Похідні 2,3-діарилпіразолідину з активністю щодо нейротензину, спосіб їх одержання, фармацевтична композиція на їх основі
Номер патенту: 77775
Опубліковано: 15.01.2007
Автори: Ланге Йозефус Г.М., Прас-Равес Марія Л., Крузе Корнеліс Г., Тьойнстра Тинка, Фенстра Рулоф В., ван Стьойвенберг Герман Г., Кейзер Гіскіас Г.






Формула / Реферат
1. Сполука формули (1)
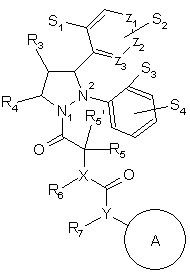 ,
,
де
S1 - водень, галоген, гідрокси або алкокси (1-3С);
S2 - водень або галоген;
S3 - водень, галоген, гідрокси або алкокси (1-3С);
S4 - водень, галоген або алкіл (1-6С), який може бути заміщений гідрокси, алкокси (1-3С), аміно, моно- або діалкіламіногрупою з 1-3 атомами С в алкільній групі (групах), SH або алкіл (1-3С);
Х - азот або вуглець;
Y - азот або кисень, якщо Х - азот, або азот, коли Х - вуглець;
R3 та R4 незалежно один від одного - водень або алкіл (1-3С);
R5 - водень або алкіл (1-6С), який може бути заміщений галогеном, CN, СF3, гідрокси, алкокси (1-3С), сульфонілалкілом (1-3С), аміно, моно- або діалкіламіногрупою з 1-3 атомами С в алкільній групі (групах), коли Х - вуглець чи азот, або R5 - алкокси (1-6С), SH або S-алкіл (1-3С), коли Х – вуглець;
R'5 - водень або алкіл (1-3С);
R6 - водень або алкіл (1-3С);
R7 - водень або алкіл (1-3С);
R5 та R6 разом або R5' та R6 разом можуть утворювати 3-7-членну циклічну групу, яка може бути заміщена нижчим алкілом, галогеном, CN або СF3, a R5+R5' разом можуть утворювати 3-7-членне кільце, а
Z1, Z2 та Z3 - вуглець, або Z1 - азот, a Z2 та Z3 - вуглець, або Z1 та Z3 - вуглець, a Z2 - азот, або Z1 та Z2 - вуглець, а Z3 - азот;
А - (полі)циклоалкільна система, що складається з 4-10-членних кілець, які можуть бути заміщені галогеном, СF3, алкілом або алкокси (1-3С), CN, ОН або SH, та її фармакологічно прийнятні солі.
2. Спосіб одержання сполуки, заявленої в п. 1, який відрізняється тим, що сполуку одержують за наступною схемою:
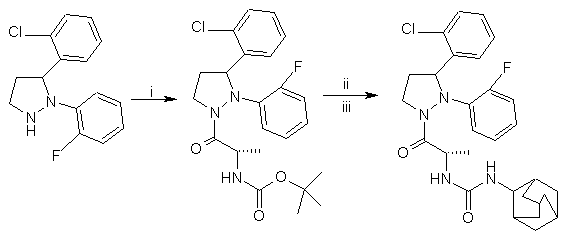 ,
,
причому після етапу і утворюються два діастереомери, які після етапу ііі можуть бути розділені колонковою хроматографією на енантіомерно чисті діастереомери.
3. Фармацевтична композиція, що містить принаймні одну сполуку за п. 1 як діючу речовину.
4. Застосування сполуки, заявленої в п. 1, для приготування фармацевтичної композиції для лікування станів та хвороб, спричинених розладами у медійованій нейротензином передачі імпульсів.
5. Застосування сполуки за п. 1 для приготування фармацевтичної композиції для лікування психозів.
6. Застосування сполуки за п. 1 для приготування фармацевтичної композиції для лікування хвороби Паркінсона.
7. Застосування сполуки за п. 1 для приготування фармацевтичної композиції для лікування депресій.
8. Застосування сполуки за п. 1 для приготування фармацевтичної композиції для лікування станів збентеженості.
