Похідні (1-арацил-3-арил-3-піперидилетил)піперидину, спосіб їх отримання та фармацевтична композиція, що має у своєму складі такі похідні
Номер патенту: 70986
Опубліковано: 15.11.2004
Автори: ЕМОН-АЛЬТ Ксав'є, Пройетто Вінченцо, Гель Патрік, Дюку Жан Філіпп








Формула / Реферат
1. Сполука формули
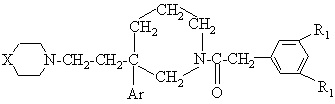 , (I)
, (I)
в якій:
X представляє групу ![]() або
або ![]() ;
;
Аr являє собою феніл, монозаміщений або дизаміщений атомом галогену; (С1-С3)алкіл;
R1 являє собою атом хлору, атом брому, (С1-С3)алкіл або трифлуорметил;
R2 являє собою групу -CR3R4CONR5R6;
R3 та R4 репрезентують однаковий радикал, вибраний з метилу, етилу, н-пропілу або
н-бутилу;
або альтернативно R3 та R4, разом з атомом карбону, до якого вони приєднані, утворюють (С3-С6)циклоалкіл;
R5 та R6, кожний незалежно, репрезентують гідроген; (С1-С3)алкіл;
або альтернативно R5 та R6, разом з атомом нітрогену, до якого вони приєднані, утворюють гетероциклічний радикал, вибраний з 1-азетидинілу, 1-піролідинілу, 1-піперидилу, 4-морфолінілу, 4-тіоморфолінілу або пергідро-1-азепінілу;
а також її солі з органічними або неорганічними кислотами, та їх сольвати та/або гідрати.
2. Сполука за п. 1, яка відрізняється тим, що Ar - 3,4-дихлорфеніл або 3,4-диметилфеніл.
3. Сполука за п. 1, яка відрізняється тим, що замісник R1 являє собою атом хлору, метил, етил або трифлуорметил.
4. Сполука за п. 1, яка відрізняється тим, що X являє собою групу ![]() ,
,
в якій R2 являє собою групу -CR3R4CONR5R6.
5. Сполука за п. 4, яка відрізняється тим, що кожний з R3 та R4 - метил або, разом з атомом карбону, до якого приєднані, вони складають циклогексил.
6. Сполука за п. 1, яка відрізняється тим, що X являє собою групу ![]() ,
,
в якій R2 являє собою групу -CR3R4CONR5R6.
7. Сполука за п. 6, яка відрізняється тим, що кожний з R3 та R4 - метил або, разом з атомом карбону, до якого приєднані, вони складають циклогексил або циклопропіл.
8. Сполука за п. 4 або 6, яка відрізняється тим, що кожний з R5 та R6 - гідроген або метил.
9. Сполука за п. 1, яка відрізняється тим, що має формулу
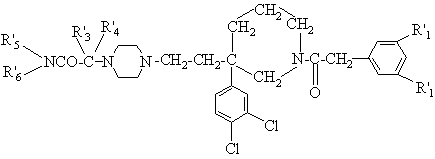 (I'),
(I'),
в якій:
R'1 - атом хлору, метил, етил або трифлуорметил;
кожний з R'3 та R'4 - метил, або альтернативно, разом з атомом карбону, до якого приєднані, вони складають циклогексил;
кожний з R'5 та R'6 - гідроген або метил;
а також її солі з органічними або неорганічними кислотами, та їх сольвати та/або гідрати.
10. Сполука за п. 1, яка відрізняється тим, що має формулу
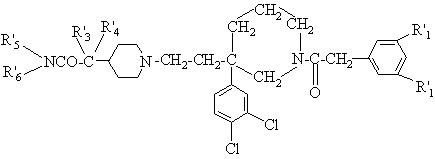 (I''),
(I''),
в якій:
R'1 - атом хлору, метил, етил або трифлуорметил;
кожний з R'3 та R'4 - метил, або альтернативно, разом з атомом карбону, до якого приєднані, вони складають циклогексил або циклопропіл;
кожний з R'5 та R'6 - гідроген або метил;
а також її солі з органічними або неорганічними кислотами, та їх сольвати та/або гідрати.
11. Сполука за будь-яким з пп. 1–10, яка відрізняється тим, що має формулу (I), (I') або (I") у оптично чистій формі.
12. 3-[2-[4-(1-карбамоїл-1-метилетил)-1-піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5-диметилфеніл)ацетил]піперидин, (-) ізомер, його солі та їх сольвати та/або гідрати.
13. 3-[2-[4-(1-N,N-диметилкарбамоїл-1-метилетил)-1-піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5-диметилфеніл)ацетил]піперидин, (-) ізомер, його солі та їх сольвати та/або гідрати.
14. 3-[2-[4-(1-карбамоїл-1-метилетил)-1-піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5-діетилфеніл)ацетил]піперидин, (-) ізомер, його солі та їх сольвати та/або гідрати.
15. 3-[2-[4-(1-карбамоїл-1-метилетил)-1-піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-[3,5-біс(трифлуорметил)феніл]ацетил]піперидин, (+) ізомер, його солі та їх сольвати та/або гідрати.
16. Cполука за будь-яким з пп. 1–15 або одна з її фармацевтично прийнятних солей, їх сольватів та/або гідратів, яка відрізняється тим, що її використовують для виготовлення медичних продуктів, призначених для лікування будь-якої патології, при якій залучено субстанцію Р та рецептори NK1 людини.
17. Сполука за п. 16 або одна з її фармацевтично прийнятних солей, їх сольватів та/або гідратів, яка відрізняється тим, що її використовують для виготовлення медичних продуктів, призначених для лікування патологій респіраторної, шлунково-кишкової, сечової, імунної або серцево-судинної систем або центральної нервової системи, а також болю, мігрені, запалень, нудоти та блювоти, і захворювань шкіри.
18. Сполука за п. 17 або одна з її фармацевтично прийнятних солей, їх сольватів та/або гідратів, яка відрізняється тим, що її використовують для виготовлення медичних продуктів, призначених для лікування обструктивного хронічного бронхіту, астми, нетримання сечі, синдрому подразненого кишечнику, хвороби Крона, виразкового коліту, депресії та тривожності.
19. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що:
1a) сполуку формули:
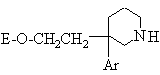 (II),
(II),
в якій Ar визначено для сполуки формули (I) за п. 1, а E - гідроген або
О-протектувальна група, обробляють функціональним похідним кислоти формули:
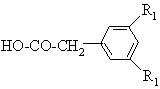 (III),
(III),
в якій R1 визначено для сполуки формули (I) за п. 1, з утворенням сполуки формули:
 IV);
IV);
2а) як варіант, коли E являє собою протектувальну групу, її видаляють дією кислоти або основи для отримання спирту формули:
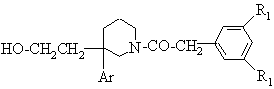 (IV, E = H);
(IV, E = H);
3a) спирт, отриманий на етапі 1a) або на етапі 2a) формули (IV, E = H) обробляють сполукою формули:
Y-SO2-Cl (V),
в якій Y - метил, феніл, толіл або трифлуорметил, для утворення сполуки формули:
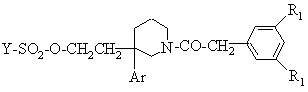 (VI);
(VI);
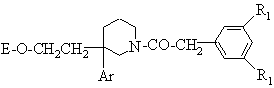
4a) сполуку формули (VI) вводять у взаємодію зі сполукою формули:
![]() (VII),
(VII),
в якій X визначено для сполуки формули (I) за п. 1;
5a) і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою.
20. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що:
1b) сполуку формули:
(II),
в якій Ar визначено для сполуки формули (I) за п. 1, а E - гідроген або
О-протектувальна група, обробляють функціональним похідним кислоти формули:
(III),
в якій R1 визначено для сполуки формули (I) за п. 1, для утворення сполуки формули:
(IV);
як варіант, коли E - протектувальна група, її видаляють дією кислоти або основи для отримання спирту формули:
(IV, E = H);
2b) отриману таким чином сполуку формули (IV, E = H) окиснюють для отримання сполуки формули:
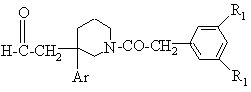 (VIII);
(VIII);
3b) сполуку формули (VIII) вводять у взаємодію зі сполукою формули:
![]() (VII),
(VII),
в якій X визначено для сполуки формули (I) за п. 1, у присутності кислоти, з наступним відновленням утвореної проміжної солі імінію відновником;
4b) і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою.
21. Стереоспецифічний спосіб отримання сполук формули (I) за п. 1, що мають (S)-конфігурацію, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що:
1d) (S)-ізомер сполуки формули:
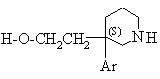 (II*, Е=Н),
(II*, Е=Н),
в якій Ar визначено для сполуки формули (I) за п. 1, обробляють функціональним похідним кислоти формули:
(III),
в якій R1 визначено для сполуки формули (I) за п. 1, для утворення сполуки формули:
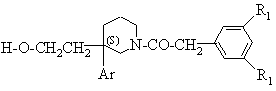 (IV*, Е=Н);
(IV*, Е=Н);
2d) сполуку формули (IV*) окиснюють для утворення сполуки формули:
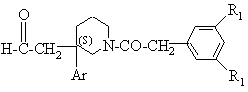 (VIII*);
(VIII*);
3d) сполуку формули (VIII*) вводять у взаємодію зі сполукою формули:
![]() (VII),
(VII),
в якій X визначено для сполуки формули (I) за п. 1, у присутності кислоти, з наступним відновленням утвореної проміжної солі імінію відновником;
4d) і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою.
22. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що:
сполуку формули:
(VI),
в якій:
Ar - феніл, монозаміщений або дизаміщений атомом галогену; (С1-С3)алкіл;
Y - метил, феніл, толіл або трифлуорметил;
R1 являє собою атом хлору, атом брому, (С1-С3)алкіл або трифлуорметил;
вводять у взаємодію зі сполукою формули:
![]() (VII),
(VII),
в якій X визначено для сполуки формули (I) за п. 1,
і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою.
23. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що:
сполуку формули:
(VIII),
в якій Ar та R1 визначено для сполуки формули (I) за п. 1, вводять у взаємодію зі сполукою формули:
![]() (VII),
(VII),
в якій X визначено для сполуки формули (I) за п. 1, у присутності кислоти з наступним відновленням утвореної проміжної солі імінію відновником,
і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою.
24. Стереоспецифічний спосіб отримання сполук формули (I) за п. 1, що мають (S)-конфігурацію, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що:
сполуку формули:
(VIII*),
в якій Ar та R1 визначено для сполуки формули (I) за п. 1, вводять у взаємодію зі сполукою формули:
![]() (VII),
(VII),
в якій X визначено для сполуки формули (I) за п. 1, у присутності кислоти з наступним відновленням утвореної проміжної солі імінію відновником,
і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою.
25. Сполука формули:
![]() (VII),
(VII),
в якій:
X являє собою групу ![]() або
або ![]() ;
;
R2 являє собою групу -CR3R4CONR5R6;
R3 та R4 репрезентують однаковий радикал, вибраний з метилу, етилу, н-пропілу або н-бутилу;
або R3 та R4 разом з атомом карбону, до якого вони приєднані, утворюють
(С3-С6)циклоалкіл;
R5 та R6, кожний незалежно, репрезентують гідроген; (С1-С3)алкіл;
або альтернативно R5 та R6, разом з атомом нітрогену, до якого вони приєднані, утворюють гетероциклічний радикал, вибраний з 1-азетидинілу, 1-піролідинілу, 1-піперидилу, 4-морфолінілу, 4-тіоморфолінілу або пергідро-1-азепінілу,
та її солі з органічними або неорганічними кислотами.
26. Фармацевтична композиція, що містить як активний інгредієнт сполуку за будь-яким з пп. 1–15 або одну з її фармацевтично прийнятних солей, їх сольватів та/або гідратів.
27. Фармацевтична композиція за п. 26, яка відрізняється тим, що містить 0,1–1000 мг активного інгредієнта у формі дозованої одиниці, в якій активний інгредієнт змішаний з щонайменше одним фармацевтичним ексципієнтом.
