Спосіб одержання силденафілу
Номер патенту: 99447
Опубліковано: 27.08.2012
Автори: Стропнік Тадей, Зю Йі, Шен Жінкшам, Лью Зенг, Бомбек Сергея, Ванг Зен, Тіан Гюанхю










Формула / Реферат
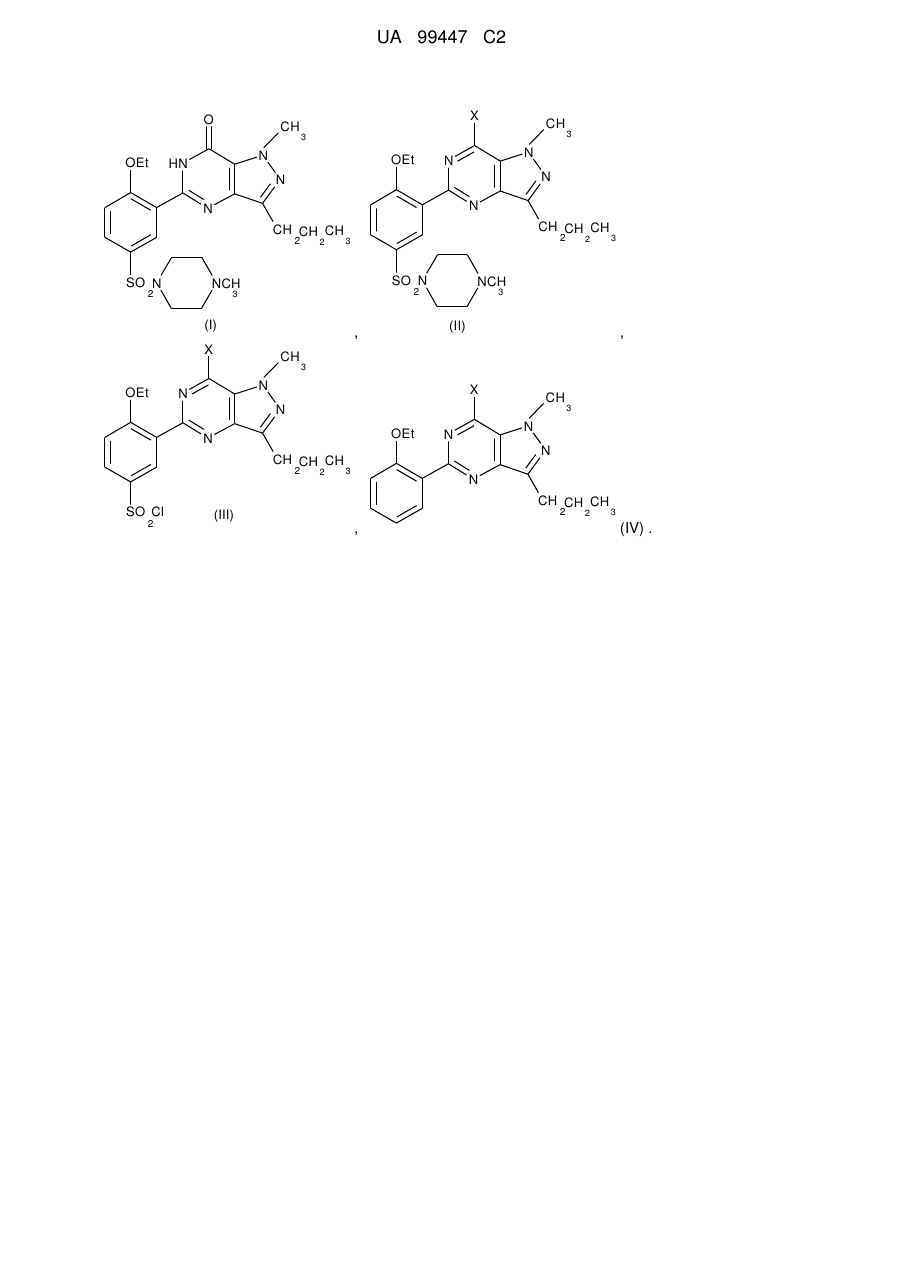
1. Спосіб одержання сполуки формули (І):
, (I)
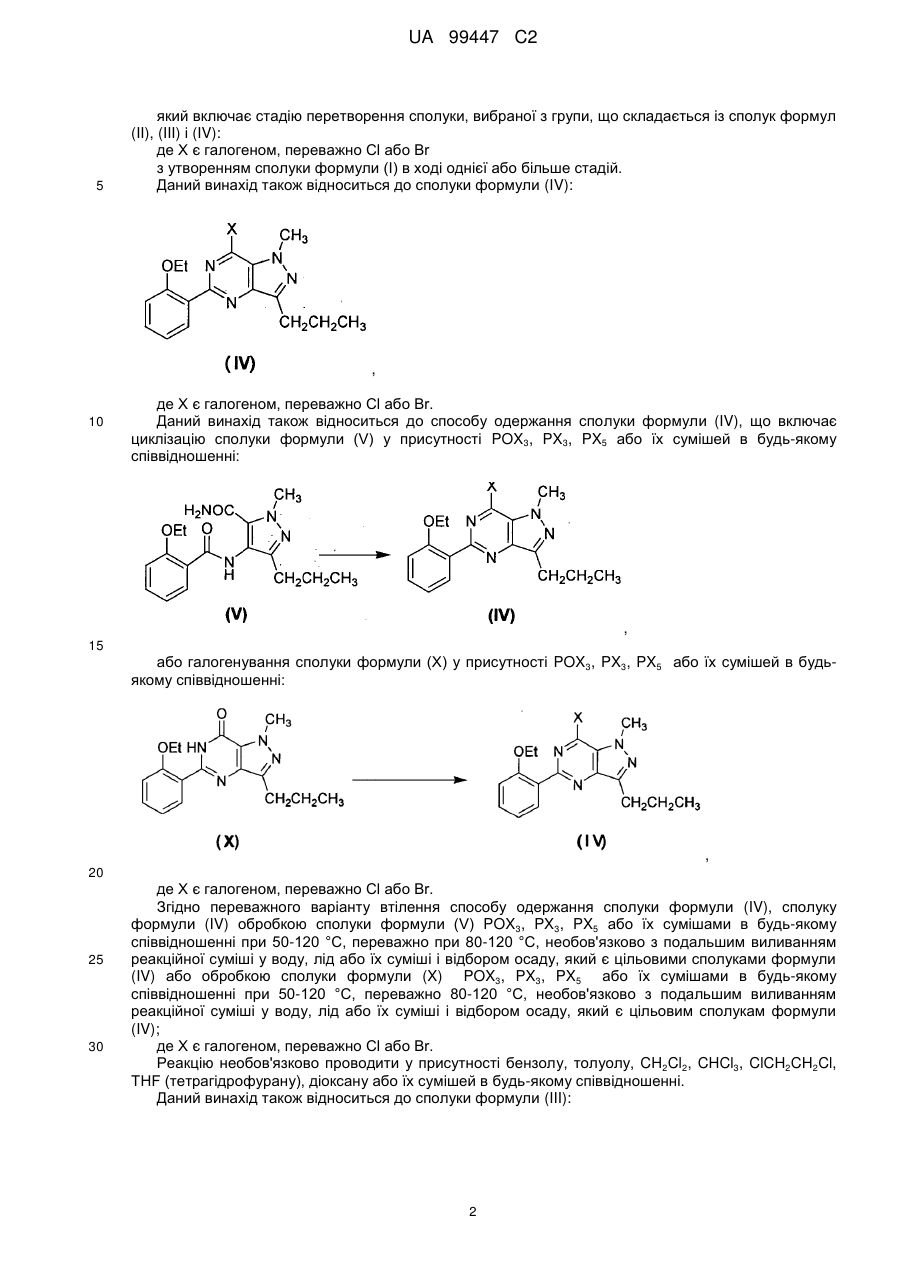
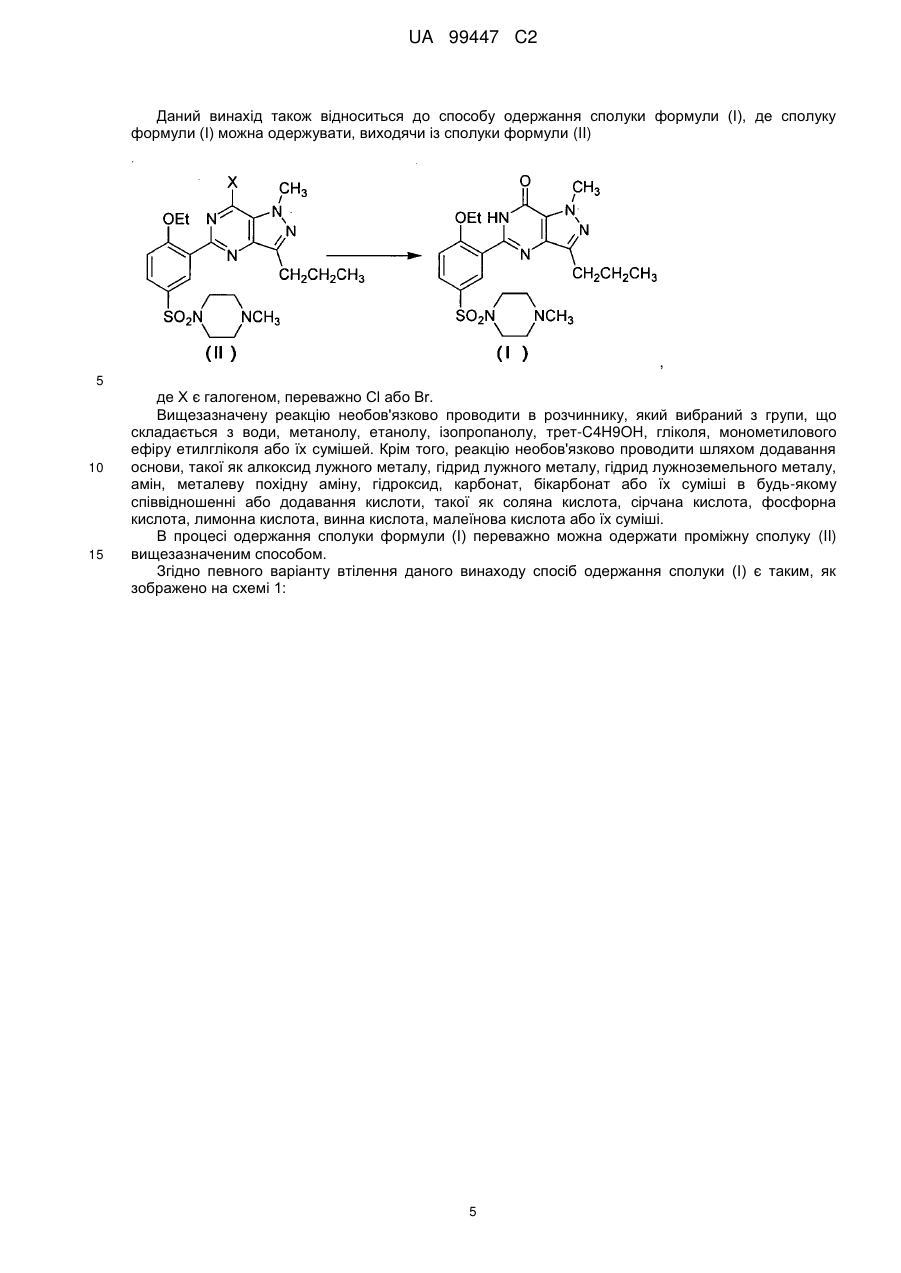
в якому здійснюють стадію перетворення сполуки, яка вибрана з групи, що складається із сполук формул (II), (III) і (IV):
де X є галогеном,
в ході однієї або більше стадій з утворенням сполуки формули (І).
2. Спосіб за п. 1, де X є Сl або Вr.
