Спосіб одержання плевромутилінів
Номер патенту: 96775
Опубліковано: 12.12.2011
Автори: Махер Інгольф, Бергер Андреас, Декрістофоро Мартін






Формула / Реферат
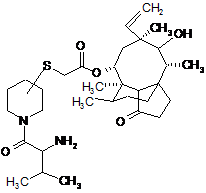
1. Спосіб одержання піперидиніл-сульфаніл-ацетилмутилінів формули:
 , I
, I
де атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває або в (S)-конфігурації, або в (R)-конфігурації, а 2-аміно-3-метилбутирильна група, зв'язана з піперидиновим кільцем, перебуває або в (S)-конфігурації, або в (R)-конфігурації, в якому здійснюють наступні етапи:
а) зняття захисту з N-захищеного піперидинил-сульфаніл-ацетил-мутиліну формули:
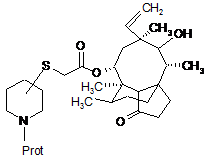 Prot - захисна група II
Prot - захисна група II
і виділення сполуки формули:
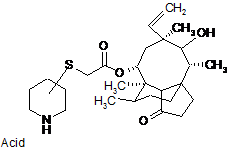 , Acid - кислота III
, Acid - кислота III
де атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває або в (S)-конфігурації, або в (R)-конфігурації, у вільній формі або у формі солі приєднання кислоти, у кристалічній формі,
б) ацилювання зазначеної сполуки формули III (R)- або (S)-валіном, захищеним як енамін і активованим як змішаний ангідрид карбонової кислоти, з утворенням сполуки формули:
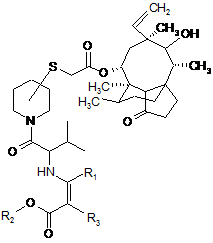 , IV
, IV
де R1 і R2 означають С1-4алкіл, a R3 означає водень або С1-4алкіл,
в) зняття захисту зі сполуки формули IV і виділення сполуки формули І.
2. Спосіб за п. 1, у якому сполуку формули І виділяють у формі фармацевтично прийнятної солі.
3. Спосіб за п. 1 або 2, у якому сполуку формули І виділяють у формі гідрохлориду.
4. Спосіб за п. 1, у якому зазначена сполука формули III являє собою сіль приєднання метансульфонової кислоти.
5. Спосіб за пп. 1-4, у якому N-захисна група являє собою трет-бутоксикарбонільну групу.
6. Спосіб за п. 1, у якому зазначений N-захищений піперидиніл-сульфаніл-ацетилмутилін одержують шляхом реакції плевромутилін-22-О-сульфонату (наприклад, мезилату, безилату або тозилату) з N-захищеним піперидин-тіолом.
7. Спосіб за п. 6, у якому N-захищений піперидиніл-сульфаніл-ацетилмутилін одержують шляхом реакції плевромутилін-22-О-сульфонату з N-захищеним піперидин-тіолом, у якому атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває або в (S)-конфігурації, або в (R)-конфігурації.
8. Спосіб за будь-яким з пп. 1-7, у якому виділена сполука формули І являє собою 3-заміщений піперидиніл-сульфаніл-ацетилмутилін.
9. Спосіб за будь-яким з пп. 1-8, у якому зазначений 3-заміщений піперидиніл-сульфаніл-ацетилмутилін являє собою 14-О-[N-3-метил-2-(R)-амінобутирилпіперидин-3(S)-іл)сульфанілацетил]-мутиліну гідрохлорид.
10. Сполука формули IV, як визначено в п. 1.
11. Сполука за п. 10, у якому R1 і R2 означають метил, а R3 означає водень.
12. Сполука за п. 11 у кристалічній формі.
13. Сіль приєднання кислоти сполуки формули III у кристалічній формі.
14. Кристалічний гідрохлорид 14-О-[(N-3-метил-2-(R)-амінобутирилпіперидин-3(S)-іл)сульфаніл-ацетил]мутиліну.
15. Сполука за п. 14, що характеризується 3-(S)-діастереомірним надлишком ³97 %.