Текст
1. Сполука формули CH2 CH2 CH 2 R1 3 R' 5 R' 6 CH 2 R'3 R'4 NCO-C CH 2 N -CH2-CH 2-C CH2 CH2 R' 1 N-C-CH 2 O R' 1 70986 4 в якій Ar визначено для сполуки формули (I) за п. 1, а E - гідроген або О-протектувальна група, обробляють функціональним похідним кислоти формули: R1 Cl Cl (I''), в якій: R'1 - атом хлор у, метил, етил або трифлуорметил; кожний з R'3 та R'4 - метил, або альтернативно, разом з атомом карбону, до якого приєднані, вони складають циклогексил або циклопропіл; кожний з R'5 та R'6 - гідроген або метил; а також її солі з органічними або неорганічними кислотами, та їх сольвати та/або гідрати. 11. Сполука за будь-яким з пп. 1–10, яка відрізняється тим, що має формулу (I), (I') або (I") у оптично чистій формі. 12. 3-[2-[4-(1-карбамоїл-1-метилетил)-1піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5диметилфеніл)ацетил]піперидин, (-) ізомер, його солі та їх сольвати та/або гідрати. 13. 3-[2-[4-(1-N,N-диметилкарбамоїл-1-метилетил)1-піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5диметилфеніл)ацетил]піперидин, (-) ізомер, його солі та їх сольвати та/або гідрати. 14. 3-[2-[4-(1-карбамоїл-1-метилетил)-1піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5діетилфеніл)ацетил]піперидин, (-) ізомер, його солі та їх сольвати та/або гідрати. 15. 3-[2-[4-(1-карбамоїл-1-метилетил)-1піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-[3,5біс(трифлуорметил)феніл]ацетил]піперидин, (+) ізомер, його солі та їх сольвати та/або гідрати. 16. Cполука за будь-яким з пп. 1–15 або одна з її фармацевтично прийнятних солей, їх сольватів та/або гідратів, яка відрізняється тим, що її використовують для виготовлення медичних продуктів, призначених для лікування будь-якої патології, при якій залучено субстанцію Р та рецептори NK1 людини. 17. Сполука за п. 16 або одна з її фармацевтично прийнятних солей, їх сольватів та/або гідратів, яка відрізняє ться тим, що її використовують для виготовлення медичних продуктів, призначених для лікування патологій респіраторної, шлунковокишкової, сечової, імунної або серцево-судинної систем або центральної нервової системи, а також болю, мігрені, запалень, нудоти та блювоти, і захворювань шкіри. 18. Сполука за п. 17 або одна з її фармацевтично прийнятних солей, їх сольватів та/або гідратів, яка відрізняє ться тим, що її використовують для виготовлення медичних продуктів, призначених для лікування обструктивного хронічного бронхіту, астми, нетримання сечі, синдрому подразненого кишечнику, хвороби Крона, виразкового коліту, депресії та тривожності. 19. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що: 1a) сполуку формули: NH E-O-CH2 CH2 Ar (II), HO-CO-CH2 R1 (III), в якій R1 визначено для сполуки формули (I) за п. 1, з утворенням сполуки формули: R1 N -CO-CH2 E-O-CH2CH2 R 1 (IV); 2а) як варіант, коли E являє собою протектувальну груп у, її видаляють дією кислоти або основи для отримання спирту формули: Ar R1 HO-CH2 CH2 N -CO-CH2 Ar R1 (IV, E=H); 3a) спирт, отриманий на етапі 1a) або на етапі 2a) формули (IV, E = H) обробляють сполукою формули: Y-SO 2-Cl (V), в якій Y - метил, феніл, толіл або трифлуорметил, для утворення сполуки формули: R1 N -CO-CH 2 Y-SO2 -O-CH 2CH2 Ar R1 (VI); 4a) сполуку формули (VI) вводять у взаємодію зі сполукою формули: X NH (VII), в якій X визначено для сполуки формули (I) за п. 1; 5a) і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою. 20. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що: 1b) сполуку формули: E-O-CH2CH2 NH Ar (II), в якій Ar визначено для сполуки формули (I) за п. 1, а E - гідроген або О-протектувальна група, обробляють функціональним похідним кислоти формули: R1 HO-CO-CH2 R1 (III), в якій R1 визначено для сполуки формули (I) за п. 1, для утворення сполуки формули: 5 70986 6 R1 N-CO-CH 2 E-O-CH 2CH2 R1 N-CO-CH 2 Ar R1 (IV, E=H); 2b) отриману таким чином сполуку формули (IV, E = H) окиснюють для отримання сполуки формули: R1 O N-CO-CH2 H-C-CH2 R1 Ar (VIII); 3b) сполуку формули (VIII) вводять у взаємодію зі сполукою формули: X N-CO-CH2 Ar R1 (VIII*); 3d) сполуку формули (VIII*) вводять у взаємодію зі сполукою формули: X NH (VII), в якій X визначено для сполуки формули (I) за п. 1, у присутності кислоти, з наступним відновленням утвореної проміжної солі імінію відновником; 4d) і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою. 22. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що: сполуку формули: R1 N -CO-CH 2 Y-SO2 -O-CH 2CH2 Ar NH (VII), в якій X визначено для сполуки формули (I) за п. 1, у присутності кислоти, з наступним відновленням утвореної проміжної солі імінію відновником; 4b) і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою. 21. Стереоспецифічний спосіб отримання сполук формули (I) за п. 1, що мають (S)-конфігурацію, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що: 1d) (S)-ізомер сполуки формули: (S) H-O-CH 2CH 2 NH Ar (II*, Е=Н), в якій Ar визначено для сполуки формули (I) за п. 1, обробляють функціональним похідним кислоти формули: R1 в якій R1 визначено для сполуки формули (I) за п. 1, для утворення сполуки формули: R1 Ar X NH (VII), в якій X визначено для сполуки формули (I) за п. 1, і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою. 23. Спосіб отримання сполук формули (I) за п. 1, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що: сполуку формули: R1 O N -CO-CH2 H-C-CH2 R1 (VIII), в якій Ar та R1 визначено для сполуки формули (I) за п. 1, вводять у взаємодію зі сполукою формули: R1 (III), (S) R1 (VI), в якій: Ar - феніл, монозаміщений або дизаміщений атомом галогену; (С1-С3)алкіл; Y - метил, феніл, толіл або трифлуорметил; R1 являє собою атом хлору, атом брому, (С1С3)алкіл або трифлуорметил; вводять у взаємодію зі сполукою формули: Ar HO-CO-CH2 H-O-CH2CH 2 (S) H-C-CH2 Ar R1 (IV); як варіант, коли E - протектувальна група, її видаляють дією кислоти або основи для отримання спирту формули: HO-CH 2CH2 R1 O N -CO-CH2 R1 (IV*, Е=Н); 2d) сполуку формули (IV*) окиснюють для утворення сполуки формули: X NH (VII), в якій X визначено для сполуки формули (I) за п. 1, у присутності кислоти з наступним відновленням утвореної проміжної солі імінію відновником, і, як варіант, отриману таким чином сполуку перетворюють у одну з її солей з органічною або неорганічною кислотою. 24. Стереоспецифічний спосіб отримання сполук формули (I) за п. 1, що мають (S)-конфігурацію, їх солей та їх сольватів та/або гідратів, який відрізняється тим, що: сполуку формули: 7 70986 8 R3 та R4 репрезентують однаковий радикал, вибO раний з метилу, етилу, н-пропілу або н-бутилу; (S) N-CO-CH2 або R3 та R4 разом з атомом карбону, до якого H-C-CH2 вони приєднані, утворюють Ar R1 (С3-С6)циклоалкіл; (VIII*), R5 та R6, кожний незалежно, репрезентують гідров якій Ar та R1 визначено для сполуки формули (I) ген; (С1-С3)алкіл; за п. 1, вводять у взаємодію зі сполукою формули: або альтернативно R5 та R6, разом з атомом нітроX гену, до якого вони приєднані, утворюють гетероNH (VII), циклічний радикал, вибраний з 1-азетидинілу, 1в якій X визначено для сполуки формули (I) за п. 1, піролідинілу, 1-піперидилу, 4-морфолінілу, 4у присутності кислоти з наступним відновленням тіоморфолінілу або пергідро-1-азепінілу, утвореної проміжної солі імінію відновником, та її солі з органічними або неорганічними кислоі, як варіант, отриману таким чином сполуку перетами. творюють у одну з її солей з органічною або неор26. Фармацевтична композиція, що містить як акганічною кислотою. тивний інгредієнт сполуку за будь-яким з пп. 1–15 25. Сполука формули: або одну з її фармацевтично прийнятних солей, їх сольватів та/або гідратів. X NH 27. Фармацевтична композиція за п. 26, яка відрі(VII), зняється тим, що містить 0,1–1000 мг активного в якій: інгредієнта у формі дозованої одиниці, в якій активний інгредієнт змішаний з щонайменше одним R2-CH R2 -N X являє собою гр упу або ; фармацевтичним ексципієнтом. R2 являє собою групу -CR3R4CONR5R6; R1 Представлений винахід стосується нових піперидинових похідних, способу їх виго товлення та фармацевтичних композицій, що містять їх як активний інгредієнт. Переважно, представлений винахід стосується нових піперидинових похідних для терапевтичного використання при патологічних станах, в яких залучено систему тахікінінів, наприклад, без обмеження: біль (L. Urban et al., TINS, 1994, 17 , 432438; L.Seguin et al.. Pain, 1995, 61 , 325-343; S.H. Buck, 1994, The Tachykinin Receptors, Humana Press, Totowa, New-Jersey), алергія та запалення (S.H. Buck, 1994, The Tachykinin Receptors, Humana Press, Totowa, New-Jersey), шлунковокишкові розлади (P. Holzer та U. Holzer-Petsche, Pharmacol. Ther., 1997, 73, 173-217 та 219-263), респіраторні розлади (J. Mizrahi et al., Pharmacology, 1982, 25, 39-50; С Advenier et al., Eur. Respir. J., 1997, 10, 1892-1906; С Advenier та X. EmondsAlt, Pulmonary Pharmacol., 1996., 9, 329-333), сечові розлади (S.H. Buck, 1994, The Tachykinin Receptors, Humana Press, Totowa, New-Jersey; C.A. Maggi, Progress у Neurobiology, 1995, 45, 1-98), неврологічні та нейропсихіатричні розлади (C.A. Maggi et al., J. Autonomic Pharmacol., 1993, 13, 2393; M. Otsuka та К. Yoshioka, Physiol. Rev. 1993, 73 , 229-308). В останні роки було проведено багато досліджень на тахікінінах та їх рецепторах. Тахікініни розподілені як у центральній нервовій системі, так і у периферійній нервовій системі. Рецептори тахікінінів розпізнано та розподілено на 3 типи: NK1 NK2 та NK3. Субстанція Ρ (SP) є ендогенним лігандом рецепторів NK1 нейрокінін A (NKa) є лігандом рецепторів NK2, а нейрокінін В (NKB) є лігандом рецепторів NK3. Рецептори NK1 NK2 та NK3 виявлено у різних видах. В оглядах, зроблених C.A. Maggi et al. (J. Autonomic Pharmacol., 1993, 13 , 23-93) і D. Regoli et al. (Pharmacol. Rev., 1994, 46 , 551-599), розглянуто рецептори тахікінінів та їх антагоністи, а також представлено фармакологічне дослідження і застосування їх у терапії людини. Багато патентів і заявок на винаходи описують сполуки, що є активними на рецепторах тахікінінів. Отже, Європейська заявка на винахід 0 512 901 стосується сполук формули: в якій, зокрема: Q' представляє атом оксигену або два атоми гідрогену, Τ=-С(О)- або -СН2-, а Υ, Аr', Ζ', m', н', р' та q мають різні значення. Заявка на винахід ЕР 0 714 891 стосується сполук формули: в якій: - р"=1, 2 або 3; - m" та n" є незалежно 0-6; - W, Ra , Rb , Rc, Rd та Re мають різні значення. Зараз виявлено нові сполуки, що мають дуже сильну спорідненість і високу селективність до рецепторів людини ΝΚ1 субстанції Р, і які є антагоністами вказаних рецепторів. Крім того, сполуки згідно з представленим винаходом мають гарну біопридатність при пероральному застосуванні. 9 70986 10 Ці сполуки можна використовувати для виготовлення медичних продуктів, придатних для лікування будь-якої патології, при якій залучено субстанцію Ρ та рецептори NK1, зокрема для лікування патологій респіраторної, шлунковокишкової, сечової, імунної, серцево-судинної та центральної нервової систем а також для лікуванв якій X та R1 визначено для сполуки формули ня болю, мігрені, запалень, нудоти та блювоти, захворювань шкіри. (І), а також їх солі з органічними або неорганічниОтже, згідно з одним з аспектів винаходу зами кислотами, та їх сольвати та/або гідрати. Згідно з представленим винаходом кращими пропоновано сполуки формули: сполуками формули (I) є сполуки, в яких Аг представляє 3,4-дихлорфеніл або 3,4-диметилфеніл. Згідно з представленим винаходом кращими сполуками формули (І) є сполуки, в яких замісники R1 репрезентують атом хлор у, метил, етил або в якій: трифлуорметил. Аr представляє феніл, монозаміщений або диЗгідно з представленим винаходом кращими заміщений атомом галогену; (С1 -С3) алкіл; сполуками формули (І) є сполуки, в яких X представляє груп у R2-N=, в якій R2 представляє групу X представляє груп у гр упу ; CR3R4CONR5R6. Зокрема, кращими сполуками є сполуки, в яких груп у кожний з R3 та R4 репрезентує метил або альтерR1 представляє атом хлору, атом брому, (С1нативно, разом з атомом карбону, до якого приєдС3) алкіл або трифлуорметил; нані, вони складають циклогексил. Зокрема, споR2 представляє групу -CR3R4CONR5R6; луки, що також є кращими, є сполуками, в яких R3 та R4 репрезентують однаковий радикал, кожний з R6 та R6 репрезентує гідроген або метил. вибраний з метилу, е тилу, н-пропілу або н-бутилу; Згідно з представленим винаходом кращими або альтернативно R3 та R4, разом з атомом сполуками формули (І) є сполуки, в яких X предкарбону, до якого вони приєднані, утворюють (С3ставляє груп у R2-CH=, в якій R2 представляє групу С6) циклоалкіл; -CR3R4CONR5R6. R5 та R6, кожний незалежно, репрезентують гіЗокрема, кращими сполуками є сполуки, в яких дроген; (С1 –С3) алкіл; кожний з R3 та R4 репрезентує метил або альтерабо альтернативно R5 та R6, разом з атомом нативно, разом з атомом карбону, до якого приєднітрогену, до якого вони приєднані, утворюють нані, вони складають циклопропіл або циклогекгетероциклічний радикал, вибраний з 1сил. Зокрема, сполуки, що також є кращими, є азетидинілу, 1-піролідинілу, 1-піперидилу, 4сполуками, в яких кожний з R5 та R6 репрезентує морфолінілу, 4-тіоморфолінілу або пергідро-1гідроген або метил. азепінілу; а також їх можливі солі з органічними Згідно з представленим винаходом сполуки, або неорганічними кислотами, та їх сольвати що є кращими, є сполуками формули: та/або гідрати. Сполуки формули (І) згідно з винаходом включають оптично чисті ізомери та їх суміші у будьякому співвідношенні. Можна утворювати солі сполук формули (І). Ці солі включають солі з органічними або з неорганічними кислотами, що дозволяють придатне розділення або кристалізацію сполук формули (І), наприклад, пікринова або щавлева кислота, або в якій: оптично активною кислотою, наприклад, мигдальR'1 представляє атом хлору, метил, етил або трифлуорметил; ною або камфорсульфоновою кислотою, та кислокожний з R'3 та R'4 репрезентує метил, або тами, що утворюють фармацевтично прийнятні альтернативно, разом з атомом карбону, до якого солі, наприклад, гідрохлорид, гідробромід, сульфат, гідросульфат, дигідрофосфат, метансульфоприєднані, вони складають циклогексил; кожний з R'5 та R' 6 репрезентує гідроген або метил; нат, метилсульфат, оксалат, малеат, фумарат, а також їх солі з органічними або неоргасукцинат, 2-нафталінсульфонат, глкжонат, цитрат, нічними кислотами, та їх сольвати та/або гідрати. бензолсульфонат або пара-толуолсульфонат. Згідно з представленим винаходом кращими споТермін "галоген" означає атом хлору, брому, флуору чи йоду. луками є сполуки формули: У представленому описі, алкілгрупи є лінійними чи розгалуженими. Згідно з представленим винаходом кращими сполуками є сполуки формули: в якій: 11 70986 12 R'1 представляє атом хлору, метил, етил або три3-[2-[4-(1-карбамоїлциклопропіл)-1флуорметил; кожний з R'3 та R'4 репрезентує мепіперидил]етил]3-(3,4-дихлорфеніл)-1-[2-[3,5тил, або альтернативно, разом з атомом карбону, біс(трифлуорметил)-феніл]ацетил]піперидин, (+) до якого приєднані, вони складають циклогексил ізомер; або циклопропіл; кожний з R'5 та R'6 репрезентує 3-[2-[4-(1-карбамоїлциклопропіл)-1гідроген або метил; а також їх солі з органічними піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5або неорганічними кислотами, та їх сольвати дихлорфеніл) ацетил] піперидин, (+) ізомер; та/або гідрати. 