Процес виготовлення вуглеводневих продуктів
Номер патенту: 91042
Опубліковано: 25.06.2010
Автори: Ходжес Майкл Г., Кеклер Кеннет Пол, Корма Авеліно, Нокс Томас, Гріноу Пол












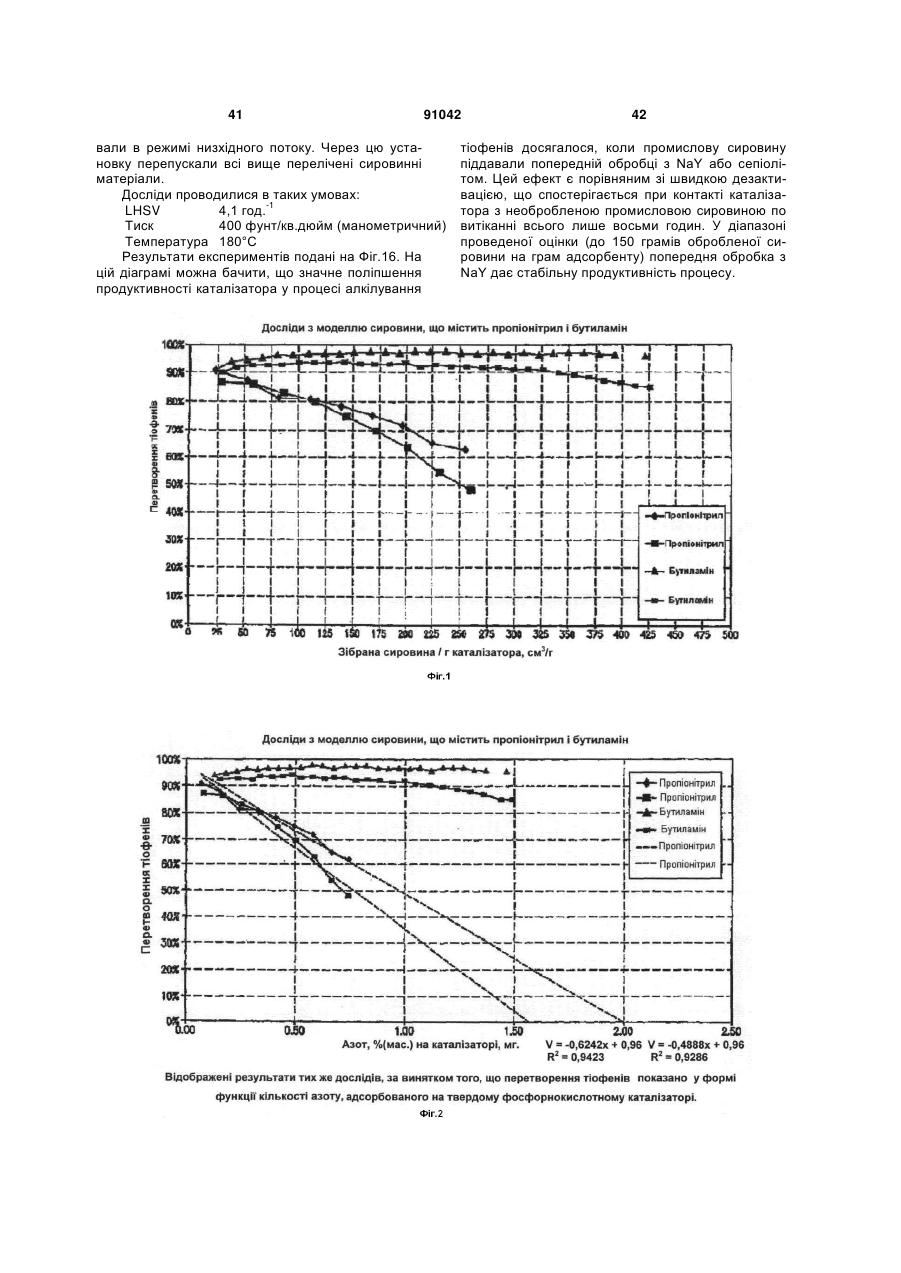
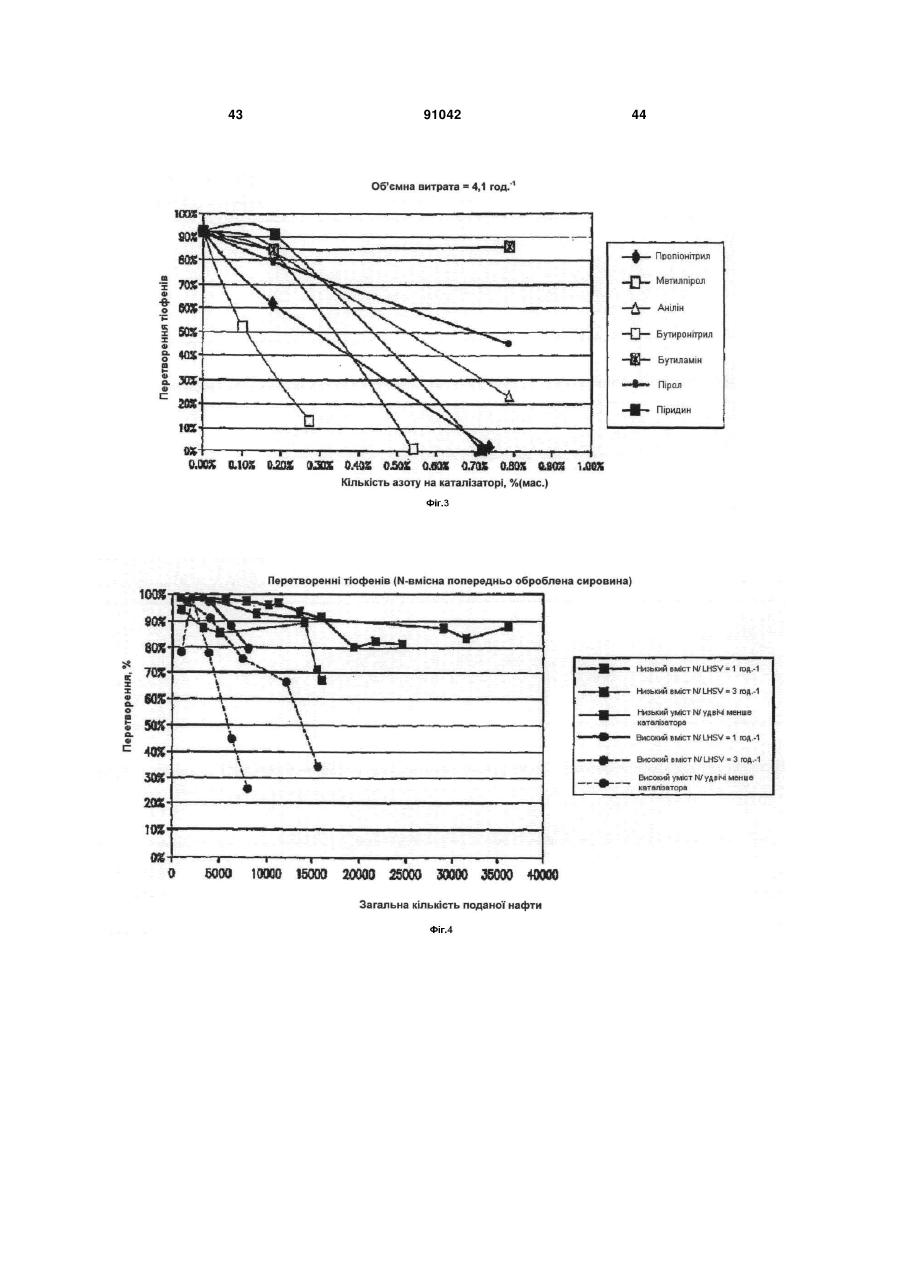
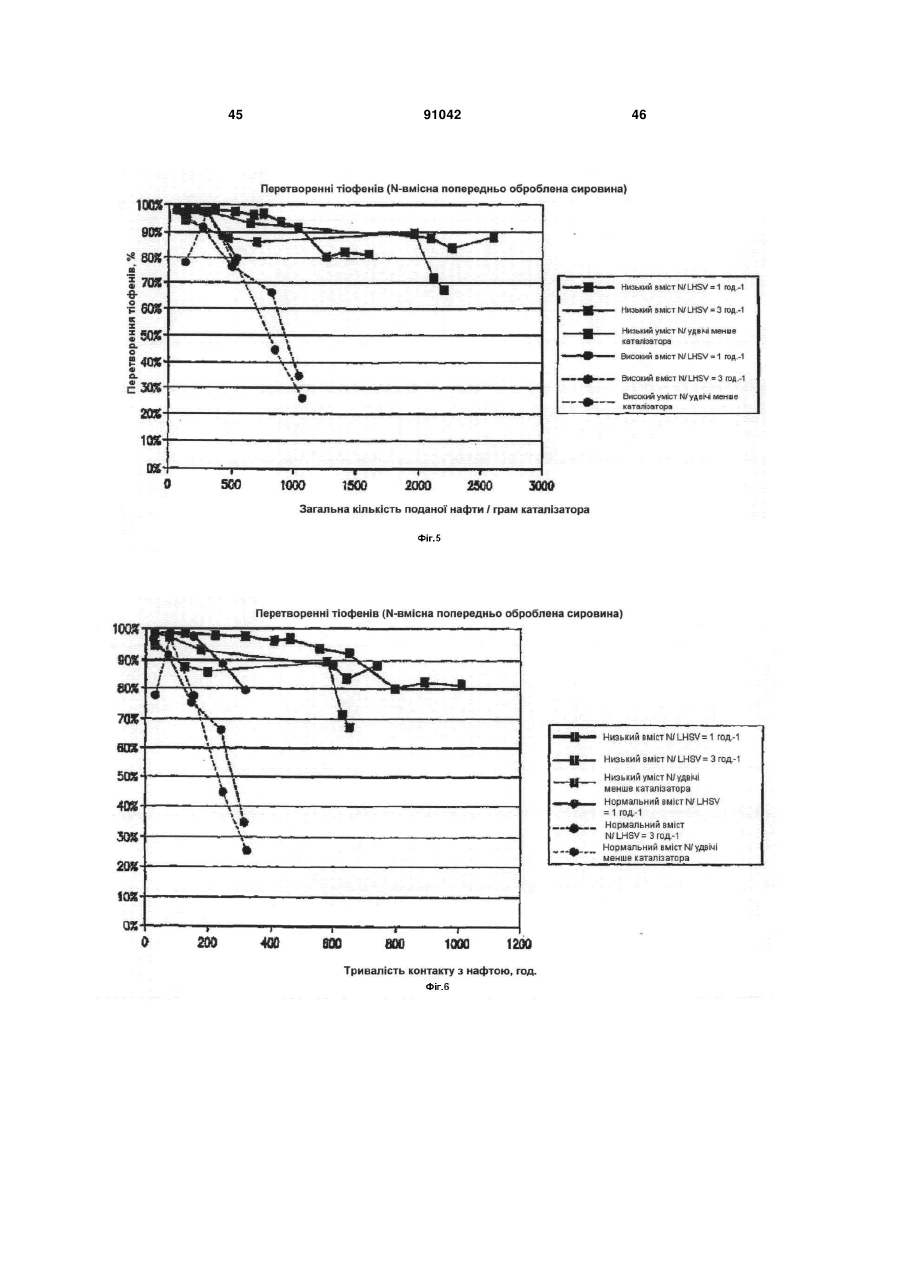
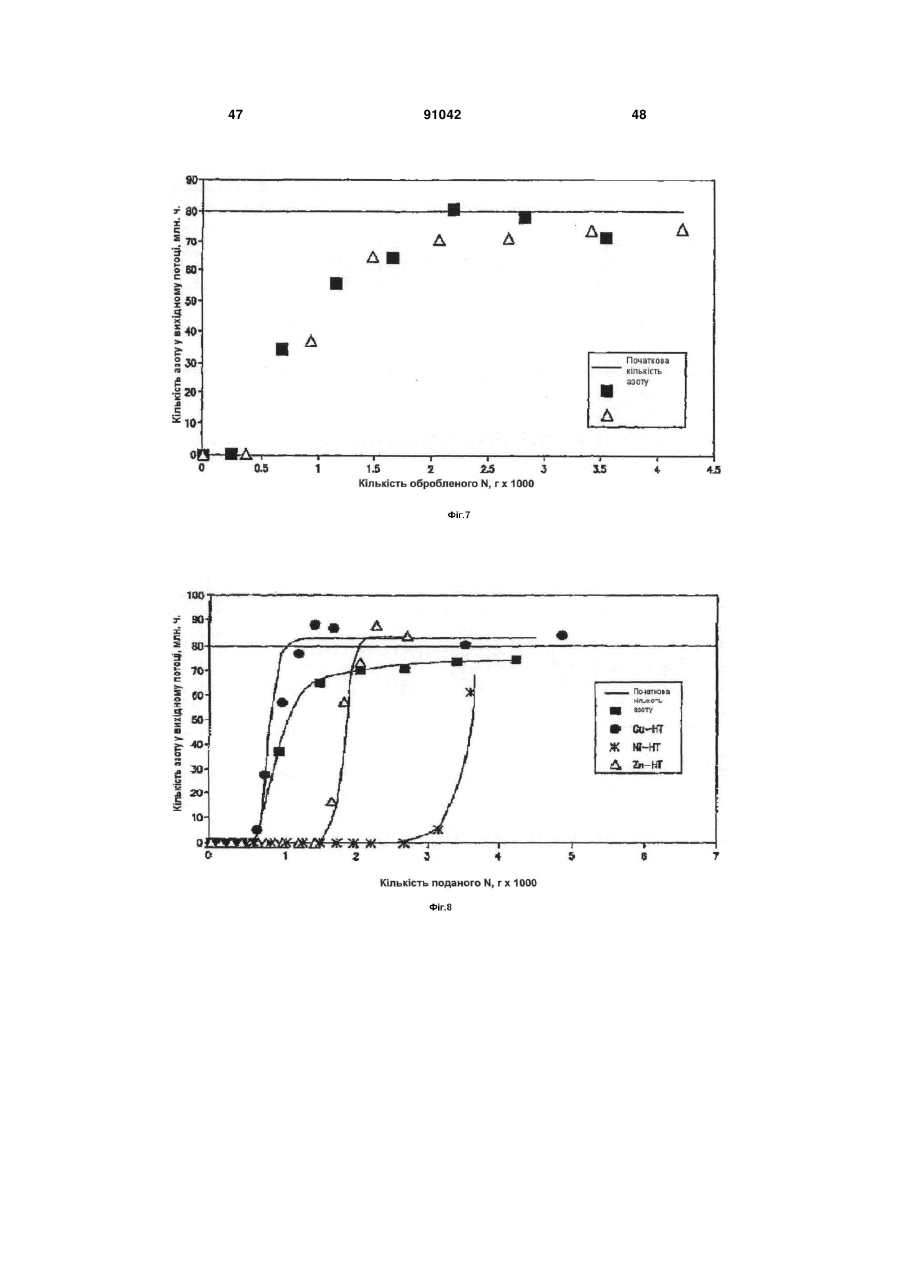
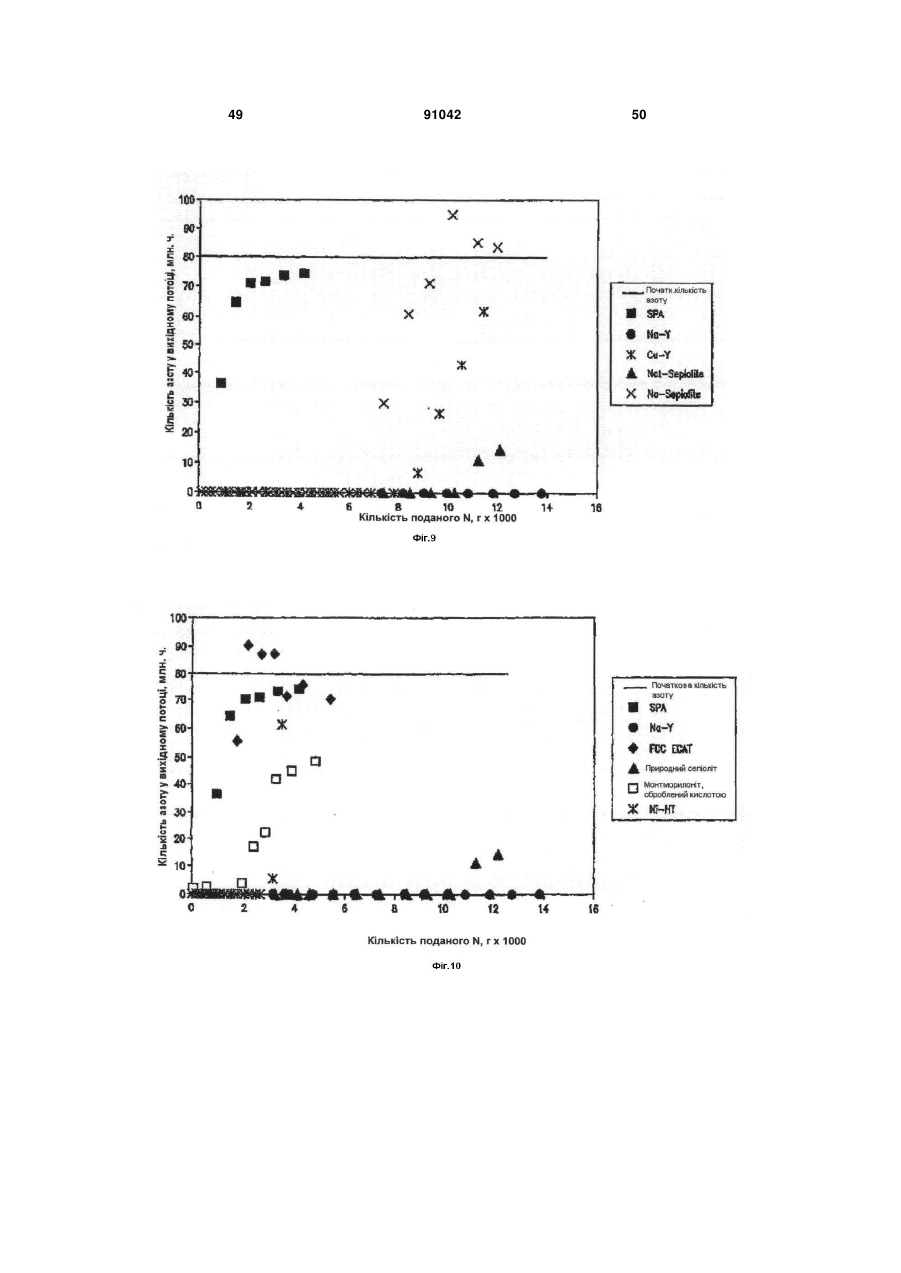
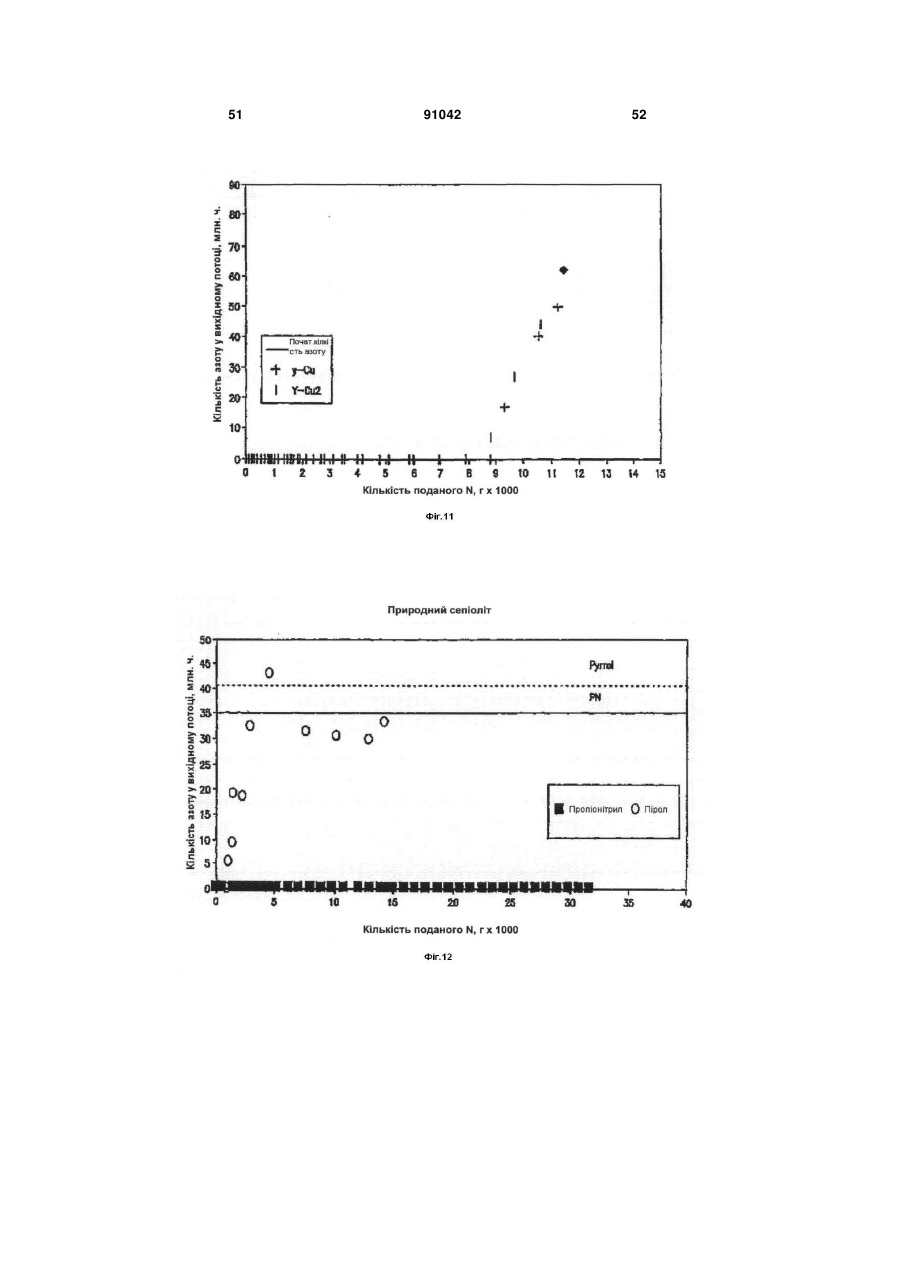
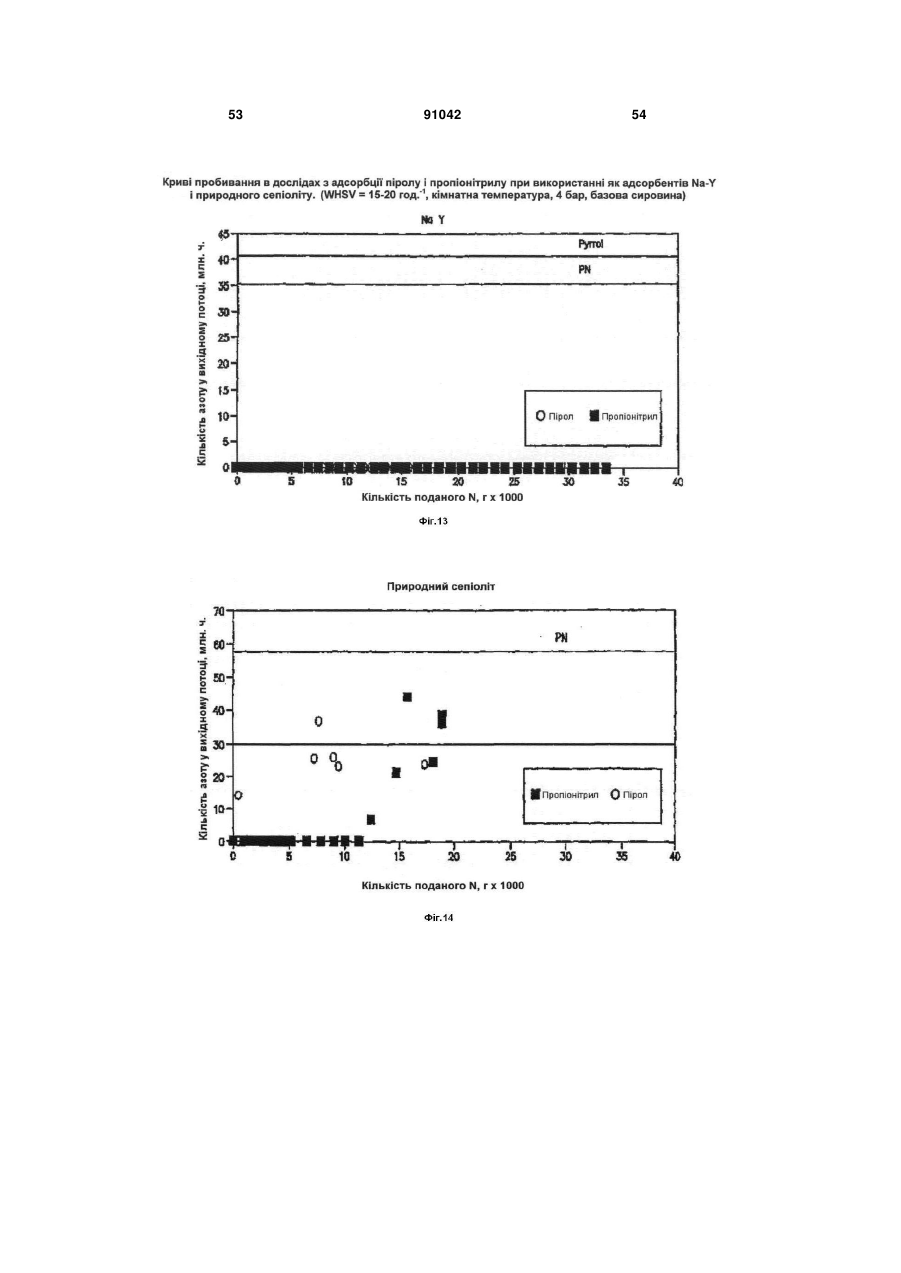
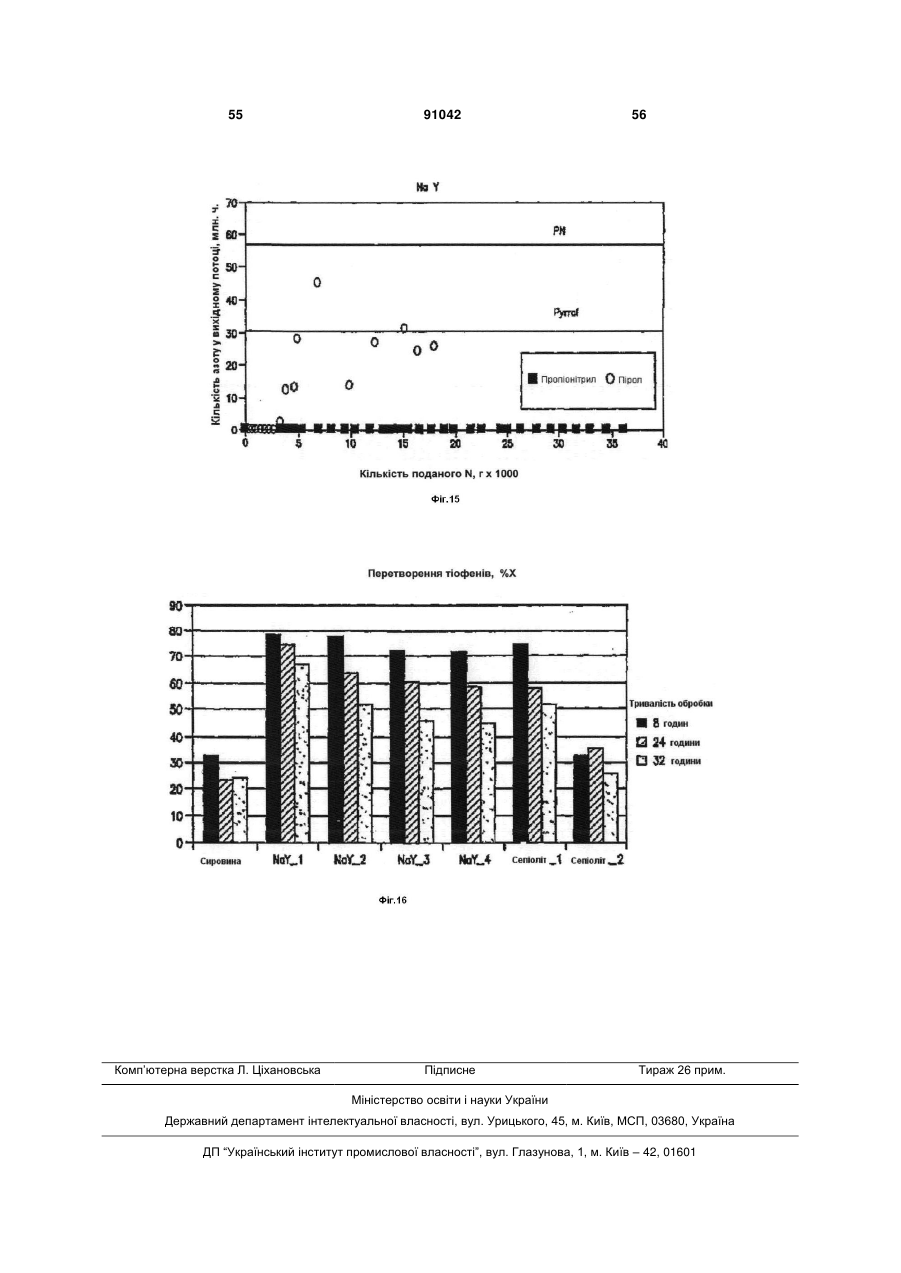
Формула / Реферат
1. Процес виготовлення вуглеводневих продуктів, що є рідкими у навколишніх умовах і містять органічні сполуки сірки більшої молекулярної маси, ніж органічні сполуки сірки у вуглеводневій сировині, який включає:
- постачання вуглеводневої сировини, що містить речовину, яка кипить при температурах в інтервалі приблизно від 60 до 425 °С і яка містить сірковмісні сполуки з рівнем вмісту сірки до 5000 частин на млн. і азотовмісні сполуки з рівнем вмісту азоту до 2000 частин на млн., включаючи неосновні азотовмісні органічні сполуки з рівнем вмісту азоту до 200 частин на млн., і має високий вміст олефінів до 60 % мас.,
- вилучення із вуглеводневої сировини неосновних азотовмісних сполук за допомогою процесу адсорбції з одержанням вихідного потоку, що містить зменшену кількість неосновних азотовмісних сполук, і
