Спосіб одержання похідної 4-(2-оксопіперидин-1-іл)масляної кислоти як проміжної сполуки для синтезування медикаменту
Номер патенту: 107721
Опубліковано: 10.02.2015
Автори: Кім Кюу Юн, Лі Хе Бон, Кім Бон Чан, Лі Кюу Вон, Ань Цзи Ень







Формула / Реферат
1. Спосіб одержання сполуки формули (2), який відрізняється тим, що сполуку формули (4) піддають реакції зі сполукою формули (5):
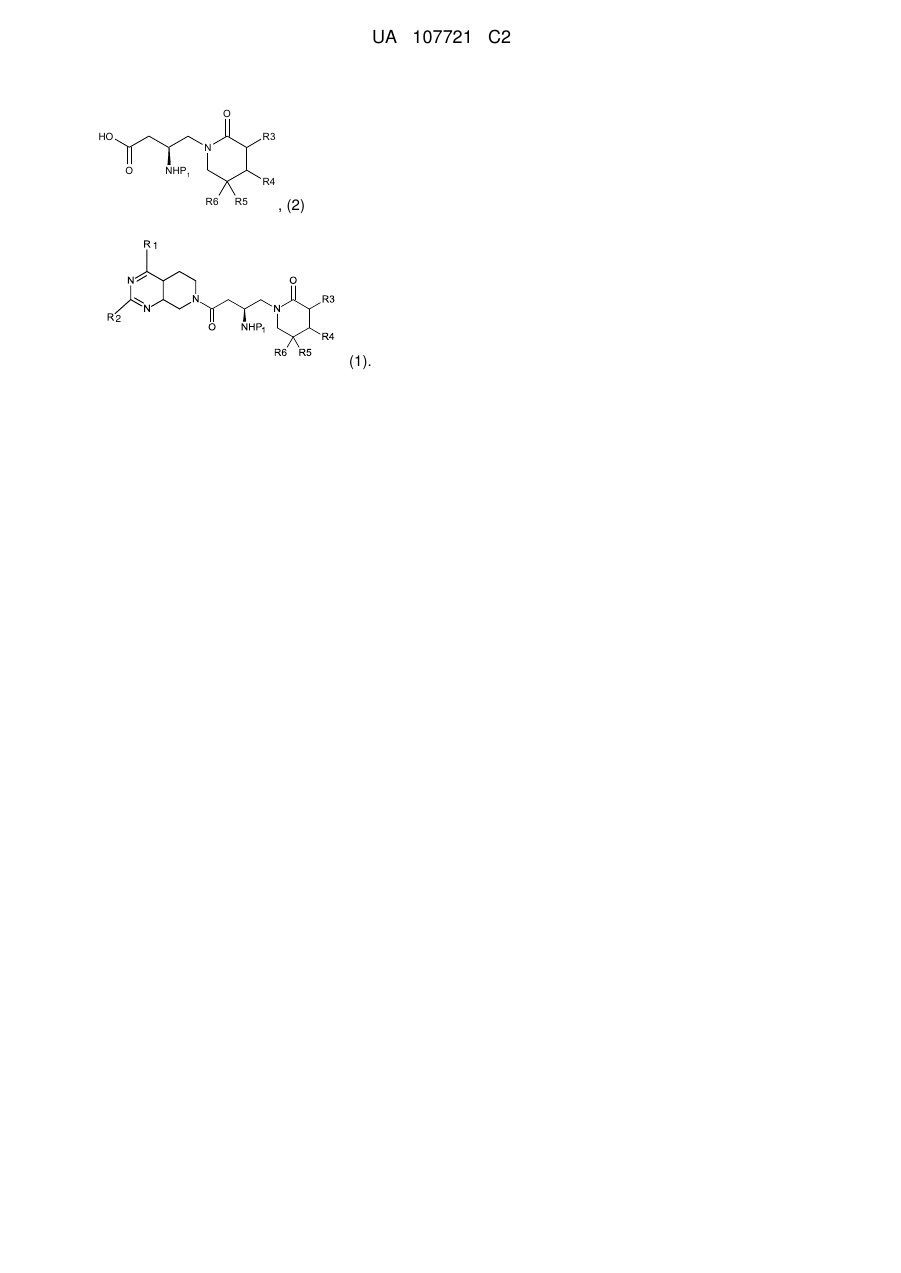
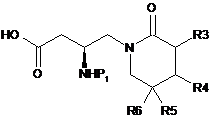 , (2)
, (2)
 , (4)
, (4)
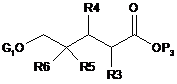 , (5)
, (5)
де кожний з R3, R4, R5 і R6 є незалежно воднем, галогеном або заміщеним або незаміщеним С1-С4алкілом; Р1 є захисною групою аміну; кожний з Р2 і Р3 є незалежно бензильною групою, метильною групою, етильною групою, і-пропiльною групою або t-бутильною групою; і G1O є заміщуваною групою.
2. Спосіб за пунктом 1, при якому здійснюють:
(a) етап реакції сполучення з додаванням основи до сполуки формули (4) і сполуки формули (5);
(b) етап циклізації з додаванням кислоти з одержанням сполуки формули (2а); і
(c) етап гідролізу одержаної сполуки формули (2а) з одержанням сполуки формули (2):
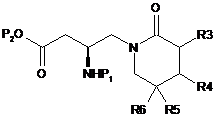 , (2a)
, (2a)
де R3, R4, R5, R6, Р1 і Р2 є такими, як визначено в пункті 1.
3. Спосіб за пунктом 1 або 2, який відрізняється тим, що Р2 є t-бутильною групою, а Р3 є метильною або етильною групою.
4. Спосіб за пунктом 1 або 2, який відрізняється тим, що G1O є трифлатом, мезилатом, тозилатом, безилатом або нонафлатом.
5. Спосіб за пунктом 1 або 2, який відрізняється тим, що R3 і R4 є воднем, a R5 і R6 є фтором.
6. Спосіб за пунктом 2, який відрізняється тим, що на етапі (а) як основу використовують С1-С4триалкіламін.
7. Спосіб за пунктом 2, який відрізняється тим, що на етапі (b) як кислоту використовують оцтову кислоту.
8. Спосіб за пунктом 2, який відрізняється тим, що у випадку сполуки формули (2а), де Р1 є Вос і Р2 є t-бутил, гідроліз на вказаному етапі (с) проводять в основних умовах, щоб вибірково видалити тільки Р2 з-поміж захисних груп Р1 і Р2 і отримати сполуку формули (2).
9. Спосіб за пунктом 8, який відрізняється тим, що як основу використовують водний розчин гідроокису натрію.
10. Спосіб одержання сполуки формули (5), як визначено в пункті 1, при якому здійснюють:
(a) етап відновлення сполуки формули (7) з одержанням сполуки у вигляді первинного спирту; і
(b) етап реакції спиртової сполуки, одержаної на попередньому етапі, зі сполукою G1, яка відповідає частині G1O сполуки формули (5), з одержанням сполуки формули (5):
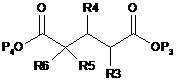 , (7)
, (7)
де R3, R4, R5, R6 і Р3 є такими, як визначено вище в пункті 1, а Р4 є бензильною групою, метильною групою, етильною групою, і-пропільною групою або t-бутильною групою.
11. Спосіб за пунктом 10, який відрізняється тим, що на етапі (а) відновлення проводять з використанням NaBH4.
12. Спосіб за пунктом 10, який відрізняється тим, що на етапі (b) сполуку G1 вибирають з групи, яка складається з ангідриду трифторметансульфонової кислоти (Tf2O), трифторметансульфонілхлориду (TfCl), метансульфонілхлориду (MsCl), толуолсульфонілхлориду (TsCl), бромбензолсульфонілхлориду (BsCl), (CF3(CF2)3SO2)F і (CF3(CF2)3SO2)2O.
13. Спосіб одержання сполуки формули (4), як визначено в пункті 1, при якому здійснюють:
(a) етап перетворення карбонової кислоти сполуки формули (8) на ефірну групу шляхом введення групи Р2 з одержанням сполуки формули (9);
(b) етап вибіркового відновлення ефірної групи Р5, присутньої в сполуці формули (9), з одержанням сполуки формули (10);
(c) етап введення заміщуваної групи G2O в сполуку формули (10) з одержанням сполуки формули (11);
(d) етап реакції сполуки формули (11) з азидною сполукою з одержанням сполуки формули (12); і
(e) етап піддавання сполуки формули (12) гідрогенізації з одержанням сполуки формули (4):
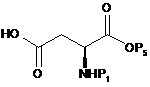 , (8)
, (8)
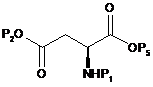 , (9)
, (9)
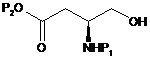 , (10)
, (10)
 , (11)
, (11)
 , (12)
, (12)
де Р1 і Р2 є такими, як визначено в пункті 1, Р5 є метильною групою, етильною групою, і-пропільною групою або t-бутильною групою, a G2O є заміщуваною групою.
14. Спосіб за пунктом 13, який відрізняється тим, що Р1 є Вос, Р2 є і-пропільною групою або t-бутильною групою, a G2O є трифлатом або нонафлатом.
