Похідні фторопіридинону, що мають антибактеріальну активність
Номер патенту: 107423
Опубліковано: 25.12.2014
Автори: Монтґомері Юстін Ян, Браун Меттью Френк, Ке І, Рейллі Юса, Прайс Лорен Майкл, Плюммер Марк Стівен, Мелнік Майкл Джозеф








Формула / Реферат
1. Сполука формули:
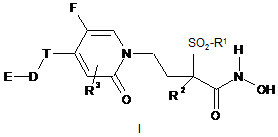
або її фармацевтично прийнятна сіль,
де:
R1 є (С1-С3)алкілом;
R2 є воднем або (С1-С3)алкілом;
R3 є воднем, галогеном, гідрокси, ціано, (С1-С3)алкілом, (С1-С3)алкокси, трифторметилом або трифторметилокси;
Т є етинілом, вибірково заміщеним (С6-С10)арилом або вибірково заміщеним гетероарилом;
D є відсутнім або являє собою -(СН2)r-, -(СН2)n-О-(СН2)р- або зв'язок;
r є цілим числом 1, 2, або 3;
n та р незалежним чином являють собою ціле число 0, 1 або 2;
Е є відсутнім або являє собою замісник, вибраний з групи, що містить:
і) (С3-С10)циклоалкіл, вибірково заміщений;
іі) (С6-С10)арил, вибірково заміщений;
ііі) гетероарил, вибірково заміщений; та
iv) гетероцикл, вибірково заміщений;
за умови, що:
1) якщо Е є відсутнім, то D є також відсутнім;
2) Т не є незаміщеним фенілом; коли Е та D разом є відсутніми, то R3 є воднем та кожен з радикалів R1 та R2 є метилом;
де кожен "(С6-С10)арил, вибірково заміщений" або "вибірково заміщений (С6-С10)арил" є вибірково заміщеним від 1 до 4 замісниками, кожен з яких є незалежно вибраним з групи, що містить галоген, ціано, нітро, гідрокси, вибірково заміщений (С1-С6)алкіл, вибірково заміщений (С1-С6)алкокси, фосфат, -SO2NR4R5, -(CH2)m-NR5-C(О)-R4, -(CH2)m-C(О)-N-R4R5, -C(О)-R4,-C(О)-О-R4, -SR4, -SO2R4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або (С1-С3)алкіл, та m незалежним чином являє собою ціле число від 0 до 4;
де кожен "гетероарил, вибірково заміщений" або "вибірково заміщений гетероарил" являє собою 5-10-членний моноциклічний або біциклічний гетероарильний фрагмент, що містить від одного до чотирьох гетероатомів, вибраних з кисню, азоту та сірки, та є вибірково заміщеним від 1 до 4 замісниками, кожен з яких є незалежно вибраним з групи, що містить галоген, ціано, нітро, гідрокси, вибірково заміщений (С1-С6)алкіл, вибірково заміщений (С1-С6)алкокси, фосфат, -SО2NR4R5, -(CH2)m-N-C(О)-R4, -(CH2)m-C(О)-N-R4R5, -C(О)-R4, -C(О)-О-R4, -SR4, -SО2R4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або С1-С3алкіл, та m незалежним чином являє собою ціле число від 0 до 4;
де кожен "вибірково заміщений (С3-С10)циклоалкіл" являє собою насичений або частково насичений моноциклічний, біциклічний, біциклічний з містковими зв'язками або трициклічний алкільний радикал, де кожен циклічний фрагмент має 3-10 атомів карбону, та є вибірково заміщеним від 1 до 4 замісниками, кожен з яких є незалежно вибраним з групи, що містить галоген, ціано, нітро, гідрокси, вибірково заміщений (С1-С6)алкіл, вибірково заміщений (С1-С6)алкокси, фосфат, оксо, -SО2NR4R5, -(CH2)m-NR5-C(О)-R4, -(CH2)m-C(О)-N-R4R5, -C(О)-R4, -C(О)-О-R4, -SR4, -SО2R4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або (С1-С3)алкіл, та m незалежним чином являє собою ціле число від 0 до 4;
де кожен "гетероцикл, вибірково заміщений" являє собою 3-10-членне моноциклічне або біциклічне кільце, яке містить від одного до трьох гетероатомів, вибраних з кисню, азоту та сірки, та є вибірково заміщеним від 1 до 4 замісниками, кожен з яких є незалежно вибраним з групи, що містить галоген, ціано, нітро, гідрокси, вибірково заміщений (С1-С6)алкіл, вибірково заміщений (С1-С6)алкокси, пентафторосульфоніл, фосфат, оксо, -SО2NR4R5, -(CH2)m-N-C(О)-R4, -(CH2)m-C(О)-N-R4R5, -C(О)-R4, -C(О)-О-R4, -SR4, -SО2R4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або (С1-С3)алкіл, та m незалежним чином являє собою ціле число від 0 до 4;
де кожен "вибірково заміщений (С1-С6)алкіл" є вибірково заміщеним від 1 до 3 замісниками, кожен з яких є незалежно вибраним з групи, що містить галоген, ціано, сульфонамід, іміно, -OR4, -SR4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або (С1-С3)алкіл;
де кожен "вибірково заміщений (С1-С6)алкокси" є вибірково заміщеним від 1 до 3 замісниками, кожен з яких є незалежно вибраним з групи, що містить галоген, ціано, сульфонамід, іміно, -OR4, -SR4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або С1-С3 алкіл.
2. Сполука за п. 1 або її фармацевтично прийнятна сіль, де R1 та R2 є метилом.
3. Сполука за п. 1 або 2 або її фармацевтично прийнятна сіль, де R3 є воднем.
4. Сполука за п. 2 або 3 або її фармацевтично прийнятна сіль, де вказана сполука є істотно чистим R-енантіомером.
5. Сполука за будь-яким з пп. 1, 2, 3 або 4 або її фармацевтично прийнятна сіль, де Т являє собою феніл, який може бути вибірково заміщеним від 1 до 4 замісниками, де кожен замісник є незалежно вибраним з групи, що містить галоген, ціано, нітро, гідрокси, вибірково заміщений (С1-С6)алкіл, вибірково заміщений (С1-С6)алкокси, фосфат, -SО2NR4R5, -(CH2)m-NR5-C(О)-R4, -(CH2)m-C(О)-N-R4R5, -C(О)-R4, -C(О)-О-R4, -SR4, -SО2R4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або (С1-С3)алкіл, та m незалежним чином являє собою ціле число від 0 до 4.
6. Сполука за будь-яким з пп. 1, 2, 3, 4 або 5 або її фармацевтично прийнятна сіль, де D та Е є разом відсутніми.
7. Сполука за будь-яким з пп. 1-5 або її фармацевтично прийнятна сіль, де D є зв'язком.
8. Сполука за п. 7 або її фармацевтично прийнятна сіль, де Е являє собою циклогексил, піримідиніл, триазоліл, піридиніл, ізоксазоліл або циклопропіл, де будь-що з вищенаведеного може бути вибірково заміщено від 1 до 4 замісниками, де кожен замісник є незалежно вибраним з групи, що містить галоген, ціано, нітро, гідрокси, вибірково заміщений (С1-С6)алкіл, вибірково заміщений (С1-С6)алкокси, фосфат, -SО2NR4R5, -(CH2)m-NR5-C(О)-R4, -(CH2)m-C(О)-N-R4R5, -C(О)-R4, -C(О)-О-R4, -SR4, -SО2R4 та -NR4R5, де кожен радикал R4 та R5 незалежним чином являє собою водень або (С1-С3)алкіл, та m незалежним чином являє собою ціле число від 0 до 4.
9. Сполука формули:
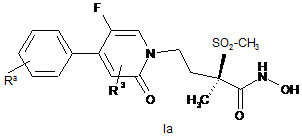
або її фармацевтично прийнятна сіль,
де:
R3 являє собою водень, галоген, гідрокси, ціано, (С1-С3)алкіл, (С1-С3)алкокси, трифторометил або трифторометилокси;
Ra являє собою один або декілька замісників, вибраних з групи, що містить (С1-С3)алкіл, (С1-С3)алкокси, фтор, хлор, гідрокси, трифторометил та трифторометилокси.
10. (2R)-4-{5-фторо-2-оксо-4-[4-(2Н-1,2,3-триазол-2-іл)феніл]піридин-1(2Н)-іл}-N-гідрокси-2-метил-2-(метилсульфоніл)бутанамід або його фармацевтично прийнятна сіль.
11. (2R)-4-[5-фторо-4-(2-фторо-4-метоксифеніл)-2-оксопіридин-1(2Н)-іл]-N-гідрокси-2-метил-2-(метилсульфоніл)бутанамід або його фармацевтично прийнятна сіль.
12. Фармацевтична композиція, що містить сполуку за будь-яким з пп. 1-11 або її фармацевтично прийнятну сіль у суміші з принаймні одним фармацевтично прийнятним наповнювачем.
13. Спосіб лікування бактеріальних інфекцій, за яким вводять пацієнту у разі потреби терапевтично ефективну кількість сполуки за будь-яким з пп. 1-11 або її фармацевтично прийнятної солі.
14. Застосування сполуки за будь-яким з пп. 1-11 або її фармацевтично прийнятної солі у виробництві лікарського препарату для лікування бактеріальних інфекцій.
Текст
Реферат: Заявлений винахід стосується нового класу похідних гідроксамової кислоти, їх застосування як інгібіторів LpxC та, зокрема, їх застосування у лікуванні бактеріальних інфекцій. UA 107423 C2 (12) UA 107423 C2 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 Цей винахід стосується нових похідних гідроксамової кислоти. Винахід також стосується способів застосування таких сполук у лікуванні бактеріальних інфекцій (особливо грамнегативних інфекцій) та фармацевтичних композицій, що містять такі сполуки. Інфекції, спричинені грамнегативними бактеріями, як-то Pseudomonas aeruginosa, Entgerobacteriaceae, що виробляють β-лактамазу розширеного спектру (ESBL), Entgerobacteriaceae та Acinetobacter baumannii є важливою проблемою у галузі охорони здоров'я, особливо у випадку внутрішньолікарняних інфекцій. Крім того, зростає рівень стійкості до звичайної терапії антибіотиками, що сильно обмежує варіанти лікування. Наприклад, у 2002 році, 33 % інфекцій з відділень інтенсивної терапії, спричинених Pseudomonas aeruginosa були стійкими до фторохінолонів, у той час, як стійкість до іміпенему сягала 22 % (CID 42: 657-68, 2006). На додачу до цього, у світі також зросла кількість інфекцій зі стійкістю до багатьох ліків (MDR); у випадку з Pseudomonas aeruginosa, кількість MDR зросла від 4 % у 1992 р. до 14 % у 2002 р. (Biochem Pharm 71: 991, 2006). Грамнегативні бактерії є унікальними завдяки тому, що їх зовнішня мембрана містить ліпополісахарид (LPS), який є критичним для підтримання мембранної цілісності та важливим для життєздатності бактерій (розглянуто у Ann. Rev. Biochem 76: 295-329, 2007). Головним ліпідним компонентом LPS є ліпід A та інгібування біосінтезу ліпідів є летальним для бактерій. Синтез ліпіду А відбувається на цитоплазматичній поверхні бактеріальної внутрішньої мембрани та до цього процесу залучено дев'ять різних ферментів, які є високо консервативними у більшості грамнегативних бактерій. LpxC [UDP-3-O-(R-3- гідроксиміристойл)N- ацетилглюкозамін деацетилаза] є ферментом, що каталізує перший етап біосинтезу ліпіду А, усунення N- ацетильної групи UDP-3-O-(R-3-гідроксиміристойл)-N-ацетилглюкозаміну. LpxC є 2+ Zn -залежним ферментом, який не має гомологів у ссавців, що робить його гарним об'єктом для розробки нових антибіотиків. Також були знайдені окремі інгібітори LpxC з низькою наномолярною спорідненістю (Biochemistry 45: 7940-48, 2006). Винахідники відкрили новий клас інгібіторів LpxC. Ці сполуки або їх фармацевтично прийнятні солі можуть бути зображені у вигляді нижченаведеної Формули I: де: 1 R являє собою C1-C3 алкіл; 2 R являє собою водень або C1-C3 алкіл; 3 R являє собою водень, галоген, гідрокси, ціано, C 1-C3 алкіл, C1-C3 алкокси, трифторометил або трифторометилокси; T являє собою етиніл, вибірково заміщений (C6-C10) арилом або вибірково заміщений гетероарилом; D є відсутнім або являє собою -(CH2)r-, -(CH2)n-O-(CH2)p- або зв'язок; r є цілим числом 1, 2 або 3; n та p є незалежними одне від одного цілими числами 0, 1 або 2; E є відсутнім або являє собою замісник, вибраний з групи, що охоплює: i) (C3-C10) циклоалкіл, вибірково заміщений; ii) (C6-C10) арил, вибірково заміщений; iii) гетероарил, вибірково заміщений; та iv) гетероцикл, вибірково заміщений; за умови, що: 1) якщо E є відсутнім, то D є також відсутнім; 2) якщо E та D є разом відсутніми, то T не є незаміщеним фенілом; R3 є воднем та кожен з радикалів R1 та R2 є метилом. Сполуки Формули I проявляють антибактеріальну активність, особливо проти грамнегативних організмів та їх можна буде застосувати для лікування бактеріальних інфекцій у ссавців, особливо людини. Також ці сполуки можуть бути застосовані у ветеринарії, як-то для лікування інфекцій домашньої худоби та свійських тварин. 1 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 60 Сполуки за Формулою I є корисними у лікуванні численних, особливо грамнегативних інфекцій, що охоплюють госпітальну (нозокоміальну) пневмонію, інфекції сечовивідних шляхів, системні інфекції (бактеріємію та сепсис), інфекції шкіри та м'яких тканин, хірургічні інфекції, внутрішньочеревні інфекції, легеневі інфекції (в тому числі інфекції пацієнтів з муковісцидозом), Helicobacter pylori (та сукупність пов'язаних з ним шлункових ускладнень, як-то виразкова хвороба чи рак шлунку тощо), ендокардит, синдром діабетичної стопи, остеомієліт та інфекції центральної нервової системи. Для спрощення застосування, сполуки звичайно можна змішати з принаймні одним наповнювачем та приготувати у вигляді фармацевтичної лікарської форми. Приклади таких лікарських форм охоплюють таблетки, капсули, розчини / суспензії для ін'єкцій, аерозолі для інгаляцій, креми / мазі для місцевого, вушного або офтальматичного застосування та розчини / суспензії для перорального прийому. Заголовки в цьому документі застосовані тільки для прискорення їх перегляду читачем. Вони не повинні бути витлумачені в якості будь-якого обмеження винаходу або пунктів Формули Винаходу. Як застосовано у даній заявці, включаючи також Формулу Винаходу, наступні терміни мають зазначені нижче визначення, якщо тільки в тексті навмисно не вказано іншого. Однину та множину, крім ознак числа слід вживати взаємозамінним чином. a. "C1-C3 алкіл ” стосується розгалуженої або прямої ланцюгової алкільної групи, що містить 1-3 атоми карбону, як-то метил, етил, n-пропіл або ізопропіл тощо. b. "C1-C3 алкокси" стосується прямої або розгалуженої ланцюгової алкокси групи, що містить 1-3 атоми карбону, як-то метокси, етокси, n-пропокси, ізопропокси тощо. c. “ галоген" стосується атому хлору, фтору, йоду чи брому. d. "C1-C6 алкіл ” стосується розгалуженої або прямої ланцюгової алкільної групи, що містить 1-6 атомів карбону, як-то метил, етил, n-пропіл, ізопропіл, n- бутил, бутил, пентил тощо. e. “ Вибірково заміщений C1-C6 алкіл ” стосується розгалуженої або прямої ланцюгової алкільної групи, що містить 1-6 атомів карбону, як-то метил, етил, n-пропіл, ізопропіл, n- бутил, ізобутил, пентил тощо. Така алкільна група може бути вибірково заміщеною, де до трьох атомів водню є заміщеними замісником, вибраним з групи, що охоплює галоген, ціано, сульфонамід, іміно, -OR4, -SR4, та –NR4R5, де кожен з радикалів R4 та R5 незалежним чином являють собою водень або C1-C3 алкіл. f. “ C1-C6 алкокси " стосується прямої або розгалуженої ланцюгової алкокси групи, що містить 1-6 атомів карбону, як-то метокси, етокси, n-пропокси, ізопропокси, n- бутокси, ізобутокси, пентокси тощо. g. “ Вибірково заміщений C1-C6 алкокси ” стосується прямої або розгалуженої ланцюгової алкоксигрупи, що містить 1-6 атомів карбону, як-то метокси, етокси, n-пропокси, ізопропокси, nбутокси, ізобутокси, пентокси тощо. Така алкоксигрупа може бути вибірково заміщеною, де в такому разі до трьох атомів водню буде заміщено замісником, вибраним з групи, що охоплює галоген, ціано, сульфонамід, іміно, -OR4, -SR4, та –NR4R5, де раді кали R4 та R5 незалежним чином являють собою водень або C1-C3 алкіл. h. “(C6-C10) арил ” має відношення до циклічного, ароматичного вуглеводу, що містить 6-10 атомів карбону. Приклади такої арильної групи охоплюють феніл, нафтил тощо. i. “ Вибірково заміщений (C6-C10) арил ” має відношення до циклічного, ароматичного вуглеводу, як визначено вище, де арильна функціональна група може бути вибірково заміщена замісниками, що не містять водень (до 4 замісників), де кожен з них є незалежно вибраним з групи, що охоплює галоген, ціано, нітро, гідрокси, вибірково заміщений (C1-C6) алкіл, вибірково заміщений (C1-C6) алкокси, трифторометил, трифторометокси, фосфат, -SO2NR4R5, -(CH2)mNR5-C(O)-R4, -(CH2)m-C(O)-N-R4R5, -C(O)-R4,-C(O)-O-R4, -SR4, -SO2R4 та -NR4R5, де раді кали R4 та R5 є визначеними вище та кожною буквою m тут позначено ціле число (незалежно від інших) від 0 до 4. Ці замісники можуть бути однаковими або різними та можуть бути розміщені у будь-якому хімічно прийнятному положенні кільця. “ Вибірково заміщений феніл ” стосується фенільного кільця, заміщеного, як описано вище. j. “ Гетероарил ” має відношення до ароматичного кільця, що має один чи більше гетероатомів, вибраних з кисню, азоту та сірки. Більш докладно це стосується 5- або 6-членного кільця, що містить 1, 2, 3, або 4 атоми азоту; 1 атом кисню; 1 атом сірки; 1 атом азоту та 1 атом сірки; 1 атом азоту та 1 атом кисню; 2 атоми азоту та 1 атом кисню; або 2 атоми азоту та 1 атом сірки. 