Текст
1. Сполука формули (1) 2 (19) 1 3 77775 4 причому після етапу і утворюються два діастерео5. Застосування сполуки за п. 1 для приготування мери, які після етапу ііі можуть бути розділені кофармацевтичної композиції для лікування психолонковою хроматографією на енантіомерно чисті зів. діастереомери. 6. Застосування сполуки за п. 1 для приготування 3. Фармацевтична композиція, що містить принайфармацевтичної композиції для лікування хвороби мні одну сполуку за п. 1 як діючу речовину. Паркінсона. 4. Застосування сполуки, заявленої в п. 1, для 7. Застосування сполуки за п. 1 для приготування приготування фармацевтичної композиції для ліфармацевтичної композиції для лікування депрекування станів та хвороб, спричинених розладами сій. у медійованій нейротензином передачі імпульсів. 8. Застосування сполуки за п. 1 для приготування фармацевтичної композиції для лікування станів збентеженості. Винахід стосується групи нових похідних 2,3діарилпіразолідину, які мають інгібувальну активність щодо ферментів, які розкладають нейропептид нейротензин. Встановлено, що сполуки формули (1) де - S1 - водень, галоген, гідроксі або алкоксі (13С) - S2- водень або галоген - S3 - водень, галоген, гідроксі або алкоксі (1 3С) - S4 - водень, галоген або алкіл (1-6С), який може бути заміщений гідроксі, алкоксі (1-3С), аміно, моно- або діалкіламіногрупою з 1-3 атомами С в алкільній групі (групах), SH або алкіл (1-3С) - X - азот або вуглець - Υ - азот або кисень, якщо X - азот, або азот, коли X - вуглець - R3 та R4 незалежно один від одного - водень або алкіл (1-3С) - R5 - водень або алкіл (1-6С), який може бути заміщений галогеном, CN, CF3, гідроксі, алкоксі (13С), сульфонілалкілом (1-3С), аміно, моно- або діалкіламіногрупою з 1-3 атомами С в алкільній групі (група х), коли X - вуглець чи азот, або R5 алкоксі (1-6С), SH або S-алкіл (1-3С), коли X - вуглець - R'5 - водень або алкіл (1-3С) - R6 - водень або алкіл (1-3С) - R7 - водень або алкіл (1-3С) - R5 та R6 разом або R'5 та R6 разом можуть утворювати 3-7-членну циклічну групу, яка може бути заміщена нижчим алкілом, галогеном, CN або CF3, a R5+R' 5 разом можуть утворювати 3-7-членне кільце, а - Z1, Z2 та Z3 - вуглець, або Ζ1 - азот, a Z2 та Z3 - вуглець, або Ζ1 та Z3 - вуглець, a Z2 - азот, або Ζ1 та Ζ2 - вуглець, a Z3 - азот - А - (полі)циклоалкільна система, що складається з 4-10-членних кілець, які можуть бути заміщені галогеном, CF3, алкілом або алкоксі (1-3С), CN, ОН або SH, та їх солі мають інгібувальну активність проти розкладу нейротензину. Зокрема, ці сполуки інгібують ферменти Тімет олігопептидазу EC 3.4.24.15 та Нейролізин EC 3.4.24.16, які розщеплюють нейропептид нейротензин. Завдяки інгібуванню розкладу нейротензину тими ферментами збільшуються рівні ендогенного нейротензину, що сприяє лікуванню хвороб, пов'язаних з рівнями нейротензину. Сполуки згідно з винаходом активно інгібують зазначені ферменти у діапазоні значень рІС 50 5,08,0 при випробуваннях за методикою, описаною у [Biochem. J 280, 421-426 та Eur. J. Biochem. 202, 269-276]. Сполуки згідно з винаходом можна використовувати для лікування станів та хвороб, спричинених розладами у медійованій нейротензином передачі імпульсів, наприклад, периферійних розладів кров'яного тиску або спорожнення шлунку, нервових розладів, як хвороба Паркінсона, та розладів центральної нервової системи, наприклад, бентежність, депресія, психози та інші психічні розлади. Сполуки формули (1) можна одержувати одним з чотирьох наступних способів А, В, С, D. Вихідними сполуками для здійснення цих чотирьох способів є заміщені 2,3-діарилпіразолідини, що мають одну зі структур, наведених на Фіг.1. Частина R.7-Y-A сполук формули (1) може мати стр уктури груп, зазначених на Фіг.2. Вихідні похідні піразолідину за Фіг.1 можна одержувати способом, що наведений на схемі 1: 5 77775 як описано у прикладі 5. Спосіб А: Сполуки, зазначені у таблиці А, можна синтезувати шля хом, наведеним для сполук А23/А24. Після етапу і утворюються два діастереомери, які після етапу ііі можна розділити колонковою хроматографією на енантіомерно чисті діастереомери