Піперидинонкарбоксамідазаіндани – антагоністи рецептора cgrp
Номер патенту: 109463
Опубліковано: 25.08.2015
Автори: Зартман С. Блейр, Джиннетті Ентоні, Ван Чен, Мітчелл Хелен Дж., Пеуан Деніел В., Стівенсон Хітер Е., Фрелі Марк Е., Стейєс Доннетт, Белл Ян М., Галліккіо Стівен Н.






Формула / Реферат
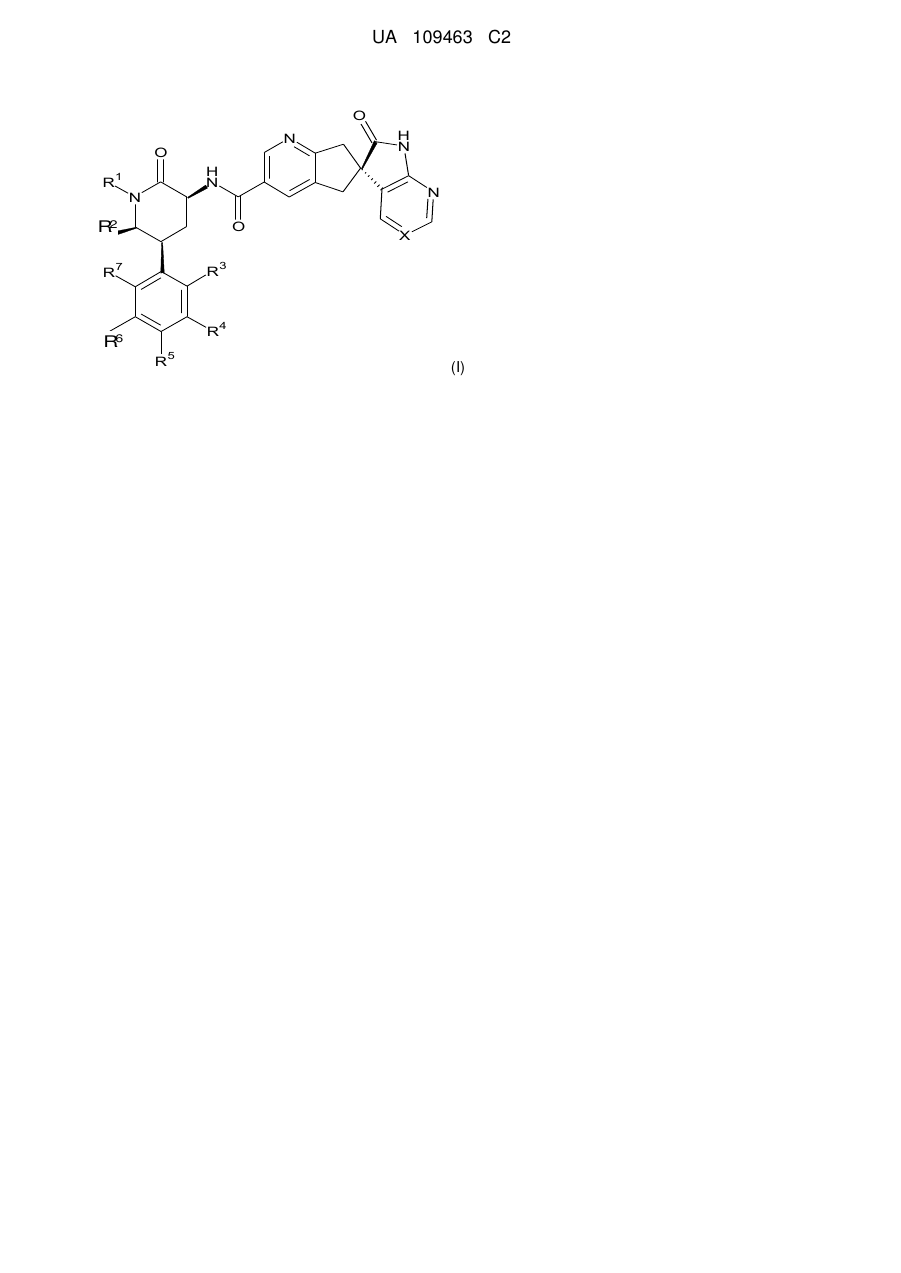
1. Сполука формули І
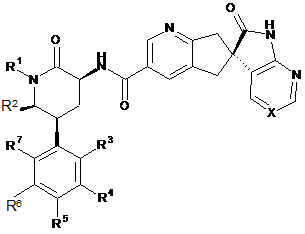 I
I
або її фармацевтично прийнятна сіль, де:
X вибирають з -C(R8)= або -N=, де R8 являє собою водень, F або CN;
R1 вибирають із групи, що складається з: С1-4алкілу, циклопропілметилу, циклобутилметилу і [1-(трифторметил)циклопропіл]метилу, кожний з яких необов'язково заміщений одним або більше із замісників, якщо допускають валентності, незалежно вибраних із групи, що складається з: F і гідрокси;
R2 вибирають з водню і метилу;
якщо R2 являє собою водень, тоді
R3 вибирають з водню, F або Сl;
R4 вибирають з водню, F або Сl;
R5 являє собою водень;
R6 вибирають з водню або F; і
R7 вибирають з водню, F або Сl;
за винятком того, що щонайменше два з R3, R4, R6 і R7 повинні являти собою F або Cl, якщо тільки R3 не являє собою F, і в цьому випадку R4, R6 і R7 усі можуть являти собою водень; і, якщо R4 являє собою Cl, тоді R7 не може являти собою Cl;
якщо R2 являє собою метил, тоді
R3 вибирають з водню, метилу, F, Cl або Вг;
R4 вибирають з водню, метилу, F або Cl;
R5 вибирають з водню або F;
R6 вибирають з водню або F; і
R7 вибирають з водню, метилу, F або Cl;
за винятком того, що, якщо R5 являє собою F, тоді щонайменше три з R3, R4, R6 і R7 повинні являти собою F; і, якщо R4 являє собою метил або Cl, тоді R7 не може являти собою метил або Cl.
2. Сполука за п. 1 або її фармацевтично прийнятна сіль, де X являє собою -N=.
3. Сполука за п. 1 або її фармацевтично прийнятна сіль, де X являє собою -СН=.
4. Сполука за п. 1 або її фармацевтично прийнятна сіль, де X являє собою -C(CN)=.
5. Сполука за п. 1 або її фармацевтично прийнятна сіль, де X являє собою -C(F)=.
6. Сполука за п. 1 або її фармацевтично прийнятна сіль, де R1 являє собою С1-4алкіл, необов'язково заміщений 1-3 F або гідрокси, або і тим, і іншим.
7. Сполука за п. 6 або її фармацевтично прийнятна сіль, де R1 вибирають з: ізопропілу, 2,2,2-трифторетилу, 2,2-дифторетилу, 2-метилпропілу, 3,3,3-трифторпропілу і 3,3,3-трифтор-2-гідроксипропілу.
8. Сполука за п. 7 або її фармацевтично прийнятна сіль, де R1 являє собою 2,2,2-трифторетил.
9. Сполука за п. 7 або її фармацевтично прийнятна сіль, де R2 являє собою водень.
10. Сполука за п. 9 або її фармацевтично прийнятна сіль, де щонайменше два з R3, R4, R6 і R7 являють собою F або Cl, за винятком того, що, якщо R4 являє собою Cl, тоді R7 не може являти собою Cl.
11. Сполука за п. 9 або її фармацевтично прийнятна сіль, де R3 являє собою F і R4, R6 і R7 являють собою водень.
12. Сполука за п. 1 або її фармацевтично прийнятна сіль, де R2 являє собою метил.
13. Сполука за п. 12 або її фармацевтично прийнятна сіль, де R5 являє собою F і щонайменше три з R3, R4, R6 і R7 являють собою F.
14. Сполука за п. 12 або її фармацевтично прийнятна сіль, де R5 являє собою водень і, якщо R4 являє собою метил або Cl, тоді R7 не може являти собою метил або Cl.
15. Сполука за п. 12 або її фармацевтично прийнятна сіль, де R3 вибирають з водню, метилу, F або Cl; R4 вибирають з водню, метилу, F або Cl; R5 являє собою водень; R6 вибирають з водню або F; і R7 вибирають з водню, метилу, F або Cl; за винятком того, що, якщо R4 являє собою метил або Cl, тоді R7 не може являти собою метил або Cl.
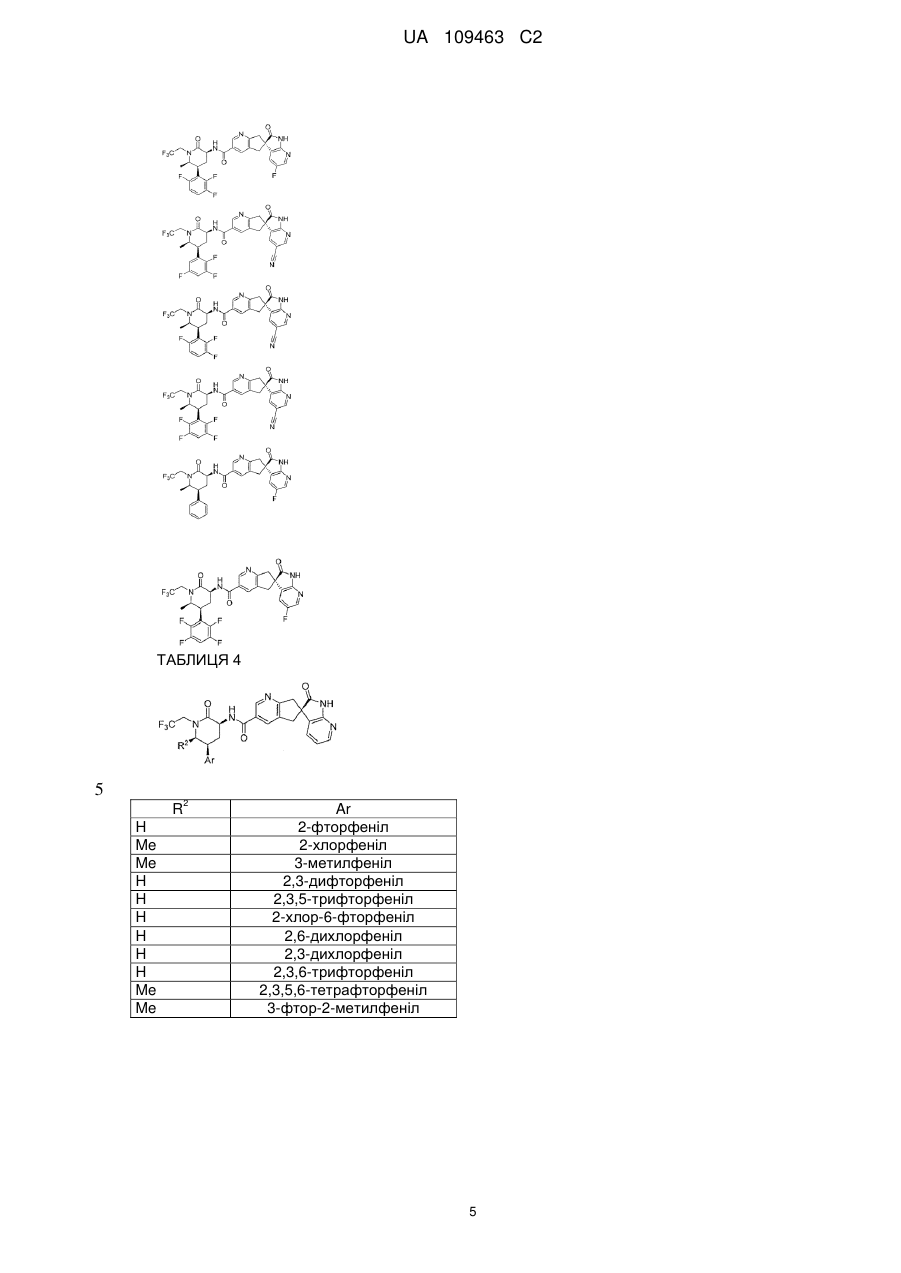
16. Сполука за п. 1, яку вибирають з наступних:
 ,
,
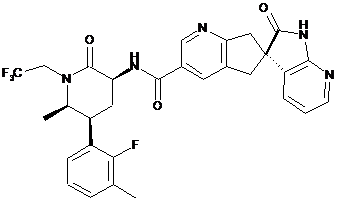 ,
,
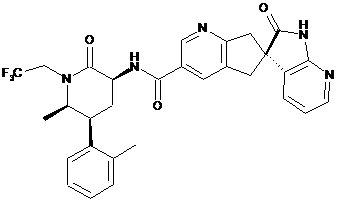 ,
,
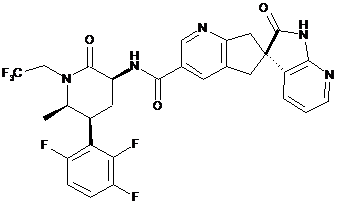 ,
,
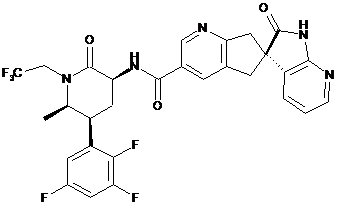 ,
,
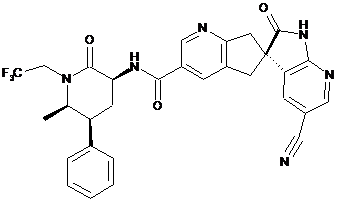 ,
,
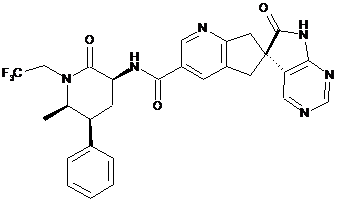 ,
,
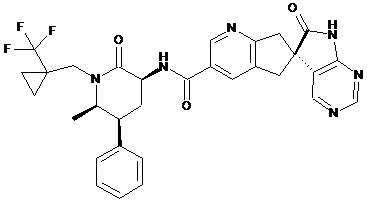 ,
,
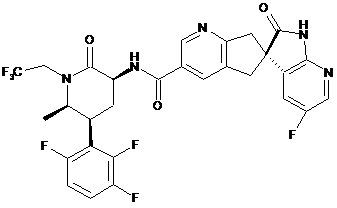 ,
,
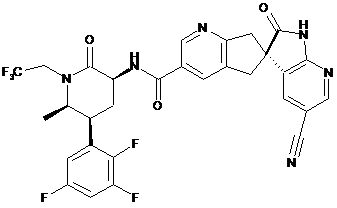 ,
,
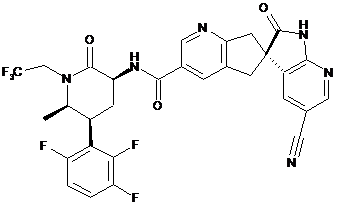 ,
,
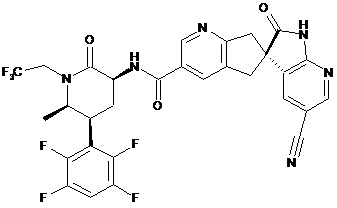 ,
,
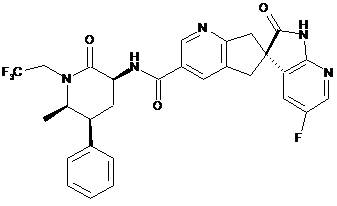 ,
,
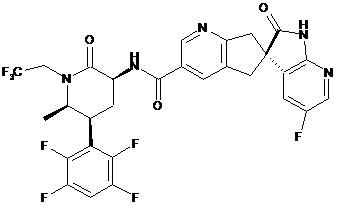 ,
,
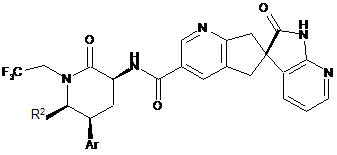 , де
, де
R2
Аr
Н
2-фторфеніл
Me
2-хлорфеніл
Me
3-метилфеніл
H
2,3-дифторфеніл
H
2,3,5-трифторфеніл
Н
2-хлор-6-фторфеніл
Н
2,6-дихлорфеніл
Н
2,3-дихлорфеніл
Н
2,3,6-трифторфеніл
Me
2,3,5,6-тетрафторфеніл
Me
3-фтор-2-метилфеніл
 , де
, де
R1
Аr
циклобутилметил
2,3-дифторфеніл
2-метилпропіл
2-фторфеніл
циклобутилметил
2-фторфеніл
ізопропіл
2-фторфеніл
(2S)-3,3,3-трифтор-2-гідроксипропіл
2,3-дифторфеніл
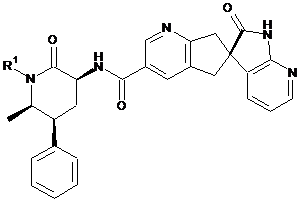 , де
, де
R1
3,3,3-трифторпропіл
2-метилпропіл
(2S)-3,3,3-трифтор-1-гідроксипропіл
циклопропілметил
[1-(трифторметил)циклопропіл]метил
2,2-дифторетил
[(1R)-2,2-дифторциклопропіл]метил
[(1S)-2,2-дифторциклопропіл]метил
або її фармацевтично прийнятна сіль.
