Спосіб одержання похідних пірролідіну
Номер патенту: 18263
Опубліковано: 25.12.1997


Формула / Реферат
Способ получения производных пирролидина общей формулы:
в или -форме,
где - прямая связь или
- группа формулы:
где и каждый независимо представляет собой кислород или равно 1 или 2;
отличающийся тем, что производное пирролидина общей формулы:
где имеет вышеуказанные значения, подвергают взаимодействию с соединением общей формулы:
где и имеют вышеуказанные значения - атом галогена в присутствии акцептора кислоты, такого как карбонат щелочного металла, или, в случае необходимости, соединение 1, где и дигидробензофуран-5-ил.
Текст
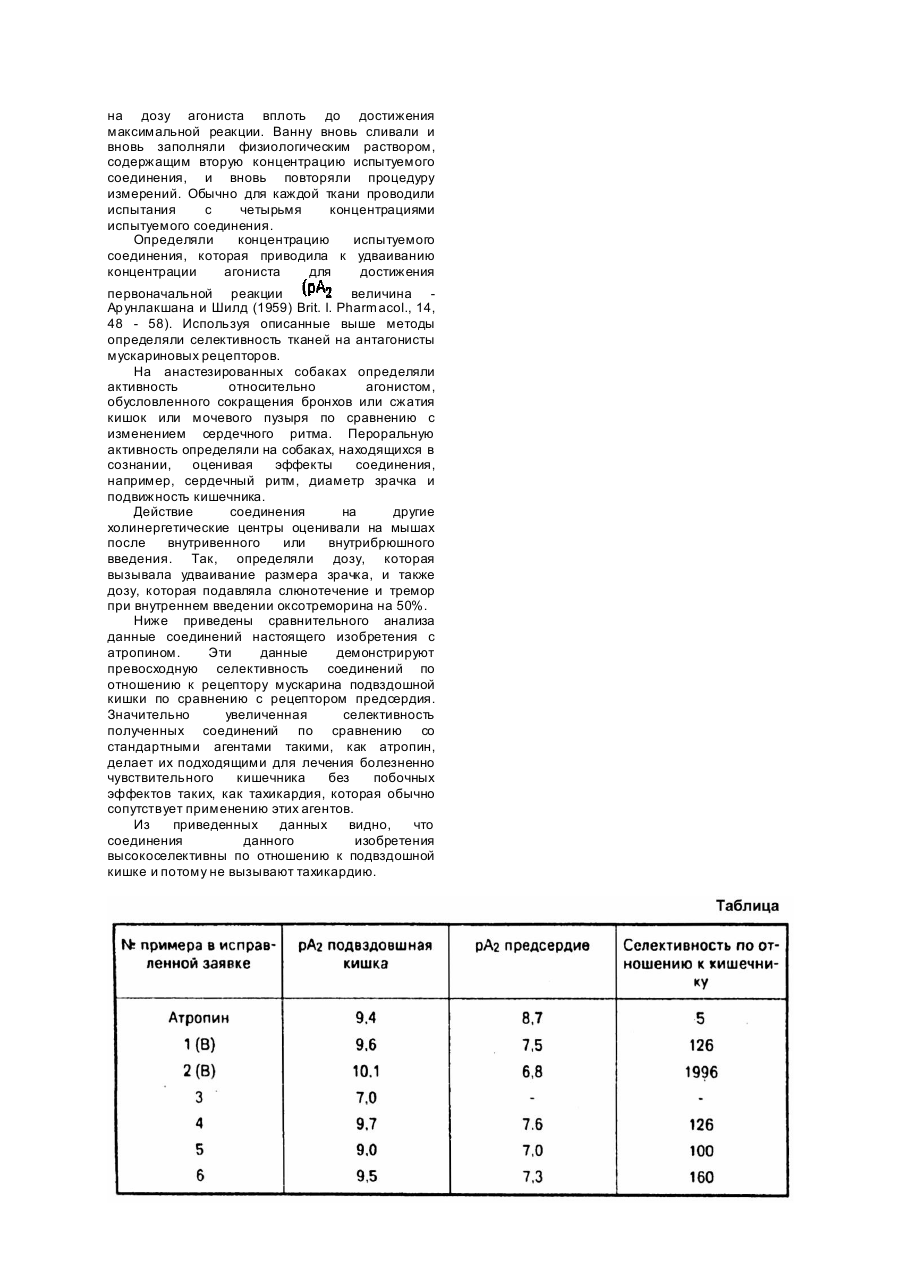
Изобретение относится к области получения новых производных пирролидина общей формулы в где или -форме, - прямая связь или ил)этил/пирролидин в виде пены. Выход 1,9г / (C) Следуя аналогичной процедуре, из -карбамоил-1,1-дифенилметил/ пирролидина (2,8г), получили карбамоил-1,1-дифенилметил-/-1-/2-(2,3дигидробензофуран-5-ил)этил/пирролидин в виде пены. Выход 1,7г - гр уппа формулы: где и - каждый независимо представляет собой кислород или или 2, являющихся антагонистами мускариновых рецепторов. Целью изобретения является разработка, на основе известных методов, способа получения новых соединений, обладающих ценным фармакологическим свойством, а именно селективностью в отношении мускариновых рецепторов гладких мышц над мускариновыми кардиорецепторами, не проявляя при этом какойлибо значительной антигистаминной активностью. Изобретение иллюстрируется следующими примерами. Пример 1 (A). Получение карбамоил-1,1-дифенилметил/-1-/2-(2,3дигидробензофуран-5-ил)этил/-пирролидина. В течение двух часов кипятили с обратным холодильником смесь, содержащую 0,33г -карбамоил-1,1-дифенил-метил/пирролидина, 0,25г 5-/2-бромэтил/-2,3дигидробензофурана, 0,3г безводного карбоната калия и 10мл ацетонитрила. Смесь экстрагировали 50мл дихлорметана и 10мл 10% го водного карбоната калия, слои разделяли, и водный слой экстрагировали трижды дихлорметаном (по 20мл каждый раз). Объединенные дихлорметановые экстракты сушили над сульфатом магния и концентрировали в вакууме, получая камедь, которую очищали на хроматографической колонке (силикагель) с использованием в качестве элюирующего растворителя дихлорметана, содержащего 0 - 8% метанола, фракция, содержащие продукт, объединили и концентрировали в вакууме, в результате чего получали масло. Последнее кристаллизировали в диизопропиловом простом эфире и получали целевое соединение в виде бесцветного порошка с выходом 0,17г. Точка плавления 131 - 132°C. Элементный состав: Найдено, %: Вычислено, %: (B) Аналогичной процедурой, используя в качестве исходного реагента карбамоил-1,1-дифенилметилпирролидин (1,95г), получали -карбамоил-1,1дифенилметил/-1-/2-(2,3-дигидробензофуран-5 Пример 2 (A). Получение карбамоил-1,1-дифенилметил-1-/2-(индан-5ил)этил/пирролидина. В течение 1,25 часов кипятили с обратным холодильником смесь 0,6г карбамоил-1,1-дифенилметил/пирролидина 0,49г 5-/2-бромэтил/-индана, 0,6г безводного карбоната калия и 20мл ацетонитрила. Смесь фракционировали между 10% - ным