А23 та А24. Див. схему А.1. Спосіб В: Сполуки, зазначені у таблиці В, можна синтезувати шляхом, зображеним на схемі В.1. Етапи і та іі реакції на схемі В.1 ідентичні етапам і та іі відповідно на схемі А.1. Спосіб С Сполуки, зазначені у таблиці С, можна синтезувати шляхом, наведеним для сполук С2 та С8, як зображено на схемі С.1: Спосіб D Сполуки, зазначені у таблиці D, можна синтезувати шляхом, наведеним для сполук С2 та С8, як зображено на схемі С.1: 6 Етапи реакції і та іі схеми D є ідентичні етапам і та іі відповідно реакції, наведеної на схемі С.1, етапи реакції ііі та iv відповідно. Далі приготування сполук формули (1) танизки проміжних сполук за способами докладно описується у наступних прикладах. Приклад 1 Етап і (схема А.1) До 50мл сухого ацетонітрилу додають при кімнатній температурі в атмосфері азоту при перемішуванні: 4г (14,5ммоля) II, 2,7г (14,3ммоля) N-BocL-аланину та 3,8г (18,4ммоля) DCC (діциклогексилкарбодііміду). Утворюється осад, який продовжують перемішувати усю ніч. Тонкошарова хроматографія реакційної суміші показує схожу на вісімку подвійну пляму, що містить два можливі діастереомери. Осад відфільтровують. До фільтрату додають близько 20г кремнезему та концентрують у вакуумі. Одержаний порошок уміщують на верхівку сухої колонки (SiO2) та виконують елюювання (елюент СН 2СІ2/МеОН 98:2). Частину колонки, що містить два діастереомери, збирають та вмішують до МеОН. Суспензію фільтрують, осад ще раз промивають МеОН. Об'єднані фракції МеОН концентрують у вакуумі й одержаний осад вміщують до СН2СІ2, після чого сушать над MgSO4. Осушний агент видаляють фільтрацією, а розчинник випарюють у вакуумі, виділяючи близько 5г (80%) сирого продукту. Етап іі (схема А.1): 5г (близько 10ммолів) сполуки з етапу і розчиняють при перемішуванні у 100мл розчину трифтороцтової кислоти/СН2СІ2/Н2О 70/25/5. Перемішування триває 2 години. Далі реакційну суміш концентрують у вакуумі, а осад вміщують до СН2СІ2. Одержаний розчин обробляють насиченим водним розчином К2СО3, промивають водою та розсолом і суша ть над MgSO4 . Осушний агент видаляють фільтрацією, а розчинник випарюють у вакуумі, виділяючи близько 4г (майже 100%) сирого аміну. Етап iiі (схема A.1): При кімнатній температурі в атмосфері азоту 0,50г (1,44ммоля) сирого аміну з етапу іі суспендують у 10мл ацетонітрилу при перемішуванні. Далі додають 0,26г (1,44ммоля) 2адамантилізоціанату. Реакцію продовжують ще 2 години. До реакційної суміші додають близько 2г кремнезему та концентрують у вакуумі. Одержа 7 77775 8 ний порошок уміщують до верху сухої колонки (SiO2) та виконують елюювання (елюент ЕtOАс/петролейний етер 1/1). Частину колонки, що містить два діастереомери, збирають окремо та вмішують до МеОН. Одержані дві суспензії фільтрують нарізно, кожний з одержаних двох осадів промивають МеОН один раз. Для кожного діастереомеру відповідні фракції МеОН зливають разом і концентрують у вакуумі, після чого кожний осад вміщують до СН 2СІ2 і потім сушать кожний осад над MgSO4 . Після видалення осушного агента та розчинника у вакуумі одержують дві тверді фази, кожна з яких містить один діастереомер: 0,16г А23 Розділення енантіомерів проміжної сполуки пі(21%), точка топлення 140-143°С, та 0,22г А24 сля етапу іі (схема А.1) виконують на колонці Хіра(29%), точка топлення 145-148°С. лсел CD (25x5см 2, 20мкм, елюент гексан/етанол Примітка: 4:1). Сполука А12 одержана енантіомерно чистою. Сполуки таблиці А одержують таким само чиПроміжну сполуку після етапу іі (схема А.1) роздіном: ляють на енантіомери, після чого провадять етап (схема А.2). Енантіомер (+)- А12 являє собою евтомер. Таблиця А R3, R4, R6, R 7, S 2, S 4 = Η Χ, Υ = Ν Сполука піразолідин Α1 І Α2 II A3 II Α4 III Α5 III Α6 II Α7 II Α8 II Α9 III Α10 IV A11 III Α12 III Α13 II А14 II Α15 III Α16 I Α17 II Α18 V Α19 I Α20 III Α21 III Α22 I Α23 II Α24 II Α25 II Α26 II Α27 II Α28 II Α29 II Α30 VI А31 X А32 VIII А33 II Приклад 2 Етап iiі (схема В.