17. Сполука за п. 1, яка являє собою
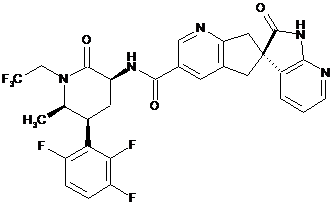 ,
,
або її фармацевтично прийнятна сіль.
18. Сполука за п.1, яка являє собою
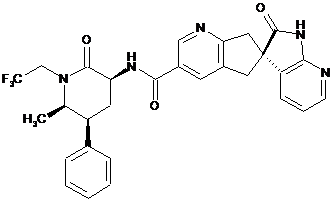 ,
,
або її фармацевтично прийнятна сіль.
19. Фармацевтична композиція, яка містить інертний носій і сполуку за п. 1 або її фармацевтично прийнятну сіль.
20. Застосування сполуки за будь-яким з пп. 1-18 або її фармацевтично прийнятної солі і фармацевтично прийнятного носія для виготовлення лікарського засобу для лікування головного болю.
21. Застосування за п. 20, де вказаний головний біль являє собою мігрень.
Текст
Реферат: Даний винахід стосується похідних піперидинонкарбоксамідазаіндану формули І, які є антагоністами рецепторів CGRP і які корисні при лікуванні або для запобігання захворюванням, у яких беруть участь CGRP, таким як мігрень. Даний винахід стосується також фармацевтичних композицій, що містять зазначені сполуки, і застосування зазначених сполук і композицій для профілактики або лікування таких захворювань, у яких бере участь CGRP. UA 109463 C2 (12) UA 109463 C2 O N O H N 1 R H N N N O 2 R X 3 7 R R 4 6 R R 5 R (I) UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 CGRP (кальцитонін-ген-зв'язаний пептид) являє собою природний пептид, що містить 37 амінокислот, який виробляється в результаті тканиноспецифічного альтернуючого процесингу іРНК кальцитоніну і широко розповсюджений у центральній і периферичній нервовій системі. CGRP розташовується переважно в сенсорних аферентних і центральних нейронах і опосередковує деякі біологічні функції, включаючи вазодилатацію. CGRP експресуються в альфа- і бета-формах, що відрізняються однією і трьома амінокислотами у щурів і людей, відповідно. CGRP-альфа і CGRP-бета демонструють аналогічні біологічні властивості. Після виділення з клітини, CGRP ініціює її біологічну реакцію за рахунок зв'язування з CGRPрецептором, що являє собою гетеродимер, який складається з G-білка, зв'язаного з кальцитоніноподібним рецептором (CLR) в асоціації з одним трансмембранним білком, відомим як білок 1, модифікуючим рецепторну активність (RAMP1). CGRP-рецептори переважно зв'язані з активацією аденілілциклази і були виявлені і фармакологічно оцінені в деяких тканинах і клітинах, включаючи клітини і тканини мозку, серцево-судинної системи, ендотелію і гладкої мускулатури. CGRP являє собою ефективний нейромодулятор, що бере участь у патології цереброваскулярних порушень, таких як мігрень і кластерний головний біль. У клінічних дослідженнях було виявлено, що підвищені рівні CGRP у яремній вені спостерігаються під час нападів мігрені (Goadsby et al. (1990) Ann. Neurol. 28, 183-187), підвищені рівні CGRP спостерігаються в слині пацієнтів, що страждають мігренню між нападами (Bellamy et al. (2006) Headache 46, 24-33) і під час нападів (Cady et al. (2009) Headache 49, 1258-1266), і було показано, що самі CGRP запускають механізм мігреневого головного болю (Lassen et al. (2002) Cephalalgia 22, 54-61). У клінічних дослідженнях було показано, що антагоніст рецептора CGRP BIBN4096BS ефективний при лікуванні гострих нападів мігрені (Olesen et al. (2004) New Engl. J. Med. 350, 1104-1110) і здатний запобігти головному болю, викликаному введенням CGRP контрольній групі (Petersen et al. (2005) Clin. Pharmacol. Ther. 77, 202-213). Було також показано, що біодоступний при пероральному введенні антагоніст рецептора CGRP телкагепант також виявляє протимігреневу ефективність у фазі III клінічних досліджень (Ho et al. (2008) Lancet 372, 2115-2123; Connor et al. (2009) Neurology 73, 970-977). CGRP-опосередкована активація тригеменоваскулярної системи може відігравати ключову роль у патогенезі мігрені. Крім того, CGRP активує рецептори гладкої мускулатури внутрішньочерепних судин, приводячи до посиленої вазодилатації, яка, як вважають, додає внесок у головний біль під час нападів мігрені (Lance, Headache Pathogenesis: Monoamints, Neuropeptides, Purines and Nitric Oxide, Lippincott-Raven Publishers, 1997, 3-9). Середня менінгеальна артерія, основна артерія твердої оболонки головного мозку, іннервується сенсорними волокнами тригемінального ганглія, що містить деякі нейропептиди, включаючи CORP. Стимуляція тригемінального ганглія у кішок викликає підвищені рівні CGRP, і у людей активація тригемінальної системи викликає почервоніння обличчя і підвищені рівні CGRP у зовнішній яремній вені (Goadsby et al. (1988) Ann. Neurol. 23, 193-196). Електрична стимуляція твердої оболонки мозку у щурів збільшує діаметр середньої менінгеальної артерії, ефект, що блокується попереднім введенням CGRP(8-37), пептидного антагоніста рецептора CGRP (Williamson et al. (1997) Cephalalgia 17, 525-531). Стимуляція тригемінального ганглія збільшує приплив крові до морди у щурів, що можна інгібувати, використовуючи CGRP(8-37) (Escott et al. (1995) Brain Res. 669, 93-99). Електрична стимуляція тригемінального ганглія у мавп викликає посилення припливу крові до морди, що можна блокувати, використовуючи непептидний антагоніст рецептора CGRP BIBN4096BS (Doods et al. (2000) Br. J. Pharmacol. 129, 420-423). Таким чином, судинний ефект CGRP можна послабити, запобігти або повернути назад, використовуючи антагоніст рецептора CGRP. Було показано, що CGRP-опосередкована вазодилатація середньої менінгеальної артерії у щурів сенсибілізує нейрони каудального ядра трійчастого нерва (Williamson et al, The CGRP Family: Calcitonin Gene-Related Peptide (CGRP), Amylin, and Adrenomedullin, Landes Bioscience, 2000, 245-247). Аналогічно, розширення кровоносних судин твердої оболонки головногомозку під час мігреневого головного болю може сенсибілізувати тригемінальні нейрони. Деякі із пов'язаних з мігренню симптомів, включаючи позачерепний біль і лицьову алодинію, можуть бути результатами сенсибілізованих тригемінальних нейронів (Burstein et al. (2000) Ann. Neurol. 47, 614-624). Антагоніст CGRP може бути сприятливим засобом для ослаблення, запобігання або повороту назад нейрональної сенсибілізації. Здатність сполук даного винаходу діяти як антагоністи рецептора CGRP робить їх корисними фармакологічними агентами при лікуванні порушень, таких як ті, що включають CGRP у людей і тварин, але особливо у людей. Такі порушення включають мігрень і кластерний головний біль (Doods (2001) Curr. Opin. Invest. Drugs 2, 1261-1268; Edvinsson et al. (1994) 1 UA 109463 C2 35 Cephalalgia 14, 320-327); хронічний головний біль напруження (Ashina et al. (2000) Neurology 14, 1335-1340); біль (Yu et al. (1998) Eur. J. Pharmacol. 347, 275-282); хронічний біль (Hulsebosch et al. (2000) Pain 86, 163-175); нейрогенні запалення і біль при запаленнях (Holzer (1988) Neuroscience 24, 739-768; Delay-Goyet et al. (1992) Acta Physiol. Scanda. 146, 537-538; Salmon et al. (2001) Nature Neurosci. 4, 357-358); очний біль (May et al. (2002) Cephalalgia 22, 195-196), зубний біль (Awawdeh et al. (2002) Int. Endocrin. J. 35, 30-36), інсулінонезалежний цукровий діабет (Molina et al. (1990) Diabetes 39, 260-265); судинні порушення; запалення (Zhang et al. (2001) Pain 89, 265); артрит, бронхіальну гіперактивність, астму (Foster et al. (1992) Ann. NY Acad. Sci. 657, 397-404; Schini et al. (1994) Am. J. Physiol. 267, H2483-H2490; Zheng et al. (1993) J Virol. 67, 5786-5791); шок, сепсис (Beer et al. (2002) Crit. Care Med. 30, 1794-1798); синдром відмови від наркотиків (Salmon et al. (2001) Nature Neurosci. 4, 357-358); морфінову толерантність (Menard et al. (1996) J. Neurosci. 16, 2342-2351); припливи крові у чоловіків і жінок (Chen et al. (1993) Lancet 342, 49; Spetz et al. (2001) J. Urology 166, 1720-1723); алергійний дерматит (Wallengren (2000) Contact Dermatitis 43, 137-143); псоріаз; енцефаліт, травми мозку, ішемію, удар, епілепсію і нейродегенеративні захворювання (Rohrenbeck et al. (1999) Neurobiol. Dis. 6, 15-34); шкірні хвороби (Geppetti і Holzer, Eds., Neurogenic Inflammation, 1996, CRC Press, Boca Raton, FL), нейрогенні почервоніння шкіри, шкірні порозовіння і еритему; тінітус (Herzog et al. (2002) J. Membr. Biol. 189, 225); тучність (Walker et al. (2010) Endocrinology 151, 4257-4269); запальні захворювання кишечнику, синдром подразненого кишечнику (Hoffman et al. (2002) Scand. J. Gastroenterol. 37, 414-422) і цистит. Особливо важливим є екстрене або профілактичне лікування головного болю, включаючи мігрень і кластерний головний біль. У патенті США № 7390798, виданому 24 червня 2008, і в патентній публікації США № US 2010/0179166, опублікованій 15 липня 2010, розкриті карбоксамідні антагоністи рецептора CGRP. Даний винахід належить до класу більш високоефективних антагоністів рецепторів CGRP у порівнянні з розкритими раніше аналогами, до фармацевтичних композицій, що їх включають, і їх використання в терапії. СУТЬ ВИНАХОДУ Даний винахід стосується похідних піперидинонкарбоксамідазаіндану, що являють собою високоефективні антагоністи рецепторів CGRP і корисні при лікуванні або для профілактики захворювань, у яких беруть участь CGRP, таких як мігрень. Даний винахід стосується також фармацевтичних композицій, що включають зазначені сполуки, і використання зазначених сполук і композицій для профілактики або лікування таких захворювань, у яких бере участь CGRP. ДОКЛАДНИЙ ОПИС ВИНАХОДУ Даний винахід стосується групи сполук формули I: 40 або її фармацевтично прийнятної солі, де: 8 8 X вибирають з -C(R )= або -N=, де R являє собою водень, F або CN; 1 R вибирають із групи, що складається з: С1-4алкілу, циклопропілметилу, циклобутилметилу і [1-(трифторметил)циклопропіл]метилу, кожний з який необов'язково заміщений одним або 5 10 15 20 25 30 2 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 більше із замісників, якщо допускають валентності, незалежно вибраних із групи, що складається з: F і гідрокси; 2 R вибирають з водню і метилу; 2 якщо R являє собою водень, тоді 3 R вибирають з водню, F або Cl; 4 R вибирають з водню, F або Cl; 5 R являє собою водень; 6 R вибирають з водню або F; і 7 R вибирають з водню, F або Cl; 3 4 6 7 за винятком того, що щонайменше два з R , R , R і R повинні являти собою F або Cl, якщо 3 4 6 7 4 тільки R не являє собою F, і в цьому випадку R , R і R усі можуть бути воднями; і, якщо R 7 являє собою Cl, тоді R не може являти собою Cl; 2 якщо R являє собою метил, тоді 3 R вибирають з водню, метилу, F, Cl або Br; 4 R вибирають з водню, метилу, F або Cl; 5 R вибирають з водню або F; 6 R вибирають з водню або F; і 7 R вибирають з водню, метилу, F або Cl; 5 3 4 6 7 за винятком того, що, якщо R являє собою F, тоді щонайменше три з R , R , R і R повинні 4 7 являти собою F; і, якщо R являє собою метил або Cl, тоді R не може являти собою метил або Cl. Усередині зазначеної групи даний винахід охоплює першу підгрупу сполук формули I або їх фармацевтично прийнятну сіль, де X являє собою -N=. Також усередині зазначеної групи даний винахід включає другу підгрупу сполук формули I або їх фармацевтично прийнятну сіль, де X являє собою -CH=. Також усередині зазначеної групи даний винахід включає третю підгрупу сполук формули I або їх фармацевтично прийнятну сіль, де X являє собою -C(CN)=. Також усередині зазначеної групи даний винахід включає четверту підгрупу сполук формули 1 I або їх фармацевтично прийнятну сіль, де R являє собою С1-4алкіл, необов'язково заміщений 1-3 F або гідрокси, або і тим, і іншим. У четвертій підгрупі сполук даний винахід включає перший клас сполук формули I або їх 1 фармацевтично прийнятну сіль, де R вибирають з: ізопропілу, 2,2,2-трифторетилу, 2,2дифторетилу, 2-метилпропілу, 3,3,3-трифторпропілу і 3,3,3-трифтор-2-гідроксипропілу. У першому класі сполук даний винахід включає перший підклас сполук формули I або їх 1 фармацевтично прийнятну сіль, де R являє собою 2,2,2-трифторетил. Також усередині зазначеної групи сполук даний винахід включає п'яту підгрупу сполук 2 формули I або їх фармацевтично прийнятну сіль, де R являє собою водень. У п'ятій підгрупі сполук даний винахід включає другий клас сполук формули I або їх 3 4 6 7 фармацевтично прийнятну сіль, де щонайменше два з R , R , R і R являють собою F або Cl, за 4 7 винятком того, що, якщо R являє собою Cl, тоді R не може являти собою Cl. Також у п'ятій підгрупі сполук даний винахід включає третій клас сполук формули I або їх 3 4 6 7 фармацевтично прийнятну сіль, де R являє собою F і R , R і R являють собою водень. Також усередині зазначеної групи, даний винахід включає шосту підгрупу сполук формули I 2 або їх фармацевтично прийнятну сіль, де R являє собою метил. У шостій підгрупі сполук даний винахід включає четвертий клас сполук формули I або їх 5 3 4 6 7 фармацевтично прийнятну сіль, де R являє собою F і щонайменше три з R , R , R і R являють собою F. Також у шостій підгрупі сполук даний винахід включає п'ятий клас сполук формули I або їх 5 5 фармацевтично прийнятну сіль, де R являє собою водень і, якщо R являє собою метил або Cl, 7 тоді R не може бути метилом або Cl. Також у шостій підгрупі сполук даний винахід включає шостий клас сполук формули I або їх 3 4 фармацевтично прийнятну сіль, де R вибирають з водню, метилу, F або Cl; R вибирають з 5 6 7 водню, метилу, F або Cl; R являє собою водень; R вибирають з водню або F; і R