15. Спосіб одержання сполуки формули (4), як визначено в пункті 1, при якому здійснюють:
(a) етап перетворення сполуки карбонової кислоти формули (13) на активований складний ефір, який потім піддають реакції зі сполукою вторинного аміну, з одержанням амідної сполуки формули (14);
(b) етап відновлення амідної групи сполуки формули (14), з одержанням сполуки третинного аміну формули (15); і
(c) етап піддавання сполуки третинного аміну формули (15) реакції дебензилювання, з одержанням сполуки формули (4):
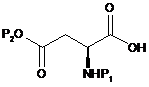 , (13)
, (13)
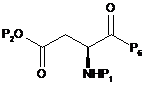 , (14)
, (14)
 , (15)
, (15)
де Р1 і Р2 є такими, як визначено в пункті 1, Р6 є монобензиламіном, дибензиламіном, моноаліламіном або діаліламіном.
16. Спосіб одержання сполуки формули (4), як визначено в пункті 1, при якому здійснюють:
(a) етап перетворення сполуки карбонової кислоти формули (13) на активований складний ефір, який потім піддають реакції зі сполукою, що є джерелом азоту, з одержанням амідної сполуки формули (16);
(b) етап відновлення амідної групи сполуки формули (16) з одержанням нітрильної сполуки формули (17); і
(c) етап піддавання нітрильної сполуки формули (17) реакції гідрогенізації з одержанням сполуки формули (4):
, (13)
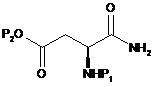 , (16)
, (16)
 , (17)
, (17)
де Р1 і Р2 є такими, як визначено в пункті 1.
17. Спосіб за пунктом 16, який відрізняється тим, що Р1 є Вос, а Р2 є і-пропіл або t-бутил.
18. Спосіб за пунктом 16, який відрізняється тим, що на етапі (а) як активуючий агент використовують хлорформат або Вос2О.
19. Спосіб за пунктом 16, який відрізняється тим, що на етапі (а) як сполуку, що є джерелом азоту, використовують газ аміак або сіль амонію.
20. Спосіб за пунктом 16, який відрізняється тим, що на етапі (b) відновлення проводять з використанням ангідриду трифторметансульфонової кислоти і Et3N або хлорангідриду ціанурової кислоти і диметилформаміду (DMF).
21. Спосіб за пунктом 16, який відрізняється тим, що на етапі (с) гідрогенізацію проводять з використанням металу, вибраного з паладію, нікелю (І) хлориду, платини (ІV) оксиду і паладію гідрооксиду.
22. Спосіб за пунктом 16, який відрізняється тим, що на етапі (с) гідрогенізацію проводять з використанням паладію гідрооксиду як металу, оцтової кислоти і водню.
Текст
Реферат: Даний винахід стосується нового способу одержання сполуки формули (2) як проміжного продукту, який може бути ефективно використаний для одержання сполуки формули (1), що демонструє хорошу інгібіторну активність проти ферменту дипептидилпептидаза IV. UA 107721 C2 (12) UA 107721 C2 O HO R3 N O NHP1 R4 R6 R5 , (2) 1 2 (1). UA 107721 C2 5 10 15 20 25 30 Галузь техніки Даний винахід стосується нового способу приготування сполуки формули (2) як головного проміжного продукту для синтезування сполуки формули (1), яка демонструє хорошу інгібіторну активність у відношенні дипептидилпептидази-IV (DPP-IV) і отже може використовуватись як лікарський засіб. Рівень техніки Сполука формули (1), як сполука, що була описана в Міжнародній патентній публікації WO 06/104356, демонструє хорошу інгібіторну активність у відношенні ферменту дипептидилпептидаза-IV і тому може ефективно використовуватись в лікуванні і профілактиці захворювань, викликаних дією дипептидилпептидази-IV, включаючи діабет (зокрема, діабет ІІ типу), ожиріння і т.п. Способи приготування сполуки формули (1) за допомогою сполуки формули (2) в якості проміжного продукту були описані в публікації WO 06/104356. У відповідності до цього джерела, сполуки формул (1) і (2) можуть бути одержані такими способами, як, наприклад, наступна схема реакції 1: Схема реакції 1 Однак в масовому виробництві вказаним відомим способом важко одержати сполуку формули (2), що має високу оптичну чистоту, через рацемізацію стереогенного центру, на якому в сполуці формули (2) є якоюсь мірою присутньою аміногрупа; відповідно, важко також одержати сполуку формули (1) високої оптичної чистоти. Суть винаходу Метою цього винаходу є створення нового способу приготування сполуки формули (2) високої оптичної чистоти в якості головного проміжного продукту для приготування сполуки формули (1), яка може бути використана в медичній сфері як агент для пригнічення DPP-IV. Відповідно, даний винахід пропонує новий спосіб приготування сполуки формули (2) в якості головного проміжного продукту, який може бути ефективно використаний для приготування сполуки формули (1) як агенту для пригнічення DPP-IV. У вищенаведеній формулі: R1 - це водень або CF3, R2 вибирається з групи, яка складається з водню, заміщеного або незаміщеного C 1-C10 алкілу, заміщеного або незаміщеного C3-C10 циклоалкілу, заміщеного або незаміщеного C4-C8 арилу і заміщеного або незаміщеного C3-C7 гетероарилу; і 1 UA 107721 C2 кожний з R3, R4, R5 і R6 - це, незалежно, водень, галоген або заміщений або незаміщений C1-C4 алкіл. 5 10 15 20 25 30 У вищенаведеній формулі R3, R4, R5 і R6 є такими, як визначено вище, а P 1 - це захисна група для аміну. Переважно, P1 є Boc (бутилоксикарбоніл), Cbz (бензилоксикарбоніл) або Fmoc (9-фторенілметилоксикарбоніл), а краще Boc. У вищенаведених формулах (1) і (2), коли C 1-C4 алкіл є заміщеним, переважно заміщенням може бути галоген, а краще - фтор. 1. Приготування сполуки формули (2) Спосіб приготування сполуки формули (2) у відповідності до цього винаходу відрізняється тим, що сполука формули (4) вступає в реакцію зі сполукою формули (5), і додатково включає етап видалення групи, що захищає карбонову кислоту, похідну від сполуки формули (4), після реакції вказаних двох сполук. У вищенаведених формулах: P1, R3, R4, R5 і R6 є такими, як визначено вище; кожний з P2 і P3 є незалежно бензольною групою, метильною групою, етильною групою, iпропильною групою або t-бутильною групою; G1 функціонує як хороша заміщувана група разом з киснем. G1O є трифлатом (трифторметансульфонат), мезилатом, тозилатом, безилатом або нонафлатом (нонафторбутансульфонат), а краще трифлатом або нонафлатом. Спосіб за цим винаходом дає сполуку формули (2) зі сполуки формули (4) і сполуки формули (5) через сполуку формули (2а) і конкретно включає: (а) етап реакції сполучення при додаванні основи до сполуки формули (4) і сполуки формули (5), (б) етап циклізації при додаванні кислоти для одержання сполуки формули (2а), і (в) етап видалення захисної групи карбонової кислоти шляхом гідролізу одержаної сполуки формули (2а) з одержанням сполуки формули (2). Спосіб за цим винаходом може бути представлений наступними схемами реакції 2 і 3: Схема реакції 2 Схема реакції 3 2 UA 107721 C2 5 10 15 20 25 30 35 40 45 50 На вищенаведених схемах: a - це основа, така як Et3N, основа Хуніга (диізопропилетиламін) і т.п.; b - це кислота, така як AcOH і т.п., і органічний розчинник, такий як CH 2Cl2 і т.п.; c - змінюється в залежності від захисної групи і типово вибирається з такого (1) сильна кислота, така як H2SO4 і т.