Інгібітори металопротеїнази, фармацевтична композиція на їх основі, їх застосування








Формула / Реферат
1. Сполука формули І або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер
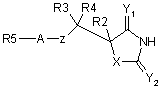 , І
, І
де
Х є NН;
Y1 та Y2 є О,
Z вибрано з О, S;
А - безпосередній зв'язок;
R2 вибрано з Н, алкілу, гетероалкілу, циклоалкілу, гетероциклоалкілу, фенілу, гетероарилу, фенілалкілу, гетероарилалкілу, циклоалкілалкілу і гетероциклоалкілалкілу;
R3 та R4 незалежно вибрані з Н, (С1-3)алкілу;
R2 може бути, як варіант, незалежно заміщений однією або більше групами, вибраними з алкілу, гало, галогеналкілу, гідрокси, алкокси, аміно, N-алкіламіно, N,N-діалкіламіно, N-алкіламідо, ціано та алкілсульфонаміно;
R5 представляє біциклічну або трициклічну групу, що містить дві або три кільцеві структури, кожна з них має до 7 кільцевих атомів, що незалежно вибрані з циклоалкілу, фенілу, гетероциклоалкілу або гетероарилу, кожна кільцева структура незалежно, як варіант, заміщена одним чи більше замісниками, незалежно вибраними з галогену, гідрокси, алкілу, алкокси, галогеналкокси, ціано і алкілсульфонілу, де будь-який радикал алкілу у будь-якому заміснику може бути сам, як варіант, заміщений одною чи більше групами, вибраними з галогену, гідрокси і алкокси;
коли R5 представляє біциклічну або трициклічну групу, кожна кільцева структура об'єднана з наступною кільцевою структурою безпосереднім зв'язком, -О- або (С1-6)алкілом;
за умови, що
коли Z представляє О, R2 представляє метил, R3 представляє Н і R4 представляє Н, тоді R5 не представляє р-бензилоксифеніл.
2. Сполука формули І за п. 1 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де R2 представляє Н, алкіл, гідроксіалкіл, алкоксіалкіл, арилоксіалкіл, аміноалкіл, циклоалкілалкіл, фенілалкіл, гетероалкіл, гетероциклоалкілалкіл або гетероарилалкіл.
3. Сполука формули I за будь-яким з попередніх пунктів або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де кожний з R3 та R4 незалежно вибраний з Н і метилу.
4. Сполука формули I за будь-яким з попередніх пунктів або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де R5 містить два або три, як варіант, заміщені, арильні або гетероарильні 5- або 6-членні кільця.
5. Сполука формули II за п. 1 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер
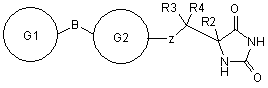 , ІІ
, ІІ
де
кожний з G1 та G2 є моноциклічною кільцевою структурою, кожна з яких містить до 7 кільцевих атомів, незалежно вибраних з циклоалкілу, фенілу, гетероциклоалкілу або гетероарилу, кожна кільцева структура незалежно, як варіант, заміщена одним чи двома замісниками, незалежно вибраними з галогену, гідрокси, галогеналкокси, ціано, алкілу, алкокси, алкілсульфонілу і ацетамідо, де будь-який радикал алкілу у будь-якому заміснику може бути сам, як варіант, заміщений одною чи більше групами, що вибрані з галогену, гідрокси і алкокси;
Z представляє О або S;
В вибрано з безпосереднього зв'язку, О і (С1-6)алкілу;
R2 вибрано з Н, (С1-6)алкілу, гідроксіалкілу, алкоксіалкілу, аміноалкілу, (N-алкіламіно)алкілу, (N,N-діалкіламіно)алкілу, амідоалкілу, або R2 представляє групу формули III
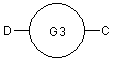 , III
, III
де С та D незалежно вибрані з безпосереднього зв'язку, Н, (С1-С6)алкілу, (С1-С6)галогеналкілу або (С1-С6)гетероалкілу, що містить один або два гетероатоми, що вибрані з N, О або S так, що, коли два гетероатоми присутні, вони розділені щонайменше двома атомами карбону;
G3 є моноциклічною кільцевою структурою, що містить до 7 кільцевих атомів, незалежно вибраних з циклоалкілу, фенілу, гетероциклоалкілу або гетероарилу, що, як варіант, заміщені одним чи двома замісниками, незалежно вибраними з галогену, гідрокси, ціано, алкілу і алкокси;
R3 та R4 незалежно вибрані з Н або (С1-3)алкілу.
6. Сполука формули II за п. 5 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де В є безпосереднім зв'язком або представляє О.
7. Сполука формули II за п. 5 або п. 6 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де R2 вибрано з (С1-6)алкілу, феніл-(С1-6)алкілу або гетероарил-(С1-6)алкілу, причому кожний, як варіант, заміщений гало, галогеналкілом, гідрокси, алкокси або ціано.
8. Сполука формули II за будь-яким з пп. 5-7 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де кожний з R3 та R4 представляє Н.
9. Сполука формули II за будь-яким з пп. 5-8 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де кожний з G1 та G2 є, як варіант, заміщеною моноциклічною групою, кільцева структура кожної з яких містить до 6 кільцевих атомів, незалежно вибраних з фенілу або гетероарилу.
10. Сполука формули II за п. 9 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де G1 заміщено галогеном, гідрокси, амідо, ціано, алкілом, галогеналкілом або алкокси.
11. Фармацевтична композиція, яка містить сполуку формули І за п. 1 або сполуку формули II за п. 5, або її фармацевтично прийнятну сіль чи здатний до гідролізу in vivo естер та фармацевтично прийнятний носій.
12. Спосіб лікування опосередкованих металопротеїназою хвороби або стану, який полягає в уведенні теплокровній тварині терапевтично ефективної кількості сполуки формули І або II, або її фармацевтично прийнятної солі чи здатного до гідролізу in vivo естеру.
13. Застосування сполуки формули І або II або її фармацевтично прийнятної солі чи здатного до гідролізу in vivo попередника у виробництві медикаменту для застосування при лікуванні хвороби або стану, опосередкованих одним чи більше металопротеїназними ферментами.
Текст
1. Сполука формули І або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер R5 A R4 R2 Y1 z NH X Y2 ,І де Х є NН; Y1 та Y2 є О, Z вибрано з О, S; А - безпосередній зв'язок; R2 вибрано з Н, алкілу, гетероалкілу, циклоалкілу, гетероциклоалкілу, фенілу, ге тероарилу, фенілалкілу, гетероарилалкілу, циклоалкілалкілу і гетероциклоалкілалкілу; R3 та R4 незалежно вибрані з Н, (С1-3)алкілу; UA R3 (19) (21) 2003098170 (22) 13.03.2002 (24) 15.01.2007 (86) PCT/SE02/00473, 13.03.2002 (31) 0100902-6 (32) 15.03.2001 (33) SE (46) 15.01.2007, Бюл. № 1, 2007 р. (72) Мунк Аф Росенскельд, SE (73) АСТРАЗЕНЕКА АБ, SE (56) STN International, File CAPLUS, CAPLUS accession number 1974:463633, Document number 81:63633, Blaha, Ludvik et al:"5-Meth yl-5phenoxymethyl-hydantoins" US 3529019 A, 15.09.1970 STN International , File CAPLUS, CAPLUS accession number 1989:173366, Document number 110:173366, Oh, Chang Hyun et al:"Synthesis of new hydantoin-3-acetic acid derivatives" 2 77667 1 (11) (54) ІНГІБІТОРИ МЕТАЛОПРОТЕЇНАЗИ, ФАРМАЦ ЕВТИЧНА КОМПОЗИЦІЯ НА ЇХ ОСНОВІ, ЇХ ЗАСТОСУВАННЯ (13) C2 ДО ПАТЕНТУ НА ВИНАХІД 3 77667 4 R2 може бути, як варіант, незалежно заміщений R2 вибрано з Н, (С1-6)алкілу, гідроксіалкілу, алкооднією або більше групами, вибраними з алкілу, ксіалкілу, аміноалкілу, (N-алкіламіно)алкілу, (N,Nгало, галогеналкілу, гідрокси, алкокси, аміно, Nдіалкіламіно)алкілу, амідоалкілу, або R2 предстаалкіламіно, N,N-діалкіламіно, N-алкіламідо, ціано вляє груп у формули III та алкілсульфонаміно; R5 представляє біциклічну або трициклічну групу, D C G3 що містить дві або три кільцеві структури, кожна з , III них має до 7 кільцевих атомів, що незалежно вибде С та D незалежно вибрані з безпосереднього рані з циклоалкілу, фенілу, гетероциклоалкілу або зв'язку, Н, (С1-С6)алкілу, (С1-С6)галогеналкілу або гетероарилу, кожна кільцева структура незалежно, (С1-С6)гетероалкілу, що містить один або два геяк варіант, заміщена одним чи більше замісникатероатоми, що вибрані з N, О або S так, що, коли ми, незалежно вибраними з галогену, гідрокси, два гетероатоми присутні, вони розділені щонайалкілу, алкокси, галогеналкокси, ціано і алкілсульменше двома атомами карбону; фонілу, де будь-який радикал алкілу у будь-якому G3 є моноциклічною кільцевою структурою, що заміснику може бути сам, як варіант, заміщений містить до 7 кільцевих атомів, незалежно вибраодною чи більше групами, вибраними з галогену, них з циклоалкілу, фенілу, гетероциклоалкілу або гідрокси і алкокси; гетероарилу, що, як варіант, заміщені одним чи коли R5 представляє біциклічну або трициклічну двома замісниками, незалежно вибраними з галогруп у, кожна кільцева структура об'єднана з настугену, гідрокси, ціано, алкілу і алкокси; пною кільцевою структурою безпосереднім зв'язR3 та R4 незалежно вибрані з Н або (С1-3)алкілу. ком, -О- або (С1-6)алкілом; 6. Сполука формули II за п. 5 або її фармацевтичза умови, що но прийнятна сіль чи здатний до гідролізу in vi vo коли Z представляє О, R2 представляє метил, R3 естер, де В є безпосереднім зв'язком або предстапредставляє Н і R4 представляє Н, тоді R5 не вляє О. представляє р-бензилоксифеніл. 7. Сполука формули II за п. 5 або п. 6 або її фар2. Сполука формули І за п. 1 або її фармацевтично мацевтично прийнятна сіль чи здатний до гідролізу прийнятна сіль чи здатний до гідролізу in vivo есin vivo естер, де R2 вибрано з (С1-6)алкілу, фенілтер, де R2 представляє Н, алкіл, гідроксіалкіл, ал(С1-6)алкілу або гетероарил-(С1-6)алкілу, причому коксіалкіл, арилоксіалкіл, аміноалкіл, циклоалкілакожний, як варіант, заміщений гало, галогеналкілкіл, фенілалкіл, гетероалкіл, лом, гідрокси, алкокси або ціано. гетероциклоалкілалкіл або гетероарилалкіл. 8. Сполука формули II за будь-яким з пп. 5-7 або її 3. Сполука формули I за будь-яким з попередніх фармацевтично прийнятна сіль чи здатний до гідпунктів або її фармацевтично прийнятна сіль чи ролізу in vivo естер, де кожний з R3 та R4 предстаздатний до гідролізу in vi vo естер, де кожний з R3 вляє Н. та R4 незалежно вибраний з Н і метилу. 