Похідні 2-(імінометил)амінофенілу, спосіб їх одержання (варіанти) та фармацевтична композиція, що містить ці сполуки
Номер патенту: 70921
Опубліковано: 15.11.2004
Автори: Овен Серж, Шабріе де Лассон'єр П'єр-Етьєн, Арнетт Джеремі, БІГГ Денніс
































Формула / Реферат
1. Похідні 2-(імінометил)амінофенілу загальної формули (І)
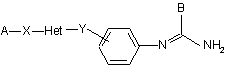 , (І)
, (І)
де А являє собою ароматичну сполуку, що відповідає структурам:
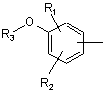 ,
,
де R1 та R2 являють собою, незалежно, водень, галоген, групу ОН, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, лінійний або розгалужений алкокси-радикал, що містить від 1 до 6 атомів вуглецю,
R3 являє собою водень, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, або радикал -COR4, R4 являє собою алкіл-радикал, що містить від 1 до 6 атомів вуглецю,
або
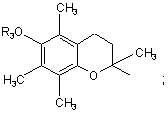
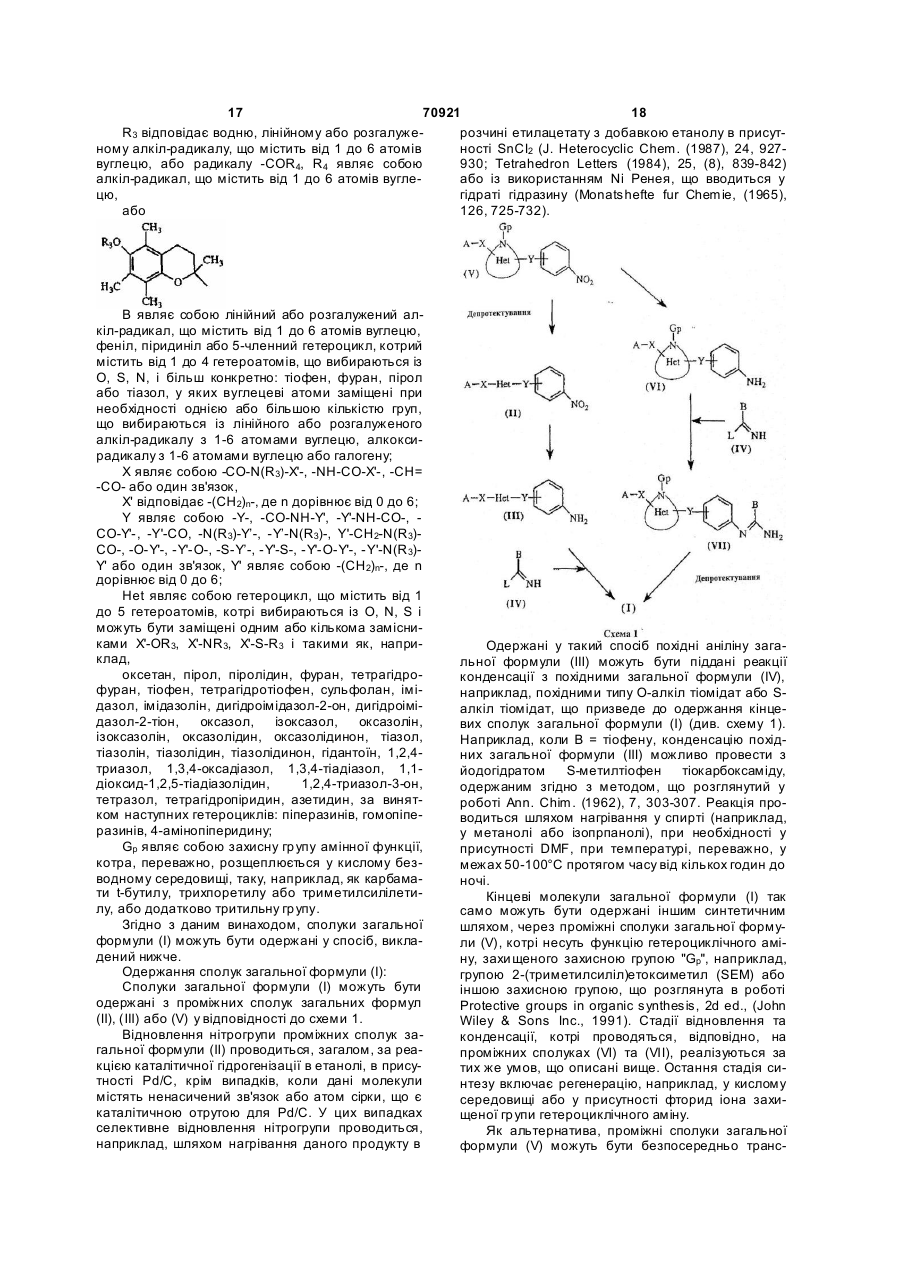
В являє собою лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, феніл, піридиніл або 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибирають з O, S, N, і, більш конкретно, тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибирають з лінійного або розгалуженого алкіл-радикала з 1-6 атомами вуглецю, алкокси-радикала з 1-6 атомами вуглецю або галогену,
Х являє собою -CO-N(R3)-X’-, NH-CO-X’-, -CH=, -CO- або один зв’язок,
X’ являє собою -(CH2)n-, де n дорівнює від 0 до 6,
Y являє собою -Y’-, -CO-NH-Y’, -Y’-NH-CO-, -CO-Y’-, -Y’-CO, -N(R3)-Y’-, -Y’-N(R3)-, Y’-CH2-N(R3)-CO-, -O-Y’-, -Y’-O-, -S-Y’-, -Y’-S-, -Y’-O-Y’-, -Y’-N(R3)-Y’ або один зв’язок,
Y’ являє собою -(СН2)n-, де n дорівнює від 0 до 6,
Het являє собою гетероцикл, вибраний з групи, що містить оксетан, пірол, піролідин, фуран, тетрагідрофуран, тіофен, тетрагідротіофен, сульфолан, імідазол, імідазолін, дигідроімідазол-2-он, дигідроімідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізоксазолін, оксазолідин, оксазолідинон, тіазол, тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4-триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1-діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, тетразол та тетрагідропіридин,
та адитивні солі з кислотами зазначеної сполуки загальної формули (I).
2. Сполука за п. 1, яка відрізняється тим, що А являє собою ароматичну сполуку, що відповідає структурі
,
де R1 та R2 являють собою, незалежно, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, або лінійний або розгалужений алкокси-радикал, що містить від 1 до 6 атомів вуглецю,
R3 являє собою водень або лінійний чи розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю,
В являє собою 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибирають з O, S, N, і, більш конкретно, тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибирають з лінійного або розгалуженого алкіл-радикала з 1-6 атомами вуглецю, алкокси-радикала з 1-6 атомами вуглецю або галогену,
Х являє собою -NH-CO-X’-, -CH=, -CO- або один зв’язок,
X’ являє собою -(CH2)n-, де n дорівнює від 0 до 6,
Y являє собою -Y’-, -Y’-NH-CO-, -Y’-CO, -Y’-O-, -Y’-O-Y’-, -Y’-N(R3)-Y’ або один зв’язок,
Y’ являє собою -(СН2)n-, де n дорівнює від 0 до 6,
Het являє собою гетероцикл, вибраний з групи, що містить оксетан, пірол, піролідин, фуран, тетрагідрофуран, тіофен, тетрагідротіофен, сульфолан, імідазол, імідазолін, дигідроімідазол-2-он, дигідроімідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізоксазолін, оксазолідин, оксазолідинон, тіазол, тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4-триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1-діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, тетразол та тетрагідропіридин,
та адитивні солі з кислотами зазначеної сполуки загальної формули (I).
3. Сполука за п. 2, яка відрізняється тим, що В являє собою тіофеновий цикл, у якому вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибирають із лінійного або розгалуженого алкілу з 1-6 атомами вуглецю, алкокси-радикала з 1-6 атомами вуглецю або галогену.
4. Сполука за пп. 1-3, яка відрізняється тим, що вона є однією з наступних сполук:
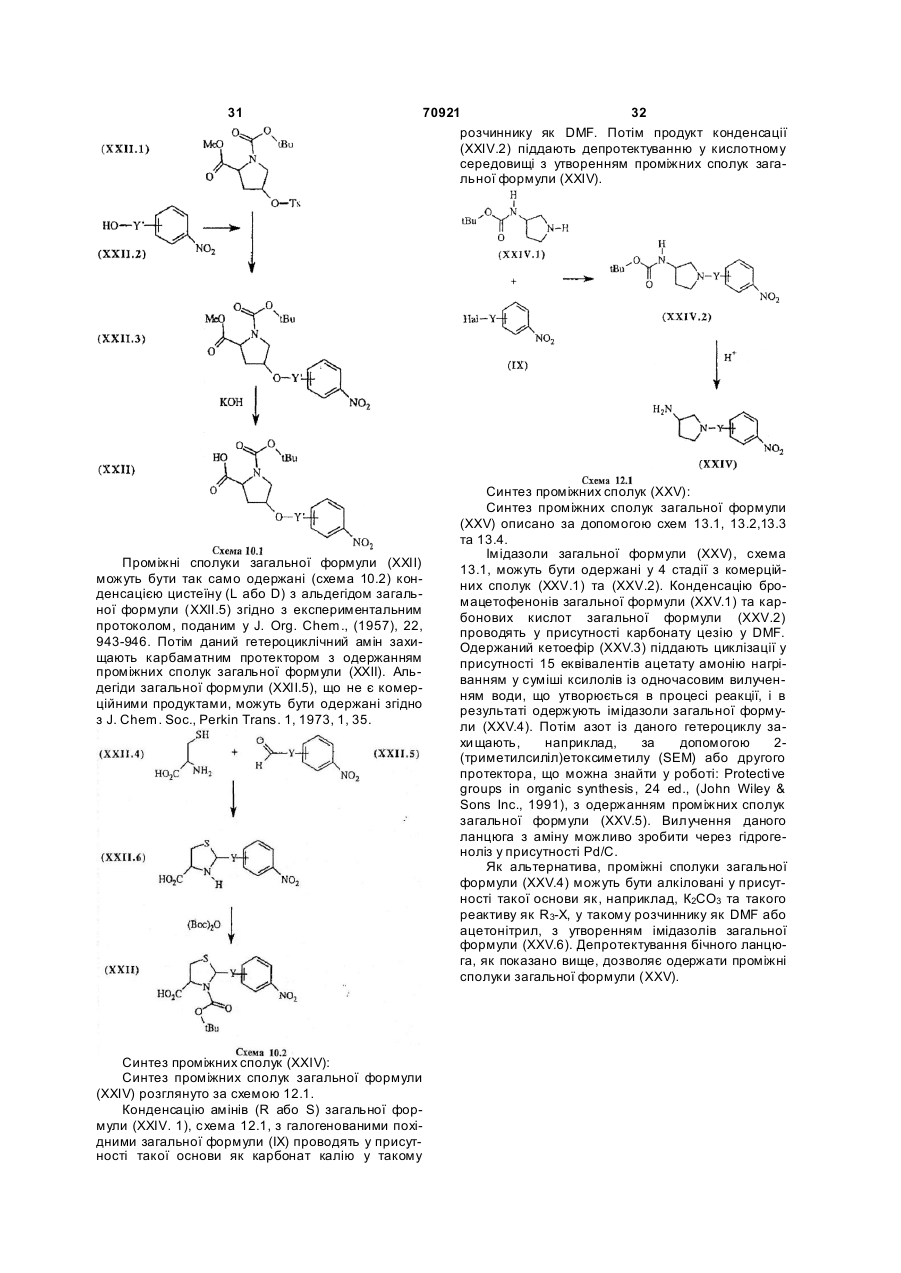
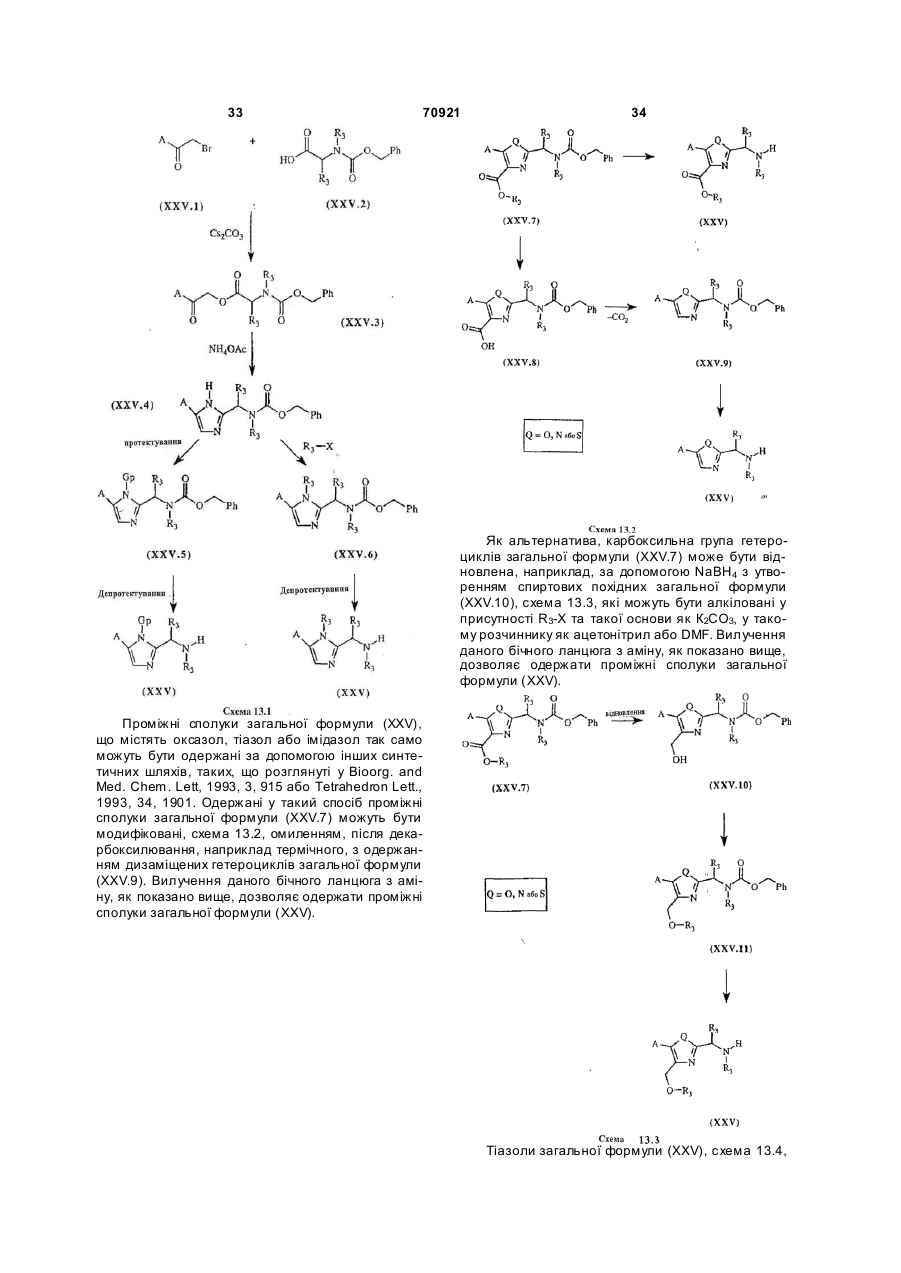
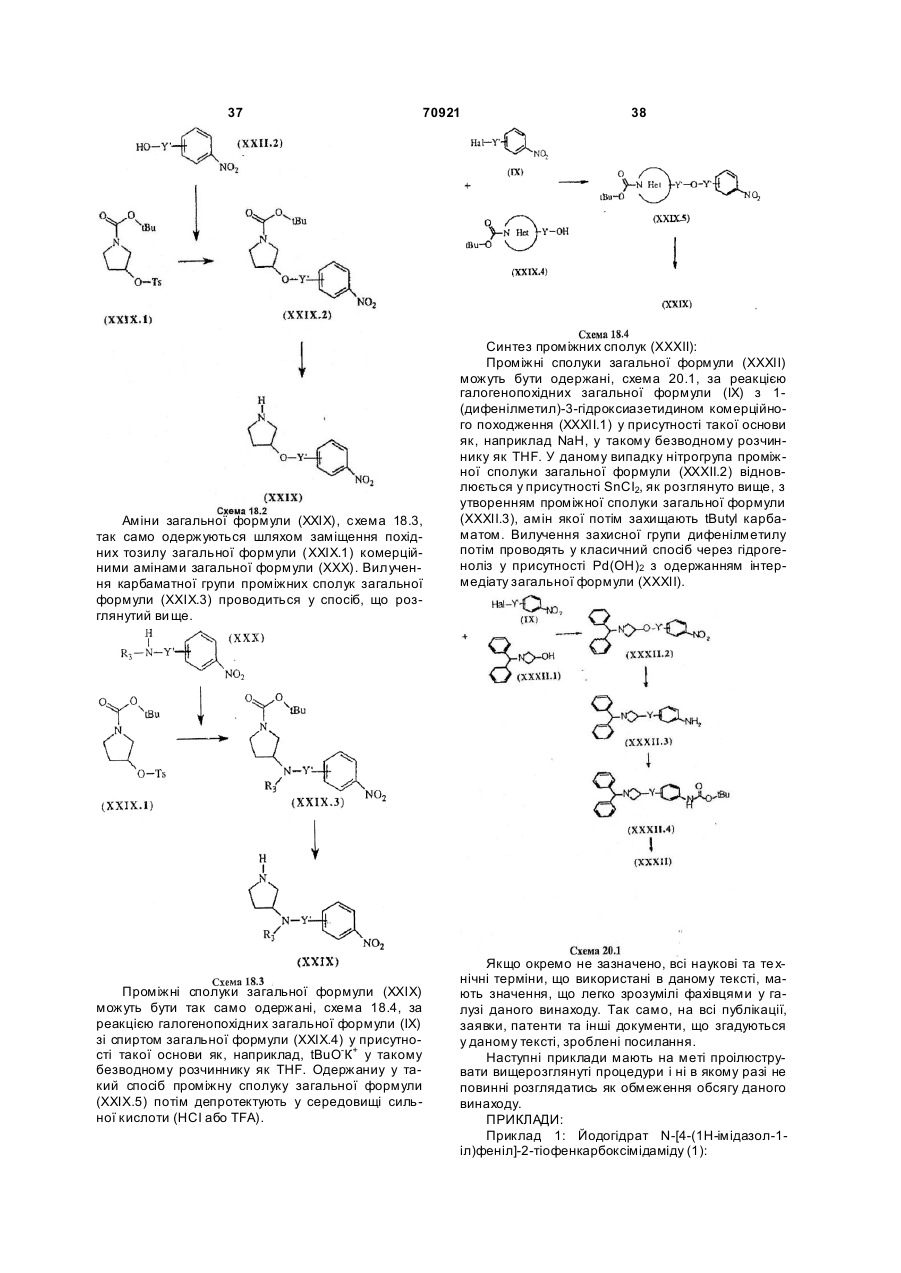
- гідройодид N-(3,5-ди-t-бутил-4-гідроксифеніл)-5-[4-{іміно(2-тієніл)метиламіно}феніл]-2–фуранкарбоксаміду;
- гідрохлорид 3-(3,5-ди-t-бутил-4-гідроксифеніл)-1-[4-{іміно(2-тієніл)метиламіно}феніл]-2,5–імідазолідиндіону;
- гідрохлорид 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3-[4-{іміно(2-тієніл)метиламіно}феніл]-4–тіазолідинону;
- фумарат 5-[(3,5-ди-t-бутил-4-гідроксифеніл)метилен]-1-метил-3-[4-{іміно(2-тієніл) метиламіно}феніл]-2,4–імідазолідиндіону;
- гідрохлорид 2-(S)-4-(S)-N-[4-гідрокси-3,5-біс-(1,1-диметилетил)-феніл]-4-{4-[(іміно(2-тієніл)метил)аміно]фенокси}-пролінаміду;
- фумарат N-[4-гідрокси-3,5-біс-(1,1-диметилетил)-феніл]-2-(R,S)-{4-[(іміно(2-тієніл) метил)аміно]феніл}-4-(R)-тіазолідин карбоксаміду;
- гідройодид N-[4-(4-феніл-1,2,3,6-тетрагідропіридин-1-іл)-феніл]-2-тіофен карбоксімідаміду;
- гідрохлорид N-[4-гідрокси-3,5-біс-(1,1-диметил)етилфеніл]-2-{4-[(іміно(2-тієніл)метил) аміно]феніл}-4-тіазолкарбоксаміду;
- гідрохлорид метил 1-[(3,4-дигідро-6-гідрокси-2,5,7,8-тетраметил-2-Н-[1]-бензопіран-2-іл)карбоніл]-4-(S)-{4-[(іміно(2-тієніл)метил)аміно]фенокси}-піролідин-2-(S)-карбоксилату;
- гідрохлорид 1-[(3,4-дигідро-6-гідрокси-2,5,7,8-тетраметил-2-Н-[1]-бензопіран-2-іл)карбоніл]-3-(S)-{4-[(іміно(2-тієніл)метил)аміно]фенокси}-піролідину;
- 3-{[(3,4-дигідро-6-гідрокси-2,5,7,8-тетраметил-2Н-[1]-бензопіран-2-іл)карбоніл]аміно}-1-{4-[(іміно(2-тієніл)метил)аміно]феніл}піролідин;
- гідрохлорид 4-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-N-{4[(іміно(2-тієніл)метил)аміно]бензоїл}-N-метил-1Н-імідазол-2-метанаміну;
- гідройодид N-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-1-{4-[(іміно(2-тієніл)метил)аміно]феніл}-1Н-пірол-2-карбоксаміду;
- гідройодид 1-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-3-{4-[[іміно(2-тієніл)метил]аміно]феніл]карбоніл}-2-імідазолідинону;
- гідройодид 3-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-4,5-дигідро-N-{4-[[іміно(2-тієніл)метил]аміно]феніл}-5-ізоксазолацетаміду;
- гідрохлорид 4-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-N-{4-[[іміно(2-тієніл)метил)аміно]феніл}-N-метил-2-тіазолметанаміну;
- гідрохлорид 4-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-N-{4-[(іміно(2-тієніл)метил)аміно]феніл}-N-метил-1Н-імідазол-2-метанаміну;
- 3-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-4,5-дигідро-5-{2-{4-[(іміно(2-тієніл)метил)аміно]феноксі)етил}-ізоксазол;
- гідрохлорид 1-{[3,5-біс(1,1-диметилетил)-4-гідроксифеніл]аміно}карбоніл}-3-{4-[(іміно(2-тієніл)метил)аміно]феноксі}-азетидину;
- гідрохлорид 1-(2-гідрокси-5-метоксибензоїл)-3-{4-[(іміно(2-тієніл)метил)аміно]феноксі}-азетидину;
- гідрохлорид 1-[(3,4-дигідро-6-гідрокси-2,5,7,8-тетраметил-2Н-[1]-бензопіран-2-іл)карбоніл]-4-[4-[(іміно(2-тієніл)метил)аміно]фенокси]-піперидину;
- гідрохлорид 1-[(3,4-дигідро-6-гідрокси-2,5,7,8-тетраметил-2Н-[1]-бензопіран-2-іл)карбоніл]-3-{4-[(іміно(2-тієніл)метил)аміно]феноксі}-азетидину;
або їх солі чи енантіомери.
5. Сполука за будь-яким з пп. 1-4, яка відрізняється тим, що вона є однією з наступних сполук:
- гідрохлорид 3-(3,5-ди-t-бутил-4-гідроксифеніл)-1-[4-{іміно(2-тієніл)метиламіно}феніл]-2,5–імідазолідиндіону;
- гідрохлорид 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3-[4-{іміно(2-тієніл)метиламіно}феніл]-4–тіазолідинону;
- фумарат 5-[(3,5-ди-t-бутил-4-гідроксифеніл)метилен]-1-метил-3-[4-{іміно(2-тієніл)метиламіно}феніл]-2,4–імідазолідиндіону;
- гідрохлорид 2-(S)-4-(S)-N-[4-гідрокси-3,5-біс-(1,1-диметилетил)-феніл]-4-{4-[(іміно(2-тієніл)метил)аміно]фенокси}-пролінаміду;
- гідрохлорид N-[4-гідрокси-3,5-біс-(1,1-диметил)етил-феніл]-2-{4-[(іміно(2-тієніл)метил)аміно]феніл}-4-тіазол карбоксаміду;
або їх солі чи енантіомери.