- приведення в контакт вихідного потоку з кислотним каталізатором в умовах алкілування, ефективних для перетворення частини сірковмісних сполук на сірковмісні сполуки більшої молекулярної маси і вищої температури кипіння, за допомогою алкілування олефінами з одержанням рідкого вихідного потоку, що містить сірковмісні сполуки більшої молекулярної маси і вищої температури кипіння.
2. Процес за п. 1, який відрізняється тим, що адсорбент, який використовують в процесі адсорбції, містить цеоліти, які мають структуру фожазиту.
3. Процес за п. 1, який відрізняється тим, що адсорбент, який використовують в процесі адсорбції, вибирають з групи: лужні фожазити, лужноземельні фожазити, лужні фожазити, частково обмінені з Н+ або перехідними металами IB, IIB, IV, VIII груп та їх сумішами, лужноземельні фожазити, частково обмінені з Н+ або перехідними металами IB, IIB, IV, VIII груп та їх сумішами, кристалічні силікати магнію та лужні обмінні кристалічні силікати магнію.
4. Процес за п. 2, який відрізняється тим, що адсорбент, який використовують в процесі адсорбції, регенерують органічним розчинником.
5. Процес за п. 4, який відрізняється тим, що органічний розчинник містить одне ароматичне кільце.
6. Процес за п. 5, який відрізняється тим, що розчинник вибирають з групи: бензол і алкілбензоли, в яких загальна кількість атомів вуглецю є не більше одинадцяти.
7. Процес за п. 4, який відрізняється тим, що розчинником є аліфатичний спирт, який має не більше дванадцяти атомів вуглецю.
8. Процес за п. 3, який відрізняється тим, що адсорбентом є сепіоліт у природній формі або в лужній обмінній формі.