3-[2-[4-(1-карбамоїл-1-метилетил)-1Згідно з представленим винаходом кращими піперазиніл]-етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5сполуками є сполуки формул (І), (I') та (І") у оптичдіетилфеніл)-ацетил]піперидин, (-) ізомер; но чистій формі. 3-[2-[4-(1-карбамоїл-1-метилетил)-1Нижченаведені сполуки: піперидил]-етил]-3-(3,4-диметилфеніл)-1-[2-(3,53-[2-[4-(1-карбамоїл-1-метилкарбамоїл)-1дихлорфеніл) ацетил]піперидин; піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,53-[2-[4-(1-карбамоїл-1-метилетил)-1диметилфеніл)ацетил]піперидин, (-) ізомер; піперазиніл]-етил]-3-(3,4-диметиПалфеніл)-1-[23-[2-[4-(1-карбамоїл-1-метилетил)-1(3,5-дихлорфеніл)-ацетил]піпериди; а також їх сопіперазиніл]-етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5лі, та їх сольвати та/або гідрати, є особливо диметилфеніл)ацетил]піперидин, (-) ізомер; Згідно з ще одним аспектом представлений 3-[2-[4-(1-N,N-диметилкарбамоїл-1винахід стосується способу виготовлення сполук метилетил)-1-піперазиніл]етил]-3-(3,4формули (І), їх солей та їх сольватів та/або гідрадихлорфеніл)-1 -[2-(3,5тів, який відрізняється тим, що: 1а) сполука фордиметилфеніл)ацетил]піперидин, (-) ізомер; мули: 3-[2-[4-(1-карбамоїл-1-метилетил)-1піперидил]-етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5дихлорфеніл)ацетил]піперидин, (-) ізомер; 3-[2-[4-(1-карбамоїл-1-метилетил)-1піперидил]-етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5біс(трифлуорметил)феніл]ацетил]піперидин, (+) в якій Аr визначено для сполуки формули (І), а ізомер; Е представляє гідроген або О-протектувальну гру3-[2-[4-(1-карбамоїлциклогексил)-1пу, обробляють функціональним похідним кислоти піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5формули: диметилфеніл)ацетил]-піперидин, (-) ізомер; 3-[2-[4-(1-карбамоїлциклогексил)-1піперазиніл]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5диметилфеніл)ацетил]-піперидин, (-) ізомер; 3-[2-[4-(1-карбамотциклогексил)піперидин-1іл]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5дихлорфеніл)ацетил]-піперидин, (-) ізомер; в якій R1 визначено для сполуки формули (І), з 3-[2-[4-(1-карбамоїл-1-метилетил)піперазин-1утворенням сполуки формули: іл]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5дихлорфеніл)ацетил]піперидин, (+) ізомер; 3-[2-[4-(1-N,N-диметилкарбамоїл-1метилетил)-1-піперазиніл]етил]-3-(3,4дихлорфеніл)-1-[2-(3,5дихлорфеніл)ацетил]піперидин, (+) ізомер; 3-[2-[4-(1-карбамоїлциклогексил)-12а) як варіант, коли Ε представляє протектупіперазиніл]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5вальну груп у, її видаляють дією кислоти або оснодихлорфеніл)ацетил]-піперидин, (+) ізомер; ви, з одержанням спирту формули: 3-[2-[4-(1-карбамоїлциклогексил)-1піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-[3,5біс(трифлуорметил)-феніл]ацетил]піперидин, (+) ізомер; 3-[2-[4-(1-карбамоїлциклогексил)-1піперазиніл]етил]-3-(3,4-дихлорфеніл)-1-[2-[3,5За) отриманий на етапі 1а) або на етапі 2а) біс(трифлуорметил)-феніл]ацетил]піперидин, (+) спирт формули (IV, Е=Н ) обробляють сполукою ізомер; формули: 3-(2-[4-(1-N,N-диметилкарбамоїл-1Y-SO 2-CI (V) метилетил)-1-піперидил]етил]-3-(3,4в якій Υ представляє метил, феніл, толіл або дихлорфеніл)-1-[2-(3,5трифлуорметил, з одержанням сполуки формули: диметилфеніл)ацетил]піперидин, (-) ізомер; 3-[2-[4-(1-карбамоїл-1-метилетил)-1піперидил]-етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5діетилфеніл)-ацетил]піперидин, (-) ізомер; 3-[2-[4-(1-карбамоїлциклопропіл)-1піперидил]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5диметилфеніл)ацетил)-піперидин, (-) ізомер; 13 70986 14 4а) сполука формули (VI) реагує зі сполукою етапі 4а) зі сполукою формули (VII). Реакцію проформули: водять в такому інертному розчиннику, як Ν,Νдиметилформамід, ацетонітрил, метиленхлорид, толуол або ізопропанол, та у присутності або відсутності основи. Коли використовують основу, її вибирають з органічних основ, як-то триетиламін, в якій X визначено для сполуки формули (І); Ν,Ν-діізопропілетиламін або N-метилморфолін, та 5а) і, як варіант, отриману таким чином сполуку з карбонатів або гідрокарбонатів лужних металів, перетворюють у одну з її солей з органічною або як-то карбонат калію, карбонат натрію або гідроканеорганічною кислотою. рбонат натрію. У відсутності основи реакцію проКоли Ε представляє О-протектувальну гр упу, водять, використовуючи надлишок сполуки форцю груп у вибирають зі звичайних Омули (VII), та у присутності йодиду лужного протектувальних гр уп, що добре відомі фахівцям, металу, як-то йодиду калію або йодиду натрію. наприклад, 2-тетрагідропіраніл, бензоїл або (С1 Реакцію проводять при температурі між кімнатною С4)алкілкарбоніл. температурою та 100°С. На етапі 1а), функціональне похідне кислоти Згідно з одним варіантом способу: (III), що використовують, само є кислотою, або 1b) його здійснюють як на етапі 1а) та, як варіальтернативно, одним з її функціональних похідант, як на етапі 2а); них, які реагують з амінами, наприклад, ангідри2b) отриману таким чином сполуку формули дом, змішаним ангідридом, хлорангідридом або (IV, Ε=Н ) окиснюють для виготовлення сполуки активованим естером, наприклад, паранітрофеніловий естер. Коли кислоту формули (III) використовують як таку, спосіб здійснюють у присутності використовуваного у хімії пептидів сполучального засобу, формули: наприклад, 1,3-дициклогексилкарбодіімід або гек3b) сполука формули (VIII) реагує зі сполукою сафлуорфосфа т бензотриазол-1ілокситвизначеної вище формули (VII) у присутності кисрис(диметиламіно)фосфонію у присутності основи, лоти, з наступним відновленням відновником наприклад, триетиламін або Ν,Νутвореної проміжної солі імінію; діізопропілетиламін, в такому інертному розчинни4b) і, як варіант, отриману таким чином сполуку, як дихлорметан або Ν,Ν-диметилформамід, ку перетворюють у одну з її солей з органічною при температурі від 0°С до кімнатної. або неорганічною кислотою. Коли використовують хлорангідрид, реакцію Згідно з варіантом способу, на етапі 2b), спирт проводять в такому інертному розчиннику, як дихформули (IV, Ε=Η ) окиснюють, одержавши альделорметан або бензол, у присутності основи, нагід формули (VIII). Реакцію окиснення проводять, приклад, триетиламін або N-метилморфолін, при використовуючи, наприклад, оксалілхлорид, диметемпературі від -60°С до кімнатної. тилсульфоксид та триетиламін у такому розчинниОтриману таким чином сполуку формули (IV), ку, як дихлорметан при температурі від -78°С до як варіант, депротектують на етапі 2а) згідно з вікімнатної. домими фахівцям способами. Наприклад, коли Ε Далі, на етапі 3b), сполука формули (VII) реапредставляє 2-тетрагідропіраніл, депротектування гує з альдегідом формули (VIII) у присутності такої проводять кислотним гідролізом, використовуючи кислоти, як оцтова кислота, в такому інертному гідрохлоридну кислоту у такому розчиннику, як розчиннику, як метанол або дихлорметан, з утвоефір, метанол або суміш цих розчинників, або виренням in situ проміжного іміну, який відновлюють користовуючи п-толуолсульфонат піридинію у тахімічно, використовуючи, наприклад, ціаноборогідкому розчиннику, як метанол, або альтернативно, рид натрію або триацетоксиборогідрид натрію, або використовуючи смолу Amberlyst® у такому розвідновлюють каталітично, використовуючи водень чиннику, як метанол. Реакцію проводять при темта каталізатор, як-то паладій-на-вугіллі або Нікель пературі між кімнатною та температурою кипіння Ренея (Raney®). розчиннику. Коли Ε представляє бензоїл або (С1– Під кінець, отримують сполуки формули (І) згіС4 алкілкарбонільну групу, депротектування продно з винаходом. водять гідролізом у лужному середовищі, викорисОтримані таким чином сполуки формули (І) товуючи, наприклад, гідроксид лужного металу, виділяють у формі вільної основи або у формі солі наприклад, гідроксид натрію, гідроксид калію або звичайними способами. гідроксид літію, в такому інертному розчиннику, як Коли сполуки формули (І) отримують у формі вода, метанол, етанол, діоксан або суміш цих розвільної основи, сіль одержують обробкою вибрас чинників, при температурі від 0 С до температури ною кислотою в органічному розчиннику. Обробка кипіння розчиннику. вільної основи у розчині, наприклад, у е фірі, як-то На етапі За), реакцію спирту формули (IV, діетиловий етер, у спирті, як-то 2-пропанол, у ацеΕ=Η ) з сульфонілхлоридом формули (V) провотоні, у ди хлорметані, або у етилацетаті розчином дять у присутності основи, наприклад, триетилавибраної кислоти у одному з вищезазначених розмін, піридин, Ν,Ν-діізопропілетиламін або Nчинників дає відповідну сіль, яку виділяють звиметилморфолін, в такому інертному розчиннику, як чайними способами. дихлорметан, бензол або толуол, при температурі Отже, наприклад, виготовляютьгідрохлорид, від -20°С до температури кипіння розчиннику. гідробромід, сульфат, гідросульфат, дигідрофосОтримана таким чином сполука (VI) реагує на фат, метансульфонат, метилсульфат, оксалат, 15 70986 16 малеат, сукцинат, фумарат, 2-нафталінсульфонат, бензолсульфонат, пара-толуолсульфонат або глюконат. Після закінчення реакції, сполуки формули (І) можна виділити у формі одної з її солей, наприклад, гідрохлориду або оксалату; в цьому випадку, якщо необхідно, вільну основу можна виготовити нейтралізацією вказаної солі органічною або неорганічною основою, як-то гідроксид натрію або триетиламін, або карбонатом або гідрокарбонатом лужного металу, як-то карбонат або гідрокарбонат натрію чи калію. Сполуки формули (II) виготовляють відомими способами, зокрема, описаними у заявках на винаходи ЕР-А-0 512 901, ЕР-А-0 591 040 або ЕР-А-0 714 891. На етапі a2 схеми 2 сполука 1 реагує з кетоСполуки формули (III) комерційно доступні або ном формули (XVI), у присутності 2їх виготовляють згідно з відомими способами. гідроксіізобутиронітрилу згідно зі способом, описаОтже, наприклад, сполуки формули (III) вигоним у Eur. J. Med. Chem., 1990, 25 , 609-615. товляють згідно з нижченаведеною схемою 1. Отримане таким чином нітрильне похідне формули (XVII) гідролізують на етапі Ь2 згідно зі способами, що відомі фахівцям, для одержання кислотного похідного формули (XVIII). Кислота (XVIII) реагує на етапі c 2 з аміном формули (XIX) згідно зі звичайними способами сполучення пептидів, для одержання похідного (XXI). Альтернативно, на етапі d2, нітрильне похідне формули (XVII) гідролізують згідно з відомими способами, одержавши карбоксамідне похідне формули (XX), яке, як варіант, депротектують на етапі е2, згідно зі звичайними способами, одержаЕтапи a1 та b1 схеми 1 проводять згідно зі вши сполуку (VII), в якій R5=R6=Η. способами, описаними у J. Am. Chem. бос, 1941, На етапі f2 реакцією сполуки формули (XX) у присутності сильної основи, відповідно, з (С163 , 3280-3282. С3)алкілгалогенідом, або послідовно з двома (С1На етапі с1 естер формули (XII) виготовляють С3)алкілгалогенідами, або з дигалогенідом формуз кислоти формули (XI) згідно з відомими фахівли Hal-R 5-R6-Hal згідно зі звичайними способами цям способами. алкілування виготовляють сполуку формули (XXI), Отриманий таким чином естер (XII) відновлюв якій, відповідно, R5 представляє (С1-С3)алкіл, a ють на етапі d1 до спирту формули (XIII) згідно зі R6=Η, або кожний з R5 та R6 незалежно репрезенспособами, що відомі фахі вцям. тують (С1 -С3)алкіл, або R5 та R6, разом з атомом Етапи е1 та f1 проводять згідно зі способами, нітрогену, до якого вони приєднані, складають геописаними у J. Med. Chem,, 1973, 16, 684-687. тероцикп. Отримані таким чином фенілацетонітрильні Отриману таким чином сполуку (XXI) депротепохідні формули (XV) гідролізують на етапі g1 у ктують на етапі g2, згідно з відомими способами, сполуки формули (III) згідно зі способами, описадля одержання очікуваної сполуки (VII). ними у J. Org. Chem., 1968, 33, 4288 або у ЕР-А-0 Сполуки формули (VII), в яких X представляє 714 891. груп у R2-CH=, в якій R2 представляє групу Бромопохідні формули (IX) відомі або їх вигоCR3R4CONR5R6, виготовляють згідно з нижченатовляють згідно з відомими способами, як-то опиведеною схемою 3. саними у J. Org.Chem., 1971, 36(1) , 193-196, або у J. Am. Chem. Soc, 1941, 63 , 3280-3282. Сполуки формули (VII), в яких X представляє груп у R2-N=, в якій R2 представляє групу CR3R4CONR5R6, виготовляють згідно з нижченаведеною схемою 2: 17 70986 На етапі a3 схеми 3, реакція сполуки 2 у присутності сильної основи, як-то гідрид натрію або амід натрію, з, відповідно, лінійним (С1С4)алкілгалогенідом, або з дигалогенідом формули Hal(CH2)m-Hal, в якому m=2-5, a Hal представляє атом галогену, в такому інертному розчиннику, як Ν,Ν-диметилформамід або дихлорметан, при температурі від 0°С до кімнатної, згідно зі звичайними способами алкілування дає сполуку формули (XXII), в якій, відповідно, кожний з R3 та R4 репрезентує лінійний (С1-С4)алкіл, або, разом з атомом карбону, до якого приєднані, вони складають (С3С6)циклоалкіл. Отримане таким чином нітрильне похідне (XXII) гідролізують на етапі b3, згідно зі способами, що відомі фахівцям, одержавши карбоксамідне похідне (ХХІІІ). Як варіант, на етапі сЗ, піридинове кільце гідрують у присутності каталізатору, як-то оксид платини, згідно з відомими способами, з утворенням сполуки формули (VII), в якій R5 та R6= Η. На етапі d5 реакція алкілування сполуки формули (ХХІІІ) згідно зі звичайними способами, що описані попередньо, з наступним відновленням засобами звичайного каталітичного гідрування отриманої таким чином сполуки (XXIV) дає сполуку формули (VII), в якій R5 та/або R6¹Η. Сполуки формули (VII), в яких X представляє груп у –CH-CR3R4CONTR5R6 можна також отримати згідно з нижченаведеною схемою 4. Схема 4 На етапі a4 схеми 4, реакція сполуки 3 з придатним органолітієвим або органомагнієвим похідним, наприклад, метиллітієм, хлоридом етилмагнію, хлоридом пропілмагнію або хлоридом пентан-1,5-ди(магнію), згідно зі способами, що описано у ЕР-А-0 625 509, дає спирт формули (XXV). Отриманий таким чином спирт (XXV) окиснюють на етапі b4 у кислоту формули (XXVI) згідно зі способом, що описано у Helvetica Chimica Acta, 1972, 55 (7), 2439. Кислота (XXVI) реагує на етапі c 4 з аміном формули (XIX) згідно зі звичайними способами сполучення пептидів, для одержання сполуки (XXVII). Сполуку (XXVII) депротектують на етапі d4 , згідно з відомими способами, одержуючи очікувану сполуку (VII). 