9. Сполука формули II за будь-яким з пп. 5-8 або її 4. Сполука формули I за будь-яким з попередніх фармацевтично прийнятна сіль чи здатний до гідпунктів або її фармацевтично прийнятна сіль чи ролізу in vivo естер, де кожний з G1 та G2 є, як здатний до гідролізу in vivo естер, де R5 містить варіант, заміщеною моноциклічною групою, кільдва або три, як варіант, заміщені, арильні або гецева структура кожної з яких містить до 6 кільцетероарильні 5- або 6-членні кільця. вих атомів, незалежно вибраних з фенілу або ге5. Сполука формули II за п. 1 або її фармацевтичтероарилу. но прийнятна сіль чи здатний до гідролізу in vi vo 10. Сполука формули II за п. 9 або її фармацевтиестер чно прийнятна сіль чи здатний до гідролізу in vivo R3 R4 O R2 B естер, де G1 заміщено галогеном, гідрокси, амідо, G1 G2 z NH ціано, алкілом, галогеналкілом або алкокси. N 11. Фармацевтична композиція, яка містить сполуH O , ІІ ку формули І за п. 1 або сполуку формули II за де п. 5, або її фармацевтично прийнятну сіль чи здакожний з G1 та G2 є моноциклічною кільцевою тний до гідролізу in vivo естер та фармацевтично структурою, кожна з яких містить до 7 кільцевих прийнятний носій. атомів, незалежно вибраних з циклоалкілу, фені12. Спосіб лікування опосередкованих металопролу, ге тероциклоалкілу або гетероарилу, кожна кітеїназою хвороби або стану, який полягає в увельцева структура незалежно, як варіант, заміщена денні теплокровній тварині терапевтично ефектиодним чи двома замісниками, незалежно вибравної кількості сполуки формули І або II, або її ними з галогену, гідрокси, галогеналкокси, ціано, фармацевтично прийнятної солі чи здатного до алкілу, алкокси, алкілсульфонілу і ацетамідо, де гідролізу in vivo естер у. будь-який радикал алкілу у будь-якому заміснику 13. Застосування сполуки формули І або II або її може бути сам, як варіант, заміщений одною чи фармацевтично прийнятної солі чи здатного до більше групами, що вибрані з галогену, гідрокси і гідролізу in vivo попередника у виробництві медиалкокси; каменту для застосування при лікуванні хвороби Z представляє О або S; або стану, опосередкованих одним чи більше меВ вибрано з безпосереднього зв'язку, О і (С1талопротеїназними ферментами. 6)алкілу; 5 77667 Даний винахід стосується сполук, корисних при інгібуванні металопротеїназ і, зокрема, фармацевтичних композицій, що їх містять, а також їх застосування. Сполуки цього винаходу є інгібіторами одного чи більше металопротеїназних ферментів. Ме талопротеїнази є надродиною протеїназ (ферментів), число яких в останні роки різко зросло. На основі структурних та функціональних досліджень ці ферменти розподілені на родини та підродини, [як описано N.M. Hooper (1994) FEES Letters 354:16]. Приклади металопротеїназ включають матричні металопротеїнази (ММР), як-то колагенази (ММР1, ММР8, ММР13), желатинази (ММР2, ММР9), стромелізини (ММРЗ, ММР10, ММР11), матрилізин (ММР7), металоеластаза (ММР12), енамелізин (ММР19), МТ-ММР (ММР14, ММР15, ММР16, ММР17); репролізин або адамалізин або MDC родина, яка включає секретази та шедази, як-то перетворюючі TNF ферменти (ADAM 10 та ТАСЕ); астацинова родина, що включає ферменти, як-то перетворююча проколаген протеїназа (РСР); та інші металопротеїнази, як-то агреканаза, родина перетворюючих ендотелій ферментів та родина перетворюючих ангіотензин ферментів. Металопротеїнази, можна вважати важливими при гіперволемії фізіологічних хворобливих процесів, до яких залучено корекцію тканини, як-то розвиток ембріону, утворення кісток та корекцію матки при менструації. Це базується на здатності металопротеїназ розщеплювати багато матричних субстратів, як-то колаген, протеоглікан та фібронектин. Металопротеїнази, можна також вважати важливими при перетворенні або секреції важливих клітинних посередників, як-то фактор некрозу пухлин (TNF); та посттрансляційне протеолізне перетворення, або втрата біологічно важливих мембранних білків, як-то рецептор CD23 з низькою спорідненістю до ІgЕ [для повнішого огляду див. N.М.Hooper et al., (1997) Biochem J. 321:265-279]. Металопротеїнази пов'язані з багатьма хворобами або станами. Інгібування активності одної чи більше металопротеїназ може бути дуже корисним при цих хворобах або станах, наприклад: різних запальних та алергічних хворобах, як-то, запалення суглобів (особливо ревматоїдний артрит, остеоартрит та подагра), запалення шлунковокишкового тракту (особливо запальна хвороба кишечнику, виразковий коліт та гастрит), запалення шкіри (особливо псоріаз, екзема, дерматит); при метастазах або інвазії пухлин; при хворобі, пов'язаній з нерегульованою деградацією екстрацелюлярної матриці, як-то остеоартрит; при резорбтивній хворобі кісток (як-то остеопороз та хвороба Педжета); при хворобах, пов'язаних з порушеним ангіогенезом; пов'язана з діабетом посилена корекція колагену, хвороба зубів (як-то гінгівіт), покриття виразками роговиці, покриття виразками шкіри, постоперативні стани (як-то анастамоз товстої кишки) та загоєння поранень шкіри; демієлінізуючі хвороби центральної та периферійної нервових систем (як-то розсіяний склероз); хвороба Альцгеймера; корекція екстрацелюлярної матриці, яку спостерігають при серцево-судинних хворобах, як-то рестеноз та атеросклероз; астма, 6 риніт; та хронічні обструктивні хвороби легенів (COPD). ММР12 відома також як еластаза або металоеластаза макрофагів, була спочатку клонована у мишах за допомогою [Shapiro et.al. 1992, J. Biol. Chem. 267: 4664] та людей за допомоги тієї ж групи у 1995. ММР-12 преференційно експресується в активованих макрофагах, і було виявлено, що вона секретується з альвеолярних макрофагів курців [Shapiro et. al., 1993, J. Biol. Chem., 268:23824], а також у пінних клітинах в атеросклеротичних ураженнях [Matsumoto et. al., 1998, Am J Pathol 153: 109]. Мишача модель COPD базується на контрольному зараженні мишей сигаретним димом протягом 6 місяців, двома сигаретами 6 діб на тиждень Дикі миші формують емфізему легенів після цієї обробки. Коли уражених ММР12 мишей тестували у цій моделі, вони не формували значної емфіземи, чітко вказуючи, що ММР-12 є ключовим ферментом у патогенезі COPD. Роль ММР, як-то ММР12 у COPD (емфізема та бронхіт) [обговорено Anderson та Shinagawa, 1999, Current Opinion in Anti-inflammatory and Immunomodulatory Investigational Drugs 1(1): 29-38]. Зараз виявлено, що паління збільшує інфільтрацію макрофагів та похідну від макрофагів експресію ММР-12 у бляшках Кангаварі сонної артерії людини [Matetzky S, Fishbein MC et. al., Circulation 102:(18), 36-39 Suppl. S. Oct 31, 20001]. MMP13, або колагеназа 3, була спочатку клонована з похідної з пухлин молочної залози бібліотеки кДНК [J.M.P. Freije et. al. (1994) J. Biol. Chem. 269(24): 16766-16773]. ПЛР-РНК-аналіз РНК з великого числа тканин показав, що експресія ММР13 була обмежена карциномами молочної залози, оскільки вона не була виявлена у фіброаденомах молочної залози, нормальних або спочиваючих грудних залозах, плаценті, печінці, яєчнику, матці, простаті або заушній залозі або у лініях клітин раку молочної залози (T47-D, MCF-7 та ZR75-1). На додаток до цього спостереження ММР13 було виявлено у трансформованих епідермальних кератиноцитах [N. Johansson et. al., (1997) Клітин Growth Differ. 8(2):243-2501]. сквамозно-клітинній карциномі [Ν. Johansson et al., (1997) Am. J. Pathol. 151(2):499-5081] та епідермальних пухлинах [K. Airola et. al., (1997) J. Invest. Dermatol. 109(2):225231]. Ці результати підтверджують, що ММР13 секретується у трансформованих епітеліальних клітинах та може бути включеною у деградацію екстрацелюлярної матриці та клітино-матричну взаємодію, пов'язану з метастазом, як зокрема, спостерігали при ураженнях раком молочної залози та при злоякісному рості епітелію при карциногенезі шкіри. Нещодавно опубліковані дані свідчать, що ММР 13 грає роль в обороті інших сполучних тканин. Наприклад, сумісна з ММР13-субстратною специфічністю та перевагою стосовно розкладання колагену типу II [P. G. Mitchell et. al., (1996) J. Clin. Invest 97(3):761-76S; V. Knauper et. al., (1996) The Biochemical Journal 271:1544-15501]. MMP13, як припущено, грає роль під час первинного утворення кісток та корекції скелета [М. StahleBackdahl et. al., (1997) Lab. Invest. 76(5):717-728: N. 7 77667 8 Johansson et. al., (1997) Dev. Dyn. 208(31:387-397], Resp. Клітин & Моль. Biol., (Nov 1997) 17 (5):583при деструктивних хворобах суглобів, як-то ревма591]. Також, збільшену експресію ММР9 виявлено тоїдний та остео-артрит [D. Wemicke et. al., (1996) у деяких інших патологічних станах, що свідчить J. Rheumatol. 23:590-595; P. G. Mitchell et. al., про залучення ММР9 у хворобливі процеси, як-то (1996) J. Clin. Invest 97(3V.761-768: 0. Lindy et. al., COPD, артрит, метастаз пухлин, хвороба Альц(1997) Artritis Rheum 40(8):1391-13991]; та при геймера, розсіяний склероз, та руйнування тромасептичному ослабленні заміни стегна [S. Imai et. боцитів при атеросклерозі, призводячи до гострих al., (1998) J. Bone Суглоб Surg. Br, 80(4):701-710]. коронарних станів, як-то інфаркт міокарду. ММР13 також залучено у хронічний пародонтоз ММР-8 (колагеназа-2, нейтрофільна колагенадорослих, оскільки вона локалізована в епітелії за) є ферментом з розміром 53кДа родини матрихронічно запаленої слизової тканини ясен людини чних металопротеїназ, що преференційно експре[V. J. Uitto et. al., (1998) Am. J. Pathol 152(6): 1489сується у нейтрофілах. Останні дослідження 14991] та при корекції колагенної матриці при хросвідчать, що ММР-8 експресується також в інших нічних пораненнях [М. Vaalamo et. al., (1997) J. клітинах, як-то остеоартритні хондроцити. [Shlopov Invest Dermatol. 