Текст
1. Процес виготовлення вуглеводневих продуктів, що є рідкими у навколишніх умовах і містять органічні сполуки сірки більшої молекулярної маси, ніж органічні сполуки сірки у вуглеводневій сировині, який включає: - постачання вуглеводневої сировини, що містить речовину, яка кипить при температурах в інтервалі приблизно від 60 до 425 °С і яка містить сірковмісні сполуки з рівнем вмісту сірки до 5000 частин на млн. і азотовмісні сполуки з рівнем вмісту азоту до 2000 частин на млн., включаючи неосновні азотовмісні органічні сполуки з рівнем вмісту азоту до 200 частин на млн., і має високий вміст олефінів до 60 % мас., - вилучення із вуглеводневої сировини неосновних азотовмісних сполук за допомогою процесу адсорбції з одержанням вихідного потоку, що містить зменшену кількість неосновних азотовмісних сполук, і - приведення в контакт вихідного потоку з кислотним каталізатором в умовах алкілування, ефективних для перетворення частини сірковмісних спо C2 2 UA 1 3 ки твердим адсорбентом для видалення неосновних азотовмісних сполук, хімічне перетворення сірковмісної домішки або домішок на продукти з більш високою температурою кипіння шляхом алкілування при використанні стадії контактування з кислотним каталізатором при підвищених температурах. Після цього алкіловані сполуки сірки концентрують шляхом дистиляції і піддають гідруванню для видалення сірки. Отримувані в результаті продукти можна безпосередньо використовувати як моторні палива та компоненти для змішування й одержання таким чином палив, що є менш шкідливими для навколишнього середовища. Рівень техніки Добре відомо, що двигуни внутрішнього згоряння після їх винайдення зробили справжню революцію в царині транспортних засобів упродовж останніх десятиліть XIX століття. У той час як одні винахідники, Бенц і Готліб Вільгельм Даймлер, винаходили та розробляли двигуни з електричним запаленням палива, наприклад бензину, Рудольф Дизель винайшов і побудував двигун, названий в його ім'я дизельним, в якому використовується принцип самозапалення палива при стисканні останнього і який дав можливість використовувати в транспортних засобах дешеві органічні палива. Так само, якщо не більш важливі, розробки все більш досконалих двигунів з іскровим запаленням для транспортних засобів йшли пліч-о-пліч з вдосконаленням складів бензинового палива. Сучасні бензинові двигуни з високими робочими показниками висувають все більш жорсткі умови до складів використовуваних у них палив, не знижуючи при цьому вимог щодо їхніх вартісних характеристик. Сьогодні більшість моторних палив отримується із природної нафти. І це є цілком зрозумілим, оскільки нафта до тих пір в усьому світі залишається головним джерелом вуглеводнів, що використовуються як палива і як нафтохімічна сировина. Поряд з тим, що різні сорти природної нафти, або сирої нафти, дуже різняться своїми складами, усі вони містять сполуки сірки, а більшість з них містять сполуки азоту, до котрих може входити також кисень, але вміст останнього у переважної кількості сортів сирої нафти є низьким. У загальному випадку концентрація сірки в сирій нафті є меншою, ніж приблизно 8 %, і у більшості сортів вона лежить у межах приблизно від 0,5% до 1,5%. Вміст азоту звичайно складає менше 0,2%, але у деяких сортів він може становити 1,6%. Сира нафта рідко використовується в тій формі, в котрій вона видобувається зі свердловини, - і переробляється на нафтоочисних заводах на широкий спектр палив і нафтохімічних сировинних матеріалів. Звичайно палива для транспортних засобів виготовляють шляхом обробки і змішування дистильованих фракцій сирої нафти таким чином, щоб задовольнити технічним умовам їх конкретного застосування. Оскільки більшість наявних сьогодні в розпорядженні в достатніх кількостях сортів сирої нафти мають великий вміст сірки, дистильовані фракції повинні позбавлятися сірки для того, щоб давати продукти, які відповідають технічним вимогам і/або нормам охорони навколишньо 91042 4 го середовища. Сірковмісні органічні сполуки в паливах до тих пір залишаються головним джерелом забруднення навколишнього середовища. Під час згоряння ці сполуки перетворюються на оксиди сірки, котрі у свою чергу стають джерелом виникнення оксикислот сірки і, крім того, дають викиди часток золи в атмосферу. Перед лицем невпинного зростання вимог щодо вмісту сірки в моторних паливах видалення сірки із нафтової сировини та її продуктів буде найближчими роками набирати все більшої важливості. Ще зовсім недавно законодавствами Європи, Японії та США вміст сірки в дизельному паливі був обмежений максимальним рівнем 0,05%(мас), і ось вже є ознаки того, що незабаром цей рівень буде знижений ще більше. У бензині кількість сірки сьогодні для всіх нафтоочисних підприємств США обмежена середнім рівнем 30 мільйонних частин (млн. ч). У 2006 р. і наступних роках цей середній рівень замінюватися максимальною межею 80 млн. ч. Сьогодні одним із основних процесів очистки нафти, використовуваним для її переробки на бажані палива, наприклад на бензин і дизельне паливо, є каталітичний крекінг-процес у псевдозрідженому шарі. У цьому процесі високомолекулярна вуглеводнева сировина переробляється на продукти меншої молекулярної маси шляхом приведення її в контакт з гарячими, тонко здрібненими, твердими частками каталізатора у псевдозрідженому або диспергованому стані. Підходяща для цього вуглеводнева сировина, як правило, має температури кипіння в інтервалі приблизно від 205°С до 650°С і приводиться в контакт з каталізатором при температурах в інтервалі приблизно від 450°С до 650°С. До числа підходящих сировинних матеріалів входять різноманітні фракції мінеральних масел, наприклад легких газойлів, важких газойлів, ширококиплячих газойлів, вакуумних газойлів, керосинів, декантованих масел, залишкових фракцій, слабо крекінгованої нафти, і рециклові газойлі, що отримуються із будь-яких із цих сировин, а також фракції, одержані із сланцевих масел, процесів обробки бітумінозних пісків і процесів зрідження вугілля. Продуктами крекінг-процесу у псевдозрідженому шарі каталізатора, які зазвичай поділяються за їхніми температурами кипіння, є легкий лігроїн (з температурами кипіння приблизно від 10°С до 221°С), важкий лігроїн (з температурами кипіння приблизно від 10°С до 249°С), керосин (з температурами кипіння приблизно від 180°С до 300°С), легкий рецикловий газойль (з температурами кипіння приблизно від 221°С до 345°С) і важкий рецикловий газойль (з температурами кипіння вище, ніж приблизно 345°С). Крекінг-процес у псевдозрідженому шарі каталізатора дає не тільки значну частину всієї кількості бензину, що виробляється у Сполучених Штатах, але також велику частину сірки, що міститься в цьому бензині. Сірка в рідких продуктах цього процесу є у формі її органічних сполук і перетворюється на оксиди сірки, коли ці продукти використовуються в паливах. Ці оксиди сірки є шкідливими атмосферними забрудненнями. Крім того, вони 5 дезактивують багато видів каталізаторів, розроблених для каталітичних конвертерів, що використовуються в автомобілях для каталізу перетворення шкідливих викидів з відпрацьованими газами двигунів на гази, що є менш шкідливими. Отже, є бажаним зменшувати вміст сірки у продуктах каталітичного крекінгу до якомога менших рівнів. Сірковмісні домішки бензинів прямої гонки, які одержуються шляхом простої перегонки сирої нафти, звичайно дуже відрізняються від бензинів крекінг-процесу. Перші містять, головним чином, меркаптани і сульфіди, у той час як другі багаті на тіофен, бензотіофен та похідні тіофену та бензотіофену. Продукти з низьким вмістом сірки зазвичай отримуються за допомогою каталітичного крекінгпроцесу шляхом гідрообробки вхідної сировини процесу або його вихідних продуктів. Гідрообробка полягає в обробці продуктів крекінг-процесу воднем при наявності каталізатора і дозволяє перетворювати сірку в сірковмісних домішках на сульфід водню, який можна відокремлювати і перетворювати на елементарну сірку. Але обробка цього типу є доволі витратною, оскільки вона потребує джерела водню, технологічного обладнання високого тиску, коштовних каталізаторів гідрообробки та установки з відновлення сірки для перетворення отримуваного сульфіду водню на елементарну сірку. Крім того, процес гідрообробки може призводити до небажаного розкладання олефінів у сировині внаслідок перетворення їх на насичені вуглеводні шляхом гідрування. Це розкладання олефінів гідруванням зазвичай є небажаним, оскільки потребує застосовувати коштовний водень, а також тому, що олефіни є цінними високооктановими компонентами бензину. Наприклад, один з продуктів крекінг-процесу, лігроїн з діапазоном температур кипіння бензину, має відносно високе октанове число, зумовлене високим вмістом у ньому олефінів. Гідрообробка такого матеріалу разом з бажаною десульфурацією знижує вміст у ньому олефінів, внаслідок чого октанове число продукту гідрокрекінгу знижується разом зі зростанням ступеню десульфурації. Для видалення головної частини сірки із нафтових дистилятів, призначених для змішування компонентів моторних палив з виходу нафтоочистки, можна використовувати звичайні каталізатори гідродесульфурації, але вони не є ефективними для видалення сірки зі сполук, у котрих доступ до атома сірки утруднений просторовою структурою, що його оточує, як це має місце, наприклад, у багато кільцевих ароматичних сполуках сірки. Це особливо стосується тих сполук, де гетероатом сірки має подвійне оточення (як, наприклад, у 4,6диметилдибензотіофені). Застосування звичайних каталізаторів гідродесульфурації в умовах високих температур знижує вихід процесу, прискорює коксування каталізатора і руйнує якість продукту (наприклад, колір). Застосування ж високого тиску потребує великих капіталовкладень. Отже, існує необхідність у невитратному процесі ефективного видалення сірковмісних домішок із рідких вуглеводневих дистилятів. Існує також потреба в такому 91042 6 процесі, який би давав можливість видаляти сірковмісні домішки із рідких вуглеводневих дистилятів, що є, наприклад, продуктами каталітичного крекінг-процесу у псевдозрідженому шарі, що є високоолефіновими і містять як тіофенові, так і бензотіофенові сполуки, котрі є небажаними домішками. Для того, щоб задовольняти більш жорстким технічним умовам у майбутньому, такі закриті сполуки сірки також повинні видалятися із дистилятної сировини і продуктів перегонки. При цьому видалення сірки на нафтоочисних заводах із моторних палив і особливо із компонентів для бензинової суміші повинно здійснюватися економно. Царина техніки видалення сірки із дистилятної сировини та її продуктів переповнена різноманітними процесами, які для цього призначені. У патентні США № 2,448,211 на ім'я Філіпа Д. Цезара (Philip D. Caesar) стверджується, що тіофен та його похідні можуть піддаватися алкілуванню за допомогою реакції з олефіновими вуглеводнями при температурах приблизно від 140°С до 400°С і наявності каталізатора, яким може бути, наприклад, активована природна глина або синтетичний композитний адсорбент із двоокису кремнію і принаймні одного амфотерного оксиду металу. Підходящими каталізаторами на основі природних активованих глин є, наприклад, глиняні каталізатори з осадженими на них хлоридом цинку або фосфорною кислотою. Підходящими каталізаторами на основі системи двоокис кремніюамфотерний оксид металу є комбінації двоокису кремнію з такими матеріалами, як оксид алюмінію, двоокис цирконію, двоокис церію та оксид торію. У патентні США № 2,469,823 на ім'я Роланда К. Хенсфорда і Філіпа Д. Цезара (Rowland C. Hansford and Philip D. Caesar) вказується на те, що трифторид бору може використовуватися для каталізу процесів алкілування тіофену та алкілтіофенів такими алкілувальними агентами, як олефіни, галоїдні алкіли, спирти і меркаптани. Крім того, у патенті США № 2,921,081 на ім'я Цимершіда (Zimmerschied et al.) описані тверді кислотні каталізатори, які можуть виготовлятися шляхом об'єднання сполуки цирконію, вибраної із групи, що складається із двоокису цирконію і галоїдів цирконію, з кислотою, вибраною із групи, що складається із ортофосфорної кислоти, пірофосфорної кислоти і трифосфорної кислоти. Цимершід також цитує публікації, де тіофен алкілують пропіленом при температурі 227°С і наявності такого каталізатора. У патентні США № 2,563,087 на ім'я Джерома А. Веслі (Jerome A. Vesely) пропонується видаляти тіофен із ароматичних вуглеводнів шляхом селективного алкілування тіофену і відокремлення утворюваного алкілтіофену шляхом дистиляції. Селективне алкілування при цьому проводять шляхом змішування забрудненого тіофеном ароматичного вуглеводню з алкілувальним агентом і приведення цієї суміші в контакт з каталізатором алкілування в умовах ретельно контрольованої температури в інтервалі приблизно від -20°С до 85°С. Вказується також на те, що підходящими для цього алкілувальними агентами є меркаптани, естери неорганічних кислот і алкоксидні сполуки, 7 наприклад, аліфатичні спирти, етери й естери карбонових кислот. Вказується також на те, що підходящими каталізаторами алкілування є: (1) металогалогеніди Фріделя-Крафтса (Friedel-Crafts), які бажано використовувати у безводній формі; (2) фосфорна кислота, краще, якщо це є пірофосфорна кислота, або суміш такого матеріалу із сірчаною кислотою, в котрій об'ємне відношення сірчаної кислоти до фосфорної кислоти є менше, ніж приблизно 4:1; і (3) суміш фосфорної кислоти, наприклад ортофосфорної або пірофосфорної кислоти, з кремнієвмісним адсорбентом, таким, як кізельгур або кремнієвмісна глина, кальцинована до температури приблизно від 400 до 500°С з утворенням силікофосфорної кислоти, яка звичайно зветься твердим фосфорнокислотним каталізатором. У патенті США № 4,775,462 на ім'я Тамоцу Імаїта Джефрі К. Брикера (Tamotsu Imai and Jeffery C. Bricker) описаний неокисний процес нейтралізації кислої вуглеводневої фракції, за допомогою якого меркаптани перетворюються на тіоетери, про котрі говориться, що вони можуть використовуватися в паливах. Цей процес здійснюють шляхом приведення в контакт меркаптановмісної вуглеводневої фракції з каталізатором, що складається із кислого неорганічного оксиду, полімерної смоли сульфонової кислоти, шаруватої сполуки, твердого кислотного каталізатора, розподіленого на оксиді алюмінію галогеніду бору або розподіленого на оксиді алюмінію галогеніду алюмінію при наявності ненасиченого вуглеводню в кількості, до дорівнює молярній кількості меркаптанів, зазвичай приблизно від 0,01%(мас.) до 20%(мас). У той час як отримуваний за допомогою цього процесу продукт практично не містив меркаптанів, рівень елементарної сірки в ньому не знижувався. У патенті США № 5,171,916 на ім'я Квані Н. Ле і Майкла С. Сарлі (Quany N. Le and Michael S. Sarli) описаний процес підвищення якості легкого рециклового газойлю шляхом (А) алкілування гетероароматичних сполук цього рециклового газойлю аліфатичним вуглеводнем, що містить від 14 до 24 атомів вуглецю і принаймні один аліфатичний подвійний зв'язок, при наявності кристалічного металосилікатного каталізатора, і (В) відокремлення висококиплячого продукту алкілування з температурою кипіння в діапазоні кипіння мастильних матеріалів від неперетвореного легкого рециклового газойлю шляхом фракційної перегонки. Повідомляється також, що неперетворений легкий рецикловий газойль має знижений вміст сірки й азоту, а висококиплячий продукт алкілування може використовуватися як сировинна суміш синтетичних алкілованих ароматичних мастил. У