Спосіб отримання похідних 2-азадигідроксибіцикло[2.2.1]гептану і їх солей l-винної кислоти
Номер патенту: 52702
Опубліковано: 15.01.2003
Автори: Лєон Патрік, Дюран Т'єррі, Пауерз Меттью, Ларго Дені, О'брайєн Майкл











Формула / Реферат
1. Спосіб отримання похідної 2-азадигідроксибіцикло[2.2.1]гептану формули:
 або
або  ,
,
де * позначає R-хіральність, *' позначає S-хіральність, R представляє водень або, відповідно, групу формули
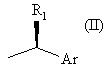 або
або 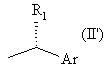 ,
,
де R1 позначає алкіл і Аr представляє необов'язково заміщений арил, що включає бісгідроксилювання похідної біцикло[2.2.1]гептену формули
 або
або 
при від приблизно 0,1 мол. % до приблизно 5 мол. осмату металу або від приблизно 0,06 мол. % до приблизно 0,07 мол. тетроксиду осмію й окислювача, спроможного регенерувати тетроксид осмію.
2. Спосіб по п. 1, де R являє собою групу формули
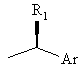 або
або 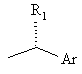 .
.
3. Спосіб по п. 2, де R1 представляє метил або етил і Аr представляє необов'язково заміщений феніл, що, коли заміщений, то заміщений одним або декількома метилом або метокси.
4. Спосіб по п. 3, де R1 представляє метил і Аr представляє феніл.
5. Спосіб по п. 1, де бісгідроксилювання здійснюють із використанням тетроксиду осмію в кількості від приблизно 0,06 мол. % до приблизно 0,07 мол. %.
6. Спосіб по п. 5, де тетроксид осмію присутній у кількості приблизно 0,06 мол. %.
7. Спосіб по п. 1, де бісгідроксилювання здійснюють, використовуючи осмат металу в кількості від приблизно 0,1 мол. % до приблизно 5 мол. %.
8. Спосіб по п. 7, де осмат металу присутній у кількості від приблизно 0,2 мол. % до приблизно 0,5 мол. %.
9. Спосіб по п. 1, де як осмат металу використовують К2ОsО4.2Н2О.
10. Спосіб по п. 1, де як окислювач, спроможний регенерувати тетроксид осмію, використовують N- метилморфоліноксид.
11. Спосіб отримання солі L-винної кислоти (1R) діастереомера похідної 2-азадигідроксибіцикло[2.2.1]гептану по п. 1, який відрізняється тим, що (1R) діастереомер похідної 2-азадигідроксибицикло[2.2.1]гептану по п. 1, де R є групою формули
,
обробляють L-винною кислотою.
12. Спосіб по п. 11, де R1 представляє метил і Аr представляє феніл.
13. Спосіб по п. 11, в якому додатково отримують L-виннокислу сіль (1R) діастереомеру похідної 2-азадигідроксибіцикло[2.2.1]гептану, переважно в енантіомерно очищеному стані в присутності (1S) діастереомеру похідної 2-азадигідроксибіцикло[2.2.1]гептану.
14. Спосіб по п. 11, де обробку здійснюють у суміші водно-органічного розчинника.
15. Спосіб по п. 14, де органічним розчинником є ізопропанол.
16. Спосіб по п. 14, де обробку здійснюють у суміші вода-ізопропанол, що має об'ємне співвідношення компонентів від приблизно 30:70 до приблизно 15:85.
17. Спосіб по п. 16, де об'ємне співвідношення складає приблизно 25:75.
18. Сполука формули
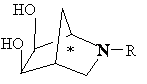 • L-винна кислота,
• L-винна кислота,
де R визначений у п. 11.
19. Спосіб отримання сполуки формули
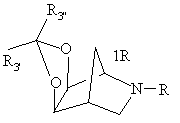 ,
,
де R визначений у п. 1, Rз' і Rз" представляють водень, алкіл або феніл, або Rз' і Rз", узяті разом з атомом вуглецю, до якого вони приєднані, утворюють циклоалкіл, що включає кислотно-каталізуючу ацеталізацію або кеталізацію сполуки формули
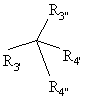 ,
,
де R4' і R4" представляють алкокси або, узяті разом з атомом вуглецю, до якого вони приєднані, утворюють карбоніл, із (1R) діастереомером похідної 2-азадигідроксибіцикло-[2.2.1]гептану по п. 1 або його сіллю в ізопропанолі.
20. Спосіб по 19, де R4' і R4" представляють метокси і Rз' і Rз" представляють метил.
21. Спосіб по п.19, де кислотний каталіз здійснюють, використовуючи трифтороцтову кислоту.
22. Спосіб по п. 19, де R представляє групу формули
.
23. Спосіб по п. 22, де R1 представляє метил і Аr представляє феніл.
24. Спосіб по п. 19, де (1R) діастереомер похідної 2-азадигідроксибіцикло[2.2.1]гептану є у вигляді його солі L-винної кислоти.
25. Спосіб отримання похідної лактаму формули
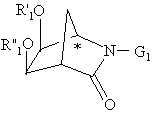 ,
,
де R'1 і R"1 незалежно представляють ацил або ароїл або, узяті разом, утворюють необов'язково заміщений метилен і G1 представляє водень або амінозахисну групу, в якому окислюють похідну біс-O-захищеного 1R-2-азадигідроксибіцикло[2.2.1]гептану формули
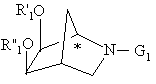
за допомогою RuO2 або його гідрату, узятих у кількості від приблизно 0,1 мол. % до приблизно 1 мол. %, в присутності приблизно 3 еквівалентів окислювача з утворенням похідної лактаму з енантіомерним надлишком більшим або рівним 95 %.
26. Спосіб по п. 25, де RuO2 беруть у кількості приблизно 0,5 мол. %.
27. Спосіб по п. 25, де похідну лактаму одержують з енантіомерним надлишком більшим або рівним 99 %.
Текст
1 Спосіб отримання похідної 2азадипдроксибіцикло[2 2 1]гептану формули НО НО і\ НО но U W ^ T _ P де або * позначає R-хіральність, 00 l / _ j ^ J i * / N-R (Г) *' позначає S хіральність, R представляє водень або, ВІДПОВІДНО, групу формули (П) (ІГ) "Аг R, "Аг або де Ri позначає алкіл і Аг представляє необов'язково заміщений арил, що включає біспдроксилювання похідної біцикло[2 2 1]гептену формули (Ш) або при від приблизно 0,1 мол % до приблизно 5мол осмату металу або від приблизно О.Обмол % до приблизно 0,07мол тетроксиду осмію й окислювача, спроможного регенерувати тетроксид ОСМІЮ 2 Спосіб по п 1, де R являє собою групу формули або 3 Спосіб по п 2, де Ri представляє метил або етил і Аг представляє необов'язково заміщений феніл, що, коли заміщений, то заміщений одним або декількома метилом або метокси 4 Спосіб по п 3, де Ri представляє метил і Аг представляє феніл 5 Спосіб по п 1 , де біспдроксилювання здійснюють із використанням тетроксиду осмію в КІЛЬКОСТІ від приблизно О.Обмол % до приблизно 0,07мол % 6 Спосіб по п 5, де тетроксид осмію присутній у КІЛЬКОСТІ приблизно О.Обмол % 7 Спосіб по п 1, де біспдроксилювання здійснюють, використовуючи осмат металу в КІЛЬКОСТІ від приблизно 0 , 1 м о л % до приблизно 5мол % 8 Спосіб по п 7, де осмат металу присутній у КІЛЬКОСТІ від приблизно 0,2мол % до приблизно 0,5мол % 9 Спосіб по п 1, де як осмат металу використовують K 2 O S O 4 2H 2 O 10 Спосіб по п 1 , де як окислювач, спроможний регенерувати тетроксид осмію, використовують Nметилморфоліноксид 11 Спосіб отримання солі L-винноі кислоти (1R) діастереомера похідної 2азадипдроксибіцикло[2 2 1]гептану по п 1, який відрізняється тим, що (1R) діастереомер похідної 2-азадипдроксибицикло[2 2 1]гептану по п 1 , де R є групою формули R, Аг обробляють L-винною кислотою 12 Спосіб по п 11, де Ri представляє метил і Аг представляє феніл 13 Спосіб поп 11, в якому додатково отримують L-виннокислу сіль (1R) діастереомеру похідної 2азадипдроксибіцикло[2 2 1]гептану, переважно в енантюмерно очищеному стані в присутності (1S) О о ю діастереомеру похідної 2азадипдроксибіцикло[2 2 1]гептану 14 Спосіб по п 11, де обробку здійснюють у суміші водно-органічного розчинника 15 Спосіб по п 14, де органічним розчинником є ізопропанол 16 Спосіб по п 14, де обробку здійснюють у суміші вода-ізопропанол, що має об'ємне співвідношення компонентів від приблизно ЗО 70 до приблизно 15 85 17 Спосіб по п 16, де об'ємне співвідношення складає приблизно 25 75 18 Сполука формули 52702 4 21 Спосіб по п 19, де кислотний каталіз здійснюють, використовуючи трифтороцтову кислоту 22 Спосіб по п 19, де R представляє групу формули N-R • L-винна кислота, де R визначений у п 11 19 Спосіб отримання сполуки