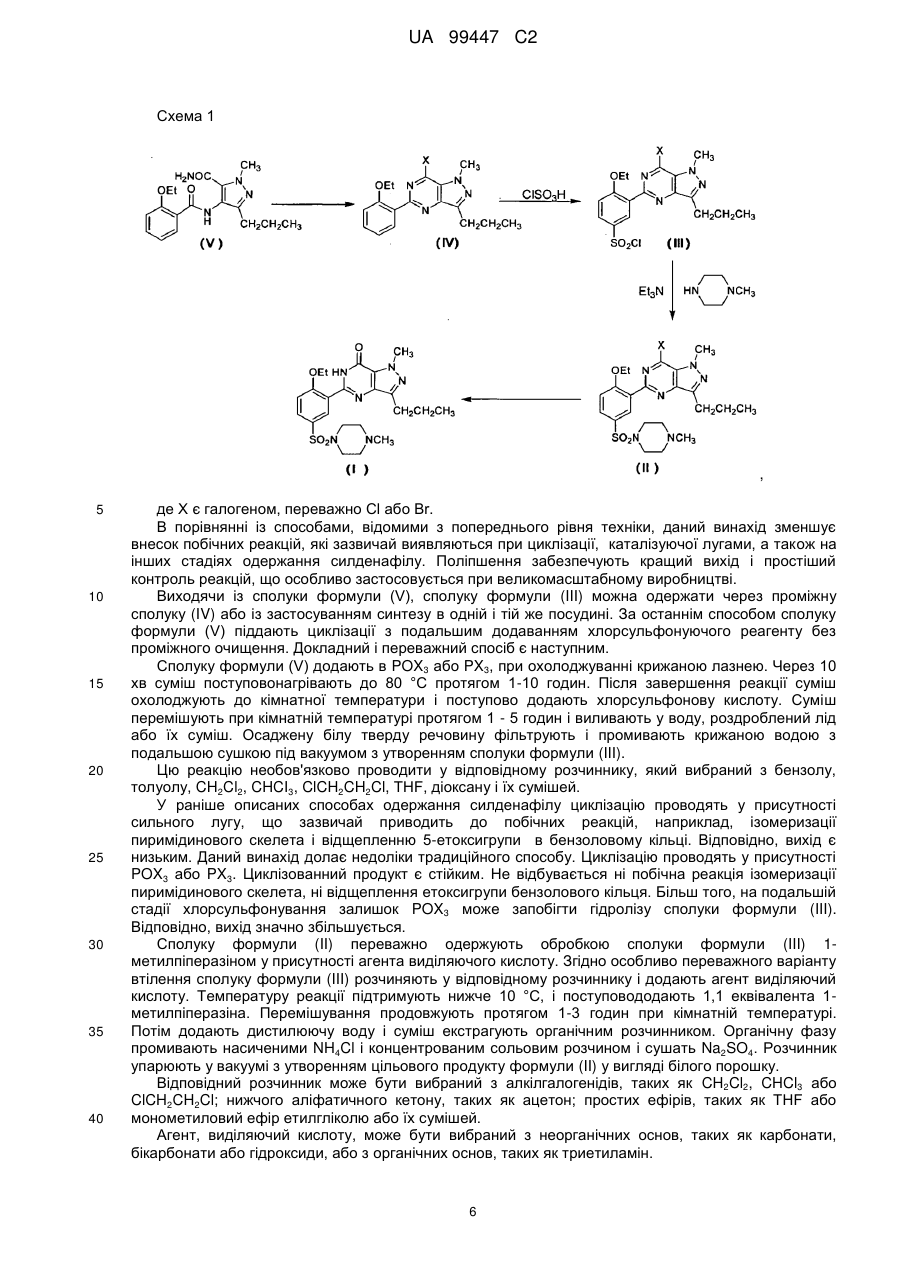
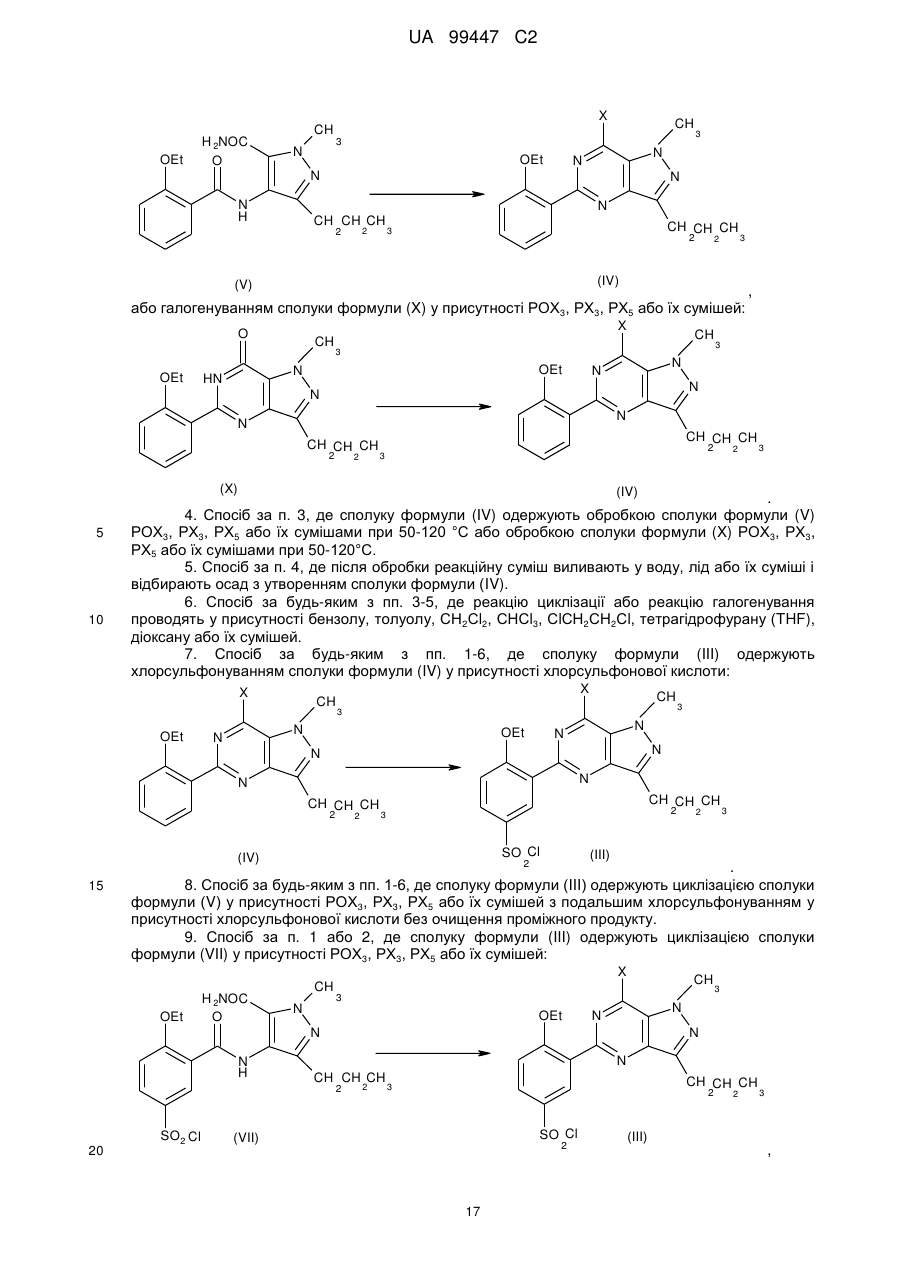
3. Спосіб за п. 1 або 2, де сполуку формули (IV) одержують циклізацією сполуки формули (V) у присутності РОХ3, РХ3, РХ5 або їх сумішей:
,
або галогенуванням сполуки формули (X) у присутності РОХ3, РХ3, РХ5 або їх сумішей:
.
4. Спосіб за п. 3, де сполуку формули (IV) одержують обробкою сполуки формули (V) РОХ3, РХ3, РХ5 або їх сумішами при 50-120 °С або обробкою сполуки формули (X) РОХ3, РХ3, РХ5 або їх сумішами при 50-120°С.
5. Спосіб за п. 4, де після обробки реакційну суміш виливають у воду, лід або їх суміші і відбирають осад з утворенням сполуки формули (IV).
6. Спосіб за будь-яким з пп. 3-5, де реакцію циклізації або реакцію галогенування проводять у присутності бензолу, толуолу, СН2Сl2, СНСl3, СlСН2СН2Сl, тетрагідрофурану (THF), діоксану або їх сумішей.
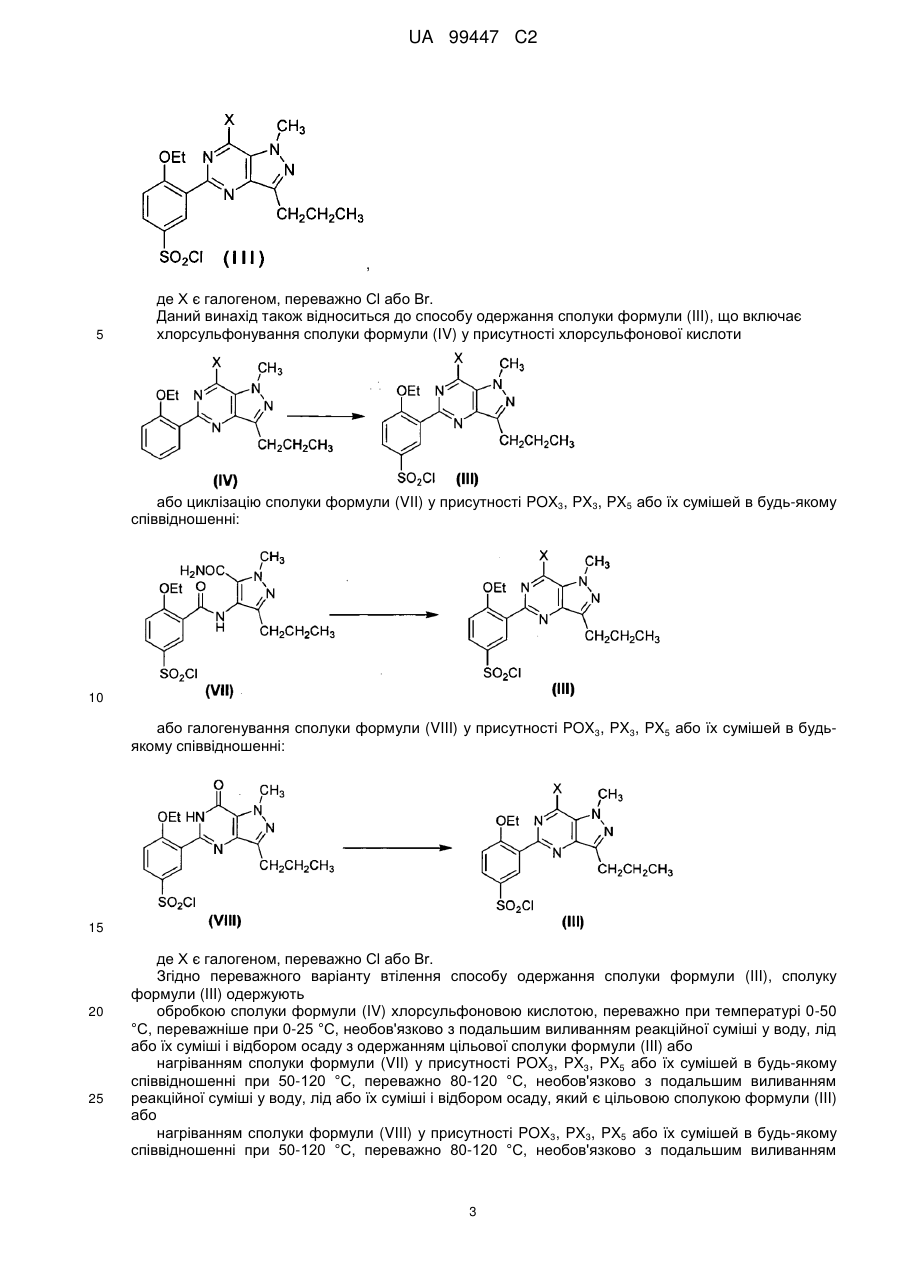
7. Спосіб за будь-яким з пп. 1-6, де сполуку формули (III) одержують хлорсульфонуванням сполуки формули (IV) у присутності хлорсульфонової кислоти:
.
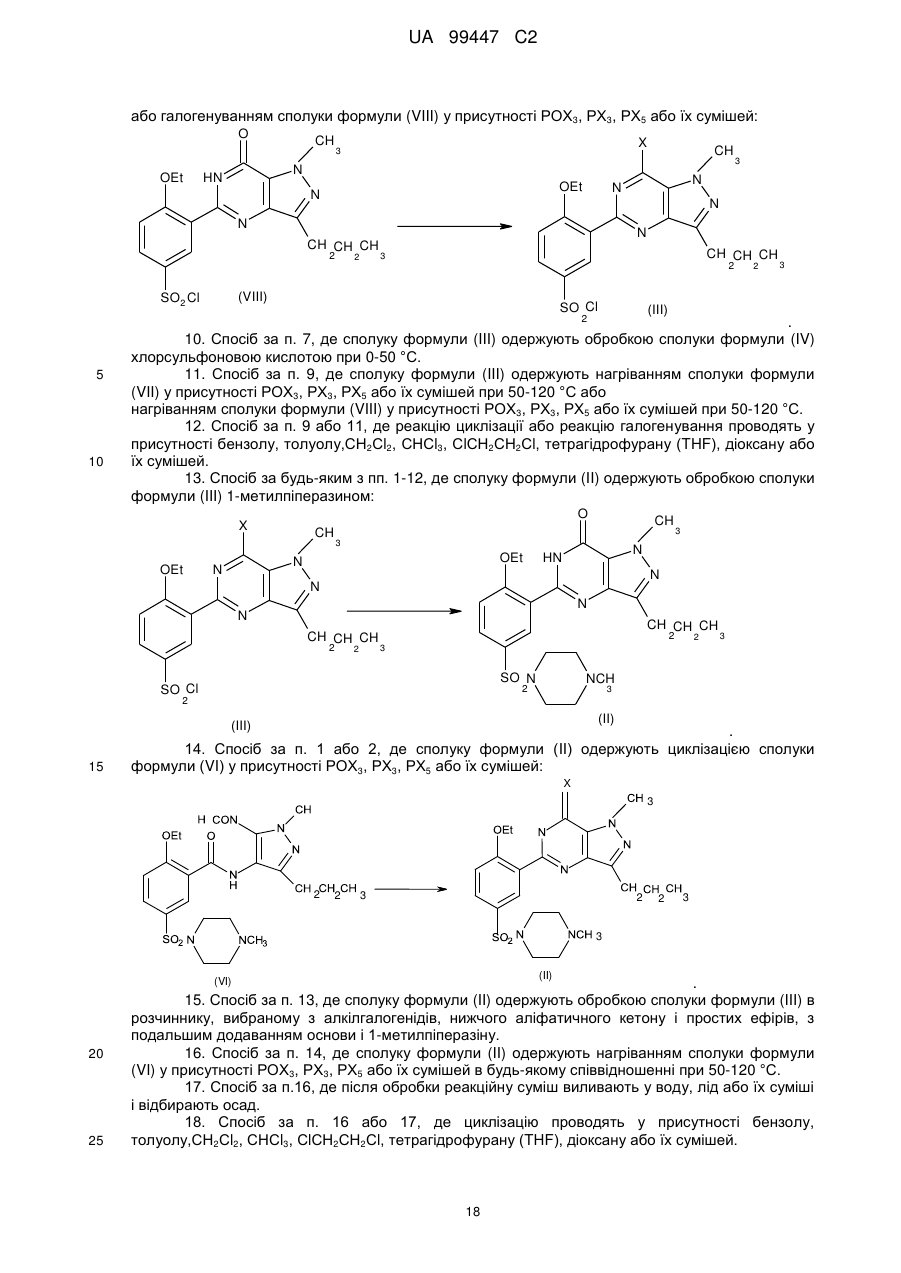
8. Спосіб за будь-яким з пп. 1-6, де сполуку формули (III) одержують циклізацією сполуки формули (V) у присутності РОХ3, РХ3, РХ5 або їх сумішей з подальшим хлорсульфонуванням у присутності хлорсульфонової кислоти без очищення проміжного продукту.