І) R5 Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Me Me Η Me Me Et Et nBut iBut Η Η Η Η nPr R5' Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η Η YR7 A 1 1 2 3 4 5 4 6 6 7 7 8 8 9 8 7 7 7 8 8 8 10 8 8 8 8 8 8 10 8 8 8 8 примітка [a] +94 діастереомери діастереомери діастереомери Точка топлення Див. додаток 1 Див. додаток 2 Див. додаток 3 153-5 >220 185-8 120-5 130-3 195-8 241-2 >280 164-5 135-40 105-10 168-71 208-210 115-120 Див. додаток 4 140-5 125-145 132-150 Див. додаток 5 140-3 145-8 145-8 155-8 122-5 122-5 Див. додаток 6 221-3 208-210 145-165 110-130 0,20г (0,67ммоля) трифосгену розчиняють у 10мл сухого ді хлорметану. До суміші додають роз 9 77775 10 чин 0,70г (2,0ммоля) похідного піразолідину та шаровій колонковій хроматографії (SiO2, елюент 0,42мл (2,4ммоля) діізопропилетиламіну протягом СН2СІ2/МeОН 99:1). Збирають фракції, що містять 45 хвилин. Реакційну суміш безперервно переміпродукт, видаляють елюент у вакуумі та одержушують. Далі до неї додають розчин 0,33г ють олію, яка кристалізується при перемішуванні в (2,0ммоля) метил-2-адамантиламіну та 0,42мл діізопропилетері. Після фільтрації та сушки одер(2,4ммоля) діізопропилетиламіну в 5мл сухого діхжують 0,69г (вихід 64%) твердої сполуки В2 (точка лорметану протягом 5 хвилин. Реакційну суміш топлення 184-186°С). залишають на ніч, після чого випарюють розчинПримітка: Застосований метил-2ник у вакуумі. Осад занурюють до етилацетату, адамантиламін легко одержати стандартним меодержаний розчин обробляють 5% водним розчитодом відновлювального амінування, починаючи з ном NaHCO3 та розсолом відповідно. Органічну 2-адамантанону та метиламінгідрохлориду, прифазу відокремлюють і сушать над MgSO4. Фільтчому відновлювачем слугує NaВН(ОАс) 3. рацією осушного агента та видаленням розчинниСполуки з таблиці В готують таким само чика у вакуумі одержують олію, яку піддають тонконом. Таблиця В R3, R4, R5, R 5' , S2, S4 = Η Сполука піразолідин В1 III В2 III В3 III В4 III В5 III В6 II В7 II В8 І В9 І X N N N N N N N N N Υ N N N N O O O O O R6 Η Η Me Η Η Η Η Η Η R7 nPr Me Η Me YR7 A 11 12 4 13 14 15 14 14 15 Точка топлення 132-4 184-6 222-4 140-2 110-2 142-4 135-8 141-3 151-4 Примітка: Потрібну проміжну сполуку після етапу іі (схема В.1) у випадку В3 (R6=Me) можна приготувати подібно до етапів і та іі у схемі А.1. Приклад 3 Етап і (схема С.1) 16г (160ммолів) бурштинового ангідриду розчиняють у сухому діетилетері. Потім додають з перемішуванням до розчину бурштинового ангідриду 44г (160ммолів) II, розчиненого у діетилетері. По закінченні додання реакційну суміш доводять до температури флегми й залишають на ніч. Утворений осад відфільтровують, осад двічі промивають діетилетером. Сушать на відкритому повітрі й одержують 45,6г (вихід 75%) потрібної проміжної сполуки. Етап іі (схема С.1) У атмосфері азоту 4,5г (12ммолів) проміжної сполуки з етапу і та 7,9г (61ммоль, 5г-екв) діізопропилетиламіну розчиняють у 50мл сухого СН 2СІ2. Одержаний розчин доводять при перемішуванні до 4°С. Далі додають 0,90г (7,0ммолів) 1-гідроксі-7аза-бензтриазолу та 4,20г (15ммолів) 2-хлор-1,3діметилімідазолінійгексафторфосфату. Потім до реакційної суміші додають 2,19г (15ммолів) 2аміноадамантану і дають реагувати 1 годину при кімнатній температурі. До реакційної суміші додають біля 4г кремнезему та концентрують у вакуумі. Одержаний порошок вміщують на суху колонку (SiO2) й виконують елюювання (елюент EtOAc/петролейний етер 1:1). Частину колонки, що містить продукт, збирають і вміщують до МеОН. Одержану суспензію фільтрують, осад промивають один раз МеОН. Фракції МеОН зливають і концентрують у вакуумі, осад вміщують до СН 2СІ2, одержаний розчин су шать над MgSO4 . Після видалення осушного агента та розчинника у вакуумі одержують 2,0г твердого С2 (вихід 32%), точка топлення 192-195°С. Етап Ні (схема С.1) При перемішуванні в атмосфері азоту 6,0г (60ммолів) бурштинового ангідриду суспендують у 35мл толуолу. Потім додають 2,07г (18ммолів) Nгідроксісукциніміду, 0,73г (6ммолів) 4діметиламінпіридіну, 13,3г (18ммолів) сухого третбутанолу та 1,82г (18ммолів) триетиламіну. С уміш доводять до температури флегми й залишають на ніч. Охолоджують реакційну суміш та додають ЕtOАс. Одержаний розчин обробляють відповідно 10% водним розчином цитринової кислоти та