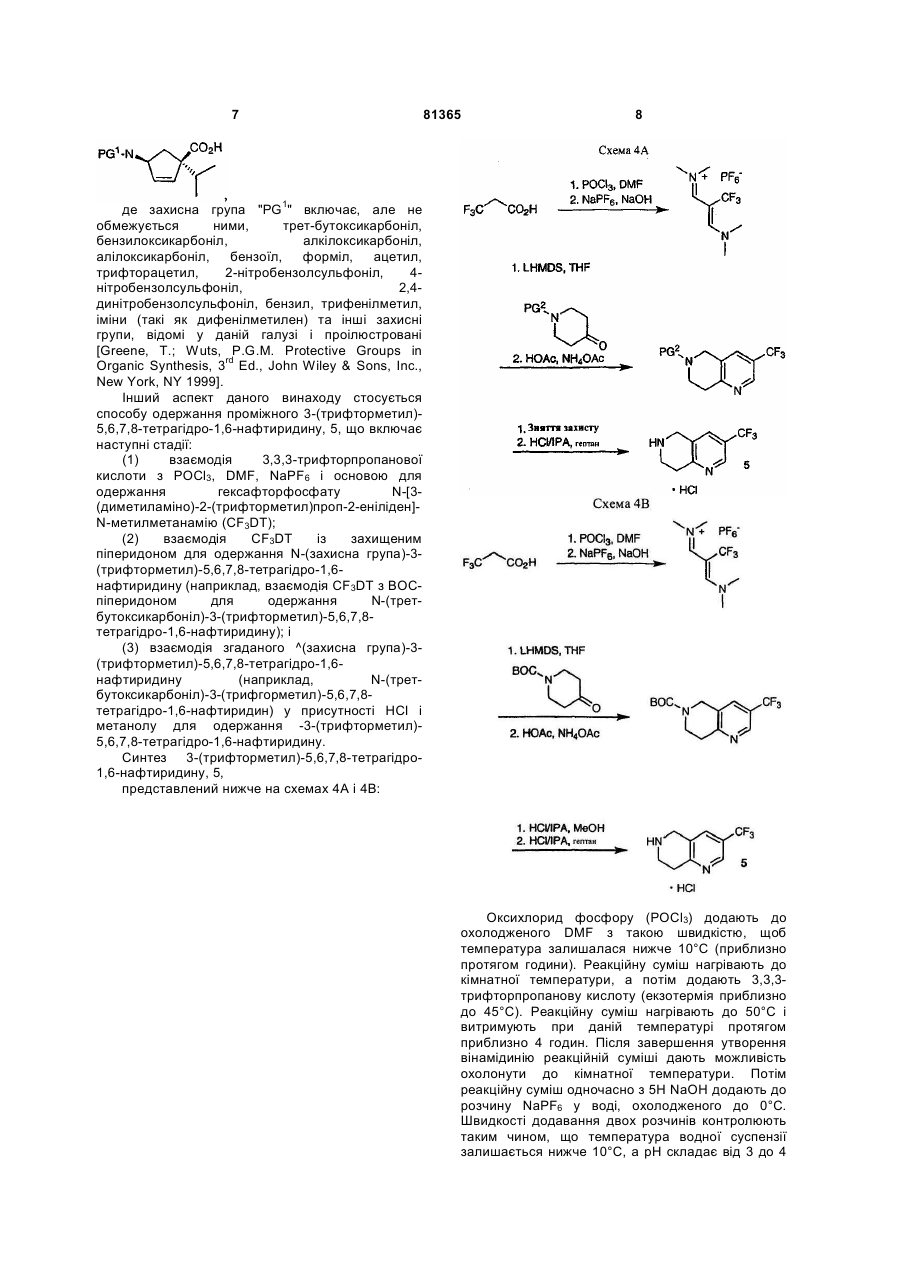
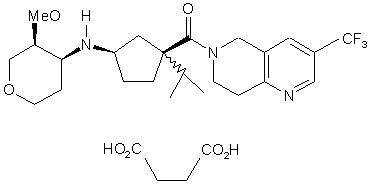
1. Сполука O MeO H C2 2 (19) 1 3 декількох дорогих реагентів, що знижує загальну ефективність процесу синтезу. Подібним чином синтез циклопентенового структурного блоку приводить до високого рівня вмісту небажаних стереоізомерів. Інші аспекти синтезу також є неефективними, дорогими і/або невідповідними для промислового виробництва. Більше того, синтезовані раніше гідрохлориди сполуки 1 виявляють далеко не ідеальні властивості при їх розчиненні, обробці (наприклад, гідроскопічність) і т.п. Таким чином, зберігається потреба у розробці вдосконаленого синтетичного способу одержання сполуки 1, придатного для великомасштабного виробництва, зберігання і розповсюдження. Також зберігається потреба у розробці вдосконалених сольових форм сполуки 1. Даний винахід стосується ефективного синтезу для одержання ((1R,3S)-3-ізопропіл-3-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну і його сукцинату. Даний винахід також стосується ефективного синтезу для одержання проміжних сполук (ЗІІ)-3-метокситетрагідро-4Н-піран-4-ону; (1S,4S)-4-(2,5-диметил-1Н-пірол-1-іл)-1ізопропілциклопент-2-ен-1-карбонової кислоти і 3(трифторметил)-5,6,7,8-тетрагідро-1,6нафтиридину; а також для одержання попередника (3S,4S)-N-((1S,4S)-4-ізопропіл-4-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопент-2-ен-1-іл)-3метокситетрагідро-2Н-піран-4-аміну. Крім того, даний винахід базується на чудових властивостях сукцинату ((1R,3S)-3-ізопропіл-3-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну. Відповідно до одного з аспектів, даний винахід стосується способу одержання (3S,4S)-N-((1S,4S)4-ізопропіл-4-{[3-(трифторметил)-7,8-дигідро-1,6нафтиридин-6(5Н)-іл]карбоніл}циклопент-2-ен-1іл)-3-метокситетрагідро-2Н-піран-4-аміну, 2: що включає наступні стадії: (1) взаємодія (1S,4S)-4-(2,5-диметил-1Н-пірол1-іл)-1-ізопропілциклопент-2-ен-1-карбонової кислоти з 3-(трифторметил)-5,6,7,8-тетрагідро-1,6нафтиридином для одержання 6-{[(1S,4S)-4-(2,5диметил-1Н-пірол-1-іл)-1-ізопропілциклопент-2-ен1-іл]карбоніл}-3-(трифторметил)-5,6,7,8тетрагідро-1,6-нафтиридину; (2) обробка 6-{[(1S,4S)-4-(2,5-диметил-1Нпірол-1-іл)-1-ізопропілциклопент-2-ен-1іл]карбоніл}-3-(трифторметил)-5,6,7,8-тетрагідро1,6-нафтиридину гідроксиламіном для одержання (1S,4S)-4-ізопропіл-4-{[3-(трифторметил)7,8дигідро-1,6-нафтиридин-6(5Н)іл]карбоніл}циклопент-2-ен-1-аміну; і (3) поєднання (1S,4S)-4-ізопропіл-4-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин 81365 4 6(5Н)-іл]карбоніл}циклопент-2-ен-1-аміну з (3R)-3мeтoкcитeтpaгiдpo-4Н-піран-4-оном шляхом відновного амінування. Відповідно до наступного аспекту, даний винахід стосується способу одержання ((1R,3S)-3ізопропіл-3-{[3-(трифторметил)-7,8-дигідро-1,6нафтиридин-6(5Н)іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну, 1: що включає наступні стадії: (4) гідрогенізація (3S,4S)-Н-{(1S,4S)-4ізопротл-4-{[3-(трифторметил)-7,8-дигідро-1,6нафтиридин-6(5Н)-іл]карбоніл}циклопент-2-ен-1іл)-3-метокситетрагідро-2Н-піран-4-аміну. Відповідно до іншого аспекту даного винаходу, що більш детально описується нижче, стадію гідрогенізації здійснюють перед одержанням сполуки 2. Синтези (3S,4S)-N-((1S,4S)-4-ізопропіл-4-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопент-2-ен-1-іл)-3метокситетрагідро-2Н-піран-4-аміну, 2, і ((1R,3S)-3ізопропіл-3-{[3-(трифторметил)-7,8-дигідро-1,6нафтиридин-6(5Н)іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну, 1, представлені на схемі 1: N-((1R,3S)-3-ізопропіл-3-{[3-(трифторметил)7,8-дигідро-1,6-нафтиридин-6(5H)іл]карбоніл}циклопентил)-N-[(цис-3S,4S)-3метокситетрагідро-2H-піран-4-іл]амін, 1, синтезують, послідовно поєднуючи три сполукиструктурних блоки, 4, 5 і 6 (наведені нижче схеми 3, 4 і 5). Реакцію амідування між 3(трифторметил)-5,6,7,8-тетрагідро-1,6нафтиридином, 5, і (1S,4S)-4-(2,5-диметил-1Нпірол-1-іл)-1-ізопропілциклопент-2-ен-1 5 81365 6 карбоновою кислотою, 4, здійснюють у