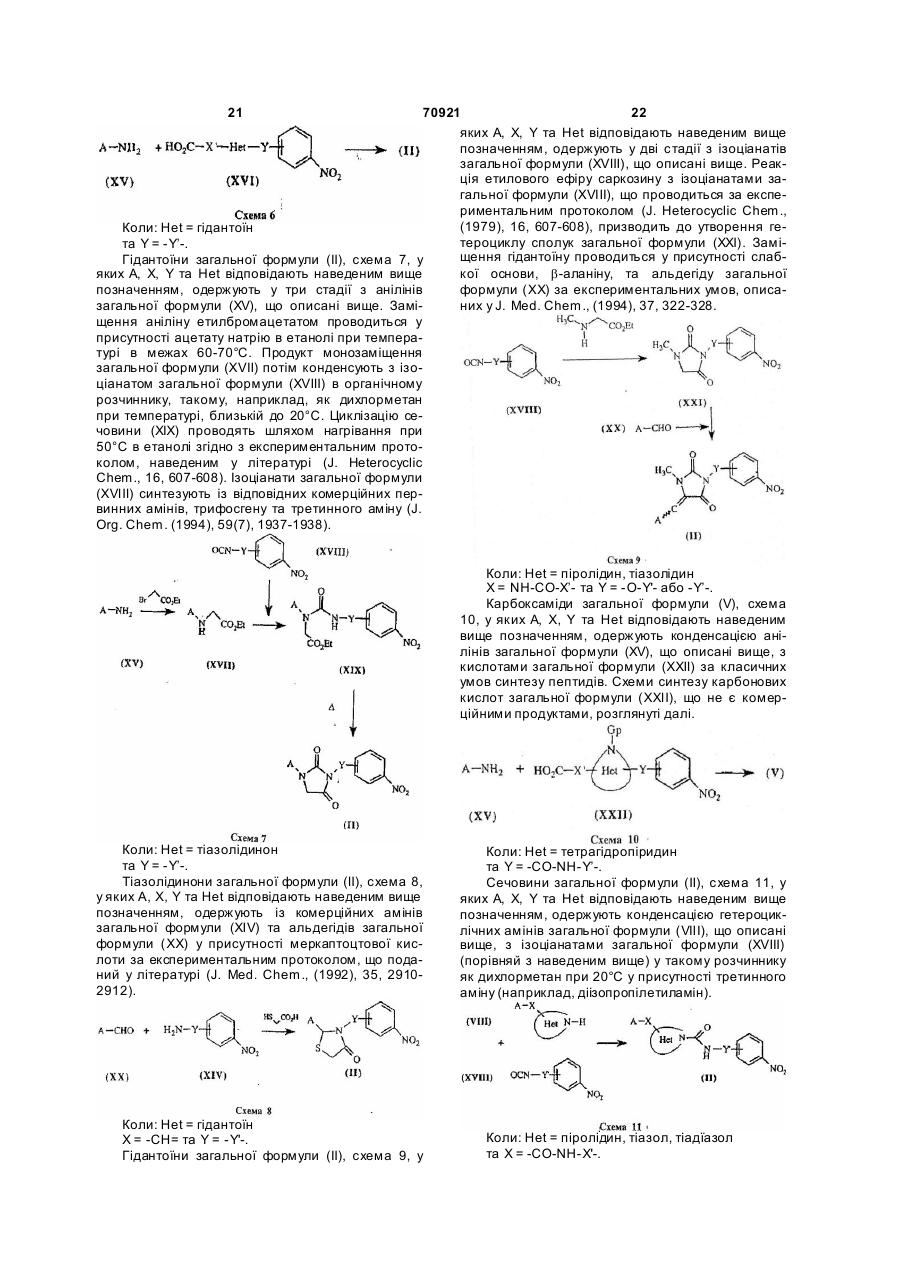
6. Спосіб одержання сполук загальної формули (І) за п.1, який відрізняється тим, що включає конденсацію, краще у суміші ізопропанолу та DMF при кімнатній температурі, похідних аніліну загальної формули (ІІІ)
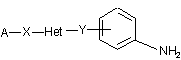 (ІІІ)
(ІІІ)
з похідними S-алкілтіоімідату загальної формули (IV)
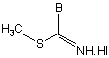
з одержанням кінцевих сполук загальної формули (І),
причому сполуки загальних формул (I), (III) та (ІV) є такими, де
А являє собою ароматичну сполуку, що відповідає структурам
або
В являє собою лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, феніл, піридиніл або 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибирають із O, S, N, і, більш конкретно, тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибирають із лінійного або розгалуженого алкілу з 1-6 атомами вуглецю, алкокси-радикала з 1-6 атомами вуглецю або галогену,
Х являє собою -CO-N(R3)-X’-, -NH-CO-X’-, -CH=, -CO- або один зв’язок,
X’ являє собою -(CH2)n-, де n дорівнює від 0 до 6,
Y являє собою -Y’-, -CO-NH-Y’, -Y’-NH-CO-, -CO-Y’-, -Y’-CO, -N(R3)-Y’-, -Y’-N(R3)-, Y’-CH2-N(R3)-CO-, -O-Y’-, -Y’-O-, -S-Y’-, -Y’-S-, -Y’-O-Y’-, -Y’-N(R3)-Y’ або один зв’язок,
Y’ являє собою -(СН2)n-, де n дорівнює від 0 до 6,
Het являє собою гетероцикл, вибраний з групи, що містить оксетан, пірол, піролідин, фуран, тетрагідрофуран, тіофен, тетрагідротіофен, сульфолан, імідазол, імідазолін, дигідроімідазол-2-он, дигідроімідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізоксазолін, оксазолідин, оксазолідинон, тіазол, тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4-триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1-діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, тетразол, тетрагідропіридин.
7. Спосіб одержання сполук загальної формули (І) за п.1, у яких гетероцикл Het містить принаймні один атом азоту, який відрізняється тим, що включає дві наступні стадії:
конденсацію, краще у суміші ізопропанолу та DMF при кімнатній температурі, сполуки загальної формули (VІ)
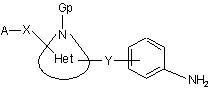 (VІ)
(VІ)
зі сполукою загальної формули (IV)
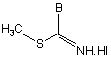 (IV)
(IV)
з одержанням сполуки загальної формули (VII)
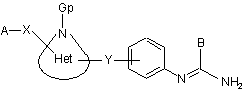 (VII),
(VII),
відщеплення захисної групи Gp сполуки загальної формули (VII), що визначена вище, з одержанням сполуки загальної формули (І),
причому сполуки загальних формул (I), (IV), (VI) та (VII) є такими, де
А являє собою ароматичну сполуку, що відповідає структурам
,
де R1 та R2 являють собою, незалежно, водень, галоген, групу ОН, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, лінійний або розгалужений алкокси-радикал, що містить від 1 до 6 атомів вуглецю,
R3 являє собою водень, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, або радикал -COR4,
R4 являє собою алкіл-радикал, що містить від 1 до 6 атомів вуглецю,
або
В являє собою лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, феніл, піридиніл або 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибирають із O, S, N, і, більш конкретно, тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибирають із лінійного або розгалуженого алкілу з 1-6 атомами вуглецю, алкокси-радикала з 1-6 атомами вуглецю або галогену,
Х являє собою -CO-N(R3)-X’-, -NH-CO-X’-, -CH=, -CO- або один зв’язок,
X’ являє собою -(CH2)n-, де n дорівнює від 0 до 6,
Y являє собою -Y’-, -CO-NH-Y’, -Y’-NH-CO-, -CO-Y’-, -Y’-CO, -N(R3)-Y’-, -Y’-N(R3)-, Y’-CH2-N(R3)-CO-, -O-Y’-, -Y’-O-, -S-Y’-, -Y’-S-, -Y’-O-Y’-, -Y’-N(R3)-Y’ або один зв’язок,
Y’ являє собою -(СН2)n-, де n дорівнює від 0 до 6,
Het являє собою гетероцикл, вибраний з групи, що містить пірол, піролідин, імідазол, імідазолін, дигідроімідазол-2-он, дигідроімідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізоксазолін, оксазолідин, оксазолідинон, тіазол, тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4-триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1-діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, тетразол, тетрагідропіридин,
Gp являє собою захисну групу амінної функції, котра, переважно, розщеплюється у кислому безводному середовищі, таку, наприклад, як карбамати t-бутилу, трихлоретилу або триметилсилілетилу, або додатково тритильну групу.
8. Сполука загальної формули (І) за будь-яким з пп. 1-5 або її фармацевтично прийнятна сіль, яка відрізняється тим, що використовується як медикамент.
9. Фармацевтична композиція, що містить як активний інгредієнт принаймні одну сполуку за п. 8.
10. Сполука загальної формули (І) за будь-яким з пп. 1-5 або її фармацевтично прийнятна сіль, яка відрізняється тим, що використовується для виготовлення медикаменту, призначеного для інгібування NO-синтази.
11. Сполука загальної формули (І) за будь-яким з пп. 1-5 або її фармацевтично прийнятна сіль, яка відрізняється тим, що використовується для виготовлення медикаменту, призначеного для інгібування ліпідного переокислення.
12. Сполука загальної формули (І) за будь-яким з пп. 1-5 або її фармацевтично прийнятна сіль, яка відрізняється тим, що використовується для виготовлення медикаменту, що водночас виявляє інгібуючу активність щодо NO-синтази та інгібуючу активність щодо ліпідного переокислення.
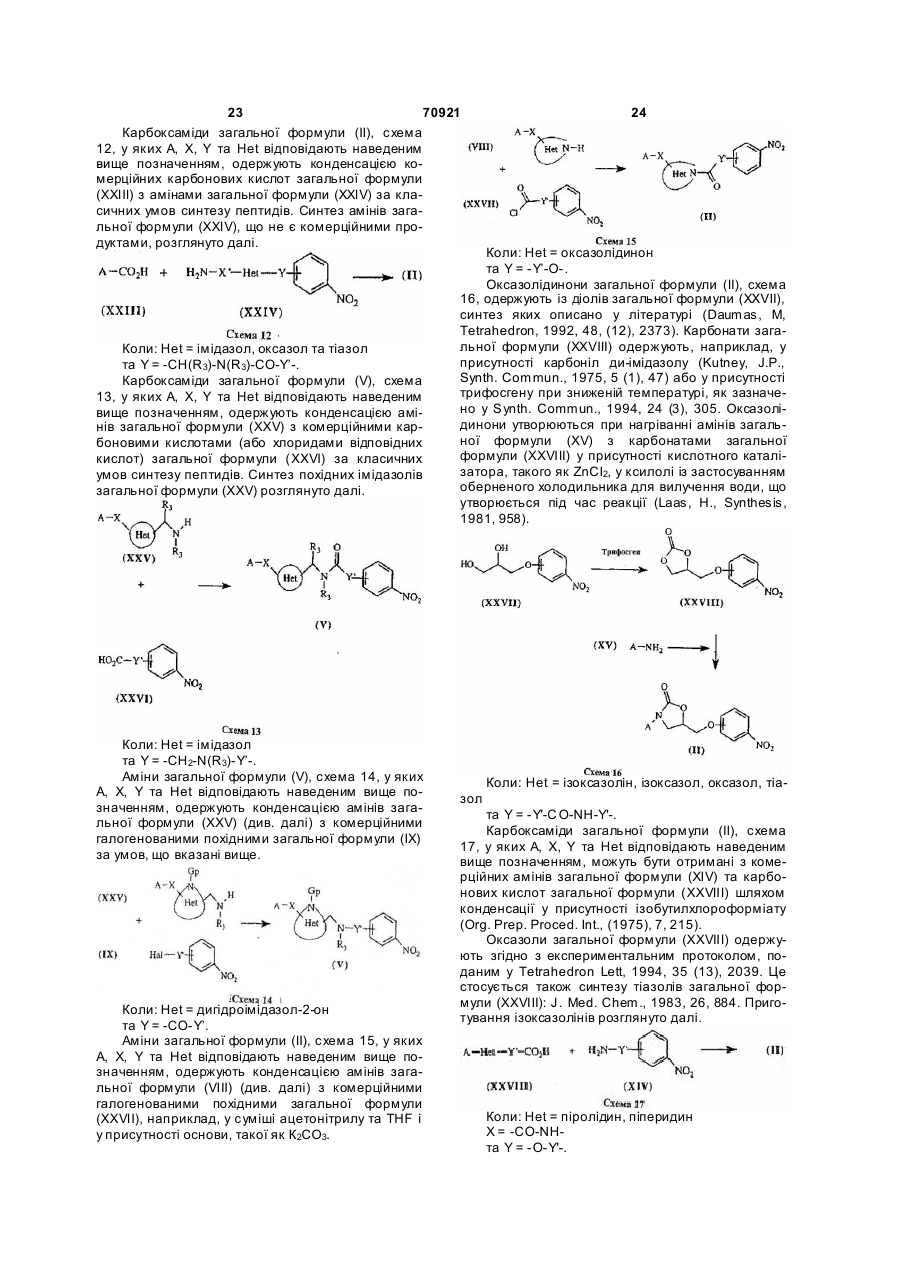
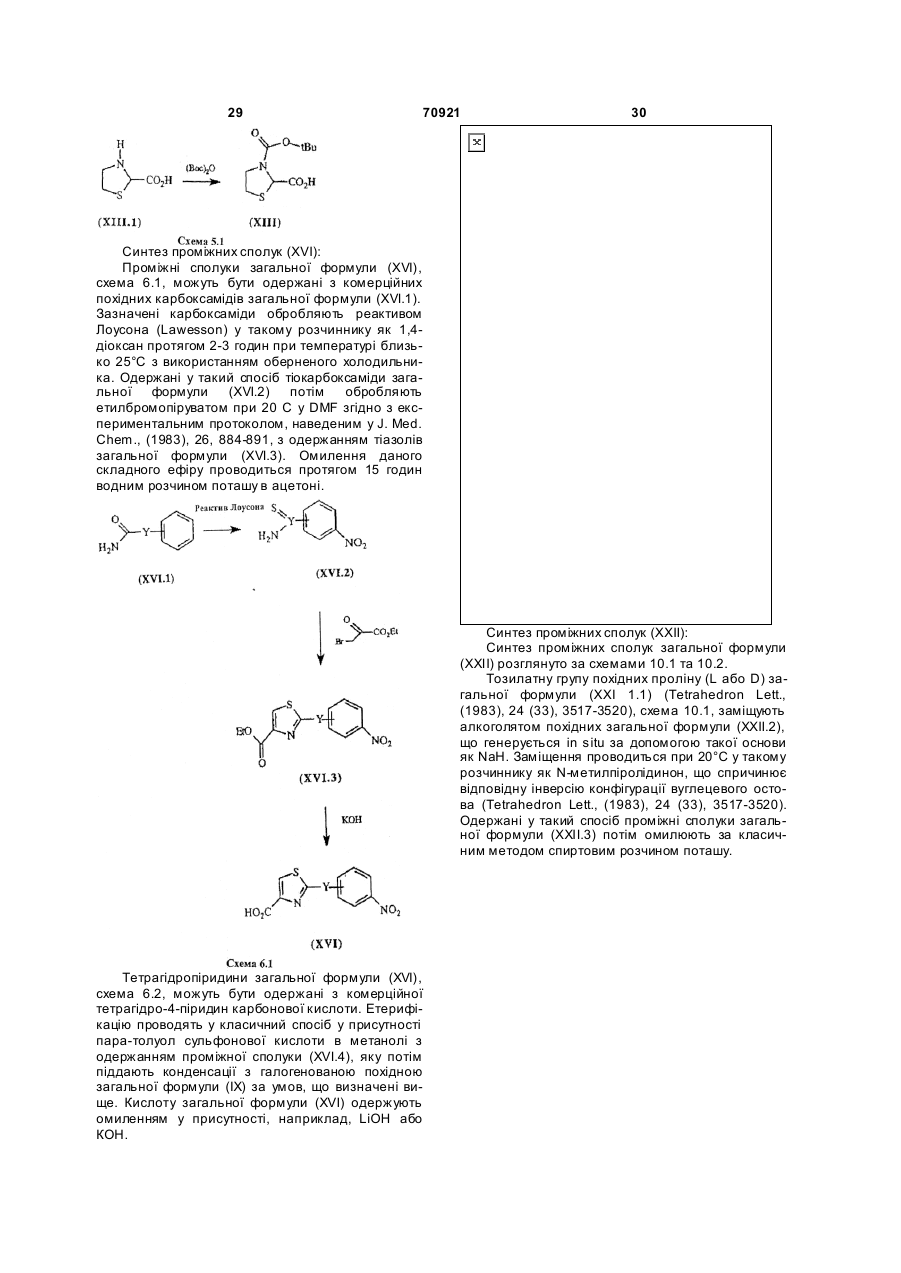
Текст
1. Похідні 2-(імінометил)амінофенілу загальної формули (І) B 3 70921 4 - фумарат N-[4-гідрокси-3,5-біс-(1,1-диметилетил)феніл]-2-(R,S)-{4-[(іміно(2-тієніл) меO R3 тил)аміно]феніл}-4-(R)-тіазолідин карбоксаміду; гідройодид N-[4-(4-феніл-1,2,3,6тетрагідропіридин-1-іл)-феніл]-2-тіофен карбоксімідаміду; R2 гідрохлорид N-[4-гідрокси-3,5-біс-(1,1, диметил)етилфеніл]-2-{4-[(іміно(2-тієніл)метил) де R1 та R2 являють собою, незалежно, лінійний аміно]феніл}-4-тіазолкарбоксаміду; або розгалужений алкіл-радикал, що містить від 1 - гідрохлорид метил 1-[(3,4-дигідро-6-гідроксидо 6 атомів вуглецю, або лінійний або розгалуже2,5,7,8-тетраметил-2-Н-[1]-бензопіран-2ний алкокси-радикал, що містить від 1 до 6 атомів іл)карбоніл]-4-(S)-{4-[(іміно(2вуглецю, тієніл)метил)аміно]фенокси}-піролідин-2-(S)R3 являє собою водень або лінійний чи розгалужекарбоксилату; ний алкіл-радикал, що містить від 1 до 6 атомів - гідрохлорид 1-[(3,4-дигідро-6-гідрокси-2,5,7,8вуглецю, тетраметил-2-Н-[1]-бензопіран-2-іл)карбоніл]-3-(S)В являє собою 5-членний гетероцикл, котрий міс{4-[(іміно(2-тієніл)метил)аміно]фенокси}тить від 1 до 4 гетероатомів, що вибирають з O, S, піролідину; N, і, більш конкретно, тіофен, фуран, пірол або - 3-{[(3,4-дигідро-6-гідрокси-2,5,7,8-тетраметил-2Нтіазол, у яких вуглецеві атоми заміщені при необ[1]-бензопіран-2-іл)карбоніл]аміно}-1-{4-[(іміно(2хідності однією або більшою кількістю гр уп, що тієніл)метил)аміно]феніл}піролідин; вибирають з лінійного або розгалуженого алкілгідрохлорид 4-[3,5-біс-(1,1-диметилетил)-4радикала з 1-6 атомами вуглецю, алкоксигідроксифеніл]-N-{4[(іміно(2радикала з 1-6 атомами вуглецю або галогену, тієніл)метил)аміно]бензоїл}-N-метил-1Н-імідазолХ являє собою -NH-CO-X’-, -CH=, -CO- або один 2-метанаміну; зв’язок, гідройодид N-[3,5-біс-(1,1-диметилетил)-4X’ являє собою -(CH2)n-, де n дорівнює від 0 до 6, гідроксифеніл]-1-{4-[(іміно(2Y являє собою -Y’-, -Y’-NH-CO-, -Y’-CO, -Y’-O-, -Y’тієніл)метил)аміно]феніл}-1Н-пірол-2O-Y’-, -Y’-N(R 3)-Y’ або один зв’язок, карбоксаміду; Y’ являє собою -(СН2)n-, де n дорівнює від 0 до 6, гідройодид 1-[3,5-біс-(1,1-диметилетил)-4Het являє собою гетероцикл, вибраний з групи, що гідроксифеніл]-3-{4-[[іміно(2містить оксетан, пірол, піролідин, фуран, тетрагідтієніл)метил]аміно]феніл]карбоніл}-2рофуран, тіо фен, тетрагідротіофен, сульфолан, імідазолідинону; імідазол, імідазолін, дигідроімідазол-2-он, дигідрогідройодид 3-[3,5-біс-(1,1-диметилетил)-4імідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізогідроксифеніл]-4,5-дигідро-N-{4-[[іміно(2ксазолін, оксазолідин, оксазолідинон, тіазол, тіатієніл)метил]аміно]феніл}-5-ізоксазолацетаміду; золін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4гідрохлорид 4-[3,5-біс-(1,1-диметилетил)-4триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1гідроксифеніл]-N-{4-[[іміно(2діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, тієніл)метил)аміно]феніл}-N-метил-2тетразол та тетрагідропіридин, тіазолметанаміну; та адитивні солі з кислотами зазначеної сполуки гідрохлорид 4-[3,5-біс-(1,1-диметилетил)-4загальної формули (I). гідроксифеніл]-N-{4-[(іміно(23. Сполука за п. 2, яка відрізняє ться тим, що В тієніл)метил)аміно]феніл}-N-метил-1Н-імідазол-2являє собою тіофеновий цикл, у якому вуглецеві метанаміну; атоми заміщені при необхідності однією або біль- 3-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]-4,5шою кількістю груп, що вибирають із лінійного або дигідро-5-{2-{4-[(іміно(2розгалуженого алкілу з 1-6 атомами вуглецю, алтієніл)метил)аміно]феноксі)етил}-ізоксазол; кокси-радикала з 1-6 атомами вуглецю або галогегідрохлорид 1-{[3,5-біс(1,1-диметилетил)-4ну. гідроксифеніл]аміно}карбоніл}-3-{4-[(іміно(24. Сполука за пп. 1-3, яка відрізняється тим, що тієніл)метил)аміно]феноксі}-азетидину; вона є однією з наступних сполук: - гідрохлорид 1-(2-гідрокси-5-метоксибензоїл)-3-{4- гідройодид N-(3,5-ди-t-бутил-4-гідроксифеніл)-5[(іміно(2-тієніл)метил)аміно]феноксі}-азетидину; [4-{іміно(2-тієніл)метиламіно}феніл]-2– - гідрохлорид 1-[(3,4-дигідро-6-гідрокси-2,5,7,8фуранкарбоксаміду; тетраметил-2Н-[1]-бензопіран-2-іл)карбоніл]-4-[4- гідрохлорид 3-(3,5-ди-t-бутил-4-гідроксифеніл)-1[(іміно(2-тієніл)метил)аміно]фенокси]-піперидину; [4-{іміно(2-тієніл)метиламіно}феніл]-2,5– - гідрохлорид 1-[(3,4-дигідро-6-гідрокси-2,5,7,8імідазолідиндіону; тетраметил-2Н-[1]-бензопіран-2-іл)карбоніл]-3-{4- гідрохлорид 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3[(іміно(2-тієніл)метил)аміно]феноксі}-азетидину; [4-{іміно(2-тієніл)метиламіно}феніл]-4– або їх солі чи енантіомери. тіазолідинону; 5. Сполука за будь-яким з пп. 1-4, яка відрізняфумарат 5-[(3,5-ди-t-бутил-4ється тим, що вона є однією з наступних сполук: гідроксифеніл)метилен]-1-метил-3-[4-{іміно(2- гідрохлорид 3-(3,5-ди-t-бутил-4-гідроксифеніл)-1тієніл) метиламіно}феніл]-2,4–імідазолідиндіону; [4-{іміно(2-тієніл)метиламіно}феніл]-2,5– - гідрохлорид 2-(S)-4-(S)-N-[4-гідрокси-3,5-біс-(1,1імідазолідиндіону; диметилетил)-феніл]-4-{4-[(іміно(2тієніл)метил)аміно]фенокси}-пролінаміду; R1 5 70921 6 - гідрохлорид 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3-O-Y’-, -Y’-O-, -S-Y’-, -Y’-S-, -Y’-O-Y’-, -Y’-N(R 3)-Y’ [4-{іміно(2-тієніл)метиламіно}феніл]-4– або один зв’язок, тіазолідинону; Y’ являє собою -(СН2)n-, де n дорівнює від 0 до 6, фумарат 5-[(3,5-ди-t-бутил-4Het являє собою гетероцикл, вибраний з групи, що гідроксифеніл)метилен]-1-метил-3-[4-{іміно(2містить оксетан, пірол, піролідин, фуран, тетрагідтієніл)метиламіно}феніл]-2,4–імідазолідиндіону; рофуран, тіо фен, тетрагідротіофен, сульфолан, - гідрохлорид 2-(S)-4-(S)-N-[4-гідрокси-3,5-біс-(1,1імідазол, імідазолін, дигідроімідазол-2-он, дигідродиметилетил)-феніл]-4-{4-[(іміно(2імідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізотієніл)метил)аміно]фенокси}-пролінаміду; ксазолін, оксазолідин, оксазолідинон, тіазол, тіагідрохлорид N-[4-гідрокси-3,5-біс-(1,1золін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4диметил)етил-феніл]-2-{4-[(іміно(2триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1тієніл)метил)аміно]феніл}-4-тіазол карбоксаміду; діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, або їх солі чи енантіомери. тетразол, тетрагідропіридин. 6. Спосіб одержання сполук загальної формули (І) 7. Спосіб одержання сполук загальної формули (І) за п.1, який відрізняється тим, що включає конза п.1, у яких гетероцикл Het містить принаймні денсацію, краще у суміші ізопропанолу та D MF при один атом азоту, який відрізняється тим, що кімнатній температурі, похідних аніліну загальної включає дві наступні стадії: формули (ІІІ) конденсацію, краще у суміші ізопропанолу та DMF при кімнатній температурі, сполуки загальної формули (VІ) A X Het Y Gp NH2 (ІІІ) A X N з похідними S-алкілтіоімідату загальної формули (IV) He t Y B NH2 (VІ) H3C зі сполукою загальної формули (IV) S B NH.HI з одержанням кінцевих сполук загальної формули H3C (І), S причому сполуки загальних формул (I), (III) та (ІV) NH.HI (IV) є такими, де з одержанням сполуки загальної формули (VII) А являє собою ароматичну сполуку, що відповідає Gp структурам R1 A X N O B R3 Het Y N R2 або CH3 R3O CH3 H3C O ; CH3 В являє собою лінійний або розгалужений алкілрадикал, що містить від 1 до 6 атомів вуглецю, феніл, піридиніл або 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибирають із O, S, N, і, більш конкретно, тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибирають із лінійного або розгалуженого алкілу з 1-6 атомами вуглецю, алкокси-радикала з 1-6 атомами вуглецю або галогену, Х являє собою -CO-N(R3)-X’-, -NH-CO-X’-, -CH=, CO- або один зв’язок, X’ являє собою -(CH2)n-, де n дорівнює від 0 до 6, Y являє собою -Y’-, -CO-NH-Y’, -Y’-NH-CO-, -COY’-, -Y’-CO, -N(R 3)-Y’-, -Y’-N(R3)-, Y’-CH2-N(R3)-CO-, NH2 (VII), відщеплення захисної групи Gp сполуки загальної формули (VII), що визначена вище, з одержанням сполуки загальної формули (І), причому сполуки загальних формул (I), (IV), (VI) та (VII) є такими, де А являє собою ароматичну сполуку, що відповідає структурам R1 O R3 R2 , де R1 та R2 являють собою, незалежно, водень, галоген, групу ОН, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, лінійний або розгалужений алкокси-радикал, що містить від 1 до 6 атомів вуглецю, R3 являє собою водень, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, або радикал -COR4, R4 являє собою алкіл-радикал, що містить від 1 до 6 атомів вуглецю, або 7 70921 8 нон, гідантоїн, 1,2,4-триазол, 1,3,4-оксадіазол, CH3 1,3,4-тіадіазол, 1,1-діоксид-1,2,5-тіадіазолідин, R3O 1,2,4-триазол-3-он, тетразол, тетрагідропіридин, Gp являє собою захисну груп у амінної функції, котCH3 ра, переважно, розщеплюється у кислому безводному середовищі, таку, наприклад, як карбамати tO ; H3C бутилу, три хлоретилу або триметилсилілетилу, CH3 або додатково тритильну груп у. 8. Сполука загальної формули (І) за будь-яким з В являє собою лінійний або розгалужений алкілпп. 1-5 або її фармацевтично прийнятна сіль, яка радикал, що містить від 1 до 6 атомів вуглецю, відрізняє ться тим, що використовується як медифеніл, піридиніл або 5-членний гетероцикл, котрий камент. містить від 1 до 4 гетероатомів, що вибирають із 9. Фармацевтична композиція, що містить як актиO, S, N, і, більш конкретно, тіофен, фуран, пірол вний інгредієнт принаймні одну сполуку за п. 8. або тіазол, у яких вуглецеві атоми заміщені при 10. Сполука загальної формули (І) за будь-яким з необхідності однією або більшою кількістю груп, пп. 1-5 або її фармацевтично прийнятна сіль, яка що вибирають із лінійного або розгалуженого алківідрізняє ться тим, що використовується для вилу з 1-6 атомами вуглецю, алкокси-радикала з 1-6 готовлення медикаменту, призначеного для інгібуатомами вуглецю або галогену, вання NO-синтази. Х являє собою -CO-N(R3)-X’-, -NH-CO-X’-, -CH=, 11. Сполука загальної формули (І) за будь-яким з CO- або один зв’язок, пп. 1-5 або її фармацевтично прийнятна сіль, яка X’ являє собою -(CH2)n-, де n дорівнює від 0 до 6, відрізняє ться тим, що використовується для виY являє собою -Y’-, -CO-NH-Y’, -Y’-NH-CO-, -COготовлення медикаменту, призначеного для інгібуY’-, -Y’-CO, -N(R 3)-Y’-, -Y’-N(R3)-, Y’-CH2-N(R3)-CO-, вання ліпідного переокислення. -O-Y’-, -Y’-O-, -S-Y’-, -Y’-S-, -Y’-O-Y’-, -Y’-N(R 3)-Y’ 12. Сполука загальної формули (І) за будь-яким з або один зв’язок, пп. 1-5 або її фармацевтично прийнятна сіль, яка Y’ являє собою -(СН2)n-, де n дорівнює від 0 до 6, відрізняє ться тим, що використовується для виHet являє собою гетероцикл, вибраний з групи, що готовлення медикаменту, що водночас виявляє містить пірол, піролідин, імідазол, імідазолін, дигіінгібуючу активність щодо NO-синтази та інгібуючу дроімідазол-2-он, дигідроімідазол-2-тіон, оксазол, активність щодо ліпідного переокислення. ізоксазол, оксазолін, ізоксазолін, оксазолідин, оксазолідинон, тіазол, тіазолін, тіазолідин, тіазоліди Даний винахід стосується нових похідних 2(імінометил)аміно-фенілу, які виявляють інгібуючу активність щодо ферментів NO-синтаз, котрі продукують моноксид азоту NO, та/або активність щодо захоплення активних форм кисню (скорочення ROS у подальшому викладі відповідає виразу "частки активного кисню"). Даний винахід стосується похідних згідно з загальною формулою (І), що визначена нижче, способів їх одержання, фармацевтичних композицій, що містять ці похідні, та їх застосування у терапії, зокрема, їх застосування як інгібіторів NO-синтаз та захоплювачів активних форм кисню у селективний або неселективний спосіб. З ура хуванням потенційної ролі NO та ROS у патофізіології описані нові похідні, що відповідають загальній формулі (І), можуть виявляти сприятливий ефект у лікуванні патологій, де приймають участь зазначені хімічні речовини. А саме: - розладів серцево-судинної системи та мозкового кровообігу, що включають, наприклад, атеросклероз, мігрень, артеріальну гіпертензію, септичний шок, серцеві або церебральні інфаркти ішемічного або геморагічного походження, ішемії та тромбози. - розладів центральної або периферичної нервової системи, таких як, наприклад, невродегенеративні захворювання, серед яких можна відмітити мозковий інфаркт, субарахноїдальний крововилив, старіння, старечі деменеції, включаючи хворобу Альцгеймера, хорею Гентінгтона, хворобу Паркінсона, хворобу Кпейцфельда Якоба, бічний аміотрофічний склероз, і також невралгічні болі, травми головного або спинного мозку, опіоманію, хронічний алкоголізм, привикання до інших наркотичних засобів, розлади ерекції та репродукції, розлади розумових здібностей, енцефалопатїї, енцефалопатії вірусного або токсичного походження. - розладів скелетної мускулатури та нервовом'язових синапсів (міопатія, міозит) і також хвороб шкіри. - проліферативних та запалювальних хвороб, таких як, наприклад, атеросклероз, легенева гіпертензія, утр уднене дихання при гломерулонефриті, портальна гіпертензія, псоріаз, ревматоїдний артроз та артрит, фібрози, амілоїдози, запалення шлунково-кишкової системи (коліт, хвороба Крона) або легеневої системи та дихальних шляхів (астма, синусити, риніти). - патологій, пов'язаних із трансплантацією органів. - аутоімунних та вірусних захворювань, таких як, наприклад, вовчак, сіда, паразитарні та вірусні інфекції, діабет, тромботичний склероз. - раку. - невралгічних захворювань, що пов'язані з інтоксикаціями (отруєння кадмієм, вдихання nгексану, пестицидів, гербіцидів), з лікуванням (радіотерапія) або з розладами генетичного походження (хвороба Вільсона). 