109(1):96-101]. et al, (1997) Artritis Rheum, 40:2065]. Вироблена MMP-9 (Желатиназа В; 92кДа Колагеназа типу нейтрофілами ММР може викликати корекцію ткаIV; 92кДа Желатиназа) є секретованим білком, нини, а тому блокування ММР-8 повинно мати поякий спочатку очищали, потім клонували та секвезитивний вплив при фіброзних хворобах, напринсували, у 1989 [S.M. Wilhelm et. al. (1989) J. Biol клад, легенів, та при дегратативних хворобах типу Chem. 264(29): 17213-17221; опублікована помилемфіземи легенів. ММР-8 була також виявлена як ка у J. Biol Chem. (1990) 265(36):22570]. Нещодавзверхрегульована при остеоартриті, свідчачи тим ній огляд ММР9 пропонує чудове джерело детасамим, що блокування ММР-8 може також бути льної інформації та посилань на цю протеазу: Т.Н. корисним при цій хворобі. Vu & Ζ. Werb (1998) [In: Matri x Metalloproteinases. ММР-3 (стромеліназа-1) є ферментом з розмі1998. Edited by W.C. Parks & R.P. Mecham. ppl 15 ром 53кДа родини матричних металопротеїназ. 148. Academic Press. ISBN 0-12-545090-7]. НаступАктивність ММР-3 продемонстрована у фіброблані відомості взяті з цього огляду Т.Н. Vu & Ζ. Werb стах, виділених із запалених ясен [Uitto V. J. et. аl., (1998). (1981) J. РеrіоdontаІ Res., 16:417-424], та рівні феЕкспресія ММР9 звичайно обмежена кількома рменту скорельовані з суворістю хвороби ясен типами клітин, включаючи трофобласти, остеокла[Overall С. Μ. et. al., (1987) J. Periоdontal Res., сти, нейтрофіли та макрофаги. Однак, її експресію 22:81-88]. ММР-3 продукуються також у базальних можна індукувати у тих же самих клітинах та у інкератиноцитах при багатьох хронічних виразках ших типах клітин кількома посередниками, вклю[Saarialho-Kere U. K. et. al., (1994) J. Clin. Invest, чаючи обробку клітин факторами росту або цито94:79-88]. MMP-3 mKNA та білок виявлені у базакінами. Вони є посередниками, часто залученими у льних кератиноцитах, межуючих, але на відстані початкову запальну реакцію. Як інші секретовані від краю поранення, в якому можливо представляє ММР, ММР9 вивільняється як неактивний профеділянки проліферуючого епідермісу. ММР-3 може рмент, який далі розщеплюється з утворенням тим самим заважати загоєнню епідермісу. Кілька активного ферменту. Потрібні для цієї активації дослідників продемонстрували стійке підвищення протеази in vivo невідомі. Баланс активної ММР9 ММР-3 у синовіальних рідинах від пацієнтів з реввідносно неактивного ферменту далі регулюється матоїдним та остеоартритом порівняно з контроin vivo взаємодією з ТЇМР-1 (Інгібітор тканинної лем [Walakovits L. A. et. аl., (1992) Artritis Rheum., металопротеїнази-1), природно існуючим білком. 35:35-42; Zafarullah M. et. al., (1993) J. Rheumatol., ТІМР-1 приєднується до С-термінального регіону 20:693-697]. Ці дослідження дають основу для дуММР9, призводячи до інгібування каталітичного мки, що інгібітор ММР-3 лікуватиме хвороби, при домену ММР9. Баланс індукованої експресії прояких залучено руйнування екстрацелюлярної матММРЭ, розщеплення про- в активну ММР9 та нариці, що призводить до обумовленого інфільтраціявність ТІМР-1 комбінують для визначення кількоєю лімфоцитів запалення або втрати необхідної сті каталітично активної ММР9, яка є присутньою для функції органу структурної цілісності. на локальній ділянці. Протеолітично активна Відомо ряд інгібіторів металопротеїнази [див., ММР9 атакує субстрати, що включають желатин, наприклад, огляд інгібіторів ММР Beckett R.P. та еластин, та природні колагени типу IV та тип у V; Whittaker Μ., 1998, Exp. Opin. Ther. Patents, вона не має активності проти природного колагену 8131:25 9-282]. Відмінні класи сполук можуть мати типу І, протеогліканів або ламінінів. відмінні ступені потужності та селективності інгібуЗ'являється багато даних стосовно ролі ММР9 вання різних металопротеїназ. у різних фізіологічних та патологічних процесах. [Whittaker Μ. et. al., 1999, Chemical Reviews Фізіологічні ролі включають інвазію ембріональних 99(9):2735-2776] розглядають багато відомих спотрофобластів через епітелій матки на ранніх еталук інгібіторів ММР. Вони констатують, що ефекипах ембріональної імплантації; деяку роль у рості вний інгібітор ММР потребує зв'язувальної цинк та розвитку кісток; та міграцію запальних клітин з групи або ZBG (функціональної групи, здатної хесудинної системи у тканини. латувати активну ділянку з іоном цинку (II)), щоВивільнення ММР-9, виміряне з використаннайменше одної функціональної групи, яка забезням ферментного імунодослідження, було значно печує водневий зв'язок з основою ферменту, та підвищеним у тканинних рідинах та в АМодного чи більше бічних ланцюгів, які забезпечунадосадкової рідиниах від нелікованих астматиків ють ефективну ван-дер-ваальсівську взаємодію з порівняно з астматиками з інших популяцій [Am. J. ділянками ферменту. Зв'язувальні цинк групи у 9 77667 10 відомих інгібіторах ММР включають карбоксильні тенційного протиракового засобу: групи, гідроксамові групи, сульфгідрильні групи або меркаптогрупи, тощо. Наприклад, Whittaker Μ. et. аl., обговорюють такі інгібітори ММР: Вищенаведена сполука створена для клінічної розробки. Вона має меркаптоацильну зв'язувальну цинк групу, триметилгідантоїнілетильну груп у в положенні Р1 та лейциніл-трет-бутилгліциніловий скелет. [Патенти Чехії №№ 151744 (19731119) та 152617 (1974022)] описують синтез та антиконвульсивну активність наступних сполук: R=4-NO2, 4-ОМе, 2-NO2, [Патент США №3529019 (19700915)] описує наступні сполуки як інтермедіати: Вищенаведена сполука має меркаптоацильну зв'язувальну цинк групу та імідну груп у в положенні Р1. Вищенаведена сполука була розроблена для лікування артриту. Вона має непептидну сукцинілгідроксаматну зв'язувальну цинк групу та триметилгідантоїнілетильну груп у в положенні Р1. Вищенаведена сполука є фталімідним похідним, що інгібує колагенази. Вона має непептидну сукциніл-гідроксаматну зв'язувальну цинк групу та циклічну імідну гр упу в положенні Р1. Whittaker Μ. el. аl., також обговорюють інші інгібітори ММР, що мають циклічну імідну груп у в положенні Р1 та різні зв'язувальні цинк групи (сукцинілгідроксаматну, карбоксильну, тіолову, груп у на основі фосфору). Вищенаведені сполуки виявлені як гарні інгібітори ММР8 та ММР9 [заявки РСТ WO9858925, WO9858915]. Вони мають піримідин-2,3,4трионову зв'язувальну цинк групу. Наступні сполуки невідомі як інгібітори ММР: [Lora-Tamayo, Μ et. al., 1968, An. Quim 64C6: 591-606] описують синтез наступних сполук як по [Заявка РСТ WO 00/09103] описує сполуки, корисні для лікування порушень зору, включаючи наступні (сполуки 81 і 83, Таблиця А, стр.47): Ми розкрили новий клас сполук, що є інгібіторами металопротеїназ та представляють особливий інтерес стосовно інгібування ММР, як-то ММР12. Сполуки є інгібіторами метал опротеїнази, що мають зв'язувальну метал груп у, якої нема у відомих інгібіторах метал опротеїнази. Зокрема, ми відкрили сполуки, що є потужними інгібіторами ММР12 та мають потрібні профілі активності. Сполуки цього винаходу мають корисну потужність, селективність та/або фармакокінетичні властивості. Сполука інгібіторів металопротеїназ мають зв'язувальну метал групу та одн у чи більше функціональних груп або бічних ланцюгів, характерних тим, що зв'язувальна метал група має формулу (k) де X вибрано з групи: NR1, О, S; Y1 та Y2 незалежно вибрані з групи: О, S; R1 вибрано з групи: Н, алкіл, галогеналкіл; Будь-які вищенаведені алкілгрупи можуть бути лінійними чи розгалуженими; будь-яка вищенаведена алкілгрупа представляє переважно (С17)алкіл, а найпереважніше (С1-6)алкіл. Сполука інгібітору металопротеїнази є сполукою, що інгібує активність ферменту металопротеїнази (наприклад, ММР). Як необмежувальний приклад, сполука інгібітору може виявляти величини ІК50 in vivo в межах 0.1-10000 наномоль, переважно 0.1-1000 наномоль. 11 77667 12 Зв'язувальна метал група є функціональною трициклічну групу, що містить одну, дві або три групою, здатною приєднувати іон металу на активкільцеві структури, кожна з них має до 7 кільцевих ній ділянці ферменту. Наприклад, зв'язувальна атомів, що незалежно вибрані з групи: циклоалкіл, метал група буде зв'язувальною цинк групою в арил, гетероциклоалкіл або гетероарил, кожна інгібіторах ММР, хелатуючи активну ділянку з іокільцева структура незалежно, як варіант, заміщеном цинку(ІІ). Зв'язувальна метал група формули на одним чи більше замісниками, незалежно виб(k) базується на п'яти-членній кільцевій структурі раними з групи: галоген, гідроксил, алкіл, алкокта є переважно гідантоїновою групою, найперевасил, галогеналкоксил, аміногрупа, Nжніше 5-заміщеним 1-Н,3-Н-імідазолідин-2,4алкіламіногрупа, Ν,Ν-діалкіламіногрупа, алкілсудіоном. льфонаміногрупа, алкілкарбоксіаміногрупа, ціаноЗгідно з першим аспектом винаходу запропогрупа, нітрогрупа, тіол, алкілтіол, алкілсульфоніл, новано сполуки формули І галогеналкілсульфоніл, алкіламіносульфоніл, карбоксилат, алкілкарбоксилат, амінокарбоксил, Nалкіламіно-карбоксил, Ν,Ν-діалкіламіно-карбоксил, де будь-який алкіл у будь-якому заміснику може l бути сам, як варіант, заміщеним одною чи більше групами, вибраними з групи: галоген, гідроксил, алкоксил, галогеналкоксил, аміногрупа, Nалкіламіногрупа, Ν,Ν-діалкіламіногрупа, Nде алкілсульфонаміногрупа, NX вибрано з групи: NR1, О, S; алкілкарбоксіаміногрупа, ціаногрупа, нітрогрупа, Y1 та Y2 незалежно вибрані з групи: О, S; тіол, алкілтіол, алкілсульфоніл, NΖ вибрано з групи: О, S; алкіламіносульфоніл, карбоксилат, алкілкарбокА вибрано з групи: безпосередній зв'язок, (С1сил, амінокарбоксил, N-алкіламінокарбоксил, Ν,Ν6)алкіл, (С1-6)галогеналкіл, або діалкіламінокарбоксил, карбамат; (С1-6)гетероалкіл, що містить гетерогрупу, виколи R5 представляє дициклічну або трициклібрану з групи: Ν, О, S, SO, SO2, або містить дві чну груп у, кожна кільцева структура об'єднана з гетерогрупи, вибрані з групи: N, О, S, SO, SO2, та наступною кільцевою структурою безпосереднім розділені щонайменше двома атомами карбону; зв'язком, -О-, (С1-6)алкілом, (С1-6)галогеналкілом, R1 