присутності метансульфонілхлориду, одержуючи 6-{[(1S,4S)-4(2,5-диметил-1Н-пірол-1-іл)-1-ізопропілциклопент2-ен-1-іл]карбоніл}-3-(трифторметил)-5,6,7,8тетрагідро-1,6-нафтиридин. Зняття захисту з групи піролу здійснюють на даній стадії, використовуючи гідроксиламін як прямий процес для одержання солі аміну. Сіль аміну і (3R)-3-метокситетрагідро4Н-піран-4-он, 6, поєднують шляхом відновного амінування у присутності трибутиламінового буфера, і з NaBH(OAc)3, одержуючи (3S,4S)-N((1S,4S)-4-ізопропіл-4-{[3-(трифторметил)-7,8дигідро-1,6-нафтиридин-6(5H)іл]карбоніл}циклопент-2-ен-1-іл)-3метокситетрагідро-2Н-піран-4-амін, 2. Циклопентеновий фрагмент сполуки 2 потім гідрогенізують, одержуючи сполуку 1 у вигляді вільної основи. Інший аспект даного винаходу включає спосіб одержання сукцинату ((1R,3S)-3-ізопропіл-3-{[3(трифторметал)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбонїл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну, 3: (2,5-диметил-1Н-пірол-1-іл)циклопент-2-ен-1карбоксилату; (3) взаємодія згаданого метил (1R,4S)-4-(2,5диметилi-1Н-пірол-1-іл)циклопент-2-ен-1карбоксилату з 2-йодпропаном для одержання метил (1S,4S)-4-(2,5-диметил-1Н-пірол-1-іл)-1ізопропілциклопент-2-ен-1-карбоксилату; і (4) взаємодія згаданого метил-(1S,4S)-4-(2,5диметил-1Н-пірол-1-іл)-1-ізопропілциклопент-2-ен1-карбоксилату з NaOH і МеОН для одержання (1S,4S)-4-(2,5-диметил-1Н-пірол-1-іл)-1ізопропілциклопент-2-ен-1-карбонової кислоти. Синтез (1S,4S)-4-(2,5-диметил-1Н-пірол-1-іл)1-ізопропілциклопент-2-ен-1-карбонової кислоти, 4, представлений на схемі 3: що включає додаткову стадію: (5) контактування ((1R,3S)-3-ізопропіл-3-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну, 1, з бурштиновою кислотою. Перед стадією (5) сполука 1 може бути, необов'язково, очищена за допомогою кристалізації або у вигляді сукцинату, або іншої солі, такої як бензолсульфонат, з подальшим руйнуванням солі. Очищення таким способом описане у наступних прикладах. Синтез сполуки 3, сукцинату, представлений на схемі 2: Відповідно до даного синтезу, (1R,4S)-4аміноциклопентен-2-ен-1-карбонову кислоту перетворюють у відповідний складний метиловий ефір шляхом обробки тіонілхлоридом у метанолі. Метил (1R,4S)-4-(2,5-диметил-1Н-пірол-1іл)циклопент-2-ен-1-карбоксилат потім синтезують взаємодією метил-(1R,4S)-4-аміноциклопент-2-ен1-карбоксилату з 2,4-пентандіоном у присутності DIEA. Подальше алкілування метил-(1R,4S)-4-(2,5диметил-1Н-пірол-1-іл)циклопент-2-ен-1карбоксилату йодпропаном здійснюють, використовуючи біс-(триметилсиліл)амід літію як основу. Одержаний алкілований продукт потім гідролізують для одержання (1S,4S)-4-(2,5диметил-1Н-пірол-1-іл)-1-ізопропілциклопент-2-ен1-карбонової кислоти 4. Незважаючи на те, що наведений вище опис передбачає гідрогенізацію циклопентенового фрагмента на останній стадії для одержання циклопентану, фактично, багато сполук, представлених на схемах 1 ІЗ, можуть бути гідрогенізовані на проміжній стадії з одержанням аналога циклопентану. (Завдяки тому, що піролозахисна група, при її наявності, також відновлюється до піролідину під час такого процесу, може бути використана інша захисна група). Більш того, відповідно до іншої відмітної ознаки цього аспекту даного винаходу, можуть бути синтезовані проміжні сполуки, що мають захисні групи, відмінні від 2,5-диметил-1Н-пірол-1ілу. Таким чином, досвідчені хіміки прагнуть одержати проміжні сполуки загальної формули: Інший аспект даного винаходу стосується способу одержання проміжної (1S,4S)-4-(2,5диметил-1Н-пірол-1-іл)-1-ізопропілциклопент-2-ен1-карбонової кислоти, 4, що включає наступні стадії: (1) взаємодія (1R,4S)-4-аміноциклопентен-2ен-1-карбонової кислоти з МеОН у присутності тіонілхлориду для одержання метил (1R,4S)-4аміноциклопент-2-ен-1-карбоксилату; (2) взаємодія згаданого метил (1R,4S)-4аміноциклопент-2-ен-1-карбоксилату з ацетилацетоном для одержання метил (1R,4S)-4 7 81365 8 де захисна група "PG1" включає, але не обмежується ними, трет-бутоксикарбоніл, бензилоксикарбоніл, алкілоксикарбоніл, алілоксикарбоніл, бензоїл, форміл, ацетил, трифторацетил, 2-нітробензолсульфоніл, 4нітробензолсульфоніл, 2,4динітробензолсульфоніл, бензил, трифенілметил, іміни (такі як дифенілметилен) та інші захисні групи, відомі у даній галузі і проілюстровані [Greene, Т.