Спосіб одержання дезоксирибофуранозних сполук






Формула / Реферат
1. Спосіб одержання сполуки (2)
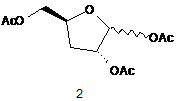 ,
,
при якому здійснюють:
(і) реакцію сполуки (4) з алкілкетенацеталем і каталітичною кислотою
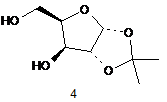 ,
,
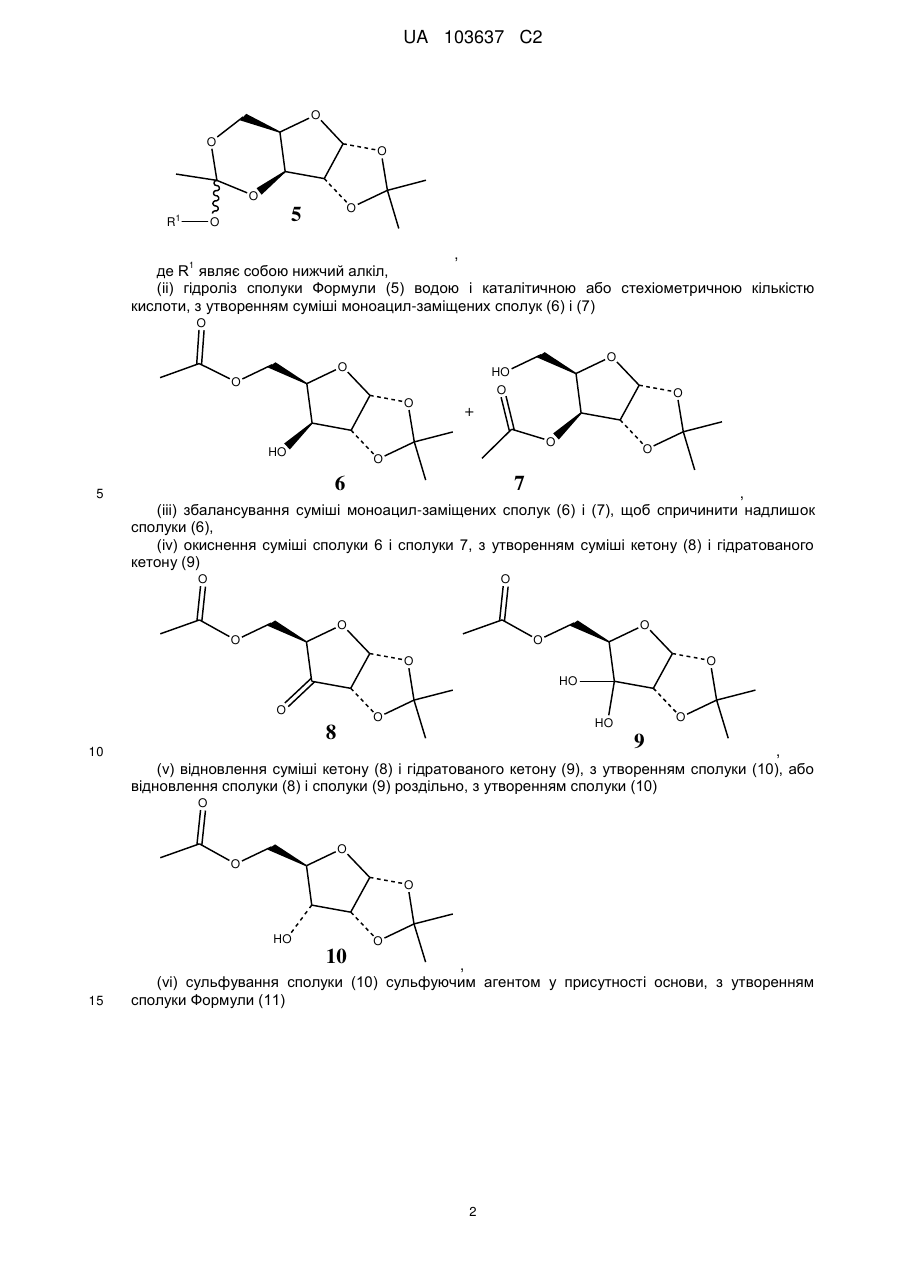
з утворенням циклічної сполуки Формули (5)
 ,
,
де R1 являє собою нижчий алкіл,
(іі) гідроліз сполуки Формули (5) водою і каталітичною або стехіометричною кількістю кислоти, з утворенням суміші моноацилзаміщених сполук (6) і (7)
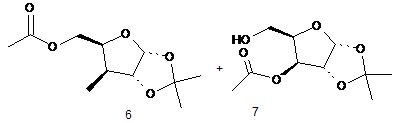 ,
,
(ііі) збалансування суміші моноацилзаміщених сполук (6) і (7), щоб спричинити утворення надлишку сполуки (6),
(iv) окиснення суміші сполуки 6 і сполуки 7, з утворенням суміші кетону (8) і гідратованого кетону (9)
 ,
,
(v) відновлення суміші кетону (8) і гідратованого кетону (9), з утворенням сполуки (10), або відновлення сполуки (8) і сполуки (9) роздільно, з утворенням сполуки (10)
 ,
,
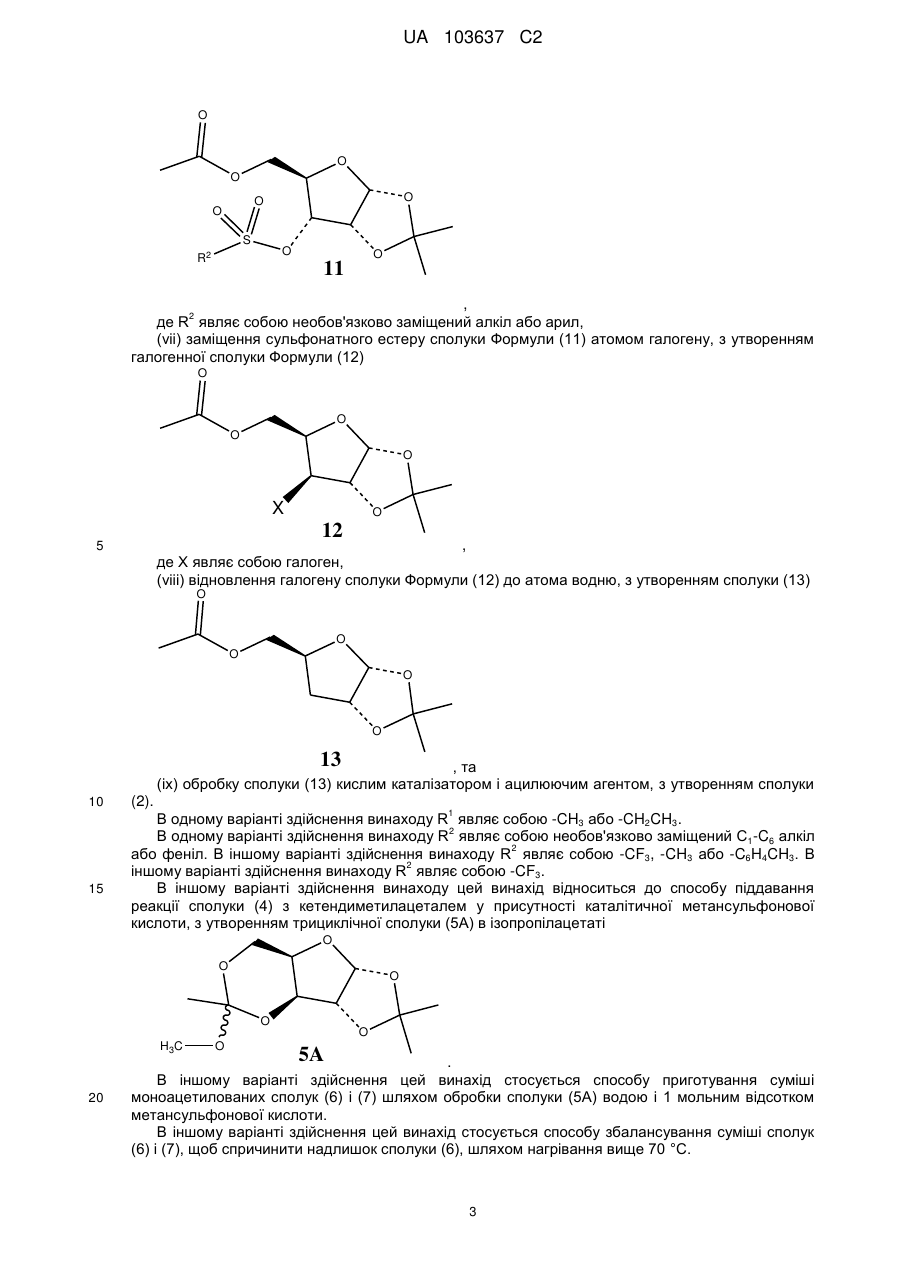
(vi) сульфування сполуки (10) сульфуючим агентом у присутності основи, з утворенням сполуки Формули (11)
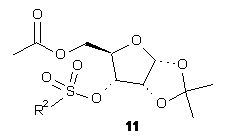 ,
,
де R2 являє собою необов'язково заміщений алкіл або арил,
(vii) заміщення сульфонатного естеру сполуки Формули (11) атомом галогену, з утворенням галогенної сполуки Формули (12)
 ,
,
де X являє собою галоген,
(viii) відновлення галогену сполуки Формули (12) до атома водню, з утворенням сполуки (13)
 , та
, та
(іх) обробку сполуки (13) кислим каталізатором і ацилюючим агентом, з утворенням сполуки (2).