формули N-R де R визначений у п 1, R3 і R3 представляють водень, алкіл або феніл, або R3 і R3, узяті разом з атомом вуглецю, до якого вони приєднані, утворюють циклоалкіл, що включає кислотнокаталізуючу ацеталізацію або кеталізацію сполуки формули де R4 і R4 представляють алкокси або, узяті разом з атомом вуглецю, до якого вони приєднані, утворюють карбоніл, із (1R) діастереомером похідної 2азадипдроксибіцикло-[2 2 1]гептану по п 1 або його сіллю в ізопропанолі 20 Спосіб по 19, де R4 і R4 представляють метокси і R3 і R3 представляють метил Даний винахід відноситься до способу отримання похідної 2-азадипдроксибіцикло [2 2 1] гептану Винахід відноситься також до L - виннокислої солі (1R) діастереомера похідної 2 - азадипдроксибіцикло [2 2 1] гептану і її одержанню Крім того, винахід відноситься до способу біс О - захисту (1R) діастереомера похідної 2 - азадипдроксибіцикло [2 2 1] гептану і способу окислювання похідних (1R) діастереомера похідної 2- азадипдроксибіцикло [2 2 1] гептану до ВІДПОВІДНОГО ПОХІДНОЇ лакта му У патенті США №5284769 описується, що по Аг представляє метил і Аг 23 Спосіб по п 22, де представляє феніл 24 Спосіб по п 19, де (1R) діастереомер похідної 2-азадипдроксибіцикло[2 2 1]гептану є у вигляді його солі L-винноі кислоти 25 Спосіб отримання похідної лактаму формули R\O N-G, де R'i і R"i незалежно представляють ацил або ароїл або, узяті разом, утворюють необов'язково заміщений метилен і Gi представляє водень або амінозахисну групу, в якому окислюють похідну біс-О-захищеного 1R-2азадипдроксибіцикло[2 2 1]гептану формули N-G, за допомогою RuCb або його гідрату, узятих у КІЛЬКОСТІ від приблизно 0,1мол% до приблизно 1мол%, в присутності приблизно 3 еквівалентів окислювача з утворенням похідної лактаму з енантюмерним надлишком більшим або рівним 95 % 26 Спосіб по п 25, де RuCb беруть у КІЛЬКОСТІ приблизно 0,5мол % 27 Спосіб по п 25, де похідну лактаму одержують з енантюмерним надлишком більшим або рівним 99% хідна лактаму, що охоплюється похідної лактаму, отриманим ВІДПОВІДНО до винаходу, може використовуватися як синтон для отримання фармацевтичне активних способів J Chen et al , Tet Lett, ЗО 5543 (1989) описують похідне лактаму, що охоплюється похідним лактаму, отриманим ВІДПОВІДНО до винаходу, як використовуване для отримання речовини, що є агоністом аденозину С К -F Chui, Syn Comm , 26(3), 577 (1996) описує поділ суміші діастереомерів похідних біциклогептенамину формули (і) і (і і) 52702 (0 + ' (") використовуючи L - дибензоілвинну кислоту, наприклад методом фракційної кристалізації У Chui не описані засоби поділу бісгідроксилированих продуктів діастереомірної суміші S J С Taylor et al , Tetrahedron Asymmetry, 4(6), 1117 (1993) описують ферментативний поділ лактаму формули (Ні) О, (їм) с отриманням енантиомерів формули (iv) і (v) .О Н (IV) І (V) SJ С Taylor et al не описують ніяких способів поділу бісгідроксилированих продуктів лактаму (III) У патенті США №5284769 описаний ферментативний поділ лактаму формули (vi) О. (IV) с отриманням енантиомерів лактаму У патенті США №5284769 не описані НІЯКІ засоби поділу бісгідроксилированих продуктів лактаму (vi) Спосіб ВІДПОВІДНО до винаходу відноситься до отримання похідної 2 - азадипдроксибіцикло [2 2 1] гептану формули де * означає хіральність R, *' позначає хіральність S, R представляє водень або, ВІДПОВІДНО, групу формули R. R, (II) або (IIі) де R1 представляє алкіл і Аг представляє необов'язково заміщений арил, що полягає в біспдроксилируванні похідної біцикло [2 2 1 ] гептену формули ^ , де (III) або (ІІГ) *' і R зазначені вище, в присутності від приблизно 0,1 мол % до приблизно 5мол % осмату металу чи від приблизно О.Обмол до приблизно 0,07мол % тетроксиду осмію та окислювача, який може регенерувати тетроксид осмію Даний винахід відноситься також до обробки (1R) діастереомеру похідної 2азадипдроксибіцикло [2 2 1] гептану (І), де R представляє групу формули II, L - винною кислотою і до нього L - виннокислої солі Крім того, винахід відноситься до застосування (1R) діастереомера похідної 2 азадипдроксибіцикло [2 2 1] гептану або солі його при що каталізує кислотою реакції ацеталізацм або кеталізацм, що призводить до захисту дипдрокси частин його в ізопропанолі Крім того, винахід відноситься до окислювання біс О - захищеного похідної (1R) діастереомеру похідної 2азадипдроксибіцикло [2 2 1] гептану у ВІДПОВІДНОГО похідної лактаму при від приблизно 0,01 мол % до приблизно 1мол % RuO2 або його гідрату, приблизно 3 еквівалентам окислювача, з отриманням похідної лактаму з енантиомірним надлишком ("еі"), більш або дорівнює 95% Варто врахувати, що, як тут використовується, слідуючи терміни, використовувані вище і протягом всього опису, якщо не обговорено особливо, мають такі значення "Алкіл" означає аліфатичну вуглеводневу групу, що може бути прямою або розгалуженою, що має від 1 до 4 атомів вуглецю Приклади алкільних груп включають метил, етил, ізопропіл і третбутил "Необов'язково заміщений метилен" означає СНг - або таку групу, де атоми водню замінені, незалежно, одною або двома групами, що можуть бути однаковими або різноманітними, обраними з алкілу або фенілу, або замінені таким чином, що разом із вуглецевим атомом метилену утворюють циклоалкіл "Арил" означає необов'язково заміщений феніл або необов'язково заміщений а або а - нафтил Заміщений арил заміщений одним або декількома заступниками арильної групи, що можуть бути однаковими або різними і включають галоген, алкіл, алкокси або нітро "Алкокси" означає алкіл - О - групу, де алкільна група описана вище Приклади алкоксігрупи включають метокси, етокси, ізопропокси і трет бутокси "Циклоалкіл" означає аліфатичне циклічне кільце з 5-6 атомів вуглецю Прикладом циклоалкільної групи є циклогексил "Ацил" означає алкіл - СО - групу, де алкільна група описана вище Приклади ацильних груп включають ацетил і пропаноїл "Ароїл" означає арил - СО - групу, де арильна група описана вище Прикладом арильної групи є бензоїл "Галоген" означає фтор, хлор, бром або йод Кращий фтор і хлор "Окислювач, спроможний регенерувати тетроксид осмію," означає окислювач, що буде окисляти осмій в осматі металу (Os+6) до тетроксиду осмію (Os+6) або знову окисляти тетроксид осмію, що відновлюється в процесі біспдроксилірування в тетроксид осмію (Os+ ) Приклади окислювачів, 52702 спроможних регенерувати тетроксид осмію, включають N-N-метилморфолиноксид або триетиламіноксид і ферицианід калію Кз[Ре(СІЧ)б], кращим є N-N-метилморфолиноксид "Осмат металу" означає сіль з'єднання, утворену М п+ , катіоном металу, де п дорівнює 1 або 2, і комплексом аніона оксиду осмію [OSO4]2, або гідрати солі Кращими осматами металів є осмати лужних або лужноземельних металів, включаючи осмати натрію, калію, рубідію, цезію, кальцію і барію, більш кращий K2OSO4- 2H2O Приклади способів, використовуваних для отримання осматів металів, описані Б Н Івановим-Єміним та ш , Ж неорг хім , 31(5) 1238 (1986), Н С Jewiss, J C S Dalton Trans , 199 (1985), Б Н Івановим-Єміним та ш , Ж неорг хім, 29(4), 1241 (1984), Б Н ІвановимЄміним та ш,, Ж неорг хім , 28(5) 1246 (1983) "Його сіль" означає сполучення з основною частиною, нейтралізованою кислотою з утворенням відповідної солі приєднання кислоти Кислоти, що можна використовувати для отримання солей приєднання кислот, включають, переважно, кислоти, що дають при взаємодії з вільною підставою фармацевтично прийнятної солі, тобто солі, аніони яких нетоксичні для пацієнта, і тому наступне використання солі приєднання кислоти не обмежується через прояв ХІМІЧНО реакційної активності солі Сіль