5-членне кільце має 2 подвійних зв'язки та 6- членне кільце має 3 подвійних зв'язки (“ далі зазначено, як “ 5-6- членний гетероарил ”). Термін “ гетероарил ” також охоплює біциклічні групи, де гетероарильне кільце є злитим з бензеновим кільцем, гетероциклічним кільцем, циклоалкіловим кільцем або іншим гетероариловим кільцем. Приклади таких гетероарильних 2 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 60 кільцевих систем охоплюють, але без обмеження, піроліл, фураніл, тіеніл, імідазоліл, оксазоліл, індоліл, тіазоліл, піразоліл, піридиніл, піримідиніл, пуриніл, хинолініл, бензофуран, тетразол, ізохинолініл, оксидіазоліл, тіадіазоліл, ізотіадіазоліл, ізоксазоліл, тріазоліл, бензо[b]тіеніл, 2-, 4-, 5-, 6- або 7-бензоксазоліл, 7-бензімідазоліл або бензотіазоліл. k. "Вибірково заміщений гетероарил ” стосується гетероарильної функціональної групи, як було безпосередньо визначено вище, у якої до 4 атомів карбону гетероарильної функціональної групи може бути заміщено на замісник, де кожен замісник є незалежно вибраним з групи, що охоплює галоген, ціано, нітро, гідрокси, вибірково заміщений (C1-C6) алкіл, вибірково заміщений (C1-C6) алкокси, трифторометил, трифторометокси, фосфат, -SO2NR4R5, -(CH2)m-N-C(O)-R4, (CH2)m-C(O)-N-R4R5, -C(O)-R4, -C(O)-O-R4, -SR4, -SO2R4 та -NR4R5, де m, R4 та R5 визначені вище. Ці замісники можуть бути однаковими або різними та можуть розміщуватися у будь-якому хімічно прийнятному положенні кільця. Будь-яке посилання на “ вибірково заміщений 5-6- членний гетероарил ” стосується, як зазначено у визначенні, 5-6- членного гетероарильного кільця, що має заміщення, безпосередньо описані вище. l. “(C3-C10) циклоалкіл ” стосується насиченого або частково насиченого моноциклічного, біциклічного, біциклічного з містковими зв'язками або трициклічного алкільного радікалу, де кожна циклічна функціональна група має 3-10 атомів карбону. Приклади таких циклоалкільних радикалів охоплюють циклопропіл, циклобутил, циклопентил, циклогексил, циклооктил тощо. m. Вибірково заміщений “ (C3-C10) циклоалкіл ” стосується (C3-C10) циклоалкільної функціональної групи, як описано вище. Така циклоалкільна група може бути вибірково мати заміщення до 4 атомів водню на замісник, вибраний з групи, що охоплює галоген, ціано, нітро, гідрокси, вибірково заміщений (C1-C6) алкіл, вибірково заміщений (C1-C6) алкокси, трифторометил, трифторометокси, фосфат, оксо, -SO2NR4R5, -(CH2)m-NR5-C(O)-R4, -(CH2)mC(O)-N-R4R5, -C(O)-R4, -C(O)-O-R4, -SR4, -SO2R4 та -NR4R5, де m, R4 та R5 визначені вище… Ці замісники можуть бути однаковими або різними та можуть розміщуватися у будь-якому хімічно прийнятному положенні кільця. n. “(C3-C6) циклоалкіл ” стосується циклопропільної, циклобутильної, циклопентильної або циклогексильної функціональної групи, де будь-яка з них може бути вибірково заміщена, як описано вище, за умов хімічної прийнятності. o. “ Гетероцикл ” або “ гетероциклічне кільце ” має відношення до будь-якого 3- або 4членного кільця, що містить гетероатом, вибраний з кисню, азоту та сірки; або 5-, 6-, 7-, 8-, 9-, або 10- членне кільце, що містить 1, 2, або 3 атоми азоту; 1 атом кисню; 1 атом сірки; 1 атом азоту та 1 атом сірки; 1 атом азоту та 1 атом кисню; 2 атоми кисню у несуміжних положеннях; 1 кисень та 1 атом сірки у несуміжних положеннях; або 2 атоми сірки у несуміжних положеннях. 5членне кільце має 0-1 подвійних зв'язки, 6- та 7- членні кільця мають 0-2 подвійних зв'язки та 8-, 9- або 10 - членні кільця можуть мати 0, 1, 2, або 3 подвійних зв'язки. Термін “ гетероциклічний ” також охоплює біциклічні групи, у яких будь-які вищевказані гетероциклічні кільця злиті з бензеновим, циклогексановим або циклопентановим кільцем або з іншим гетероциклічним кільцем (наприклад, це може бути індоліл, хиноліл, ізохиноліл, тетрагідрохиноліл, бензофурил, дигідробензофурил або бензотіеніл тощо). Гетероциклічні сполуки охоплюють: піперидиніл, піперазиніл, азепан, азокан, морфолініл, ізохроміл, хінолін, тетрагідротріазин, тетрагідропіразол, дигідро-оксатіол-4-іл, дигідро-1H-ізоіндол, тетрагідро-оксазоліл, тетрагідрооксазиніл, тіоморфолініл, тетрагідропірімідініл, діоксолініл, октагідробензофураніл, октагідробензімідазоліл та октагідробензотіазоліл. p. “ Вибірково заміщена гетероциклічна сполука ” стосується гетероциклічної функціональної групи, як безпосередньо описано вище, де до 4 атомів карбону гетероциклічної функціональної групи може бути заміщено замісником, де кожен замісник є незалежно вибраним з групи, що охоплює галоген, ціано, нітро, гідрокси, вибірково заміщений (C1-C6) алкіл, вибірково заміщений (C1-C6) алкокси, трифторометил, трифторометокси, пентафторосульфоніл, фосфат, оксо, SO2NR4R5, -(CH2)m-N-C(O)-R4, -(CH2)m-C(O)-N-R4R5, -C(O)-R4, -C(O)-O-R4, -SR4, -SO2R4 та NR4R5, де m, R4 та R5 визначені вище. Ці замісники можуть бути однаковими або різними та можуть розміщуватися у будь-якому хімічно прийнятному положенні кільця. Будь-який атом азоту у такому гетероциклічному кільці вибірково може бути заміщений (C1-C6) алкілом або будь-яким іншим замісником з перелічених вище за умов хімічної прийнятності такого заміщення. Крім того, будь-який атом сірки у кільці може бути додатково заміщений одним або двома атомами кисню також за умов хімічної прийнятності такого заміщення. q. “ терапевтично ефективна кількість ” має відношення до кількості сполуки Формули I, яке при введенні пацієнту забезпечить отримання бажаного ефекту; тобто зменшення тяжкості 3 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 симптомів, пов'язаних з бактеріальною інфекцією, зниження кількості бактерій в уражених тканинах та/або запобігання зростання кількості бактерій в уражених тканинах (локалізованого або системного). r. “ пацієнт ” стосується теплокровних тварин, як-то наприклад, сільськогосподарських тварин, морських свинок, мишей, щурів, піщанок, кішок, кролів, собак, мавп,шимпанзе та людини. s. “ лікування ” стосується здатності сполуки усувати, полегшувати або сповільнювати прогресування бактеріальної інфекції пацієнта (або стану) або будь-якого пошкодження тканини, пов'язаного з хворобою. t. Вираз “ фармацевтично прийнятний ” вказує на те, що речовина або композиція повинна бути хімічно або токсикологічно сумісною з іншими інгредієнтами препарату та/або з твариною, що підлягає лікуванню. u. “ Ізомер ” має відношення до “ стереоізомеру ” та до "геометричного ізомеру ”, як визначено нижче. v. Під “ стереоізомером ” маються на увазі сполуки, що мають один або більше хіральних центрів та кожен з таких центрів може існувати у R або S конфігурації. Стереоїзомери охоплюють всі діастереомерні, енантіомерні та епімерні форми, а також рацемати та їх суміші. w. Під “ геометричним ізомером ” маються на увазі сполуки, що можуть існувати у цис-, транс-, анти-, проти-, E- (entgegen – навпроти), Z –(zusammen – поруч) формах, а також їх суміші. x. Сполуки за “ Формулою I ”, “ формулою I ”, “ формулою (I) ” та “ сполуки винаходу ” у цій заявці застосовують взаємозамінне та їх слід розглядати у якості синонімів. y. Терміни “ піридин ” та “ піридинон ” також мають бути застосовані у цій заявці взаємозамінним чином без жодних відмінностей або вийнятковостей, якщо не вказано іншого. Будь-який пересічний фахівець у цій галузі зможе легко це зрозуміти. Застосована тут фраза “ фармацевтично прийнятна сіль (солі) ”, якщо тільки в тексті не зазначено іншого, охоплює солі кислих або основних груп, що можуть бути присутніми у сполуках заявленого винаходу. Сполуки заявленого винаходу, які є основними за природою здатні утворювати велику різноманітність солей з різними неорганічними та органічними кислотами. Кислоти, що можуть бути застосовані для отримання фармацевтично прийнятних солей приєднання кислоти таких основних сполук здатні утворювати нетоксичні солі приєднання кислоти, тобто солі, що містять фармакологічно прийнятні аніони, як-то солі гідрохлориду, гідроброміду, гідроіодіду, нітрату, сульфату, бісульфату, фосфату, кислого фосфату, ізонікотинату, ацетату, лактату, саліцилату, цитрату, кислого цитрату, тартрату, пантотенату, бітартрату, аскорбату, сукцинату, малеату, гентізінату, фумарату, глюконату, глюкуронату, сахарату, форміату, бензоату, глутамату, метансульфонату, етансульфонату, бензолсульфонату, р-толуолсульфонату та памоату [тобто 1,1’-метилен-біс-(2- гідрокси3нафтоату)]. Сполуки заявленого винаходу, що містять основну функціональну групу, як-то аміногрупу, на додачу до зазначених вище кислот також можуть утворювати фармацевтично прийнятні солі з різними амінокислотами. Винахід також стосується солей приєднання основи сполук винаходу. Хімічні основи, що можуть бути застосовані у якості реагентів для отримання цих фармацевтично прийнятних основних солей утворюють з такими сполуками нетоксичні основні солі. Такі нетоксичні основні солі охоплюють, але без обмеження, солі, отримані з таких фармакологічно прийнятних катіонів, як-то катіонів лужних металів (наприклад, калію та натрію) та катіонів лужноземельних металів (наприклад, кальцію та магнію), солі приєднання амонію або водорозчинного аміну, як-то Nметилглюкамін (меглумін), нижчий алканол - амоній та інші основні солі фармацевтично прийнятних органічних амінів. Прийнятні основні солі отримують з основ, що утворюють нетоксичні солі. Необмежені приклади таких солей охоплюють солі алюмінію, аргініну, бензатину, кальцію, холіну, діетиламіну, діоламіну, гліцину, лізину, магнію, меглуміну, оламіну, калію, натрію, трометаміну та цинку. Також можуть утворюватися напівсолі кислот та основ, наприклад, гемісульфатних та гемікальцієвих солей. Огляд прийнятних солей див. у книзі Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (WileyVCH, 2002). Способи отримання фармацевтично прийнятних солей сполук винаходу відомі фахівцям у цей галузі. Деякі сполуки за Формулою (I) можуть існувати у вигляді геометричних ізомерів. Сполуки Формули (I) можуть мати один або декілька асиметричних центрів, таким чином існуючі у вигляді двох або більше стереоізомерних форм. Заявлений винахід стосується всіх окремих стереоізомерів та геометричних ізомерів сполук за Формулою (I) та їх сумішей. Окремі 4 UA 107423 C2 5 10 15 20 25 30 35 40 енантіомери можна отримати шляхом хірального розділення або за допомогою застосування відповідного енантіомеру у синтезі. На додачу, сполуки заявленого винаходу можуть існувати у несольватованій та у сольватованій формах з фармацевтично прийнятнимирозчинниками, як-то водою та етанолом тощо. Для цілей заявленого винаходу, сольватовані форми головним чином вважаються еквівалентними несольватованим. Також сполуки можуть існувати у одному або у декількох кристалічних станах, тобто бути поліморфічними або вони також можуть існувати у вигляді аморфної твердої речовини. Слід зазначити, що всі такі форми охоплюються Формулою Винаходу. Винахід також стосується проліків сполуки винаходу. Отже, певні похідні сполуки винаходу, що можуть самі по собі мати невелику фармакологічну активність або зовсім її не мати, можуть при введенні у (на) тіло пацієнта бути перетворені, наприклад, шляхом гідролітичного розщеплення на сполуки винаходу, що мають бажану активність. Такі похідні позначають, як “ проліки ”. Додаткову інформацію про отримання та застосування проліків можна знайти у Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association). Цей винахід також охоплює сполуки, що містять захисні групи. Будь-якому досвідченому фахівцю у цей галузі буде зрозуміло, що сполуки винаходу також можуть бути отримані разом з певними захисними групами, корисними для очищення або зберігання, які можуть бути вилучені перед введенням пацієнту. Про захист та зняття захисту функціональних груп описано у книгах "Protective Groups in Organic Chemistry", edited by J.W.F. McOmie, Plenum Press (1973) and "Protective Groups in Organic Synthesis", 3rd edition, T.W. Greene and P.G.M. Wuts, WileyInterscience (1999). Заявлений винахід також охоплює мічені ізотопами сполуки, ідентичні наведеним у Формулі I, але що фактично мають один або більше атомів, замінених атомом, що має атомну масу або масове число, відмінні від атомної маси або масового числа, зазвичай існуючого у природі. Приклади ізотопів, що можуть бути включені у сполуки винаходу охоплюють ізотопи водню, 2 3 13 14 15 вуглецю, азоту, кисню, фосфору, фтору та хлору, як-то, але без обмеження, H, H, C, C, N, 17 18 31 32 35 18 36 O, O, P, P, S, F та Cl, відповідно. Сполуки заявленого винаходу, їх проліки та фармацевтично прийнятні солі вказаних сполук або вказаних проліків, що містять вищезазначені ізотопи