водным карбонатом калия (10мл) и дихлорметаном (50мл), слои разделяли, и водный слой экстрагировали трижды дихлорметаном (по 100мл). Объединенные дихлорметановые экстракты сушили над сульфатом магния и концентрировали в вакууме, получая камедь, которую очищали на хроматографической колонке с силикагелем, используя в качестве элюирующего растворителя дихлорметан с 0 - 6% метанола. Продуктовые фракции объединяли и концентрировали в вакууме, получая целевой продукт в виде пены. Выход 0,29г. Элементный анализ для Найдено, %: Вычислено, %: (B) Следуя аналогичной методике, из -карбамоил-1,1-дифенилметил /пирролидина (0,64г) получали карбамоил-1,1-дифенилметил/-1-/2-(индан-5ил)этил /пирролидин. Выход 0,34г Пример 3. Получение карбамоил-1,1-дифенилметил/-1-/3,4метилендиоксибензил/пирролидина. Смесь 0,75г -карбамоил-1,1,дифенилметил/-пирролидина, 0,51г 3,4метилендиоксибензилхлорида, 0,75г безводного карбоната калия и 30мл ацетонитрила перемешивали при комнатной температуре в течение получаса. Смесь фракционировали между 10% - ным водным карбонатом калия (20мл) и дихлорметаном (50мл), слои делили, и водный слой экстрагировали трижды дихлорметаном (по 50мл). Объединенные дихлорметановые экстракты сушили над сульфатом магния и концентрировали в вакууме, получая пену, которую очищали на силикагельной хроматографической колонке, элюируя дихлорметаном с 0 - 10% метанола. Продуктовые фракции объединяли и концентрировали в вакууме, получая целевой продукт в виде пены с выходом 0,5г. Элементный состав для Найдено, %: Вычислено, %: Пример 4. Получение карбамоил-1,1-дифенилметил/-1-/2-/3,4метилендиоксифенил/этил/пирролидина. В течение трех часов с обратным холодильником кипятили смесь 0,3г карбамоил-1,1-дифенилметил/пирролидина, 0,247г 3,4-метилендиоксифенэтилбромида, 0,4г безводного карбоната калия и 10мл ацетонитрила. Смесь самопроизвольно принимала комнатную температуру, добавляли 6мл воды, и новую смесь трижды экстрагировали дихлорметаном (по 50мл). Объединенные дихлорметановые экстракты сушили над сульфатом магния и концентрировали в вакууме, получая бесцветную пену, которую очищали на силикагельной хроматографической колонке, используя в качестве элюирующего растворителя дихлорметан с 0 - 6% метанола. Продуктовые фракции объединяли и концентрировали в вакууме, получая целевое соединение в виде бесцветной пены с выходом 0,27г. Элементный анализ для Найдено, %: Вычислено, %: Пример 5. Получение карбамоил-1,1-дифенилметил/-1-/2-/1,4бензодиоксан-6-ил/этил/пирролидина. В течение трех часов кипятили с обратным холодильником смесь 0,3г карбамоил-1,1-дифенилметил/пирролидина, 0,26г 6-/2-бромэтил/-1,4-бензодиоксана, 0,4г безводного карбоната калия и 10мл ацетонитрила. После охлаждения до комнатной температуры добавляли 40мл воды, и смесь экстрагировали трижды дихлорметаном (по 30мл). Объединенные дихлорметановые экстракты сушили над сульфатом магния и концентрировали в вакууме, получая пенку, которую подвергали очистке на силикагельной хроматографической колонке с использованием для элюирования дихлорметана с 0 - 10% метанола. Продуктовые фракции объединяли и концентрировали