Спосіб отримання нуклеозіду або його фармацевтично придатних солей






Формула / Реферат
Способ получения нуклеозида общей формулы
где R - основание одной из формул
где R1 - водород, метил, фтор, или йод или их фармацевтически приемлемых солей, отличающийся тем, что соединение общей формулы
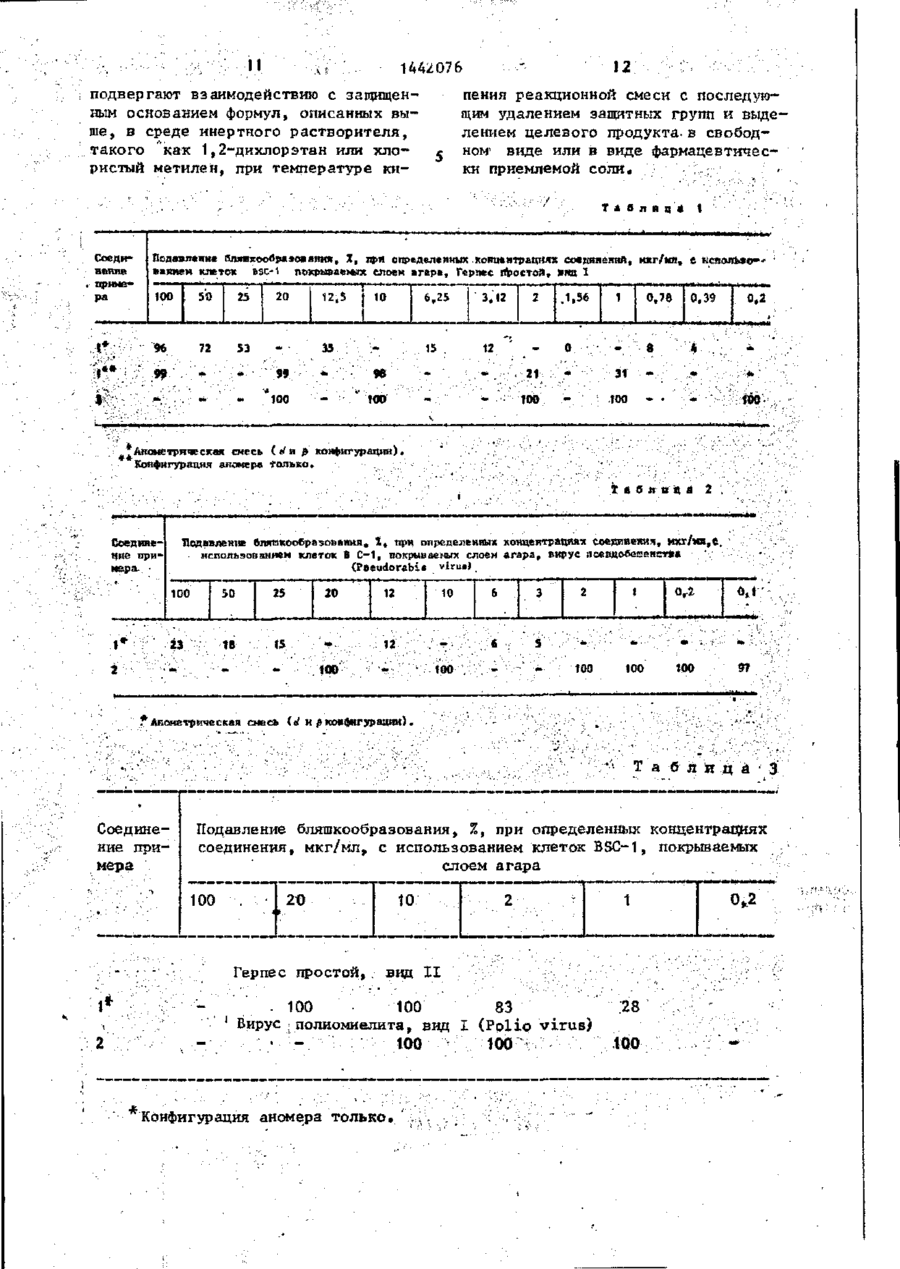
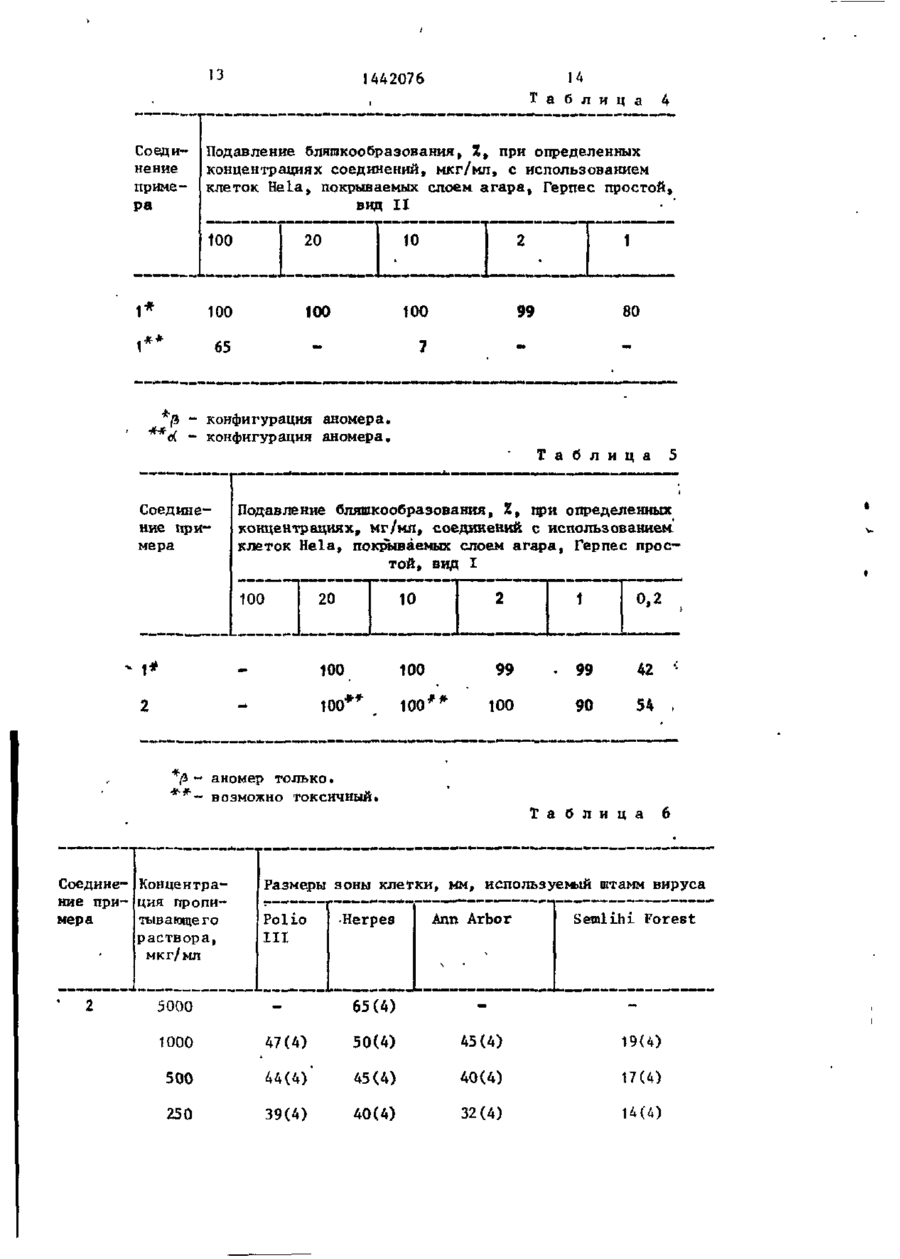
где Q - группа СНХ, где Х - отщепляемая группа, J1 и J2 - независимо гидроксизащитная группа, повергают взаимодействию с защищенным основанием формул, описанных выше, в среде инертного растворителя, такого как 1,2-дихлорэтан или хлористый метилен, при температуре кипения реакционной смеси с последующим удалением защитных групп и выделением целевого продукта в свободном видеили в виде фармацевтически приемлемой соли.
Текст
Изобретение относится к нуклеозидамі в частности к получению нуклеозида формулы (HOH2C)CH-O-CH(R)-C(F)^-(OH)CH, где R - основание одной из формул ЬРПФ-К -N-C(0)-N = C(NH2)-CR1 = СН, гце R, - водород, метил, фтор или йод, или их фармацевтически приемлемых солей, обладающих противовирусной активностью. Цель изобретения выявление новых более активных соединений. Получение их ведут взаимодействием соединения формулы (J 1 OH 2 C)CH-O-Q-C(F) 1 -(J 1 O)CH, где Q - группа СНХ, где X - отщепляем