18 Сполуку 3 виготовляють реакцією етилізоніпекотату з бензилбромідом, у присутності основи, згідно зі звичайними способами алкілування. Сполуки формули (VII) є новими та охоплені рамками винаходу. Отже, згідно з ще одним аспектом винаходу, предметом винаходу є сполука формули: в якій: X представляє R2-N=, R 2-CH=, R2 представляє групу -CR3R4CONR5R6 R3 та R4 репрезентують однаковий радикал, вибраний з метилу, е тилу, н-пропілу або н-бутилу; або R3 та R4, разом з атомом карбону, до якого вони приєднані, складають (С3-С6)циклоалкіл; кожний з R5тa R6 незалежно, репрезентує гідроген; (С1-С3)алкіл; або альтернативно, R5 та R6, разом з атомом нітрогену, до якого вони приєднані, складають гетероциклічний радикал, вибраний з 1-азетидинілу, 1-піролідинілу, 1-піперидилу, 4-морфолінілу, 4тіоморфолінілу або пергідро-1-азепінілу; та їх солі з органічними або неорганічними кислотами. Розділення рацемічних сумішей сполук формули (І) робить можливим виділити енантіомери формули в якій: позначка "*" означає, що помічений нею атом карбону має певну абсолютну конфігурацію (S) або (R); X, Аr та R1 визначено для сполуки формули (І); а також їх можливі солі з органічними або неорганічними кислотами, та їх сольвати та/або гідрати. Однак, краще проводити розділення рацемічних сумішей з інтермедіату формули (II, Е=Н), який є корисним для виготовлення сполук формули (І), як описано в заявках на винаходи ЕР-А-0 512 901, ЕР-А-0 612 716 та ЕР-А-0 591 040. Згідно з ще одним аспектом представлений винахід стосується стереоспецифічного способу виготовлення сполуки формули (І), що має (З)конфігурацію, її солей та їх сольватів та/або гідратів, який відрізняється тим, що: Id) (S)-iзoмep сполуки формули: в якій Аr визначено для сполуки формули (І), обробляють функціональним похідним кислоти формули: в якій R1 визначено для сполуки формули (І), з утворенням сполуки формули: 19 70986 20 2d) сполуку формули (IV*) окиснюють з утвовивільнення інгібується пероральним або інтраперенням сполуки формули: ритональним застосуванням сполуки згідно з представленим винаходом. Цей тест було проведено адаптованим способом, описаним R. Steinberg et al., J. Neurochemistry, 1995, 65 , 25432548. 3d) сполука формули (VIII*) реагує зі сполукою Ці результати свідчать, що сполуки формули формули: (І) є активними перорально, що вони перетинають бар'єр кров-мозок, і що вони здатні блокувати специфічну стосовно рецепторів NH1 дію у центральв якій X визначено для сполуки формули (I), у ній нервовій системі. присутності кислоти, з наступним відновленням Сполуки формули (І) оцінювали у тесті на звуутвореної проміжної солі імінію відновником; ження бронхів у морських свинок згідно зі спосо4d) і, як варіант, отриману таким чином сполубом, описаним X. Emonds-Alt et al., European Jourку перетворюють у одну з її солей з органічною або неорганічною кислотою. nal of Pharmacology, 1993, 250 , 403-413. Сполуки Вищезазначені сполуки формули (І) також формули (І), що застосовували внутрішньовенно, включають сполуки, в яких один або більше атомів сильно антагонізували звуження бронхів, індукогідрогену або карбону замінено їх радіоактивними ване внутрішньовенним застосуванням септиду до ізотопами, наприклад, тритієм або карбоном-14. морських свинок в умовах цього експерименту. Такі мічені сполуки є корисними при дослідженні Фармакологічну активність in vivo сполук форметаболізму або фармакокінетики, у біохімічних мули (І) оцінювали також у моделі гіпотензії у сотестах як ліганди рецепторів. бак згідно зі способом, описаним X. Emonds-Alt et Сполуки згідно з винаходом піддавали біохіміal., Eur. J. Pharmacol., 1993, 250 , 403-413. Сполучним тестам. ки формули (І), що застосовували вн утрішньовенСпорідненість сполук до рецепторів тахікінінів но, сильно інгібують гіпотензію, індуковану внутріоцінювали in vitro кількома біохімічними тестами, шньовенним застосуванням [Sar9, використовуючи радіоліганди: 11 Меt(О 2) ]субстанції Ρ у анестезованих собак в цих 1) Зв'язування [125I]BH-SP (субстанція Р, мічеумовах експерименту. на йодом-125 з використанням реагенту БолтонаЦі результати свідчать, що сполуки формули Хантера) з рецепторами NK1 клітин лімфобластів (І) блокують специфічну стосовно рецепторів ΝΚ1 людини (D.G. Payan et al., J. Immunol., 1984, 133 , дію у периферійній нервовій системі. 3260-3265). Сполуки представленого винаходу є, зокрема, 2) Зв'язування [125l]His-NKA з клонованими реактивними інгредієнтами фармацевтичних компоцепторами людини NK2, експресованими клітиназицій, чия токсичність сумісна з їх використанням ми СНО (Y. Takeda et al., J. Neurochem., 1992, 59 , як медичних продуктів. Вищезазначені сполуки формули (І) можна ви740-745). користовувати при добовій дозі 0,01-100мг/кг маси 3) Зв'язування [125l]His [MePhe7] NK3 з рецептіла лікуємого ссавця, переважно при добовій дозі торами NKB кори головного мозку щура, кори го0,1-50мг/кг. Для людини доза може знаходитися ловного мозку морської свинки та кори головного переважно в межах 0,1-4000мг на добу, краще 0,5мозку піщанки, а також з клонованими рецептора1000мг, залежно від віку лікуємого або типу лікуми людини NK3, експресованими клітинами СНО вання: профілактичного або зцілювального. (Buell et al., FEBS Letters, 1992, 299 , 90-95). Для їх використання як медичних продуктів Тести проводили згідно з X. Emonds-Alt et al., сполуки формули (І) звичайно застосовують як (Eur. J. Pharmacol., 1993, 250 , 403-413; Life Sci-, дозовані одиниці. Вказані дозовані одиниці переважно формують у фармацевтичні композиції, в 1995, 56 , PL 27-32). яких активний інгредієнт змішують з одним чи біСполуки згідно з винаходом сильно інгібують льше фармацевтичними ексципієнтами. зв'язування субстанції Ρ з рецепторами ΝΚ, клітин Отже, згідно з ще одним аспектом представлімфобластів людини ІМ9. Константа інгібування лений винахід стосується фармацевтичних компоКi, стосовно рецепторів ΝΚ1 клітин лімфобластів зицій, що містять, як активний інгредієнт, сполуку людини має порядок 1011 М. формули (І) чи одну з її фармацевтично прийнятКонстанти інгібування Кi, стосовно клонованих них солей, їх сольватів та/або гідратів. -8 рецепторів людини ΝΚ2 мають порядок 10 М, а У фармацевтичних композиціях представленоконстанти інгібування Кi, стосовно клонованих рего винаходу для перорального, сублінгвального, -7 цепторів людини ΝΚ3 є більшими за 10 М. інгаляційного, підшкірного, внутрішньом'язового, Сполуки формули (І) є потужними та селективнутрішньовенного, трансдермального, місцевого вними антагоністами субстанції Ρ стосовно рецепабо ректального застосування, активні інгредієнти торів ΝΚ1 людини. можна застосовувати у одиничних формах для Тому сполуки формули (І) оцінювали також in застосування, у суміші зі звичайними фармацевvi vo на тваринних моделях. тичними носіями, тваринами та людиною. ПрийняУ смугастому тілі морської свинки локальне тні форми одиниць для застосування включають застосування агоністу, що є специфічним стосовно форми для перорального застосування, напри9 11 рецепторів NK1 наприклад, [Sar , Met(O2) ] субклад, таблетки, желатинові капсули, порошки, грастанції Ρ, збільшує вивільнення ацетилхоліну. Це нули та розчини або суспензії для перорального 21 70986 22 застосування, форми для під'язичного та защічновикористовува ти такий співрозчинник, як наприго застосування, аерозолі, форми для локального клад, спирт, як-то етанол або такий гліколь, як позастосування, імплантати, форми для підшкірного, ліетиленгліколь чи пропіленгліколь, і гідрофільну внутрішньом'язового, внутрішньовенного, інтранаПАР, як-то полісорбат 80 або поліоксамер 188. зального та інтраокулярного застосування та фоДля виготовлення придатного для ін'єкцій маслярми для ректального застосування. ного розчину для внутрішньом'язового застосуКоли тверду композицію виготовляють у формі вання, активний інгредієнт можна розчиняти у тритаблеток або желатинових капсул, суміш фармагліцериді або естері гліцерину. цевтичних ексципієнтів, що можуть бути розріджуДля застосування інгаляцією використовують вачами, як-то наприклад, лактозою, мікрокристаліаерозоль, що містить, наприклад, триолеат сорбіту чною целюлозою, крохмалем, дикальційабо олеїнову кислоту, а також трихлорфлуормефосфа том, зв'язуючими, як-то, наприклад, полівітан, дихлорфлуорметан, дихлортетрафлуорпеннілпіролідоном, гідроксипропілметилцелюлозою, тан або будь-який інший біологічно сумісний газузасобами для кришення, як-то перехресно зшитим ватий пропелент; можливо також використовувати полівінілпіролідоном, перехресно зшитою карбоксистему, що містить активний інгредієнт поодинці симетилцелюлозою, реологічними засобами, як-то або у комбінації з ексципієнтом, у формі порошку. оксид силіцію або тальк, та змащувачами, як-то Активний інгредієнт може також бути у формі стеарат магнію, стеаринова кислота, гліцерилкомплексу з циклодекстрином, наприклад, a-, bтрибегенат або стеарилфумарат натрію, додають або g-циклодекстрином, 2-гідроксипропіл-bдо мікронізованого чи немікронізованого активного циклодекстрином або метил-b-циклодекстрином. інгредієнту. Активний інгредієнт можна також виготовити у До композиції можна додавати змочувальні формі мікрокапсул або мікросфер, як варіант, з засоби або ПАР, як-то лаурилсульфат натрію, поодним чи більше носіями або добавками. лісорбат 80 або поліоксамер 188. Серед форм для тривалого вивільнення, що Таблетки можна виготовити різними способакорисні у випадку хронічного лікування, можливо ми: безпосереднє пресування, сухе гранулювання, використовува ти імплантати. їх можна виготовити вологе гранулювання, або плавлення. у формі масляної суспензії або у формі суспензії Таблетки можуть бути непокритими, покритимікросфер у ізотонічному середовищі. ми цукром, наприклад сахарозою, різними полімеУ кожній дозованій одиниці, активний інгредірами або іншими прийнятними матеріалами. єнт формули (І) присутній у кількості, придатній Таблетки можуть мати швидке, довготривале для передбачуваної добової дози. Взагалі, кожну чи затримане вивільнення активної речовини дозовану одиницю прийнятно пристосовують до при застосуванні для виготовлення полімерних дози та типу передбачуваного застосування, наматриць або специфічних плівкоутворюючих приклад, таблеток, желатинових капсул тощо, паполімерів. кетиків, ампул, сиропів тощо, або крапель, так, Желатинові капсули можуть бути твердими чи щоб дозована одиниця, що потребує застосовум'якими, покритими плівкою або ні, з тим, щоб мавання 1-4 рази на добу, містила 0,1-1000мг активти швидку, подовжену чи затриману активність ного інгредієнту, переважно 0,5-250мг. (наприклад, для кишкових форм). Хоча такі дози є прикладами для середніх сиВони можуть містити не тільки тверду компотуацій, в окремих випадках можливо придатними є зицію, створену, як вищезазначено, для таблеток, вищі чи нижчі дози, такі дози також входять до але також рідку або напівтверду композицію. рамок представленого винаходу. Згідно зі звичайПрепарат у формі сиропу чи еліксиру може міною практикою, дозу, що є придатною для кожного стити активний інгредієнт разом з замінником цукпацієнта, визначає лікар згідно зі способом застору, переважно малокалорійним замінником цукру, сування та віком, масою та чутливістю вказаного метилпарабеном та пропілпарабеном як антисеппацієнта. тиком, а також смаковим засобом та придатним Згідно з ще одним аспектом, представлений барвником. винахід стосується використання сполуки формуПорошки чи гранули для диспергування у воді ли (І), одної з її фармацевтично прийнятних солей, можуть містити активну речовину як суміш з дисїх сольватів та/або гідратів, для виготовлення мепергаторами, змочувальними речовинами, або дичних продуктів призначених для лікування будьсуспендувальними засобами, наприклад, полівініяких патологій, при яких залучено субстанцію Ρ та лпіролідоном, а також з замінниками цукру або рецептори ΝΚ1 людини. посилювачами смаку. Згідно з ще одним аспектом, представлений Для ректального застосування використовувинахід стосується використання сполуки формують супозиторії, які виготовляють зі зв'язуючими, ли (І), одної з її фармацевтично прийнятних солей, що плавляться при ректальній температурі, наприїх сольватів та/або гідратів, для виготовлення меклад, масло какао або поліетиленгліколі. дичних продуктів, призначених для лікування паДля парентерального, інтраназального чи інтологій респіраторної, шлунково-кишкової, сечової, траокулярного застосування використовують водні імунної або серцево-судинної системи та центрасуспензії, ізотонічні сольові розчини або придатні льної нервової системи, а також болю, мігрені, для ін'єкцій стерильні розчини, що містять фармазапалень, нудоти та блювоти, та за хворювань кологічно сумісні диспергатори та/або солюбілізашкіри. тори, наприклад, пропіленгліколь. Наприклад, але без обмеження, сполуки форОтже, для виготовлення водного розчину, мули (І) є корисними: який можна ін'єктува ти вн утрішньовенно, можна 23 70986 24 - як аналгетики, зокрема при лікуванні болю - при лікуванні змін проникності бар'єру кровпри травмах, наприклад, болю після операції; немозок при запальних та автоімунних процесах у вралгії брахіального сплетіння; хронічного болю, центральній нервовій системі, наприклад, при спонаприклад, болю при артриті, викликаного остеоріднених зі СНІД інфекціях; артритом, ревматоїдним артритом або псоріатич- як релаксант м'язів та антиспазматичний ним артритом; невропатичного болю, наприклад, засіб; постгерпетичної невралгії, невралгії трійчастого - при лікуванні гострої або тривалої та переднерва, сегментальної або міжреберної невралгії, бачуваної нудоти та блювоти, наприклад, нудоти фіброміалгії, каузалгії, периферійної невропатії, та блювоти, викликаної ліками, наприклад, викодіабетичної невропатії, викликаної хіміотерапією, ристовуваними у хіміотерапії при лікуванні раку невропатії, спорідненої зі СН1Д невропатії, потизасобів; радіаційною терапією при опромінюванні личної невралгії, колінної невралгії, або глосогрудної клітини або черева при лікуванні раку або фарингеальної невралгії; ілюзорного болю після карциноїдозу; поглинанням отрути; токсинами, ампутацій; різні форми головного болю, наприспричиненими метаболічними або інфекційними клад, хронічної або гострої