патенті США № 5,599,441 на ім'я Ніка А. Колінза і Джефрі К. Тревела (Nick A. Collins and Jeffrey C. Trewella) описаний процес видалення тіофенових сполук сірки із крекінг-лігроїну шляхом (А) приведення в контакт лігроїну з кислотним каталізатором для алкілування тіофенових сполук олефінами, наявними в лігроїні, як алкілувальним агентом; (В) видалення вихідного потоку із зони алкілування; і (С) відокремлення алкілованих тіо 91042 8 фенових сполук від вихідного потоку зони алкілування шляхом фракційної перегонки. Стверджується також, що до крекінг-лігроїну можуть додаватися олефіни як додатковий алкілувальний для здійснення цього процесу. Трохи пізніше у патенті США № 6,024,865 на ім'я Брюса Д. Александера зі співроб. (Bruce D. Alexander, George A. Huff, Vivek R. Pradhan, William J. Reagan and Roger H. Cayton) був описаний продукт зі зниженим вмістом сірки, отримуваний із сировини, що складається із суміші вуглеводнів і включає у себе як небажані домішки сірковмісні ароматичні сполуки. Цей процес включає у себе відокремлення сировини шляхом фракційної перегонки у фракцію з більш низькою температурою кипіння, що містить більш леткі сірковмісні ароматичні домішки, і принаймні одну висококиплячу фракцію, що містить менш леткі сірковмісні ароматичні домішки. Після цього обидві фракції окремо одна від одної піддають реакціям, які дозволяють ефективно перетворювати принаймні частину їхнього вмісту сірковмісних ароматичних домішок на висококиплячі сірковмісні продукти шляхом алкілування алкілувальним агентом при наявності кислотного каталізатора. Сірковмісні продукти з більшими температурами кипіння видаляють шляхом фракційної перегонки. Повідомляється також, що алкілування може здійснюватися постадійно за умови, що режим алкілування буде менш жорстким на початковій стадії, ніж на вторинній стадії, наприклад, шляхом застосування більш низьких температур на першій стадії порівняно з більш високими температурами на другій стадії. У патенті США № 6,059,962 на ім'я Брюса Д. Александера зі співроб. (Bruce D. Alexander, George A. Huff, Vivek R. Pradhan, William J. Reagan and Roger H. Clayton) описаний продукт зі зниженим вмістом сірки, який отримують за допомогою багатостадійного процесу із сировини, що складається із суміші вуглеводнів і включає у себе сірковмісні ароматичні сполуки, що є небажаними домішками. На першій стадії цього процесу (1) сировину піддають алкілуванню, яке дозволяє ефективно перетворити частину домішок на сірковмісні сполуки з більш високими температурами кипіння, і (2) відокремлення шляхом фракційної перегонки на фракцію з нижчими температурами кипіння і фракцію з вищими температурами кипіння. Фракція з нижчими температурами кипіння складається із вуглеводнів і має знижений вміст сірки порівняно із сировиною. Фракція з вищими температурами кипіння складається із вуглеводнів, містить неперетворені сірковмісні ароматичні домішки і також сірковмісні продукти з вищими температурами кипіння. На кожній наступній стадії (1) фракцію з вищими температурами кипіння з попередньої стадії піддають алкілуванню в умовах ефективного перетворення принаймні частини її вмісту сірковмісних ароматичних сполук на сірковмісні продукти з вищими температурами кипіння і (2) відокремлюють утворювані продукти шляхом фракційної перегонки у фракцію вуглеводнів з нижчими температурами кипіння і фракцію з вищими температурами кипіння, яка містить сірковмісні продукти алкілування з вищими температурами 9 кипіння. Отриманий у результаті цього процесу загальний вуглеводневий продукт зі зниженим вмістом сірки складається із фракцій з нижчими температурами кипіння, отриманих на різних стадіях. Авторами цього патенту також стверджується, що алкілування може здійснюватися по стадіях за умови, що режим алкілування буде менш жорстким на початковій стадії, ніж на вторинній стадії, наприклад, шляхом застосування нижчої температури на першій стадії і на противагу їй - вищої температури на другій стадії. У даній галузі визнана також необхідність видалення деяких сполук азоту наприкінці різноманітних процесів. Наприклад, у патенті США № 6,602,405 В2 (Pradhan et al.) описаний процес виготовлення продуктів, що мають знижений вміст сірки, де основні домішки, що містять азот, видаляють із сировини перед надсиланням її в олефіномодифікаційну зону реакції, використовуючи для цього твердий фосфорнокислотний каталізатор або кислотний каталізатор із полімерної смоли. У патенті США № 6,599,417 В2 (Pradhan et al.) також описане видалення основних азотовмісних домішок із сировини перед надсиланням її в олефіномодифікаційну зону реакції. У патенті США № 6,736,660 В2 (Pradhan et al.) також описане видалення основних азотовмісних органічних сполук після процесу з кислотним каталізатором. Поряд з усвідомленням необхідності видаляти азотовмісні молекули після процесу на основі кислотного каталізатора попередніми роботами не визнавався той факт, що від'ємний вплив неосновних азотовмісних органічних сполук в отруєнні каталізатора є ще більшим, ніж основних або нейтральних сполук азоту. Описані у відомих технічних рішення типові процеси видалення азотовмісних молекул шляхом застосування, наприклад, стадій промивки кислотою або методів кислотного запобіжного шару, не дозволяють видаляти ці дуже шкідливі неосновні сполуки азоту. Отже, існує нагальна потреба у процесах виготовлення продуктів зі зниженим вмістом сірки із сировини, яка містить обмежені кількості сірковмісних і неосновних азотовмісних органічних сполук і з якої шкідливі неосновні сполуки азоту можуть за допомогою таких процесів легко видалятися до того, як вони зможуть вчиняти отрутну дію щодо каталізатора. Отже, у світлі вищевикладеного даний винахід спрямований на подолання зазначених проблем з метою одержання компонентів для готування із них на нафтоочисних заводах сумішей моторних палив, менш шкідливих для навколишнього середовища. Суть винаходу Даним винаходом пропонуються економічно ефективні процеси виготовлення компонентів для готування із них на нафтоочисних заводах сумішей моторних палив, де зазначені процеси є інтегрованими, багатостадійними і включають у себе обробку легкої фракції нафтоочистки для видалення неосновних азотовмісних сполук, хімічне 91042 10 перетворення однієї чи більше сірковмісних домішок на продукти з вищими температурами кипіння шляхом алкілування олефінами, та ефективне видалення продуктів з вищими температурами кипіння шляхом фракційної перегонки. Даним винаходом передбачена обробка вуглеводневих матеріалів різноманітних типів і, зокрема, вуглеводневих масел із нафти, що містять сірку. У загальному випадку вміст сірки в цих маслах є не менше 1% і лежить в інтервалі приблизно від 2% до 3%. Процеси за винаходом є особливо ефективними в обробці після нафтоочистки потоку сировини, що містить бензин, керосин, легкий лігроїн, важкий лігроїн і легкий рецикловий газойль, а в кращому варіанті також лігроїн з виходу каталітичних процесів і/або термічних крекінг-процесів. Даним винаходом в одному з варіантів його здійснення пропонується процес виготовлення продуктів, що є рідкими при навколишніх умовах і містять органічні сполуки сірки більшої молекулярної маси, ніж відповідні сірковмісні сполуки в сировині, де запропонований процес включає у себе: (а) постачання сировини, що містить суміш вуглеводнів, до якої входять олефіни, сірковмісні органічні сполуки і неосновні азотовмісні органічні сполуки, де зазначена сировина є матеріалом, що містить вуглеводні, кипить при температурах в інтервалі приблизно від 60°С до 425°С і має вміст сірки в інтервалі приблизно від 4000 до 5000 млн. ч. і вміст азоту приблизно до 200 млн. ч., включаючи вміст неосновних сполук азоту до 200 млн. ч.; (b) перепускання сировини через шар твердого адсорбенту, що містить лужний або лужноземельний цеоліт типу фожазиту, або лужні чи лужноземельні цеоліти типу фожазиту, частково обмінні з Н+ або перехідними металами IB, IIB, IVB і VIII груп, кристалічні силікати магнію і лужні обмінні кристалічні силікати магнію, або суміші всього вищепереліченого, в умовах, підходящих для поглинання в шарі, для здійснення селективної адсорбції і/або комплексоутворення принаймні частини наявних неосновних азотовмісних органічних сполук з адсорбентом і в результаті цього - отримання із зазначеного шару вихідного потоку, що містить менше азотовмісних органічних сполук, ніж сировина; (с) на стадії контактування при підвищених температурах приведення в контакт вихідного потоку з кислотним каталізатором в умовах, ефективних для перетворення частини домішок, наприклад, тіофенів на сірковмісний матеріал більшої молекулярної маси шляхом алкілування олефінами і в результаті цього - утворення потоку продукту. Підходящими сировинними матеріалами, в тому числі, є продукти нафтоочисних крекінгпроцесів, що складаються головним чином із матеріалу з температурами кипіння в інтервалі приблизно від 60°С до 425°С. У кращому варіанті такий потік нафтоочистки складається головним чином із матеріалу, що кипить при температурах в інтервалі приблизно від 60°С до 400°С, і ще краще - в інтервалі приблизно від 90°С до 375°С. У тому випадку, коли сировиною для обробки є лігроїн з виходу крекінг-процесу нафтоочистки, ця сировина складається головним чином із матеріалу, який 11 має температури кипіння в інтервалі приблизно від 20°С до 250°С. У кращому варіанті сировиною є потік лігроїну, що складається головним чином із матеріалу, який має температури кипіння в інтервалі приблизно від 40°С до 225°С, а ще краще - в інтервалі приблизно від 60°С до 200°С. Кращою сировиною для обробки за допомогою процесів за даним винаходом є лігроїн, вироблений за допомогою процесу каталітичного крекінгу. Бажано, щоб молярний вміст олефінів у сировині принаймні дорівнював їх молярному вмісту у сірковмісних органічних сполуках. Для кислотного каталізу на стадії контактування при алкілуванні тіофену вигідно використовувати твердий фосфорнокислотний каталізатор. Підвищені температури на стадії контактування при алкілуванні тіофену лежать в інтервалі приблизно від 90°С до 250°С, краще - в інтервалі приблизно від 100°С до 235°С, і ще краще - в інтервалі приблизно від 140°С до 220°С. Даний винахід є особливо корисним у зменшенні кількості сірковмісних органічних сполук у сировині, в котрих доступ до атому сірки утруднений його атомним просторовим оточенням, як, наприклад, у багатокільцевих ароматичних сполуках сірки. Зазвичай такими сірковмісними органічними сполуками є принаймні сульфіди, гетероатомні сульфіди і/або сполуки, вибрані із групи, що складається із заміщених бензотіофенів і дибензотіофенів. З метою більш повного висвітлення суті винаходу нижче як приклади розглянуто деякі варіанти його здійснення з поясненнями на супровідних фігурах креслення. Перелік фігур креслення На Фіг.1 показані графіки перетворення тіофенів у різних варіантах сировинних матеріалів, що містять сполуки азоту з різними ступенями кислотності. На Фіг.2 показані графіки перетворення тіофенів у тих же варіантах процесу, що й на Фіг.1, але в залежності від кількості азоту, адсорбованого на каталізаторі. На Фіг.3 показані графіки перетворення тіофенів в залежності від кількості азоту, в %(мас), осадженого на каталізаторі в додаткових варіантах сировинних матеріалів, що містять сполуки азоту з різними ступенями кислотності. На Фіг.4, 5 і 6 показані графіки, що ілюструють перетворення тіофенів із різноманітних сировинних матеріалів, оброблюваних і не оброблюваних в різних умовах реакції. На Фіг.7-11 показана ємність адсорбції різних адсорбентів щодо бутиронітрилу. На Фіг.12 і 13 показані криві прориву в адсорбції піролу і пропіонітрилу. На Фіг.14 і 15 показані криві прориву в адсорбції пропіонітрилу і піролу і при наявності ароматичних сполук у сировині. На Фіг.16 показані результати перетворення тіофенів із різноманітних сировин, що піддавалися обробці протягом різних періодів часу різними адсорбентами. Загальний опис винаходу Сировинними матеріалами, підходящими для 91042 12 застосування до них даного винаходу, є такі, що отримуються із нафтових дистилятів і в загальному випадку містять більшість фракцій нафтоочистки, які складаються головним чином із вуглеводневих сполук, що є рідкими при навколишніх умовах. Нафтові дистиляти є рідинами, які киплять у широких або вузьких температурних інтервалах у діапазоні приблизно від 10°С до 345°С. Такі рідини зустрічаються також при очищанні продуктів процесів зрідження вугілля та переробки бітумінозних глин і пісків. Ці дистилятні сировинні матеріали можуть містити до 2,5%(мас.) елементарної сірки, але в загальному випадку вміст останньої в них становить приблизно від 1,0%(мас.) до 0,9%(мас). Більш високий вміст сірки в загальному випадку мають дистиляти прямої гонки, отримувані із високочистої сирої нафти, кубові дистиляти і каталітичні рециклові газойлі із крекінг-апаратів із завислим каталізатором, де обробляються сировинні матеріали з відносно високим вмістом сірки. Вміст азоту в дистилятних сировинних матеріалах за винаходом у загальному випадку також є функцією вмісту азоту в сирій нафті, гідрогенізаційної потужності нафтоочисної установки на барель сирої нафти і альтернативного розташування сировинних компонентів гідрування дистиляту. Дистилятними сировинними матеріалами з більш високим вмістом азоту є, як правило, кубовий дистилят і каталітичні рециклові газойлі. Ці дистилятні сировинні матеріали можуть мати загальну концентрацію азоту до 2000 млн. ч., але як правило вміст азоту в них становить приблизно від 5 млн. ч. до 900 млн. ч. Підходящі фракції нафтоочистки мають густину в градусах АРІ, як правило, в діапазоні приблизно від 10° АРІ до 100° АРІ, в кращих варіантах - в діапазоні від 10° АРІ до 75° АРІ або 100 АРІ, а в найкращих - в діапазоні приблизно від 15° АРІ до 50° АРІ. До числа цих фракцій належать, наприклад, лігроїн каталітичного процесу, лігроїн рідкого або затриманого процесу, легкий лігроїн, лігроїн процесу гідрокрекінгу, лігроїни процесів гідрування, ізомеризовані нафтопродукти і продукти каталітичного реформінгу, а також їх комбінації. Продукти каталітичного реформінгу і лігроїни процесу каталітичного крекінгу часто поділяються на фракції більш вузького діапазону кипіння, наприклад, легкі та важкі каталітичні лігроїни, і легкі та важкі продукти каталітичного реформінгу, які можуть вироблятися по спеціальних замовленнях для використання як сировинного матеріалу за даним винаходом. Кращими фракціями є легкий лігроїн прямої гонки, лігроїни процесу каталітичного крекінгу, включаючи легкі і важкі лігроїни каталітичного крекінгу, продукти каталітичного реформінгу, включаючи легкі і важкі продукти каталітичного реформінгу та похідні таких вуглеводневих фракцій нафтоочистки. До числа більш підходящих сировинних матеріалів для процесів за даним винаходом належать будь-які складні суміші вуглеводнів, отримувані із дистилятних фракцій очистки нафти, які в загальному випадку мають температури кипіння приблизно від 60°С до 425°С. Такі сировинні матеріали, як правило, являють собою суміші вуглеводнів, 13 але містять малу кількість сірковмісних органічних домішок, включаючи ароматичні домішки, наприклад, тіофенові сполуки і бензотіофенові сполуки. Кращі сировинні матеріали мають початкову температуру кипіння нижче, ніж приблизно 79°С, і кінцеву точку дистиляції - не більше, ніж приблизно 345°С, а краще - не більше, ніж приблизно 249°С. У разі потреби сировина може