розсолом, після чого органічну фракцію сушать над MgSO4 . Після видалення осушного агента та розчинника вакуум-випарюванням залишається коричнева олія. Кристалізація з діетилетеру/гексану дає 4,4г (42%) цільового моноефіру. Етап iv (схема С.1) Реакцію виконують за методикою, описаною у [Synthesis (2000), p.1369-1371]. Монотретбутилефір бур штинової кислоти метилюють у позиції 2 шляхом реакції з діізопропиламідом літію та метилйодидом у те трагідрофурані при -78°С. Вихід монотретбутилефір у 2-метилбурштинової кислоти становить до 60%. Етап ν (схема С.1) При перемішуванні 1,8г (9,8ммоля) монотретбутиле фіру 2-метилбурштинової кислоти з етапу iv розчинюють у 45мл сухого СН 2СІ2, після чого доводять розчин до 4°С. До одержаного розчину до 11 77775 12 дають 0,9г (6,4ммоля) 1-гідроксі-7-азаУ атмосфері азоту 2,17г (5,3ммоля) проміжної бензтриазолу та 4,0г (15ммолів) 2-хлор-1,3сполуки з етапу vi та 4,7мл (27ммолів, 5,1г-екв) діметилімідазолінійгексафторфосфату. діізопропилетиламіну розчинюють у 25мл сухого Наступне додання 4,1г (14ммолів) III не підвиСН2СІ2; одержаний розчин доводять до 4°С при щує температури. Реакцію залишають продовжуперемішуванні. Після того додають 0,42г ватися всю ніч при кімнатній температурі. До реак(3,1ммоля) 1-гідроксі-7-аза-бензтриазолу та 1,85г ційної суміші додають біля 3г силікагелю (SiO2) і (6,6ммолів) 2-хлор-1,3концентрують її у вакуумі. Одержаний порошок діметилімідазолінійгексафторфосфату. Далі до вміщують на суху колонку (SiO2) й виконують реакційної суміші додають 1,0г (6,6ммолів) 2елюювання (елюент ЕtOАс/петролейний етер 1:4). аміноадамантину і дають реагувати 1 годину при Частину колонки, що містить продукт, збирають і кімнатній температурі. вміщують до МеОН. Одержану суспензію фільтДо реакційної суміші додають біля 4г кремнерують, осад промивають один раз МеОН. Фракції зему та концентрують у вакуумі. Одержаний поМеОН зливають і концентрують у вакуумі, осад рошок вміщують на суху колонку (SiO2) й виконувміщують до СН 2СІ2, одержаний розчин сушать ють елюювання (елюент ЕtOАс/петролейний етер над MgSO4 . Після видалення осушного агента та 1:2). Частини колонки, що містять діастереомерні розчинника випарюванням у вакуумі одержують 3г рацемати, збирають нарізно і вміщують до МеОН. (вихід 66%) потрібної проміжної сполуки. Одержані дві суспензії фільтрують нарізно, кожний Етап vi (схема С.1) з двох осадів промивають МеОН один раз. Для Гідроліз третбутилефір у проміжної сполуки з кожного діастереомерного рацемату зливають етапу ν виконують наступним чином: 3г (6,4ммоля) відповідні фракції МеОН, концентрують у вакуумі, третбутилефіру розчинюють у 30мл сухого СН 2СІ2, кожний осад окремо вміщують до СН 2СІ2 і сушать після чого додають по краплинах 10мл трифторовідповідні розчини над MgSO4. Після видалення цтової кислоти. За 2 години реакція закінчується, осушного агента та розчинника у вакуумі одержуреакційну суміш концентрують у вакуумі, осад розють дві тверді фази, кожна з яких містить один з чиняють у невеличкій кількості діетилетеру, вміможливих діастереомерних рацематів: 1,08г С8 щують до верху короткої колонки (сухий SiO2) та (вихід 37%) - активний рацемат, точка топлення елююють діетилетером. Елюат, що містить про238-240°С, та 1,09г (вихід 37%) другого, фармакодукт, концентрують у вакуумі, осад перемішують логічно не активного рацемату, точка топлення усю ніч у петролейному етері. Кристали відфільт125-130°С (у таблиці С не зазначений). ровують, сушать на відкритому повітрі й одержуСполуки за таблицею С одержують аналогічють 2,1г (ви хід 80%) потрібної проміжної сполуки. ним чином: Етап vii (схема С.1) Таблиця С R3, R4, R5, R 5' , S2, S4 = Η Х=С, Y=N Сполука піразолідин С1 III С2 II C3 II С4 І С5 І С6 VII С7 VII С8 III С9 IX R Η Η Η Η Η Η Η Me Η Приклад 4 Етап iiі (схема D.1) У атмосфері азоту 0,92г (4,9ммоля) проміжної сполуки з етапу іі та 4,4мл (25ммолів, 5,1г-екв) діізопропилетиламіну розчинюють у 15мл сухого СН2СІ2. Одержаний розчин доводять при перемішуванні до 4°С. Потім додають 0,45г (3,3ммоля) 1гідроксі-7-аза-бензтриазолу та 2,1г (7,5ммолів) 