вибирають з 4 7 водню, метилу, F або Cl; за винятком того, що, якщо R являє собою метил або Cl, тоді R не може бути метилом або Cl. Також усередині зазначеної групи сполук даний винахід включає сьому підгрупу сполук формули I або їх фармацевтично прийнятну сіль, де X являє собою -C(F)=. Даний винахід також включає сполуки, вибрані з наступних: 3 UA 109463 C2 4 UA 109463 C2 ТАБЛИЦЯ 4 5 R H Me Me H H H H H H Me Me 2 Ar 2-фторфеніл 2-хлорфеніл 3-метилфеніл 2,3-дифторфеніл 2,3,5-трифторфеніл 2-хлор-6-фторфеніл 2,6-дихлорфеніл 2,3-дихлорфеніл 2,3,6-трифторфеніл 2,3,5,6-тетрафторфеніл 3-фтор-2-метилфеніл 5 UA 109463 C2 ТАБЛИЦЯ 5 1 R циклобутилметил 2-метилпропіл циклобутилметил ізопропіл (2S)-3,3,3-трифтор-2гідроксипропіл 5 Ar 2,3-дифторфеніл 2-фторфеніл 2-фторфеніл 2-фторфеніл 2,3-дифторфеніл ТАБЛИЦЯ 6 1 R 3,3,3-трифторпропіл 2-метилпропіл (2S)-3,3,3-трифтор-2-гідроксипропіл циклопропілметил [1-(трифторметил)циклопропіл]метил 2,2-дифторетил [(1R)-2,2-дифторциклопропіл]метил [(1S)-2,2-дифторциклопропіл]метил 10 15 20 25 30 або їх фармацевтично прийнятну сіль. Даний винахід також включає фармацевтичні композиції, що містять інертний носій і сполуку формули I або її фармацевтично прийнятну сіль. Даний винахід також включає спосіб лікування головного болю у потребуючого такого лікування пацієнта-ссавця, що включає введення пацієнту терапевтично ефективної кількості сполуки формули I або її фармацевтично прийнятної солі. У конкретному варіанті даного винаходу головний біль являє собою мігреневий головний біль. Даний винахід також включає застосування сполуки формули I або її фармацевтично прийнятної солі і фармацевтично прийнятного носія для виготовлення лікарського засобу для лікування головного болю. У конкретному варіанті даного винаходу головний біль являє собою мігреневий головний біль. Даний винахід стосується також лікарських засобів або фармацевтичних композицій для лікування захворювань або порушень, у яких беруть участь CGRP, таких як мігрень, які містять сполуку формули I або її фармацевтично прийнятну сіль і фармацевтично прийнятний носій. Даний винахід стосується також застосування сполуки формули I для лікування захворювань або порушень, у яких бере участь CGRP, таких як мігрень. Даний винахід далі стосується способу виготовлення лікарського засобу або композиції для лікування захворювань або порушень, у яких беруть участь CGRP, таких як мігрень, що включає об'єднання сполук формули I з одним або більше із фармацевтично прийнятних носіїв. Сполуки даного винаходу можуть містити один або більше асиметричних центрів і тому можуть існувати у вигляді рацематів і рацемічних сумішей, окремих енантіомерів, 6 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 діастереоізомерних сумішей і індивідуальних діастереоізомерів. Можуть бути присутніми додаткові асиметричні центри в залежності від природи різних замісників у молекули. Кожен такий асиметричний центр приведе незалежно до двох оптичних ізомерів, і варто розуміти, що усі з можливих оптичних ізомерів і діастереоізомерів у сумішах і як чисті або частково очищені сполуки включені в обсяг даного винаходу. Якщо не зазначена конкретна стереохімія, у даному винаході мається на увазі, що всі такі ізомерні форми зазначеної сполуки включені в обсяг даного винаходу. Незалежний синтез діастереоізомерів або їх хроматографічний поділ можна здійснити відомими фахівцям у даній галузі способами, використовуючи відповідні модифікації розкритих в описі методів. Абсолютну стереохімію зазначених сполук можна визначити, використовуючи рентгенівську кристалографію кристалічних продуктів або кристалічних похідних, що дериватизують при необхідності, використовуючи реагент, що містить асиметричний центр із відомою асиметричною конфігурацією. При бажанні рацемічні суміші зазначених сполук можна розділити таким чином, щоб виділити індивідуальні енантіомери. Зазначений поділ можна здійснити, використовуючи способи, добре відомі фахівцям у даній галузі, такі як приєднання рацемічної суміші сполук до енантіомерно чистої сполуки для утворення суміші діастереоізомерів, з наступним поділом на індивідуальні діастереоізомери стандартними способами, такими як фракційна кристалізація або хроматографія. У результаті реакції приєднання часто утворюють солі, використовуючи енантіомерно чисту кислоту або основу. Діастереоізомерні похідні потім можна перетворити в чисті енантіомери, відщеплюючи доданий хіральний залишок. Рацемічні суміші сполук можна також розділити безпосередньо, використовуючи хроматографічні методики, використовуючи хіральні стаціонарні фази, причому зазначені способи добре відомі фахівцям у даній галузі. Альтернативно, будь-які енантіомери сполуки можна отримати в результаті стереоспецифічного синтезу, використовуючи оптично чисті вихідні матеріали або реагенти з відомою конфігурацією способами, добре відомими фахівцям у даній галузі. У сполуках формули I, атоми можуть мати свій природний ізотопний склад або один або більше з атомів можуть бути штучно збагачені конкретним ізотопом, з тим же самим атомним номером, але з атомною масою або масовим числом, що відрізняються від атомної маси або масового числа, що переважно спостерігаються в природі. Мають на увазі, що даний винахід включає всі ізотопні варіанти сполук загальної формули I. Наприклад, різні ізотопні форми 1 2 водню (H) включають протій ( H) і дейтерій ( H). Протій являє собою переважний ізотоп водню, що існує в природі. Збагачення дейтерієм може забезпечити деякі терапевтичні переваги, такі як підвищення in vivo напівжиття або зменшення необхідних доз, або може запропонувати сполуку, яку можна використовувати як стандарт для характеризації біологічних зразків. Ізотопно збагачені сполуки загальної формули I можна отримати без небажаних експериментів, використовуючи звичайні способи, добре відомі фахівцям у даній галузі, або способами, аналогічними способам, розкритим на схемах і в прикладах, використовуючи відповідні ізотопно збагачені реагенти і/або проміжні сполуки. Таутомери сполук, визначених у формулі I, також включені в обсяг даного винаходу. Наприклад, сполуки, що включають карбоніл-CH2C(О)-групи (кетоформи), можуть зазнавати таутомеризм з утворенням гідроксил-CH-C(OH)-груп (енольні форми). Як кето-, так і енольні форми включені в обсяг даного винаходу. У тому значенні, як тут використано, термін "алкіл" означає лінійну або розгалужену структуру, що не містить вуглець-вуглецевих подвійних або потрійних зв'язків. Так, C1-4алкіл визначає групу як таку, що містить 1, 2, 3 або 4 атоми вуглецю в лінійному або розгалуженому ланцюгу, таку як С1-4алкіл, такі як спеціально включені, але ними не обмежуючи, метил, етил, нпропіл, ізопропіл, н-бутил, ізобутил і трет-бутил. Як зовсім очевидно фахівцям у даній галузі, "F" означає фтор, "Cl" означає хлор і "Br" означає бром. Вираз "фармацевтично прийнятний" використовують в описі відносно тих сполук, матеріалів, композицій і/або дозованих форм, що з погляду фахівців-медиків придатні для використання в контакті з людськими тканинами і тканинами тварин, без надлишкової токсичності, подразнення, алергійних реакцій або інших проблем і ускладнень, з відповідним розумним співвідношення користь/ризик. У тому значенні, як тут використано, термін "фармацевтично прийнятна сіль" стосується похідних, де вихідну сполуку модифікують, отримуючи її солі з кислотами або основами. Солі у твердій формі можуть існувати в більш ніж одній кристалічній структурі і можуть також існувати у формі гідратів. Приклади фармацевтично прийнятних солей включають, але ними не обмежуються, солі мінеральних або органічних кислот основних залишків, таких як аміни; солі 7 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 лужних металів або органічних сполук кислотних залишків, таких як карбонові кислоти; і т. п. Фармацевтично прийнятні солі включають звичайні нетоксичні солі або солі четвертинного амонію, утворені з вихідними сполуками, наприклад з нетоксичними неорганічними або органічними кислотами. Наприклад, такі звичайні нетоксичні солі включають солі, отримані з неорганічними кислотами, такими як хлористоводнева, бромистоводнева, сірчана, сульфамінова, фосфорна, азотна і т. п.; і солі, отримані з органічними кислотами, такими як оцтова, пропіонова, бурштинова, гліколева, стеаринова, молочна, яблучна, винна, лимонна, аскорбінова, памоєва, малеїнова, гідроксималеїнова, фенілоцтова, глутамінова, бензойна, саліцилова, сульфанілова, 2-ацетоксибензойна, фумарова, толуолсульфонова, метансульфонова, етандисульфонова, щавлева, ізетіонова, і т. п. Солі, отримані з неорганічними основами, включають солі алюмінію, амонію, кальцію, міді, заліза (2- і 3валентного), літію, магнію, марганцю (2- і 3-валентного), калію, натрію, цинку і т. п. Якщо сполука даного винаходу є основною, її солі можна отримати з фармацевтично прийнятними нетоксичними кислотами, включаючи неорганічні і органічні кислоти. Такі кислоти включають оцтову, бензолсульфонову, бензойну, камфорсульфонову, лимонну, етансульфонову, фумарову, глюконову, глутаміновую, бромистоводневу, хлористоводневу, ізетіонову, молочну, малеїнову, яблучну, мигдальну, метансульфонову, муцинову, азотну, памоєву, пантотенову, фосфорну, бурштинову, сірчану, винну, п-толуолсульфонову кислоту і т. п. В одному з аспектів даного винаходу використовують солі лимонної, бромистоводневої, хлористоводневої, малеїнової, фосфорної, сірчаної, фумарової і винної кислот. Варто розуміти, що в тому значенні, як тут використано, посилання на сполуки формули I означає також, що включено фармацевтично прийнятні солі. Ілюстрацією даного винаходу є застосування сполук, розкритих у розділі приклади. Конкретні сполуки даного винаходу включають сполуки, які можна вибрати з групи, що складається з розкритих у наступних прикладах сполук і їх фармацевтично прийнятних солей і їх індивідуальних діастереоізомерів. Цільові сполуки можна використовувати в способі антагонізму рецепторів CGRP у пацієнтів, таких як ссавці, що потребують такого антагонізму, який включає введення ефективної кількості сполуки даного винаходу. Даний винахід стосується застосування розкритих в описі сполук як антагоністів рецепторів CGRP. Крім приматів, особливо людей, різних інших ссавців також можна лікувати у відповідності зі способом даного винаходу. Інший варіант даного винаходу стосується способу лікування, контролю, полегшення або зменшення ризику захворювань або порушень у пацієнта, у яких беруть участь рецептори CGRP, який включає введення пацієнту терапевтично ефективної кількості сполуки, що являє собою антагоніст рецепторів CGRP. Даний винахід далі стосується способу виготовлення лікарського засобу для антагоністичної активності відносно рецепторів CGRP у людей і тварин, що включає об'єднання сполуки даного винаходу з фармацевтичним носієм або розріджувачем. Суб'єктом, для лікування якого використовують способи даного винаходу, звичайно є ссавець, люди, чоловіки або жінки, для яких бажаний антагонізм активності рецепторів CGRP. Термін "терапевтично ефективна кількість" означає таку кількість цільової сполуки, що викликає біологічну або медичну реакцію тканин, систем у тварин або людей, що необхідна досліднику, ветеринару, лікарю або іншим клініцистам. У тому значенні, як тут використаний, термін "лікування" стосується як лікування, так і запобігання або профілактичного лікування зазначених станів, особливо у пацієнтів, що схильні до таких захворювань або порушень. Термін "композиція", у тому значенні, як тут використаний, включає продукт, що містить специфіковані компоненти в специфікованих кількостях, так само як будь-які продукти, що безпосередньо або побічно поліпшують результат комбінації специфічних інгредієнтів у специфічних кількостях. Такий термін відносно фармацевтичних композицій означає продукт, який включає вищевказаний активний інгредієнт (інгредієнти) і інертний інгредієнт (інгредієнти), який складає носій, також як будь-який продукт, який поліпшує прямо або побічно за рахунок комбінації, комплексоутворення або агрегації будь-яких двох або більше інгредієнтів, або за рахунок дисоціації одного або більше з інгредієнтів, або за рахунок інших типів реакцій або взаємодій одного або більше з інгредієнтів. Відповідно, фармацевтичні композиції даного винаходу включають будь-які композиції, отримані шляхом змішування сполуки даного винаходу і фармацевтично прийнятного носія. Під виразом "фармацевтично прийнятний" мають на увазі носій, розріджувач або ексципієнт, які повинні бути сумісними з іншими інгредієнтами фармацевтичної композиції і не повинні робити шкідливого впливу на їхнього реципієнта. Даний винахід включає у свій обсяг проліки сполук розглянутого винаходу. Звичайно такі проліки є похідними сполук даного винаходу, які легко перетворювані in vivo у потрібні сполуки. 