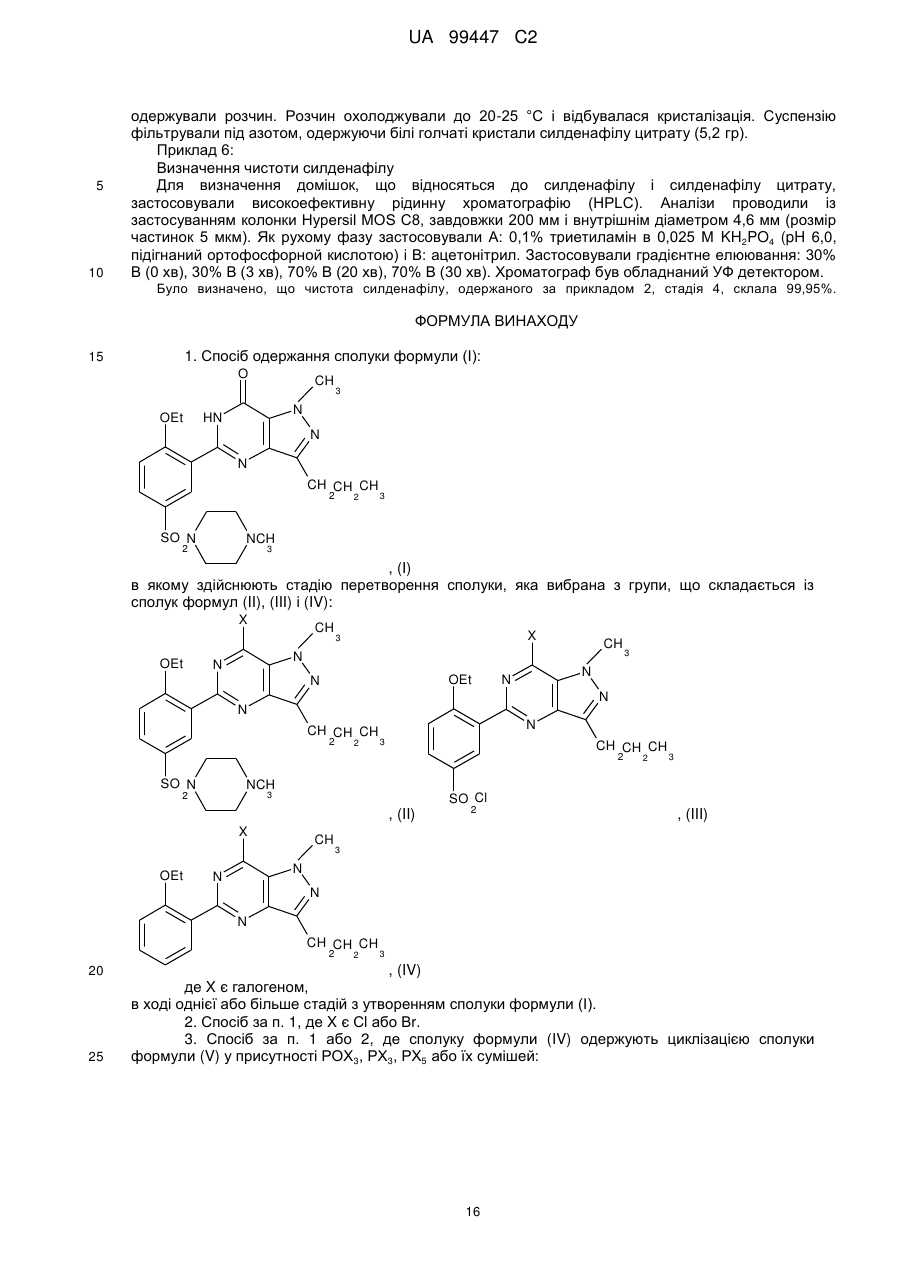
9. Спосіб за п. 1 або 2, де сполуку формули (III) одержують циклізацією сполуки формули (VII) у присутності РОХ3, РХ3, РХ5 або їх сумішей:
,
або галогенуванням сполуки формули (VIII) у присутності РОХ3, РХ3, РХ5 або їх сумішей:
.
10. Спосіб за п. 7, де сполуку формули (III) одержують обробкою сполуки формули (IV) хлорсульфоновою кислотою при 0-50 °С.
11. Спосіб за п. 9, де сполуку формули (III) одержують нагріванням сполуки формули (VII) у присутності РОХ3, РХ3, РХ5 або їх сумішей при 50-120 °С або
нагріванням сполуки формули (VIII) у присутності РОХ3, РХ3, РХ5 або їх сумішей при 50-120 °С.
12. Спосіб за п. 9 або 11, де реакцію циклізації або реакцію галогенування проводять у присутності бензолу, толуолу,СН2Сl2, СНСl3, СlСН2СН2Сl, тетрагідрофурану (THF), діоксану або їх сумішей.
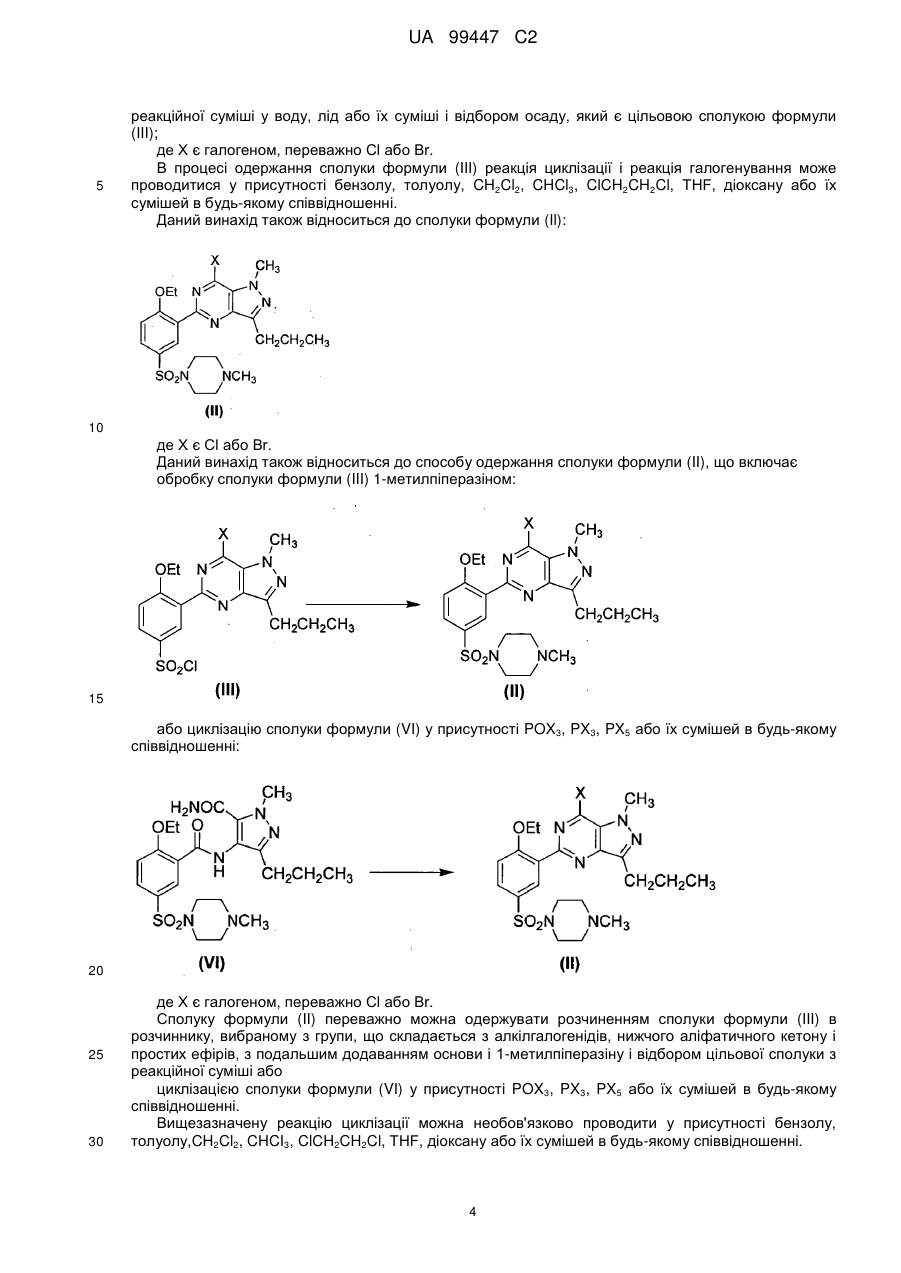
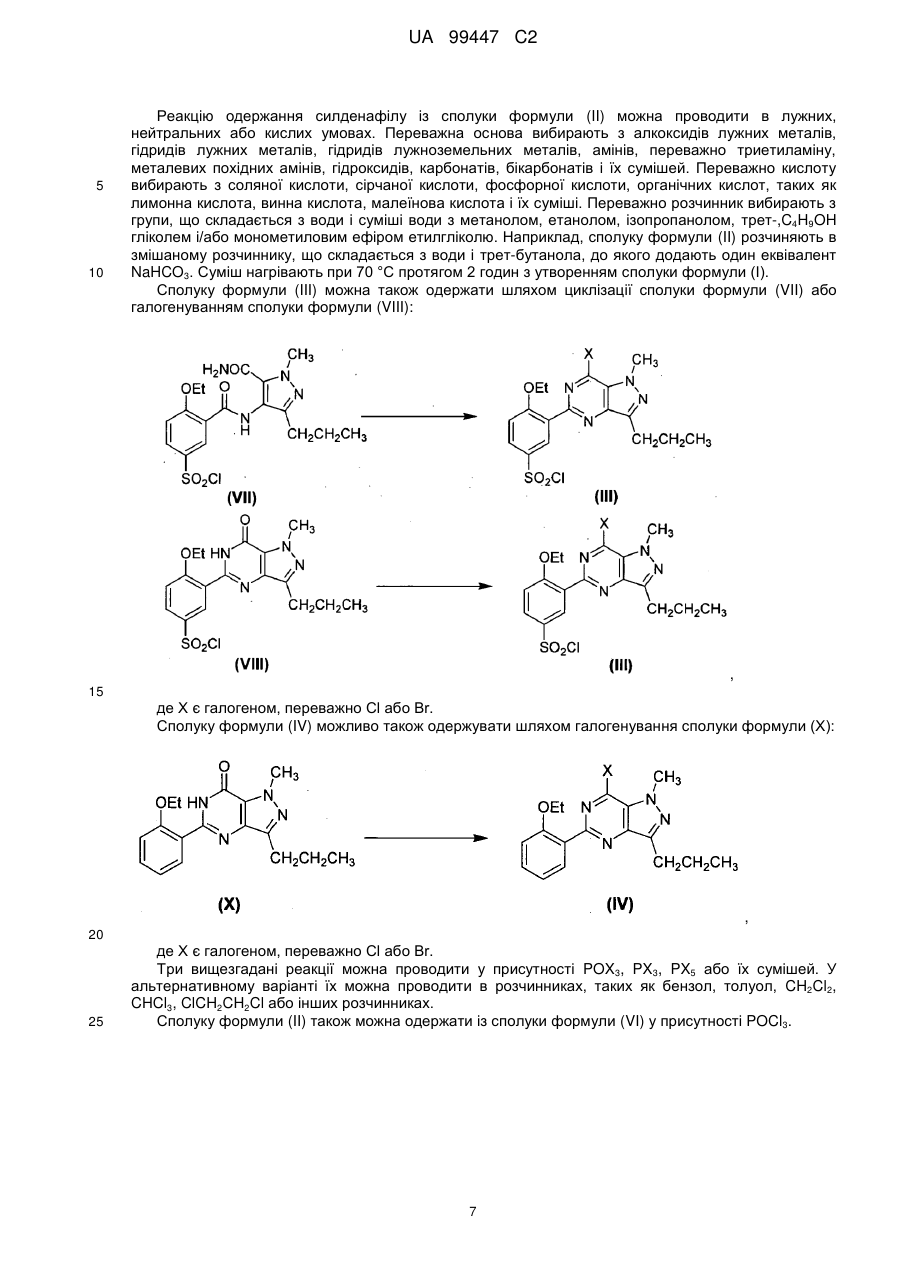
13. Спосіб за будь-яким з пп. 1-12, де сполуку формули (II) одержують обробкою сполуки формули (III) 1-метилпіперазином:
.
14. Спосіб за п. 1 або 2, де сполуку формули (II) одержують циклізацією сполуки формули (VI) у присутності РОХ3, РХ3, РХ5 або їх сумішей:
.
15. Спосіб за п. 13, де сполуку формули (II) одержують обробкою сполуки формули (III) в розчиннику, вибраному з алкілгалогенідів, нижчого аліфатичного кетону і простих ефірів, з подальшим додаванням основи і 1-метилпіперазіну.
16. Спосіб за п. 14, де сполуку формули (II) одержують нагріванням сполуки формули (VI) у присутності РОХ3, РХ3, РХ5 або їх сумішей в будь-якому співвідношенні при 50-120 °С.
17. Спосіб за п.16, де після обробки реакційну суміш виливають у воду, лід або їх суміші і відбирають осад.
18. Спосіб за п. 16 або 17, де циклізацію проводять у присутності бензолу, толуолу,СН2Сl2, СНСl3, СlСН2СН2Сl, тетрагідрофурану (THF), діоксану або їх сумішей.