2хлор-1,3-діметилімідазолінійгексафторфосфату. Далі до реакційної суміші додають 1,08г (7,2ммоля) 2-аміноадамантину і дають реагувати 1 годину при кімнатній температурі. Цю реакційну суміш використовують у наступному етапі iv. Етап iv (схема D.1) До реакційної суміші з етапу ііі додають при перемішуванні 45мл сухого СН 2СІ2 та 11мл R7 Η Η Η Η Η Η Η Η Η YR7 A 8 8 7 8 7 7 8 8 8 Точка топлення 210-2 90-4 230-2 160-4 198-202 208-210 215-7 238-240 147-150 (143ммоля) трифтороцтової кислоти. Перемішування триває 24 години. Реакційну суміш концентрують у вакуумі, осад розчиняють у невеличкій кількості діетилетеру, вміщують до верху короткої колонки (сухий SiO2) та елююють діетилетером. Елюат, що містить продукт, концентрують у вакуумі й одержують 0,87г (вихід 67% у два прийоми) потрібної проміжної сполуки. Етап ν (схема D.1) 0,87г (3,28ммоля) моноаміду метилбурштинової кислоти з етапу iv розчинюють у 15мл сухого СН2СІ2 і доводять розчин до 4°С. До розчину додають 0,3г (2,2ммоля) 1-гідроксі-7-азабензтриазолу та 1,40г (5,0ммолів) 2-хлор-1,3діметилімідазолінійгексафторфос-фату. Наступне додання 1,33г (4,80ммолів) II не підвищує темпе 13 77775 14 ратури. Реакцію залишають продовжуватися всю відповідні розчини над MgSO4. Після видалення ніч при кімнатній температурі. До реакційної суміші осушного агента та розчинника у вакуумі одержудодають біля 3г силікагелю (SiO2) і концентрують її ють дві тверді фази, кожна з яких містить один з у вакуумі. Одержаний порошок вміщують на суху можливих діастереомерних рацематів: 0,31г (вихід колонку (SiO2) й виконують елюювання (елюент 18%) неактивного рацемату (у таблиці D не зазнаЕtOАс/петролейний етер 1:1). чений), який плавиться при 90-95°С, твердне при Частини колонки, що містять діастереомерні 130°С і знову плавиться при 160-165°С, та 0,40г рацемати, збирають нарізно і вміщують до МеОН. (вихід 23%) активного рацемату D1, який плавитьОдержані дві суспензії фільтрують нарізно, кожний ся при 80-82°С, твердне при 100°С і знову плаз двох осадів промивають МеОН один раз. Для виться при 125-128°С. кожного діастереомерного рацемату зливають Сполуки за таблицею D одержують аналогічвідповідні фракції МеОН, концентрують у вакуумі, ним чином: кожний осад окремо вміщують до СН 2СІ2 і сушать Таблиця D R3, R4, R5' , S2 , S4 = Η X=C, Y=N Сполука піразолідин D1 II D2 II D3 II D4 II D5 II D6 II R5 Me nBut nBut iBut Et Et Приклад 5 2,3-діарилпіразолідини l-Х, що слугували вихідними матеріалами у вищенаведених прикладах 1-4, одержують наступним способом: Етап і (схема 1) Суміш 16,9мл оцтової кислоти та 2,3мл води охолоджують крижаною водою, після чого обережно додають 6,8мл концентрованої сірчаної кислоти. До охолодженого розчину додають з енергійним перемішуванням у атмосфері азоту 13,3г (82ммоля) 2-фторфенілгідразину малими порціями. До одержаного розчину додають порціями суміш 10,0г (82ммоля) 2-фторстиролу та 2,46г (82ммоля) параформальдегіду, утримуючи темпе R6 Η Η Η Η Η Η YR7 A 8 8 8 8 8 8 примітка діастереомери діастереомери Точка топлення 80-2/125-8 80-1/150-5 210-2 155-8 90-2/125-8 90-2/155-7 ратур у нижче 25°С. Реакція займає тривалий час. Енергійне перемішування продовжують усю ніч при кімнатній температурі. При охолодженні доливають 50мл води, після чого двічі провадять екстракцію діетилтером. Водну фракцію підлужують 50% водним розчином NaOH, після чого двічі екстрагують діетилетером. Етерну фракцію тричі промивають водою та один раз розсолом, а за потреби сушать над MgSO 4. Після фільтрації осушного агента та видалення розчинника у вакуумі одержують 16г (вихід 75%) сирої сиропоподібної олії. Олію не очищують, а зберігають у атмосфері азоту при -20°С, щоб запобігти окисленню піролідинового ядра. 15 Комп’ютерна в ерстка О. Гапоненко 77775 Підписне 16 Тираж 26 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійською2,3 diaril-pyparazolidine derivatives inhibiting enzymes degrading neurotensin, pharmaceutical conmposition
Автори англійськоюVeenstra Roelof W., Lange Josephus H. M., Pras-Raves Maria L., Kruse, Cornelis G., Kruse Cornelis G., van Stuivenberg Herman H., Tuinstra Tinka, Keizer Hiskias G.