приєднання кислоти може використовуватися, наприклад, як джерело для регенерації вихідної основи шляхом обробки основою, таким як луг, із метою очищення і/або ідентифікації або з метою взаємоперетворення в іншу сіль приєднання кислоти шляхом іонообмінних методів Приклади солей приєднання кислот включають охоплюючи наступні кислоти мінеральні кислоти, такі як бромистоводнева кислота, хлористоводнева кислота, сірчана кислота, фосфорна кислота і сульфамшова кислота, і органічні кислоти, такі як оцтова кислота, лимонна кислота, молочна кислота, винна кислота, дибензоілвинка кислота, малонова кислота, бурштинова кислота, 2,3 - диметиоксиянтарна кислота, метансульфонова кислота, етансульфонова кислота, бензол сульфокислота, п - толуол сул ьфон о ва кислота, циклогексилсульфамінова кислота, хінна кислота і тому подібне Конкретним здійсненням способу біспдроксилірування ВІДПОВІДНО до винаходу є такий, коли R представляє групу формули II або IIі Кращим здійсненням способу біспдроксилірування ВІДПОВІДНО до винаходу є такий, коли Ri представляє метил або етил і Аг представляє необов'язково заміщений феніл, що, коли заміщений, те заміщений одним або декількома метилом або мето кс и Більш кращим варіантом здійснення способу біспдроксилірування ВІДПОВІДНО ДО винаходу є та кий, коли Ri представляє метил і Аг представляє феніл По кращому варіанту здійснення біспдроксилірування використовується тетроксид осмію в КІЛЬКОСТІ від приблизно О.Обмол % до приблизно 0,07мол % більш переважно приблизно О.Обмол % У іншому кращому варіанті здійснення біспд 8 роксилірування використовують осмат металу в КІЛЬКОСТІ від приблизно 0,1 мол % до приблизно 5мол , більш переважно від приблизно 0,2мол % до приблизно 0,5мол % Ще у одному кращому варіанті здійснення біспдроксилірування в якості осмату металу використовують осмат лужного або лужноземельного металу, більш кращий K2OSO4 2Й2О Конкретним прикладом отримання солі L винної кислоти (1R) діастереомера похідної 2 азадипдроксибіцикло [2 2 1] гептану, тобто N—R • сіль L-винної кислоти ВІДПОВІДНО до винаходу є такий, коли R являє собою групу формули II Кращим варіантом здійснення способу отримання солі L-винноі кислоти (1R) діастереомера похідної 2 - азадипдроксибіцикло [2 2 1] гептану є такий, коли R1 являє собою метил і Аг представляє феніл Інший конкретний варіант здійснення винаходу полягає в одержанні солі L -винної кислоти (1R) діастереомеру похідної 2-азадипдроксибіцикло [2 2 1] гептану, по суті, у енантиомірно чистому виді при (1S) діастереомера похідної 2 - азадипдроксибіцикло [2 2 1] гептану Кращим варіантом отримання солі L - винної кислоти (1R) діастереомера похідної 2 - азадипдроксибіцикло [2 2 1] гептану, ВІДПОВІДНО ДО винаходу, є такий, коли отримання прпроводять у суміші водно-органічного розчинника Більш кращим варіантом отримання солі L винної кислоти (1R) діастереомера похідної 2 азадипдроксибіцикло [2 2 1] гептану є такий, коли органічним розчинником є ізопропанол (ІП) Ще одним кращим варіантом отримання солі L -винної кислоти (1R) діастереомера похідної 2 - азадипдроксибіцикло [2 2 1] гептану є такий, коли отримання проводять у суміші вода-ІП з об'ємним співвідношенням від приблизно 30 70 до приблизно 15 85, більш переважно приблизно 25 75 Конкретний варіант здійснення способу кислотно-каталізуючої реакції ацеталізацм або кеталізацм ВІДПОВІДНО до винаходу відноситься до способу отримання з'єднання формули IV (IV) де R зазначений вище, R3 і R3 представляють водень, алкіл або феніл, або R3 і F?3, узяті разом з атомом вуглецю, до якого вони приєднаю, утворюють циклоалкіл, який полягає в що кислотнокаталізуючій ацеталізацм або кеталізацм з'єднання формули V Rg. R. (V) R, де R4 і R4 представляють алкокси або, узяті 52702 разом з атомом вуглецю, до якого вони приєднані, утворять карбоніл, здійснюваної з (1R) діастереомером 2 - азадипдроксибіцикло -[2 2 1] гептану або його сіллю в ІП Кращий варіант здійснення кислотнокаталізуючої реакції ацеталізацм або кеталізаци є такий, при котрому R4 і F 4 являють собою метокси ? і R3 і F 4 являють собою метил ? Іншим кращим варіантом здійснення кислотнокаталізуючої реакції ацеталізацм або кеталізаци є такий, при якому кислотний каталіз виконують із використанням трифтороцтової кислоти (ТФО) Ще одним кращим варіантом здійснення кислотно-каталізуючої реакції ацеталізацм або кеталізацм є такий, при котрому R позначає групу формули II Більш кращим варіантом здійснення кислотнокаталізуючої реакції ацеталізацм або кеталізаци є такий, при котрому Ri представляє метил, Аг представляє феніл Ще одним кращим варіантом здійснення кислотно-каталізуючої реакції ацеталізацм або кеталізацм є такий, при котрому (1R) діастереомер похідної 2-азадипдроксибіцикло [2 21] гептану поданий у виді його солі L - винної кислоти Конкретний варіант здійснення препаративного лактамного методу по винаходу відносить до способу отримання похідної лактаму формули VI R',0 R'tO Н_ f (VI) де R'i і R"i незалежно представляють ацил або ароїл, або, узяті разом, утворять необов'язково заміщений метилен і d представляє водень або амінозахисну групу, що полягає в окислюванні біс О - захищеного (1R) діастереомеру похідної 2 азадипдроксибіцикло [2 2 1] гептану формули VII R",0 (VII) в присутності від приблизно 0,1 мол % до приблизно 1мол % RuO2 або його гідрату приблизно 3 еквівалентами окислювача з утворенням похідної лактаму з и більш чи дорівнює 95% Кращий варіант здійснення способу отримання лактаму є такий, при якому RuO2 є присутнім у КІЛЬКОСТІ приблизно 0,5- мол % Інший кращий варіант здійснення методу отримання лактаму є такий, при якому з'єднання лактаму одержують із и більш або рівним 99% Основні параметри препаративних методів зазначені вище і далі Звичайно, біспдроксилірування здійснюють в умовах, описаних V VanRheenen et al, Tetrahedron Letters, voI 23,1973-1976(1976) Окислювач повинний здійснювати біспдроксилірування в екзоформі Більш конкретно, окислювання може бути виконане за допомогою перманганату калію або тетроксиду осмію або осмату металу і проводиться при N - метилморфолин-оксиду або оксиду триетиламіку або ферицианіду калію Кз [Fe(CH)6] ВІДПОВІДНО ДО винаходу тетроксид осмію вико 10 ристовують у каталітичній КІЛЬКОСТІ, ЩО призводить до більш ефективного контролю за залишками осмію в продукті Реакція за участю осмію може проходити вже при такому його невеличкому складі, як від приблизно О.Обмол % до приблизно 0,1мол %, що займає, ВІДПОВІДНО ВІД приблизно 21 до приблизно 5 годин Переважно, взаємодія проходить в присутності приблизно О.