вибрано з групи: Н, (С1-З)алкіл, галогенал(С1-6)гетероалкілом, (С1-6)алкенілом, (С1кіл; 6)алкінілом, сульфоном, CO, S, або її конденсоваR2 та R3 незалежно вибрані з групи: Н, галоно з наступною кільцевою структурою; ген (переважно F), алкіл, гетероалкіл, циклоалкіл, Будь-яка гетероалкілгрупа, визначена вище, є гетероциклоалкіл, арил, гетероарил, алкіларил, заміщеним гетероатомом алкілом, що містить одну алкіл-гетероарил, гетероалкіл-арил, гетероалкілчи більше гетерогруп, незалежно вибраних з гругетероарил, арил-алкіл, арил-гетероалкіл, гетеропи: N, О, S, SO, SO2 , (а гетерогр упою є гетероатом арил-алкіл, гетероарил-гетероалкіл, арил-арил, або група атомів); арил-гетероарил, гетероарил-арил, гетероарилБудь-який гетероциклоалкіл або гетероарил, гетероарил, циклоалкіл-алкіл, гетероциклоалкілвизначений вище, містить одну чи більше гетероалкіл, алкіл-циклоалкіл, алкіл-гетероциклоалкіл; груп, незалежно вибраних з групи: N, О, S, SO, R4 вибрано з групи: Н, галоген, (переважно F), SO2; (С1-3)алкіл або галогеналкіл; Будь-який алкіл, алкеніл або алкініл, визначені Кожний з радикалів R2 та R3 може бути, як вище, можуть бути лінійними чи розгалуженими; варіант, незалежно заміщеним однією або більше якщо не встановлено інше, будь-яка вищенаведе(краще однією) групами, вибраними з групи: алкіл, на алкілгрупа представляє переважно (С1-7)алкіл, гетероалкіл, арил, гетероарил, галоген, галогенала найпереважніше (С1-6)алкіл; кіл, гідроксил, алкоксил, галогеналкоксил, тіол, За умови, що алкілтіол, арилтіол, алкілсульфон, галогеналкілсуколи X представляє NR1, R1 представляє Η, льфон, арилсульфон, аміносульфон, NΥ1 представляє О, Y2 представляє О, Ζ представалкіламіносульфон, Ν,Ν-діалкіламіносульфон, ляє О, R2 представляє метил, R3 представляє Н, ариламіносульфон, аміногрупа, N-алкіламіногрупа, R4 представляє Н, А є безпосереднім зв'язком, Ν,Ν-діалкіламіногрупа, амідогрупа, Nтоді R5 не представляє р-хлор-феніл, оалкіламідогрупа, Ν,Ν-діалкіламідогрупа, ціаногруметоксифеніл, р-метоксифеніл, 3,4-дихлорфеніл, па, сульфонаміногрупа, алкілсульфонаміногрупа, о-нітрофеніл, р-нітрофеніл, 2-метокси-4арилсульфонаміногрупа, амідиногрупа, Nамінофеніл, 2-метокси-5-флуорфеніл або раміносульфон-амідиногрупа, гуанідиногрупа, Nбензилоксифеніл; ціано-гуанідиногрупа, тіогуанідиногрупа, 2-нітроколи X представляє NR1, R1 представляє Η, етен-1,1-діамін, карбоксил, алкіл-карбоксил, нітроΥ1 представляє О, Υ2 представляє О, Ζ представгрупа, карбамат; ляє О, R2 представляє феніл, R3 представляє Н, Як варіант, R2 та R3 можуть бути об'єднані R4 представляє Н, А є безпосереднім зв'язком, для утворення кільця, що містить до 7 кільцевих тоді R5 не представляє р-хлор-феніл. атомів, або R2 та R4 можуть бути об'єднані для Переважними сполуками формули І є ті, де виутворення кільця, що містить до 7 кільцевих атокористано будь-що з одного чи більше з наступномів, або R3 та R4 можуть бути об'єднані для утвого: рення кільця, що містить до 7 кільцевих атомів; X представляє NR1; R5 представляє моноциклічну, дициклічну або Щонайменше один з Υ1 та Υ2 представляє О; 13 77667 14 зокрема, обидва Υ1 та Υ2 представляють О; або гетероарил, що, як варіант, заміщено одним R1 представляє Н, (С1-3)алкіл, (С1чи двома замісниками, незалежно вибраними з 3)галогеналкіл; зокрема, R1 представляє Н; групи: галоген, гідроксил, аміногрупа, NR2 представляє Н, алкіл, гідроксіалкіл, алкокалкіламіногрупа, Ν,Ν-діалкіламіногрупа, ціаногрусіалкіл, арилоксіалкіл, аміноалкіл, циклоалкілпа, нітрогрупа, алкіл, алкоксил, алкілсульфон, гаалкіл, алкіл-циклоалкіл, арилалкіл, алкіларил, аллогеналкілсульфон, або алкіл заміщений одною чи кіл-гетероарил, гетероалкіл, гетероциклоалкілбільше групами, що вибрані з групи: галоген, гідалкіл, алкіл-гетероциклоалкіл, гетероарил-алкіл, роксил, аміногрупа, N-алкіламіногрупа, Ν,Νгетероалкіл-арил, зокрема, R2 представляє алкіл, діалкіламіногрупа, ціаногрупа, нітрогрупа, алкокR3 та/або R4 представляє Н; сил, галогеналкоксил; R3 та/або R4 представляє метил; Як варіант, R2 заміщено замісником, вибраним R5 містить один, два або три, як варіант, заз групи: галоген, галогеналкіл, гідроксил, алкоксил, міщені, арильні або гетероарильні 5 або 6-членні галогеналкоксил, аміногрупа, аміноалкіл, Nкільця; алкіламіногрупа, Ν,Ν-діалкіламіногрупа, (NR5 представляє дициклічну або трициклічну алкіламіно)алкіл, (N,N-діалкіламіно)алкіл, алкілсугруп у, що містить дві або три, як варіант, заміщені льфон, аміносульфон, N-алкіламіносульфон, Ν,Νкільцеві структури. діалкіламіносульфон, амідогрупа, NЗокрема, особливими сполуками формули І є алкіламідогрупа, Ν,Ν-діалкіламідогрупа, ціаногруті, де R5 представляє дициклічну або трициклічну па, сульфонаміногрупа, алкіл-сульфонаміногрупа, груп у, що містить дві або три, як варіант, заміщені амідиногрупа, N-аміносульфон-амідиногрупа, гуакільцеві структури. нідиногрупа, N-ціано-гуанідиногрупа, тіогуанідиноІншими кращими сполуками винаходу є сполугрупа, 2-нітрогуанідиногрупа, алкоксикарбоніл, ки формули II: карбоксил, алкілкарбоксил, карбамат; R3 та R4 незалежно вибрані з групи: Η або (С1-3)алкіл; ll Як варіант, R2 та R3 можуть бути об'єднані для утворення кільця, що містить до 7 кільцевих атомів, або R2 та R4 можуть бути об'єднані для де утворення кільця, що містить до 7 кільцевих атокожний з G1, та G2 є моноциклічною кільцевою мів, або R3 та R4 можуть бути об'єднані для утвоструктурою, кожна з яких містить до 7 кільцевих рення кільця, що містить до 7 кільцевих атомі; атомів, незалежно вибраних з групи: циклоалкіл, Будь-яка гетероалкілгрупа, визначена вище, є арил, гетероциклоалкіл або гетероарил, кожна заміщеним гетероатомом алкілом, що містить одну кільцева структура незалежно, як варіант, заміщечи більше гетерогруп, незалежно вибраних з груна одним чи двома замісниками, незалежно вибпи: Ν, О, S, SO, SO2 , (а гетерогр упою є гетероатом раними з групи: галоген, гідроксил, галогеналкокабо група атомів); сил, аміногрупа, N-алкіламіногрупа, Ν,ΝБудь-який гетероциклоалкіл або гетероарил, діалкіламіногрупа, ціаногрупа, нітрогрупа, алкіл, визначений вище, містить одну чи · більше гетероалкоксил, алкілсульфон, галогеналкілсульфон, груп, незалежно вибраних з групи: N, О, S, SO, алкілкарбамат, алкіламід, де будь-який алкіл у SO2; будь-якому заміснику може бути сам, як варіант, Будь-який алкіл, алкеніл або алкініл, визначені заміщеним одною чи більше групами, що вибрані з вище, можуть бути лінійними чи розгалуженими; групи: галоген, гідроксил, аміногрупа, Nякщо не встановлено інше, будь-яка вищенаведеалкіламіногрупа, Ν,Ν-діалкіламіногрупа, ціаногруна алкілгрупа представляє переважно (С1-7)алкіл, па, нітрогрупа, алкоксил, галогеналкоксил, арилока найпереважніше (С1-6)алкіл; сил, гетероарилоксил, карбамат; Переважними сполуками формули II є ті, де Ζ представляє О або S; використано будь-що з одного чи більше з наступВ вибрано з групи: безпосередній зв'язок, О, ного: (С1-6)алкіл, (С1-6)гетероалкіл; В є безпосереднім зв'язком або представляє R2 вибрано з групи: Н, (С1-6)алкіл, галогеналО; кіл, гідроксіалкіл, алкоксіалкіл, аміноалкіл, (NR2 вибрано з групи: Н, (С1-6)алкіл, арил-(С1алкіламіно)алкіл, (N,N-діалкіламіно)алкіл, амідоал6)алкіл або гетероарил-(С1-6)алкіл, як варіант, кіл, тіоалкіл, або R2 представляє групу формули III заміщений замісником, вибраним з групи: галоген, галогеналкіл, гідроксил, алкоксил, галогеналкокIII сил, аміногрупа, аміноалкіл, N-алкіламіногрупа, Ν,Ν-діалкіламіногрупа, (N-алкіламіно)алкіл, (N,NС та D незалежно вибрані з групи: безпосередіалкіламіно)алкіл, алкілсульфон, аміносульфон, дній зв'язок, Н, (С1-С6)алкіл, N-алкіламіно-сульфон, Ν,Ν-діалкіламіно-сульфон, (С1-С6)галогеналкіл, або (С1-С6)гетероалкіл, амідогрупа, N-алкіламідогрупа, Ν,Νщо містить один або два гетеро атоми, вибрані з діалкіламідогрупа, ціаногрупа, сульфонаміногрупа, групи: N, О або S так, що, коли два гетероатоми алкіл-сульфонаміногрупа, амідиногрупа, Nприсутні, вони розділені щонайменше двома атоаміносульфон-амідиногрупа, карбоксил, алкілкармами карбону; боксил, алкоксикарбоніл, карбамат; G3 є моноциклічною кільцевою структурою, що Кожний з R3 та R4 представляє Н; містить до 7 кільцевих атомів, яку незалежно вибКожний з G1 та G2 є, як варіант, заміщеною рано з групи: циклоалкіл, арил, гетероциклоалкіл моноциклічною групою, кільцева структура кожної 15 77667 16 з яких містить до 6 кільцевих атомів, незалежно вибраних з групи: арил або гетероарил, зокрема, G1 заміщено замісником, вибраним з групи: галоген, гідроксил, галогеналкоксил, аміногрупа, аміногрупа, N-алкіламіногрупа, Ν,Ν-діалкіламіногрупа, ціаногрупа, алкіл, галогеналкіл, алкоксил, де будьякий алкіл у будь-якому заміснику може бути сам, як варіант, заміщеним одною чи більше групами, що вибрано з групи: галоген, гідроксил, аміногрупа, N-алкіламіногрупа, Ν,Ν-діалкіламіногрупа, алкоксил, галогеналкоксил, ціаногрупа, карбамат. Наприклад, особливими сполуками винаходу є сполуки формули II, де В є безпосереднім зв'язком або представляє О; і Ζ представляє О або S; a R2 вибрано з групи: Н, (С1-6)алкіл, арил-(С1-6)алкіл або гетероарил-(С1-6)алкіл, як варіант, заміщений замісником, вибраним з групи: галоген, галогеналкіл, гідроксил, алкоксил, галогеналкоксил, аміногрупа, аміноалкіл, N-алкіламіногрупа, Ν,Νдіалкіламіногрупа, і кожний з R3 та R4 представСлід відмітити, що певні замісники та ряд заляє Н; а кожний з G1 та G2 є моноциклічною грумісників у сполуках формули І вибирають так, щоб пою, кільцева структура кожної з яких містить до 6 попередити стерично небажані комбінації. кільцевих атомів, незалежно вибраних з групи: Кожна сполук-приклад представляє особливий арил або гетероарил, зокрема, G1 заміщено заміта незалежний аспект винаходу. сником, вибраним