9 70921 10 - усіх патологій, що характеризуються надлишCO-Y'-, -Y'-CO, -N(R 3)-Y'-, -Y'-N(R 3)-, Y'-CH2-N(R3)ковим продукуванням або дисфункцією NO та/або CO-, -Ο-Υ'-, -Υ'-Ο-, -S-Y'-, -Y'-S-, -Υ'-Ο-Υ'-, -Υ'-Ν(R3)ROS. Υ' або один зв'язок, Υ' являє собою -(СН2)n-, де n Експериментальні свідчення участі NO або дорівнює від 0 до 6; ROS у перелічених патологіях містяться у J. Med. Het являє собою гетероцикл, що містить від 1 Chem. (1995) 38, 4343-4362; Free Radic. Biol. Med. до 5 гетероатомів, котрі вибираються із О, N, S і (1996) 20, 675-705; The Neuroscientist (1977) 3, можуть бути заміщені одним або кількома замісни327-333. ками X'-OR3, X'-NR3, X'-S-R3 і такими як, наприКрім того, автори даного винаходу у попереклад, дніх патентах вже дали опис інгібіторів NO синтаз оксетан, пірол, піролідин, фуран, тетрагідрота їх застосування (патенти US 5081148; US фуран, тіофен, тетрагідротіофен, сульфолан, імі5360925) і пізніше встановили зв'язок цих інгібітодазол, імідазолін, дигідроімідазол-2-он, дигідрів із продуктами, що мають властивості антиокроiмідазол-2-тіон, оксазол, ізоксазол, оксазолін, сидантів або інгібіторів активних радикалів (заявка ізоксазолін, оксазолідин, оксазолідинон, тіазол, на патент не опублікована). тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4Предметом даного винаходу є нові похідні 2триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1(імінометил)аміно-фенілу, їх одержання та застодіоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, сування у терапії. тетразол, тетрагідропіридин, азетидин, за винятСполуки даного винаходу відповідають загаком наступних гетероциклів: піперазинів, гомопіпельній формулі (І): разинів, 4-амінопіперидину; за умови, якщо А являє атом водню, Het не є таким радикалом як піперидин, піролідин або морфолін. Сполуки загальної формули (І), що містять один або кілька асиметричних центрів, являють собою ізомерні форми. Рацемати та енантіомери у якій: зазначених сполук так само входять в обсяг даноА являє собою атом водню або, краще, аромаго винаходу. Сполуки даного винаходу також мотичну сполуку, що відповідає структурі: жуть існувати у вигляді основ або кислотноадитивних солей. Ще один аспект даного винаходу стосується сполук загальної формули (І), у яких: А являє собою атом водню або, краще, ароматичну сполуку, що відповідає структурі: у якій: R1 та R2 являють собою, незалежно, водень, галоген, групу ОН, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, лінійний або розгалужений алкокси-радикал, що у якій: містить від 1 до 6 атомів вуглецю, R1 та R2 являють собою, незалежно, лінійний R3 відповідає водню, лінійному або розгалужеабо розгалужений алкіл-радикал, що містить від 1 ному алкіл-радикалу, що містить від 1 до 6 атомів до 6 атомів вуглецю, або лінійний або розгалужевуглецю, або радикалу -COR4, R4 являє собою ний алкокси-радикал, що містить від 1 до 6 атомів алкіл-радикал, що містить від 1 до 6 атомів вуглевуглецю, цю, R3 відповідає водню або лінійному або розгаабо луженому алкіл-радикалу, що містить від 1 до 6 атомів вуглецю; В являє собою 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибираються із О, S, N, і більш конкретно: тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, В являє собою лінійний або розгалужений алщо вибираються із лінійного або розгалуженого кіл-радикал, що містить від 1 до 6 атомів вуглецю, алкілу з 1-6 атомами і вуглецю, алкокси-радикалу з феніл, піридиніл або 5-членний гетероцикл, котрий 1-6 атомами вуглецю або галогену; містить від 1 до 4 гетероатомів, що вибираються із X являє собою -NH-CO-X'-, -CH=, -CO- або О, S, N, і більш конкретно: тіофен, фуран, пірол один зв'язок, або тіазол, у яких вуглецеві атоми заміщені при X' відповідає -(СН2)n-, де n дорівнює від 0 до 6; необхідності однією або більшою кількістю груп, Υ являє собою -У-, -Y'-NH-CO-, -Y'-CO, -Y’-О-, що вибираються із лінійного або розгалуженого Y'-O-Y'-, -Y’-N(R 3)-Y' або один зв'язок, Y' являє соалкілу з 1-6 атомами вуглецю, алкокси-радикалу з бою -(СН2)n-, де n дорівнює від 0 до 6; 1-6 атомами вуглецю або галогену; Het являє собою гетероцикл, що містить від 1 X являє собою -CO-N(R3)-X'-, NH-CO-X'-, -СН=, до 5 гетероатомів, які вибираються із О, N, S і мо-CO- або один зв'язок, жуть бути заміщені одним або кількома замісникаX' відповідає -(СН2)n-, де n дорівнює від 0 до 6; ми X'-OR3, X'-NR3, X'-S-R3 і такими як, наприклад, Υ являє собою -Υ'-, -CO-NH-Y', -Y'-NH-CO-, оксетан, пірол, піролідин, фуран, тетрагідро 11 70921 12 фуран, тіофен, тетрагідротіофен, сульфолан, імітіофенкарбоксімідаміду; дазол, імідазолін, дигідроімідазол-2-он, дигідроімі- фумарату Ν-[4-(1,2,3,6-тетрагідропіридин-1дазол-2-тіон, оксазол, ізоксазол, оксазолін, ізоксаіл)феніл]-2-тіофенкарбоксімідаміду; золін, оксазолідин, оксазолідинон, тіазол, тіазолін, хлорогідрату N-[4-(1Н-імідазол-1тіазолідин, тіазолідинон, гідантоїн, 1,2,4-триазол, ілметил)феніл]-2-тіофенкарбоксімідаміду; 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1-діоксид-1,2,5N-[4-(2-тіазолідиніл)етил)феніл]-2тіадіазолідин, 1,2,4-триазол-3-он, тетразол, тетратіофенкарбоксімідаміду; гідропіридин, азетидин, за винятком наступних йодогідрату Ν-(4-[2-(1Η-імідазол-1гетероциклів: піперазинів, гомопіперазинів, 4іл)етил]феніл]-2-тіофенкарбоксімідаміду; амінопіперидину; - фумарату N-(4-[2-(1,2,3,6-тетрагідропіридинза умови, якщо А являє атом водню, Het не є 1-іл)етил]феніл)-2-тіофенкарбоксімідаміду; таким радикалом як піперидин, піролідин або - N-[4-(3-тіазолідинілкарбонілметил)феніл]-2морфолін. тіофенкарбоксімідаміду; Особливо ж даний винахід стосується сполук фумарату N-(4-{[2загальної формули (І), у яких: А являє собою атом тіазолідиніл]карбоніламінометил}феніл)-2водню або, краще, ароматичну сполуку, що відпотіофенкарбоксімідаміду; відає структурі: йодогідрату N-(3,5-ди-t-бутил-4гідроксифеніл)-5-[4-{іміно(2тієніл)метиламіно}феніл]-2-фуранкарбоксаміду; хлорогідрату 3-(3,5-ди-t-бутил-4гідроксифеніл)-1-[4-{іміно(2тієніл)метиламіно}феніл]-2,5-імідазолідиндіону; у якій: хлорогідрату 2-(3,5-ди-t-бутил-4R1 та R2 являють собою, незалежно, лінійний гідроксифеніл)-3-[4-{іміно(2або розгалужений алкіл-радикал, що містить від 1 тієніл)метиламіно}феніл]-4-тіазолідинону; до 6 атомів вуглецю, або лінійний або розгалужефумарату 5-[(3,5-ди-t-бутил-4ний алкокси-радикал, що містить від 1 до 6 атомів гідроксифеніл)метилен]-1-метил-3-[4-{іміно(2вуглецю, тієніл)метиламіно}феніл]-2.4-імідазолідиндіону; R3 відповідає водню або лінійному або розга- хлорогідрату 2-(S)-4-(S)-N-[4-гідрокси-3,5-біслуженому алкіл-радикалу, що містить від 1 до 6 (1,1-диметилетил)-феніл]-4-{4-[(іміно(2атомів вуглецю; тієніл)метил)аміно]фенокси}-пролінаміду; В являє собою тіофеновий цикл, у якому вугхлорогідрату 5,6-дигідро-N-{4-[(іміно(2лецеві атоми заміщені при необхідності однією або тієніл)метил)аміно]феніл}-1-(2Н)-піридин карбокбільшою кількістю груп, що вибираються із лінійносаміду; го або розгалуженого алкілу з 1-6 атомами вуглефумарату N-[4-пдрокси-3,5-біс-(1,1цю, алкокси-радикалу з 1-6 атомами вуглецю або диметилетил)-феніл]-2-(R,S)-{4-[(iмінo(2галогену; тієніл)метил)аміно]феніл}-4-(R)-тіазолідин карбокX являє собою -NH-CO-X'-, -CH=, -CO- або саміду; один зв'язок, - йодогідрату N-[3,5-біс-(1,1-диметилетил)-4X' відповідає -(СН2)n-, де n дорівнює від 0 до 6; гідроксифеніл]-2-{4-[(іміно(2Υ являє собою -Υ’-, -Y'-NH-CO-, -Y'-CO, -Y'-O-, тієніл)метил)аміно]феніл}-4-тіазолкарбоксаміду -Y'-O-Y’-, -Y'-N(R 3)-Y' або один зв'язок, Υ' являє - дихлорогідрату N-[3,5-біс-(1,1-диметалетил)собою -(СН2)n-, де n дорівнює від 0 до 6; Het являє 4-гідроксифеніл]-4(S)-{4-[(іміно(2собою гетероцикл, що містить від 1 до 5 гетеротієніл)метил)аміно]фенокси}-піролідин-2-(R)атомів, котрі вибираються із О, N, S і можуть бути карбоксаміду заміщені одним або кількома замісниками Х'-ОR3, - хлорогідрату метил-1-[(3,4-дигідро-6-гідроксиX'-NR3, X'-S-R3 і такими як, наприклад, 2,5,7,8-тетраметил-2-Н-[1]-бензопіран-2оксетан, пірол, піролідин, фуран, тетрагідроіл)карбоніл]-4-(S)-{4-[(іміно(2фуран, тіофен, тетрагідротіофен, сульфолан, імітієніл)метил)аміно]фенокси}-піролідин-2-(S)дазол, імідазолін, дигідроімідазол-2-он, дигідроімікарбоксилату дазол-2-тіон, оксазол, ізоксазол, оксазолін, хлорогідрату 1-[(3,4-дигідро-6-гідроксиізоксазолін, оксазолідин, оксазолідинон, тіазол, 2,5,7,8-тетраметил-2-Н-[1]-бензопіран-2тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4іл)карбоніл]-3-(S)-{4-[(іміно(2триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1тієніл)метил)аміно]фенокси}-піролідину діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, 3-{[(3,4-дигідро-6-гідрокси-2,5,7,8тетразол, тетрагідропіридин, азетидин, за виняттетраметил-2Н-[1]-бензопіран-2-іл)карбоніл]аміно}ком наступних гетероциклів: піперазинів, гомопіпе1-{4-[(іміно(2-тієніл)метил)аміно]феніл}піролідину разинів, 4-амінопіперидину; - хлорогідрату 4-[3,5-біс-(1,1-диметилетил)-4за умови, якщо А являє атом водню, Het не є гідроксифеніл]-N-{4[(іміно(2таким радикалом як піперидин, піролідин або тієніл)метил)аміно]бензоїл}-N-метил-1Н-імідазолморфолін. 2-метанаміну Даний винахід стосується, переважно, наступ- йодогідрату N-[3,5-біс-(1,1-диметилетил)-4них сполук: гідроксифеніл]-1-{4-[(іміно(2- йодогідрату N-[4-(1Н-імідазол-1-іл)феніл]-2тієніл)метил)аміно]феніл}-1Н-пірол-2-карбоксаміду тіофенкарбоксімідаміду; - йодогідрату 1-[3,5-бю-(1,1-диметилетил)-4N-[4-(3-тіазолідинілметил)феніл]-2гідроксифеніл]-3-{4-[[іміно(2 13 70921 14 тієніл)метил]аміно]феніл]карбоніл}-2Так само, предметом даного винаходу є опиімідазолідинону сані вище сполуки загальної формули (І) або їх - йодогідрату 3-[3,5-б|'с-(1,1-диметилетил)-4фармацевтично прийнятні солі як медикаменти. гідроксифеніл]-4,5-дигідро-N-{4-[[іміно(2Винахід стосується також фармацевтичних компотієніл)метил]аміно]феніл}-5-ізоксазолацетаміду зицій, що містять зазначені сполуки або їх фарма- хлорогідрату 4-[3,5-біс-(1,1-диметилетил)-4цевтично прийнятні солі, і застосування цих сполук гідроксифеніл]-N-{4-[[іміно(2або фармацевтично прийнятних солей для виготієніл)метил)аміно]феніл}-N-метил-2товлення медикаментів, призначених для інгібутіазолметанаміну вання NО синтаз, інгібування процесу ліпідного - хлорогідрату 4-[3,5-біс-(1,1-диметилетил)-4переокислення або для забезпечення подвійної гідроксифеніл]-N-{4-[[іміно(2функції інгібування NО синтаз та процесу ліпідного тієніл)метил)аміно]феніл}-N-метил-1Н-імідазол-2перокислення. метанаміну Під фармацевтично прийнятною сіллю мають- 3-[3,5-біс-(1,1-диметилетил)-4-гідроксифеніл]ся на думці солі приєднання неорганічних кислот, 4,5-дигідро-5-{2-{4-[[іміно(2такі як хпорогідрати, сульфати, фосфати , дифостієніл)метил)аміно]фенокси)етил}ізоксазолу фати, бромогідрати, йодогідрати та нітрати, або - хлорогідрату 1-{[3,5-біс(1,1-диметилетил)-4органічних кислот, такі як ацетати, малеати, фугідроксифеніл]-аміно}карбоніл}-3-{4-[(іміно(2марати, тартрати, сукцинати, цитрати, лактати, тієніл)метил)аміно]фенокси}азетидину метан сульфонати, р-толуолсульфонати, памоати, - хлорогідрату 1-(2-гідрокси-5-метоксибензоїл)оксалати та стеарати. Так само, в обсяг даного 3-{4-[(іміно(2винаходу входять, якщо можуть бути використані, тієніл)метил)аміно]фенокси}азетидину солі, що утворені з таких основ як гідроксиди нахлорогідрату 1-[(3,4-дигідро-6-гідрокситрію або калію. Інші приклади фармацевтично 2,5,7,8-тетраметил-2Н-[1]-бензопіран-2прийнятних солей можна знайти у "Pharmaceutical іл)карбоніл]-4-[4-[(іміно(2Salts", J. Pharm. Sci. 66: 1 (1977). тіеніл)метил)аміно]фенокси}-піперидину Фармацевтична композиція може бути у формі хлорогідрату 1-[(3,4-дигідро-6-гідрокситвердої речовини, наприклад, порошків, гранул, 2,5,7,8-тетраметил-2Н-[1]-бензопіран-2таблеток, капсул, ліпосом або супозиторіїв. Відпоіл)карбоніл]-3-{4-[(іміно(2відними твердими носіями можуть бути, напритієніл)метил)аміно]фенокси}-азетидину клад, фосфат кальцію, стеарат магнію, тальк, цукабо їх солей чи енантіомерів. ри, лактоза, декстрин, амідон, желатин, целюлоза, Більша перевага у даному винаході віддається метилцелюлоза, натрій карбоксиметилцелюлоза, наступним сполукам: полівінілпіролідон та віск. - йодогідрату N-[4-(1Н-імідазол-1-іл)феніл]-2Фармацевтичні композиції, що містять сполуки тіофенкарбоксімідаміду; даного винаходу, можуть також бути у рідкій фор- фумарату N-[4-(1,2,3,6-тетрагідропіридин-1мі, наприклад, розчинів, емульсій, суспензій та іл)феніл]-2-тіофенкарбоксімідаміду; сиропів. Відповідними рідкими носіями можуть йодогідрату N-(4-[2-(1H-імідазол-1бути, наприклад, вода, органічні розчинники, такі іл)етил]феніл]-2-тіофенкарбоксімідаміду; як гліцерин або гліколі, так само як і їх суміші у - фумарату N-(4-[2-(1,2,3,6-тетрагідропіридинрізних пропорціях у воді. 1-іл)етил]феніл)-2·-тіофенкарбоксімідаміду; Призначення ліків, згідно з даним винаходом, - N-[4-(3-тіазолідинілкарбонілметил)феніл]-2може бути місцевим, оральним, парентеральним, тіофенкарбоксімідаміду; шляхом внутрішньом'язової ін'єкції і таке інше. хлорогідрату 3-(3,5-ди-t-бутил-4Доза призначення, передбачена для ліків згідгідроксифеніл)-1-[4-{іміно(2но з даним винаходом, складає від 0,1мг до 10г, у тієніл)метиламіно}феніл]-2,5-імідазолідиндіону; залежності від застосованої активної сполуки. хлорогідрату 2-(3,5-ди--t-бутил-4Крім того, даний винахід пропонує, як нові гідроксифеніл)-3-[4-{іміно(2промислові продукти, проміжні сполуки синтезу тієніл)метиламіно}феніл]-4-тіазолідинону; продуктів загальної формули (І), а саме, сполуки фумарату 5-[(3,5-ди-t-бутил-4загальних формул (II), (III), (V), (VI) та (VII). гідроксифеніл)метилен]-1-метил-3-[4-{іміно(2тієніл)метиламіно}феніл]-2,4-імідазолідиндіону; - хлорогідрату 2-(S)-4-(S)-N-[гідрокси-3,5-біс(1,1-диметилетил)-феніл]-4-{4-[(іміно(2тієніл)метил)аміно]фенокси}-пролінаміду; хлорогідрату 5,6-дигідро-N-{4-[(іміно(2тієніл)метил)аміно]феніл}-1-(2Н)-піридин карбоксаміду; хлорогідрату N-[4-гідрокси-3,5-біс-(1,1диметил)етил-феніл]-2-{4-[(іміно(2тієніл)метил)аміно]феніл}-4-тіазол карбоксаміду; або їх солям чи енантіомерам. Нарешті, особлива увага в даному винаході віддається фумарату N-[4-(1,2,3,6тетрагідропіридин-1-іл)феніл]-2тіофенкарбоксімідаміду або його солям. 15 70921 16 ми як, наприклад, оксетан, пірол, піролідин, фуран, тетрагідрофуран, тіофен, тетрагідротіофен, сульфолан, імідазол, імідазолін, дигідроімідазол-2-он, дигідроімідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізоксазолін, оксазолідин, оксазолідинон, тіазол, тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, тетразол, тетрагідропіридин, азетидин, за винятком наступних гетероциклів: піперазинів, гомопіперазинів, 4-амінопіперидину; Gp являє собою захисну гр упу амінної функції, яка, переважно, розщеплюється у кислому безводному середовищі, таку, наприклад, як карбамати t-бутилу, три хлоретилу або триметилсилілетилу, або додатково тритильну груп у. Нарешті, даний винахід пропонує способи одержання сполук загальної формули (І), що визначені вище, і які включають, наприклад, реакцію у нижчому спирті, такому як метанол, етанол, ізоу яких: пропіловий спирт або t-бутанол, краще, в ізопропіА являє собою ароматичну сполуку, що відполовому спирті, при температурі у межах від 20 до відає структурам: 90°С, наприклад, при 50°С, та протягом 48 годин, краще, від 15 до 24 годин, при необхідності у присутності DMF, сполуки загальної формули (III) у яких: R1 та R2 являють собою, незалежно, водень, галоген, групу ОН, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, лінійний або розгалужений алкокси-радикал, що містить від 1 до 6 атомів вуглецю, R3 відповідає водню, лінійному або розгалуженому алкіл-радикалу, що містить від 1 до 6 атомів вуглецю, або радикалу -COR4, R4 являє собою алкіл-радикал, що містить від 1 до 6 атомів вуглецю, або В являє собою лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, феніл, піридиніл або 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибираються із О, S, N, і більш конкретно: тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибираються із лінійного або розгалуженого алкілу з 1-6 атомами вуглецю, алкокси-радикалу з 1-6 атомами вуглецю або галогену; X являє собою -CO-N(R3)-X'-, -NH-CO-X'-, CH=, -CO- або один зв'язок, X' відповідає -(СН2)n-, де n дорівнює від 0 до 6; Υ являє собою -Y’-, -CO-NH-Y', -Y'-NH-CO-, CO-Y'-, -Y'-CO, -N(R 3)-Y'-, -Y'-N(R 3)-, Y'-CH2-N(R3)CO-, -O-Y'-, -Y'-O-, -S-Y'-, -Y'-S-, -Y’-О-Y’-, -Υ'-Ν(R3)Υ' або один зв'язок, Υ' являє собою -(СН2)n-, де n дорівнює від 0 до 6; Het являє собою гетероцикл, що містить від 1 до 5 гетероатомів, котрі вибираються із О, N, S і можуть бути заміщені одним або кількома замісниками Х'-O 3, X'-NR3, X'-S-R3 і таки зі сполукою загальної формули (IV) вказана сполука загальної формули (IV) може бути при необхідності переведена у сіль за допомогою мінеральної кислоти G, В визначена вище, і L є відщеплюваною групою, що являє собою алкокси-радикал, тіоалкіл-радикал, сульфонову кислоту, галоїдну сполуку, ариловий спирт або тазил (інші відщеплювані групи, добре відомі фахівцям у даній галузі, котрі можуть бути при необхідності використані в даному винаході, розглянуті у роботі Ad vanced Organic Chemistry, J. March, 3rd Edition (1985), Me Graw-Hill, p.315). Краще, коли G являє собою НСІ, НВr або НІ. Інші передбачені способи одержання сполук можна знайти в літературі (наприклад, The Chemistry of Amidines and Imidates, Vol.2, Saul Patai and Zvi Rappoport, John Wiley & Sons, 1991). У зазначених вище способах сполуки загальних формул (І), (III), (IV), (VI) та (VII) є такими, де: А являє собою атом водню або ароматичну сполуку, що відповідає структурам: у яких: R1 та R2 являють собою, незалежно, водень, галоген, групу ОН, лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, лінійний або розгалужений алкокси-радикал, що містить від 1 до 6 атомів вуглецю, 17 70921 18 R3 відповідає водню, лінійному або розгалужерозчині етилацетату з добавкою етанолу в присутному алкіл-радикалу, що містить від 1 до 6 атомів ності SnCI2 (J. Heterocyclic Chem. (1987), 24, 927вуглецю, або радикалу -COR4, R4 являє собою 930; Tetrahedron Letters (1984), 25, (8), 839-842) алкіл-радикал, що містить від 1 до 6 атомів вуглеабо із використанням Ni Ренея, що вводиться у цю, гідраті гідразину (Monatshefte fur Chemie, (1965), або 126, 725-732). В являє собою лінійний або розгалужений алкіл-радикал, що містить від 1 до 6 атомів вуглецю, феніл, піридиніл або 5-членний гетероцикл, котрий містить від 1 до 4 гетероатомів, що вибираються із О, S, N, і більш конкретно: тіофен, фуран, пірол або тіазол, у яких вуглецеві атоми заміщені при необхідності однією або більшою кількістю груп, що вибираються із лінійного або розгалуженого алкіл-радикалу з 1-6 атомами вуглецю, алкоксирадикалу з 1-6 атомами вуглецю або галогену; X являє собою -CO-N(R3)-X'-, -NH-CO-X'-, -СН= -CO- або один зв'язок, X' відповідає -(СН2)n-, де n дорівнює від 0 до 6; Υ являє собою -Υ-, -CO-NH-Y', -Y'-NH-CO-, CO-Y'-, -Y'-CO, -N(R 3)-Y’-, -Y’-N(R 3)-, Y'-CH2-N(R3)CO-, -O-Y'-, -Y'-O-, -S-Y’-, -Y'-S-, -Y'-O-Y'-, -Y'-N(R 3)Y' або один зв'язок, Υ' являє собою -(СН2)n-, де n дорівнює від 0 до 6; Het являє собою гетероцикл, що містить від 1 до 5 гетероатомів, котрі вибираються із О, N, S і можуть бути заміщені одним або кількома замісниками X'-OR3, X'-NR3, X'-S-R3 і такими як, наприклад, оксетан, пірол, піролідин, фуран, тетрагідрофуран, тіофен, тетрагідротіофен, сульфолан, імідазол, імідазолін, дигідроімідазол-2-он, дигідроімідазол-2-тіон, оксазол, ізоксазол, оксазолін, ізоксазолін, оксазолідин, оксазолідинон, тіазол, тіазолін, тіазолідин, тіазолідинон, гідантоїн, 