та/або інші ізотопи інших атомів також охоплюються обсягом заявленого винаходу. Деякі мічені ізотопом сполуки заявленого винаходу, наприклад, мічені 3 14 радіоактивними ізотопами, як-то H та C, є корисними для застосування у аналізі розподілення ліків та/або субстратної тканини. Особливо бажаними через легкість їх отримання та виявлення 3 14 є ізотопи, що містять тритій, тобто H; та карбон-14, тобто C. Додатковим чином, заміщення 2 більш важкими ізотопами, як-то дейтерієм, тобто H, може надати певні терапевтичні переваги, обумовлені більшою метаболічною стабільністю, наприклад підвищеним періодом напіврозпаду in vivo або зменшеним дозуванням та, отже, за деяких обставин воно може бути бажаним. Мічені ізотопами сполуки цього винаходу та їх проліки головним чином можуть бути отримані шляхом проведення процедур, наведених у Схемах або прикладах нижче за допомогою заміщення неміченого ізотопом реагенту на мічений ізотопом реагент, який можна легко отримати. Всі сполуки Формули I містять сульфонільну функціональну групу, як показано нижче: 45 50 Ця сульфонільна функціональна група буде завжди заміщеною на функціональну групу нижчого алкілу, звичайно метилу. Суміжний з сульфонілом атом карбону теж вибірково може 2 1 та 2 бути заміщено, як наведено у вигляді R . Звичайно, обидва раді кали R R будуть метилом. Як буде зрозуміло фахівцям, карбон, суміжний з сульфонільною функціональною групою являє собою хіральний центр. Отже, сполуки можуть існувати у вигляді рацемату, як S 5 UA 107423 C2 енантіомер або як R- енантіомер. У додатковому втіленні, сполуки можуть бути отримані та застосовані у вигляді R- енантіомеру, як показано нижче: 5 10 Як буде зрозуміло фахівцям, сполуки, подібні до синтезованої рідко будуть присутніми тільки у вигляді єдиного енантіомеру. Протилежно орієнтований енантіомер (тобто Sенантіомер) також може бути присутнім у малих кількостях (тобто продукт є “ істотно чистим ”). Ця мала кількість може сягати до 10 % у ваговому співвідношенні, більш звичайно не більш, ніж 5 % у ваговому співвідношенні та у додатковому втіленні не більш, ніж 1 % у ваговому співвідношенні або, більш докладно, не більш, ніж 0.5 % у ваговому співвідношенні. Всі сполуки за Формулою I містять функціональну групу піридинону, як показано нижче: 15 20 25 30 35 40 Це піридинонове кільце буде приєднано до залишку молекули через 1- та 4- положення, як зображено вище. Положення 3 завжди буде заміщено функціональною групою фтору, як зображено вище. Функціональна група піридинону може бути вибірково заміщена, як зображено 3 3 у вигляді функціональної групи R .Радикал R може являти собою один замісник, що не містить водню, як визначено вище. Цей замісник, що не містить водню може бути розміщений або у 3 положенні 2 або у положенні 5 піридинонового кільця. Звичайно радикалом R є водень. Функціональна група, позначена, як T буде завжди присутньою у молекулі. Вона буде присутньою у вигляді етинілу, арилу або гетероарилу (або кільцева система може бути заміщена, як визначено вище) Звичайно, вона буде присутньою у вигляді фенілу, який може бути вибірково заміщеним. Якщо T є гетероарилом, то він буде пов'язаний з піридиноном через карбон - карбоновий зв'язок (тобто гетероатом(атоми) не буде зв'язаним з піридиноном). Якщо E є присутнім та D являє собою зв'язок, то він може являти собою будь-який хімічно прийнятний зв'язок, тобто карбон-карбон, карбон-азот тощо. Присутність D та E є вибірковою. При наявності, D звичайно буде являти собою зв'язок та E буде присутнім у вигляді або 5-6- членного гетероарилу або (C3-C6) циклоалкілу, кожен з яких може бути вибірково заміщеним, як визначено вище. Більш специфічні втілення винаходу охоплюють сполуки Формули I, у яких: a) R1 є метилом; b) R2 є метилом; c) R3 є воднем; d) сполука присутня у вигляді R- енантіомеру (тобто є істотно чистою); e) T є фенілом, що може бути вибірково заміщеним та D та E разом відсутні; та f) T є фенілом, D є зв'язком та E є або C3-C6 циклоалкілом або 5-6- членним гетероарилом та кожен з них може бути вибірково заміщеним. Додаткове втілення винаходу стосується сполуки Формули I, яка є істотно чистою та у якої: a) R1 та R2 є метилом, R3 є воднем та T, D та E визначені вище; b) R1 та R2 є метилом, R3 є воднем, T є вибірково заміщеним фенілом та E та D разом відсутні; 6 UA 107423 C2 5 10 15 c) R1 та R2 є метилом, R3 є воднем, T є вибірково заміщеним фенілом, D є зв'язком та E є 5-6- членним гетероарилом, що може бути вибірково заміщеним; та d) R1 та R2 є метилом, R3 є воднем, T є вибірково заміщеним фенілом, D є зв'язком та E є C3-C6 циклоалкілом, що може бути вибірково заміщеним. У додатковому втіленні, винахід стосується підвиду, наведеного у вигляді Формули la нижче, де молекула являє собою R- енантіомер (тобто S- енантіомер також вибірково може бути 1 2 3 присутнім у малих кількостях). Як показано нижче, R та R є метилом, R є воднем, E та D a разом відсутні та T є заміщеним фенілом. Більш докладно, R є одним або декількома замісниками, вибраними з групи, що охоплює C1-C3 алкіл, C1-C3 алкокси, фтор, хлор, гідрокси, трифторометил та трифторометилокси. У додатковому втіленні, винахід стосується підвиду, наведеного у вигляді Формули Ib нижче, де молекула зображена у вигляді R- енантіомеру (тобто S- енантіомер може вибірково бути 1 2 3 присутнім у мінімальній кількості). Як показано нижче, R та R є метилом, R є воднем, T є фенілом, D є зв'язком та E є 5-6- членним гетероарилом, що може бути вибірково заміщеним. 20 У більш специфічному втіленні винаходу, інгібітор LpxC є наступною сполукою або її фармацевтично прийнятною сіллю: 25 У більш специфічному втіленні винаходу, інгібітор LpxC є наступною сполукою або її фармацевтично прийнятною сіллю: 7 UA 107423 C2 O O S O H N N F OH O F O 5 10 15 20 25 Сполуки Формули I можуть бути отримані за допомогою ряду способів, аналогічних відомим у цій галузі. Наведені нижче схеми реакції ілюструють два альтернативних способи отримання цих сполук. Інші способи, в тому числі їх модифікації, також відомі фахівцям у цей галузі. Синтез сполук Формули I зображено нижче у Схемі A. Першим його етапом є проведення Nалкілування, зображеного на етапі A (Step A). Піридинон структури 1 вступає в реакцію з сульфонільною похідною структури 2, створеної проміжним продуктом структури 3. Структура 3 може бути додатково дериватизована для отримання сполуки Формули I. На схемі наведені два альтернативних шляхи синтезу (Option А або Option B), але читач легко зауважить, що вони є варіаціями одного й того ж синтезу, відрізняючись лише порядком проведення його етапів. На початку синтезу, позначеного, як Option A, галоїд, позначений, як X, у 4- му положенні 1 1 піридинонової структури 3 заміщують на бажану кінцеву функціональну групу E-D-T-M , де M є металом, як-то похідною бору, прийнятним для проведення типової реакції крос-сполучення, якто реакції Судзукі-Міяура. Гідроліз або усунення етилової захисної групи (або інших прийнятних захисних груп) на етапі C веде до утворення сполуки структури 5. Тоді кінцева карбонова кислота структури 5 перетворюється на захищену похідну гідроксамової кислоти, позначену, як структура 8. Зняття захисту захищеної похідної гідроксамової кислоти (структури 8), як зображено на етапі H веде до утворення кінцевого продукту Формули I. Хоча ці реакції є добре відомі фахівцям, вони більш детально розглянуті нижче. На початку синтезу, позначеного, як Option B Схеми A, етилову захисну групу (або інші звичайні захисні групи) відокремлюють від піридинонової структури 3 з отриманням сполуки структури 6, як зображено у етапі E. На етапі F, кінцева карбонова кислота структури 6 перетворюється на захищену похідну гідроксамової кислоти структури 7 через умови амідування. На етапі G, функцію галоїду у 4 положенні піридинонової функціональної групи 1 потім безпосередньою заміщують бажаною кінцевою функціональною групою E-D-T-M за допомогою реакції сполучення з отриманням похідних захищеної гідроксамової кислоти (структури 8). Як і раніше, зняття захисту захищеної похідної гідроксамової кислоти, як зображено на етапі H, веде до утворення сполуки Формули I. 30 8 UA 107423 C2 5 10 15 N- алкілування, що зображене вище на етапі А може бути проведено за допомогою добре відомих фахівцям способів. Одним з початкових матеріалів є похідна 2- піридинону структури 1. 3 У цьому піридиноні, X являє собою галоїд та R являє собою таку ж функціональну групу, яка є бажаною у кінцевому продукті. Багато цих піридинонових похідних відомі у цій галузі та залишок можна отримати за допомогою також відомих у цій галузі способів синтезу, опис яких читач зможе знайти у Tet. Lett. (2005) Vol 46, 7917. Отримання подібних речовин також зображено у розділі Отримання 2 нижче. Інший реактант N - алкілування, зображений на Етапі А є захищеним алкілсульфонатом 1 2 структури 2, де раді кали R та R існують у вигляді тієї ж самої функціональної групи, яка є бажаною у кінцевому продукті. Також там зображена етильна захисна група, але будь-яка стандартна захисна група може бути заміщеною. Ці алкільні сульфонати також відомі у цей галузі та опис їх отримання можна знайти у Journal of Organic Chemistry, (1980) Vol 45, 8, 14861489. Отримання подібних речовин також зображено у розділі Отримання 1 нижче. N - алкілування може бути проведено таким саме чином, як відомо у цей галузі. Звичайно еквівалентні кількості сполук структури 1 та 2 взаємодіють у суміші апротонних та протонних 9 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 розчинників, як-то тетрагідрофуран та t- бутанол у присутності основи, як-то карбонату калію, карбонату цезію, карбонату натрію, гідриду натрію тощо. Також при бажанні можна застосувати агенти перенесення, як-то тетрабутил амонію бромід. Звичайно реактанти нагрівають та дають можливість реакції відбуватися до її завершення. Бажаний продукт структури 3 можна отримати також за допомогою відомих у цій галузі способів. При бажанні продукт структури 3 може бути очищено або альтернативним чином у наступному етапі реакції може бути застосовано неочищений продукт. Таке N- алкілування зображено у розділі Отримання 2 нижче. На схемі показано шлях включення до молекул функціональної групи гідроксамової кислоти. Спочатку від карбонової кислоти відокремлюють захисну групу з отриманням проміжної сполуки зі структурою 5 та 6, як зображено на етапі C (Option A) та етапі E (Option B) відповідно. Спосіб, яким чином це досягається буде змінюватися в залежності від фактичної захисної групи та добре відомий фахівцям у цій галузі. Обговорення потенційних захисних груп та способів їх усунення можна знайти у McOmie або Greene supra. Спосіб відокремлення етильної функціональної групи, зображений на Схемі A наведено у розділі Отримання 2 нижче… На етапах F та D, функціональна група гідроксамової кислоти, як зображено, є включеною до складу молекули. Можна застосувати захищене джерело гідроксаміну з наступною реакцією зняття захисту або, альтернативним чином, гідроксамін можна включити безпосереднім чином для уникнення етапів зняття захисту. У будь-якому випадку, гідроксамову кислоту включають до складу молекули за допомогою стандартних реакцій амідування. Наприклад, сполука структури 5 (Option A) або 6 (Option B) може контактувати з надлишком оксалілхлориду у апротонному розчиннику, як-то у дихлорметані протягом періоду часу, достатнього для утворення хлориду відповідної кислоти з наступним додаванням або надлишку гідроксиламіну або захищеного гідроксиламіну. Далі реакції дозволяють проходити до її завершення та з реакційного середовища виділяють захищені проміжні сполуки структури 7 (Option B) або 8 (Option A) та проводять їх очищення за допомогою способів, відомих у цій галузі. Як зазначено вище, також за допомогою способів, відомих у цій галузі може бути проведено будь-яке зняття захисту (див.Greene або McOmie supra). Альтернативним чином, як відомо у цей галузі, можна отримати амід за допомогою реагенту зв'язування аміду, 1,1'-карбонілдіімідазолу (CDI), 2-хлоро-4,6диметокси-1,3,5-тріазину (CDMT) або 1-етил-3-(3-диметиламінопропіл) карбодііміду (EDCI). У схемі також зображений спосіб введення до складу молекули кінцевої функціональної групи, E-D-T. Незважаючи на вибраний шлях (Option або Option B), в кінці процесу проводять реакцію зв'язування для прикріплення кінцевої функціональної групи, E-D-T до 4- положення 1 піридинонової проміжної сполуки. На Схемі A спів- реактант зображено, як E-D-T-M , де E-D-T1 M є такою ж самою функціональною групою, яка є бажаною у кінцевому продукті за виключенням того, що вона буде заміщена на метал (або металоїд), як-то магній, мідь, борний ефір / кислота тощо в потрібній точці приєднання до піридинонової проміжної сполуки структури 3 або 7 (тобто до іншого реактанту). Кінцеві групи, що охоплюються Формулою I, тобто E-D-T або вже є відомими у цій галузі або їх можна отримати за допомогою способів, аналогічних відомим у цій галузі. Реакція зв'язування може бути проведена за допомогою численних технічних прийомів. Наприклад, для отримання карбон-карбонового зв'язку можна застосувати реакцію Судзукі1 Міяура. У такій реакції, M буде існувати у вигляді борної кислоти / ефіру. Еквівалентні молярні кількості реактантів будуть взаємодіяти у розчиннику, як-то тетрагідрофуран, 2метилтетрагідрофуран, 1,4-діоксан, вода, толуол або у їх суміші у присутності переходного металу - каталізатору, як-то вільного або зв'язаного зі смолою паладію або видів нікелю, разом з основою, як-то карбонат натрію, карбонат калію, фторид цезію, карбонат цезію тощо. Реакційна суміш може бути нагріта до досягнення адекватного перетворення або мікрохвильовим шляхом або за допомогою іншого прийнятного способу. По завершенні бажаний продукт може бути виділено, відновлено з реакції та додатково очищено за допомогою відомих у цей галузі способів. Аналогічним чином для досягнення цієї мети можна застосувати методологію Кастро-Стевенса чи Соногашіра-Хагінара. Функціональна група Т буде належати до прийнятних видів кінцевого ацетилену, що вступає в реакцію