2. Спосіб за п. 1, де R1 являє собою -СН3 або -СН2СН3.
3. Спосіб за п. 1, де R2 являє собою -CF3, -CH3 або -С6Н4СН3.
4. Спосіб за п. 1, де на стадії (і) сполуку (4) піддають реакції з кетендиметилацеталем у присутності каталітичної метансульфонової кислоти, з утворенням трициклічної сполуки (5А), в ізопропілацетаті.
5. Спосіб за п. 1, де на стадії (ііі) здійснюють збалансування суміші сполук (6) і (7), щоб спричинити надлишок сполуки (6), шляхом нагрівання вище 70 °C.
6. Спосіб за п. 5, де збалансування суміші сполук (6) і (7) спричиняє утворення надлишку більше ніж у 90 % сполуки (6) над сполукою (7).
7. Спосіб за п. 1, де на стадії (iv) суміш сполук (6) і (7) окиснюють за допомогою гіпохлориту натрію у присутності TEMPO і ацетату натрію у дві фази з ізопропілацетатом.
8. Спосіб за п. 1, де на стадії (v) сполуку (10) утворюють як єдиний ізомер, використовуючи триацетоксиборогідрид натрію.
9. Спосіб за п. 1, де сполуку (8) виділяють із суміші сполук (8) і (9).
10. Спосіб за п. 9, в якому здійснюють відновлення сполуки (8), з утворенням сполуки (10) як єдиного ізомеру.
11. Спосіб за п. 10, де відновлення виконують, використовуючи платину на вуглеці як каталізатор в присутності водню.
12. Спосіб за п. 1, де на стадії (vi) основа являє собою DMAP.
13. Спосіб за п. 1, де на стадії (viii) при відновленні сполуки Формули (12) використовують гідроксид паладію на вуглеці (каталізатор Перлмана) в присутності водню.
14. Спосіб одержання сполуки Формули (11), при якому здійснюють:
(і) окиснення сполуки (6) з утворенням сполуки (8)
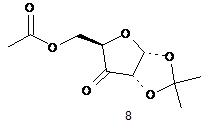 ,
,
(ii) відновлення сполуки (8), з утворенням сполуки (10)
, та
(ііі) сульфування сполуки (10) сульфуючим агентом у присутності основи, з утворенням сполуки Формули (11)
,
де R2 являє собою необов'язково заміщений алкіл або арил.
15. Спосіб за п. 14, де R2 являє собою -CF3, -СН3 або -С6H4СН3.
16. Спосіб за п. 14, де сполуку (6) окиснюють за допомогою гіпохлориту натрію у присутності TEMPO і ацетату натрію у дві фази з ізопропілацетатом.
17. Спосіб за п. 14, де відновлення сполуки (8) утворює єдиний ізомер сполуки (10).
18. Спосіб за п. 15, де відновлення виконують, використовуючи платину на вуглеці як каталізатор у присутності водню.
19. Спосіб за п. 14, де сульфуючий агент являє собою трифторометансульфоновий ангідрид, а основа являє собою DMAP.
20. Спосіб за п. 14, в якому далі здійснюють заміщення сульфонатного естеру сполуки Формули (11) атомом галогену, з утворенням галогенної сполуки Формули (12)
,
де Х являє собою галоген,
відновлення галогену сполуки Формули (12) до атома водню, з утворенням сполуки (13)
, та
обробку сполуки (13) кислим каталізатором і ацилюючим агентом, з утворенням сполуки (2)
.
21. Спосіб за п. 20, де при відновленні сполуки Формули (12) використовують гідроксид паладію на вуглеці (каталізатор Перлмана) в присутності водню.
22. Спосіб за п. 20, де кислий каталізатор являє собою сірчану кислоту, а ацилюючий агент являє собою оцтовий ангідрид.
23. Сполука, вибрана з групи, що складається з:
 ,
, 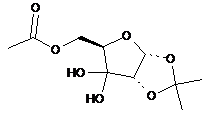 ,
,
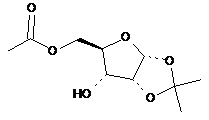 та
та 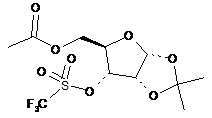 .
.
Текст
Реферат: Винахід стосується способів одержання дезоксирибофуранозних сполук, таких як сполука (2) UA 103637 C2 (12) UA 103637 C2 AcO O OAc OAc 2 , які є корисними проміжними сполуками в одержанні фармацевтичних сполук, таких як 5-аміно3-(2'-О-ацетил-3'-дезокси--D-рибофуранозил)-3Н-тіазоло[4,5-d]піримідин-2-он і тому подібні. UA 103637 C2 5 10 15 20 25 30 35 ГАЛУЗЬ ТЕХНІКИ Цей винахід відноситься до способів одержання дезоксирибофуранозних сполук, які є корисними проміжними сполуками для одержання фармацевтичних сполук, таких як 5-аміно-3(2’-O-ацетил-3’-дезокси-β-D-рибофуранозил)-3H-тіазоло[4,5-d]піримідин-2-он і тому подібних. ПОПЕРЕДНІЙ РІВЕНЬ ТЕХНІКИ Нуклеозидні аналоги - це важливий клас сполук, корисних при лікуванні захворювань. Наприклад, нуклеозидні аналоги використовувалися при лікуванні різних видів раку і вірусних інфекцій. Після входження в клітину нуклеозидні аналоги часто фосфорилюються шляхами нуклеозидного порятунку, в яких ці аналоги фосфорилюються до відповідних моно-, ди- і трифосфатів. Серед інших внутрішньоклітинних призначень трифосфориловані нуклеозидні аналоги часто служать субстратами ДНК- або РНК-полімераз і захоплюються у склад ДНК і/або РНК. Коли трифосфориловані нуклеозидні аналоги є сильними інгібіторами полімерази, тоді вони можуть індукувати передчасну термінацію молекул нуклеїнової кислотної, що знаходяться на стадії виникнення. Коли трифосфориловані нуклеозидні аналоги захоплюються у реплікати або транскрипти нуклеїнової кислоти, тоді може виникнути генна експресії або руйнування функції. Деякі нуклеозидні аналоги можуть бути ефективними завдяки їх здатності інгібувати аденозинкіназу. Аденозинкіназа каталізує фосфорилування аденозину до аденозин 5'монофосфату (AMP). Інгібування аденозинкінази може ефективно збільшувати позаклітинний рівень аденозину у людей і таким чином служити лікувальним засобом при ішемічних станах, таких як удар, запалення, артрит, судоми і епілепсія. Протягом декількох останніх десятиріч спостерігалися значні зусилля, витрачені на дослідження терапевтичних застосувань нуклеозидних аналогів. Наприклад, певні піримідо[4,5d]піримідинові нуклеозиди, описані в патенті США № 5041542 на ім'я Robins та ін., є ефективними при лікуванні проти L1210 у мишей BDF1. Крім того, 3-β-Dрибофуранозилтіазоло[4,5-d]піримідини, що демонструють значну імуноактивність, включаючи проліферацію клітин селезінки миші та in vivo активність проти вірусу Semliki Forest, описані в патентах США №№. 5041426 і 4880784 на ім'я Robins та ін. В ряді публікацій також описані неглікозильні похідні тіазоло[4,5-d]піримідинової складової. Дивіться, наприклад, патенти США №№ 5994321 і 5446045; Revankar et al., J. Het. Chem., 30, 1341-49 (1993); і Lewis et al., J. Het. Chem., 32, 547-56 (1995). Було продемонстровано, що сполуки 3,5-дизаміщеного-3H-тіазоло[4,5-d]піримідин-2-ону мають імуномодуляторну активність. Одержання та придатність цього класу сполук обговорюється в публікації патентної заявки США № US2006/0160830 (заявка США № 11/304,691) та заявці США № 11/873,202, які обидві в їх повному обсязі включені в цей опис шляхом посилання. СУТЬ ВИНАХОДУ Цей винахід спрямований на спосіб одержання сполуки (2), O AcO OAc 2 40 OAc , при якому здійснюють: (i) реакцію сполуки (4) з алкілкетенацеталем і каталітичною кислотою O HO O HO O 4 , з утворенням циклічної сполуки Формули (5) 1 UA 103637 C2 O O O O 1 R O 5 O , 1 де R являє собою нижчий алкіл, (ii) гідроліз сполуки Формули (5) водою і каталітичною або стехіометричною кількістю кислоти, з утворенням суміші моноацил-заміщених сполук (6) і (7) O O O HO O O O O HO 5 O + O O 6 7 , (iii) збалансування суміші моноацил-заміщених сполук (6) і (7), щоб спричинити надлишок сполуки (6), (iv) окиснення суміші сполуки 6 і сполуки 7, з утворенням суміші кетону (8) і гідратованого кетону (9) O O O O O O O O HO O O 10 O HO 8 9 , (v) відновлення суміші кетону (8) і гідратованого кетону (9), з утворенням сполуки (10), або відновлення сполуки (8) і сполуки (9) роздільно, з утворенням сполуки (10) O O O O HO O 10 15 , (vi) сульфування сполуки (10) сульфуючим агентом у присутності основи, з утворенням сполуки Формули (11) 2 UA 103637 C2 O O O O O O S O R2 O 11 , 2 де R являє собою необов'язково заміщений алкіл або арил, (vii) заміщення сульфонатного естеру сполуки Формули (11) атомом галогену, з утворенням галогенної сполуки Формули (12) O O O O X O 12 , де X являє собою галоген, (viii) відновлення галогену сполуки Формули (12) до атома водню, з утворенням сполуки (13) 5 O O O O O 13 , та (ix) обробку сполуки (13) кислим каталізатором і ацилюючим агентом, з утворенням сполуки 10 15 (2). 