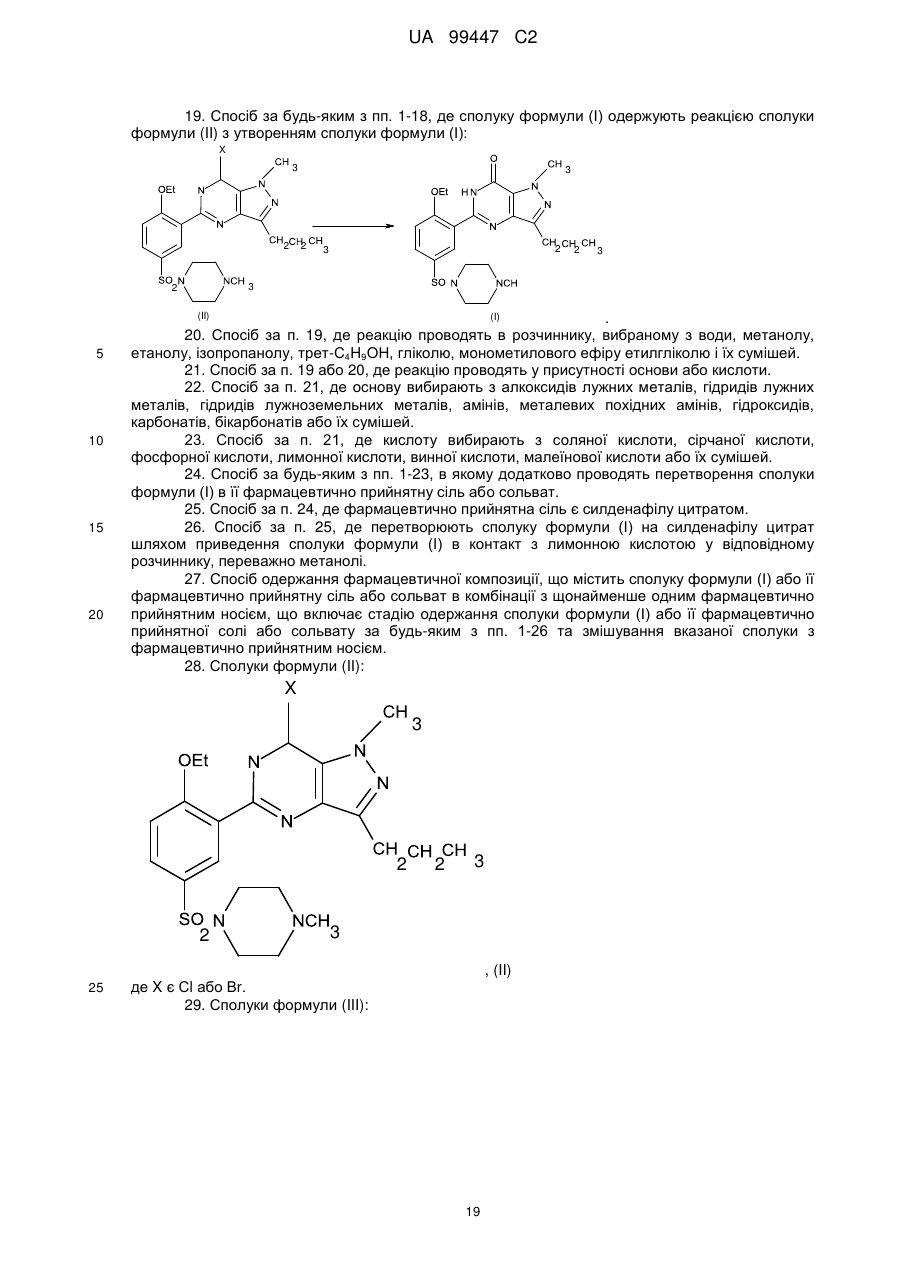
19. Спосіб за будь-яким з пп. 1-18, де сполуку формули (І) одержують реакцією сполуки формули (II) з утворенням сполуки формули (І):
.
20. Спосіб за п. 19, де реакцію проводять в розчиннику, вибраному з води, метанолу, етанолу, ізопропанолу, трет-С4H9ОН, гліколю, монометилового ефіру етилгліколю і їх сумішей.
21. Спосіб за п. 19 або 20, де реакцію проводять у присутності основи або кислоти.
22. Спосіб за п. 21, де основу вибирають з алкоксидів лужних металів, гідридів лужних металів, гідридів лужноземельних металів, амінів, металевих похідних амінів, гідроксидів, карбонатів, бікарбонатів або їх сумішей.
23. Спосіб за п. 21, де кислоту вибирають з соляної кислоти, сірчаної кислоти, фосфорної кислоти, лимонної кислоти, винної кислоти, малеїнової кислоти або їх сумішей.
24. Спосіб за будь-яким з пп. 1-23, в якому додатково проводять перетворення сполуки формули (І) в її фармацевтично прийнятну сіль або сольват.
25. Спосіб за п. 24, де фармацевтично прийнятна сіль є силденафілу цитратом.
26. Спосіб за п. 25, де перетворюють сполуку формули (І) на силденафілу цитрат шляхом приведення сполуки формули (І) в контакт з лимонною кислотою у відповідному розчиннику, переважно метанолі.
27. Спосіб одержання фармацевтичної композиції, що містить сполуку формули (І) або її фармацевтично прийнятну сіль або сольват в комбінації з щонайменше одним фармацевтично прийнятним носієм, що включає стадію одержання сполуки формули (І) або її фармацевтично прийнятної солі або сольвату за будь-яким з пп. 1-26 та змішування вказаної сполуки з фармацевтично прийнятним носієм.
28. Сполуки формули (II):
, (II)
де X є Сl або Вr.
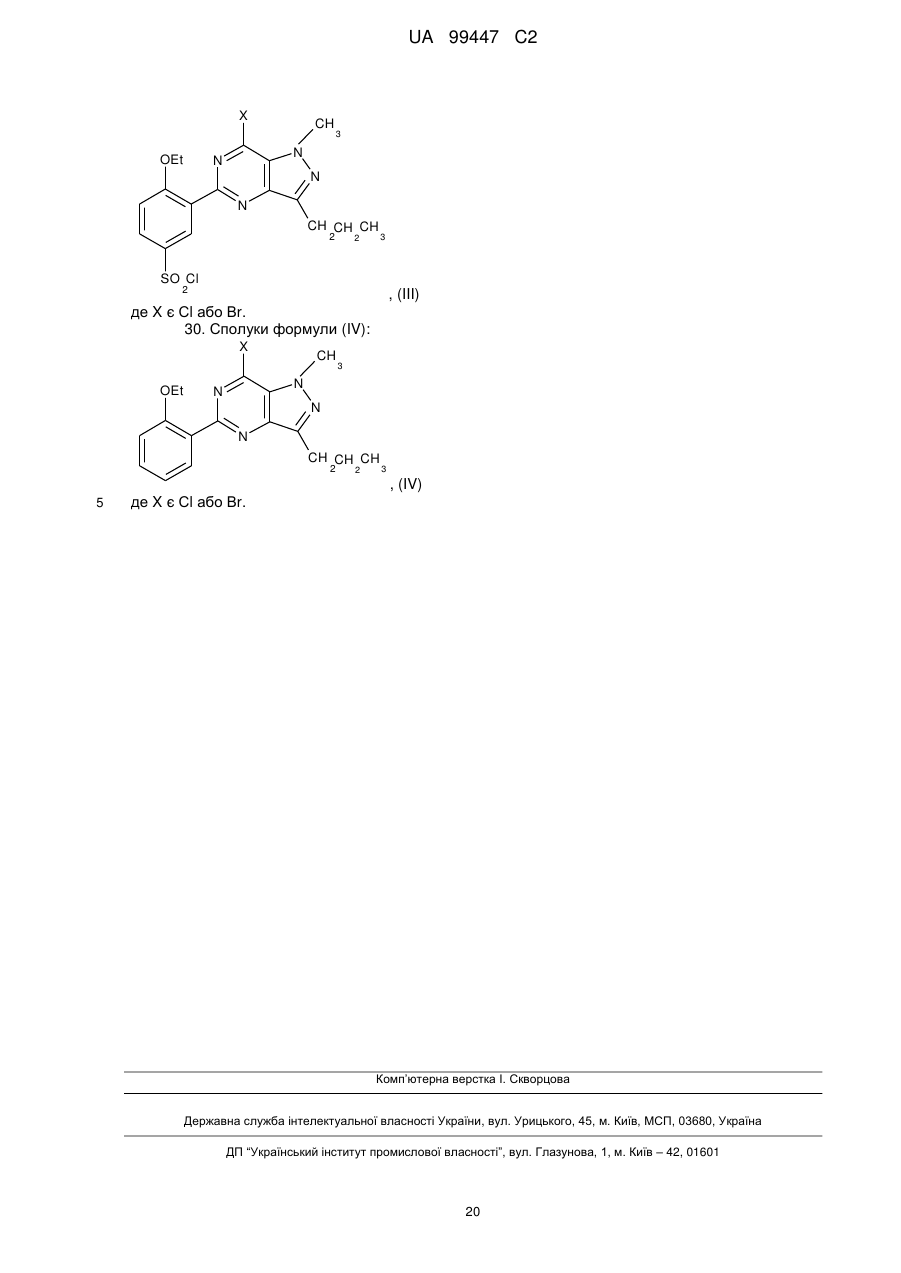
29. Сполуки формули (III):
, (III)
де X є Сl або Вr.
30. Сполуки формули (IV):
Текст
Реферат: Даний винахід стосується способу одержання сполуки формули (І), що включає стадію перетворення сполуки, яку вибрано з групи, що складається із сполук формул (II), (III) або (IV), де X є галогеном, в ході однієї або більше стадій з утворенням сполуки формули (І). Винахід також стосується способу одержання фармацевтичної композиції, що включає сполуку формули (І) або її фармацевтично прийнятну сіль або сольват. Більше того, винахід стосується проміжних продуктів, придатних для застосування у вищезазначених способах, а також способів їх одержання. UA 99447 C2 (12) UA 99447 C2 O OEt CH X N HN CH 3 OEt N N N N N N CH CH CH 2 SO N 2 CH CH CH SO N CH 3 , 3 N N X CH N OEt N CH CH CH 2 SO Cl 2 3 (II) , X 2 NCH 2 3 (I) OEt 2 3 NCH 2 3 2 3 N N N 3 N CH CH CH 2 (III) , 2 3 (IV) . UA 99447 C2 5 10 15 20 25 30 Даний винахід відноситься до способу одержання силденафілу і його фармацевтично прийнятних солей і сольватів. Винахід також направлений на спосіб одержання фармацевтичної композиції, що включає силденафіл або його фармацевтично прийнятну сіль або сольват. Більш того, винахід відноситься до проміжних сполук, придатних для застосування у вищезгаданих способах, а також до способів їх одержання. Сполука формули (I), що має хімічну назву 5-[2-етокси-5-(4-метилпіперазин-1-ілсульфоніл) феніл]-1-метил-3-нпропіл-1,6-дигідро-7Н-піразоло[4,3-альфа]піримідин-7-он, також відомо під родовою назвою силденафіл. Первинну сполуку застосовували для лікування серцево-судинних захворювань, таких як стенокардія, гіпертензія, серцева недостатність, атеросклероз і так далі. Пізніше було виявлено, що дану сполуку, зокрема, можна застосовувати для лікування еректильної дисфункції у чоловіків. Силденафіл є селективним інгібітором фосфодіестерази типу 5. Дана сполука і її одержання були спочатку описані в EP-A-O 463756 (відповідає CN1057464A), а також було виявлено, що сполука може застосовуватися при лікуванні деяких серцево-судинних захворювань. Її застосування при лікуванні еректильної дисфункції у чоловіків було вперше описане в WO-A94/28902 (відповідає CN1124926A). Покращуваний спосіб одержання силденафілу був описаний в EP-A-O 812845 (відповідає CN1168376A). CN1208337C описує спосіб одержання силденафілу за участю неорганічного окислювача. CN1176081C і CN1281851A описують два способи одержання силденафілу і його проміжних сполук. Bioorg. Med. Chem. Lett. 2000, 10, 1983-1986 описує конвергентний спосіб із застосуванням реагентів на полімерній підкладці в багатостадійній реакції, що приводить до чистого і ефективного препарату без необхідності застосування традиційних способів очищення. US-B-6 204383 описує спосіб, при якому використовується менше основної проміжної сполуки при одержані силденафілу. WO-A-2001/019827 описує дешевший спосіб одержання силденафілу метилуванням. Більшість способів одержання силденафілу в попередньому рівні техніки супроводжуються побічними реакціями. Тому вихід кінцевого продукту зменшується, що обмежує застосування способів попереднього рівня техніки у фармацевтичній промисловості. Даний винахід пропонує новий спосіб одержання силденафілу і його проміжних сполук, що має багато переваг в порівнянні із способами попереднього рівня техніки, таких як зменшення побічних реакцій і збільшення виходу продукту. Даний винахід відноситься до способу одержання сполуки формули (I): 35 1 UA 99447 C2 5 який включає стадію перетворення сполуки, вибраної з групи, що складається із сполук формул (II), (III) і (IV): де X є галогеном, переважно Cl або Br з утворенням сполуки формули (I) в ході однієї або більше стадій. Даний винахід також відноситься до сполуки формули (IV): , 10 де X є галогеном, переважно Cl або Br. Даний винахід також відноситься до способу одержання сполуки формули (IV), що включає циклізацію сполуки формули (V) у присутності POX3, PX3, PX5 або їх сумішей в будь-якому співвідношенні: , 15 або галогенування сполуки формули (X) у присутності POX3, PX3, PX5 або їх сумішей в будьякому співвідношенні: , 20 25 30 де X є галогеном, переважно Cl або Br. Згідно переважного варіанту втілення способу одержання сполуки формули (IV), сполуку формули (IV) обробкою сполуки формули (V) POX3, PX3, PX5 або їх сумішами в будь-якому співвідношенні при 50-120 °C, переважно при 80-120 °C, необов'язково з подальшим виливанням реакційної суміші у воду, лід або їх суміші і відбором осаду, який є цільовими сполуками формули (IV) або обробкою сполуки формули (X) POX3, PX3, PX5 або їх сумішами в будь-якому співвідношенні при 50-120 °C, переважно 80-120 °C, необов'язково з подальшим виливанням реакційної суміші у воду, лід або їх суміші і відбором осаду, який є цільовим сполукам формули (IV); де X є галогеном, переважно Cl або Br. Реакцію необов'язково проводити у присутності бензолу, толуолу, CH2Cl2, CHCl3, ClCH2CH2Cl, THF (тетрагідрофурану), діоксану або їх сумішей в будь-якому співвідношенні. Даний винахід також відноситься до сполуки формули (III): 2 UA 99447 C2 , 5 де X є галогеном, переважно Cl або Br. Даний винахід також відноситься до способу одержання сполуки формули (III), що включає хлорсульфонування сполуки формули (IV) у присутності хлорсульфонової кислоти або циклізацію сполуки формули (VII) у присутності POX3, PX3, PX5 або їх сумішей в будь-якому співвідношенні: 10 або галогенування сполуки формули (VIII) у присутності POX3, PX3, PX5 або їх сумішей в будьякому співвідношенні: 15 20 25 де X є галогеном, переважно Cl або Br. Згідно переважного варіанту втілення способу одержання сполуки формули (III), сполуку формули (III) одержують обробкою сполуки формули (IV) хлорсульфоновою кислотою, переважно при температурі 0-50 °C, переважніше при 0-25 °C, необов'язково з подальшим виливанням реакційної суміші у воду, лід або їх суміші і відбором осаду з одержанням цільової сполуки формули (III) або нагріванням сполуки формули (VII) у присутності POX3, PX3, PX5 або їх сумішей в будь-якому співвідношенні при 50-120 °C, переважно 80-120 °C, необов'язково з подальшим виливанням реакційної суміші у воду, лід або їх суміші і відбором осаду, який є цільовою сполукою формули (III) або нагріванням сполуки формули (VIII) у присутності POX3, PX3, PX5 або їх сумішей в будь-якому співвідношенні при 50-120 °C, переважно 80-120 °C, необов'язково з подальшим виливанням 3 UA 99447 C2 5 реакційної суміші у воду, лід або їх суміші і відбором осаду, який є цільовою сполукою формули (III); де X є галогеном, переважно Cl або Br. В процесі одержання сполуки формули (III) реакція циклізації і реакція галогенування може проводитися у присутності бензолу, толуолу, CH2Cl2, CHCl3, ClCH2CH2Cl, THF, діоксану або їх сумішей в будь-якому співвідношенні. Даний винахід також відноситься до сполуки формули (Il): 10 де X є Cl або Br. Даний винахід також відноситься до способу одержання сполуки формули (II), що включає обробку сполуки формули (III) 1-метилпіперазіном: 15 або циклізацію сполуки формули (VI) у присутності POX3, PX3, PX5 або їх сумішей в будь-якому співвідношенні: 20 25 30 де X є галогеном, переважно Cl або Br. Сполуку формули (II) переважно можна одержувати розчиненням сполуки формули (III) в розчиннику, вибраному з групи, що складається з алкілгалогенідів, нижчого аліфатичного кетону і простих ефірів, з подальшим додаванням основи і 1-метилпіперазіну і відбором цільової сполуки з реакційної суміші або циклізацією сполуки формули (VI) у присутності POX3, PX3, PX5 або їх сумішей в будь-якому співвідношенні. Вищезазначену реакцію циклізації можна необов'язково проводити у присутності бензолу, толуолу,CH2Cl2, CHCI3, ClCH2CH2Cl, THF, діоксану або їх сумішей в будь-якому співвідношенні. 4 UA 99447 C2 Даний винахід також відноситься до способу одержання сполуки формули (I), де сполуку формули (I) можна одержувати, виходячи із сполуки формули (II) , 5 10 15 де X є галогеном, переважно Cl або Br. Вищезазначену реакцію необов'язково проводити в розчиннику, який вибраний з групи, що складається з води, метанолу, етанолу, ізопропанолу, трет-C4H9OH, гліколя, монометилового ефіру етилгліколя або їх сумішей. Крім того, реакцію необов'язково проводити шляхом додавання основи, такої як алкоксид лужного металу, гідрид лужного металу, гідрид лужноземельного металу, амін, металеву похідну аміну, гідроксид, карбонат, бікарбонат або їх суміші в будь-якому співвідношенні або додавання кислоти, такої як соляна кислота, сірчана кислота, фосфорна кислота, лимонна кислота, винна кислота, малеїнова кислота або їх суміші. В процесі одержання сполуки формули (I) переважно можна одержати проміжну сполуку (II) вищезазначеним способом. Згідно певного варіанту втілення даного винаходу спосіб одержання сполуки (I) є таким, як зображено на схемі 1: 5 UA 99447 C2 Схема 1 , 5 10 15 20 25 30 35 40 де X є галогеном, переважно Cl або Br. В порівнянні із способами, відомими з попереднього рівня техніки, даний винахід зменшує внесок побічних реакцій, які зазвичай виявляються при циклізації, каталізуючої лугами, а також на інших стадіях одержання силденафілу. Поліпшення забезпечують кращий вихід і простіший контроль реакцій, що особливо застосовується при великомасштабному виробництві. Виходячи із сполуки формули (V), сполуку формули (III) можна одержати через проміжну сполуку (IV) або із застосуванням синтезу в одній і тій же посудині. За останнім способом сполуку формули (V) піддають