Спосіб отримання силденафілу та проміжні сполуки





Формула / Реферат
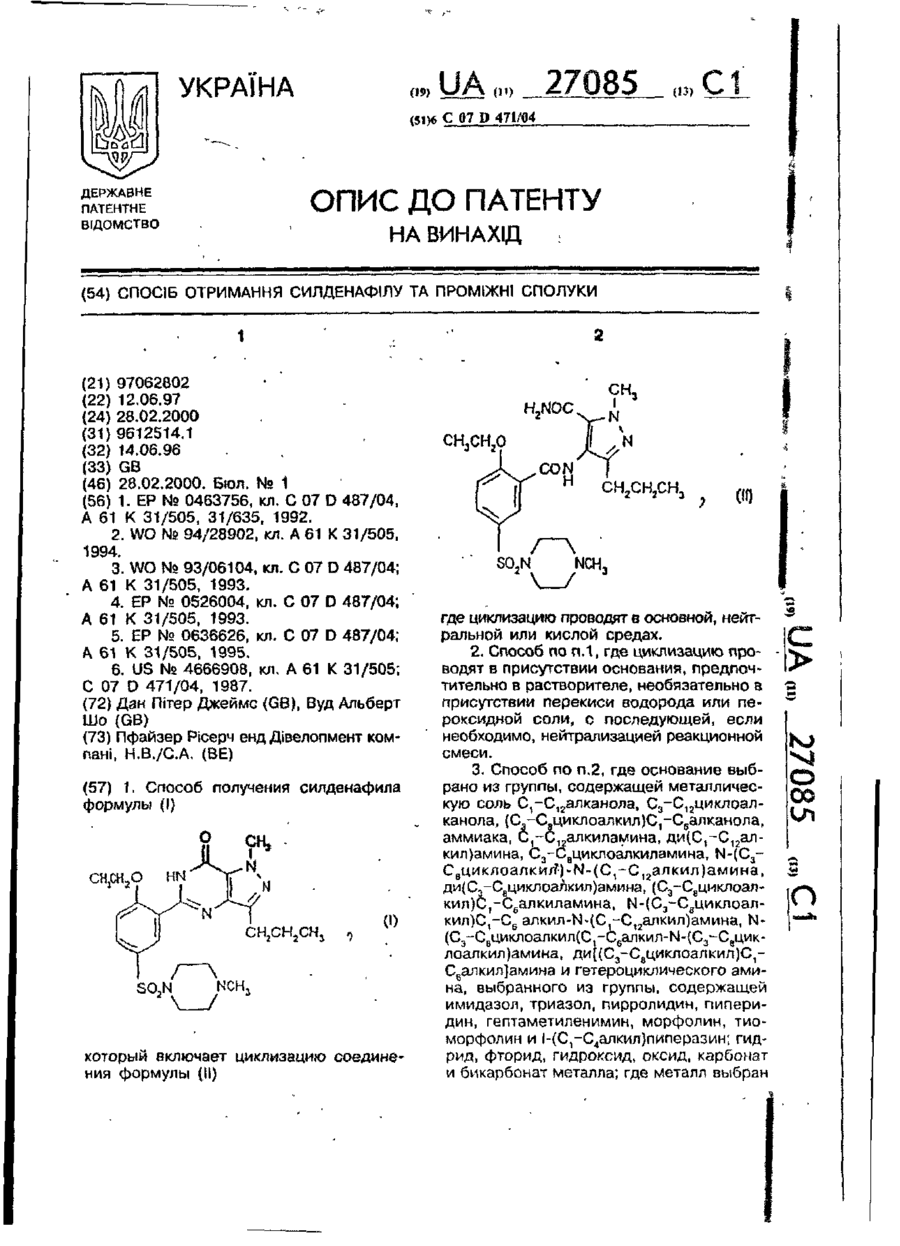
1. Способ получения силденафила формулы (I)
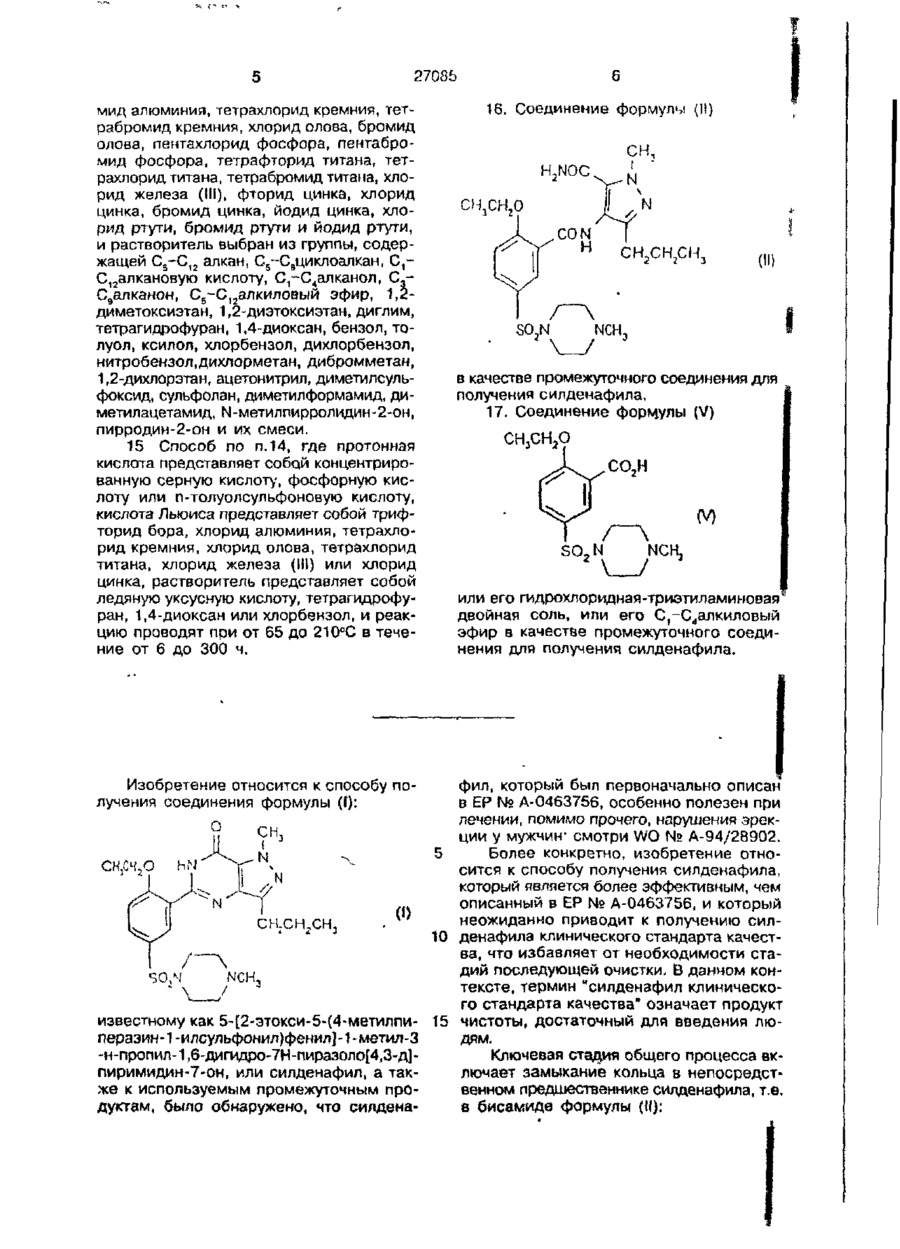
который включает циклизацию соединения формулы (II)
где циклизацию проводят в основной, нейтральной или кислой средах.
