Сполуки фосфінової амінокислоти і фармацевтична композиція, яка їх містить
Номер патенту: 91214
Опубліковано: 12.07.2010
Автори: Макарітіс Анастасіос, Діве Вінсен, Жюльєн Ніколя, Їотакіс Атанасіос, Скальбер Елізабет





Формула / Реферат
1. Сполука формули (І):
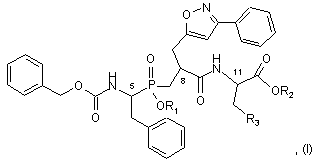
в якій:
R1 являє собою атом водню або групу, яку вибирають з (С1-С6)алкілкарбонілоксі(С1-С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою, і (С1-С6)алкілкарбонілтіо-(С1-С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою,
R2 являє собою атом водню або групу, яку вибирають з (С1-С6)алкілкарбонілоксі(С1-С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою, арилкарбонілтіо-(С1-С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою, і арил(С1-С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою і арильна частина якого необов'язково заміщена (С1-С6)алкілкарбонілоксигрупою,
R3 являє собою фенільну групу, необов'язково заміщену гідроксигрупою, або R3 являє собою 3-індолільну групу,
або
R1 може також являти собою лінійну або розгалужену (С1-С6)алкільну групу і R2 являє собою лінійну або розгалужену (С1-С6)алкільну групу, коли R3 являє собою фенільну групу, заміщену гідроксигрупою,
її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною основою і її гідрати і сольвати.
2. Сполука формули (І) за п. 1, яка відрізняється тим, що R1 і R2 кожний являє собою атом водню, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною основою.
3. Сполука формули (І) за пп. 1 або 2, яка відрізняється тим, що R1 являє собою атом водню і R3 являє собою фенільну групу, заміщену гідроксигрупою, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною основою.
4. Сполука формули (І) за будь-яким з пп. 1-3, яка відрізняється тим, що R2 являє собою атом водню і R3 являє собою фенільну групу, заміщену гідроксигрупою, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною основою.
5. Сполука формули (І) за п. 1, яка вибрана з групи:
(5R,8R,11S)-5-бензил-6-гідрокси-11-(1Н-індол-3-ілметил)-3,9-діоксо-1-феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6-фосфадодекан-12-ової кислоти 6-оксид,
(5R,8R,11S)-5,11-дибензил-6-гідрокси-3,9-діоксо-1-феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6-фосфадодекан-12-ової кислоти 6-оксид,
(5R,8R,11S)-5-бензил-6-гідрокси-11-(4-гідроксибензил)-3,9-діоксо-1-феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6-фосфадодекан-12-ової кислоти 6-оксид,
її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною основою.
6. Фармацевтична композиція, яка містить як активний інгредієнт щонайменше одну сполуку за будь-яким з пп. 1-5 в поєднанні з одним або більше фармацевтично прийнятними, інертними, нетоксичними наповнювачами або носіями.
7. Фармацевтична композиція за п. 6 для застосування у приготуванні лікарських засобів для лікування артеріальної гіпертензії і її ускладнень, включаючи легеневу артеріальну гіпертензію, ішемію міокарда, стенокардію, серцеву недостатність, васкулопатію, нефропатію, діабетичну ретинопатію, атеросклероз і постангіопластичний рестеноз, гостру або хронічну ниркову недостатність, церебрально-васкулярні захворювання, включаючи інсульт і субарахноїдальну кровотечу, і периферійну ішемію.
Текст
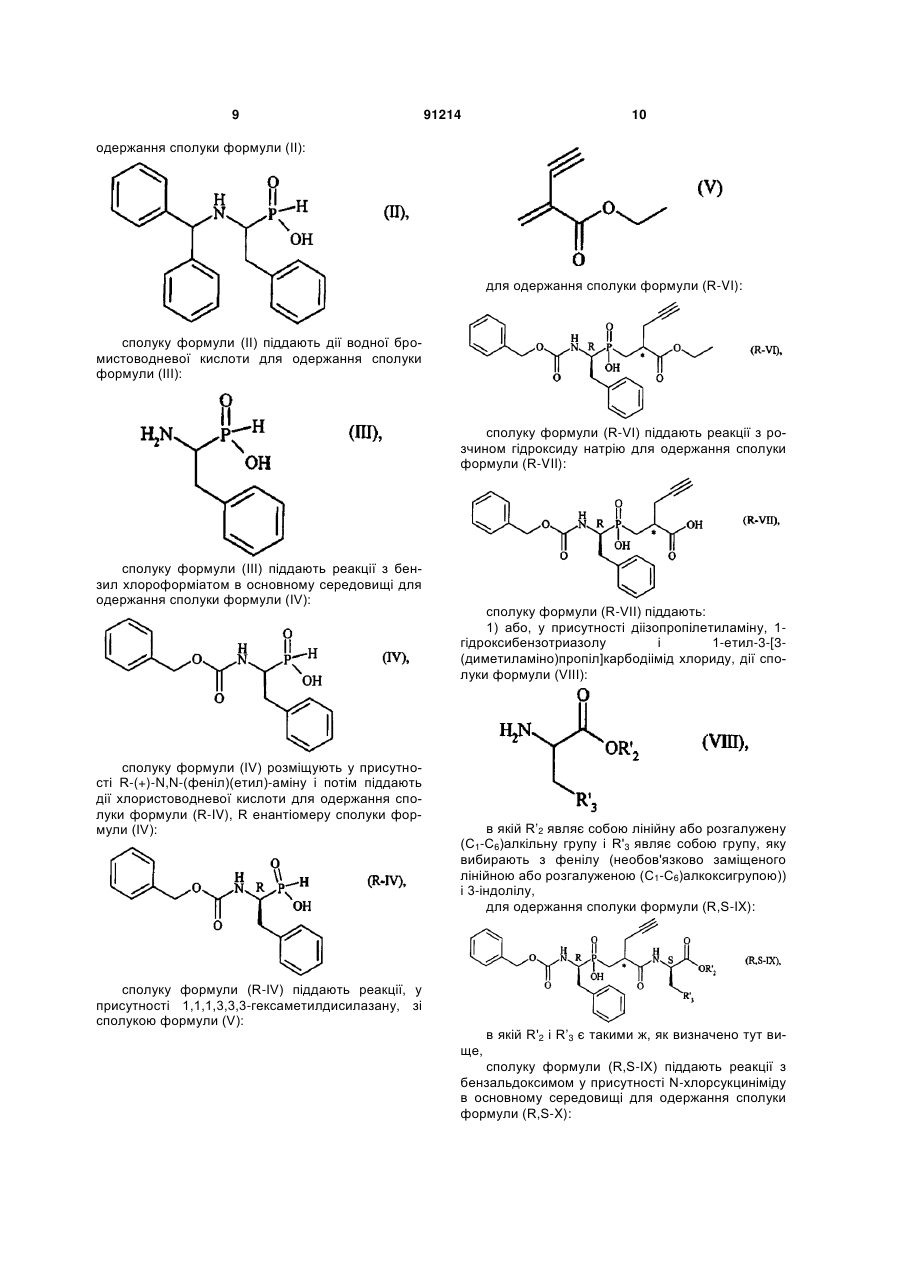
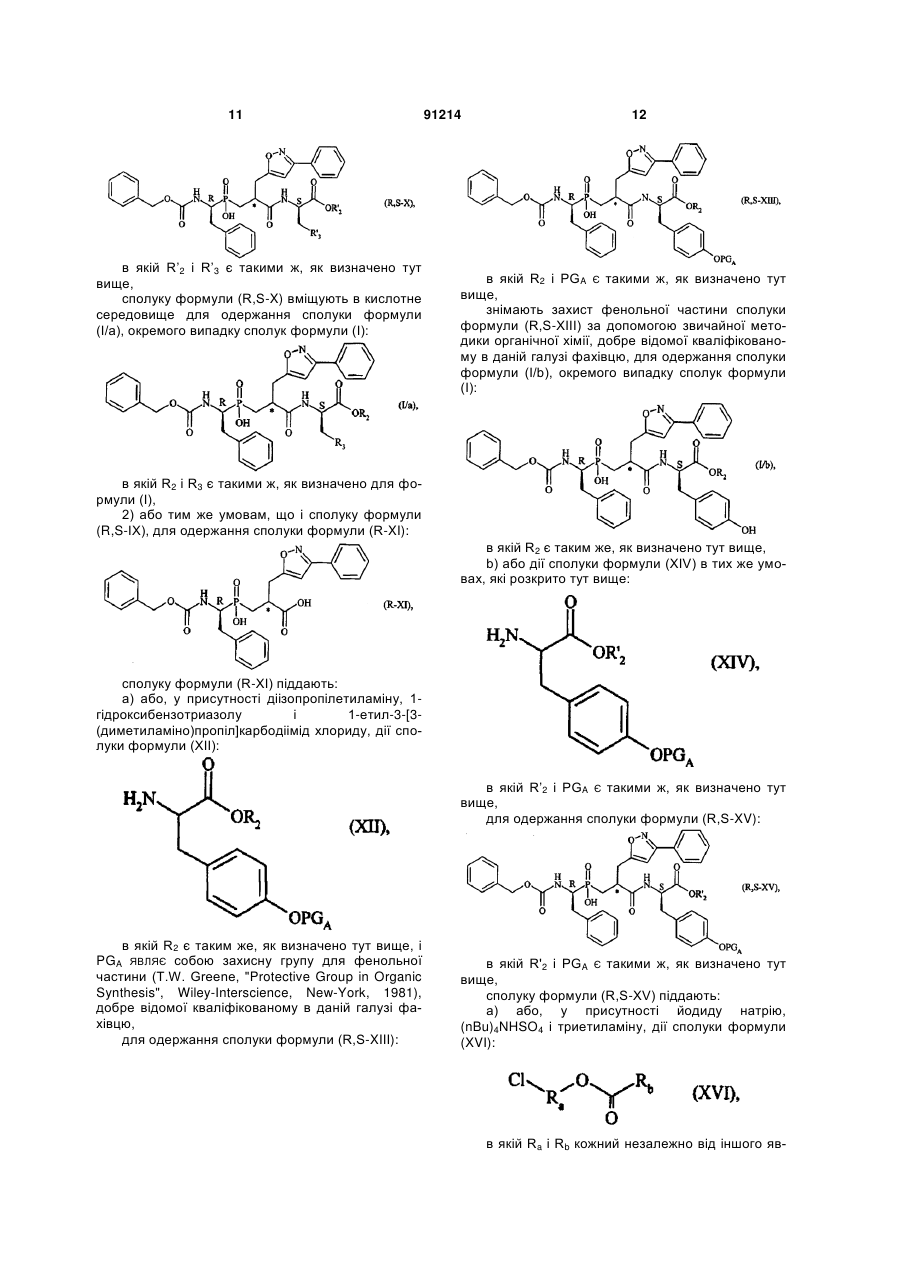
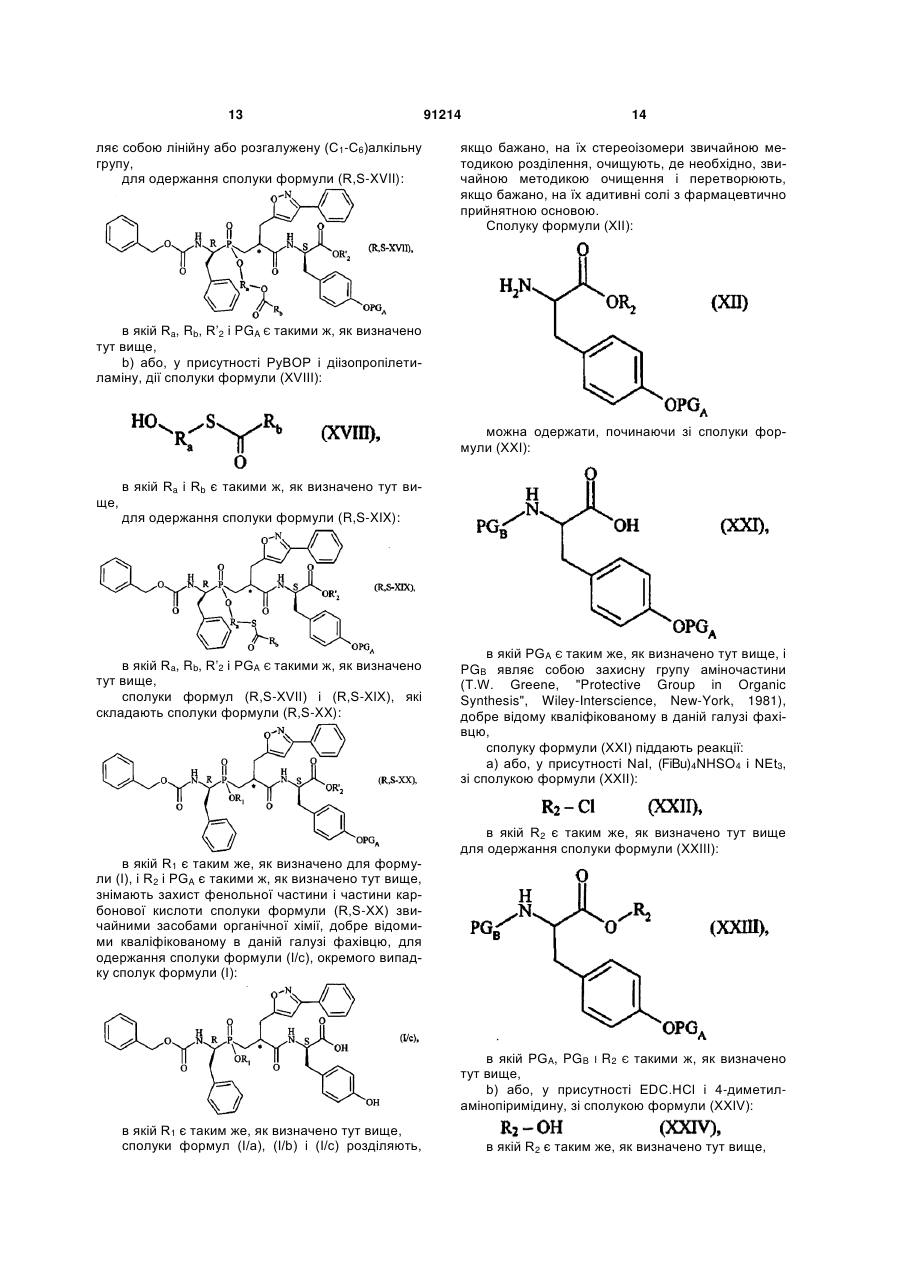
1. Сполука формули (І): C2 2 (19) 1 3 91214 4 вання артеріальної гіпертензії і її ускладнень, включаючи легеневу артеріальну гіпертензію, ішемію міокарда, стенокардію, серцеву недостатність, васкулопатію, нефропатію, діабетичну ретинопатію, атеросклероз і постангіопластичний рестеноз, гостру або хронічну ниркову недостатність, церебрально-васкулярні захворювання, включаючи інсульт і субарахноїдальну кровотечу, і периферійну ішемію. Даний винахід стосується нових сполук фосфінової амінокислоти, способу їх одержання і фармацевтичних композицій, які їх містять. Нові сполуки фосфінової амінокислоти відповідно до даного винаходу мають чудову властивість одночасного інгібування ангіотензин І - перетворювального ферменту (АСЕ) і ендотелінперетворювального ферменту (ЕСЕ). Баланс, який існує між вазоактивними пептидами - вазоконстрикторами (ангіотензин II, ендотелій-1), з одного боку, і вазодилататорами (натрійуретичними факторами, брадикінін), з іншого боку, - складає важливий елемент в регулюванні артеріального тиску. Синтез і розщеплення цих пептидів знаходяться під контролем різних ферментів, серед яких три цинкові металлопротеази, мабуть, відіграють переважаючу роль: - ангіотензин І - перетворювальний фермент (АСЕ), який, з одного боку, перетворює ангіотензин І, неактивний декапептид, в ангіотензин II, активний октапептид, і, з іншого боку, розщеплює брадикінін на неактивні пептиди, ендотелін-перетворювальний фермент (ЕСЕ), який розщеплює великий ендотелій-1, щоб утворити ендотелій-1, і, мабуть, бере участь у меншій мірі в розщепленні брадикініну, - нейтральна ендопептидаза (NEP), яка інактивує атріальний натрійуретичний пептид (ANP) і брадикінін, щоб утворити неактивні пептиди. Ангіотензин II являє собою вазоконстриктор і антинатрійуретичний октапептид. Ендотеліни являють собою вазоконстрикторний і антинатрійуретичний поліпептиди з близько двадцяти амінокислот, які містять два дисульфідні місточки, які зв'язують цистеїнові залишки. Брадикінін являє собою вазодилататорний і натрійуретичний нонапептид. Ангіотензин II, ендотелій і брадикінін являють собою найбільш важливі поліпептиди, який досі вважають залученими в регулювання судинного тонусу, серцево-судинну реконструкцію і гідроелектролітичний гомеостаз. їх метаболізм суттєво контролюється трьома ферментами: АСЕ, ЕСЕ і NEP. Артеріальна гіпертензія, а також інші серцевосудинні патології характеризуються порушенням рівноваги в балансі пептидів на користь вазоконстрикторних пептидів, які чинять всеохоплюючу несприятливу дію в нирково-серцевосудинній сфері (вологозатримування, затримування натрію, серцево-судинна гіпертрофія і т.