Похідні бензімідазолону та хіназолінону як агоністи людських рецепторів orl1
Номер патенту: 83868
Опубліковано: 26.08.2008
Автори: Ден Гарток Якобус А.Й., Тьойнстра Тинка, ван Шаренбург Густав Й.М., Стьойвенберг Герман Г., Терпстра Ян-Вілем




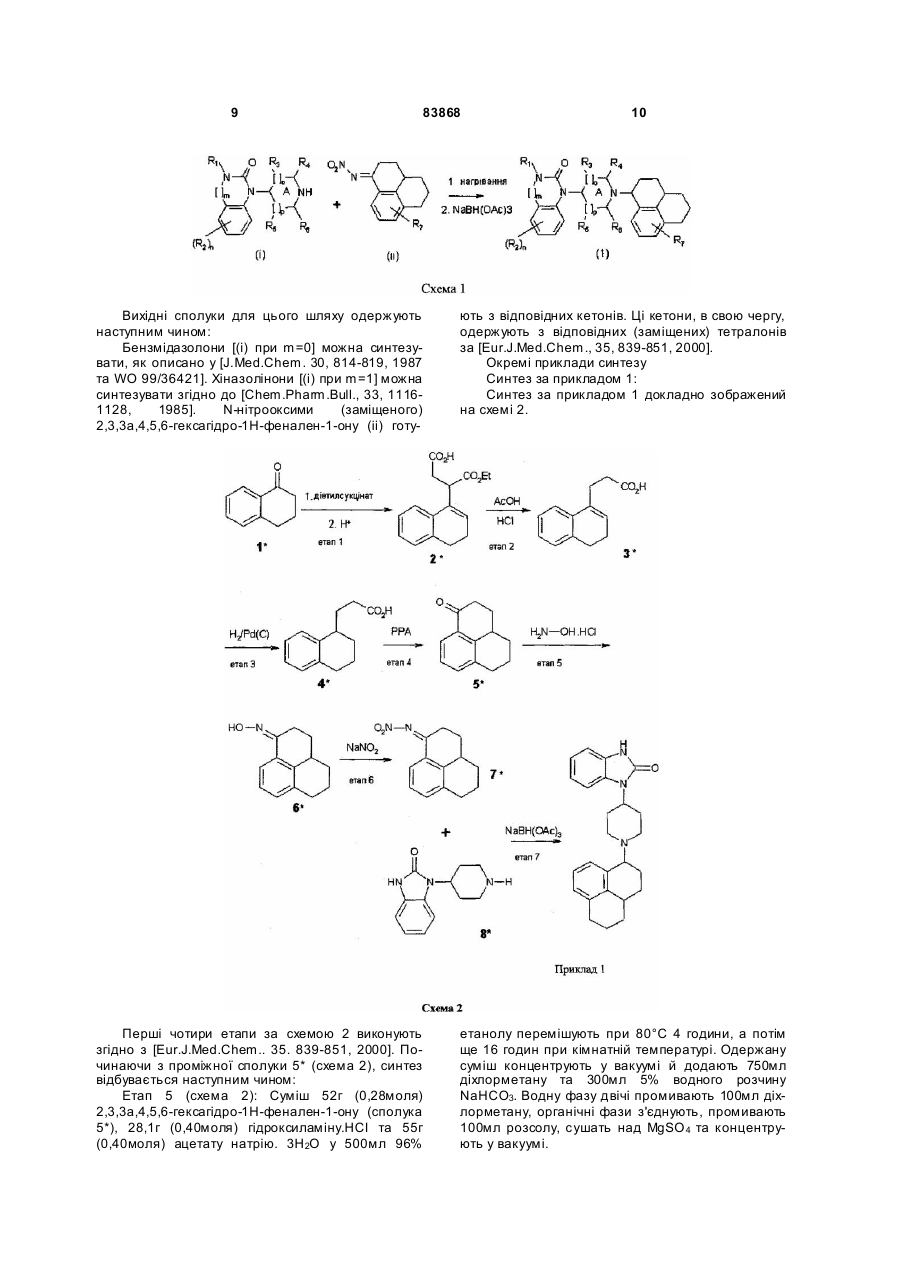


Формула / Реферат
1.Сполуки загальної формули (1)
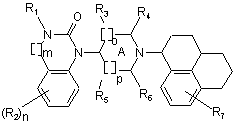 , (1)
, (1)
де:
R1 представляє Н, (1-6С)алкіл, (1-3С)алкіл(3-6С)циклоалкіл, (2-7С)карбалкокси або (2-7С)ацил,
[]m позначає -(СH2)m, де m - 0 або 1,
R2 представляє галоген, СF3, (1-6С) алкіл, (1-3С)алкіл(3-6С)циклоалкіл, феніл, амін, (1-3С)аміноалкіл, алкіл(1-3C)амін, діалкіл(1-3C)амін, ціан, (1-3C)ціаноалкіл, гідрокси, (1-3C)гідроксіалкіл, (1-3C)алкокси, ОСF3, (2-7С)ацил, трифторацетил, амінокарбоксил, (1-3C)алкілсульфоніл або трифторметилсульфоніл,
n - ціле число, що дорівнює 0-4, за умови, що, коли n дорівнює 2, 3 або 4, замісники можуть бути однакові або різні,
А - насичене або частково насичене кільце,
[]о-[]р представляють відповідно -(СН2)о та -(СН2)p за умови, що таке значення -СН- є можливим, коли А - частково насичене кільце, а о та р незалежно один від одного дорівнюють 0, 1 або 2,
R3, R4, R5 та R6 незалежно один від одного представляють водень, (1-3C)алкіл, (1-3C)алкіл(3-6С)циклоалкіл, СН2ОН, або R3 та R5 або R3 та R6, або R4 та R5, або R4 та R6 разом утворюють алкіленовий місток з 1-3 атомами вуглецю за умови, що, коли о = 2, R3 - водень, а коли р = 2, R5 - водень,
R7 представляє Н, галоген, СF3, (1-6С)алкіл, (1-3C)алкіл(3-6С)циклоалкіл, амін, (1-3C)аміноалкіл, алкіл(1-3C)амін, діалкіл(1-3C)амін, гідрокси, (1-3C)гідроксіалкіл, (1-3C)алкокси, ОСF3, (2-7С)ацил, амінокарбоксил або (1-3C)алкілсульфоніл,
усі стереоізомери, а також фармацевтично прийнятні солі та проліки, що є похідними сполук формули (1), де присутня група, яка легко відщеплюється після введення до організму, наприклад, такі як амідин, енамін, основа Манніха, похідне гідроксилметилену, похідне О-(ацилоксиметиленкарбамату), карбамат або енамінон.
2. Сполуки за п. 1 загальної формули (1), у яких: А - насичене кільце, R1 представляє водень, (1-3C)алкіл або (2-4С)ацил, R3, R4, R5 та R6 незалежно один від одного представляють водень або (1-3C)алкіл, або R3 та R5 або R3 та R6, або R4 та R5 або R4 та R6 разом можуть утворювати алкіленовий місток з 1-3 атомами вуглецю, за умови, що при о = 2 R3 - водень, при р=2 R5 - водень, R7 представляє Н, галоген, СF3, (1-3C)алкіл, амін, (1-3C)аміноалкіл, алкіл(1-3C)амін, діалкіл(1-3C)амін, гідрокси, (1-3C)алкокси, ОСF3, а R2, m, n, о та р мають значення, наведені вище.