мігрені, скроневорозладами, наприклад, гастрит, або продукованинижньощелепного болю, болю верхньощелепного ми при бактеріальних чи вірусних шлунковосинусу, лицьової невралгії або одонталгії; болю, кишкових інфекціях; при вагітності; при вестибулящо відчувають страждаючі від раку; болю вісцерарних розладах, як-то морська хвороба, запаморольного походження; шлунково-кишкового болю; чення або хвороба Меньєра; при постоперативних викликаного ущемленням нервів болю, викликанозахворюваннях; нудоти та блювоти, викликаної го інтенсивним спортивним тренуванням болю; діалізом або простагландинами; шлунководисменореї; болю при менструаціях; викликаного кишковою непрохідністю; при зменшеній шлункоменінгітом чи арахноїдитом болю; болю скелетних во-кишковій рухливості; при вісцеральному болю, м'язів; викликаного спінальним стенозом, випавикликаного інфарктом міокарду або перитонітом; данням дисків або ішіасом болю у нижній частині при мігрені; при гірській хворобі; прийняттям опіоїспини; болю, що відчувають страждаючі від ангіни; дного аналгетику, як-то морфіну; при шлункововикликаного анкілозним спондилітом болю; пов'ястравохідному рефлюксі; при кислотній диспепсії заного з подагрою болю; пов'язаного з пологами, або надлишковому споживанні їжі та питва, при рубцеванням або дерматозом при свербці болю; підвищеній кислотності, відрижці та печії, наприталамічного болю; клад, епізодичній або нічній печії або печії, викли- як анти-запальні засоби, зокрема для лікуканої їжею, та диспепсії; вання запалень при астмі, грипі, хронічному брон- при лікуванні хвороб шлунково-кишкової сисхіті (зокрема обструктивному хронічному бронхіті теми, як-то синдрому подразненого кишечнику, та COPD (хронічне обструктивне захворювання шлункових та кишкових виразок, виразки страволегень)), кашлі, алергії, бронхоспазмі та ревматоїходу, діареї, гіперсекреції, лімфоми, гастриту, дному артриті; запальних захворювань шлунковошлунково-стравохідного рефлюксу, невтримання кишкової системи, наприклад, хвороби Крона, викалу, хвороби Гіршспрунга та алергії до їжі; разкового коліту, панкреатиту, гастриту, запалення - при лікуванні захворювань шкіри, як-то псорікишок, викликаного нестероїдними антизапальниаз, свербіж та опіки, зокрема сонячні опіки; ми засобами розладу, викликаних бактеріальною при лікуванні хвороб серцево-судинної систеінфекцією запальних та секреторних наслідків, ми, як-то гіпертензії, судинних аспектів мігрені, наприклад, викликаних Clostridium difficile] запальнабряку, тромбозу, стенокардії, спазмів судин, них захворювань шкіри, наприклад, герпесу та захворювань кровообігу, викликаних розширенням екземи; запальних захворювань сечового міхура, судин, хвороби Рейнольда, фіброзу, колагенових наприклад, циститу та невтримання сечі; офтальзахворювань та атеросклерозу; мологічних запалень, наприклад, кон'юнктивіт та - при лікуванні мало-клітинного раку легенів; вітреоренопатія; дентальних запалень, наприклад, пухлин мозку та аденокарциноми урогенітальної гінгівіт та парадонтоз; сфери; - при лікуванні алергічних захворювань, зок- демієлінаційних захворювань, як-то розсіянорема шкіри, наприклад, кропивниці, контактного го склерозу або бічного аміотрофічного склерозу; дерматиту, атопічного дерматиту та респіраторних - при лікуванні хвороб імунної системи, пов'язахворювань, наприклад, риніту; заних з пригніченням або стимуляцією функції іму- при лікуванні хвороб центральної нервової нних клітин, наприклад, ревматоїдного артриту, системи, зокрема психозів, наприклад, шизофрепсоріазу, хвороби Крона, діабету, во вчаку та реакнії, маній та деменції; розладу пізнавальної здатції відторгнення після трансплантації; ності, наприклад, хвороби Альцгеймера, тривож- при лікуванні розладів сечовипускання, зокності, спорідненої зі СНІД деменції, діабетичної рема полакурії; невропатії; депресії; хвороби Паркінсона; залеж- при лікуванні гістіоцитозного ретикулезу, наності від ліків; зловживання речовинами; розладу приклад, у лімфатичних тканинах; свідомості, розладів сну, розладів циркадного ри- як засіб зменшення апетиту; тму, розладів настрою та епілепсії; синдрому Дау- при лікуванні емфіземи; хвороби Рейтера; на; хореї Хантингтона; споріднених зі стресом согеморою; матичних розладів; нейродегенеративних - при лікуванні очних захворювань, як-то глаузахворювань, наприклад, хвороби Піка або хворокоми, очної гіпертензії, міозу та надлишкової секби Крейфельдта-Якоба; асоційовані з панічністю, реції сліз; фобіями або стресом розлади; 25 70986 26 - при лікуванні або попередженні епілептичних конвульсій, черепної травми, травми спинного хребта, викликаних судинною атакою або оклюзією ішемічних уражень мозку; A) 2-(3,4-диметилфеніл)-4-(2- при лікуванні розладів частоти пульсу та сететрагідропіранілокси)-бутанонітрил рцевого ритму, зокрема викликаних болем або 6,6г 60% гідриду натрію у маслі додають порстресом розладів; ціями при кімнатній температурі до розчину 20г - при лікуванні чутливої до подразнення шкіри 3,4-диметилфенілацетонітрилу у 100мл безводнота для попередження або лікування подразнень го ТГФ і суміш залишають перемішуватися при шкіри або слизових мембран, лупи, еритеми або кімнатній температурі протягом 2 годин. Далі досвербежу. дають краплями 29г 1-бром-2-(2- при лікуванні неврологічних розладів шкіри, тетрагідропіранілокси)етану та суміш залишають як-то лишаїв, свербежу, викликаючої свербіж токперемішува тися при кімнатній температурі протясидермії та суворої сверблячки неврологічного гом 2 діб. Реакційну суміш виливають на лід та походження; екстрагують етилацетатом, органічну фазу проми- при лікуванні виразок та усі х викликаних вають водою та насиченим розчином NaCI і суHelicobacter pylori або уреазо-позитивними грамшать над Na2SO4, а розчинник випарюють у вакунегативними бактеріями захворювань; умі. Залишок хроматографують на силікагелі, при лікуванні викликаних ангіогенезом хвороб елююючи толуолом, а потім сумішшю толуабо тих, в яких ангіогенез є симптомом; ол/етилацетат з градієнтом від (99/1; за об'ємом) - при лікуванні очної та/або палпебральної до (90/10; за об'ємом). Отримують 17г очікуваного алгії (algia) та/або очної або палпебральної продукту. дизестезії; B) Метил 4-ціано-4-(3,4-диметилфеніл)-6-(2- як засоби проти виділення поту. тетра-гідропіранілокси)гексаноат Згідно з представленим винаходом запропо0,3мл 40% розчин гідроксиду бензилтриметиновано також спосіб лікування вказаних хвороб ламонію (Triton®B) у МеОН додають до Суміші 17г вищезазначеними дозами. отриманої на попередньому етапі сполуки та 11мл Фармацевтичні композиції згідно з представметилакрилату у 30мл діоксану, і суміш залишаленим винаходом можуть також містити інші актиють перемішува тися при кімнатній температурі вні продукти, що корисні при лікуванні вищезазнапротягом 48 годин. Реакційну суміш концентрують чених захворювань або розладів, наприклад, у вакуумі, залишок переносять у водний 0,5Η розбронхорозширюючі засоби, засоби проти кашлю, чин НСІ та екстрагують ефіром, органічну фазу антигістамінові засоби, анти-запальні засоби, анпромивають водним 10% розчином тиблювальні засоби та хіміотерапевтичні засоби. Na2CO3 та сушать над Na2SO4, а розчинник У виготовленнях та у прикладах: використано випарюють у вакуумі. Отримують 23г очікуваного такі скорочення продукту. ДМФ: диметилформамід C) 5-(3,4-диметилфеніл)-5-[2-(2ДМСО: диметилсульфоксид тетрагідропіранілокси)етил]-2-піперидон. ДХМ: ди хлорметан 40мл 20% водного розчину аміаку додають до ТГФ: тетрагідрофуран розчину 23г отриманої на попередньому етапі ефір: діетиловий етер сполуки у 250мл 95% ЕЮН, з наступним додавангідрохлоридний ефір: насичений розчин гідроням нікелю Ренея (Raney®). Цю суміш далі гідрухлоридної кислоти у діетиловому етері ють протягом 24 годин при 40°С та при тиску 16 ВОР: гексафлуорфосфат бензотриазол-1 бар. Каталізатор відфільтровують на целіті ілокситрис(диметиламіно)фосфонію (Celite®) та концентрують фільтрат у вакуумі. Темп.плавл.: температура плавлення. Отримують 22г очікуваного продукту. Темп.кип.: температура кипіння. D) 3-(3,4-диметилфеніл)-3-[2-(2оксид силіціюН: силікагель 60Н, постачальник тетрагідропіранілокси)-етил]піперидин. - Merck (Darmstadt). 22г отриманої на попередньому етапі сполуки ЯМР: ядерний магнітний резонанс. Спектри додають до суспензії 10г алюмогідриду літію у ЯМР зареєстровані при 200МГц у диметилсульфо200мл тетрагідрофурану, з наступним кип'ятінням ксиді - ДМСО-d6, використовуючи пік ДМСО-d6 як під зворотним холодильником протягом 2 годин. внутрішній стандарт. Хімічні зсуви d є визначені у Після охолодження до кімнатної температури, дочастинах на мільйон (чнм). Спостережені сигнали дають 10мл води та 80мл тетрагідрофурану, а позначені так: s: синглет; d: дублет; t: триплет; q: потім 10мл 4Η NaOH та 30мл води. Мінеральні квартет; m: мультиплет. солі відфільтровують на целіті (Celite®) та конценВиготовлення 1,1 трують фільтрат у вакуумі. Отримують 15г очікува3-(3, 4-дихлорфеніл)-3-(2ного продукту. гідроксіетил)піперидин, (-) ізомер, Виготовлення 2,1 3,5-дихлорфенілоцтова кислота. (lll):R1=CI. A) 3,5-ди хлорбензилхлорид. Виготовлення цієї сполуки описано у заявці на Розчин 12,5г тіонілхлориду у 20мл хлорофорвинахід ЕР-А-0 591 040. Виготовлення 1,2 3-(3,4му додають краплями при кімнатній температурі диметилфеніл)-3-[2-(2-тетрагідропіранілокси)до розчину 14,5г 3,5-дихлорбензилового спирту у етил]піперидин 150мл хлороформу, з наступною витримкою при 27 70986 28 40-50°С протягом 8 годин та перемішуванням при юючи циклогексаном/етилацетатом суміш (95/5; кімнатній температурі протягом ночі. Суміш концеза об'ємом). Отримують 12г очікуваного продукнтрують під вакуумом, одержавши 16 г очікуваного ту. продукту, який використовують без подальшої C) 3,5-діетилбензойна кислота. обробки. Розчин 22г КОН у 15мл води додають до розB) 3,5-дихлорфенілацетонітрил. чину 12г отриманої на попередньому етапі сполуки Розчин 6,5г ціаніду калію у 50мл води додають у 60мл ЕtOН, з наступним кип'ятінням під зворотдо розчину 16г отриманої на попередньому етапі ним холодильником протягом 24 годин. Реакційну сполуки у 50мл ЕЮН, та суміш кип'я тять під зворосуміш концентрують у вакуумі, залишок екстрагутним холодильником протягом 4 годин. Утворену ють водою, водну фазу промивають ефіром та суміш концентрують у вакуумі, залишок перенопідкислюють до рН=2 додаванням концентрованої сять у воді та екстрагують ефіром, органічну фазу НСІ і утворений осад сушать при перемішуванні, промивають водою та сушать Na2SO4, а розчинник промивають водою та сушать під вакуумом. Отривипарюють у вакуумі. Залишок хроматографують мують 13г очікуваного продукту. на оксиді силіцію Н, елююючи сумішшю гепD) Метил 3,5-діетилбензоат. тан/толуол (50/50; за об'ємом), а потім толуолом. Суміш 13г отриманої на попередньому етапі Отримують 7г очікуваного продукту, продукт викосполуки у 90мл МеОН та 10 крапель H2SO4 кип'я ристовують без подальшої обробки. тять під зворотним холодильником протягом 48 C) 3,5-дихлорфенілоцтова кислота. годин. Реакційну суміш концентрують у вакуумі, Розчин 8,4г КОН у 10мл води додають до роззалишок переносять у воду, нейтралізують додачину 7г отриманої на попередньому етапі сполуки ванням 10% розчину NaHCO3 та екстрагують ефіу 50мл ЕtOН, з наступним кип'ятінням під зворотром, органічну фазу промивають 10% розчином ним холодильником протягом 5 годин. Цю суміш NaHCO3, водою та суша ть над Na2SO4, а розчинконцентрують у вакуумі, залишок переносять у ник випарюють у вакуумі. Отримують 12г очікуваводі та водну фазу промивають ефіром, підкисного продукту. люють до рН=1 додаванням концентрованої НСІ E) 3,5-діетилбензиловий спирт. та залишають перемішуватися при кімнатній темСуспензію 2,5г алюмогідриду літію у 50мл тетпературі протягом ночі. Утворений кристалічний рагідрофурану охолоджують до 0°С, краплями допродукт сушать при перемішуванні, промивають дають розчин 12г отриманої на попередньому етаводою та сушать під вакуумом при 60°С. Отримупі сполуки у 50мл тетрагідрофурану і суміш ють 7г очікуваного продукту; Темп.плавл.=112залишають перемішуватися протягом 30 хвилин. 114,5°С. Реакційну суміш гідролізують додаванням 2,5мл Виготовлення 2,2 води, 2,5мл 4Η NaOH та 7,5мл води. Мінеральні 3,5-діетилфенілоцтова кислота. (Ill): Ri=Et. солі відфільтровують та концентрують фільтрат у 3,5-діетилбромбензол. вакуумі. Отримують 10,9г очікуваного продукту, Суміш 20г 4-бром-2,6-діетиланіліну, 160мл оцпродукт використовують без подальшої обробки. тової кислоти, 100мл концентрованого розчину ІСІ, F) 3,5-діетилбензил метансульфонат. 30мл води та 100мл ЕtOН охолоджують до -5°С, Розчин 8,4г метансульфонілхлориду у 50мл додають краплями розчин 6,6г нітриту натрію у дихлорметану додають краплями при кімнатній 25мл води та суміш залишають перемішуватися температурі до розчину 10,9г отриманої на попепри кімнатній температурі протягом 30 хвилин. редньому етапі сполуки та 7,4г триетиламіну у Реакційну суміш виливають у 170мл 50% охоло100мл дихлорметану і суміш залишають перемідженої до 0°С Н3РО2 та залишають перемішува тишуватися протягом 30 хвилин. Реакційну суміш ся протягом 2 годин при 0°С, а потім протягом 48 концентрують у вакуумі, залишок переносять у годин при кімнатній температурі. Реакційну суміш воду та екстрагують ефіром, органічну фазу проекстрагують ефіром, органічну фазу промивають мивають водою та сушать Na2SO4 , а розчинник водою, 1Η розчином NaOH, водою та сушать над випарюють у вакуумі. Отримують 16г очікуваного Na2SO4, a розчинник випарюють у вакуумі. Запродукту, продукт використовують без подальшої лишок хроматографують на силікагелі, елюююобробки. чи циклогексаном. Отримують 18г очікуваного G) 3,5-діетилфенілацетонітрил. продукту. Розчин 5,15г ціаніду калію у 20мл води додаB) 3,5-діетилбензонітрил. ють до розчину 16г отриманої на попередньому Суміш 24,7г отриманої на попередньому етапі етапі сполуки у 100мл диметилформаміду та сусполуки та 12г ціаніду купруму у 70мл диметилфоміш гріють при 80°С протягом 1 години. Реакційну рмаміду кип'ятять під зворотним холодильником суміш концентрують у вакуумі, залишок перенопротягом 15 годин. Після охолодження до кімнатсять у воду та екстрагують ефіром, органічну фазу ної температури, реакційну суміш виливають у промивають водою та сушать над Na2SO4, a роз50мл води та залишають перемішуватися при кімчинник випарюють у вакуумі. Залишок хроматогнатній температурі доки не утворюється гумоподірафують на силікагелі Н, елююючи дихлорметабна речовина. Суміш охолоджують на льодяній ном. Отримують 3г очікуваного продукту. бані, додають 150мл етилендіаміну та this суміш Н) 3,5-діетилфенілоцтова кислота. залишають перемішуватися при кімнатній темпеРозчин 7,8г КОН у 10мл води додають до розратурі протягом 2 годин. Суміш екстрагують етичину 3г отриманої на попередньому етапі сполуки лацетатом, органічну фаз у промивають водою та у 50мл ЕtOН з наступним кип'ятінням під зворотсушать Na2SO4 та розчинник випарюють у вакууним холодильником протягом 5 годин. Цю суміш мі. Залишок хроматографують на силікагелі, елюконцентрують у вакуумі, залишок переносять у 29 70986 30 воду і водн у фазу промивають ефіром, підкислюють до рН=1 додаванням концентрованої НСІ та Реакційну суміш виливають на лід та залишазалишають перемішуватися при кімнатній темпеють перемішуватися протягом ЗО хвилин. Суміш ратурі протягом ночі. Утворений кристалічний проекстрагують ефіром, органічну фазу кількаразово дукт сушать при перемішуванні, промивають вопромивають водою та сушать Na2SO4, а розчинник дою та сушать під вакуумом. Отримують 2,5г випарюють у вакуумі. Отримують 13г очікуваного очікуваного продукту. продукту. 