д.). Незважаючи на головні терапевтичні досягнення, одержані у 1980-х за допомогою селективних інгібіторів АСЕ, виявилась необхідною розробка нових сполук для того, щоб удосконалити ще більш контроль кров'яного тиску у пацієнтів з гіпертензією, а також середню тривалість їх життя (переважна дія на головні серцево-судинні фактори ризику). Три металлопротеази АСЕ, ЕСЕ і NEP, відповідно, виявляються, багатообіцяючими мішенями для лікування серцево-судинних захворювань. Таким чином, для того, щоб боротись з побічними вазоконстрикторними впливами ангіотензину II і ендотеліну-1 і щоб підвищити захисні вазодилататорні впливи ANP і брадикініну, були розроблені інгібітори АСЕ, NEP і ЕСЕ. Відомі численні сполуки, які мають ту чи іншу із зазначених активностей. Численні патентні заявки описують сполуки амінокислот, які корисні як інгібітори АСЕ і ЕСЕ і корисні як змішані інгібітори ACE/NEP і ECE/NEP. Селективні інгібітори АСЕ, які використовувались як антигіпертонічні засоби протягом декількох років, розкриті в описі патенту US 4 396 772. Формули розкритих сполук включають: Описи патентів WO 97/32874 і US 5 476 847 розкривають сполуки, які являють собою інгібітори ЕСЕ. Формули розкритих сполук включають: - в описі патенту WO 97/32874 - в описі патенту US 5 476 847 Відповідно до патентної заявки WO 95/35302, відомі деякі сполуки фосфінової кислоти, які володіють інгібуючою активністю відносно ЕСЕ, корисною у лікуванні серцево-судинних захворювань. Формули розкритих сполук включають: Змішані інгібітори ACE/NEP (клас "інгібіторов 5 91214 6 вазопептидази") також були синтезовані і клінічно досліджені. Змішані інгібітори ACE/NEP розкриті в описах до патентів WO 97/24341, WO 96/22998, WO 93/08142 і EP 0 723 974. Розкриті сполуки являють собою сульфуровані похідні пептидів. У Bioorganic and Medical Chemistry Letters, 1996, 6(11), 1257-1260, розкриті сполуки фосфінової кислоти, які володіють змішаною інгібуючою активністю відносно ACE/NEP, корисною у лікуванні серцево-судинних захворювань. Формули цих сполук включають: Ці сполуки мають антигіпертонічну ефективність, яка є більшою, ніж ефективність селективних інгібіторів АСЕ. Проте, вони мають головні побічні ефекти, особливо по відношенню до ангіоневротичного набряку, можливо пов'язані з надлишком брадикініну (Trends in Pharmacological ScL, 2001, 22, 106-109; The Lancet, 2001, 358, 1525-1532). Це призвело до затримки клінічної розробки подальших удосконалених змішаних інгібіторів ACE/NEP, таких як омапатрилат (Curr Opin Investig Drugs, 2001, 2, 1414-1422). Фармакологічні властивості змішаних інгібіторів ACE/NEP, які розкриті у попередньому рівні техніки, не враховують головну серцево-судинну роль системи ендотеліну (Journal of Hypertension, 1998, 16(8), 1081-1098), а також залучення NEP в розщеплення ендотеліну-1 (J. Biol Chem., 1990, 265, 14150-14155). Відповідно, лікування із застосуванням змішаних інгібіторів ACE/NEP має наслідком збільшення рівня ендотеліну-1, який в довгостроковому плані може спричинити шкідливий ефект, а не очікувану терапевтичну користь. Нарешті, змішані інгібітори ECE/NEP розкриті в Life Sciences, 2000, 67(9), 1025-1033 і NaunynSchmiedeberg's Arch. Pharmacol, 358 (1, suppl 1): R 513-514 (Abstr). Формули цих сполук є наступними є: - в Life Sciences - в Naunyn-Schmiedeberg's Arch. Pharmacol. Зацікавленість у наявності змішаних інгібіторів ECE/NEP проявляється через зменшення рівня ендотеліну, в той час збільшуючи рівень натрійуретичних пептидів, і, відповідно одержання адитивного або синергічного ефекту, який є переважним для лікування серцево-судинних і ниркових захворювань. Проте, через те що NEP є одним з найбільш важливих учасників у розщепленні брадикініну in vivo, припинення клінічних досліджень інгібіторів ACE/NEP, також очевидно звело нанівець розробку інших множинних інгібіторів вазопептидаз, включаючи поєднання інгібування NEP, а саме змішаних ECE/NEP або потрійних ECE/ACE/NEP інгібіторів. З іншого боку, альтернатива змішаних інгібіторів АСЕ/ЕСЕ залишається дуже обіцяючою, дозволяючи очікувати підвищену серцево-судинну ефективність і хорошу безпечність застосування. Такі сполуки повинні зробити можливим зменшення утворення двох потужних вазоконстрикторних пептидів, ангіотензину II і ендотеліну-1, і збільшення рівнів брадикініну до прийнятної міри. Крім того цікаво відмітити, що хоча системи ангіотензину і ендотеліну функціонують незалежно, вони також функціонують на основі взаємодії. Цей "взаємний вплив" між двома системами був досліджений на експериментальному рівні, а також на клінічному рівні. Особливо досліджували роль ендотеліну-1 як посередника певних серцевосудинних ефектів ангіотензину II (Hypertension, 1997, 30, 29-34; Cardiovasc. Res., 1999, 43, 300307; Hypertension, 2002, 40, 840-846; Clin. Exp. Pharmacol Physiol, 2003, 30, 278-283; Hypertension, 2002, 39, 715-720; Hypertension, 2003, 42, 825-830; Bioorg. Med. Chem. Letters, 2003, 13, 1093-1096). В цілому, одержані дані говорять про те, що інгібування однієї з цих систем призводить до гіперактивності іншої системи, що сприяє підходу "змішаного інгібування" для того, щоб підсилити терапевтичну потужність кожної з цих властивостей, в той чай уникаючи протилежного регулювання. Нарешті, доказ на користь концепції змішаного інгібування АСЕ/ЕСЕ, тобто терапевтична перевага, яка очікується від цього підходу, досі аналізували на експериментальному рівні, у трьох терапевтичних напрямках: артеріальна гіпертензія, серцева недостатність і нирковий захист. Підтрим 7 ка на користь концепції загалом була забезпечена за допомогою об'єднання двох селективних сполук: з одного боку, селективний інгібітор АСЕ або блокатор АТ1-рецептора ангіотензину II для блокування системи ангіотензину і, з іншого боку, блокатор ЕТA-рецептора ендотеліну-1 або блокатор ЕТА/ЕТВ рецептора ендотеліну-1 змішаного типу або, в рідкісних випадках, інгібітор ЕСЕ для блокування ендотеліну. Література широко підтримує цю концепцію. Терапевтичні переваги були продемонстровані на тваринах або на структурному рівні, або на функціональному рівні при артеріальній гіпертензії (J Cardiovasc Pharmacol, 2000, 36, S337-S341; Clin Sci., 2002, 103, 363S-366S; Am J Hypertens., 2003, 16, 324-328) і серцевій недостатності (Cardiovasc. Res., 2002, 54, 85-94; Circulation, 2002, 106, 11591164). Рішуче передбачається захист конкретних цільових органів, таких як нирка і мозок (J. Am. Soc. Nephrol, 2001, 12,2572-2584). Клінічним відображенням всіх цих передклінічних результатів могло б бути більше число пацієнтів, стан яких нормалізувався за допомогою змішаного лікування. Можливо, що користь від „тиску крові" буде, у тривалому періоді, доповнена кращим захистом цільових органів при артеріальній гіпертензії (тобто запобігання „пошкодженню цільових органів" на серцево-судинному, нирковому і мозковому рівнях), а також сприятливими впливами на певні фактори ризику, таким чином запобігаючи ускладненням, які виникають при артеріальній гіпертензії. Даний винахід має за мету забезпечення нових сполук, які поводять себе як змішані інгібітори АСЕ/ЕСЕ без будь-якого інгібування NEP. Сполуки за даним винаходом є, відповідно, дуже