з групи: галоген, гідроксил, гаКоли у сполуках формули І наявні оптично аклогеналкоксил, амідогрупа, аміногрупа, Nтивні центри, ми розкриваємо всі індивідуальні алкіламіногрупа, Ν,Ν-діалкіламіногрупа, ціаногруоптично активні форми та їх комбінації як певні па, алкіл, галогеналкіл, алкоксил, де будь-який індивідуальні втілення винаходу, а також їх відпоалкіл у будь-якому заміснику може бути сам, як відні рацемати. Рацемати можна розділити на інваріант, заміщеним одною чи більше групами, видивідуальні оптично активні форми з використанбраними з групи: галоген, гідроксил, аміногрупа, Nням відомих способів (cf. Ad vanced Organic алкіламіногрупа, Ν,Ν-діалкіламіногрупа, алкоксил, Chemistry: 3rd Edition: author J March, p.104-107), галогеналкоксил, ціаногрупа, карбамат. Придатні включаючи, наприклад, утворення діастереомерзначення для R2 включають наступні: них похідних, що мають звичайні оптично активні допоміжники, а потім їх розділення та розщеплення. Слід відмітити, що сполуки згідно з винаходом можуть містити один чи більше асиметрично заміщених атомів карбону. Наявність одного чи більше цих асиметричних центрів (хіральних центрів) у сполуці формули І може давати стереоізомери, та у кожному випадку, як виявляється, поширюватися на всі такі стереоізомери, включаючи енантіомери та діастереомери, та суміші, включаючи їх рацемічні суміші. Коли у сполуках формули І наявні таутомери, ми розкриваємо всі індивідуальні таутомерні форми та їх комбінації як певні індивідуальні втілення винаходу. Як визначено вище, сполуки винаходу є інгібіторами металопротеїнази, зокрема, вони є інгібіторами ММР12. Кожне з вищенаведених визначень для сполук формули І представляє незалежне та особливе втілення винаходу. Деякі сполуки винаходу мають особливе використання як інгібітори ММР13 та/або ММР9 та/або ММР8 та/або ММРЗ. Сполуки даного винаходу виявляють сприятливий профіль селективності. Не вдаючись до теоретичних міркувань, сполуки винаходу, можна вважати такими, що виявляють селективне інгібуПридатні значення для R5 включають насвання для будь-якого з вищенаведених визначень тупні: стосовно будь-якої інгібіторної активності відносно ТАСЕ, як необмежувальний приклад, вони можуть виявляти 100-1000-кратно більшу селективність по 17 77667 18 відношенню до інгібіторної активності відносно стану згідно з відомими у рівні техніки принципами. ТАСЕ. Звичайно одиничні дозовані форми містять Сполуки даного винаходу можуть бути забезприблизно 1мг-500мг сполуки цього винаходу. печені як фармацевтично прийнятні солі. Вони Тому, згідно з наступним аспектом, запроповключають кислотно-адитивні солі, як-то гідрохлоновано сполуку формули І або її фармацевтично рид, гідробромід, цитрат та мале та, та солі, утвоприйнятну сіль або здатний до гідролізу in vi vo рені з фосфатною та сульфатною кислотами. Згідестер для застосування у способі терапевтичного но з наступним аспектом, придатними солями є лікування людини або тварин, або для застосусолі основ, як-то сіль лужного металу, наприклад, вання як терапевтичного засобу. Ми розкриваємо натрію або калію, сіль лужноземельного металу, її застосування при лікуванні хвороби або стану, · наприклад, кальцію або магнію, або сіль органичопосередкованих одним чи більше металопротеїного аміну, наприклад, триетиламіну. назних ферментів. Зокрема, ми розкриваємо заВони також можуть бути забезпечені як здатні стосування при лікуванні хвороби або стану, оподо гідролізу in vivo естери. Ними є фармацевтично середкованих ММР12 та/або ММР13 та/або ММР9 прийнятні естери, що гідролізуються у тілі людини, та/або ММР8 та/або ММР3; зокрема, застосування утворюючи вихідн у сполуку. Такі естери можна при лікуванні хвороби або стану, опосередкованих ідентефікувати уведенням, наприклад внутрішньоММР12 або ММР9; а найкраще, застосування при венно тестованій тварині, досліджуваної сполуки, лікуванні хвороби або стану, опосередкованих а далі дослідженням рідин з організму тестММР12. тварини. Придатні здатні до гідролізу in vivo естеЗгідно з наступним аспектом нами запропонори для карбоксилу включають метоксиметил та вано спосіб лікування опосередкованих металодля гідроксилу включають форміл та ацетил, зокпротеїназою хвороби або стану, який полягає в рема ацетил. уведенні теплокровній тварині терапевтично ефеДля застосування сполуки інгібітору металоктивної кількості сполуки формули І або її фармапротеїнази винаходу (сполуки формули І або II) цевтично прийнятної солі або здатного до гідроліабо її фармацевтично прийнятної солі або здатнозу in vivo естеру. Ми також описуємо застосування го до гідролізу in vi vo естеру для терапевтичного сполуки формули І або її фармацевтично прийнятлікування (включаючи профілактичне лікування) ної солі або здатного до гідролізу in vi vo поперессавців, включаючи . людину, її звичайно формуднику у виробництві медикаменту для застосуванють згідно зі стандартною фармацевтичною пракня при лікуванні хвороби або стану, тикою як фармацевтичну композицію. опосередкованих одним чи більше металопротеїТому, згідно з наступним аспектом запропононазних ферментів. вано фармацевтичну композицію, яка містить споНаприклад, запропоновано спосіб лікування луку винаходу (сполука формули І або формули II) опосередкованих металопротеїназою хвороби або або її фармацевтично прийнятну сіль або здатний стану, який полягає в уведенні теплокровній твадо гідролізу in vivo естер та фармацевтично пририні терапевтично ефективної кількості сполуки йнятний носій. формули II (або її фармацевтично прийнятної солі Фармацевтичні композиції винаходу можна або здатного до гідролізу in vivo естеру). Ми також уводити стандартним чином при хворобі або стані, описуємо застосування сполуки формули II (або її що треба лікувати, наприклад пероральним, локафармацевтично прийнятної солі або здатного до льним, парентеральним, букальним, назальним, гідролізу in vivo попереднику) у виробництві медивагінальним або ректальним уведенням або інгакаменту для застосування при лікуванні хвороби ляцією. Для цього, сполуки винаходу можна форабо стану, опосередкованих одним чи більше мемувати відомими у рівні техніки засобами, наприталопротеїназних ферментів. клад, у таблетки, капсули, водні або масляні Опосередковані металопротеїназою хвороби розчини, суспензії, емульсії, креми, мазі, гелі, наабо стани включають астму, риніт, хронічні обзальні спреї, супозиторії, високодисперсні порошки структивні хвороби легенів (COPD), артрит (як-то або аерозолі для інгаляції, а для парентерального ревматоїдний артрит та остеоартрит), атеросклезастосування (включаючи внутрішньовенне, внутроз та рестеноз, рак, інвазію та метастаз, хвороби, рішньом'язове або вливанням) стерильні водні або при яких залучено деструкцію тканин, послабленмасляні розчини або суспензії або стерильні емуня заміни суглобу стегна, хворобу зубів, фіброзну льсії. хворобу, інфаркт та хворобу серця, фіброз печінки На додаток до сполук даного винаходу фарта нирок, ендометріоз, хвороби, пов'язані з ослабмацевтична композиція цього винаходу може таленням екстрацелюлярної матриці, серцеву некож містити, або бути уведеною разом (одночасно стачу, аневрізми аорти, хвороби центральної нерабо послідовно) з одним чи більше потрібними вової системи (CNS), як-то хвороба Альцгеймера фармакологічними засобами при лікуванні одної та розсіяний склероз (MS), гематологічні розлади. чи більше хвороб або станів, згаданих вище. Отримання сполук винаходу Фармацевтичні композиції цього винаходу Згідно з наступним аспектом винахід стосуєтьзвичайно вживатимуться людиною так, щоб, нася способу отримання сполуки формули І або II, приклад, отримати добову дозу 0.5-75мг/кг від маабо її фармацевтично прийнятної солі або здатноси тіла (та переважно 0.5-30мг/кг від маси тіла). го до гідролізу in vi vo естеру, як описано нижче. Цю добову доз у можна давати поділеними дозами, (a) Сполуки формули І, в якій кожний з Y1 та якщо необхідно, точна кількість отриманої сполуки Y2 представляє О, Ζ представляє О, а X та R5 є та шля х уведення залежить від маси, віку та ста ті такими, як описано для формули І, можна отримапацієнта, якого лікують, та від певної хвороби або ти реакцією сполуки формули VI, в якій К є відще 19 77667 20 плювалювальною групою (наприклад, естер хлоРекомбінантний рrоММРІЗ людини можна ексриду або сульфоналу), a R5 як описано для форпресувати та очищати, [як описано Knauper et al., мули І, V. Knauper et. al., (1996) The Biochemical Journal 271:1544-1550 (1996)]. Очищений фермент можна використовува ти для контролю активності інгібіторів таким чином: очищений рrоММРІЗ активують з використанням 1мМ амінофенілртутної кислоти (АРМА), 20 годин при 21°С; активований ММР13 (11,25нг на аналіз) інкубують протягом 4-5 годин зі сполукою формули VII, в якій G є сульфгідпри 35°С у буфері для аналізу (0,1Μ Tris-HCI, pH рильною групою (SH) або гідроксильною групою, а 7,5, що містить 0,1Μ NaCI, 20мМ СаСІ2, 0,02мМ X таким, як описано для формули І. Реакцію переZnCI та 0,05% (за масою/об'ємом) Brij 35), викориважно проводять у присутності основи, як-то діестовуючи синтетичний субстрат 7тилізопропіламін або карбонат цезію, та у присутметоксикумарин-4-іл)ацетил. Рrо.Lеu.Сlу.Lеu.N-3ності придатного розчиннику, наприклад, ДМФ. (2,4-динітрофеніл)-L-2,3-діамінопропіоніл. Альтернативно, сполуки можна отримати таАІа.