2. Способ по п.1, где циклизацию проводят в присутствии основания, предпочтительно в растворителе, необязательно в присутствии перекиси водорода или пероксидной соли, с последующей, если необходимо, нейтрализацией реакционной смеси.
3. Способ по п.2, где основание выбрано из группы, содержащей металлическую соль С1-С12 алканола, С3-С12 циклоалканола, (С3-С8-циклоалкил)С1-С6-алканола, аммиака, С1-С12алкиламина, ди(С1-С12алкил)амина, С3-С8циклоалкиламина, N-(C3-С8циклоалкил)-N-(С1-С12алкил)амина, ди(С3-С8циклоалкил)амина, (С3-С8циклоалкил)С1-С6алкиламина, N-(С3-С8циклоалкил)С1-С6 алкил-N-(С1-С12алкил)амина, N-(С3-С8циклоалкил),С1-С6алкил-N-(С3-С8циклоалкил)амина, ди[(С3-С8циклоалкил)С1-С6алкил]амина и гетероциклического амина, выбранного из группы, содержащей имидазол, триазол, пирролидин, пиперидин, гептаметиленимин, морфолин, тиоморфолин и I-(С1-С4алкил)пиперазин; гидрид, фторид, гидроксид, оксид, карбонат и бикарбонат металла; где металл выбран из группы, содержащей литий, натрий, калий, рубидий, цезий, бериллий, магний, кальций, стронций, барий, алюминий, индий, таллий, титан, цирконий, кобальт, медь, серебро, цинк, кадмий, ртуть и церий; и С7-С12 бициклический амидин; и растворитель может быть выбран из группы, содержащей С1-С12алканол, С3-, С12циклоалканол, (С3-С8циклоалкил)С1-С6алканол, С1-С9алканон, С4-С10циклоалканон, С5-С12алкиловый эфир, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, бензол, толуол, ксилол, хлорбензол, дихлорбензол, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, диметилацетамид, N-метилпирролидин-2-он, пирролидин-2-он, пиридин и вода и их смеси.
4. Способ по п.3, где основание выбрано из группы, содержащей соль щелочного или щелочноземельного металла С1-С12алканола, С3-С12циклоалканола и (С3-С8циклоалкил)С1-С6алканола; соль щелочного металла и аммиака, N-(вторичный или третичный С3-С6алкил)-N-(первичный, вторичный или третичный С3-С6алкил)амина, С3-С8циклоалкиламина, N-(С3-С8циклоалкил)-N-(первичный, вторичный или третичный С3-С6алкил)амина, ди(С3-С8циклоалкил)амина и 1-метилпиперазина; гидрид, гидроксид, оксид, карбонат и бикарбонат щелочного или щелочноземельного металла; 1,5-диазабицикло[4,3,0]нон-5-ен и 1,8-диазабицикло[5,4,0]ундец-7-ен, и растворитель выбран из группы, содержащей этанол, 2-пропанол, вторичный или третичный С4-С12алканол, С3-С12циклоалканол, третичный С4-С12циклоалканол, вторичный или третичный (С3-С7циклоалкил)С2-С6алканол, С3-С9алканон, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, толуол, ксилол, хлорбензол, 1,2-дихлорбензол, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, N-метилпирроли-дин-2-он, пиридин и вода и их смеси.
5. Способ по любому из пп.2 - 4, где реакцию проводят при от 50 до 170°С в течение от 3 до 170ч.
6. Способ по п.5, где количество применяемого основания составляет от 1,0 до 5,0 молекулярных эквивалентов.
7. Способ по п.6, где основание выбрано из группы, содержащей литиевые, натриевые и калиевые соли С1-С12алканола, С4-С12циклоалканола, аммиака, циклогексиламина и 1-метилпиперазина; гидридные соли лития, натрия и калия; карбонат цезия и оксид бария, растворитель выбирают из группы, содержащей этанол, третичный С4-С10спирт, третичный С6-С8циклоалканол, тетрагидрофуран, 1,4-диоксан, и ацетонитрил, реакцию проводят при от 60 до 105°С, и количество применяемого основания составляет от 1,1 до 2,0 молекулярных эквивалентов.
8. Способ по п.7, где основание выбрано из группы, содержащей С1-С12алкоксидные и гидридные соли лития, натрия и калия, амид натрия, циклогексиламид натрия и карбонат цезия, растворитель выбирают из группы, содержащей этанол, трет-бутанол, трет-амиловый спирт, 1-метилциклогексанол, тетрагидрофуран и 1,4-диоксан, реакцию проводят в течение от 3 до 60ч.
9. Способ по п.8, где основание выбрано из группы, содержащей этоксид натрия, трет-бутоксид натрия, трет-бутоксид калия и гидрид натрия, и растворитель выбирают из группы, содержащей этанол, трет-бутанол, трет-амиловый спирт и тетрагидрофуран.
10. Способ по п.1, где циклизацию проводят путем нагревания соединения формулы (II), необязательно в присутствии растворителя и/или необязательно в присутствии дегидратирующего агента, и/или механической водоудаляющей системы.
11. Способ по п.10, где растворитель выбран из группы, содержащей 1,2-дихлорбензол, диметилсульфоксид, сульфолан, Т-метилпирролидон-2-он и пирролидин-2-он и их смеси, и дегидратирующий агент выбран из группы, содержащей безводный карбонат калия, безводный карбонат натрия, безводный сульфат магния, безводный сульфат натрия, пентоксид фосфора и молекулярные сита.
12. Способ по п.11, где растворитель представляет собой 1,2-дихлорбензол, сульфолан или N-метилпирролидин-2-он, дегидратирующий агент представляет собой молекулярные сита, и реакцию проводят при от 180 до 220°С в течение от 0,5 до 72ч.
13. Способ по п.1, где циклизацию проводят в присутствии протонной кислоты или кислоты Льюиса, необязательно в присутствии растворителя.
14. Способ по п.13, где протонная кислота выбрана из группы, содержащей неорганическую кислоту, органосульфоновую кислоту, органофосфоновую кислоту и органическую карбоновую кислоту, кислота Льюиса выбрана из группы, содержащей трифторид бора, трихлорид бора, трибромид бора, хлорид алюминия, бромид алюминия, тетрахлорид кремния, тетрабромид кремния, хлорид олова, бромид олова, пентахлорид фосфора, пентабромид фосфора, тетрафторид титана, тетрахлорид титана, тетрабромид титана, хлорид железа (III), фторид цинка, хлорид цинка, бромид цинка, йодид цинка, хлорид ртути, бромид ртути и йодид ртути, и растворитель выбран из группы, содержащей С5-С12алкан, С5-С8циклоалкан, С1-С12алкановую кислоту, С1-С4алканол, С1-С9алканон, С5-С12алкиловый эфир, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, бензол, толуол, ксилол, хлорбензол, дихлорбензол, нитробензол,дихлорметан, дибромметан, 1,2-дихлорэтан, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, диметилацетамид, N-метилпирролидин-2-он, пирродин-2-он и их смеси.