ефективними у лікуванні артеріальної гіпертензії і її ускладнень, включаючи легеневу артеріальну гіпертензію, ішемію міокарда, стенокардію, серцеву недостатність, васкулопатію, нефропатію, діабетичну ретинопатію, атеросклероз і постангіопластичний рестеноз, гостру або хронічну ниркову недостатність, церебрально-васкулярні захворювання, включаючи інсульт і субарахноїдальну кровотечу, і периферійну ішемію. Більш конкретно, даний винахід стосується сполук формули (І): в якій: R1 являє собою атом водню або групу, яку вибирають з (С1-С6)алкіл-карбонілоксі(С1-С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою, і (С1-С6)алкілкарбонілтіо-(С1-С6)алкілу, при цьому кожна алкіль 91214 8 на частина може бути лінійною або розгалуженою, R2 являє собою атом водню або групу, яку вибирають з (С1-С6)алкіл-карбонілоксі(С1-С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою, арилкарбонілтіо-(С1С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою, і арил-(С1С6)алкілу, при цьому кожна алкільна частина може бути лінійною або розгалуженою і арильна частина якого необов'язково заміщена (С1-С6)алкілкарбонілоксигрупою, R3 являє собою фенільну групу, необов'язково заміщену гідроксигрупою або R3 являє собою 3індолільну групу, або R1 може також являти собою лінійну або розгалужену (С1-С6)алкільну групу і R2 являє собою лінійну або розгалужену (С1-С6)алкільну групу, коли R3 являє собою фенільну групу, заміщену гідроксигрупою, їх енантіомерів і діастереоізомерів, а також їх адитивних солей з фармацевтично прийнятною основою, і їх гідратів і сольватів. Серед фармацевтично прийнятних основ можуть бути згадані, без будь-якого обмеження, гідроксид натрію, гідроксид калію, триетиламін, третбутиламін і т.д. Відповідно до переважного втілення даного винаходу переважні сполуки являють собою сполуки, в яких R1 і R2 кожний являє собою атом водню. Відповідно до другого переважного втілення даного винаходу переважні сполуки являють собою сполуки, в яких R1 являє собою атом водню і R3 являє собою фенільну групу, заміщену гідроксигрупою. Відповідно до третього переважного втілення даного винаходу переважні сполуки являють собою сполуки, в яких R2 являє собою атом водню і R3 являє собою фенільну групу, заміщену гідроксигрупою. Серед переважних сполук можуть бути згадані більш конкретно: (5R,8R,11S)-5-бензил-6-гідрокси-11-(1Ніндол-3-ілметил)-3,9-діоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-ової кислоти 6-оксид - (5R,8R, 11S)-5,11-дибензил-6-гідрокси-3,9діоксо-1-феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2окса-4,10-діаза-6-фосфадодекан-12-ової кислоти 6-оксид, - (5R,8R, 11S)-5-бензил-6-гідрокси-11 -(4гідроксибензил)-3,9-діоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан- 12-ової кислоти 6-оксид. Енантіомери, діастереоізомери і адитивні солі з фармацевтично прийнятною основою переважних сполук утворюють невід'ємну частину даного винаходу. Даний винахід стосується також способу одержання сполук формули (І), який відрізняється тим, що як вихідний матеріал використовують дифенілметанамін хлорид, який піддають реакції з фенілацетальдегідом у присутності фосфінової кислоти, H3PO2, і хлористоводневої кислоти для 9 91214 10 одержання сполуки формули (II): для одержання сполуки формули (R-VI): сполуку формули (II) піддають дії водної бромистоводневої кислоти для одержання сполуки формули (III): сполуку формули (R-VI) піддають реакції з розчином гідроксиду натрію для одержання сполуки формули (R-VII): сполуку формули (III) піддають реакції з бензил хлороформіатом в основному середовищі для одержання сполуки формули (IV): сполуку формули (IV) розміщують у присутності R-(+)-N,N-(феніл)(етил)-аміну і потім піддають дії хлористоводневої кислоти для одержання сполуки формули (R-IV), R енантіомеру сполуки формули (IV): сполуку формули (R-VII) піддають: 1) або, у присутності діізопропілетиламіну, 1гідроксибензотриазолу і 1-етил-3-[3(диметиламіно)пропіл]карбодіімід хлориду, дії сполуки формули (VIII): в якій R’2 являє собою лінійну або розгалужену (С1-С6)алкільну групу і R'3 являє собою групу, яку вибирають з фенілу (необов'язково заміщеного лінійною або розгалуженою (С1-С6)алкоксигрупою)) і 3-індолілу, для одержання сполуки формули (R,S-IX): сполуку формули (R-IV) піддають реакції, у присутності 1,1,1,3,3,3-гексаметилдисилазану, зі сполукою формули (V): ще, в якій R'2 і R’3 є такими ж, як визначено тут ви сполуку формули (R,S-IX) піддають реакції з бензальдоксимом у присутності N-хлорсукциніміду в основному середовищі для одержання сполуки формули (R,S-X): 11 в якій R’2 і R’3 є такими ж, як визначено тут вище, сполуку формули (R,S-X) вміщують в кислотне середовище для одержання сполуки формули (І/а), окремого випадку сполук формули (І): 91214 12 в якій R2 і PGA Є такими ж, як визначено тут вище, знімають захист фенольної частини сполуки формули (R,S-XIII) за допомогою звичайної методики органічної хімії, добре відомої кваліфікованому в даній галузі фахівцю, для одержання сполуки формули (І/b), окремого випадку сполук формули (І): в якій R2 і R3 є такими ж, як визначено для формули (І), 2) або тим же умовам, що і сполуку формули (R,S-IX), для одержання сполуки формули (R-XI): в якій R2 є таким же, як визначено тут вище, b) або дії сполуки формули (XIV) в тих же умовах, які розкрито тут вище: сполуку формули (R-XI) піддають: а) або, у присутності діізопропілетиламіну, 1гідроксибензотриазолу і 1-етил-3-[3(диметиламіно)пропіл]карбодіімід хлориду, дії сполуки формули (XII): в якій R’2 і PGA Є такими ж, як визначено тут вище, для одержання сполуки формули (R,S-XV): в якій R2 є таким же, як визначено тут вище, і PGA ЯВЛЯЄ собою захисну групу для фенольної частини (Т.W. Greene, "Protective Group in Organic Synthesis", Wiley-Interscience, New-York, 1981), добре відомої кваліфікованому в даній галузі фахівцю, для одержання сполуки формули (R,S-XIII): в якій R'2 і PGA Є такими ж, як визначено тут вище, сполуку формули (R,S-XV) піддають: a) або, у присутності йодиду натрію, (nBu)4NHSO4 і триетиламіну, дії сполуки формули (XVI): в якій Ra і Rb кожний незалежно від іншого яв 13 ляє собою лінійну або розгалужену (С1-С6)алкільну групу, для одержання сполуки формули (R,S-XVII): 91214 14 якщо бажано, на їх стереоізомери звичайною методикою розділення, очищують, де необхідно, звичайною методикою очищення і перетворюють, якщо бажано, на їх адитивні солі з фармацевтично прийнятною основою. Сполуку формули (XII): в якій Ra, Rb, R’2 і PGA Є такими ж, як визначено тут вище, b) або, у присутності PyBOP і діізопропілетиламіну, дії сполуки