п., і CH2Cl2, водний NaOH, Boc2O, і (2) NaOH, EtOH, H2O, рефлюкс, коли P1 є Boc і P2 є t-бутильною групою або є умови гідролізу з використанням основи, вказаної в пункті (2), коли P1 є Boc і P2 є бензильна група, метильна група, етильна група та i-пропильна група. R3, R4, R5, R6, P1, P2, P3 і G1 є такими, як визначено вище. Більш конкретно, на етапі (а) незахищений первинний амін сполуки формули (4) сполучається з атомом вуглецю, що має заміщувану групу в сполуці формули (5), в основних умовах і -OG1 видаляється. Ця реакція в якості основи використовує C 1-C4 триалкіламін, переважно триетиламін або диізопропилетиламін. В якості розчинника для цієї реакції можуть використовуватись звичайні органічні розчинники, такі як дихлоретан або дихлорметан, або циклічні ефіри (наприклад, тетрагідрофуран (THF) або діоксан). Для сприяння цій реакції використовувана основа альтернативно слугує розчинником. Реакція може проводитись при будь-якій температурі від 0° до температури дефлегмації. На етапі (b) синтезується сполука формули (2а) шляхом циклізації вторинної аміногрупи сполуки, одержаної на попередньому етапі (а), з внутрішньою групою ефіру в кислотних умовах. В цій реакції в якості кислоти можуть використовуватись неорганічні кислоти, такі як соляна кислота, сірчана кислота, азотна кислота, фосфорна кислота і т.п., або органічні кислоти, такі як мурашина кислота, оцтова кислота, винна кислота і т.п., причому оцтова кислота є найбільш доцільною. На цьому етапі можуть використовуватись такі ж розчинник і температурні умови, як описані для попереднього етапу (а). Вказані етапи (а) і (b) проводяться у безперервному режимі. На етапі (с) сполука формули (2а), одержана на етапі (b), гідролізується з одержанням сполуки формули (2). Більш конкретно, у випадку, коли в сполуці формули (2а) P1 є Boc і P2 є tбутильною групою, спочатку може використовуватись сильна кислота, така як сірчана кислота, соляна кислота, фосфорна кислота, ТФО (трифтороцтова кислота) і т.п., для видалення обох захисних груп, після чого захисна група Вос може знову приєднуватись до аміногрупи за основних умов, щоб дати бажану сполуку формули (2). Як варіант, гідроліз за основних умов, швидше ніж за кислотних умов, може привести до вибіркового видалення тільки P 2 серед захисних груп P1 і P2, щоб забезпечити одержання сполуки формули (2), і цей спосіб є більш ефективним. Переважно, в якості основи використовується розчин гідроокису натрію. Після завершення реакції сполука формули (2) може одержуватись як твердий продукт шляхом підкислення з використанням кислоти. У випадку, коли в сполуці формули (2а) P1 є Boc, а P2 є бензильною групою, метильною групою, етильною групою або і-пропильною групою, гідроліз може проводитись з використанням основи. Реакція видалення захисних груп проводиться з використанням H 2/Pd-C, коли P1 є Cbz, + або з використанням Bu4N F , коли P1 є Fmoc. Переважно, сполука формули (2) може одержуватись з високим виходом, коли P 2 є tбутильною групою або i-пропильною групою, а краще t-бутильною групою, і P3 є метильною групою або етильною групою. Додатково, даний винахід пропонує спосіб приготування сполук формул (4) і (5) як вихідних матеріалів, використовуваних для приготування сполуки формули (2). 2. Приготування сполуки формули (5) Сполука формули (5) як один з вихідних матеріалів, використовуваних для приготування сполуки формули (2), може бути приготовлена з відомої сполуки формули (7), яку можна одержати зі сполуки формули (6) способом, показаним на наступній схемі реакції 4, як описано в WO 06/104356. Спосіб приготування сполуки формули (5) включає: (а) етап відновлення сполуки формули (7) з одержанням сполуки у вигляді первинного спирту; і (б) етап реакції одержаного на етапі (а) спирту зі сполукою G 1, яка відповідає частині G1O 3 UA 107721 C2 сполуки формули (2), з одержанням сполуки формули (5). Цей спосіб може бути представлений, як показано на наступній схемі реакції 5. Схема реакції 4 5 10 15 20 25 30 35 40 45 На вищенаведеній схемі: a - це етил акрилат (де P4 є етилом), порошок Cu, TMEDA (тетраметил-етилендіамін) або THF; X - це галоген, такий як Br, F або Cl і т.п.; P4 - це бензильна група, метильна група, етильна група, i-пропильна група або t-бутильна група; R3, R4, R5, R6 і P3 є такими, як раніше було визначено. Схема реакції 5 На вищенаведеній схемі: a - це NaBH4, а також EtOH або MeOH або i-PrOH; b - це ангідрид трифторметан сульфонової кислоти (Tf 2O), трифторметан сульфоніл хлорид (TfCl), метансульфоніл хлорид (MsCl), толуолсульфоніл хлорид (TsCl), бромбензолсульфоніл хлорид (BsCl), (CF3(CF2)3SO2)F або (CF3(CF2)3SO2)2O, піридин або триалкіламін, і CH2Cl2; R3, R4, R5, R6, P3, P4 і G1 є такими, як раніше було визначено. Більш конкретно, на вищеозначеному етапі (а) борогідрид натрію (NaBH 4) використовується для того, щоб вибірково відновити тільки ефірну групу Р 4, що захищає карбонову кислоту, щоб одержати сполуку у вигляді первинного спирту, яка згодом на етапі (b) реагує зі сполукою G 1, яка відповідає частині G1O сполуки формули (2), тобто зі сполукою G 1, вибраною з групи, яка складається з ангідриду трифторметан сульфонової кислоти (Tf 2O), трифторметан сульфоніл хлориду (TfCl), метансульфоніл хлориду (MsCl), толуолсульфоніл хлориду (TsCl), бромбензолсульфоніл хлориду (BsCl), (CF 3(CF2)3SO2)F і (CF3(CF2)3SO2)2O, в CH2Cl2 в якості розчинника в присутності піридину або триалкіламіну, з одержанням сполуки формули (5). Тільки в якості прикладу, коли G1O бажаної сполуки формули (2) є трифлатом, ця реакція проводиться з використанням ангідриду трифторметан сульфонової кислоти, щоб одержати сполуку формули (5). 3. Приготування сполуки формули (4) Тим часом, сполука формули (4) як останній з вихідних матеріалів для приготування сполуки формули (2) може бути одержана за будь-яким з наступних способів. Перший спосіб приготування сполуки формули (4) включає: (а) етап перетворення групи карбонової кислоти сполуки формули (8) на ефірну групу шляхом введення групи Р2 з одержанням сполуки формули (9); (b) етап вибіркового відновлення ефірної групи Р 5, присутньої в сполуці формули (9), з одержанням сполуки формули (10); (с) етап введення заміщуваної групи G 2O в сполуку формули (10) з одержанням сполуки формули (11); (d) етап реакції сполуки формули (11) з азидною сполукою з одержанням сполуки формули (12); і (е) етап піддавання сполуки формули (12) гідрогенізації з одержанням сполуки формули (4). Цей перший спосіб приготування сполуки формули (4), як описано вище, включає процедури для введення аміногрупи в атом вуглецю, до якого приєднана ефірна група в сполуці формули (8), і може бути представлений так, як показано на наступній схемі реакції 6. Схема реакції 6 4 UA 107721 C2 5 10 15 20 25 30 35 40 45 На вищенаведеній схемі: a - це DMAP, Boc2O (де P2 є t-бутильною групою), і t-BuOH або THF; b - це NaBH4, і MeOH або EtOH; c - це Tf2O, MsCl, TsCl, (CF3(CF2)3SO2)F або (CF3(CF2)3SO2)2O і т.п., піридин або триалкіламін, CH2Cl2; d - це NaN3, DMF, або NMP, або DMAc, або DMAc/EtOAc, або DMAc/H2O, або DMAc/MeOH, нагрівання; e - вибирається з таких умов (1) H2, Pd/C, MeOH або EtOH, (2) NaBH 4, Pd/C, MeOH, (3) PPh3, H2O, THF, і (4) триалкіл фосфін або триалкіл фосфіт, H2O, THF; P5 - це метильна група, етильна група, i-пропильна група або t-бутильна група; G2 є разом з киснем хорошою заміщуваною групою, включаючи трифлат, мезилат, тозилат, безилат, нонафлат і т.