Назва патенту російськоюПроизводные 2,3-диарилпиразолидина с активностью в отношении нейротензина, способ их получения, фармацевтическая композиция на их основе
Автори російськоюФенстра Рулоф В., Ланге Йозефус Г.М., Прас-Равес Мария Л., Крузе Корнелис Г., ван Стёйвенберг Герман Г., Тёйнстра Тынка, Кейзер Гискиас Г.
МПК / Мітки
МПК: C07D 401/04, A61P 25/22, A61K 31/415, C07D 231/04, A61P 25/00, A61P 25/24, A61P 25/16, A61P 43/00
Мітки: композиція, нейротензину, одержання, фармацевтична, активністю, спосіб, 2,3-діарилпіразолідину, основі, похідні
Код посилання
<a href="https://ua.patents.su/8-77775-pokhidni-23-diarilpirazolidinu-z-aktivnistyu-shhodo-nejjrotenzinu-sposib-kh-oderzhannya-farmacevtichna-kompoziciya-na-kh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Похідні 2,3-діарилпіразолідину з активністю щодо нейротензину, спосіб їх одержання, фармацевтична композиція на їх основі</a>
Похідні 4,5-дигідро-1н-піразолу з антагоністичною активністю щодо рецепторів канабіс-1, спосіб їх одержання, фармацевтична композиція та спосіб лікування

Номер патенту: 74066
Опубліковано: 17.10.2005
Автори: Гогендорн Ян, Ланге Йозефус, Крузе Корнеліс, Тіпкер Якобус
МПК: A61P 25/08, A61P 25/22, A61P 1/00, C07D 231/06, A61P 3/04, A61K 31/4439, A61K 31/415, A61P 25/04, A61P 25/14, C07D 231/08, A61P 25/24, A61P 25/16, A61P 9/10, A61P 25/18, A61P 43/00, A61P 25/00, C07D 401/04, A61P 25/28
Мітки: похідні, 4,5-дигідро-1н-піразолу, спосіб, фармацевтична, рецепторів, активністю, лікування, антагоністичною, канабіс-1, одержання, композиція
Формула / Реферат:
1. Енантіомер, що має S-конфігурацію у 4-й позиції його 4,5-дигідропіразольного кільця сполуки формули (І), (I)де- R та R1 однакові або різні й являють собою 3-піридил або 4-піридил, або феніл, що може бути заміщений галогеном або метоксигрупою,- R2 та R3 однакові або різні й являють собою водень, алкіл (1-3 С) або диметиламін,- R4 являє...
Похідні 4,5-дигідро-1н-піразолу з сильнодіючою антагоністичною активністю щодо рецепторів св-1, спосіб їх одержання (варіанти), проміжні сполуки, фармацевтична композиція та спосіб її одержання

Номер патенту: 77441
Опубліковано: 15.12.2006
Автори: Герреманс Арнольдус Г.Й., Тіпкер Якобус, Ланге Йозефус Х.М., Крузе Корнеліс Г., ван Стюйвенберг Герман Г.
МПК: A61P 43/00, A61P 27/06, A61K 31/4155, A61P 9/10, A61K 31/194, A61P 31/12, A61P 3/04, A61P 25/00, A61K 31/4439, A61P 25/14, A61P 25/30, A61K 31/551, A61P 25/02, A61P 9/00, A61P 25/24, A61P 25/16, A61P 31/04, A61K 31/541, C07D 409/04, A61P 3/10, A61P 1/12, A61P 35/00, A61P 25/28, A61K 31/4545, A61K 31/496, C07D 403/12, A61P 1/14, A61P 1/04, C07D 401/12, A61K 31/454, A61P 11/06, A61K 31/55, A61K 31/5377, A61P 1/08, A61K 31/415, C07D 231/06, A61K 31/4725, A61P 25/22, A61P 29/00, C07D 401/04, A61P 25/18, A61P 25/08, C07D 403/06
Мітки: св-1, сильнодіючою, 4,5-дигідро-1н-піразолу, спосіб, похідні, проміжні, композиція, антагоністичною, активністю, одержання, варіанти, фармацевтична, сполуки, рецепторів
Формула / Реферат:
1. Сполуки загальної формули (Iа) або (Іb) (Iа), , (Іb)де- R та R1 незалежно являють собою феніл, тієніл або піриділ, групи яких можуть бути заміщені 1, 2 або 3 однаковими або різними замісниками Y з групи С1-3-алкіл або алкокси, гідрокси, галоген, трифторметил, трифторметилтіо,...