Обмол % тетроксида осмію Окислювання може проходити в середовищі водно-органічного розчинника, такого як вода - трет-бутанол або вода-ацетон, більш краща вода-ацетон Ефірний розчинник, такий як трет-бутилметиловий ефір або диїзопропіловий ефір, можуть додатково бути присутнім, коли окислювання проходить у середовищі розчинника вода-ацетон Кращий діапазон об'ємних кількостей у суміші ефір ацетон вода складає від 1,9 16,7 1 до 11,17,4 1, більш краще від 11,116,7 1 до 16,7 16,7 1 Біспдроксилірування також може бути виконано тим же засобом із використанням суміші діастереомерів (І) і (Г), тобто без необхідності їхнього поділу перед проведенням біспдроксилірування (1R) діастереомер формули І, де R є групою формули II, може бути виділений у виді солей оптично активних органічних кислот і, більш конкретно, із суміші діастереомерних з'єднань формул І і Г шляхом діастереоселективної кристалізації з використанням таких оптично активних органічних кислот Однієї з використовуваних оптично активних органічних кислот є L - диметоксиянтарна кислота Отримання солі з використанням L - диметоксиянтарної кислоти проводять у підхожому органічному розчиннику, такому як кетон або аліфатичний спирт, особливо кращий ІП ВІДПОВІДНО до винаходу, L - винна кислота представляє іншу підхожу оптично активну органічну кислоту Одержання солі з використанням L -винної кислоти проводять у розчиннику, такому як водноорганічний розчинник, де органічним розчинником може бути аліфатичний спирт, такий як ІП Дякуючи використанню L - винної кислоти досягають підвищений вихід і енантиомірна чистота бажаного діастереомера (І) Д и гідро кс и часті з'єднання формули І, де R являє собою водень або групу формули II, можуть бути захищені у виді складного ефіру або ацеталя/кеталя з отриманням прпродукту формули VIII R',0 (VIII) де R являє собою водень або групу формули II і R'i і R"i, такі, як зазначені вище Звичайно захист пдроксигруп досягає або етерифікацією, або ацеталізацією / кеталізацією Наприклад, етерифікація проходить шляхом взаємодії ациловміщуваної групи, такої як оцтова кислота або пропюнова кислота, при п - толуолсульфоновоі кислоти в органічному розчиннику, такому як ароматний вуглеводень, наприклад бензол або толуол, при поступовому видаленні води в міру и утворення, наприклад азеотропним відгоном Наприклад, ацеталізацією / кеталізацією здійснюють шляхом взаємодії з альдегідом або кетоном, можливо у виді кеталя, в присутності кислоти, такої як 11 52702 ТФО, в органічному розчиннику, такому як аліфатичний спирт, наприклад ІП, ароматний вуглеводень, наприклад бензол або толуол або простий ефір, наприклад трет-бутилметиловий ефір або диїзопропіловий ефір, при температурі від приблизно 50°С до приблизно температури кипіння реакційної суміші Коли використовують ефірний розчинник, оцтова кислота також може бути присутнім, що призводить до утворення солі сполучення IV, що може бути екстрагована водою Краще середовище для кеталізацм, ВІДПОВІДНО ДО винаходу, включає використання 2,2 - диметоксипропану, ТФО і ІП, що дає поліпшений вихід і енантиомірний надлишок прпродукту Реакція проходить при температурі приблизно 70°С Продукт формули VIII, де R представляє групу формули II, можна перетворити в продукт формули VIII, де R представляє водень, шляхом гідрування Звичайно, гідрування проводять воднем, що необов'язково знаходиться під збільшеним тиском, в присутності каталізатора, такого як паладій на вугіллі, в органічному розчиннику, такому як спирт, наприклад метанол, етанол або ІП, при від приблизно 0°С до приблизно 50°С Продукт формули VIII, де R представляє водень, також одержують, застосовуючи ті ж самі реагенти гідрування й умови стосовно до солі похідної формули IV, де R представляє групу формули II Продукт формули VIII, де R представляє водень, може бути перетворений у продукт формули IX R'.O |\ (IX) де R'i і R"i зазначені вище і G2 представляє амінозахисну групу, шляхом селективного введення підхожої захисної групи Захисні групи обрані з тих, що пізніше можуть бути селективно віддалені Ці захисні групи включають наступні, особливо добре ПІДХОЖІ групи трет-бутоксикарбоніл, хлорацетил, метоксиметил, трихлор-2,2,2 - етоксикарбоніл, трет-бутил, бензил, п-нітробензил, п-метоксибензил, дифенілметил, триалкилсиліл, алилоксикарбоніл і бензилоксикарбоніл, де фенільне кільце необов'язково заміщене галогеном, алкілом або алкокси Серед особо зручних захисних груп можна зазначити такі, що описані Т W Greene and P G M Wuis, "Protecting Groups in Organic Synthesis", Chapter 7, 2nd edition, J'&bn Wiley & Sons (1991) Особливий інтерес представляє трет-бутоксикарбонільна група Продукт формули IX, де G2 представляє третбутоксикарбоніл, може бути отриманий безпосередньо з продукту формули VIII, де R представляє групу формули II, шляхом одночасного гідрування і трет-бутоксикарбонілювання Наприклад, реакцію здійснюють шляхом одночасно взаємодії продукту формули VIII, де R представляє групу формули II, із воднем при каталізатора, такого як паладій на вуплл, і ди-третбутилкарбонатом в органічному розчиннику, такому як спирт, наприклад метанол, етанол або ІП, при від приблизно 0°С до приблизно 50°С Цю реакцію, зокрема, використовують, коли R'i і R"i узяті 12 разом, утворять необов'язково заміщений метилен Альтернативно, продукт формули IX, де G2 представляє трет-бутоксикарбоніл, може бути отриманий у дві стадії з продукту формули VIII, де R представляє групу формули II, здійснюючи спочатку видалення групи формули II гідруванням, з отриманням ВІДПОВІДНОГО продукту, де R представляє водень, а потім здійснюючи третбутоксикарбонілювання цього продукту Видалення гідруванням здійснюють, як описано вище, і трет-бутоксикарбонілювання здійснюють у воді в лужних умовах, використовуючи (Вос)2О Продукт формули IX потім окисляють у продукт формули X R%0 (X) де R'i, R''21 Сгвизначені вище Звичайно окислювання проводять за допомогою оксиду рутенію (RUO4), що, необов'язково, може бути отриманий in situ із попередника, такого як RuO2 або RuCb, в присутності окислювача, обраного з перюдату, такого як перюдат натрію, ппогалогеніту, такого як гіпохлорит або ппоброміт натрію, або бромату, такого як бромат натрію, або оксиду органічного третинного аміну, такого як Nметилморфолин-оксид або триетиламшоксид Реакція протікає в розчиннику, такому як вода, або в гомогенної або гетерогенному водно-органічному середовищі, наприклад у суміші вода-ЕЮАс Окислювання також може бути проведене з використанням тільки гіпохлориту натрію або з використанням перманганату калію або вольфрамату натрію при окислювача, такого як гіпохлорит натрію, пероксид водню або алкілпдропероксид Продукт формули X може бути також отриманий шляхом окислювання продукту формули VIII, де R представляє водень, в умовах, описаних вище, із наступним захистом атома азоту лактаму формули XI R'O K (XI) де R'i і R"i визначені вище, шляхом вступу захисної групи, як зазначено вище Продукти формул X и XI, зокрема, можуть використовуватися для отримання карбоцукру формули XII с*") де R2 подає карбокси, алкоксикарбоніл, N - алкіламинокарбоніл, пдроксиметил або алкоксиметил, і R' і R", що можуть бути однаковими або різними, представляють водень, ацил або ароїл, або R'i і R"i, узяті разом з атомом вуглецю, до якого вони приєднані, утворять необов'язково заміщену метиленову групу, атом вуглецю которої необов'язково заміщений однією або двома групами, що можуть бути однаковими або різними, обраними з 13 алкілу або фенілу, або два алкіли, узяті разом, можуть утворювати циклоалкіл, і d представляє водень або амінозахисну групу G2 Більш конкретно, F 2 представляє етиламінокарбонільну групу ? або пдроксиметильну групу, і R' і R", узяті разом, утворять ізопропілиденову групу Продукт формули X може бути перетворений у продукт формули XII в умовах, що відповідають природі заступника F?2, що повинний бути введений Продукт (формули XII, де F 2 представляє кар? бокси, може бути отриманий шляхом взаємодії мінеральної основи, такого як NaOH, із продуктом формули X с наступною заміною захисної групи Сз на водень і, необов'язково, груп R'i і R"i на водень Продукт формули XII, де R2 представляє карбокси, може бути отриманий заміною захисної групи G2 формули X на атом водню з наступною дією мінеральної основи, такого як карбонат натрію, і, необов'язково, заміною радикалів R'1 і R"i на водень Продукт формули XII, де R2 представляє алкоксикарбоніл, може бути отриманий шляхом взаємодії алкоксиду лужного металу з продуктом формули X с наступною заміною захисної групи G2 на водень і, необов'язково, груп Ri і R"i на водень Продукт