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, Inc., New York, NY 1999]. Інший аспект даного винаходу стосується способу одержання проміжного 3-(трифторметил)5,6,7,8-тетрагідро-1,6-нафтиридину, 5, що включає наступні стадії: (1) взаємодія 3,3,3-трифторпропанової кислоти з РОСl3, DMF, NaPF6 і основою для одержання гексафторфосфату N-[3(диметиламіно)-2-(трифторметил)проп-2-еніліден]N-метилметанамію (CF3DT); (2) взаємодія CF3DT із захищеним піперидоном для одержання N-(захисна група)-3(трифторметил)-5,6,7,8-тетрагідро-1,6нафтиридину (наприклад, взаємодія CF3DT з ВОСпіперидоном для одержання N-(третбутоксикарбоніл)-3-(трифторметил)-5,6,7,8тетрагідро-1,6-нафтиридину); і (3) взаємодія згаданого ^(захисна група)-3(трифторметил)-5,6,7,8-тетрагідро-1,6нафтиридину (наприклад, N-(третбутоксикарбоніл)-3-(трифгорметил)-5,6,7,8тетрагідро-1,6-нафтиридин) у присутності НСI і метанолу для одержання -3-(трифторметил)5,6,7,8-тетрагідро-1,6-нафтиридину. Синтез 3-(трифторметил)-5,6,7,8-тетрагідро1,6-нафтиридину, 5, представлений нижче на схемах 4А і 4В: Оксихлорид фосфору (РОСl3) додають до охолодженого DMF з такою швидкістю, щоб температура залишалася нижче 10°С (приблизно протягом години). Реакційну суміш нагрівають до кімнатної температури, а потім додають 3,3,3трифторпропанову кислоту (екзотермія приблизно до 45°С). Реакційну суміш нагрівають до 50°С і витримують при даній температурі протягом приблизно 4 годин. Після завершення утворення вінамідинію реакційній суміші дають можливість охолонути до кімнатної температури. Потім реакційну суміш одночасно з 5Η NaOH додають до розчину NaPF6 у воді, охолодженого до 0°С. Швидкості додавання двох розчинів контролюють таким чином, що температура водної суспензії залишається нижче 10°С, а рН складає від 3 до 4 9 (додавання здійснюють протягом приблизно 2 годин). Завершивши додавання, одержану жовту суспензію піддають старінню протягом години при 0°С, а потім фільтрують, відділяючи тверді речовини. Фільтрувальний осад промивають льодяною водою (2 рази) і сушать азотом у вакуумі. Типовий вихід становить 85%. Розчин захищеного піперидону, наприклад, захищеного ВОС піперидону, у ТГФ потім додають до охолодженого (-20°С) розчину гексаметилдисиліламіду літію (LiN(TMS)2) У ТГФ, підтримуючи температуру нижче 10°С для утворення еноляту літію (приблизно 45 хвилин). Після нагрівання до кімнатної температури розчин еноляту переносять в охолоджену (-20°С) суспензію CF3DT у ТГФ з такою швидкістю, що внутрішня температура залишається на рівні нижче -10°С (протягом приблизно 45 хвилин). Одержану суміш піддають старінню протягом 2 годин при температурі -20°С, потім додають оцтову кислоту і суміш нагрівають до кімнатної температури протягом 30 хвилин. Потім додають ацетат амонію і суміш нагрівають до 65°С. Після двох часів витримування при температурі 65°С реакційну суміш охолоджують до кімнатної температури. Потім додають воду і гептани і шари розділяють. Органічний шар промивають 2Μ водною лимонною кислотою, а потім аналізують на ВОС нафтиридин. Типовий вихід становить 65%. Після цього органічний шар зі стадії утворення нафтиридину концентрують, а розчинник замінюють метанолом. Потім додають НСI в ІРА і суміш нагрівають при 60°С до завершення зняття захисту (близько години). Після охолоджування до кімнатної температури додають воду, а рН доводять приблизно до 10,5. Додають ІРАс і шари розділяють. Водний шар знову додатково двічі екстрагують ізопропілацетатом (ІРАс). Об'єднані органічні шари концентрують, а розчинник замінюють ІРА. Одержаний розчин ІРА фільтрують, видаляючи неорганічні солі, осад на фільтрі промивають ІРА, а об'єднаний фільтрат і промивні