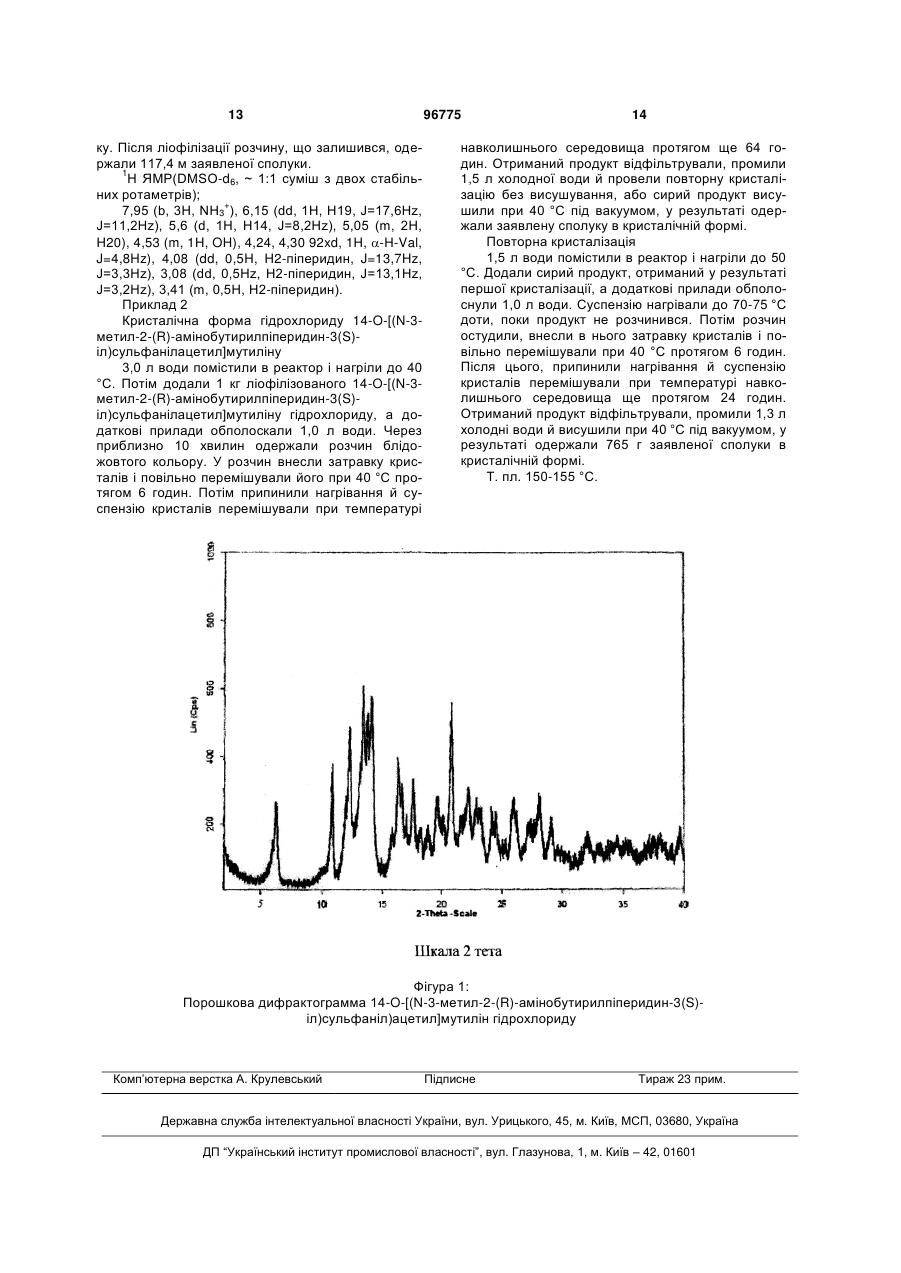
16. Сполука за п. 14, що характеризується рентгенографічними піками при наступних значеннях кута 2-тета: 6,2±0,2; 10,9±0,2; 12,3±0,2; 13,4±0,2; 14,1±0,2; 20,8±0,2.
17. Спосіб одержання кристалічної сполуки за п. 14, в якому здійснюють наступні етапи:
розчинення й нагрівання гідрохлориду 14-О-[(N-3-метил-2-(R)-амінобутирилпіперидин-3(S)-іл)сульфанілацетил]мутиліну у водному середовищі;
необов'язкове затравлювання розчину кристалами й перемішування при підвищеній температурі;
охолодження отриманої суспензії й перемішування при температурі навколишнього середовища;
виділення кристалічного продукту й необов'язкове повторення процедури.
18. Фармацевтична композиція, що містить кристалічний 14-O-[(N-3-мeтил-2-(R)-амінобутирилпіперидин-3(S)-іл)сульфаніл-ацетил]мутиліну гідрохлорид.
Текст
1. Спосіб одержання піперидиніл-сульфанілацетилмутилінів формули: CH2 3 96775 CH2 CH3 OH O S CH3 O H3C H3C N O O HN R2 R1 O R3 O , IV де R1 і R2 означають С1-4алкіл, a R3 означає водень або С1-4алкіл, в) зняття захисту зі сполуки формули IV і виділення сполуки формули І. 2. Спосіб за п. 1, у якому сполуку формули І виділяють у формі фармацевтично прийнятної солі. 3. Спосіб за п. 1 або 2, у якому сполуку формули І виділяють у формі гідрохлориду. 4. Спосіб за п. 1, у якому зазначена сполука формули III являє собою сіль приєднання метансульфонової кислоти. 5. Спосіб за пп. 1-4, у якому N-захисна група являє собою трет-бутоксикарбонільну групу. 6. Спосіб за п. 1, у якому зазначений N-захищений піперидиніл-сульфаніл-ацетилмутилін одержують шляхом реакції плевромутилін-22-О-сульфонату (наприклад, мезилату, безилату або тозилату) з Nзахищеним піперидин-тіолом. 7. Спосіб за п. 6, у якому N-захищений піперидиніл-сульфаніл-ацетилмутилін одержують шляхом реакції плевромутилін-22-О-сульфонату з Nзахищеним піперидин-тіолом, у якому атом вуглецю піперидинового кільця, зв'язаний з атомом сір Дійсний винахід стосується нового способу одержання 14-O-[(N-(3-метил-2-амінобутирилпіперидиніл)сульфаніл)ацетил]-мутилінів. Плевромутилін, сполука формули: являє собою природний антибіотик і продукується, зокрема, базидиоміцетами Pleurotus mutilus і P. Passeckerianus, див., наприклад, The Merck Index, 12-го видання, пункт 7694. 4 ки, перебуває або в (S)-конфігурації, або в (R)конфігурації. 8. Спосіб за будь-яким з пп. 1-7, у якому виділена сполука формули І являє собою 3-заміщений піперидиніл-сульфаніл-ацетилмутилін. 9. Спосіб за будь-яким з пп. 1-8, у якому зазначений 3-заміщений піперидиніл-сульфанілацетилмутилін являє собою 14-О-[N-3-метил-2-(R)амінобутирилпіперидин-3(S)-іл)сульфанілацетил]мутиліну гідрохлорид. 10. Сполука формули IV, як визначено в п. 1. 11. Сполука за п. 10, у якому R1 і R2 означають метил, а R3 означає водень. 12. Сполука за п. 11 у кристалічній формі. 13. Сіль приєднання кислоти сполуки формули III у кристалічній формі. 14. Кристалічний гідрохлорид 14-О-[(N-3-метил-2(R)-амінобутирилпіперидин-3(S)-іл)сульфанілацетил]мутиліну. 15. Сполука за п. 14, що характеризується 3-(S)діастереомірним надлишком 97 %. 16. Сполука за п. 14, що характеризується рентгенографічними піками при наступних значеннях кута 2-тета: 6,2±0,2; 10,9±0,2; 12,3±0,2; 13,4±0,2; 14,1±0,2; 20,8±0,2. 17. Спосіб одержання кристалічної сполуки за п. 14, в якому здійснюють наступні етапи: розчинення й нагрівання гідрохлориду 14-О-[(N-3метил-2-(R)-амінобутирилпіперидин-3(S)іл)сульфанілацетил]мутиліну у водному середовищі; необов'язкове затравлювання розчину кристалами й перемішування при підвищеній температурі; охолодження отриманої суспензії й перемішування при температурі навколишнього середовища; виділення кристалічного продукту й необов'язкове повторення процедури. 18. Фармацевтична композиція, що містить кристалічний 14-O-[(N-3-мeтил-2-(R)амінобутирилпіперидин-3(S)-іл)сульфанілацетил]мутиліну гідрохлорид. Був відкритий ряд нових плевромутилінів, що мають базову кільцеву структуру плевромутиліну, заміщених у положенні гідроксильної групи, наприклад, як антимікробних засобів. Група похідних плевромутиліну, валіл-заміщених піперидинилсульфаніл-ацетилмутилінів, викликає особливо великий інтерес у зв'язку з їх яскраво вираженою антимікробною активністю, як описано в публікації WO 02/22580. Для одержання по суті чистих ізомерів сполук цієї групи, існує потреба в розробці способу, що був би підходящим для використання в промисловому масштабі, і не вимагав би застосування дорогих вихідних матеріалів, шкідливих для навколишнього середовища