15. Способ по п.14, где протонная кислота представляет собой концентрированную серную кислоту, фосфорную кислоту или n-толуолсульфоновую кислоту, кислота Льюиса представляет собой трифторид бора, хлорид алюминия, тетрахлорид кремния, хлорид олова, тетрахлорид титана, хлорид железа (III) или хлорид цинка, растворитель представляет собой ледяную уксусную кислоту, тетрагидрофуран, 1,4-диоксан или хлорбензол, и реакцию проводят при от 65 до 210°С в течение от 6 до 300ч.
16. Соединение формулы (II)
в качестве промежуточного соединения для получения силденафила.
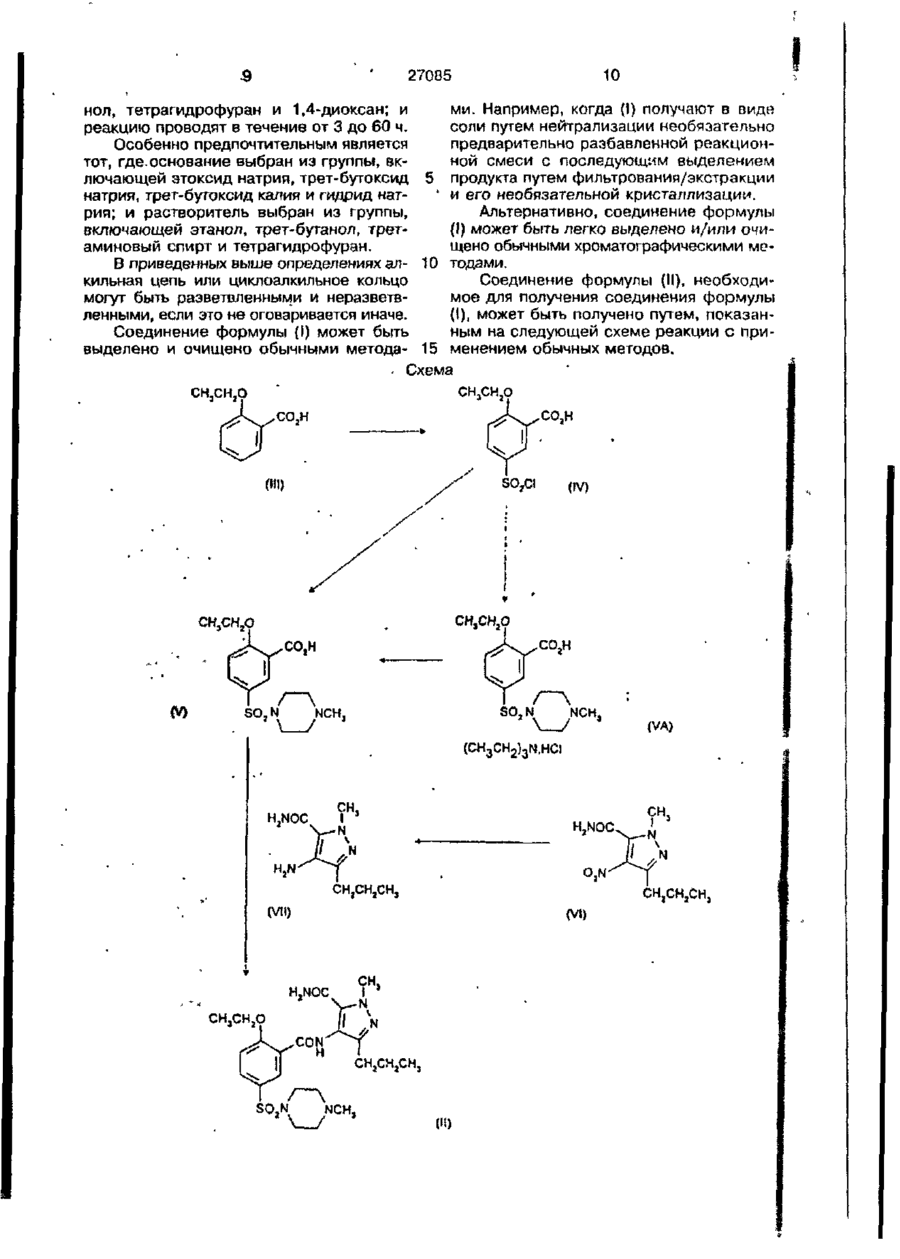
17. Соединение формулы (V)
или его гидрохлоридная-триэтиламиновая двойная соль, или его С1-С4алкиловый эфир в качестве промежуточного соединения для получения силденафила.
Текст
1. Способ получения силденафила формулы (I) (I) SO2N NCH, который включает циклизацию соединения формулы (II) сн э сн 3 о сн 2 сн 2 сн 3 (И) S02N где циклизацию проводят в основной, нейтральной или кислой средах. 2. Способ по п.1, где циклизацию проводят в присутствии основания, предпочтительно в растворителе, необязательно в присутствии перекиси водорода или пероксидной соли, с последующей, если необходимо, нейтрализацией реакционной смеси. 3. Способ по п.2, где основание выбрано из группы, содержащей металлическую соль С^С^алканола, С 3 -С 12 циклоалканола, (Сз-СдЦИклоалкил)С,-С6алканола, аммиака, С,-С12алкиламина, AH(Ct-C12aJiкил)амина, С3-Свциклоалкиламина, N-(C3~ С 8 циклоалкил*-)-Ы-(С 1 -С 12 алкил)амина, ди(Сз-Сециклоалкил)амина, (С3-Сециклоалкил)6,-С 6 алкиламина, Ы-(С3-Свциклоалкил)С,-С 6 алкил-Ы^С^С^алкилЭамина, N(С3-Свциклоалкил(С1-С6алкил-М-(С3-С8циклоалкил)амина, ди[(С 3 -С 8 циклоалкил)С 1 С6алкил]амина и гетероциклического амина, выбранного из группы, содержащей имидазол, триазол, пирролидин, пиперидин, гептаметиленимин, морфолин, тиоморфолин и 1-(С,-С4алкил)пиперазин; гидрид, фторид, гидроксид, оксид, карбонат и бикарбонат металла; где металл выбран С 27085 из группы, содержащей литий, натрий, калий, рубидий, цезий, бериллий, магний, кальций, стронций, барий, алюминий, индий, таллий, титан, цирконий, кобальт, медь, серебро, цинк, кадмий, ртуть и церий; и С7~С12 бициклический амидин; и растворитель может быть выбран из группы, содержащей С,-С12алканол, С 3 -, С12циклоалканол, (С 3 -С в циклоалкил)С,С6алканол, С--С9алканон, С4-С10циклоалканон, С 5 -С 1 г алкиловый эфир, 1,2диметоксиэтаи, 1,2-диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, бензол, толуол, ксилол, хлорбензол, дихлорбензол, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, дим етил ацетамид, Ы-метилпирролидин-2-он, пирролидин-2он, пиридин и вода и их смеси. 4. Способ по п.З, где основание выбрано из группы, содержащей соль щелочного или щелочноземельного металла С,С12алканола, С3-С]2циклоалканола и (С 3 Свциклоалкил^-С^алканола; соль щелочного металла и аммиака, Ы-(вторичный или третичный С3-С6алкил)-М-(п9рвичный, вторичный или третичный С3~С6алкил)амина, С3-Свциклоалкиламина, Ы-(С3-Сециклоалкил)-Ы~ (первичный, вторичный или третичный С3-С6алкил)амина, ди(С3-Сациклоалкил)амина и 1-метилпиперазина; гидрид, гидроксид, оксид, карбонат и бикарбонат щелочного или щелочноземельного металла; 1,5-диазабицикло[4,3,0]нон-5-ен и 1,8-диазабицикло[5,4,0]ундец-7-ен, • и растворитель выбран из группы, содержащей этанол, 2-пропанол,вторичный или третичный С4-С1гэлканол, Сэ~С12циклоалканол, третичный С^-С^циклоалканол, вторичный или третичный (С3-С7циклоалкиЛ)С 2 -С Б алканол, С 3 -С 9 алканон, 1,2диметоксиэтан, 1,2-диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, толуол, ксилол, хлорбензол, 1,2-дихлорбензол, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, N-метилпирролидин-2-он, пиридин и вода и их смеси. 5. Способ по любому из пп.2-4, где реакцию проводят при от 50 до 170°С в течение от 3 до 170 ч. 6. Способ по п 5, где количество применяемого основания составляет от 1,0 до 5,0 молекулярных эквивалентов. 7. Способ по п.6, где основание выбрано из группы, содержащей литиевые, натриевые и калиевые соли Сг-С,2алканола, С4'С,2циклоалканола, аммиака, циклогексиламина и 1-метилпиперазина; гидридные соли лития, натрия и калия; карбонат цезия и оксид бария, растворитель выбирают из группы, содержащей этанол, третичный С в -С 1О спирт, третичный С 6 СдЦиклоалканол, тетрагидрофуран, 1,4диоксан, и ацетонитрил, реакцию проводят при от 60 до 105°С, и количество применяемого основания составляет от 1,1 до 2,0 молекулярных эквивалентов. 8. Способ по п.7, где основание выбрано из группы, содержащей С,-С,2алкоксидные и гидридные соли лития, натрия и калия, амид натрия, циклогексиламид натрия и карбонат цезия, растворитель выбирают из группы, содержащей этанол, трет-бутанол, трет-амиловый спирт, 1-метилциклогексанол, тетрагидрофуран и 1,4-диоксан, реакцию проводят в течение от 3 до 60 ч. , 9. Способ по п.8, где основание выбрано из группы, содержащей этоксид натрия, трет-бутоксид натрия, трет-бутоксид калия и гидрид натрия, и растворитель выбирают из группы, содержащей этанол, трет-бутанол, трет-амиловый спирт и тетрагидрофуран. 10. Способ по п.1, где циклизацию проводят путем нагревания соединения формулы (II), необязательно в присутствии растворителя и/или необязательно в присутствии дегидратирующего агента, и/ или механической водоудаляющей системы. 11. Способ по п. 10, где растворитель выбран из группы, содержащей -1,2-дихлорбензол, диметилсульфоксид, сульфолан, М-метилпирролидон-2-он ипирролидин-2-он и их смеси, и дегидратирующий агент выбран из группы, содержащей безводный карбонат калия, безводный карбонат натрия, безводный сульфат магния, безводный сульфат натрия, пентоксид фосфора и молекулярные сита. 12. Способ по п.11, где растворитель представляет собой 1,2-дихлорбензол, сульфолан или Ы-метилпирролидин-2-он, дегидратирующий агент представляет собой молекулярные сита, и реакцию проводят при от 180 до 220°С в течение от 0,5 до 72 ч. 13. Способ по п.1, где циклизацию проводят в присутствии протонной кислоты или кислоты Льюиса, необязательно в присутствии растворителя. 