п.; P1 і P2 є такими, як раніше було визначено. На етапі (а) вказаної реакції група карбонової кислоти формули (8) шляхом введення Р2 перетворюється на ефірну групу, щоб дати сполуку формули (9). В цій реакції в якості розчинника використовується t-BuOH або THF, а також використовується каталітична кількість (0,5 моль % ~ 30 моль %) 4-ди(метиламіно)піридину (DMAP). Наприклад, коли Р2, що вводиться, являє собою t-бутильну групу, використовується еквівалент Boc 2O, і реакцію проводять в межах між кімнатною температурою і 40°С, щоб одержати бажану ефірну сполуку формули (9). На етапі (b) вказаної реакції ефірна група, яка є початково присутньою в сполуці формули (9), тобто ефірна група, що знаходиться в позиції Р5, вибірково відновлюється борогідридом натрію (NaBH4) з утворенням сполуки формули (10) у вигляді первинного спирту. В цій реакції в якості розчинника використовується метиловий спирт або етиловий спирт. На етапі (с) вказаної реакції заміщувана група G 2O вводиться через реакцію з ангідридом трифторметан сульфонової кислоти (Tf 2O), трифторметан сульфоніл хлоридом (TfCl), метансульфоніл хлоридом (MsCl), толуолсульфоніл хлоридом (TsCl), бромбензолсульфоніл хлоридом (BsCl), (CF3(CF2)3SO2)F або (CF3(CF2)3SO2)2O в CH2Cl2 в якості розчинника в присутності піридину або триалкіламіну, щоб одержати сполуку формули (11). На етапі (d) вказаної реакції сполука формули (11) вступає в реакцію з 1,0-2,0 еквівалентами азиду натрію в умовах нагрівання (60-80°С), для одержання сполуки формули (12). Азидну групу одержаної у такий спосіб сполуки формули (12) можна перетворити на аміногрупу, скориставшись реакцією гідрогенізації в різних умовах реакції (е), для одержання сполуки формули (4). Зокрема, коли P1 є Boc, P2 є i-пропильна група або t-бутильна група і G2O є трифлатом або нонафлатом, сполука формули (4) може бути одержана з високим виходом. Другий спосіб приготування сполуки формули (4) включає: (а) етап перетворення сполуки карбонової кислоти формули (13) на активований складний ефір, який потім вступає в реакцію зі сполукою вторинного аміну з утворенням сполуки аміду формули (14); (b) етап відновлення амідної групи сполуки формули (14) з одержанням третинної сполуки аміну формули (15); і (с) етап піддавання сполуки третинного аміну формули (15) реакції дебензилювання, для одержання сполуки формули (4). Цей спосіб можна представити так, як показано на наступній схемі реакції 7. Схема реакції 7 5 UA 107721 C2 5 10 15 20 25 30 35 40 45 50 На вищенаведеній схемі: a вибирається з умов (1) i-BuOCOCl, NMM (N-метилморфолін), Bn2NH, або BnNH2, або диаліламін, або аліламін; і (2) i-BuOCOCl, NMM, диаліламін; b вибирається з умов (1) Os(CO)12, Ru(CO)12, RuCl2(CO)2(PPh3)2 або RuH2(CO)2(PPh3)2 в якості каталізатору реакції, Et3SiH, толуол, рефлюкс, (2) RuH(CO)(PPh3)3, Ru3(CO)12 або RuCl(PPh3)3 в якості каталізатору реакції, Ph2SiH2, PMHS (полідиметилсилоксан), THF або 2-Me THF, 1,4-діоксан, етиловий ефір, толуол, (3) 9-BBN (9-Борабіцикло[3,3,1]нонан), THF, рефлюкс, і (4) BH3DMS або BH3THF, толуол, нагрівання; P6 - це монобензиламін, або дибензиламін, або моноаліламін, або диаліламін, P1 і P2 є такими, як раніше було визначено. На етапі (а) вказаної реакції амідна сполука формули (14) може бути одержана у зручний спосіб шляхом перетворення сполуки карбонової кислоти формули (13) на активований складний ефір під дією ізобутилу хлорформату і основи, а потім реакції з вторинним аміном, таким як Bn2NH, диаліламін і т.п. На етапі (b) вказаної реакції амідна група амідної сполуки формули (14) може бути відновлена різними способами, відомими у відповідній технічній галузі, щоб одержати третинну сполуку аміну формули (15). Наприклад, відомими є наступні способи перетворення амідної групи на амін: Спосіб b-1: дивись, наприклад, Tetrahydron Lett. 2001, 42, 1945; Спосіб b-2: дивись, наприклад, Tetrahydron Lett. 1998, 39, 1017; Спосіб b-3: дивись, наприклад, Org. Lett. 1999, 1, 799, і Tetrahydron Lett. 1999, 40, 3673; Спосіб b-3: дивись, наприклад, Bioorg. Med. Chem. 2006, 14, 6586, і Chem. Eur. J. 2006, 12, 6910, і Synthesis 2005, 2281. Тільки в якості прикладу, у випадку сполуки формули (14), коли P 1 є Boc, а P2 є t-бутил, бажана сполука формули (15) може бути одержана в різних каталітичних умовах b-1. До того ж, каталізатори і умови, описані у вищенаведеному b-2, можуть бути використані для одержання бажаної сполуки формули (15). Зокрема, коли Ph2SiH2 використовується під каталізатором Ru3(CO)12, реакція може проводитись з використанням 0,5 моль % ~ 30 моль % Ru 3(CO) і 5,0 еквівалентів Ph2SiH2 в присутності розчинника THF при 80°С, щоб одержати бажану сполуку формули (15). Швидкість прогресування відновлення в умовах b-3, як визначено вище, є дещо низькою (максимум 25% швидкість прогресування). В умовах реакції b-4, як визначено вище, найкращий результат з точки зору виходу може бути одержаний тоді, коли реакція проводиться з використанням 2,0 еквівалентів BH3DMS в розчиннику толуолі при 50°С (14: 15: 15a = 11,4: 61,2: 10,7). В тому числі, сполука формули (15а) може бути дебензильована шляхом дегідрогенізації в присутності каталізаторів на основі Pd/C, щоб одержати сполуку формули (4), як визначено вище. На етапі (с) вказаної реакції сполука формули (15) може бути дебензильована, наприклад, шляхом реакції дебензилювання з використанням H2 і Pd/C для бензильної захисної групи або реакції деарилювання з використанням PdCl2/1,3-диметилбарбітурової кислоти, для одержання сполуки формули (4). Третій спосіб приготування сполуки формули (4) включає: (а) етап перетворення сполуки карбонової кислоти формули (13) на активований складний ефір, який потім вступає в реакцію зі сполукою, що є джерелом азоту, для одержання амідної сполуки формули (16); (b) етап відновлення амідної групи амідної сполуки формули (16), для одержання нітрильної сполуки формули (17); і (с) етап піддавання нітрильної сполуки формули (17) реакції гідрогенізації, для одержання сполуки формули (4). Цей спосіб може бути представлений так, як показано на наступній схемі реакції 8. Схема реакції 8 6 UA 107721 C2 5 10 15 20 25 30 35 40 45 На вищенаведеній схемі: a вибирається з умов (1) EtOCOCl, NMM, NH 3(g), і (2) Boc2O, NH4HCO3, піридин, DMF (диметилформамід); b вибирається з умов (1) (CF3CO)2O, Et3N, і (2) ціанурова кислота, DMF; c вибирається з умов (1) Pd/C, H 2, AcOH, 310 кПа, (2) NiCl6H2O, NaBH4, (3) CF3CO2H, NaBH4, (4) PtO2, H2, AcOH, (5) PtO2, H2, EtOH, CHCl3, (6) Pd(OH)2, H2, MeOH: AcOH (1:1) або AcOH:толуол (1:1), і (7) Pd(OH)2, H2, AcOH; P1 і P2 є такими, як було визначено раніше. Більш конкретно, на етапі (а) група карбонової кислоти вихідної сполуки формули (13) перетворюється на активовану ефірну групу з використанням хлорформату або Boc 2O в якості активуючого агента в основних умовах і наступної реакції зі сполукою, що є джерелом азоту, такою як газ аміак або сіль амонію (наприклад, бікарбонат амонію або карбонат амонію і т.п.), для одержання амідної сполуки формули (16). В цьому випадку, коли в сполуці формули (13) P 1 є Boc, а P2 є i-пропильною групою або t-бутильною групою, результат реакції буде кращим з точки зору виходу. На етапі (b) амідна група сполуки формули (16), одержана у такий спосіб, вступає в реакцію з ангідридом трифторметан сульфонової кислоти/Et 3N або ціануровою кислотою/DMF, для одержання сполуки формули (17), яка має нітрильну групу (-CN). На етапі (с) гідрогенізація може проводитись з використанням металу, вибраного з паладію, нікелю(І) хлориду, платини(IV) оксиду, платини гідрооксиду, для одержання сполуки первинного аміну формули (4). Даний винахід ілюструється більш докладно за допомогою наступних Способів приготування і Прикладів. Однак ці Способи приготування і Приклади жодним чином не обмежують об’єм цього винаходу. Докладний опис винаходу Спосіб за цим винаходом може дати сполуку формули (2), що має високу оптичну чистоту, в якості проміжного продукту для приготування сполуки формули (1) з високою оптичною чистотою, яка може використовуватись як медикамент для лікування або профілактики захворювань, включаючи діабет, спричинених дією дипептидилпептидази IV. Спосіб приготування 1: Синтез диметил 2,2-дифторпентандиоату До розчину етилу бромдифторацетату (33,2 г) в тетрагідрофурані (94,0 г) додали етил акрилат (8,2 г) і порошок міді (10,9 г). Після нагрівання до 50°С додали краплями TMEDA (9,5 г), після чого реакційну суміш перемішували впродовж 3 годин при тій самій температурі. Після зникнення етил акрилату як вихідного матеріалу, до реакційного розчину додали метил tбутиловий ефір (MTBE, 73,7 г), після чого краплями додали 10% водний розчин амонію хлориду (49,8 г), і потім суміш перемішували 30 хвилин. Залишок міді, що залишився, видалили фільтрацією через целіт, і для розділення шарів додали метил t-бутиловий ефір (MTBE, 66,3 г). Відділений органічний шар послідовно промили 10% водним розчином NH 4Cl (66,3 г) і 3N водним розчином соляної кислоти (99,6 г) належним чином, після чого перегнали під зниженим тиском, щоб одержати 55,0 г бажаної титульної сполуки. 