води знову концентрують до загального об'єму близько 5мг/л. Після цього розчин вільної основи/ІРА додають протягом 30 хвилин до 60°С НСI в ІРА. Тверді речовини стають помітними після додавання приблизно 50% розчину. Закінчивши додавання НСI/ІРА, додають гептан для завершення кристалізації і суспензію охолоджують до кімнатної температури. Тверді речовини виділяють фільтрацією і промивають ІРА/гептаном (3 рази по 1мл/г), а потім сушать. Типовий вихід з ВОС нафтиридину становить 75%. Як вказано вище, можуть бути використані піперидони, що містять захисні групи ("PG2"), відмінні від трет-бутоксикарбонілу. Інші захисні групи, які можуть бути корисні у синтезі нафтиридину, включають, але не обмежуються ними, бензилоксикарбоніл, алкілоксикарбоніл, алілоксикарбоніл, бензоїл, ацетил, форміл, трифторацетил, 2-нітробензолсульфоніл, 4нітробензолсульфоніл, 2,4динітробензолсульфоніл, N-бензил, трифенілметил та інші захисні групи, відомі у даній 81365 10 галузі і проілюстровані [Greene, Т.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, Inc., New York, NY 1999]. Інший аспект даного винаходу стосується способу одержання проміжного (3R)-3метокситетрагідро-4H-піран-4-ону, 6, що включає наступні стадії: (1) взаємодія тетрагідро-4Н-піран-4-ону з трипропілортоформіатом і хлорбензолом для одержання 4-пропокситетрагідро-2Н-піранетену; (2) взаємодія вказаного 4-пропокситетрагідро2Н-піранетену у присутності ацетону, води, простого діефіру гідрохінідин-1,4-фталазиндіїлу (DHQD2PHAI), дегідрату осмату калію і моногідрату 4-метилморфолін-N-оксиду (NMO) для одержання натрієвої солі 3,4-дигідрокситетрапдро2Н-піран-4-сульфонової кислоти; (3) взаємодія вказаної натрієвої солі 3,4дигідрокситетрагідро-2Н-піран-4-сульфонової кислоти з метанолом і триметилортоформіатом у присутності кислоти для одержання 4,4диметокситетрагідро-2Н-піран-3-олу; і (4) взаємодія вказаного 4,4диметокситетрагідро-2Н-піран-3-олу у присутності ТГФ, NaOt-Bu, Me2SO4 і кислоти для одержання (3R)-3-метокситетрагідро-4H-піран-4-ону. Синтез (3R)-3-метокситетрагідро-4Н-піран-4ону, 6, наведений на схемі 5: Вихідний піранон перетворюють в його дипропілкеталь шляхом обробки трипропілортоформіатом у присутності кислотного каталізатора. Потім необроблений кеталь нагрівають у присутності хлорбензолу. У таких умовах видалення пропанолу приводить до одержання простого пропіленольного ефіру. Реакцію направляють, видаляючи пропанол шляхом дистиляції у ході реакції. Потім необроблений простий енольний ефір окиснюють у модифікованих умовах асиметричного дигідроксилювання Sharpless. У даній реакції як стехіометричний окисник використовують оксид Nметилморфоліну. Звичайно реакція приводить до одержання такого продукту, як α-гідроксикетон у кількості приблизно від 60 до 85% енантіомерного надлишку (ее). α-Гідроксикетон не виділяють безпосередньо, а додають водний розчин Na2S2O5 для одержання бісульфітного аддукту кетону. З суміші ацетону і води кристалізується рацемічний бісульфітний аддукт. Рацемат видаляють 11 фільтрацією, при цьому одержуваний маточний розчин звичайно складає від 95 до 99% енантіомерного надлишку (ее). Ацетон видаляють у вакуумі і додають ізопропанол, одержуючи аддукт кристалічного бісульфіту з великим енантіомерним надлишком. Його обробляють НСI і метанолом з триметилортоформіатом як акцептор води для одержання диметилкеталю. Потім гідроксильну групу метилюють, використовуючи NaOt-Bu і Me2SO4. Додаючи воду і НСI до реакційної суміші, одержують цільовий αметоксипіранон з енантіомерним надлишком, що складає близько 96%. Нижче наведені значення деяких абревіатур, акронімів та інших скорочень. Незважаючи на те, що наведені терміни відомі фахівцям у даній галузі, нижче представлена таблиця, підсумовуюча такі терміни: Приклад 1 ((1R,3S)-Ізопропіл-3-{[3-(трифторметил)-7,8дигідро-1,6-нафтиридин-6(5Н)іл]карбоніл)циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]амін Стадія 1 - 