мати кінцеву точку дистиляції не більше 221°С. Передбачається також, що для використання їх як сировини одна чи більше вищезазначених дистилятних фракцій можуть об'єднуватися. У багатьох випадках робочі характеристики отримуваних шляхом прямої очистки нафти моторних палив або компонентів очистки різноманітних альтернативних сировинних матеріалів для готування сумішей моторних палив, можуть бути порівняними між собою. У цих випадках чинниками, визначальними в тому, яку сировинну фракцію використовувати, можуть бути локальні диспозиційні умови постачання сировини і зокрема, наприклад, об'ємний доступ до фракції, розташування найближчих місць підключення і короткотермінова економічна ефективність. Особливо підходящими сировинними матеріалами для процесів за даним винаходом є продукти каталітичного крекінгу. Сировинні матеріали цього типу є рідинами, що киплять при температурах нижче приблизно 345°С, тобто це є, наприклад, легкий лігроїн, важкий лігроїн і легкий рецикловий газойль. Але цілком зрозуміло також, що сировиною в реалізації даного винаходу може служити весь вихідний потік летких продуктів процесу каталітичного крекінгу. Бажаність застосування продуктів каталітичного крекінгу зумовлена тим, що вони, як правило, містять відносно велику кількість олефінів, що зазвичай позбавляє необхідності додавати додатковий алкілувальний агент на першій стадії алкілування у процесі за винаходом. Окрім сірковмісних органічних сполук, наприклад меркаптанів і сульфідів, часто головним компонентом сірковмісних домішок у продуктах каталітичного крекінгу є сірковмісні ароматичні сполуки, наприклад, тіофен, бензотіофен і похідні тіофену та бензотіофену, а такі домішки легко видаляються за допомогою процесу за даним винаходом. Наприклад, типовий легкий лігроїн процесу каталітичного крекінгу у псевдозрідженому шарі із отримуваного з нафти газойлю може містити приблизно до 60%(мас.) олефінів і приблизно до 0,5%(мас.) сірки, де більша частина сірки є у формі тіофенових і бензотіофенових сполук. Кращою для обробки за допомогою процесу за даним винаходом є сировина, котра складається із продуктів каталітичного крекінгу і містить, крім того, принаймні 1%(мас.) олефінів. Особливо підходящою при цьому є сировина, яка складається із продуктів каталітичного крекінгу і містить, крім того, принаймні 5%(мас.) олефінів. Такі сировинні матеріали можуть бути частиною летких продуктів процесу каталітичного крекінгу, які відокремлюються шляхом перегонки. У практичному застосуванні даного винаходу оброблювана сировина містить домішки у формі сірковмісних ароматичних сполук. В одному з варі 91042 14 антів здійснення винаходу сировина містить домішки у формі як тіофенових, так і бензотіофенових сполук. У разі потреби принаймні приблизно 50% і більше цих сірковмісних ароматичних сполук можуть перетворюватися на сірковмісний матеріал з більш високою температурою кипіння. В іншому варіанті здійснення винаходу сировина містить бензотіофен із якого за допомогою процесу за винаходом принаймні приблизно 50% перетворюється на сірковмісний матеріал з більш високою температурою кипіння шляхом алкілування, який видаляється шляхом фракційної перегонки. Як каталізатор у зоні алкілування тіофену за винаходом може використовуватися будь-який кислотний матеріал, котрий володіє здатністю активувати алкілування сірковмісних ароматичних сполук олефінами або спиртами. Поряд з рідкими кислотами, наприклад із сірчаною кислою, при цьому особливо підходящими є тверді кислотні каталізатори, які складаються із рідких кислот, утримуваних на твердому носії. Тверді кислотні каталізатори, як правило, є більш підходящими, ніж рідкі каталізатори, оскільки приводити сировину в контакт з таким матеріалом набагато легше. Наприклад, при використанні твердого каталізатора потік сировини може просто перепускатися через один чи більше стаціонарних шарів із твердих часток кислотного каталізатора при відповідній температурі. У разі потреби, на різних стадіях процесу за винаходом можуть використовуватися різні кислотні каталізатори. Наприклад, жорсткі умови алкілування, що використовувалися на одному етапі процесу, можуть на наступному етапі бути пом'якшені шляхом використання на ньому менш активного каталізатора, в той час каталізатор з більшою активністю може використовуватися на початковому етапі стадії алкілування. Каталізаторами, підходящими для застосування в даному винаході, є наприклад кислотні матеріали, такі, як кислотні полімерні смоли, кислоти на носіях і кислотні неорганічні оксиди. Підходящими кислотними полімерними смолами є, наприклад, полімерні сульфокислотні смоли, які є добре відомими в даній галузі і в широкому асортименті випускаються промисловістю. Типовим прикладом такого матеріалу є амберліст (Amberlyst® 35), що виробляється фірмою Rohm and Haas Co. Підходящими кислотами на носіях для застосування в каталізаторах є, наприклад, кислоти Бренстеда (Bronsted) (зокрема, фосфорна кислота, сірчана кислота, борна кислота, HP, фторсульфокислота, трифторметансульфокислота і дигідроксифторборна кислота), а також кислоти Л'юїса (наприклад, BF3, BCI3, AICI3, AIBr3, FeCI2, FeCI3, ZnCI2, SbF5, SbCI5 і комбінації АІСІ3 та НСІ), що утримуються на твердих носіях, котрими можуть служити, наприклад, двоокис кремнію, оксид алюмінію, системи двоокис кремнію-оксиди алюмінію, двоокис цирконію або глини. Каталізатори на носіяхвиготовляють, як правило, шляхом об'єднання вибраної рідкої кислоти з відповідним носієм і наступної сушки. Особливо підходящими є каталізатори на носіях, котрі виготовляють шляхом об'єднання фосфорної кислоти з носієм. Ці каталізатори фігурують в даному описі 15 під назвою „твердих фосфорнокислотних каталізаторів". Вони є кращими завдяки їхній високій ефективності і водночас - низькій вартості. У патенті США № 2,921,081 (Zimmerschied et al.), включеному тут в усій його повноті шляхом посилання, описаний процес виготовлення твердих фосфорнокислотних каталізаторів шляхом об'єднання цирконієвої сполуки, вибраної із групи, що складається із двоокису цирконію і галоїдів цирконію, з кислотою, вибраною із групи, що складається із ортофосфорної кислоти, пірофосфорної кислоти і трифосфорної кислоти. У патенті США № 2,120,702 (Ipatieff et al.), включеному тут в усій його повноті шляхом посилання, описаний процес виготовлення твердих фосфорнокислотних каталізаторів шляхом об'єднання фосфорної кислоти з кремнієвмісним матеріалом. У британському патенті № 863,539, включеному тут в усій його повноті шляхом посилання, також описаний процес виготовлення твердих фосфорнокислотних каталізаторів шляхом осадження фосфорної кислоти на твердий кремнієвмісний матеріал, наприклад, інфузорну землю або кізельгур. Твердий фосфорнокислотний каталізатор, виготовлений шляхом осадження фосфорної кислоти на кізельгур, містить: (і) вільну фосфорну кислоту або кислоти, тобто ортофосфорну кислоту, пірофосфорну кислоту або трифосфорну кислоту, і (іі) фосфати кремнію, що утворюються шляхом хімічної реакції цієї кислоти або кислот з кізельгуром. Як показано у цитованому вище патенті, безводні фосфати кремнію, що є неактивними як каталізатори алкілування, можуть бути зроблені каталітично активними шляхом їх гідролізу з утворенням суміші ортофосфорної і поліфосфорної кислот. Точний склад цієї суміші залежить від кількості води, з якою контактує каталізатор. Для підтримання твердого фосфорнокислотного каталізатора алкілування на задовільному рівні активності, коли він використовується для обробки практично безводної вуглеводневої сировини, зазвичай у сировину додають невелику кількість води або спирту, наприклад, ізопропілового спирту, для підтримання каталізатора на достатньому рівні гідратації. Вважається, що спирт, який входить у контакт з каталізатором, дегідратується і що утворювана в результаті цього вода гідратує каталізатор. Якщо каталізатор містить занадто мало води, то він стає схильним до набуття дуже високої кислотності, що призводить до його швидкої дезактивації внаслідок коксування, і, крім того, каталізатор не буде мати хорошої фізичної цілісності. Гідратація ж каталізатора знижує його кислотність і схильність до швидкої дезактивації через коксування. Але, з іншого боку, надмірна гідратація такого каталізатора може викликати його розм'якшення, фізичну агломерацію і високі перепади тиску в реакторах зі стаціонарним шаром. Таким чином, слід дотримуватися певного оптимального рівня гідратації для твердих фосфорнокислотних каталізаторів, котрий є функцією умов реакції, використовуваного матеріалу носія й алкілувального агента. У кращих варіантах здійснення винаходу із застосуванням твердих фосфорнокислотних каталі 91042 16 заторів агент гідратації застосовується в кількості, достатній для підвищення робочих характеристик каталізатора. Агентом гідратації зазвичай служить принаймні один матеріал із групи, що складається із води та алканолів, які мають приблизно від 2 до 5 атомів вуглецю. Зазвичай задовільною є кількість агента гідратації, що забезпечує концентрацію води в сировині в інтервалі приблизно від 50 до 1000 млн. ч. Цю воду постачають як правило у формі спирту, яким може служити, наприклад, ізопропіловий спирт. Підходящими для застосування в каталізаторах кислотними неорганічними оксидами можуть бути, наприклад, оксиди алюмінію, системи двоокис кремнію-оксиди алюмінію, природні і синтетичні таблетовані глини і природні та синтетичні цеоліти, наприклад, фожазити, морденіти, L-, омега-, Х-, Y-, бета- і ZSM цеоліти. Серед цеолітів особливо підходящими є бета-, Y-, ZSM-3, ZSM-4, ZSM-5, ZSM-18 і ZSM-20 цеоліти. Цеоліти бажано вбудовувати в матрицю із неорганічного оксиду, наприклад, системи двоокис кремнію-оксид алюмінію. Дійсно, каталізатор рівноважного крекінгу може при застосуванні даного винаходу використовуватися у формі кислотного каталізатора. Каталізатори можуть являти собою суміші різних матеріалів, наприклад кислоти Л'юїса (наприклад, BF3, BCI3, SbF5, AICI3 та інші), нецеолітного твердого неорганічного оксиду (наприклад, двоокису кремнію, оксиду алюмінію і системи двоокис кремнію-оксид алюмінію і т.п.) і крупнопористого кристалічного молекулярного сита (наприклад, цеолітів, таблетованих глин та алюмофосфатів). Бажано, щоб твердий каталізатор мав фізичну форму, яка б дозволяла здійснювати швидкий та ефективний контакт з реагентами в даному процесі. При цьому в кращому варіанті твердий каталізатор має форму часток з максимальним розміром, середня величина якого лежить в інтервалі приблизно від 0,1мм до 2см. Наприклад, може використовуватися каталізатор у формі практично сферичних бусинок середнім діаметром приблизно від 0,1мм до 2см. В альтернативному варіанті каталізатор може мати форму стрижнів діаметром приблизно від 0,1мм до 1см і завдовжки в інтервалі приблизно від 0,2мм до 2см. Як зазначалося вище, передбачається, що даний винахід буде застосовуватися до сировинних матеріалів, які будуть містити, окрім сірковмісних органічних домішок, також домішки у формі азотовмісних органічних сполук. Багато серед типових азотовмісних домішок є органічними основами і в деяких випадках вони можуть викликати дезактивацію кислотного каталізатора або каталізаторів за винаходом. Було знайдено, що більшість отруйних для каталізаторів азотовмісних органічних молекул не є основними. Було виявлено також, що типові промислові сировинні матеріали, що піддаються алкілуванню тіофенів, містять переважну частину, часто більше 70%(мол.), неосновних, тобто нейтральних або трохи кислотних сполук азоту. Цими сполуками є ацетонітрили, пропіонітрили, бутиронітрили та піроли. Не звертаючись до теоретичних роз'яснень, 17 можна вважати, що неосновні сполуки азоту перетворюються на основні сполуки на активних ділянках кислотного каталізатора. Ці неосновні сполуки азоту зазвичай не видаляються на етапах промивки кислотою, на етапах промивки водою або на етапах застосування запобіжного шару, на котрих повинні видалятися основні сполуки азоту перед їх приведенням у контакт з кислотним каталізатором. Вважається, що неосновні сполуки азоту селективно отруюють активні ділянки на кислотному каталізаторі, оскільки це є ділянки, що перетворюють неосновні сполуки азоту на основні. Сировинна фракція бензину діапазону від легкої до середньої каталітичного крекінг-процесу у псевдозрідженому шарі зазвичай має 10-25 млн. ч. (мас.) неосновних сполук азоту, в той час як більш важка сировина може мати більше 50 млн. ч. (мас.) неосновних сполук азоту. У відповідності до процесу за даним винаходом ці неосновні сполуки азоту потрібно видаляти, і вони можуть видалятися за допомогою комбінації із таких заходів: промивки основою з наступною за нею промивкою кислотою; адсорбування за допомогою адсорбенту або комбінації адсорбентів, які можуть регенеруватися; взаємодії з кислотним матеріалом, який у кращому варіанті реагує з неосновними сполуками азоту з утворенням основних сполук азоту, котрі після цього можуть адсорбуватися. Низка із зазначених вище трьох процесів може також застосовуватися для видалення неосновних сполук азоту. У відповідності до даного винаходу комбінація операцій промивки основою і промивки кислотою може здійснюватися при температурах в інтервалі приблизно від 0 до 100°С, краще - в інтервалі приблизно від 20 до 50°С, і під тиском в інтервалі приблизно від 0 до 100 фунт./кв.дюйм, а краще - в інтервалі приблизно від 1 до 25 фунт./кв.дюйм. До числа підходящих основних розчинів входять розчини неорганічних основ, наприклад, гідроксиду натрію або гідроксиду калію в концентрації приблизно від 5 до 50%(мас), краще - від 10 до 20%(мас). Промивка основою може здійснюватися в одну-три стадії контактування, а найкраще - в одну стадію контактування. Основний розчин рециркулюють, забезпечуючи відношення контактування від 10 до 100 об'ємів розчину на об'єм нафтової сировини, а найкраще - при відношенні приблизно від 50 до 100 об'ємів розчину на об'єм сировини. Підходящими кислотними розчинами можуть бути розчини неорганічних кислот, наприклад, сірчаної кислоти в концентраціях в інтервалі приблизно від 5 до 25%(мас), а в кращому варіанті - в інтервалі приблизно від 10 до 20%(мас). Кислотна промивка може здійснюватися в 1-3 стадії контактування, а найкраще - в одну стадію контактування. Кислотні розчини рециркулюють, забезпечуючи відношення контактування від 10 до 100 об'ємів розчину на об'єм нафтової сировини, а найкраще при відношенні приблизно від 50 до 100 об'ємів розчину на об'єм сировини. Промивка основою повинна проводитися першою, а слідом за нею - промивка кислотою для захисту кислотного каталізатора від переходу на 91042 18 нього будь-якої основи з етапу промивки основою. Відповідно до даного винаходу ефективними адсорбентами неосновних сполук азоту можуть бути, наприклад, лужні або лужноземельні цеоліти типу фожазиту, або частково обмінні лужні або лужноземельні цеоліти типу фожазиту з Н+ або перехідними металами IB, IIB, IVB і VIII груп, кристалічні силікати магнію, лужні обмінні кристалічні силікати магнію або суміші всього вищепереліченого. Адсорбентом може бути також фізична суміш сепіоліту, Na-X і Na-Y цеолітів, де ці компоненти входять у суміш в кількостях в інтервалі приблизно від 5 до 95%(об.) кожний. Адсорбція може здійснюватися при температурах в інтервалі приблизно від 0 до 100°С, краще - в інтервалі приблизно від 10 до 40°С, під тиском в інтервалі приблизно від 0 до 300 фунт./кв.дюйм, а краще - від 100 до 150 фунт./кв.