Аrg.NН2 за наявності чи відсутності інгібіторів. ким же чином, тобто, реакцією сполук формули VI Активність визначають вимірюванням флуоресцета VII, але К у сполуці VI є сульфгідрильною грунції при λекс 328нм та λем 393нм. Процент інгібупою (SH) або гідроксигрупою, a G у формулі VIII є вання розраховують таким чином: відщеплюваною групою. % Інгібування дорівнює [Флуоресценціяз інгібіто(b) Сполуки формули І, в якій Υ1 та Υ2, кожром -Флуоресценція фонова] поділені на [Флуоресний, представляють О, X представляє NR1 (R1=H), ценція без інгібітору-Флуоресценція фонова]. Ζ представляє S або О, a R2, R3, R4, R5 є такими, Подібний протокол можна застосовувати для як описано для формули І, можна отримати реакінших експресованих та очищених рrоММР з викоцією сполуки формули VII, в якій R2, R3, R4, R5 та ристанням субстратів та буферів, оптимальних А є такими, як описано для формули І, для певних ММР, наприклад, [як описано С. Graham Knight et. al., (1992) FEES Lett. 296(3):263VIIl 266]. Адамалізинова родина, включаючи, наприз солями амонію та ціаніду у протонних розклад, TNF-конвертазу чинниках, переважно у присутності надлишку карЗдатність сполук інгібувати фермент proTNFaбонату амонію та ціаніду калію в етанолі у гермеконвертази можна визначити з використанням тичній посудині при 40-80°С протягом 4-24 годин. аналізу частково очищеного, виділеного ферменКетони формули VIII звичайно отримують обту, фермент отримано з мембран ТНР-1, [як опиробкою спиртів або тіолів формули IX, в якій R5 і А сано K. М. Mohler et. al., (1994) Nature 370:218є такими, як описано для формули І, галогенкето220]. Активність очищеного ферменту та його інгінами формули X, в якій R2 є таким, як описано для бування визначають інкубуванням частково очиформули І, і надлишком основи. щеного ферменту за наявності чи відсутності тестсполук з використанням субстрату 4',5'-диметоксифлуорецеїнілSеr.Рrо.Lеu.АІа.Сln.АІа.\/аІ.Аrg.Sеr.Sе r.Sеr.Аrg.С уs (4-(3-сукцинімід-1-іл)-флуорецеїн)NН2 у буфері для аналізу (50мМ Tris-HCI, pH7,4, що містить 0,1% (за масою/об'ємом) Triton Х-100 Сполуки винаходу можна оцінювати, наприта 2мМ СаСІ2), при 26°С протягом 18 годин. Стуклад, у таких аналізах: пінь інгібування визначають як для ММР13 за виАналізи виділених Ферментів нятком того, що використовували λекс 490нм та Родина матричних металопротеїназ. включаλем 530нм. Субстрат синтезували таким чином. ючи наприклад ММР12. ММР13. Пептидну частину субстрату сажали на Fmoc-N-HРекомбінантний каталітичний домен ММР12 Rink-MBHA-полістирольну смолу вручну або на людини можна експресувати та очищати, [як опиавтоматичному синтезаторі пептидів стандартнисано Parkar A.A. et al, (2000), Protein Expression ми способами, в яких залучено використання and Purification, 20:152]. Очищений фермент можFmoc-амінокислот та гексафлуорфосфату 0на використовувати для контролю активності інгібензотриазол-1-іл-N-,N,N',N'-тетраметилуронію біторів таким чином: ММР12 (50нг/мл кінцева кон(HBTU) як засобу сполучення з щонайменше 4центрація) інкубують протягом 30 хвилин при або 5-кратним надлишком Fmoc-амінокислоти та кімнатній температурі у буфері для аналізу (0,1Μ HBTU. Ser1 та Pro2 були подвійно сполучені. ЗаTris-HCI, pH7,3, що містить 0,1Μ NaCI, 20мМ Састосовували наступну стратегію захисту бічного СІ2, 0,040мМ ZnCl та 0,05% (за масою/об'ємом) Brij ланцюга; Ser1 (But), Ghi 5(Trityl), Arg 8,12(Pmc або 35) з використанням синтетичного субстрату MacPbf), Ser9,10,11(Trityl), Cys 13(Trityl). Після приєднання Pro-Cha-G1y-Nva-His-Ala-Dpa-NH2 за наявності чи N-термінальну Fmoc-захисну групу видаляли обвідсутності інгібіторів. Активність визначають виміробкою Fmoc-пептидильної смоли у ДМФ. Отрирюванням флуоресценції при λекс 328нм та λем ману так аміно-пептидильну смолу активували 393нм. Процент інгібування розраховують таким обробкою протягом 1,5-2 годин при 70°С з 1,5-2 чином: еквавалентами 4',5'-диметокси-флуорецеїн-4(5)% Інгібування дорівнює [Флуоресценціяз інгібітокарбонової кислоти [Khanna & Ullman, (1980) Anal ром -Флуоресценція фонова] поділені на [ФлуоресBiochem. 108:156-161), яка була попередньо актиценція без інгібітору-Флуоресценція фонова]. 21 77667 22 вована діізопропілкарбодіімідом та 1аналізу концентрації TNFα за допомогою ELISA. гідроксибензотриазолом у ДМФ]. ДиметоксифлуоТест як засіб інгібування in vitro деградації рецеїніл-пептид далі одночасно позбавляли захихряща сту та відщеплювали від смоли обробкою трифлуЗдатність сполук цього винаходу інгібува ти деороцтовою кислотою, що містить по 5% кожного з градацію агреканового або колагенового компоневоди та триетилсилану. Диметоксифлуорецеїнілнтів хряща можна визначити, [як описано по суті K. пептид виділяли випарюванням, розтиранням з М. Bottomley et, al., (1997) Biochem J. 323:483-488]. діетиловим етером та фільтруванням. Виділений Фармакодинамічний тест пептид реагував з 4-(N-малеїнімідо)-флуорецеїном Для оцінки здатності до виведення та біозасу ДМФ, що містить діізопропілетиламін, продукт воюваності сполук цього винаходу ex vi vo застосоочищали за допомогою ОФ-ВЕРХ та під кінець вують фармакодинамічний тест, який використовиділяли сублімацією з водної оцтової кислоти. вує вищенаведені аналізи з синтетичним Продукт характеризували за допомогою MALDIсубстратом або альтернативно ВЕРХ або масTOF MS та амінокислотного аналізу. спектрометричний аналіз. Це є загальним тестом, Природні субстрати який можна використовувати для оцінки швидкості Активність сполук винаходу як інгібіторів девиведення сполук через ряд видів. Тварин (наприградації агрекану можна аналізувати з викорисклад, щурів, мавп) дозують внутрішньовенно або танням способів, основаних, [наприклад, на відперорально розчинною композицією сполуки (як-то критті Е. С Arner et. al., (1998) Osteoarthritis and 20% за масою/об'ємом ДМСО, 60% за маCartilage 6:214-228; (1999) J. Biol. Chem., 274 (10), сою/об'ємом PEG400) та у наступний момент часу 6594-6601] та описаних там антитілах. Потужність (наприклад, 5, 15, 30, 60, 120, 240, 480, 720, 1220 сполук як інгібіторів проти колагенази можна вихвилин) зразки крові переносять з прийнятної позначити, [як описано Т. Cawston та A. Barrett (1979) судини у 10U гепарину. Фракції плазми отримують, Anal. Biochem. 99:340-345]. центрифугують та білки плазми осаджують ацетоІнгібування активності металопротеїнази в акнітрилом (80% за масою/об'ємом кінцева конценттивності на базі клітин/тканин рація). Через 30 хвилин при -20°С білки плазми Тест як засіб інгібування мембранних шедаз, осаджують центрифугуванням та надосадкову як-то TNF-конвертази фракцію випарюють досуха з використанням апаЗдатність сполук цього винаходу інгібувати рату Savant speed vac. Осад відтворюють у буфері клітинне перетворення продукування TNFα можна для аналізу досліджуваної сполуки, а далі аналівизначити у клітинах ТНР-1 з використанням зують на вміст сполуки аналізом з синтетичним ELISA для детектування вивільненого TNF, [як субстратом. Коротше, для оцінюваних сполук буописано по суті К. М. Mohler et. al., (1994) Nature дують криву концентрація сполуки - реакція. Се370:218-220]. Подібним чином перетворення або рійні розбавлення екстрактів відтвореної плазми втрату інши х мембранних молекул, [як-то описааналізують на активність, та кількість сполуки, них у N. М. Hooper et. al., (1997) Biochem. J. представленої у ви хідному зразку плазми, розра321:265-279] можна тестува ти, застосовуючи приховують з використанням кривої концентрація йнятні лінії клітин та придатні антитіла для визнасполуки - реакція, зважаючи на фактор розбавчення відкинутого білку. лення загальної плазми. Тест як засіб інгібування інвазії на базі клітин Дослідження in vivo Здатність сполуки цього винаходу інгібувати Тест як засобу проти TNF міграцію клітин при аналізі інвазії можна визначиЗдатність сполук цього винаходу як ex vivo ти, [як описано A. Albinie де al., (1987) Cancer ReTNFα інгібіторів визначають на щурах. Коротше, search 47:3239-3245]. групи самців щурів Wistar Alderley Park (АР) (180Тест як засіб інгібування шедазної активності 210г) дозують сполукою (6 щурів) або носієм ліків TNF всієї крові (10 щурів) прийнятним шляхом наприклад, пероЗдатність сполук цього винаходу інгібувати ральним, інтраперитональним, підшкірним. Через продукування TNFα визначають при аналізі всієї 90 хвилин щурів їх вбивали з використанням збікрові людини, де для стимуляції вивільнення TNFα льшеної концентрації СО2 та позбавляли крові використовують LPS. Гепаризовану через сідничну вену у 5 одиниць натрій(10одиниць/мл) кров людини, отриману від волонгепарину/мл крові. Зразки крові негайно поміщатерів, розбавляють 1:5 середовищем (RPMI1640+ ють на лід та центрифугують при 2000об/хвил прогідрокарбонат, пеніцилін, стрептоміцин та глутатягом 10 хвилин при 4°С та зібрані плазми замомін) та інкубують (160мкл) з 20мкл тест-сполуки рожують при -20°С для наступного аналізу їх дії на (при потроєнні), у ДМСО або прийнятному носії, продукування TNFα стимульованою LPS кров'ю протягом 30 хвилин при 37°С у зволоженому людини. Зразки плазми щурів розтоплюють та (5%СО2/95% повітря) інкубаторі, перед додаван175мкл кожного зразку додають у 96-комірковий ням 20мкл LPS (Е. соlі. 0111:В4; кінцева концентпланшет. П'ятдесят мкл гепаризованої крові людирація 10мкг/мл). Кожний аналіз включає контролі ни далі додають до кожної комірки, змішують та розбавленої крові, інкубованої із одним середовипланшет інкубують протягом 30 хвилин при 37°С щем (6 комірок/планшет) або з відомим інгібітором (зволожений інкубатор). LPS (25мкл; кінцева конTNFα як стандартом. Планшети далі інкубують центрація 10мкг/мл) додають до комірок та інкубупротягом 6 годин при 37°С (зволожений інкубатор), вання продовжують ще 5,5 годин. Контрольні коміцентрифугують (2000об/хвил протягом 10 хвилин; рки інкубують з 25мкл одного середовища. 4°С), плазму збирають (50-100мкл) та зберігають у Планшети далі центрифугують протягом 10 хви96-коміркових планшетах при -70°С до наступного лин при 2000об/хвил та 200мкл надосадкової ріди 23 77667 24 ни переносять у комірковий планшет та заморо(112мг, 0.36ммоль) твердий продукт білого кольожують при -20°С для наступного аналізу концентру, з виходом 72%. 