3. Сполуки за п. 1 загальної формули (1), у яких: А - насичене кільце, m = 0, n = 0 або 1, р = 1, q = 0, R1 - Н або ацетил, R2 представляє галоген, СF3, (1-3C)алкіл, амін, ціан, ОСН3 або ОСF3, R3, R4, R5 та R6 незалежно один від одного представляють водень або (1-2С)алкіл, або R4 та R6 разом можуть утворювати алкіленовий місток з 1-2 атомами вуглецю, а R7 - Н, галоген, СF3, (1-3C)алкіл, амін, гідрокси або ОСF3.
4. Сполуки за п. 1 загальної формули (2) та їх стереоізомери
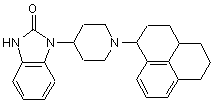 . (2)
. (2)
5. Сполуки загальної формули (3)
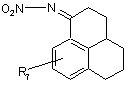 , (3)
, (3)
де R7 має значення за п. 1,
придатні для синтезу сполук загальної формули (1).
6. Сполука, як заявлено в будь-якому з пп. 1-4, або її сіль для застосування у лікарських засобах.
7. Фармацевтична композиція, яка містить фармакологічно дієву кількість принаймні однієї зі сполук, як заявлено в будь-якому з пп. 1-4, як діючу речовину.
8. Застосування сполуки, як заявлено в будь-якому з пп. 1-4, для приготування фармацевтичної композиції для лікування розладів, пов'язаних з рецепторами ORL1, або таких, що їх можна лікувати дією на ці рецептори.
9. Застосування за п. 8, яке відрізняється тим, що зазначені розлади включають: гострі та хронічні больові стани, розлади центральної нервової системи, як-от симптоми бентежності та стресу, депресії, різні форми епілепсії, інсульту, розлади, які характеризуються пошкодженням розуму та пам'яті, такі як хвороба Альцгеймера, хвороба Крейцфельда-Якоба, хвороба Гантінгтона, хвороба Паркінсона, нейрореабілітація (травматичні пошкодження мозку), гострі травми мозку або хребта, розлади хімічного походження, в тому числі пов'язані із зловживанням певними речовинами (наркозалежність, алкоголізм) або спричинені таким зловживанням (синдром відвикання), розлади харчування (нервова анорексія, нервова булімія, опасистість); шлунково-кишкові розлади, зокрема синдром подразнення кишечнику, запальні хвороби кишечнику (хвороба Крона) та виразковий коліт, запалення сечового тракту, ниркові розлади, що характеризуються дисбалансом вживання-видалення води або видалення солей; серцево-судинні захворювання, як інфаркт міокарда, аритмія, гіпертензія, тромбоз, анемія, атеросклероз, грудна жаба, захворювання шкіри, як кропивниця, вовчак та сверблячка; офтальмологічні розлади, як глаукома; респіраторні захворювання, включаючи хронічну обструкцію легенів, бронхіт, цистофіброз; захворювання імунної системи та вірусні інфекції.
Текст
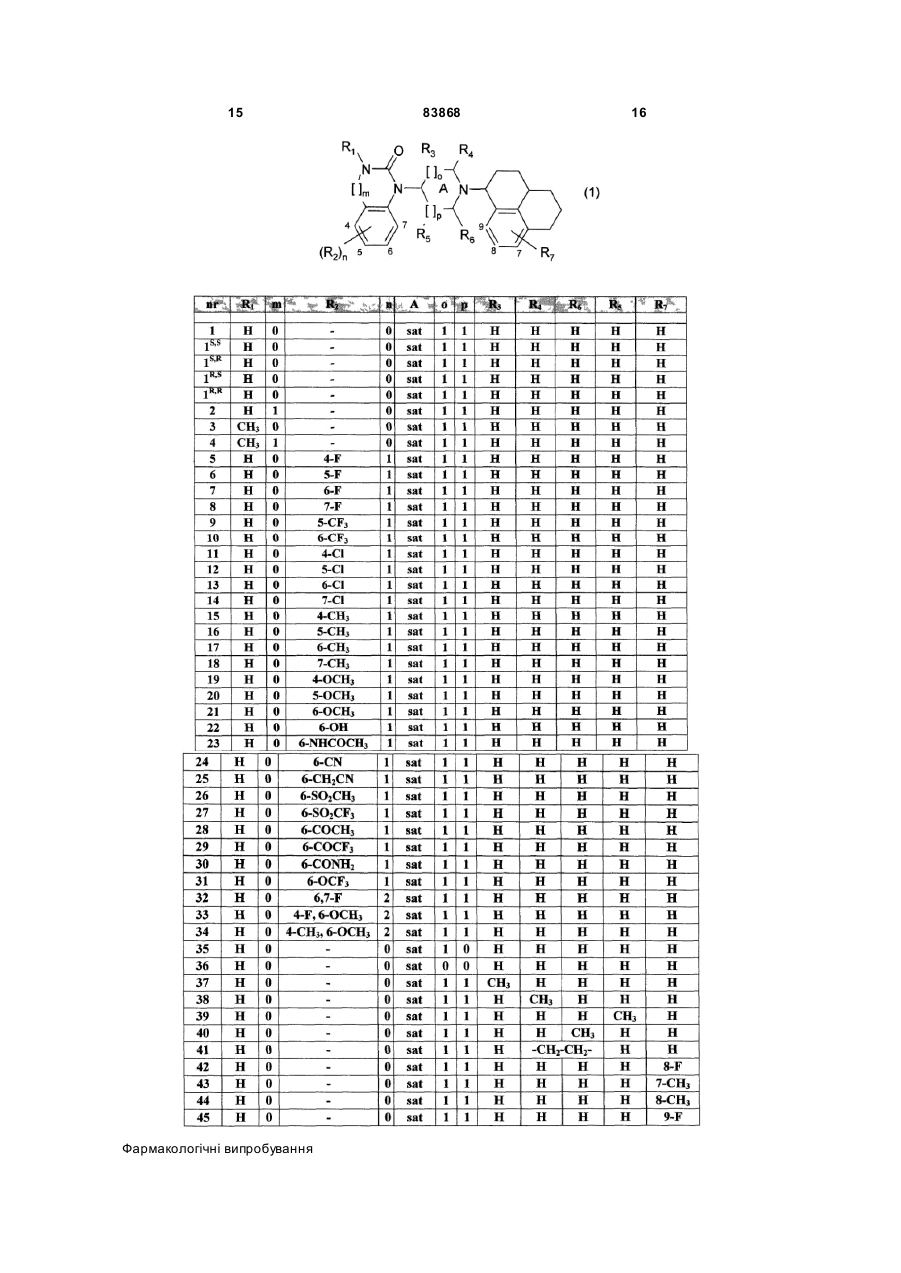
1.Сполуки загальної формули (1) R1 R3 R4 O C2 2 (19) 1 3 83868 3. Сполуки за п. 1 загальної формули (1), у яких: А - насичене кільце, m = 0, n = 0 або 1, р = 1, q = 0, R1 - Н або ацетил, R2 представляє галоген, СF3, (13C)алкіл, амін, ціан, ОСН3 або ОСF3, R3, R4, R5 та R6 незалежно один від одного представляють водень або (1-2С)алкіл, або R4 та R6 разом можуть утворювати алкіленовий місток з 1-2 атомами вуглецю, а R7 - Н, галоген, СF3, (1-3C)алкіл, амін, гідрокси або ОСF3. 4. Сполуки за п. 1 загальної формули (2) та їх стереоізомери O HN N N 5. Сполуки загальної формули (3) N O2 N (2) . (3) R7 , де R7 має значення за п. 1, придатні для синтезу сполук загальної формули (1). 6. Сполука, як заявлено в будь-якому з пп. 1-4, або її сіль для застосування у лікарських засобах. 7. Фармацевтична композиція, яка містить фармакологічно дієву кількість принаймні однієї зі сполук, як заявлено в будь-якому з пп. 1-4, як діючу речовину. Винахід стосується групи нових похідних бензімідазолону та хіназолінону, які є агоністами людських рецепторів ORL1 (ноціцептину). Винахід стосується також одержання цих сполук, фармаційних композицій, які містять фармакологічно дійову кількість принаймні одного з цих похідних бензімідазолону та хіназолінону в якості діючої речовини, а також застосування цих фармаційних композицій для лікування розладів, пов'язаних з рецепторами ORL1. Винахід стосується застосування сполуки, що описана в цьому винаході, для ви готовлення лікарського засобу, що дає корисний ефект. Корисний ефект роз'яснюється в описі або є очевидним для фа хівця на підставі змісту опису та професійного досвіду. Винахід також стосується застосування сполуки за винаходом для одержання засобу для лікування або профілактики певного захворювання або стану. Зокрема, винахід стосується нового застосування для лікування хвороби або стану, який роз'яснюється в описі або є очевидним для фахівця на підставі змісту опису та професійного досвіду. У варіантах здійснення винаходу окремі сполуки, що описуються тут, використовуються для виготовлення лікарського засобу. 4 8. Застосування сполуки, як заявлено в будьякому з пп. 1-4, для приготування фармацевтичної композиції для лікування розладів, пов'язаних з рецепторами ORL1, або таких, що їх можна лікувати дією на ці рецептори. 9. Застосування за п. 8, яке відрізняється тим, що зазначені розлади включають: гострі та хронічні больові стани, розлади центральної нервової системи, як-от симптоми бентежності та стресу, депресії, різні форми епілепсії, інсульту, розлади, які характеризуються пошкодженням розуму та пам'я ті, такі як хвороба Альцгеймера, хвороба Крейцфельда-Якоба, хвороба Гантінгтона, хвороба Паркінсона, нейрореабілітація (травматичні пошкодження мозку), гострі травми мозку або хребта, розлади хімічного походження, в тому числі пов'язані із зловживанням певними речовинами (наркозалежність, алкоголізм) або спричинені таким зловживанням (синдром відвикання), розлади харчування (нервова анорексія, нервова булімія, опасистість); шлунково-кишкові розлади, зокрема синдром подразнення кишечнику, запальні хвороби кишечнику (хвороба Крона) та виразковий коліт, запалення сечового тракту, ниркові розлади, що характеризуються дисбалансом вживаннявидалення води або видалення солей; серцевосудинні захворювання, як інфаркт міокарда, аритмія, гіпертензія, тромбоз, анемія, атеросклероз, грудна жаба, захворювання шкіри, як кропивниця, вовчак та сверблячка; офтальмологічні розлади, як глаукома; респіраторні захворювання, включаючи хронічну обструкцію легенів, бронхіт, цистофіброз; захворювання імунної системи та вірусні інфекції. "Подібний до рецептора опіатів 1" (ORL1) рецептор був виділений з бібліотеки людської цДНК. Встановлено, що цей "рецептор-одинак" за гомологією нагадує рецептори m-, k- та d-опіатів [Mollereau et al., FEBS Lett. 341. 