у присутності солей міді, як-то 1 йодиду міді. У такій реакції M може бути присутнім у вигляді купратних видів, отриманих in situ. Еквівалентні молярні кількості реактантів будуть взаємодіяти у розчиннику, як-то тетрагідрофуран, 2- метилтетрагідрофуран, диметилформамід або у їх суміші у присутності переходного металу - каталізатору, як-то вільного або зв'язаного зі смолою паладію або видів нікелю, разом з прийнятною основою, як-то з прийнятною органічною основою, наприклад з N, N-діізопропілетиламіном. Реакційна суміш може бути нагріта до досягнення адекватного перетворення або мікрохвильовим шляхом або за допомогою іншого прийнятного способу. По 10 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 завершенні бажаний продукт може бути виділено, відновлено з реакції та додатково очищено за допомогою відомих у цей галузі способів. Зображені вище схеми реакції отримання сполук Формули I мають виключно ілюстративний характер та, як буде зрозуміло фахівцям, їх можна модифікувати у залежності від конкретної сполуки, наявності реагентів тощо. Сполуки винаходу можна застосувати у лікуванні або у запобіганні інфекційних розладів, особливо розладів, спричинених сприйнятливими та стійкими до багатьох ліків (MDR) грамнегативними бактеріями. Приклади таких грамнегативних бактерій охоплюють Acinetobacter baumannii, Acinetobacter spp., Achromobacter spp., Aeromonas spp., Bacteroides fragilis, Bordetella spp., Borrelia spp., Brucella spp., Campylobacter spp., Citrobacter diversus (koseri), Citrobacter freundii, Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Francisella tularensis, Fusobacterium spp., Haemophilus influenzae (які виділяють та не виділяють β-лактамазу), Helicobacter pylori, Klebsiella оксиtoca, Klebsiella pneumoniae (в тому числі ті, що кодують βлактамази розширеного спектру дії (далі у тексті позначені, як " ESBLs "), Legionella pneumophila, Moraxella catarrhalis (які виділяють та не виділяють β-лактамазу), Morganella morganii, Neisseria gonorrhoeae, Neisseria meningitidis, Proteus vulgaris, Porphyromonas spp., Prevotella spp., Mannheimia haemolyticus, Pasteurella spp., Proteus mirabilis, Providencia spp., Pseudomonas aeruginosa, Pseudomonas spp., Salmonella spp., Shigella spp., Serratia marcescens, Treponema spp., Burkholderia cepacia, Vibrio spp., Yersinia spp. та Stenotrophomonas mulophilia. Приклади інших грамнегативних організмів охоплюють членів родини Enterobacteriaceae, здатних до експресії ESBLs; KPCs (карбапенемаз бактерії Klebsiella pneumoniae), CTX-M (βлактамаз типу CTX-M), всіх o-β-лактамаз (як-то, наприклад, NDM-1) та β-лактамаз типу AmpC, наприклад, тих, що наділяють стійкістю до наявних в даний час цефалоспоринів, цефаміцинів, карбапенемів та комбінацій β-лактам / β-лактамазних інгібіторів. У більш специфічному втіленні, грамнегативні бактерії є вибраними з групи, що охоплює Acinetobacter baumannii, Acinetobacter spp., Citrobacter spp., Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Klebsiella оксиtoca, Klebsiella pneumoniae, Serratia marcescens, Stenotrophomonas maltophilia, Pseudomonas aeruginosa та членів родин Enterobacteriaceae та Pseudomonas, здатних до експресії ESBLs, KPCs, CTX-M, всіх o-β-лактамаз (як-то, наприклад, NDM-1) та β-лактамаз типу AmpC, наприклад, тих, що наділяють стійкістю до наявних в даний час цефалоспоринів, цефаміцинів, карбапенемів та комбінацій β-лактам / β-лактамазних інгібіторів. Приклади інфекцій, прийнятних для лікування сполуками Формули I охоплюють нозокоміальну пневмонію, інфекції сечовивідних шляхів, системні інфекції (бактеріємію та сепсис), інфекції шкіри та м'яких тканин, хірургічні інфекції, внутрішньочеревні інфекції, інфекції легенів у пацієнтів з кістозним фіброзом, лікування пацієнтів, що страждають від інфекцій легенів, ендокардиту, синдрому діабетичної стопи, остеомієліту та інфекцій центральної нервової системи. На додачу, сполуки винаходу можна застосувати у лікуванні інфекцій шлунково-кишкового тракту людини (та інших ссавців), спричинених Helicobacter pylori. Знищення цих бактерій веде за собою поліпшення стану здоров'я, в тому числі зменшення диспепсичних явищ, зниження появи рецидивів виразкової хвороби та рецидивів кровотечі та знижує ризик виникнення раку шлунка тощо. Більш детальне обговорення щодо знищення бактерій H. pylori та його впливу на шлунково - кишкові захворювання можна знайти тут: www.informahealthcare.com, Expert Opin. Drug Saf. (2008) 7(3). Для проявлення такої антибактеріальної активності, сполуки повинні бути застосовані у терапевтично ефективній кількості. Вираз " терапевтично ефективна кількість " застосовують для опису кількості сполуки, достатньої для лікування інфекції з розумним співвідношенням користі до ризику та прийнятної для будь-якого медичного лікування. Проте зрозуміло, що загальну добову дозу сполуки буде визначати практикуючий лікар в рамках здорового медичного судження. Певний терапевтично ефективний рівень дози для будь-якого окремого пацієнта буде залежати від численних та добре відомих у медицині факторів, що охоплюють розлади, що підлягають лікуванню, тяжкість захворювання, активність та властивості певної застосованої сполуки; вік, вагу тіла, загальний стан здоров'я, стать та дієту пацієнта; час та шлях введення, швидкість виведення певних застосованих сполук; тривалість лікування; а також від інших ліків, застосованих у комбінації або одночасно з застосуванням певної сполуки тощо. Проте, в якості загальної рекомендації, загальна добова доза буде звичайно знаходитися у діапазоні від близько 0.1 мг/кг/добу до 5000 мг/кг/добу у вигляді одиничних або розділених форм 11 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 60 дозування. Звичайно дозування для людини знаходиться у діапазоні біля 10-3000 мг на добу у вигляді одиничних або багаторазових доз. Для лікування інфекційного захворювання звичайно застосовують будь-який шлях введення сполуки винаходу, в тому числі пероральне, парентеральне, місцеве, ректальне, трансмукозальне та інтестинальне. Парентеральні введення охоплюють ін'єкції для отримання системного ефекту або ін'єкції безпосередньо у постраждалу ділянку. Прикладами видів парентерального введення є підшкірні, внутрішньовенні, внутрішньом'язові, внутрішньошкірні, інтратекальні, внутрішньоочні, інтраназальні, інтравентрикулярні ін'єкції або способи вливань (інфузії). Види місцевого введення охоплюють лікування легко досяжних для місцевого застосування ділянок, як-то, наприклад, очей, вух, в тому числі лікування інфекцій зовнішнього та середнього вуха, піхви, відкритих ран шкіри, включаючи поверхню шкіри та підшкірні структури або нижній частини кишечника. Трансмукозальне введення охоплює введення за допомогою інгаляцій або назальних аерозолів. Сполуки за даним винаходом можуть бути отримані для введення будь-яким способом для застосування в медицині людини або ветеринарії за аналогією з іншими біологічно активними агентами, як-то антибіотиками. Такі способи відомі в даній галузі та підсумовані нижче. Композиція може бути отримана для введення будь-яким відомим у цей галузі шляхом, як-то підшкірним, пероральним, місцевим, парентеральним або шляхом інгаляції. Композиції можуть існувати у будь-якому відомому у цій галузі вигляді, в тому числі, але без обмеження, у вигляді таблеток, капсул, порошків, гранул, пастилок, кремів або рідких препаратів, як-то пероральні або стерильні парентеральні розчини або суспензії. Препарати для місцевого застосування за даним винаходом можуть бути представлені у вигляді, наприклад, мазей, кремів чи лосьйонів, очних мазей / крапель та вушних крапель, просочених пов'язок та аерозолів та можуть містити відповідні традиційні добавки, як-то консерванти, розчинники, що сприяють проникненню лікарського засобу, пом'якшувальні речовини тощо. Такі композиції для місцевого застосування також можуть містити звичайні носії, як-то основи кремів та мазей, а також етанол або олеіловий спирт для примочок. Такі носії можуть складати, наприклад, від біля 1 % до 98 % вмісту препарату. Таблетки та капсули для перорального введення можуть бути надані у формі одиничної дози та можуть містити звичайні наповнювачі, як-то зв'язувальні агенти, наприклад камедь, желатин, сорбіт, трагакант, полівінілпіролідон; наповнювачі, наприклад лактозу, цукор, кукурудзяний крохмаль, фосфат кальцію, сорбіт або гліцин; змащувальні речовини для таблетування, наприклад стеарат магнію, тальк, поліетиленгліколь або діоксид кремнію; дезінтегруючі агенти, наприклад картопляний крохмаль або прийнятні агенти змочування, як-то лаурилсульфат натрію. Таблетки можуть бути вкриті покриттям у відповідності зі способами, добре відомими у звичайній фармацевтичній практиці. Рідкі препарати для перорального застосування можуть існувати у вигляді, наприклад, водних або масляних суспензій, розчинів, емульсій, сиропів або еліксирів або можуть бути представлені у вигляді сухого продукту для відновлення водою або іншим відповідним носієм перед застосуванням. Такі рідкі препарати можуть містити загальноприйнятні домішки, як-то агенти суспендування, наприклад сорбіт, метилцелюлозу, сироп глюкози, желатин, гідроксиетилцелюлозу, карбоксиметилцелюлозу, гель стеарату алюмінію або гідровані їстівні жири, емульгатори, наприклад лецитин, моноолеат сорбіту або гуміарабік, неводні носії (що можуть включати харчові масла), наприклад мигдальне масло, жирні складні ефіри, як-то гліцерин, пропіленгліколь або етиловий спирт; консерванти, наприклад метил- або пропіл- ргідроксибензоат або сорбінову кислоту та, при бажанні, звичайні смакові агенти або барвники. Для парентерального введення, рідкі стандартні лікарські форми отримують шляхом застосування сполуки та стерильного транспортного засобу, типовим з яких є вода. Сполука, в залежності від такого транспортного засобу та застосованої концентрації, може бути або суспендованою або розчиненою у ньому або у іншому прийнятному розчиннику. Для отримання розчинів, сполука може бути розчинена у воді для ін'єкцій та стерилізована шляхом фільтрації перед розподіленням у відповідні флакони або ампули та запечатуванням. Переважним чином, у прийнятному транспортному засобі можуть бути розчинені різні відповідні агенти, як-то місцеві анестетики, консерванти та буферизуючі агенти. Для посилення стійкості, після розподілення по флаконам та вилучення води у вакуумі, композицію можна заморозити. Порошок у сухому ліофілізованому стані зберігають у запечатаних флаконах в комплекті з флаконами з водою для ін'єкцій, яку можна потім додати для відновлення рідини перед застосуванням. Суспензії для парентерального застосування отримують істотно однаковим чином за виключенням випадку, коли замість розчинення та стерилізації, що не може бути здійснена шляхом фільтрації, сполуку суспендують у транспортному засобі. Сполука може бути стерилізована шляхом дії оксирану 12 UA 107423 C2 5 10 15 20 25 30 35 40 45 (етиленоксиду) перед суспендуванням у стерильному носії. Переважним чином, для забезпечення рівномірного розподілу сполуки, до складу композиції включають поверхневоактивну речовину або агент змочування. В залежності від способу введення, вміст активного матеріалу у композиції може дорівнювати, наприклад, біля 0.1 % - 100 % за вагою. Якщо композиції містять дозовані одиниці, то кожна така одиниця буде містити, наприклад, біля 0.51000 мг активного інгредієнту. Дозування, що застосовують для лікування дорослої людини знаходиться у діапазоні, наприклад, біля 10-3000 мг на добу в залежності від шляху та частоти введення. При бажанні, сполуки винаходу можуть бути застосовані у комбінації з одним або декількома додатковими антибактеріальними агентами ("додаткові активні агенти"). Таке застосування сполуки винаходу у комбінації з додатковим активним агентом може бути прийнятним для одночасного, роздільного або послідовного застосування. Наведені нижче приклади та процедури додатково ілюструють та наводять зразки сполук заявленого винаходу та способів їх отримання. Треба розуміти,що вказані приклади та процедури жодним чином не обмежують обсяг заявленого винаходу. Молекули з одиничним хіральним центром, якщо не вказано іншого, існують в наступних прикладах у вигляді рацемічної суміші. Молекули з двома або більше хіральними центрами, якщо не вказано іншого, існують у вигляді рацемічною суміші діастереомерів. Окремі енантіомери / діастереомери можуть бути отримані способами, відомими фахівцям у даній галузі. Приклади. Головним чином, експерименти проводили у інертній атмосфері (азот або аргон), зокрема у випадках застосування реагентів або проміжних сполук, чутливих до кисню або вологості. Наявні у продажу розчинники та реагенти головним чином застосовували без додаткового очищення, в том числі, у разі необхідності, безводні розчинники (головним чином, продукти TM Sure-Seal від Aldrich Chemical Company, Milwaukee, Wisconsin). Дані мас-спектрометрії отримували або за допомогою рідкісної хромато – мас спектрометрії (РХ-МС; LСМS) або хімічної іонізації під атмосферним тиском (APCI). Дані хімічних зсувів для ядерного хімічного резонансу (ЯМР; NMR) наведені у вигляді кількості частин на мільйон (ppm, δ) з посиланням на залишкові піки застосованих мічених дейтерієм розчинників. Температури плавлення наведені без урахування поправок. Мас-спектри низької роздільної здатності (LRMS)були отримані або шляхом застосування хімічної іонізації (aмоній) за допомогою приладу Hewlett Packard 5989® або за допомогою платформи для дослідження хімічної іонізації під атмосферним тиском (APCI) Fisons (або Micro Mass) з застосуванням 50/50 суміші ацетонітрилу/води з 0.1 % мурашиною кислотою у якості іонізуючого агента. Температура навколишнього середовища дорівнювала 2025 °C. Умови реакцій процесів синтезу (тривалість реакції та температура), на які зроблені посилання, у інших прикладах можуть відрізнятися. Головним чином, реакції супроводжувалися тонкошаровою хроматографією або мас-спектрометрією та отримані результати при необхідності піддавали додатковій обробці. Результати очищення