8 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 Так, у способах лікування даного винаходу, термін "уведення" сполуки включає лікування різних описаних станів сполуками, специфічно розкритими, або сполуками, які можуть не бути специфічно розкриті, але які перетворюються в специфіковані сполуки in vivo після введення пацієнту. Звичайні процедури вибору і отримання придатних пролікарських похідних розкриті, наприклад, у "Design of Prodrugs, " ed. H. Bundgaard, Elsevier, 1985. Метаболіти зазначених сполук включають активні зразки, отримані після введення сполук даного винаходу в біологічні середовища. Здатність сполук даного винаходу діяти як антагоністи рецепторів CGRP робить їх корисними фармакологічними агентами для лікування порушень, що включають CGRP, у людей і тварин, але особливо у людей. Сполуки даного винаходу знаходять застосування при лікуванні, профілактиці, полегшенні, контролі або для зменшення ризику одного або більше з наступних станів або захворювань: головного болю; мігрені; кластерного головного болю; хронічного головного болю напруження; болю; хронічного болю; нейрогенних запалень і болю при запаленнях; нейропатичного болю; очного болю; зубного болю; діабету; інсулінонезалежного цукрового діабету; судинних порушень; запалень; артриту; бронхіальної гіперреактивності, астми; шоку; сепсису; синдрому відмовлення від наркотиків; морфінізму; припливів крові у чоловіків і у жінок; алергійних дерматитів; псоріазу; енцефаліту; при травмах головного мозку; при епілепсії; нейродегенеративних захворювань; хвороб шкіри; нейрогенного почервоніння шкіри, rosaceousness шкіри і еритеми; тучності; запального захворювання кишечнику, синдрому подразненого кишечнику, циститу, і інших станів, які можна лікувати або попереджувати за рахунок антагонізму рецепторів CGRP. Особливу важливість являє лікування гострого головного болю або профілактичне лікування головного болю, включаючи мігрень і кластерний головний біль. Цільові сполуки даного винаходу далі можна застосовувати в способі профілактики, лікування, контролю, полегшення або зменшення ризику зазначених вище захворювань, порушень і станів. Цільові сполуки, крім того, можна застосовувати в способі профілактики, лікування, контролю, полегшення або зменшення ризику виникнення вищевказаних захворювань, порушень і станів у комбінації з іншими агентами. Сполуки даного винаходу можна застосовувати в комбінації з одним або більше з інших лікарських засобів для лікування, профілактики, контролю, полегшення або зменшення ризику виникнення вищевказаних захворювань або станів, для яких можна використовувати сполуки формули I або інші лікарські засоби, де комбінація лікарських засобів виявляється більш безпечною або більш ефективною, ніж використання одного тільки лікарського засобу. Такий інший лікарський засіб (засоби) можна вводити способом і в кількості, які звичайно для них використовують, одночасно або послідовно із сполукою формули I. Якщо сполуку формули I використовують одночасно з одним або більше з інших лікарських засобів, переважна фармацевтична композиція в одиничній дозованій формі, що містить такі інші лікарські засоби і сполуку формули I. Однак, комбінована терапія може також включати способи лікування, у яких сполука формули I і один або більше з лікарських засобів вводять відповідно до різних схем, що перекриваються. Варто також враховувати, що, якщо їх використовують у комбінації з одним або більше з інших активних інгредієнтів, сполуки даного винаходу і інші активні інгредієнти можна використовувати в більш низьких дозах, ніж, якщо кожне використовують окремо. Відповідно, фармацевтичні композиції даного винаходу включають такі, які містять один або більше з інших активних інгредієнтів на додаток до сполуки формули I. Наприклад, сполуки даного винаходу можна використовувати разом із протимігреневим агентом, таким як ерготамін і дигідроерготамін, або з іншими агоністами серотоніну, особливо агоністами 5-HT1B/1D, наприклад суматриптаном, наратриптаном, золмітриптаном, елетриптаном, алмотриптаном, фроватриптаном, донітриптаном і ризатриптаном, з агоністами 5-HT1D, такими як PNU-142633, і агоністами 5-HT1F такими як LY334370; з інгібіторами циклооксигенази, такими як селективний інгібітор циклооксигенази-2, наприклад рофекоксиб, еторикоксиб, целекоксиб, валдекоксиб або паракоксиб; з нестероїдними протизапальними агентами або цитокінпригнічувальними протизапальними агентами, наприклад зі сполуками, такими як ібупрофен, кетопрофен, фенопрофен, напроксен, індометацин, суліндак, мелоксикам, пріоксикам, теноксикам, лорноксикам, кеторолак, етодолак, мефенамінова кислота, меклофенамінова кислота, флуфенамінова кислота, толфенамінова кислота, диклофенак, оксапрозин, апазон, німесулід, намбуметон, тенідап, етанерцепт, толметин, фенілбутазон, оксифенбутазон, дифлунісал, салсалат, олсалазин або сульфасалазин і т. п.; або глюкокортикоїди. Аналогічно, сполуки даного винаходу можна вводити з анальгетиками, такими 9 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 як аспірин, ацетамінофен, фенацетин, фентаніл, суфентаніл, метадон, ацетилметадол, бупренорфін або морфін. Крім того, сполуки даного винаходу можна використовувати разом з інгібіторами інтерлейкіну, такими як інгібітор інтерлейкіну-1; антагоністами рецептора NK-1, наприклад апрепітантом; антагоністами NMDA; антагоністами NR2B; антагоністами рецептора брадикініну1; агоністами рецептора аденозин-A1; із блокаторами натрієвих канальців, наприклад ламотригіном; з агністами опіатів, такими як ацетат левометадилу або ацетат метадилу; з інгібіторами ліпоксигенази, такими як інгібітор 5-ліпоксигеназа; з антагоністами альфарецепторів, наприклад індораміном; з агоністами альфа-рецепторів; з антагоністами рецепторів ванілоїду; з інгібіторами реніну; з інгібіторами гранзиму B; з антагоністами речовини Р; з антагоністами ендотеліну; з попередниками норепінефрину; з агентами проти страху (занепокоєння), такими як діазепам, алпразолам, хлордіазепоксид і хлоразепат; з антагоністами рецептора серотоніну 5HT2; з антагоністами опоїдів, такими як кодеїн, гідрокодеїн, трамадол, декстропропоксифен і фебтаніл; з агоністами, антагоністами або потенціаторами mGluR5; з модуляторами рецептора GABA А, наприклад кальційакампросатом; з нікотиновими антагоністами або агоністами, включаючи нікотин; з мускариновими агоністами або антагоністами; із селективними інгібіторами повторного захоплення серотоніну, наприклад флуоксетином, пароксетином, сертраліном, дулоксетином, есциталопрамом або циталопрамом; з антидепресантами, наприклад амітрифіліном, нортрифіліном, кломіпраміном, іміпраміном, венлафаксином, доксепіном, протриптиліном, десипраміном, триміпраміном або іміпраміном; з антагоністами лейкотриєну, наприклад монтелукастом або зафірлукастом; з інгібіторами оксиду азоту або з інгібіторами синтезу оксиду азоту. Крім того, сполуки даного винаходу можна використовувати разом з інгібіторами щілинного контакту; з інгібіторами нейронових кальцієвих каналів, такими як цивамід; з антагоністами AMPA/KA, такими як LY293558; з агоністами сигма-рецепторів; і з вітаміном B2. Крім того, сполуки даного винаходу можна використовувати разом з алкалоїдами ріжка, відмінними від ерготаміну і дигідроерготаміну, наприклад з ергоновіном, метилергоновіном, метерголіном, мезилатами ерголоїду, дигідроергокорніном, дигідроергокристином, дигідроергокриптином, дигідро-α-ергокриптином, дигідро-β-ергокриптином, ерготоксином, ергокорніном, ергокристином, ергокриптином, α-ергокриптином, β-ергокриптином, ергосином, ергостаном, бромкриптином або метисергідом. Крім того, сполуки даного винаходу можна використовувати разом з β-адренергічними антагоністами, такими як тимолол, пропанолол, атенолол, метопролол або надолол, і т. п.; з інгібіторами MAO, наприклад фенелзином; із блокаторами кальцієвих канальців, наприклад флунаризином, дилтіаземом, амлодипіном, фелодипіном, нізоліпіном, ізрадипіном, німодипіном, ломеризином, верапамілом, ніфедипіном або прохлорперазином; з нейролептиками, такими як оланзапін, дроперидол, прохлорперазин, хлорпромазин і кветіапін; з антикоагулянтами, такими як топірамат, зонізамід, тонаберсат, караберсат, леветирацетам, ламотригін, тіагабін, габапентин, прегабалін або дивалпроекснатрій; з антигіпертензивними препаратами, такими як антагоністи ангіотензину II, наприклад лозартан, ірбесартин, валсартан, епросартан, телмісартан, олмесартан, медоксоміл, кандесартан і кандесартанцилексетил, з антагоністами ангіотензину I, з інгібіторами ферменту перетворення ангіотензину, такими як лізиноприл, еналаприл, каптоприл, беназеприл, квінаприл, периндоприл, раміприл і трандолаприл; або з токсином ботуліну типу A або B. Сполуки даного винаходу можна використовувати разом із потенціюючими засобами, такими як кофеїн, з H2-антагоністами, симетиконом, гідроксидами алюмінію або магнію; із протинабряковими агентами, такими як оксиметазолін, епінефрин, нафазолін, ксилометазолін, пропілгекседрин або леводезоксіефедрин; із протикашлевими засобами, такими як караміфен, карбетапентан або декстрометорфан; з діуретиками; із прокінетичними агентами, такими як метоклопромід або домперідон; седативними або неседативними антигістамінами, такими як акривастин, азатадин, бромдифенгідрамін, бромфенірамін, карбіноксамін, хлорфенірамін, клемастин, дексбромфенірамін, дексхлорфенірамін, дифенгідрамін, доксиламін, лоратадин, феніндамін, фенірамін, фенілтолоксамін, прометазин, піриламін, терфенадин, трипролідин, фенілеприн, фенілпропаноламін або псевдоефедрин. Сполуки даного винаходу також можна використовувати разом зі сполуками, що запобігають блюванню. У варіантах даного винаходу сполуки даного винаходу використовують разом із протимігреневими агентами, такими як: ерготамін або дигідроерготамін; з агоністами 5-HT1, особливо агоністами 5-HT1B/1D, зокрема суматриптаном, наратриптаном, золмітриптаном, елетриптаном, алмотриптаном, фроватриптаном, донітриптаном, авітриптаном і ризатриптаном, і іншими агоністами серотоніну; і з інгібіторами циклооксигенази, такими як 10 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 селективний інгібітор циклооксигенази-2, зокрема рофекоксиб, еторикоксиб, целекоксиб, валдекоксиб або паракоксиб. Вищевказані комбінації включають комбінації сполук даного винаходу не тільки з однією іншою активною сполукою, але також із двома або більше іншими активними сполуками. Аналогічно, сполуки даного винаходу можна використовувати в комбінації з іншими лікарськими засобами, що використовують для профілактики, лікування, контролю, послаблення або зменшення ризику захворювань або станів, для лікування яких придатні сполуки даного винаходу. Такі інші лікарські засоби можна вводити способами і у кількостях, що звичайно використовують для таких цілей, одночасно або послідовно зі сполукою даного винаходу. Якщо сполуку даного винаходу використовують одночасно з одним або більше з інших лікарських засобів, переважними є фармацевтичні композиції, що містять такі інші лікарські засоби у доповнення до сполуки даного винаходу. Відповідно, фармацевтичні композиції даного винаходу включають такі, які також містять один або більше з інших активних інгредієнтів у доповнення до сполуки даного винаходу. Масове відношення сполуки даного винаходу до іншого активного інгредієнта (інгредієнтів) може варіюватися і буде залежати від ефективної дози кожного з інгредієнтів. Звичайно використовують ефективну дозу кожного з інгредієнтів. Так, наприклад, якщо сполуку даного винаходу поєднують з іншим агентом, масове відношення сполуки даного винаходу до іншого агента звичайно знаходиться в інтервалі від близько 1000:1 до близько 1:1000 або від близько 200:1 до близько 1:200. Комбінації сполук даного винаходу і інших активних інгредієнтів звичайно також входять у вищевказаний інтервал співвідношень, але в кожному випадку повинна бути використана ефективна доза кожного з активних інгредієнтів. У таких комбінаціях сполуку даного винаходу і інші активні агенти можна вводити окремо або разом. Крім того, введення одного елемента можна здійснити до, одночасно або після введення іншого агента (агентів) і використовуючи однакові або різні способи введення. Сполуки даного винаходу можна вводити перорально, парентерально (наприклад, внутрішньом'язово, внутрішньоочеревинно, внутрішньовенно, ICV, використовуючи інтрацистернальні ін'єкції або вливання, підшкірні ін'єкції або імпланти), використовуючи спреї для інгаляцій, назальний, вагінальний, ректальний, під язик, букальний або місцевий способи введення, і вони можуть бути приготовлені окремо або разом у придатних одиничних дозованих композиціях, що містять звичайні, нетоксичні фармацевтично прийнятні носії, ад'юванти і засоби доставки, придатні для кожного способу введення. Крім лікування теплокровної тварин, сполуки даного винаходу ефективні при використанні для людей. Фармацевтичні композиції для введення сполук даного винаходу зручно представляти в одиничній дозованій формі, і отримати їх можна будь-яким із способів, добре відомих фахівцям у даній галузі фармацевтики. Усі способи включають стадію об'єднання активного інгредієнта з носієм, що складає один або більше з допоміжних інгредієнтів. Звичайно зазначені фармацевтичні композиції готують, використовуючи рівномірне і ретельне змішування активного інгредієнта з рідким носієм або з тонкоподрібненим твердим носієм, або з обома, і потім, за необхідності, формуючи продукт у необхідну композицію. У фармацевтичну композицію активну сполуку включають у