33-38, 1994; Bunzow et al., FEBS Lett.. 347. 284-288, 1994]. Незважаючи на близькість рецепторів ORL1 за послідовностями та структурою до рецепторів опіатів, класичні ліганди рецепторів опіатів з ними не взаємодіють. У 1995р. з екстрактів мозку був виділений чистий нейропептид із 17 амінокислот, який, як доведено пізніше, був природним лігандом пов'язаного з G-протеїном рецептора ORL1 [Reinscheid et al., Science. 270. 792-794,1995; Meunier et al., Nature. 377. 532-535, 1995]. Цей пептид назвали орфанином FQ або ноціцептином, і він не зв'язується з жодним з трьох традиційних рецепторів опіатів. Це відкриття породило значний обсяг досліджень функціональної ролі рецептора ORL1 та пошук його нових лігандів. З'явилося кількасот публікацій, включаючи кілька оглядів [наприклад, Grond et al., Anaesthesist 51, 996-1005, 2002], та кількадесят патентних заявок, де описуються пептидні та непептидні ліганди. Описані сполуки суттєво відрізняються за потужністю щодо рецепторів ORL1 та вибірковістю (ORL1 на тлі m-опіатів). Оскільки рецептори m-опіатів розсіяні по всьому орга 5 83868 нізму, недостатня вибірковість може викликати низку небажаних бічних яви щ, схожих на дію опіатів, наприклад, сонливість, респіраторну депресію, переносність та залежність ["Drug News Perspect, 14. 335, 2001]. Фармакодинамічні та фармакокінетичні властивості описаних сполук in vivo також є дуже різними. Низка пов'язаних з ORL1 патентних заявок стосуються похідних бензімідазолону: наприклад, [WO 98/54158, WO 99/36421, WO 00/006545, WO 00/08013, WO 01/39775 та US 20020128288]. Найближчим прототипом цього винаходу є заявка WO 01/39775. Втім, описані там похідні бензімідазолону навряд чи відповідають загальновизнаним суттєвим критеріям для терапевтичних засобів на базі ORL1. Вони характеризуються тим, що: 1) не досить потужні (спорідненість до рецепторів ORL1 у межах 166-1252нМ); 2) є маловибірковими щодо рецепторів mопіатів (спорідненість у межах 19-457нМ); 3) непридатні для орального введення; 4) не засвоюються ЦНС. Найпотужнішим агоністом ORL1 є Ro 64-6198. Ця сполука не містить бензімідазолонову частину, а замість неї має спіроядро [EP 0856514; Eur.J.Med.Chem., 35 (2000), 839-851 та Proc.Natl.Acad.Sci.USA, 2000, 97, 4938]. Ro 646198 - це могутня і вибіркова сполука, яка легко долає бар'єр між мозком та кров'ю. Однак, незважаючи на гарне зв'язування in vitro, поведінка цього ліганду in vivo пов'язана з певними вадами: 1) у моделях бентежності він виявляється менш ефективним, ніж прогнозувалося на підставі даних in vitro; 2) терапевтичне вікно між бажаною ефективністю агоніста ORL1 та небажаними бічними діями опіатів in vivo виявляється вужчим, ніж прогнозувалося на підставі даних in vitro. З опублікованих даних про похідні бензімідазолону та Ro 64-6198, на які ми посилалися вище, не можна визначити шляхів поліпшення фармакологічних характеристик найкращих із описаних сполук. В огляді на цю тему [Zaveri et al., Characterization of opiates, neurolepics and synthetic analogs at ORL1 and opioid receptors, Eur.J.Pharmacol., 428, 29-36, 2001] стверджується: "За відсутності моделі малої молекули в активному сайті або структури кристалу зв'язаного з малою молекулою рецептора ORL1 треба бути дуже обережним при оцінці серед різних класів лігандів рецепторів ORL1, навіть близьких за структурою". Нами несподівано встановлено, що група сполук серед похідних бензімідазолону та хіназолінону з новими комбінаціями замісників посідає дуже високу спорідненість до людських рецепторів ORL1. Більш того, ці сполуки є напрочуд вибірковими до рецепторів ORL1 відносно до рецепторів m-опіатів, придатні для орального введення та долають бар'єр між кров'ю та мозком. Фармакологічні характеристики in vitro та in vivo кількох із цих сполук є значно кращі, ніж у Ro 64-6109, особливо щодо терапевтичного вікна між бажаною ефективністю агоніста ORL1 та небажаними бічними діями 6 опіатів in vivo. Винахід стосується сполук загальної формули (1): де: R1 представляє H, (1-6C) алкіл, (1-3С)алкіл(36С)циклоалкіл, (2-7С)карбалкокси або (2-7С)ацил, []m позначає -(CH2)m, де m - 0 або 1, R2 представляє галоген, CF3, (1-6C) алкіл, (13С)алкіл(3-6С)циклоалкіл, феніл, амін, (13С)аміноалкіл, алкіл(1-3С)амін, діалкіл(1-3С)амін, ціан, (1-3С)ціаноалкіл, гідрокси, (13С)гідроксиалкіл, (1-3С)алкокси, OCF3, (27С)ацил, трифторацетил, амінокарбоксил, (13C)алкілсульфоніл або трифторметилсульфоніл, n - ціле число, що дорівнює 0-4, за умови, що при n=2, 3 або 4 замісники можуть бути однакові або різні, А - насичене або частково насичене кільце, []o [] p представляють відповідно -(СН2)о та (СН2)р за умови, що таке значення -CH- є можливим, коли А - частково насичене кільце, а о та р незалежно один від одного дорівнюють 0, 1 або 2, R3, R4, R5 та R6 незалежно один від одного представляють водень, (1-3C)алкіл, (1-3С)алкіл(36С)циклоалкіл. CH2OH, або (R3 та R5), або (R3 та R6), або (R4 та R5), або (R4 та R6) разом утворюють алкіленовий місток з 1-3 атомами вуглецю за умови, що, коли о=2, R3 - водень, а коли р=2, R5 - водень, R7 представляє H, галоген, CF3, (1-6C)алкіл, (1-3С)алкіл(3-6С)циклоалкіл, амін, (13С)аміноалкіл, алкіл(1-3С)амін, діалкіл(1-3С)амін, гідрокси, (1-3С)гідроксиалкіл, (1-3С)алкокси, OCF3, (2-7С)ацил, амінокарбоксил або (13C)алкілсульфоніл, та їх фармаційно прийнятні солі та проліки. Винахід охоплює усі сполуки формули (1), рацемати, суміші діастереоізомерів та окремі стереоізомери. Отже, сполуки, у яких замісники потенційно асиметричних атомів вуглецю знаходяться у Rконфігурації або у S-конфігурації, належать