1 В одному варіанті здійснення винаходу R являє собою -CH3 або -CH2CH3. 2 В одному варіанті здійснення винаходу R являє собою необов'язково заміщений C1-C6 алкіл 2 або феніл. В іншому варіанті здійснення винаходу R являє собою -CF3, -CH3 або -C6H4CH3. В 2 іншому варіанті здійснення винаходу R являє собою -CF3. В іншому варіанті здійснення винаходу цей винахід відноситься до способу піддавання реакції сполуки (4) з кетендиметилацеталем у присутності каталітичної метансульфонової кислоти, з утворенням трициклічної сполуки (5A) в ізопропілацетаті O O O O O H3C 20 O 5A . В іншому варіанті здійснення цей винахід стосується способу приготування суміші моноацетилованих сполук (6) і (7) шляхом обробки сполуки (5A) водою і 1 мольним відсотком метансульфонової кислоти. В іншому варіанті здійснення цей винахід стосується способу збалансування суміші сполук (6) і (7), щоб спричинити надлишок сполуки (6), шляхом нагрівання вище 70 °C. 3 UA 103637 C2 5 10 15 20 25 30 В іншому варіанті здійснення цей винахід стосується способу збалансування суміші сполук (6) і (7), щоб спричинити надлишок більше ніж у 90 % сполуки (6) над сполукою (7). В іншому варіанті здійснення цей винахід стосується способу збалансування суміші сполук (6) і (7) на користь сполуки (6) шляхом нагрівання вище 70 °C у присутності ізопропілацетату і води. В іншому варіанті здійснення цей винахід стосується способу окиснення суміші сполук (6) і (7), з утворенням сполуки (8) і її гідратованої форми (9). В іншому варіанті здійснення цей винахід стосується способу окиснення суміші сполук (6) і (7), з утворенням сполуки (8) і її гідратованої форми (9), за допомогою використання гіпохлориту натрію у присутності 2,2,6,6-тетраметилпіперидин 1-оксилу (TEMPO) і ацетату натрію у дві фази з ізопропілацетатом. В іншому варіанті здійснення цей винахід стосується способу відновлення сполук (8) або (9) або їх суміші, з утворенням сполуки (10) як єдиного ізомеру. В іншому варіанті здійснення цей винахід стосується способу відновлення суміші сполук (8) і (9), з утворенням сполуки (10) як єдиного ізомеру, використовуючи триацетоксиборогідрид натрію. В іншому варіанті здійснення цей винахід стосується способу відновлення суміші сполук (8) і (9), з утворенням сполуки (10) як єдиного ізомеру, використовуючи триацетоксиборогідрид натрію у вологому ізопропілацетаті. В іншому варіанті здійснення цей винахід стосується способу відділення сполуки (8) від сполуки (9). В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (8), з утворенням сполуки (10) як єдиного ізомеру. В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (8), з утворенням сполуки (10) як єдиного ізомеру, використовуючи платину на вуглецю як каталізатор в присутності водню. В іншому варіанті здійснення цей винахід стосується способу сульфування сполуки (10) сульфуючим агентом у присутності 4-(N, N-диметиламіно)піридину (DMAP), з утворенням сполуки Формули (11). В іншому варіанті здійснення цей винахід стосується способу сульфування сполуки (10) трифторметансульфоновим ангідридом у присутності DMAP, з утворенням сполуки (11A), без використання галогенованих розчинників або температур нижче 0 °C O O O S F3C 35 O O O O O 11A . В іншому варіанті здійснення цей винахід стосується способу сульфування сполуки (10) трифторметансульфоновим ангідридом у присутності DMAP, з утворенням сполуки (11A), в суміші ізопропілацетату і диметоксіетану при 5-10 °C. В іншому варіанті здійснення цей винахід стосується способу заміщення сульфонілзаміщеної сполуки (11A) йодидом при менше ніж 60 °C у низькокиплячих органічних розчинниках, з утворенням сполуки (12A) O O O O I 40 O 12A . В іншому варіанті здійснення цей винахід стосується способу заміщення сульфоніл 4 UA 103637 C2 5 10 15 20 25 30 35 40 45 50 55 60 заміщеної сполуки (11A) йодидом натрію у вологому ізопропілацетаті і диметоксіетані при 55 °C, з утворенням сполуки (12A). В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (12A), з утворенням водневої сполуки (13). В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (12A), з утворенням водневої сполуки (13), використовуючи каталітичне гідрогенування. В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (12A), з утворенням водневої сполуки (13), використовуючи гідроксид паладію на вуглецю (каталізатор Перлмана (Pearlman)) в присутності водню. В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (12A), з утворенням водневої сполуки (13), використовуючи водень і каталітичний гідроксид паладію на вуглецю (каталізатор Перлмана (Pearlman)) у присутності амінної основи, такої як діізопропілетиламін або триетиламін. В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (12A), з утворенням водневої сполуки (13), використовуючи водень і каталітичний гідроксид паладію на вуглецю (каталізатор Перлмана (Pearlman)) у присутності діізопропілетиламіну в етанолі і ізопропілацетату. В іншому варіанті здійснення цей винахід стосується способу відновлення сполуки (12A), з утворенням водневої сполуки (13), використовуючи водень і каталітичний паладій на вуглецю у присутності триетиламіну в етилацетаті. В іншому варіанті здійснення цей винахід стосується способу обробки сполуки (13) каталітичною кількістю сірчаної кислоти і додаванням протягом 12 годин оцтового ангідриду як ацилюючого агента в оцтовій кислоті, з утворенням сполуки (2). Спосіб за цим винаходом є особливо корисним для масштабного комерційного виробництва описаних тут сполук. Способи є простими в здійсненні, надійними і ефективними. Зокрема, способи особливо корисні для великомасштабного виробництва дезоксицукрів. До того ж, способи рентабельні і демонструють ефективну продуктивність і значно вищий загальний вихід у порівнянні зі способами одержання, які використовуються у цій галузі. ДОКЛАДНИЙ ОПИС ВИНАХОДУ Термін "що включає" (та його граматичні варіанти), як використовується тут, використовується у сенсі, що включає значення "що має" або "включаючий", "маючий", а не у виключному сенсі "що складається тільки з". Терміни з неозначеним артиклем "a" і означеним артиклем "the", як використовується тут, треба розуміти так, що містять в собі як множину, так і однину. Як використовується тут, термін "галогенід" або "галоген" відноситься до фторидів, хлоридів, бромідів і йодидів. Термін "галоген" відноситься до фтору, хлору, брому і йоду. Термін "алкіл", як застосовується тут, якщо інше не вказано, включає насичені одновалентні вуглеводневі радикали, що мають нерозгалужені, розгалужені або циклічні складові (включаючи сконденсовані та біциклічні складові, з'єднані місточковим зв'язком, та спіроциклічні складові), або комбінацію вищезгаданих складових. Стосовно алкільної групи, щоб вона мала циклічні складові, то ця група повинна мати принаймні три атоми вуглецю. Термін "арил", як застосовується тут, якщо інше не вказано, включає