Оксимпохідні піперазину з антагоністичною активністю до рецептора nk-1, їх застосування, фармацевтична композиція на їх основі, спосіб її одержання та спосіб одержання проміжних сполук

Номер патенту: 75425
Опубліковано: 17.04.2006
Автори: Тульп Мартінус Т.М., МакКрірі Ендрю К., Герреманс Арнольдус Г.Й., Коолен Гейн К.А.К., ван Маарсевейн Ян Г., ван ден Гоогенбанд Адріанус, ван Шарренбург Густаф Й.М., Івема Баккер Воутер І.
МПК: C07D 241/00, A61K 31/495, C07D 403/14, C07D 403/06, C07D 209/00, C07D 265/00, A61P 1/00, C07D 413/14
Мітки: застосування, сполук, нк-1, спосіб, рецептора, проміжних, одержання, композиція, антагоністичною, оксимпохідні, піперазину, фармацевтична, активністю, основі
Формула / Реферат:
1. Сполуки загальної формули (1), (1)де:Х означає феніл чи піридил, заміщений 1 або 2 замісниками з групи СН3, СF3, ОСН3, галоген, ціано та 5-СF3-тетразол-1-іл;Y означає 2- або 3-індоліл, феніл, 7-азоіндол-3-іл або 3-індазоліл, 2-нафтіл, 3-бензо[b]тіофеніл або 2-бензофураніл, причому ці групи можуть бути заміщеними одним або більше галогеном...
Сульфонамідні похідні, спосіб їх одержання та фармацевтична композиція на їх основі

Номер патенту: 63990
Опубліковано: 16.02.2004
Автори: Макдауелл Роберт, Кнолле Йохен, Бодарі Сара Кетрін, Катбертсон Роберт Ендрю, Вілл Девід Вільям, Карніато Дені, Шойнеманн Карлхайнц, Гурвест Жан-Франсуа, Пейман Ануширван, Гадек Томас
МПК: A61P 43/00, C07D 401/12, A61P 29/00, A61P 13/12, C07D 409/12, A61P 9/00, A61P 19/10, A61K 31/506, C07C 311/19, C07D 239/16, A61K 31/505, A61K 31/197, A61P 27/02, A61P 9/10, A61P 35/00
Мітки: фармацевтична, похідні, основі, одержання, композиція, сульфонамідні, спосіб
Формула / Реферат:
1. Сульфонамідні похідні формули І:, (I)в якійR1 і R2, незалежно один від одного, означають водень чи (С1-С6)-алкіл, незаміщений чи заміщений радикалом R3,чи в якій радикали R1 і R2, спільно, являють собою насичений чи ненасичений двовалентний (С2-С9)-алкіленовий радикал, що є незаміщеним чи заміщеним однією чи більше групами з числа наступних:...
Похідні 4,5-дигідро-1н-піразолу, що виявляють антагоністичну активність щодо св1, спосіб їх одержання, фармацевтична композиція та спосіб її виготовлення, спосіб лікування захворювань (варіанти)

Номер патенту: 74367
Опубліковано: 15.12.2005
Автори: ван Фліт Бернард Ж., Тульп Мартінус Т.М., Крузе Корнеліс Г., Тіпкер Якобус, Ланге Йозефус Г.М.
МПК: A61P 9/00, C07D 401/12, A61P 1/04, C07D 401/04, A61P 43/00, A61P 25/28, A61P 25/18, A61K 31/4439, A61P 25/14, A61P 25/04, A61P 25/24, C07D 231/06, A61P 25/22, C07D 231/08, A61P 25/16, C07D 409/04, A61P 25/08, A61P 9/10, A61P 3/04, A61K 31/415
Мітки: активність, св1, фармацевтична, виявляють, 4,5-дигідро-1н-піразолу, спосіб, антагоністичну, лікування, одержання, варіанти, похідні, композиція, виготовлення, захворювань
Формула / Реферат:
1. Похідні 4,5-дигідро-1Н-піразолу формули (І), (І)деR та R1 є однакові або різні групи, представлені фенілом, тієнілом або піридилом, які можуть бути заміщені 1, 2 або 3 замісниками Y, які можуть бути однаковими або різними, вибраними з: (С1-3)-алкіл- або алкокси, гідрокси, галоген, трифторметил, трифторметилтіо, трифторметокси, нітро, аміно, моно-...
Попередній патент: Розсадосадильна машина
Наступний патент: Сільськогосподарська машина на самохідному шасі з кількома робочими органами
Випадковий патент: Спосіб оцінки ступеня тяжкості нефропатії вагітних