можуть варіювати між експериментами та головним чином, розчинники та співвідношення розчинників, застосовані для елюентів / градієнтів обирали для забезпечення прийнятних значень R fs або часу утримування. Абревіатури, застосовані у обговоренні вище та у наведених нижче Прикладах мають наступні значення. Якщо скорочення не визначено, то воно має своє загальноприйнятне розуміння. Ac ACN AC2O APCI Aq. 9-BBN bd bm bs; br. S. BOC o C CBZ CDI CDMT = = = = = = = = = = = = = = ацетат ацетонітрил оцтовий ангідрид хімічна іонізація під атмосферним тиском водний 9-борабіцикло [3.3.1]нонан широкий дублет широкий мультиплет широкий синглет терт- бутоксикарбоніл градусів Цельсію бензилоксикарбоніл 1,1’-карбонілдіімідазол 2-хлоро-4,6-диметокси -1,3,5-тріазин 13 UA 107423 C2 см d DCC D СМ dd ddd DIAD DME DMF DMA DMAP DMSO dq dt EDCI Ее eq. EtO Et2O EtOAc g G СМ S h 1 H HATU = = = = = = = = = = = = = = = = = = = = = = = = = HCl H2N-OTHP HOBT HPLC Гц IPA J KOAc K3PO4 L LСМS LDA LG LiHMDS m M M% max mCPBA MeOH meq MeTHF mg MgSO4 мГц хв. mL мМ мМol MS MTBE m/z N NaHCO3 = = = = = = = = = = = = = = = = = = = = = = = = = = = = = = = = = = сантиметр дублет 1,3-дициклогексилкарбодіімід дихлорметан дублет дублетів дублет дублетів дублетів діізопропілазодикарбоксилат диметиловий ефір диметилформамід диметилацетамід 4- диметиламінопіридин диметилсульфоксид дублет квартетів дублет триплетів 1- етил -3-(3-диметиламінопропіл)карбодіімід енантіомерний надлишок еквівалент етокси діетиловий ефір етилацетат грам газова хромато – мас спектрометрія годин протон (2-(7-аза-1H-бензотріазол-1-іл)-1,1,3,3-тетраметилуронію гексафторофосфат) соляна кислота O-тетрагідро-2H-піран-2-іл-гідроксиламін гідроксибензотріазол високоефективна рідинна хроматографія герц ізопропанол константа взаємодії ацетат калію фосфат калію літр рідкісна хромато – мас спектрометрія іізопропіламід літію група, що заміщується гексаметилдисилазид літію / біс(триметилсиліл)амід літію мультиплет молярна концентрація мольний відсоток максимум мета-хлоропербензойна кислота метанол міліеквівалент 2- метилтетрагідрофуран міліграм сульфат магнію мегагерц хвилин мілілітр міліметр мілімоль мас-спектрометрія метил- трет- бутиловий ефір співвідношення маси \ заряду нормальність бікарбонат натрію 14 UA 107423 C2 Na2SO4 NH4Cl NММ NMP NMR Pd TM Pd EnCat Pd(dppf)Cl2 10 15 20 25 = Pd(PPh3)4 ppt p-TLC PyBop q Rf rt s sat. t або tr TBAB TBS TFA THF THP TLC TMS TPP TPPO µL 5 = = = = = = = = = = = = = = = = = = = = = = = = = = = сульфат натрію хлорид амонію N- метилморфолін 1- метил -2-піролідон ядерний магнітний резонанс паладій ацетат паладію та BINAP, мікроінкапсульований у полісечовинній матриці (0.39 мМ / г). Pd- завантаження BINAP 0.25, Pd 1.0 комплекс біс (дифенілфосфіно) фероценпаладію(II) хлориду з дихлорметаном тетракис (трифенілфосфін)паладій(0) осад препаративна тонкошарова хроматографія бензотріазол-1-іл-окси-триспіролідинофосфон-гексафторо фосфат квартет коефіцієнт утримання кімнатна температура синглет насичений триплет бромід тетрабутиламонію трет- бутил-диметилсиліл трифторооцтова кислота тетрагідрофуран тетрагідропіраніл тонкошарова хроматографія триметілсиліл триметілфосфін триметілфосфін оксид мікролітр Етап отримання початкових матеріалів. Отримання 1. Синтез базового зразка 1 (Template 1; T1): Етил 4-бромо-2-метил-2-(метилсульфоніл)бутаноат та окремі енантіомери (R) та (S). Етап A) Етил 2-(метилсульфоніл) пропаноат: Метансульфонат натрію (103 г, 937 мМ) об'єднали з етил 2-хлоропропіонатом (109 г, 892 мМ) у етанолі (350 мл) у 500 мл круглодонній одногорлій колбі. Реакцію проводили при температурі 77 °C протягом 20 год. та потім реакційну суміш залишили охолоджуватися до кімнатної температури. Тверду фазу відокремили шляхом фільтрації через целіт та матеріал фільтру промили етанолом. Об'єднані фільтрати концентрували in vacuo. Потім неочищений продукт суспендували у діетиловому ефірі (250 мл) та тверду фазу відокремили шляхом фільтрації, після чого фільтрат концентрували in vacuo з отриманням зазначеної сполуки у 1 вигляді блідо-жовтого масла (51 г., 73 %). H ЯМР (400 мГц, хлороформ-d) δ ppm 1.32 (t, J=7.05 Гц, 3 H) 1.67 (d, J=7.47 Гц, 3 H) 3.05 (s, 3 H) 3.83-3.92 (m, 1 H) 4.18-4.37 (m, 2 H). Етап B) Етил 4-бромо-2- метил -2-(метилсульфоніл)бутаноат: Гідрид натрію (60 % дисперсія у мінеральній оливі, 2.33 г., 58.3 мМ) промили гексаном (2 × 10 мл) у 100 мл круглодонній колбі з двома горловинами у азотному середовищі з наступним суспендуванням у DMF (30 мл). До суспензії додали по краплям етил 2-(метилсульфоніл) пропаноат (10.0 г, 55.49 мМ) у DMF (10 мл), потім суміш перемішували 30 хв. при кімнатній температурі, охолодили до 0 °C та додали по краплям 1,2- дибромоетан (5.17 мл, 58.8). Суміш залишили нагріватися на ніч до кімнатної температури з перемішуванням. Потім реакцію у суміші зупинили насиченим водним розчином хлориду амонію (100 мл) та провели екстрагування з діетиловим ефіром (4 × 50 мл). Об'єднані органічні сполуки промили 50 % насиченим хлоридом натрію (4 × 50 мл), висушили (MgSO 4), та концентрували in vacuo. 15 UA 107423 C2 5 10 15 Неочищений матеріал очистили за допомогою хроматографії з силікагелем (350 г., 230-400 мкм) та елюювали розчином EtOAc у гексані (10-20 %) з отриманням зазначеної сполуки у вигляді 1 блідо-жовтого масла (7.9 г, 50 %). H ЯМР (400 мГц, хлороформ-d) δ ppm 1.33 (t, J=7.05 Гц, 3 H) 1.64 (s, 3 H) 2.49-2.59 (m, 1 H) 2.78 (ddd, J=13.89, 10.16, 6.64 Гц, 1 H) 3.05 (s, 3 H) 3.33-3.41 (m, 1 H) 3.46-3.54 (m, 1 H) 4.22-4.37 (m, 2 H). Етап C) Хіральне розділення етил 4-бромо-2-метил-2-(метилсульфоніл)бутаноату. Неочищений етил 4-бромо-2-метил-2-(метилсульфоніл)бутаноат (1.82 кг) очистили за допомогою флеш-хроматографії на колонці LP-600 з толуолом у якості елюенту з отриманням чистого етил 4-бромо-2-метил-2-(метилсульфоніл)бутаноату (1.63 кг). Очищений матеріал розчинили у етанолі (75 г/л) та розділили шляхом хіральної колоночної хроматографії на мікрокапілярній колонці MCC-2 (умови перелічені у Таблиці 1) з отриманням енантіомеру #1 20 o (738.4 г., час утримування = 4.719 хв., [α]589 = +14.1 ) з енантіомерним надлишком у 99 % та енантіомеру #2 (763.8 г, час утримування t=4.040 хв.) з енантіомерним надлишком у 95 %. Чистоту енантіомерів визначали за допомогою хіральної ВЕРХ-хроматографії, 4.6 × 250 мм колонка Chiralpak AD, 10 мкм, довжина хвилі 215 нм, мобільна фаза: етанол, ізократичне елюювання зі швидкістю 1 мл/ хв. при температурі навколишнього середовища. Таблиця 1 Нерухома фаза розмір колонки / температура рухома фаза концентрація завантаження швидкість завантаження швидкість елюенту швидкість очищеного продукту швидкість екстрагування швидкість переробки період часу 20 25 Chiralpak AD AD, 20μ 5 × 10 см /30 °C 100 % етанол 75 г/л у рухом фаз 4.0 мл / хв. 90.5 мл / хв. 35.6 мл / хв. 58.9 мл / хв. 262 мл / хв. 1.0 хв. Було визначено, що енантіомер #1 являє собою базовий зразок 1 (T1), тобто етил (2R)-4бромо-2-метил-2-(метилсульфоніл)бутаноат. Отримання 2. На Схемі B зображено отримання етил(2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2метил-2-(метилсульфоніл)бутаноату (T2) та (2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду (T3) та відповідних рацемічних та діастереомерних сумішей етил 4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2метил-2-(метилсульфоніл)бутаноату (T4) та 4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2-метил2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду (T5). 30 16 UA 107423 C2 Синтез базового зразка 3 (T3): (2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2- метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. 5 10 15 20 25 30 35 40 45 50 Етап A) Сполука III: 5-фторо-4-йодопіридин-2(1H)-один. Концентровану HCl (50 мл) додали до суміші 2,5-дифторо-4-йодопіридину (2.0 г., 8.3 мМ) у 1,4-діоксані (350 мл) та воді (100 мл). Суміш нагріли до конденсації та інкубували з цією температурою та струшуванням протягом ночі. Потім реакційну суміш піддали концентрації до сухого стану та залишок розтерли у воді (20 мл). Тверду фракцію зібрали за допомогою фільтрації та промили водою (2 × 30 мл) та гексаном (3 × 30 мл), після чого її висушили у вакуумі з отриманням зазначеної сполуки у вигляді жовтої твердої речовини (1.0 г., 50 %). MS 1 (LСМS) m/z 240.0 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 7.02 (d, J=5.07 Гц, 1 H) 7.68 (d, J=2.34 Гц, 1 H) 11.50 (br. s., 1 H). Етап B) Базовий зразок 2 (T2): Етил(2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2- метил -2-(метилсульфоніл)бутаноат. Карбонат цезію (1.77 г., 5.44 мМ) додали до суспензії 5-фторо-4-йодопіридин-2(1H)-один (1.00 г., 4.2 мМ) та етил (2R)-4-бромо-2-метил-2-(метилсульфоніл)бутаноату (1.56 г., 5.44 мМ) у безводному THF (45 мл). Реакційну суміш нагріли до 70 °C та інкубували її з перемішуванням протягом ночі з цією температурою. Реакцію згасили водою (100 мл) та екстрагували з EtOAc (2 × 100 мл). Об'єднані органічні сполуки промили розсолом (100 мл), висушили (MgSO 4), відфільтрували та концентрували. Неочищений продукт очистили за допомогою флешхроматографії на колонці Varian SF15-24g та елюювали EtOAc у n-гептані (30-80 %) з отриманням зазначеної сполуки у вигляді жовтого залишку (691 мг, 37 %). MS (LСМS) m/z 446.0 1 (M+1). H ЯМР (400 мГц, хлороформ -d) δ ppm 1.36 (t, 3 H) 1.75 (s, 3 H) 2.37-2.57 (m, 2 H) 3.10 (s, 3 H) 3.83-4.02 (m, 1 H) 4.16-4.37 (m, 3 H) 7.15 (d, 1 H) 7.20 (d, J=3.32 Гц, 1 H). Етап C) Сполука IV: (2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2-метил-2(метилсульфоніл)бутанова кислота. Гідроксид калію (669 мг, 7.7 мМ) додали до розчину етил (2R)-4-(5-фторо-4-йодо-2оксопіридин-1(2H)-іл)-2-метил-2-(метилсульфоніл)бутаноату (691 мг, 1.55 мМ) у 2метилтетрагідрофурані з водою (2:1 22.5 мл) та розчин перемішували протягом 2 год. при 70 °C. Реакційну суміш розвели 1 N водним розчином NaOH (50 мл). Органічні сполуки відокремили, водний шар промили EtOAc (2 × 50 мл) та підкислили pH до 3 за допомогою 3 M водного розчину HCl. Водний шар екстрагували EtOAc (3 × 60 мл), висушили (MgSO4), відфільтрували та піддали концентруванню з отриманням зазначеної сполуки у вигляді жовто-білої твердої 1 речовини (290 мг, 44.8 %). MS (LСМS) m/z 418.0. H ЯМР (400 мГц, DMSO-d6) δ ppm 1.53 (s, 3 H) 2.08-2.20 (m, 1 H) 2.36-2.48 (m, 1 H) 3.13 (s, 3 H) 3.79-4.02 (m, 2 H) 7.03 (d, J=6.05 Гц, 1 H) 7.96 (d, J=4.29 Гц, 1 H) 13.82 (br. s., 1 H). Етап D) Базовий зразок 3 (T3): (2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2-метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. N- метилморфолін (120 мкл, 1.1 мМ) додали до розчину CDMT (178 мг, 1.01 мМ) та (2R)-4(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2-метил-2-(метилсульфоніл) бутанової кислоти (280 мг, 0.762 мМ) у 2-метилтетрагідрофурані (7.60 мл) та реакційну суміш перемішували при кімнатній температурі протягом 1 год. Далі до реакційної суміші додали THP- гідроксиламін (117 мг, 1.00 мМ) та реакційну суміш знов перемішували при кімнатній температурі протягом ночі. Реакцію згасили водою (50 мл) та водний шар екстрагували EtOAc (3 × 50 мл). Об'єднані органічні сполуки промили розсолом (50 мл), висушили (MgSO 4), відфільтрували та піддали концентруванню з отриманням зазначеної сполуки у вигляді білуватої твердої речовини (399.8 мг) MS (LСМS) 515.0 (M-1). Приклад 1 (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. 17 UA 107423 C2 5 10 15 20 25 30 35 40 45 Етап A) 2-[4-(4,4,5,5-тетраметил -1,3,2-діоксоборолан-2-іл) феніл] -2H-тетразол. Pd(dppf)Cl2 (70.2 мг, 0.10 мМ) додали до суспензії 4,4,4',4',5,5,5',5'-октаметил -2,2'-бі-1,3,2діоксоборолану (291 мг, 1.15 мМ), 2-(4-бромфеніл)-2H-тетразолу (215 мг, 0.96 мМ) та ацетату калію (191 мг, 1.91 мМ) у 1,4-діоксані (4.78 мл). Отриману суспензію нагріли до 80 °C та інкубували з перемішуванням протягом ночі з цією ж температурою. Далі реакційну суміш охолодили, відфільтрували через целіт та концентрували in vacuo. Неочищений продукт очистили за допомогою флеш-хроматографії з 40г силікагелю на колонці Redisep та елюювали його за допомогою EtOAc у n-гептані (0-50 %) з отриманням зазначеної сполуки у вигляді світло1 жовтої твердої речовини (258 мг, 99 %). MS (LСМS) m/z 273.2 (M+1). H ЯМР (400 мГц, хлороформ-d) δ ppm 1.36 (s, 12 H) 7.66-7.73 (m, 2 H) 7.96-8.02 (m, 2 H) 9.01 (s, 1 H). Етап B) (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2- метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Pd EnCat™ (317 мг, 0.10 мМ) додали до суміші карбонату калію (393 мг, 2.84 мМ), 2-[4(4,4,5,5-тетраметил-1,3,2-діоксоборолан-2-іл) феніл] -2H-тетразолу (258.4 мг, 0.95 мМ), та (2R)4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран2-ілокси)бутанаміду (490 мг, 0.95 мМ), T3, у розчині 1,4-діоксану з водою (4:1, 10 мл). Реакційну суміш нагріли до 80 °C та інкубували з перемішуванням протягом ночі з цією температурою. Потім реакційну суміш відфільтрували через целіт, та матеріал фільтру промили метанолом (250 мл). Об'єднані фільтрати концентрували в умовах зниженого тиску та отриманий неочищений матеріал очистили за допомогою флеш-хроматографії з застосуванням у якості елюенту EtOAc у n-гептані (20-100 %) та метанолу у EtOAc (0-10 %) з отриманням зазначеної сполуки у вигляді світло-коричневої твердої речовини (500 мг, 98 %). MS (LСМS) m/z 534.4 (M1). 