6-{[(1S,4S)-4-(2,5-диметил-1Н-пірол1-іл)-1-ізопропілциклопент-2-ен-1-іл]карбоніл}-3(трифторметил)-5,6,7,8-тетрагідро-1,6-нафтиридин Піролциклопентенову кислоту (730г, 90,4% мас, 2,67ммоль) і діізопропілетиламін (0,93л, 5,34моль) змішують у ТГФ (6,6л). Одержаний темний розчин (KF=195мг/мл) охолоджують до 0°С, потім протягом 1 хвилини додають метансульфонілхлорид (228мл, 2,94моль), після чого внутрішня температура протягом декількох 81365 12 хвилин підвищується до 13°С. Охолоджувальну ванну видаляють, а суміш витримують при кімнатній температурі протягом 4 годин. Реакційний розчин охолоджують до ~15°С і додають НСI сіль тетрагідронафтиридину (670г, 73,2% мас. у вигляді еквівалента вільної основи, 2,42моль). Температуру підвищують до 23°С і додають іншу порцію діізопропілетиламіну (0,93л, 5,34моль) з охолоджуванням при ~25°С протягом 15 хвилин. Суміш витримують протягом >1 години, а потім розбавляють 5% NaHCO3 (16л). Продукт екстрагують етилацетатом (ЕtOАс) (16л). Органічну фазу промивають водою (10л) і концентрують у вакуумі, заміщаючи ЕtOАс метанолом до об'єму 10л. Вихід аміду, що підтверджується аналізом, є кількісним (1,055кг, 2,45моль). Стадія 2 (1S,4S)-4-ізопропіл-4-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопент-2-ен-1-амін Амід (1,055кг, 2,45моль) у метанолі (10л) додають до гідроксиламіну-НСІ (1кг, 14,4моль), 50% гідроксиламіну у воді (1л, 16,3моль) і воді (5л). Одержану суспензію нагрівають до дефлегмації (71°С) і підтримують при даній температурі протягом 6 годин. Реакційний розчин охолоджують до кімнатної температури, а рН доводять до 11,0 за допомогою 10Η NaOH. Реакційний розчин розбавляють водою (12л) і продукт тричі екстрагують хлорбензолом (14л, 13л і 13л). Кожний органічний шар один раз промивають цією ж водою (10л). Органічні шари, що містять продукт (відповідно до аналізу: 0,79кг, 2,24моль, вихід 92%), об'єднують і концентрують. Розчинник замінюють ізопропанолом (8л). Додають безводну НСI в ІРА (4,3Н, 1,4л, 6,0моль). Суміш концентрують до ~2л. Одержану суспензію нагрівають до 70°С і повільно додають н-гептан (8л). Суспензію охолоджують і витримують при кімнатній температурі протягом ночі (16 годин). Тверді речовини відфільтровують, промивають сумішшю 20% ІРА/гептан (1,5л) і сушать в атмосфері азоту, одержуючи 1,12кг солі НСI (0,76кг еквіваленту вільної основи), при цьому загальний вихід становить 91%. Стадія 3 - (3S,4S)-N-((1S,4S)-4-ізопропіл-4-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопент-2-ен-1-іл)-3метокситетрагідро-2Н-піран-4-амін 13 Аміндигідрохлорид (777г, 552г у вигляді вільної основи, 1,56моль) суспендують в IPAc (3л). Суміш охолоджують на льодяній бані і додають hBu3N (860мл, 3,61моль) з подальшим додаванням ізопропанолу (260мл, 3,40моль). При 5°С додають триацетоксиборгідрид натрію (724г, 3,42моль). Через годину до суміші при 1°С додають розчин метоксипіранону в ІРАс (1,76л, 160г/л розчину, 2,17моль). Через 6 годин суміш розподіляють між насиченим водним NaHCO3 (3л), водою (8л) і ЕtOАс (10л). Водну фазу додатково екстрагують EtOAc (15л). Об'єднані органічні фази екстрагують насиченим NaHCO3 (4л), сушать над MgSO4 (600г), а потім концентрують. Одержане масло розчиняють в CH3CN (15л) і екстрагують гептаном (3´4л). Ацетонітрильна фаза містить 654г (1,4моль, 90%) (3S,4S)-N-((1S,4S)-4-ізопропіл-4-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопент-2-ен-1-іл)-3метокситетрагідро-2Н-піран-4-аміну. Розчинник видаляють у вакуумі, одержуючи масло, яке використовують на кінцевій стадії гідрогенізації. Стадія 4 ((1R,3S)-3-ізопропіл-3-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніп}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]амін Необроблений зв'язаний циклопентен зі стадії 3 (640г) розбавляють метанолом (3,2л) і розчин концентрують до масла. Розбавлення метанолом (3,2л) і концентрування додатково повторюють двічі. Після остаточного концентрування масло розбавляють метанолом (6,4л) і завантажують в автоклав. Каталізатор, 5% Pd/C (256г), завантажують в автоклав