формули XII, де R2 представляє алкоксикарбоніл, може бути отриманий заміною захисної групи G2 продукту формули X на водень із наступною дією алкоголяту лужного металу і, необов'язково, заміною радикалів R'i і R"i на водень Продукт формули XII, де R2 представляє N алкіламшокарбоніл, може бути отриманий шляхом взаємодії алкіламшу з продуктом формули X с наступною заміною захисної групи G2 на водень, і, необов'язково, груп R'i і R"i на водень Продукт формули XII, де R2 представляє N алкіламшо - карбоніл, може бути отриманий заміною захисної групи G2 продукту формули X на водень із наступною дією алкіламшу і, необов'язково, заміною радикалів R'i і R"i на водень Продукт формули XII, де R2 представляє пдроксиметильну групу, можна одержати взаємодією відновлювана, такого як борпдрид, наприклад борпдрид натрію або калію, із продуктом формули X с наступною заміною захисної групи G2 на водень і, необов'язково, груп R'i і R"i на водень Продукт формули XII, де R2 представляє пдроксиметильний радикал, можна одержати заміною захисної групи G2 продукту формули X на атом водню з наступною дією відновлювача, такого як борпдрид натрію або калію, і, необов'язково, заміною радикалів R'i і R"i на водень Даний винахід також включає виділення (1S) діастереомера похідної формули Г, використовуючи оптично-активні органічні кислоти протилежної конфігурації стосовно тих, що описані для виділення (1R) діастереомера похідної формули І В І Д ПОВІДНО до винаходу, (1S) діастереомер сполучення формули Г може бути потім переведений у ВІДПОВІДНІ (1S) діастереомери сполучень формул IV, VI, VII, VIII, IX і X, із використанням способів, застосовуваних для отримання (1R) діастереомірних сполучень цих формул ВИХІДНІ продукти і проміжні продукти одержу 52702 14 ють за допомогою відомих методів або їхньої адаптації Сполучення формул XIII і ХІІГ де *, *', R1 і Аг визначені вище, можуть бути отримані реакцією Дильса-Альдера із суміші гомохіральних амінів формул де *, *', RI і Аг визначені вище, у виді солі, краще з мінеральною кислотою, такий як НСІ, формальдегідом і циклопентадієном, в умовах, описаних S D Larsen і Р A Gneco у J Am Chem S o c , vol 107, 1768-1769 (1985) Цей спосіб призводить до суміші двох діастереомерів Діастереомери можуть бути розділені, використовуючи L - дибензоілвинну кислоту, як описано С, К -F Chiu y S y n Comm , 26(3), 577 (1996) Сполучення формул III і III', де R представляє водень, можуть бути отримані гідруванням сполучень формул XIII і ХІІГ двостадійним засобом Спочатку сполучення опрацьовують 2,2,2 - трихлоретоксикарбоніл (Тгос) хлоридом або (3- (триметил-силіл ) етоксикарбоніл (Теос) хлоридом з отриманням ВІДПОВІДНИХ Тгос -або Теос-похідних (карбаматів), потім опрацьовують Zn у спиртовому розчиннику, наприклад етанолі, при нагріванні, або Zn у розчиннику органічній кислоті, такий як оцтова кислота, при кімнатній температурі Даний винахід далі ілюструється, але не обмежується, слідуючими прикладами отримання сполучень, ВІДПОВІДНО до даного винаходу У спектрах ядерного магнитного резонансу (ЯМР) ХІМІЧНІ зсуви виражені в м д ( р р т ) (МІЛЬ ЙОННІ частки) щодо тетраметилсилану Скорочення мають наступні значення с = синглет, д = дублет, т = триплет, м = мультиплет, дд = дублет дублетів, ддд = дублет дублетів, дт = дублет триплетів, ш = широкий ПРИКЛАД 1а Одержання 2 - (а - S - метилбензил) - 2 - азабицикло -[2 2 1] гепт - 5 - єну У 2л реактор загружають 255 р (S) - (-) - а метилбензіламшу і ЗООмл води Суспензію охолоджують до -5°С і протягом однієї години при перемішуванні добавляють розчин 185мл концентрованої НСІ у ЮОмл води рН суміші встановлюють між 5 і 6,5 Перемішування продовжують протягом ЗО хвилин і потім загружають 242 мл 3 7 % розчину формальдегіду Після перемішування протягом ще 40 хвилин переганяють (~ 270мл) циклопентадіен прямо в реакційну суміш Отриману суміш енергійно перемішують протягом ночі при -5°С Завершення реакції визначають за допомогою високоефективної рідинної хроматографії (ВЕРХ) Два прошарки, що утворилися, розділяють і водяний прошарок промивають 250мл гептану перед тим як підлужити до рН 11 за допомогою 168мл 5 0 % ного розчину NaOH і колотого льоду Органічну суміш потім екстрагують 2х500мл і 2х300мл порціями EtOAc Об'єднані екстракти промивають 200мл холодної води, потім 200мл насиченого розчину NaCI, висушують над безводним Na2SO4 і 15 фільтрують Прозорий фільтрат концентрують на роторному випарнику з отриманням 408,4г (97,4%) жовтої олії, 2 - (a -S- метилбензил) -2- азабицикло [2 2 1] гепт-5-ену, у діастереомірному співвідношенні 77,1 22,9 із перевагою бажаного ізомеру 1 Н ЯМР (500МГц, CDC1 3 ) 5 1,35(д,2Н), 1,46(д, 1Н), 1,62 (д,1Н), 2,89(д,1Н), 3,05(м,1Н), 4,13(с, 1Н), 6,11(д, 1Н), 6,32 (м, 1Н), 7,26(д, 2Н), 7,33(д, 2Н), МС (ЕІ, 70 ев) m/z (відносна інтенсивність) 199(М+,70) ПРИКЛАД 1Ь Одержання 2-(а -S- метилбензил) -2- азабицикло -[2 2 1] гепт - 5 - єну У 250мл трьохгорлу колбу, постачену зворотнім холодильником і мішалкою, в атмосфері аргону загружають розчин, що складається з 20г (S) - () -а - метилбензиламину (165ммоль) у 60мл води, рН розчину якого доведений до 6,1 за допомогою 17мл 36% НСІ (в/об) Після охолодження до 5°С добавляють 20мл 37% водяного розчину (в/об) формальдегіду Розчин перемішують протягом 5 хвилин при 5°С, потім добавляють 21,8г (ЗЗОммоль) циклопентадієну Суміш перемішують протягом 16 годин при температурі від -5 до 0°С Водяну фазу відокремлюють декантацією і промивають 50мл пентану Нейтралізують концентрованим розчином NaOH до рН 8 Потім проводять 2 екстракції ЕЮАс, кожна по 70мол рН водяної фази встановлюють рівним 11 шляхом додавання концентрованого розчину NaOH Потім проводять ще 2 екстракції ЕЮАс по 70мол Органічні фази об'єднують і потім промивають два рази по 50мл води і висушують над Na2SO4 Після фільтрації і відгону розчинника до сухого стану при зниженому тиску одержують 33,1г 2-(а -S- метилбензил) - 2 азабицикло [2 2 1] гепт - 5 - єну у виді світложовто і олії ПРИКЛАД 2 Одержання 5R, 6S - дипдрокси - 2 - (а - S - метилбензил) -2 - азабицикло [2 2 1] гептану У 500мл трьохгорлу колбу, з зворотнім холодильником і мішалкою, що містить розчин 20г 2- (а -S- метилбензил) -2- азабицикло [2 2 1] гепт -5- єну (75,34ммоль) у 220мл трет-бутанолу, добавляють 12г N-N-метилморфолиноксиду в 32мл води при температурі приблизно 25°С и потім повільно добавляють 6,3мл 25% (в/об) розчину тетроксиду осмію (OslU) трет-бутанолу Перемішування продовжують протягом 2-х годин при температурі приблизно 20°С, після чого ще 3 години при 65°С Після розпарювання трет-бутанолу при зниженому тиску залишок знову розчиняють у 350мл ІП Після концентрування до сухого стану при зниженому тиску одержують 24г цис -5,6- дипдрокси -2-(а -Sметилбензил) - 2-азабицикло [2 2 1] гептану у виді олії Шляхом кристалізації в циклогексані одержують 14г 5R, 6S - дипдрокси -2 - (a -S- метилбензил) -2- азабицикло [2 2 1] гептану з ізомерної чистотою більш 95% ЯМР спектр, визначений у дейтерохлорофоромі, показує такі ХІМІЧНІ зсуви (5) 1,21(ЗН, д), 1,38 (1Н, д), 1,59 (1Н, д), 2,22 (2Н, м), 2,45(1 Н, дд), 2,95(1 Н, с), 3,99 (1Н, к), 3,78 (1Н, д), 3,90 (1Н, д), 7,28 (5Н, м), ПРИКЛАД За Одержання 5R, 6S- дипдрокси 2- (a -S- метилбензил) -2-азабицикло [2 2 1] гептану 52702 16 Розчин 0,5ммоль суміші (78/22 у моль) 5R.6Sдипдрокси -2- (а - S - метилбензил) -2- азабицикло [2 2 1 ] гептану і 5S, 6R- дипдрокси -2- (а -3метилбензил) -2- азабицикло [2 2 1 ] гептану і 0,5ммоль L - диметиоксиянтарної кислоти в 1мл ІП перемішують протягом 24 годин при температурі в межах від 25°С в початку до 