до обсягу винаходу. Також до нього належать проліки, тобто сполуки, які, потрапляючи до організму будь-яким відомим шляхом, метаболізуються у сполуки формули (1). Проліки - це біозворотні похідні молекул лікарських засобів, здатні долати певні бар'єри на користь молекули вихідного засобу. До таких бар'єрів належать, зокрема, розчинність, проникність, стабільність, досистемний метаболізм та обмеження прицільності [J.Stella, Prodrugs as therapeutics, Expert Opin.Ther.Patents, 14 (3), 277-280, 2004]. Зокрема, це стосується сполук з первинними або вторинними аміно-або гідроксигрупами. Такі сполуки здатні реагувати з органічними кислотами, даючи сполуки формули (1), де присутня додаткова група, яка легко відщеплю 7 83868 ється після введення до організму, наприклад, такі, як амідин, енамін, основа Манніха, похідне гідроксилметилену, похідне О(ацілоксіметиленкарбамату), карбамат, ефір, амід або енамінон. Проліки- це неактивна сполука, яка набуває активної форми після засвоєння організмом [Medicinal Chemistry: Principles та Practice, 1994, ISBN 0-85186-494-5, Ed.: F. D. King, p.216]. Зокрема, винахід стосується сполук формули (1), у яких: А - насичене кільце, R1 представляє водень, (1-3С)алкіл або (2-4С)ацил, R3, R4, R5 та R6 незалежно один від одного представляють водень, або (1-3С)алкіл, або (R3 та R5), або (R3 та R6), або (R4 та R5), або (R4 та R6) разом можуть утворювати алкіленовий місток з 1-3 атомами вуглецю, за умови, що при о=2 R 3 - водень, при р=2 R5 - водень, R7 представляє H, галоген, CF 3, (1-3С) алкіл, амін, (1-3С)аміноалкіл, алкіл(1-3С)амін, діалкіл(1-3С)амін, гідрокси, (1-3С)алкокси, OCF3, a R2, m, n, о, та р мають значення, наведені вище. Зокрема, винахід стосується сполук формули (1), у яких: А - насичене кільце, m=0, n=0 або 1, р=1, q=0, R1 - H або ацетил, R2 представляє галоген, CF3, (1-3С)алкіл, амін, ціан, OCH3 або OCF3, R3, R4, R5 та R6 незалежно один від одного представляють водень або (1-2С)алкіл, або (R4 та R6) разом можуть утворювати алкіленовий місток з 1-2 атомами вуглецю, a R7 - H, галоген, CF3, (1-3С) алкіл, амін, гідрокси або OCF3. Найприйнятнішою є сполука формули (2) та її стереоізомери. Надалі ця сполука називатиметься "приклад 1". Сполукам за винаходом можна надавати форм, придатних для введення до організму, за допомогою відомих способів з використанням допоміжних речовин та/або рідких або твердих носіїв. Фармаційно прийнятні солі можна одержувати звичайними способами, добре відомими фахівцям з рівня техніки, наприклад, змішуванням сполуки за винаходом з відповідною кислотою. Придатні кислі адуктні солі можна одержувати з неорганічними кислотами, як от соляною, або з органічними кислотами, наприклад, фумаровою. Сполуки за винаходом загальної формули (1), а також їхні солі, посідають агоністичну активність щодо ORL1. Вони придатні для лікування розладів, пов'язаних з рецепторами ORL1 або таких, що 8 їх можна лікувати, діючи на ці рецептори. Наприклад, це стосується гострих та хронічних больових станів, розладів центральної нервової системи, як от послаблення симптомів бентежності, депресії, різних форм епілепсії, інсульту, розладів, які характеризуються пошкодженням розсудку та пам'яті, хвороби Альцгеймера, хвороби КрейцфельдаЯкоба, хвороби Гантінгтона, хвороби Паркінсона, нейрореабілітації (травматичні пошкодження мозку), гострих травм мозку або хребта, розладів хімічного походження, в тому числі пов'язаних із зловживанням певних речовин (наркозалежність, алкоголізм) або спричинених таким зловживанням (синдром відвикання), розладів харчування (нервова анорексія, нервова булімія, опасистість); шлунково-кишкових розладів, зокрема, синдрому подразнення кишковика, запалювальних хвороб кишковика (хвороба Крона, виразковий коліт), запалення сечового тракту, ниркові розлади, що характеризуються дисбалансом вживаннявидалення води або видалення солей; серцевосудинних захворювань, таких як інфаркт міокарду, аритмія, гіпертензія, тромбоз, анемія, атеросклероз, грудна жаба, захворювань шкіри, таких як кропивниця, вовчанка та сверблячка; офтальмологічних розладів, таких як глаукома; респіраторних захворювань, включаючи хронічну обструкцію легенів, бронхіт, цистофіброз; захворювань імунної системи та вірусних інфекцій. Сполуки за винаходом звичайно вводять у складі фармаційних композицій, які становлять важливу частину винаходу й посідають новизну завдяки наявності сполук, зокрема, окремих сполук за цим винаходом. Серед видів фармаційних композицій можна навести, зокрема, таблетки, жувальні таблетки, капсули, розчини, парентеральні розчини, супозиторії, суспензорії та інші, описані тут або очевидні для фахівця після ознайомлення з описом на підставі загальних професійних знань з рівня техніки. У варіантах здійснення винаходу передбачається фармаційний набір в одному або кількох упакуваннях, заповнених одним або кількома компонентами фармаційної композиції згідно з винаходом. До таких упакувань можуть додаватися різноманітні друковані матеріали, наприклад, інструкції з застосування, або повідомлення за формою, встановленою державним органом, який регулює виробництво, застосування або продаж фармаційної продукції, що містить дозвіл цього органу на виготовлення, застосування або продаж препарату для споживання людьми або тваринами. Загальна характеристика синтезу Сполуки за винаходом та їх солі можна одержати загальним шляхом, зображеним на схемі 1. 9 83868 10 Вихідні сполуки для цього шляху одержують наступним чином: Бензмідазолони [(і) при m=0] можна синтезувати, як описано у [J.Med.Chem. 30, 814-819, 1987 та WO 99/36421]. Хіназолінони [(і) при m=1] можна синтезувати згідно до [Chem.Pharm.Bull., 33, 11161128, 1985]. N-нітрооксими (заміщеного) 2,3,3а,4,5,6-гексагідро-1Н-фенален-1-ону (іі) готу ють з відповідних кетонів. Ці кетони, в свою чергу, одержують з відповідних (заміщених) тетралонів за [Eur.J.Med.Chem., 35, 839-851, 2000]. Окремі приклади синтезу Синтез за прикладом 1: Синтез за прикладом 1 докладно зображений на схемі 2. Перші чотири етапи за схемою 2 виконують згідно з [Eur.J.Med.Chem.. 35. 