органічний радикал, що походить від ароматичного вуглеводню завдяки видаленню одного водню, такий як феніл або нафтил. "Алкільні" і "арильні" групи необов'язково заміщені 1-5 замісниками, вибраними з -OH, галогену, -CN, C1-C6 алкілу, арилалкілу, C1-C6 алкокси, C1-C6 алкенілу, C1-C6 гідроксилу, C1-C6 гідроксіалкілу, аміно, C1-C6 алкіламіну, C1-C6 діалкіламіну, де алкільні групи можуть бути далі заміщені одним або більше галогенами. Термін "Ac" означає ацетил. Термін "алкілкетенацеталь" означає 1,1-діалкоксіетен. Термін "каталітичний" означає залучення або дію як каталізатора. Термін "стехіометричний" означає еквівалентну кількість. Сполуки цього опису можуть існувати як єдині стереоізомери, рацемати та/або різноманітні суміші енантіомерів та/або діастереомерів. Всі такі єдині стереоізомери, рацемати та/або різноманітні суміші енантіомерів та/або діастереомерів знаходяться в обсязі розкриття цього винаходу. Як використовується тут, термін "окиснювальний агент" відноситься до речовини або різновидів часток, молекул, радикалів тощо, що набувають електрони в хімічній реакції, а термін "відновлювальний агент" відноситься до речовини, що втрачає електрони в хімічній реакції. Термін "імуномодулятор" означає природні або синтетичні продукти, спроможні 5 UA 103637 C2 5 10 15 20 модифікувати нормальну або порушену імунну систему шляхом стимуляції або пригнічення. Терміни "R" і "S" означають специфічну стереохімічну конфігурацію замісника при асиметричному атомі вуглецю в хімічній структурі, яку наведено. Сполуки винаходу можуть демонструвати явище таутомерії. Незважаючи на те, що наведені вище формули не можуть виразно показати усі можливі таутомерні форми, слід розуміти, що наведені вище формули призначені для того, щоб представити будь-яку таутомерну форму зображеної сполуки, і не повинні обмежуватися лише специфічною формою сполуки, зображеної на структурних формулах. Як взагалі зрозуміло фахівцям у цій галузі, оптично чиста сполука, що має один хіральний центр (тобто, один асиметричний атом вуглецю), є такою, що складається по суті з одного з двох можливих енантіомерів (тобто є енантіомерно чистою), та оптично чиста сполука, що має більш ніж один хіральний центр, є такою, що вона є як діастереомерно чистою, так і енантіомерно чистою. Переважно, сполуки цього винаходу застосовуються у формі, яка є принаймні на 90 % вільною від інших енантіомерів або діастереомерів цих сполук, тобто у формі, що містить принаймні 90 % єдиного ізомеру (80 % надлишку енантіомеру ("е.е.") або надлишку діастереомеру ("d.e.")), більш переважно принаймні 95 % (90 % надлишку енантіомеру або надлишку діастереомеру), навіть більш переважно принаймні 97,5 % (95 % надлишку енантіомеру або надлишку діастереомеру) та найбільш переважно принаймні 99 % (98 % надлишку енантіомеру або надлишку діастереомеру). Сполука (2) корисна як проміжна сполука при приготуванні фармацевтичних сполук, таких як 5-аміно-3-(2’-O-ацетил-3’-дезокси-β-D-рибофуранозил)-3H-тіазоло[4,5-d]піримідин-2-он (3), і їх фармацевтично прийнятних солей. Як описано в заявці США № 11/873,202, дезоксирибофуранозну сполуку (2) сполучають з 5-аміно-3H-тіазоло[4,5-d]піримідин-2-оном (1), з утворенням сполуки (3) S S O N O + N H N OAc O NH2 1 30 35 40 45 50 N N NH2 AcO OAc 25 N O AcO 2 3 OAc . Сполука (3) використовується у способах лікування або профілактики хвороб. Наприклад, сполука (3) використовується у способах лікування або профілактики початку утворення та/або розвитку пухлин або різних видів раку. Також описані способи лікування або профілактики інфекцій патогенних збудників, таких як, наприклад, віруси, зокрема вірус гепатиту B або вірус гепатиту C. Сполука (3) також використовується в способах модуляції активності імунних цитокінів. ПРИКЛАДИ Наступні приклади призначені тільки для ілюстративних цілей і не призначені обмежувати обсяг формули винаходу. У схемах синтезу, наведених нижче, якщо не вказано інше, усі температури позначаються у градусах Цельсію, а усі частини та відсотки вказано за масою. Реактиви були придбані у комерційних постачальників, таких як Aldrich Chemical Company або Lancaster Synthesis Ltd., і використовувалися без подальшого очищення, якщо інше не вказано. Всі розчинники були придбані у комерційних постачальників, таких як Aldrich, EMD Chemicals або Fisher, та їх застосовували у тому вигляді, в якому їх отримали. Реакції, наведені нижче, виконували взагалі в умовах позитивного тиску азоту або аргону при температурі навколишнього середовища (якщо не вказано інше) у безводних розчинниках, а до реакційних колб пристосували гумові перегородки для введення субстратів та реактивів шприцом або додатковою лійкою для рідин або лійкою для порошків у випадку твердих речовин. Реакції проаналізували шляхом тонкошарової хроматографії (TLC, ТШХ) та/або РХ-МС (LCMS) та їх припиняли, звертаючи увагу на витрату початкового матеріалу. Аналітичну тонкошарову хроматографію (TLC) виконували на скляних планшетах, попередньо покритих силікагелем 60 F254, розміром 0,25 мм (EMD Chemicals), та за нею спостерігали за допомогою ультрафіолетового випромінювання (254 нм), та/або з йодом на силікагелі, та/або нагріванням з барвником для TLC, таким як етанолова фосфомолібденова кислота, розчин пара-анісальдегіду з кислотою, розчин нінгідрину, розчин перманганату калію або розчин сульфату церію. Препаративну тонкошарову хроматографію (prepTLC) виконували на скляних планшетах, попередньо покритих силікагелем 60 F254, розміром 0,5 мм (20 × 20 см, від Thomson Instrument 6 UA 103637 C2 5 10 15 20 25 30 35 40 45 Company), та за нею спостерігали за допомогою ультрафіолетового випромінювання (254 нм). 1 13 Спектри Н-ЯМР та С-ЯМР реєстрували на апараті Varian Mercury-VX400 при 400 МГц. -1 Спектри ЯМР отримали стосовно розчинів CDCl3 (надані у ppm (млн )), застосовуючи -1 -1 хлороформ як контрольний стандарт (7,27 млн для протона та 77,00 млн для вуглецю), -1 -1 -1 CD3OD (3,4 та 4,8 млн для протонів та 49,3 млн для вуглецю), ДМСО-d6 (2,49 млн для -1 протона) або тетраметилсилан як внутрішній стандарт (0,00 млн ), коли він був доречним. Інші розчинники для ЯМР застосовувалися за необхідністю. Для позначення піків мультиплетності застосовуються наступні абревіатури: с (синглет), д (дублет), т (триплет), к (квартет), м (мультиплет), шир. (розширений), шир. с (широкий синглет), дд (дублет дублетів), дт (дублет триплетів). Константи взаємодії, якщо наводяться, визначаються у герцах (Гц). Інфрачервоні (IR) спектри реєструвалися на спектрометрі ATR FT-IR Spectrometer як чисті -1 оливи або тверді речовини, та коли їх наведено, вони визначаються у хвильових числах (см ). Мас-спектри, які наведено, являють собою (+)-ES або APCI (+) LC/MS (рідинна хроматографія/мас-спектроскопія), яку проводив відділ аналітичної хімії фірми Anadys Pharmaceuticals, Inc. Елементний аналіз був проведений Atlantic Microlab, Inc., з м. Норкросс, штат Джорджія. Точки плавлення (Т. пл.) (mp) визначали на відкритому капілярному апараті, та вони не корегувалися. В наданих схемах синтезу та в описаних експериментальних процедурах застосовано багато звичайних хімічних абревіатур: DME (1,2-диметоксіетан), MTBE (метил трет-бутиловий етер), TEMPO (2,2,6,6-тетраметилпіперидин 1-оксил), 2,2-DMP (2,2-диметоксипропан), Ac (ацетил), ACN (ацетонітрил), Bn (бензил), BOC (трет-бутоксикарбоніл), Bz (бензоїл), DBU (1,8діазабіцикло[5,4,0]ундец-7-ен), DCC (N, N’-дициклогексилкарбодіімід), DCE (1,2-дихлоретан), DCM (дихлорметан), DEAD (діетилазодикарбоксилат), DIEA (діізопропілетиламін), DMA (N, Nдиметилацетамід), DMAP (4-(N, N-диметиламіно)піридин), DMF (N, N-диметилформамід), DMSO (ДМСО) (диметилсульфоксид), EDC (1-(3-диметиламінопропіл)-3-етилкарбодііміду гідрохлорид), Et (етил), EtOAc (етилацетат), EtOH (етанол), HATU (O-(7-азабензотриазол-1-іл)-1,1,3,3тетраметилуронію гексафторфосфат), HBTU (O-бензотриазол-1-іл-N, N,N",N’-тетраметилуронію гексафторфосфат), HF (фтороводень), HOBT (1-гідроксибензотриазолу гідрат), HPLC (рідинна t хроматографія високого розрізнення), IPA (ізопропіловий спирт), KO Bu (трет-бутоксид калію), LDA (діізопропіламід літію), MCPBA (3-хлорпероксибензойна кислота), Me (метил), MeCN (ацетонітрил), MeOH (метанол), NaH (гідрид натрію), NaOAc (ацетат натрію), NaOEt (етоксид натрію), Phe (фенілаланін), PPTS (p-толуолсульфонат піридинію), PS (з полімерним носієм), Py (піридин), pyBOP (бензотриазол-1-ілокси)трипіролідинoфосфонію гексафторфосфат), TEA (триетиламін), TFA (трифтороцтова кислота), TFAA (трифтороцтовий ангідрид), THF (тетрагідрофуран), TLC (тонкошарова хроматографія), Tol (толуоїл), Val (валін) тощо. Приклад 1: Одержання сполуки (6) (основної) і сполуки (7) (другорядної) (a) Етап 1: Утворення трициклічної Сполуки (5A) 4-Літрову 4-горлову колбу, споряджену впуском для азоту, додатковою лійкою, термометром і механічною мішалкою, завантажили моноацетонксилозою (152,16 г, 800 ммоль) і ізопропілацетатом (1200 мл) і перемішували доти, поки тверді речовини розчинилися, утворюючи трохи мутний розчин. Додали кетендиметилацеталь (3,36 мл, 35,5 ммоль) і реакційну суміш охолодили до 3 °C, використовуючи льодяну баню. Додали метансульфонову кислоту (0,52 мл, 8 ммоль), а потім краплями додавали кетендиметилацеталь (80 мл, 844,5 ммоль) протягом 45 хвилин. Температура реакційної суміші досягла 10 °C впродовж додавання. Коли додавання завершилося, тоді тонкошарова хроматографія з використанням 80 % MTBE у гексані показала повне чисте перетворення на значно швидше утворюваний трицикл 5A. Льодяну баню видалили. (b) Етап 2: Гідроліз сполуки 5A до суміші моноацетатів 50 7 UA 103637 C2 5 10 15 20 25 30 35 40 45 До зазначеної вище реакційної суміші додали воду (72 мл, 4000 ммоль), усю й одразу, і суміш перемішували при температурі навколишнього середовища протягом 90 хвилин. Тонкошарова хроматографія реакційної суміші із застосуванням 80 % MTBE у гексані показала, що утворилися два нові середньополяризовані продукти, причому з повільнішою швидкістю утворення з цих двох був головний продукт. Реакційну суміш перенесли до 2-літрової розділювальної лійки і струшували з 120 мл водного розчину (60 мл 1,0M NaHCO3, 60 мл 30 % NaCl), фази розділили і органічну фазу перенесли до круглодонної колби і леткі речовини видалили у вакуумі. (c) Етап 3: Збалансування до Сполуки (6). Речовину, виділену при випарюванні, розчинили у щойно приготованому ізопропілацетаті (1200 мл) і воді (72 мл) і нагрівали до 77 °C протягом 12 годин, потім охолодили до температури навколишнього середовища. Аналіз за допомогою тонкошарової хроматографії із застосуванням 80 % MTBE у гексані показав, що швидше утворюваним з цих двох продуктів є головний продукт при присутності тільки слідів повільніше утворюваного продукту. 0,2 мл зразка реакційної суміші випарили до сухості, в результаті чого одержали 37 мг 1 твердої речовини. H ЯМР підтвердив, що бажаний ацетат сполуки (6) є продуктом, присутнім у 1 значно більшій кількості. H ЯМР (400 МГц, CDCl3) : 5,92 (1H, д, J=3,3 Гц), 4,51-4,56 (2H, м), 4,24-4,28 (1H, м), 4,13-4,19 (2H, м), 2,98 (1H, д, J=4,0 Гц), 2,11 (3H, с), 1,51 (3H, с), 1,33 (3H, с). Приклад 2: Одержання сполук (8) і (9) 4-Літрову колбу, що вже містить приблизно 0,8 моль сполуки (6) у вологому ізопропілацетаті з попереднього етапу, спорядили впуском для азоту, додатковою лійкою, термометром і механічною мішалкою. Додали TEMPO (800 мг) і суміш перемішували і охолодили на льодяній бані. В окремій колбі водний розчин, що містив 64,3 г броміду натрію, 98,4 г ацетату натрію, розчинили в 320 мл деіонізованої води, охолодили до 5 °C. Коли температура реакційної суміші досягла 5 °C, тоді додали до неї цей попередньо охолоджений водний розчин, з утворенням двофазної реакційної суміші. До холодного розчину додавали краплями 735 мл водного розчину гіпохлориту натрію (титрованого безпосередньо перед використанням, 10,15 % або 1,36M, 1,002 моль, 1,25 еквівалента) протягом 2 годин, підтримуючи екзотермічне додавання при 7 °C або нижче. Коли додавання завершили, тоді перемішування продовжували протягом 30 хвилин, і TLC (80 % MTBE-гексан) показала повне перетворення на повільніше утворюваний кетон. Реакційну суміш перенесли до 4-літрової розділювальної лійки і фази розділилися. Темну органічну частину промили один раз 160 мл водного 2,5 % розчину тіосульфату натрію. Одержану блідо-жовту органічну частину промили 160 мл 30 % розчину хлориду натрію. Водні фази об'єднали і додали 44,1 г твердого хлориду натрію і перемішували до тих пір, поки вся соль розчинилася. Одержаний водний розчин екстрагували двічі 400 мл порціями ізопропілацетату, органічні екстракти об'єднали і промили один раз 50 мл 30 % розчину хлориду натрію. Всі органічні порції об'єднали, в результаті чого отримали трохи мутний розчин. Порцію у 0,25 мл цього розчину випарили, в результаті чого отримали 14 мг твердої 1 речовини. H ЯМР підтвердив присутність як кетону, так і гідрату у вигляді суміші приблизно 1:1. 1 H ЯМР (400 МГц, CDCl3) : 6,09 (1H, д, J=4,4 Гц, сполука 8), 5,84 (1H, д, J=3,9 Гц, сполука 9), 4,61 (1H, дд, J1=11,7 Гц, J2=6,3 Гц, сполука 9), 4,56 (1H, т, J=3,3 Гц, сполука 8), 4,36-4,42 (2H, м, Сполуки 8 і 9), 4,20-4,24 (2H, м, Сполуки 8 і 9), 4,06-4,15 (2H, м, Сполуки 8 і 9), 2,11 (3H, с, сполука 9), 2,05 (3H, с, сполука 8), 1,58 (3H, с, сполука 9), 1,50 (3H, с, сполука 8), 1,43 (3H, с, сполука 8), 1,36 (3H, с, сполука 9). Приклад 3: Одержання сполуки (10) 8 UA 103637 C2 5 10 15 20 25 30 35 40 45 50 55 60 4-Літрову 4-горлову колбу, споряджену впуском для азоту, додатковою лійкою, термометром і механічною мішалкою, завантажили мутним органічним розчином кетону (8) і його гідрату (9). Його охолодили до 4 °C при перемішуванні, використовуючи льодяну баню. До цього холодного розчину додали з 15 хвилинними інтервалами чотири порції у 42,4 г твердого триацетоксиборогідриду натрію. Після останнього додавання реакційну суміш перемішували при 5 °C протягом 60 хвилин. Продовжуючи перемішування при 5 °C, швидко додали 1,0 M водний розчин карбонату натрію (800 мл). Температура реакційної суміші піднялася до 12 °C, і виникло виділення невеликої кількості газу. Суміш значною мірою густішала. Після перемішування протягом 15 хвилин реакційну суміш перенесли до 4-літрової розділювальної лійки, і фази розділили, водна частина містила певну кількість твердої речовини. Органічну частину перемішували з 2,0 M водним розчином карбонату натрію (400 мл) протягом 10 хвилин, фази розділилися, і обидві водні фази об'єднали. Тверду речовину у водній фазі відфільтрували, а потім розчинили у воді (600 мл) і додали назад до одержаної гомогенної водної фази. Водну фазу екстрагували двома 200 мл порціями ізопропілацетату і органічні частини об'єднали. Загальна маса органічної фази становила 2370,5 г. Порцію у 5 г органічної фази випарили, в результаті чого отримали 243 мг оливи, що кристалізувалася під вакуумом. Розрахований вихід: 2370,5 г розчину x 0,243 г продукту/5 г 1 розчину = 115,2 г (496,15 ммоль, 62 %) сполуки 10. H ЯМР показав, що це є дуже чистий 1 зразок. H ЯМР (400 МГц, CDCl3) : 5,82 (1H, д, J=3,9 Гц), 4,58 (1H, т, J=4,3 Гц), 4,43 (1H, дд, J1=12,4 Гц, J2=2,4 Гц), 4,16-4,20 (1H, м), 3,93-3,97 (1H, м), 3,81-3,87 (1H, м), 2,45 (1H, д, J=10,8 Гц), 2,10 (3H, с), 1,58 (3H, с), 1,38 (3H, с). 