0 Етап C) (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-N- гідрокси-2метил-2-(метилсульфоніл)бутанамід. Соляну кислоту (4.0 M у 1,4-діоксані, 1.7 мл, 6.63 мМ) додали до розчину (2R)-4-{5-фторо-2оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2-метил-2-метилсульфоніл)-N-(тетрагідро2H-піран-2-ілокси)бутанаміду (500 мг, 0.94 мМ) у суміші дихлорметан: метанол (5:1,6 мл) при кімнатній температурі. Далі реакційну суміш перемішували протягом 1 год., після чого концентрували в умовах зниженого тиску з отриманням осаду, який потім розтирали протягом ночі у суміші діетилового ефіру з пентаном (1:1). Тверду фракцію зібрали за допомогою фільтрації та висушили в умовах зниженого тиску з отриманням зазначеної сполуки у вигляді 1 твердої речовини (340 мг, 76 %). MS (LСМS) m/z 451.0 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.56 (s, 3 H) 2.09-2.21 (m, 1 H) 2.42-2.45 (m, 1 H) 3.09 (s, 3 H) 3.78 (m, J=11.80, 11.80, 5.20 Гц, 1 H) 3.97-4.10 (m, 1 H) 6.63 (d, J=7.61 Гц, 1 H) 7.84 (dd, J=8.68, 1.66 Гц, 2 H) 8.00-8.15 (m, 3 H) 10.16 (s, 1 H) 11.08 (br. s., 1 H). Приклад 2 (2R)-4-[5-фторо-4-(2-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил -2(метилсульфоніл)бутанамід. Етап A) (2R)-4-[5-фторо-4-(2-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-2- метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (470 мг, 48.7 %) отримали у вигляді твердої речовини з (2-фторо-3метилфеніл)борної кислоти (388 мг, 2.52 мМ) за допомогою процедури, аналогічної тій, що 18 UA 107423 C2 5 10 15 20 25 30 35 40 45 описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2-метил-2метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) 1 m/z 499 (M+1). H ЯМР (400 мГц, метанол-d4) δ ppm 1.49-1.57 (m, 3 H) 1.59 (d, J=3.71Гц, 3 H) 1.64-1.74 (m, 3 H) 2.16-2.26 (m, 1 H) 2.27-2.31 (m, 3 H) 3.10 (d, J=5.66Гц, 3 H) 3.31 (s, 1 H) 3.473.55 (m, 1 H) 3.72-3.88 (m, 1 H) 3.90 (s, 1 H) 3.99-4.15 (m, 2 H) 4.94-4.99 (m, 1 H) 6.47 (d, J=7.22 Гц, 1 H) 7.20-7.26 (m, 1 H) 7.26-7.32 (m, 1 H) 7.40-7.47 (m, 1 H) 8.01 (dd, J=11.90, 5.85 Гц, 1H) 11.52 (d, J=3.51Гц, 1 H). Етап B) (2R)-4-[5-фторо-4-(2-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2метил -2-(метилсульфоніл)бутанамід. Зазначену сполуку (185 мг, 46.6 %) отримали у вигляді твердої речовини з (2R)-4-[5-фторо4-(2-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N-(тетрагідро2H-піран-2-ілокси)бутанаміду (477 мг, 0.957 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-N-гідрокси-21 метил-2-(метилсульфоніл)бутанаміду, Приклад 1, Етап C. MS (LСМS) m/z 415 (M+1) H ЯМР (400 мГц, DMSO-d6) δ ppm 1.54 (s, 3 H) 2.13 (ddd, J=13.03, 11.07, 4.78 Гц, 1 H) 2.40-2.45 (m, 1 H) 3.08 (s, 3 H) 3.76 (td, J=11.81, 5.07 Гц, 1 H) 4.02 (td, J=11.85, 5.17 Гц, 1 H) 6.53 (d, J=7.61 Гц, 1 H) 7.45-7.64 (m, 4 H) 8.02 (d, J=6.63 Гц, 1 H) 9.11 -` 9.26 (m, 1 H) 11.00 – 11.13 (m, 1 H). Приклад 3 (2R)-4-[4-(4-хлорфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. Етап A) (2R)-4-[4-(4-хлорфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід Зазначену сполуку (870 мг, 59.8 %) отримали у вигляді твердої речовини з (4хлорфеніл)борної кислоти (610 мг, 4.36 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2- метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, етап B. MS (LСМS) 1 m/z 502 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.17-1.28 (m, 2 H) 1.45-1.53 (m, 3 H) 1.56 (d, J=3.71 Гц, 3 H) 1.60-1.72 (m, 3 H) 2.08-2.23 (m, 1 H)3.07 (d, J=6.44 Гц, 3 H) 3.48 (d, J=11.12 Гц, 1 H) 3.67-3.85 (m, 1 H) 3.96-4.12 (m, 2 H) 4.88-4.97 (m, 1 H) 6.53 (d, J=7.61 Гц, 1 H) 7.50-7.62 (m, 3 H) 8.00 (dd, J=13.07, 6.63 Гц, 1 H) 11.50 (s, 1 H). Етап B) (2R)-4-[4-(4-хлорфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2- метил-2(метилсульфоніл)бутанамід. Зазначену сполуку (340 мг, 47.0 %) отримали у вигляді твердої речовини з (2R)-4-[4-(4хлорфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2Hпіран-2-ілокси)бутанамідом (870 мг, 1.74 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл] піридин-1(2H)-іл}-N-гідрокси-21 метил-2-(метилсульфоніл)бутанаміду, Приклад 1, Етап C. MS (LСМS) m/z 417 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.54 (s, 3 H) 2.13 (ddd, J=13.12, 11.27, 5.07 Гц, 1 H) 2.37-2.45 (m, 1 H) 3.08 (s, 3 H) 3.76 (td, J=11.90, 5.07 Гц, 1 H) 3.93-4.12 (m, 1 H) 6.53 (d, J=7.61 Гц, 1 H) 7.49-7.66 (m, 4 H) 8.02 (d, J=6.63 Гц, 1 H) 9.21 (s, 1 H) 10.95-11.17 (m, 1 H). Приклад 4. (2R)-4-[5-фторо-4-(2-фторфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. 19 UA 107423 C2 5 10 15 20 Етап A) (2R)-4-[5-фторо-4-(2-фторфеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (230 мг, 81.7 %) отримали у вигляді твердої речовини з (2-фторо феніл)борної кислоти (122 мг, 0.871 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) 1 m/z 485 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.21-1.28 (m, 2 H) 1.48-1.56 (m, 3 H) 1.58 (d, J=3.71 Гц, 3 H) 1.63-1.74 (m, 3 H) 2.15-2.26 (m, 1 H) 3.10 (d, J=5.66 Гц, 3 H) 3.34 (br. s., 1 H) 3.51 (d, J=10.73 Гц, 1 H) 3.71-3.88 (m, 1 H) 3.99-4.15 (m, 2 H) 4.94-4.99 (m, 1 H) 6.50 (d, J=7.02 Гц, 1 H) 7.31-7.40 (m, 2 H) 7.46-7.53 (m, 1 H) 7.56 (m, J=7.76, 7.76, 5.56, 1.76 Гц, 1 H) 8.01 (dd, J=11.90, 5.85 Гц, 1 H) 11.51 (d, J=3.32 Гц, 1 H). Етап B) (2R)-4-[5-фторо-4-(2-фторфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2- метил-2(метилсульфоніл)бутанамід. Зазначену сполуку (68 мг, 36.0 %) отримали у вигляді твердої речовини з (2R)-4-[5-фторо-4(2-фторфеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2ілокси)бутанаміду (230 мг, 0.475 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл] піридин-1(2H)-іл}-N-гідрокси-2- метил -21 (метилсульфоніл)бутанаміду, Приклад 1, Етап C. MS (LСМS) m/z 401 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.58 (s, 3 H) 2.18 (td, J=12.15, 4.98 Гц, 1 H) 2.47 (m, 1 H) 3.12 (s, 3 H) 3.79 (td, J=11.85, 5.17 Гц, 1 H) 4.07 (td, J=11.81, 4.68 Гц, 1 H) 6.50 (d, J=7.02 Гц, 1 H) 7.30-7.44 (m, 2 H) 7.46-7.64 (m, 2 H) 8.04 (d, J=6.05 Гц, 1 H) 9.20-9.32 (m, 1 H) 10.99-11.17 (m, 1 H). Приклад 5. (2R)-4-[4-(2,3-дигідро-1-бензофуран-5-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2метил -2-(метилсульфоніл)бутанамід. 25 30 35 40 45 50 Етап A) (2R)-4-[4-(2,3-дигідро-1-бензофуран-5-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-2- метил 2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (198 мг, 61.0 %) отримали у вигляді твердої речовини з 2,3-дигідро-1бензофуран-5-іл борної кислоти (153 мг, 0.871 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл) феніл]піридин-1(2H)-іл}-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) 1 m/z 509 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.46-1.56 (m, 2 H) 1.58 (d, J=4.10 Гц, 3 H) 1.631.76 (m, 3 H) 2.12-2.27 (m, 1 H) 2.40-2.48 (m, 1 H) 3.10 (d, J=6.05 Гц, 3 H) 3.23 (t, J=8.78 Гц, 2 H) 3.35 (br. s, 1 H) 3.51 (d, J=12.10 Гц, 1 H) 3.67-3.88 (m, 1 H) 3.98-4.15 (m, 2 H) 4.59 (t, J=8.78 Гц, 2 H) 4.96 (d, J=2.73 Гц, 1 H) 6.46 (d, J=7.81 Гц, 1 H) 6.81-6.92 (m, 1 H) 7.28-7.39 (m, 1 H) 7.47 (s, 1 H) 7.96 (dd, J=12.78, 6.73 Гц, 1 H) 11.55 (s, 1 H). Етап B) (2R)-4-[4-(2,3-дигідро-1-бензофуран-5-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-Nгідрокси-2-метил-2-(метилсульфоніл)бутанамід. Зазначену сполуку (165 мг, 53.0 %) отримали у вигляді твердої речовини з (2R)-4-[4-(2,3дигідро-1-бензофуран-5-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N(тетрагідро-2H-піран-2-ілокси)бутанаміду (198 мг, 0.389 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин1(2H)-іл}-N-гідрокси-2-метил-2-(метилсульфоніл)бутанаміду, Приклад 1, Етап C. MS (LСМS) m/z 1 425 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.57 (s, 3 H) 2.16 (dd, J=5.56, 1.07 Гц, 1 H) 2.362.49 (m, 1 H) 3.11 (s, 3 H) 3.23 (t, J=8.59 Гц, 2 H) 3.66-3.86 (m, 1 H) 4.04 (dd, J=6.15, 0.88 Гц, 1 H) 4.59 (t, J=8.78 Гц, 2 H) 6.45 (d, J=7.81 Гц, 1 H) 6.87 (d, J=8.39 Гц, 1 H) 7.34 (dd, J=8.20, 1.95 Гц, 1 H) 7.46 (s, 1 H) 7.98 (d, J=6.83 Гц, 1 H) 9.15-9.31 (m, 1 H) 11.01-11.19 (m, 1 H). Приклад 6. (2R)-4-[4-(3,4-дифторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. 20 UA 107423 C2 5 10 15 20 Етап A) (2R)-4-[4-(3,4-дифторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (760 мг, 52.1 %) отримали у вигляді твердої речовини з (3,4дифторфеніл)борної кислоти (596 мг, 3.78 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) m/z 503 (M+1). Етап B) (2R)-4-[4-(3,4-дифторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. Зазначену сполуку (350 мг, 55.0 %) отримали у вигляді твердої речовини з (2R)-4-[4-(3,4дифторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2Hпіран-2-ілокси)бутанаміду (760 мг, 1.51 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-N-гідрокси-21 метил-2-(метилсульфоніл)бутанаміду, Приклад 1, етап C. MS (LСМS) m/z 419 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.57 (s, 3 H) 2.06 – 2.25 (m, 1 H) 2.38 – 2.48 (m, 1 H) 3.11 (s, 3 H) 3.68 – 3.87 (m, 1 H) 3.96 – 4.18 (m, 1 H) 6.60 (d, J=7.61 Гц, 1 H) 7.37 – 7.52 (m, 1 H) 7.52 – 7.65 (m, 1 H) 7.65 – 7.84 (m, 1 H) 8.06 (d, J=6.63 Гц, 1 H) 9.13 – 9.39 (m, 1 H) 11.08 (s, 1 H). Приклад 7. (2R)-4-{5-фторо-2-оксо-4-[4-(2,2,2-трифтороетокси)феніл]піридин-1(2H)-іл}-N-гідрокси-2метил -2-(метилсульфоніл)бутанамід. 25 30 35 40 45 Етап A) (2R)-4-{5-фторо-2-оксо-4-[4-(2,2,2-трифтороетокси)феніл]піридин-1(2H)-іл}-2-метил2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (860 мг, 78.6 %) отримали у вигляді твердої речовини з [4-(2,2,2трифтороетокси)феніл]борної кислоти (554 мг, 2.52 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) m/z 565 (M+1). Етап B) (2R)-4-{5-фторо-2-оксо-4-[4-(2,2,2-трифтороетокси)феніл]піридин-1(2H)-іл}-Nгідрокси-2-метил-2-(метилсульфоніл)бутанамід. Зазначену сполуку (310 мг, 42.3 %) отримали у вигляді твердої речовини з (2R)-4-{5-фторо2-оксо-4-[4-(2,2,2-трифтороетокси)феніл]піридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)-N(тетрагідро-2H-піран-2-ілокси)бутанаміду (860 мг, 1.52 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин1(2H)-іл}-N-гідрокси-2-метил-2-(метилсульфоніл)бутанаміду, Приклад 1, Етап C. MS (LСМS) m/z 1 419 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.54 (s, 3 H) 2.06-2.22 (m, 1 H) 2.37-2.45 (m, 1 H) 3.08 (s, 3 H) 3.74 (td, J=11.76, 4.98 Гц, 1 H) 3.93-4.10 (m, 1 H) 4.81 (q, J=8.98 Гц, 2 H) 6.49 (d, J=7.61 Гц, 1 H) 7.16 (d, J=8.98 Гц, 2 H) 7.49-7.62 (m, 2 H) 7.98 (d, J=6.63 Гц, 1 H) 9.21 (br. s., 1 H) 11.07 (s, 1 H). Приклад 8. (2R)-4-[4-(3,4-дигідро-2H-хромен-6-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. 21 UA 107423 C2 5 10 15 20 ЕтапA) (2R)-4-[4-(3,4-дигідро-2H-хромен-6-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-2- метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (500 мг, 82.3 %) отримали у вигляді твердої речовини з 3,4-дигідро-2Hхромен-6-іл борної кислоти (228 мг, 1.28 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-ілфеніл]піридин-1(2H)-іл}-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) m/z 523 (M+1). Етап B) (2R)-4-[4-(3,4-дигідро-2H-хромен-6-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-N- гідрокси-2метил-2-(метилсульфоніл)бутанамід. Зазначену сполуку (240 мг, 57.1 %) отримали у вигляді твердої речовини з (2R)-4-[4-(3,4дигідро-2H-хромен-6-іл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфо ніл)-N(тетрагідро-2H-піран-2-ілокси)бутанаміду (500 мг, 0.957 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин1(2H)-іл}-N-гідрокси-2-метил-2-(метилсульфоніл)бутанаміду, Приклад 1, Етап C. MS (LСМS) m/z 1 439 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.52 (s, 3 H) 1.83-1.97 (m, 2 H) 2.12 (ddd, J=13.03, 11.27, 5.17 Гц, 1 H) 2.34-2.44 (m, 1 H) 2.75 (t, J=6.34 Гц, 2 H) 3.07 (s, 3 H) 3.71 (td, J=11.76, 5.17 Гц, 1 H) 3.92-4.07 (m, 1 H) 4.08-4.20 (m, 2 H) 6.42 (d, J=7.61 Гц, 1 H) 6.79 (d, J=8.39 Гц, 1 H) 7.207.32 (m, 2 H) 7.94 (d, J=6.83 Гц, 1 H) 9.20 (br. s., 1 H) 11.07 (s, 1 H). Приклад 9. (2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. 25 30 35 40 45 50 Етап A) Етил(2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-2- метил-2(метилсульфоніл)бутаноат. 1,4-діоксан (10 мл) та 3 M водний розчин K3PO4 (1.12 мл, 3.3 мМ) додали до колби з [4(метилтіо)феніл]борною кислотою (0.283 г., 1.68 мМ), Pd(dppf)Cl 2 (82 мг, 0.112 мМ) та етил(2R)4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2-метил-2-(метилсульфоніл) бутано атом, T2, (500 мг, 1.12 мМ) попередньо продутим азотом. Реакційну суміш нагріли до 60 °C та перемішували при цій температурі протягом 1 год. Далі реакційну суміш розвели EtOAc та промили водою. Органічну фракцію висушили (MgSO4), відфільтрували та піддали концентруванню. Неочищений продукт очистили за допомогою флеш-хроматографії на колонці з силікагелем (40 г) з застосуванням EtOAc у n-гептані (0-100 %) у якості елюенту та отриманням зазначеної 1 сполуки у вигляді смоли (492 мг, 99 %). MS (LСМS) m/z 442.1 (M+1). H ЯМР (400 мГц, хлороформ-d) δ ppm 1.17-1.27 (m, 3 H) 1.74 (s, 3 H) 2.38-2.61 (m, 5 H) 3.09 (s, 3 H) 3.88-4.02 (m, 1 H) 4.17-4.32 (m, 3 H) 6.57 (d, J=7.61 Гц, 1 H) 7.21-7.34 (m, 3 H) 7.37-7.48 (m, 2 H). Етап B) (2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-2-метил-2(метилсульфоніл)бутанова кислота. Моногідрат гідроксиду літію (165 мг, 6.68 мМ) додали до розчину етил (2R)-4-{5-фторо-4-[4(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)бутаноату (0.492 г., 1.12 мМ) у суміші THF: вода (1:1, 14 мл) та реакційну суміш перемішували при кімнатній температурі протягом 18 год., після чого її підкислили шляхом додавання 4 M водного розчину HCl з отриманням осаду. Тверду фракцію зібрали за допомогою фільтрації та висушили у вакуумі з отриманням зазначеної сполуки у вигляді твердої речовини (339 мг, 73 %). MS (LСМS) m/z 414.1 1 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.54 (s, 3 H) 2.17 (ddd, J=13.42, 10.00, 5.07 Гц, 1 H) 2.41-2.45 (m, 1 H) 2.49 (s, 3 H), 3.14 (s, 3 H) 3.78-4.15 (m, 2 H) 6.46 (d, J=7.81 Гц, 1 H) 7.28-7.39 (m, 2 H) 7.43-7.55 (m, 2 H) 8.01 (d, J=6.83 Гц, 1 H). 