839-851, 2000]. Починаючи з проміжної сполуки 5* (схема 2), синтез відбувається наступним чином: Етап 5 (схема 2): Суміш 52г (0,28моля) 2,3,3а,4,5,6-гексагідро-1Н-фенален-1-ону (сполука 5*), 28,1г (0,40моля) гідроксиламіну.НСІ та 55г (0,40моля) ацетату натрію. 3Н2О у 500мл 96% етанолу перемішують при 80°C 4 години, а потім ще 16 годин при кімнатній температурі. Одержану суміш концентрують у вакуумі й додають 750мл діхлорметану та 300мл 5% водного розчину NaHCO3. Водну фазу двічі промивають 100мл діхлорметану, органічні фази з'єднують, промивають 100мл розсолу, сушать над MgSO 4 та концентрують у вакуумі. 11 83868 12 Етап 6 (схема 2): 58,8г (вихід 100%) одержаного білястого твердого оксиму (сполука 6*) суспендують у 600мл трет-бутилметилетеру. При кімнатній температурі до цієї суспензії додають 230мл розчину 41,4г (0,6моля) нітриту натрію у воді, а потім 290мл 2N розчину сірчаної кислоти. Після перемішування 16 годин при 40°C суміш охолоджують до кімнатної температури та додають 300мл насиченого водного розчину NaHCO3. Водну фазу двічі екстрагують 300мл третбутилметилетеру, органічні фази з'єднують, промивають 150мл розсолу, сушать над MgSO 4 та концентрують у вакуумі. Одержану коричневу олію очищують колонковою хроматографією (силікагель) з елюентом діхлорметаном. Одержану олію після концентрування у вакуумі розтирають з циклогексаном, одержаний осад відфільтровують та сушать. Одержують 34г (0,148моля) білястого твердого чистого NO2-імінy (сполука 7*) (вихід 52%) з точкою топлення 64-69°C. Етап 7 (схема 2): Суміш 6,51г (30ммолів) 4-(1бензімідазолон)піперидину (сполука 8* фірми ACROS), 6,9г (30ммолів) NO2-імінy (сполука 7*) та 5,25мл диізопропилетиламіну у 450мл 1,2діхлоретану нагрівають при 50°C та перемішують 16 годин у атмосфері азоту. Після охолодження до кімнатної температури додають 12,7г (60ммолів) NaBH(OАс)3 і одержану суміш перемішують 24 години в атмосфері азоту. Після концентрування реакційної суміші у вакуумі додають при перемішуванні 500мл діхлорметану та 500мл 5% водного розчину NaHCO3. Водну фазу двічі промивають 100мл діхлорметану, органічні фази з'єднують, промивають 100мл розсолу, сушать над MgSO4 та концентрують у вакуумі. Сирий продукт очищують колонковою хроматографією (силікагель) з суміш шю діхлорметан-метанол-аміак (92:7,5:0,5) у ролі елюента. Після концентрування у вакуумі одержують 8,09г (21ммоль) чистого продукту (ви хід 70%), точка топлення якого 155-158°C. До 8,09г (21ммоль) чистого продукту додають 60мл етанолу та 2,44г (21ммоль) фумарової кислоти. Концентруванням одержаного розчину у вакуумі одержують 10,53г (21ммоль) фумарату сполуки за прикладом 1 (вихід 100%) у вигляді білої піни з M+ 358 та точкою топлення 232-234°C. Синтез за прикладом 2: Суміш 2,31г (10ммолів) 4-(1хіназолинон)піперидину та 2,3г (10ммолів) NO2імінy (сполука 7*) у 100мл 1,2-діхлоретану в атмосфері азоту нагрівають з перемішуванням 7 годин при 50°C і потім 16 годин при кімнатній температурі. Після того додають 4,2г (20ммолів) NaBH(OАс)3 та перемішують суміш при кімнатній температурі в атмосфері азоту 24 години. Одержану суміш концентрують у вакуумі й додають 200мл діхлорметану та 200мл 5% водного розчину NaHCO3. Водну фазу двічі промивають 40мл діхлорметану, органічні фази з'єднують, промивають 40мл розсолу, сушать над MgSO4 та концентрують у вакуумі. Сирий продукт очищують колонковою хроматографією (силікагель) з сумішшю діхлорметан-метаноламіак (94,5:5:0,5) у ролі елюента. Після концентрування у вакуумі 2,5г (6,2ммоля) очищеного продукту розчиняють у 50мл розчину HCl в етанолі. Концентруванням у вакуумі одержують 2,05г (4,7ммоля) солі HCl за прикладом 2 (вихід 47%) у вигляді білої аморфної маси з M+ 402 та точкою топлення 172-180°C. Синтез за прикладом 13: Синтез за прикладом 13 детально показаний на схемі 3: Етап 1 (схема 3): Розчин 3,84г (20ммолів) 2,4діхлорнітробензолу (сполука 9* фірми Aldrich), 4,1мл (20ммолів) 4-амін-1-бензилпіперидину (сполука 10* фірми Aldrich), 4,46г (32ммоля) К2СО3 у 50мл діметилформаміду перемішують 18 годин при 95°C в атмосфері азоту. Після охолодження до кімнатної температури суміш виливають у 150мл води-250мл діхлорметану. Водну фазу двічі 13 83868 екстрагують 50мл діхлорметану, органічні фази з'єднують, двічі промивають 50мл води, сушать над MgSO4 та концентрують у вакуумі. Отриманий сирий продукт очищують колонковою хроматографією (силікагель) з сумішшю діхлорметан-метанол (97:3) у ролі елюента. Після концентрування у вакуумі одержують 4,8г (13,8ммоля) чистого продукту у вигляді жовтої олії (вихід 69%). Етап 2 (схема 3): частину скелетного нікелевого каталізатора гідрування (біля 500мг R 2800 фірми Aldrich [7440-02-0]) двічі промивають 10мл 96% етанолу, після чого додають в атмосфері азоту до розчину 4,8г (13,8ммолів) сполуки 11* у 200мл 96% етанолу. Розчин гідрують при кімнатній температурі та атмосферному тиску 2,5 години. Далі суміш фільтрують крізь Hyflo, промивають 300мл 96% етанолу, фільтрат концентрують у вакуумі та одержують 4,36г (13,8ммоля) сполуки 12* у вигляді барвистої олії (ви хід 100%). Етап 3 (схема 3): до розчину 4,36г (13,8ммолів) сполуки 12*продукту за попереднім етапом у 200мл ацетонітрилу додають при перемішуванні в атмосфері азоту при кімнатній температурі 3,36г (20,7ммолів) 1,1-карбонілдиімідазолу (CDI фірми ACROS). Через 10 хвилин починає утворюватися осад і росте 3 години, після чого його відфільтровують, промивають 200мл ацетонітрилу та сушать у вакуумі, одержуючи 3,30г (9,7ммолів) майже чистої сполуки 13* (вихід 70%). Етап 4 (схема 3): до суспензії 3,30г (9,7ммолів) сполуки 13* у 90мл 1,2-діхлоретану додають по краплинах в атмосфері азоту при охолодженні до 0°C та перемішуванні частину (1,17мл, 10,7ммолів) 1-хлоретилхлорформіату. Після перемішування протягом 30 хвилин при 0°C та 90 хвилин при 80°C суміш знов охолоджують до 0°C та додають по краплинах другу частин у (1,17мл, 