1 H ЯМР (400 Мгц, CDCl3) 1,26 (t, J=7,2 Гц, 3H), 1,37 (t, J=7,2 Гц, 3H), 2,37-2,49 (m, 2H), 2,55 (t, J=7,2 Гц, 2H), 4,16 (q, J=7,2 Гц, 2H), 4,29 (q, J=7,2 Гц, 2H). Спосіб приготування 2: Синтез етил 4,4-дифтор-5-гідроксипентаноату 7 UA 107721 C2 5 10 15 20 25 30 35 40 45 14,8 г сполуки, одержаної під час Способу приготування 1, розвели етиловим спиртом (20,4 г) і тетрагідрофураном (69,1 г), а потім охолодили до 0°С. До цього розчину повільно частинами додали борогідрид натрію (NaBH4, 3,5 г), підтримуючи внутрішню температуру нижче 30°С. 1 Після підтвердження завершення реакції за допомогою Н ЯМР, реакційний розчин охолодили до температури 10°С, після чого повільно додали 10% водний розчин амонію хлориду (77,7 г). Сполуку бору, що залишилась, відфільтрували через целіт, і фільтрат перегнали під зниженим тиском, щоб видалити тетрагідрофуран. Потім додали етилацетат (105,2 г), щоб розділити шари, і органічний шар перегнали під зниженим тиском, для одержання 10,8 г титульної сполуки. 1 H ЯМР (400 МГц, CDCl3) 1,23 (t, J=7,2 Гц, 3H), 2,15-2,29 (m, 2H), 2,49 (t, J=7,2 Гц, 2H), 3,69 (t, J=12,0 Гц, 2H), 4,12 (q, J=4,0 Гц, 2H). Приклад 1: Синтез етил 4,4-дифтор-5-{[(трифторметил)сульфоніл]окси}-пентаноату До розчину 10,8 г сполуки, одержаної під час Способу приготування 2, розчиненої в дихлорметані (100, 2 г), додали піридин (7,0 г), після чого одержану суміш охолодили до -5°С. Після завершення охолодження повільно краплями додали ангідрид трифторметан сульфонової кислоти (20,1 г), підтримуючи температуру реакції нижче 6,3°С. Після перемішування реакційного розчину впродовж 30 хвилин додали краплями 1,5N розчин соляної кислоти при 0°С, щоб розділити шари. Водний шар відділили і двічі піддали зворотній екстракції дихлорметаном (33,4 г), об’єднали екстракти з відділеним органічним шаром, а потім перегнали під зниженим тиском, щоб одержати 19,7 г титульної сполуки у вигляді жовтої олії. 1 H ЯМР (500 МГц, CDCl3) 1,27 (t, J=7,2 Гц, 3H), 2,29-2,39 (m, 2H), 2,59 (t, J=7,6 Гц, 2H), 4,18 (q, J=7,2 Гц, 2H), 4,55 (t, J=11,6 Гц, 2H). Приклад 2-1: Синтез етил 4,4-дифтор-5-{[(нонафторбутил)сульфоніл]-окси} пентаноату До розчину 100,0 г одержаної під час Способу приготування 2, розчиненої в дихлорметані (300,0 мл), додали піридин (65,7 г), і охолодили одержану суміш до -10°С. Після завершення охолодження повільно краплями додали нонафторбутансульфоновий ангідрид (477,4 г). Після перемішування реакційного розчину впродовж 3 годин, додали краплями 1,0N розчин соляної кислоти (300,0 мл), щоб розділити шари. Водний шар відділили і піддали зворотній екстракції дихлорметаном (500,0 мл), об’єднали екстракт з відділеним органічним шаром, а потім перегнали під зниженим тиском, щоб одержати 177,5 г титульної сполуки. 1 H ЯМР (500 МГц, CDCl3) 1,26 (t, 3H, J=7,3 Гц), 2,30-2,36 (m, 2H), 2,58 (t, 2H, J=7,4 Гц), 4,16 (q, 2H, J=7,3 Гц), 4.57 (t, 2H, J=11 Гц). Приклад 2-2: Синтез етил 4,4-дифтор-5-{[(нонафторбутил)сульфоніл]-окси}пентаноату До розчину 500,0 г одержаної під час Способу приготування 2, розчиненої в дихлорметані (1000,0 мл), додали триетиламін (389,0 г), і охолодили одержану суміш до 0°С. Після завершення охолодження повільно краплями додали перфторбутансульфоніл хлорид (948,80 г). Реакційний розчин перемішували впродовж 3 годин при кімнатній температурі, перегнали під зниженим тиском, розчинили в метил t-бутиловому ефірі (MTBE, 3000,0 мл), після чого тричі промили водою. Одержаний у такий спосіб органічний шар зневоднили магнію сульфатом, профільтрували через целіт, а потім перегнали під зниженим тиском, щоб одержати 960,0 г титульної сполуки. Приклад 3: Синтез метил (2S)-2-[(tert-бутоксикарбоніл)аміно]-4-оксо-пентаноату До 25,0 г вихідного матеріалу, (3S)-3-[(t-бутоксикарбоніл)аміно]-4-оксо-пентанової кислоти, додали t-бутанол (96,9 г), а потім Boc2O (25,4 г) і диметиламінопіридин (DMAP, 62,0 г, 0,5 моль 8 UA 107721 C2 5 10 15 20 25 30 35 40 45 50 %) при кімнатній температурі, після чого реакційну суміш перемішували впродовж 23 годин при 40°С. Після завершення реакції додали етилен дихлорид (62,3 г) в t-бутанолі, а суміш потім перегнали під зниженим тиском, щоб одержати 30,7 г титульної сполуки. 1 H ЯМР (400 МГц, CDCl3) 1,45 (s, 9H), 1,47 (s, 9H), 2,71 (dd, J=4,8; 16,4 Гц, 1H), 2,88 (dd, J=4,4; 16,4 Гц, 1H), 3,75 (s, 3H), 4,53 (m, 1H), 5,44 (br d, J=8,0 Гц, 1H). Приклад 4: Синтез tert-бутил (3S)-3-[(tert-бутоксикарбоніл)аміно]-4-гідрокси-бутаноату 30,7 г сполуки, одержаної в Прикладі 3, розчинили в етиловому спирті (112,3 г) і, після зниження внутрішньої температури до 10,5°С, повільно краплями додали натрію борогідрид (NaBH4, 5,7 г). Одержаний реакційний розчин перемішували, підтримуючи температуру нижче 1 22°С. Після підтвердження завершення реакції за допомогою H ЯМР і ТШХ, до реакційного розчину повільно додали краплями 3,0N розчин соляної кислоти (30,7 г) при внутрішній температурі 10°С, після чого додали розведений 0,2% розчин соляної кислоти (100,0 г). Реакційний розчин відрегулювали до pH 3~4 шляхом додавання 9,0% водного розчину соляної кислоти, а потім двічі піддали зворотній екстракції етилацетатом (100,0 г) і толуолом (44,0 г). Одержаний у такий спосіб органічний шар перегнали під зниженим тиском, щоб одержати 25,1 г титульної сполуки. 1 H ЯМР (500 МГц, CDCl3) 1,44 (s, 9H), 1,45 (s, 9H), 2,48-2,57 (m, 2H), 3,69 (d, J=4,9 Гц, 1H), 3,97 (m, 1H), 5,22 (bs, 1H). Приклад 5: tert-бутил (3S)-[(tert-бутоксикарбоніл)аміно]-4-[(метилсульфоніл)окси]-бутаноат До 25,1 г сполуки, одержаної в Прикладі 4, додали дихлорметан (133,0 г) і триетиламін (148,0 г), після чого одержану суміш охолодили до 0°С. До цього реакційного розчину повільно краплями впродовж 50 хвилин додали метансульфоніл хлорид (11,8 г), розведений дихлорметаном (39,9 г), підтримуючи внутрішню температуру нижче 12°С. Після завершення реакції, реакційний розчин промили 0,5N водним розчином соляної кислоти (120,0 г) і водою (100,4 г), а потім перегнали під зниженим тиском, щоб одержати 31,5 г титульної сполуки. 1 H ЯМР (500 МГц, CDCl3) 1,44 (s, 9H), 1,46 (s, 9H), 2,62 (d, J=6,0 Гц, 2H), 3,04 (s, 3H), 4,21 (m, 1H), 4,30 (d, J=5,2 Гц, 2H), 5,16 (br d, J=7,2 Гц, 1H). Приклад 6: Синтез tert-бутил (3S)-4-азидо-3-[(tert-бутоксикарбоніл)аміно]-бутаноату Натрію азид (NaN3, 11,6 г) розвели диметилацетамідом (DMAc, 260,0 г). Після підвищення внутрішньої температури до 80°С, до цього додали розчин 31,5 г сполуки, одержаної в Прикладі 5, розведеної диметилацетамідом (DMAc, 45,0 г). Реакція здійснювалась при 80°С впродовж 2 годин. До реакційного розчину додали толуол (251,0 г) і воду (320,0 г), щоб розділити шари. Одержаний у такий спосіб органічний шар перегнали під зниженим тиском, щоб одержати 24,0 г титульної сполуки. 