14. Способ по п.13, где протонная кислота выбрана из группы, содержащей неорганическую кислоту, органосульфоновую кислоту, органофосфоновую кислоту и органическую карбоновую кислоту, кислота Льюиса выбрана из группы, содержащей трифторид бора, трихлорид бора, трибромид бора, хлорид алюминия, бро 1708b мид алюминия, тетрахлорид кремния, тетрабромид кремния, хлорид олова, бромид олова, пентахлорид фосфора, пентабромид фосфора, тетрафторид титана, тетрахлорид титана, тетрабромид титана, хлорид железа (Hi), фторид цинка, хлорид цинка, бромид цинка, йодид цинка, хлорид ртути, бромид ртути и йодид ртути, и растворитель выбран из группы, содержащей С5~С12 алкан, С5~Свциклоалкан, С,С12алкановую кислоту, СрС^алканол, С,С9алканон, С5-С,2алкиловый эфир, 1,2диметоксиэтан, 1,2-диэтоксиэтан, диглим, тетраги дрофу ран, 1,4-диоксан, бензол, толуол, ксилол, хлорбензол, дихлорбензол, нитробензол,дихлорметан, дибромметан, 1,2-дихлорэтан, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, диметилацетамид, ІЧ-метилпирролидин-2-он, пирродин-2-он и их смеси. 15 Способ по п. 14, где протонная кислота представляет собой концентрированную серную кислоту, фосфорную кислоту или п-толуолсульфоновую кислоту, кислота Льюиса представляет собой трифторид бора, хлорид алюминия, тетрахлорид кремния, хлорид олова, тетрахлорид титана, хлорид железа (111) или хлорид цинка, растворитель представляет собой ледяную уксусную кислоту, тетрагидрофуран, 1,4-диоксан или хлорбензол, и реакцию проводят при от 65 до 210°С в течение от б до 300 ч. Изобретение относится к способу получения соединения формулы (Г): 16. Соединение формулы (И) H 2 NOC сн 3 сн 2 о сн 2 сн 2 сн 3 (II) NCH, в качестве промежуточного соединения для получения силденафила. 17. Соединение формулы (V) сн 3 сн 2 о СО 2 Н (V} NCH, или его гидрохлоридная-триэтиламиновая двойная соль, или его С,-Слалкиловый эфир в качестве промежуточного соединения для получения силденафила. фил, который был первоначально описан в ЕР № А-0463756, особенно полезен при лечении, помимо прочего, нарушения эрекции у мужчин' смотри WO № А-94/28902. 5 Более конкретно, изобретение отноСН,СЧ2О сится к способу получения силденафила, который является более эффективным, чем описанный в ЕР № А-0463756, и который (0 неожиданно приводит к получению сил10 денафила клинического стандарта качества, что избавляет от необходимости стадий последующей очистки. В данном конSO,N NCR тексте, термин "силденафил клинического стандарта качества" означает продукт известному как 5-[2-этокси-5-(4-метилпи- 15 чистоты, достаточный для введения люпераэин-1-илсульфонил)фенил]-1-метил-3 дям. Ключевая стадия общего процесса вк-н-пропил-116-дигидро-7Н-пиразоло[4,3-Д]лючает замыкание кольца в непосредстпиримидин-7-он, или силденафил, а таквенном предшественнике силденафила, т.е. же к используемым промежуточным пров бисамидв формулы (II): дуктам, было обнаружено, что силдена 27085 7 сн 3 сн г о 5 CON H CHXHXK (ID 10 Таким образом, изобретение относится к способу получения соединения формулы (I), который заключается в циклизации соединения формулы (II). В предпочтительном осуществлении циклизацию проводят в присутствии основания, предпочтительно в растворителе, необязательно, в присутствии перекиси водорода или соли пероксида, с последующей нейтрализацией реакционной смеси, где это необходимо. Подходящее основание может быть выбрано из группы, включающей соль металла и С,-С]2алканола, С3~С)2циклоалканола, (С--С циклоалкил^-С^алканола, аммиака, С,-С,2алкиламина, д и ^ - С ^ а л кил)амина, С3-С6циклоалкиламина, N-(C3С в циклоалкил)-И-(С 1 -С 1 2 алкил}амина, ди(С3-Свциклоалкил)амина, {С3-Сациклоалкил)С,-С5алкиламина, Ы-(С3-С8циклоалкил)С,-С6алкил-Ы-(С1-С12алкил)амина1 Nлоалкил)-амина, ди[(С3-Свциклоалкил)С,С6алкил]амина и гетероциклического амина, выбранного из группы, содержащей аимидазол, триазол, пирролидин, пиперидин, гептаметиленимин, морфолин, тиоморфолин и 1-(С,-Сдалкил)пиперазин; гидрид, фторид, гидроксил, оксид, карбонат и бикарбонат металла; где металл выбран из группы, включающей литий, натрий, калий, рубидий, цезий, бериллий, магний, кальций, стронций, барий, алюминий, индий, таллий, титан, цирконий, кобальт, медь, серебро, цинк, кадмий, ртуть и церий, и С7-С12бициклический амидин. Предпочтительно, основание выбрано из группы, содержащей соль щелочного или щелочноземельного металла и С,С^алканола, С3-С,гциклоалканола и (С 3 Свциклоалкил)С,~С6алканола; соль щелочного металла и аммиака, Ы-(вторичный или третичный С3-С6алкил)-1М-(первичный, вторичный или третичный С3-С6алкил)-амина, С3~С?циклоалкиламина, М-(С3-С8циклоалкил)-Ы-(первичный, вторичный или тре 15 20 25 30 35 40 45 50 55 8 тичный С3-Сеалкил)амина, ди(С 3 -С 8 циклоалкил)амина и 1-метилпиперазина; и гидрид, гидроксид, оксид, карбонат и бикарбонат щелочного или щелочноземельного металла; 1,5-диазабицикло[4,3,0]нон5-ен и 1,8-диазабицикло[5,4,0]ундец-7-ен. Подходящий растворитель может быть выбран из группы, содержащей С,-С,2алканол, С3-С]2циклоалканол, (С3-С8циклоалкил)С,-С 6 алканол, С 3 -С б д алканон, С 4 С^циклоалканон, С5~С,2алкиловый эфир, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, бензол, толуол, ксилол, хлорбензол, дихлорбензол, ацетонитрил, ди метил сульфоксид, сульфолан, диметилформамид, диметилацетамид, Ы-мегилпирролидин-2-он, пирролидин-2-он, пиридин и вода и их смеси. Предпочтительно, растворитель выбран из группы, содержащей этанол, 2пропанол, вторичный или третичный С 4 С12алканол, С3-С12циклоалканол, третичный С4-С12циклоалканол, вторичный или третичный (С3-С7циклоалкил)С2-С6алканол, С 3 -С д алканон, 1,2-диметоксиэтан, 1,2диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, толуол, ксилол, хлорбензол, 1,2-дихлорбензол, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, п-метилпирролидин-2-он, пиридин и вода и их смеси. Далее, предпочтительными признаками является то, что количество применяемого основания составляет от 1,0 до 5,0 молекулярных эквивалентов и что реакцию