5°С Отримані кристали відокремлюють фільтрацією і висушують У такий спосіб одержують 110мг 5R.6S - дипдрокси -2- (а -S-метилбензил) - 2 - азабицикло [2 2 1] гептану з енантиомірним надлишком 97% Суміш 5R.6S - дипдрокси -2- (a -S- метилбензил) - 2- азабицикло- [2 2 1]-гептану і 5S, 6R- дипдрокси -2- (a -S- метилбензил) -2- азабицикло [2 2 1] гептану (78/22 у моль) може бути отримана в такий спосіб у 250мл круглодону трьохгорлу колбу, з зворотним холодильником і мішалкою, що містить розчин 7г 2- (a -S- метилбензил) -2азабицикло [2 2 1] гепт -5- єну (35ммоль) у 70мл трет-бутанолу, добавляють при температурі приблизно 25°С 4,12г N -метилморфолиноксиду в 11мл води і потім повільно добавляють 360мл 2,5% рОЗЧИНу (В/Об) ТетрОКСИДУ ОСМІЮ (OSO4) У трет-бутанолі Суміш перемішують протягом 1 години при температурі приблизно 20°С и потім протягом 4 годин при 65°С Після розпарювання третбутанолу при зниженому тиску залишок опрацьовують 150мл ІП Після концентрування до сухого стану при зниженому тиску одержують 8,27г продукту, протонний ЯМР спектр якого показує, що він складається із суміші (78/22 у моль) 5R, 6S- дипдрокси -2- (a -S- метилбензил) -2-азабицикло 1 2 2 1] -гептану і 5S, 6R- дипдрокси -2- (a -S- метилбензил) -2-азабицикло -[2 2 1] гептану ПРИКЛАД ЗЬ Одержання 5R, 6S - дипдрокси 2- (a -S- метилбензил) -2-азабицикло [2 2 1] гептану У 50мл одногору круглодону колбу, з магнітною мішалкою й зворотним холодильником, загружають 1г 2- (a-S- метилбензил) -2-азабицикло [2 2 1] гепт -5-ену (5ммоль), 2,5мл диїзопропілового ефіру, 2,5мл ацетону, 0,9мл 58% мас водяні NN-метилморфолиноксиди і 0,15мл води Суміш перемішують протягом 5 хвилин і відразу загружають 9мг твердого K2OSO4 2НгО і продовжують перемішування при кімнатній температурі протягом 25 хвилин Суміш потім перемішують при КИП'ЯТІННІ ЗІ зворотним холодильником протягом 7,5 годин Після закінчення цього часу ВЕРХ показує 95%-не завершення реакції окислювання До коричневої суміші, охолодженої до кімнатної температури, добавляють розчин 630мг сульфіту натрію в 4мл води Двохфазну суміш перемішують при кімнатній температурі протягом 1 години Велику частину органічних розчинників упарюють при зниженому тиску, добавляють 5мл диїзопропілового ефіру Водяну фазу відокремлюють декантацією і повторно екстрагують 2 х 5мл диїзопропілового ефіру Об'єднані органічні фази промивають водяним насиченим розчином хлориду натрію, висушують над Na2SO4, фільтрують і переганяють при зниженому тиску з отриманням 1,04г (89%, скоригований вихід -86%) 5R.6S - дипдрокси -2- (а -3метилбензил) -2-азабицикло [2 2 1] гептана у виді олії, що кристалізується при стоянні Чистота про 18 17 52702 1 дуісгу складає 95мол % ВІДПОВІДНО даним Н ЯМР дендиокси -2- (а -S-метилбензил) -2- азабицикло в CDCI3 (містить 4МОЛ % N - метилморфоліну + [2 2 1] гептану О.бмол % вихідного продукту) У 2л чотирьохгорлий циліндричний реактор із ПРИКЛАД Зс Отримання L - тартрату 5R, 6S сорочкою, з термопарою, мішалкою і зворотним дипдрокси - 2 - (а - S - метилбензил) - 2 - азабицихолодильником загружають 223г L - тартрату кло [2 2 1] гептану 5R.6S - дипдрокси -2- (a -S- метилбензил) -2- азабицикло [2 2 1] гептану і потім 1200мл ІП ПочинаУ 2л реактор загружаюсь 210г 2-(a-S- метилють перемішування і добавляють 286мл 2,2- дибензил) -2- азабицикло [2 21] гепт - 5 - єну, метоксипропану і 44,6мл ТФО Суспензію 1200мл 2 - метил - 2 - пропану і 182мл 4нагрівають до 720°С до повного розчинення тверметилморфоліну N - оксиду До цієї суміші по крадих речовин Через 5 годин реакційну масу охолоплях добавляють 8мл 2,5% розчину тетроксиду джують до 65°С и вміст переносять у Зл круглодоосмію в трет-бутанолі У атмосфері азоту суміш ну колбу Приблизно 1100мл розчинника нагрівають до 62°С протягом 22 годин при енервідганяють при 48°С и тиску 124 мбар У вихідний гійному перемішуванні Реакційну суміш концент2 - літровий чотирьохгорлий циліндричний реактор рують за допомогою роторного випарника при із сорочкою добавляють 1,2л 2М розчину NaOH 60°С Добавляють ЗООмл ІП і розчин знову конценпри перемішуванні при 25°С У розчин NaOH дотрують при 60°С с отриманням 246г 5R.6S - дипдбавляють залишок від перегонки, описаної вище (~ рокси - 2 - (a- S - метилбензил) - 2 -азабицикло 700мл розчину) Рудувато-коричневий розчин охо[2 2 1] гептану у виді темно-коричневого сиропу лоджують до 25°С протягом більш 40 хвилин ТвеНеочищений продукт суспендирують у 1,8л 75% рдий продукт починає осаджуватися з розчину при ного ІП при 40°С До цієї суспензії добавляють при 28°С Суспензію перемішують декілька годин і поенергійному перемішуванні 158,2г L - винної кистім фільтрують за допомогою лійки Бюхнера діалоти Перемішування продовжують при 40°С прометром 11см, з фільтрувальною бумагою Ватман тягом 2,5 годин Суміш прохолоджують до 30°С, №1 Корж на фільтрі промивають ЗООмл води Не фільтрують, промивають 500 мл 75% - ного ІП і зовсім білі тверді продукти перекладають у взвісь 200мл ІП, потім висушують при 70°С в вакуумі у воді протягом 13 годин і повторно фільтрують, протягом 16 годин з отриманням 269,5г бажаної промивають водою і висушують на повітрі Потім солі L - тартрату у вигляді твердого продукту кретверді прородукти сушать у вакуумі при 50°С с мового кольору (т пл 143-145°С, співвідношення отриманням 112г 5R, 6S - ізопропілідендюкси -2діастереомерів 94,2 5 8) (a-S-метилбензил) -2- азабицикло [2 2 1] гептану у 1 Н ЯМР (500 МГц, CDC13) 5 1,3 (д, ЗН), 2,5 (м, вигляді білого твердого продукту, що за даними 2Н), 4,18 (із, 2Н), 7,36 (т, 2Н), 7,4 (т, 2Н) ВЕРХ є диастереомерно чистим МС (ЕІ.70 ев) m/z (відносна інтенсивність) 1 233(М+13) Н ЯМР (500Мгц, CDC13) 5 1,28 (с, ЗН), 1,27 (д, ЗН), 1,39 (с, ЗН), 1,63 (с, 1Н), 2,27 (д, 1Н), 2,4 (д, ПРИКЛАД 4а Одержання 5R, 6S - ізопропілі1Н), 2,51 (дд, 1Н), 3,12 (с, 1Н), 3,46 (до, 1Н) , 4,2 дендиокси - 2 - (а - S - метилбензил) - 2 - азабици(дд, 2Н), 7,28 (м, 5Н), кло [2 2 1] гептану МС (ЕІ, 70ев) m/z (відносна інтенсивність) 273 У 500мл трьохгорлу колбу, з зворотним холо(М+, 8,4) дильником і мішалкою, що містить розчин 18,4г 5R.6S - дипдрокси -2- (a -S- метилбензил) -2- азаПРИКЛАД 5а Одержання 5R, 6S - ізопропілібицикло [2 2 1] гептану (76ммоль) у 130мл толуодендиокси - 2 - (трет-бутоксикарбоніл) - 2 - азабилу, повільно добавляють 31,7г 2,2 - диметоксипроцикло [2 2 1] гептану пану (304ммоль) і потім 13г (114ммоль) ТФО У 250мл трьохгорлу колбу, з мішалкою, поміСуміш нагрівають протягом 4 годин 10 хвилин при щають 0,5г 5% каталізатора паладій на вугіллі, 5г 65°С Після охолодження до 30°С и концентруван5R, 6S - ізопропілідендиокси -2- (a - S ня за допомогою роторного випарника для видаметилбензил) - 2 -азабицикло [2 21]- гептану, лення толуолу, надлишку 2,2 - диметиоксипропану 3,98г ди-трет-бутилдикарбонату і 36 мл метанол і, частково, ТФО реакційну суміш опрацьовують Апарат продувають аргоном, потім воднем і потім дихлорметаном, потім нейтралізують шляхом допідтримують атмосферу водню при 25°С Реакція давання ЮОмл 2н NaOH Після декантації, висупродовжується 5 годин з продуванням воднем кошування органічної фази над Na2SO4, фільтруванжні 15 хвилин для видалення дюксиду вуглецю, ня, обробляння для знебарвлення активованого що утворився вугілля (30г) протягом ЗО хвилин при температурі Після фільтрації через Сіагсеїо і концентрукипіння дихлорметану, фільтрування через Сіагвання до сухого стану при зниженому тиску вихід сеіо і концентрування до сухого стану при знижескладає 4,84г 5R.6S - ізопропілідендиокси -2-(третному тиску вихід складає 18,8г 5R, 6S - ізопропілибутоксикарбоніл) - 2 - азабицикло [2 2 