1 H ЯМР (500 МГц, CDCl3) 1,47 (s, 9H), 1,49 (s, 9H), 2,49 (d, J=6,0 Гц, 2H), 3,44-3,55 (m, 2H), 4,09 (br s, 1H), 5,14 (br s, 1H). Приклад 7: Синтез tert-бутил (3S)-4-аміно-3-[(tert-бутоксикарбоніл)аміно]-бутаноату До 21,0 г сполуки, одержаної в Прикладі 6, додали тетрагідрофуран (93,3 г), а потім трифенілфосфін (PPh3, 21,0 г) при 40°С. Одержану суміш перемішували впродовж 2 годин при тій самій температурі, після чого додали воду (3,8 г). Реакційний розчин перегнали під зниженим тиском, а одержаний твердий трифенілфосфін оксид розвели толуолом (26,0 г) і n-гексаном (41,0 г), а потім відфільтрували. Фільтрат відрегулювали до pH 2~3 1,0N водним розчином соляної кислоти (110,0 г), після чого піддали розділенню шарів. Щоб видалити будь-який залишковий твердий трифенілфосфін оксид, одержаний вище водний шар промили дихлорметаном (100,0 г), а потім відрегулювали до pH 8~9 28% водним розчином аміаку (7,6 г). Одержаний у такий спосіб водний розчин екстрагували дихлорметаном (100,0 г) і перегнали під зниженим тиском, щоб одержати 8,5 г титульної сполуки у вигляді білої твердої речовини. 9 UA 107721 C2 H ЯМР (500 МГц, CDCl3) 1,44 (s, 9H), 1,45 (s, 9H), 2,45 (d, J=6,1 Гц, 2H), 2,77 (d, J=5,5 Гц, 2H), 3,87 (br s, 1H), 5,22 (br s, 1H). Приклад 8: Синтез N,N-дибензил-L-N(Boc)-аспартамід 4-tert-бутилового ефіру 1 5 10 15 20 25 30 35 40 45 N-Boc-L-аспарагінової кислоти 4-t-бутиловий ефір (29,0 г; 0,10 моль) додали до THF (200 мл). Після охолодження до температури нижче -5°С, до реакційного розчину додали ізобутилхлорформат (13,0 мл; 0,10 моль), а потім краплями N-метил морфолін (12,0 мл; 0,10 моль), і перемішували реакційну суміш понад 30 хвилин. До реакційної суміші краплями додали дибензиламін (21,1 мл; 0,11 моль), після чого реакційну суміш перемішували понад 3 години, контролюючи протікання реакції за допомогою ТШХ (EtOAc: Гексан=1:4). Після завершення реакції реакційний розчин перемішували при додаванні етилацетату (300,0 мл) і 1N соляної кислоти, щоб розділити шари, з подальшою перегонкою під зниженим тиском для осадження твердої речовини. Тверду речовину відфільтрували і промили етилацетатом (100 мл), після чого промивки концентрувались перегонкою знову під зниженим тиском. Залишок було піддано очистці на силікагелевій колонці, щоб одержати очищений бажаний продукт (41,7 г; 0,89 моль). 1 H ЯМР (400 МГц, CDCl3) : 7,32 (m, 5H), 7,20 (m, 5H), 5,39 (d, J=7,2 Гц, 1H), 5,30 (m, 1H), 4,87-4,77 (m, 2H), 4,48-4,39 (m, 2H), 2,72 (dd, J=15,8 Гц, J=8,0 Гц, 1H), 2,56 (dd, J=15,8 Гц, J=6,4 Гц, 1H), 1,43 (s, 9H), 1,37 (s, 9H). Мас (ESI, m/z): 491 (M+Na), 469 (M+H), 413 (M-55). Приклад 9: Синтез N, N-диаліл-L-N(Boc)-аспартамід 4-tert-бутилового ефіру L-N(Boc)-аспарагінової кислоти 4-t-бутиловий ефір (5,00 г; 17,3 моль) додали до THF (50 мл). Після охолодження до температури нижче -5°С, до реакційного розчину додали спочатку ізобутилхлорформат (2,26 мл; 17,3 моль), а потім краплями N-метил морфолін (1,90 мл; 17,3 моль), і реакційну суміш перемішували понад 30 хвилин. До реакційної суміші додали краплями диаліламін (2,35 мл; 19,0 моль), після чого суміш перемішували понад 3 години і контролювали прогресування реакції за допомогою ТШХ (EtOAc: Гексан=1:4). Після завершення реакції реакційний розчин перемішували при додаванні етилацетату (60 мл) і 1N соляної кислоти і, після розділення шарів, концентрували перегонкою під зниженим тиском. Залишок потім піддали очистці на силікагелевій колонці, щоб одержати очищений бажаний продукт (6,0 г; 16,3 моль). 1 H ЯМР (400 МГц, CDCl3) : 5,78 (m, 2H), 5,30 (m, 1H), 5,23-5,11 (m, 1H), 5,30 (m, 1H), 4,93 (m, 1H), 4,11-3,84 (m, 4H), 2,68 (dd, J=15,8 Гц, J=8,0 Гц, 1H), 2,51 (dd, J=15,8 Гц, J=8,0 Гц, 1H), 1,44 (s, 9H), 1,42 (s, 9H). Мас (ESI, m/z): 391 (M+Na), 369 (M+H), 313 (M-55). Приклад 10: Синтез N,N-дибензил-4-аміно-3(S)-N(Boc)-амінобутанової кислоти 4-tertбутилового ефіру 10,0 г сполуки, одержаної в Прикладі 8, Ru3(CO)12 (136 мг, 1 моль %), і дифенілсилан (19,7 мл, 106,7 ммоль) додали до тетрагідрофурану (50 мл), і перемішували реакційний розчин зі зворотним холодильником впродовж більше ніж 40 годин. Реакційний розчин екстрагували етилацетатом (200 мл) і концентрували перегонкою під зниженим тиском. Потім залишок піддали очистці на силікагелевій колонці, щоб одержати очищений бажаний продукт (4,7 г; 10,5 ммоль). 1 H ЯМР (400 МГц, CDCl3) : 7,31-7,20 (m, 10H), 5,12 (bs, 1H), 3,90 (bs, 1H), 3,63 (d, J=12,0 Гц, 2H), 3,48 (d, J=12,0 Гц, 2H), 3,24 (m, 1H), 3,16 (bs, 1H), 2,42 (m, 2H), 1,81 (m, 1H), 1,59 (m, 9H), 1,46 (s, 9H), 1,06 (s, 9H). Мас (ESI, m/z): 455 (M+H), 441 (M-13). Приклад 11: Синтез tert-бутил (3S)-4-аміно-3-[(tert-бутоксикарбоніл)аміно]-4-оксобутаноату 10 UA 107721 C2 5 10 15 20 25 30 35 40 360,0 г вихідного матеріалу, N-Boc-Asp(O-t-Bu)OH, разом з Boc2O (353,0 г) і бікарбонатом амонію (NH4HCO3, 123,9 г), додали до диметилформаміду (1174,6 г) і до цього додали краплями піридин (61,0 г) при кімнатній температурі, після чого реакційну суміш перемішували приблизно впродовж 3 годин. Після завершення реакції, до реакційного розчину додали воду (1440 мл) і толуол (1800 мл) і перемішували 30 хвилин, щоб розділити шари. Одержаний у такий спосіб органічний шар перегнали під зниженим тиском, щоб видалити t-бутанол і толуол і одержати титульну сполуку, яку було безпосередньо використано в наступній реакції. Приклад 12: Синтез (S)-tert-бутил 3-(tert-бутоксикарбоніламіно)-3-ціанопропаноату До сполуки, одержаної в Прикладі 11, додали диметилформамід (1019,5 г), а потім краплями хлорангідрид ціанурової кислоти (112,0 г) впродовж 1,5 години при температурі нижче 25°С. Реакційний розчин перемішували впродовж однієї години при кімнатній температурі, а потім до цього додали 0,1N водний розчин гідрооксиду натрію (1850,0 г) і толуол (1860 мл), щоб розділити шари. Одержаний у такий спосіб органічний шар промили ще раз водою (700 мл), а потім перегнали під зниженим тиском, щоб одержати 318,3 г титульної сполуки. 1 H ЯМР (500 МГц, CDCl3) : 1,44 (s, 9H), 1,45 (s, 9H), 2,45 (d, J=6,1 Гц, 2H), 2,77 (d, J=5,5 Гц, 2H), 3,87 (br s, 1H), 5,22 (br s, 1H). Приклад 13: Синтез tert-бутил (3S)-4-аміно-3-[(tert-бутоксикарбоніл)аміно]-бутаноату До 212,1 г сполуки, одержаної в Прикладі 12, додали оцтову кислоту (4000 мл), а потім 20 ваг. % Pd(OH)2 (1,1 г) при 40°С. Суміш перемішували впродовж 8 годин, підтримуючи внутрішню температуру нижче 45°С і тиск водню 3 атмосфери. Після завершення реакції реакційний розчин перегнали під зниженим тиском, щоб видалити оцтову кислоту, розвели толуолом (640 л), а потім профільтрували через целіт. До фільтрату додали 0,25N водний розчин соляної кислоти (1060 мл), щоб розділити шари. Одержаний у такий спосіб водний шар зробили основним за допомогою водного розчину аміаку (543,1 г), після чого екстрагували метил tбутиловим ефіром (MTBE, 1000 мл). Одержаний у такий спосіб органічний шар перегнали під зниженим тиском, щоб одержати 185,0 г титульної сполуки. Приклад 14: Синтез 3-t-бутоксикарбоніламіно-4-(5,5-дифтор-2-оксо-піперидин-1-іл)-масляної кислоти t-бутилового ефіру Триетиламін (13,2 г) додали до 16,0 г сполуки, одержаної в Прикладі 1 або 2-1 або 2-2, і 14,1 г сполуки, одержаної в Прикладі 7 або 13, після чого суміш перемішували впродовж 21 години при 40°С. Потім додали дихлорметан (154,8 г) і оцтову кислоту (18,3 г), і суміш перемішували впродовж 5 годин при кімнатній температурі. До одержаного реакційного розчину додали 0,5N водний розчин соляної кислоти (116,8 г), після чого суміш перемішували впродовж 30 хвилин, щоб розділити шари. Одержаний у такий спосіб органічний шар перегнали під зниженим тиском, щоб одержати 23,6 г титульної сполуки. 