дюйм. Постачання сировини на адсорбент здійснюється з погодинною витратою маси (WHSV: weight hourly space velocity) в інтервалі приблизно від 0,5 до 50 год."1, а найкраще - в інтервалі приблизно від 10 до 15 год."1 Кількість адсорбенту може братися такою, щоб вона була достатньою для безперервної роботи протягом від 0,5 до 15 днів протягом від 0,5 до 15 днів між регенераціями, а краще - протягом від 1 до 15 днів між регенераціями. Регенерація відпрацьованого адсорбенту може проводитися шляхом термообробки, промивки розчинником або методом десорбції коливним тиском. Одним із цих методів може бути, наприклад, високотемпературне окислювання при температурах приблизно від 100 до 1000°С, краще - при температурах приблизно від 100 до 500°С, під тиском у межах приблизно від 0 до 100 фунт./кв.дюйм (абсолютний), а краще - в межах приблизно від 0 до 50 фунт./кв.дюйм (абсолютний), при наявності кисневмісного газу. Високотемпературний піроліз проводять при температурах приблизно від 0 до 1000°С, краще від 100 до 500°С, під тиском в інтервалі приблизно від 0 до 100 фунт./кв.дюйм (абсолютний), а краще - в інтервалі приблизно від 0 до 50 фунт./кв.дюйм (абсолютний). Високотемпературну гідрообробку проводять при температурах приблизно від 500 до 700°С і під тиском в інтервалі приблизно від 25 до 40 атмосфер при наявності газу, що містить водень. Ефективним розчинником для промивки є толуол, оскільки він видобувається в самому процесі очистки нафти. Вважається також, що ефективними розчинниками для регенерації є багато інших фракцій процесу очистки нафти. Регенерацію шляхом промивання розчинником зазвичай проводять при температурах в інтервалі приблизно від 50 до 40°F і під тиском приблизно від 0 до 300 фунт./кв.дюйм (манометричний), а краще - при температурах в інтервалі приблизно від 50 до 150°F і під тиском приблизно від 0 до 50 фунт./кв.дюйм (манометричний). Крім того, регенерація каталізатора методом коливного тиску може проводитися в умовах тем 19 ператури приблизно від 0 до 500°F і тиску приблизно від 0 до 50 фунт./кв.дюйм (абсолютний) при використанні омивного газу, наприклад, азоту. Раніше для видалення основних азотовмісних домішок як правило застосовували обробку кислотним матеріалом. Ці методи включали у себе такі процедури, як промивання водним розчином кислоти і використання запобіжного шару, який розташовується перед кислотним каталізатором. Ефективними матеріалами для запобіжного шару є, наприклад, А-цеоліт, Y-цеоліт, L-цеоліт, морденіт, фторований оксид алюмінію, свіжий каталізатор крекінг-процесу, каталізатор рівноважного крекінгу і кислотні полімерні смоли. При застосуванні методу запобіжного шару часто бажано використовувати два запобіжні шари таким чином, щоб один з них міг бути регенерований, у той час як інший використовувався для попередньої обробки сировини і захисту кислотного каталізатора. При застосуванні каталізатора крекінг-процесу для видалення основних азотовмісних домішок регенерація каталізатора може здійснюватися в регенераторі блоку каталітичного крекінгу, коли він втратив свою активність і, отже, здатність видаляти такі забруднення. Якщо для видалення основних азотовмісних сполук застосовується промивання кислотою, то сировину змішують з водним розчином відповідної кислоти. Підходящими для цього кислотами є, наприклад, соляна кислота, сірчана кислота й оцтова кислота. Концентрація кислоти у водному розчині не є критичною, але зазвичай вибирається таким чином, щоб лежати в інтервалі приблизно від 0,1 до 30%(мас). Наприклад, для видалення основних азотовмісних сполук із важкого лігроїну каталітичного крекінгпроцесу може використовуватися 2%(мас.) розчин сірчаної кислоти у воді. При практичному застосуванні даного винаходу після видалення неосновних сполук азоту сировину на стадії алкілування приводять у контакт з кислотним каталізатором при температурі і протягом часу, ефективних у досягненні бажаного ступеню перетворення вибраних сірковмісних органічних домішок на сірковмісний матеріал з більш високою температурою кипіння. При цьому бажано, щоб температура процесу контактування була вище приблизно 50°С, краще - вище приблизно 85°С, і ще краще - вище приблизно 100°С. Контактування проводять при температурі, зазвичай, в інтервалі приблизно від 50 до 260°С, в кращому варіанті - в інтервалі приблизно від 85 до 220°С, а в ще в кращому варіанті - в інтервалі приблизно від 100 до 200°С. Цілком зрозуміло, що оптимальна температура цього процесу залежить від використовуваного кислотного каталізатора, вибраного алкілувального агента або агентів, концентрації алкілувального агента або агентів і природи сірковмісних ароматичних домішок, які потрібно видаляти. Вихідний потік стадії контактування з кислотним каталізатором далі може розділятися на принаймні одну низькокиплячу фракцію, що являє собою фракцію, збіднілу на сірку, і одну висококиплячу фракцію, яка містить частину висококиплячих сірковмісних матеріалів, як про це сказано, 91042 20 наприклад, у патенті США № 6,736,963, вміст якого включений тут шляхом посилання. Даним винаходом запропонований інтегрований багатостадійний процес для концентрування сірковмісних ароматичних домішок вуглеводневої сировини у відносно малому об'ємі матеріалу з високою температурою кипіння. У результаті цього концентрування сірка може бути видалена легше і меншим коштом, а для цього видалення може бути застосований будь-який відомий процес. Наприклад, цей матеріал може бути примішаний до важких палив, де вміст сірки є менш небажаним. В іншому варіанті може бути ефективно застосований процес гідрування за порівняно низький кошт завдяки його зменшеному об'єму порівняно з об'ємом вхідної сировини. В іншому варіанті може застосовуватися видалення неосновних сполук азоту за допомогою адсорбентів за винаходом, що також збільшує продуктивність інших процесів, де використовуються тверді кислотні каталізатори, наприклад, процес каталітичної конденсації або полімеризації у виготовленні полібензину із легких олефінів. У різноманітних промислових хімічних і нафтохімічних процесах реакція конденсації олефінів або суміші олефінів над кислотним каталізатором використовується, для одержання високомолекулярних продуктів. Ці процеси звуться тут процесами полімеризації, а їхніми продуктами можуть бути низькомолекулярні олігомери або високомолекулярні полімери. Олігомери утворюються шляхом конденсації двох, трьох або чотирьох олефінових молекул одна з одною, а полімери утворюються шляхом конденсації п'яти чи більше олефінових молекул одна з одною. Використовуваний тут термін "полімеризація" означає процес утворення олігомерів і/або полімерів. Низькомолекулярні олефіни (наприклад, пропен, метилпропен, 1-бутен і 2-бутен) можуть перетворюватися шляхом полімеризації над твердим кислотним каталізатором (наприклад, твердим фосфорнокислотним каталізатором) на продукт, що складається із олігомерів і є цінним як компонент для високооктанової бензинової суміші і як сировинний матеріал для одержання хімічних проміжних продуктів і кінцевих продуктів, включаючи спирти, детергенти і пластмаси. Такий процес проводять, як правило, над стаціонарним шаром твердого кислотного каталізатора при підвищених температурах і тиску. Такого типу процеси полімеризації більш докладно описані в патенті США № 5,932,778, вміст якого включений тут шляхом посилання. ПРИКЛАД 1 На Фіг.1 у вигляді графіків подані результати чотирьох дослідів, проведених на експериментальній напівпромисловій установці із застосуванням процесу алкілування тіофенів з кислотним каталізатором. Досліди проводилися із сировинними матеріалами, із котрих два як неосновну сполуку азоту містили пропіонітрил, а два інші як неосновну сполуку азоту містили бутиламін. На графіку перетворення тіофенів по осі Y відкладений молярні відсотки перетвореного тіофену, а по осі X кумулятивне завантаження каталізатора сирови 21 91042 ною. На Фіг.2 показаний аналогічний графік перетворення тіофенів у функції кількості в %(мас.) азоту на каталізаторі. На цих графіках можна добре бачити, що наявність неосновних сполук азоту в сировині призво 22 дить до значного зниження активності перетворення тіофенів, де сировини мали лише основні сполуки азоту. Склади експериментальних сировинних матеріалів, використовуваних у дослідах, ілюстрованих на Фіг.1 і 2, подані нижче, в Табл.1. Таблиця 1 Склади експериментальних сировинних матеріалів 2-Метил-2-бутен 4-Метил-1-пентен Гексан Тіофен 2-Метилтіофен 3-Метилтіофен Пропіонітрил Досліди з пропіонітрилом 20%(мас.) 20%(мас.) 60%(мас.) 160 млн. ч. 160 млн. ч. 160 млн. ч. 45 млн. ч. (азоту) До дослідної напівпромислової установки, на котрій були отримані результати експериментів, подані на Фіг.1 і 2, завантажували 54см3 твердого фосфорнокислотного каталізатора (С84-5-02 фірми Sud Chemie, Inc. Louisville, Ky., USA), який роздрібнювали до розмірів -12 +20 меш сита Тайлера (W.S. Tyler) для типових випробувань за стандартом США. Використовуваний напівпромисловий реактор являв собою трубу із нержавіючої сталі завдовжки 34 дюйми, зовнішнім діаметром ¾ дюйма, внутрішнім діаметром 0,620 дюйма і товщиною стінок 0,065 дюйма. Температури реактора підтримували чотирма секціями стінок реактора з електричним нагрівом усередині ізольованої печі. Температури цих секцій контролювали за допомогою програмованого комп'ютера та одноточкових термопар на кожній секції стінок реактора. Реактор також мав термоколодязь із нержавіючої сталі зовнішнім діаметром ⅛ дюйма, який проходив зверху через центральну частину реактора. Для моніторингу температур по реактору у цьому термоколодязі розміщувалася багатоточкова термопара (3 триточкова термопара з 2-дюймовою відстанню між точками). Описаний реактор дослідної установки мав зону попереднього нагріву (температура зони 1), яку заповняли просіяною через сито Тайлера (W.S. Tyler) крихтою оксиду алюмінію розміром -12 +20 меш (сито для типових випробувань за стандартом США). Другу і третю нагріті зони завантажували 54см3 твердого фосфорнокислотного каталізатора (С84-5-02 виробництва фірми Sud Chemie, Inc. Louisville, Ky., USA), який роздрібнювали до розмірів -12 +20 меш сита Тайлера (W.S. Tyler) для типових випробувань за стандартом США. Решту об'єму реактора, температурну зону 4, яка служила зоною охолодження та опорою для каталізатора, заповняли просіяною через сито Досліди з бутиламіном 20% (мас.) 20%(мас.) 60%(мас.) 153 млн. Ч. (мас.) 153 млн. Ч. (мас.) 150 млн. Ч. (мас.) 51,6 млн. ч. (азоту) Тайлера (W.S. Tyler) крихтою оксиду алюмінію розміром -12 +20 меш (сито для типових випробувань за стандартом США). Потік сировини для обробки постачали в реактор за допомогою прецизійного інжекторного дозувального насоса (ISCO). Сировину попередньо нагрівали до температури реакції в зоні попереднього нагріву, температуру виміряли уздовж центральної лінії термопарами в різних точках, і по результатам вимірювань відповідними чином регулювали зони нагріву. Вихідний рідкий продукт реактора спрямовували в охолоджений сепараторприймач високого тиску, де для підтримування вихідного тиску реактора на потрібному робочому рівні використовували азот. Тиск регулювали по вихідному газу із сепаратора-приймача за допомогою регулювального клапана виробництва фірми Badger Research. Рідкі зразки зливали із приймача-сепаратора високого тиску і піддавали аналізу на багатоколонковому газовому хроматографі на вміст сірки, вміст азоту та вміст олефінів. Для проведення цих експериментів у реактор завантажували 54см3 каталізатора. Швидкість потоку сировини регулювали таким чином, щоб погодинна об'ємна витрата рідини (LHSV: liquid hydraulic space velocity (стандартний об'єм сировини в см3/годину, розділена на завантажений об'єм каталізатора в см3) становила 3,0год.-1. Температуру в зоні реакції підтримували на рівні 350°F +/-5°F, а тиск - на рівні 400 фунт./кв.дюйм +/-10 фунт./кв.дюйм (манометричний). В усіх дослідах використовували такі умови обробки: LHSV 3,0 год.-1 Тиск 400 фунт/кв.дюйм (манометричний) Температура 350°F Нижче в Табл.2 наведені фактичні результати, ілюстровані графічно на Фіг.1 і 2. 23 91042 24 Таблиця 2 Пропіонітрил. Дослід №1 Тривалість процесу Перетворення тіофену Азот на каталізаторі год. 0,0 6,7 14,7 22,7 30,7 38,7 46,7 54,7 62,7 70,7 % 0 90,8% 87,6% 81,3% 80,8% 78,5% 75,1% 71,6% 64,7% 62,2% %(мас.) 0,00% 0,07% 0,16% 0,24% 0,33% 0,42% 0,50% 0,58% 0,67% 0,74% Загальна кількість сировини/каталізатора см3 сировини/г каталіз. 0,0 24,0 52,7 81,5 110,3 139,0 167,8 196,5 225,3 254,1 Пропіонітрил. Дослід №2 Тривалість процесу Перетворення тіофену Азот на каталізаторі год. 0,0 8,0 16,0 24,0 32,0 40,0 48,0 56,0 64,0 72,0 % 0 86,8% 86,3% 83,0% 80,1% 74,6% 69,1% 63,1% 54,0% 47,7% %(мас.) 0,00% 0,09% 0,17% 0,26% 0,34% 0,43% 0,51% 0,59% 0,67% 0,74% Загальна кількість сировини/каталізатора см3 сировини/г каталіз. 0,0 28,8 57,6 86,3 115,1 143,9 172,7 201,5 230,3 259,0 Бутиламін Дослід № 1 Тривалість процесу Перетворення тіофену Азот на каталізаторі год. 10,7 14,7 18,7 22,7 26,7 30,7 34,7 38,7 42,7 46,7 50,7 54,7 58,7 62,7 66,7 70,7 74,7 78,7 82,7 86,7 90,7 94,7 98,7 102,7 % 94,1% 94,8% 95,4% 96,6% 96,2% 96,8% 96,7% 96,9% 97,1% 97,6% 97,7% 97,0% 97,5% 97,6% 97,5% 96,9% 96,7% 96,8% 96,3% 96,8% 96,0% 96,8% 96,7% 96,8% %(мас.) 0,13% 0,18% 0,23% 0,28% 0,33% 0,38% 0,43% 0,48% 0,53% 0,58% 0,63% 0,68% 0,73% 0,77% 0,82% 0,87% 0,92% 0,97% 1,02% 1,07% 1,12% 1,17% 1,22% 1,27% Загальна кількість сировини/каталізатора см3 сировини/г каталіз. 37,8 52,0 66,2 80,4 94,6 108,8 123,0 137,2 151,4 165,5 179,7 193,9 208,1 222,3 236,5 250,7 264,9 279,1 293,3 307,4 321,6 335,8 350,0 364,2 25 91042 26 Бутиламін Дослід № 1 (продовження) Тривалість процесу Перетворення тіофену Азот на каталізаторі год. 106,7 110,7 114,7 118,7 % 96,1% 96,1% 96,1% 95,5% %(мас.) 1,32% 1,37% 1,42% 1,47% Загальна кількість сировини/каталізатора см3 сировини/г каталіз. 378,4 392,6 406,8 421,0 Бутиламін Дослід № 2 Тривалість процесу Перетворення тіофену Азот на каталізаторі год. 8,0 12,0 16,0 20,0 24,0 28,0 32,0 36,0 40,0 44,0 48,0 52,0 56,0 60,0 64,0 68,0 72,0 76,0 80,0 84,0 88,0 92,0 96,0 100,0 104,0 108,0 112,0 116,0 120,0 % 90,2% 92,1% 92,9% 92,8% 92,9% 93,6% 93,5% 93,6% 93,8% 93,1% 93,1% 92,9% 93,3% 92,0% 92,6% 92,1% 92,0% 91,6% 91,7% 91,1% 90,9% 90,3% 89,7% 89,0% 88,2% 87,1% 86,2% 85,2% 84,8% %(мас.) 0,10% 0,15% 0,20% 0,25% 0,30% 0,35% 0,40% 0,44% 0,49% 0,54% 0,59% 0,64% 0,69% 0,74% 0,79% 0,84% 0,89% 0,94% 0,99% 1,04% 1,09% 1,14% 1,19% 1,24% 1,29% 1,33% 1,38% 1,43% 1,48% ПРИКЛАД 2 На Фіг.3 графічно подані результати додаткових експериментів на дослідній напівпромисловій установці щодо здійснення процесу алкілування тіофенів з кислотним каталізатором. Досліди проводили з моделями сировин, що містили 80 млн. ч. (мас.) семи сполук азоту, як неосновних, так і основних. На Фіг.3 графіки перетворення тіофенів показані у формі залежності відкладених по осі Y молярних відсотків перетвореного тіофену від масових відсотків адсорбованого на каталізаторі азоту. Із цих графіків ясно видно, що чим більш основною є сполука азоту, тим більш плоскою є відповідна їй крива лінія, тобто тим меншою є дезактивація каталізатора. Наявність неосновної сполуки азоту у сировині призводить до значного зниження активності перетворення тіофенів порівняно із сировинами, які містять тільки основні сполуки азоту. Загальна кількість сировини/каталізатора см3 сировини/г каталіз. 