1,2,4триазол, 1,3,4-оксадіазол, 1,3,4-тіадіазол, 1,1діоксид-1,2,5-тіадіазолідин, 1,2,4-триазол-3-он, тетразол, тетрагідропіридин, азетидин, за винятком наступних гетероциклів: піперазинів, гомопіперазинів, 4-амінопіперидину; Gp являє собою захисну гр упу амінної функції, котра, переважно, розщеплюється у кислому безводному середовищі, таку, наприклад, як карбамати t-бутилу, трихпоретилу або триметилсилілетилу, або додатково тритильну гр упу. Згідно з даним винаходом, сполуки загальної формули (І) можуть бути одержані у спосіб, викладений нижче. Одержання сполук загальної формули (І): Сполуки загальної формули (І) можуть бути одержані з проміжних сполук загальних формул (II), (III) або (V) у відповідності до схеми 1. Відновлення нітрогрупи проміжних сполук загальної формули (II) проводиться, загалом, за реакцією каталітичної гідрогенізації в етанолі, в присутності Pd/C, крім випадків, коли дані молекули містять ненасичений зв'язок або атом сірки, що є каталітичною отрутою для Pd/C. У цих випадках селективне відновлення нітрогрупи проводиться, наприклад, шляхом нагрівання даного продукту в Одержані у такий спосіб похідні аніліну загальної формули (III) можуть бути піддані реакції конденсації з похідними загальної формули (IV), наприклад, похідними типу О-алкіл тіомідат або Sалкіл тіомідат, що призведе до одержання кінцевих сполук загальної формули (І) (див. схему 1). Наприклад, коли В = тіофену, конденсацію похідних загальної формули (III) можливо провести з йодогідратом S-метилтіофен тіокарбоксаміду, одержаним згідно з методом, що розглянутий у роботі Ann. Chim. (1962), 7, 303-307. Реакція проводиться шляхом нагрівання у спирті (наприклад, у метанолі або ізопрпанолі), при необхідності у присутності DMF, при температурі, переважно, у межах 50-100°С протягом часу від кількох годин до ночі. Кінцеві молекули загальної формули (І) так само можуть бути одержані іншим синтетичним шляхом, через проміжні сполуки загальної формули (V), котрі несуть функцію гетероциклічного аміну, захи щеного захисною групою "Gp", наприклад, групою 2-(триметилсиліл)етоксиметил (SEM) або іншою захисною групою, що розглянута в роботі Protective groups in organic synthesis, 2d ed., (John Wiley & Sons Inc., 1991). Стадії відновлення та конденсації, котрі проводяться, відповідно, на проміжних сполуках (VI) та (VII), реалізуються за тих же умов, що описані вище. Остання стадія синтезу включає регенерацію, наприклад, у кислому середовищі або у присутності фторид іона захищеної гр упи гетероциклічного аміну. Як альтернатива, проміжні сполуки загальної формули (V) можуть бути безпосередньо транс 19 70921 20 формовані в проміжні сполуки загальної формули сутності такого реактиву сполучення як дициклоге(II) через виділення гетероциклічного аміну шляксилкарбодіімід (DCC), 1,1’-карбонілдиімідазол хом обробки, наприклад, у кислому середовищі (CDI) (J. Med. Chem. (1992), 35 (23), 4464-4472) або у присутності фторид іона. або хлорогідрату 1-(3-диметиламінопропіл)-3Одержання сполук загальних формул (II), (III) етилкарбодиіміду (EDC або WSCI) (John Jones, та (V): The chemical synthesis of peptides, 54 (Clarendon Проміжні сполуки загальних формул (II), (III) та Press, Oxford, 1991)). Синтез карбонових кислот (V) можуть бути одержані різними синтетичними загальної формули (Х.2) розглянуто далі. Проміжні шляхами, що ілюструються нижче. сполуки загальної формули (XII) потім піддають Коли: Неt=імідазол, тетрагідропіридин, тіазолідепротектуванню у кислому середовищі за доподин, дигідроімідазол-2-он могою, наприклад, трифтороцтової кислоти або та Υ = -Υ'-. розчину соляної кислоти в органічному розчинниАміни загальної формули (II), схема 2, у яких ку. А, X, Υ та Het відповідають вище наведеним позначенням, можуть бути одержані нуклеофільним заміщенням комерційних галогенованих похідних загальної формули (IX) гетероциклічним аміном загальної формули (VIII). Реакція проводиться в ацетонітрилі, THF або DMF у присутності основи, такої як К2СО3, у температурному інтервалі 20110°С. Синтез некомерційних гетероциклічних похідних загальної формули (VIII) розглянуто далі. дин Коли: Неt=імідазол, тіазолідин, тетрагідропіри та Υ = -Υ'-. Гетероциклічні аміни загальної формули (III), схема 3, у яких А, X, Υ та Неt відповідають наведеним вище позначенням, одержують у дві стадії з амінів загальної формули (VIII) (див. далі). Змішування бром-похідної загальної формули (X), синтез якої деталізований далі, з аміном загальної формули (VIII) у такому розчиннику як ацетонітрил або DMF у присутності основи дає проміжні сполуки загальної формули (XI). Депротектування амінної функції у середовищі органічної кислоти дає сполуки загальної формули (III). Коли: Неt=тіазолідин та Υ = -CO-Y’Карбоксаміди загальної формули (III), схема 4, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією амінів загальної формули (VIII), описаних ви ще, з карбоновими кислотами загальної формули (Х.2). Карбоксамідні зв'язки утворюються за класичних умов синтезу пептидів (М. Bodanszky et A. Bodanszky, The Practice of Peptide Synthesis, 145 (SpringerVerlag, 1984)) у THF, дихлорметані або DMF у при Коли: Het = тіазолідин та Υ = -CO-NH-Y'-. Карбоксаміди загальної формули (V), схема 5, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією карбонових кислот загальної формули (XIII) з комерційними амінами загальної формули (XIV) за класичних умов синтезу пептидів. Синтез карбонових кислот загальної формули (XIII) розглянуто далі. Коли: Het = тіазол, фуран, пірол, тетрагідропіридин, піролідин та X = -NH-CO-X'-. Карбоксаміди загальної формули (II), схема 6, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією анілінів загальної формули (XV) з карбоновими кислотами загальної формули (XVI) за класичних умов конденсації пептидів. Аніліни загальної формули (XV) одержують гідрогенізацією, у присутності деякої кількості каталізатора Pd/C, відповідних нітробензольних похідних, котрі синтезуються у спосіб, поданий в роботі J. Org. Chem. (1968), 33 (1), 223226). Кислоти загальної формули (XVI), схема 6, що не є комерційними, одержують відомими із літератури способами. Синтез піролів розглянуто у Chem. Heterocycl. Compd., 1982, 18, 375. Заміщені проліни одержують із комерційних гідроксипролінів за методами, розглянутими у J. Org. Chem., 1991, 56, 3009. Синтез похідних тіазолу та тетрагідропіридину розглянуто далі. 21 70921 22 яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують у дві стадії з ізоціанатів загальної формули (XVIII), що описані вище. Реакція етилового ефіру саркозину з ізоціанатами загальної формули (XVIII), що проводиться за експериментальним протоколом (J. Heterocyclic Chem., (1979), 16, 607-608), призводить до утворення геКоли: Het = гідантоїн тероциклу сполук загальної формули (XXI). Заміта Υ = -Υ’-. щення гідантоїну проводиться у присутності слабГідантоїни загальної формули (II), схема 7, у яких А, X, Υ та Het відповідають наведеним вище кої основи, b-аланіну, та альдегіду загальної позначенням, одержують у три стадії з анілінів формули (XX) за експериментальних умов, описазагальної формули (XV), що описані вище. Заміних у J. Med. Chem., (1994), 37, 322-328. щення аніліну етилбромацетатом проводиться у присутності ацетату натрію в етанолі при температурі в межах 60-70°С. Продукт монозаміщення загальної формули (XVII) потім конденсують з ізоціанатом загальної формули (XVIII) в органічному розчиннику, такому, наприклад, як дихлорметан при температурі, близькій до 20°С. Циклізацію сечовини (XIX) проводять шляхом нагрівання при 50°С в етанолі згідно з експериментальним протоколом, наведеним у літературі (J. Heterocyclic Chem., 16, 607-608). Ізоціанати загальної формули (XVIII) синтезують із відповідних комерційних первинних амінів, трифосгену та третинного аміну (J. Org. Chem. (1994), 59(7), 1937-1938). Коли: Het = піролідин, тіазолідин X = NH-CO-X’- та Υ = -Ο-Υ'- або -Υ’-. Карбоксаміди загальної формули (V), схема 10, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією анілінів загальної формули (XV), що описані вище, з кислотами загальної формули (XXII) за класичних умов синтезу пептидів. Схеми синтезу карбонових кислот загальної формули (XXII), що не є комерційними продуктами, розглянуті далі. Коли: Het = тіазолідинон та Y = -Υ’-. Тіазолідинони загальної формули (II), схема 8, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують із комерційних амінів загальної формули (XIV) та альдегідів загальної формули (XX) у присутності меркаптоцтової кислоти за експериментальним протоколом, що поданий у літературі (J. Med. Chem., (1992), 35, 29102912). Коли: Het = гідантоїн X = -СН= та Υ = -Υ'-. Гідантоїни загальної формули (II), схема 9, у Коли: Het = тетрагідропіридин та Υ = -CO-NH-Y’-. Сечовини загальної формули (II), схема 11, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією гетероциклічних амінів загальної формули (VIII), що описані вище, з ізоціанатами загальної формули (XVIII) (порівняй з наведеним вище) у такому розчиннику як дихлорметан при 20°С у присутності третинного аміну (наприклад, діізопропілетиламін). Коли: Het = піролідин, тіазол, тіадїазол та X = -CO-NH-X'-. 23 70921 Карбоксаміди загальної формули (II), схема 12, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією комерційних карбонових кислот загальної формули (XXIII) з амінами загальної формули (XXIV) за класичних умов синтезу пептидів. Синтез амінів загальної формули (XXIV), що не є комерційними продуктами, розглянуто далі. Коли: Het = імідазол, оксазол та тіазол та Υ = -CH(R 3)-N(R3)-CO-Y’-. Карбоксаміди загальної формули (V), схема 13, у яких А, Χ, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією амінів загальної формули (XXV) з комерційними карбоновими кислотами (або хлоридами відповідних кислот) загальної формули (XXVI) за класичних умов синтезу пептидів. Синтез похідних імідазолів загальної формули (XXV) розглянуто далі. Коли: Het = імідазол та Υ = -CH2-N(R3)-Y’-. Аміни загальної формули (V), схема 14, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією амінів загальної формули (XXV) (див. далі) з комерційними галогенованими похідними загальної формули (IX) за умов, що вказані вище. Коли: Het = дигідроімідазол-2-он та Y = -СО-Y’. Аміни загальної формули (II), схема 15, у яких А, X, Υ та Het відповідають наведеним вище позначенням, одержують конденсацією амінів загальної формули (VIII) (див. далі) з комерційними галогенованими похідними загальної формули (XXVII), наприклад, у суміші ацетонітрилу та THF і у присутності основи, такої як К2СО3. 24 Коли: Het = оксазолідинон та Υ = -Y’-О-. Оксазолідинони загальної формули (II), схема 16, одержують із діолів загальної формули (XXVII), синтез яких описано у літературі (Daumas, Μ, Tetrahedron, 1992, 48, (12), 2373). Карбонати загальної формули (XXVIII) одержують, наприклад, у присутності карбоніл ди-імідазолу (Kutney, J.P., Synth. Commun., 1975, 5 (1), 47) або у присутності трифосгену при зниженій температурі, як зазначено у Synth. Commun., 1994, 24 (3), 305. Оксазолідинони утворюються при нагріванні амінів загальної формули (XV) з карбонатами загальної формули (XXVIII) у присутності кислотного каталізатора, такого як ZnCI2, у ксилолі із застосуванням оберненого холодильника для вилучення води, що утворюється під час реакції (Laas, H., Synthesis, 1981, 958). Коли: Het = ізоксазолін, ізоксазол, оксазол, тіазол та Υ = -Y'-C O-NH-Y'-. Карбоксаміди загальної формули (II), схема 17, у яких А, X, Υ та Het відповідають наведеним вище позначенням, можуть бути отримані з комерційних амінів загальної формули (XIV) та карбонових кислот загальної формули (XXVIII) шляхом конденсації у присутності ізобутилхлороформіату (Org. Prep. Proced. Int., (1975), 7, 215). Оксазоли загальної формули (XXVIII) одержують згідно з експериментальним протоколом, поданим у Tetrahedron Lett, 1994, 35 (13), 2039. Це стосується також синтезу тіазолів загальної формули (XXVIII): J. Med. Chem., 1983, 26, 884. Приготування ізоксазолінів розглянуто далі. Коли: Het = піролідин, піперидин X = -CO-NHта Υ = -Ο-Υ'-. 25 70921 Карбоксаміди загальної формули (II), схема 18, у яких А, X, Υ та Het відповідають наведеним вище позначенням, можуть бути одержані конденсацією комерційних карбонових кислот загальної формули (ХХІІІ) з амінами загальної формули (XXIX) за класичних умов синтезу пептидів. Синтез амінів загальної формули (XXIX) розглянуто нижче. зол 26 Коли: Het = ізоксазолін, оксазол, тіазол, іміда та Υ = -Υ'-Ο-Υ’- або -Y'-N(R3)-Y'-. Прості ефіри загальної формули (II), схема 19, у яких А, X, Υ та Het відповідають наведеним вище позначенням, можуть бути одержані зі складних ефірів загальної формули (XXVIII.4), схема 17.1, за реакцією з гідридами, наприклад, LiAIH4, у такому розчиннику як, наприклад, безводний THF. Одержані у такий спосіб первинні спирти потім піддають конденсації з галогенопохідними загальної формули (IX) за допомогою такої основи як, наприклад, КОН в органічному середовищі та у присутності такого міжфазового каталізатора як Алікат 336 (I'Aliquat 336). Первинні спирти (XXXI) можуть бути так само активовані у формі сульфонат-похідних тозилхлоридом у присутності піридину, що призведе до утворення проміжних сполук загальної формули (XXXII). Потім конденсацію спиртів загальної формули (ХХІІ.2) проводять у присутності такої сильної основи як, наприклад, NaH, в апротонному розчиннику (THF або DMF) при температурі у межах 20-80°С з одержанням простих ефірів загальної формули (II). Так само, аміни загальної формули (II), схема 19, одержують заміщенням тозилатної групи проміжних сполук загальної формули (XXXII), що отримані класичним способом зі спиртів загальної формули (XXXI) та тозилхлориду у присутності піридину, комерційними амінами загальної формули (XXX) за реакцією у такому розчиннику як, наприклад, ацетонітрил або DMF у присутності основи (К2СО3) при температурі в межах 20-85°С. Коли: Het = азетидин X = -CO-NH та Υ = -Ο-Υ’-. Карбоксаміди загальної формули (III), схема 20, у яких А, X, Υ та Het відповідають наведеним вище позначенням, можуть бути одержані конденсацією комерційних карбонових кислот загальної формули (XXIII) з амінами загальної формули (XXXII) .за класичних умов пептидного синтезу. Синтез амінів загальної формули (XXXII) розглянутий далі. Депротектування даного аніліну проводиться за допомогою такої сильної кислоти як, наприклад, трифтороцтова, при необхідності у присутності триетилсілану. Коли: Het = азетидин X = -NH-CO-X'та Υ = -Ο-Υ’-. Сечовини загальної формули (III), схема 21, у яких А, X, Υ та Het відповідають наведеним вище позначенням, можуть бути одержані приєднанням амінів загальної формули (XXXII) до ізоціанатів (XXXIV), одержаних за реакцією амінів загальної формули (XV) з три фосгеном у присутності такого третинного аміну як, наприклад, діізопропілетиламін у такому нейтральному розчиннику як дихлорметан (J. Org. Chem. (1994), 59 (7), 1937-1938). Одержані у такий спосіб сечовини загальної формули (XXXV) піддають депротектуванню обробкою у середовищі сильної кислоти як розглянуто вище. 27 70921 28 Синтез амінів загальної формули (XXXII) описаний у 2 стадії з анілінів загальної формули (XV) (див. нижче. вище), котрі конденсують з 2-хлоретил ізоціанатом у DMF при 20°С з утворенням сечовин загальної формули (VIII.4). Циклізація для одержання (VIII) потім проводиться шляхом обробки в основному середовищі, наприклад, у tBuOK у D MF. Коли: Het = тіазол та Y = -CH2-N(R3)-Y’-. Аміни загальної формули (II), схема 22, у яких А, X, Υ та Het відповідають наведеним вище позначенням, можуть бути одержані конденсацією амінів загальної формули (XXV) (див. далі) з комерційними галогенованими похідними загальної формули (IX) за умов, що визначені вище. Одержання різних проміжних сполук синтезу: Синтез проміжних сполук (VIII): Синтез проміжних сполук загальної формули (VIII) ілюструється схемами 2.1 та 2.2. Проміжні сполуки загальної формули (VIII), схема 2.1, можуть бути одержані, наприклад, у три стадії з 4-імідазол карбонової кислоти. Протектування азоту даного гетероциклу проводять за допомогою (Вос)2О у присутності такої основи як К2СО3 у DMF. Конденсацію з амінами загальної формули (XV) (див. вище) реалізують у класичний спосіб за умов синтезу пептидів з одержанням проміжних сполук загальної формули (VIII.3). Даний гетероциклічний амін регенерують обробкою у кислотному середовищі, зокрема, у три фтороцтовій кислоті, з одержанням проміжних сполук загальної формули (VIII). Синтез проміжних сполук (X): Проміжні сполуки загальної формули (X), схема 3.1, можуть бути одержані з карбонових кислот загальної формули (Х.1) комерційного походження. Після захисту амінної групи протектором у формі карбамату проводиться селективне відновлення функції карбокислоти літійалюмінієвим гідридом у такому розчиннику як THF при 20°С. Потім проміжну сполуку (Х.3) бромують у присутності тетрабромовуглецю та трифенілфосфін у в такому розчиннику як дихлорметан. Синтез проміжних сполук (ХНІ): Проміжні сполуки загальної формули (XIII), схема 5.1, можуть бути одержані з похідних (R або S) тіазолідин карбонових кислот у присутності (Вос)2О за класичних умов. ДигідроімІдазол-2-они загальної формули (VIII), схема 2.2, можуть бути одержані, наприклад, 29 70921 30 Синтез проміжних сполук (XVI): Проміжні сполуки загальної формули (XVI), схема 6.1, можуть бути одержані з комерційних похідних карбоксамідів загальної формули (XVI.1). Зазначені карбоксаміди обробляють реактивом Лоусона (Lawesson) у такому розчиннику як 1,4діоксан протягом 2-3 годин при температурі близько 25°С з використанням оберненого холодильника. Одержані у такий спосіб тіокарбоксаміди загальної формули (XVI.2) потім обробляють етилбромопіруватом при 20 С у DMF згідно з експериментальним протоколом, наведеним у J. Med. Chem., (1983), 26, 884-891, з одержанням тіазолів загальної формули (XVI.3). Омилення даного складного ефіру проводиться протягом 15 годин водним розчином поташу в ацетоні. Синтез проміжних сполук (XXII): Синтез проміжних сполук загальної формули (XXII) розглянуто за схемами 10.1 та 10.2. Тозилатну групу похідних проліну (L або D) загальної формули (XXI 1.1) (Tetrahedron Lett., (1983), 24 (33), 3517-3520), схема 10.1, заміщують алкоголятом похідних загальної формули (XXII.2), що генерується in situ за допомогою такої основи як NaH. Заміщення проводиться при 20°С у такому розчиннику як N-метилпіролідинон, що спричинює відповідну інверсію конфігурації вуглецевого остова (Tetrahedron Lett., (1983), 24 (33), 3517-3520). Одержані у такий спосіб проміжні сполуки загальної формули (XXII.3) потім омилюють за класичним методом спиртовим розчином поташу. Тетрагідропіридини загальної формули (XVI), схема 6.2, можуть бути одержані з комерційної тетрагідро-4-піридин карбонової кислоти. Етерифікацію проводять у класичний спосіб у присутності пара-толуол сульфонової кислоти в метанолі з одержанням проміжної сполуки (XVI.4), яку потім піддають конденсації з галогенованою похідною загальної формули (IX) за умов, що визначені вище. Кислоту загальної формули (XVI) одержують омиленням у присутності, наприклад, LiOH або КОН. 31 70921 32 розчиннику як DMF. Потім продукт конденсації (XXIV.2) піддають депротектуванню у кислотному середовищі з утворенням проміжних сполук загальної формули (XXIV). Проміжні сполуки загальної формули (XXII) можуть бути так само одержані (схема 10.2) конденсацією цистеїну (L або D) з альдегідом загальної формули (XXII.5) згідно з експериментальним протоколом, поданим у J. Оrg. Сhem., (1957), 22, 943-946. Потім даний гетероциклічний амін захищають карбаматним протектором з одержанням проміжних сполук загальної формули (XXII). Альдегіди загальної формули (XXII.5), що не є комерційними продуктами, можуть бути одержані згідно з J. Chem. Soc., Perkin Trans. 