реагентів і розчинників або стадій, що вимагають більших витрат часу, й трудомістких стадій очищення. В одному аспекті дійсний винахід представляє спосіб одержання сполуки формули: 5 де атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває або в (S)конфігурації, або в (R)-конфігурації, і 2-аміно-3метилбутирильна група, яка зв'язана з піперидиновим кільцем, перебуває або в (S)-конфігурації, або в (R)-конфігурації, що включає наступні етапи: а) зняття захисту з N-захищеного піперидинілсульфаніл-ацетилмутиліну формули: і виділення сполуки формули: де атом вуглецю піпиридинового кільця, зв'язаний з атомом сірки, перебуває або в (S)конфігурації, або в (R)-конфігурації, у вільній формі або у формі солі приєднання кислоти, б) ацилювання зазначеної сполуки формули III або (R)- або (S)-валіном, захищеним як енамін і активованим як змішаний ангідрид карбонової кислоти, з одержанням сполуки формули: 96775 6 де R1 і R2 означають С1-4алкіл, a R3 означає водень або С1-4алкіл, с) зняття захисту зі сполуки формули IV і виділення сполуки формули І. У кращому варіанті здійснення, сполуку формули І виділяють у формі фармацевтично прийнятної солі. У ще більш кращому варіанті здійснення, сполуку формули І виділяють у формі гідрохлориду. У сполуці формули І атом вуглецю піперидинового кільця зв'язаний з атомом сірки. Цей зв'язок може перебувати в будь-якім положенні піперидинового кільця, наприклад, в -, - або -положенні, переважно, в -положенні стосовно атома азоту піперидинового кільця. Зняття захисту з N-захищеної сполуки формули II здійснюється шляхом кислотного гідролізу захисної групи з утворенням солі приєднання кислоти (додаткової солі кислоти) у вигляді суміші ізомерів стосовно конфігурації атома вуглецю піперидинового кільця, зв'язаного з атомом сірки в сполуці формули II. Переважно, конфігурація мутилінівого кільця в сполуці формули II є такою ж, як і конфігурація природного мутиліна. Суміш ізомерів легко розділяється за допомогою кристалізації, у результаті чого виходять по суті чисті ізомери, у яких атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває або в (S)конфігурації, або в (R)-конфігурації. Стадія кристалізації забезпечує високий ступінь чистоти, тому сполука формули III у кристалічній формі є дуже придатною як проміжна сполука, особливо в промислових масштабах, у способі одержання піперидиніл-сульфаніл-ацетилмутилінів. Додатковою перевагою застосування сполуки формули III у кристалічній формі є те, що інші стадії очищення, такі як хроматографія, можна не проводити, оскільки чистота сполуки формули III у кристалічній формі цілком достатня для виробництва валілзаміщених піперидиніл-сульфанілацетилмутилінів. Для зняття захисту зі сполуки формули II можна використати неорганічну або органічну кислоти. У кращому варіанті здійснення, для одержання відповідного мезилату у вигляді (додаткової солі кислоти) солі приєднання кислоти використовують метансульфонову кислоту. Як захисні групи, називаних також «Prot», у сполуках формули II можуть 7 бути використані традиційні N-захисні групи. Переважно, коли використовують для цих цілей третбутоксикарбонільну групу. Установлено, що шляхом підбору підходящого агента для зняття захисту, розчинність (S)конфігурації або (R)-конфігурації може бути селективно поліпшена, у тому розумінні, що ізомерна (S)-конфігурація може бути відділена від (R)конфігурації. Наприклад, при використанні як агент для зняття захисту метансульфонової кислоти, продукт, що викристалізувався, являє собою чистий (S)-ізомер, у той час як (R)-ізомер залишається в розчині (див. Приклад 1Е). На наступному етапі, здійснюється введення ванільної групи в практично чистий ізомер сполуки формули III шляхом ацилювання зазначеної сполуки за допомогою (R)-або (S)-валіну, захищеного як енамін, для одержання сполуки формули IV. Захищений валін одержують шляхом реакції (R)або (S)-валіну зі складним -кетоефіром формули R1-CO-CH(R3)-COOR2, де R1, R2 і R3 такі, як визначено вище. Переважно, використовується метиловий ефір ацетоуксусної кислоти. Переважно, коли валін активований по методу змішаних ангідридів карбонових кислот. Змішаний ангідрид одержують in situ, наприклад, при додаванні хлориду триметилуксусной кислоти. Після додавання сполуки Формули III, отриманої на етапі а), утворюється сполука формули IV. Ці сполуки мають чудові кристалізаційні властивості, у порівнянні зі сполуками, що містять інші захисні групи, наприклад, третбутоксикарбоніл. Сполуки, що володіють такими чудовими кристалізаційними властивостями