28,4 42,6 56,7 70,9 85,1 99,3 113,5 127,7 141,9 156,1 170,2 184,4 198,6 212,8 227,0 241,2 255,4 269,6 283,7 297,9 312,1 326,3 340,5 354,7 368,9 383,1 397,2 411,4 425,6 Для визначення об’ємної витрати рідини, за котрої б забезпечувалося 95% перетворення тіофену в підлуженій сировині (50% 1-гексен, 50% nгептан, 200 млн. ч. S у формі тіофену) були проведені попередні експерименти, які дозволили в цих дослідах ясно і швидко визначати отруйну дію азотних забруднень сировини. По результатам цих експериментів для решти даної дослідної програми була встановлена об’ємна витрата рідини (WHSV) 4,1год.-1. В експериментах даного прикладу використовували базову сировину такого складу: 50% 1-гексен 50% n-гептан 200 млн. ч. сірки (у формі тіофену) 80 млн. ч. (мас.) N (у формі різноманітних азотних домішок) Як домішки до базової сировини в дослідженнях оцінювалися такі азотні забруднення: 27 91042 Без домішок Пропіонітрил (неосновний) Метилпірол (неосновний) Анілін (основний) Бутиламін (основний) Пірол (неосновний) Піридин (основний) Бутиронітрил (неосновний) Отримані результати показали, що різноманітні сполуки азоту за їхньою отруйною дією на процес алкілування тіофенів із застосуванням кислотного каталізатора можна поділити на такі групи: (і) високоотруйні для такого процесу сполуки, до котрих належать, зокрема, піридин, метилпірол, пропіонітрил і бутиронітрил; (іі) сполуки, що є помірно отруйними для даного процесу - анілін і пірол; і (ііі) низькоотруйні сполуки для процесу алкілування тіофенів - бутиламін. В умовах реакції, що використовувалися в даному прикладі, більшість сполук азоту протягом перших днів реакції всі разом утримувалися на каталізаторі. У подальшому триванні експерименту адсорбція азоту трохи і особливо стосовно нітрилових сполук. У даних експе 28 риментах на дослідній напівпромисловій установці використовувався твердий фосфорнокислотний каталізатор, який виробляється фірмою Sud Chemie Inc. Louisville, KY, USA під маркою С-84-05. Дослідну установку завантажували 300мг твердого фосфорнокислотного каталізатора, який роздрібнювали до розміру 0,4-0,6мм комірок сита Тайлера (Tyler). Перед використанням каталізатор сушили в потоку азоту протягом 2 годин при 200°С. Дослідна напівпромислова установка складалася із паралельно включених реакторів зі стаціонарним шаром, кожний з яких міг вміщати від 50 до 1000мг каталізатора, і мала конструкцію, аналогічну описаній вище у Прикладі 1. Реактори працювали в режимі низхідного потоку. Усі досліди проводилися в таких умовах реакції: LHSV 4,1 год.-1 Тиск 400 фунт/кв.дюйм (манометричний) Температура 180°С Графічні дані на Фіг.3 подані в цифрах у Табл.3. Таблиця 3 Кількість Адсорбція Перетворення Адсорбція Сполука азоазоту на азоту через тіофенів че- азоту через ту каталізаторі 8год. рез 8год. 32год. через 8год. 0,00 92% Пропіонітрил 98% 0,18% 62% 89% Метилпірол 100% 0,18% 84% 38% Анілін 100% 0,18% 83% 100% Бутиламін 100% 0,18% 85% 100% Пірол 100% 0,18% 79% 100% Піридин 100% 0,18% 91% 82% Бутиронітрил 55% 0,10% 52% 15% ПРИКЛАД 3 У Табл.4 подані результати контрольних аналізів промислової первинної сировини, що піддається процесу алкілування тіофенів з кислотним каталізатором і містить усі природні неосновні сполуки азоту, а також сировинних фракцій, підданих обробці з метою видалення азоту по-перше промивкою кислотою і по-друге обробкою смолою. Цим промисловим сировинним матеріалом є бензинова фракція каталітичного крекінгу у псевдозрідженому шарі (FCC: fluidized catalytic cracking) діапазону легких фракцій. Можна бачити, що ця сировина, котра є типовою для сировинних матеріалів, оброблених за допомогою процесу алкілування тіофенів з кислотним каталізатором, містить Кількість Середня Перетворення азоту на адсорбція тіофенів чекаталізаторі азоту рез 32год. через 32год. 92% 94% 0,74% 2% 69% 0,54% 1% 100% 0,79% 23% 100% 0,79% 86% 100% 0,79% 45% 91% 0,72% 1% 35% 0,28% 13% великий набір різноманітних сполук азоту. Ці сполуки можна поділити на три загальні типи: (1) основні азотовмісні речовини, до числа котрих належать бутиламін, гексамін і піридин; (2) нейтральні сполуки, включаючи ацетонітрил, пропіонітрил і бутиронітрил; і (3) трохи підкислені сполуки азоту, до числа яких належать піроли. Промивка кислотою видаляє, головним чином, основні сполуки азоту і залишає невидаленими більшість неосновних сполук азоту. Це є, зокрема, бутиронітрил, який має обмежену розчинність у воді і погано піддається видаленню шляхом промивки кислотою. Обробка цієї сировини смолою дозволяє видаляти додаткові кількості неосновних сполук азоту. Таблиця 4 Результати контрольних аналізів вмісту азоту в стандартній сировині, підданій обробці на дослідній напівпромисловій установці Промислова сировина Загальний вміст азоту (ASTM D4629) Ацетонітрил Азот, млн. ч. 21,0 0,29 Після промивки кислоПісля обробки смолою тою Азот, млн. ч. Азот, млн. ч 8,4 3,9 29 91042 30 Продовження таблиці 4 Результати контрольних аналізів вмісту азоту в стандартній сировині, підданій обробці на дослідній напівпромисловій установці Промислова сировина Пропіонітрил Ізобутиронітрил Бутиламін Бутиронітрил Піридин 1-Метилпірол Пірол Диметилформамід Валеронітрил 2-Метилпіридин Гексиламін 3-Метилпіридин Азот, млн. ч. 6,35 0,65 0,16 0,16 1,42 0,22 0,18 0,24 0,07 0,13 0,42 0,10 2,83 0,48 0,10 0,38 0,64 0,13 0,20 0,22 0,41 0,12 1,82 1,29 0,31 0,33 0,15 Була проведена серія експериментів з необробленою промисловою сировиною і промисловою сировиною, підданою обробці кислотою і смолою для видалення більшості наявного в ній азоту. На Фіг.4, 5 і 6 подані графіки, на котрих по осі Y відкладений рівень перетворення тіофенів у молярних відсотках, а по горизонтальній осі - загальна кількість сировини в грамах, що постачалася в дослідний напівпромисловий реактор, поділеної на кількість каталізатора в грамах, що завантажувався в цей реактор у плині часу як для необробленої сировини, так і для сировини, підданої промивці кислотою при різних величинах погодинної об'ємної витрати і завантаження каталізатора, зазначених на цих графіках. Дослідні установки працювали в таких режимах: (і) з погодинною об'ємною витратою 1,5год.-1, (іі) з погодинною об'ємною витратою 3,0год.-1 за такої ж самої кількості каталізатора, що й у режимі (і) і зі збільшеною вдвічі об'ємною витратою сировини; і (ііі) з погодинною об'ємною ви-1 тратою 3,0год. за зниженої вдвічі кількості каталізатора, завантаженого в реактор, порівняно з режимом (і). Різні величини об'ємної витрати сировини дозволяють спостерігати більш чіткий ефект видалення азоту із сировини. Як можна бачити по цих даних, продуктивність каталізатора (в грамах обробленої сировини на грам каталізатора) при Після промивки кислоПісля обробки смолою тою Азот, млн. ч. Азот, млн. ч 5,79 0,37 0,41 0,31 0,13 0,16 1,13 0,55 0,20 0,25 0,07 0,14 0,16 0,19 0,18 0,38 0,01 0,35 0,11 0,24 0,17 0,13 0,09 0,08 0,09 0,12 високому відсотку перетворення тіофенів (>80%) зростає більше, ніж у 3 рази, коли із сировини на попередній обробці була видалена більша частина азотовмісних речовин. Ця більшість видаленого азоту була у формі неосновних його сполук у даній промисловій сировині. У цьому експерименті використовувалася та ж сама дослідна установка, що й у Прикладі 1. При обробці сировини в даному прикладі застосовувалися такі умови процесу (за винятком тих, що зазначені на Фіг.4, 5 і 6): LHSV: зазначено на Фіг.4, 5 і 6 Температура: 180°С Тиск: 400 фунт/кв.дюйм (манометричний) На поданих графіках можна ясно спостерігати значне зниження активності перетворення тіофенів у реакції алкілування тіофенів з кислотним каталізатором при обробці промислових сировин, що містять неосновні сполуки азоту. ПРИКЛАД 4 У Табл.5 наведені результати двох титрувань, проведених з двома основами, піридином і 2,6дитрет-бутилпіридином, на твердому фосфорнокислотному каталізаторі промислового виготовлення - продукт фірми Sud-Chemie Inc. Титрування проводили за такою методикою. 31 91042 Зразки каталізатора роздрібнювали і просіювали. До реактора дослідної напівпромислової установки зі стаціонарним шаром каталізатора завантажували агломерати цих зразків діаметрами в інтервалі 180-355мм. Зразки (50мг) піддавали обробці в потоку Не (1,33см3/с) при температурі 453К протягом 1год., після чого проводили титрувальні вимірювання. Для проведення експерименту готували рідкі суміші n-гексану (Fluka, 99,5%, 4,5мл) з піридином (Fischer, 99,9%, 20мл) або з 2,6-дитрет-бутилпіридином (Aldrich, 97%, 50мл). Приготовану суміш уводили в потік Не (1,33см3/с) з погодинною об'ємною витратою рідини 0,09см3/с, у результаті чого одержували суміші, що складалися із n-гексану з тиском 0,3кПа і піридину з тиском 5,3Па або 2,6-дитрет-бутилпіридином з тиском 4,7Па. Температура шару каталізатора становила 453К. Кількість титрованого розчину, адсорбованого на каталізаторі, обчислювали за даними його концентрації у вихідному потоці, виміряної методом газової хроматографії (капілярна колонка на метилсиліконі Hewlett-Packard 6890 GC, 30м НР-1, детектор іонізації полум'я). 32 Не звертаючись до теоретичних роз'яснень, із Табл.5 можна зробити очевидний висновок про існування двох типів кислотних ділянок на твердих фосфорнокислотних (SPA) каталізаторах: одними із них є сильнокислотні ділянки (ті, що були титровані 2,6-дитрет-бутилпіридином), а іншими - слабокислотні ділянки (різниця між ділянками, титрованими піридином, і ділянками, титрованими 2,6дитрет-бутилпіридином). Можна вважати, що сильнокислотні ділянки відіграють головну роль у перетворенні тіофенів. Сильнокислотні ділянки піддаються селективному отруєнню неосновними сполуками азоту, оскільки ці сполуки не адсорбуються на слабокислотних ділянках, але все ж реагують на сильнокислотних ділянках з утворенням продуктів, що є основними, і потім міцно адсорбуються на сильнокислотних ділянках. У результаті ця реакція неосновних сполук азоту на сильнокислотних ділянках зменшує кількість дієздатних ділянок, потрібних для алкілування тіофенів, і таким чином є відповідальною за дуже велику здатність неосновних сполук азоту отруювати реакцію алкілування тіофенів. Таблиця 5 Титрування твердих фосфорнокислотних каталізаторів Титрант Піридин 2,6-дитрет-бутилпіридин Молекулярна маса 78 192 Титрант, мкмоль/г 612,5 214,4 Таким чином, із наведених вище прикладів стає очевидним, що існує нагальна потреба у процесі ефективного видалення широкого різноманіття сполук азоту і, зокрема, неосновних сполук азоту із промислової сировини, що піддається алкілуванню тіофенів. У прикладах, наведених нижче, розглядаються необов'язкові параметри процесу за даним винаходом, що є підходящими для видалення неосновних сполук азоту із промислової сировини, що піддається алкілуванню тіофенів. ПРИКЛАД 5 Експерименти, описані в даному Прикладі, проводилися у стаціонарній багатошаровій адсорбційній укладці. До спільного входу постачання сировини було підключено чотири реактори із нержавіючої сталі зі стаціонарним шаром каталізатора. Рідку сировину вводили в реактори за допомогою здвоєного поршневого насоса, здатного підтримувати постійний потік сировини при мінімальних поршневих пульсаціях. Конструкція цієї установки дозволяла перепускати сировину крізь шар адсорбенту низхідним або висхідним потоком. Зразки накопиченої рідини відбиралися на виході Титрант, г/г каталіз. 0,047775 0,041165 N, г/г каталіз. N %(мас.) 0,008575 0,003002 0,86% 0,30% труб реакторів через певні інтервали часу і піддавалися газово-хроматографічному аналізу. Для аналізу вихідного потоку використовувався прилад Varian-3380 GC, обладнаний детектором іонізації полум'я (FID) та імпульсним полуменево-фотометричним детектором (PFPD: Pulsed Flame Photometric Detector), що працював у режимі азоту. PFPD детектором аналізувалися пропіонітрил, бутиронітрил, пірол і тіофен, a FID детектором - гептан і гексен-1, причому детектори працювали паралельно один одному. Різні сполуки розділялися в колонці CP-Sil 24-CB. Експерименти з адсорбції проводили при кімнатній температурі, під тиском 3,5 бар і величині погодинної об'ємної витрати рідини (WHSV) в інтервалі 15-20год.-1 Кількість використовуваного адсорбенту становила 2г. Адсорбент в усіх випадках висушували (протягом 2 год. при 200°С у потоці 100мл N2) і компактували з n-гептаном, після чого вводили азотовмісну сировину з постійною витратою приблизно 1,0мл/хв. Об'ємну витрату рідини в усіх випадках визначали за кількістю обробленої рідини, відновленої на виходах реакторів. Адсорбція бутиронітрилу 33 91042 34 Таблиця 6 Адсорбенти, використовувані для видалення бутиронітрилу Адсорбент Na-Y цеоліт Природний силікат магнію Промислова тверда фосфорна кислота Каталізатор рівноважного крекінгу у псевдозрідженому шарі Н-Монтморилоніт Сu-Гідротальцит Zn-Гідротальцит Ni-Гідротальцит Cu-Na Y цеоліт Na-обмінний сепіоліт Характеристики CBV-1 00 (фірми Zeolyst Intl.), Si/AI=2,6 Природні волокнисті силікати магнію з кристалічною структурою Твердий фосфорнокислотний каталізатор виробництва фірми Sud Chemie FCC ECAT, 800 млн. ч. Ni, 2000 млн. ч. V, розмір елементарної комірки = 24,32 Å Монтморилоніт, оброблений кислотою (6год., 0,2М розчин НСІ,при кімнатній температурі) AI3+/(AI3++Cu2++Mg2+)=0,25; Cu2+/(Cu2++Mg2+=0,5 (молярні відношення) AI3+/(AI3++Zn2++Mg2+)=0,25; Zn2+/(Zn2++Mg2+)=0,5 (молярні відношення) AI3+/(AI3++Ni2++Mg2+)=0,25; Ni2+/(Ni2++Mg2+)=0,13 (молярні відношення) Cu-обмінний CBV-100 (2,7%(мас.) Na2O, 15,8%(мас.) CuO) Na-обмінний природний сепіоліт (4,4%(мас.) Na2O) Перелік використовуваних адсорбентів та їхні головні характеристики подані в Табл.6. У дослідах, де використовувався промисловий твердий фосфорнокислотний каталізатор, первинні таблетки каталізатора роздрібнювали і просіювали до розміру часток 0,4-0,6мм, а адсорбцію проводили двічі з метою контролю відтворюваності протоколу експерименту. Решту адсорбентів так само пресували, роздрібнювали і просіювали до такого ж розміру часток (0,4-0,6мм), за винятком каталізатора рівноважного крекінгу у псевдозрідженому шарі (FCC ECAT), який вже мав первинну форму мікросфер. Базова модель сировини, яка містила n-гептан у кількості 50%(мас), гексен-1 у кількості 50%(мас.) і тіофен (200 мас. млн. ч. S), була доповнена домішками 80 мас. млн. ч. азоту у формі бутиронітрилу. Цю сировину перепускали через адсорбенти, описані вище в Табл.6. Результати експериментів з адсорбції Результати експериментів, проведених з промисловим твердим фосфорнокислотним каталізатором, подані у формі графіка на Фіг.7 (крива проривання) і в Табл.7. Ці результати свідчать про добру відтворюваність експерименту. Наведені в Табл.7 величини ємності адсорбції визначали як кількість адсорбованого N на 100г адсорбенту до того моменту, коли у вихідному потоку починав появлятися N. Звідси можна бачити, що ємність адсорбції для бутиронітрилу в цих умовах є відносно низькою. Порівняні результати для решти адсорбентів також наведені в Табл.7 та ілюстровані на Фіг.911. На Фіг.8 промисловий твердий фосфорнокислотний каталізатор порівнюється з різними адсорбентами на основі гідротальциту. На Фіг.9 дано порівняння промислового твердого фосфорнокислотного каталізатора з двома Y-цеолітами, промисловим Na-Y (CBV-100 виробництва фірми Zeolyst Intl.), Cu-обмінним Y, двома сепіолітами - природним сепіолітом і Na-обмінним сепіолітом. На Фіг.10 дано порівняння промислового твердого фосфорнокислотного каталізатора з каталізатором рівноважного крекінгу у псевдозрідженому шарі (FCC ECAT), кислотно-обмінним монтморилонітом і з адсорбентами на основі гідротальцитів, Y-цеоліту і сепіоліту. Після перепускання 173г сировини через 2г шар NaX-адсорбенту в описаних вище умовах у вихідному потоці бутиронітрил не виявлявся. Отже, мідь-обмінний Na-Y мав мінімальну ємність адсорбції азоту, що становила 0,69г N/100г адсорбенту. Мідь-обмінний Y-цеоліт також показав значну ємність адсорбції. Як можна бачити в Табл.7 і на Фіг.11, повторне проведення даного експерименту підтвердило ці результати. І нарешті, адсорбенти на основі сепіоліту також дають високі ємності адсорбції бутиронітрилу. Таблиця 7 Ємності адсорбції у різноманітних адсорбентів Адсорбент ВР каталізатор ВР каталізатор FCC ECAT Н-Монтморилоніт Cu-Гідротальцит Zn-Гідротальцит Ni-Гідротальцит Cu-Y-цеоліт WHSV, год.