1] - гептану, дендиокси -2- (a -S- метилбензил) -2- азабицикло структура якого підтверджена ЯМР спектром, що [2 2 1] гептану, структура якого підтверджена провизначається в диметилсульфоксиді -d6 і має такі тонним ЯМР спектром, що при визначенні в дейХІМІЧНІ зсуви (5) 1,16 (с, ЗН), 1,28 (с, ЗН), 1,32 (с, терохлороформі показав такі ХІМІЧНІ зсуви 1Н), 1,34 (с, ЗН), 1,65 (д, 1Н), 2,38 (м, 1Н), 2,65 (д, (5) 1,22 (ЗН, д), 1,23 (6Н, с), 1, 31 (1Н, д), 1, 57 (1Н, д), 2,08 (1Н, д), 2,34 (1Н, шс), 2,45 (1Н, дд), 3,06 (1Н, с), 3,40 (1Н, к), 4,09 (1Н, д), 4,19 (1Н, д), 7,26 (5Н, м) ПРИКЛАД 4Ь Одержання 5R, 6S - ізопропілі 1Н), 2,99 (м, 1Н), 3,84 (м, 1Н), 3,94 (д, 1Н), 4 16 (д, 1Н) ПРИКЛАД 5Ь Одержання 5R, 6S - ізопропілідендиокси - 2 - (трет-бутоксикарбонил) - 2 - азабицикло [2 2 1] - гептану У 2 літровий чотирьохгорлий циліндричний 19 52702 реактор із сорочкою, з термопарою, мішалкою, трубкою для подачі газу, перегородкою для входу азоту і водню, послідовно загружають 140 г 5R, 6S - ізопропілідендиокси -2- (a-S-метилбензил) -2азабицикло [2 2 1] гептану, 13,4г 10% Pd /С і 900мл метанолу У суспензію що перемішується барботирують азот протягом 10 хвилин і потім водень протягом 10 хвилин при 25°С Цю процедуру повторюють кожні ЗО хвилин і за реакцією стежать за допомогою ТСХ (силікагель, ЕЮАс, проявник йод) Через 3 годин, ВІДПОВІДНО до ТСХ, реакці завершена на 50% До цього частково відновленого розчину добавляють 56г ди-третбутилдикарбоксилату протягом 10 хвилин і потім барботирують азот / водень, як описано вище Кожні ЗО хвилин добавляють додаткові 10г дитрет-бутилдикарбоксилату, барботирують азот / водень доти, поки не буде додано усього 112г дитрет-бутилдикарбоксилату (56г плюс порції по 10г) Реакційну суміш перемішують усю ніч при 25°С Суспензію Pd / С фільтрують на ЛІЙЦІ БЮХнера діаметром 9см, з фільтрувальною бумагою № 54 і прошарком 5г це литу, і реактор і корж на фільтрі промивають ЮОмл метанолу Фільтрат поміщають у 2-х літрову одногорлу круглодону колбу і 750мл розчинника відганяють при 40°С і 105 мбар (залишається ~ 250мл світло-жовтого розчину) У вихідну реакційну судину поміщають 1л води, що охолоджують до 10°С Жовтий залишок від описаної вище відгону добавляють у холодну воду фактично одною порцією 5R, 6S - ізопропілдендиокси -2- (трет-бутоксикарбонил) -2азабицикло -[2 2 1] гептан осаджується з розчину у виді білого твердого продукту Взвісь перемішують протягом ЗО хвилин при 6°С і потім фільтрують і промивають водою Отриманий білий твердий продукт висушують у вакуумі при 60°С з отриманням 129,6г білого твердого продукту, що ВІДПОВІДНО до хіральної ВЕРХ є енантиомірно чистим 1 Н ЯМР (500Мгц, CDC13) 5 1,28 (с, ЗН), 1,4 (с, ЗН), 1,45 (с, 9Н) , 1,87 (д, 1Н), 2,53 (с, 1Н), 2,82 (д,1Н), 3,17 (дц, 1Н), 4,09 (м, 2Н), 4,2 (м, 2Н), МС (FAB-LRP) m/z (відносна інтенсивність) 270 (М+Н) +, 9,4) ПРИКЛАД 6а Одержання 5R, 6S - ізопропілідендиокси - 2 - (трет-бутоксикарбоніл) - 2 - азабицикло - [2 2 1] гептан - 3 - ону У 30-мл пробірку поміщають 270мг 5R.6S - ізопропілідендиокси -2- (трет-бутоксикарбоніл) -2азабицикло - [2 2 1] гептану (Іммоль) і 40мг RuO2 Н2О (О.Зекв) Добавляють Юмл ЕЮАс і 720мг води (40екв) Потім добавляють 2,14г перюдату натрію (Юекв) і пробірку герметично закривають Перемішування продовжують протягом 16 годин при 50°С Реакційну суміш фільтрують через СІагсеіо і проводять дві екстракції ЕЮАс, кожна по 20мол Органічні фази висушують над N32SO4 Органічні фази висушують над N32SO4 Після фільтрації і концентрування до сухого стану при зниженому тиску одержують 245г твердої речовини, що містить 68% 5R,6S-i3onpomnifleHflHOKCH -2(трет-бутоксикарбоніл) -2- азабицикло - [2 2 1] гептан -3-ону і 32% вихідного продукту Структура отриманого продукту підтверджена ЯМР спектром, що визначають у диметилсульфоксиді - d6 і має такі ХІМІЧНІ зсуви (5) 1,38 (9Н, с), 1,23 (ЗН, с), 1,33 20 (ЗН, с), 1,85 (1Н, д), 1,93 (1Н, д), 2,69 (1Н, с), 4,24 (1Н, с), 4,41 (1Н, д), 4,51 (1Н, д) ПРИКЛАД 6Ь Одержання 5R, 6S - ізопропілідендиокси - 2 - (трет-бутоксикарбоніл) -2- азабицикло - [2 2 1] гептан-3- ону У 2-х літровий чотирьохгорлий циліндричний реактор із сорочкою, з термопарою, мішалкою і холодильником, послідовно загружають при перемішуванні 120г 5R, 6S- ізопропілідендиокси -2(трет-бутоксикарбоніл) -2-азабицикло [2 21] гептану, 0,3г RuO2, 201,2г бромату натрію, 960мл ЕЮАс і ЮООмл води Реакційну суміш нагрівають до 45°С и перемішують при цій температурі протягом 15 годин Перемішування переривають і водяний прошарок відкидають У реакційну судину добавляють насичений розчин NaCI (500мл) і суспензію перемішують протягом 10 хвилин Перемішування знову переривають, дають можливість розслоіться і водяний прошарок видаляють У реакційну судину загружають 33% розчин динатрієвої солі малеїнової кислоти (500мл), суспензію перемішують 5 хвилин і прошарки знову розділяють Органічний прошарок потім фільтрують через прошарок целіту для відділення каталізатора, а розчинник видаляють у вакуумі Отриманий твердий продукт висушують у вакуумній печі з отриманням 117г 5R, бЗ-ізопропілідендиокси -2- (третбутоксикарбоніл) -2-азабицикло - [2 2 1] гептан -3ону у вигляді білого твердого продукту, засміченного 5% вихідного Продукту 115г зразка цього продукту розчиняють у 350мл гептану при 85°С и лишають прохолоджуватися до 25°С протягом більш 3-х годин, потім охолоджують до 5°С, після чого фільтрують і висушують у вакуумі при 60°С Одержують 92г (74%) 5R/ 6S -ізопропілідендиокси -2- (трет-бутоксикарбоніл) -2-азабицикло -[2 2 1] гептан -3-ону у вигляді білого кристалічного твердого продукту 1 Н ЯМР 5 1,32 (м, ЗН), 1,48 (м, 12Н), 1,82 (м, 1Н), 2,1 (м, 1Н), 4,43 (м, 1Н), 4,48 (м, 1Н), 4,6 (м, 1Н), МС (FAB - LRP у нітробензиловому спирті) 284 (М+Н) + 10%) ПРИКЛАД 7а Одержання 2R, 3S - ізопропілідендиокси - 4R - аміно -1S - етилокшокарбонілцизелопентан бензоату У пробірку Бергофа поміщають 568г 5R, 6S изопропіліден-дюкси -2-(трет-бутоксикарбоніл) -2азабицикло - [2 2 1] гептан -3- ону і Юмл 70% ного (по масі) водяного розчину етиламшу Суміш нагрівають при перемішуванні протягом 4 годин при температурі 60°С Після охолодження надлишок етиламіну і води видаляють при зниженому тиску Після висушування при зниженому тиску одержують 650мг (вихід 98%) 2R.3S - ізопропілідендиоксі - 4R -трет-бутоксикарбониламино - 1S етиламшокарбонілциклопентана, структура якого підтверджена протонним ЯМР спектром, кут обертання [ а ] % = 15,0 (с=1, метанол) У розчин 200мг 2R, 3S - ізопропілідендиоксі 4R - трет-бутоксикарбоніламшо -1S- етиламшокарбонілциклопентану в 1,6мл безводного дихлорметану добавляють 275мл ТФО Суміш перемішують усю ніч при температурі приблизно -5°С Реакційну масу виливають у 4мл 2,5н водяного розчину карбонату натрію Органічний прошарок 22 21 52702 концентрують при зниженому тиску при темпера0,85г ТФО Після 6 годин перемішування і концентурі нижче 25° С У такий спосіб одержують 125мг трування до сухого стану вихід складає 1,16г 2R, продукту, що розчиняють у 0,5мл тетрапдрофура3S - ізопропілідендиокси - 4R - аміно - 1S ну У цей розчин добавляють 70мг бензойної кисетиламшокарбоніл циклопентан трифторацетату, лоти Після охолодження отриманого розчину приструктура якого підвладна ЯМР спектром, що виблизно до 0°С отримані кристали відокремлюють значався в диметилсульфоксиді -do і показав такі фільтруванням і промивають у пентані У такий ХІМІЧНІ зсуви спосіб одержують 138мг 2R, 3S - ізопропілідендио0,79 (т, ЗН), 1,03 (с, ЗН), 1,19 (с, ЗН), 1,42 (м, кси 4R аміно 131Н), 2,05 (м, 1Н), 2,52 (м, 1Н), 2,89 (до,), 3,04 (м, етиламшокарбонілпиклопентан бензоату 1Н), 