зручні в обігу в процесі одержання й виділення й мають переваги при додатковому очищенні, наприклад у порівнянні з 3-заміщеними піперидиніл-сульфанілацетилмутилінами формули І, енамін-захищену сполуку формули IV виділяють і очищають за допомогою кристалізації, у той час як відповідне трет-бутоксикарбоніл-захищене похідне необхідно очищати з використанням трудомістких і тривалих стадій хроматографічного очищення. Захисну групу сполук формули IV видаляють шляхом кислотного гідролізу. Після видалення складного ефіру ацетоуксусной кислоти й екстракції, сполука формули І може бути виділена у вільній формі, або у випадку додавання кислоти, що дає фармацевтично прийнятну сіль і після ліофілізації відповідної водної фази, в аморфній формі фармацевтично прийнятної солі, наприклад, у формі гідрохлориду. У кращому варіанті здійснення, атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває в -положенні щодо атома азоту піперидинового кільця, що означає 3-заміщений піперидиніл-сульфаніл-ацетилмутилін формули І. Більш переважно, дійсний винахід стосується до 14-O-[(N-3-метил-2-(R)-амінобутирилпіперидин3(S)-іл)сульфаніл-ацетил]мутилін гідрохлориду. Дійсний винахід також стосується до нової кристалічної форми 14-O-[((N-3-метил-2-(R)амінобутирил-піперидин-3(S)іл)сульфаніл)ацетил]мутилін гідрохлориду. Ліофілізована аморфна сполука 14-O-[((N-3-метил-2(R)-амінобутирилпіперидин-3(S) 96775 8 іл)сульфаніл)ацетил]-мутилін гідрохлориду, отриману за допомогою послідовності реакцій, описаних вище, можна перевести в кристалічну форму з використанням процесу кристалізації у водному середовищі. Процес можна поліпшити й прискорити за допомогою використання затравки кристалів. За допомогою перекристалізації кристалічний 14O-[(N-3-метил-2-(R)-амінобутирил-піперидин-3(S)іл)сульфаніл-ацетил]мутилін гідрохлорид може бути приведений у форму, що має бажану консистенцію, а також хімічну й оптичну чистоту. Одержують по суті чисті ізомери, що характеризуються діастереомерним надлишком 97% стосовно 3-(S)положення. Кристалічний 14-O-[(N-3-метил-2-(R)амінобутирил-піперидин-3-(S)-іл)сульфанілацетил]мутилін гідрохлорид характеризується порошковою рентгенограмою з піками при значеннях кутів 2-тета, рівних 6,2±0,2; 10,9±0,2; 12,3±0,2; 13,4±0,2; 14,1±0,2; 20,8±0,2 градусам (2-тета, CuK ). Він також може бути охарактеризований за допомогою інфрачервоного спектру, що має характеристичні смуги при приблизно 2927, 1721, 1645, -1 1462,1403,1142 см . Кристалічний 14-O-[(N-3-метил-2-(R)амінобутирил-піперидин-3(S)-іл)сульфанілацетил]мутилін гідрохлорид характеризується високою чистотою й кращою стабільністю, чим аморфна ліофілізована форма, що більше краща при виготовленні фармацевтичних композицій, що містять кристалічний 14-O-[((N-3-метил-2-(R)амінобутирил-піперидин-3(S)іл)сульфаніл)ацетил]мутиліну гідрохлорид як активний інгредієнт. N-захищені піперидинилсульфанілацетилмутиліни формули II можуть бути отримані шляхом реакції 14-О-меркаптоацетил-мутиліну з Nзахищеним гідроксипіперидином, що має групу, що йде, в , або -положенні стосовно атома азоту піперидинового кільця, наприклад, Н-ВОС-3(R)метилсульфоніл-оксипіперидин, як описано в WO 02/22580. Більш переважно, і як інший аспект дійсного винаходу, N-захищені піперидиніл-сульфанілмутиліни можуть бути отримані шляхом реакції плевромутилін-22-O-сульфонату (наприклад, мезилату, безилату або тозилату) з N-захищеним піперидин-тіолом. Захисні групи включають підходящі захисні групи, наприклад, традиційно використовувані захисні групи. Переважно, як N-захисну групу використовують трет-бутоксикарбонільну групу, а в іншому кращому варіанті здійснення, використають Nзахищений піперидин, що має тіольную групу в положенні 3 піперидинового кільця. Може бути використаний рацемічно або знантіомірно чистий N-захищений піперидин-тіол, що має або (R)- або (S)-конфігурацію при атомі вуглецю, що несе тіольную групу. Переважно, для того щоб уникнути використання дорогих хіральних вихідних матеріалів, використають N-захищений піперидин-тіол. Nзахищений піперидин-тіол можна одержати починаючи з підходящого гідроксипіперидину шляхом додавання N-захисної групи (наприклад, трет 9 бутоксикарбонілу) і реакцією із хлоридом або ангідридом сульфонової кислоти (наприклад, метансульфонілхлорид). Тіольная група вводиться шляхом реакції із сірковмісним нуклеофільним реагентом, наприклад, тіоацетатом, і основного гідролізу відповідного складного тіоефіру (наприклад, N-BOC-3-(R,S)-ацетилтіопіперидину). У випадку, коли гідроксипіперидин використають у