циклізації з подальшим додаванням хлорсульфонуючого реагенту без проміжного очищення. Докладний і переважний спосіб є наступним. Сполуку формули (V) додають в POX3 або PX3, при охолоджуванні крижаною лазнею. Через 10 хв суміш поступовонагрівають до 80 °C протягом 1-10 годин. Після завершення реакції суміш охолоджують до кімнатної температури і поступово додають хлорсульфонову кислоту. Суміш перемішують при кімнатній температурі протягом 1 - 5 годин і виливають у воду, роздроблений лід або їх суміш. Осаджену білу тверду речовину фільтрують і промивають крижаною водою з подальшою сушкою під вакуумом з утворенням сполуки формули (III). Цю реакцію необов'язково проводити у відповідному розчиннику, який вибраний з бензолу, толуолу, CH2Cl2, CHCI3, ClCH2CH2Cl, THF, діоксану і їх сумішей. У раніше описаних способах одержання силденафілу циклізацію проводять у присутності сильного лугу, що зазвичай приводить до побічних реакцій, наприклад, ізомеризації пиримідинового скелета і відщепленню 5-етоксигрупи в бензоловому кільці. Відповідно, вихід є низьким. Даний винахід долає недоліки традиційного способу. Циклізацію проводять у присутності POX3 або PX3. Циклізованний продукт є стійким. Не відбувається ні побічна реакція ізомеризації пиримідинового скелета, ні відщеплення етоксигрупи бензолового кільця. Більш того, на подальшій стадії хлорсульфонування залишок POX3 може запобігти гідролізу сполуки формули (III). Відповідно, вихід значно збільшується. Сполуку формули (II) переважно одержують обробкою сполуки формули (III) 1метилпіперазіном у присутності агента виділяючого кислоту. Згідно особливо переважного варіанту втілення сполуку формули (III) розчиняють у відповідному розчиннику і додають агент виділяючий кислоту. Температуру реакції підтримують нижче 10 °C, і поступовододають 1,1 еквівалента 1метилпіперазіна. Перемішування продовжують протягом 1-3 годин при кімнатній температурі. Потім додають дистилюючу воду і суміш екстрагують органічним розчинником. Органічну фазу промивають насиченими NH4Cl і концентрованим сольовим розчином і сушать Na2SO4. Розчинник упарюють у вакуумі з утворенням цільового продукту формули (II) у вигляді білого порошку. Відповідний розчинник може бути вибраний з алкілгалогенідів, таких як CH2Cl2, CHCl3 або ClCH2CH2Cl; нижчого аліфатичного кетону, таких як ацетон; простих ефірів, таких як THF або монометиловий ефір етилгліколю або їх сумішей. Агент, виділяючий кислоту, може бути вибраний з неорганічних основ, таких як карбонати, бікарбонати або гідроксиди, або з органічних основ, таких як триетиламін. 6 UA 99447 C2 5 10 Реакцію одержання силденафілу із сполуки формули (II) можна проводити в лужних, нейтральних або кислих умовах. Переважна основа вибирають з алкоксидів лужних металів, гідридів лужних металів, гідридів лужноземельних металів, амінів, переважно триетиламіну, металевих похідних амінів, гідроксидів, карбонатів, бікарбонатів і їх сумішей. Переважно кислоту вибирають з соляної кислоти, сірчаної кислоти, фосфорної кислоти, органічних кислот, таких як лимонна кислота, винна кислота, малеїнова кислота і їх суміші. Переважно розчинник вибирають з групи, що складається з води і суміші води з метанолом, етанолом, ізопропанолом, трет-,C4H9OH гліколем і/або монометиловим ефіром етилгліколю. Наприклад, сполуку формули (II) розчиняють в змішаному розчиннику, що складається з води і трет-бутанола, до якого додають один еквівалент NaHCO3. Суміш нагрівають при 70 °C протягом 2 годин з утворенням сполуки формули (I). Сполуку формули (III) можна також одержати шляхом циклізації сполуки формули (VII) або галогенуванням сполуки формули (VIII): , 15 де X є галогеном, переважно Cl або Br. Сполуку формули (IV) можливо також одержувати шляхом галогенування сполуки формули (X): , 20 25 де X є галогеном, переважно Cl або Br. Три вищезгадані реакції можна проводити у присутності POX3, PX3, PX5 або їх сумішей. У альтернативному варіанті їх можна проводити в розчинниках, таких як бензол, толуол, CH2Cl2, CHCl3, ClCH2CH2Cl або інших розчинниках. Сполуку формули (II) також можна одержати із сполуки формули (VI) у присутності POCl3. 7 UA 99447 C2 , 5 де X є Cl. Інформацію про одержання сполук формул (VIII) і (X) можна також знайти в EP-A-O 463756 (відповідає CN1028758C). Інформацію про одержання сполуки формули (VI), можна також знайти в EP-A-O 812845 (відповідає CN1106399C). Проміжну сполуку (V) за даним винаходом можна одержати за допомогою традиційних способів, відомих в літературі. Наприклад, сполуку можна одержати обробкою 2-етоксибензоілхлориду 1-метил-3-н-пропіл-4-амінопіразол-5-карбоксамідом (XI): 10 15 20 25 30 35 40 Сполуку формули (VII) можна одержати із сполуки формули (V) традиційними способами, відомими в літературі. Сполуки, одержані способом за даним винаходом, можуть знаходитися в різних кристалічних формах. Кристалічна форма сполуки формули (I), тобто вільна основа силденафілу, описується в Melnikov et al., Journal of Pharmaceutical Sciences, 2003, 92, 2140- 2143. Спосіб за даним винаходом може додатково включати перетворення сполуки формули (I) в його фармацевтично прийнятну сіль або сольват. Переважно прийнятною сіллю сполуки формули (I) є силденафілу цитрат. Способи одержання фармацевтично прийнятних солей або сольватів сполуки формули (I) в цілому відомі фахівцям в даній області техніки. Дані способи, а також різні солі силденафілу і їх поліморфні форми в основному описані в Badwan et al., Analytical Profiles of Drug Substances and Excipients, Vol. 27, Academic Press, 2001; Admour et al., "Solid State Modification and Transformation in Sildenafil and Terfenadine Salts", MSc. Thesis, Jordan University of Science and Technology, Irbid, Jordan, January 1999; 46th APV Congress, Berlin, 3 to 6 April 2000, pages 639-640; WO-A2004/072079; WO-A-2005/067936; WO-A-2007/080362; WO-A-2007/110559; і Journal of Shenyang Pharmaceutical University, 2002, 19, 173-175. Згідно переважного варіанту втілення сполуку формули (I) перетворюють на силденафілу цитрат шляхом приведення сполуки формули (I) в контакт з лимонною кислотою у відповідному розчиннику, переважно метанолі. Особливо переважний спосіб одержання силденафілу цитрату включає стадії: (1) суспендування вільної основи силденафілу в метанолі і нагрівання суспензії до 45-65 °C; (2) додавання лимонної кислоти, переважно безводної лимонної кислоти і метанолу до суспензії, одержаної на стадії (1), переважно при температурі 50-60 °C і додатково нагріву суміші до кипіння з утворенням розчину; (3) концентрації розчину з метою зменшення об'єму суміші, переважно менш ніж на 50 %, переважніше менш ніж на 35 % її первинного об'єму з метою одержання суспензії; (4) охолоджування суспензії до кімнатної температури і (5) витягання одержаних кристалів, переважно фільтруванням. 8 UA 99447 C2 5 10 15 20 25 30 35 40 45 50 55 Винахід також відноситься до способу одержання фармацевтичної композиції, що містить сполуку формули (I) або його фармацевтично прийнятну сіль або його сольват в комбінації з щонайменше одним фармацевтично прийнятним носієм, який включає стадію одержання сполуки формули (I) або його фармацевтично прийнятної солі або його сольвату згідно способів даного винаходу, як описано вище. Фармацевтичні препарати силденофілу і силденафілу цитрату можна одержати способами, які добре відомі фахівцям в даній області техніки. Склади, що застосовуються, описані наприклад, в EP-A-0 941075, EP-A-0 960621, JP-A-10298062, WO-A-2004/017976, WO-A-2004/072079 і IPCOM000146068D. Далі винахід пояснюється в приведених нижче прикладах без обмеження винаходу вказаними прикладами. Приклади Температури плавлення визначали у відкритих капілярах в апараті BUCHI-510 для визначення температури плавлення і були невідрегульовані. EI-MS (мас-спектрометрія електронного удару) спектри вимірювали на спектрометрі Finnigan MAT-95 при 70 еВ і температурі іонного джерела 200; 1H ЯМР спектри визначали в розчині CDCI3 на спектрометрі Varian Mercury 300. Всі результати по спектрах знаходилися в хорошій відповідності з очікуваними результатами. Для позначення основних піків застосовували традиційні скорочення: наприклад, s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет. В рамках даних прикладів кімнатна температура означає 20-25 °C. Приклад 1 Приклад одержання 1: 1-метил-3-н-пропіл-4-(2-етоксибензамідо)-піразол-5-карбоксамід (V) При охолоджуванні на крижаній лазні, в трехгорловій колбі місткістю 250 мл сполука (XI) (20 гр, 0,11 моль) розчиняли в дихлорметані (100 мл) і триетиламіні (22,2 гр, 0,22 моль) з одержанням розчину. До розчину при температурі нижче 5 °С додавали 2-етоксибензоіл хлорид і суміш перемішували при кімнатній температурі протягом 2 годин. Реакцію зупиняли додаванням води (40 мл) і розділяли шари. Органічну фазу промивали концентрованим сольовим розчином (30 мл) і насиченим водним розчином гідрокарбонату натрію, сушили над безводним сульфатом натрію і потім концентрували. Залишок, що утворився, очищали перекристалізацією з суміші етилацетат/петролейний ефір, одержуючи сполуку (V) (31,5 гр, вихід 87%) у вигляді білої твердої 1H речовини, температура плавлення (Т. пл.) 153-154 °C. ЯМР (CDCl3, 300 Мгц) д: 0.93 (3H, t), 1.54 (3H, t), 1.65 (2H, m), 2.54 (2H, t), 4.06 (3H, s), 4.31 (2H, q), 5.62 (IH, br s), 7.05 (1H, d), 7.13 (1H, t), 7.54 (1H, t), 7.91 (1H, br s), 8.27 (1H, dd), 9.47 (1H, s). Приклад одержання 1a: 1-метил-3-н-пропіл-4-(2-етоксибензамідо)-піразол-5-карбоксамід (V) При охолоджуванні на крижаній лазні, в трехгорловій колбі місткістю 250 мл сполуку (XI) (21,8 гр, 0,10 моль) розчиняли в етилацетаті (200 мл) і триетиламіні (32 мл, 0,23 моль) з одержанням о розчину. До розчину при температурі нижче 5 С додавали 2-етоксибензоіл хлорид (17 мл, 0,11 моль) і суміш перемішували при кімнатній температурі протягом 2 годин. Реакцію зупиняли додаванням води (170 мл) і петролейного ефіру (100 мл) і суміш перемішували ще 0,5 год. Фільтрували тверду речовину і поміщали у воду (170 мл), перемішували протягом 0,5 год., оС фільтрували і сушили (70 , 12 ч) з утворенням сполуки (V) у вигляді білої твердої речовини (32,0 гр, вихід 95%) . Т. пл. 153-154 °C. Залишок, що утворився, очищали перекристалізацією з суміші 1H етилацетат/петролейний ефір. ЯМР (CDCl3, 300 Мгц) д: 0.93 (3H, t), 1.54 (3H, t), 1.65 (2H, m), 2.54 (2H, t), 4.06 (3H, s), 4.31 (2H, q), 5.62 (IH, br s), 7.05 (1H, d), 7.13 (1H, t), 7.54 (1H, t), 7.91 (1H, br s), 8.27 (1H, dd), 9.47 (1H, s) Приклад одержання 2: 4-[2-етокси-5-(хлорсульфоніл)бензамідо]-1-метил-3-н-пропіл-піразол-5-карбоксамід (VII) При охолоджуванні на крижаній лазні сполуку формули (V) (5 гр, 0,015 моль) порціями додавали до хлорсульфоновой кислоти (10 мл). Суміш перемішували протягом 30 хв і потім прибирали крижану лазню. Реакція продовжувалася ще 2 години. Залишок поміщали в крижану воду. Тверду речовину, що утворилася, фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі з одержанням (VII) (3,5 гр) у вигляді білої твердої речовини з 1H виходом 54%. ЯМР (CDCl3, 300 Мгц) д: 0.94 (3H, t), 1.62 (3H, t), 1.66 (2H, m), 2.53 (2H, t), 4.06 (3H, s), 4.46 (2H, q), 5.72 (1H, s), 7.25 (1H, t), 7.62 (1H, s), 