4,16 (м, 1Н) ПРИКЛАД 7Ь Одержання 2R, 3S - ізопропіліПРИКЛАД 7с Одержання 2R, 3S - ізопропілідендиокси 4R амшо-ISдендиокси -4R -аміно -1S етиламшокарбонілциклопентан трифторацетату етиламшокарбоніл циклопентану У 25мл-овий автоклав, з магнітною мішалкою, До розчину 167мг 5R, 6S - изопропілідендиоквводять 1,47г 5R, 6S -ізопропілідендиокси - 2 си - 2 - (трет-бутоксикарбоніл) -2- азабицикло (трет-бутоксикарбонил) - 2 - азабицикло [2 21] [2 2 1] гептан -3- ону в 1 мл дихлорметана, охологептан -3-ону в розчині в Юмл безводного толуолу дженого до 0°С, добавляють 90мкл ТФО Темпеі потім приблизно 0,7мл етиламшу Автоклав заратурі дають піднятися до 23°С протягом більш 40 кривають і потім нагрівають при температурі 90хвилин, потім перемішують 22 години при цій тем100°С протягом 21 години Після охолодження пературі Добавляють ще 90 кл ТФО і потім змітолуол упарюють і розчиняють залишок у Юмл шують протягом ще однієї години при 23°С Після дихлорметану і Юмл води Після декантації оргарозпарювання при зниженому тиску одержують нічну фазу промивають Юмл води Об'єднані во123мг 5R, 6S - ізопропілідендиокси - 2 - азабицикдяні прошарки промивають Юмл дихлорметану ло - [2 2 1] гептан - 3 - ону, чистота якого, визначеОб'єднані органічні фази промивають Юмл насина за допомогою ВЕРХ, складає приблизно 92%, і ченого розчину хлориду натрію і потім сушать над структура якого підтверджена протонним ЯМР Na2SO4 Після фільтрування і концентрування до спектром сухого стану при зниженому тиску вихід складає Розчин Юг 5R, 6S - ізопропілідендиокси - 2 1,58г продукту, що містить 95% 2R, 3S - ізопропіліазабицикло-[2 2 1] гептан - 3 - ону в ЮОмл 70% дендиокси - 4R -трет-бутоксикарбоніламшо - 1S ного (по масі) водяного розчину етиламшу нагріетиламшокарбонілцикло-пентану, структура якого вають до 110°С протягом 20 годин при об'ємному підвладна ЯМР спектром у диметилсульфоксидітиску Після охолодження надлишок етиламшу сіб, що показав такі ХІМІЧНІ зсуви видаляють при зниженому тиску, потім промивають дихлорметаном для видалення вихідного не0,95 (т, ЗН), 1,14 (с, ЗН), 1,31 (с, 12Н), 1,55, прореагованого продукту Водяний прошарок потім 1Н), 2,11 (м, 1Н), 2,64(м, 1Н), 3,00 (до, 2Н), 3,77 (м, концентрують і сушать У такий спосіб одержують 1Н), 4,23 (м, 1Н), 4,54 (м, 1Н), 7,07 (д, 1Н), 8,12 (т, 10,54г 2R, ЗЗ-ізопропілідендиокси - 4R - аміно - 1S 1Н) - етиламшокарбонілциклопентану У 25мл-ову колбу поміщають 1,22г 2R, 3S ізопропілідендиокси 4R -третДійсний винахід може бути здійснений в інших бутоксикарбоніламшо - 1S - етиламшокарбонілциконкретних формах, не виходячи за рамки суті або клопентану і Юмл дихлорметану При температурі суттєвих ознак приблизно 25°С добавляють при перемішуванні ТОВ "Міжнародний науковий комітет" вул Артема, 77, м Київ, 04050, Україна (044)236-47-24
ДивитисяДодаткова інформація
Назва патенту англійськоюA method for preparation of a 2-azadihydroxy-bicyclo[2.2.1]heptane derivatives and l-tartaric acid salt thereof
Назва патенту російськоюСпособ получения производных 2-азадигидроксибицикло [2.2.1.] гептана и l-виннокислой соли производного
МПК / Мітки
МПК: C07D 209/52, C07D 209/02
Мітки: похідних, отримання, солей, l-винної, кислоти, 2-азадигідроксибіцикло[2.2.1]гептану, спосіб
Код посилання
<a href="https://ua.patents.su/11-52702-sposib-otrimannya-pokhidnikh-2-azadigidroksibiciklo221geptanu-i-kh-solejj-l-vinno-kisloti.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання похідних 2-азадигідроксибіцикло[2.2.1]гептану і їх солей l-винної кислоти</a>
Спосіб отримання похідних 7-[2-(2-амітіазоліл-4)2-оксііміноацетамідо]-3-ацетоксіметил-3-цефем-4карбонової кислоти чи її солей з лужними металами

Номер патенту: 4796
Опубліковано: 28.12.1994
Автори: Рене Ейме, Андре Лютц
МПК: C07D 501/00, A61K 31/545
Мітки: спосіб, лужними, кислоти, солей, похідних, 7-[2-(2-амітіазоліл-4)2-оксііміноацетамідо]-3-ацетоксіметил-3-цефем-4карбонової, отримання, металами
Спосіб одержання похідних (1н-імідазол-1-ілметіл)замішаного бензімідазола або їх фармацевтично прийнятих солей кислоти, або солей металів, або стереоізомерів

Номер патенту: 2706
Опубліковано: 26.12.1994
Автори: Жерар Шарль Санз, Едді Жан Едгард Фрейн, Альфонс Герман Маргарета Реймакерс
МПК: A61K 31/44, A61K 31/4433, A61K 31/47, A61P 17/00, A61P 35/00, C07D 409/14, A61K 31/443, A61K 31/4427, C07D 521/00, C07D 403/14, C07D 417/14, A61K 31/425, A61P 19/06, A61K 31/415, C07D 403/06, C07D 401/14, A61P 43/00, A61P 5/00, C07D 405/14
Мітки: металів, 1н-імідазол-1-ілметіл)замішаного, фармацевтично, кислоти, стереоізомерів, спосіб, прийнятих, бензімідазола, похідних, солей, одержання
Формула / Реферат:
Способ получения производных (1Н-имидазол-1-илметил)-замещенного бензимидазола общей формулы где R2 — водород, С1— С6-алкил, С3— С7-цикло-алкил, фенил, необязательно замещенный двумя заместителями, выбранными из гало-, С1— С4-алкила, С1— С4-алкилоксикарбонила, карбоксила или С1— С4-алкилокси, тиенилфуранил, галофуранил, имидазолил или пиридинил, R1 — водород, С3— С7 - циклоалкил, фенил, С4 - С6-алкил, необязательно замещенный...
Спосіб одержання похідних 2-тієнілоксіуксусної кислоти чи їх фармацевтично прийнятних солей

Номер патенту: 4227
Опубліковано: 27.12.1994
Автори: Франц Ровенсцкі, Дітер Біндер, Хуберт Петер Фербер
МПК: A61K 31/381, A61P 43/00, C07D 333/32, A61P 9/00, A61K 31/38, A61P 7/02, A61P 11/00, A61P 29/00
Мітки: фармацевтично, солей, прийнятних, одержання, спосіб, похідних, кислоти, 2-тієнілоксіуксусної
Формула / Реферат:
1. Способ получения производных 2-тиенил-оксиуксусной кислоты общей формулы где R - незамещенный или замещенный галогеном фенил, или их фармацевтически применимых солей, отличающийся тем, что соединение общей формулыгде R имеет указанные значения, подвергают окислению окисью серебра в водно-щелочной среде с последующим выделением целевого продукта в свободном виде или в виде фармацевтически применимой если....
Спосіб отримання похідних простацикліну чи їх солей

Номер патенту: 5164
Опубліковано: 28.12.1994
Автори: Герда Маннесман, Хельмут Форбрюгген, Бернд Радюхель, Вольфганг Лозерт, Хорке Казальс, Вернер Скубалла
МПК: A61P 43/00, C07D 309/12, A61P 25/02, C07C 45/67, C07C 62/00, C07D 319/00, C07C 405/00, C07D 317/72, A61K 31/557, C07C 401/00, C07C 51/00, C07C 67/343, C07C 45/71, C07C 69/732, A61K 31/5575, C07C 45/59, C07C 67/00, A61P 7/02, C07C 51/347, C07C 59/00
Мітки: простацикліну, похідних, солей, отримання, спосіб
Формула / Реферат:
Способ получения производных простациклина общей формулы где R1 - водород или низший алкил; А - этилен, транс-винилен или СºС-группа;W - свободный или защищенный тетрагидропира-нильной группой оксиметиленовый радикал или радикалгде гидроксил может находиться в а - или b-положении;D и Е вместе - прямая связь, или D-прямая или разветвленная алкиленовая группа с 1-6 атомами углерода,...
Спосіб отримання алкенових похідних чи їх солей

Номер патенту: 5558
Опубліковано: 28.12.1994
Автори: Арто Йоханнес Карьялайнен, Лаурі Вейкко Матті Кангас, Гуіллєрмо Луіс Бланко, Ханну Каллєрво Сундівіст, Мар'я-Лііса Сьодервалл, Кауоко Ойва Антеро Куркела, Реійо Юхані Тоівола
МПК: C07C 11/00
Мітки: похідних, солей, спосіб, алкенових, отримання
Формула / Реферат:
Способ получения алкеновых производных общей формулыгде R1 и R2 - водород или гидроксил; R3 - водород,гидроксил,метоксил или 2-(N,N-диметиламино) этоксил; R4 - хлор или бром, n = 1 или 2,или их солей отличающийся тем, что соединение общей формулыгде R5 - тетращдропиран-2-илокси- или бензилокси; R6 - водород или тетрагидропиран-2-илокси; R7 - водород, метокси или...