1, 1973, 1, 35. Синтез проміжних сполук (XXIV): Синтез проміжних сполук загальної формули (XXIV) розглянуто за схемою 12.1. Конденсацію амінів (R або S) загальної формули (XXIV. 1), схема 12.1, з галогенованими похідними загальної формули (IX) проводять у присутності такої основи як карбонат калію у такому Синтез проміжних сполук (XXV): Синтез проміжних сполук загальної формули (XXV) описано за допомогою схем 13.1, 13.2,13.3 та 13.4. Імідазоли загальної формули (XXV), схема 13.1, можуть бути одержані у 4 стадії з комерційних сполук (XXV.1) та (XXV.2). Конденсацію бромацетофенонів загальної формули (XXV.1) та карбонових кислот загальної формули (XXV.2) проводять у присутності карбонату цезію у DMF. Одержаний кетоефір (XXV.3) піддають циклізації у присутності 15 еквівалентів ацетату амонію нагріванням у суміші ксилолів із одночасовим вилученням води, що утворюється в процесі реакції, і в результаті одержують імідазоли загальної формули (XXV.4). Потім азот із даного гетероциклу захи щають, наприклад, за допомогою 2(триметилсиліл)етоксиметилу (SEM) або другого протектора, що можна знайти у роботі: Protecti ve groups in organic synthesis, 24 ed., (John Wiley & Sons Inc., 1991), з одержанням проміжних сполук загальної формули (XXV.5). Вилучення даного ланцюга з аміну можливо зробити через гідрогеноліз у присутності Pd/C. Як альтернатива, проміжні сполуки загальної формули (XXV.4) можуть бути алкіловані у присутності такої основи як, наприклад, К2СО3 та такого реактиву як R3-X, у такому розчиннику як DMF або ацетонітрил, з утворенням імідазолів загальної формули (XXV.6). Депротектування бічного ланцюга, як показано вище, дозволяє одержати проміжні сполуки загальної формули (XXV). 33 70921 34 Як альтернатива, карбоксильна група гетероциклів загальної формули (XXV.7) може бути відновлена, наприклад, за допомогою NaBH4 з утворенням спиртових похідних загальної формули (XXV.10), схема 13.3, які можуть бути алкіловані у присутності R3-X та такої основи як К2СО3, у такому розчиннику як ацетонітрил або DMF. Вилучення даного бічного ланцюга з аміну, як показано вище, дозволяє одержати проміжні сполуки загальної формули (XXV). Проміжні сполуки загальної формули (XXV), що містять оксазол, тіазол або імідазол так само можуть бути одержані за допомогою інших синтетичних шляхів, таких, що розглянуті у Bioorg. and Med. Chem. Lett, 1993, 3, 915 або Tetrahedron Lett., 1993, 34, 1901. Одержані у такий спосіб проміжні сполуки загальної формули (XXV.7) можуть бути модифіковані, схема 13.2, омиленням, після декарбоксилювання, наприклад термічного, з одержанням дизаміщених гетероциклів загальної формули (XXV.9). Вилучення даного бічного ланцюга з аміну, як показано вище, дозволяє одержати проміжні сполуки загальної формули (XXV). Тіазоли загальної формули (XXV), схема 13.4, 35 70921 36 можуть бути так само одержані у 4 стадії з комерприсутності, наприклад, борогідриду натрію у таційного саркозинамід хлорогідрату. Спочатку дакому розчиннику як, наприклад, безводний THF, ний амін захищають у класичний спосіб за допомодозволяє отримати проміжну сполуку загальної гою tBu карбамату, і карбоксамідну групу формули (XXIX), що несе функцію первинного трансформують у тіокарбоксамід у присутності спирту, не торкаючись нітрогрупи (Rao, А. V. R., J. реактива Лоусона. Утворення тіазольного циклу Chem. Soc. Chem. Commun., 1992, 11, 859). здійснюється за реакцією тіокарбоксаміду з проміжною сполукою загальної формули (XXV.1) за експериментальним протоколом (J. Org. Chem., (1995), 60, 5638-5642). Амінну групу регенерують обробкою проміжної сполуки загальної формули (XXV.12) у середовищі сильної кислоти, наприклад, трифтороцтової. Ізоксазоліни та ізоксазоли загальної формули (XXVIII), схема 17.1, одержують за реакцією комерційних альдегідів загальної формули (XX) з гідроксиламін хлорогідратом. Одержаний у такий спосіб оксим загальної формули (XXVIII. 1) спочатку активують у формі хлориду оксиму загальної формули (XXVIII.2) за реакцією з N-хлорсукцинімідом у DMF і потім піддають реакції зі складними ефірами загальної формули (XXVIII.3) для одержання похідних ізоксазолінів або зі складними ефірами загальної формули (XXVIII.4) для одержання похідних ізоксазолів згідно з експериментальним протоколом (Tetrahedron Lett., 1996, 37 (26), 4455; J. Med. Chem., 1997, 40, 50-60 тa 2064-2084). Наступне омилення ізоксазолінів або ізоксазолів загальної формули (XXVIII.5) здійснюється у класичний спосіб за умов, що визначені вище. Складні ненасичені ефіри загальних формул (XXVIH.3) та (XXVIII.4), що не є комерційними продуктами, можуть бути .одержані за методами, що викладені у літературі (J. Med. Chem., 1987, 30, 193; J. Org. Chem., 1980, 45, 5017). Синтез проміжних сполук (XXIX): Синтез проміжних сполук загальної формули (XXIX) розглянуто за схемами 18.1, 18.2, 18.3 та 18.4. Проміжні сполуки загальної формули (XXIX) можуть бути одержані, схема 18.1, із проміжних сполук загальної формули (XXII.3), що розглянуті вище, обробкою у середовищі сильної кислоти для регенерування функції гетероциклічного аміну. Селективне відновлення карбоксильної групи у Проміжні сполуки загальної формули (XXIX) можуть бути так само одержані, схема 18.2, виходячи із проміжних сполук загальної формули (XXIX.1) (R або S), які готуються аналогічно отриманню сполук загальної формули (XXII.1). Конденсація спиртових похідних загальної формули (XXII.2) з проміжними сполуками загальної формули (XXIX.1) так само описана вище. Вилучення гетероциклічного аміну проводиться у присутності розчину сильної кислоти, наприклад трифтороцтової, в органічному розчиннику. 37 70921 Аміни загальної формули (XXIX), схема 18.3, так само одержуються шляхом заміщення похідних тозилу загальної формули (XXIX.1) комерційними амінами загальної формули (XXX). Вилучення карбаматної групи проміжних сполук загальної формули (ХХІХ.3) проводиться у спосіб, що розглянутий ви ще. Проміжні сполуки загальної формули (XXIX) можуть бути так само одержані, схема 18.4, за реакцією галогенопохідних загальної формули (IX) зі спиртом загальної формули (ХХІХ.4) у присутності такої основи як, наприклад, tBuO-К+ у такому безводному розчиннику як THF. Одержаниу у такий спосіб проміжну сполуку загальної формули (ХХІХ.5) потім депротектують у середовищі сильної кислоти (НСІ або TFA). 38 Синтез проміжних сполук (XXXII): Проміжні сполуки загальної формули (XXXII) можуть бути одержані, схема 20.1, за реакцією галогенопохідних загальної формули (IX) з 1(дифенілметил)-3-гідроксиазетидином комерційного походження (XXXII.1) у присутності такої основи як, наприклад NaH, у такому безводному розчиннику як THF. У даному випадку нітрогрупа проміжної сполуки загальної формули (XXXII.2) відновлюється у присутності SnCI2, як розглянуто вище, з утворенням проміжної сполуки загальної формули (XXXII.3), амін якої потім захищають tButyl карбаматом. Вилучення захисної групи дифенілметилу потім проводять у класичний спосіб через гідрогеноліз у присутності Pd(OH)2 з одержанням інтермедіату загальної формули (XXXII). Якщо окремо не зазначено, всі наукові та те хнічні терміни, що використані в даному тексті, мають значення, що легко зрозумілі фахівцями у галузі даного винаходу. Так само, на всі публікації, заявки, патенти та інші документи, що згадуються у даному тексті, зроблені посилання. Наступні приклади мають на меті проілюструвати вищерозглянуті процедури і ні в якому разі не повинні розглядатись як обмеження обсягу даного винаходу. ПРИКЛАДИ: Приклад 1: Йодогідрат N-[4-(1Н-імідазол-1іл)феніл]-2-тіофенкарбоксімідаміду (1): 39 70921 40 1.1 1-(4-нітрофеніл)-1Н-імідазол: холодильника. 9г (64,5ммоль) карбонату калію та 5г (3,75мл, Осад, що утворився, відфільтровували, мато35,2ммоль) 1-фтор-4-нітробензолу додавали у чний розчин випаровували, і залишок обробляли розчин 2г імідазолу (29,4ммоль) у 14мл DMF. Реа50мл дихлорметану та промивали 3 рази 50мл кційну суміш перемішували протягом 1,5 годин при води. Органічні фази осушували, випарювали та 110°С. Етилацетат (50мл) додавали до середовиочищали на силікагелі у суміші етилацетат/гептан ща, котре промивали 3 рази 50мл води. Органічні (1/2). Потрібний продукт одержали у вигляді безфази осушували над сульфатом магнію та конценбарвного масла (1,5г, 72%). трували під вакуумом. 4,4г одержаного у такий ЯМР 1Н (CDCI3, 100МГц, d): 3,05 (m, 4Н, 2СН2), спосіб продукту (ви хід 80%) у вигляді безбарвного 3,68 (s, 2Н, CH2-S), 4,04 (s, 2Н, СН2), 7,53-7,62масла використовували без додаткового очищен8,17-8,26 (4s, 4Н, Η аром.). ня на наступних стадіях. 2.3 3-(4-амінобензил)-тіазолідин: 3-(4-нітробензил)-тіазолідин (1,1г, 5ммоль) ЯМР 1Н (CDCI3, 100МГц, d): 6,92 (t, 1H, Η аром. імідазол), 7,16 (s, 1Н, Η аром. імідазол), 7,24-7,32змішували з 10мл концентрованої соляної кислоти 8,18-8,27 (4s, 4Н, Η аром.), 7,59 (s, 1 Η, Η аром. при 0°С. Порціями додавали двохводне хлористе імідазол). олово (7,7г, 34ммоль). Дану суміш нагрівали із 1.2 1-(4-амінофеніл)-1Н-імідазол: оберненим холодильником протягом 2 годин, і До безводного метанолу (140мл) додавали 1кислоту випарювали під зниженим тиском. Потім (4-нітрофеніл)-1Н-імідазол (4,4г, 23,5ммоль), і у залишок розводили 20мл води та нейтралізували даний розчин додавали паладій на вуглеці (0,44г). 2Ν розчином соди (разом 100мл). До суміші додаСуміш витримували в атмосфері водню протягом 4 вали 100мл дихлорметану і потім фільтрували годин. Каталізатор відфільтровували, і розчинник через целіт для вилучення із суспензії солей. Орвипарювали до сухого стану. Одержали потрібний ганічну фазу екстрагували, промивали 3 рази 50мл квазічистий продукт з виходом 89% (3,3г). води, осушували, фільтр ували та випарювали до сухого стану за умов зниженого тиску. Потрібний ЯМР 1Н (CDCI3, 100МГц, d): 6,61-6,69-6,95-7,05 продукт очищали на силікагелі у суміші дихлорме(4s, 4H, Η аром.), 6,88 (t, 1H, Η аром. імідазол), тан/метанол (98/2) і одержували у вигляді порошку 7,07 (s, 1Н, Η аром. імідазол), 7,52 (s, 1Н, Η аром. бежевого кольору (0,6г, 63%). Т пл=73-74°С. імідазол). 1.3 Йодогідрат N-[4-(1Н-імідазол-1-іл)феніл]-2ЯМР 1Н (CDCI3, 100МГц, d): 3,02 (m, 4Н, 2СН2), тіофенкарбоксімідаміду (1): 3,44 (s, 2H, СH2), 3,66 (s широка, 2Н, NH2), 4,07 (s, 1-(4-амінофеніл)-1Н-імідазол (0,3г, 1,7ммоль) 2Н, СН2), 6,62-6,71-7,10-7,27 (4s, 4Н, Η аром.). та йодогідрат 5-метил-2-тіофентіокарбоксіміду 2.4 [4-(3-тіазолідинілметил)феніл]-2(0,5г, 1,75ммоль) додавали у розчин 1мл ізопропатіофенкарбоксімідамід (2): нолу та 1мл DMF, і реакційну суміш перемішували 3-(4-амінобензил)-тіазолідин (0,6г, 3ммоль) та протягом 18 годин при 25°С. Утворений осад відйодогідрат S-метил-2-тіофентіокарбоксімід (1,14г, фільтровували та промивали 15мл дихлорметану 4ммоль) переводили у розчин з 7мл суміші ізопрота 15мл етанолу. Одержали потрібний продукт панол/DMF (2/5). Дан у суміш перемішували протя(0,48г, 73%) у сольовій формі (йодогідрат). гом 18 годин при кімнатній температурі. Потім доТпл=252-253°С (розклад). давали 10мл етилацетату, і продукт реакції екстрагували 3 рази 10мл води. Водну фазу збиЯМР 1Н (D MSO, 400МГц, d): 7,24 (s, 1Н, Η рали та підлуговували насиченим розчином гідроаром.), 7,38 (t, 1Н, Η аром.), 7,55-7,57-7,85-7,87 (4s, 4Н, Η аром.), 7,89 (s, 1Н, Η аром.), 8,10 (m, 2Н, карбонату натрію, і потім продукт екстрагували 3 рази 10мл етилацетату. Продукт очищали на силіΗ аром.), 8,50 (s, 1Н, Η аром.). кагелі у суміші дихлорметан/метанол (95/5) і одеІЧ: n C=N (амідин): 1585см -1. ржали у вигляді білого порошку (0,6г, 65%). Приклад 2: N-[4-(3-тіазолідинілметил)феніл1Тпл=161,5-163,5°С. 2-тіофенкарбоксімідамід (2): 2.1 1-бромометил-4-нітробензол: ЯМР 1Н (CDCI3, 400МГц, d): 2,98 (t, 2H, CH2), 3,14 (t, 2Н, СН2), 3,54 (s, 2H, СН2), 4,10 (s, 2Н, 4-нітробензиловий спирт (6г, 39ммоль) змішуСН2), 4,85 (s широка, 2Н, NH2), 6,98 (s, 1Н, Η вали з дихлорметаном (100мл) і додавали чотириаром.), 7,00 (s, 1H, Η аром.), 7,10 (t, 1H, тіофен), бромистий вуглець (14,9г, 45ммоль). При 0°С пор7,34 (s, 1Н, Η аром.), 7,36 (s, 1Н, Η аром.), 7,42 (t, ціями додавали трифенілфосфін (11,8г, 45ммоль). 1H, тіофен), 7,45 (m, 1H, тіофен). Потім дану суміш перемішували при кімнатній температурі протягом 2 годин. Розчин випарювали, і IК: n C=N (амідин): 1593см -1. одержаний продукт очищали на силікагелі у суміші Приклад 3: Фумарат N-[4-(1,2,3,6етилацетат/гептан (1/2). В результаті одержали тетрагідропіридин-1-іл)феніл]-2голчасті кристали білого кольору (7,2г, 85%). тіофенкарбоксімідаміду (3): Тпл=97-98°С. 3.1 1-(4-нітрофеніл)-1,2,3,6-тетрагідропіридин: Одержання потрібного продукту проводилось ЯМР 1Н (CDCI3, 100МГц, d): 4,53 (s, 2H, CH2), за тим самим експериментальним протоколом, що 7,53-7,61-8,18-8,27 (4s, 4H, Η аром.). й для проміжної сполуки 1.1. Замість імідазолу 2.2 3-(4-нітробензил)-тіазолідин: Суміш тіазолідину (0,9г, 10ммоль) та карбонавикористовували 1,2,3,6-тетрагідропіридин. Безбарвне масло. ту калію (2,5г, 18ммоль) в ацетонітрилі (10мл) витримували при 70°С. Потім додавали по краплях ЯМР 1Н (CDCI3, 100МГц, d): 2,33 (m, 2Н, СН2), 1-бромометил-4-нітробензол (2г, 9,2ммоль) у роз3,59 (t, 2H, СН2), 3,90 (m, 2H, СН2), 5,90 (m, 2Н, чині ацетонітрилу (25мл), і реакцію проводили СН=СН), 6,75-6,82-8,07-8,18 (m, 4Н, Η аром.). протягом 2 годин з використанням оберненого 3.2 1-(4-амінофеніл)-1,2,3,6-тетрагідропіридин: 41 70921 42 Одержання потрібного продукту проводилось Пара-амінофенілоцтову кислоту (3г, 20ммоль) за тим самим експериментальним протоколом, що розчиняють у 60мл суміші THF/H2O (2/1). Додають й для проміжної сполуки 2.3. Замість 3-(411мл 10% соди, потім 6г ди-t-бутил дикарбонату нітробензил)-тіазолідину використовували 1-(4(28ммоль) у розчині 50мл суміші THF/H2O (2/1). нітрофеніл)-1,2,3,6-тетрагідропіридин. Безбарвне Перемішують протягом 18 годин при кімнатній темасло. мпературі, потім випаровують THF за умов зниженого тиску. Далі даний розчин підкислюють (рН=2) ЯМР 1Н (CDCI3, 100МГц, d): 2,31 (m, 2H, CH2), 3,21 (t, 2Н, СН2), 3,43 (m, 2H, NH2), 3,56 (m, 2Н, 10% розчином кислого сульфату калію (разом 45мл), і продукт реакції екстрагують триразовим СН2), 5,84 (m, 2Н, СН=СН), 6,75 (m, 4Н, Η аром.). промиванням етилацетатом (3 рази по 50мл). Ор3.3 Фумарат Ν-[4-(1,2,3,6-тетрагідропіридин-1ганічні фази осушують та випарюють з одержаніл)феніл]-2-тіофенкарбоксімідаміду (3): ням 4,32г (87%) чистої 4-(-t-бутоксикарбоніламіно)Одержання потрібного продукту проводилось за тим самим експериментальним протоколом, що бензолоцтової кислоти у вигляді бежевого порошку. Т пл=149-150°С. й для проміжної сполуки 1.3. Замість 1-(4амінофеніл)-1Н-імідазолу використовували 1-(4ЯМР 1Н (CDCI3, 100МГц, d): 1,52 (s, 9Н, tBu), амінофеніл)-1,2,3,6-тетрагідропіридин. Бежевий 3,60 (s, 2H, CH2), 4,12 (s широка, 1Н, СООН), 6,55 порошок. Тпл=193-194°С. (s, 1Н, NH), 7,21 (m, 4Н, Η аром.). 5.2 (t-бутоксикарбоніламіно)-бензол етанол: ЯМР 1Н (D MSO, 400МГц, d): 2,23 (m, 2Н, СН2), 4-(t--бутоксикарбоніламіно)-бензолоцтову кис3,29 (m, 2H, СН2), 3,61 (m, 2H, СН2), 5,84 (m, 2Н, лоту (2,9г, 11,4ммоль) розчиняють у 10мл безводСН=СН ), 6,56 (s, 1Н, фумарова кислота), 6,89 (m, ного THF при 0°С і додають суспензію LіАІН4 4Н, Η аром.), 7,13 (m, 1Н, Η аром.), 7,67 (m, 1Н, Η (0,52г, 13,6ммоль) у 30мл THF. Реакційну суміш аром.), 7,77 (m, 1Н, Η аром.). перемішують протягом 1,5 години при кімнатній ІЧ: n C=N (амідин): 1560см -1. температурі. До суміші додають 50мл етилацетаПриклад 4: Хлорогідрат N-[4-(1Н-імідазол-1ту, потім 20мл 2N соди. Потрібний продукт екстраілметил)феніл]-2-тіофенкарбоксімідаміду (4): гують із органічної фази і потім промивають 3 рази 4.1 1-(4-нітробензил)-1Н-імідазол: 15мл води. Органічну фазу осушують, і розчинник Одержання потрібного продукту проводилось випарюють під зниженим тиском, потім продукт за тим самим експериментальним протоколом, що реакції очищають на силікагелі у суміші дихлормей для проміжної сполуки 1.1. Замість 1-фтор-4тан/метанол (95/5). Одержують 1,1г (40%) у виглянітробензолу використовували 1-бромометил-4ді безбарвного масла. нітробензол. Безбарвне масло. ЯМР 1Н (CDCI3, 100МГц, d): 1,53 (s, 9H, tBu), ЯМР 1Н (CDCI3, 100МГц, d): 5,26 (s, 2H, CH2), 2,82 (t, 2H, CH 2), 3,83 (q, 2H, CH2-ОН), 6,47 (s, 1H, 6,92 (m, 1Н, Η імідазол), 7,16 (m, 1Н, Η імідазол), NH), 7,23 (m, 4H, Η аром.). 7,59 (m, 1Н, Η імідазол), 7,24-7,32-8,18-8,27 (4s, 5.3 (2-бромоетил-444Н, Η аром.). бутоксикарбоніламіно)бензол: 4.2 1-(4-амінобензил)-1Н-імідазол: 4-(t-бутоксикарбоніламіно)-бензол етанол Одержання потрібного продукту проводилось (0,75г, 3,1ммоль) та чотирибромистий вуглець за тим самим експериментальним протоколом, що (1,2г, 3,6ммоль) розчиняли у 20мл дихпорметану й для проміжної сполуки 1.2. Замість 1-(4при 0°С. Порціями додавали трифенілфосфін амінофеніл)-1Н-імідазолу використовували 1-(4(0,94г, 3,6ммоль), і суміш перемішували протягом нітробензил)-1Н-імідазол. Блідо-жовтий порошок. 1 години при кімнатній температурі. Розчинник Тпл=121-122°С. випарювали під зниженим тиском, і одержаний ЯМР 1Н (CDCI3 , 100МГц, d): 2,87 (s широка, 2H, продукт очищали на силікагелі у суміші етилацеNH2), 4,98 (s, 2H, CH2), 6,88 (m, 1Н, Η імідазол), тат/гептан (1/2). 1-(2-бромоетил-4-t7,06 (m, 1Н, Η імідазол), 7,52 (m, 1Н, Η імідазол), бутоксикарбоніламіно)бензол одержували у ви6,60-6,69-6,95-7,05 (4s, 4Н, Η аром.). гляді білого порошку (0,8г, 84%). Тпл=129-130°C. 4.3 Хлорогідрат N-[4-(1Н-імідазол-1ЯМР 1Н (CDCI3, 100МГц, d): 1,52 (s, 9H, tBu), ілметил)феніл]-2-тіофенкарбоксімідаміду (4): 3,11 (t, 2H, CH 2), 3,54 (t, 2H, CH 2Br), 6,45 (s, 1H, Одержання потрібного продукту проводилось NH), 7,22 (m, 4H, Η аром.). за тим самим експериментальним протоколом, що 5.4 3-{2-[4-(ій для проміжної сполуки 2.4. Замість 3-(4бутоксикарбоніламіно)феніл]етил}тіазолідин: амінобензил)-тіазолідину використовували 1-(4Одержання потрібного продукту проводилось амінобензил)-1Н-імідазол. Після солеутворення за за тим самим експериментальним протоколом, що допомогою молярного розчину НСІ у безводному й для проміжної сполуки 2.2. Замість 1діетиловому ефірі одержали бежевий порошок. бромометил-4-нітробензолу використовували (2Тпл=261-263°С. бромоетил-4-t-бутоксикарбоніламіно)бензол. БезЯМР 1Н (DMSO, 400МГц, d): 5,12 (s, 2Н, СН2), барвне масло. 6,46 (s широка, 2Н, NH2), 6,83-6,85-7,22-7,24 (4s, ЯМР 1Н (CDCI3, 100МГц, d): 1,52 (s, 9Н, tBu), 4Н, Η аром.), 6,90 (s, 1Н, Η аром.), 7,09 (t, 1Н, Η 2,90 (m, 8Н, 4СН2), 4,10 (s, 2H, N-CH2-S), 6,46 (s, аром.), 7,20 (s, 1Н, Η аром.), 7,60 (d, 1Н, Η аром.), 1Н, NH), 7,25 (m, 4H, Η аром.). 