у вигляді суспензії у метанолі (1,4л). Гідрогенізацію (40 фунтів на кв. дюйм) здійснюють протягом ночі (18 годин) при 25°С. Партію фільтрують через solka floe (глибина -1,5) у 8-л лійці із загартованого скла. Автоклав промивають метанолом (5,0л) і цю ж рідину для промивання використовують для промивання осаду на фільтрі. Осад знову промивають метанолом (1,3л). Послідовне промивання додатково повторюють чотири рази. Аналіз за допомогою РХ показує, що об'єднаний фільтрат і промивні води (загальна маса =17,1кг) містять.633г (вихід 98,5%) вільної основи. Фільтрат і промивні води концентрують до масла 81365 14 (підтверджено аналізом 770г, 1,634моль). Масло розчиняють в ІРА (3,1л) і розчин концентрують до коричневого масла. Розбавлення ІРА (3,1л) і концентрування додатково повторюють двічі. Приклад 2 Сукцинат((1R,3S)-3-ізопропіл-3-([3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл)циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл1аміну Синтез 1: Очищення від бензолсульфонату: Одержане масло з прикладу 1 розчиняють в ІРА (1,54л) і переносять у колбу для кристалізації. До партії додають промивальний ІРА (2´385мл). Розчин нагрівають до 56°С, і у цей момент додають бензолсульфонову кислоту (283г, 1,79моль), що приводить до підвищення температури до 71°С. Розчин охолоджують до 60°С і додають затравку бензолсульфонату (1г). Розбавлену суспензію витримують протягом 30 хвилин, одержуючи густий шар затравки, після чого протягом 50 хвилин додають гептан (4,62л). Після витримування при температурі від 55 до 65°С протягом 2 годин, суспензію охолоджують протягом ночі до температури навколишнього середовища. Суспензію фільтрують і вологий осад промивають сумішшю 2:1 гептан/ІРА (2´2,3л). Не зовсім білу тверду речовину сушать в. атмосфері N2/вакуум, одержуючи бензолсульфонат (875г, 98,3% мас.) у вигляді жовтувато-коричневої твердої речовини (точно встановлений вихід становить 84%). Воду (11,1л), ІРАс (32л) і бензолсульфонат ((1R,3S)-3-ізопропіл-3-{[3(трифторметил)-7,8-дигідро-1,6-нафтиридин6(5Н)-іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну (1,645кг) додають до розчину K2СО3 (6,58кг) у воді (18,3л). Суміш витримують протягом 15 хвилин. Шари розділяють і органічний шар промивають водою (16л). Розчин в ІРАс вільної основи ((1R,3S)-3ізопропіл-3-{[3-(трифторметил)-7,8-дигідро-1,6нафтиридин-6(5Н)іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну (60,5кг, що містить 1,16кг вільної основи) концентрують до масла. Одержане масло розбавляють етанолом (3,785л) і знову концентрують до масла. Розбавлення і концентрування повторюють ще тричі. Синтез солі бурштинової кислоти: Після остаточного концентрування масло розбавляють етанолом (4,055л). Потім розчин етанолу нагрівають до 65°С. Однією порцією додають бурштинову кислоту (294г, 2,48моль) з подальшим додаванням гептану (530мл) протягом 10 хвилин. Потім розчин затравлюють сукцинатом ((1R,3S)-3ізопропіл-3-{[3-(трифторметил)-7,8-дигідро-1,6нафтиридин-6(5Н)іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну (5,8г) у вигляді суспензії у гептані (50мл). Одержану суспензію витримують протягом години при 65°С, протягом якої вона помітно густіє. У кінці витримування затравкового шару протягом 1,5 години додають гептан (7,54л). Суспензію 15 витримують протягом 2 годин при 65°С, а потім їй дають можливість охолонути до кімнатної температури протягом ночі (~9 годин). Тверді речовини відфільтровують, промивають 2:1 гептаном/етанолом (2´2,3л, 2мл/г вільної основи) і сушать у вакуумі потоком Ν2 протягом 2 годин. Осад на фільтрі розбивають і додатково сушать в N2/вакуумі протягом -48 годин, одержуючи 1,328кг сукцинату (91,6%). Висушену партію сукцинату ((1R,3S)-3-ізопропіл-3-{[3-(трифторметил)-7,8дигідро-1,6-нафтиридин-6(5Н)іл]карбоніл}циклопентил)[(3S,4S)-3метокситетрагідро-2Н-піран-4-іл]аміну (1,328г) пропускають через Соmil, використовуючи сито розміром 460мкм, одержуючи 1,313г. Синтез 2 Утворення сукцинату: Вільну основу (3034,2г) завантажують через 1-мкм вбудований фільтр у 72-л колбу, обладнану термопарою, підвісною мішалкою, впускним отвором для N2/вакууму і концентратором періодичної дії, при цьому роблять відмітку на колбі на рівні 11,2л. Об'єднані концентрати додатково концентрують до ~1л, замінюючи розчинник на EtOH при постійному об'ємі, використовуючи 17л EtOH. Розчин розбавляють за допомогою ЕtOН до 11,2-л відмітки на колбі. Вакуум випускають, а концентратор періодичної дії замінюють наливною лійкою. Додають 839г бурштинової кислоти і партію нагрівають до 65°С. Протягом 7 хвилин додають гептан (1,4л). 5г затравки розчиняють у 100мл гептану і додають до партії. Розбавлену суспензію витримують протягом 60 хвилин, одержуючи густий шар затравки, після чого протягом 120 хвилин додають 34,9л гептану. Після витримування при температурі 65°С протягом 2 годин суспензію охолоджують протягом ночі до температури навколишнього середовища. Аналіз маточних розчинів за допомогою РХ показує втрату 6,5мг/мл. Суспензію фільтрують і вологий осад промивають 2´8 л 3/1 гептану/EtOH з подальшою заміною рідини для промивання 4л 3/1 гептану/EtOH. Не зовсім білу тверду речовину сушать в атмосфері N2/вакуум на фільтрувальній ємності, одержуючи 3404г жовтувато-коричневої твердої речовини, 96,7% мас, при цьому точно встановлений вихід становить 86% з підтвердженою аналізом 10,3% втратою. Перекристалізація сукцинату (необов'язкова): 12,1л EtOH завантажують у візуально чисту 50-л колбу, обладнану впускним отвором для N2/вакууму, механічною мішалкою, термопарою і впускним адаптером. Додають сукцинат і промивають 4,7л ЕtOН. Суспензію нагрівають до 65°С, після чого розчин переносять в 72-л колбу в умовах статичного вакууму через вбудований 1мкм фільтр. Завершивши перенесення, 50-л колбу промивають 1л EtOH, а рідину для промивання переносять через вбудований фільтр до партії у 72-л колбі. Фільтрований розчин EtOH концентрують при температурі 38-44°С і тиску 5 дюймів Hg до
ДивитисяДодаткова інформація
Назва патенту англійськоюCcr-2 antagonist salt, method for modulation of chemokine receptor activity, method for treating (variants) and pharmaceutical composition
Автори англійськоюJensen Mark, Larsen Robert, Sidler Daniel Richard
Назва патенту російськоюСоль антагониста ccr-2, способ модуляции активности рецептора хемокина, способ лечения (варианты) и фармацевтическая композиция
Автори російськоюДженсен Марк, Ларсен Роберт, Сидлер Дениэл Ричард
МПК / Мітки
МПК: A61K 31/4375, A61P 37/00, C07D 471/04
Мітки: варіанти, рецептора, хемокіну, активності, ccr-2, сіль, фармацевтична, модуляції, композиція, спосіб, антагоніста, лікування
Код посилання
<a href="https://ua.patents.su/11-81365-sil-antagonista-ccr-2-sposib-modulyaci-aktivnosti-receptora-khemokinu-sposib-likuvannya-varianti-ta-farmacevtichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">Сіль антагоніста ccr-2, спосіб модуляції активності рецептора хемокіну, спосіб лікування (варіанти) та фармацевтична композиція</a>
Похідні піримідину, їх застосування, спосіб їх одержання (варіанти), фармацевтична композиція, яка їх містить, спосіб її одержання, спосіб лікування раку та спосіб модуляції активності рецептора інсулінподібног

Номер патенту: 78259
Опубліковано: 15.03.2007
Автори: Папе Ендрю, Томас Ендрю, Барлаам Бернард
МПК: A61P 43/00, C07D 409/14, A61P 35/00, C07D 405/14, C07D 413/14, C07D 401/14, C07D 403/14, A61K 31/506
Мітки: модуляції, піримідину, інсулінподібног, одержання, яка, варіанти, рецептора, застосування, спосіб, містить, похідні, активності, лікування, раку, фармацевтична, композиція
Формула / Реферат:
1. Похідні піримідину формули (I):, (I)в якійR1 являє собою 5- або 6-членне гетероароматичне кільце, що містить принаймні один кільцевий гетероатом, вибраний з азоту, кисню та сірки, при цьому кільце є необов'язково заміщеним принаймні одним замісником, вибраним з C1 - C6алкілу, C1 - C6алкокси (кожний з яких може бути необов'язково заміщеним принаймні...