в вакууме, получая целевой продукт в виде бесцветной пены. Выход продукта 0,21г. Элементным анализом для Найдено, %: Вычислено, %: Пример 6. Получение карбамоил-1,1-дифенилметил/-1-/2-/бензофуран5-ил/этил/пирролидина. В течение получаса кипятили с обратным холодильником смесь 1,79г /1карбамоил-1,1-дифенилметил/пирролидина, 1,2г 5-/2-бромэтил/-бензо/2,3-в/фурана, 3г безводного карбоната калия и 30мл ацетонитрила. Смесь фракционировали между 30мл этилацетата и 30мл 10% - го водного карбоната калия, слои разделяли и водный слой экстрагировали трижды этилацетатом (по 50мл). Объединенные этилацетатные экстракты сушили над сульфатом магния и концентрировали в вакууме, получая коричневую камедь, которую подвергали очистке на силикагельной хроматографической колонке, используя для элюирования дихлорметан с 2% метанола. Продуктовые фракции объединяли и концентрировали в вакууме, получая целевое соединение в виде пены. Выход 1,4г. Элементным анализом для Найдено, %: Вычислено, %: Пример 7. Получение карбамоил-1,1-дифенилметил дигидробензофуран-5-ил/этил/пирролидина (альтернативный примеру I (B)). К раствору 0,1 г -карбамоил-1,1дифенилметил/-1-/2-/бензофуран-5ил/этил/пирролидина (по примеру 6) в 2мл уксусной кислоты добавляли 10мг 10% - ного палладий на угле, и смесь гидрировали при 40°C и атмосферном давлении в течение шести часов. Катализатор отфильтровывали и промывали водой (20мл). Объединенный фильтрат и промывки переводили в делительную воронку, добавляли 20мл дихлорметана, и смесь подщелачивали добавлением 10% водной гидроокиси натрия. Слои разделяли, и водный слой дополнительно экстрагировали трижды дихлорметаном (по 30мл). Объединенные дихлорметановые экстракты сушили над сульфатом магния и концентрировали в вакууме, получая бесцветное твердое вещество, которое подвергали очистке на силикагельной хроматографической колонке, осуществляя элюирование дихлорметаном с 4% метанола. Продуктовые фракции объединяли и концентрировали в вакууме, получая целевое соединение в виде бесцветной стекловидной массы с выходом 0,048г. Спектр продукта идентичен продукту по примеру 1 (B). Избирательность соединений по настоящему изобретению как антагонистов мускариновых рецепторов может быть определена следующим образом. Умерщвляли самцов морских свинок, брали кишки, трахею, мочевой пузырь и правое предсердие и суспендировали их в физиологическом растворе при напряжении покоя 1г при 32°C и аэрировании 95% кислорода и 5% углекислого газа. Записывали сокращение кишок, мочевого пузыря и трахеи с помощью изотонического (кишки) или изометрического преобразователя (трахея, мочевой пузырь). Частоту сокращения самопроизвольно бьющегося правого предсердия определяют по изометрически записываемых сокращений. Определяли зависимость реакции от дозы либо на ацетилхолин (кишки) или карбахол (трахея, предсердие, мочевой пузырь), используя 1 - 5 минутный контакт для каждой дозы агониста, вплоть до достижения максимальной реакции. Сливали ванну с органами и вновь заполняли физиологическим солевым раствором, содержащим наименьшую дозу испытуемого соединения. Испытуемому соединению давали возможность