проводят при от 50 до 170°С в течение от 3 до 170 ч. В более предпочтительном способе основание выбирают из группы, содержащей литиевые, натриевые и калиевые соли С,~С12алканола, С^-С^циклоалканола, аммиака, циклогексиламина и 1-метилпиперазтина, гидридные соли лития, натрия и калия; карбонат цезия, и оксид бария; растворитель выбирают из группы, содержащей этанол, третичный С 4 -С 10 спирт, третичный С6~С8циклоалканол, тетрагидрофуран, 1,4-диоксан и ацетонитрил, реакцию проводят при от 60 до 105°С, и количество применяемого основания составляет от 1,1 до 2,0 молекулярных эквивалентов. Даже более предпочтительным является способ, где основание выбран из группы, содержащей С,-С,2алкоксидные и гидридные соли лития, натрия и калия, амид натрия, циклогексиламид натрия и карбонат цезия; растворитель выбран из группы, включающей этанол, трет-бутанол, трет-амиловый спирт, 1-метилциклогекса 27085 10 ми. Например, когда (1) получают в виде нол, тетрагидрофуран и 1,4-диоксан; и соли путем нейтрализации необязательно реакцию проводят в течение от 3 до 60 ч. предварительно разбавленной реакционОсобенно предпочтительным является ной смеси с последующем выделением тот, где. основание выбран из группы, вкпродукта путем фильтрования/экстракции лючающей этоксид натрия, трет-бутоксид и его необязательной кристаллизации. натрия, трет-бутоксид калия и гидрид натАльтернативно, соединение формулы рия; и растворитель выбран из группы, (I) может быть легко выделено и/или очивключающей этанол, трет-бутанол, третщено обычными хроматографическими меаминовый спирт и тетрагидрофуран. В приведенных выше определениях ал- 10 тодами. Соединение формулы (II), необходикильная цепь или циклоалкильное кольцо мое для получения соединения формулы могут быть разветвленными и нераэветв(I), может быть получено путем, показанленными, если это не оговаривается иначе. ным на следующей схеме реакции с приСоединение формулы (I) может быть выделено и очищено обычными метода- 15 менением обычных методов. • Схема СН,СН,0 сн э сн,о со,н SO,C1 (III) си,сн2о сн,сн2о (V) со г н SO, N NCH, NCH, 99% по данным анализов ВЭЖХ и ТСХ получали, применяя процедуру, сходную с описанной в примере 1, когда в качестве растворителя использовали 1,4-диоксан и реакцию проводили * при 100°С в течение 4 ч. П р и м е р 14. Целевое соединение (85%) чистоты > 99% по данным анализов ВЭЖХ и ТСХ получали, применяя процедуру, сходную с описанной в примере 1, когда в качестве растворителя использовали 1,2-диметоксиэтан и реакцию проводили в течение 30 ч. П р и м е р 15. Целевое соединение (83%) чистоты > 99% по данным анализов ВЭЖХ и ТСХ получали, применяя процедуру, сходную с описанной в примере 1, когда в качестве растворителя использовали 3,7-диметилоктан-З-ол и реакцию проводили при 100°С в течение 16 ч. П р и м е р 16. Целевое соединение (74%) чистоты > 99% по данным анализов ВЭЖХ и ТСХ получали, применяя процедуру, сходную с описанной в примере 1, когда в качестве основания использовали н-декоксид натрия, в качестве растворителя использовали 1,4-диоксан и реакцию проводили при 100°С в течение 20 ч. П р и м е р 17. Целевое соединение (85%) чистоты > 99% по данным анализов ВЭЖХ и ТСХ получали, применяя процедуру, сходную с описанной в примере 1, когда в качестве основания использовали амид натрия, в качестве растворителя использовали 1,4-диоксан и реакцию проводили при 100°С в течение 18 ч. П р и м е р 18. Целевое соединение (91%) чистоты > 99% по данным анализов ВЭЖХ и ТСХ получали, применяя процедуру, сходную с описанной в примере 1, когда в качестве основания ислользовали циклогексиламид натрия, в качестве растворителя использовали 1,4-диоксан и реакцию проводили при 100°С в течение 6,5 ч. П р и м е р 19. Целевое соединение (84%) чистоты > 99% по данным анализов ВЭЖХ и ТСХ получали, применяя процедуру, сходную с описанной в примере 1, когда в качестве основания использовали 4-метилпиперазид натрия, в качестве растворителя использовали 1,4-диоксан и реакцию проводили при 100°С в течение 8 ч. П р и м е р ы 20-21. В реакционных условиях, сходных с описанными в примере 1, использование метоксида натрия ' 17 27085 в метаноле в течение 32 ч давало четырехкомпонентную смесь, из которой целевое соединение выделяли путем хроматографии с выходом 34,5%, тогда как использование трет-бутоксида калия в метаноле в течение 40 ч позволяло получить смесь продуктов, которая, по данным спектроскопических анализов ТСХ и ЯМР, имеет установленный выход 69% целевого соединения. П р и м е р 22. В реакционных условиях, сходных с описанными в примере 1, применение трет-бупжсида калия в безводном ди метил сульфоксиде при 100°С в течение 50 ч позволяло получить сырой продукт (выход 88 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имеет установленный выход 24% целевого соединения. П р и м е р 23. В реакционных условиях, сходных с описанными в примере 1, использование этоксида магния в пиридине при температуре возгонки в течение 96 ч давало сырой продукт (выход 79 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 16% целевого соединения. П р и м е р 24. В реакционных условиях, сходных с описанными в примере 1, использование этоксида бария (в виде 10% м/о раствора в этаноле) в трет-амиловом спирте при 100°С в течение 20 ч позволяло получить сырой продукт (выход 76,5 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 75,5% целевого соединения. П р и м е р 25, В реакционных условиях, сходных с описанными в примере 1, использование зтоксида титана в пиридине при 100°С в течение 90 ч давало сырой продукт (выход 82 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 32% целевого соединения. П р и м е р 26. В реакционных условиях, сходных с описанными в примере 1, использование этоксида меди в пиридине при 100°С в течение 98% ч позволяло получить сырой продукт (выход 89,5 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 18,5 целевого соединения. П р и м е р 27. В реакционных условиях, сходных с описанными в примере 1, использование три-трет-бутоксида алюминия в пиридине при 100°С в течение 72 ч позволяло получить сырой продукт, который, по данным анализов ТСХ и ВЭЖХ, имел максимальный (благодаря загрязне 5 10 15 20 25 30 35 40 45 50 55 18 нию солью алюминия) установленный в\ | ход 66% целевого соединения. П р и м е р 28. В реакционных условиях, сходных с описанными в примере 1, использование диизопропиламида лития (в виде 1,5М раствора моно(тетрагидрофуранового) комплекса в циклогексане) общим количеством 3,6 мол.