4-Літрову 4-горлову колбу, споряджену короткою дистиляційною насадкою, температурним зондом і механічною мішалкою, завантажили 2370,5 г органічної фази. Її нагрівали до видалення 2400 мл дистиляту при атмосферному тиску. Додали до колби щойно приготований ізопропілацетат (1500 мл) і 1500 мл видалили шляхом перегонки. Реакційну суміш у колбі потім розвели 920 мл ізопропілацетату, в результаті чого отримали трохи мутний розчин. Цей розчин був одразу готовий для використання на наступному етапі. Приклад 4: Альтернативне одержання сполук (8) і (10) (a) Етап 1: Одержання сполуки (8) Колбу, що містила сполуку 6 (приблизно 0,2 моль) у вологому ізопропілацетаті з Прикладу 1, спорядили впуском для азоту, термометром, додатковою лійкою і магнітною мішалкою. Додали TEMPO (200 мг) і суміш перемішували і охолодили на льодяній бані з температурою 0 °C. В окремій колбі водний розчин, що містив бромід натрію (16,08 г) і ацетат натрію (24,6 г), розчинені в деіонізованій воді (80 мл), охолодили до 5 °C. Коли температура реакційної суміші досягла 5 °C, тоді до неї додали попередньо охолоджений водний розчин, щоб утворити двофазну реакційну суміш. До цієї холодної суміші додавали краплями водний розчин гіпохлориту натрію (з поміткою 10-15 %; 180 мл) протягом 1 години, підтримуючи екзотермічне додавання при 7 °C або нижче. Коли додавання завершили, тоді TLC (80 % MTBE-гексани) показала повне перетворення на кетон нижчого Rf. Охолоджувальну баню видалили і додали твердий NaCl (25 г). Після перемішування протягом 30 хвилин, суміш перенесли до 1-л розділювальної лійки і фази потім розділилися. Темну органічну частину струшували з 1,0 M NaHCO3 (25 мл), а потім додали 2,0 M Na2SO3 (30 мл) і струшування продовжували доти, поки усі кольори зникли (виникало деяке виділення газу). Отриману чисту органічну частину промили один раз 15 % водним NaCl (20 мл). Чисту органічну фазу перенесли до 1-л колби, спорядженої температурним зондом, дистиляційною насадкою і магнітною мішалкою. Температуру довели до 85 °C, щоб відігнати розчинник. Коли відгонку зупинили, тоді температуру підняли до 105 °C, щоб завершити відгонку. Перегонну колбу охолодили до температури навколишнього середовища і суміш розвели ізопропілацетатом (100 мл). Додали активований вуглець (Darco G60; 5 г) і суміш перемішували при температурі навколишнього середовища протягом 90 хвилин. Цю суміш профільтрували, використовуючи Celite, і тверді речовини промили ізопропілацетатом (2 × 30 мл). Блідо-жовтий фільтрат мав масу 220,5 г. 2,0 мл цього розчину (маса = 1,826 г) випарили, в результаті чого одержали 0,189 г блідо-жовтої оливи. Розрахунок показав концентрацію розчину в 0,41 M 1 сполуки 8 і загальний вихід 22,86 г (49,6 % від моноацетонксилози). H ЯМР (400 МГц, CDCl3) : 1,43 (3H, с), 1,50 (3H, с), 2,05 (3H, с), 4,21 (1H, дд, J1=11,9 Гц, J2=3,9 Гц), 4,37 (1H, д, J=4,7 Гц), 1 4,40 (1H, дд, J1=12,5 Гц, J2=3,2 Гц), 4,56 (1H, т, J=3,1 Гц), 6,09 (1H, д, J=3,8 Гц). H-ЯМР показав, що присутня тільки сполука 8 (сполука 9 відсутня). b. Етап 2: Одержання сполуки (10) 9 UA 103637 C2 O O O O H2 3% Pt-C O O O O i-PrOAc O HO O O 10 8 5 10 250-мл тригорлову круглодонну колбу, споряджену температурним зондом, балоном, заповненим газоподібним воднем, і магнітною мішалкою, завантажили 62 мл 0,41 M розчину сполуки 8, одержаної вище, і вологим 3 % Pt-C (2,05 г, Johnson Matthey типу B101018-3, лот C9264, 58,25 % води). Температуру встановили на рівні 26 °C, суміш дегазували шляхом поміщення у вакуум і три рази промили газоподібним воднем, а потім суміш енергійно перемішували в атмосфері водню протягом 16 годин. GC-аналіз показав повне перетворення на сполуку 10. Розчин профільтрували через допоміжний фільтрувальний матеріал Celite, тверді речовини промили ізопропілацетатом (2 × 30 мл), і прозорий безбарвний фільтрат потім 1 випарили, в результаті чого отримали 5,74 г оливи, що кристалізувалася. H-ЯМР підтвердив, що сполука (10) була єдиним продуктом. Приклад 5: Одержання сполуки (11A) O O O O O S S O O F 3C O O O CF3 O O HO DMAP i-propOAc-DME O S 15 20 25 30 35 O F 3C 10 O O O 11A O 4-Літрову колбу, що вже містила приблизно 496,15 ммоль сполуки 10 в сухому ізопропілацетаті з Прикладу 3, спорядили впуском для азоту, термометром, резиновою мембраною і механічною мішалкою. В окремій колбі DMAP (90,92 г, 744,23 ммоль, 1,5 екв.) розчинили в 255 мл гарячого DME. Гарячий розчин додали до реакційної суміші у колбі і реакційну суміш охолодили на льодяній бані до 5 °C. Трифторометансульфоновий ангідрид (104,34 мл, 620,19 ммоль, 1,25 екв.) додали при 1,17 мл/хвилину, використовуючи шприц-насос. Максимальна температура, досягнута протягом додавання, становила 7 °C. Коли додавання завершили, а температура реакційної суміші повернулася до 5 °C, тоді TLC (20 % EtOAcтолуол) показала завершення, чисте перетворення до швидше утворюваного трифлату. До реакційної суміші, що мала 5 °C, додали 1,0M HCl (745 мл), викликаючи екзотермічне підвищення температури до 9 °C. Після перемішування протягом 5 хвилин реакційну суміш перенесли до розділювальної лійки, і фази розділили. Органічну фазу промили двома порціями 1,0 M HCl (300 мл) і один раз 240 мл водного розчину (120 мл 1,0 M NaHCO 3, 120 мл 30 % хлориду натрію). Усі водні фази об'єднали і екстрагували один раз 500 мл ізопропілацетату. Екстракт промили двома 100 мл порціями 1,0 M HCl і один раз 80 мл водного розчину (40 мл 1,0 M NaHCO3, 40 мл 30 % хлориду натрію). Всі органічні фази об'єднали, в результаті чого одержали трохи мутний розчин трифлату 11A. 1 Порцію у 0,25 мл цього розчину випарили, в результаті чого одержали 22 мг оливи. H ЯМР 1 показав, що це був дуже чистий зразок з невеликою кількістю залишкового ізопропілацетату. H ЯМР (400 МГц, CDCl3) : 5,85 (1H, д, J=3,9 Гц), 4,85 (1H, дд, J1=8,6 Гц, J2=4,6 Гц), 4,77 (1H, т, J=4,3 Гц), 4,37-4,42 (2H, м), 4,22-4,26 (1H, м), 2,11 (3H, с), 1,61 (3H, с), 1,40 (3H, с). Приклад 6: Одержання сполуки (12A) O O O O O O NaI O S F3 C O O O O O i-propOAc-DME 11A I O 12A 4-Літрову 4-горлову колбу, споряджену впуском для азоту, температурним зондом, конденсатором і механічною мішалкою, завантажили ізопропілацетатним розчином трифлату 10 UA 103637 C2 5 10 15 (повинно було бути 496,15 ммоль) і 255 мл DME. Додали твердий йодид натрію (111,55 г, 744,23 ммоль, 1,5 екв.) і суміш перемішували при 55 °C протягом 17 годин. TLC (10 % EtOAc-толуол) показала повне перетворення на йодид. Додали воду (400 мл) і суміш швидко перемішували протягом п'яти хвилин. Суміш перенесли до розділювальної лійки, і фази розділили. Органічну фазу промили один раз 400 мл водного розчину (200 мл 1,0M NaHCO3 і 200 мл 30 % NaCl). Водні фази об'єднали і екстрагували один раз ізопропілацетатом (400 мл). Екстракт промили один раз водою (100 мл) і один раз 100 мл водного розчину (50 мл 1,0M NaHCO 3 і 50 мл 30 % NaCl). Усі органічні фази об'єднали. Розчин сполуки 12A перенесли до 3-літрової круглодонної колби, спорядженої короткою дистиляційною насадкою. Два літри розчинника видалили шляхом простої перегонки. Суміш охолодили до температури навколишнього середовища і залишковий об'єм мав становити 500 мл. До цього додали 183 мл ізопропілацетату і 208 мл 200 пробного етанолу, щоб створити 0,5M розчин сполуки 12A в 20 % розчині етанол/ізопропілацетат. 