1 рації TNF за допомогою ELISA. Н ЯМР (300МГц, ДМСО-d6): δ 10.57 (1Н, bs); Результати аналізу розраховують за допомо8.00 (1Н, s); 7.63-7.58 (4Н, m); 7.43 (2Н, m); 7.01 гою програмного забезпечення для кожної сполу(2Н, d); 4.07 (2Н, dd); 1.67 (2Н, m); 0.86 (3Н, t). ки/дози: LC-MS (APCI) m/z 311.1 (MH+). Процент інгібування TNFα= Приклад 2 Сполуки загальної формули Значення T NFa (Контролі - ЗначенняT NFa (оброблені ´ 100 ) ) Значення T NFa (Контролі ) Тест як засобу проти артриту Активність сполуки як засобу проти артриту тестують в індукованому колагеном артриті (СІА) [як визначено D. Е. Trentham et al., (1977) J. Exp. Med. 146:857]. У цій моделі кислотний розчинний природний колаген типу II викликає поліартрит у щурів при застосуванні у неповному ад'юванті Фрейнда. Подібні умови можна використовувати для виклику артриту у мишей та приматів. Тест як засобу проти раку Активність сполуки як засобу проти раку можна визначити, [як описано по суті І. J. Fidler (1978) Methods у Cancer Research 15:399-439], застосовуючи, наприклад, лінію клітин В16 [описано у В. Hibner et. al, Abstract 283 p75 10th NCI-EORTC Symposium, Amsterdam June 16-19 1998]. Тест як засобу проти емфіземи. Активність сполуки як засобу проти емфіземи можна визначити, [як описано по суті Hautamaki et. al., (1997) Science, 277:2002]. Винахід далі буде ілюстровано, але без обмеження, наступними прикладами: Загальні аналітичні способи: спектри 1Н-ЯМР реєстрували на приладі Varian VmtyJnova 400MHz або Varian Mercury-VX300MH z. Центральний пік розчиннику хлороформ-d (δн 7,27млн -1), диметилсульфоксид-d6 (δн 2,50млн-1) або метанол-d4 (δн 3,31 25млн -1) використовували як внутрішні стандарти. Мас-спектри низького розділення отримували на системі MS-LC (з електророзпилювальним інтерфейсом) Agilent 1100 з іонізаційною камерою АРСІ (хімічна іонізація при атмосферному тиску). Приклад 1 5-(Дифеніл-4-ілоксиметил)-5-етилімідазолідин-2,4-діон 4-Гідрокси-дифеніл (84мг, 0.5ммоль) додавали до 1-бром-2-бутанону (0.055мл, 0.55ммоль) і безводн. карбонату калію (95мг, 0.69ммоль) в сухому ацетоні (2.5мл). Суміш перемішували протягом 2 годин при температурі навколишнього середовища, тоді розбавляли етилацетатом (2.5мл). Осад, що утворювався зверху, випаровувався. Стриману олію перемішували при 75°С усю ніч, у герметизованому флаконі, разом з карбонатом амонію (290мг, 3.0ммоль) та цианідом калію (79мг, 1.2ммоль) в 50% етанолі (3мл). Отриманий . розчин виливали у розчин етилацетату (20мл), етеру (10мл) та води (15мл), разом з насиченим хлоридом амонію (вільна величина, 2мл). Органічну фазу промивали додатково один раз водою (10 мл), тоді випарювали разом гептаном, одержуючи названу сполуку було синтезовано способом, описаним в Прикладі 1 25 1) Для даних ЯМР див. експериментальну час тину. 5-[1-(Дифеніл-4-ілокси)-етил]-5-метилімідазолідин-2,4-діон LC-MS (APCI) m/z 311.2 (МН+). 5-(4'-Ціано-дифеніл-4-ілоксиметил)-5-етилімідазолідин-2.4-дiон LC-MS (АРСІ) m/z 336.2 (МН+). 5-(4'-Хлор-дифеніл-4-ілоксиметил)-5-метилімідазолідин-2,4-діон 77667 26 LC-MS (АРСІ) m/z 331.2 (МН+). 5-(4'-Ціано-дифеніл-4-ілоксиметил)-5-метилімідазолідин-2,4-діон LC-MS (АРСІ) m/z 322.2 (МН+). 5-(4'-Ціано-дифеніл-4-ілоксиметил)-5-третбутил-імідазолідин-2,4-діон LC-MS (АРСІ) m/z 364 (МН+). 5-(4'-Ціано-дифеніл-4-ілоксиметил)-5-фенілімідазолідин-2,4-діон LC-MS (АРСІ) m/z 384 (МН+). 5-Метил-5-[4-(4-трифлуорметил-фенокси)феноксиметил]-імідазолідин-2.4-діон LC-MS (АРСІ) m/z 381.4 (МН+). 5-(4-Ціано-феноксиметил)-5-(3-метоксифеніл)-імідазолідин-2.4-діон LC-MS (АРСІ) m/z 33S.2 (МН+). 5-(4-Ціано-феноксиметил)-5-(3-6ром-феніл)імідазолідин-2,4-діон LC-MS (АРСІ) m/z 386.1 (МН+). 5-(4-Ціано-феноксиметил)-5-фенілімідазолідин-2.4-діон LC-MS (АРСІ) m/z 308.1 (МН+). 5-(4-Бром-феноксиметил)-5-(3-метокси-феніл)імiдазолідин-2,4-діон LC-MS (АРСІ) m/z 393.1 (МH+). 5-(4-Бром-феноксиметил)-5-(3-бром-феніл)імідазолідин-2,4-діон LC-MS (АРСІ) m/z 442.9 (МН+). 5-(4-Бром-феноксиметил)-5-фенілімідазолідин-2,4-діон LC-MS (APCI) m/z 363.1 (МН+). 5-(4-Метокси-феноксиметил)-5-(3-метоксифеніл)-імідазолідин-2,4-діон LC-MS (APCI) m/z 343.2(MH+). 5-(4-Метокси-феноксиметил)-5-(3-бромфеніл)-імідазолідин-2.4-діон LC-MS (APCI) m/z 393.2 (MH+). 5-(4-Метокси-феноксиметил)-5-феніл)імідазолідин-2,4-діон LC-MS (APCI) m/z 313.2 (MH+). 5-(4-Метил-феноксиметил)-5-(3-метоксифеніл)-імідазолідин-2,4-діон LC-MS (APCI) m/z 327.1 (MH+). 5-(4-Метил-феноксиметил)-5-(3-бром-феніл)імідазолідин-2.4-діон LC-MS (APCI) m/z 377.1 (MH+). 5-(4-Метил-феноксиметил)-5-феніл)імідазолідин-2,4-діон LC-MS (APCI) m/z 297.1 (MH+). 5-Феноксиметил-5-(3-метокси-феніл)імідазолідин-2,4-діон LC-MS (APCI) m/z 313.2 (MH+). 5-Феноксиметил-5-(3-бром-феніл)імідазолідин-2,4-діон LC-MS (APCI) m/z 363 (MH+). 5-Феноксиметил-5-феніл-імідазолідин-2,4-діон LC-MS (APCI) m/z 283.2 (MH+). 6-(4-Хлор-фенокси)-1,3-діаза-спіро[4,4]нонан2,4-діон LC-MS (APCI) m/z 281 (MH+). 5-Метил-5-[(4-тіофен-2-іл-феноксиметил)імідазолідин-2,4-діон 1-(4-Тієн-2-ілфенокси)ацетоне (114мг, 0.49ммоль), цианід натрію (40мг, 0.81ммоль), карбонат амонію (222мг, 2.85ммоль), воду (5мл) та етанол змішували нагрівали при 80°С протягом 10 27 77667 28 годин. Після охолодження, реакційну суміш обро(2H, d); 7.08 (1H, m); 7.12 (1H, d); 7.30 (1H, t); 7.53 бляли водою, твердий осад відфільтровували і (2H, d). висушували для отримання 105мг продукту. 5-Етил-5-(4'-метокси-дифеніл-4-ілоксиметил)LC-MS (APCI) m/z 303 (MH+). імідазолідин-2,4-діон 1 H ЯМР (ДМСО-d 6): δ 1.31 (3H, s); 3.95, 4.10 LC-MS (APCI) m/z 341 (MH+). 1 (2H, abq, J=9.8 Hz); 6.95 (2H, d); 7.08 (1H, dd); 7.37 H ЯМР (ДМСО-d6): δ 0.48 (3Н, t); 1.56-1.74 (1H, d); 7.45 (1H, d); 7.55 (2H, d); 8.03 (1H, s). (2H, m); 3.77 (3H, s); 3.97, 4.11 (2H, abq, J=10.0 Hz); Вихідні матеріали було приготовлено таким 6.94-7.00 (4H, m); 7.49-7.54 (4H, m); 7.97 (1H, s); чином: 10.71 (1H, brs) 1-(4-Йодфенокси)ацетон 5-Етил-5-(4'-(трифлуорметил-дифеніл-44-Йодфенол (4.9г, 22ммоль) перемішували раілоксиметил)-імідазолідин-2,4-діон зом з карбонатом калію (4.7г, 33ммоль), хлорацеLC-MS (APCI) m/z 378 (MH+). 1 тоном (4.5мл, 55ммоль) та ацетоном, нагріваючи H ЯМР (ДMCO-d6): δ 0.83 (3Н, t); 1.66 (2H, під зворотним холодильниоком протягом 18 годин. oct); 4.01, 4.14 (2H, abq, J=9.8 Hz); 7.04 (2H, d); Реакційну суміш виливали у воду (100мл), екстра7.67 (2H, d); 7.75 (2H, d); 7.84 (2H, d); 8.01 (1H, s); 10.75 (1H, bs). гували етилацетатом (3´50мл), екстракти проми5-Етил-5-(3'-('метокси-дифеніл-4-ілоксиметил)вали розсолом, висушували над сульфатом натрію та випарювали. Залишок очищали флешімідазолідин-2,4-діон LC-MS (APCI) m/z 340 (MH+). хроматографією, елюючи з дихлорметаном. 1 H ЯМР (ДМСО-d6): δ 0.83 (3Н, t); 1.65 (2H, LC-MS (APCI) m/z 275 (МН+). 1 oct); 3.76 (3H, s); 3.97, 4.10 (2H, abq, J=9.7 Hz); H ЯМР (CDCI3): δ 2.26 (3Н, s); 4.51 (2Н, s); 6.93-6.99 (3H, m); 7.49-7.53 (3H, m); 7.99 (1H, s); 6.65 (2Н, d); 7.57 (2Н, d). 1-(4-Тієн-2-ілфенокси)ацетон 10.74 (1H, bs). 5-Етил-5-(4'-(трифлуорметокси-дифеніл-41-(4-Йодфенокси)ацетон (192мг, 0.69ммоль) ілоксиметил)-імідазолідин-2,4-діон обробляли тіофен-2-бороновою кислотою (102мг, LC-MS (APCI) m/z 395 (MH+). 0.79ммоль), комплексом [1,1'1 H ЯМР (ДМСО-d6): δ 0.84 (3Н, t); 1.56-1.74 біс(дифенілфосфіно)фероцен]дихлорпаладію (II) з дихлорметаном (1:1) (36мг), диметилформамід (2H, m); 4.00, 4.13 (2H, abq, J=10.9 Hz); 7.01 (2H, d); 7.40 (2H, d); 7.61, 7.72 (4H, abq, J=8.9 Hz); 7.79 (12мл) та ацетатом амонію (135мг), і перемішува(1H, s); 10.72 (1H, bs). ли разом при 80°С протягом 3 годин. Після охоло5-Етил-5-[(4-тіофен-2-іл-феноксиметил)дження, реакційну суміш обробляли розбавленою мідазолідин-2,4-діон гідрохлоридною кислотою та екстрагували етилацетатом. Продукт очищали флеш-хроматографією LC-MS (APCI) m/z 317 (MH+). 1 Н ЯМР (ДМСО-d6): δ 0.82 (3Н, t); 1.54-1.74 на діоксиді силіцію, елюючи сумішшю 50% етила(2Н, m); 3.97, 4.12 (2Н, abq, J=10.0 Hz); 6.95 (2H, цетат:ізогексан для отримання 114мг продукту. d); 7.08 (1H, dd); 7.37 (1H, dd); 7.44 (1H, dd); 7.55 LC-MS (APCI) m/z 232 (МН+). (2H, d); 7.98 (1H, s); 10.67 (1H,s). Наступні сполуки було отримано як описано для синтезу 5-метил-5-[(4-тієн-25-Феніл-5-(4'-(трифлуорметил-дифеніл-4ілоксиметил)-iмідазолідин-2.4-діон ілфенокси)метил]імідазолідин-2,4-діону LC-MS (АРСІ) m/z 426 (МН+). 5-Метил-5-(4'-(трифлуорметил-дифеніл-41 Н ЯМР (ДМСО-d6): 54.21,4.62 (2Н, abq, J=10.1 ілокси метил)-імідазолідин-2,4-діон Hz); 7.10 (2Н, d); 7.38-7.47 (3Н, m); 7.61-7.69 (4Н, LC-MS (APCI) m/z 365 (МН+). 1 Н ЯМР (ДMCO-d 6): δ 1.46 (3Н, s); 4.05, 4.22 m); 7.76, 7.84 (4Н, abq, J=8.8 Hz); 8.76 (1H, s); 10.92 (1H, bs). (2Н, ABq, J=9.9 Hz); 7.04 (2Н, d); 7.61 (2Н, d); 7.04, 5-трет-Бутил-5-(4-піридин-3-іл-феноксиметил)7.61 (4Н, ABq, J=9.8 H z). імідазолідин-2,4-діон 5-(4'-(Метокси-дифеніл-4-ілоксиметил)-5LC-MS (APCI) m/z 340 (MH+). метил-імідазолідин-2,4-діон 1 LC-MS (APCI) m/z 326 (MH+). H ЯМР (ДМСО-d 6): δ 1.02 (9H, s); 4.15, 4.36 (2H, abq, J=9.9 Hz); 7.10 (2H, d); 7.70-7.75 (3H, m); 5-(4'-(Флуор-дифеніл-4-ілоксиметил)-5-метил8.08 (1H, s); 8.39 (1H, dd); 8.65 (1H, dd); 9.00 (1H, імідазолідин-2,4-діон s). LC-MS (АРСІ) m/z 315 (МН+). 1 5-трет-Бутил-5-(4'-метокси-дифеніл-4Н ЯМР (ДМСО-d 6): δ 1,45 (3Н, s); 4.02, 4.20 (2Н, abq, J=9.9 Hz); 6.99 (2H, d); 7.12 (2H, t); 7.50 ілоксиметил)-імідазолідин-2,4-діон LC-MS (APCI) m/z 368 (MH+). (2H, d); 7.55 (2H, dd). 1 H ЯМР (ДMCO-d6): δ 1.01 (9H, s); 3.76 (3H, s); N'-(4'-(4-Метил-2,5-діоксо-імідазолідин-44.10, 4.31 (2H, abq, J=9.7 Hz); 6.95-7.01 (4H, dd); ілметокси)-дифеніл-3-іл]-ацетамід 7.48-7.55 (4H, dd); 8.05 (1H, s); 10.59 (1H, bs). LC-MS (АРСІ) m/z 354 (MH+). 