1 ЯМР: δ (чнм): 1,1: t: 6H; 2,4: q: 4H; 3,4: s: 2H; B) Дигідрохлорид 2-(4-бензил-16,8: m: 3H; 12,2: bs: 1H. піперазиніл)ізобутираміду Виготовлення 3,1 Суміш 13г отриманої на попередньому етапі Гідрохлорид 2-(4-піперидил)ізобутираміду сполуки та 130мл 90% розчину H2SO4 швидко грі(VII), НСІ: X-=CH-C(CH 3)2-CONH2 ють при 110°С протягом 30 хвилин. Після охолоА) 2-Метил-2-(4-піридил)пропіонітрил. дження до кімнатної температури, реакційну суміш Суміш 3г гідрохлориду 4-піридилацетонітрилу виливають на лід та підлужують до рН=10 додау 50мл диметилформаміду охолоджують до 0°С, ванням концентрованого розчину NH4OH та судодають порціями 2,6г 60% гідриду натрію у маслі шать при перемішуванні утворений кристалічний та суміш залишають перемішуватися при кімнатній продукт. Продукт розчиняють у дихлорметані, ортемпературі протягом 2 годин. Реакційну суміш ганічну фазу сушать MgSO4 та випарюють розохолоджують на льодяній бані, додають краплями чинник у вакуумі. Продукт переносять у гідро6г метилйодиду і суміш залишають перемішува тихлоридний ефір і утворений осад сушать при ся при кімнатній температурі протягом ночі. Реакперемішуванні. Отримують 9,5г очікуваного ційну суміш виливають у суміш льоду з водою та продукту. екстрагують ефіром, органічну фазу промивають С) Дигідрохлорид 2-(1насиченим розчином NaCI, сушать над MgSO 4 та піперазиніл)ізобутираміду. фільтрують, а розчинник випарюють у вакуумі. Суміш 1,3г отриманої на попередньому етапі Залишок хроматографують на силікагелі Н, елюсполуки та 0,18г 10% паладію-на-вугіллі у 30мл юючи дихлорметаном, а потім сумішшю ди хло95% ЕtOН гідрують протягом ночі при кімнатній рметан/МеОН (98/2; за об'ємом). Отримують температурі та при атмосферному тиску. Каталіза2,39г очікуваного продукту у формі масла, яке тор відфільтровують на целіті (Celite®), а фільтрат кристалізується. концентрують у вакуумі. Отримують 0,6г очікуваноB) Гідрохлорид 2-(4-піридил)ізобутирамід. го продукту. Суміш 2,39г отриманої на попередньому етапі Виготовлення 3,3 сполуки та 10мл концентрованого розчину H2SO4 Дигідрохлорид 1-(1гріють при 100°С протягом 15 хвилин. Реакційну піперазиніл)циклогексанкарбоксаміду суміш о холоджують до кімнатної температури, 50г додають льоду, цю суміш підлужують до рН=14 додаванням концентрованого розчину NaOH, мінеральні солі відфільтровують, фільтрат екстраA) 1 -(4-бензил-1 гують етилацетатом, а потім дихлорметаном, попіперазиніл)циклогексанкарбонітрил, єднані органічні фази сушать MgSO 4 та 5,7г циклогексанону, 20г сухо го MgSO 4, 10г фільтрують, а розчинник випарюють під вакуумом Ν,Ν-диметилацетаміду, 10г 1-бензилпіперазину та (Темп.плавл.=134°С, основа). Отриманий продукт 9,5мл 2-гідроксіізобутиронітрилу змішують разом розчиняють у ацетоні та підкислюють до рН=1 дота гріють при 45°С протягом 48 годин з інтенсивдаванням гідрохлоридного ефіру, а утворений ним перемішуванням. Реакційну суміш виливають осад сушать при перемішуванні. Отримують 2,9г на лід та залишають перемішува тися протягом 30 очікуваного продукту. хвилин. Суміш екстрагують ефіром, органічну фаC) Гідрохлорид 2-(4-піперидил)ізобутираміду. зу кількаразово промивають водою та сушать Суміш 2,9г отриманої на попередньому етапі Na2SO4, та розчинник випарюють у вакуумі. Отрисполуки, 1г ° РtO2 та 50мл МеОН гідрують протямують 15г очікуваного продукту. гом 3 діб при 60°С під тиском 60 бар. Каталізатор B) Дигідрохлорид 1-(4-бензил-1відфільтровують на целіті (Celite®) та промивають піперазиніл)циклогексанкарбоксаміду. МеОН і концентрують фільтрат у вакуумі. Залишок Цю сполуку отримують згідно зі способом, що переносять у ацетонітрил і утворений осад сушать описано у етапі В з виготовлення 3,2, починаючи з при перемішуванні та промивають ацетонітрилом, 15г отриманої на попередньому етапі сполуки та а потім ефіром. Отримують 2,5г очікуваного проду50мл 90% розчину H2SO4. Отримують 5,5г очікувакту; Темп.плавл. >260°С. ного продукту. Виготовлення 3,2 C) Дигідрохлорид 1-(1Дигідрохлорид 2-(1-піперазиніл)ізобутираміду. піперазиніл)циклогексанкарбоксаміду. (VII), 2НСІ: X-=N-C(CH 3)2-CONH2 Цю сполуку отримують згідно зі способом, що A) 2-(4-бензил-1-піперазиніл)-2описано у етапі С з виготовлення 3,2, починаючи з метилпропіонітрил. 2,3г отриманої на попередньому етапі сполуки та 4,5мл ацетону, 20г сухого MgSO4 , 10г Ν,Ν0,3г 10% паладію-на-вугіллі у 30мл 95% ЕtOН. диметилацетаміду, 10г 1-бензилпіперазину та Отримують 1,6г очікуваного продукту. 9,5мл 2-гідроксіізобутиронітрилу змішують разом Виготовлення 3,4 та гріють при 45°С Протягом 48 годин з інтенсивДиформіат М,ІМ-диметил-2-(1ним перемішуванням. 31 70986 32 птеразиніл)ізобутираміду. (VII), 2НСО2Н: X-=N-C(CH 3)2-CON(CH3)2 C) Гідрохлорид 1-(4А) N,N-диметил-2-(4-бензил-1піперидил)циклогексанкарбоксаміду. піперазиніл)ізобутирамід. Суміш 2,9г отриманої на попередньому етапі 1,44г 60% гідриду натрію у маслі додають порсполуки, 0,5г PtO2 та 50мл МеОН гідрують протяціями до суміші 2,6г отриманої на етапі В виготовгом 3 діб при 60°С, при тиску 80 бар. Каталізатор лення 3,2 сполуки (вільна основа) у 50мл безводвідфільтровують на целіті (Celite®) та концентруного тетрагідрофурану. ють фільтрат у вакуумі. Залишок переносять у 1,3мл метилйодиду далі додають краплями та ацетонітрил та залишають перемішуватися при цю суміш залишають перемішуватися при кімнаткімнатній температурі протягом 1 години, а утвоній температурі протягом 4 годин. Реакційну суміш рений осад сушать при перемішуванні. Отримують виливають у воду та екстрагують ефіром, органіч2,7г очікуваного продукту; Темп.плавл.=235°С. ну фазу сушать MgSO 4 та розчинники випарюВиготовлення 3,6 ють під вакуумом. Отримують 1,8г очікуваного Гідрохлорид N,N-диметил-2-(4продукту. піперидил)ізобутираміду. В) Диформіат N,N-диметил-2-(1(VII), НСІ: X-=CH-C(CH 3)2-CON(CH3)2 піперазиніл)ізобутираміду. A) Етил 1-бензил-4-піперидинкарбоксилат. 2г форміату амонію та 0,5г 5% паладію-на30г бензилброміду додають краплями до сувугіллі додають до розчину 1,8г отриманої на поміші 25г етилізоніпекотату та 25г КаСО3 у 125мл передньому етапі сполуки у 30мл МеОН та залидиметилформаміду, підтримуючи температуру шають суміш перемішуватися при кімнатній темреакційної суміші між 25 та 30°С, і утворену суміш пературі протягом 4 годин. Каталізатор перемішують далі при кімнатній температурі провідфільтровують на целіті (Celite®) і концентрують тягом 1 години. Реакційну суміш виливають у 1л фільтрат у вакуумі. Залишок переносять у етилальодяної води та екстрагують двічі ефіром, органіцетат і утворений осад сушать при перемішуванні, чну фазу промивають водою та суша ть MgSO4 , а промивають етилацетатом та сушать. Отримують розчинник випарюють у вакуумі. Утворене масло 1,2г очікуваного продукту. переганяють під зниженим тиском. Отримують Виготовлення 3,5 29,2г очікуваного продукту; Темп.кип.=120-122°С Гідрохлорид 1 -(4при 2,7Па. піперидил)циклогексанкарбоксаміду B) 2-(1-бензил-4-піперидил)-2-пропанол. Розчин 24,73г отриманої на попередньому етапі сполуки у 100мл бензолу додають краплями, підтримуючи температур у середовища між 25 та 30°С, до 200мл 1,5Μ розчину метиллітію, як комA) 1 -(4-піридил)циклогексанкарбонітрил. плекс з бромідом літію, у ефірі в атмосфері аргону Суміш 3г гідрохлориду 4-піридилацетонітрилу з наступним кип'ятінням під зворотним холодильу 50мл диметилформаціду охолоджують до 0°С, ником протягом 48 годин. Реакційну суміш охолододають порціями 2,6г 60% гідриду натрію у маслі джують до кімнатної температури і далі виливають та суміш залишають перемішуватися при кімнатній у 400мл насиченого розчину хлориду амонію у температурі протягом 1 години 30 хвилин. Реакводі, що перед тим охолоджений на льодяній бані. ційну суміш о холоджують на льодяній бані, 2,7 мл Суміш екстрагують три рази ефіром, поєднані ор1,5-дибромпентану додають краплями та цю суміш ганічні фази сушать над MgSO4 та концентрують залишають перемішуватися при кімнатній темперозчинник у вакуумі. Залишок розчиняють у 100мл ратурі протягом 48 годин. Реакційну суміш вилиацетону, о холоджують до 10°С та підкислюють до вають у насичений розчин NH4C1 та екстрагують рН=1 додаванням гідрохлоридного ефіру, а утвоефіром, органічну фазу тричі промивають водою рений осад сушать при перемішуванні та промита суша ть MgSO4, а розчинник випарюють у вакувають сумішшю ацетон/ефір (50/50; за об'ємом). умі. Залишок хроматографують на силікагелі Н, Отримують 24,5г очікуваного продукту у формі елююючи дихлорметаном, а потім сумішшю дихгідрохлориду; Темп.плавл.=204°С. лорметан/МеОН (98/2; за об'ємом). Отримують Для вивільнення основи, гідрохлорид перено2,5г очікуваного продукту; Темп.плавл.=79°С. сять у концентрований розчин NaOH, екстрагують B) Гідрохлорид 1-(4ефіром та сушать над MgSO 4, а розчинник випапіридил)циклогексанкарбоксаміду. рюють у вакуумі. Отримують 21г очікуваного проСуміш 2,5г отриманої на попередньому етапі дукту; Темп.плавл.=66°С. сполуки та 15мл концентрованого розчину K2SO4 C) 2-(1-бензил-4-піперидил)-2-метилпропанова гріють при 100°С протягом 15 хвилин. Реакційну кислота. суміш охолоджують до кімнатної температури, Суміш 5,98г 95% сульфатної кислоти та 4,42г виливають на лід та підлужують до рН=14 додадимлячої сульфатної кислоти, що містить 30% SO3 ванням концентрованого розчину NaOH, і утвореохолоджують до 3°С і додають краплями розчин 2г ний осад сушать при перемішуванні, промивають отриманої на попередньому етапі сполуки у 1,55г водою та сушать. Отриманий продукт розчиняють 100% мурашиної кислоти, підтримуючи темперау ацетоні, підкислюють до рН=1 додаванням гідротуру нижче 10°С. Суміш залишають перемішувахлоридного ефіру та залишають перемішуватися тися протягом 2 годин при 3-5°С та потім дають при кімнатній температурі протягом 30 хвилин, а повернутися до кімнатної температури і витримуутворений осад сушать при перемішуванні. Отриють протягом ночі при кімнатній температурі. Реамують Зг очікуваного продукту; Темп.плавл.=224°С кційну суміш виливають на лід, рН доводять до 6,5 (з розкладанням). 33 70986 34 додаванням концентрованого розчину NaOH та мивають водою і сушать. Осад розчиняють у додаванням концентрованого розчину NH4OH та дихлорметані, підкислюють до рН=1 додаванням екстрагують три рази дихлорметаном, поєднані гідрохлоридного ефіру і утворений осад сушать органічні фази сушать над MgSO4 і розчинник конпри перемішуванні. Отримують 1,8г очікуваного центрують у вакуумі. Залишок переносять у ацепродукту. тоні та осад сушать при перемішуванні і сушать. С) Гідрохлорид 1-(4Отримують 1,22г очікуваного продукту; піперидил)циклопропанкарбоксаміду. Темп.плавл.=195°С. Суміш 1,8г отриманої на попередньому етапі D) Гідрохлорид N,N-диметил-2-(1-бензил-4сполуки та 0,6г РtO2 у 50мл МеОН гідрують протяпіперидил)ізобутираміду. гом 15 годин при 80°С та при тиску 100 бар. КатаСуміш 1,2г отриманої на попередньому етапі лізатор відфільтровують на целіті (Celite®), концесполуки, 0,8мл триетиламіну, 2,8мл 2Μ розчину нтрують фільтрат у вакуумі до об'єму 5мл та диметиламіну у те трагідрофурані та 2,5г ВОР у додають ацетонітрил поки не відбувається криста20мл дихлорметану перемішують протягом 1 голізація. Після сушки з перемішуванням, а потім дини при кімнатній температурі. Реакційну суміш сушки отримують 1,7г очікуваного продукту. концентрують у вакуумі, залишок переносять у Виготовлення 3,8 ефір, органічну фазу промивають водою, 1Η розГідрохлорид 2-Метил-1-(4-морфолініл)-2-(4чином NaOH, насиченим розчином NaCI та сушать піперидил)-1-пропанону. над MgSO4, а розчинник концентрують у вакуумі. Залишок хроматографують на силікагелі Н, елююючи дихлорметаном, а потім сумішшю дихлорметан/МеОН з градієнтом від (99/1; за об'ємом) до А.) Гідрохлорид 2-(1-бензил-4-піперидил)-2(95/5; за об'ємом). Отриманий продукт розчиняють метил-1-(4-морфолініл)-1-пропанону. у ацетоні та підкислюють до рН=-1 додаванням Суміш 1г отриманої на етапі С з виготовлення гідрохлоридного ефіру і утворений осад сушать 3,6 сполуки та 1,2мл тіонілхлориду у 20мл 1,2при перемішуванні та сушать. Отримують 0,8г очідихлорпентану гріють при 80°С протягом 3 годин. куваного продукту; Темп.плавл.=229°С. Реакційну суміш концентрують у вакуумі, отримаE) Гідрохлорид N,N-диметил-2-(4ний так хлорангідрид розчиняють у 20мл дихлорпiперидил)ізобутираміду. метану, цей розчин додають до суміші 0,7г морСуміш 0,8г отриманої на попередньому етапі фоліну та 1,6мл триетиламіну у 20мл сполуки та 0,2г 10% паладію-на-вугіллі у 20мл охолодженого перед тим до to 0°C дихлорметану, і МеОН гідрують протягом ночі при атмосферному утворену суміш перемішують при кімнатній темпетиску та при кімнатній температурі. Каталізатор ратурі протягом 24 годин. Реакційну суміш конценвідфільтровують на целіті (Ceiite®) і фільтрат контрують у вакуумі, залишок екстрагують ефіром, центрують у вакуумі. Залишок розчиняють у ацеорганічну фазу промивають 1Η розчином NaOH, тонітрилі, додають ефір та утворений осад сушать водою та сушать MgSO4 , а розчинник випарюють у при перемішуванні та сушать. Отримують 0,51г вакуумі. Отриманий продукт розчиняють у ацетоні очікуваного продукту; Темп.плавл.=258°С. та підкислюють до рН=1 додаванням гідрохлоридВиготовлення 3,7 ного ефіру, осад 3, що утворився, суша ть при пеГідрохлорид 1-(4ремішуванні та сушать Отримують. 