формули (XVIII): можна одержати, починаючи зі сполуки формули (XXI): ще, в якій Ra і Rb є такими ж, як визначено тут видля одержання сполуки формули (R,S-XIX): в якій Ra, Rb, R’2 і PGA Є такими ж, як визначено тут вище, сполуки формул (R,S-XVII) і (R,S-XIX), які складають сполуки формули (R,S-XX): в якій R1 є таким же, як визначено для формули (І), і R2 і PGA є такими ж, як визначено тут вище, знімають захист фенольної частини і частини карбонової кислоти сполуки формули (R,S-XX) звичайними засобами органічної хімії, добре відомими кваліфікованому в даній галузі фахівцю, для одержання сполуки формули (І/с), окремого випадку сполук формули (І): в якій PGA Є таким же, як визначено тут вище, і PGB являє собою захисну групу аміночастини (Т.W. Greene, "Protective Group in Organic Synthesis", Wiley-Interscience, New-York, 1981), добре відому кваліфікованому в даній галузі фахівцю, сполуку формули (XXI) піддають реакції: а) або, у присутності NaI, (FiBu)4NHSO4 і NEt3, зі сполукою формули (XXII): в якій R2 є таким же, як визначено тут вище для одержання сполуки формули (XXIII): в якій PGA, PGB І R2 Є такими ж, як визначено тут вище, b) або, у присутності EDC.HCl і 4-диметиламінопіримідину, зі сполукою формули (XXIV): в якій R1 є таким же, як визначено тут вище, сполуки формул (І/а), (І/b) і (І/с) розділяють, в якій R2 є таким же, як визначено тут вище, 15 для одержання сполуки формули (XVIII), як визначено тут вище, знімають захист аміночастини сполуки формули (XXIII) відповідно до звичайної методики органічного синтезу, добре відомої кваліфікованому в даній галузі фахівцю. Даний винахід стосується також фармацевтичних композицій формули (І), які містять сполуки в поєднанні з одним або більше фармацевтично прийнятним наповнювачем. Серед фармацевтичних композицій відповідно до даного винаходу можуть бути згадані більш конкретно ті, які прийнятні для орального, парентерального, назального, під- або черезшкірного, ректального, перлінгвального, очного або респіраторного введення, і особливо таблетки або драже, під'язикові таблетки, саше, порошки, желатинові капсули, коржики, супозиторії, креми, мазі, шкірні гелі і придатні для пиття та ін'єкцій ампули. Корисне дозування змінюється в залежності від статі, віку і ваги пацієнта, шляху ведення, природи терапевтичного призначення і будь-якого пов'язаного лікування, і знаходиться в діапазоні від 0,1мг до 1г на 24 години, за одне або більше введень. Приклади, які слідують, ілюструють винахід, без обмеження його жодним чином. Приготування, йдуть далі, приводять до одержання сполук за даним винаходом або до синтезу проміжних сполук, які застосовують у приготуваннях за даним винаходом. Використані вихідні матеріали являють собою комерційні продукти або їх одержують відповідно до відомих методів приготування. Структури сполук, які описують у Прикладах, були визначені відповідно до звичайної спектрофотометричної методики (інфрачервоний, ЯМР, мас-спектрометрія). (5R,8*,11S) сполука означає рацемічну суміш двох діастереоізомерів, які мають абсолютні конфігурації (5R,8S,11S) і (5R,8R,11S). Приготування 1: Етил 2-метилен-4-пентиноат 34,9г діетил малонату повільно додають до розчину EtONa, приготованого шляхом розчинення 5,0г натрію в 280мл абсолютного етанолу. Реакційну суміш нагрівають при 50 C протягом 1 години, і потім краплями до суміші додають 38,8мл 3бром-1-пропіну в 100мл абсолютного етанолу. Реакційну суміш перемішують протягом 12 годин при цій температурі. Після випаровування під зниженим тиском, залишок додають в діетиловий ефір і екстрагують водою. Органічну фазу висушують над сульфатом натрію, фільтрують і потім випаровують під зниженим тиском. Олію, яку одержують, додають в 100мл абсолютного етанолу і потім краплями додають розчин KOH (12,2г в 340мл етанолу). Після перемішування при температурі навколишнього середовища протягом 1,5 годин, етанол випаровують під зниженим тиском, і залишок розчиняють у воді і екстрагують діетиловим ефіром. Водну фазу охолоджують за допомогою льодяної ванни, окислюють 2М HCl до рН~1 і екстрагують діетиловим ефіром. Органічну фазу висушують над сульфатом натрію, фільтрують і потім випаровують під зниженим тиском. До одер 91214 16 жаного залишку додають 35,0мл піридину, 13,4г параформальдегіду і 1,85мл піперидину, і реакційну суміш перемішують при 100-105 C протягом 3 годин. Після охолодження до температури навколишнього середовища, суміш розбавляють 400мл діетилового ефіру і органічну фазу послідовно промивають водою, 2М HCl, 5% розчином NaHCO3 і насиченим розчином NaCl. Органічну фазу висушують над сульфатом натрію, фільтрують і потім випаровують під зниженим тиском. Хроматографія над силікагелем (петролейний ефір (4060°С)/діетиловий ефір: від 9,5/0,5 до 8/12) дозволяє виділити очікуваний продукт. Rf=0,31 (петролейний ефір (4060°С)/діетиловий ефір: 9,5/0,5) Приготування 2: 2-{[(1R)-1{[(Бензилокси)карбоніл]аміно}-2-фенілетил)(гідрокси)фосфорил]метил}-4-пентинова кислота Стадія А: 1-(Бензгідриламіно)-2фенілетилфосфінова кислота 51,5мл 50% водного розчину H3PO2 додають до суспензії 109,5г хлориду дифенілметанаміну в 750мл 90% етанолу, і потім реакційну суміш нагрівають до 85-90 C. При цій температурі протягом 3 годин додають 58,4мл фенілацетальдегіду в 175мл етанолу і нагрівання продовжують протягом більше 3 годин, з наступним перемішуванням протягом 16 годин при температурі навколишнього середовища. Утворений осад відфільтровують, промивають холодним етанолом і діетиловим ефіром і потім висушують, таким чином дозволяючи виділити очікуваний продукт. Стадія В: 1-Аміно-2-фенілетилфосфінова кислота 181,1г сполуки вказаної вище Стадії А в 500мл бромистоводневої кислоти нагрівають при 110120 C протягом 2 годин. Суміш потім концентрують під зниженим тиском, і залишок розбавляють водою і екстрагують діетиловим ефіром. Водну фазу концентрують і до залишку додають 600мл абсолютного етанолу. До реакційної суміші повільно додають 60мл заздалегідь охолодженого оксиду пропілену, температуру якої підтримують на рівні 0 С. Очікуваний продукт, який осаджується при охолодженні, відфільтровують, промивають діетиловим ефіром і потім висушують. Стадія С: 1-{[(Бензилокси)карбоніл]аміно}-2фенілетилфосфінова кислота 110мл 4М розчину гідроксиду натрію додають до суспензії 49,4г сполуки вказаної вище Стадії В в 120мл води. Температуру реакційної суміші доводять 0 C, і потім протягом 1 години додають 45,5мл бензилхлорформіату. Суміш перемішують при 0 C протягом 1 години і при температурі навколишнього середовища протягом 4 годин, в той час доводячи рН розчину до 9-10 