8.18 (1H, dd), 8.95 (1H, d), 9.20 (1H, s). Приклад одержання 3: 7-хлор-1-метил-5-(2-етоксифеніл)-3-н-пропіл-1H-піразоло[4,3-альфа]піримідин (IV) (X=Cl) Спосіб 1 9 UA 99447 C2 5 10 15 20 25 30 35 40 45 50 55 При охолоджуванні на крижаній лазні в трехгорлову колбу місткістю 50 мл додавали сполуку (V) (5 гр 0,015 моль). Для одержання розчину в колбу по краплях додавали POCl3 (20 мл). Розчин нагрівали при 80 °C протягом 2 годин, щоб зупинити реакцію суміш виливали в крижану воду і потім суміш екстрагували дихлорметаном (3 x 30 мл). Органічну фазу промивали концентрованим сольовим розчином (2 x 10 мл) і сушили над безводим сульфатом натрію (2 гр ), з подальшою 1 концентрацією у вакуумі з одержанням сполуки (IV) (X=Cl, 4,1 гр, вихід 82%). H ЯМР (CDCl3, 300 Мгц) д: 1.03 (3H, t), 1.38 (3H, t), 1.90 (2H, m), 3.10 (2H, t), 4.15 (2H, q), 4.38 (3H, s), 7.03 (1H, d), 7.07 (1H, t), 7.42 (1H, t), 7.79 (1H, dd) . EI-MS m/z 330 (M+, 76), 332(25), 315(52), 317 (15), 294(100), 296(32), 279(60), 261(28), 159(20). Спосіб 2 При охолоджуванні на водяній лазні в трехгорлову колбу місткістю 50 мл додавали сполуку (X) (5 гр, 0,015 моль). Для одержання розчину в колбу по краплях додавали POCl3 (20 мл). Розчин нагрівали при 80 °C протягом 2 годин, для зупинки реакції суміш виливали в крижану воду і потім суміш екстрагували дихлорметаном (3 x 30 мл). Органічну фазу промивали концентрованим сольовим розчином (2 x 10 мл) і сушили над безводним сульфатом натрію (2 гр), далі концентрували у вакуумі, одержуючи сполуку (IV) (X=Cl, 4,4 гр, вихід 83%) у вигляді білої твердої речовини. Спосіб 3 Сполуку (V) (5 гр, 0,015 моль) розчиняли в бензолі (20 мл). При охолоджуванні на крижаній лазні в розчин по краплях додавали розчин POCI3 (2,8 мл) в бензолі і потім прибирали крижану лазню. Реакційну суміш нагрівали при 80 °C протягом 3 годин і потім бензол видаляли перегонкою при зниженому тиску. Залишок виливали в крижану воду (20 мл) і розчин екстрагували дихлорметаном (3 x 30 мл). Органічну фазу промивали концентрованим сольовим розчином (2 x 10 мл), сушили над безводним сульфатом натрію (2 гр ), далі концентрували у вакуумі, одержуючи сполуку (IV) (X=Cl, 3,6 гр, вихід 72%) у вигляді білої твердої речовини. Приклад одержання 4: 7-хлор-1-метил-5-[2-етокси-5-(хлорсульфоніл)феніл]-3-н-пропіл-1Н-піразоло[4, 3альфа]піримідин (III) (X=Cl) Спосіб 1 При охолоджуванні на крижаній лазні в трехгорлову колбу місткістю 50 мл додавали сполуку (V) (5 гр, 0,015 моль) і потім додавали POCI3 (10 мл). Реакційну суміш нагрівали при 80 °C протягом 2 годин і розчин, що утворився, охолоджували до температури нижче 5 °C. Через 30 хв до реакційної суміші додавали хлорсульфонову кислоту (10 мл), прибирали крижану лазню і далі реакційну суміш при кімнатній температурі перемішували протягом 2 годин. Суміш виливали в крижану воду (20 мл). Утворену тверду речовину фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі протягом 3 годин, одержуючи сполуку (III) (X=Cl, 5,7 гр ) з 1H виходом 88%, Т. пл. 155-156 °C. ЯМР (CDCl3, 300 Мгц) д: 1.04 (3H, t), 1.43 (3H, t), 1.89 (2H, m), 3.09 (2H, t), 4.27 (2H, q), 4.42 (3H, s), 7.19 (1H, d), 8.10 (1H, dd), 8.46 (1H, d) . EI-MS m/z 430(31), 428 (M+, 41), 413(45), 415(34), 393(20), 357(24), 328(25), 292(100). Спосіб 2 При охолоджуванні на крижаній лазні в колбу місткістю 50 мл додавали сполуку (IV) (X=Cl, 3 гр) і потім для одержання розчину хлорсульфонову кислоту (6 мл). Через 30 хв прибирали крижану лазню. Далі реакційну суміш перемішували при кімнатній температурі протягом 2 годин і потім суміш виливали в крижану воду (15 мл). Утворену тверду речовину фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі при 35 °С протягом 3 годин, одержуючи сполуку (III) (X=Cl, 2,8 гр, вихід 72%). Спосіб 3 У колбу місткістю 50 мл додавали сполуку (VII) (5 гр, 0,012 моль) і потім для одержання розчину поступово додавали POCl3 (10 мл). Реакційну суміш нагрівали при 80 °C протягом 2 годин і потім виливали в крижану воду (20 мл), утворену тверду речовину фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі, одержуючи сполуку (III) (X=Cl, 3,2 гр, вихід 64%). Спосіб 4 У колбу місткістю 50 мл додавали сполуку (VIII) (5 гр, 0,012 моль) і потім для одержання розчину поступово додавали POCl3 (10 мл), реакційну суміш нагрівали при 80 °C протягом 2 годин і потім виливали в крижану воду (20 мл). Утворену тверду речовину фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі, одержуючи сполуку (III) (X=Cl, 4,8, вихід 92%) у вигляді білої твердої речовини. Спосіб 5 10 UA 99447 C2 5 10 15 20 25 30 35 40 45 50 55 У колбу місткістю 50 мл додавали сполуку (VIII) (5 гр, 0,012 моль) і потім для одержання розчину поступово додавали POCl3 (10 мл), реакційну суміш нагрівали при 70 °C протягом 2 годин і потім виливали в крижану воду (20 мл). Утворену тверду речовину фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі, одержуючи сполуку (III) (X=Cl, 4,8 гр, вихід 92%) у вигляді білої твердої речовини. Спосіб 6 Сполука (VIII) (5 гр, 0,012 моль) розчиняли в бензолі (40 мл). При охолоджуванні на крижаній лазні в розчин додавали PCl5. Через 30 хв крижану лазню прибирали, реакційну суміш нагрівали при 90 °C протягом 2 годин і потім виливали в крижану воду (20 мл). Утворену тверду речовину фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі, одержуючи сполуку (III) (X=Cl, 3,5 гр, вихід 67%) у вигляді білої твердої речовини. Приклад одержання 5: 7-хлор-1-метил-5-[2-етокси-5-(4-метилпіперазінілсульфоніл)феніл]-3-н-пропіл-1Н-піразоло[4,3альфа]піримідин (II) (X=Cl) Спосіб 1 Сполуку (III) (X=Cl, 2 гр ) розчиняли в дихлорметані (20 мл) і триетиламіні (0,94 гр, 9,3 моль) для одержання розчину. Потім, при охолоджуванні на крижаній лазні по краплях додавали розчин 1-метипіперазину (0,51 гр, 5,1 моль) в дихлорметані (5 мл). Крижану лазню прибирали і суміш перемішували при кімнатній температурі протягом 2 годин. Додавали воду і розділяли шари. Органічну фазу промивали насиченим водним розчином хлориду амонія (2 х 5 мл) і концентрованим сольовим розчином (2 х 5 мл) і сушили над безводним Na2SO4 (1 гр ) протягом 30 хв. Розчинник видаляли шляхом перегонки за зниженого тиску з утворенням неочищеної сполуки (II) у вигляді білої твердої речовини. Неочищений продукт очищали перекристалізацією з суміші дихлорметан/петролейний ефір з одержанням сполуки (II) (X=Cl, 1,95 гр, вихід 85 %) у вигляді 0 1 білих голчатих кристалів, Т. пл 157 - 159 C. H ЯМР (CDCl3, 300 МГц) δ: 1.03 (3H, t), 1.41 (3H, t), 1.90 (2H, m), 2.41 (3H, s), 2.69 (4H, s), 3.04 (2H, t), 3.23 (4H, s), 4.19 (2H, q), 4.37 (3H, s), 7.10 (1H, d), + 7.78 (1H, dd), 8.17 (1H, s) EI-MS m/z 494 (2), 492 (M , 6), 424 (2), 422(7), 330(7), 99(100). Спосіб 2 У колбу місткістю 50 мл додавали сполуку (VI) (2 гр, 0,004 моль). Для одержання розчину поступово додавали POCl3 (10 мл). Реакційну суміш нагрівали при 80 °C протягом 2 годин і потім виливали в крижану воду (8 мл). Білу тверду речовину, що утворилася, фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі з утворенням сполуки (II) (X=Cl, 1,5 гр, вихід 75%) у вигляді білої твердої речовини. Спосіб 3 Сполуку (VI) (2 гр, 4 моль) розчиняли в бензолі (10 мл). До розчину поступово додавали розчин POCI3 (0,65 мл) в бензолі (2 мл), реакційну суміш нагрівали при 80 °C протягом 2 годин і потім виливали в крижану воду (8 мл). Білу тверду речовину, що утворилася, фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили під вакуумом з одержанням сполуки (II) (X=Cl, 1,4 гр, вихід 70 %) у вигляді білої твердої речовини. Приклад одержання 6: 1-метил-5-[2-етокси-5-(4-метилпіперазінілсульфоніл)феніл]-3-н-пропіл-1,6-дигідро-7Нпіразоло[4,3- альфи]піримідин-7-он (I) Спосіб 1 Сполуку (II) (X=Cl, 1 g) розчиняли в трет-бутиловому спирті (3 мл) і воді (3 мл) для одержання розчину. До розчину додавали NaHCO3 (0,17 гр, 2,1 моль) і суміш нагрівали до утворення флегми протягом 2 годин. Трет-бутиловий спирт видаляли перегонкою при зниженому тиску. При охолоджуванні на водяній лазні pH розчину підганяли до значень 8,5-9,5 додаванням водного 1 моль/л розчину HCl. Тверду білу речовину, що утворилася, фільтрували і очищали шляхом перекристалізації з етанолу, одержуючи сполуку (I) (0,87 гр, вихід 90 %) у вигляді білих голчатих 1 кристалів, Т. пл. 186-188 °C; H ЯМР (CDCl3, 300 МГц) δ: 1.01 (3H, t), 1.63 (3H, t), 1.85 (2H, m), 2.27 (3H, s), 2.50 (4H, t), 2.92 (2H, t), 3.10 (4H, t), 4.27 (3H, s), 4.37 (2H, q), 7.14 (1H, d), 7.83 (1H, dd), 8.83 + (1H, d), 10.81 (1H, s) EI-MS m/z 474 (M , 4), 410 (8), 404 (58), 312 (7), 99(100). Спосіб 2 Сполуку (II) (X=Cl, 1 гр) розчиняли у воді (5 мл) і суміш нагрівали до утворення флегми протягом 4 годин. До розчину додавали дихлорметан (15 мл) і шари розділяли. Органічну фазу промивали концентрованим сольовим розчином (2 x 5 мл) і сушили над безводим Na2SO4 протягом 30 хв. Дихлорметан видаляли перегонкою при зниженому тиску з утворенням сполуки (I) (0,80 гр, вихід 83 %) у вигляді білої твердої речовини. Спосіб 3 11 UA 99447 C2 5 10 15 20 25 30 35 40 45 50 55 60 Сполуку (II) (X=Cl, 1 гр) розчиняли в 1 моль/л водному розчині HCl (5 мл) і реакційну суміш нагрівали при 60 °C протягом 2 годин. При охолоджуванні на крижаній лазні pH розчину підганяли до значень 8,5-9,5 додаванням NaHCO3. Білу тверду речовину, що утворилася, фільтрували і очищали перекристалізацією з етанолу з утворенням сполуки (I) (0,76 гр, вихід 79%) у вигляді білого порошку. Приклад одержання 7: 7-бром-1-метил-5-(2-етоксифеніл)-3-н-пропіл-1H-піpазоло[4,3-альфа]піримідин (IV) (X=Br) Сполуку, вказану в заголовку, одержували, слідуючи процедурі способу 2 Прикладу одержання 3, замінивши POCI3 на PBr3 з утворенням сполуки (IV) (X=Br, 4,0 гр) з виходом 67 %. Приклад одержання 8: 7-бром-1-метил-5-[2-етокси-5-(хлорсульфоніл)феніл]-3-н-пропіл-1Н-піразоло[4,3альфи]піримідин (III) (X=Br). Спосіб 1 Сполуку, вказану в заголовку, одержували, слідуючи процедурі способу 1 Прикладу одержання 1 4, замінивши POCI3 на PBr3 з утворенням сполуки (III) (X=Br, 5,0 гр) з виходом 70%. H ЯМР (CDCl3, 300 МГц) δ: 1.03 (3H, t), 1.43 (3H, t), 1.88 (2H, m), 3.04 (2H, t), 4.23 (2H, q), 4.40 (3H, s), 7.15 (1H, d), + 8.07 (1H, dd), 8.45 (1H, d) EI-MS m/z 475(8), 473(M +1, 8), 382(24), 380(24), 292(76), 82(98), 80(100), 79(40) . Спосіб 2 Сполуку, вказану в заголовку, одержували, слідуючи процедурі способу 3 Прикладу одержання 4, замінивши POCI3 на POBr3 з утворенням сполуки (III) (X=Br, 3,4 гр) з виходом 59 %. Спосіб 3 Сполуку, вказану в заголовку, одержували, слідуючи процедурі способу 6 Прикладу одержання 4, замінивши PCI5 на PBr5 з утворенням сполуки (III) (X=Br, 3,0 гр) з виходом 52%. Приклад одержання 9: 7-бром-1-метил-5-[2-етокси-5-(4-метилпіперазінілсульфоніл)феніл]-3-н-пропіл-1H-піразоло[4,3альфи]піримідин (II) (X=Br) Сполуку, вказану в заголовку, одержували з 7-бром-1-метил-5-[2-етокси-5(хлорсульфоніл)феніл]-3-н-пропіл-1H-піразоло[4,3-альфа]піримідину ((III), X=Br), слідуючи процедурі за способом 1 Прикладу одержання 5 з утворенням сполуки (II) (X=Br, 0,8 гр) з виходом 1 72%. H ЯМР (CDCl3, 300 МГц) δ: 1.03 (3H, t), 1.43 (3H, t), 1.87 (2H, m), 2.51 (3H, s), 2.83(4H, s), 3.04 (2H, t), 3.50 (4H, s), 