22 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 Етап C) (2R)-4-{5-фторо-4-[4-(метилтіо) феніл] -2-оксопіридин-1(2H)-іл}-2-метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. N, N-діізопропілетиламін (450 мкл, 2.45 мМ), O-(тетрагідро-2H-піран-2-іл) гідроксиламін (192 мг, 1.64 мМ) та HATU (447 мг, 1.23 мМ) додали до розчину (2R)-4-{5-фторо-4-[4(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)бутанової кислоти (339 мг, 0.82 мМ) у DMF (10 мл). Потім реакційну суміш перемішували при кімнатній температурі протягом 18 год., після чого її розвели EtOAc та промили розсолом. Органічну фракцію висушили (MgSO4), відфільтрували та піддали концентруванню. Неочищений залишок очистили шляхом флеш-хроматографії на колонці з силікагелем (40 г) з застосуванням EtOAc у n-гептані (0-100 %) у якості елюенту з отриманням зазначеної сполуки(420 мг, 100 %). MS (LСМS) m/z 511.4 (M-1). Етап D) (2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-N-гідрокси-2- метил-2(метилсульфоніл)бутанамід. Водний розчин HCl (4 M, 3 мл) додали до розчину 2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2оксопіридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2ілокси)бутанамід(437 мг, 0.852 мМ) у THF (10 мл) та реакційну суміш перемішували при кімнатній температурі протягом 3 годин, після чого піддали концентруванню та кілька раз довели до азеотропного стану з EtOAc та n-гептаном до отримання зазначеної сполуки у 1 вигляді білуватої твердої речовини (233 мг, 64 %). MS (LСМS) m/z 429.1 (M+1). H ЯМР (400 мГц, метанол-d4) δ ppm 1.69 (s, 3 H) 2.37 (ddd, J=13.51, 10.59, 5.17 Гц, 1 H) 2.51 (s, 3 H) 2.57-2.74 (m, 1 H) 2.82 (s, 3 H) 3.09 (s, 3 H) 3.97 (ddd, J=13.12, 10.59, 5.56 Гц, 1 H) 4.27 (ddd, J=13.03, 10.49, 5.17 Гц, 1 H) 6.68 (d, J=7.22 Гц, 1 H) 7.25-7.40 (m, 2 H) 7.47-7.59 (m, 2 H) 7.90 (d, J=6.05 Гц, 1 H). Приклад 10. (2R)-4-[4-(4-етоксифеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. Етап A) Етил (2R)-4-[4-(4-етоксифеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил -2(метилсульфоніл)бутаноат. Зазначену сполуку (320 мг, 64 %) отримали у вигляді смоли з (4-етоксифеніл)борної кислоти (280 мг, 1.68 мМ) за допомогою процедури, аналогічної тій, що описана для етил(2R)-4-{5фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)бутаноату, 1 Приклад 9, Етап A. MS (LСМS) m/z 440.3 (M+1). H ЯМР (400 мГц, хлороформ-d) δ ppm 1.22 (t, J=7.12 Гц, 3 H) 1.40 (t, J=7.02 Гц, 3 H) 1.73 (s, 3 H) 2.36-2.61 (m, 2 H) 3.09 (s, 3 H) 3.85-3.98 (m, 1 H) 4.07 (dd, J=14.93, 7.12 Гц, 2 H) 4.25 (m, 3 H) 6.51-6.58 (m, 1 H) 6.93 (d, J=8.98 Гц, 3 H) 7.227.31 (m, 1 H) 7.44 (d, J=7.02 Гц, 1 H). Етап B) (2R)-4-[4-(4-етоксифеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2(метилсульфоніл)бутанова кислота. Гідроксид літію (108 мг, 4.37 мМ) додали до розчину етил (2R)-4-[4-(4-етоксифеніл)-5-фторо2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)бутаноату (320 мг, 0.728 мМ) у суміші тетрагідрофурану з водою (1:1, 20 мл) та реакційну суміш перемішували при кімнатній температурі до повного розчинення. Реакційну суміш підкислили 4 M водним розчином HCl та екстрагували з EtOAc. Об'єднані органічні шари висушили (MgSO 4), відфільтрували та піддали концентруванню з отриманням зазначеної сполуки у вигляді твердої речовини (220 мг, 73 %). 1 MS (LСМS) m/z 412.2 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.26-1.40 (m, 3 H) 1.54 (s, 3 H) 2.07-2.26 (m, 1 H) 2.43 (d, J=6.24 Гц, 1 H) 3.14 (s, 3 H) 3.83-4.15 (m, 4 H) 6.42 (d, J=7.81 Гц, 1 H) 6.93-7.07 (m, 2 H) 7.50 (dd, J=8.68, 1.85 Гц, 2 H) 7.97 (d, J=6.83 Гц, 1 H). Етап C) (2R)-4-[4-(4-етоксифеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (273 мг, 100 %) отримали з (2R)-4-[4-(4-етоксифеніл)-5-фторо-2оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)бутанової кислоти (220 мг, 0.535 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-4-[4- (метилтіо)феніл]-2оксопіридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 9, Етап C. MS (LСМS) m/z 509.4 (M+1). 23 UA 107423 C2 5 10 Етап D) (2R)-4-[4-(4-етоксифеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2- метил -2(метилсульфоніл)бутанамід. Зазначену сполуку (205 мг, 83 %) отримали у вигляді твердої речовини з (2R)-4-[4-(4етоксифеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N(тетрагідро-2Hпіран-2-ілокси)бутанаміду (297 мг, 0.582 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-N-гідрокси-2-метил-21 (метилсульфоніл)бутанаміду, Приклад 9, Етап D. MS (LСМS) m/z 427.2 (M+1). H ЯМР (400 мГц, метанол-d4) δ ppm 1.40 (t, J=6.93 Гц, 3 H) 1.69 (s, 3 H) 2.28-2.42 (m, 1 H) 2.54-2.69 (m, 1 H) 3.09 (s, 3 H) 3.86-3.99 (m, 1 H) 4.09 (q, J=7.02 Гц, 2 H) 4.18-4.32 (m, 1 H) 6.60 (d, J=7.42 Гц, 1 H) 7.01 (d, J=8.98 Гц, 2 H) 7.54 (dd, J=8.78, 1.56 Гц, 2 H) 7.82 (d, J=6.24 Гц, 1 H). Приклад 11 (2R)-4-[5-фторо-2-оксо-4-(4-пропілфеніл)піридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. 15 20 25 30 35 Етап A) (2R)-4-[5-фторо-2-оксо-4-(4-пропілфеніл)піридин-1(2H)-іл]-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (75.5 мг, 21 %) отримали у вигляді твердої речовини з (2R)-4-(5-фторо-4йодо-2-оксопіридин-1(2H)-іл)-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2ілокси)бутанаміду, T3, (360 мг, 0.697 мМ) та (4-пропілфеніл)борної кислоти (171 мг, 1.04 мМ) за допомогою процедури, аналогічної тій, що описана для етил(2R)-4-{5-фторо-4-[4(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)бутаноату, Приклад 9, Етап A. MS (LСМS) m/z 507.4(M-1). Етап B) (2R)-4-[5-фторо-2-оксо-4-(4-пропілфеніл)піридин-1(2H)-іл]-N-гідрокси-2- метил-2(метилсульфоніл)бутанамід. Зазначену сполуку (58 мг, 94 %) отримали у вигляді твердої речовини з (2R)-4-[5-фторо-2оксо-4-(4-пропілфеніл)піридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2ілокси)бутанаміду (75 мг, 0.15 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-N-гідрокси-2-метил-21 (метилсульфоніл)бутанаміду, Приклад 9, Етап D. MS (LСМS) m/z 425.2 (M+1). H ЯМР (400 мГц, МЕТАНОЛ -d4) δ ppm 0.89-1.00 (m, 3 H) 1.57-1.76 (m, 5 H) 2.29-2.45 (m, 1 H) 2.55-2.69 (m, 3 H) 3.09 (s, 3 H) 3.86-4.04 (m, 1 H) 4.17-4.34 (m, 1 H) 6.63 (d, J=7.02 Гц, 1 H) 7.31 (d, J=8.00 Гц, 2 H) 7.44-7.55 (m, 2 H) 7.85 (d, J=5.85 Гц, 1 H). Приклад 12. (2R)-4-{5-фторо-2-оксо-4-[4-(пентафторо-6λ-сульфаніл)феніл]піридин-1(2H)-іл}-N- гідрокси-2метил-2-(метилсульфоніл)бутанамід. 40 45 50 Етап A) (2R)-4-{5-фторо-2-оксо-4-[4-(пентафторо-6λ-сульфаніл)феніл]піридин-1(2H)-іл}-2метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. 1,4-діоксан додали до 1-бромо-4-(пентафторо-6λ-сульфаніл)бензену (500 мг, 1.77 мМ), 4,4,4',4',5,5,5',5'-октаметил-2,2'-бі-1,3,2-діоксоборолану (628 мг, 2.47 мМ), ацетату калію (347 мг, 3.53 мМ) та Pd(dppf)Cl2 (130 мг, 0.177 мМ). Суміш нагріли до 80 °C та перемішували при цій температурі протягом 3 год., після чого до неї додали (2R)-4-(5-фторо-4-йодо-2-оксопіридин1(2H)-іл)-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід, T3, (456 мг, 0.883 мМ) та водний розчин Na2CO3 (2.0 N, 1.77 мл, 3.53 мМ) та реакційну суміш перемішували при 80 °C протягом ночі. Суміш розвели EtOAc та промили розсолом. Органічну фракцію висушили (MgSO4), відфільтрували та піддали концентруванню. Неочищений залишок очистили 24 UA 107423 C2 5 10 15 20 25 30 35 за допомогою флеш-хроматографії на колонці з силікагелем (40г) з застосуванням EtOAc у nгептані (0-100 %) у якості елюенту з отриманням зазначеної сполуки (205 мг, 39.2 %). MS(LСМS) m/z 591.4 (M-1). Етап B) (2R)-4-{5-фторо-2-оксо-4-[4-(пентафторо-6λ-сульфаніл)феніл]піридин-1(2H)-іл}-Nгідрокси-2-метил-2-(метилсульфоніл)бутанамід. Водний розчин HCl (1.0 M) додали до розчину (2R)-4-{5-фторо-2-оксо-4-[4-(пентафторо-6λсульфаніл)феніл]піридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2ілокси)бутанаміду (81мг, 0.14мМ) та реакційну суміш перемішували при кімнатній температурі протягом ночі з наступним концентруванням in vacuo з отриманням зазначеної сполуки у вигляді 1 твердої речовини (70мг, 100 %). MS(LСМS) m/z 509.1 (M+1). H ЯМР (400мГц, DMSO-d6) δ ppm 1.58 (s, 3 H) 2.06-2.26 (m, 1 H) 3.11 (s, 3 H) 3.70-3.89 (m, 1 H) 3.97-4.14 (m, 1 H) 6.64 (d, J=7.41 Гц, 1 H) 7.81 (d, 2 H) 8.04 (d, J=8.78 Гц, 2 H) 8.11 (d, J=6.44 Гц, 1 H). Приклад 13 (2R)-4-[5-фторо-4-(3-метилфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. Етап A) (2R)-4-[5-фторо-4-(3-метилфеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (510 мг, 55 %) отримали у вигляді смоли з (3-метилфеніл)борної кислоти (384 мг, 2.82 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-2оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)-N-(тетрагідро2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) m/z 479.4 (M-1). Етап B) (2R)-4-[5-фторо-4-(3-метилфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2- метил-2(метилсульфоніл)бутанамід. Зазначену сполуку (255 мг, 61 %) отримали у вигляді твердої речовини з (2R)-4-[5-фторо-4(3-метилфеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N-(тетрагідро-2H-піран-2ілокси)бутанаміду (510 мг, 1.06 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-N-гідрокси-2-метил-21 (метилсульфоніл)бутанаміду, Приклад 9, Етап D. MS (LСМS) m/z 397.1 (M+1). H ЯМР (400 мГц, метанол-d4) δ ppm 1.70 (s, 3 H) 2.28-2.44 (m, 4 H) 2.62 (dd, J=10.44, 5.37 Гц, 1 H) 3.09 (s, 3 H) 3.96 (ddd, J=12.98, 10.63, 5.46 Гц, 1 H) 4.19-4.35 (m, 1 H) 6.64 (d, J=7.22 Гц, 1 H) 7.27-7.45 (m, 4 H) 7.88 (d, J=5.85 Гц, 1 H). Приклад 14 (2R)-4-[5-фторо-4-(4-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. 40 45 50 Етап A) (2R)-4-[5-фторо-4-(4-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-2-метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. Зазначену сполуку (360 мг, 38 %) отримали у вигляді смоли з (4-фторо-3-метилфеніл)борної кислоти (434 мг, 2.82 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5фторо-2-оксо-4-[4-(2H-тетразол-2-іл)феніл]піридин-1(2H)-іл}-2-метил-2-(метилсульфоніл)-N(тетрагідро-2H-піран-2-ілокси)бутанаміду, Приклад 1, Етап B. MS (LСМS) m/z 497.0 (M-1). Етап B) (2R)-4-[5-фторо-4-(4-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2метил-2-(метилсульфоніл)бутанамід. Зазначену сполуку (271 мг, 91 %) отримали у вигляді білої твердої речовини з (2R)-4-[5фторо-4-(4-фторо-3-метилфеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N 25 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 (тетрагідро-2H-піран-2-ілокси)бутанаміду (360мг, 0.722 мМ) за допомогою процедури, аналогічної тій, що описана для (2R)-4-{5-фторо-4-[4-(метилтіо)феніл]-2-оксопіридин-1(2H)-іл}-Nгідрокси-2-метил-2-(метилсульфоніл)бутанаміду, Приклад 9, Етап D. MS (LСМS) m/z 415.1 1 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.54 (s, 3 H) 2.13 (ddd, J=13.12, 11.17, 4.98 Гц, 1 H) 2.26 (s, 3 H) 2.40-2.45 (m, 1 H) 3.08 (s, 3 H) 3.75 (td, J=11.85, 5.17 Гц, 1 H) 4.02 (td, J=11.85, 4.98 Гц, 1 H) 6.49 (d, J=7.61 Гц, 1 H) 7.20-7.28 (m, 1 H) 7.38-7.45 (m, 1 H) 7.50 (d, J=7.41 Гц, 1 H) 8.01 (d, J=6.63 Гц, 1 H). Приклад 15 5-фторо-1-[(3R)-3-(гідроксиаміно)-3-(метилсульфоніл)бутил]-4-[4-(окситан-3-ілокси) феніл]піридин-2(1H)-один. Етап A) 3-(4-бромфенокси)окситан. 4-бромфенол (2.5г., 14.5 мМ) та K2CO3 (5.45г., 39.4 мМ) додали до розчину окситан-3-іл 4метилбензенсульфонату (3.00г., 13.1 мМ) у DMF (10 мл) у запечатаній пробірці. Реакційну суміш нагріли до 100 °C та перемішували при цій температурі протягом 24 год., після чого її розвели EtOAc та промили водою. Органічний шар висушили (MgSO 4), відфільтрували та піддали концентруванню з отриманням зазначеної сполуки..MS (G СМ S) m/z 228. Цей матеріал застосували у наступних етапах без додаткового очищення. Етап B) 4,4,5,5-тетраметил-2-[4-(окситан-3-ілокси) феніл] -1,3,2-діоксоборолан. Зазначену сполуку (3.11г., 86 %) отримали з 3-(4-бромофенокси)окситану (3.00г., 13.1 мМ) за допомогою процедури, аналогічної тій, що описана для 2-[4-(4,4,5,5-тетраметил -1,3,21 діоксоборолан-2-іл) феніл]-2H-тетразолу, Приклад 1, Етап A. MS (G СМ S) m/z 276. H ЯМР (400 мГц, хлороформ-d) δ ppm 1.31 (s, 12 H) 4.70-4.79 (m, 2 H) 4.91-5.00 (m, 2 H) 5.18-5.27 (m, 1 H) 6.67 (d, J=8.78 Гц, 2 H) 7.73 (d, J=8.78 Гц, 2 H). Етап C) 5-фторо-1-{(3R)-3-(метилсульфоніл)-3-[(тетрагідро-2H-піран-2-ілокси)аміно] бутил}4-[4-(окситан-3-ілокси) феніл]піридин-2(1H)-один. 1,4-діоксан (20 мл) та воду (5 мл) додали до попередньо продутої азотом колби, що містила (2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2-метил-2-(метилсульфоніл) -N-(тетрагідро-2Hпіран-2-ілокси)бутанамід, T3 (287 мг, 0.56 мМ), 4,4,5,5-тетраметил-2-[4-(окситан-3-ілокси) феніл]-1,3,2-діоксоборолан (301 мг, 1.09 мМ), Pd(PPh3)4 (65 мг, 0.06 мМ) та карбонат калію (230мг, 1.67 мМ). Потім реакційну суміш нагріли до 80 °C та перемішували при цій температурі протягом ночі, після чого її піддали концентруванню та очистили шляхом флеш-хроматографії за допомогою двох колонок з 12 г силікагелю. Першу колонку елюювали метанолом у дихлорметані (0-20 %), а другу колонку елюювали EtOAc у n-гептані (0-100 %) з наступним елююванням метанолом у дихлорметані (0-20 %) з отриманням зазначеної сполуки у вигляді твердої речовини (164 мг, 54 %). MS (LСМS) m/z 537.4 (M-1). Етап D) 5-фторо-1-[(3R)-3-(гідроксиаміно)-3-(метилсульфоніл)бутил]-4-[4-(окситан-3-ілокси) феніл] піридин-2(1H)-один. Трифторооцтову кислоту (1 мл) додали до розчину 5-фторо-1-{(3R)-3-(метилсульфоніл)-3[(тетрагідро-2H-піран-2-ілокси)аміно]бутил}-4-[4-(окситан-3-ілокси)феніл] піридин-2(1H)-один (164 мг, 0.304 мМ) у DСМ (10 мл). Реакційну суміш перемішували при кімнатній температурі протягом ночі та потім піддали концентруванню у вакуумі. Залишок повторно розчинили у дихлорметані та n-гептані та знов піддали концентруванню з отриманням зазначеної сполуки у 1 вигляді твердої речовини. (113 мг, 82 %). MS (LСМS) m/z 455.1 (M+1). H ЯМР (400 мГц, метанол-d4) δ ppm 1.69 (s, 3 H) 2.35 (ddd, J=13.66, 10.63, 5.17 Гц, 1 H) 2.53-2.67 (m, 1 H) 3.09 (s, 3 H) 3.91 (ddd, J=12.98, 10.63, 5.27 Гц, 1 H) 4.16-4.30 (m, 1 H) 4.69 (dd, J=7.51, 4.78 Гц, 2 H) 5.02 (t, J=6.73 Гц, 2 H) 5.33 (t, J=5.46 Гц, 1 H) 6.58 (d, J=7.42 Гц, 1 H) 6.85-6.91 (m, 2 H) 7.55 (dd, J=8.68, 1.85 Гц, 2 H) 7.80 (d, J=6.24 Гц, 1 H). Приклад 16 (2R)-4-[4-(4-хлоро-2-фторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил-2(метилсульфоніл)бутанамід. 