за допомогою додавання 2М розчину гідроксиду натрію. Суміш потім перемішують при температурі навколишнього середовища протягом ночі і потім екстрагують діетиловим ефіром. Водну фазу підкислюють додаванням 6М хлористоводневої кислоти. Очікуваний продукт осаджується, його відфільтровують, промивають водою і діетиловим ефіром і потім висушують над P2O5. Стадія D: (1R)-1 17 {[(Бензилокси)карбоніл]аміно}-2фенілетилфосфінова кислота Розчин 83,4г сполуки вказаної вище Стадії C в 1000мл абсолютного етанолу нагрівають зі зворотним холодильником. Розчин 34,4мл R-(+)-N,N(феніл)(етил)аміну в 170мл етанолу повільно додають до реакційної суміші. Через 15 хвилин температуру реакційної суміші доводять до температури навколишнього середовища і охолоджують при 4 С протягом ночі. Утворений осад відфільтровують і промивають абсолютним етанолом і діетиловим ефіром. Тверду речовину перекристалізовують з 445мл абсолютного етанолу. Сіль, яку одержують, суспендують в 300мл 6М хлористоводневої кислоти і перемішують протягом 2-3 годин. Тверду речовину відфільтровують, промивають H2O і Et2O, і потім висушують над P2O5. Оптичне обертання [ ]20D=: -46,7 (1% в абсолютному етанолі) Стадія Е: (1R)-1-{[(Бензилокси}карбоніл]аміно}2-фенілетил-[2-(етокси-карбоніл}-4-пентил] фосфінова кислота 1,3 ммоль сполуки Приготування 1 додають краплями до суміші 1 ммоль сполуки вказаної вище Стадії D і 5 ммоль HMDS, нагрітої при 110 C протягом 1 години в атмосфері аргону. Реакційну суміш нагрівають при 100-105 C протягом ще 3 годин. її потім охолоджують до 70 С, і додають абсолютний етанол невеликими порціями досі в атмосфері аргону. Перемішування продовжують протягом наступних 15 хвилин при цій температурі. Розчинник потім випаровують під зниженим тиском. Хроматографія над силікагелем (хлороформ/метанол/оцтова кислота: 7/0,3/0,3) дозволяє виділити очікуваний продукт. Стадія F: 2-{[(1R)-1{[(Бензилокси)карбоніл]аміно}-2фенілетил)(гідрокси)-фосфорил]метил}-4пентинова кислота Розчин 1 ммоль сполуки вказаної вище Стадії E в 9мл етанолу охолоджують до 0 С. 5-6 ммоль 1M гідроксиду натрію додають невеликими порціями, і потім реакційну суміш перемішують при температурі навколишнього середовища протягом 68 годин. Після ацилювання з 2М HCl, етанол випаровують і залишок додають у воду і екстрагують етилацетатом. Органічну фазу промивають водою і насиченим розчином хлориду натрію і потім висушують над сульфатом натрію і випаровують під зниженим тиском. Очікуваний продукт одержують шляхом осаджування з суміші діетиловий ефір/петролейний ефір. Приготування 3: 3-[((1R)-1{[(Бензилокси)карбоніл]аміно}-2-фенілетил)(гідрокси)фосфорил]-2-[(3-феніл-5ізоксазоліл)метил]пропанова кислота Стадія A: 2-{[((1R)-1{[(Бензилокси)карбоніл]аміно}-2фенілетил}(гідрокси)-фосфорил]метил}-4пентинова кислота Цю сполуку одержують відповідно до методики, яка розкрита в літературі (А. Makaritis et al, Chem. Eur. J., 2003, 9, 2079-2094). Стадія В: 3-[((1R)-1-{[(Бензилокси)карбоніл] аміно}-2-фенілетил)(гідрокси)-фосфорил]-2-[(3 91214 18 феніл-5-ізоксазоліл)метил]пропанова кислота 1,3-Диполярне циклоприєднання проводять на сполуці вказаної вище Стадії А, використовуючи бензальдегід оксим, відповідно до методики, розкритої в літературі (A. Makaritis et al, Chem. Eur. J., 2003, 9, 2079-2094). ПРИКЛАД 1: (5R,8R,11S)-Бензил-6-гідрокси-11-(1Н-індол-3ілметил)-3,9-діоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-онової кислоти 6-оксид Стадія А: (1R)-1-{[(Бензилокси)карбоніл]аміно}2-фенілетил-[2-({[(1S)-1-(1H-індол-3-ілметил)-2метокси-2-оксоетил]аміно}карбоніл)-4феніл]фосфінова кислота До суспензії 1 ммоль сполуки Приготування 2 в 20мл дихлорметану додають 3 ммоль діізопропілетиламіну, 1 ммоль хлориду 1-триптофан метилового ефіру, 1 ммоль HOBt і 4 ммоль EDC.HCl. Реакційну суміш перемішують протягом 2 годин при температурі навколишнього середовища і потім розбавляють дихлорметаном. Потім додають 1M хлористоводневу кислоту, щоб утворити дві фази. Органічну фазу промивають 1M HCl, висушують над сульфатом натрію і потім концентрують під зниженим тиском. Очікуваний продукт одержують шляхом осадження з суміші діетиловий ефір/петролейний ефір. Стадія В: (1R)-1{[(Бензилокси)карбоніл]аміно}-2-фенілетил-{3-{[(1S)-1-(1Hіндол-3-ілметил)-2-метокси-2-оксоетил]аміно}-3оксо-2-[(3-феніл-4ізоксазоліл)метил)пропіл}фосфінова кислота 6 ммоль бензальдоксиму розчиняють в 5мл хлороформу, і до розчину додають 2 краплі піридину. Потім додають 6 ммоль N-хлорсукциніміду і після перемішування протягом 10 хвилин при температурі навколишнього середовища реакційну суміш перемішують при 45 C протягом 3-4 годин. Потім додають 1 ммоль сполуки вищевказаної Стадії А, також як і 7 ммоль триетиламіну. Реакційну суміш перемішують при 45 C протягом 96 годин і потім концентрують під зниженим тиском, і залишок додають в етилацетат і промивають 1M HCl і насиченим розчином хлориду натрію. Органічну фазу висушують над сульфатом натрію і концентрують під зниженим тиском. Очікуваний продукт одержують шляхом осадження з суміші діетиловий ефір/петролейний ефір. Стадія С: (5R,8*,11S)-5-Бензил-6-гідрокси-11(1Н-індол-3-ілметил)-3,9-діоксо-1-феніл-8-[(3феніл-4-ізоксазоліл}метил)]-2-окса-4,10-діаза-6фосфадлодекан-12-ової кислоти 6-оксид Продукт одержують відповідно до методики Стадії F Приготування 1, використовуючи сполуку вказаної вище Стадії В замість сполуки Стадії Е. Стадія D: (5R,8R,11S)-5-Бензил-6-гідрокси-11(1Н-індол-3-ілметил)-3,9-діоксо-1-феніл-8-[(3феніл-4-ізоксазоліл)метил)]-2-окса-4,10-діаза-6фосфадодекан-12-ової кислоти 6-оксид (R,R,S)діастереоізомер одержують очищенням сполуки вказаної вище Стадії C в ізократичному режимі з буфером, який складається з 40% ацетонітрилу і 60% 83,3мМ форміату амонію при рН 6,4, використовуючи 250х30мм AIT колонку (нерухома 19 фаза Kromasil Q18 5мкм, пори 100Å). Mac-спектрометрія (ES/MS) = 733,2Да ПРИКЛАД 2: (5R,8R,11S)-5,11-Дибензил-6-гідрокси-3,9діоксо-1-феніл-8-[(3-феніл-5-ізоксазоліл)]-2-окса4,10-діаза-6-фосфадодекан-12-ової кислоти 6оксид Стадія А: 2-({[(18)-1-Бензил-2-трет-бутокси-2оксоетил]аміно}карбоніл)-4-пентиніл-((1R)-1{[(бензилокси)карбоніл]аміно}-2фенілетил)фосфінова кислота Цю сполуку одержують відповідно до методики, розкритої на Стадії А Прикладу 1, використовуючи хлорид L-фенілаланін О-ди-трет-бутилового ефіру замість хлориду 1-триптофан метилового ефіру. Стадія В: 3-({[(1S)-1-Бензил-2-трет-бутокси-2оксоетил]аміно}-3-оксо-2-[(3-феніл-5ізоксазоліл}метил]пропіл((1R)-1{[(бензилокси}карбоніл]аміно}-2-фенілетил)фосфінова кислота Цю сполуку одержують відповідно до методики, розкритої на Стадії В