0,7г очікуванопіперидил)циклопропанкарбоксаміду. го продукту. В) Гідрохлорид 2-Метил-1-(4-морфолініл)-2-(4піперидил)-1-пропанону. Суміш 0,7г отриманої на 3 попередніх етапах A) 1-(4-піридил)циклопропанкарбонітрил. сполуки, 0,7г форміату амонію та 0,2г 10% пала3,5г 4-піридилацетонітрилуу додають до сумідію-на-вугіллі у 10мл МеОН перемішують при кімші 2,5г аміду натрію у 80мл дихлорметану, а потім натній температурі протягом 4 годин. Каталізатор 2,6мл 1,2-диброметану, і суміш перемішують провідфільтровують на целіті (Celite®) та фільтрат тягом ночі при кімнатній температурі. Реакційну концентрують у вакуумі. Залишок розчиняють у суміш виливають у воду та екстрагують етилацеацетонітрилі, додають ефір та утворений осад татом, органічну фазу промивають водою та сусушать при перемішуванні і сушать. Отримують шать Na2SO4, а розчинник випарюють під вакуу0,46г очікуваного продукту; Темп.плавл.=225°С. мом. Залишок хроматографують на силікагелі, Приклад 1 елююючи дихлорметаном, а потім сумішшю дихМоногідрат гідрохлориду 3-[2-[4-(1 -карбамоїллорметан/МеОН від (99/1; за об'ємом) до (95/5; за 1-метилетил)-1 -піперидил]етил]-3-(3,4об'ємом). Отримують 2,5г очікуваного продукту. дихлорфеніл)-1-[2-(3,5B) Гідрохлорид 1-(4диметилфеніл)ацетил]піперидину, (-) ізомер. піридил)циклопропанкарбоксаміду. Суміш 2,5г отриманої на попередньому етапі сполуки та 20мл 96% розчину H2SO4 нагрівають швидко до 100°С та залишають перемішуватися протягом 1 години при 100°С. А) 3-(3,4-Дихлорфеніл)-3-(2-гідроксиетил)-1-[2Після охолодження до кімнатної температури, (3,5-диметилфеніл)ацетил]піперидин, одиничний реакційну суміш виливають на лід та нейтралізуізомер. ють до рН=7 додаванням 20% розчину NH4OH і 2,3мл триетиламіну додають до суміші 2,0г сушать утворений осад при перемішуванні, про 35 70986 36 3,5-диметилфенілоцтової кислоти у 100мл дихлорметану при кімнатній температурі, а потім 3г отриманої у Виготовленні 1 сполуки та 5,3г ВОР, і цю суміш перемішують протягом 1 години 0,23г отриманої у Виготовленні 3,2 сполуки при кімнатній температурі. Реакційну суміш конце(вільна основа) додають при кімнатній температурі нтрують у вакуумі, залишок екстрагують ефіром, в атмосфері азоту до розчину 0,5г отриманої на органічну фазу промивають водою, 2Η розчином етапі В прикладу 1 сполуки у 20мл дихлорметану, НСІ, водою, водним 10% розчином NaOH, сушать а потім 0,1мл оцтової кислоти, та суміш перемінад Na2SO4 та фільтрують, а фільтрат концентрушують при кімнатній температурі протягом 30 хвиють у вакуумі. Залишок хроматографують на силілин. Далі додають 0,55г триацетоксиборогідриду кагелі Н, елююючи дихлорметаном, а потім сумінатрію та суміш залишають перемішуватися при шшю ди хлорметан/МеОН (98/2; за об'ємом). кімнатній температурі протягом ночі. Водний 10% Отримують 3,9г очікуваного продукту, продукт вирозчин Na2CO3 додають та реакційну суміш перекористовують на наступному етапі без подальшої мішують протягом 15 хвилин при кімнатній темпеобробки. ратурі. Реакційну суміш екстрагують дихлорметаВ) 3-(3,4-дихлорфеніл)-3-(формілметил)-1-[2ном, органічну фаз у промивають водним 10% (3,5-диметилфеніл)ацел]піперидин, одиничний розчином Na2CO3, суша ть над Na2SO4 та фільтруізомер. ють, а фільтрат концентрують у вакуумі. Залишок Розчин 0,25мл оксалілхлориду у 3мл дихлорхроматографують на силікагелі, елююючи дихлометану охолоджують до -70°С, в атмосфері азоту, рметаном, а потім сумішшю дихлорметан/МеОН з додають краплями розчин 0,35мл диметилсульфоградієнтом від (99/1; за об'ємом) до (95/5;· за об'ксиду у 3мл дихлорметану, а потім розчин у 5мл ємом). Отриманий продукт розчиняють у дихлордихлорметану 0,5г отриманої на попередньому метані та підкислюють до рН=1 додаванням гідроетапі сполуки і суміш перемішують протягом 15 хлоридного ефіру, а утворений осад сушать при хвилин при -50°С. Далі додають 0,9мл триетиламіперемішуванні, промивають ефіром та сушать під ну та суміш залишають перемішуватися, даючи їй вакуумом. Отримують 0,4г очікуваного продукту. повернутися до кімнатної температури. Реакційну a D20=-37° (с=1, МеОН) 1 суміш промивають водою, 1Η розчином НСІ та Н ЯМР: δ (чнм): 0,6-2,3: гл: 18Н; 2,3-4,7: m: 10% розчином NaHCO3, органічну фазу сушать 16Н; 6,4-8,0: m: 8Н. Na2SO4 та фільтрують, та концентрують фільтрат Приклад 3 у вакуумі. Отримують 0,5г очікуваного продукту, Дигідрохлорид 3-[2-[4-(1-N,Nякий використовують на наступному етапі без подиметилкарбамот-1-метилетил)-1дальшої обробки. пiперазиніл]етил]-3-(3,4-дихлорфеніл)-1-[2-(3,5-) С) Моногідрат гідрохлориду 3-[2-[4-(1диметилфеніл)ацетил]піперидину 1,25 Н2О, (-) карбамоїл-1-метилетил)-1-піперидил]-етил-3-(3,4ізомер. дихлорфеніл)-1-[2-(3,5-диметилфеніл)ацетил]піперидину, (-) ізомер. 0,08мл оцтової кислоти додають при кімнатній температурі в атмосфері азоту до розчину 0,24 г До 20мл МеОН додають при кімнатній темпеотриманої у Виготовленні 3,1 сполуки (вільна осратурі 0,6г отриманої на етапі В сполуки прикладу нова) у 3мл МеОН, а потім розчин 0,5г отриманої 1, 0,3г отриманої у Виготовленні 3,4 сполуки, 0,1 на попередньому етапі сполуки у 5мл МеОН. мл оцтової кислоти, а потім 0,12г ціаноборогідриду Через 5 хвилин додають 0,08г ціаноборогідринатрію, та суміш перемішують протягом ночі при ду натрію та суміш залишають перемішуватися кімнатній температурі. До реакційної суміші додапри кімнатній температурі протягом ночі. Реакційють водний 10% розчин Na2CO3 i суміш залишають ну суміш виливають у водний 10% розчин NaHCO3 перемішува тися протягом 15 хвилин. Суміш екстта екстрагують ефіром, органічну фазу тричі прорагують етилацетатом, органічну фазу промивамивають водою, сушать Na2SO 4 та фільтр ують і ють водним 10% розчином Nа2СО3, водою, насиконцентрують фільтрат у вакуумі. Залишок хромаченим розчином NaCI та сушать над Na2SO4, а тографують на силікагелі Н, елююючи дихлормерозчинник випарюють у вакуумі. Залишок хроматотаном, а потім сумішшю дихлорметан/МеОН з граграфують на силікагелі, елююючи дихлорметаном, дієнтом від (99/1; за об'ємом) до (90/10; за а потім сумішшю дихлорметан/МеОН з градієнтом об'ємом). Отриманий продукт розчиняють у дихвід (99/1; за об'ємом) до (95/5; за об'ємом). Отрилорметані, підкислюють до рН=1 додаванням гідманий продукт розчиняють у дихлорметані та підрохлоридного ефіру та концентрують під вакуукислюють до рН=1 додаванням гідрохлоридного мом. Після розтирання з ефіром, сушки з ефіру, а утворений осад сушать при перемішуванперемішуванням та сушки під вакуумом отримують ні, промивають ефіром та сушать під вакуумом. 0,5г очікуваного продукту. Отримують 0,4г очікуваного продукту. 20 a D =-27,7° (с=1, МеОН) 1 a D20=-28,4°(c=1, MeOH) Н ЯМР: δ (чнм): 0,7-1,2; bs: 6Н; 1,2-2,4; m: 1 Н ЯМР: δ (чнм): 0,7 to 2,3; m: 18Н; 2,35 to 4,7; 16Н; 2,5-4,8: m: 12Н; 6,5-8,0: m: 8Н; 3 10,2: bs: 1H. m: 22Н; 6,5 to 7,8: m: 6H; 10,3: s: 1H. Приклад 2 Приклад 4 Дигідрохлорид 3-[2-[4-(1-карбамоїл-1Півторагідрат гідрохлориду 3-[2-[4-(1метилетил)-1-піперазиніл]етил-3-(3,4карбамоїл-1-метилетил)-1-піперидил]етил]-3-(3,4дихлорфеніл)-1-[2-(3,5-диметилфеніл)ацетил]дихлорфеніл)-1-[2-(3,5піперидину -2,7 Н2О, (-) ізомер. 37 дихлорфеніл)ацетил]піперидину, (-) ізомер. 70986 :А) 3-(3,4-дихлорфеніл)-3-(2-гідроксіетил)-1-[2(3,5-дихлорфеніл)ацетил]піперидин, одиничний ізомер. 4,75г отриманої у Виготовленні 1 сполуки, 3,55г отриманої у Виготовленні 2,1 сполуки, 3,6мл триетиламіну, а потім 8,4г ВОР додають при кімнатній температурі до 150мл дихлорметану і суміш залишають перемішуватися при кімнатній температурі протягом 2годин. Реакційну суміш концентрують у вакуумі, залишок екстрагують етилацетатом, органічну фазу промивають 1Η розчином НСІ, водою, 1Η розчином NaOH, водою, насиченим розчином NaCI та сушать над Na2SO4, а розчинник випарюють у вакуумі. Отримують 8г очікуваного продукту, продукт використовують без подальшої обробки. B) 3-(3,4-дихлорфеніл)-3-(формілметил)-1-[2(3,5-дихлорфеніл)ацетил]піперидин, одиничний ізомер. Цю сполуку виготовляють згідно зі способом, описаним у етапі В прикладу 1, починаючи з 0,25мл оксалілхлориду у 6мл дихлорметану, 0,38мл диметилсульфоксиду у 3мл дихлорметану, 1г отриманої на попередньому етапі сполуки у 6мл дихлорметану, а потім 1,5мл триетиламіну. Отримують 1,0г очікуваного продукту, продукт використовують без подальшої обробки. C) Півторагідрат гідрохлориду 3-[2-[4-(1карбамоїл-1-метилетил)-1-піперидил]-етил]-3-(3,4дихлорфеніл)-1-[2-(3,5-дихлорфеніл)ацетил]піперидину, (-) ізомер. Цю сполуку виготовляють згідно зі способом, що описано у етапі С прикладу 1, починаючи з 0,25г отриманої у Виготовленні 3,1 сполуки (вільна основа) у 3мл МеОН, 0,08мл оцтової кислоти, 0,5г отриманої на попередньому етапі сполуки у 5мл МеОН, а потім 0,08г ціаноборогідриду натрію. Отримують 0,52г очікуваного продукту. a D20=-0,6°(с=1, МеОН) Приклад 5 Моногідрат гідрохлориду 3-[2-[4-(1-карбамоїл1-метилетил)-1-піперидил]етил]-3-(3,4дихлорфеніл)-1-[2-[3,5-біс(трифлуорметил)феніл]ацетил]піперидину, (+) ізомер. А) 3-(3,4-дихлорфеніл)-3-(2-гідроксіетил)-1-[2[3,5 біс(трифлуорметил)феніл]ацетил]-піперидин, одиничний ізомер. 1,2г отриманої у Виготовленні 1 сполуки, 1,2г 3,5-біс(трифлуорметил)фенілоцтової кислоти, 1,7мл триетиламіну, а потім 2,16г ВОР додають при кімнатній температурі до 50мл дихлорметану та суміш перемішують протягом 15 хвилин. Реакційну суміш концентрують у вакуумі, залишок переносять у 0,1Η розчин НСІ та екстрагують ефіром, органічну фазу промивають 1Η розчином НСІ, водою, 1Η розчином NaOH, водою та сушать над Na2SO4, та розчинник випарюють у вакуумі. Отримують 2,1г очікуваного продукту, продукт використовують без подальшої обробки. 38 В)3-(3,4-дихлорфеніл)-3-(формілметил)-1-[2[3,5-біс(трифлуорметил)чЬеніл]ацетил]-піперидин, одиничний ізомер. 20мл дихлорметану охолоджують до -78°С, 1,5г отриманої на попередньому етапі сполуки, 0,45мл диметилсульфоксиду, а потім 0,3мл оксалілхлориду додають в атмосфері азоту і суміш далі залишають перемішуватися при -78°С протягом 30 хвилин. Потім додають 2мл триетиламіну і суміш перемішують, даючи їй повернутися до кімнатної температури. До реакційної суміші додають 1Η розчин НСІ, утворену суміш екстрагують дихлорметаном, органічну фазу промивають 1 Η розчином НСІ, водою, водним 10% розчином Nа2СO3та сушать над Na2SO4, а розчинник випарюють у вакуумі. Отримують 1,5г очікуваного продукту, продукт використовують без подальшої обробки. С) Моногідрат гідрохлориду 3-[2-[4-(1карбамоїл-1-метилетил)-1-піперидил]-етил]-3-(3,4дихлорфеніл)-1-[2-[3,5біс(трифлуорметил)феніл]ацетил]піперидину, (+) ізомер. Суміш 0,35г отриманої у Виготовленні 3,1 сполуки та 0,4г К2СО 3 у 10мл ацетонітрилу кип'ятять під зворотним холодильником протягом 3 годин. Нерозчинний матеріал відфільтровують та концентрують фільтрат у вакуумі. Продукт з виготовлення 3,1 у формі отриманої так вільної основи розчиняють у 3мл МеОН, додають 0,08мл оцтової кислоти, а потім розчин 0,5г отриманої на попередньому етапі сполуки у 5мл МеОН, і суміш залишають перемішуватися при кімнатній температурі протягом 5 хвилин. Далі додають 0,08г ціаноборогідриду натрію і суміш залишають перемішуватися протягом ночі при кімнатній температурі. Реакційну суміш виливають у водний 10% розчин NaHCO3 та екстрагують ефіром, органічну фазу промивають водою та сушать Na2SO4, а розчинник випарюють у вакуумі. Залишок хроматографують на силікагелі Н, елююючи дихлорметаном, а потім сумішшю дихлорметан/МеОН з градієнтом від (99/1; за об'ємом) до (90/10; за об'ємом). Отриманий продукт розчиняють у дихлорметані та підкислюють до pH=1 додаванням гідрохлоридного ефіру, утворений осад сушать при перемішуванні. Це дає після сушки під вакуумом 0,54г продукту. aD20=+28,2°(c=1,MeOH) 1 Н ЯМР: δ (чим): 0,6-2,2: m: 16Н; 2,3-4,2:m: 12Н; 6,6-8,0: m: 8Н; 10,3: s: 1H. Приклад 6 Напівгідрат гідрохлориду 3-[2-[4-(1-N,Nдиметилкарбамот-1-метилетил)-1-піперидил]етил]3-(3,4-дихлорфеніл)-1-[2-(3,5диметилфеніл)ацетил]піперидину, (-) ізомер. Суміш 0,35г отриманої у Виготовленні 3,6 сполуки та 0,4г К2СО 3 у 10мл ацетонітрилу кип'ятять під зворотним холодильником протягом 3 годин. Нерозчинний матеріал відфільтровують та концентрують фільтрат у вакуумі. Продукт з виготовлення 3,6 у формі отриманої так вільної основи розчиняють у 3мл МеОН, додають 0,1мл оцтової 39 70986 40 кислоти, а потім розчин 0,6г отриманої на етапі В B) 3-(3,4-дихлорфеніл)-3-(формілметил)-1-[2прикладу 1 сполуки у 5мл МеОН, та суміш зали(3,5-діетилфеніл)ацетил]піперидин, одиничний шають перемішуватися при кімнатній температурі ізомер. протягом 5 хвилин. Далі додають 0,1г ціаноборогіРозчин 0,5г отриманої на попередньому етапі дриду натрію та суміш залишають перемішуватися сполуки у 10мл дихлорметану охолоджують до при кімнатній температурі протягом ночі. Реакцій78°С в атмосфері азоту, додають 0,23мл диметилну суміш виливають у водний 10% розчин NaHCO3 сульфоксиду, а потім 0,16мл оксалілхлориду, і та екстрагують ефіром, органічну фазу промивасуміш перемішують при -78°С протягом 30 хвилин. ють водою та сушать MgSO 4, а розчинник випаДодають 0,95мл триетиламіну далі та суміш залирюють у вакуумі. Залишок хроматографують на шають перемішуватися, даючи їй повернутися до силікагелі Н, елююючи дихлорметаном, а потім кімнатної температури. До реакційної суміші досумішшю дихлорметан/МеОН з градієнтом від дають 1Η розчин НСІ, утворену суміш екстрагують (99/1; за об'ємом) до (90/10; за об'ємом). Отримадихлорметаном, органічну фазу промивають 1Η ний продукт розчиняють у дихлорметані та підкисрозчином НОІ, водою, 10% розчином Na2CO3Ta люють до рН=1 додаванням гідрохлоридного ефісушать над Na2SO4, а розчинник випарюють у вару, а розчинник випарюють під вакуумом. Після куумі. Отримують 0,5г очікуваного продукту, пророзтирання з ефіром, сушки з перемішуванням та дукт використовують без подальшої обробки. сушки отримують 0,68г очікуваного продукту; C) Напівгідрат гідрохлориду 3-[2-[4-(1Темп.плавл.=202°С. карбамоїл-;1-метилетил)-1-піперидил]-етил]-3-(3,4aD20=-27,17° (с=1, МеОН) дихлорфеніл)-1-[2-(3,5-діетилфеніл)1 Н ЯМР: 6 (чнм): 0,6-2,5: m: 23Н; 2,5-4,6: m: ацетил]піперидину, (-) ізомер. 