7,74 (s, 2Н, Η аром.). 5.5 3-{2-[4-амінофеніл]етил}тіазолідин: ІЧ: n C=N (амідин): 1599см -1. У колбу на 100мл, що містить розчин 616мг Приклад 5: N-[4-{2-(3-тіазолідиніл)етил}феніл](2ммоль) проміжної сполуки 5.4 у 10мл дихлорме2-тіофенкарбоксімідамід (5): тану, додають 2,3г (20ммоль) трифтороцтової кис5.1 4-(t-бутоксикарбоніламіно)-бензолоцтова лоти. Після перемішування протягом 1 години при кислота: 43 70921 44 20°С реакційну суміш концентрують під вакуумом. Тпл=214-215°С. Залишок розводять сумішшю 20мл дихпорметану ЯМР 1Н (DMSO, 400МГц, d: 3,11 (t, 2Н, СН2), та 20мл 4Ν соди. Після декантації органічну фазу 4,33 (t, 2Н, СН2), 7,29 (m, 6Н, Η аром), 7,99 (m, 2Н, промивають послідовно 3x20мл води та 20мл розΗ аром.), 8,70 (s широка, 2Н, NH2). чину солі. Органічний розчин осушують над сульІЧ: n C=N (амідин): 1597см -1. фатом натрію, фільтрують, і розчинник випарюють Приклад 7: Фумарат N-{4-[2-(1,2,3,6під зниженим тиском. Одержують безбарвне мастетрагідропіридин-1-іл)етил]феніл}-2ло з виходом 72%. тіофенкарбоксімідаміду (7): ЯМР 1Н (CDCI3, 100МГц, d): 2,85 (m, 8Н, 4СН2), 7.1 1-{2-[4-(t4,15 (s, 2H, N-CH 2-S), 7,25 (m, 4Н, Η аром.). бутоксикарбоніламіно)феніл]етил}-1,2,3,65.6 [4-{2-(3-тіазолідінил)етил}феніл]-2тетрагідропіридин: тіофенкарбоксімідамід (5): Одержання потрібного продукту проводилось Одержання потрібного продукту проводилось за тим самим експериментальним протоколом, що за тим самим експериментальним протоколом, що й для проміжної сполуки 6.1. Замість тіазолідину й для проміжної сполуки 2.4. Замість 3-(4використовували 1,2,3,6-тетрагідропіридин. Безамінобензил)-тіазолідину використовували 3-(2[4барвне масло. амінофеніл]етил}тіазолідин. Бежевий порошок. ЯМР 1Н (CDCI3, 100МГц, d): 1,57 (s, 9H, tBu), Тпл=60,5-61,5°С. 2,10 (m, 2H, CH2), 2,70 (m, 6H, 3CH2), 3,00 (m, 2H, ЯМР 1Н (DMSO, 400МГц, d): 2,65 (t, 2Н, СН2), CH2), 5,72 (m, 2H, CH=CH), 6,48 (s, 1H, NH), 7,10 2,82 (t, 2H, СН2), 2,91 (t, 2H, СН2), 3,13 (t, 2Н, СН2), (m, 4H, Η аром.). 4,13 (в, 2Н, N-CH2-S), 6,93-6,95-7,19-7,21 (4s, 4Н, Η 7.2 1-[2-(4-амінофеніл)етил]-1,2,3,6аром.), 7,09 (t, 1Н, Η тіофен), 7,44 (m, 2Н, Η тіотетрагідропіридин: фен). Одержання потрібного продукту проводилось ІЧ: n C=N (амідин): 1591см -1. за тим самим експериментальним протоколом, що Приклад 6: Йодогідрат N-{4-[2-(1Н-імідазол-1й для проміжної сполуки 5.5. Замість 3-{2-[4іл)етил]феніл}-2-тіофенкарбоксімідаміду (6): амінофеніл]етил}тіазолідину використовували 16.1 1-{2-[4-(t{2-[4-(t-бутоксикарбоніламіно)феніл]етил}-1,2,3,6бутоксикарбоніламіно)феніл]етил}-1Н-імідазол: тетрагідропіридин. Безбарвне масло. У колбі на 100мл змішують 2,5г (18ммоль) ЯМР 1Н (CDCI3, 100МГц, d): 3,20 (m, 2H, CH2), К2СО3 з 680мг (10ммоль) імідазолу, розведеного у 3,80 (m, 6Н, 3СН2), 4,10 (m, 2Н, СН2), 4,57 (s широ10мл ацетонітрилу. Реакційну суміш нагрівають до ка, 2Н, NH2), 6,90 (m, 2Н, СН=СН ), 8,00 (m, 4Н, Η 70°С, потім додають по краплях розчин 2г аром.). (9,2ммоль) 1-бромометил-4-нітробензолу у 25мл 7.3 Фумарат Ν-{4-[2-(1,2,3,6-тетрагідропіридинацетонітрилу. Після 2 годин перемішування при 1-іл)етил]феніл}-2-тіофенкарбоксімідаміду (7): вказаній температурі реакційну суміш охолоджуОдержання потрібного продукту проводилось ють і фільтрують для вилучення нерозчинних реза тим самим експериментальним протоколом, що човин. Фільтрат концентрують під вакуумом, і зай для проміжної сполуки 1.3. Замість 1-(4лишок розчиняють у 50мл дихлорметану. амінофеніл)-1Н-імідазолу використовували 1-[2-(4Органічний розчин послідовно промивають 3х50мл амінофеніл)етил]-1,2,3,6-тетрагідропіридин. Білий води та 50мл розчину солі. Після осушення над порошок. Тпл=128-129°С. сульфатом натрію та фільтрування органічну фазу ЯМР 1Н (D MSO, 400МГц, d): 2,19 (m, 2Н, СН2), концентрують під вакуумом, і залишок піддають 2,83 (m, 6Н, 3СН2), 3,25 (m, 2Н, СН2), 5,72 (m, 2H, очищенню на колонці з кремнеземом (елюент: СН=СН ), 6,58 (s, 3H, фумарова кислота), 6,81дихлорметан/метанол: 95/5). Каштанове масло. 6,83-7,18-7,20 (4s, 4Н, Η аром.), 7,10 (t, 1Н, Η тіоЯМР 1Н (CDCI3, 100МГц, d): 1,50 (s, 9H, tBu), фен), 7,63 (m, 1Н, Η тіофен), 7,75 (m, 1Н, Η тіо2,90 (t, 2H, CH 2), 4,10 (t, 2H, CH 2), 6,50 (s, 1H, NH), фен). 7,05 (m, 4H, Η аром.), 6,85 (m, 1H, Η імідазол), 7,03 ІЧ: n C=N (амідин): 1620см -1. (s, 1H, Η імідазол), 7,32 (m, 1H, Η імідазол). Приклад 8: N-[4-(36.2 1-[2-(4-амінофеніл)етил]-1Н-імідазол: тіазолідинілкарбонілметил)феніл]-2Одержання потрібного продукту проводилось тіофенкарбоксімідамід (8): за тим самим експериментальним протоколом, що 8.1 3-[{4-(tй для проміжної сполуки 5.5. Замість 3-{2-[4-(tбутоксикарбоніламібутоксикарбоніламіно)феніл]етил}тіазолідину вино)феніл}метилкарбоніл]тіазолідин: користовували 1-{2-[4-(t-бутоксикарбоніламіно) 4-(t-бутоксикарбоніламіно)-бензолоцтову кисфеніл]етил}-1Н-імідазол. Безбарвне масло. лоту (1,4г, 5,6ммоль), проміжну сполуку 5.1 та карЯМР 1Н (CDCI3, 100МГц, d): 2,90 (t, 2H, CH2), бонілімідазол (0,9г, 5,6ммоль) розчиняли у 15мл 3,35 (s широка, 2Н, NH2), 4,10 (t, 2Н, СН2), 6,70 (m, THF. Реакцію проводили протягом 1 години при 4Н, Η аром.), 6,85 (m, 1H, Η імідазол), 7,03 (s, 1H, кімнатній температурі, потім додавали тіазолідин Η імідазол), 7,32 (m, 1H, Η імідазол). (0,5г, 5,6ммоль) у розчині THF (5мл). Дану суміш 6.3 Йодогідрат N-{4-[2-(1H-імідазол-1знов перемішували протягом 2 годин при кімнатній іл)етил]феніл}-2-тіофенкарбоксімідаміду (6): температурі. Розчинники випарювали під знижеОдержання потрібного продукту проводилось ним тиском, потім залишок обробляли 25мл дихза тим самим експериментальним протоколом, що лорметану та промивали 3 рази 15мл води. Оргай для проміжної сполуки 1.3. Замість 1-(4нічну фазу осушували та концентрували під амінофеніл)-1Н-імідазолу використовували 1-[2-(4зниженим тиском. 3-[{4-(tамінофеніл)етил]-1Н-імідазол. Бежевий порошок. бутоксикарбоніламі 45 70921 46 но)феніл}метилкарбоніл]тіазолідин одержували у масла з виходом 80% (1,25г). вигляді білого порошку (1,43г, 79%) і використовуЯМР 1Н (CDCI3, 100МГц, d): 1,45 (s, 9H, tBu), вали на наступних стадіях без додаткової очистки. 3,09 (m, 3H, CH 2-S, CH-S), 3,86 (m, 2H, CH 2-CH2-N), Тпл=223-224°С. 4,57 (m, 2H, CH2-NH), 6,60 (s широка, 1Н, NH), ЯМР 1Н (CDCI3, 100МГц, d): 1,51 (s, 9H, tBu), 7,41-7,50-8,14-8,23 (4s, 4H, Η аром.) 3,00 (m, 2H, CH2-S), 3,67 (s, 2H, N-CH 2-S), 3,88 (m, 9.3 (4-амінобензил)-3-(12H, CH2-N), 4,52 (d, J=16Гц, 2Н, CH2-CO), 6,52 (s бутоксикарбоніл)тіазолідин-2-карбоксамід: широка, 1Н, NH), 7,26 (m, 4H, Η аром.). До розчину 1,25г (3,4ммоль) N-(4-нітробензил)8.2 3-[(4-амінофеніл)метилкарбоніл]тіазолідин: 3-(t-бутоксикарбоніл)тіазолідин-2-карбоксаміду у 3-[(4-амінофеніл)метилкарбоніл]тіазолідин 2,5мл метанолу додають на кінчику шпателя Нікеодержували у вигляді безбарвного масла з вихоля - Ренея. Під оберненим холодильником до судом 44% згідно зі способом, описаним для проміміші додають по краплям гідрат гідразину (1,75мл), жної сполуки 5,5. і реакція протікає протягом 1 години. Перед охолодженням до кімнатної температури каталізатор ЯМР 1Н (CDCI3 , 100МГц, d): 1,62 (s широка, 2Н, відфільтровують і промивають надлишком метаNH2), 2,98 (m, 2Н, CH2-S), 3,61 (s, 2H, N-CH 2-S), нолу. Розчинник випарюють під зниженим тиском, 3,80 (m, 2H, CH2-N), 4,52 (d, J=16Гц, 2Н, CH2-CO), потім залишок обробляють дихпорметаном (20мл) 6,61-6,69-7,01-7,09 (4s, 4H, Η аром.). та промивають 3 рази 15мл води. Органічну фазу 8.3 [4-(3-тіазолідинілкарбонілметил)феніл-2осушують, і розчинник випарюють під зниженим тіофенкарбоксімідамід (8): тиском. N-(4-амінобензил)-3-(tЗастосовувався той самий спосіб, що й для бутоксикарбоніл)тіазолідин-2-карбоксамід одерпроміжної сполуки 2.4. Замість 3-(4жують у вигляді інертної твердої речовини жовтого амінобензил)тіазолідину використовували 3-[(4кольору (0,815г, 71%), яку використовують на наамінофеніл)метилкарбоніл]тіазолідин. Вільну осступних стадіях без додаткової очистки. нову одержували з виходом 64%. Тпл=163,0163,5°С. ЯМР 1Н (CDCI3, 100МГц, d): 1,43 (s, 9H, tBu), 3,08 (m, 2H, CH 2-S), 3,67 (m, 3H, CH 2-CH2-N, CH-S), ЯМР 1Н (CDCI3, 400МГц, d): 3,01 (m, 2Н, CH2S), 3,69 (d, J=6Гц, 2Н, N-CH2-S), 3,75-3,88 (2t, 2H, 4,36 (m, 2H, CH2-NH), 6,05 (s широка, 1Н, NH), 6,60-6,69-7,04-7,12 (4s, 4H, Η аром.). CH2-N), 4,55 (d, 2H, CH2-CO), 4,87 (s, 2H, NH 2), 9.4 [4-{[3-(t-бутоксикарбоніл)-26,95-6,97-7,22-7,24 (4s, 4H, Η аром.), 7,08 (t, 1H, тіазолідиніл]карбоніламінометил}феніл]-2тіофен), 7,43 (m, 2H, тіофен). тіофенкарбоксімідоамід: ІЧ: n C=N (амід): 1630см -1; n C=N (амідин): -1 Одержання потрібного продукту проводилось 1577см . за тим самим експериментальним протоколом, що Приклад 9: Фумарат N-(4-{[2й для проміжної сполуки 2.4. Замість 3-(4тіазолідиніл]карбоніламінометил}феніл)-2амінобензил)-тіазолідину використовували N-(4тіофенкарбоксімідаміду (9): амінобензил)-3-(t-бутоксикарбоніл)тіазолідин-29.1 3-(t-бутоксикарбоніл)тіазолідин-2карбоксамід. Потрібну сполуку одержали з вихокарбонова кислота: дом 77%. Тіазолідин-2-карбонову кислоту (2г, 15ммоль) ЯМР 1Н (CDCI3, 100МГц, d): 1,45 (s, 9H, tBu), перемішують у присутності ди-t-бутил дикарбонату 3,14 (m, 3H, CH 2-S, CH-S), 3,84 (m, 2H, CH 2-CH2-N), згідно зі способом, застосованим для проміжної 4,46 (m, 2H, CH2-NH), 4,83 (s широка, 2Н, NH2), сполуки 5.1. 3-(t-бутоксикарбоніл)тіазолідин-26,27 (s широка, 1H, NH), 7,22 (m, 7H, Η аром.). карбонову кислоту одержують у вигляді блідо9.5 Фумарат N-(4-{[2жовтого масла з виходом 97% (3,4г) і використотіазолідиніл]карбоніламінометил}феніл)-2вують на подальших стадіях без додаткової очисттіофенкарбоксімідаміду (9): ки. Одержання потрібного продукту проводилось ЯМР 1Н (CDCI3, 100МГц, d): 1,46 (s, 9H, tBu), за тим самим експериментальним протоколом, що 3,10 (m, 3H, CH 2-S, CH-S), 3,85 (m, 2H, CH 2-N). й для проміжної сполуки 5.5. Замість 3-{2-[49.2 (4-нітробензил)-3-(tамінофеніл]етил}тіазолідину використовували [4бутоксикарбоніл)тіазолідин-2-карбоксамід: {[3-(t-бутоксикарбоніл)-23-(t-бутоксикарбоніл)тіазолідин-2-карбонову тіазолідиніл]карбоніламінометил}феніл]-2кислоту (1г, 4,3ммоль) та карбонілімідазол (0,7г, тіофенкарбоксімідамід. Потрібний продукт одер4,3ммоль) розчиняють у THF (10мл). Дану суміш жали у вигляді вільної основи з виходом 34%, яку перемішують протягом 1 години при кімнатній тепереводили у сольову форму за допомогою еквімпературі. Потім додають хлорогідрат 4валента фумарової кислоти шляхом нагрівання з нітробензиламіну (0,81г, 4,3ммоль) та триетиламін оберненим холодильником у етанолі. Тпл=167(0,6мл, 0,43г, 4,3ммоль) у суспензії у 10мл суміші 168°С. THF та D MF (1/1) та нагрівають із оберненим хоЯМР 1Н (DMSO, 400МГц, d): 2,78 (t, 2Н, CH 2-S), лодильником протягом 5 годин. Потім зазначені 3,06 (m, 2H, CH2-CH2-N), 3,28 (s широка, 1H, CHрозчинники випарюють під зниженим тиском. ЗаS), 4,26 (m, 2H, CH2-NH), 4,86 (s широка, 1Н, NH), лишок обробляють 25мл етилацетату та промивають 3 рази 15мл води. Органічну фазу осушу6,45 (s широка, 2H, NH2), 6,81-6,83-7,19-7,21 (4s, 4H, Η аром.), 7,10 (t, 1H, тіофен), 7,61 (d, 1H, тіоють, і розчинник випарюють під зниженим тиском. фен), 7,74 (m, 1H, тіофен), 8,53 (t, 1H, NH-CO). Одержаний продукт очищають на силікагелі у суміші дихлорметан/метанол (95/5). N-(4ІЧ: n C=N (амід): 1624см -1; n C=N (амідин): -1 нітробензил)-3-(t-бутоксикарбоніл)тіазолідин-21584см . карбоксамід одержують у ви гляді блідо-жовтого Приклад 10: Йодогідрат N-(3,5-ди-t-бутил-4 47 70921 48 гідроксифеніл)-5-[4-{-іміно(2-тієніл)вигляді сольової форми з виходом 27%. Тпл=273метиламіно}феніл]-2-фуран карбоксаміду (10): 274°С. 10.1 2,6-ди-t-бутил-4-нітрофенол: ЯМР 1Н (DMSO, 400МГц, d): 1,40 (s, 18H, 2tBu), 2,6-ди-t-бутилфенол (8г, 39ммоль) розчиняють 6,90 (s, 1H, OH), 7,45 (m, 5H, Η аром.), 7,54 (s, 2H, у 25мл циклогексану при 10°С. До даного реакційΗ аром.), 8,15 (m, 4H, Η аром.), 9,05-9,90 (2s широного середовища при вказаній температурі додакі, 2Н, NH2), 10,01 (s, 1H, NH-CO), 11,57 (s, 1H, HI). ють по краплях суміш (1/1) азотної та оцтової кисІЧ: n ОН: 3423-3242см -1; n С= О (амід): 1646см -1; лот (5мл). Потім суміш перемішують протягом 15 n С=N (амідин): 1554см -1. хвилин при кімнатній температурі, відфільтровуПриклад 11: Хлорогідрат 3-(3,5-ди-t-бутил-4ють утворений осад та промивають водою та пенгідроксифеніл)-1-[4-{іміно(2-тієніл)таном. Одержаний 2,6-ди-t-бутил-4-нітрофенол метиламіно}феніл]-2,5-імідазолідиндіону (11): (6,34г, 65%) осушують у сушильній шафі і викорис11.1 Етил (3,5-ди-t-бутил-4товують на наступних стадіях без додаткової очисгідроксифеніл)аміноацетат: тки. Блідо-жовтий порошок. Тпл=167-168°С. 1г (4,5ммоль) 2,6-ди-t-бутил-4-амінофенолу ЯМР 1Н (CDCI3, 100МГц, d): 1,48 (s, 18H, 2tBu), (проміжна сполука 10.2) та 0,65г ацетату натрію 5,93 (s, 1H, OH), 8,13 (s, 2H, Η аром.). (7,9ммоль) суспендували у 1мл етанолу. Потім до 10.2 2,6-ди-t-бутил-4-амінофенол: суміші додавали етилбромоацетат (0,94г, 2,6-ди-t-бутил-4-нітрофенол (6,3г, 25ммоль) 5,65ммоль), і суміш нагрівали при 65°С протягом 2 розчиняють у метанолі (100мл). Додають 0,6г пагодин. Реакційну суміш виливали у 20мл льодяної ладію на вуглеці (10%), і суміш розміщують в атводи, і продукт реакції екстрагували дихпорметамосфері водню під тиском 2бар. Каталізатор відном (3 рази 15мл). Органічні фази осушували, і фільтровують, і розчинник випарюють під розчинник випарювали під зниженим тиском. Зазниженим тиском. Залишок обробляють гептаном лишок перепускали через силікагель у ди хпормета фільтр ують. У такий спосіб одержують 2,6-ди-tтані. Одержали безбарвне масло, що складається бутил-4-амінофенол (2,7г, 48%), котрий викорисіз двох сполук: продукту моно- та подвійного замітовується на наступних стадіях без додаткової щення. Суміш вказаних двох сполук використовуочистки. Рожевий порошок. Тпл=123-124°С. вали без додаткової очистки на наступній стадії. ЯМР 1Н (CDCI3, 100МГц, d): 6,60 (s, 2H, Ph), 11.2 Етил-(3,5-ди-t-бутил-4-гідроксифеніл)-(44,65 (s широка, 1Н, ОН), 3,15 (s широка, 2Н, NH2), нітрофенілкарбамоїл)аміноацетат: 1,42 (s, 18H, 2tBu). 1,13г (4,2ммоль) проміжної сполуки 11.1 та 10.3 N-(3,5-ди-t-бутил-4-гідроксифеніл)-5-(40,69г (4,23ммоль) 4-нітрофенілізоціанату розчинянітрофеніл)-2-фуран карбоксамід: ють у 8мл дихлорметану. Реакційну суміш переміОдержання потрібного продукту проводилось шують протягом 2,5 годин при кімнатній темпераза тим самим експериментальним протоколом, що турі. Розчинник випарюють під зниженим тиском, і й для проміжної сполуки 8.1. Замість тіазолідину залишок перепускають через силікагель у дихлорта 4-(t-бутоксикарбоніламіно)-бензолоцтової кисметані. У такий спосіб виділяють 0,66г етил-(3,5лоти використовували, відповідно, 2,6-ди-t-бутилди-t-бутил-4-гідроксифеніл)-(44-амінофенол та 5-(4-нітрофеніл)-2-фураннітрофенілкарбамоїл)аміноацетату у вигляді безкарбонову кислоту. Потрібну сполуку одержали у барвного масла. (Вихід для двох стадій складає вигляді безбарвного масла з виходом 56%. 31%). ЯМР 1Н (DMSO, 100МГц, d): 1,41 (s, 18Н, 2tBu), ЯМР 1Н (CDCI3, 100МГц, d): 1,30 (t, 3Н, СН3), 6,91 (s, 1H, ОН), 7,42 (m, 4H, Η аром.), 7,54 (s, 2Н, 1,46 (s, 18H, 2tBu), 4,23 (q, 2H, CH2-СН3), 4,38 (s, Η аром.), 8,30 (m, 4Н, Η аром.), 10,11 (s, 1Н, NH). 2H, СН2-СO), 5,50 (s, 1Н, ОН), 6,75 (s широка, 1H, 10.4 N-(3,5-ди-t-бутил-4-гідроксифеніл)-5-(4NH), 7,28 (s, 2H, Η аром.), 7,40-7,50-8,10-8,20 (4s, амінофеніл)-2-фуран карбоксамід: 4H, Η аром.) Одержання потрібного продукту проводилось 11.3 (3,5-ди-t-бутил-4-гідроксифеніл)-1-(4за тим самим експериментальним протоколом, що нітрофеніл)-2,5-імідазолідиндіон: й для проміжної сполуки 1.2. Замість 1-(40,66г (1,4ммоль) проміжної сполуки 11.2 рознітрофеніл)-1Н-імідазолу використовували N-(3,5чиняють у 10мл етанолу при 50°С, і суміш нагріди-t--бутил-4-гїдроксифеніл)-5-(4-нітрофеніл)-2вають при даній температурі протягом 2 годин. фуран карбоксамід. Потрібну сполуку одержали у Утворений осад відфільтровують та промивають вигляді безбарвного масла з виходом 59%. холодним етанолом. Одержану сполуку викорисЯМР 1Н (DMSO, 100МГц, d): 1,41 (s, 18Н, 2tBu), товують безпосередньо на наступній стадії без 4,70 (s широка, 2Н, NH2), 6,91 (s, 1H, ОН), 7,50 (m, додаткової очистки. 4Н, Η аром.), 7,54 (s, 2Н, Η аром.), 8,20 (m, 4Н, Η ЯМР 1Н (CDCI3, 100МГц, d): 1,47 (s, 18H, 2tBu), аром.). 4,51 (s, 2H, N-CH2-CO), 5,27 (s, 1H, OH), 7,33 (s, 10.5 Йодогідрат N-(3,5-ди-t-бутил-42H, Η аром.), 7,77-7,86-8,32-8,41 (4s, 4H, Η аром.). гідроксифеніл)-5-[4-{іміно(211.4 (3,5-ди-t-бутил-4-гідроксифеніл)-1-(4тієніл)метиламіно}феніл]-2-фуран карбоксаміду амінофеніл)-2,5-імідазолідиндіон: (10): Одержання потрібного продукту проводилось Одержання потрібного продукту проводилось за тим самим експериментальним протоколом, що за тим самим експериментальним протоколом, що й для проміжної сполуки 1.2. Замість 1-(4й для проміжної сполуки 1.3. Замість 1-(4нітрофеніл)-1Н-імідазолу використовували 3-(3,5амінофеніл)-1Н-імідазолу використовували N-(3,5ди-t-бутил-4-гідроксифеніл)-1-(4-нітрофеніл)-2,5ди-t-бутил-4-гідроксифеніл)-5-(4-амінофеніл)-2імідазолідиндіон. Потрібну сполуку одержали у фуран карбоксамід. Потрібну сполуку одержали у вигляді білого осаду з виходом 87%. Її використо 49 70921 50 вували на наступній стадії без додаткової очистки. (s, 1H, CH-S), 5,91 (s, 1H, OH), 6,51-6,59-6,78-6,86 (4s, 4H, Η аром.), 7,04 (s, 2H, Η аром.). ЯМР 1Н (CDCI3, 100МГц, d): 1,47 (s, 18H, 2tBu), 4,45 (s, 2H, N-CH2-CO), 5,18 (s, 1H, OH), 6,70-6,8012.3 Хлорогідрат 2-(3,5-ди-t-бутил-4гідроксифеніл)-3-[4-{іміно(27,16-7,23 (4s, 4H, Η аром.) 7,39 (s, 2H, Η аром.). тієніл)метиламіно}феніл]-4-тіазолідинону (12): 11.5 Хлорогідрат 3-(3,5-ди-t-бутил-4Одержання потрібного продукту проводилось гідроксифеніл)-1-[4-{іміно(2за тим самим експериментальним протоколом, що тієніл)метиламіно}феніл]-2,5-імідазолідиндіону (11): й для проміжної сполуки 2.4. Замість 3-(4амінобензил)-тіазолідину використовували 2-(3,5Одержання потрібного продукту проводилось ди-t-бутил-4-гідроксифеніл)-3-(4-амінофеніл]-4за тим самим експериментальним протоколом, що тіазолідинон. Потрібну сполуку одержують у сой для проміжної сполуки 2.4. Замість 3-(4льовій формі (хлоропдрату) обробкою вільної осамінобензил)-тіазолідину використовували 3-(3,5ди-t-бутил-4-гідроксифеніл)-1-(4-амінофеніл]-2,5нови 1N розчином хлористого етилу з ви ходом 43%. Тпл=58-61°С. імідазолідиндіон. Вільну основу переводять у сольову форму обробкою 1N розчином хлористого ЯМР 1Н (DMSO, 400МГц, d): 1,32 (s, 18Н, 2tBu), етилу. Хлорогідрат одержали з виходом 53%. 3,93 (m, 2H, CH-S), 6,57 (s, 1H, CH-S), 7,08 (s, 2H, Тпл=258-265°С. Η аром.), 7,41 (m, 5H, Η аром.), 8,15 (m, 2H, Η аром.), 9,10-9,90 (2s широкі, 2H, NH2), 11,45 (s шиЯМР 1Н (DMSO, 400МГц, d): 1,40 (s, 18Н, 2tBu), рока, 1H, НСІ). 4,65 (s, 2H, CH2), 7,08 (s, 1H, ОН), 7,40 (m, 3H, Η аром.), 7,61 (s, 4H, Η аром.), 8,21 (m, 2H, Η аром.), ІЧ: n ОН: 3624-3423см-1; n С= О (тіазолідинон); 9,20-9,95 (2s широкі, 2H, NH 2), 11,75 (s, 1H, HCI). 