1 H ЯМР (500 МГц, CDCl3) : 1,42 (s, 9H), 1,46 (s, 9H), 2,27 (m, 2H), 2,40-2,64 (m, 4H), 3,20 (dd, J=4,3; 13,5 Гц, 1H), 3,56-3,70 (m, 2H), 3,76-3,91 (m, 2H), 4,16 (m, 1H), 5,20 (d, J=8,6 Гц, 1H). Приклад 15: Синтез 3-t-бутоксикарбоніламіно-4-(5,5-дифтор-2-оксо-піперидин-1-іл)-масляної кислоти 11 UA 107721 C2 5 10 15 20 25 30 35 40 45 50 23,6 г сполуки, одержаної в Прикладі 14, додали до дихлорметану (20,0 г), після чого додали H3PO4 (30,0 г), і суміш перемішували впродовж 16 годин при кімнатній температурі. Після підтвердження від’єднання всіх t-бутильних груп і t-бутилоксикарбонільних груп, реакційний розчин відрегулювали до pH 7,0~8,0 за допомогою 10N водного перекису водню, і до цього додали Boc2O (16,0 г). Після завершення додавання, 10N водний перекис водню використовувався для підтримування pH реакційного розчину на рівні 8,0~9,0. Після перемішування впродовж 3 годин натрію фосфат, що утворився, відфільтрували, після чого фільтрат відрегулювали до pH 2,0~3,0 за допомогою 3,0N водного розчину соляної кислоти. Одержану тверду речовину відфільтрували і висушили в атмосфері азоту, щоб одержати 14,5 г титульної сполуки. 1 H ЯМР (500 МГц, CDCl3) : 1,32 (s, 9H), 2,20-2,43 (m, 6H), 3,26-3,31 (m, 2H), 3,61 (m, 1H), 3,81 (m, 1H), 4,02 (m, 1H), 6,73 (d, J=8,6 Гц, 1H), 12,16 (s, 1H). Для титульної сполуки, одержаної як описано вище, за допомогою ВЕРХ (високоефективної рідинної хроматографії) були визначені кількісно її енантіомерні ізомери, тобто S-форма і Rформа, після чого був обчислений надлишок цих енантіомерних ізомерів (S проти R форми) (ee = енантіомерний надлишок), який склав ee > 99%. З іншого боку, у випадку Порівняльного Прикладу, приготовленого у відповідності до відомого способу на основі WO 06/104356, як описано далі, надлишок (ee) енантіомерних ізомерів (S проти R форми) становив 80%. З цього можна зробити висновок, що сполука формули (2), яка має високу оптичну чистоту, може одержуватись згідно способу за цим винаходом. Порівняльний Приклад 1: Синтез 3-t-бутоксикарбоніламіно-4-(5,5-дифтор-2-оксо-піперидин1-іл)-масляної кислоти t-бутилового ефіру Порівняльний Приклад 1-1: Синтез метил 5-аміно-4,4-дифтор-пентаноату HCl До 10,0 г сполуки, одержаної в Прикладі 1, додали 40 мл безводного розчину аміаку (7M розчин в метиловому спирті), і суміш перемішували впродовж 3 годин. Реакційний розчин перегнали і до цього краплями додали 30 мл розчину соляної кислоти, насиченого метиловим спиртом. Реакційну суміш перемішували при кімнатній температурі, після чого перегнали, щоб одержати 7,2 г титульної сполуки у вигляді білої твердої речовини. 1 H ЯМР (500 МГц, CD3OD) : 2,35 (m, 2H), 2,59 (t, J=7,6 Гц, 2H), 3,49 (t, J=15,3 Гц, 2H), 3,68 (s, 3H). Порівняльний Приклад 1-2: Синтез 3-t-бутоксикарбоніламіно-4-(5,5-дифтор-2-оксопіперидин-1-іл)-масляної кислоти t-бутилового ефіру До розчину сполуки (1,93 г), одержаної в Прикладі 4, розчиненої в дихлорметані (20,0 г) і H2O (4,0 г), додали NaBr (0,8 г) і TEMPO (11 мг, 1 моль %). До цього реакційного розчину повільно краплями додали розчин 5% NaOCl (11,5 г) і NaHCO 3 (1,7 г), розчинений в H2O (12,0 г) впродовж приблизно 2 годин, підтримуючи температуру нижче 5°С. Після завершення додавання краплями, реакційний розчин перемішували впродовж 30 хвилин, щоб розділити шари. До одержаного у такий спосіб органічного шару додали сполуку (1,6 г), одержану в Порівняльному Прикладі 1-1. Після перемішування впродовж 15 хвилин при кімнатній температурі до реакційного розчину додали NaBH(OAc) 3 (2,23 г). Після перемішування впродовж близько 19 годин до реакційного розчину додали краплями 10% водний розчин NaHCO3 (20,0 г) і 0,5N водний розчин соляної кислоти (20,0 г), щоб розділити шари. Одержаний у такий спосіб органічний шар піддали зневодненню під безводним MgSO 4, щоб одержати 2,0 г (вихід 73%) тієї ж титульної сполуки, що й в Прикладі 14, у вигляді жовтої твердої речовини. Для цієї титульної сполуки, одержаної як описано вище, були кількісно визначені її енантіомерні ізомери, тобто S-форма і R-форма, за допомогою ВЕРХ (високоефективної рідинної хроматографії), після чого було обчислено надлишок (ee) енантіомерних ізомерів (S проти R форми), який становив ee = 80%. 12 UA 107721 C2 ФОРМУЛА ВИНАХОДУ 1. Спосіб одержання сполуки формули (2), який відрізняється тим, що сполуку формули (4) піддають реакції зі сполукою формули (5): 5 O HO R3 N O NHP1 R4 R6 P2O NHP1 R4 , (4) O G1O OP3 R6 15 , (2) NH2 O 10 R5 R5 R3 , (5) де кожний з R3, R4, R5 і R6 є незалежно воднем, галогеном або заміщеним або незаміщеним С1-С4алкілом; Р1 є захисною групою аміну; кожний з Р2 і Р3 є незалежно бензильною групою, метильною групою, етильною групою, і-пропiльною групою або t-бутильною групою; і G1O є заміщуваною групою. 2. Спосіб за пунктом 1, при якому здійснюють: (a) етап реакції сполучення з додаванням основи до сполуки формули (4) і сполуки формули (5); (b) етап циклізації з додаванням кислоти з одержанням сполуки формули (2а); і (c) етап гідролізу одержаної сполуки формули (2а) з одержанням сполуки формули (2): O P2O R3 N O NHP1 R4 R6 R5 , (2a) 20 25 30 35 де R3, R4, R5, R6, Р1 і Р2 є такими, як визначено в пункті 1. 3. Спосіб за пунктом 1 або 2, який відрізняється тим, що Р2 є t-бутильною групою, а Р3 є метильною або етильною групою. 4. Спосіб за пунктом 1 або 2, який відрізняється тим, що G1O є трифлатом, мезилатом, тозилатом, безилатом або нонафлатом. 5. Спосіб за пунктом 1 або 2, який відрізняється тим, що R3 і R4 є воднем, a R5 і R6 є фтором. 6. Спосіб за пунктом 2, який відрізняється тим, що на етапі (а) як основу використовують С 1С4триалкіламін. 7. Спосіб за пунктом 2, який відрізняється тим, що на етапі (b) як кислоту використовують оцтову кислоту. 8. Спосіб за пунктом 2, який відрізняється тим, що у випадку сполуки формули (2а), де Р1 є Вос і Р2 є t-бутил, гідроліз на вказаному етапі (с) проводять в основних умовах, щоб вибірково видалити тільки Р2 з-поміж захисних груп Р1 і Р2 і отримати сполуку формули (2). 9. Спосіб за пунктом 8, який відрізняється тим, що як основу використовують водний розчин гідроокису натрію. 10. Спосіб одержання сполуки формули (5), як визначено в пункті 1, при якому здійснюють: (a) етап відновлення сполуки формули (7) з одержанням сполуки у вигляді первинного спирту; і (b) етап реакції спиртової сполуки, одержаної на попередньому етапі, зі сполукою G1, яка відповідає частині G1O сполуки формули (5), з одержанням сполуки формули (5): 13 UA 107721 C2 O R4 O P4O OP3 R6 5 10 15 20 R5 R3 , (7) де R3, R4, R5, R6 і Р3 є такими, як визначено вище в пункті 1, а Р4 є бензильною групою, метильною групою, етильною групою, і-пропільною групою або t-бутильною групою. 11. Спосіб за пунктом 10, який відрізняється тим, що на етапі (а) відновлення проводять з використанням NaBH4. 