10,7ммолів) 1-хлоретилхлорформіату. Суміш знову перемішують протягом 30 хвилин при 0°C та 16 годин при 80°C. Після охолодження до кімнатної температури суміш концентрують у вакуумі й до 14 дають до осаду 75мл метанолу. Одержаний розчин перемішують 1 годину при 65°C та концентрують у вакуумі. Після додання 75мл діхлорметану одержана бура напівтверда маса затвердіває при перемішуванні протягом 1 години. Осад відфільтровують, промивають 100мл діхлорметану та сушать. Сирий продукт очищують колонковою хроматографією (силікагель) з сумішшю діхлорметанметанол-аміак (92:7,5:0,5) у ролі елюента. Після концентрування у вакуумі одержують 1,61г (6,4ммоля) білої твердої сполуки 14* (вихід 66%). Етап 5 (схема 3): суміш 1,61г (6,4ммоля) сполуки 14* та 1,47г (6,4ммоля) NO2-імінy (сполука 7*) у 200мл 1,2-діхлоретану в атмосфері азоту нагрівають з перемішуванням при 50°C 16 годин. Після охолодження до кімнатної температури до одержаної суміші додають 2,76г (13ммолів) NaBH(OАс)3 та перемішують в атмосфері азоту при кімнатній температурі протягом 24 годин. Трохи забарвлений розчин виливають до суміші 300мл діхлорметану, 100мл води та 50мл 5% водного розчину NaHCO3. Водну фаз у двічі промивають 70мл діхлорметану, органічні фази з'єднують, сушать над MgSO4 та концентрують у вакуумі. Сирий продукт очищують колонковою хроматографією (силікагель) з сумішшю діхлорметан-метаноламіак (92:7,5:0,5) у ролі елюента. Чистий продукт концентрують у вакуумі та стверджують випарюванням разом з ацетонітрилом. Після перемішування у 100мл диізопропилетеру осад відфільтровують та сушать, одержуючи 1,35г (3,2ммоля) сполуки за прикладом 13 (вихід 50%) у вигляді трохи забарвленої чистої твердої маси з M+ 422 та точкою топлення 185-188°C. Цими та подібними способами синтезують наступні сполуки. Вони призначені для більш детальної ілюстрації винаходу й ніяким чином не обмежують обсяг винаходу. Дані про структур у цих сполук, представлену загальною формулою (1), наведені у наступній таблиці. 15 Фармакологічні випробування 83868 16 17 83868 Агоністичні властивості сполук за винаходом щодо рецепторів ORL1 in vitro та in vivo, a також їх активність (або відсутність такої) щодо m-опіатів визначають за наведеними нижче методиками. Спорідненість до людських рецепторів ORL1 Спорідненість до людських рецепторів ORL1 визначають методом зв'язування рецепторів in vitro, який описаний [Ardati et al., Mol.Pharmacol., 51, 816, 1997]. Коротко кажучи, препарати мембран одержують з клітин ЯКХ (яєчників китайського хом'яка), у яких людський рецептор ORL1 завжди стабільно виражений. Мембрани інкубують з [3H]-ноціцептином у присутності дослідних сполук у різних концентраціях, розбавлених у відповідному буфері або ж без них. Неспецифічним вважається зв'язування, яке залишається у присутності 10-6M ноціцептину. Зв'язану радіоактивність відокремлюють від вільної фільтрацією крізь скловолоконні фільтри Packard GF/B з кількома промивками крижаним буфером з використанням харвестера клітин Packard. Зв'язану радіоактивність вимірюють лічильником сцинтиляцій (Topcount фірми Packard), використовуючи рідку сцинтиляційну суміш (Microscint 0 фірми Packard). Виміряну радіоактивність накладають на графік проти концентрації витискувальної дослідної сполуки та розраховують криві витискування за допомогою чотирьохпараметричної логістичної регресії, одержуючи величини ІС50, тобто такої концентрації дослідної сполуки, при якій витискується 50% радіоліганду. Величини спорідненості рKi вираховують, коригуючи значення ІС 50 для концентрації радіоліганду та його спорідненості до людського рецептора ORL1 згідно з рівнянням Ченга-Прусофа рKi=-Ig[IC 50/(1+S/K d) де ІС 50 - величина, визначена вище, S - концентрація [3Н]-ноціцептину, використаного у тесті, у молях/л (як правило, 0,2нМ), a Kd - константа рівноваги дисоціації [3H]-ноціцептину для людських рецепторів ORL1 (0,4нМ). Сполуки за винаходом посідають високу спорідненість до рецепторів ORL1 в описаному вище тесті на зв'язування. Ця властивість робить їх корисними при лікуванні розладів, з якими пов'язані рецептори ORL1, або таких, що їх можна лікувати, діючи на ці рецептори. Спорідненість до людських рецепторів mопіатів Спорідненість до рецепторів m-опіатів визначають методом зв'язування рецепторів in vitro, який описаний у [Childers et al., Eur.J.Pharm., 55, 11, 1979]. Коротко кажучи, препарати мембран одержують з клітин ЯКХ (яєчників китайського хом'яка), у яких людський рецептор ORL1 завжди є стабільно виражений, та інкубують з [3H]налоксоном у присутності дослідних сполук в інтервалі концентрацій від 10мкМ до 0,1нМ, розбавлених у відповідному буфері або ж без них. Неспецифічним вважається зв'язування, яке залишається у присутності 10-7M левалорфантартрату. Зв'язану радіоактивність відокремлюють від вільної, як описано вище, і так само вираховують спорідненість сполук, використовуючи концентрацію (S) 1нМ [ 3Н]-налоксону при значенні рKi 1,3нМ. 18 Сполуки за винаходом здебільшого посідають низьку спорідненість до рецепторів m-опіатів за методом зв'язування рецепторів, описаним вище. Отже, вони навряд чи здатні спричинити небажані бічні явища, характерні для опіатів типу морфію. Агонізм до рецепторів ORL1 in vitro Активація пов'язаних з G-протеїном рецепторів ORL1 інгібує активність аденилатциклази та знижує внутрішньоклітинну концентрацію другого месенджера цАМФ. Активність сполук щодо рецепторів ORL1 визначають за методикою, описаною [Jenck et al, Ргос. Natl. Acad. Sci. USA, 97, 49384943, 2000]. Показано, що вони є потужними агоністами. Агонізм до рецепторів ORL1 in vivo Після внутрішньочеревного або орального введення сполуки за винаходом виявляють високу активність при тесті з ультразвуковою подразнювальною вокалізацією за [Van der Poel et al., Psychopharmacology, 97, 147-148, 1989]. Це є доказом не лише доброї біозасвоюваності після орального введення, але й подолання бар'єру між мозком та кров'ю. Пептидний ноціцептин у цьому тесті також виявляє