формі окремого знантіомера (наприклад, 3(R)-гідроксипіперидина), то вищеописана послідовність реакцій, що включає нуклеофільне заміщення з використанням тіоцетату, здійснюється керованим способом (вальденовський обіг) для одержання відповідного N-захищеного піперидинтіолу, де атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває або в (S)конфігурації, або в (R)-конфігурації (наприклад, 3(S)-піперидин-тіол). Наступна реакція із плевромутилін-22-Осульфонатом приводить до утворення Nзахищених піперидинил-сульфанілацетилмутилінів формули II, де атом вуглецю піперидинового кільця, зв'язаний з атомом сірки, перебуває або в (S)-конфігурації, або в (R)конфігурації. У наведені нижче прикладах, які ілюструють дійсний винахід, посилання на температуру наведені в градусах Цельсія. У тексті використаються наступні абревіатури: N-BOC = N-бутоксикарбоніл, RT = кімнатна температура, MTBE = метил-трет-бутиловый ефір. Нумерація мутилінових циклів, на які даються посилання в прикладах, наведена в наступній нижче формулі: Приклади Приклад 1 14-O-[(N-3-метил-2-(R)амінобутирилпіперидин-3(S)-іл)сульфанілацетил]мутиліну гідрохлорид A. N-BOC-3-(R,S)-гідроксипіперидин 202,4 г 3-(R,S)-гідроксипіперидину розчинили в 4,5 л деіоніизованої води в 10-літровому реакторі. Додали 336 г гідрокарбонату натрію, розчинених в 1,1 л води. При енергійному перемішуванні до розчину при кімнатній температурі додали 534 г дитрет-бутилдикарбонату. Після перемішування протягом ночі суміш екстрагували СН2СІ2(3х). Змішані 96775 10 екстракти відмили деіонізованою водою, а розчинник видалили дистиляцією. Отриманий осад знову розчинили в СН2СІ2, а розчин випарили досуха. У результаті було отримано 423 г N-ВОС-3-(R,S)гідроксипіперидину, що міг бути використаний у наступних етапах без додаткового очищення. Б. N-ВОС-3-(R,S)-метил-сульфонілоксипіперидин До розчину 216 г N-ВОС-3-(R,S)гідроксипіперидину в 5 л CH2CI2 додали 222 мл триетиламіну при температурі 0-5 °С. Утримуючи температуру на рівні 0-5 °С протягом 45 хвилин по краплях додавали 137 г метансульфохлориду, розчинених в 300 мл CH2CI2. Після додаткового перемішування протягом 70 хвилин додали 2 л деіонізованої води й рН суміші довели до 5,9 шляхом додавання приблизно 90 мл 2 N розчину НСІ. Водну фазу відокремили, а органічну фазу відмили водою. Розчин випарили досуха й одержали 270-280 u маслянистого осаду. Після обробки 1 л н-гептаном відбулася кристалізація. Кристали виділили й висушили під вакуумом. У результаті було отримано 250 г N-BOC-3-(R,S)-метилсульфоніл-оксипіперидину. Т. Пл. 69 °C. 1 H ЯMP(CDCl3); 4,71 (m, 1H, CHOSO2CH3), 3,23,6 (m, 4H, CHN), 3,05 (s, 3H, CH3SO2), 1,94 (m, 2H, H4), 1,83, 1,54 (2xm, 2H, H5), 1,46 (m, 9H, трет.бутил). В. N-ВОС-3-(R,S)-ацетилтіо-піперидин 2,7 л диметилформаміду помістили в реактор з інертною атмосферою. При нагріванні додали 251,4 г N-ВОС-3-(R,S)-метил-сульфонілоксипіперидину. При досягненні внутрішньої температури 50°C додали тіоацетат калію. Після перемішуванні протягом 90 хвилин при температурі 95°C реакційну суміш перенесли в реактор, що містить 4 л води. Додали 4,2 л петролейного ефіру. Після енергійного перемішування протягом 5 хвилин водну фазу видалили. Після повторного додавання води рН довели до значення >8 шляхом додавання гідроксиду натрію. Органічну фазу відокремили, відмили водою й обробили активованим вугіллям. Розчин випарили досуха. Після проведення флеш-хроматографії на диоксиді кремнію з використанням петролейного ефіру, толуолу й етилацетату, і концентрування розчину, що містить продукт, при охолодженні відбулася кристалізація, у результаті було отримано 110 г NВОС-3-(R,S)-ацетилтіопіперидину. Т. пл. 46-48 °C (н-гептан). 1 H ЯMP(CDCl3); 3,79 (dd, 1Н, Н2), 3,5-3,6 (m, 2H, H3, H6), 3,17-3,27 (m, 2H, H3, H6), 2,32 (s, 3H, CH3SO2), 1,99 (m, 1H, H4), 1,55, 1,72 (m, H4, H5), 1,47 (s, 9H, трет.-бутил). Г. N-BOC-піперидин-3-(R,S)-тіол 200 г N-BOC-3-(R,S)-ацетилпіперидину помістили в 10-літровий реактор, що містить 3,41 л метанолу в інертній атмосфері. До цього розчину додавали 42 г метоксиду натрію в метанолі протягом 15 хвилин. Після додаткового перемішування додали 170 мл 5 N НСІ, у результаті чого значення рН змінилося до 2,6-3. Розчин сконцентрували з використанням випарника. Отримана двофазна суміш містила 1,7 л метил-трет-бутилового ефіру 11 (МТБЕ) і 1,7 л води. Після перемішування, відділення водної фази й відмивання, фазу МТБЕ виділили й випарили, вихід масла склав 170 г. 1 H ЯMP(DMSO-d6); 3,92 (b, 1Н, Н6), 3,69 (d, 1H, H2), 32,7-2,9 (m, 3H, H2, H3, H6), 2,61 (d, 1H, SH2), 2,00 (m, 1Н, Н4), 1,64 (m, 1Н, Н5), 1,45-1,31 (m, 2H, H4, H5), 1,39 (s, 9H, трет.