1 Узяли аліквоту у 0,2 мл і випарили, в результаті чого одержали 42 мг оливи. H ЯМР 1 показав, що це був дуже чистий зразок сполуки 12A. H ЯМР (400 МГц, CDCl3) : 6,02 (1H, д, J=2,9 Гц), 5,04 (1H, д, J=2,9 Гц), 4,35 (1H, д, J=3,1 Гц), 4,15-4,24 (2H, м), 3,77-3,80 (1H, м), 2,10 (3H, с), 1,52 (3H, с), 1,33 (3H, с). Приклад 7: Одержання сполуки (13) O O O O O H2, Pd(OH)2/C O O DIEA I O 13 12A 20 25 30 35 40 45 O i-propOAc-EtOH O 3-Літрову круглодонну колбу, споряджену великою магнітною перемішувальною пластиною, завантажили розчином сполуки 12A (повинно бути 496,15 ммоль у вигляді 0,5 M розчину в 20 % суміші етанол/ізопропілацетат), діізопропілетиламіном (112,34 мл, 644,8 ммоль, 1,3 екв.) і 20,37 г 20 % Pd(OH)2/C (каталізатор Перлмана (Pearlman)). Під час швидкого перемішування реакційну суміш дегазували за допомогою легкого вакууму і потім заповнювали газоподібним воднем три рази. Реакційну суміш потім перемішували в атмосфері водню протягом 18 годин. TLC (10 % EtOAc-толуол) показала чисте повне перетворення на повільніше утворювану водневу сполуку. Реакційну суміш профільтрували через Celite і темну тверду речовину промили двома 200 мл порціями ізопропілацетату. Фільтрат перенесли до 4-літрової розділювальної лійки і промили один раз 1,0 M HCl (645 мл), один раз 200 мл водного розчину (100 мл 2,5 % тіосульфату натрію, 100 мл 1,0M NaHCO3) і один раз 200 мл 30 % NaCl. Всі водні фази об'єднали і екстрагували двома 200 мл порціями ізопропілацетату. Екстракти об'єднали і промили один раз 80 мл водного розчину (40 мл 2,5 % тіосульфату натрію, 40 мл 1,0M NaHCO3) і один раз 80 мл 30 % NaCl. Органічні частини об'єднали, перенесли до 3-літрової круглодонної колби і 1,5 літри розчинника видалили шляхом атмосферної відгонки. Охолоджений залишок мав об'єм у 450 мл. Додали 50 мл ізопропілацетату, щоб утворити розчин ближче до 1,0 M, додали 10 г вугілля Norit і суміш перемішували дві години при температурі навколишнього середовища. Цю суміш потім профільтрували через Celite, в результаті чого отримали прозорий фільтрат золотистого кольору. Фільтрат концентрували у вакуумі, в результаті чого отримали 103,47 г 1 (478,52 ммоль) прозорої оливи золотистого кольору. H ЯМР показав дуже високу чистоту 1 сполуки 13. H ЯМР (400 МГц, CDCl3) : 5,83 (1H, д, J=3,7 Гц), 4,74 (1H, т, J=4,2 Гц), 4,39-4,45 (1H, м), 4,28 (1H, дд, J1=11,8 Гц, J2=3,1 Гц), 4,08 (1H, дд, J1=12,5 Гц, J2=6,2 Гц), 2,07-2,12 (4H, м), 1,62-1,69 (1H, м), 1,52 (3H, с), 1,33 (3H, с). Сполука 13 може бути далі очищена шляхом вакуумної перегонки, якщо необхідно. BP=70 °C при 0,025 мм Hg. Приклад 8: Одержання сполуки (2) 11 UA 103637 C2 O O O O O Ac2O O O O O H2SO4-AcOH 13 2 O O O 5 10 15 20 25 мл круглодонну колбу, споряджену магнітною мішалкою і резиновою мембраною, завантажили сполукою 13 (640 мг, 2,96 ммоль) і 5 мл оцтової кислоти. У окремій колбі оцтовий ангідрид (0,562 мл, 6 ммоль, 2 екв.) розвели оцтовою кислотою до загального об'єму у 2,0 мл і 0,1 мл цього розчину оцтового ангідриду додали до реакційної суміші. Додали сірчану кислоту (0,15 мл 1,0M розчину в оцтовій кислоті, 0,15 ммоль, 0,05 екв.) до реакційної суміші, а потім решту цього розчину оцтового ангідриду (1,9 мл) додавали протягом 12 годин, використовуючи шприц-помпу. TLC (30 % EtOAc-гексан) показала дуже чисте перетворення на бажану сполуку 2. Реакційну суміш розвели толуолом і випарили у вакуумі. Залишок розчинили в MTBE, перемішували з 10 % карбонатом натрію протягом 15 хвилин, і фази розділилися. Органічну частину висушили (MgSO4), профільтрували через маленький шар силікагелю і випарили, в 1 результаті чого одержали 680 мг (2,61 ммоль) прозорої оливи. H ЯМР показав, що це була чиста суміш обох аномерів. Важливо відзначити, що побудова і впорядкування способів і етапів, наведених в варіантах здійснення за прикладами, є тільки ілюстративними. Незважаючи на те, що тільки декілька варіантів здійснення цього розкриття винаходу були описані детально, фахівці у цій галузі легко зрозуміють, що можна здійснити багато модифікацій без суттєвого відходу від нових ідей і переваг предмету цього винаходу, викладеного в формулі винаходу. Відповідно, усі такі модифікації повинні включатися в обсяг цього розкриття винаходу, визначеного в доданій формулі винаходу. Порядок або послідовність будь-якого способу або етапів способів можуть варіюватися або переставлятися згідно з альтернативними варіантами здійснення. Інші заміщення, модифікації, зміни і вилучення можуть бути зроблені в побудові, робочих умовах та режимах і впорядкуванні варіантів здійснення без відходу від духу цього розкриття винаходу, визначеного в доданій формулі винаходу. 25 ФОРМУЛА ВИНАХОДУ 1. Спосіб одержання сполуки (2) O AcO OAc OAc 30 2 , при якому здійснюють: (і) реакцію сполуки (4) з алкілкетенацеталем і каталітичною кислотою O HO O HO O 4 , з утворенням циклічної сполуки Формули (5) 12 UA 103637 C2 O O O O R 1 O 5 O , 1 де R являє собою нижчий алкіл, (іі) гідроліз сполуки Формули (5) водою і каталітичною або стехіометричною кількістю кислоти, з утворенням суміші моноацилзаміщених сполук (6) і (7) O O O O O HO O O + O O O 5 7 6 , (ііі) збалансування суміші моноацилзаміщених сполук (6) і (7), щоб спричинити утворення надлишку сполуки (6), (iv) окиснення суміші сполуки 6 і сполуки 7, з утворенням суміші кетону (8) і гідратованого кетону (9) O O O O O O O O 10 15 O O HO , O HO 9 8 , (v) відновлення суміші кетону (8) і гідратованого кетону (9), з утворенням сполуки (10), або відновлення сполуки (8) і сполуки (9) роздільно, з утворенням сполуки (10) 10 , (vi) сульфування сполуки (10) сульфуючим агентом у присутності основи, з утворенням сполуки Формули (11) 11 , 2 де R являє собою необов'язково заміщений алкіл або арил, (vii) заміщення сульфонатного естеру сполуки Формули (11) атомом галогену, з утворенням галогенної сполуки Формули (12) 13 UA 103637 C2 O O O O X O 12 , де X являє собою галоген, (viii) відновлення галогену сполуки Формули (12) до атома водню, з утворенням сполуки (13) O O O O O 5 10 15 20 25 30 13 , та (іх) обробку сполуки (13) кислим каталізатором і ацилюючим агентом, з утворенням сполуки (2). 1 2. Спосіб за п. 1, де R являє собою -СН3 або -СН2СН3. 2 3. Спосіб за п. 1, де R являє собою -CF3, -CH3 або -С6Н4СН3. 4. Спосіб за п. 1, де на стадії (і) сполуку (4) піддають реакції з кетендиметилацеталем у присутності каталітичної метансульфонової кислоти, з утворенням трициклічної сполуки (5А), в ізопропілацетаті. 5. Спосіб за п. 1, де на стадії (ііі) здійснюють збалансування суміші сполук (6) і (7), щоб спричинити надлишок сполуки (6), шляхом нагрівання вище 70 °C. 6. Спосіб за п. 5, де збалансування суміші сполук (6) і (7) спричиняє утворення надлишку більше ніж у 90 % сполуки (6) над сполукою (7). 