экв. (1,2 мол.экв., добавленных в три стадии) в безводном 1,4-диоксане сначала при 0°С в течение 15 мин, затем при комнатной температуре в течение 1 ч и в последующем при 100°С в течение суммарно 140 ч давало сырой продукт (выход 60,5 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход в 55,5% целевого соединения. П р и м е р 29. В реакционных условиях, сходных с описанными в примере 1, применение 2,0 мол.экв. 1,8-диазабицикло[5,4,0]ундец-7-ена в пиридине при 100°С в течение 44 ч позволяло получить сырой продукт (выход 6,5 мас.%), который по данным анализов ТСХ и ВЭЖХ имел установленный выход в 3,3% целевого соединения. П р и м е р 30. В реакционных условиях, сходных с описанными в примере 1, применение фторида калия в трет-амиловом спирте при 100°С в течение 44 ч позволяло получить сырой продукт (выход 85 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход в 3,5% целевого соединения. П р и м е р 31. К перемешиваемой суспензии целевого соединения из получения 4 (9,85 г, 0,02 моль) в этаноле (30 мл) добавляли гранулы 85% гидроксида калия с последующим добавлением воды (30 мл), что давало прозрачный раствор. Реакционную смесь нагревали с обратным холодильником в течение 5 ч и затем этанольную фазу удаляли путем выпаривания при пониженном давлении. Полученную смесь разбавляли водой (60 мл), рН доводили до 7, используя разбавленную серную кислоту и осажденный продукт гранулирования в течение 30 мин. Твердое вещество собирали путем фильтрования, промывали водой и сушили а вакууме, что давало продукт (7,96 г), 96,4%, который, по данным анализа ВЭЖХ, представляет собой целевое соединение. П р и м е р ы 32-34. В реакционных условиях, сходных с описанными в примере 1, использование оксида бария в течение 52 часов давало целевое соединение (89%) с чистотой > 99% по данным анализоз ВЭЖХ и ТСХ. 19 27085 Повтор с использованием диметилформамида в качестве растворителя при 100°С в течение 31 ч давало сырой продукт (выход 75,5 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 54% целевого соединения. Дальнейший повтор с использованием пиридина в качестве растворителя при 100°С в течение 16 ч давало сырой продукт, который, по данным анализов ТСХ и ВЭЖХ, имел максимальный (ввиду загрязнения солью бария) установленный выход 90% целевого соединения. П р и м е р 35. В реакционных условиях, сходных с описанными в примере 1, использование карбоната цезия в 4метилпентан-2-оне (метилизобутиловом кетоне) при 100°С в течение 96 ч позволяло получить сырой продукт (массовый выход 18,5%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 13% целевого соединения. П р и м е р 36. В реакционных условиях, сходных с описанными в примере 1, использование бикарбоната калия в третамиловом спирте при 100°С в течение 115 ч давало сырой продукт (выход 82,5 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 20% целевого соединения. П р и м е р 37. Целевое соединение из получения 4 (12,32 г, 0,025 моль) нагревали при 215-220°С в течение 40 минут и полученному расплаву давали остыть до комнатной температуры. Смолообразный сырой продукт растворяли в дихлорметанё (25 мл) и затем очищали путем хроматографии на силикагеле, применяя в качестве элюента смеси метанола в дихлорметане с возрастающей полярностью. Выпаривание в вакууме подходящих однокомпонентных фракций давало чистое (по данным анализа 'Н ЯМР) целевое соединение (1,76 г, 14,8%), тогда как порцию менее чистого целевого соединения (0,87 г, 7,3%) получали из следующих фракций. Дальнейшая хроматографическая обработка последнего давала дополнительное количество (0,48 г) чистого целевого соединения, причем общий выход составил 2,24 г, 18,8%. 5 10 15 „ 20 25 30 35 40 45 50 П р и м е р ы 38-40. Перемешиваемую смесь целевого соединения из получения 4 (12,32 г, 0,025 моль) и 1,2-дихлорбензола (61 мл) нагревали с обрат- 55 ным холодильником в течение 72 ч. Полученной темно-коричневой реакционной смеси давали остыть, разбавляли дихлор- . метаном (60 мл) и фильтровали. Выпаривание фильтрата при пониженном давле 20 нии давало, содержащее растворитель, темно-коричневое масло (17,51 г), 28,2% не содержащего растворитель продукта, который, по данным анализов ТСХ и ВЭЖХ, представлял собой целевое соединение. Повтор с применением сульфолана в качестве растворителя при около 205°С в течение 5 ч давало сырой продукт (выход 14 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 12% целевого соединения. Дальнейший повтор с применением Nметилпирролидин-2-она в качестве растворителя при 205-210°С в течение 3 ч давало сырой продукт (выход 21,5 мас.%), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 6,5% целевого соединения П р и м е р 41. В реакционных условиях, сходных с описанными в примере 38, за исключением того, что реакцию пдоводили в течение 24 ч в присутствии 4А молекулярных сит, получали содержащий растворитель продукт, 6,0% не содержащего растворитель материала, который, по данным анализов ВЭЖХ, представляет собой целевое соединение. П р и м е р 42. В перемешиваемой суспензии целевого соединения из получения 4 (12,32 г, 0,025 моль) в хлорбензоле (61 мл) добавляли концентрированную серную кислоту (1,0 мл, 1,84 г, 18,75 моль) и затем полученную смесь нагревали до тех пор, пока растворитель не начинал отгоняться. Когда дистиллят переставал .