-бутил). Д. 14-O-[(N-ВОС-піперидин-3-(R,S)іл)сульфаніл ацетил] мутилін 359,7 г 22 О-плевромутилінтозилату суспендували в 3,2 л метил-трет-бутилового ефіру (МТБЭ). Додали 1350 мл 1 N розчину гідроксида натрію і 21,1 г хлориду бензил-трибутиламмонию. Суміш охолодили до 15 °С и додали по краплях розчин, що містить 161,6 г N-BOC-піперидин-3-(R,S)-тіолу в 800 мл МТБЭ. Двофазну суміш перемішали протягом однієї години при температурі 20°С. Після завершення реакції фази були розділені. Органічну фазу висушили й випарили з одержанням на виході 521,5 г 14-O-[(N-ВОС-піперидин-3-(R,S)іл)сульфаніл-ацетил]мутиліну у вигляді маслянистої рідини, що використали на наступному етапі без додаткового очищення. 1 H ЯMP(CDCl3); 6,48 (dd, 1Н, H19, J=17,4Hz, J=11,2Hz), 5,77 (d, 1H, H14, J=8,4Hz), 5,34, 5,20 (2xdd, 2H, H20, J=11,2Hz, J=1,3Hz J=17,4Hz, J=1,3Hz), 4,0, 3,75, 2,96, 2,01 (b, 6H, піперидин). 3,37 (dd, 1H, H11, J=10,5Hz, J=6,6Hz), 3,19 (m, 2H, SCH2), 2,85 (m, 1H, CHS), 2,36 (dq, 1H, H10, J=6,6Hz, J=6,5Hz), 2,11 (b, 1H, H4) 1,47 (s, 12H, (CH3)15), 1,18 (s, 3H, (CH3)18), 0,89 (d, 3H, (CH3)17 J=6,9Hz), 0,79 (d, 3H, (CH3)16, J=6,6Hz). E. 14-O-[(піперидин-3-(S)-тіоацетил]мутилін метансульфонат 521 г 14-O-[(N-ВОС-піперидин-3-(R,S))іл)сульфанілацетил]-мутиліну розчинили в 4 л 2пропанолу й нагріли до 55 °С. Після додавання 165 мл метансульфонової кислоти розчин перемішували протягом 5 годин при тій же температурі. Після завершення реакції гідролізу ВОС-групи реакційну суміш остудили до 0 °C и перемішували ще протягом двох годин. Продукт, що викристалізувався, отфільтрували, промили 2-пропанолом і висушили під вакуумом. У результаті було отримано 159,8 г 14-O-[(піперидин-3-(S)тіоацетил]мутилін метан-сульфонату. Т. пл. 250-255 °С. 1 + H ЯMP(DMSO-d6); 8,58 (m, 2Н, NH2 ), 6,15 (dd, 1Н, Н19, J=17,2Hz, J=11,5Hz), 5,57 (d, 1Н, Н14, J=8,2Hz), 5,07 (m, 2H, H20), 4,5 (b, 1Н, ОН), 3,41 (s, 2H, SCH2), 2,4 (b, 1Н, Н4) 2,32 (s, 3H, CH3SO3), 1,37 (s, 3H, (CH3)15), 1,07 (s, 3H, (CH3)18), 0,83 (d, 3H, (CH3)17 J=7,0Hz), 0,64 (d, 3H, (CH3) 16, J=6,6Hz). Ж. Калієва сіль N-(3-метокси-1-метил-3-оксо-1пропенил)-R-валіну (датська сіль R-валіну) 36,6 г твердого KOH розчинили в 1250 мл 2пропанолу при невеликому нагріванні. Потім до розчину додали 65 г R-валіну і 65,9 мл метилового ефіру ацетоуксусної кислоти. Суміш перемішували й нагрівали зі зворотним холодильником протягом 2 годин. Зворотний холодильник замінили на конденсатор Клайзена й коротку колону, а воду, що утворилася в ході реакції конденсації видаляли відгоном з використанням приблизно 1 л 2 96775 12 пропанола. Після цього, додали 500 мл 2пропанолу й знову відігнали 500 мл. Нагрітий розчин перелили в 3 л МТБЕ й перемішували при охолодженні льодом протягом приблизно 3 годин. Отриману суспензію залишили на ніч при 4 °С (без доступу вологи), потім її отфільтрували, промили 500 мл МТБЕ й висушили, одержавши в результаті калієву сіль N-(3-метокси-1-метал-3-оксо-1пропенил)-11-валіну. З. 14-O-[((N(3-метил-2-(R)-N-(3-метокси-1метил-3-оксо-1-пропениламіно)бутирил)піперидин3-(S)-іл)сульфанілацетил]мутилін (датська сполука) До суспензії 88,2 г датської солі R-валіну в 2175 мл МТБЕ додали 14,5 мл води і суміш перемішували протягом 10 хвилин при кімнатній температурі. Потім суміш остудили до 0 °С, додали до неї 3,5 мл 4-метилморфоліну й 41 мл хлорангидриду триметилуксусної кислоти й перемішували суміш протягом 1 години. Потім до отриманої суміші додали 2175 мл попередньо охолодженої води (0-4 °C) і 166,4 г метансульфонату 14-O[(піперидин-3(S)-тіоацетил)]мутиліну. рН суміші підтримували при значенні 7 шляхом додавання приблизно 210 мл 2 N розчину NaOH. Охолоджену суміш (0 °C) перемішували протягом 30 хвилин, що привело до утворення кристалів датської сполуки. Для завершення реакції, суспензію нагріли до 30 °C и перемішували протягом 1 години. Після цього, установили значення рН на рівні 9,5 шляхом додавання приблизно 85 мл 2 N розчину NaOH. Після охолодження до 0 °C и перемішування протягом ще 2 годин, кристали відфільтрували, промили охолодженою водою і МТБЕ, а потім висушили під вакуумом. У результаті було отримано 195,4 г датські сполуки у вигляді МТБЕ-сольвату. Т.