кількості, достатній для досягнення необхідного ефекту у відношенні процесу або ступеня захворювань. У тому значенні, як тут використаний, термін "композиція" включає продукт, що містить специфічні інгредієнти в специфічних кількостях, а також будь-який продукт, що поліпшує прямо або побічно комбінацію специфікованих інгредієнтів у специфічних кількостях. Фармацевтичні композиції, що містять активний інгредієнт, можуть бути у формі таблеток для перорального введення, наприклад у вигляді таблеток, коржів, пастилок, водних або масляних суспензій, диспергованих порошків або гранул, емульсій, розчинів, твердих або м'яких капсул або сиропів, або еліксирів. Композиції, призначені для перорального введення, можна отримати способами, відомими фахівцям в галузі виготовлення фармацевтичних композицій, і такі композиції можуть містити один або більше з агентів, вибраних із групи, що складається з підсолоджуючих агентів, смакових агентів, барвних агентів, і консервантів для отримання фармацевтично елегантних і смачних препаратів. Таблетки містять активний інгредієнт у суміші з нетоксичними, фармацевтично прийнятними ексципієнтами, які придатні для виготовлення таблеток. Зазначені ексципієнти можуть бути, наприклад, інертними розріджувачами, такими як карбонат кальцію, карбонат натрію, лактоза, фосфат кальцію або фосфат натрію; агентами, що сприяють гранулюванню, або дезінтегрантами, наприклад кукурудзяним крохмалем або альгіновою кислотою; сполучними агентами, наприклад крохмалем, желатином або смолою акації; і лубрикантами, наприклад стеаратом магнію, стеариновою кислотою або тальком. Такі таблетки можуть бути без покриття, або вони можуть бути покриті з використанням відомих 11 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 способів для затримки руйнування і абсорбції в шлунково-кишковому тракті і, таким чином, забезпечити уповільнену дію протягом більш тривалого періоду. Наприклад, можна використовувати уповільнюючий матеріал, такий як гліцеринмоностеарат або гліцериндистеарат. Можна також використовувати методики нанесення покриттів, розкриті в патентах США 4256108, 4166452 і 4265874, для отримання осмотичних таблеток для контрольованого вивільнення. Таблетки для перорального введення можна також приготувати для негайного вивільнення, такі як таблетки або вафлі, що швидко розплавляються, таблетки, що швидко розчиняються, або плівки, що швидко розчиняються. Композиції для перорального введення можуть бути також у вигляді твердих желатинових капсул, де активний інгредієнт змішаний з інертним твердим розріджувачем, наприклад карбонатом кальцію, фосфатом кальцію або каоліном, або у вигляді м'яких желатинових капсул, де активний інгредієнт змішаний з водним або масляним середовищем, наприклад з арахісовою олією, рідким парафіном або оливковою олією. Водні суспензії містять активні матеріали в суміші з ексципієнтами, придатними для виготовлення водних суспензій. Такі ексципієнти являють собою суспендуючі агенти, наприклад натрійкарбоксиметилцелюлозу, метилцелюлозу, гідроксипропілметилцелюлозу, альгінат натрію, полівінілпіролідон, смолу трагаканту і смолу акації; диспергуючі або змочувальні агенти можуть бути природними фосфатидами, наприклад лецитином, або продуктами конденсації алкіленоксиду і жирних кислот, наприклад поліоксіетиленстеаратом, або продуктами конденсації етиленоксиду з довголанцюжковими аліфатичними спиртами, наприклад гептадекаетиленоксицетанолом, або продуктами конденсації етиленоксиду з частковими ефірами, отриманими з жирних кислот і гекситу, такими як поліоксіетиленсорбітолмоноолеат, або продуктами конденсації етиленоксиду з частковими ефірами, отриманими з жирних кислот і ангідридів гекситу, наприклад поліетиленсорбітану моноолеатом. Водні суспензії можуть також містити один або більше з консервантів, наприклад етил або н-пропіл, п-гідроксибензоат, один або більше з барвних агентів, один або більше із смакових агентів і один або більше з підсолоджуючих агентів, таких як сахароза або сахарин. Масляні суспензії можна приготувати, суспендуючи активний інгредієнт у рослинній олії, наприклад в арахісовій олії, оливковій олії, кунжутній олії або кокосовій олії, або в мінеральному маслі, такому як рідкий парафін. Масляні суспензії можуть містити загущувальний агент, наприклад бджолиний віск, твердий парафін або цетиловий спирт. Можуть бути додані підсолоджуючі агенти, такі як ті, що перераховані вище, і смакові агенти, для отримання смачних препаратів для перорального введення. Зазначені композиції можуть бути законсервовані за рахунок додавання антиоксидантів, таких як аскорбінова кислота. Дисперговані порошки і гранули, що придатні для отримання водних суспензій шляхом додавання води, надають активний інгредієнт у суміші з диспергованим або змочувальним агентом і одним або більше з консервантів. Приклади придатних диспергованих або змочувальних агентів представлені перерахованими вище. Додаткові ексципієнти, наприклад підсолоджуючі агенти, смакові агенти і барвні агенти, також можуть бути присутніми. Фармацевтичні композиції даного винаходу можуть також бути у формі емульсій типу маслоу-воді. Масляна фаза може бути рослинною олією, наприклад оливковою або арахісовою олією, або мінеральним маслом, наприклад рідким парафіном, або їх сумішами. Придатні емульгуючі агенти можуть бути природними смолами, наприклад смолою акації або смолою трагаканту, природними фосфатидами, наприклад соєвим лецитином, і складними ефірами або частковими ефірами, отриманими з жирних кислот і ангідридів, наприклад сорбітанмоноолеатом, і продуктами конденсації зазначених часткових ефірів з етиленоксидом, наприклад поліоксіетиленсорбітанмоноолеатом. Зазначені емульсії можуть також містити підсолоджуючі і смакові агенти. Сиропи і еліксири можна приготувати, використовуючи підсолоджуючі агенти, наприклад гліцерин, пропіленгліколь, сорбіт або сахарозу. Такі композиції можуть також містити пом'якшувачі, консерванти і смакові, і барвні агенти. Фармацевтичні композиції можуть бути у формі стерильних водних або масляних суспензій для ін'єкцій. Такі суспензії можна приготувати відомими фахівцями у даній галузі способами, використовуючи придатні диспергуючі або змочувальні агенти, що були згадані вище. Стерильні препарати для ін'єкцій можуть також бути стерильними розчинами або суспензіями для ін'єкцій у нетоксичних парентерально прийнятних розріджувачах або розчинниках, наприклад, у вигляді розчину в 1,3-бутандіолі. Серед прийнятних носіїв і розчинників, які можна використовувати, вода, розчин Рінгера і ізотонічний розчин хлориду натрію. Крім того, стерильні нелеткі масла звичайно використовують як розчинник або суспендуюче середовище. Для зазначених цілей можна використовувати будь-які звичайні масла, включаючи синтетичні моно- або дигліцериди. 12 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 Крім того, жирні кислоти, такі як олеїнова кислота, знаходять застосування при виготовленні препаратів для ін'єкцій. Сполуки даного винаходу можна також вводити у формі супозиторіїв для ректального введення лікарських засобів. Такі композиції можна отримати, змішуючи лікарський засіб із придатним ексципієнтом, що не викликає подразнення, який є твердим при звичайній температурі, але стає рідким при ректальній температурі, і тому плавиться в ректумі з виділенням лікарського засобу. Такими матеріалами є масло какао і поліетиленгліколі. Для місцевого застосування використовують креми, мазі, желе, розчини або суспензії і т. п., що містять сполуки даного винаходу. Аналогічно, можна також використовувати трансдермальні пластири для місцевого введення. Фармацевтичні композиції і спосіб даного винаходу можуть також включати інші терапевтично активні сполуки, як тут зазначено, які звичайно використовують при лікуванні вищевказаних патологічних станів. При лікуванні, профілактиці, контролі, ослабленні або зменшенні ризику виникнення станів, що вимагають протидії активності рецептора CGRP, придатні рівні доз звичайно складають від близько 0,01 до 500 мг на кг маси тіла пацієнта в день, які можна вводити у вигляді однієї або декількох доз. Придатний рівень доз може складати від близько 0,01 до 250 мг/кг у день, від близько 0,05 до 100 мг/кг у день або від близько 0,1 до 50 мг/кг у день. Усередині зазначеного інтервалу доза може складати від 0,05 до 0,5, від 0,5 до 5 або від 5 до 50 мг/кг у день. Для перорального введення можна приготувати композиції у формі таблеток, що містять від 1,0 до 1000 мг активного інгредієнта, зокрема 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 і 1000,0 мг активного інгредієнта, для симптоматичного підбирання дози для підлягаючого лікуванню пацієнта. Сполуки можна вводити за схемою від 1 до 4 разів в день або можна вводити один або два рази в день. При лікуванні, профілактиці, контролі, полегшенні або зменшенні ризику виникнення головного болю, мігрені, кластерного головного болю або при інших захворюваннях, при яких запропоновані сполуки даного винаходу, звичайно задовільних результатів досягають при введенні денних доз від близько 0,1 мг до близько 100 мг на кг маси тіла тварини, що вводяться у вигляді однієї денної дози або у вигляді розділених доз від двох до шести разів у день, або у формі з уповільненим вивільненням. Для більшості великих ссавців повна денна доза складає від близько 1,0 мг до близько 1000 мг або від близько 1 мг до близько 50 мг. У випадку дорослої людини масою 70 кг повна денна доза звичайно складає від близько 7 мг до близько 350 мг. Таку схему прийому доз можна модифікувати для досягнення оптимальної терапевтичної реакції. Варто враховувати, однак, що конкретний рівень доз і частота введення можуть змінюватися для кожного окремого пацієнта і будуть залежати від різних факторів, включаючи активність конкретної використовуваної сполуки, метаболічну стабільність і тривалість дії вибраної сполуки, віку, маси тіла, загального стану здоров'я, статі, харчування, способу і часу введення, швидкості виділення, комбінації лікарських засобів і тяжкості конкретного стану отримуючого лікування пацієнта. Корисність сполук даного винаходу як антагоністів активності рецепторів CGRP можна I25 продемонструвати способами, відомими фахівцям у даній галузі. Інгібування зв'язування ICGRP з рецепторами і функціональний антагонізм рецепторів CGRP визначають наступним чином. 125 АНАЛІЗ ЗВ'ЯЗУВАННЯ ПРИРОДНИХ РЕЦЕПТОРІВ. Зв'язування I-CGRP з рецепторами в мембранах клітин SK-N-MC здійснюють практично за способом (Edvinsson et al. (2001) Eur. J. Pharmacol. 415, 39-44). Коротше, мембрани (25 мкг) інкубують у 1 мл зв'язувального буфера [10 мМ HEPES, pН 7,4, 5 мМ MgCl2 і 0,2 % альбуміну бичачої сироватки (BSA)], що містить 10 пM 125 I-CGRP і антагоніст. Після інкубування при кімнатній температурі протягом 3 годин, аналіз закінчують, використовуючи фільтрацію через GFB скловолоконні фільтрувальні пластини (PerkinElmer), які були блоковані 0,5 % поліетиленіміном протягом 3 годин. Фільтри тричі промивають охолодженим льодом аналітичним буфером (10 мМ HEPES, pН 7,4, і 5 мМ MgCl 2), потім пластини сушать повітрям. Додають сцинтиляційну рідину (50 мкл) і радіоактивність визначають, використовуючи прилад Topcount (Packard Instrument). Аналіз результатів здійснюють, використовуючи Prism, і Ki визначають, використовуючи рівняння Ченга-Пруссова (Cheng & Prusoff (1973) Biochem. Pharmacol. 