1679-1658см-1; n С=N (амідин): 1568см -1. -1 Приклад 13: Фумарат 5-[(3,5-ди-t-бутил-4ІЧ: n ОН: 3637-3437см ; n C=O (імідазолідиндіон): гідроксифеніл)метилен]-1-метил-3-[4-{іміно(21712см -1; n C=O (амідин): 1598см-1. тієніл)метиламіно}феніл]-2,4-імідазолідиндіону Приклад 12: Хлорогідрат 2-(3,5-ди-t-бутил-4(13): гідроксифеніл)-3-[4-{іміно(213.1 1-метил-3-(4-нітрофеніл)-2,4тієніл)метиламіно}феніл]-4-тіазолідинону (12): імідазолідиндіон: 12.1 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3-(40,47г етилового ефіру саркозину, НСІ (3ммоль) нітрофеніл)-4-тіазолідинон: розчиняють у 5мл дихлорметану та додають 5г 3,5-ди-t-бутил-4-гідроксибензальдегіду 0,42мл (3ммоль) триетиламіну. До вказаної суміші (21ммоль) та 2,95г паранітроаніліну (21ммоль) по краплям додають 0,5г 4-нітрофенілізоціанату розчиняють у 50мл безводного толуолу. Додають (3ммоль) у 5мл дихлорметану, і суміш витримують 0,5мл льодяної оцтової кислоти, і суміш витриму30 хвилин при кімнатній температурі. Потім органіють під оберненим холодильником протягом 24 чний розчин промивають водою (3 рази 10мл), годин. Потім додають 1,96г меркаптооцтової кисосушують, і розчинник випарюють під зниженим лоти (21ммоль) і продовжують реакцію під обертиском. неним холодильником ще протягом 24 годин. ПісЗалишок обробляють 10мл етанолу, і суміш ля охолодження до кімнатної температури нагрівають із оберненим холодильником протягом реакційну суміш промивають водою (3 рази 30мл), 2 годин. Після охолодження до кімнатної темпераі після декантації органічну фазу осушують над тури утворений осад відфільтровують. У такий сульфатом натрію, і розчинник випарюють під спосіб одержують 1-метил-3-(4-нітрофеніл)-2,4зниженим тиском. Залишок очищають на силікагеімідазолідиндіон з виходом 72% (0,5г), котрий вилі у суміші етилацетат/гептан (1/4) і одержують користовують на наступній стадії без додаткової 1,33г чистого 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3очистки. (4-нітрофеніл-4-тіазолідинону у вигляді безбарвного масла (15%). ЯМР 1Н (CDCI3, 100МГц, d): 3,11 (s, 3H, CH3), 1 4,09 (s, 2Н, СН2), 7,70-7,79-8,27-8,37 (4s, 4Н, Η ЯМР Н (CDCI3, 100МГц, d): 1,36 (s, 18H, 2tBu), аром.). 3,91 (s, 2H, CH-S), 5,28 (s, 1H, CH-S), 6,20 (s, 1H, 13.2 5-[(3,5-ди-t-бутил-4OH), 7,03 (s, 2H, Η аром.), 7,38-7,48-8,11-8,20 (4s, гідроксифеніл)метилен]-1-метил-3-(4-нітрофеніл)4H, Η аром.). 2,4-імідазолідиндіон: 12.2 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3-(4амінофеніл)-4-тіазолідинон: Проміжну сполуку 13.1 (0,5г, 2,13ммоль), 3,5ди-t-бутил-4-гідроксибензальдегід (0,5г, 1,3г 2-(3,5-ди-t-бутил-4-гідроксифеніл)-3-(4нітрофеніл)-4-тіазолідинону (3ммоль) та 3,4г 2,13ммоль) та b-аланін (0,123г, 1,4ммоль) розчи(15ммоль) двохводного хлористого олова розчиняють в оцтовій кислоті (10мл). З використанням няють у 25мл етилацетату. Реакцію підтримують оберненого холодильника реакцію проводять пропротягом 2 годин при 70°С. Після охолодження до тягом 24 годин. Після охолодження до кімнатної кімнатної температури суміш виливають у насичетемператури до суміші додають 40мл води, і суміш ний розчин гідрокарбонату натрію. Потрібний проперемішують протягом 1 години. Утворений осад дукт потім екстрагують із органічної фази та провідфільтровують та промивають водою. Фільтрат мивають 3 рази 10мл води. 2-(3,5-ди-t-бутил-4концентрують під вакуумом, і залишок після випагiдроксифеніл)-3-(4-амінофеніл)-4-тіазолідинон рювання очищають на силікагелі (елюент: гепочищають на силікагелі у суміші етилацетат/гептан тан/етилацетат: 4/1). Чисті фракції збирають та (1/1) і одержують у вигляді бежевого масла з виконцентрують до сухого стану, і одержують потрібходом 69% (0,82г). ний продукт з виходом 32% (0,3г). ЯМР 1Н (CDCI3, 100МГц, d): 1,37 (s, 18H, 2tBu), ЯМР 1Н (CDCI3, 100МГц, d): 1,49 (s, 18H, 2tBu), 3,64 (s широка, 2H, NH2), 3,89 (s, 2H, CH2-S), 5,22 3,35 (s, 3H, CH3), 5,59 (s, 1H, OH), 6,40 (s, 1H, 51 70921 52 CH=C), 7,75-7,84-8,31-8,40 (4s, 4H, Η аром.), 7,92 осушення одержують безбарвні кристали з вихо(s, 2H, Η аром.). дом 63%. Тпл=155-157°С. 13.3 5-[(3,5-ди-t-бутил-4ЯМР 1Н (ДМСО, 400МГц, d): 1,34-1,40 (2s, 9Н, гідроксифеніл)метилен]-1-метил-3-(4-амінофеніл)tBu), 2,45 (m, 2H, CH 2), 3,60 (m, 2Н, CH2-N), 3,582,4-імідазолідиндіон: 3,63 (2s, 3H, O-CH3), 4,40 (m, 1Н, СН-СО2), 5,22 (m, Одержання потрібного продукту проводилось 1Н, НС-О), 7,63 (m, 4Н, Рh). за тим самим експериментальним протоколом, що 14.2 2-(S)-4-(S)-1-[(1,1й для проміжної сполуки 12.2. Замість 2-(З,5-ди-tдиметилетокси)карбоніл]-4-(4-нітрофенокси)бутил-4-гідроксифеніл)-3-(4-нітрофеніл)-4пролін: тіазолідинону використовували 5-[(3,5-ди-t-бутилУ колбу ємністю 100мл, що містить 2,87г 4-гідроксифеніл)метилен]-1-метил-3-(4(7,84ммоль) сполуки 14.1 у 40мл етанолу, додають нітрофеніл)-2,4-імідазолідиндіон. Потрібну сполуку при 20°С 730мг (разом 16ммоль) поташу, розвеодержують з виходом 45%. деного у 5мл води. Після 15 годин перемішування ЯМР 1Н (CDCI3, 100МГц, d): 1,47 (s, 18H, 2tBu), реакційну суміш розводять 100мл етилацетату, 3,30 (s, 3H, CH3), 5,51 (s, 1H, ОН), 6,28 (s, 1H, підкислюють при 0°С 12N розчином соляної кислоCH=C), 6,69-6,78-7,12-7,21 (4s, 4Н, Η аром.), 7,91 ти та декантують. Органічну фазу промивають (s, 2Н, Η аром.). 50мл води та 50 л розчину солі. Після осушування 13.4 Фумарат 5-[(3,5-ди-t-бутил-4над сульфатом натрію органічний розчин фільтгідроксифеніл)метилен]-1-метил-3-[4-{іміно(2рують та концентрують до сухого стану під вакуутієніл)метиламіно}феніл]-2,4-імідазолідиндіону мом. Одержують 2,67г білого порошку, котрий ви(13): користовується безпосередньо на наступній стадії Одержання потрібного продукту проводилось без додаткової очистки. за тим самим експериментальним протоколом, що ЯМР 1Н (CDCI3, 100МГц, d): 1,50 (s, 9Н, tBu), й для проміжної сполуки 2.4. Замість 3-(42,60 (m, 2H, CH2), 3,80 (m, 2H, CH 2-N), 4,60 (m, 1H, амінобензил)-тіазолідину використовували 5-[(3,5CH-CO2), 5,07 (m, 1H, НС-О), 7,58 (m, 4H, Ph), 8,95 ди-t-бутил-4-гідроксифеніл)метилен]-1-метил-3-(4(s широка, 1 Η, СО2Н). амінофеніл)-2,4-імідазолідиндіон. Потрібну сполу14.3 2-(S)-4-(S)-1-[(1,1ку одержують у сольовій формі (фумарату) обробдиметилетокси)карбоніл]-N-[4-гідрокси-3,5-біс-(1,1кою вільної основи еквівалентом фумарової кисдиметилетил)феніл]-4-(4-нітрофенокси)лоти у теплому етанолі з виходом 35%. Тпл=54,5пролінамід: 57,5°С. До розчину 1,99г (5,64ммоль) проміжної сполуЯМР 1Н (ДМСО, 400МГц, d): 1,40 (s, 18Н, 2tBu), ки 14.2, 1,25г (5,64ммоль) проміжної сполуки 10.2 3,22 (s, 3H, CH3), 6,59 (s, 1H, СН =С), 6,61 (s, фута 845мг (6,20ммоль) гідроксибензотриазолу у марова кислота), 6,97-6,99-7,30-7,32 (4s, 4Н, Η 25мл DMF при 0°С додають 1,28г (6,20ммоль) диаром.), 7,11 (t, 1Н, тіофен), 7,64 (d, 1Н, тіофен), циклогексилкарбодііміду. Після 24 годин перемі7,79 (m, 1Н, тіофен), 7,96 (s, 2Н, Η аром.). шування при 20°С реакційну суміш фільтрують, і осад промивають етилацетатом. Фільтрат розвоІЧ: n ОН: 3618-3433см-1; n С= О (імідазолідиндіон): дять 100мл етилацетату і послідовно промивають 1711см -1; n C=N (амідин): 1585см -1. 2x40мл 1Ν соди, 2x40мл води та 40мл розчину Приклад 14: Хлорогідрат 2-(S)-4-(S)-N-[4солі. Після осушення над сульфатом натрію оргагідрокси-3,5-бic-(1,1-диметилетил)-феніл]-4-{4нічний розчин фільтрують та концентрують до су[(іміно(2-тієніл)метил)аміно]фенокси}-пролінаміду хого стану під вакуумом і одержують каштанове (14): масло, що очищають на колонці з кремнеземом 14.1 Метиловий ефір 2-(S)-4-(S)-1-[(1,1(елюент: гептан/етилацетат: 1/1). Чисті фракції диметилетокси)карбоніл]-4-(4-нітрофенокси)збирають і після концентрування під вакуумом проліну: одержують 1,35г (43%) бежевого порошку. До охолодженої до 0°С суспензії 1,23г Тпл=117-120°С. (30,7ммоль) NaH при 60% у 30мл безводного NЯМР 1Н (CDCI3,100МГц, d): 1,20-1,70 (m, 27Н, 3 метил-2-піролідинону, в інертному середовищі, x tBu), 2,68 (m, 2H, CH 2), 3,80 (m, 2H, CH2-N), 4,58 повільно додають розчин 4,37г (30,7ммоль) 4(m, 1H, CH-CO2), 5,10 (m, 2H, OH, HC-O), 7,25-7,28 нітрофенолу у 30мл безводного N-метил-2(2s, 2H, Ph-OH), 7,51 (m, 4H, Ph-NO2), 8,00 (s шипіролідинону. Після перемішування протягом 1 рока, 1Н, NHCO). години при 0°С однією порцією додають похідну 14.4 2-(S)-4-(S)-1-[(1,1проліну (6г, 15ммоль). Реакційну суміш перемішудиметилетокси)карбоніл]-N-[4-гідрокси-3,5-біс-(1,1ють при 20°С протягом 15 годин з наступним надиметилетил)феніл]-4-(4-амінофенокси)гріванням при 80°С протягом 2 годин для заверпролінамід: шення реакції. Після охолодження до 20°С до В автоклаві з магнітною мішалкою розчиняють даної суміші додають 200мл етилацетату та 100мл 1,35г (2,4ммоль) проміжної сполуки 14.3 у 30мл 1N соди. Після декантації органічну фазу промиетанолу в присутності 1/2 шпателя 10% Pd/C. Реавають послідовно розведеними 1N розчинами сокційну суміш перемішують у атмосфері водню під ди до повної екстракції фенольної похідної, що не тиском 1,5бар протягом 3 годин. Після фільтрупрореагувала, 2x100мл води та 100мл розчину вання через целіт фільтрат концентрують під васолі. Органічний розчин осушують над сульфатом куумом. Залишок обробляють сумішшю 1/1 етилонатрію, фільтрують та концентрують під зниженим вого ефіру та гептану, і після кристалізації тиском і одержують світло-жовте масло, що спонфільтрують та промивають гептаном. Одержують танно кристалізується на повітрі. Кристали збирабежевий порошок з виходом 60%. Тпл=112-113°С. ють та промивають 3x50мл етилового ефіру. Після 53 70921 54 15.2 N-(4-амінофеніл)-5,6-дигідро-1-(2Н)ЯМР 1Н (CDCI3 , 100МГц, d): 1,20-1,70 (m, 27Н, піридин карбоксамід: 3 x tBu), 2,55 (m, 2H, CH2), 3,50 (s широка, 2Н, NH2), 3,75 (m, 2Н, CH2-N), 4,48 (m, 1H, CH-СО2), Одержання потрібного продукту проводилось за тим самим експериментальним протоколом, що 4,80 (m, 1Н, НС-О), 5,10 (s, 1Н, ОН), 6,65 (т, 4Н, й для проміжної сполуки 12.2. Замість 2-(3,5-ди-tPh-NH 2), 7,28 (m, 2Н, Ph-OH), 8,00 (s широка, 1Н, бутил-4-гідроксифеніл)-3-(4-нітрофеніл)-4NHCO). тіазолідинону використовували 5,6-дигідро-N-(414.5 Хлорогідрат 2-(S)-4-(S)-N-[4-гідрокси-3,5біс-(1,1-диметилетил)феніл]-4-{4-[(іміно(2нітрофеніл)-1-(2Н)-піридин карбоксамід. Одержали коричневе масло з виходом 36%. тіеніл)метил)зміно]фенокси}-пролінаміду (14): При температурі 50°С протягом 48 годин наЯМР 1Н (CDCI3 + D2O, 400МГц, d): 2,20 (m, 2Η, грівають суміш 694мг (1,32ммоль) проміжної спо=СН-СН2), 3,59 (m, 2H, CH2-N), 3,95 (m, 2Н, =CHлуки 14.4 у присутності 376мг (1,32ммоль) йодогіCH2-N), 5,84 (m, 2H, CH=CH), 6,90 (m, 4H, Ph), 9,32 драту S-метил-2-тіофентіокарбоксіміду у розчині (s широка, 1H, NHCO). 15мл ізопропанолу. Потім реакційну суміш концен15.3 Хлорогідрат 5,6-дигідро-N-{4-[(іміно(2трують під вакуумом, і залишок після випарювання тієніл)метил)аміно]феніл}-1-(2Н)-піридин карбоксуспендують у 50мл етилацетату. Після додання саміду (15): 50мл насиченого розчину Na2CO3 органічну фазу Одержання потрібного продукту проводилось декантують та послідовно промивають 25мл насиза тим самим експериментальним протоколом, що ченого розчину Na2CO3, 50мл води та 50мл розчий для проміжної сполуки 14.5. Замість 2-(S)-4-(S)ну солі. Після осушення над сульфатом натрію 1-[(1,1-диметилетокси)карбоніл]-N-[4-гідрокси-3,5органічний розчин відфільтровують та концентрубіс-(1,1-диметилетил)феніл]-4-(4-амінофенокси)ють до сухого стан у під вакуумом з одержанням пролінаміду використовували N-(4-амінофеніл)жовтого порошку, який очищають на колонці з 5,6-дигідро-1-(2Н)-піридин карбоксамід. Після пекремнеземом (елюент: етилацетат). Чисті фракції реведення у сольову форму за допомогою 1М збирають, і після концентрування під вакуумом розчину НСІ в е тиловому ефірі одержали блідоодержують 686мг (82%) бежевого порошку, котрий жовтий порошок з виходом 55%. Тпл=230-231°С. негайно розчиняють у 5мл 4М розчину НСІ у 1,4ЯМР 1Н (ДМСО, 400МГц, d): 2,16 (m, 2Н, =СНдіоксані. Після перемішування протягом 15 годин СН2), 3,59 (m, 2H, CH 2-N), 3,98 (m, 2Н, =CH-CH 2-N), при 20°С до реакційної суміші додають 20мл без5,80 (m, 2H, CH=CH), 7,52 (m, 4Н, Ph), 7,38 (s, 1H, водного етилового ефіру. Утворений осад також тіофен), 8,16 (m, 2Н, тіофен), 8,78 (s широка, 1Н, відфільтровують, промивають 2x25мл безводного NH+), 8,81 (s, 1Н, CONH), 9,73 (s широка, 1Н, NH+), етилового ефіру та висушують у сушильній шафі з 11,41 (s широка, 1Н, NH+). одержанням 270мг бежевого порошку. Тпл=233,5ІЧ: n С=О (сечовина): 1637см -1; n C=N (амідин): 235°С. 1583см -1. 1 ЯМР Н (ДМСО, 400МГц, d): 1,37 (s, 18Н, 2 x Приклад 16: Фумарат N-[4-пдрокси-3,5-бic-(1,1tBu), 2,61 (m, 2H, CH 2), 3,60 (m, 2Н, CH2-N), 4,56 диметилетил)феніл]-2-(R,S)-{4-[(іміно(2(m, 1H, CH-CO2), 5,25 (m, 1Н, НС-О), 6,92 (s, 1Н, тієніл)метил)аміно]феніл}-4-(R)-тіазолідин карбокОН), 7,21 (m, 4Н, Ph-N), 7,38 (m, 1Н, тіофен), 7,45 саміду (16): (s, 2Н, Ph-OH), 8,18 (m, 2Н, тіофен), 8,78 (s широ16.1 2-(R,S)-(4-нітрофеніл)-4-(R)-тіазолідин кака, 1Н, NH+), 9,09 (s широка, 1Н, NH+), 9,80 (s ширбонова кислота: рока, 1Н, NH+), 10,68 (с, 1Η, CONH), 11,42 (s широУ 75мл води розчиняють 3г (17,08ммоль) хпока, 1Η, ΝΗ+). рогідрату L-цистеїну та 2,18г (22,2ммоль) ацетату ІЧ: n ОН: 3624-3420см -1; n С= О (амід); 1653см -1; натрію. В процесі додавання до цього розчину 3,10г (20,5ммоль) 4-нітробензальдегіду у 80мл n C=N (амідин): 1610см -1. 95% етанолу (однією порцією) суміш енергійно Приклад 15: Хлорогідрат 5,6-дигідро-N-{4перемішують. В останньому розчині блідо-жовтого [(іміно(2-тієніл)метил)аміно]феніл}-1-(2Н)кольору швидко з'являється білий осад, котрий піридинкарбоксаміду (15): утворюється у надлишку. Перемішування продов15.1 5,6-дигідро-N-(4-нітрофеніл)-1-(2Н)жують протягом 1 години, потім реакційну суміш піридин карбоксамід: охолоджують до 0°С та фільтрують. Осад послідоУ тригорловому реакторі місткістю 100мл в вно промивають 200мл води, 100мл охолодженого атмосфері аргону розчиняють 900мг (5ммоль) 4етанолу та 100мл етилового ефіру. Після осушеннітрофенілізоціанату у 17мл безводного DMF. До ня одержують білий порошок з виходом 87%. цього розчину одноразово додають 0,45мл Тпл=120-121°С. (5ммоль) 1,2,3,6-тетрагідропіридину, і перемішування продовжують протягом 15 годин. Потім реаЯМР 1Н (ацетон D6, 100Мгц, d): 3,50 (m, 2Н, кційну суміш концентрують до сухого стану під CH2-S), 4,25 (m, 1Н, СН-СО), 4,75 (босс, 2Н, СО2Н вакуумом, і залишок після випарювання вводять у + NH), 5,86 (s, 1Н, N-CH-S), 8,20 (m, 4Н, Ph). колонку з кремнеземом. Після елюювання суміш16.2 3-[(1,1-диметилетокси)карбоніл]-2-(R,S)шю гептан/етилацетат 4/6 чисті фракції збирають (4-нітрофеніл)-4-(R)-тіазолідин карбонова кислота: та концентрують під зниженим тиском з одержанОдержання потрібного продукту проводилось ням 860мг (70%) яскраво-жовтого порошку. за тим самим експериментальним протоколом, що Тпл=169-170°С. й для проміжної сполуки 5.1. Замість 4-(t-бутоксикарбоніламіно)-бензолоцтової кислоти використоЯМР 1Н (ДМСО, 100МГц, d): 2,29 (m, 2Н, =СНвували 2-(R,S)-(4-нітрофеніл)-4-(R)-тіазолідин карСН2), 3,69 (m, 2H, CH 2-N), 4,10 (m, 2Н, =CH-CH 2-N), бонову кислоту. Одержали блідо-жовтий порошок 5,91 (m, 2H, CH=CH), 8,09 (m, 4Н, Ph), 9,32 (s широка, 1Н, NHCO). з виходом 59%. Тпл=145-146°С. 55 70921 56 тієніл)метил)аміно]феніл}-4-тіазолкарбоксаміду ЯМР 1Н (CDCI3, 100МГц, d): 1,35 (m, 9H, tBu), (17): 3,40 (m, 2H, CH2-S), 4,95 (m, 1H, CH-CO), 6,10 (m, 1H, N-CH-S), 8,00 (m, 4H, Ph), 10,00 (s широка, 1Н, 17.1 4-нітробензол-карботіоамід: До розчину 4,15г (25ммоль) 4-нітробензаміду у CO2H). 100мл діоксану-1,4 додають 6,06г (15ммоль) реак16.3 3-[(1,1-диметилетокси)карбоніл]-N-(4тиву Лоусона. Реакційну суміш нагрівають із обергідрокси-3,5-біс-(1,1-диметилетил)феніл]2-(R,S)-(4неним холодильником протягом 2 годин. Після нітрофеніл)-4-(R)-тіазолідин карбоксамід: Одержання потрібного продукту проводилось охолодження до кімнатної температури даний розчин виливають у 150мл води та екстрагують 5 раза тим самим експериментальним протоколом, що зів 100мл етилацетату. Органічний розчин осушуй для проміжної сполуки 14.3. Замість 2-(S)-4-(S)ють над сульфатом магнію, відфільтровують та 1-[(1,1-диметилетокси)карбоніл]-4-(4концентрують під вакуумом з одержанням жовтого нітрофенокси)-проліну використовували 3-[(1,1диметилетокси)карбоніл]-2-(R,S)-(4-нітрофеніл)-4масла, яке очищають на колонці з кремнеземом (елюент: гептан/етилацетат 1/1). Чисті фракції (R)-тіазолідин карбонову кислоту. Одержали білий збирають та концентрують під вакуумом з одерпорошок з виходом 41%. Тпл=226-227°С. жанням 3,26г жовтого порошку при виході 72%. ЯМР 1Н (CDCI3, 100МГц, d): 1,45 (m, 27Н, 3 x Тпл=165-167°С. tBu), 3,52 (m, 2H, CH 2-S), 5,00 (m, 1H, CH-CO), 5,15 17.2 Етил-2-(4-нітрофеніл)-4(s, 1H, OH), 6,10 (s широка, 1H, N-CH-S), 7,30 (s, тіазолкарбоксилат: 2H, Ph-OH), 7,92 (m, 4H, Ph-NO2), 8,60 (s широка, У колбу, що містить 100мл DMF, вводять по1Η, CONH). слідовно 3,26г (17,9ммоль) проміжної сполуки 17.1 16.4 3-[(1,1-диметилетокси)карбоніл]-N-[4та 2,26мл (18ммоль) етилбромпїрувату. Після пегідрокси-3,5-біс-(1,1-димeтилeтил)фeнiл]2-(R,S)-(4ремішування реакційної суміші при 23°С протягом aмiнoфeнiл)-4-(R)-тiaзoлiдин карбоксамід: 1 години розчин концентрують під вакуумом. ЗаОдержання потрібного продукту проводилось лишок після випарювання розчиняють у 150мл за тим самим експериментальним протоколом, що дихлорметану та промивають послідовно 100мл й для проміжної сполуки 12.2. Замість 2-(3,5-ди-tводи та 100мл розчину солі. Після осушування над бутил-4-гідроксифеніл)-3-(4-нітрофеніл)-4сульфатом магнію та фільтрування органічний тіазолідинону використовували 3-[(1,1розчин концентрують під вакуумом. Одержаний диметилетокси)карбоніл]-N-[4-гідрокси-3,5-біс-(1,1порошок потім перемішують у присутності 100мл диметилетил)феніл]-2-(R,S)-(4-нітрофеніл)-4-(R)суміші (3/1) толуолу та етанолу, відфільтровують тіазолідин карбоксамід. Потрібний продукт одерта промивають 25мл тіє ж суміші розчинників. жали у вигляді блідо-жовтого порошку з виходом Одержують 3,2г (60%) бежевого порошку. Тпл=15621%. Тпл=196-198°С. 158°С. ЯМР 1Н (CDCI3, 100МГц, d): 1,40 (m, 27Н, 3 x 17.3 2-(4-нітрофеніл)-4-тіазолкарбонова кисtBu), 3,50 (m, 4H, CH 2-S+NH2), 5,00 (m, 1H, CHлота: CO), 5,10 (s, 1H, OH), 6,01 (s широка, 1H, N-CH-S), До розчину проміжної сполуки 17.2 (2,15г, 6,98 (m, 4H, Ph-NH2), 7,25 (s, 2H, Ph-OH), 8,50 (s 7,25ммоль) у 100мл ацетону при 23°С додають по широка, 1Η, CONH). краплях розчин 0,82г (14,5ммоль) КОН у 5мл води. 16.5 Фумарат N-[4-гідрокси-3,5-біс-(1,1Після перемішування протягом ночі утворений диметилетил)феніл]-2-(R,S)-{4-[(іміно(2осад відфільтровують та промивають 10мл ацетотієніл)метил)аміно]феніл}-4-(R)-тіазолідин карбокну. Даний осад обробляють сумішшю 100мл етисаміду (16): лацетату та 100мл 1М розчину НСІ. Після деканОдержання потрібного продукту проводилось тації водну фазу піддають повторному за тим самим експериментальним протоколом, що екстрагуванню 25мл етилацетату. Органічні фази й для проміжної сполуки 14.5. Замість 2-(S)-4-(S)збирають та промивають послідовно 25мл води та 1-[(1,1-диметилетокси)карбоніл]-N-[4-гідрокси-3,550мл розчину солі. Органічний розчин осушують біс-(1,1-диметилетил)феніл]-4-(4-амінофенокси)над сульфатом натрію, фільтрують та концентрупролінаміду використовували проміжну сполуку ють під вакуумом з одержанням жовтого порошку з 16.4. Сполуку 16.5 одержують у вигляді вільної виходом 93%. Тпл=250-252°С. основи і потім переводять у сольову форму в при17.4 N-[3,5-біс(1,1-ділметилетил)-4сутності фумарової кислоти шляхом нагрівання в гідроксифеніл]-2-(4-нітрофеніл)-4етанолі з оберненим холодильником протягом 30 тіазолкарбоксамід: хвилин. Одержують жовтий порошок із загальним Одержання потрібного продукту проводилось виходом 30%. Тпл=201-204°С. 1 за тим самим експериментальним протоколом, що ЯМР Н (ДМСО, 400МГц, d): 1,37 (s, 18Н, 2 x й для проміжної сполуки 14.3. Замість проміжної tBu), 3,17 (m, 2H, CH 2-S), 3,29 (s широка, 1H, NH сполуки 14.2 використовували проміжну сполуку тіазолідин), 3,91 (m, _H, CH-CO), 4,31 (m, _Н, СН17.3. Потрібну сполуку одержали у вигляді жовтого СО), 5,59 (s, _Н, N-CH-S), 5,67 (s, _Н, N-CH-S), порошку з ви ходом 51%. Тпл=262-264°С. 6,61 (s, 2Н, фум.), 6,74 (m, 2Н, NH2 амідин), 7,11 ЯМР 1Н (ацетон d6, 100Мгц, d): 1,60 (s, 18Н, 2 (m, 1Н, тіофен), 7,19 (m, 4Н, Ph-N), 7,42 (s, 2Н, PhtBu), 6,12 (s, 1Н, ОН), 8,21 (m, 2H, Η аром.), 8,50 (s, OH), 7,62 (m, 1Н, тіофен), 7,73 (s широка, 1Н, тіо4H, Η аром.), 8,60 (s, 1H, тіазол), 9,93 (s широка, фен), 9,69 (s, _H, CONH), 9,95 (s, _H, CONH). 