4.21 (2H, q), 4.39 (3H, s), 7.14 (1H, d), 7.77 (1H, dd), 8.20 (1H, s) EI-MS m/z 538 + (M , 6), 536(6), 468 (6), 466(6), 456(20), 99(100). Приклад одержання 10: 1-метил-5-[2-етокси-5-(4-метилпіперазінілсульфоніл)феніл]-3-н-пропіл-1,6-дигідро-7Нпіразоло[4,3- альфи]піримідин-7-он (I) Сполуку, вказану в заголовку, було одержано з 7-бром-1-метил-5-[2-етокси-5-(4метилпіперазінілсульфоніл) феніл]-3-н-пропіл-1,6-дигідро-7Н-піpазоло[4,3-альфа]піримідину, слідуючи процедурі за способом 1 Прикладу одержання 6 з утворенням сполуки (I) (0,85 гр) з виходом 75 %. Приклад 2 Одержання сполука формули (I):Получение соединения формулы (I): Стадія 1. У колбу місткістю 50 мл додавали сполуку (V) (5 гр, 0,015 моль) і для одержання розчину поступово додавали POCI3 (10 мл). Суміш нагрівали при 120 °C протягом 1 години, далі реакційну суміш виливали в крижану воду (30 мл) для зупинки реакції, після цього суміш екстрагували дихлорметаном (3 x 30 мл). Органічну фазу промивали концентрованим сольовим розчином (2 x 10 мл) і сушили над безводим сульфатом натрію протягом 30 хвилин, далі шляхом концентрації одержували сполуку (IV) (X=Cl, 4,1 гр ) у вигляді білої твердої речовини. Стадія 2. При охолоджуванні на крижаній лазні сполука (IV) (X=Cl, 5 гр, 0,015 моль) додавали порціями до хлорсульфонової кислоти (10 мл). Суміш перемішували на крижаній лазні протягом 30 хвилин, потім крижану лазню прибирали. Реакцію продовжували ще дві години. Залишок виливали у воду. Тверду речовину, що утворилася, фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі, одержуючи сполуку (III) (X=Cl) у вигляді білої твердої речовини. Стадія 3. Сполуку (Ill) (X=Cl, 2 гр ) розчиняли в дихлорметані (20 мл) і триетиламіні (0,94 гр, 9,3 моль) для одержання розчину. Потім, при охолоджуванні на крижаній лазні по краплях додавали розчин 1-метилпіперазіну (0,51 гр, 5,1 моль) в дихлорметані (5 мл). Крижану лазню прибирали і суміш перемішували при кімнатній температурі протягом 2 годин. Додавали воду і розділяли шари. Органічну фазу промивали насиченим розчином хлориду амонія (2 x 10 мл), концентрованим сольовим розчином (2 x 10 мл) і сушили над безводним Na2SO4 (1 гр ) протягом 30 хвилин. 12 UA 99447 C2 5 10 15 20 25 30 35 Розчинник видаляли шляхом перегонки при зниженому тиску з утворенням неочищеної сполуки (II) (X=Cl) у вигляді білої твердої речовини. Неочищений продукт очищали перекристалізацією з суміші дихлорметан/петролейний ефір з одержанням сполуки (II) (X=Cl) у вигляді білої кристалічної твердої речовини. Стадія 4. Сполуку (II) (X=Cl, 1 гр) розчиняли в трет-бутиловому спирті (3 мл) і у відповідній кількості води для одержання розчину. До розчину додавали NaHCO3 (2,1 моль) і суміш нагрівали при 90 °C протягом 1,5 години. Трет-бутиловий спирт видаляли перегонкою при зниженому тиску при 40 °C. pH розчину підганяли до значень 8,5-9,5 і тверду речовину, що утворилася, фільтрували і очищали перекристалізацією з етанолу з одержанням сполуки (I) (0,87 гр) у вигляді білих голчатих кристалів. Альтернатива A: Стадія 2 може проводитися безпосередньо після стадії 1 без очищення проміжного продукту, докладна процедура виглядає таким чином: У трехгорлову колбу місткістю 50 мл додавали сполуку (V) (5 гр, 0,015 моль). При охолоджуванні на крижаній лазні додавали POCl3 (10 мл) для одержання розчину і реакційну суміш нагрівали при 120 °C протягом 1 години, потім одержаний розчин охолоджували до температури нижче 0 °C. У реакційну суміш додавали хлорсульфонову кислоту (10 мл). Через 30 хвилин крижану лазню прибирали і реакційну суміш перемішували при кімнатній температурі протягом 2 годин. Суміш виливали в крижану воду (30 мл), білу тверду речовину, що утворилася, фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі протягом 3 годин при 35 °C з одержанням сполука (III) (X=Cl, 5,7 гр ). Приклад 3: При охолоджуванні на крижаній лазні в трехгорлову колбу місткістю 50 мл додавали сполуку (X) (5 гр, 0,016 моль). Для одержання розчину в колбу по краплях додавали POCl3 (10 мл). Розчин нагрівали при 50 °C протягом 4 годин, для зупинки реакції суміш виливали на лід і потім суміш екстрагували дихлорметаном (3 x 30 мл). Органічну фазу промивали концентрованим сольовим розчином (2 x 10 мл) і сушили над безводним сульфатом натрію (2 гр) протягом 30 хвилин з подальшим упарюванням, одержуючи сполука (IV) (X=Cl, 4,4 гр ) у вигляді білої твердої речовини. У колбу місткістю 50 мл додавали сполуку (VIII) (5 гр, 0,012 моль) і для одержання розчину поступово додавали POCl3 (10 мл). Реакційну суміш нагрівали при 60 °C протягом 3 годин і потім виливали у воду (20 мл). Тверду речовину, що утворилася, фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі протягом 2 годин при 35 °C, одержуючи сполуку (III) (X=Cl, 4,8 г) у вигляді білої твердої речовини. 13 UA 99447 C2 5 У колбу місткістю 50 мл додавали сполуку (VI) (2 гр, 0,004 моль). Для одержання розчину поступово додавали POCl3 (10 мл). Реакційну суміш нагрівали при 100 °C протягом 1,5 годин і потім виливали на роздроблений лід. Білу тверду речовину, що утворилася, фільтрували, промивали крижаною водою до нейтральної реакції фільтрату і сушили у вакуумі протягом 2 годин при 35 °C, одержуючи сполуку (II) (X=Cl, 1,5 g) у вигляді білої твердої речовини. Приклад 4: 10 (III) (X=Br) Сполуку формули (III) (X=Br) одержували за способом альтернативи А Прикладу 2, замінюючи POCI3 на PBr3, з одержанням сполуки (III) (X=Br, 5,0 гр). 15 20 25 30 (II) (X=Br) Сполуку формули (II) (X=Br) одержували із сполуки (III) (X=Br, 1,0 гр), слідуючи стадії 3 в Прикладі 2, одержуючи сполуку (II) (X=Br, 0,8 гр ). Сполуку формули (I) одержували із сполуки (II) (X=Br, 0,5 гр), слідуючи стадії 4 в Прикладі 2, одержуючи сполуку (I) (0,36 гр) у вигляді білих голчатих кристалів. Приклад 5 Одержання силденафілу цитрату Приклад 5.1: Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівали до 45-65 °C. До суспензії додавали лимонну кислоту, моногідрат (2,10 гр, 10 моль), при цьому одержували розчин. Через короткий проміжок часу утворився осад. Осад охолоджували, фільтрували і промивали метанолом, одержуючи 6,6 гр силденафілу цитрату. Осад перекристалізовували з метанолу (160 мл). Одержували білі голчаті кристали силденафілу цитрату (4,5 гр ). Приклад 5.2. Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівали до 45-65 °C. До суспензії додавали лимонну кислоту, моногідрат (2,10 гр, 10 моль), при цьому одержували розчин. Через короткий проміжок часу утворився осад. Потім осад охолоджували і додавали метанол (150 мл). Суспензію, що утворилася, нагрівали до утворення флегми для одержання розчину. Розчин 14 UA 99447 C2 5 10 15 20 25 30 35 40 45 50 55 60 охолоджували, і відбувалася кристалізація. Кристали фільтрували і промивали метанолом. Одержували білі голчаті кристали силденафілу цитрату (5,2 гр ). Приклад 5.3: Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівали до 45-65 °C. До суспензії додавали безводну лимонну кислоту (1,90 гр, 10 моль), при цьому одержували розчин. Через короткий проміжок часу утворився осад. Потім осад охолоджували, фільтрували і промивали метанолом, одержуючи 6,4 гр силденафілу цитрату. Осад перекристалізовували з метанолу (160 мл). Одержували білі голчаті кристали силденафілу цитрату (4,6 гр). Приклад 5.4: Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівали до 45-65 °C. До суспензії додавали безводну лимонну кислоту (1,90 гр, 10 моль), при цьому одержували розчин. Через короткий проміжок часу утворився осад. Потім осад охолоджували і додавали метанол (150 мл). Суспензію, що утворилася, нагрівали до утворення флегми для одержання розчину. Розчин охолоджували, і відбувалася кристалізація. Кристали фільтрували і промивали метанолом. Одержували білі голчаті кристали силденафілу цитрату (5,2 гр ). Приклад 5.5: Суспензію силденафілу (4,75 гр, 10 моль) і лимонної кислоти (2,1 гр ) в метанолі (20 мл) нагрівали до утворення флегми протягом 1 години і потім охолоджували до 20-25 °C. Відбувалося осадження, і осад фільтрували і промивали метанолом. Осад перекристалізовували з метанолу. Одержували білі голчаті кристали силденафілу цитрату (5,7 гр). Приклад 5.6: Спосіб підвищення виходу при кристалізації упарюванням розчину або суспензії Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівали до 45-65 °C. До суспензії при 50-60 °С додавали безводну лимонну кислоту (1,90 гр, 10 моль) і метанол (150 мл), і при кип’ячені із зворотним холодильником відбувалося розчинення. Потім розчин упарювали до об'єму 50-60 мл. Під час упарювання відбувалося утворення осаду. Суспензію охолоджували до кімнатної температури, кристали фільтрували і промивали метанолом. Продукт сушили у вакуумній сушильній печі при 60 °C. Одержували білі голчаті кристали силденафілу цитрату (6,3 гр). Приклад 5.7: Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівали до 45-65 °C. До суспензії при 50-60 °С додавали безводну лимонну кислоту (1,90 гр, 10 моль), при цьому відбувалося розчинення. Через короткий проміжок часу утворився осад. Додавали метанол (150 мл) і суспензію нагрівали до утворення флегми, одержуючи розчин. Розчин потім упарювали і охолоджували до 20-25 °C, одержуючи білі голчаті кристали силденафілу цитрату (5,8 гр). Приклад 5.8: Кристалізація білих голчатих кристалів силденафілу цитрату з малим розміром частинок. Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівали до 45-65 °C. До суспензії додавали безводну лимонну кислоту (1,90 гр, 10 моль) і метанол (150 моль) і одержували розчин. Розчин швидко охолоджували до 20-25 °C і відбувалася кристалізація. Кристали фільтрували і промивали метанолом. Одержували білі голчаті кристали силденафілу цитрату (5,2 гр) малого розміру. Приклад 5.9: Кристалізація білих голчатих кристалів силденафілу цитрату з малим розміром частинок. Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівають до 45-65 °C. До суспензії додавали безводну лимонну кислоту (1,90 гр, 10 моль) і метанол (150 моль) і одержували розчин. Розчин упарювали, швидко охолоджували до 20-25 °C, що приводило до кристалізації. Кристали фільтрували і промивали метанолом. Одержували білі голчаті кристали силденафілу цитрату малого розміру (5,8 гр). Приклад 5.10: Кристалізація білих голчатих кристалів силденафілу цитрату з великим розміром частинок. Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівають до 45-65 °C. До суспензії додавали безводну лимонну кислоту (1,90 гр, 10 моль) і метанол (150 моль) і одержували розчин. Розчин упарювали, охолоджували до 20-25 °C і відбувалася кристалізація. По-іншому суспензію нагрівали до 55 °C і охолоджували до 20 °C кілька разів. Отримані кристали фільтрували і промивали метанолом. Одержували білі голчаті кристали силденафілу цитрату великого розміру (5,2 гр). Приклад 5.11: Одержання силденафілу цитрату Суспензію силденафілу (4,75 гр, 10 моль) в метанолі (20 мл) нагрівають до 45-65 °C у атмосфері азоту. До суспензії додавали лимонну кислоту (1,90 гр, 10 моль) і метанол (150 мл) і 15 UA 99447 C2 5 10 одержували розчин. Розчин охолоджували до 20-25 °C і відбувалася кристалізація. Суспензію фільтрували під азотом, одержуючи білі голчаті кристали силденафілу цитрату (5,2 гр). Приклад 6: Визначення чистоти силденафілу Для визначення домішок, що відносяться до силденафілу і силденафілу цитрату, застосовували високоефективну рідинну хроматографію (HPLC). Аналізи проводили із застосуванням колонки Hypersil MOS C8, завдовжки 200 мм і внутрішнім діаметром 4,6 мм (розмір частинок 5 мкм). Як рухому фазу застосовували A: 0,1% триетиламін в 0,025 M KH2PO4 (pH 6,0, підігнаний ортофосфорной кислотою) і B: ацетонітрил. Застосовували градієнтне елюювання: 30% B (0 хв), 30% B (3 хв), 70% B (20 хв), 70% B (30 хв). Хроматограф був обладнаний УФ детектором. Було визначено, що чистота силденафілу, одержаного за прикладом 2, стадія 4, склала 99,95%. ФОРМУЛА ВИНАХОДУ 1. Спосіб одержання сполуки формули (І): 15 O OEt CH 3 N HN N N CH CH CH 2 SO N 2 3 NCH 2 3 , (I) в якому здійснюють стадію перетворення сполуки, яка вибрана з групи, що складається із сполук формул (II), (III) і (IV): X OEt CH X 3 CH N N OEt N 3 N N N N CH CH CH 2 2 N 3 CH CH CH 2 SO N 3 NCH 2 3 SO Cl , (II) X OEt 2 CH 2 , (III) 3 N N N N CH CH CH 2 20 25 2 3 , (IV) де X є галогеном, в ході однієї або більше стадій з утворенням сполуки формули (І). 2. Спосіб за п. 1, де X є Сl або Вr. 3. Спосіб за п. 1 або 2, де сполуку формули (IV) одержують циклізацією сполуки формули (V) у присутності РОХ3, РХ3, РХ5 або їх сумішей: 16 UA 99447 C2 X OEt H 2NOC O CH CH N OEt N N N N H 3 3 N N CH CH CH 2 2 CH CH CH 3 2 2 3 (IV) (V) , або галогенуванням сполуки формули (X) у присутності РОХ3, РХ3, РХ5 або їх сумішей: X O OEt CH OEt N HN CH 3 N N N N N CH CH CH 2 2 CH CH CH 2 3 (X) 5 10 3 . 4. Спосіб за п. 3, де сполуку формули (IV) одержують обробкою сполуки формули (V) РОХ3, РХ3, РХ5 або їх сумішами при 50-120 °С або обробкою сполуки формули (X) РОХ3, РХ3, РХ5 або їх сумішами при 50-120°С. 5. Спосіб за п. 4, де після обробки реакційну суміш виливають у воду, лід або їх суміші і відбирають осад з утворенням сполуки формули (IV). 6. Спосіб за будь-яким з пп. 3-5, де реакцію циклізації або реакцію галогенування проводять у присутності бензолу, толуолу, СН2Сl2, СНСl3, СlСН2СН2Сl, тетрагідрофурану (THF), діоксану або їх сумішей. 7. Спосіб за будь-яким з пп. 1-6, де сполуку формули (III) одержують хлорсульфонуванням сполуки формули (IV) у присутності хлорсульфонової кислоти: OEt CH X N N CH 3 OEt 3 N N N N N N CH CH CH 2 2 CH CH CH 2 3 SO Cl (IV) 2 3 (III) 2 . 8. Спосіб за будь-яким з пп. 1-6, де сполуку формули (III) одержують циклізацією сполуки формули (V) у присутності РОХ3, РХ3, РХ5 або їх сумішей з подальшим хлорсульфонуванням у присутності хлорсульфонової кислоти без очищення проміжного продукту. 9. Спосіб за п. 1 або 2, де сполуку формули (III) одержують циклізацією сполуки формули (VII) у присутності РОХ3, РХ3, РХ5 або їх сумішей: X OEt H 2NOC O CH N CH 3 OEt N H N N N CH CH CH 2 SO2 Cl 3 N N 20 2 (IV) X 15 3 N 2 CH CH CH 3 2 SO Cl (VII) 2 17 2 3 (III) , UA 99447 C2 або галогенуванням сполуки формули (VIII) у присутності РОХ3, РХ3, РХ5 або їх сумішей: O OEt CH X 3 HN OEt N N CH CH CH 2 2 N CH CH CH 3 2 (VIII) SO2 Cl SO Cl 2 3 (III) 2 10 3 N N N 5 CH N . 10. Спосіб за п. 7, де сполуку формули (III) одержують обробкою сполуки формули (IV) хлорсульфоновою кислотою при 0-50 °С. 11. Спосіб за п. 9, де сполуку формули (III) одержують нагріванням сполуки формули (VII) у присутності РОХ3, РХ3, РХ5 або їх сумішей при 50-120 °С або нагріванням сполуки формули (VIII) у присутності РОХ3, РХ3, РХ5 або їх сумішей при 50-120 °С. 12. Спосіб за п. 9 або 11, де реакцію циклізації або реакцію галогенування проводять у присутності бензолу, толуолу,СН2Сl2, СНСl3, СlСН2СН2Сl, тетрагідрофурану (THF), діоксану або їх сумішей. 13. Спосіб за будь-яким з пп. 1-12, де сполуку формули (II) одержують обробкою сполуки формули (III) 1-метилпіперазином: O X OEt CH 3 OEt N N CH 3 N HN N N N N CH CH CH 2 2 CH CH CH 2 2 3 3 SO N SO Cl NCH 2 3 2 (II) (III) 15 . 14. Спосіб за п. 1 або 2, де сполуку формули (II) одержують циклізацією сполуки формули (VI) у присутності РОХ3, РХ3, РХ5 або їх сумішей: X 3 2 2 3 2 2 3 3 (II) (VI) 20 25 . 15. Спосіб за п. 13, де сполуку формули (II) одержують обробкою сполуки формули (III) в розчиннику, вибраному з алкілгалогенідів, нижчого аліфатичного кетону і простих ефірів, з подальшим додаванням основи і 1-метилпіперазіну. 16. Спосіб за п. 14, де сполуку формули (II) одержують нагріванням сполуки формули (VI) у присутності РОХ3, РХ3, РХ5 або їх сумішей в будь-якому співвідношенні при 50-120 °С. 17. Спосіб за п.16, де після обробки реакційну суміш виливають у воду, лід або їх суміші і відбирають осад. 18. Спосіб за п. 16 або 17, де циклізацію проводять у присутності бензолу, толуолу,СН2Сl2, СНСl3, СlСН2СН2Сl, тетрагідрофурану (THF), діоксану або їх сумішей. 18 UA 99447 C2 19. Спосіб за будь-яким з пп. 1-18, де сполуку формули (І) одержують реакцією сполуки формули (II) з утворенням сполуки формули (І): X 3 2 3 2 2 3 (II) 10 15 20 (I) . 20. Спосіб за п. 19, де реакцію проводять в розчиннику, вибраному з води, метанолу, етанолу, ізопропанолу, трет-С4H9ОН, гліколю, монометилового ефіру етилгліколю і їх сумішей. 21. Спосіб за п. 19 або 20, де реакцію проводять у присутності основи або кислоти. 22. Спосіб за п. 21, де основу вибирають з алкоксидів лужних металів, гідридів лужних металів, гідридів лужноземельних металів, амінів, металевих похідних амінів, гідроксидів, карбонатів, бікарбонатів або їх сумішей. 23. Спосіб за п. 21, де кислоту вибирають з соляної кислоти, сірчаної кислоти, фосфорної кислоти, лимонної кислоти, винної кислоти, малеїнової кислоти або їх сумішей. 24. Спосіб за будь-яким з пп. 1-23, в якому додатково проводять перетворення сполуки формули (І) в її фармацевтично прийнятну сіль або сольват. 25. Спосіб за п. 24, де фармацевтично прийнятна сіль є силденафілу цитратом. 26. Спосіб за п. 25, де перетворюють сполуку формули (І) на силденафілу цитрат шляхом приведення сполуки формули (І) в контакт з лимонною кислотою у відповідному розчиннику, переважно метанолі. 27. Спосіб одержання фармацевтичної композиції, що містить сполуку формули (І) або її фармацевтично прийнятну сіль або сольват в комбінації з щонайменше одним фармацевтично прийнятним носієм, що включає стадію одержання сполуки формули (І) або її фармацевтично прийнятної солі або сольвату за будь-яким з пп. 1-26 та змішування вказаної сполуки з фармацевтично прийнятним носієм. 28. Сполуки формули (II): X 3 2 2 2 3 3 , (II) 25 3 3 2 5 2 де X є Сl або Вr. 29. Сполуки формули (III): 19 UA 99447 C2 X OEt CH 3 N N N N CH CH CH 2 2 3 SO Cl 2 , (III) де X є Сl або Вr. 30. Сполуки формули (IV): X OEt CH 3 N N N N CH CH CH 2 2 3 , (IV) 5 де X є Сl або Вr. Комп’ютерна верстка І. Скворцова Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 20
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of sildenafil
Автори англійськоюTian, Guanghui, Zhu, Yi, Liu, Zheng, Wang, Zhen, Shen, Jingsham, Bombek, Sergeja, Stropnik, Tadej
Назва патенту російськоюСпособ получения силденафила
Автори російськоюТиан Гюанхю, Зю Йи, Лью Зенг, Ванг Зен, Шен Жинкшам, Бомбек Сергея, Стропник Тадей
МПК / Мітки
МПК: C07D 487/04
Мітки: одержання, спосіб, силденафілу
Код посилання
<a href="https://ua.patents.su/22-99447-sposib-oderzhannya-sildenafilu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання силденафілу</a>
Спосіб одержання циталопраму (варіанти), s-циталопраму, проміжні кетони та спосіб одержання рацемічних сполук

Номер патенту: 72238
Опубліковано: 15.02.2005
Автори: Рок Майкл Харольд, Петерсен Ханс, Еллегор Петер
МПК: C07C 255/56, C07D 307/87, C07C 253/30
Мітки: циталопраму, проміжні, рацемічних, кетони, одержання, сполук, варіанти, спосіб, s-циталопраму
Формула / Реферат:
1. Спосіб одержання циталопраму, згідно з яким здійснюють реакцію сполуки формули IV, IVде R являє собою ацил, з 3-(N,N-диметиламіно)пропілмагнійгалогенідом, переважно з 3-(N,N-диметиламіно)пропілмагнійхлоридом, з одержанням циталопраму формули I, Iякий виділяють у вигляді основи або її фармацевтично прийнятної солі.2. Спосіб за п. 1, який відрізняється тим, що проміжну сполуку формули IV одержують...
Спосіб отримання силденафілу та проміжні сполуки
Номер патенту: 27085
Опубліковано: 28.02.2000
Автори: Дан Пітер Джеймс, Вуд Альберт Шо
МПК: C07D 231/40, A61K 31/519, C07D 487/04, C07B 61/00, C07D 231/14, C07D 295/26, C07D 295/22, A61P 13/02, A61K 31/505, A61P 15/00
Мітки: спосіб, проміжні, силденафілу, отримання, сполуки
Формула / Реферат:
1. Способ получения силденафила формулы (I)который включает циклизацию соединения формулы (II)где циклизацию проводят в основной, нейтральной или кислой средах.2. Способ по п.1, где циклизацию проводят в присутствии основания, предпочтительно в растворителе, необязательно в присутствии перекиси водорода или пероксидной соли, с последующей, если необходимо, нейтрализацией реакционной смеси.3. Способ по...
Похідні циклопептидів, спосіб їх одержання, фармацевтична композиція, спосіб її одержання

Номер патенту: 55439
Опубліковано: 15.04.2003
Автори: ХЬОЛЬЦЕМАНН Гюнтер, ГОДМАН Сімон, ФІТТШЕН Клаус
МПК: C07K 7/56
Мітки: композиція, фармацевтична, циклопептидів, спосіб, одержання, похідні
Формула / Реферат:
1. Сполуки формули Іцикло(Arg-X-Asp-R1), (I)в якійХ являє собою Gly, Ala або NH-NH-CO,причому можуть братися до уваги також похідні вказаних амінокислот, і залишки амінокислот з'єднані один з одним через -аміно- і -карбоксигрупи по типу пептидного зв'язку,R1 являє собою залишок формули II(II),R2, R3, R4 кожний незалежно один від одного являє собою Н, А, Аr, R5-Ar, Het абоR5-Het,А...
Тієнопіримідини, спосіб їх одержання, фармацевтична композиція та спосіб її одержання

Номер патенту: 66856
Опубліковано: 15.06.2004
Автори: Клюхен Франс-Вернер, Джонас Рохус, Шеллінг П'єр, Крістадлер Марія
МПК: C07D 495/04, A61P 43/00, A61K 31/519, A61P 9/00, A61P 15/10
Мітки: одержання, спосіб, композиція, тієнопіримідини, фармацевтична
Формула / Реферат:
1. Тієнопіримідини формули І, Iв якійR1, R2 кожний незалежно один від одного означає Н, А, ОА або Hal,R1 і R2 разом означають також алкілен з 3-5 С-атомами, -О-СН2-СН2-, -СН2-О-СН2-,-О-СН2-О- або -О-СН2-СН2-О-,Х означає R4, R5 або R6, одноразово заміщений R7,R4 означає прямий чи розгалужений алкілен з 1-10 С-атомами, де одна або...
Глікокон’югати камптотецину, спосіб їх одержання (варіанти) та лікарський засіб

Номер патенту: 61110
Опубліковано: 17.11.2003
Автори: Баумгартен Йорг, фон ДЕМ БРУХ Карстен, ШПЕРЦЕЛЬ Міхаель, ЛЕРХЕН Ханс-Георг
МПК: C07K 5/068, C07K 9/00, A61P 35/00, C07K 5/078, C07H 15/26, A61K 38/00, C07K 5/072
Мітки: камптотецину, глікокон'югати, спосіб, одержання, варіанти, лікарський, засіб
Формула / Реферат:
1. Глікокон’югати камптотецину формули (I)в якійR1 є повністю стереометричним неполярним бічним ланцюгом амінокислотиіR2 є основним бічним ланцюгом амінокислоти,а також їх солі, стереоізомери і суміші стереоізомерів.2. Сполуки формули (I) за п. 1, деR1 є розгалуженим алкільним радикалом, що має до 4 атомів вуглецюіR2 є радикалом формули -(CH2)n-R3, деR3 є...
Попередній патент: Шоколад або його аналог, який містить плоди
Наступний патент: Гетероариламідні похідні
Випадковий патент: Привід до енергозрошувального комплексу