26 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 Етап A) (2R)-4-[4-(4-хлоро-2-фторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2- метил -2(метилсульфоніл)бутанова кислота. У колбу (3 л) з механічною мішалкою додали етил(2R)-4-(5-фторо-4-йодо-2-оксопіридин1(2H)-іл)-2-метил-2-(метилсульфоніл)бутаноат, T2 (100г., 225 мМ), (4-хлоро-2-фторфеніл)борну кислоту (25.5 г., 146 мМ) та Pd(dppf)2Cl2 (4.93 г.,6.74 мМ). Колбу продули N2, потім дегазували 2метилтетрагідрофураном (1 л) та додали 3 M водним розчином K 3PO4 (225 мл, 674 мМ). Реакційну суміш нагріли до 75 °C та перемішували при цій температурі протягом 30 хв., після чого додали (4-хлоро-2-фторфеніл)борної кислоти (25.5 г., 146 мМ) та залишили нагріватися ще протягом 1.5 год., після чого її охолодили до кімнатної температури та відокремили водний шар. Органічний шар пропустили крізь шар целіту та вернули назад до реакційної посудини, до якої додали гідроксид літію (28 г., 667 мМ) у воді (700 мл), Суміш потім нагріли до 50 °C та перемішували при цій температурі протягом 1 год., після чого її охолодили та знов відокремили водний шар. До відокремленого водного шару додали целіт та суміш відфільтрували через целітову пробку. Далі фільтрат помістили у колбу з верхньоприводною мішалкою, уважно підкислили 4 M водним розчином HCl та нагріли до 50 °C з перемішуванням до отримання осаду. Тверду фракцію зібрали за допомогою фільтрації та висушили у вакуумі з отриманням зазначеної сполуки у вигляді коричневої твердої речовини (68.7 г., 74 %). MS (LСМS) m/z 420.6 1 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.54 (s, 3 H) 2.18 (ddd, J=13.17, 10.24, 5.07 Гц, 1 H) 2.41-2.45 (m, 1 H) 3.10-3.19 (s, 3 H) 3.87-4.08 (m, 2 H) 6.47 (d, J=7.02 Гц, 1 H) 7.42 (dd, J=8.39, 1.95 Гц, 1 H) 7.48-7.56 (m, 1 H) 7.60 (dd, J=9.95, 1.95 Гц, 1 H) 8.06 (d, J=6.05 Гц, 1 H). Етап B) (2R)-4-[4-(4-хлоро-2-фторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. N- метилморфолін (54 мл, 491 мМ) та 2-хлоро-4,6-диметокси-1,3,5-триазин (43.1 г., 245 мМ) додали до розчину (2R)-4-[4-(4-хлоро-2-фторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2(метилсульфоніл)бутанової кислоти (68.7г., 164мМ) у 2-метилтетрагідро фурані (1л) та реакційну суміш перемішували при кімнатній температурі протягом 2 годин, після чого додали O-(тетрагідро-2H-піран-2-іл)гідроксиламін (28.8 г., 245 мМ) та реакційну суміш перемішували при кімнатній температурі протягом 1 год. Далі суміш відфільтрували та фільтрат піддали концентруванню. Неочищений залишок очистили за допомогою колоночної хроматографії з силікагелем та елюювали 40 % EtOAc у n-гептані (4 л) та EtOAc (6 л). Необхідні фракції об'єднали та піддали концентруванню з отриманням зазначеної сполуки у вигляді білої клейкої твердої речовини (74.82г., 88 %). MS (LСМS) m/z 517.9 (M-1). Етап C) (2R)-4-[4-(4-хлоро-2-фторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-N- гідрокси-2метил-2-(метилсульфоніл)бутанамід. Воду (312 мл) та 1 N водний розчин HCl (23.9 мл, 23.9 мМ) додали до розчину (2R)-4-[4-(4хлоро-2-фторфеніл)-5-фторо-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N(тетрагідро-2H-піран-2-ілокси)бутанаміду (74.7 г.,144.1 мМ) у етанолі (126 мл). Реакційну суміш нагріли до 70 °C та інкубували її з перемішуванням протягом ночі з цією температурою. Потім реакційну суміш охолодили, тверду фракцію зібрали шляхом фільтрації та промили водою до досягнення значення pH фільтрату ~5. Далі тверду фракцію висушили у вакуумі з отриманням зазначеної сполуки у вигляді білої твердої речовини (46.48 г., 74 %). MS (LСМS) m/z 435.6 1 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.56 (s, 3 H) 2.09-2.21 (m, 1 H) 2.44 (d, J=5.27 Гц, 1 H) 3.10 (s, 3 H) 3.77 (td, J=11.90, 5.07 Гц, 1 H) 4.04 (td, J=11.95, 4.98 Гц, 1 H) 6.51 (d, J=7.02 Гц, 1 H) 7.44 (dd, J=8.29, 2.05 Гц, 1 H) 7.50-7.56 (m, 1 H) 7.62 (dd, J=9.95, 1.95 Гц, 1 H) 8.04 (d, J=5.85 Гц, 1 H) 9.22 (s, 1 H) 11.05 (s, 1 H). Приклад 17. (2R)-4-[5-фторо-4-(2-фторо-3-метоксифеніл)-2-оксопіридин-1(2H)-іл]-N-гідрокси-2-метил -2(метилсульфоніл)бутанамід. 27 UA 107423 C2 5 10 15 20 25 30 35 40 45 50 55 Етап A) (2R)-4-[5-фторо-4-(2-фторо-3-метоксифеніл)-2-оксопіридин-1(2H)-іл]-2- метил-2(метилсульфоніл)бутанова кислота. Комплекс 1,1'-біс(дифенілфосфін)фероцен-паладій(II)дихлорид дихлорметану (5.90 г., 7.22 мМ) додали до суміші етил (2R)-4-(5-фторо-4-йодо-2-оксопіридин-1(2H)-іл)-2- метил-2(метилсульфоніл)бутаноату, T2 (29.63 г., 66.55 мМ), (2-фторо-3-метоксифеніл)борної кислоти (18.50 г.,108.9 мМ) та триосновного фосфату калію (54.5 г.,205 мМ) у 2-метилтетрагідрофурані (450 мл) та деіонізованій воді (225 мл). Суміш нагріли до 60 °C та інкубували її з перемішуванням протягом ночі з цією температурою, після чого реакцію охолодили до кімнатної температури. Від фракції органічних сполук відокремили водний шар, промили її водою (200 мл) та розсолом (200 мл), висушили (MgSO4) та відфільтрували. Потім до фільтрату додали порошок активованого вугілля Darco® G-60 (меш 100) та суміш перемішували протягом 1 год. Тверду фракцію відокремили шляхом фільтрації через целіт та фільтрувальний матеріал промили EtOAc (~300 мл). Об'єднані фільтрати піддали концентруванню з отриманням червоної маслянистої речовини (30.62 г), яку розчинили у 2-метилтетрагідрофурані (450 мл) та деіонізованій воді (225 мл). Далі до суміші додали гідроксид калію (26.1 г., 465 мМ), суміш нагріли до 50 °C та інкубували її з перемішуванням протягом ночі з цією температурою. Потім реакцію охолодили до кімнатної температури, водний шар відокремили та промили діетиловим ефіром (2 × 200 мл). Далі водний шар повільно підкислили концентрованою соляною кислотою до pH ~ 1, перемішуючи суспензію протягом 1 год. Суспензію відфільтрували та тверду фракцію промили водою (3 × 100 мл) та гексаном (3 × 300 мл). Фракцію твердих частин висушили у вакуумі з отриманням зазначеної сполуки у вигляді коричневої твердої речовини (26.49 г., 1 94.54 %). MS (LСМS) m/z 416.0 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.57 (s, 3 H) 2.14-2.28 (m, 0 H) 2.42-2.54 (m, 1 H) 3.18 (s, 3 H) 3.88 (s, 3 H) 3.90-4.09 (m, 2 H) 6.44 (d, J=7.03 Гц, 1 H) 6.92-7.04 (m, 1 H) 7.20-7.36 (m, 2 H) 8.06 (d, J=5.95 Гц, 1 H) 13.90 (br. s.,1 H). Етап B) (2R)-4-[5-фторо-4-(2-фторо-3-метоксифеніл)-2-оксопіридин-1(2H)-іл]-2- метил -2(метилсульфоніл)-N-(тетрагідро-2H-піран-2-ілокси)бутанамід. N- метилморфолін (11 мл, 96.2 мМ) додали до розчину CDMT (13.5 г., 116 мМ) та (2R)-4-[5фторо-4-(2-фторо-3-метоксифеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)бутанової кислоти (26.49 г., 63.77 мМ) у 2-метилтетрагідрофурані (640 мл) та реакційну суміш перемішували протягом 2 год., після чого до неї додали THP- гідроксил амін (13.5 г., 116 мМ) та реакційну суміш перемішували при кімнатній температурі протягом ночі. Реакцію згашували насиченим водним розчином бікарбонату калію (500 мл). Органічний шар відокремили, промили водою (300 мл) та розсолом (300 мл), висушили (MgSO 4) та відфільтрували. Потім до фільтрату додали порошок активованого вугілля Darco® G-60 (меш 100) та суміш перемішували протягом 1 год. Вугілля відокремили шляхом фільтрації через целіт та фільтрувальний матеріал промили EtOAc (1 л). Фільтрат піддали концентруванню з отриманням зазначеної сполуки у вигляді жовтувато-білої твердої речовини (30.49, 92.93 %). MS (LСМS) m/z 513.9 (M-1). Етап C) (2R)-4-[5-фторо-4-(2-фторо-3-метоксифеніл)-2-оксопіридин-1(2H)-іл]-N- гідрокси-2метил-2-(метилсульфоніл)бутанамід. p-толуолсульфонат піридину (190 мг, 0.76 мМ) додали до розчину (2R)-4-[5-фторо-4-(2фторо-3-метоксифеніл)-2-оксопіридин-1(2H)-іл]-2-метил-2-(метилсульфоніл)-N-(тетра- гідро-2Hпіран-2-ілокси)бутанаміду (777 мг, 1.51 мМ) у етанолі (15 мл). Реакційну суміш нагріли до 50 °C та інкубували її з перемішуванням протягом ночі з цією температурою, після чого до розчину додали ще певну кількість p-толуолсульфонату піридину (118 мг, 0.47 мМ) та реакційну суміш інкубували з температурою 60 °C протягом 3 год., після чого її охолодили до кімнатної температури та осад зібрали шляхом фільтрації. Тверду фракцію промили етанолом (15 мл) та гексаном (15 мл) та висушили у вакуумі з отриманням зазначеної сполуки у вигляді білої твердої 1 речовини (413 мг, 63.5 %). MS (LСМS) m/z 431.0 (M+1). H ЯМР (400 мГц, DMSO-d6) δ ppm 1.57 (s, 1 H) 2.08-2.25 (m, 0 H) 2.41-2.56 (m, 1 H) 3.02-3.19 (m, 3 H) 3.71-3.85 (m, 1 H) 3.88 (s, 3 H) 3.984.13 (m, 1 H) 6.47 (d, J=7.02 Гц, 1 H) 6.93-7.08 (m, 1 H) 7.19-7.36 (m, 2 H) 8.04 (d, J=5.66 Гц, 1 H) 9.24 (d, J=1.56 Гц, 1 H) 11.07 (d, J=1.56 Гц, 1 H). Приклад 18. 28
ДивитисяДодаткова інформація
Назва патенту англійськоюFluoro-pyridinone derivatives useful as antibacterial agents
Автори англійськоюBrown, Matthew Frank, Che, Ye, Melnick, Michael Joseph, Montgomery, Justin Ian, Plummer, Mark Stephen, Price, Loren Michael, Reilly, Usa
Автори російськоюБраун Меттью Фрэнк, Ке И, Мелник Майкл Джозеф, Монтгомери Юстин Ян, Плюммер Марк Стивен, Прайс Лорен Майкл, Рейлли Юса
МПК / Мітки
МПК: C07D 401/04, C07D 405/12, A61K 31/4439, C07D 417/12, C07D 413/10, A61K 31/4433, A61K 31/4427, C07D 401/10, A61K 31/4412, C07D 213/69, C07D 405/04, C07D 213/64
Мітки: мають, антибактеріальну, активність, похідні, фторопіридинону
Код посилання
<a href="https://ua.patents.su/71-107423-pokhidni-ftoropiridinonu-shho-mayut-antibakterialnu-aktivnist.html" target="_blank" rel="follow" title="База патентів України">Похідні фторопіридинону, що мають антибактеріальну активність</a>
О-метильні похідні азитроміцину а, що мають антибактеріальну активність, спосіб їх отримання та проміжні сполуки

Номер патенту: 27105
Опубліковано: 28.02.2000
Автори: Кобрехел Габріела, ДЬОКІС Слободан, Лазаревскі Горяна
МПК: A61K 31/7042, C07H 17/00, A61K 31/70, C07H 17/08, A61P 31/04
Мітки: проміжні, активність, сполуки, азитроміцину, похідні, спосіб, антибактеріальну, о-метильні, отримання, мають
Формула / Реферат:
1. О-Метильные производные азитромицина А общей формулы (I)обладающие антибактериальной активностью.2. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=CH3, R4=R5=H.3. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=R4=CH3, R5=H.4. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5, R3=R5=H, R4=CH3.5. Соединение по п.1, отличающееся тем, что R1=R2=CO2CH2C6H5,...
Похідні син-ізомеру цефему, що мають антибактеріальну активність, спосіб їх отримання (варіанти) і фармацевтичний склад

Номер патенту: 37180
Опубліковано: 15.05.2001
Автори: ГУЕН Д'АМБРІЕР Соланж, Асзоді Жозеф, ФОБО Патрік, Шанто Жан-Франсуа
МПК: C07C 205/00, C07C 311/27, C07D 519/00, C07C 45/71, C07C 239/00, C07C 229/40, C07D 501/00, C07D 277/42, C07C 323/56, C07C 59/00, C07C 311/35, C07C 47/52, A61K 31/546, C07C 255/57, C07C 45/63, C07D 277/20, A61P 31/04, C07C 291/00, C07C 69/734, C07C 255/56, A61K 31/545, C07C 45/67, C07C 323/52
Мітки: варіанти, фармацевтичний, склад, отримання, син-ізомеру, активність, цефему, мають, спосіб, похідні, антибактеріальну
Формула / Реферат:
1. Производные син-изомера цефема общей формулы (I):,(І)гдеR1, R2, R3, R5 = Н, ОН, ОС1-С4-алкил, ОСОС1-С4-алкил, галоген, CN, NО2, независимо друг от друга, причем если R3 = ОН или ОСОС1-С4-алкил, то R1, R2, R5 отличны от Н,R4 = ОН, ОСОС1-С4-алкил,А и А1 = Н или эквивалент щелочного или щелочноземельного металла, магния, аммония,...
Похідні хінолонкарбонової кислоти, суміш їх ізомерів або окремі ізомери, їх фармакологічно прийнятні гідрати та солі, що мають антибактеріальну активність, та проміжні сполуки
Номер патенту: 27309
Опубліковано: 15.09.2000
Автори: Ендерманн Райнер, Цайлер Ханс-Йоахім, КРЕБС Андреас, БРЕММ Клаус Дітер, ШЕНКЕ Томас, ПЕТЕРСЕН Уве, ГРОХЕ Клаус, Хіммлер Томас, МЕТЦГЕР Карл-Георг
МПК: C07D 498/04, A61K 31/47, A61P 33/10, A61P 33/02, C07D 487/10, C07D 405/14, A61K 31/495, C07D 487/08, A61K 31/535, C07D 401/04, C07D 471/04, C07D 413/04, C07D 487/04, C07D 471/08, A61P 31/04, A61P 33/00, A61K 31/55, C07D 215/56
Мітки: активність, окремі, мають, хінолонкарбонової, кислоти, солі, сполуки, прийнятні, антибактеріальну, гідрати, ізомери, фармакологічно, похідні, проміжні, суміш, ізомерів
Текст:
...быть получены, например, взаимодействием по Дильсу-Альдеру диенов общей формулы (V), (V). где Rg имеет указанное выше значение и R21 или идентичен с Rie или является функциональной группой, с диенофилами общей формулы (VI), О (VI), где R22 является водородом или защитной группой, как триметилсилил, бензил, алкилфенилметил с 1-4 атомами углерода з алкильной части, метоксибензил или бензгидрил, с последующим восстановлением карбонильной...
Похідні карбапенему, їх фармацевтично прийнятні солі та ефіри, що гідролізують in vivo, спосіб їх одержання (варіанти), фармацевтична композиція, яка має антибактеріальну активність, похідні піролідин-4-іл-тіол

Номер патенту: 44885
Опубліковано: 15.03.2002
Автори: Свейн Майкл Лінгард, Беттс Майкл Джон, Дейвіс Гарет Морс
МПК: A61K 31/397, C07F 9/6574, C07D 403/12, C07D 477/00, A61K 31/40, C07D 207/16, C07F 9/572, C07F 9/568, A61P 31/04
Мітки: солі, композиція, активність, vivo, має, одержання, похідні, ефіри, фармацевтично, спосіб, карбапенему, антибактеріальну, яка, прийнятні, варіанти, піролідин-4-іл-тіол, гідролізують, фармацевтична
Формула / Реферат:
1. Производные карбапенема общей формулы I в которой R1 - оксиметил или 1-оксиэтил,R2 - водород или (С1-С4)-алкил,R3 - водород или (С1-С4)-алкил,R4 и R5, одинаковые или различные, представляют собой атом водорода, атом галогена, амино, цианогруппу, (С1-С4)-алкил, гидроксигруппу, карбоксигруппу, (С1-С4)алкоксигруппу, (С1-С4)алкоксикарбонил, карбамоил, (С1-С4)алкилкарбамоил,...
Похідні пенему, спосіб їх одержання (варіанти) та фармацевтична композиція, яка має антибактеріальну активність

Номер патенту: 46698
Опубліковано: 17.06.2002
Автори: КАШО Джузеппе, ПЕРРОТТА Енжо, Альтамура Маріа, ПЕСТЕЛЛІНІ Вітторіо, СБРАЧІ П'єро, АРКАМОНЕ Федеріко Маріа
МПК: A61K 31/43, C07D 499/00, A61P 31/04
Мітки: активність, має, похідні, фармацевтична, пенему, яка, одержання, композиція, варіанти, антибактеріальну, спосіб
Формула / Реферат:
1. Производные пенема общей формулы (I), (I)где R1 – C1-C6 гидроксиалкил;R2 – карбоксильная группа или этерифицированная карбоксильная группа, легко активируемая in vivo, или карбоксилат-анион;R3 – Н, C1-C4 алкил, необязательно замещенный карбоксиамидной группой;R4 – Н, C1-C6 алкил, необязательно замещенный фенилом, C1-C6 аминоалкил; илиR3 и R4, связанные вместе, образуют пирролидиновое,...
Попередній патент: Газорозподільна система двигуна внутрішнього згорання
Наступний патент: Пластометр скнара
Випадковий патент: Гравітаційний посівний апарат