быть мутным (после отбора около 20 мл), реакционной смеси давали остыть до комнатной температуры и добавляли дополнительное количество (20 мл) хлорбензола, затем нагревали с обратным холодильником в течение 20 "часов. Охлажденную реакционную смесь обрабатывали дичлорметаном {100 мл) с получением раствора с последующим добавлением воды (ЮС мл). рН полученной смеси доводили до 7, используя 5М водный раствор гидроксида натрия, затем органическую фазу отделяли, объединяли с дихлорметановым экстрактом (50 мл) водной фазы и выпаривали при пониженном давлении с получением твердого вещества (9,51 г), 5,5% который, по данным анализов ВЭЖХ, представлял собой целевое соединение. П р и м е р 43. К перемешиваемой суспензии целевого соединения из получения 4 (6,16 г, 12,5 моль) в ледяной уксусной кислоте (31 мл) добавляли концентрированную серную кислоту (1,0 мл, 1,84 г, 18,75 моль) и затем полученную 21 27085 смесь нагревали при 100°С в течение 115 ч). Растворитель удаляли путем выпаривания при пониженном давлении, остаток "азеотропировали" вместе с толуолом (2 х 50 мл) и полученное масло (10,5 г) встряхивали с водой (60 мл) с получением кристаллического твердого вещества, которое собирали, промывали водой (10 мл) и сушили. Данный сбор (2,03 г) объединяли со вторым сбором (3,48 г), полученным путем нейтрализации фильтрата 20% водным раствором гидроксида натрия с последующим сбором, промыванием и сушкой, как ранее, с получением сырого продукта (5,51 г), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход в 38% целевого соединения. П р и м е р 44. Перемешиваемую смесь целевого соединения из получения 4 (6,16 г, 12,5 ммоль) и ледяной уксусной кислоты (31 мл) нагревали при 100°С в течение 7 часов и полученному раствору давали остыть. Анализ ТСХ реакционной смеси показал, что на данной стадии целевое соединение отсутствовало. Добавляли 85% водный раствор уксусной кислоты (0,5 мл) и полученную смесь нагревали при 100°С, с перерывами,^ в сумме в течение 300 часов, затем выпаривали при пониженном давлении. Остаток "азеотропировали" вместе с толуолом и растворяли в воде (50 мл), затем рН перемешиваемого водного раствора доводили до 7 с помощью 20% водного раствора гидроксида натрия. Перемешивание продолжали в течение 2 ч, затем осадок собирали, промывали водой (20 мл) и сушили в вакууме при 50°С с получением сырого прод\та (5,21 г), который, по данным анализов ТСХ и ВЭЖХ, имел установленный выход 9,1% целевого соединения. П р и м е р 45. Перемешиваемую смесь моногидрата п-толуолсульфоновой кислоты (5,71 г, 0,030 моль) и хлорбензола (100 мл) нагревали с обратным холодильником до тех пор, пока не удалена вода, используя сепаратор Dean-Stark, и затем давали остыть до комнатной температуры. Добавляли целевое соединение из получения 4 {24,64 г, 0,050 моль) и реакционную смесь перемешивали с обратным холодильником в течение 24 ч, затем давали остыть. К полученной смеси добавляли дихлорметан (200 мл) и воду (200 мл), рН доводили до 7, используя 2М водный раствор гидроксида натрия и органическую фазу отделяли и объединяли с дихлоргиетановым экстрактом (100 мл) 5 10 15 20 25 30 35 40 45 50 55 22 водной фазы. Объединенные органические фазы промывали водой (100 мл) и выпаривали при пониженном давлении с получением не совсем белого твердого вещества (24,86 г), 7,3% которого, по данным анализов ТСХ и ВЭЖХ, представляло собой целевое соединение. П р и м е р 46. К перемешиваемой суспензии целевого соединения из получения 4 (12,32 г, 0,025 моль) в безводном 1,4-диоксане (61 мл) добавляли тетрахлорид титана (3,3 мл, 5,69 г, 0,030 моль), в процессе чего отмечалось интенсивное газообразование. Перемешиваемую реакционную смесь нагревали при около 70°С в течение 7,5 ч, давали остыть до комнатной температуры и затем обрабатывали водой (200 мл) и концентрированной соляной кислотой (50 мл) с получением прозрачного раствора. Раствор промывали дихлорметаном и рН доводили до 12, используя 40%-ный водный раствор гидроксида натрия; его затем перемешивали в течение 10 минут и его рН далее доводили до 7, используя 5М соляную кислоту. Осадок удаляли путем фильтрования и промывали дихлорметаном (2 х 200 мл), затем объединенные дихлорметановые смывы использовали для экстракции водного фильтрата и выпаривали при пониженном давлении с получением твердого вещества (11,36 г), 33,7% которого, по данным анализов ТСХ и ВЭЖХ, представляет собой целевое соединение. П р и м е р ы 47-52. В реакционных условиях, сходных с описанными в примере 46, изменение которых представлены в табл.3, альтернативные кислоты Льюиса давали регулируемые показанные выходы целевого соединения. Получение 1 5-Хлорсульфонил-2-этоксибензойная кислота. К перемешиваемой охлаждаемой на льду смеси тионилхлорида (11 мл, 0,151 моль) и хлорсульфоной кислоты (41,3 мл, 0,621 моль) добавляли расплавленную 2этоксибензойную кислоту (25,0 г, 0,150 моль) при этом температуру реакционной смеси поддерживали ниже 25"С. Полученную смеси перемешивали при комнатной температуре в течение 18 ч и затем выливали в перемешиваемую смесь льда (270 г) и воды (60 мл) с получением но совсем белого осадка Перемешивание продолжали в течение 1 часа, затем продукт собирали путем фильтрования, промывали водой и сушили в вакууме с получением целевого соединения (36,08 г). Образец сравнения, т.пл. 115-116GC, по 2ГОУ5 23 луч ал и путем кристаллизации из гексана1 толуола. Найдено, %: С 41,02; Н 3,27. C9H9CIO5S Вычислено, %: С 40,84; Н 3,43. 5 (CDCI3): 1,64 (ЗН, т), 4,45 (2Н, к), 7,26 (1Н, д), 8,20 (1Н, дд), 8,80 (1Н, д). Получение 2. 2-Этокси-5-(4-метилпиперазин-1-илсульфонил)бензойная кислота. а): одностадийная процедура К перемешиваемой суспензии целевого соединения из получения 1 (34,4 г, 0,130 моль) в воде (124 мл) при около 10°С добавляли 1-метилпиперазин (33,6 мл, 0,303 моль), в то время как температуру реакционной смеси поддерживали ниже 20°С. Полученный раствор охлаждали приблизительно до 10°С и через 5 мин приступали к кристаллизации твердого вещества. Еще через 2 ч твердое вещество собирали путем фильтрования, промывали ледяной водой и сушили в вакууме с получением сырого продукта (36,7 г). Образец (15,0 г) очищали путем перемешивания его в возгоняющемся ацетоне в течение 1 ч; полученной суспензии давали остыть до комнатной температуры и кристаллическое твердое вещество собирали путем фильтрования и сушили в вакууме с получением целевого соединения (11,7 г), т.