18Н; 6,4-7,8: m: 6Н; 10,1: s: 1H. При кімнатній температурі в атмосфері азоту Приклад 7 до розчину 0,23г отриманої у Виготовленні 3,1 Напівгідрат гідрохлориду 3-[2-[4-(1-карбамоїлсполуки (вільна основа) у 3мл МеОН додають 0,08 1-метилетил)-1 -піперидил]етил]-3-(3,4мл оцтової кислоти, а потім розчин 0,5г отриманої дихлорфеніл)-1 -[2-(3,5на попередньому етапі сполуки у 5мл МеОН. Чедіетилфеніл)ацетил]піперидин, (-) ізомер. рез 5 хвилин, додають 0,08г ціаноборогідриду натрію і суміш залишають перемішуватися протягом ночі при кімнатній температурі. Реакційну суміш виливають у водний 10% розчин NaHCO3 та екстА) 3-(3,4-дихлорфеніл)-3-(2-гідроксіетил)-1-[2рагують е фіром, органічну фазу промивають во(3,5-діетилфеніл)ацетил]піперидин, одиничний дою та суша ть MgSO 4, а розчинник випарюють у ізомер. вакуумі. Залишок хроматографують на силікагелі При кімнатній температурі 1,15г 3,5Н, елююючи дихлорметаном, а потім сумішшю діетилфенілоцтової кислоти додають до суміші дихлорметан/МеОН з градієнтом від (99/1; за об'1,64г отриманої у Виготовленні 1 сполуки у 30мл ємом) до (93/7; за об'ємом). Отриманий продукт дихлорметану, а потім 3мл триетиламіну та 3,2г розчиняють у дихлорметані та підкислюють до ВОР, і суміш перемішують при кімнатній темперарН=1 додаванням гідрохлоридного ефіру, а розтурі протягом 2 годин. Реакційну суміш концентчинник випарюють під вакуумом. Після розтирання рують у вакуумі, залишок переносять у 1Η розчин з ефіром, сушки з перемішуванням та сушки отриНСІ та екстрагують ефіром, органічну фазу промимують 0,51г очікуваного продукту. вають 1Η розчином НСІ, водою, 1Η розчином a D20=-30,5° (с=1, МеОН) 1 NaOH, водою, насиченим розчином NaCI та суН ЯМР; δ (чнм): 0,5-2,2: m: 23Н; 2,2-4,65:m: шать над Na2SO4, а розчинник випарюють у ваку16Н; 6,4-7,8: m: 8Н; 9,85: s: 1H. умі. Залишок хроматографують на силікагелі, Обробкою згідно зі способами, описаними у елююючи сумішшю ди хлорметан/МеОН з градієнвищенаведених прикладах, виготовляють сполуки том від (99/1; за об'ємом) до 95/5; за об'ємом). згідно з винаходом, які представлено у нижченаОтримують 1,1г очікуваного продукту, продукт виведеній таблиці 1. користовують без подальшої обробки. 41 70986 42 43 70986 Цю сполуку виготовляють згідно зі способом, описаним у етапі С прикладу 1, починаючи з отриманої на етапі В прикладу 1 сполуки та отриманої у Виго товленні 3,5 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у Прикладі 3, починаючи з отриманої на етапі В прикладу 1 сполуки та отриманої у Виготовленні 3,3 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у етапі С прикладу 4, починаючи з отриманої на етапі В прикладу 4 сполуки та отриманої у Виготовленні 3,5 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у Прикладі 3, починаючи з отриманої на етапі В прикладу 4 сполуки та отриманої у Виготовленні 3,2 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у Прикладі 3, починаючи з отриманої на етапі В прикладу 4 сполуки та отриманої у Виготовленні 3,4 сполуки. Цю сполуку виготовляють згідно зі способом, що описано у Прикладі 3, починаючи з отриманої на етапі В прикладу 4 сполуки та отриманої у Виготовленні 3,3 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у етапі С прикладу 5, починаючи з отриманої на етапі В сполуки прикладу 5 та отриманої у Виго товленні 3,5 сполуки. Цю сполуку виготовляють згідно зі способом, що описано у Прикладі 3, починаючи з отриманої на етапі В сполуки прикладу 5 та отриманої у Виготовленні 3,3 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у Прикладі 2, починаючи з отриманої на етапі В прикладу 1 сполуки та отриманої у Виготовленні 3,7 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у етапі С прикладу 5, починаючи з отриманої на етапі В сполуки прикладу 5 та отриманої у Виго товленні 3,7 сполуки. Цю сполуку виготовляють згідно зі способом, що описано у етапі С прикладу 4, починаючи з отриманої на етапі В прикладу 4 сполуки та отриманої у Виготовленні 3,7 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у етапі С прикладу 7, починаючи з отриманої на етапі В сполуки прикладу 7 та отриманої у Виготовленні 3,2 сполуки у формі вільної основи. Цю сполуку виготовляють згідно зі способом, що описано у етапі С прикладу 1, починаючи з отриманої на етапі В прикладу 1 сполуки та отриманої у Виготовленні 3,8 сполуки у формі вільної основи. Приклад 8: 1Н ЯМР: δ (чнм): 0,7-2,2: m: 27Н; 2,3-4,6: m: 14Н; 6,4-7,7: m: 8Н; 10,1: s: 1Н. Приклад 9:1Н ЯМР: δ (чнм): 0. 6-2,35: m: 22Н; 2,4-4,6: m: 14Н; 6,4-8,2: m: 8Н. 20 Приклад 10: 1Н ЯМР: δ (чнм): 0,7-2,25: m: 21Н; 2,3-4,4: m: 12Н; 6,7-7,8: m: 8Н; 10,1: s:1H. Приклад 11: 1Н ЯМР: δ (чнм): 0,6-2,2: m: 12Н; 2,3-4,4: m: 16Н; 6,8-8,0: m: 8Н. 25 44 Приклад 12: 1Н ЯМР: δ (чнм): 0,8-2,3: m: 12Н; 2,35-4,4: m: 22Н; 7,0-7,9: m: 6Н; 10,6: s: 1H. Приклад 13: 1Н ЯМР: δ (чнм): 0,9-2,3: m: 16Н; 2,35-4,5: m: 16Н; 7,0-7,9: m: 8Н. Приклад 14: 1Н ЯМР: δ (чнм): 0,9-2,3: m: 21Н; 2,4-4,3: m: 12Н; 6,8-8,1: m: 8Н; 10,0: s: 1H. Приклад 15: 1Н ЯМР: δ (чнм): 1,0-2,4: m: 16Н; 2,5-4,5: m: 16Н; 6,9-8,1: m: 8Н; 11,0: bs1H. Приклад 16: 1Н ЯМР: δ (чнм): 0,4-2,3: m: 20Н; 2,4-4,6: m: 13Н; 6,5-7,7: m: 8Н; 9,6: s: 1Н. Приклад 17: 1Н ЯМР: δ (чнм): 0,4-2,2: m: 14Н; 2,3-4,4: m: 13Н; 6,5-7,8: m: 8Н; 9,9: s: 1Н. Приклад 18: 1Н ЯМР: δ (чнм): 0,4-2,2: m: 14Н; 2,3-4,4: m: 13Н; 6,6-7,8: m: 8Н; 9,9: s: 1Н. Приклад 19:1Н ЯМР: δ (чнм): 0,6-2. 6: m: 22Н; 2,6-4,8: m: 16Н; 6,5-8,0: m: 10Н. Приклад 20: 1Н ЯМР: δ (чнм): 0,7-2,25: m: 22Н; 2,3-4,6: m: 21Н; 6,4-7,7: m: 6Н; 10,4: s: 1H. Приклад 21 Гідрохлорид 3-[2-[4-(1-карбамоїл-1 метилетил)-1 -піперидил]етил]-3-(3,4-диметилфеніл)-1-[2-(3,5-дихлорфеніл)ацетил]піперидину. A) 1 -[2-(3,5-дихлорфеніл)ацетил]-3-(3,4диметилфеніл)-3-[2-(2тетрагідропіранілокси)етил]піперидин. Суміш 3г отриманої у Виготовленні 1,2 сполуки, 1,3г отриманої у Виготовленні 2,1 сполуки, 3,2мл триетиламіну та 4,8г ВОР у 100мл дихлорметану перемішують протягом 2 годин при кімнатній температурі. Реакційну суміш концентрують у вакуумі, залишок переносять у 1Η розчин НСІ та екстрагують етилацетатом, органічну фазу промивають водою, 1Η розчином NaOH, насиченим розчином NaCI та сушать над Na2SO4, a розчинник випарюють у вакуумі. Отримують 4,5г очікуваного продукту. B) 1-[2-(3,5-дихпорфеніл)ацетил]-3-(3,4диметилфеніл)-3-(2-гідроксіетил)піперидин. Суміш 4,5г отриманої на попередньому етапі сполуки та 2мл концентрованого розчину НСІ у 10мл МеОН перемішують протягом 2 годин при кімнатній температурі. Реакційну суміш концентрують у вакуумі, залишок переносять у МеОН та розчинник випарюють у вакуумі. Залишок хроматографують на силікагелі, елююючи дихлорметаном, а потім сумішшю дихлорметан/МеОН з градієнтом від (99/1; за об'ємом), до (95/5; за об'ємом). Отримують 3г очікуваного продукту. C) 1-[2-(3,5-дихлорфеніл)ацетил]-3(формілметил)-3-(3,4-диметилфеніл)піперидин. 10мл дихлорметану охолоджують до -78°С, додають, в атмосфері азоту 0,5г отриманої на попередньому етапі сполуки та 0,18мл диметилсульфоксиду, а потім 0,13мл оксалілхлориду і суміш залишають перемішуватися при -78°С протягом 30 хвилин. Далі додають 0,75мл триетиламіну та суміш залишають перемішуватися, даючи їй нагрітися до кімнатної температури. До реакційної суміші додають 1Η розчин НСІ, утворену суміш екстрагують дихлорметаном, органічну фазу промивають водою, 10% розчином Na2CO3 Ta сушать над 45 70986 46 Na2SO4, а розчинник випарюють у вакуумі. Отрипідкислюють до рН=1 додаванням гідрохлоридного мують 0,5г очікуваного продукту. ефіру, а утворений осад сушать при перемішуванD) Гідрохлорид 3-[2-[4-(1-карбамоїл-1ні. Отримують 0,35г очікуваного продукту. 1 метилетил)-1-піперидил]-етил]-3-(3,4Н ЯМР: δ (чнм): 0,8-2,3: m: 22Н; 2,3-4,0: m: диметилфеніл)-1-[2-(3,5-дихлорфеніл)13Н; 6,5-7,6: m: 8Н; 9,5: s: 1H. ацетил]піперидину. Приклад 22 Суміш 0,5г отриманої на попередньому етапі Дигідрохлорид 3-[2-[4-(1-карбамоїл-1сполуки, 0,35г отриманої у Виготовленні 3,1 сполуметилетил)-1-піперазиніл]етил]-3-(3,4ки (вільна основа), 0,1мл оцтової кислоти та 0,15г диметилфеніл)-1-[2-(3,5ціаноборогідриду натрію у 30мл МеОН залишають дихлорфеніл)ацетил]піперидину, 1 Н2О. перемішува тися протягом ночі при кімнатній температурі. До реакційної суміші додають 10% розчин Na2CO3, утворену суміш залишають перемішуватися протягом 15 хвилин та екстрагують Цю сполуку виготовляють згідно зі способом, етилацетатом, органічну фазу промивають водою, що описано у етапі D прикладу 21, починаючи з насиченим розчином NaCI та сушать над Na2SO4, отриманої на етапі С прикладу 21 сполуки та а розчинник випарюють у вакуумі. Залишок хромаотриманої у Виготовленні 3,2 сполуки (вільна тографують на силікагелі, елююючи дихлорметаоснова). 1 ном, а потім сумішшю ди хлорметан/МеОН з градіН ЯМР: 5(чнм): 1,4; 1s: 6Н; 2,2: 2s: 6H; 1,3-4,0: єнтом (99/1; за об'ємом) до (95/5; за об'ємом). m: 26Н; 7,0-8,0: m: 6H. Отриманий продукт розчиняють у дихлорметані та Комп’ютерна в ерстка Д. Шев ерун Підписне Тираж 37 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійською(1-aracyl-3-aryl-3-piperidylethyl)piperidine, a method for the preparation threeof and pharmaceutical composition composed of such derivatives
Автори англійськоюEmon-Alt Ksave
Назва патенту російськоюПроизводные (1-арацил-3-арил-3-пиперидилетил)пиперидина, способ их получения и фармацевтическая композиция, которая имеет в своем составе такие производные
Автори російськоюЭМОН-АЛЬТ Ксавье
МПК / Мітки
МПК: A61P 9/00, A61P 1/00, A61P 25/04, C07D 211/34, A61P 25/06, C07D 211/26, A61P 25/00, A61P 11/08, A61P 1/04, A61P 29/00, A61K 31/4545, A61P 37/00, A61P 13/02, A61P 1/08, A61P 25/24, A61P 43/00, A61P 17/00, A61P 25/22, A61P 11/00, A61P 13/00
Мітки: похідні, має, такі, отримання, своєму, 1-арацил-3-арил-3-піперидилетил)піперидину, складі, фармацевтична, композиція, спосіб
Код посилання
<a href="https://ua.patents.su/23-70986-pokhidni-1-aracil-3-aril-3-piperidiletilpiperidinu-sposib-kh-otrimannya-ta-farmacevtichna-kompoziciya-shho-maeh-u-svoehmu-skladi-taki-pokhidni.html" target="_blank" rel="follow" title="База патентів України">Похідні (1-арацил-3-арил-3-піперидилетил)піперидину, спосіб їх отримання та фармацевтична композиція, що має у своєму складі такі похідні</a>
Похідні піперазину та піперидину, спосіб їх отримання, фармацевтична композиція, спосіб отримання фармацевтичної композиції і спосіб лікування розладів цнс

Номер патенту: 52656
Опубліковано: 15.01.2003
Автори: Лонг Стівен К., Тульп Мартінус Т.М., Крузе Конеліс Г., КЬОЙПЕРС Вільма, Фенстра Рулоф В.
МПК: A61P 25/04, C07D 333/54, A61K 31/357, A61K 31/495, A61K 31/55, C07D 409/10, C07D 311/18, C07D 209/08, A61P 43/00, C07D 409/12, C07D 263/58, C07D 327/00, C07D 215/12, A61K 31/496, C07D 405/04, C07D 307/79, A61K 31/00, A61K 31/423, C07D 215/38, C07D 319/00, C07D 265/36, C07D 317/66, C07D 413/10, A61K 31/343, C07D 407/12, A61K 31/34, A61K 31/5513, A61K 31/551, C07D 321/00, C07D 407/10, A61K 31/42, C07D 413/12
Мітки: композиції, фармацевтично, піперазину, піперидину, фармацевтична, похідні, спосіб, отримання, лікування, цнс, розладів, композиція
Формула / Реферат:
1. Сполуки загальної формули (а),,де- А являє собою гетероциклічну групу з 5-7 кільцевих атомів, у якій присутні 1-3 гетероатоми з групи О, N та S,- R1 - водень чи фтор ,- R2 - С1-4 - алкіл, С1-4-алкокси або оксогрупа, а р дорівнює 0, 1 чи 2,- Z являє собою вуглець чи азот, а пунктирною лінією позначено простий зв'язок, якщо Z - азот або простий чи подвійний зв'язок, якщо Z - вуглець,- R3 та R4...
Похідні піперидину з аналгезивною дією, спосіб їх отримання та фармацевтична композиція на їх основі

Номер патенту: 65552
Опубліковано: 15.04.2004
Автори: Робертс Едвард, Делорм Даніель, Вей Жонгйонг
МПК: A61P 29/00, A61K 31/4427, A61K 31/4409, A61K 31/4709, A61K 31/4436, C07D 211/46, C07D 401/06, A61K 31/443, A61K 31/661, C07D 211/70, C07D 409/06, C07D 405/06, C07F 9/59, C07D 211/22
Мітки: отримання, спосіб, основі, похідні, піперидину, дією, композиція, фармацевтична, аналгезивною
Формула / Реферат:
1. Сполука загальної формули (І),(I)де R1 вибирають з водню, розгалуженого або нерозгалуженого С1-С6-алкілу, С1-С6-алкенілу, С3-С8-циклоалкілу, С4-С8-(алкілциклоалкілу), де алкіл являє собою С1-С2-алкіл, і циклоалкіл являє собою С3-С6-циклоалкіл;С6-С10-арилу або гетероарилу, що має від 5 до 10 атомів, вибраних з С, S, N і О; де арил або гетероарил може бути необов'язково і незалежно заміщений 1 або 2 замісниками,...
Алкіламінозаміщені похідні бензодіоксану, бензофурану або бензопірану для релаксації фундального відділу шлунка, спосіб їх отримання (варіанти) та фармацевтична композиція на їх основі

Номер патенту: 70308
Опубліковано: 15.10.2004
Автори: Фершуерен Вім Гастон, Вігерінк Піт Том Берт Пауль, де Брюйн Марк Франс Леопольд, Шровен Марк Франсіс Жозефін
МПК: A61K 31/454, A61K 31/506, A61K 31/4166, A61P 43/00, A61P 1/00, C07D 405/14, C07D 405/12, A61K 31/443, C07D 417/12
Мітки: спосіб, бензофурану, композиція, варіанти, релаксації, похідні, отримання, алкіламінозаміщені, основі, бензодіоксану, фундального, бензопірану, шлунка, відділу, фармацевтична
Формула / Реферат:
1. Алкіламінозаміщені похідні бензодіоксану, бензофурану або бензопірану формули (І) ,їх стереохімічно ізомерна форма, їх N-оксидна форма або їх фармацевтично прийнятна кислотно-адитивна сіль, деAlk1 означає С1-4-алкілкарбоніл, С1-4-алкілкарбоніл-С1-4-алкіл, карбоніл, карбоніл-С1-4-алкіл або С1-6-алкандіїл, необов'язково заміщений гідроксильною групою, С1-4-алкоксигрупою, С1-4-алкілкарбонілоксигрупою,...
Арил- та гетероарилзаміщені гетероциклічні похідні сечовини, фармацевтична композиція та спосіб лікування захворювання, спричиненого кіназою raf

Номер патенту: 67763
Опубліковано: 15.07.2004
Автори: Сіблі Роберт, Лоуінгер Тімоті Бруно, Вуд Джілл Е., Рідл Бернд, Кхайер Юдей, Скотт Уільям Джей, Паульсен Хольгер, Думас Жак, Сміт Роджер А., Джонсон Джеффрі, Редмен Аніко, Хатоум-Мокдад Холіа
МПК: A61K 31/4427, C07D 231/40, A61P 35/00, C07D 401/12, C07D 401/04, A61K 31/4439, C07D 405/04, C07D 403/04, A61K 31/506, A61K 31/443, C07D 401/14, C07D 307/66, A61K 31/381, A61K 31/415, A61K 31/341, A61P 43/00, C07D 333/36
Мітки: лікування, композиція, гетероарилзаміщені, фармацевтична, сечовини, спосіб, арил, захворювання, похідні, гетероциклічні, спричиненого, кіназою
Формула / Реферат:
1. Сполука формули І та її фармацевтично прийнятні соліде А являє собою гетероарил, що його вибирають з групи, до якої належать, , ,де...
Похідні 5-арил-3-(8-азабіцикло[3.2.1]окт-3-іл)-1,3,4-оксадіазол-2(3н)-ону як ліганди рецептора 5-ht4, спосіб їх одержання, фармацевтична композиція та лікарський засіб на їх основі

Номер патенту: 57071
Опубліковано: 16.06.2003
Автори: Галлі Фредерік, Локхед Алістер, Жегам Самір, Неделек Ален, Самсон Аксель, Галле Т'єррі
МПК: A61P 9/00, A61P 13/00, A61P 1/00, A61P 43/00, A61P 25/00, C07D 451/04, A61K 31/46
Мітки: 5-арил-3-(8-азабіцикло[3.2.1]окт-3-іл)-1,3,4-оксадіазол-2(3н)-ону, спосіб, композиція, лікарський, одержання, фармацевтична, похідні, засіб, рецептора, ліганди, основі, 5-ht4
Формула / Реферат:
1. Похідні 5-арил-3-(8-азабіцикло[3.2.1]окт-3-іл)-1,3,4-оксадіазол-2(3Н)-ону у формі чистого геометричного чи оптичного ізомеру або суміші ізомерів загальної формули (І), (I)де R1 - (С1-С4)алкіл або (С3-С7)циклоалкілметил,Х1 - атом гідрогену чи галогену або (С1-С4)алкоксил, або інакшеOR1 та Х1 разом утворюють групу формул:-ОСН2О-, -O(СН2)2-, -O(СН2)3-, -O(СН2)2О- або -O(СН2)3О-,Х2 - атом гідрогену чи...
Попередній патент: Спосіб виготовлення гвинта
Наступний патент: Спосіб обробки гарячеоцинкованої листової сталі
Випадковий патент: Ланцюговий редуктор