-1 Тиск, бар 16,7 19,3 18,2 16,8 16,3 17,7 16,4 15,8 3,25 3,25 3,5 3,5 3,5 3,5 3,5 3,5 Ємність адсорбції, N, г/100г адсорбенту 0,012 0,018 0,063 0,074 0,027 0,071 0,13 0,44 35 91042 36 Продовження таблиці 7 Ємності адсорбції у різноманітних адсорбентів WHSV, год.-1 Cu-Y-цеоліт Na-Y-цеоліт Природний сепіоліт Na-Сепіоліт Ємність адсорбції, N, г/100г адсорбенту 0,40 0,69 0,51 0,33 Тиск, бар 16,9 16,1 17,3 17,4 Адсорбент 3,5 3,5 3,5 3,5 ПРИКЛАД 6 Адсорбція пропіонітрилу і піролу Використовувана базова модель сировини містила n-гептан у кількості 50%(мас), гексен-1 у кількості 50%(мас.) і тіофен (200 мас. млн. ч. S). До цієї базової сировини було добавлено 40 млн. ч. пропіонітрилу і 40 млн. ч. піролу. Цю сировину перепускали через Na-Y і сепіолітові адсорбенти для оцінки відносних ємностей адсорбції кожного адсорбенту щодо кожної зі сполук азоту. Отримані криві проривання адсорбції азоту наведені на Фіг.12 і 13. Дані стосовно ємності адсорбції по азоту підсумовані у Табл.8. Таблиця 8 Ємність адсорбції сполук азоту у Na-Y і сепіолітових адсорбентів WHSV15-20 год-1, кімнатна температура, тиск 4 бар (додано 40 млн. ч. N кожної сполуки азоту), N, г/100г адсорбенту Адсорбент Na-Y Сепіоліт Na-Y Сепіоліт Базова сировина Базова сировина + 30% ароматичних сполук ПРИКЛАД 7 Вплив ароматичних сполук на адсорбцію пропіонітрилу і піролу З метою оцінки впливу наявності ароматичних сполук у сировині була приготована така модель сировини: 35% n-гептану, 35% 1-гексену, 22% толуолу, 8% о-ксилену, 200 млн. ч. тіофену, 40 млн. ч. пропіонітрилу і 40 млн. ч. піролу. Приготовану сировину перепускали крізь Na-Y і сепіолітові адсорбенти. Отримані в цих експериментах криві проривання адсорбції показані на Фіг.14 і 15. Експерименти проводили при величинах WHSV в інтервалі 15-20 год.-1, при кімнатній температурі і тиску 4 бар. Вплив ароматичних сполук на ємність адсорбції по азоту можна спостерігати в Табл.8, де наведені величини ємності адсорбції. ПРИКЛАД 8 Регенерація NaY Пірол >0,52 0,05 0,1 0,017 N, г/100г адсорбенту Пропіонітрил >0,83 >0,76 >1,3 0,28 Сума >1,35 >0,84 >1,4 0,3 Експерименти з адсорбції на свіжому NaY показали, що цей матеріал мав високу ємність адсорбції щодо пропіонітрилу і піролу при перепусканні базової сировини з домішками 80 млн. ч. N у формі пропіонітрилу і 80 млн. ч. N у формі піролу крізь цеоліт у стаціонарному шарі. Регенерацію відпрацьованого NaY в одному випадку проводили шляхом кальцинування при 200°С упродовж 12 годин, а в іншому - шляхом промивання. Регенерацію NaY промиванням проводили за допомогою толуолу при температурі приблизно 20°С і витраті толуолу 5мл/хв. під атмосферним тиском протягом приблизно 20 годин. Регенерація промиванням з очевидністю дає кращі результати порівняно з регенерацією кальцинуванням; на промитому каталізаторі адсорбувалося на 10% більше пропіонітрилу (PN) і піролу (Ру). Таблиця 9 WHSV, Каталізатор Тиск, бар год.-1 13,84 NaY 4 Сировина Пропіонітрил, г адсорб. N/ 100г каталіз. Пірол, г адсорб. N/100г каталіз. Бутиронітрил, г адсорб. N/ 100г каталіз. Базова сировина + 80 млн. ч. N PN 80 млн. ч. N Ру >2,06 1,48 37 91042 38 Продовження таблиці 9 WHSV, Каталізатор Тиск, бар год.-1 25,86 NaY1 4 19,86 NaY 2 4 Пропіонітрил, г адсорб. N/ 100г каталіз. Сировина Пірол, г адсорб. N/100г каталіз. Бутиронітрил, г адсорб. N/ 100г каталіз. >1,2 0,99 >1,35 1,12 Базова сировина + 160 млн. ч. N PN 160 млн. ч. N Ру Базова сировина + 160 млн. ч. N PN 160 млн. ч. N Ру 1 - Регенерація NaY шляхом кальцинування (200°С). 2 - Регенерація NaY шляхом промивання толуолом при кімнатній температурі. Базова сировина: 50%nС7 і 50% 1С6= з додатком 200 млн. ч. S у формі тіофену. ПРИКЛАД 9 Конкурентна адсорбція В експериментах із сировиною, що використовувалася у Прикладі 7, але з добавками до неї 40 млн. ч. N у формі бутиронітрилу (BN), спостерігалося зменшення ємності адсорбції щодо піролу. При цьому спостерігалося також зменшення ємності адсорбції щодо пропіонітрилу. Це, очевидно, було зумовлено конкуренцією між пропіонітрилом і бутиронітрилом за активні ділянки цеоліту. Проте ця адсорбція двох даних нітрилів, очевидно, перешкоджає адсорбції піролу, що займає активні центри. Цей висновок з очевидністю випливає із порівняння Табл.9 з Табл.10. У Табл.10 подані результати двох ідентичних та одночасних процесів адсорбції. Таблиця 10 WHSV, Каталізатор Год.-1 70.52 NaY 80.88 NaY Сировина Базова сировина + 80 млн. ч. N PN 80 млн. ч. N Ру 40 млн. ч. N BN Базова сировина + 80 млн. ч. N PN 80 млн. ч. N Ру 40 млн. ч. N BN Пропіонітрил, г адБутиронітрил, г адсорб. N/ 100г каталісорб. N/ 100г каталіз. затора Пірол, г адсорб. N/100г каталізатора 0,8851 0,728 0,2651 1,0023 0,7439 0,2733 У Табл.11 поданий підсумок результатів стосовно ємності адсорбції сепіолітів і Y-цеоліту Y у Na-, Na-H, H-, Сu- і Cs-формах при обробці базової сировини, що містила пірол, пропіонітрил і бутиронітрил (відповідно до результатів, розглянутих вище, ці 3 сполуки є для каталізатора найбільш отруйними компонентами типової промислової сировини). У Табл.11 наведені величини кількості N, адсорбованого при прориванні першої сполуки. Таблиця 11 Каталізатор Природний сепіоліт Природний сепіоліт Базова сировина + 30% ароматичних сполук NaY NaY Базова сировина + 30% ароматичних сполук Пропіонітрил, г Бутиронітрил, г Пірол, г адсорб. адсорб. N/ 100г адсорб. N/ 100г N/100г каталіз. каталіз. каталіз. Сума Сировина без N 0,0486 0,049 0,0976 24,29 0,02256 0,017 0,03956 11,28 >0,8353 >0,5152 >1,3505 417,67 0,1965 0,1 0,2965 68,14 39 91042 40 Продовження таблиці 11 Каталізатор HY NaHY CuNaY NaX CsY Пропіонітрил, г Бутиронітрил, г Пірол, г адсорб. адсорб. N/ 100г адсорб. N/ 100г N/100г каталіз. каталіз. каталіз. 0,145 0,4646 0,0153 0,209 0,6678 0,0221 0,0961 0;3072 0,0101 0,241 0,7701 0,0254 0,3198 1,5908 0,086 Сума Сировина без N 0,6249 0,8989 0,4134 1,0365 1,9953 48,69 69,99 32,2 80,71 70,02 Затемнені комірки: перша сполука, що проривається Таблиця 12 Величини, що характеризують здатність твердих каталізаторів адсорбувати N Каталізатор Природний сепіоліт Природний сепіоліт Базова сировина + 30% ароматичних сполук NaY NaY Базова сировина + 30% ароматичних сполук HY NaHY CuNaY NaX CsY Пропіонітрил, г Бутиронітрил, г Пірол, г адсорб. адсорб. N/ 100г адсорб. N/ 100г N/ 100г каталіз. каталіз. каталіз. >0,79 0,049 WHSV 0,839 17,88 0,283 0,017 0,3 19,65 >0,8353 >0,5152 >1,3505 18,84 >1,3 0,1 >1,4 20,21 0,145 0,3 0,3203 >0,7 0,3198 0,818 0,67 0,3072 0,77 1,5908 0,0199 0,0283 0,0161 0,0584 0,0846 0,9829 0,9938 0,64 1,5248 1,9952 80,59 69,37 67,75 67,86 Із Табл.12 видно, що NaY- і CuY-цеоліти є ефективними адсорбентами для неосновних сполук азоту. Слід зауважити, що ароматичні сполуки конкурують за ділянки адсорбції, що з більшою силою виявляється у піролу. CsY-цеоліт адсорбував менше пропіонітрилу, але більше піролу, ніж NaY, у той час як адсорбція бутиронітрилу була також високою. Беручи до уваги це, а також дані в Табл.12 і 13, можна зробити висновок, що суміш адсорбентів може бути ефективним засобом попередньої обробки неосновних сполук азоту для процесу алкілування тіофенів. Як зазначалося вище, регенерація може здійснюватися, а ємність адсорбції відновлюватися шляхом нагріву в потоку повітря або шляхом промивання толуолом. Промислова сировина Сировина NaY-1 Сировина NaY-2 Сировина NaY-3 Сировина NaY-4 Сировина Sep-1 Сировина Sep-2 Сума ПРИКЛАД 10 У цьому прикладі продемонстрований ефект попередньої обробки, що проводилася з метою видалення неосновних сполук азоту із промислової сировини, призначеної для алкілування тіофенів (Табл.4). Промислову сировину для алкілування тіофенів перепускали паралельно через два стаціонарні шари каталізаторів, один з яких містив Na-Y, а другий - сепіоліт. Поглинання азоту здійснювалося в таких умовах: температура 70°F, об'ємна витрата WHSV=15год.-1, кількість матеріалу адсорбенту = 2 грами. Продукт із реактора з адсорбентом збирали кожні 80 хвилин. Для проведення оцінки в реакторі алкілування тіофенів були відібрані перелічені нижче сировинні матеріали. Без попередньої обробки Na-Y Адсорбент Na-Y Адсорбент Na-Y Адсорбент Na-Y Адсорбент Сепіолітовий адсорбент Сепіолітовий адсорбент У дослідній напівпромисловій установці в даному прикладі використовувався твердий фосфорнокислотний каталізатор марки С-84-05 виробництва фірми Sud Chemie Inc. У дослідну установку завантажували 300мг цього каталізатора, який був роздрібнений до розмірів 0,4-0,6мм комірок сита Тайлера. Перед використанням каталізатор про 0-180 хвилин (80 грамів) 180-340 хвилин (80 грамів) 340-510 хвилин (80 грамів) 510-730 хвилин (108 грамів) 0-180 хвилин (80 грамів) 510-730 хвилин (108 грамів) сушили в потоку азоту протягом 2 годин при температурі 200°С. Дослідна установка складалася із двох паралельних реакторів зі стаціонарним шаром, кожний з яких приймав завантаження каталізатора в кількості від 50 до 1000мг. Дослідна установка мала конструкцію, аналогічну тій, що використовувалася в Прикладі 1. Реактори працю 41 91042 вали в режимі низхідного потоку. Через цю установку перепускали всі вище перелічені сировинні матеріали. Досліди проводилися в таких умовах: -1 LHSV 4,1 год. Тиск 400 фунт/кв.дюйм (манометричний) Температура 180°С Результати експериментів подані на Фіг.16. На цій діаграмі можна бачити, що значне поліпшення продуктивності каталізатора у процесі алкілування 42 тіофенів досягалося, коли промислову сировину піддавали попередній обробці з NaY або сепіолітом. Цей ефект є порівняним зі швидкою дезактивацією, що спостерігається при контакті каталізатора з необробленою промисловою сировиною по витіканні всього лише восьми годин. У діапазоні проведеної оцінки (до 150 грамів обробленої сировини на грам адсорбенту) попередня обробка з NaY дає стабільну продуктивність процесу. 43 91042 44 45 91042 46 47 91042 48 49 91042 50 51 91042 52 53 91042 54 55 Комп’ютерна верстка Л. Ціхановська 91042 Підписне 56 Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of hydrocarbon products
Автори англійськоюKECKLER KENNETH P, CORMA AVELINO, KNOX THOMAS, GREENOUGH PAUL, HODGES MICHAEL G
Назва патенту російськоюПроцесс приготовления углеводородных продуктов
Автори російськоюКеклер Кеннет Пол, Корма Авелино, Нокс Томас, Гриноу Пол, Ходжес Майкл Г.
МПК / Мітки
МПК: C10G 29/00, C10G 69/00, B01J 20/34
Мітки: процес, виготовлення, продуктів, вуглеводневих
Код посилання
<a href="https://ua.patents.su/28-91042-proces-vigotovlennya-vuglevodnevikh-produktiv.html" target="_blank" rel="follow" title="База патентів України">Процес виготовлення вуглеводневих продуктів</a>
Спосіб термічної газифікації вуглеводневих продуктів та газогенератор для його здійснення

Номер патенту: 38802
Опубліковано: 15.05.2001
Автори: Зубанюк Юрій Романович, Мирний Володимир Семенович
МПК: F01K 23/10
Мітки: здійснення, продуктів, газифікації, газогенератор, термічної, спосіб, вуглеводневих
Текст:
...спосіб, що пропонується, наступним чином. Через завантажувальну шахту 5 газогенератора подають в термічну камеру 4 вуглеводневу сировину. Через патрубок 2 подають газоподібний водень, який через отвори 7 потрапляє в термічну камеру 5, де змішується з вуглеводневою сировиною і створює сировинну суміш. Сировинну суміш нагрівають нагрівачем 8 з використанням конвенційного типу, при цьому тільки такої кількості, яка необхідна для проведення...
Каталітична композиція, процес її виготовлення і процес гідрообробки вуглеводневої сировини з її використанням

Номер патенту: 89613
Опубліковано: 25.02.2010
Автори: Джонгкінд Германус, Ван Вен Йоханнес Антоніус Роберт, Домокос Ласло
МПК: B01J 37/03, B01J 21/00, B01J 23/882, C10G 45/02, C10G 67/00, B01J 23/883, B01J 23/85, B01J 23/888
Мітки: виготовлення, каталітична, гідрообробки, композиція, використанням, процес, сировини, вуглеводневої
Формула / Реферат:
1. Процес виготовлення каталітичної композиції, загальної формули в розрахунку на оксиди (Х)b(М)с(Z)d(O)е, (І)деХ - принаймні один неблагородний метал VIII групи,М - принаймні один неблагородний метал VIb групи,Z - один чи більше елементів, вибраних серед алюмінію, кремнію, магнію, титану, цирконію, бору і цинку,О - кисень,один із індексів b або с є цілим числом, що дорівнює 1, іd, е та інші...
Процес виготовлення каталізатора на волокнистому носії для окиснення газоподібних вуглеводнів

Номер патенту: 35851
Опубліковано: 10.10.2008
Автори: Михайловський Віліус Ярославович, Струтинська Любов Тимофіївна
МПК: B01J 23/76
Мітки: процес, каталізатора, окиснення, газоподібних, носії, волокнистому, вуглеводнів, виготовлення
Формула / Реферат:
1. Процес виготовлення каталізатора на волокнистому носії для глибокого окиснення газоподібних вуглеводнів шляхом просочування носія розчинами солей кобальту та хрому, наступного висушування та прожарювання, який відрізняється тим, що просочувальний розчин додатково містить сполуки платини або паладію, а висушування каталізатора після просочування здійснюється у дві стадії.2. Процес за п. 1, який відрізняється тим, що концентрація...
Жир кондитерський “оливія глазур” для виготовлення глазурі до харчових продуктів

Номер патенту: 27757
Опубліковано: 12.11.2007
Автор: Савус Анатолій Семенович
МПК: A23D 9/02
Мітки: продуктів, кондитерський, жир, глазур, харчових, оливія, глазурі, виготовлення
Формула / Реферат:
1. Жир кондитерський для виготовлення глазурі до харчових продуктів, до складу якого входять олія соняшникова та олеїн пальмовий, який відрізняється тим, що містить антиоксидант і регулятор кристалізації, при цьому олія соняшникова та олеїн пальмовий містяться в кондитерському жирі як саломас із вмістом компонентів, мас. %: олія соняшникова 60,0 - 70,0 олеїн пальмовий 30,0 -...
Шаруватий композиційний матеріал для каталізу, процес виготовлення каталізатора та виготовлення алкенілалканоатів

Номер патенту: 87878
Опубліковано: 25.08.2009
Автори: Хара Коюджі, Ванг Тао, Аоі Нобуюкі, Бойке Джеффрі, Ренде Дін, Такаяма Масао, Рекоске Джеймс, Брікер Джеффрі
МПК: B01J 37/025, C07C 67/055, B01J 23/44, B01J 23/48
Мітки: процес, алкенілалканоатів, матеріал, композиційний, каталізу, виготовлення, шаруватий, каталізатора
Формула / Реферат:
1. Шаруватий композиційний матеріал для каталізу процесів виготовлення алкенілалканоатів, який включає у себе:- внутрішнє ядро і- зовнішній шар, який включає тугоплавкий неорганічний оксид, волокнистий компонент, неорганічне сполучне, а також паладій, золото або обидва ці каталітичні компоненти, розподілені по зовнішньому шару.2. Композиційний матеріал за п. 1, де волокнистий компонент включає у себе неорганічні волокна,...
Попередній патент: Спосіб інертизації для запобігання пожежам
Наступний патент: Ліжко відкидне “екма”
Випадковий патент: Спосіб лікування первинної відкритокутової глаукоми