7. Спосіб за п. 1, де на стадії (iv) суміш сполук (6) і (7) окиснюють за допомогою гіпохлориту натрію у присутності TEMPO і ацетату натрію у дві фази з ізопропілацетатом. 8. Спосіб за п. 1, де на стадії (v) сполуку (10) утворюють як єдиний ізомер, використовуючи триацетоксиборогідрид натрію. 9. Спосіб за п. 1, де сполуку (8) виділяють із суміші сполук (8) і (9). 10. Спосіб за п. 9, в якому здійснюють відновлення сполуки (8), з утворенням сполуки (10) як єдиного ізомеру. 11. Спосіб за п. 10, де відновлення виконують, використовуючи платину на вуглеці як каталізатор в присутності водню. 12. Спосіб за п. 1, де на стадії (vi) основа являє собою DMAP. 13. Спосіб за п. 1, де на стадії (viii) при відновленні сполуки Формули (12) використовують гідроксид паладію на вуглеці (каталізатор Перлмана) в присутності водню. 14. Спосіб одержання сполуки Формули (11), при якому здійснюють: (і) окиснення сполуки (6) з утворенням сполуки (8) O O O O O O 8 , (ii) відновлення сполуки (8), з утворенням сполуки (10) 14 UA 103637 C2 10 , та (ііі) сульфування сполуки (10) сульфуючим агентом у присутності основи, з утворенням сполуки Формули (11) 5 10 15 11 , де R являє собою необов'язково заміщений алкіл або арил. 2 15. Спосіб за п. 14, де R являє собою -CF3, -СН3 або -С6H4СН3. 16. Спосіб за п. 14, де сполуку (6) окиснюють за допомогою гіпохлориту натрію у присутності TEMPO і ацетату натрію у дві фази з ізопропілацетатом. 17. Спосіб за п. 14, де відновлення сполуки (8) утворює єдиний ізомер сполуки (10). 18. Спосіб за п. 15, де відновлення виконують, використовуючи платину на вуглеці як каталізатор у присутності водню. 19. Спосіб за п. 14, де сульфуючий агент являє собою трифторометансульфоновий ангідрид, а основа являє собою DMAP. 20. Спосіб за п. 14, в якому далі здійснюють заміщення сульфонатного естеру сполуки Формули (11) атомом галогену, з утворенням галогенної сполуки Формули (12) 2 O O O O X O 12 , де Х являє собою галоген, відновлення галогену сполуки Формули (12) до атома водню, з утворенням сполуки (13) O O O O O 13 20 , та обробку сполуки (13) кислим каталізатором і ацилюючим агентом, з утворенням сполуки (2) O AcO OAc OAc 2 . 21. Спосіб за п. 20, де при відновленні сполуки Формули (12) використовують гідроксид паладію на вуглеці (каталізатор Перлмана) в присутності водню. 15 UA 103637 C2 22. Спосіб за п. 20, де кислий каталізатор являє собою сірчану кислоту, а ацилюючий агент являє собою оцтовий ангідрид. 23. Сполука, вибрана з групи, що складається з: O O O O O O O O HO O O O HO , , O O O O O O HO 5 O O S O та O O F3 C O O . Комп’ютерна верстка І. Скворцова Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 16
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod of preparing deoxyribofuranose compounds
Автори російськоюHaley, Gregory, J.
МПК / Мітки
МПК: A01N 43/90
Мітки: одержання, спосіб, дезоксирибофуранозних, сполук
Код посилання
<a href="https://ua.patents.su/18-103637-sposib-oderzhannya-dezoksiribofuranoznikh-spoluk.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання дезоксирибофуранозних сполук</a>
Похідні октагідро-6,10-діоксо-6н-піридазино[1,2-а][1,2]діазепін-1- карбонової кислоти, спосіб їх одержання та одержання терапевтично активних сполук

Номер патенту: 71913
Опубліковано: 17.01.2005
Автори: Крок Веронік, Ларкін Джон Патрік, Руссель Патрік, КОЛЛАДАН Колетт
МПК: C07D 487/04, C07K 5/06, C07K 5/078
Мітки: карбонової, сполук, спосіб, терапевтичної, активних, октагідро-6,10-діоксо-6н-піридазино[1,2-а][1,2]діазепін-1, кислоти, одержання, похідні
Формула / Реферат:
1. Сполуки загальної формули (IA1):,де R являє собою С1-4 алкіл, що мають конфігурацію SR або що знаходяться у вигляді суміші SR+SS, які являють собою рацемічну суміш.2. Сполуки загальної формули (ІА1) за п.1, де R являє собою метил.3. Сполуки загальної формули (IA1) за п.1, 2, які являють собою рацемічну суміш, що складається...
Спосіб одержання циталопраму (варіанти), s-циталопраму, проміжні кетони та спосіб одержання рацемічних сполук

Номер патенту: 72238
Опубліковано: 15.02.2005
Автори: Петерсен Ханс, Еллегор Петер, Рок Майкл Харольд
МПК: C07C 253/30, C07D 307/87, C07C 255/56
Мітки: сполук, s-циталопраму, рацемічних, циталопраму, проміжні, варіанти, спосіб, кетони, одержання
Формула / Реферат:
1. Спосіб одержання циталопраму, згідно з яким здійснюють реакцію сполуки формули IV, IVде R являє собою ацил, з 3-(N,N-диметиламіно)пропілмагнійгалогенідом, переважно з 3-(N,N-диметиламіно)пропілмагнійхлоридом, з одержанням циталопраму формули I, Iякий виділяють у вигляді основи або її фармацевтично прийнятної солі.2. Спосіб за п. 1, який відрізняється тим, що проміжну сполуку формули IV одержують...
Азольні сполуки з протигрибковою активністю, спосіб одержання цих сполук, спосіб одержання проміжних сполук та фармацевтична композиція

Номер патенту: 55405
Опубліковано: 15.04.2003
Автори: Фраіре Крістіна, Наполетано Мауро, Скіоппакассі Джованна, Альбіні Енріко
МПК: C07D 521/00, A61P 31/10, C07D 249/08, A61K 31/4164, A61K 31/41, A61K 31/415, A61K 31/4196, A61P 31/00, C07D 233/60, A61K 31/00
Мітки: проміжних, сполук, протигрибковою, композиція, активністю, спосіб, азольні, сполуки, цих, одержання, фармацевтична
Формула / Реферат:
1. Сполука формули (ІІ-А), (ІІ-А)де:R1 являє собою хлор, фтор, бром або трифторметил;R2 являє собою водень, хлор, фтор, бром або трифторметил;Z являє собою СН або N;R3, R4, R5, які можуть бути однаковими або різними, являють собою водень або С1-С4 алкіл при умові, що R4 відрізняється від R5, якщо R3 - водень;Х являє собою О, S, SO або SO2;R6 являє собою С1-С5 поліфторалкільну групу, яка...
Спосіб одержання 1-(тіометил)циклопропаноцтової кислоти та проміжних сполук для її одержання

Номер патенту: 50730
Опубліковано: 15.11.2002
Автори: Конлон Девід Е., Піпік Бренда, Кінг Стівен
МПК: C07C 319/00, C07C 327/00, C07C 253/16, C07D 327/00
Мітки: 1-(тіометил)циклопропаноцтової, сполук, одержання, спосіб, кислоти, проміжних
Формула / Реферат:
1. Спосіб одержання циклічного сульфіту 1,1-циклопропандиметанолу формули,який передбачає:а) взаємодію 1,1-циклопропандиметанолу з діалкілсульфітом в присутності кислоти або основи таb) вилучення з реакційної суміші спиртового побічного продукту реакції.2. Спосіб за п. 1, за яким реакцію проводять в присутності основи.3. Спосіб за...
Похідні індолу, фармацевтична композиція та спосіб одержання сполук (варіанти)

Номер патенту: 68388
Опубліковано: 16.08.2004
Автори: Паллук Райнер, Бєхтєль Вольф-Дітріх, Пшорн Уве, Вайзер Томас, Грауерт Маттіас, Картер Адріан, Хьонке Хрістоф
МПК: A61P 25/28, C07D 209/60, C07D 401/04, A61P 9/10, A61P 9/06, A61P 23/02, A61K 31/404, C07D 209/04
Мітки: похідні, сполук, спосіб, одержання, варіанти, композиція, фармацевтична, індолу
Формула / Реферат:
1. Похідні індолу загальної формули 1, 1деХ означає простий зв'язок, -О-, С1-С4алкіл, С1-С3алкоксигрупу, -О-СН2-СН2-О- чи -O-CH2-CH2-NH-,R1 означає водень, метил, етил чи феніл, R2 означає водень або метил,R3 означає водень, F, Сl, Br, гідрокси- чи метоксигрупу, R4 означає водень, метил чи етил, R5 означає водень, метил чи етил, R6 означає водень, метил чи етил, R7 означає...
Попередній патент: Шліфувальний верстат
Наступний патент: 2-(6-гідроксиметил-9-метил-2-(2′-метоксифеніл)-5н-піридо[4′,3′:5,6]пірано[2,3-d]піримідин-4-ілсульфаніл)ацетамід з антифунгальною активністю
Випадковий патент: Сталь