22, 3099-3108). РЕКОМБІНАНТНИЙ РЕЦЕПТОР. Людський CL-рецептор (Genbank реєстраційний номер L76380) субклонують у експресійний вектор pIREShyg2 (BD Biosciences Clontech) як 5'NheI і 3'PmeI фрагменти. Людський RAMP1 (Genbank реєстраційний номер AJ001014) субклонують у експресійний вектор pIRESpuro2 (BD Biosciences Clontech) як 5'NheI і 3'NotI фрагменти. HEK 293 13 UA 109463 C2 5 10 15 20 25 клітини (людські ембріональні клітини нирок; ATCC #CRL-1573) культивують у DMEM з 4,5 г/л глюкози, 1 мМ пірувату натрію і 2 мМ глутаміну, доповненому 10 % фетальною телячою сироваткою (FBS), 100 од./мл пеніциліну і 100 мкг/мл стрептоміцину, і витримують при 37 °C і 95 % вологості. Клітини субкультивують, обробляючи 0,25 % трипсином з 0,1 % EDTA у HBSS. Створення стабільної клітинної лінії здійснюють у результаті спільного трансфектування 10 мкг 2 ДНК із 30 мкг ліпофектаміну 2000 (Invitrogen) у 75 см флягах. CL-рецептор і експресійні конструкції RAMP1 спільно трансфектують у рівних кількостях. Через двадцять чотири години після трансфектування клітини розбавляють, і додають селективне середовище (середовище для вирощування+300 мкг/мл гігроміцину і 1 мкг/мл пуроміцину) наступного дня. Клональну клітинну лінію створюють шляхом поміщування однієї клітини, використовуючи клітинний сортер з активацією флуоресценції FACS Vantage SE (Becton Dickinson). Середовище для вирощування доводять до 150 мкг/мл гігроміцину і 0,5 мкг/мл пуроміцину для розмноження клітин. АНАЛІЗ ЗВ'ЯЗУВАННЯ РЕКОМБІНАНТНИХ РЕЦЕПТОРІВ. Клітини, експресуючі рекомбінантний людський СL-рецептор/RAMP1 промивають PBS і збирають у буфер для збору, що містить 50 мМ HEPES, 1 мМ EDTA і інгібітори протеаз Complete™ (Roche). Клітинну суспензію руйнують, використовуючи лабораторний гомогенізатор, і центрифугують при 48000 g для виділення мембран. Осад знову суспендують у буфері для збору плюс 250 мМ сахарози і зберігають при температурі -70 °C. Для аналізу зв'язування 20 мкг мембран інкубують у 1 мл зв'язувального буфера (10 мМ HEPES, pН 7,4, 5 мМ MgCl2 і 0,2 % BSA) протягом 3 годин при 125 кімнатній температурі, що містить 10 пМ I-hCGRP (GE Healthcare) і антагоніст. Аналіз закінчують, фільтруючи через 96-ямкові GFB скловолоконні фільтрувальні планшети (PerkinElmer), що були блоковані 0,05 % поліетиленіміном. Фільтри тричі промивають охолодженим льодом аналітичним буфером (10 мМ HEPES, pН 7,4, і 5 мМ MgCl2). Додають сцинтиляційну рідину, і планшети обробляють за допомогою Topcount (Packard). Визначають неспецифічне зв'язування і результати аналізу отримують, використовуючи удавану константу дисоціації (Ki), визначену за допомогою нелінійного підбирання методом найменших квадратів, підставляючи результати для зв'язаних CPM у представлене нижче рівняння: Yспост 30 35 40 45 ( Yмакс Yмін)(%Iмакс %Iмін / 100) Yмін ( Yмакс Yмін)(100 %Iмакс / 100) 1 ([ ліки] / K i (1 [радіомеч. / K d )nH ] , де Y являє собою спостережувані зв'язані CPM, Y макс являє собою повну кількість зв'язків, Yмін являє собою кількість неспецифічних зв'язків, (Yмакс-Yмін) являє собою кількість специфічних зв'язків, % Iмакс являє собою максимальний відсоток інгібування, % Iмін являє собою мінімальний відсоток інгібування, радіомеч. являє собою зонд, і Kd являє собою удавану константу дисоціації для радіоліганду для рецептора за даними експериментів з гарячим насиченням. ФУНКЦІОНАЛЬНИЙ АНАЛІЗ РЕКОМБІНАНТНОГО РЕЦЕПТОРА. Клітини знову суспендують у DMEM/F12 (Hyclone), доповненому 1 г/л BSA і 300 мкМ ізобутилметилксантину. Потім клітини висівають у 384-ямковий планшет (Proxiplate Plus 384; 509052761; Perkin-Elmer) при густині 2000 клітин/ямку і інкубують з антагоністом протягом 30 хв. при 37 °C. Потім до клітин додають людські α-CGRP при кінцевій концентрації 1,2 нМ і інкубують ще протягом 20 хв. при 37 °C. Після стимулювання агоністом клітини обробляють для визначення cAMP, використовуючи двостадійну процедуру згідно з рекомендованим протоколом виробника (набір HTRF cAMP динамічний аналіз 2; 62AM4PEC; Cisbio). Необроблені результати перетворюють у концентрації cAMP, використовуючи стандартну криву, потім будують залежні від дози криві і визначають значення точки перегину (IP). Приклади величин Ki в аналізі зв'язування рекомбінантних рецепторів сполук даного винаходу наведені в представленій нижче таблиці: 14 UA 109463 C2 Результати зв'язування CGRP-аналогів, розкритих у патенті США № 7390798 і патентній публікації США № 2010/0179166, представлені в наступній таблиці: Ki Сполука (нМ) Приклад 8 з патенту США 7,390,798 7,4 Приклад 9 з патенту США 7,390,798 1,9 Приклад 10 з патенту США 7,390,798 1,7 Приклад 8 з патенту США 2010/0179166 4,3 5 Сполука прикладу 32 із патенту США 7390798 має субнаномолярну ефективність, але структурно відрізняється від сполук, які розглядаються в описі винаходу. У тексті опису використані наступні скорочення: Me: метил, 15 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 Et: етил, t-Bu: трет-бутил, Bu: бутил, i-Pr: ізопропіл, Ar: арил, Ph: феніл, Bn: бензил, Py: піридил, Ac: ацетилат, OAc: ацетат, DCE: 1,2-дихлоретан, TFA: трифтороцтова кислота, TEA: триетиламін, Boc: трет-бутоксикарбоніл, BOP: (бензотриазол-1-ілокси)трис(диметиламіно)фосфонійгексафтор-фосфат, DIEA: N, N-діізопропілетиламін, HOBT: 1-гідроксибензотриазол, EDC: гідрохлорид N-(3-диметиламінопропіл)-N'-етилкарбодііміду, PyCIU: хлордипіролідинкарбеній, n-BuLi: н-бутиллітій, HATU: O-(7-азабензотриазол-1-іл)-N, N,N',N'-тетраметилуронійгексафтор-фосфат, EDTA: етилендіамінтетраоцтова кислота, ДМФ: N, N-диметилформамід, HMDS: гексаметилдисилазан, ТГФ: тетрагідрофуран, DMSO: диметилсульфоксид, SEM: 2-триметилсилілетоксиметил, SEMCl: 2-триметилсилілетоксиметилхлорид, PBPB: пербромід піридинійброміду, DMEM: модифіковане середовище Ігла за Дюльбеко (з високим вмістом глюкози), FBS: фетальна теляча сироватка, BSA: альбумін бичачої сироватки, PBS: сольовий розчин, буферизований фосфатом, HEPES: N-(2-гідроксіетил)піперазин-N'-(2-етансульфонова кислота), хв.: хвилини, год.: години, aq: водний, ВЕРХ: високоефективна рідинна хроматографія (ВЕРХ), LСМС: рідинна хроматографія-мас-спектрометрія, SFC: надкритична рідинна хроматографія, NMP: 1-метил-2-піролідинон, DMA: N, N-диметилацетамід, NBS: N-бромсукцинімід, dppf: 1,1'-біс(дифенілфосфіно)фероцен, dba: дибензиліденацетон, Ms: метансульфоніл, p-Ts: 4-толуолсульфоніл, trisyl: 2,4,6-триізопропілбензолсульфоніл, DMAP: 4-(диметиламіно)піридин. Способи отримання сполук даного винаходу проілюстровані на наступних схемах і в прикладах. Вихідні матеріали отримують згідно з процедурами, відомими фахівцям у даній галузі або проілюстрованими в описі способами. Сполуки даного винаходу можна легко отримати у відповідності з наступними схемами і конкретними прикладами або їх модифікаціями, використовуючи легко доступні вихідні матеріали, реагенти і звичайні способи синтезу. У зазначених реакціях можна також використовувати варіанти, які самі по собі відомі фахівцям у даній галузі, але не зазначені більш докладно. Загальні способи отримання сполук, заявлених у даному винаході, можуть легко зрозуміти і прийняти фахівці в даній галузі з урахуванням наступних схем. Хоча даний винахід було розкрито і проілюстровано з посиланням на деякі конкретні варіанти, фахівці в даній галузі зрозуміють, що різні адаптації, зміни, модифікації, заміщення, 16 UA 109463 C2 5 10 15 20 25 30 35 40 виключення або додавання процедур і протоколів можна здійснити, не виходячи за рамки сутності і обсягу даного винаходу. Наприклад, можна використовувати ефективні дози вищевказаних сполук даного винаходу, що відрізняються від конкретних, представлених далі значень, як наслідок розходжень у сприйнятливості підлягаючого лікуванню ссавця для будьякого із симптомів. Аналогічно, конкретно спостережувані фармакологічні реакції можуть варіюватися відповідно до і в залежності від конкретно вибраних активних сполук, або від того, чи присутні фармацевтичні носії, також як від типу лікарського засобу і застосовуваного способу введення, і такі очікувані варіанти або розходження в результатах розглядаються відповідно з цілями і практичним використанням даного винаходу. Тому мається на увазі, що даний винахід визначається обсягом представленої далі формули винаходу, і що зазначена формула інтерпретується настільки широко, наскільки це розумно. СХЕМИ РЕАКЦІЙ Сполуки даного винаходу можна легко отримати згідно з наступними схемами і конкретними прикладами або їх модифікаціями, використовуючи легко доступні вихідні матеріали, реагенти і звичайні способи синтезу. У зазначених реакціях, можливо також використовувати варіанти, що самі по собі відомі фахівцям у даній галузі, але не розкриті більш докладно. Загальні способи отримання сполук, заявлених у даному винаході, можуть легко зрозуміти і застосувати фахівці в даній галузі, вивчивши наступні схеми. Схема 1 ілюструє спосіб отримання похідних 3-амінопіперидонону типу 1.5, які можна використовувати для отримання сполук даного винаходу. Арилацетон 1.1 можна алкілувати, використовуючи похідне йодоаланіну 1.2 в основних умовах, до отримання кетоефіру 1.3. У результаті відновного амінування з наступною циклізацією і епімеризацією отримують переважно цис-заміщений лактам 1.4 у вигляді рацемічної суміші. Хіральний поділ з використанням, наприклад, нормальнофазової рідинної хроматографії і видалення Boc-захисної групи за допомогою HCl у EtOAc приводить до отримання 3-амінопіперидонону 1.5 у вигляді гідрохлоридної солі. СХЕМА 1 Альтернативна послідовність отримання похідних 3-амінопіперидонону типу 1.5 представлена на схемі 2. У результаті відновного амінування кетоефіру 1.3 аміаком з наступною епімеризацією отримують 2.1 у вигляді, головним чином, цис-заміщеної рацемічної суміші. Хіральний поділ енантіомерів приводить до отримання 2.2. N-алкілування з використанням, наприклад, LiHMDS як основи, і алкілгалогеніду або епоксиду дає 2.3. Видалення Boc-захисної групи за допомогою HCl потім дає 1.5 як гідрохлоридну сіль. СХЕМА 2 Третій спосіб отримання похідних 3-амінопіперидонону типу 1.5 представлений на схемі 3. N-алкілування 5-бром-6-метилпіридин-2(1H)-ону (3.1), з використанням карбонату цезію як 17 UA 109463 C2 5 10 основи, і алкілгалогеніду з наступним нітруванням приводить до отримання 3.2. Потім каталізоване паладієм перехресне сполучення з арилбороновою кислотою дає 3.3. Гідрування з використанням оксиду платини в кислотних умовах і хіральний поділ більшої частини суміші цис-заміщеного рацемічного продукту приводить до отримання 1.5 у вигляді одного енантіомера. Спосіб отримання похідних 3-амінопіперидонону типу 4.4 представлений на схемі 4. Арилацетонітрил 4.1 можна алкілувати, використовуючи похідне йодоаланіну 1.2, в основних умовах до отримання ціаноефіру 4.2. Відновна циклізація з використанням водню і гідроксиду паладію-на-вугіллі або нікелю Ренея, епімеризації і хірального поділу дає цис-лактам 4.3 у вигляді одного енантіомера. N-алкілування і видалення Boc-захисної групи приводить потім до отримання 4.4 у вигляді гідрохлоридної солі. 15 20 25 30 Схема 5 ілюструє альтернативний спосіб отримання похідних 3-амінопіперидонону типу 4.4. Арилацетонітрил 5.1 можна конденсувати з акрилатом 5.2 при підвищеній температурі, отримуючи 4-ціанобутаноатний ефір 5.3. Гідрування нітрилу 5.3 з використанням нікелю Ренея як каталізатора і етанольного розчину аміаку дає відповідне похідне аміну, що звичайно циклізують in situ, отримуючи піперидинон 5.4. N-алкілування лактаму 5.4 можна здійснити різними способами, відомими фахівцям в галузі органічного синтезу, причому на вибір 1 конкретних умов впливає природа алкілуючого агента, R X. Електрофільне азидування отриманого заміщеного лактаму 5.5 можна здійснити, використовуючи методику, аналогічну тій, яка розкрита Евансом із співробітниками (Evans et al. (1990) J. Am. Chem. Soc. 112, 4011-4030), для отримання азиду 5.6 у вигляді суміші діастереоізомерів, які можна розділити хроматографічними методами. Шуканий цис-діастереоізомер азиду 5.6 можна відновити, використовуючи каталітичне гідрування в присутності ди-трет-бутилдикарбонату, отримуючи відповідний Boc-захищений амін 5.7, і розділяючи енантіомери за допомогою хіральної ВЕРХ або SFC, отримуючи (3S, 5S)-ізомер 5.8. І нарешті, стандартне видалення захисних груп приводить до отримання шуканого проміжного 3-амінопіперидонону 4.4 у вигляді гідрохлоридної солі. 18 UA 109463 C2 5 10 15 20 Інший підхід до отримання похідних 3-амінопіперидонону, що представляють інтерес, які, зокрема, використовують для отримання 3-аміно-6-метил-5-арилпіперидин-2-онів, таких як 1.5, представлений на схемі 6. Зазначений піридин-2(1H)-он 3.1 можна перетворити в N-заміщений 1 піридинон 6.1 шляхом обробки придатним електрофілом (R X) у лужних умовах. Піридинон 6.1 можна потім піддати реакції сполучення Сузукі з бороновою кислотою 6.2, і отриманий 5арилпіридинон 6.3 можна гідрувати, використовуючи, наприклад, оксид платини(IV) як каталізатор, отримуючи відповідний 5-арилпіперидинон 6.4, що звичайно одержується як переважно цис-ізомер. Подальшу обробку піперидинону 6.4 можна здійснити, використовуючи методи, аналогічні методам, представленим на схемі 5. Конкретно, електрофільне азидування з наступним (в одному реакторі) відновленням і Boc-захистом приводить до отримання карбамату 6.6, і шуканий енантіомер можна отримати, використовуючи хіральну хроматографію. У деяких випадках, шуканий діастереоізомер азиду 6.5 можна виділити у вигляді рацемічної суміші (3S, 5S, 6R)- і (3R, 5R, 6S)-ізомерів з наступною хроматографічною обробкою на силікагелі сирого продукту, і отриману суміш можна обробити, як представлено на схемі 6. В інших випадках може виявитися вигідним перенести суміш діастереоізомерів азиду 6.5 уперед до відповідного карбамату 6.6. Суміш діастереоізомерів карбамату 6.6 можна епімеризувати у лужних умовах, таких як карбонат калію в EtOH, отримуючи суміш, що істотно збагачена шуканими (3S, 5S, 6R)і (3R, 5R, 6S)-ізомерами, причому подальше очищення можна використовувати для отримання енантіомера, що представляє інтерес, як тут представлено. 19 UA 109463 C2 5 Спосіб отримання проміжної азаоксіндолпіридинової кислоти 7.4 представлений на схемі 7. Діазотування амінопіридину 7.1, отримання якого розкрито в WO 2008/020902, з наступною обробкою йодидом калію приводить до отримання йодиду 7.2. Каталізоване паладієм карбонілування в метанолі потім дає складний ефір 7.3, який можна омилити, використовуючи гідроксид натрію, отримуючи сполуку 7.4. 10 15 20 Альтернативний спосіб отримання проміжної азаоксіндолпіридинової кислоти 7.4 представлений на схемі 8. Етерифікація дикислоти 8.1 з наступним бромуванням приводить до отримання сполуки 8.2. У результаті наступного відновлення боргідридом натрію отримують діол 8.3. Алкілування захищеного азаоксіндолу 8.4 бісмезилатом, отриманим з 8.3, дає спіроцикл 8.5. Каталізоване паладієм карбонілування в метанолі з наступним хіральним поділом приводить до отримання складного ефіру 8.6 у вигляді одного енантіомера. Видалення SEMзахисної групи в кислотних умовах і гідроліз складного ефіру з використанням гідроксиду натрію приводить потім до отримання сполуки 7.4. 