Прикладу 1, використовуючи сполуку вказаної вище Стадії А. Продукт очищують хроматографією на силікагелі (хлороформ/метанол/оцтова кислота: 7/0,3/0,3). Стадія С: (5R,8*,11S)-5,11-Дибензил-6гідрокси-3,9-діоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил] -2-окса-4,10-діаза-6фосфадодекан-12-ової кислоти 6-оксид Продукт одержують за допомогою піддавання реакції сполуки вказаної вище Стадії В з трифтороцтовою кислотою, триізопропіламіном, дихлорметаном і водою в пропорціях 85/2,5/10/2,5. Стадія D: (5R,8R,11S)-5,11-Дибензил-6гідрокси-3,9-діоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-ової кислоти 6-оксид (R,R,S)діастереоізомер одержують очищенням сполуки Стадії C вище в ізократичному режимі, використовуючи напівпрепаративну 250х10ММ AIT колонку (нерухома фаза Kromasil Q18 10мкм, пори 100Å). Ізократичне елюювання в кислотних умовах: 52% ацетонітрил; 0,1% трифтороцтова кислота. Mac-спектрометрія (ES/MS) = 694,3Да ПРИКЛАД 3: (5R,8R,11S)-5-Бензил-6-гідрокси-11-(4гідроксибензил)-3,9-діоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)]-2-окса-4,10-діаза-6-фосфадодекан12-ової кислоти 6-оксид Стадія A: (1R)-1-{ [(Бензилокси)карбоніл] аміно}-2-фенілетил-[2-({[(1S)-2-трет-бутокси-1-(4-третбутоксибензил)-2-оксоетил] аміно} карбоніл)-4пентиніл]фосфінова кислота Цю сполуку одержують відповідно до методики, розкритої на Стадії А Прикладу 1, використовуючи хлорид L-тирозин О-ди-трет-бутилового ефіру замість хлориду 1-триптофан метилового ефіру. Стадія В: (1R)-1-{[(Бензилокси)карбоніл]аміно}2-фенілетил-{3-{[(1S)-2-трет-бутокси-1-(4-третбутоксибензил)-2-оксоетил]аміно}-3-оксо-2-[(3феніл-5-ізоксазоліл)метил]пропіл} фосфінова кислота 91214 20 Цю сполуку одержують відповідно до методики, розкритої на Стадії В Прикладу 2, використовуючи сполуку вказаної вище Стадії А. Стадія С: (5R,8*,11S)-5-Бензил-6-гідрокси-11(4-гідроксибензил)-3,9-діоксо-1-феніл-8-[(3-феніл5-ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-ової кислоти 6-оксид Цю сполуку одержують відповідно до методики, розкритої на Стадії C Прикладу 3, використовуючи сполуку вказаної вище Стадії В. Стадія D: (5R,8R,11S)-5-Бензил-6-гідрокси-11(4-гідроксибензил)-3,9-діоксо-1-феніл-8-[(3-феніл5-ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-ової кислоти 6-оксид (R,R,S) діастереоізомер одержують очищенням сполуки вказаної вище Стадії C в ізократичному режимі, використовуючи напівпрепаративну 250х10ММ AIT колонку (нерухома фаза Kromasil Q18 10мкм, пори 100Å). Ізократичне елюювання в кислотних умовах: 43% ацетонітрил; 0,1% трифтороцтова кислота. Mac-спектрометрія (ES/MS) = 711,3Да ПРИКЛАД 4: (5R,8*,11S)-5-Бензил-11-(4-гідроксибензил)3,9,12,16-тетраоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил]-2,13,15-триокса-4,10-діаза17,17-диметилоктадекан-6-фосфінова кислота Стадія А: N-[(Бензилокси)карбоніл]-4-[(третбутоксикарбоніл)окси]-фенілаланін Гідроксид калію додають до розчину 10 ммоль N-[(бензилокси)карбоніл]-4-гідроксифенілаланін, поки не буде досягнуто рН 12. Реакційну суміш охолоджують в льодяній ванні, і додають 1,5 еквіваленти BoC2O, і температуру суміші потім повертали до температури навколишнього середовища, в той час підтримуючи рН розчину на рівні 12 шляхом додавання твердого гідроксиду калію. Через 2 години додають наступні 0,5 еквівалента BoC2O. Через 18 годин суміш потім концентрують під зниженим тиском і водну фазу екстрагують сумішшю (1/1) Еt2О/петролейний ефір. Водну фазу ацилюють в холодному стані з 2М HCl доти, доки не досягають рН 1, і потім екстрагують двічі етилацетатом. Хроматографія (хлороформ/метанол: 9,5/0,5) дозволяє виділити очікуваний продукт. Стадія В: [(2-{[(Бензилокси)карбоніл]аміно}-3{4-[(трет-бутоксикарбоніл)-окси]феніл} пропіноїл)окси]метил півалат До розчину 2,5 ммоль сполуки вказаної вище Стадії А в 15мл хлороформу додають (nBu)4NHSO4 (0,5 еквіваленти), триетиламін (2 еквіваленти), йодид натрію (1 еквівалент) і хлорметил півалат (2 еквіваленти). Реакційну суміш нагрівають зі зворотним холодильником протягом 18 годин і потім концентрують. Залишок розчиняють в діетиловому ефірі і екстрагують водою, двічі 0,5M HCl, двічі 5% NaHCO3 і водою. Органічну фазу концентрують і хроматографія (дихлорметан/метанол: від 9,9/0,1 до 9,5/0,5) дозволяє виділити очікуваний продукт. Стадія С: [(2-Аміно-3-{4-[(третбутоксикарбоніл)окси]феніл}пропаноїл)окси]метил-2,2-диметилпропаноатгідрохлорид 2 ммоль сполуки вказаної вище Стадії В розчиняють в суміші (2/1) етанол/вода і додають декі 21 лька крапель 0,5M HCl доти, доки не досягають рН 1. Реакційну суміш гідрогенізують за допомогою водневої колби у присутності Pd/C (300мг) як каталізатора. Через 2,5 години суміш фільтрують над Целітом і потім випаровують досуха, щоб одержати очікуваний продукт. Стадія D: (5R,8*,11S)-5-Бензил-11-{4-[(третбутоксикарбоніл)окси]бензил}-3,9,12,16-тетраоксо1 -феніл-8-[3-феніл-5-ізоксазол)метил]-2,13,15триокса-4,10-діаза-17,17-диметилоктадекан-6фосфінова кислота До суспензії 1,3 ммоль сполуки Приготування 3 в 7мл дихлорметану додають 3 еквіваленти діізопропілетиламіну, 1 еквівалент сполуки вказаної вище Стадії С, 1 еквівалент HOBt і 5 еквівалентів EDC.HCl. Реакційну суміш перемішують протягом 75 хвилин і потім концентрують під зниженим тиском. Залишок розчиняють в 50мл суміші (9/1) етилацетат/діетиловий ефір і потім екстрагують чотири рази 1M HCl, H2O, тричі 5% NH4HCO3, H2O, двічі 5% NaHCO3, H2O, 1M HCl і потім розсолом. Органічну фазу концентрують, і хроматографія (хлороформ/метанол/оцтова кислота: 9,5/0,4/0,1) дозволяє виділити очікуваний продукт. Стадія Е: (5R,8*,11S)-5-Бензил-11-(4гідроксибензил}-3,9,12,16-тетраоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2,13,15-триокса-4,10діаза-17,17-диметилоктадекан-6-фосфінова кислота 1 ммоль сполуки вказаної вище Стадії D розчиняють в 10мл мурашиної кислоти і 200мкл триізопропілсилану. Реакційну суміш перемішують при температурі навколишнього середовища протягом 50 хвилин і потім концентрують під зниженим тиском. Залишок розчиняють в етилацетаті і екстрагують двічі розсолом, двічі 5% NaHCO3, розсолом, 1M HCl і розсолом. Органічну фазу концентрують, щоб дати очікуваний продукт. Mac-спектрометрія (MS-ESI) = 826,3 (М+Н)+ ПРИКЛАД 5: (5R,8*,11S)-5-Бензил-11-(4-гідроксибензил}3,9,12-триоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил]-2,13-діокса-4,10-діазапентадекан-6-фосфінова кислота Стадія А: 2-{[(Бензилокси)карбоніл]аміно}-3-{4[(трет-бутоксикарбоніл)-окси]феніл}етил пропаноат До розчину 4 ммоль сполуки Стадії А Прикладу 4 в 5мл абсолютного етанолу додають HOBt (1,1 еквіваленти), EDC.HCl (1,1 