активність, але його дія стає помітною лише при введенні безпосередньо до мозку (шляхом вн утрішньоцеребральношлуночкової ін'єкції). Спричинене агоністом ORL1 зниження кров'яного тиску З 5-хвилинними інтервалами щурам, яких анестезують 80мг/кг внутрішньочеревно пентобарбіталу натрію, вводять внутрішньовенно дози агоністів ORL1, збільшуючи їх, що призводить до зниження кров'яного тиску. Це зниження виражається у ED80 - дозі, при якій кров'яний тиск знижується на 20% у порівнянні з контрольними зразками. У дослідженнях взаємодії за 10 хвилин до першої дози агоніста щурам вводять внутрішньовенно одну дозу 2мг/кг налоксону - антагоніста опіату - або 1мг/кг J-113397 - селективного антагоніста ORL1. Ця доза J-113397 здатна повністю нейтралізувати дію ноціпцептину. Така доза налоксону здатна протидіяти спричиненому морфіном зниженню кров'яного тиску, але не впливає на спричинене ноціцептином зниження тиску. Агоніст ORL1 та спричинена морфіном дія на засвоєння їжі Нещодавні дослідження показали, що засвоєння їжі піддається фармакологічному регулюванню за допомогою лігандів рецепторів опіатів. [Sanger and McCarthy, (Increased food and water intake produced in rats by opiate receptor agonists, Psychopharmacology, 74(3): 217-220, 1981)], показали, що системне введення морфіну призводить до збільшеного засвоєння харчу, а неселективний антагоніст опіатів налоксон робить цю дію зворотною. Більш того, [Сіссосіорро et al., (Reversal of stress- and CRF-induced anorexia in rats by the synthetic nococeptin/orphanin FQ receptor agonist Ro 64-6198, Psychopharmacology, 161(2): 113-119, 2002)], доводять, що системне введення Ro 646198 - агоніста ORL1 - також збільшує засвоєння харчу щурами. Самців щурів Вістар тримають поодинці та дають вільний доступ до харчу й води. їм вводять 19 83868 внутрішньочеревно одну дозу носія Ro 64-6198 (1, 3, 6,10мг/кг), сполуки за прикладом 1 (0,3,1, 3, 6,10мг/кг) або морфіну (1,3, 10мг/кг) і ставлять харч так, щоб тварини не могли до нього дістати. За 15 хвилин після введення ліків до клітин тварин кидають зважену кількість харчу (5-6 гранул=2530г). Через 60 та 120 хвилин після того харч знову зважують. Усі тварини беруть участь принаймні у 4 роздільних дослідах кожна з інтервалами між дослідами принаймні 5 діб. Дбають про те, щоб зайвий шум не завдавав додаткового стресу тваринам. У цих дослідах, де введення налоксону антагоніста опіатів (1, 3, 10мг/кг) - або J113397 антагоніста ORL1 (3, 10, 30мг/кг) передувало введенню агоніста (Ro 64-6198 - 6мг/кг; сполука за прикладом 1-10мг/кг або морфін 2мг/кг), антагоніст вводили внутрішньочеревно за 30 хвилин до агоніста. Усі експериментальні групи нараховували щонайменше по шість тварин. Приклад складання композиції для випробування на тваринах Композиція за прикладом 1: Для орального (р.о.) введення: До потрібної кількості (0,5-15мг) твердої маси за прикладом 1 у скляній трубці додають скляні бусинки та подрібнюють у вихровому млині протягом 2 хвилин. Після додання 1мл 1% водного розчину метилцелюлози сполуку суспендують у вихровій мішалці 10 хвилин. Для концентрацій менше та більше 1мг/мл частки, що залишилися несуспендованими, суспендують далі за допомогою ультразвукової ванни. Для внутрішньочеревного (і.р.) введення: До потрібної кількості (0,5-15мг) твердої маси за прикладом 1 у скляній трубці додають скляні бусинки та подрібнюють у вихровому млині протягом 2 хвилин. Після додання 1мл водного розчину 1% метилцелюлози та 5% манітолу сполуку суспендують у вихровій мішалці 10 хвилин. Нарешті, доводять рН до 7. Композиція за прикладом 2: Для орального (р.о.) введення: До потрібної кількості (0,5-15мг) твердої маси за прикладом 2 у скляній трубці додають скляні бусинки та подрібнюють у вихровому млині протягом 2 хвилин. Після додання 1мл 1% водного розчину метилцелюлози сполуку суспендують у вихровій мішалці 10 хвилин. Доводять рН до 7 кількома краплинами водного розчину 0,1N NaOH. Частки, що залишилися несуспендованими, суспендують далі за допомогою ультразвукової ванни. Для внутрішньочеревного (і.р.) введення: Для внутрішньочеревного введення композицію одержують так само, як і для орального, тільки замість 1% водного розчину метилцелюлози використовують 1% метилцелюлозу з 5% манітолу. Фармакологічні дані Спорідненість in vitro до рецепторів ORL1 та m-опіатів, функціональний агонізм до ORL1 Приклад Спорідненість Агонізм in vitro до до mПроба цАМФ ORL1 опіатів рKi рKi PEC50 20 (rac)-Ro 64-6198 Приклад 1 Приклад 1S,S Приклад 1S,R Приклад 1R,S Приклад 1R,R Приклад 2 Приклад 6 Приклад 7 Приклад 8 Приклад 10 Приклад 13 Приклад 15 Приклад 21 Приклад 31 Приклад 32 Приклад 35 Приклад 41 Приклад 42 Приклад 43 Приклад 45 9,0 8,4 8,8 8,5 8,7 7,5 8,2 8,4 8,2 7,8 7,3 8,1 8,6 73 7,5 7,4 7,5 8,5 7,9 9,2 8,4 7,3 7,0 7,5 7,7 6,8 6,6 7,3 6,4 6,9
ДивитисяДодаткова інформація
Назва патенту англійськоюBenzimidazolone and quinazolinone derivatives as agonists on human orl1 receptors
Автори англійськоюDen Hartog Jacobus A. J., van Scharrenburg Gustaaf J. M., Stuivenberg Herman H., Tuinstra Tinka
Назва патенту російськоюПроизводные бензимидазолона и хиназолинона в качестве агонистов человеческих рецепторов orl1
Автори російськоюДен Гарток Якобус А.Й., ван Шаренбург Густав Й.М., Стёйвенберг Герман Г., Тёйнстра Тынка
МПК / Мітки
МПК: A61K 31/46, C07D 401/04, C07C 251/20, A61P 25/00, C07D 451/04, A61K 31/517, A61K 31/454
Мітки: похідні, агоністи, людських, хіназолінону, бензимідазолону, рецепторів
Код посилання
<a href="https://ua.patents.su/11-83868-pokhidni-benzimidazolonu-ta-khinazolinonu-yak-agonisti-lyudskikh-receptoriv-orl1.html" target="_blank" rel="follow" title="База патентів України">Похідні бензімідазолону та хіназолінону як агоністи людських рецепторів orl1</a>
Похідні 1,3,5-триазину як ліганди для людських рецепторів аденозину-а3

Номер патенту: 77816
Опубліковано: 15.01.2007
Автори: Рейндерс Ян Г., ван Шаренбург Густаф Й.М., Густавсон Гері Р., Прас-Равес Марія Л., Ден Гартог Якобус А.Й.