пл. 136-142 °С. 1 H ЯMP(DMSO-d6, суміш з двох стабільних ротаметрів); 8,87, 8,83 (2xd, 1Н, NH, J=9,3Hz), 6,15 (m, 1Н, Н19), 5,60, 5,56 (2xd, 1H, Н14, J=8,3Hz), 5,04 (m, 2H, H20), 4,50 (m, 1H, -H-Val), 4,37 (s, 1H, СНенамін) 3,50 (s, 3Н, OCH3), 2,42, 2,40 (2xb, 3Н, (СН3)15), 1,07 (s, 3Н, (СН3)18), 0,96-0,77 (m, 9Н, (СН3)17, (CH3)VaI), 0,64 (m, 3Н, (СН3)16. І. 14-O-[((N-3-метил-2-(R)-амінобутирилпіперидин-3-(S)-іл)сульфаніл)ацетил]мутиліну гідрохлорид 145 г датської сполуки суспендували в суміші 1,8 л МТБЕ і 1,8 л води. Отриману суміш нагріли до 50 °C и енергійно перемішували. Значення рН суміші втримували на рівні 1 шляхом додавання по краплях 2 N розчину НСІ. Після гідролізу захисної групи енаміну провели високоефективну рідинну хроматографію. Органічну фазу відокремили, а водну фазу двічі екстрагували МТБЕ (щораз по 1,6 л) для видалення метилового ефіру ацетоуксусної кислоти. До водної фази при перемішуванні додали 1,6 л МТБЕ і значення рН утримували на рівні 10 шляхом додавання 10 N розчину NaOH. Фази розділили, після чого фазу МТБЕ двічі екстрагували водою, порціями по 1,6 л. Після додавання 1,5 л води рН суміші довели до значення 3,2 шляхом додавання приблизно 100 мл 2 N розчину НСІ. Водну фазу сконцентрували в роторному випарни 13 ку. Після ліофілізації розчину, що залишився, одержали 117,4 м заявленої сполуки. 1 H ЯMP(DMSO-d6, ~ 1:1 суміш з двох стабільних ротаметрів); + 7,95 (b, 3Н, NH3 ), 6,15 (dd, 1Н, Н19, J=17,6Hz, J=11,2Hz), 5,6 (d, 1H, Н14, J=8,2Hz), 5,05 (m, 2H, H20), 4,53 (m, 1H, OH), 4,24, 4,30 92xd, 1H, -H-Val, J=4,8Hz), 4,08 (dd, 0,5H, H2-піперидин, J=13,7Hz, J=3,3Hz), 3,08 (dd, 0,5Hz, Н2-піперидин, J=13,1Hz, J=3,2Hz), 3,41 (m, 0,5Н, Н2-піперидин). Приклад 2 Кристалічна форма гідрохлориду 14-O-[(N-3метил-2-(R)-амінобутирилпіперидин-3(S)іл)сульфанілацетил]мутиліну 3,0 л води помістили в реактор і нагріли до 40 °C. Потім додали 1 кг ліофілізованого 14-O-[(N-3метил-2-(R)-амінобутирилпіперидин-3(S)іл)сульфанілацетил]мутиліну гідрохлориду, а додаткові прилади обполоскали 1,0 л води. Через приблизно 10 хвилин одержали розчин блідожовтого кольору. У розчин внесли затравку кристалів і повільно перемішували його при 40 °C протягом 6 годин. Потім припинили нагрівання й суспензію кристалів перемішували при температурі 96775 14 навколишнього середовища протягом ще 64 годин. Отриманий продукт відфільтрували, промили 1,5 л холодної води й провели повторну кристалізацію без висушування, або сирий продукт висушили при 40 °C під вакуумом, у результаті одержали заявлену сполуку в кристалічній формі. Повторна кристалізація 1,5 л води помістили в реактор і нагріли до 50 °С. Додали сирий продукт, отриманий у результаті першої кристалізації, а додаткові прилади обполоснули 1,0 л води. Суспензію нагрівали до 70-75 °C доти, поки продукт не розчинився. Потім розчин остудили, внесли в нього затравку кристалів і повільно перемішували при 40 °C протягом 6 годин. Після цього, припинили нагрівання й суспензію кристалів перемішували при температурі навколишнього середовища ще протягом 24 годин. Отриманий продукт відфільтрували, промили 1,3 л холодні води й висушили при 40 °C під вакуумом, у результаті одержали 765 г заявленої сполуки в кристалічній формі. Т. пл. 150-155 °С. Фігура 1: Порошкова дифрактограмма 14-O-[(N-3-метил-2-(R)-амінобутирилпіперидин-3(S)іл)сульфаніл)ацетил]мутилін гідрохлориду Комп’ютерна верстка А. Крулевський Підписне Тираж 23 прим. Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of pleuromutilins
Автори англійськоюMacher, Ingolf, Berger, Andreas, Decristoforo, Martin
Назва патенту російськоюСпособ получения плевромутилинов
Автори російськоюМахер Ингольф, Бергер Андреас, Декристофоро Мартин
МПК / Мітки
МПК: A61K 31/445, A61P 31/04, C07D 211/54
Мітки: одержання, плевромутилінів, спосіб
Код посилання
<a href="https://ua.patents.su/7-96775-sposib-oderzhannya-plevromutiliniv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання плевромутилінів</a>
Похідні октагідро-6,10-діоксо-6н-піридазино[1,2-а][1,2]діазепін-1- карбонової кислоти, спосіб їх одержання та одержання терапевтично активних сполук

Номер патенту: 71913
Опубліковано: 17.01.2005
Автори: Руссель Патрік, КОЛЛАДАН Колетт, Крок Веронік, Ларкін Джон Патрік
МПК: C07K 5/078, C07K 5/06, C07D 487/04
Мітки: спосіб, терапевтичної, одержання, карбонової, сполук, похідні, октагідро-6,10-діоксо-6н-піридазино[1,2-а][1,2]діазепін-1, активних, кислоти
Формула / Реферат:
1. Сполуки загальної формули (IA1):,де R являє собою С1-4 алкіл, що мають конфігурацію SR або що знаходяться у вигляді суміші SR+SS, які являють собою рацемічну суміш.2. Сполуки загальної формули (ІА1) за п.1, де R являє собою метил.3. Сполуки загальної формули (IA1) за п.1, 2, які являють собою рацемічну суміш, що складається...