пл. 198-199°С, спектр 1Н ЯМР которого идентичен таковому, полученному для продукта из процедуры (б) ниже. б)- двухстадийная процедураа К перемешиваемо суспензии 1-метилпиперазина (20,81 мл, 0,208 моль и триэтиламина (28,9 мл, 0,207 моль) добавляли по каплям раствор целевого соединения из получения 1 (50,0 г, 0,189 моль) в ацетоне (150 мл), в-то время как температуру реакционной смеси поддерживали ниже 20°С. В процессе добавления образовывалось белое кристаллическое твердое вещество, и перемешивание продолжали в течение еще 1,5 часов. Фильтрование с последующим промыванием ацетоном и сушкой продукта в вакууме давало двойную соль гидрохлорида-триэтиламина целевого соединения (78,97 г), т.пл. 166-169°С. 5 10 15 20 25 30 35 40 45 50 Найдено.%: С 51,33; Н 8,14; N 9,06; СІ 8,02. CHINAS; С Д Д неї • Вычислено, %: С 51,55; Н 7,79; N 55 9,02; СІ 7,61. 6 (CD.SOCD,): 1,17 (9Н, т), 1,32 (ЗН, >), 2.15 (ЗН, с), 2,47 (6Н, ушир, с), 2,86 (2Н, ушир, с), 3,02 (6Н, к), 4,18
ДивитисяДодаткова інформація
Назва патенту англійськоюThe method for sildenafil obtaining and intermediate compounds
Автори англійськоюDan Piter James, Wood Albert Sho
Назва патенту російськоюСпособ получения силденафила и промежуточные соединения
Автори російськоюДан Питер Джеймс, Вуд Альберт Шо
МПК / Мітки
МПК: C07D 295/26, C07B 61/00, C07D 231/40, C07D 487/04, C07D 231/14, A61K 31/519, A61P 13/02, C07D 295/22, A61K 31/505, A61P 15/00
Мітки: спосіб, сполуки, силденафілу, отримання, проміжні
Код посилання
<a href="https://ua.patents.su/14-27085-sposib-otrimannya-sildenafilu-ta-promizhni-spoluki.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання силденафілу та проміжні сполуки</a>
О-метильні похідні азитроміцину а, що мають антибактеріальну активність, спосіб їх отримання та проміжні сполуки

Номер патенту: 27105
Опубліковано: 28.02.2000
Автори: Лазаревскі Горяна, ДЬОКІС Слободан, Кобрехел Габріела
МПК: A61K 31/7042, A61K 31/70, C07H 17/00, A61P 31/04, C07H 17/08
Мітки: отримання, проміжні, мають, похідні, спосіб, о-метильні, активність, антибактеріальну, сполуки, азитроміцину
Формула / Реферат:
1. О-Метильные производные азитромицина А общей формулы (I)обладающие антибактериальной активностью.2. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=CH3, R4=R5=H.3. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=R4=CH3, R5=H.4. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=R5=H, R4=CH3.5. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5,...
Сірковмісні похідні імідазолу, що мають гіпотензивну активність, спосіб їх отримання, проміжні сполуки і фармацевтична композиція

Номер патенту: 26130
Опубліковано: 07.06.1999
Автори: Фортен Мішель, Вевер Жан-Поль, Кай Жан-Клод, Жукей Сімон, Корб'є Ален, Амон Жіль
МПК: A61P 15/00, A61P 13/02, C07D 401/12, A61K 31/4164, C07D 233/94, A61P 9/00, C07D 233/84, A61K 31/415, A61P 9/10, A61P 21/00, C07D 233/90, A61K 31/4178, C07D 403/12, A61P 43/00, C07D 403/10, A61P 9/12
Мітки: спосіб, похідні, гіпотензивну, проміжні, сірковмісні, сполуки, активність, мають, композиція, імідазолу, отримання, фармацевтична
Формула / Реферат:
1. Серосодержащие производные имидазола общей формулы (I)где R1 - алкил;R2 - водород, гидроксиметил, карбоксигруппа, группа формулы -S(=O)2-A, где A - низший алкил или фенил,- группа формулы -S(=O)2(CH2)k-B, где k = 1 и 2, B - низшая алкокси- или гидроксигруппа,- группа -S-D, где D - незамещенный фенил или замещенный галогеном, низшей алкоксигруппой низший алкил, возможно замещенный галогеном, и...
Похідні азолу, що мають фунгіцидну або регулюючу ріст рослин активність, і проміжні сполуки для їх отримання

Номер патенту: 27101
Опубліковано: 28.02.2000
Автори: ІКЕДА Сусуму, Кумазава Сатору, САЙСОДЗІ Тосіхіде, СІМІЗУ Сусуму, ІТО Ацусі, САТО Набуо, ЄНАРІ Хіроюкі
МПК: C07C 1/00, C07C 25/00, C07C 17/00, C07D 521/00, C07C 49/657, A01N 43/50, C07C 13/00, C07C 49/697, C07D 233/60, A01N 43/653, C07C 45/00, C07D 249/08, C07C 69/716, C07C 67/00, C07C 17/26, C07D 303/00, C07C 45/67
Мітки: активність, сполуки, фунгіцидну, ріст, мають, регулюючу, отримання, рослин, азолу, похідні, проміжні
Формула / Реферат:
1. Производные азола общей формулы (I)где R1 и R2 - водород или С1-С5-алкил, при условии, что оба одновременно не могут быть водородом;X - галоген, С1-С5-алкил или фенил;n=0 - 2;А - азот или группа -СН-, обладающие фунгицидной или регулирующей рост растений активностью.2. Производные азола общей формулы (I) по п.1 отличающиеся тем, что R1 и R2 - водород или С1-С3-апкил, при условии, что оба...
Спосіб одержання гетероциклічних сполук та проміжні сполуки для їх одержання

Номер патенту: 26678
Опубліковано: 12.11.1999
Автори: Хайкала Хеймо Олаві, ПІППУРІ Айно Кюллікі, Пюстюнен Ярмо Йохан, Луіро Анне Марія, ЛЕННБЕРГ Карі Калєві, Норе Пентті Тапіо, Хонканен Ерккі Юхані
МПК: C07D 273/00, C07D 237/04, C07D 401/12, C07D 413/12, A61P 9/12, A61K 31/50, A61K 31/502, A61P 9/04, A61P 9/08, C07D 285/16, C07D 403/12, C07D 417/12, A61K 31/53, A61K 31/535, A61K 31/54, C07D 237/32, C07D 237/14, C07D 253/00
Мітки: спосіб, сполук, гетероциклічних, одержання, сполуки, проміжні
Формула / Реферат:
1. Способ получения гетероциклических соединений формулы (I)в которой Het представляет одну из следующих групп:где R11, R13 и R14 представляют независимо водород или низшую алкильную группу;Z представляет собой S, О или NH;A представляет валентную связь, -CH-CH- или CH2-CH2-группу;R1 и R2 независимо представляют нитро, циано, галогеновую, амино, карбоксамидо, фенильную, бензоильную,...
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Хермец Іштван, Балог Марія, Шіпош Юдіт, Пайор Аніко, Керестурі Геза, Вашварі Лелле, Хорват Агнеш, Рітлі Петер
МПК: C07D 401/04, C07F 5/00, C07D 215/56, A61K 31/495, A61P 31/04
Мітки: проміжні, кислоти, сполуки, солей, фармацевтично, одержання, хінолінкарбонової, похідних, спосіб, придатних
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...