Тетрагідропіранілциклопентилтетрагідропіридопіридинові модулятори активності хемокінового рецептора, фармацевтична композиція на їх основі та спосіб лікування розладу або захворювання (варіанти)
Номер патенту: 75828
Опубліковано: 15.05.2006
Автори: Морріелло Грегорі, Мойєс Крістофер, Дзіао Річард, Янг Ліху
МПК: C07D 471/04, A61P 37/04, A61P 29/00
Мітки: спосіб, розладу, хемокінового, модулятори, варіанти, тетрагідропіранілциклопентилтетрагідропіридопіридинові, активності, захворювання, лікування, композиція, основі, фармацевтична, рецептора
Формула / Реферат:
1. Сполука формули І, Iде:R3 є киснем або відсутній;R8 вибирають з:(a) водню,(b) С1-3алкілу, який є незаміщеним або заміщений 1-6 атомами фтору,(c) -O-С1-3алкілу,(d) фтору і(e) гідрокси;і її фармацевтично прийнятні солі і окремі діастереомери.2. Сполука за п.1, в якій R3 відсутній.3....
Похідне тетрагідробензимідазолу або його фармацевтично прийнятна сіль, що проявляють активність антагоніста 5-нт3-рецептора, та фармацевтична композиція на його основі

Номер патенту: 27290
Опубліковано: 15.09.2000
Автори: Мітсуакі Охта, Такесі Сузукі, Кейдзі Міята, Токуо Коіде, Дзунйа Охморі, Ісао Янагісава, Акіра Матсухіса
МПК: C07D 235/06, C07D 403/12, C01B 7/00, A61K 31/40, C07D 403/08, A61K 31/415
Мітки: фармацевтична, композиція, проявляють, сіль, фармацевтично, 5-нт3-рецептора, активність, прийнятна, похідне, антагоніста, основі, тетрагідробензимідазолу
Текст:
...%: C 71,77; H 6,13; N 15,13. Масс-спектр (Е1): m/z; 358 (M+). Примеp 17. Гидрохлорид N-[(4,5,6,7- тетрагидробензимидазол-5-ил)карбонил]-фенотиазина Физико-химические свойства: Т.пл. 268 – 270оС. Элементный анализ для C20H17N3О × HCl × 0,5 х х х H2O: Рассчитано, %: C 61,14; H 4,87; N 10,69; Cl 9,02. Найдено, %: C 61,15; H 4,64; N 10,60; Cl 8,59. Масс-спектр (Е1): m/z; 347 (M+, сво бодное соединение). Пример 18....
Спосіб лікування або профілактики відторгнення трансплантованих органів, тканин або клітин за допомогою антагоніста хемокінового рецептора в комбінації з циклоспорином та фармацевтична комбінація (варіанти)

Номер патенту: 60379
Опубліковано: 15.10.2003
Автори: Праудфут Аманда, Гроне Херманн-Йозеф, Нельсон Пітер Дж., Веллс Тімоті Н.С.
МПК: A61K 38/13, A61K 38/19
Мітки: варіанти, спосіб, циклоспорином, трансплантованих, фармацевтична, лікування, органів, комбінації, клітин, рецептора, профілактики, допомогою, комбінація, антагоніста, хемокінового, тканин, відторгнення
Формула / Реферат:
1. Спосіб лікування або профілактики відторгнення трансплантованих органів, тканин або клітин, що включає введення пацієнту фармацевтичної композиції, яка включає антагоніст хемокінового рецептора в комбінації з циклоспорином. 2. Спосіб за п. 1, який відрізняється тим, що антагоніст хемокінового рецептора і циклоспорин використовуються одночасно, роздільно або послідовно.3. Спосіб за будь-яким з попередніх пунктів, який...
Спірогетероциклічні сполуки (варіанти), фармацевтична композиція, яка їх містить, спосіб їх одержання (варіанти), спосіб лікування (варіанти) та спосіб модуляції аутоімунного захворювання

Номер патенту: 73320
Опубліковано: 15.07.2005
Автори: Томсон Девід С., Фрай Лі Л., Сперо Деніс М., Хікей Еужен Р., Еммануель Мішель Дж., Янг Ерік Р.Р., Ворд Янси Д., Сан Санхінг, Ліу Веймін, Морвік Тіна М.
МПК: C07D 401/12, C07D 207/16, A61P 25/28, A61K 31/496, A61P 17/06, C07D 211/66, A61P 21/04, A61P 17/00, A61K 31/428, A61P 5/14, A61P 25/02, A61K 31/536, A61P 13/12, C07D 211/58, C07D 205/00, A61P 9/10, C07D 409/12, C07D 451/04, A61P 3/10, C07D 413/12, A61P 1/04, A61P 25/00, A61K 31/5375, C07D 417/12, C07D 207/14, A61K 31/454, A61P 37/08, C07D 309/14, A61P 19/08, A61K 31/4468, C07D 401/04, A61P 11/06, A61P 29/00, C07D 335/00, A61K 31/4965, A61K 31/506, A61P 19/10, A61P 37/02, C07D 403/06
Мітки: захворювання, аутоімунного, яка, сполуки, модуляції, фармацевтична, одержання, містить, спірогетероциклічні, лікування, спосіб, композиція, варіанти
Формула / Реферат:
1. Спірогетероциклічні сполуки формули (I):, (I)у якійHet означає піперидиніл, піролідиніл, азетидиніл, тетрагідропіраніл, тетрагідротіопіраніл, тетрагідрофураніл, гексагідропіримідиніл, гексагідропіридазиніл, піперазиніл, 1,4,5,6-тетрагідропіримідин-2-іламін, дигідрооксазоліл, 1,2-тіазинаніл-1,1-діоксид, 1,2,6-тіадіазинаніл-1,1-діоксид,...
Попередній патент: Теплоутилізатор
Наступний патент: Спосіб охорони виїмкових виробок
Випадковий патент: Спосіб реабілітаційного лікування ювенільних сальпінгоофоритів на тлі урологічної патології