придти в равновесие с тканью в течение 20 минут, и повторяли измерение реакции на дозу агониста вплоть до достижения максимальной реакции. Ванну вновь сливали и вновь заполняли физиологическим раствором, содержащим вторую концентрацию испытуемого соединения, и вновь повторяли процедуру измерений. Обычно для каждой ткани проводили испытания с четырьмя концентрациями испытуемого соединения. Определяли концентрацию испытуемого соединения, которая приводила к удваиванию концентрации агониста для достижения первоначальной реакции величина Ар унлакшана и Шилд (1959) Brit. I. Pharmacol., 14, 48 - 58). Используя описанные выше методы определяли селективность тканей на антагонисты мускариновых рецепторов. На анастезированных собаках определяли активность относительно агонистом, обусловленного сокращения бронхов или сжатия кишок или мочевого пузыря по сравнению с изменением сердечного ритма. Пероральную активность определяли на собаках, находящихся в сознании, оценивая эффекты соединения, например, сердечный ритм, диаметр зрачка и подвижность кишечника. Действие соединения на другие холинергетические центры оценивали на мышах после внутривенного или внутрибрюшного введения. Так, определяли дозу, которая вызывала удваивание размера зрачка, и также дозу, которая подавляла слюнотечение и тремор при внутреннем введении оксотреморина на 50%. Ниже приведены сравнительного анализа данные соединений настоящего изобретения с атропином. Эти данные демонстрируют превосходную селективность соединений по отношению к рецептору мускарина подвздошной кишки по сравнению с рецептором предсердия. Значительно увеличенная селективность полученных соединений по сравнению со стандартными агентами такими, как атропин, делает их подходящими для лечения болезненно чувствительного кишечника без побочных эффектов таких, как тахикардия, которая обычно сопутствует применению этих агентов. Из приведенных данных видно, что соединения данного изобретения высокоселективны по отношению к подвздошной кишке и потому не вызывают тахикардию.
ДивитисяДодаткова інформація
Назва патенту англійськоюA process for preparation of derivatives of pyrrolidine
Автори англійськоюPeter Edward Cross
Назва патенту російськоюСпособ получения производных пирролидина
Автори російськоюПитер Едвард Кросс
МПК / Мітки
МПК: C07D 207/06
Мітки: похідних, спосіб, одержання, пірролідіну
Код посилання
<a href="https://ua.patents.su/3-18263-sposib-oderzhannya-pokhidnikh-pirrolidinu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідних пірролідіну</a>
Спосіб одержання похідних хіноліна або їх солей

Номер патенту: 4875
Опубліковано: 28.12.1994
Автори: Сатосі Мураяма, Хіросі Кога, Цутому Ірікура
МПК: C07D 401/04, C07D 215/233, C07D 215/14, C07D 215/18
Мітки: похідних, одержання, спосіб, солей, хіноліна
Формула / Реферат:
Способ получения производных хинолинагде R- 4-метил-1-пиперазинильная группа, или их солей, отличающийся тем, что соединение формулывводят в реакцию с N -метилпиперазином в инертном растворителе, например пиридине, при кипячении реакционной массы с выделением целевого продукта в свободном виде или в виде соли.