1 H ЯМР (ДМСО-d6): δ 1.46 (3Н, s); 2.14 (3Н, s); 5-трет-Бутил-5-(3'-три флуорметил-дифеніл-4ілоксиметил)-імідазолідин-2,4-діон 2.15 (1Н, s); 4.05, 4.20 (2Н, abq, J=9.6 Hz); 7.00 (2H, LC-MS (APCI) m/z 406 (MH+). d); 7.28-7.40 (3H, m); 7.46 (1H, bd); 7.53 (2H, d); 1 H ЯМР (ДMCO-d 6): δ 1.01 (9H, s); 4.14, 4.35 7.78-7.81 (1H, m). (2H, abq, J=9.6 Hz); 7.06 (2H, d); 7.65-7.69 (4H, m); 5-(3'-(Метокси-дифеніл-4-ілоксиметил)-5метил-імідазолідин-2,4-діон 7.89 (1H, s); 7.93 (1H, t); 8.08 (1H, s); 10.65 (1H, s). 5-трет-Бутил-5-(4'-три флуорметил-дифенілLC-MS (APCI) m/z 327 (MH+). 1 ілоксиметил)-імідазолідин-2,4-діон H ЯМР (ДМСО-d6): δ 1.45 (3Н, s); 3.83 (3Н, s); LC-MS (APCI) m/z 407 (MH+). 4.04, 4.20 (2H, abq, J=9.6 Hz); 6.85 (1H, dd); 6.99 29 77667 30 Н ЯМР (ДМСО-d6): δ 1.03 (9H, s); 4.15,4.36 чином: (2H, abq, J=10.0 Hz); 7.07, 7.68 (4H, abq, J=8.9 Hz); 1-(1,1'-дифеніл-4-ілтіо)пропан-2-он 7.76,7.84 (4H, abq, J=8.9 Hz); 8.08 (1H, s); 10.67 1-[(4-бромфеніл)тіо]пропан-2-он (357мг, (1H, s). 1.46ммоль) обробляли фенілбороновою кислотою 5-(Дифеніл-4-ілоксиметил)-5-піридин-4-іл(231мг, 1.89ммоль), комплексом [1,1'імідазолідин-2,4-діон біс(дифенілфосфіно)фероцен]дихлорпаладію (II) з LC-MS (APCI) m/z 360 (MH+). дихлорметаном (1:1) (36мг), толуолом (20мл), ме1 H ЯМР (CD3OD): δ 4.41, 4.71 (2H, ABq, J=9.7 танолом (7,5мл), насиченим розчином карбонату Hz); 7.02 (2H, d); 7.28 (1H, t); 7.39 (2H, t); 7.55 (2H, натрію (3,5мл), і перемішували разом при 80°С d); 8.14"(2H, d); 8.81 (2H, d). протягом 18 годин. Після охолодження, реакційну Приклад 3 суміші обробляли розбавленою гідрохлоридною Сполуки загальної формули кислотою та екстрагували етилацетатом. Продукт очищали флеш-хроматографією на діоксиді силіцію, елюючи сумішшю 25% етилацетат: ізогексан для отримання 277мг продукту. GC/MS m/z: 242 (M+). 1 H ЯМР (CDCI3): 52.33 (3H, s); 3.73 (2H, s); 7.37 було синтезовано способом, описаним в При(1H, s); 7.42-7.48 (4H, m); 7.54-7.59 (4H,m). кладі 1 Наступні сполуки було отримано, як описано для синтезу 5-[(1,1'-дифеніл-4-ілтіо)метил]-5метилімідазолідин-2,4-діону 4'-{[(4-метил-2,5-діоксоімідазолідин-4іл)метилiтіо)1,1'-дифеніл-4-карбонітрил Вихідний матеріал, 4'-[(2-оксопропіл)тіо]-1,1'дифеніл-4-карбонітрил, приготовляли, як описано для синтезу 1-(1,1'-дифеніл-4-ілтіо)пропан-2-ону. 1 Н ЯМР (ДМСО-d6): 6 1.37 (3Н, s); 3.30 (2Н, s); 7.45, 7.67 (4Н, abq, J=7.5 Hz); 7.88 (4Н, q); 7.99 (1H,s); 10.75(1Η, bs). 5-метил-5-[({4'-[(трифлуорметил)окси]-1,1'дифеніл-4-іл}тіо)метил]імідазолідин-2,4-діон Вихідний матеріал, 1-({4'[(трифлуорметил)окси]-1,1'-дифеніл-45-[(1,1'-дифеніл-4-ілтіо)метил-5іл}тіо)пропан-2-он, приготовляли, як описано для метилімідазолідин-2,4-діон синтезу 1-(1,1'-дифеніл-4-іл}тіо)пропан-2-ону. LC-MS(APCI) m/z 313 (МН+). LC-MS(APCI) m/z very weak 397 (MH+). 1 H ЯМР (ДМСО-d6): 51.36 (3H, s); 3.28 (2H, s); 1 H ЯМР (ДМСО-d6): δ 1.33 (3Н, s); 3.29 (2Н, s); 7.34 (1H, t); 7.44 (4H, t); 7.60 (2H, d) 7.64 (2H, d); 7.42-7.45 (4Н, m); 7.61 (2Н, d); 7.77 (2Н, d); 7.99 7.97 (1H, s); 10.74 (1H, bs). (1H,s); 10.75(1 Η, s). Вихідні матеріали було приготовлено таким 1 Комп’ютерна в ерстка Т. Чепелев а Підписне Тираж 26 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMetalloproteinase inhibitors, pharmaceutical composition and use thereof
Назва патенту російськоюИнгибиторы металлопротеиназы, фармацевтическая композиция на их основе и их применение
МПК / Мітки
МПК: A61P 27/02, A61P 19/00, A61K 31/438, A61P 19/10, A61P 19/08, A61P 35/00, C07D 401/04, C07D 223/00, C07D 233/78, A61P 7/02, A61K 31/496, A61K 31/454, A61P 25/02, A61P 9/00, A61P 9/10, C07D 403/04, A61P 19/04, A61K 31/4439, C07D 401/12, C07D 471/04, A61P 11/02, A61P 19/02, C07D 403/12, A61P 19/06, A61P 1/16, A61P 1/02, A61P 9/04, A61P 37/08, A61P 15/00, A61P 25/00, C07D 409/14, A61P 25/28, A61K 31/5377, C07D 409/12, A61K 31/506, A61P 13/12, A61K 31/444, C07D 403/06, A61P 17/06, A61P 11/06, A61K 31/4178, A61P 43/00, C07D 233/76, A61P 29/00, A61P 11/00, A61P 35/04, A61P 17/04, A61K 31/4166, C07D 405/12, C07D 417/14, C07D 401/14, A61K 31/427, C07D 471/10, A61P 1/04, C07D 405/14
Мітки: фармацевтична, металопротеїнази, композиція, інгібітори, основі, застосування
Код посилання
<a href="https://ua.patents.su/15-77667-ingibitori-metaloprotenazi-farmacevtichna-kompoziciya-na-kh-osnovi-kh-zastosuvannya.html" target="_blank" rel="follow" title="База патентів України">Інгібітори металопротеїнази, фармацевтична композиція на їх основі, їх застосування</a>
Інгібітори металопротеїнази, фармацевтична композиція на їх основі та їх застосування

Номер патенту: 77408
Опубліковано: 15.12.2006
Автори: Мунк Аф Росенскельд Магнус, Лепісте Матті, Лундквіст Мікаель, Златоідскі Паволь, Ерікссон Андерс
МПК: C07D 403/06, C07D 403/04, C07D 401/14, A61P 19/10, A61P 9/04, A61P 19/04, A61P 1/04, A61K 31/444, A61P 35/04, A61K 31/4178, C07D 471/04, A61P 17/04, A61P 17/06, A61P 37/08, A61P 25/02, A61P 19/08, A61P 15/00, A61P 43/00, C07D 405/12, A61P 9/10, A61P 11/00, A61P 1/02, A61K 31/4439, A61K 31/454, A61P 11/06, C07D 403/12, C07D 233/76, C07D 409/14, C07D 405/14, A61P 7/02, A61K 31/496, A61P 1/16, C07D 409/12, A61P 19/06, C07D 401/12, A61P 11/02, C07D 233/78, A61P 35/00, A61P 19/02, A61K 31/5377, C07D 223/00, A61P 13/12, A61P 25/00, A61K 31/427, A61K 31/506, A61P 9/00, A61P 29/00, A61P 19/00, C07D 401/04, A61K 31/438, A61P 27/02, C07D 417/14, A61K 31/4166, C07D 471/10, A61P 25/28
Мітки: композиція, металопротеїнази, застосування, інгібітори, основі, фармацевтична
Формула / Реферат:
1. Сполука формули І або її фармацевтично прийнятна сіль, ІдеX є NR1;Y1 та Y2 є О;Z вибрано з групи: SО2N(R6) або N(R7)SО2;m дорівнює 1;А вибрано з групи: безпосередній зв'язок або (С1-6)алкіл,R1 є Н або (С1-3)алкіл;R2 та R3 незалежно вибрані з групи: Н, (С1-6)алкіл та феніл,R4 є H;R6 вибрано з...
Інгібітори транспорту гліцину, їх застосування та фармацевтична композиція на їх основі

Номер патенту: 75081
Опубліковано: 15.03.2006
Автори: Сміт Пол Ґаррік, Мольтсен Айнер Кнуд, Бьогесьо Клаус Петер, Крог-Єнсен Крістіан
МПК: A61K 31/343, C07C 229/14, A61P 25/14, A61P 9/10, A61K 31/198, A61P 25/18, A61P 25/16, A61P 25/28, A61P 21/02, C07C 255/58, A61P 25/00, C07D 333/72, C07D 405/04, A61K 31/381, C07D 307/87
Мітки: інгібітори, основі, композиція, гліцину, застосування, транспорту, фармацевтична
Формула / Реферат:
1. Сполуки загальної формули І , (І)деR1 являє собою водень, С1-6-алкіл, циклоалкіл або циклоалкілалкіл;R2 і R3 незалежно являють собою водень, галоген, С1-6-алкіл, С3-8-циклоалкіл або С3-8-циклоалкіл-С1-6-алкіл або R2 і R3 разом утворюють С3-8-циклоалкіл;R4, R5, R6 і R7 незалежно являють собою водень, галоген, СF3, NO2, CN, С1-6-алкіл,...
Трициклічні інгібітори полімерази полі(адф-рибози) для лікування раку та інгібування нейротоксичності, їх застосування та фармацевтична композиція на їх основі

Номер патенту: 75034
Опубліковано: 15.03.2006
Автори: Веббер Стефен Іван, Кенен-Коч Стасі С., Тіхе Йаяшрі, Торезін Ларс Хенрік
МПК: C07D 471/06, A61P 3/10, C07D 519/00, A61P 29/00, A61K 31/55, A61P 35/00, A61P 25/28, A61K 31/437, A61P 9/10, A61K 31/551, C07D 487/06, A61P 43/00
Мітки: композиція, застосування, інгібування, трициклічні, фармацевтична, полі(адф-рибози, основі, лікування, нейротоксичності, полімерази, раку, інгібітори
Формула / Реферат:
1. Сполука, вибрана з групи, яка складається з:
Інгібітори реплікації респіраторного синцитіального вірусу, їх застосування, спосіб їх одержання, фармацевтична композиція на їх основі, спосіб її одержання

Номер патенту: 74551
Опубліковано: 16.01.2006
Автори: Соммен Франсуа Марія, Гюіллемонт Джером Еміль Джордж, Меерсман Катлін Петрус Марі-Хосе, Лакрампе Жан Фернан Арман, Андріес Конрад Жозеф Людовик Марсель, Янссенс Франс Едуард
МПК: C07D 401/14, A61K 31/501, C07D 417/14, A61K 31/506, C07D 401/06, C07D 471/04, A61P 31/12, A61K 31/443, C07D 413/14, C07D 405/14, A61K 31/4184, A61P 31/14, A61K 31/4545, A61K 31/4439, A61K 31/55, A61K 31/454, A61K 31/497
Мітки: застосування, спосіб, респіраторного, основі, вірусу, одержання, фармацевтична, інгібітори, синцитіального, композиція, реплікації
Формула / Реферат:
1. Застосування сполуки для виробництва медичного препарату для лікування вірусних інфекцій, де дана сполука є сполукою формули,її промедикаментом, N-оксидом, сіллю приєднання, четвертинним аміном, металічним комплексом або стереохімічною ізомерною формою, де -а1=а2-а3=а4- являє собою бівалентний радикал формули-СН=СН-СН=СН- (а-1);-N=CH-CH=CH- (a-2);-CH=N-CH=CH- (a-3);-CH=CH-N=CH- (a-4)...
Похідні арилоксифенілу та арилсульфанілфенілу як інгібітори транспорту гліцину, фармацевтична композиція на їх основі та спосіб лікування захворювань

Номер патенту: 76259
Опубліковано: 17.07.2006
Автори: Міккельсен Гітте, Андерсен Кім, Рен Стефен Пі, Сміт Гаррік Пол, Греве Даніель, Рухланд Томас
МПК: C07D 241/04, C07D 241/06, A61K 31/495, C07D 211/08, C07D 295/15, A61P 25/00
Мітки: лікування, інгібітори, спосіб, арилоксифенілу, гліцину, фармацевтична, композиція, транспорту, похідні, основі, захворювань, арилсульфанілфенілу
Формула / Реферат:
1. Сполуки загальної формули Іде Y являє собою N, С або СН;Х являє собою О або S;m являє собою 1 або 2;р являє собою 0, 1, 2, 3 або 4;q являє собою 0, 1 або 2;s являє собою 0, 1, 2 або 3;r являє собою 0, 1 або 2;Q являє собою C, P-OR5 або S=O, де R5 являє собою водень або С1-6-алкіл;А являє собою OR6,...
Попередній патент: Композиція, що полімеризується, спосіб склеювання об’єктів, спосіб нанесення на об’єкт покриття та покриття, нанесене на об’єкт
Наступний патент: Спосіб одержання потоку водневмісного газу (варіанти) та спосіб одержання аміаку
Випадковий патент: Спосіб прогнозування тяжкого перебігу нападів бронхіальної астми у дітей шкільного віку