еквіваленти), DMAP (каталітична кількість) і DIPEA (1,1 еквіваленти). Реакційну суміш перемішують при температурі навколишнього середовища протягом 18 годин і потім концентрують. Залишок розчиняють в діетиловому ефірі і екстрагують водою, двічі 0,5M HCl, двічі 5% NaHCO3 і водою. Водну фазу висушують над Na2SO4 і потім концентрують, щоб одержати очікуваний продукт. Стадія В: 2-Аміно-3-{4-[(третбутоксикарбоніл)окси]феніл}етил пропаноат гідрохлорид Продукт одержують відповідно до методики Стадії C Прикладу 4, використовуючи сполуку вказаної вище Стадії А. Стадія С: (5R,8*,11S)-5-Бензил-11-{4-[(трет 91214 22 бутоксикарбоніл)окси]бензил}-3,9,12-триоксо-1феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2,13-діокса4,10-діаза-пентадекан-6-фосфінова кислота Продукт одержують відповідно до методики Стадії D Прикладу 4, використовуючи сполуку вказаної вище Стадії В. Стадія D: (5R,8*,11S)-5-Бензил-11-(4гідроксибензил)-3,9,12-триоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2,13-діокса-4,10-діазапентадекан-6-фосфінова кислота Продукт одержують відповідно до методики Стадії E Прикладу 4, використовуючи сполуку вказаної вище Стадії C. Mac-спектрометрія (MS-ESI) = 740,2 (М+Н)+ ПРИКЛАД 6: (5R,8*,11S)-5-Бензил-11-(4-гідроксибензил)3,9,12,17-тетраоксо-1,17-дифеніл-8-[(3-феніл-5ізоксазоліл)метил]-2,13-діокса-4,10-діаза-16тіагептадекан-6-фосфінова кислота Стадія А: 2-(Бензоїлсульфаніл)-3-(4-третбутоксифеніл)-2-{[(9Н-флуорен-9ілметокси)карбоніл]аміно}етил пропаноат Суміш одержують відповідно до методики Стадії А Прикладу 5, використовуючи FmocTyr (OBut)(OH) замість сполуки Стадії А Прикладу 4 і використовуючи PhCOSCH2CH2OH (розкритий в літературі I. Lefebvre et al, J. Med. Chem., 1995, 38, 3941-3950) замість абсолютного етанолу в дихлорметані. Стадія В: 2-(Бензоїлсульфаніл)-2-аміно-3-(4трет-бутоксифеніл)етил пропаноат 1,5 ммоль сполуки вказаної вище Стадії А розчиняють в 12мл диметилформаміду і3 еквівалентів діетиламіну. Реакційну суміш перемішують при температурі навколишнього середовища протягом 1 години і потім концентрують під зниженим тиском. Залишок розчиняють в етилацетаті і екстрагують три рази водою і розсолом. Хроматографія (хлороформ/метанол: 9,5/0,5) і потім осадження з суміші (2/1) петролейний ефір/діетиловий ефір дозволяє виділити очікуваний продукт. Стадія С: (5R,8*,11S)-5-Бензил-11-(4-третбутоксибензил)-3,9,12,17-тетраоксо-1,17-дифеніл8-[(3-феніл-5-ізоксазоліл)метил]-2,13 -діокса-4,10діаза-16-тіагептадекан-6-фосфінова кислота Продукт одержують відповідно до методики Стадії D Прикладу 4, використовуючи сполуку вказаної вище Стадії В. Стадія D: (5R,8*,11S)-5-Бензил-11-(4-третбутоксибензил)-3,9,12,17-тетраоксо-1,17-дифеніл8-[(3-феніл-5-ізоксазоліл)метил]-2,13-діокса-4,10діаза-16-тіагептадекан-6-фосфінова кислота Продукт одержують відповідно до методики Стадії E Прикладу 4, використовуючи сполуку вказаної вище C. Mac-спектрометрія (MS-ESI) = 874,3 (M-H)ПРИКЛАД 7: (5R,8*,11S)-5-Бензил-11-(4-третбутоксибензил)-6-[2(етантіоат)етокси]-3,9-діоксо-1феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2-окса-4,10діаза-6-фосфадодекан-12-ової кислоти 6-оксид Стадія A: (5R,8*,11S)-5-Бензил-11-(4-третбутоксибензил)-3,9,12-триоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2,13-діокса-4,10-діаза14,14-диметилпентадекан-6-фосфінова кислота 23 Продукт одержують відповідно до методики Стадії D Прикладу 4, використовуючи сполуку Приготування 3 і НСl.НТyr(ОВut)ОВut. Стадія В: трет-Бутил (5R,8*,11S)-5-бензил-114-(трет-бутоксибензил)-6-[2(етантіоат)етокси]-3,9діоксо-1-феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2окса-4,10-діаза-6-фосфадодекан-12-оат 6-оксид 0,36 ммоль сполуки вказаної вище Стадії А розчиняють в 4мл диметилформаміду і потім додають 3 еквіваленти DIPEA, 3 еквіваленти CH3COSCH2CH2OH (I. Lefebvre et al., J. Med. Chem., 1995, 38, 3941-3950) і 3 еквіваленти РуРОВ. Реакційну суміш перемішують при температурі навколишнього середовища протягом 48 годин і потім розбавляють етилацетатом. Органічну фазу екстрагують чотири рази 1M HCl, H2O, тричі 5% NH4HCO3, двічі 1M HCl і потім розсолом. Органічну фазу висушують і потім концентрують. Хроматографія (хлороформ/метанол: 9,8/0,2) дозволяє виділити очікуваний продукт. Стадія С: (5R,8*,11S)-5-Бензил-11-4-(третбутоксибензил)-6-[2(етантіоат)-етокси]-3,9-діоксо1-феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2-окса4,10-діаза-6-фосфадодекан-12-онової кислоти 6оксид Продукт одержують відповідно до методики Стадії E Прикладу 4, використовуючи сполуку вказаної вище Стадії В. Mac-спектрометрія (MS-ESI) = 814,3 (М+Н)+ ПРИКЛАД 8: Етил (5R,8*,11S)-5-бензил-6-етокси-11-(4гідроксибензил)-3,9-діоксо-1-феніл-8-[(3-феніл-5ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-оат 6-оксид Стадія А: Етил (5R,8*,11S)-5-бензил-11-{4[(трет-бутоксикарбоніл)окси]-бензил}-6-етокси-3,9діоксо-1-феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2окса-4,10-діаза-6-фосфадодекан-12-оат 6-оксид Продукт одержують відповідно до методики Стадії В Прикладу 7, використовуючи сполуку Стадії C Прикладу 5 і етанол. Стадія В: Етил (5R,8*, 11S)-5-бензил-6-етокси11-(4-гідроксибензил)-3,9-діоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-оат 6-оксид Продукт одержують відповідно до методики Стадії E Прикладу 4, використовуючи сполуку вказаної вище Стадії А. Mac-спектрометрія (MS-ESI) = 768,3 (М+Н)+; 790,3 (M+Na+)+ ПРИКЛАД 9: Етил (5R,8*,11S)-5-бензил-11-(4гідроксибензил)-6-[2-метил-1-(пропіонілокси)пропокси]-3,9-діоксо-1 -феніл-8-[(3-феніл-5ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-оат 6-оксид Стадія А: Етил (5R,8*,11S)-5-бензил-11-{4[(трет-бутоксикарбоніл)окси]-бензил}-6-[2-метил-1(пропіонілокси)пропокси]-3,9-діоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-оат 6-оксид Продукт одержують відповідно до методики Стадії В Прикладу 7, використовуючи сполуку 91214 24 Стадії C Прикладу 5 і BrCH(CH(CH3)2)OCOEt (М. Neuenschwander et al., Helv. Chim. acta, 1978, 61, 2047-2058). Стадія В: Етил (5R,8*,11S)-5-бензил-11-(4гідроксибензил)-6-[2-метил-1(пропіонілокси)пропокси]-3,9-діоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-оат 6-оксид Продукт одержують відповідно до методики Стадії E Прикладу 4, використовуючи сполуку вказаної вище Стадії А. Mac-спектрометрія (MS-APCI) = 868,4 (М+Н)+; 885,5 (M+NH4+)+ ПРИКЛАД 10: (5R,8*,11S)-5-Бензил-11-(4-гідроксибензил)-6[2-метил-1-(пропіонілокси)-пропокси]-3,9-діоксо-1феніл-8-[(3-феніл-5-ізоксазоліл)метил]-2-окса-4,10діаза-6-фосфадодекан-12-онової кислоти 6-оксид Стадія А: трет-Бутил (5R,8*,11S)-5-бензил-11(4-трет-бутоксибензил)-6-[2-метил-1(пропіонілокси)пропокси]-3,9-діоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-оат 6-оксид Продукт одержують відповідно до методики Стадії А Прикладу 9, використовуючи сполуку Стадії А Прикладу 7 замість сполуки Стадії C Прикладу 5. Mac-спектрометрія (MS-ESI) = 974,5 (M+Na+)+ Стадія В: (5R,8*,11S)-5-Бензил-11-(4гідроксибензил)-6-[2-метил-1(пропіонілокси)пропокси]-3,9-діоксо-1-феніл-8-[(3феніл-5-ізоксазоліл)метил]-2-окса-4,10-діаза-6фосфадодекан-12-онової кислоти 6-оксид Продукт одержують відповідно до методики Стадії E Прикладу 4, використовуючи сполуку указаної вище Стадії А. Mac-спектрометрія (MS-ESI) = 838,5 (M-H)ФАРМАКОЛОГІЧНІ ДОСЛІДЖЕННЯ СПОЛУК ЗА ДАНИМ ВИНАХОДОМ ПРИКЛАД А: In vitro інгібуючий вплив на ангіотензин І - перетворювальний фермент (АСЕ) та ендотелінперетворювальний фермент (ЕСЕ) Для того, щоб порівняти специфічність, сполуки Прикладів досліджують на 2 ферментах: АСЕ (рекомбінантна людська форма) і ЕСЕ (рекомбінантна людська форма ізоформи ЕСЕ-1с). Дослідження проводять двічі на 96-ямкових планшетах. Інгібітор інкубують разом з ферментом протягом 45 хвилин перед додаванням субстрату гашеної флуоресценції. Виділену флуоресценцію визначають і вимірюють на планшет-рідері Fluoroscan Ascent (Thermo-Labsy stems). Використані флуорогенні субстрати являють собою: Mca-Arg-Pro-Pro-Gly-Phe-Ser-Pro-DpaCOOH (5мкМ) з АСЕ, і Mca-Arg-Pro-Pro-Gly-Phe-Ser-AlaPhc-Lys(Dnp)COOH (5мкМ; R&D Systems) з ЕСЕ. Сполуки за даним винаходом проявляють хороше інгібування АСЕ і ЕСЕ, зі значеннями IC50 25нМ і 8нМ, відповідно. Результати, які одержують, порівнюють у наступній Таблиці: 25 91214 26 Таблиця IC50 (нМ) Сполука АСЕ 36 3,8 1,4 Приклад 1 Приклад 2 Приклад 3 ПРИКЛАД В: Фармацевтична композиція 1000 таблеток, кожна з яких містить 5мг сполука Прикладу 1 пшеничний крохмаль маїсовий крохмаль лактоза стеарат магнію силікагель гідроксипропілцелюлоза Комп’ютерна верстка А. Крулевський ЕСЕ 8 7,7 1,4 5г 20г 20г 30г 2г 1г 2г Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюDerivatives of phosphinic amino acid and pharmaceutical composition containing same
Автори англійськоюDive Vincent, Jullien Nicolas, Scalbert Elizabeth, Yiotakis Athanasios, Makaritis Anastasios
Назва патенту російськоюСоединения фосфиновой аминокислоты и фармацевтическая композиция, которая их содержит
Автори російськоюДиве Винсен, Жюльен Николя, Скальбер Элизабет, Иотакис Атанасиос, Макаритис Анастасиос
МПК / Мітки
МПК: C07F 9/32, A61K 31/675, C07F 9/00
Мітки: фосфінової, амінокислоти, містить, сполуки, композиція, фармацевтична, яка
Код посилання
<a href="https://ua.patents.su/13-91214-spoluki-fosfinovo-aminokisloti-i-farmacevtichna-kompoziciya-yaka-kh-mistit.html" target="_blank" rel="follow" title="База патентів України">Сполуки фосфінової амінокислоти і фармацевтична композиція, яка їх містить</a>
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Міларі Банавара Лакшман, Маррі Джеррі Ентоні, Чу-Моєр Маргарет Юхуа, Зембровскі Уільям Джеймс
МПК: C07D 413/12, C07D 403/12, C07D 487/04, C07D 491/04, A61P 43/00, C07D 417/14, C07D 401/14, C07D 405/14, C07D 491/048, C07D 239/42, C07D 487/08, A61P 9/10, C07D 409/12, C07D 405/12, C07D 491/20, C07D 403/14, A61K 31/5377, C07D 401/04, C07D 521/00, C07D 451/02, C07D 513/10, A61K 31/506, C07D 409/14, C07D 471/08, C07D 451/06, C07D 401/12, A61K 31/517, C07D 491/10, C07D 498/04, C07D 417/12, C07D 471/04, A61P 3/10, C07D 403/04, A61K 31/53
Мітки: містить, композиція, сполуки, інгібітори, проміжної, фармацевтична, проміжні, спосіб, одержання, піримідини, сорбітдегідрогенази
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...
Сполуки піролідину і тіазолідину, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 84553
Опубліковано: 10.11.2008
Автори: Комбетт Мюріель, Бенуат Ален, Де Нантей Гійом, Арлей Елізабет
МПК: A61K 31/427, C07D 401/12, A61K 31/426, C07D 311/96, A61P 3/10, A61K 31/438, A61K 31/40, C07D 417/12, A61P 43/00, C07D 295/18, C07D 207/16, C07D 409/12, C07D 277/06, A61K 31/4025, C07D 207/06, C07D 405/12
Мітки: яка, сполуки, піролідину, композиція, одержання, тіазолідину, містить, спосіб, фармацевтична
Формула / Реферат:
1. Сполука формули (І):, (I) де:Х1 являє собою атом або групу, вибрану з CR4aR4b, О, S(O)q1 і NR5,де R4а і R4b, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену С1-С6алкільну групу,або R4a і R4b разом утворюють, з атомом вуглецю, який несе їх, С3-С7циклоалкільну групу,q1 являє...
Заміщені сполуки бензо[e][1,4]оксазино[3,2-g]ізоіндолу, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 79163
Опубліковано: 25.05.2007
Автори: Ренар Пьєр, Лепіфр Франк, Кудер Жерар, Краус-Бертьє Лоранс, Ікман Джон, Кеньяр Даніель-Енрі, Пьєр Ален
МПК: C07D 497/00, A61P 35/00
Мітки: бензо[e][1,4]оксазино[3,2-g]ізоіндолу, сполуки, заміщені, яка, композиція, одержання, містить, спосіб, фармацевтична
Формула / Реферат:
1. Сполуки формули (І):,де:W1 являє собою, з атомами вуглецю, до яких він прикріплений, фенільну групу або піридильну групу,Ζ являє собою групу, вибрану з водню, галогену і груп лінійного або розгалуженого (С1-С6)алкілу, арилу, арил-(С1-С6)алкілу, в якому алкільна частина може бути лінійною або розгалуженою, арилокси, арил-(С1-С6)алкокси,...
Сполуки 4-оксо-4,6,7,8-тетрагідропіроло[1,2-a]піразин-6-карбоксаміду, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 83638
Опубліковано: 11.08.2008
Автори: Пармантьє Жан-Жиль, Валлє Марі-Оділь, Бенуат Ален, ГЛОАНЕК Філіп, Верберен Тоні, Де Нантей Гійом, Рюпен Ален
МПК: A61P 43/00, C07D 487/04, A61P 9/00, A61K 31/519, A61P 7/02, A61P 9/02, C12Q 1/56, C12N 9/74
Мітки: фармацевтична, сполуки, композиція, спосіб, одержання, 4-оксо-4,6,7,8-тетрагідропіроло[1,2-a]піразин-6-карбоксаміду, містить, яка
Формула / Реферат:
1. Сполука формули (І):, (I)де: являє собою 1-оксидопіридильну групу, заміщену залишком молекули в будь-якому з положень 2, 3 і 4,m і n, які можуть бути однаковими або відрізнятись, кожний являє собою ціле число від 1 до 3,R1 являє собою атом водню або лінійну або...
Сполуки тіадіазину, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 80046
Опубліковано: 10.08.2007
Автори: Бовері Стефан, Данобер Лоранс, Піротт Бернар, Франкотт П'єр, Ренар Пьєр, Кеньяр Даніель-Енрі, Грандорж Еманюель, деТюльйо Паскаль, Лєстаж П'єр
МПК: C07D 513/04, A61P 25/18
Мітки: яка, спосіб, композиція, фармацевтична, одержання, сполуки, тіадіазину, містить
Формула / Реферат:
1. Сполуки формули (І):, (І)де:А, з двома атомами вуглецю, які до нього приєднані, утворює тієнільну, фурильну або піролільну групу, вибрану з груп А1, А2, А3:, ,