Спосіб одержання похідних (1н-імідазол-1-ілметіл)замішаного бензімідазола або їх фармацевтично прийнятих солей кислоти, або солей металів, або стереоізомерів

Номер патенту: 2706
Опубліковано: 26.12.1994
Автори: Альфонс Герман Маргарета Реймакерс, Едді Жан Едгард Фрейн, Жерар Шарль Санз
МПК: A61K 31/425, A61K 31/415, A61P 19/06, A61P 43/00, A61P 35/00, C07D 403/14, C07D 403/06, C07D 521/00, A61P 17/00, C07D 417/14, A61K 31/443, C07D 401/14, A61P 5/00, C07D 409/14, A61K 31/44, A61K 31/47, A61K 31/4433, A61K 31/4427, C07D 405/14
Мітки: 1н-імідазол-1-ілметіл)замішаного, фармацевтично, одержання, спосіб, солей, кислоти, похідних, бензімідазола, стереоізомерів, металів, прийнятих
Формула / Реферат:
Способ получения производных (1Н-имидазол-1-илметил)-замещенного бензимидазола общей формулы где R2 — водород, С1— С6-алкил, С3— С7-цикло-алкил, фенил, необязательно замещенный двумя заместителями, выбранными из гало-, С1— С4-алкила, С1— С4-алкилоксикарбонила, карбоксила или С1— С4-алкилокси, тиенилфуранил, галофуранил, имидазолил или пиридинил, R1 — водород, С3— С7 - циклоалкил, фенил, С4 - С6-алкил, необязательно замещенный...
Спосіб одержання похідних n-(2-оксо-3-оксазолідініл)ацетамідів

Номер патенту: 7033
Опубліковано: 31.03.1995
Автори: Ханспетер Шеллінг, Йост Харр, Рудольф Зандмайєр
МПК: C07D 263/26, A01N 43/76, C07D 263/22, C07D 413/12
Мітки: одержання, спосіб, n-(2-оксо-3-оксазолідініл)ацетамідів, похідних
Формула / Реферат:
(57) Способ получения производных N-{2-оксо-3-оксазолидинил)ацетамидов, общей формулыгдеотличающийся тем, что проводят внутримолекулярную конденсацию соединения общей формулы
Спосіб одержання похідних фенілгуанідину та похідні сульфокислоти як проміжні продукти в синтезі похідних фенілгуанідину

Номер патенту: 13323
Опубліковано: 28.02.1997
Автори: Петер Шаркезі, Чаба Хусар, Атілла Немет, Ференц Шпербет, Лайошне Палі, Єва Шомфаї
МПК: C07C 317/42, C07C 323/43, C07C 309/00, C07C 279/00, C07C 319/00, C07C 277/00
Мітки: спосіб, одержання, похідних, фенілгуанідину, похідні, сульфокислоти, синтезі, проміжні, продукти
Формула / Реферат:
(57) 1. Способ получения производных фенилгуанидина общей формулы I:где R - алкил с 1-4 атомами углерода; М - водород, натрий, калий или кальций; А - группа -S-, -SO- или -SO2-,отличающийся тем, что производное тиокарбамида общей формулы II:где R и М имеют вышеуказанные значения, подвергают окислению с последующей обработкой полученного при этом производного сульфокислоты общей формулы...
Спосіб отримання похідних бензаміду, або їх кислотно-адитивних солей, або оптичних ізомерів

Номер патенту: 4762
Опубліковано: 28.12.1994
Автори: Ян Ола Густав Лундстрем, Свен Ове Егрен, Геста Леннарт Фрорвалл, Стен Інгвар Ремсбі
МПК: C07D 207/09, A61P 1/08, A61P 25/18, A61K 31/40, A61K 31/60
Мітки: бензаміду, оптичних, отримання, солей, похідних, спосіб, ізомерів, кислотно-адітивних
Формула / Реферат:
Способ получения производных бензамида общей формулы (I)где R1 и R2 одинаковы или различны и означают водород, галоид, низший алкил; R3 - низший алкил или бензил, незамещенный или замещенный фтором;А1 и А2 - оба водород или каждый в отдельности водород или низший алкил,или их кислотно-аддитивных солей, или оптических изомеров, отличающийся тем, что соединение общей формулы (II) где R1, R2 и R3 имеют...
Попередній патент: Теплова труба
Наступний патент: Спосіб надання вогнестійкості матеріалу, який містить целюлозні волокна
Випадковий патент: Спосіб спалювання палива