1Н, СО-NH). ІЧ: n ОН: 3625-3421см -1; n С= О (амід): 1652см -1; 17.5 N-[3,5-бic(1,1-диметилетил)-4n C=N (амідин): 1604см -1. гідроксифеніл]-2-(4-амінофеніл)-4Приклад 17: Йодогідрат N-[3,5-бic(1,1тіазолкарбоксамід: диметилетил)-4-гідроксифеніл]-2-{4-[(іміно(2 57 70921 58 У розчин проміжної сполуки 17.4 (1,50г, вають послідовно 1N содою (до вилучення із орга3,18ммоль) в 50мл суміші етилаценічної фази надлишку 4-нітрофенолу), водою - до тат/етанол/ацетон (2/1/2) додають 3,59г (16ммоль) нейтральної реакції, та, нарешті, 100мл розчину SnCI2·2H2O. Дану реакційну суміш нагрівають із солі. Після осушення над сульфатом магнію та оберненим холодильником протягом 5 годин і пісфільтрування органічний розчин концентрують під ля охолодження концентрують наполовину під вакуумом, і одержують маслянистий коричневий вакуумом. Залишок після випарювання потім визалишок, який очищають на колонці з кремнезеливають у 50мл охолодженої води, і осад, що мом (елюент: гептан/етилацетат: 8/2). Чисті фракутворився, розводять 100мл етилацетату та 25мл ції збирають та концентрують під вакуумом з оденасиченого розчину NaHCO3. Каламутну суміш ржанням блідо-жовтого масла з виходом 83%. фільтрують через целіт, і фільтрат піддають декаЯМР 1Н (CDCI3, 100Мгц, d): 1,41 (s, 9H, tBu), нтації. Органічну фазу послідовно промивають 2,40 (m, 2H, CH2), 3,80 (s, 5H, CH3+CH2), 4,50 (m, 50мл води та 50мл розчину солі. Після осушення 1H, CH-N), 5,03 (m, 1H, CH-O), 6,95 (m, 2H, Η над сульфатом магнію та фільтрації органічний аром.), 8,22 (m, 2H, Η аром.). розчин концентрують під вакуумом і одержують 18.2 1,1-диметилетил-2-(R)-карбокси-4-(S)-(4яскраво-жовтий порошок, котрий очищають пронітрофенокси)-1-піролідинкарбоксилат: миванням сумішшю Et2O/гептан (90/10). Потрібну До розчину 7г (19ммоль) інтермедіату 18.1 у сполуку одержують у ви гляді блідо-жовтого поро100мл метанолу додають по краплях при 0°С розшку з ви ходом 55%. Тпл=267-268°С. чин 2,14г (38ммоль) КОН у 15мл води. Реакційну ЯМР 1Н (CDCI3, 100Мгц, d): 1,49 (s, 18H, 2 tBu), суміш перемішують протягом 15 годин при 23°С і 4,00 (s широка, 2Н, NH2), 5,11 (s, 1H, ОН), 6,72 (m, потім концентрують наполовину під вакуумом. Піс2Н, Η аром.), 7,60 (s, 2Н, Η аром.), 7,81 (m, 2Н, Η ля розведення 50мл етилацетату та 50мл 1N соди аром.), 8,05 (s, 1Н, тіазол), 9,10 (s широка, 1Н, COсуміш декантують. Органічну фазу вилучають, і NH). водну фазу підкислюють 1М охолодженою НСІ. 17.6 Йодогідрат N-[3,5-бic(1,1-диметилетил)-4Потім продукт екстрагують 100мл етилацетату. гідроксифеніл]-2-{4-[(іміно(2Органічний розчин промивають 50мл води та 50мл тієніл)метил)аміно]феніл}-4-тіазолкарбоксаміду розчину солі. Після осушення над сульфатом маг(17): нію та фільтрації розчин концентрують під вакууОдержання потрібного продукту проводилось мом. Одержують блідо-жовте масло з виходом за тим самим експериментальним протоколом, що 66%. й для проміжної сполуки 1.3. Замість проміжної ЯМР 1Н (CDCI3, 100Мгц, d): 1,45 (s, 9H, tBu), сполуки 1.2 використовували проміжну сполуку 2,52 (m, 2H, CH2), 3,80 (m, 2H, CH2), 4,48 (m, 1H, 17.5. Одержали жовтий порошок з виходом 27%. CH-N), 5,03 (m, 1H, CH-O), 5,92 (s широка, CO2H), Тпл=270-272°С. 6,92 (m, 2H, Η аром.), 8,20 (m, 2H, Η аром.). ЯМР 1Н (ДМСО d6, 400МГц, d): 1,40 (s, 18Н, 2 18.3 1,1-диметилетил-2-(R)-{[[3,5-біс(1,1tBu), 6,89 (s, 1H, OH), 7,41 (m, 1H, Η аром.), 7,63 диметилетил)-4-гідроксифеніл]аміно]карбоніл}-4(m, 4H, Η аром.), 8,11 (m, 1H, Η аром.), 8,20 (m, 1H, (S)-(нітрофенокси)-піролідин-1-карбоксилат: Η аром.), 8,36 (m, 2H, Η аром.), 8,48 (s, 1H, Η Одержання потрібного продукту проводилось аром.), 9,19 (s широка, 1H, NH+), 9,90 (s широка, за тим самим експериментальним протоколом, що 1H, NH+), 10,02 (s, 1H, CO-NH), 11,50 (s, 1H, NH +). й для проміжної сполуки 14.3. Замість проміжної сполуки 14.2 використовували проміжну сполуку ІЧ: n С=О (амід): 1660см -1; n C=N (амідин): 1646см -1. 18.2. Одержали бежевий порошок з виходом 43%. Тпл=140-142°С. Приклад 18: Дихпорогідрат Ν-[3,5-біс(1,1диметилетил)-4-гідроксифеніл]-4-(S)-{4-[(іміно(2ЯМР 1Н (CDCI3, 100Мгц, d): 1,45 (s, 18Н, 2 tBu), тієніл)метил)аміно]фенокси}-піролідин-2-(R)1,50 (s, 9Н, tBu), 2,30 (m, 1H, 1/2 CH 2), 2,95 (m, 1Н, карбоксаміду (18): 1/2 СН2), 3,75 (m, 2H, СН2), 4,65 (m, 1H, CH-N), 18.1 1-(1,1-диметилетил) та 2-метил-4-(S)-(45,10 (m, 2Н, СН-O + ОН), 6,98 (m, 2Н, Η аром.), нітрофенокси)-1,2-(R)-піролідиндикарбоксилат: 7,31 (s, 2Н, Η аром.), 8,22 (m, 2Н, Η аром.), 9,10 (s У тригорловому реакторі при 0°С, в інертному широка, 1Н, CO-NH). середовищі, додають по краплях 4,38г (31,5ммоль) 18.4 1,1-диметилетил-2-(R)-{[[3,5-біс(1,14-нітрофенолу із розчину в 40мл безводного Nдиметилетил)-4-гідроксифеніл]аміно]карбоніл}-4метил-2-піролідинону до суспензії 1,26г (S)-(4-амінофенокси)-піролідин-1-карбоксилат: (31,5ммоль) NaH при 60% у 60мл безводного NОдержання потрібного продукту проводилось метил-2-піролідинону. Реакція супроводжується за тим самим експериментальним протоколом, що значним виділенням водню. Після перемішування й для проміжної сполуки 14.4. Замість проміжної протягом 1 години при 0°С додають однією порцісполуки 14.3 використовували проміжну сполуку єю 6г (15ммоль) 1-(1,1-диметилетил та 2-метил-418.3. Після очищення на колонці з кремнеземом (R)-{[4-метилфеніл)сульфоніл]окси}-1,2-(R)(елюент: гептан/етилацетат: 1/1) та концентруванпіролідинкарбоксилату, і перемішування продовня чистих фракцій одержали потрібну сполуку у жується додатково протягом 15 годин при 23°С. вигляді бежевого порошку з виходом 70%. Реакція завершується після 5 годин нагрівання з Тпл=104-106°С. оберненим холодильником. Після охолодження до ЯМР 1Н (CDCI3, 100Мгц, d): 1,45 (s, 18Н, 2 tBu), 23°С реакційну суміш розводять 150мл етилацета1,50 (s, 9Н, tBu), 1,60 (s, 2H, NH 2), 2,10 (m, 1H, 1/2 ту та 100мл 1М розчину соди. Після декантації CH2), 2,80 (m, 1H, 1/2 CH 2), 3,60 (m, 2H, CH2), 4,60 водну фазу повторно екстрагують 2 рази по 50мл (m, 1H, CH-N), 4,85 (m, 1H, CH-O), 5,04 (s, 1H, OH), етилацетату. Органічні фази збирають та проми6,70 (m, 4H, Η аром.), 7,34 (s, 2H, Η аром.), 9,10 (s 59 70921 60 широка, 1H, CO-NH). масла з виходом 78%. 18.5 Дихлорогідрат N-[3,5-бiс(1,119.3 Метил-1-[(3,4-дигідро-6-гідрокси-2,5,7,8диметилетил)-4-гідроксифеніл]-4-(S)-{4-[(іміно(2тетраметил-2-Н-[1]-бензопіран-2-іл)карбоніл]-4-(S)тієніл)метил)аміно]фенокси}-піролідин-2-(R)(4-нітрофенокси)-піролідин-2-(S)-карбоксилат: карбоксаміду (18): До розчину 1,83г (7,33ммоль) Тролоксу (Trolox) Одержання потрібного продукту проводилось у 20мл безводного THF додають 1,3г (8,06ммоль) за тим самим експериментальним протоколом, що 1,1'-карбонілдиімідазолу. Після перемішування й для проміжної сполуки 2.4. Замість 3-(4протягом 1 години при 23°С по краплях додають амінобензил)-тіазолідину використовували промірозчин 1,95г (7,33ммоль) інтермедіату 19.2, розвежну сполуку 18.4. Вільну основу, одержану у виденого у 10мл безводного THF. Реакційну суміш гляді світло-жовтого порошку, піддавали депротекперемішують при 23°С протягом 15 годин і потім туванню безпосередньо у присутності 10 концентрують до сухого стану під вакуумом. Заеквівалентів безводного 4М розчину НСІ у діоксалишок розводять 100мл етилацетату, і органічний ні-1,4. Після перемішування протягом 15 годин розчин промивають 2 рази по 50мл води та 50мл утворений осад відфільтровували, кристали пророзчину солі. Після осушення над сульфатом магмивали ацетоном та етиловим ефіром. Одержали нію та фільтрації органічний розчин концентрують потрібний продукт у вигляді блідо-жовтого порошпід вакуумом. Залишок після випарювання очику з ви ходом 53%. Тпл=245-247°С. щають на колонці з кремнеземом (елюент: гептан/етилацетат: 6/4). Чисті фракції збирають та ЯМР 1Н (ДМСО d6, 400МГц, d): 1,36 (s, 18Н, 2 випарюють під вакуумом з одержанням жовтого tBu), 2,29 (m, 1H, 1/2 CH2), 2,71 (m, 1H, 1/2 CH2), порошку з ви ходом 61%. Тпл=103-105°С. 3,42 (m, 1H, 1/2 CH2), 3,77 (m, 1H, 1/2 CH2), 4,57 (m, 1H, CH-N), 5,26 (m, 1H, CH-O), 6,93 (s, 1H, OH), ЯМР 1Н (CDCI3, 400Мгц, d): 1,55-2,50 (m, 16Н, 7,17 (m, 2H, Η аром.), 7,37 (m, 1H, Η аром.), 7,42 Тролокс), 2,63 (m, 2Н, СН2), 3,60-3,71 (2s, 3Н, (m, 2H, Η аром.), 7,48 (s, 2H, Η аром.), 8,17 (m, 2H, ОСН3), 3,85 (m, 2Н, СН2), 4,70-4,88 (2m, 1Н, CH-N), Η аром.), 8,81 (s широка, 1H, NH+), 9,03 (s широка, 5,02 (m, 1Н, GH-O), 6,82 (m, 2Н, Η аром.), 8,20 (m, 1H, NH+), 9,78 (s широка, 1H, NH+), 10,70 (s, 1H, 2Н, Η аром.). CO-NH), 10,84 (s широка, 1H, NH+), 11,50 (s широ19.4 Метил-1-[(3,4-дигідро-6-гідрокси-2,5,7,8ка, 1Н, NH+). тетраметил-2-Н-[1]-бензопіран-2-іл)карбоніл]-4-(S)(4-амінофенокси)-піролідин-2-(S)-карбоксилат: ІЧ: n С=О (амід): 1681см -1; n C=N (амідин): -1 Одержання потрібного продукту проводилось 1652см . за тим самим експериментальним протоколом, що Приклад 19: Хлорогідрат метил-1-[(3,4-дигідрой для проміжної сполуки 14.4. Замість проміжної 6-гідрокси-2,5,7,8-тетраметил-2-Н-[1]-бензопіран-2сполуки 14.3 використовували проміжну сполуку іл)карбоніл]-4-(3)-{4-[(іміно(2-тієніл)метил)аміно]19.3. Потрібний продукт одержали у вигляді білого фенокси}-піролідин-2-(8)-карбоксилату (19): порошку з ви ходом 95%. Тпл=110-112°С. 19.1 1-(1,1-диметилетил та 2-метил-4-(S)-(419.5 Хлорогідрат метил-1-[(3,4-дигідро-6нітрофенокси)-1,2-(S)-піролідиндикарбоксилат: гідрокси-2,5,7,8-тетраметил-2-Н-[1]-бензопіран-2Одержання потрібного продукту проводилось іл)карбоніл]-4-(S)-{4-[(іміно(2за тим самим експериментальним протоколом, що тієніл)метил)аміно]фенокси}-піролідин-2-(S)й для проміжної сполуки 18.1. Замість похідної 1карбоксилату (19): (1,1-диметилетил) та 2-метил-4-(R)-{[(4Одержання потрібного продукту проводилось метилфеніл)сульфоніл]окси}-1,2-(R)за тим самим експериментальним протоколом, що піролідиндикарбоксилату використовували похідну й для проміжної сполуки 2.4. Замість проміжної 1-(1,1-диметилетил) та 2-метил 4-(S)-{[(4сполуки 2.3 використовували проміжну сполуку метилфеніл)сульфоніл]окси}-1,2-(R)19.4. Реакцію конденсації проводять тільки в одпіролідиндикарбоксилату. Потрібний продукт оденому 2-пропанолі. Після переведення у сольову ржували у вигляді білого порошку з виходом 63%. форму потрібний продукт одержують у вигляді Тпл=155-157°С. блідо-жовтого порошку з виходом 75%. Тпл=203ЯМР 1Н (ДМСО d6, 400МГц, d): 1,37 (2s, 9H, 206°С. tBu), 2,22 (m, 1H, 1/2 CH2), 2,62 (m, 1H, 1/2 CH2), ЯМР 1Н (ДМСО d6, 400МГц, d): 1,55-2,50 (m, 3,45 (m, 1H, 1/2 CH2), 3,62 (2s, 3H, OCH 3), 3,78 (m, 16H, Тролокс), 2,45 (m, 2H, СН2), 3,45-3,60 (2s, 3Н, 1H, 1/2 CH2), 4,42 (m, 1H, CH-N), 5,20 (m, 1H, CHO), 7,07 (m, 2H, Η аром.), 8,20 (m, 2H, Η аром.). ОСН3), 3,70 (m, 2Н, СН2), 4,51 (m, 1Н, CH-N), 5,02 (m, 1Н, СН-О), 7,00 (m, 2Н, Η аром.), 7,39 (m, 3Н, Η 19.2 Метил-4-(S)-(4-нітрофенокси)-піролідин-2аром.), 8,16 (m, 2Н, Η аром.), 8,80 (s широка, 1Н, (S)-карбоксилат: ΝΗ+), 9,75 (s широка, 1Н, NH+), 11,36 (s широка, До розчину 3,45г (9,4ммоль) проміжної сполуки 1Н, NH+). 19.1 у 15мл дихлорметану додають при 0°С 10мл (94ммоль) трифтороцтової кислоти, розведеної ІЧ: n С=О (амід): 1650см -1; n C=N (амідин): 10мл дихлорметану. Реакційну суміш потім пере1611см -1. мішують протягом 2 годин при 23°С і концентрують Приклад 20: Хлорогідрат 1-[(3,4-дигідро-6під вакуумом. Залишок після випарювання розвогідрокси-2,5,7,8-тетраметил-2Н-[1]-бензопіран-2дять 100мл дихлорметану, і органічний розчин іл)карбоніл]-3-(S)-{4-[(іміно(2послідовно промивають 3 рази по 20мл насиченотієніл)метил)аміно]фенокси}-піролідину (20): го розчину Na2CO3, 2 рази по 20мл води і, нарешті, 20.1 1,1-диметилетил-3-(R)-{[(420мл розчину солі. Після осушення над сульфатом метилфеніл)сульфоніл]окси}-1магнію та фільтрації органічний розчин концентпіролідинкарбоксилат: рують під вакуумом з одержанням блідо-жовтого 21,6г (114ммоль) р-толуолсульфонілхпориду
ДивитисяДодаткова інформація
Назва патенту англійськоюNovel derivatives of 2-(iminomethyl)amino-phenyl, preparation thereof, application as medicaments and pharmaceutical compositions containing these compounds
Назва патенту російськоюНовые производные 2-(иминометил)амино-фенила, их получение, применение как лекарств и фармацевтические композиции, которые содержат эти соединения
МПК / Мітки
МПК: C07D 207/16, A61P 43/00, A61K 31/4436, A61K 31/397, A61K 31/454, C07D 409/12, C07D 307/68, C07D 233/78, C07D 417/12, A61K 31/381, A61P 3/06, A61K 31/427, A61K 31/4178, C07D 409/14, A61K 31/44, A61K 31/4535, A61K 31/4025, A61K 31/422
Мітки: 2-(імінометил)амінофенілу, сполуки, одержання, похідні, варіанти, фармацевтична, композиція, спосіб, містить
Код посилання
<a href="https://ua.patents.su/39-70921-pokhidni-2-iminometilaminofenilu-sposib-kh-oderzhannya-varianti-ta-farmacevtichna-kompoziciya-shho-mistit-ci-spoluki.html" target="_blank" rel="follow" title="База патентів України">Похідні 2-(імінометил)амінофенілу, спосіб їх одержання (варіанти) та фармацевтична композиція, що містить ці сполуки</a>
Похідні індолу, фармацевтична композиція та спосіб одержання сполук (варіанти)

Номер патенту: 68388
Опубліковано: 16.08.2004
Автори: Паллук Райнер, Вайзер Томас, Грауерт Маттіас, Пшорн Уве, Бєхтєль Вольф-Дітріх, Картер Адріан, Хьонке Хрістоф
МПК: C07D 209/04, C07D 401/04, A61P 25/28, A61P 23/02, A61P 9/06, A61P 9/10, A61K 31/404, C07D 209/60
Мітки: фармацевтична, похідні, композиція, варіанти, сполук, індолу, спосіб, одержання
Формула / Реферат:
1. Похідні індолу загальної формули 1, 1деХ означає простий зв'язок, -О-, С1-С4алкіл, С1-С3алкоксигрупу, -О-СН2-СН2-О- чи -O-CH2-CH2-NH-,R1 означає водень, метил, етил чи феніл, R2 означає водень або метил,R3 означає водень, F, Сl, Br, гідрокси- чи метоксигрупу, R4 означає водень, метил чи етил, R5 означає водень, метил чи етил, R6 означає водень, метил чи етил, R7 означає...
Похідні 2-(імінометил)амінофенілу, їх отримання, застосування як ліків, та фармацевтичні композиції, що їх містять
Номер патенту: 56219
Опубліковано: 15.05.2003
Автори: Шабр'є де Лассон'єр П'єр-Ет'єн, Огует Мішель, Овін Серж, БІГГ Денніс
МПК: C07C 281/00, A61P 25/00, A61P 43/00, A61K 31/381, C07D 243/08, C07D 409/12, C07D 333/38, A61K 31/496, C07D 295/192, A61K 31/551, C07D 311/66, C07C 235/30, A61P 9/00, C07C 235/56, C07D 209/42, C07D 295/185, C07C 275/36, C07D 409/14
Мітки: похідні, 2-(імінометил)амінофенілу, ліків, композиції, містять, фармацевтичні, застосування, отримання
Формула / Реферат:
1. Сполука загальної формули (І)(І),яка відрізняється тим, що А являє собою або радикал ,де R1 та R2 являють собою, незалежно один від одного, атом водню, галоген, групу ОН, лінійний або розгалужений радикал алкілу або алкокси, що має від 1 до 6 атомів вуглецю, R3 являє собою атом водню, лінійний або розгалужений радикал алкілу, щомає від-1 до 6 атомів вуглецю, або радикал...
Похідні бензотіофену, спосіб їх одержання (варіанти), проміжні сполуки, фармацевтична композиція

Номер патенту: 44710
Опубліковано: 15.03.2002
Автори: Трашер Кеннет Джефф, Палковіч Алан Девід
МПК: A61K 31/4025, A61K 31/55, A61P 5/00, A61K 31/57, A61K 31/38, A61P 15/00, A61P 13/02, A61K 31/381, A61K 31/4535, A61P 43/00, A61P 19/10, C07D 333/62, C07D 333/64, A61K 31/565
Мітки: варіанти, сполуки, одержання, проміжні, фармацевтична, похідні, спосіб, бензотіофену, композиція
Формула / Реферат:
1. Бензотиофен формулы I: гдеR1 представляет -Н, -ОН, -О(С1-С4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), или –OSO2(С2-С6алкил);R2 представляет -Н, -ОН, -O(С1-С4алкил), -ОСОС6Н5, -ОСО(С1-С4алкил), -OSO2(С2-С6алкил) или галоид;R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино, диизопропиламино или...
Похідні арилгліцинамідів та фармацевтична композиція, що містить ці сполуки

Номер патенту: 46765
Опубліковано: 17.06.2002
Автори: Брім Ханс, Шпек Георг, Юнг Біргіт, Доллінгер Хорст, Шнорренберг Герд, Ессер Франц
МПК: A61P 15/00, A61P 27/14, C07D 211/64, A61P 35/00, A61P 1/00, C07D 211/58, A61K 31/535, A61P 19/02, A61K 31/451, A61P 11/06, C07D 401/12, C07D 211/62, A61P 11/00, A61P 13/02, A61P 43/00, A61P 27/02, C07D 295/14, A61P 29/00, C07D 471/10, A61K 31/4468, C07D 401/04, A61P 37/08, A61K 31/5375, A61P 25/00, A61K 31/495, A61P 37/00, C07D 295/145, C07D 211/66, A61K 31/445, A61K 31/4427
Мітки: похідні, містить, фармацевтична, композиція, сполуки, арилгліцинамідів
Формула / Реферат:
1. Похідні арилгліцинамідів загальної формули (І):або їх фармацевтично прийнятні солі, деАr означає незаміщений або від одного до п'яти разів заміщений феніл, або ж незаміщений, або від одного до двох разів заміщений нафтил, причому замісники фенільного та нафтильного фрагментів незалежно один від одного є атомами галогену (фтор, хлор, бром, йод), гідроксильною групою, алкілом з числом атомів вуглецю від одного до...
Гетероциклічна сполука, спосіб її одержання (варіанти), проміжні сполуки, фармацевтична композиція та спосіб лікування неврологічних захворювань або розладів (варіанти)

Номер патенту: 64006
Опубліковано: 16.02.2004
Автори: Палмер Майкл Джон, Сеннер Марк Аллен, Кемп Марк Ян, Уайтс Мартін Джеймс
МПК: C07D 401/14, A61P 25/16, A61P 19/00, A61P 21/04, A61P 25/00, A61P 21/00, C07D 413/14, C07D 417/14, A61P 27/16, C07D 401/04, C07D 413/04, C07D 521/00, C07D 471/14, A61K 31/454, A61K 31/4545, A61P 43/00, A61P 25/28, A61P 27/02, A61K 31/4427, A61P 37/00, A61P 25/14, A61K 31/4709, C07D 417/04, A61P 25/02
Мітки: сполуки, спосіб, композиція, лікування, розладів, сполука, варіанти, неврологічних, фармацевтична, проміжні, одержання, гетероциклічна, захворювань
Формула / Реферат:
1. Гетероциклічна похідна формули (І): , (I)або її фармацевтично прийнятна сіль, в якійA є нерозгалуженим C3-C5алкіленом, необов’язково, заміщеним C1-C6алкілом;X є O, S, NH або N(C1-C6алкіл);Y є О, S, NH або N(C1-C6алкіл);R є C-приєднаною 4-6-членною неароматичною гетероциклічною групою, що містить один атом азоту як гетероатом, згадана група, необов’язково, заміщена 1, 2 або 3 замісником(ами), кожний...
Попередній патент: Активуюча композиція сполук металоцену в каталітичній (спів)полімеризації олефінів
Наступний патент: Пристрій для безперервного виготовлення листових виробів з полімерних матеріалів
Випадковий патент: Прилад для виготовлення гістологічних блоків