20 UA 109463 C2 5 Спосіб отримання проміжноїдіазаоксіндолкарбонової кислоти 9.7 представлений на схемі 9. Після етерифікації кислоти 9.1 іде вінілування з використанням паладієвих каталізаторів до отримання дивінілпіридину 9.2. Озоноліз з відновною обробкою боргідридом приводить до отримання діолу 9.3. Після мезилування і обробки хлоридом натрію, отриману дихлорпроміжну сполуку 9.4 можна алкілувати оксіндолом 9.5 у лужних умовах до отримання спіроциклу 9.6, з наступним хіральним поділом енантіомерів. Дехлорування в буферованих умовах гідрування і видалення захисних груп кислотою приводить до отримання кислоти 9.7. 10 15 20 25 Корисні похідні розкритих в описі проміжних сполук можна отримати, використовуючи добре відомі способи. Два таких приклади проілюстровані на схемі 10, де проміжну сполуку азаоксіндол 7.4 перетворюють у відповідне похідне нітрилу 10.2 і фторпохідне 10.6, які обидва можна використовувати для отримання сполук даного винаходу. Бромування сполуки 7.4 Nбромсукцинімідом у дигідраті трифтористого бору приводить до отримання бромпохідного 10.1, яке можна перетворити в шуканий нітрил 10.2, використовуючи, як показано, ціанід цинку і паладієвий каталізатор. Альтернативно, у броміду 10.1 можна видалити захисні групи в стандартних умовах, отримуючи складний ефір 10.3, і його можна перетворити у відповідний аналог трибутилстанану 10.4, використовуючи біс(трибутилолово) і паладієвий каталізатор. Похідне трибутилстанану 10.4 можна перетворити у фторид 10.5 у каталізованій сріблом реакції, розкритій Ріттером із співробітниками (Tang et al. (2010) J. Am. Chem. Soc. 132, 1215012154), використовуючи Selectfluor® [N-хлорметил-N'фтортриетилендіамонійбіс(тетрафторборат)]. І нарешті, видалення SEM-захисної групи і омилення приводить до отримання шуканого фторазаоксіндолу 10.6. 21 UA 109463 C2 5 10 15 20 25 На схемі 11 проілюстровані умови, які можна використовувати для реакції сполучення проміжних сполук 3-амінопіперидонону, таких як 11.1, і проміжної сполуки карбонової кислоти 11.2, отримуючи в цьому випадку аміди 11.3. Зазначені стандартні умови реакції сполучення являють способи, які використовують для отримання сполук даного винаходу. У деяких випадках можна використовувати стратегії введення захисних груп, що добре відомі фахівцям в галузі органічного синтезу, щоб забезпечити отримання конкретних сполук даного винаходу. Варто розуміти, що альтернативні способи також можна використовувати в синтезі зазначених ключових проміжних сполук. Так, наприклад, можна використовувати послідовності рацемічних реакцій, з наступним хіральним поділом на відповідних стадіях до отримання сполук даного винаходу. Конкретний вибір реагентів, розчинників, температур і інших умов реакцій залежить від природи одержуваного продукту. У деяких випадках можна використовувати придатні стратегії введення захисних груп. У деяких випадках кінцевий продукт можна далі модифікувати, наприклад, маніпулюючи з замісниками. Такі маніпуляції можуть включати, але ними не обмежуються, реакції відновлення, окислювання, алкілування, ацилування і гідролізу, що добре відомі фахівцям у даній галузі. У деяких випадках порядок проведення реакцій на вищенаведених схемах можна змінювати, щоб полегшити здійснення реакцій або щоб уникнути отримання побічних продуктів реакцій. Крім того, можна використовувати різні стратегії введення захисних груп, щоб полегшити здійснення реакцій або щоб уникнути отримання побічних продуктів реакцій. Наведені далі приклади призначені для більш повного розуміння суті даного винаходу. Зазначені приклади є лише ілюстративними і їх не слід розглядати як обмежуючі даний винахід яким-небудь способом. ПРОМІЖНА СПОЛУКА 1 22 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 (6S)-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]-3карбонова кислота Зазначену в заголовку сполуку можна отримати, використовуючи або спосіб I, або спосіб II, як розкрито далі. Спосіб I: Стадія A: (6S)-3-йодо-5,7-дигідроспіро[циклопента[b]піридин-6,3'-піроло-[2,3-b]піридин]2'(1'Н)-он Розчин нітриту натрію (36,1 г, 523 ммоль) у воді (20 мл) по краплях додають протягом 5 хв. до розчину (6S)-3-аміно-5,7-дигідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]-2'(1'H)ону (отриманого за способом, розкритим в WO2008/020902, 66,0 г, 262 ммоль) і птолуолсульфонової кислоти (149 г, 785 ммоль) в ацетонітрилі (650 мл) при 23 °C. Після перемішування протягом 30 хв., розчин йодиду калію (109 г, 654 ммоль) у воді (20 мл) додають протягом 5 хв. Отриману суміш перемішують при температурі 23 °C протягом 40 хв., потім розбавляють водою (1 л) і підлуговують, додаючи твердий NaOH (33,0 г, 824 ммоль) при перемішуванні. Йодовмісний побічний продукт відновлюють, додаючи 10 % водний розчин тіосульфату натрію, і перемішують протягом ще 30 хв. Тверду речовину збирають фільтруванням, промивають водою і сушать в атмосфері азоту, отримуючи зазначену в заголовку сполуку, яку використовують без додаткового очищення. МС: m/z=363,9 (M+1). Стадія B: Метил-(6S)-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]-піридин-6,3'-піроло[2,3b]піридин]-3-карбоксилат Розчин (6S)-3-йодо-5,7-дигідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]-2'(1'Н)ону (51,0 г, 140 ммоль), ацетату натрію (23,0 г, 281 ммоль) і аддукту дихлор-1,1'біс(дифенілфосфіно)фероценпаладію(II) з дихлорметаном (2,9 г, 3,5 ммоль) у MeOH (560 мл) під тиском 827371 Па (120 psi) CO при 23 °C і нагрівають при 80 °C протягом 12 годин при перемішуванні. Реакційну суміш розбавляють водою (1 л), і осад, що випав, збирають фільтруванням, промивають водою і сушать в атмосфері азоту, отримуючи зазначену в заголовку сполуку, яку використовують без додаткового очищення. МС: m/z=296,1 (M+1). Стадія С: (6S)-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3b]піридин]-3-карбонова кислота Суміш метил-(6S)-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3b]піридин]-3-карбоксилату (30,0 г, 102 ммоль) і водний 6 н розчин гідроксиду натрію (50,8 мл, 305 ммоль) у MeOH (920 мл) нагрівають при кипінні із зворотним холодильником протягом 1 години. Отриману суміш залишають охолоджуватися до 23 °C перед тим, як її підкислюють до pН~6, використовуючи водний 1 н розчин хлористоводневої кислоти, отримуючи осад чорного кольору, що видаляють фільтруванням. Отриманий фільтрат концентрують при зниженому тиску до об'єму ~100 мл і потім розділяють між водою (500 мл) і 2-метилтетрагідрофураном (2МeTHF, 250 мл). Водний шар екстрагують, використовуючи 2-MeTHF (5×250 мл), і об'єднані органічні шари сушать над сульфатом натрію і концентрують, отримуючи зазначену в заголовку сполуку. МС: m/z=282,0 (M+1). Спосіб II: Стадія A: Диметил-5-бромпіридин-2,3-дикарбоксилат Концентровану сірчану кислоту (1 л, 18,7 моль) повільно додають протягом 10 хв. до суспензії піридин-2,3-дикарбонової кислоти (5,00 кг, 29,9 моль) в метанолі (50 л), розчиняючи суспензію. Отриману суміш нагрівають при кипінні зі зворотним холодильником протягом 48 годин, потім охолоджують до 40 °C. Бром (8,0 кг, 50 моль) повільно додають протягом 2 годин порціями по 1 кг, підтримуючи температуру нижче 55 °C. Реакційну суміш потім нагрівають при 55 °C протягом 24 годин, охолоджують до 50 °C і повільно додають додаткову кількість Br 2 (4,0 кг, 25 моль) протягом 1 години порціями по 1 кг, підтримуючи температуру нижче 55 °C. Реакційну суміш нагрівають при 55 °C протягом 24 годин, концентрують до мінімального об'єму (внутрішня температура ~30 °C, розчин може іноді пінитися), потім розбавляють ізопропілацетатом (50 л) і промивають насиченим водним розчином сульфіту натрію (3×20 л) (кінцевий екстракт має pН~8), потім водою (20 л). Отриманий органічний шар концентрують до 23 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 55 60 приблизно 15 л, потім розбавляють гептаном (40 л). Отриману суспензію перемішують протягом 24 годин при 23 °C. Тверду речовину відфільтровують, промивають гептаном (10 л) і сушать, отримуючи зазначену в заголовку сполуку. Стадія B: (5-Бромпіридин-2,3-діїл)диметанол Боргідрид натрію (15,9 г, 420 ммоль) додають порціями протягом 30 хв. до розчину диметил5-бромпіридин-2,3-дикарбоксилату (20 г, 73 ммоль) у етанолі (460 мл), попередньо охолодженому до 0 °C. Розчин хлориду кальцію (23,3 г, 209 ммоль) у 150 мл повільно додають при 0 °C, і реакційну суміш нагрівають до 23 °C і перемішують протягом ночі. Надлишок боргідриду натрію гасять, повільно додаючи водний 2 н розчин HCl (230 мл, 460 ммоль), з наступним перемішуванням при 23 °C протягом 2 годин. Отриману суміш концентрують досуха. До залишку додають насичений водний розчин бікарбонату натрію до досягнення значення pН приблизно 7. Водну суміш екстрагують 2-метилтетрагідрофураном (4×200 мл). Об'єднані органічні шари сушать над сульфатом натрію, потім обробляють розчином 4 н HCl у діоксані (25 мл, 100 ммоль). Отриману тверду речовину відфільтровують, промивають 2метилтетрагідрофураном і сушать, отримуючи зазначену в заголовку сполуку у вигляді гідрохлоридної солі. МС: m/z=218,1 (M+1). Стадія C: (5-Бромпіридин-2,3-діїл)диметандіїлдиметансульфонат Суспензію гідрохлориду (5-бромпіридин-2,3-діїл)диметанолу (12,9 г, 59,2 ммоль) у тетрагідрофурані (400 мл) при 0 °C обробляють триетиламіном (37,1 мл, 266 ммоль). До отриманої суміші порціями додають метансульфоновий ангідрид (30,9 г, 177 ммоль), підтримуючи температуру нижче 5 °C. Отриману реакційну суміш перемішують при температурі 0 °C протягом 1 години, потім розділяють між насиченим водним розчином бікарбонату натрію(500 мл) і етилацетатом (500 мл). Отриманий органічний шар промивають насиченим водним розчином бікарбонату натрію, сушать над сульфатом магнію і концентрують, отримуючи зазначену в заголовку сполуку. МС: m/z=376,0 (M+1). Стадія D: 3-Бром-1'-{[2-(триметилсиліл)етокси]метил}-5,7-дигідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]-2'(1'Н)-он (5-Бромпіридин-2,3-діїл)диметандіїлдиметансульфонат (17,0 г, 45,4 ммоль) додають до суміші 1-{[2-(триметилсиліл)етокси]метил}-1,3-дигідро-2H-піроло[2,3-b]піридин-2-ону (отриманого за способом, розкритим в WO2008/020902, 14,0 г, 53,0 ммоль) і карбонату цезію (49,0 г, 150 ммоль) у етанолі (500 мл) при 23 °C, і отриману суміш перемішують протягом 20 годин. Отриману реакційну суміш концентрують, потім розділяють між етилацетатом (500 мл) і водою (500 мл). Отриманий органічний шар сушать над сульфатом магнію і концентрують. Залишок очищають на хроматографічній колонці із силікагелем (спочатку гептан, потім градієнтне елюювання до 100 % EtOAc), отримуючи зазначену в заголовку сполуку. МС: m/z=448,1 (M+1). Стадія E: Метил-(6S)-2'-оксо-1'-{[2-(триметилсиліл)етокси]метил}-1',2',5,7тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]-3-карбоксилат Суміш 3-бром-1'-{[2-(триметилсиліл)етокси]метил}-5,7-дигідроспіро-[циклопента[b]піридин6,3'-піроло[2,3-b]піридин]-2'(1'H)-ону (22,0 г, 49,3 ммоль), PdCl2(dppf)•CH2Cl2 (2,012 г, 2,46 ммоль) і ацетату натрію (8,1 г, 99 ммоль) у метанолі (150 мл) піддають впливу високого тиску до 300 psi (2068427,1 Па) моноокису вуглецю і потім нагрівають при 85 °C протягом 72 годин. Отриману реакційну суміш залишають охолоджуватися і потім концентрують. Залишок очищають на хроматографічній колонці із силікагелем (спочатку гептан, потім градієнтне елюювання до 100 % EtOAc), отримуючи зазначену в заголовку сполуку у вигляді рацемічної суміші. МС: m/z=426,1 (M+1). У результаті поділу енантіомерів за допомогою надкритичної рідинної хроматографії (SFC), використовуючи колонку ChiralPak AD-H і елююючи 40 % етанолом у СО2 (0,05 % діетиламіну як модифікатора), отримують зазначену в заголовку сполуку у вигляді другого елюйованого енантіомера. Стадія F: (6S)-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3b]піридин]-3-карбонова кислота Розчин метил-(6S)-2'-оксо-1'-{[2-(триметилсиліл)етокси]метил}-1',2',5,7тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]-3-карбоксилату (238 г, 559 ммоль) у метанолі (2 л) насичують газоподібною HCl, дозволяючи температурі підвищитися до 55 °C. Отриману реакційну суміш охолоджують до 23 °C, перемішують протягом 20 годин, потім концентрують. Водний 10 н гідроксид натрію (400 мл, 4 моль) додають до розчину залишку в метанолі (2 л), і отриману суміш нагрівають при кипінні зі зворотним холодильником протягом 2 годин. Отриманий розчин охолоджують до 23 °C і значення pН доводять до 3, використовуючи концентровану HCl. Отриману тверду речовину відфільтровують, промивають водою, потім гептаном і сушать, отримуючи зазначену в заголовку сполуку. МС: m/z=282,2 (M+1). 