12. Спосіб за пунктом 10, який відрізняється тим, що на етапі (b) сполуку G1 вибирають з групи, яка складається з ангідриду трифторметансульфонової кислоти (Tf2O), трифторметансульфонілхлориду (TfCl), метансульфонілхлориду (MsCl), толуолсульфонілхлориду (TsCl), бромбензолсульфонілхлориду (BsCl), (CF3(CF2)3SO2)F і (CF3(CF2)3SO2)2O. 13. Спосіб одержання сполуки формули (4), як визначено в пункті 1, при якому здійснюють: (a) етап перетворення карбонової кислоти сполуки формули (8) на ефірну групу шляхом введення групи Р2 з одержанням сполуки формули (9); (b) етап вибіркового відновлення ефірної групи Р5, присутньої в сполуці формули (9), з одержанням сполуки формули (10); (c) етап введення заміщуваної групи G2O в сполуку формули (10) з одержанням сполуки формули (11); (d) етап реакції сполуки формули (11) з азидною сполукою з одержанням сполуки формули (12); і (e) етап піддавання сполуки формули (12) гідрогенізації з одержанням сполуки формули (4): O HO OP5 O NHP1 , (8) O P2O OP5 O 25 NHP1 P2O OH O NHP1 P2O NHP1 P2O , (11) N3 O 35 , (10) OG2 O 30 , (9) NHP1 , (12) де Р1 і Р2 є такими, як визначено в пункті 1, Р5 є метильною групою, етильною групою, іпропільною групою або t-бутильною групою, a G2O є заміщуваною групою. 14. Спосіб за пунктом 13, який відрізняється тим, що Р1 є Вос, Р2 є і-пропільною групою або tбутильною групою, a G2O є трифлатом або нонафлатом. 15. Спосіб одержання сполуки формули (4), як визначено в пункті 1, при якому здійснюють: (a) етап перетворення сполуки карбонової кислоти формули (13) на активований складний ефір, який потім піддають реакції зі сполукою вторинного аміну, з одержанням амідної сполуки формули (14); 14 UA 107721 C2 (b) етап відновлення амідної групи сполуки формули (14), з одержанням сполуки третинного аміну формули (15); і (c) етап піддавання сполуки третинного аміну формули (15) реакції дебензилювання, з одержанням сполуки формули (4): O P2O OH O 5 NHP1 , (13) O P2O P6 O NHP1 P2O P6 O 10 15 , (14) NHP1 , (15) де Р1 і Р2 є такими, як визначено в пункті 1, Р6 є монобензиламіном, дибензиламіном, моноаліламіном або діаліламіном. 16. Спосіб одержання сполуки формули (4), як визначено в пункті 1, при якому здійснюють: (a) етап перетворення сполуки карбонової кислоти формули (13) на активований складний ефір, який потім піддають реакції зі сполукою, що є джерелом азоту, з одержанням амідної сполуки формули (16); (b) етап відновлення амідної групи сполуки формули (16) з одержанням нітрильної сполуки формули (17); і (c) етап піддавання нітрильної сполуки формули (17) реакції гідрогенізації з одержанням сполуки формули (4): O P2O OH O 20 NHP1 , (13) O P2O NH2 O P2O 30 35 , (16) CN O 25 NHP1 NHP1 , (17) де Р1 і Р2 є такими, як визначено в пункті 1. 17. Спосіб за пунктом 16, який відрізняється тим, що Р1 є Вос, а Р2 є і-пропіл або t-бутил. 18. Спосіб за пунктом 16, який відрізняється тим, що на етапі (а) як активуючий агент використовують хлорформат або Вос2О. 19. Спосіб за пунктом 16, який відрізняється тим, що на етапі (а) як сполуку, що є джерелом азоту, використовують газ аміак або сіль амонію. 20. Спосіб за пунктом 16, який відрізняється тим, що на етапі (b) відновлення проводять з використанням ангідриду трифторметансульфонової кислоти і Et3N або хлорангідриду ціанурової кислоти і диметилформаміду (DMF). 21. Спосіб за пунктом 16, який відрізняється тим, що на етапі (с) гідрогенізацію проводять з використанням металу, вибраного з паладію, нікелю (І) хлориду, платини (ІV) оксиду і паладію гідрооксиду. 15 UA 107721 C2 22. Спосіб за пунктом 16, який відрізняється тим, що на етапі (с) гідрогенізацію проводять з використанням паладію гідрооксиду як металу, оцтової кислоти і водню. Комп’ютерна верстка А. Крулевський Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 16
ДивитисяДодаткова інформація
Назва патенту англійськоюProduction method of intermediate compound for synthesizing medicament
Автори англійськоюKim, Bong Chan, Kim, Kuy Young, Lee, Hee Bong, An, Ji Eun, Lee, Kyu Woong
Автори російськоюКим Бон Чан, Ким Кюу Юн, Ли Хе Бон, Ань Цзи Энь, Ли Кюу Вон
МПК / Мітки
МПК: C07D 471/04, C07D 211/36, A61K 31/45, A61P 3/10
Мітки: проміжної, медикаменту, 4-(2-оксопіперидин-1-іл)масляної, похідної, одержання, синтезування, кислоти, спосіб, сполуки
Код посилання
<a href="https://ua.patents.su/18-107721-sposib-oderzhannya-pokhidno-4-2-oksopiperidin-1-ilmaslyano-kisloti-yak-promizhno-spoluki-dlya-sintezuvannya-medikamentu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідної 4-(2-оксопіперидин-1-іл)масляної кислоти як проміжної сполуки для синтезування медикаменту</a>
Спосіб одержання гетероарилфенілборонової кислоти як проміжної сполуки для одержання похідних фенілпіридинсульфонаміду та сполуки, одержані за цим способом

Номер патенту: 83418
Опубліковано: 10.07.2008
Автори: Батлін Роджер Джон, Батлін Маргарет Енн, Меудт Андреас, Хоуган Філіп Джон
МПК: C07F 5/00, C07D 401/14
Мітки: способом, одержання, гетероарилфенілборонової, одержані, цим, фенілпіридинсульфонаміду, похідних, кислоти, спосіб, сполуки, проміжної
Формула / Реферат:
1. Спосіб одержання сполуки формули Іу якій,Х1 вибирають із О, NR1 або S; іХ2 вибирають із СН або N;де R1 являє собою азотозахисну групу,в якому проводять: послідовну взаємодію сполуки формули IIз(і) метил- або необов'язково заміщеним...
Поліморфні форми похідної 1-піролу, проміжної сполуки для одержання аторвастатину

Номер патенту: 76496
Опубліковано: 15.08.2006
Автори: Сент Кірайі Жужа, Барта Ференц, Надь Кальман, БАРКОЦІ Йожеф, ШІМІГ Дьюла, Верецкейне Донат Дйордьі, Котаі Нагі Петер, Грефф Зольтан
МПК: C07D 405/06
Мітки: одержання, 1-піролу, аторвастатину, похідної, проміжної, сполуки, поліморфні, форми
Формула / Реферат:
1. Кристалічна форма І трет-бутилового ефіру (4R-цис)-6-[2-[3-феніл-4-фенілкарбамоїл-2-(4-фторфеніл)-5-(1-метилетил)-пірол-1-іл]етил]-2,2-диметил-[1,3]діоксан-4-ілоцтової кислоти формули, (I)яка характеризується тим, що має порошкову рентгенограму, представлену в Таблиці 1 і на Фіг. 1, отриману за допомогою
Спосіб одержання 2-оксо-4-метилтіобутанової кислоти, її солей і похідних, а також проміжної сполуки

Номер патенту: 86848
Опубліковано: 25.05.2009
Автори: Бланшард Гілберт, Рей Патрік
МПК: C07C 323/60, C07C 319/00, C07C 323/52, A23K 1/16
Мітки: також, одержання, солей, сполуки, кислоти, похідних, спосіб, 2-оксо-4-метилтіобутанової, проміжної
Формула / Реферат:
1. Спосіб одержання 2-оксо-4-метилтіобутанової кислоти формули (І) і її солей, (І)де R є карбоксильною групою і її солями, який відрізняється тим, що він включає наступні стадії:проведення каталітичного і селективного окислення бут-3-ен-1,2-діолу (ІІ) з одержанням 2-оксо-бут-3-енової кислоти (ІІІ) згідно з наступною реакційною схемою (і):
Спосіб одержання похідної сполуки хіноліну та проміжна сполука

Номер патенту: 65627
Опубліковано: 15.04.2004
Автори: Такада Ясутака, Янагава Йошінобу, Охара Йошіо, Сузукі Мікіо
МПК: C07D 215/14, C07D 215/12
Мітки: хіноліну, похідної, одержання, проміжна, сполуки, сполука, спосіб
Формула / Реферат:
1. Проміжна сполука нітрилу формули (1). (1)2. Спосіб одержання похідного хіноліну (3), що проводять за допомогою сполуки нітрилу, одержуваної в результаті реакції сполуки альдегіду, представленої формулою (2), з діетилціанометилфосфонатом (2) (1) (3).
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Чу-Моєр Маргарет Юхуа, Зембровскі Уільям Джеймс, Маррі Джеррі Ентоні, Міларі Банавара Лакшман
МПК: C07D 451/02, C07D 403/04, C07D 487/04, C07D 513/10, A61P 3/10, C07D 403/14, A61K 31/5377, C07D 417/14, C07D 491/10, C07D 239/42, C07D 471/04, C07D 403/12, C07D 471/08, C07D 405/12, C07D 409/14, C07D 491/20, C07D 401/04, C07D 401/12, C07D 491/04, A61P 43/00, A61P 9/10, A61K 31/53, C07D 491/048, C07D 521/00, C07D 417/12, C07D 451/06, C07D 405/14, C07D 409/12, C07D 498/04, C07D 487/08, C07D 413/12, A61K 31/506, A61K 31/517, C07D 401/14
Мітки: піримідини, спосіб, інгібітори, проміжні, містить, фармацевтична, одержання, композиція, сорбітдегідрогенази, проміжної, сполуки
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...