МПК: A61P 3/10, A61P 1/04, A61P 25/00, A61P 7/06, C07D 405/12, C07D 405/06, A61P 17/06, C07D 401/12, A61P 27/06, A61P 11/02, A61P 25/28, C07D 251/50, C07D 251/42, A61P 31/12, A61P 29/00, A61P 9/12, A61P 43/00, A61P 11/06, A61P 25/14, C07D 401/04, A61P 35/00, A61P 17/04, A61P 25/16, A61P 25/08, C07D 405/04, A61P 37/00, A61P 37/08, A61P 35/02, A61P 1/00, A61P 11/00, A61P 31/04, A61P 17/00, A61K 31/53, A61P 25/18, A61K 31/5377, A61P 9/10, C07D 405/14, A61P 25/04, A61P 7/02, A61P 19/02, A61P 19/10, A61P 9/06
Мітки: людських, аденозину-а3, похідні, 1,3,5-триазину, рецепторів, ліганди
Формула / Реферат:
1. Сполуки загальної формули (1) , (1)де:R1 представляє галоген, алкіл(1-3С), О-алкіл(1-3С), СF3, NH2, N-(ді)-алкіл(1-3С), N-(ді)-алкеніл(1-3С), N-(ді)-алкініл(1-3С), N-алкіл(1-3С)алкеніл(1-3С), N-алкіл(1-3С)алкініл(1-3С), N-алкеніл(1-3С)алкініл(1-3С) або С2-С8 циклоалкіламіногрупу, можливо заміщену, R2, R3 та R4 незалежно один від одного - Н, галоген,...
Похідні піридазино[4,5-b]індол-1-оцетаміду для виготовлення препаратів для лікування хвороб, що стосуються дисфункції периферійних бензодіазепінових рецепторів

Номер патенту: 70349
Опубліковано: 15.10.2004
Автори: Бенавідес Хесус, МАРАБУ Бенуа, Севрен Мірей, Еванно Яннік, Марґе Франк, ФРУАССАН Жак, Ферзаз Бадіа, Жаньяк Філіп
МПК: A61K 31/5025, A61K 31/5377, A61P 35/00, A61P 9/00, A61P 25/00, A61P 17/00, A61P 43/00, A61P 13/00
Мітки: стосуються, лікування, хвороб, дисфункції, бензодіазепінових, виготовлення, препаратів, піридазино[4,5-b]індол-1-оцетаміду, периферійних, похідні, рецепторів
Формула / Реферат:
1. Застосування сполуки загальної формули (I)(I)в якійΧ - атом галогену,Υ - один або більше атомів або груп, які вибрано з гідрогену, галогенів і гідроксилу, метилу, метоксигрупи і нітрогрупи,R1 - (С1-С4)алкіл,R2 та R3 кожний, незалежно один від одного, - атом гідрогену чи (С1-C4)алкіл, або R2 і R3 утворюють, разом з атомом нітрогену, що їх несе, піролідиніл, піперидиніл або...
Азабіциклічні, азатрициклічні і азаспіроциклічні похідні аміноциклогексану як антагоністи рецепторів nmda, 5ht і нейронних нікотинових рецепторів

Номер патенту: 77778
Опубліковано: 15.01.2007
Автори: Хенріх Маркус, Гольд Маркус, Йіргенсонс Айгарс, Ванейєвс Максімс, Калвіньш Іварс, Парсонс Крістофер Грахам Рафаель, Каусс Валерьянс, Даниш Войцех
МПК: A61P 21/02, A61P 25/30, A61P 31/12, A61P 37/02, A61P 1/08, A61P 31/18, A61P 25/36, A61P 1/04, C07D 471/08, A61P 25/14, A61P 31/14, A61P 27/06, A61P 25/22, A61K 31/438, C07D 487/08, A61P 25/24, C07D 209/54, A61P 25/32, A61P 25/08, A61P 25/28, A61P 25/06, A61P 25/16, A61P 33/06, A61P 25/34, A61K 31/403, A61P 9/00, A61K 31/439, A61P 21/00, A61P 25/02, A61K 31/437, A61P 27/16, A61P 43/00, C07D 209/02, A61P 25/04, A61P 25/00, A61P 9/10, C07D 221/22, C07D 221/20, A61P 25/18
Мітки: рецепторів, азабіциклічні, похідні, нікотинових, nmda, азатрициклічні, антагоністи, нейронних, азаспіроциклічні, аміноциклогексану
Формула / Реферат:
1. Сполуки формули (1),де R і R1-R5, кожний незалежно, вибраний з С1-6-алкільних груп, С2-6-алкенільних груп, С2-6-алкінільних груп, С6-12-арил-С1-4-алкільних груп, необов'язково заміщених С6-12-арильних груп і у випадку R і R2-R5 - з атомів водню, за умови, що щонайменше один з R2 і R3 і щонайменше один з R4 і R5 не є воднем, або R і R1 разом являють собою...
Заміщені бензопірани як селективні агоністи b-рецепторів естрогенів

Номер патенту: 77986
Опубліковано: 15.02.2007
Автори: Норман Брайан Херст, Річардсон Тімоті Айво, Нойбауер Блейк Лі, Крішнан Венкатеш Гарі, Пфайфер Ланс Аллен, Додж Джеффрі Алан, Люгар Третій Чарлз Уілліс
МПК: A61P 19/10, A61P 11/00, A61P 1/04, A61P 25/00, A61P 43/00, A61P 25/28, A61P 15/00, A61P 9/00, C07D 311/94, C07D 311/80, A61K 31/353, A61P 35/00, A61P 13/08, A61P 13/02
Мітки: бензопірани, естрогенів, заміщені, b-рецепторів, агоністи, селективні
Формула / Реферат:
1. Сполука формули Ідекожний з R1, R2 та R3, незалежно від інших, є -Н, С1-С6-алкіл, -ОН, С1-С6-алкоксигрупа, галоген або група –СF3;R5 є водень або С1-С6-алкіл;кожний з Y1, Y2 та Y3, незалежно від інших, є -Н або С1-С6-алкіл; і G є -СН2-, -СН2-СН2- або –CH2-CH2-CH2;або її фармацевтичнo прийнятна сіль.2. Сполука за п. 1, де...
Агоністи альфа-рецепторів, активованих проліфератором пероксисом

Номер патенту: 82048
Опубліковано: 11.03.2008
Автори: Мейхью Даніель Рей, Гарсіа-Паредес Крістіна, Ментлоу Нейтан Брайан, Ю Янпінь, Колладо Кано Айвен, Джонстон Річард Дуейн, Етджен Гаррет Джей, мол., Саід Ашраф, Уан Сядон, Шмід Крістофер Рандал, Коффі Дейвід Скотт, Мартінеллі Майкл Джон, Летурно Майкл Едвард, Доміньяні Семюель Джеймз, Віченці Джеффрі Томас, Томпсон Річард Крейг
МПК: A61K 31/42, A61P 9/00, A61P 3/04, C07D 413/06, A61P 3/10, C07D 405/06, C07D 249/12, C07D 401/06, C07D 417/12, A61P 3/06, C07C 257/00, A61P 3/00, C07D 409/12, A61P 43/00, C07D 405/12, A61K 31/4196, A61K 31/4245, A61K 31/4439, A61K 31/4709, C07D 403/12, A61K 31/5377, C07C 281/00, C07D 409/06, C07D 407/06
Мітки: проліфератором, агоністи, активованих, пероксисом, альфа-рецепторів
Формула / Реферат:
1. Сполука, представлена наведеною нижче структурною формулою І:,та її фармацевтично прийнятні солі, сольвати та гідрати, де:(a) R1 вибраний із групи, яка включає водень, заміщену або незаміщену групу, вибрану із групи, яка включає C1-C8-алкіл, арил-С0-С4-алкіл, гетероарил-С0-С4-алкіл, С3-С6-циклоалкіларил-С0-С2-алкіл та -CH2-C(О)-R17-R18, де R17 - О...
Попередній патент: Похідні аміду, спосіб їх одержання (варіанти), фармацевтична композиція, способи лікування та застосування з використанням цих сполук
Наступний патент: Спосіб стикового з’єднання дротів
Випадковий патент: Корозійностійка сталь