24 UA 109463 C2 ПРОМІЖНА СПОЛУКА 2 5 10 15 20 25 30 35 40 45 50 (6S)-6'-оксо-5,6',7,7'-тетрагідроспіро[циклопента[b]піридин-6,5'-піроло[2,3-d]піримідин]-3карбонова кислота Стадія A: Трет-бутил-5-бром-6-хлорпіридин-3-карбоксилат До розчину 5-бром-6-хлорнікотинової кислоти (25,0 г, 106 ммоль) у тетрагідрофурані (1,06 л) додають ди-трет-бутилдикарбонат (69,2 г, 317 ммоль) і потім 4-диметиламінопіридин (12,9 г, 106 ммоль). Через 16 годин, отриману суміш розбавляють водою і додають водну хлористоводневу кислоту (106 мл, 1 M, 106 ммоль). Отриману суміш екстрагують етилацетатом (3×). Об'єднані органічні екстракти промивають сольовим розчином (3×), сушать над сульфатом магнію, фільтрують і концентрують. У результаті очищення за допомогою хроматографічної обробки на силікагелі (100 % дихлорметан → 10 % метанол/дихлорметан) отримують зазначену в заголовку сполуку. МС: m/z=294,1 (M+1). Стадія B: Трет-бутил-5,6-діетинілпіридин-3-карбоксилат До розчину трет-бутил-5-бром-6-хлорпіридин-3-карбоксилату (24,0 г, 82,0 ммоль) в ацетонітрилі (615 мл) і воді (205 мл) додають калійвінілтрифторборат (33,0 г, 246 ммоль) і тринатрієву сіль трифенілфосфін-3,3',3"-трисульфонової кислоти (4,20 г, 7,38 ммоль). Додають діізопропіламін (88,0 мл, 615 ммоль) потім ацетат паладію(II) (0,553 г, 2,46 ммоль). Отриману суміш нагрівають до 75 °C. Через 16 годин отриману суміш охолоджують до кімнатної температури і додають насичений розчин бікарбонату натрію. Отриману суміш промивають дихлорметаном (3×), і об'єднані органічні шари промивають водою, сольовим розчином, сушать сульфатом магнію, фільтрують і концентрують. У результаті очищення за допомогою хроматографічної обробки на силікагелі (100 % дихлорметан → 5 % метанол/дихлорметан) отримують зазначену в заголовку сполуку. МС: m/z=232,3 (M+1). Стадія C: Трет-бутил-5,6-біс(гідроксиметил)піридин-3-карбоксилат До розчину трет-бутил-5,6-діетенілпіридин-3-карбоксилату (19,0 г, 82 ммоль) у дихлорметані (821 мл) при -78 °C додають газ озон. Озон барботують через розчин до насичення (1 годину). Потім через розчин барботують газоподібний азот. Отриману суміш розбавляють метанолом (821 мл) і додають боргідрид натрію (7,77 г, 205 ммоль). Через 15 хв., отриману суміш гасять, використовуючи насичений водний розчин бікарбонату натрію, і промивають дихлорметаном (3×). Об'єднані органічні шари промивають сольовим розчином, сушать над сульфатом магнію, фільтрують і концентрують. У результаті очищення за допомогою хроматографічної обробки на силікагелі (100 % дихлорметан → 15 % метанол/дихлорметан) отримують зазначену в заголовку сполуку. МС: m/z=240,3 (M+1). Стадія D: Трет-бутил-5,6-біс(хлорметил)піридин-3-карбоксилат До розчину трет-бутил-5,6-біс(гідроксиметил)піридин-3-карбоксилату (5,87 г, 24,5 ммоль) у N, N-диметилформаміді (146 мл) при 0 °C додають триетиламін (13,7 мл, 98 ммоль), потім додають метансульфоновий ангідрид (12,8 г, 73,6 ммоль). Через 15 хв. додають воду (29,2 мл) і хлорид натрію (8,60 г, 147 ммоль), і отриману суміш нагрівають до кімнатної температури. Через 16 годин додають насичений водний розчин бікарбонату натрію, і отриману суміш екстрагують етилацетатом (3×). Об'єднані органічні шари промивають водою (3×) і потім сольовим розчином (3×), сушать над сульфатом магнію, фільтрують і концентрують. У результаті очищення за допомогою хроматографічної обробки на силікагелі (100 % дихлорметан → 10 % метанол/дихлорметан) отримують зазначену в заголовку сполуку. МС: m/z=276,2 (M+1). Стадія E: трет-бутил-(6S)-4'-хлор-6'-оксо-5,6',7,7'-тетрагідроспіро-[циклопента[b]піридин-6,5'піроло[2,3-d]піримідин]-3-карбоксилат До розчину трет-бутил-5,6-біс(хлорметил)піридин-3-карбоксилату (1,80 г, 6,52 ммоль) у N, Nдиметилформаміді (93,0 мл) додають 4-хлор-5,7-дигідро-6H-піроло[2,3-d]піримідин-6-он (1,80 г, 10,62 ммоль), карбонат цезію (3,65 г, 11,21 ммоль) і бромід натрію (0,671 г, 6,52 ммоль). Через 30 хв. додають насичений водний розчин бікарбонат натрію, і отриману суміш промивають етилацетатом (3×). Об'єднані органічні шари промивають водою (3×), сольовим розчином (3×) і сушать над сульфатом магнію, фільтрують і концентрують. У результаті очищення за допомогою хроматографічної обробки на силікагелі (100 % дихлорметан → 10 % 25 UA 109463 C2 5 10 15 20 25 30 35 40 45 50 метанол/дихлорметан) з наступною другою хроматографічною обробкою на силікагелі (100 % дихлорметан → 30 % етилацетат/дихлорметан) отримують зазначену в заголовку сполуку у вигляді рацемічної суміші. Хіральний поділ на індивідуальні енантіомери, які здійснюють за ® допомогою ВЕРХ, використовуючи 10 см колонку ChiralPak AD (60 % EtOH/гексани з 0,1 % діетиламіну), приводить до отримання зазначеної в заголовку сполуки у вигляді першого елюйованого енантіомера. МС: m/z=373,2 (M+1). Стадія F: трет-бутил-(6S)-6’-оксо-5,6',7,7'-тетрагідроспіро[циклопента[b]-піридин-6,5'піроло[2,3-d]піримідин]-3-карбоксилат До розчину трет-бутил-(6S)-4'-хлор-6'-оксо-5,6',7,7'-тетрагідроспіро-[циклопента[b]піридин6,5'-піроло[2,3-d]піримідин]-3-карбоксилату (200 мг, 0,537 ммоль) у сухому етилацетаті (5,37 мл) додають триетиламін (299 мкл, 2,15 ммоль) і паладій-на-вугіллі (571 мг, 10 %, 0,537 ммоль). Реакційну суміш поміщають в апарат Парра при тиску водню 50 psi (344737,85 Па). Через 1 ® годину отриману реакційну суміш фільтрують через Celite в атмосфері азоту, промиваючи етилацетатом. Отриманий фільтрат концентрують, отримуючи зазначену в заголовку сполуку, разом з одним еквівалентом гідрохлориду триетиламіну. МС: m/z=339,3 (M+1). Стадія G: (6S)-6'-оксо-5,6',7,7'-тетрагідроспіро[циклопента[b]піридин-6,5'-піроло[2,3d]піримідин]-3-карбонова кислота До твердого трет-бутил-(6S)-6'-оксо-5,6',7,7'-тетрагідроспіро-[циклопента[b]піридин-6,5'піроло[2,3-d]піримідин]-3-карбоксилату (255 мг, 0,536 ммоль), що містить один еквівалент гідрохлориду триетиламіну (з попередньої стадії), додають розчин хлористоводневої кислоти (20 мл, 4 M у діоксані). Через 16 годин отриману суміш концентрують, отримуючи зазначену в заголовку сполуку у вигляді солі бісхлористоводневої кислоти з одним еквівалентом 1 гідрохлориду триетиламіну. МС: m/z=283,2 (M+1). H-ЯМР (500 MГц, ДМСО) δ: 11,75 (с, 1H); 9,50 (с, 1H); 8,90 (с, 1Н); 8,80 (с, 1Н); 8,40 (с, 1Н); 8,20 (с, 1Н); 3,50-3,40 (м, 4H). ПРОМІЖНА СПОЛУКА 3 (6S)-5'-ціано-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]3-карбонова кислота Стадія A: (6S)-5'-бром-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]-піридин-6,3'-піроло[2,3b]піридин]-3-карбонова кислота До переміщуваної суміші (6S)-2'-оксо-1',2',5,7-тетрагідроспіро-[циклопента[b]піридин-6,3'піроло[2,3-b]піридин]-3-карбонової кислоти (розкритої в розділі проміжна сполука 1) (1,03 г, 3,66 ммоль) у дигідраті трифтористого бору (12 мл) додають N-бромсукцинімід (1,37 г, 7,70 ммоль), і отриману суміш нагрівають при 50 °C протягом 2 годин. Реакційну суміш охолоджують до кімнатної температури і додають N-бромсукцинімід (1,83 г, 10,3 ммоль). Отриману суміш нагрівають при 50 °C протягом 16 годин, залишають охолоджуватися до кімнатної температури, і отриману сиру суміш очищають, використовуючи ВЕРХ із оберненою фазою на колонці C-18, здійснюючи градієнтне елюювання H2О:CH3CN:TFA-95:5:0,1 до 65:35:0,1, отримуючи зазначену в заголовку сполуку. МС: m/z=361,9 (M+1). Стадія B: (6S)-5'-ціано-2'-оксо-1',2',5,7-тетрагідроспіро[циклопента[b]-піридин-6,3'-піроло[2,3b]піридин]-3-карбонова кислота Аргон барботують через перемішувану суміш (6S)-5'-бром-2'-оксо-1',2',5,7тетрагідроспіро[циклопента[b]піридин-6,3'-піроло[2,3-b]піридин]-3-карбонової кислоти (352 мг, 0,98 ммоль), ціаніду цинку (150 мг, 1,28 ммоль) і цинку (20 мг, 0,31 ммоль) у N, Nдиметилацетаміді (6 мл) протягом 10 хв. До отриманої суміші додають трис(дибензиліденацетон)дипаладій(0) (20 мг, 0,022 ммоль) і 1,1'-біс(дифенілфосфіно)фероцен (16 мг, 0,029 ммоль), і аргон барботують через отриману суміш протягом додатково 5 хв. Отриману реакційну суміш нагрівають при 120 °C протягом 18 годин, охолоджують до кімнатної температури, і очищають, безпосередньо використовуючи ВЕРХ із оберненою фазою на колонці C-18, здійснюючи градієнтне елюювання Н2О:CH3CN:TFA-95:5:0,1 до 65:35:0,1, отримуючи 1 зазначену в заголовку сполуку. МС: m/z=306,9 (M+1). H-ЯМР (400 MГц, ДМСО-d6) δ: 11,71 (с, 26 UA 109463 C2 1H), 8,91 (д, 1H, J=1,9 Гц), 8,61 (д, 1H, J=2,0 Гц), 8,14 (д, 1H, J=1,8 Гц), 8,03 (д, 1H, J=1,8 Гц), 3,49-3,34 (м, 4H). ПРОМІЖНА СПОЛУКА 4 5 10 15 20 25 30 35 40 45 50 (3S, 5S, 6R)-3-аміно-6-метил-5-феніл-1-(2,2,2-трифторетил)піперидин-2-он Стадія A: Метил-2-[(трет-бутоксикарбоніл)аміно]-4-(3-хлорфеніл)-5-оксогексаноат Суміш карбонату цезію (9,80 г, 30,1 ммоль) і метил-N-(трет-бутоксикарбоніл)-3-йодо-Dаланінату (9,90 г, 30,1 ммоль) у ДМФ (75 мл) перемішують при температурі 23 °C протягом 45 хв. перед тим, як додають 1-(3-хлорфеніл)пропан-2-он (6,09 г, 36,1 ммоль) і додаткову кількість карбонату цезію (9,80 г, 30,1 ммоль). Отриману суміш перемішують протягом 2,5 годин. Потім основну частину ДМФ видаляють при пониженому тиску при температурі бані
ДивитисяДодаткова інформація
Назва патенту англійськоюPiperidinone carboxamide azaindane cgrp receptor antagonists
Автори англійськоюBell, Ian, M., Fraley, Mark, E., Gallicchio, Steven, N., Ginnetti, Anthony, Mitchell, Helen, J., Paone, Daniel, V., Staas, Donnette, Wang, Cheng, Zartman, C. Blair, Stevenson, Heather, E.
Автори російськоюБелл Ян М., Фрэли Марк Е., Галликкио Стивен Н., Джиннетти Энтони, Митчелл Хелен Дж., Пэуан Дэниел В., Стэйес Доннетт, Ван Чен, Зартман С. Блэйр, Стивенсон Хитер Е.
МПК / Мітки
МПК: A61P 9/08, A61K 31/437, C07D 471/10, A61K 31/519
Мітки: піперидинонкарбоксамідазаіндани, рецептора, антагоністи
Код посилання
<a href="https://ua.patents.su/52-109463-piperidinonkarboksamidazaindani-antagonisti-receptora-cgrp.html" target="_blank" rel="follow" title="База патентів України">Піперидинонкарбоксамідазаіндани – антагоністи рецептора cgrp</a>
Антагоністи рецептора cgrp

Номер патенту: 82877
Опубліковано: 26.05.2008
Автори: Нгуйєн Дім Н., Уілльямс Тереза М., Бергі Крістофер С., Шо Ентоні У., Ден Чжену Цз, Пеуан Деніел В.
МПК: C07D 471/00, A61P 25/06, A61K 31/4188, C07D 471/04
Мітки: антагоністи, рецептора
Формула / Реферат:
1. Сполука формули І:, ІдеR1 вибраний з:С1-С6-алкілу, незаміщеного або заміщеного одним або декількома замісниками, незалежно один від одного вибраними з: С1-С6-алкілу, С3-С6-циклоалкілу, фенілу, гетероарилу, гетероциклу, (F)рС1-С3-алкілу, галогену, OR4, O(CH2)sOR4, CO2R4, CN, NR10R11, O(CO)R4,R2 вибраний з:арилу, незаміщеного або...
Cgrp-антагоністи, спосіб їх одержання, а також їх застосування як лікарських засобів

Номер патенту: 88163
Опубліковано: 25.09.2009
Автори: Палеарі Фабіо, Доодс Хенрі, Мюллер Штефан Георг, Сантагостіно Марко, Дрейер Александер, Лустенбергер Філіпп, Шенцле Герхард, Арндт Кірстен, Рудольф Клаус, Штенкамп Дірк
МПК: A61P 25/06, A61K 31/551, C07D 401/14, C07D 401/04
Мітки: застосування, спосіб, cgrp-антагоністи, лікарських, також, одержання, засобів
N-(2-феніл-4-амінобутил)-1-нафтаміди як антагоністи рецептора нейрокініну-1

Номер патенту: 67839
Опубліковано: 15.07.2004
Автор: Бернстейн Пітер Роберт
МПК: A61P 11/02, A61P 25/14, A61K 31/166, A61K 31/5375, C07D 295/12, A61P 11/06, A61P 25/00, A61P 1/08, A61P 19/02, A61P 25/06, A61P 37/08, A61P 9/12, A61K 31/277, A61P 1/00, A61P 25/18, A61P 17/00, A61P 25/22, C07C 255/57, A61P 35/00, C07D 295/125, A61P 11/00, A61P 29/00, A61P 25/28, A61P 25/24, A61P 43/00
Мітки: антагоністи, нейрокініну-1, n-(2-феніл-4-амінобутил)-1-нафтаміди, рецептора
Формула / Реферат:
1. Сполука формули (І)R1R2N-CH2CH2-CHAr1-CH2-NR3-CO-R4 , (I)у якій:R1 являє собою водень, С1-6алкіл, С2-6алкеніл, арил, С1-6алканоїл, С1-6алкоксикарбоніл або арилкарбоніл; причому будь-яка з таких груп є необов'язково заміщеною;R2 являє собою водень або С1-6алкіл; абоR1 або R2 пов'язані з утворенням необов'язково заміщеного морфолінового кільця;Аr1 являє собою феніл, моно- або дизаміщений...
Антагоністи ванілоїдного рецептора підтипу 1 (vr1) і їх застосування

Номер патенту: 94940
Опубліковано: 25.06.2011
Автори: Браун Брайан С., Лі Чіх-Хунг, Кеніг Джон Р., Гомтсян Артур Р.
МПК: A61K 31/352, A61K 31/416, C07D 493/00, C07D 231/56
Мітки: антагоністи, рецептора, ванілоїдного, vr1, застосування, підтипу
Формула / Реферат:
1. Сполука формули (І) або її фармацевтично прийнятна сіль, проліки, сіль проліків або їх комбінація, (I) деX1 є -(CR1aR1b)m-, -(CR1aR1b)nG1- або -(CR1aR1b)p-G1-C(R1aR1b)-; m дорівнює 1, 2, 3 або 4; n дорівнює 1, 2 або 3; р дорівнює 1 або 2; G1 являє собою О,...
Застосування антагоністів cgrp та інгібіторів вивільнення cgrp для усунення припливів у період менопаузи

Номер патенту: 73137
Опубліковано: 15.06.2005
Автори: Енгель Вольфганг, Дудс Хенрі, Еберлейн Вольфганг, Рудольф Клаус
МПК: A61K 31/00, A61P 15/12
Мітки: застосування, припливів, період, інгібіторів, антагоністів, вивільнення, менопаузи, усунення
Формула / Реферат:
1. Застосування діючої речовини, вибраної з антагоністів CGRP та інгібіторів вивільнення CGRP, для усунення припливів у період менопаузи.2. Застосування за п. 1, яке відрізняється тим, що воно являє собою монотерапію з використанням однієї діючої речовини.3. Застосування за п. 1, яке відрізняється тим, що воно являє собою доповнення до гормонзамісної терапії.4. Застосування за п. 1, яке відрізняється тим, що діюча...
Попередній патент: Спосіб та пристрій виконання фінансової операції за допомогою незахищеної відкритої телекомунікаційної інфраструктури
Наступний патент: Спірооксіндольні антагоністи mdm2
Випадковий патент: Спосіб експлуатації гальмових колодок на пасажирських вагонах