Акрилоїлзаміщенa похідна дистаміцину, спосіб її одержання (варіанти) та фармацевтична композиція на її основі

Номер патенту: 61922
Опубліковано: 15.12.2003
Автори: Каполонго Лаура, Кодзі Паоло, Францетті Крістіна, Калдареллі Маріна, Беріа Італо, Бьясолі Джованні
МПК: A61P 31/12, C07D 207/34, A61P 35/00, A61K 31/40
Мітки: композиція, фармацевтична, одержання, спосіб, акрилоїлзаміщенa, основі, похідна, варіанти, дистаміцину
Формула / Реферат:
1. Акрилоїлзаміщена похідна дистаміцину формули:,де:n означає 2, 3 або 4;R1 або R2 вибирають, кожен незалежно, з: водню, галогену і С1-С4 алкілу;R3 означає водень або галоген;В вибирають з:де R4, R5, R6, R7 і R8 означають, кожен незалежно, водень або С1-С4 алкіл, за умови, що, принаймні, один з R4, R5 і R6 означає С1-С4 алкіл, або її фармацевтичнo прийнятна сіль.2. Сполука за п....
Похідні морфоліну, спосіб їх одержання та фармацевтичні препарати, що містять вказані похідні

Номер патенту: 73931
Опубліковано: 17.10.2005
Автори: Дюку Жан Філіп, Проєтто Вінченцо, Гейоль Патрік, ЕМОН-АЛЬТ Ксав'є
МПК: A61P 25/10, A61P 25/06, A61P 37/00, A61P 29/00, C07D 413/06, A61K 31/5377, A61P 29/02, A61P 1/08, A61P 13/02, A61P 25/22, A61P 9/00, A61P 1/04, A61P 25/00, A61P 13/00, A61P 17/00, A61P 25/24, A61P 1/00, A61P 11/06, C07D 265/30, A61P 11/00
Мітки: фармацевтичні, препарати, одержання, похідні, містять, морфоліну, вказані, спосіб
Формула / Реферат:
1. Сполука формули (I): (I),в якій Аr являє собою феніл, монозаміщений або дизаміщений атомом галогену; (С1-С3)алкіл;Χ являє собою групу R2-N=; групу R2-CH=;R1 являє собою атом хлору, атом брому, (С1-С3)алкіл або трифлуорметил;R2 являє собою (С1-С6)алкіл; (С3-С6)циклоалкіл; групу –CR4R5CONR6R7;R3 являє собою групу...
Сольвати піметрозину, пестицидна композиція та спосіб її одержання

Номер патенту: 72521
Опубліковано: 15.03.2005
Автор: Гутманн Штефан
МПК: A01N 43/707, C07D 401/12, A01P 1/00, C07D 253/00
Мітки: спосіб, одержання, пестицидна, сольвати, піметрозину, композиція
Формула / Реферат:
1. Сполуки формули,деr і s незалежно один від одного означають будь-яке число від 0,00 до 12,00; іL означає метанол, етанол, пропанол, ізопропанол, бутанол, ізобутанол, трет-бутанол, циклогексанол, тетрагідрофуриловий спирт, етиленгліколь, гліцерин, метилацетат, етилацетат, етиллактат, бутиролактон, етиленкарбонат, пропіленкарбонат,...
Тієнілазолілалкоксіетанаміни, спосіб їх одержання, проміжні продукти для їх одержання та фармацевтична композиція

Номер патенту: 58589
Опубліковано: 15.08.2003
Автори: АНДАЛУЗ-МАТАРО Блас, ФРІГОЛА КОНСТАНСА Хорді, МЕРСЕ-ВІДАЛЬ Рамон
МПК: A61K 31/4155, A61K 31/381
Мітки: композиція, спосіб, одержання, продукти, фармацевтична, проміжні, тієнілазолілалкоксіетанаміни
Формула / Реферат:
1. Похідна тієнілазолілалкоксіетанаміну загальної формули (І):, (І)деR1 - атом водню або атом галогену, або алкільний радикал з 1-4 атомами вуглецю;R2, R3 та R4, незалежно, - атом водню або алкільний радикал з 1-4 атомами вуглецю; таAz являє собою азотвмісне гетероциклічне ароматичне п'ятичленне N-метил-заміщене кільце, до складу якого входять від одного до трьох атомів азоту, загальної формули...
Попередній патент: Високотемпературне мастило для гарячої обробки високоякісних і вуглецевих сталей тиском та його застосування
Наступний патент: Екстракт із рослинної сировини з високим вмістом поліфенолу, спосіб його одержання
Випадковий патент: Гідроножиці віброударні