мая группа,' Jj и Зг - независимо гидроксизащитная группа, с защищен- , ным основанием описанных формул. Процесс ведут в среде инертного растворителя, такого как 1,2-дихлорэтан > или хлористый метилен, при температуре кипения реакционной смеси с последующим удалением защитных групп и выделением целевого продукта в свободном виде или в виде фармацев- • тически приемлемой соли. 7 табл. с о I Изобретение относится к способу получения новых нуклеозидов общей формулы 1442076 вор выпаривают до сухости под вакуумом. Полученный остаток растворяют в воде и раствор дважды экстрагируют £ диэтиловым эфиром. Водный слой выпаривают. Остаток поглощают в этаноле и повторно выпаривают до полного обезвоживания. Получают 1 г неочищенного продукта, который хроматографиО Н F 10 руют на 30 г силикагеле Woelm (70где R - основание одной из формул 150 меш), при элгоировании этилацетатом с выходом 0,76 г требуемого прод у к т а . Этот продукт дополнительно очищают перекристаллизацией из этил15 ,ацетата с получением 0,37 г белого, кристаллического продукта. . о NMR(CD3OD, 90 мГц), ^ : 1,93 (S, • . ЗН), 3,5-4,67 (серии m, 4H); 4,88 где Rf - водород, метил, фтор или (bS, ЗН); 6,3 t , J = 9 Гц, 1H)i 7,47 20 (m,iH)J масс-спектр і га/е = 278 = и с йод, или их фармацевтически приемлемых ходный , солей, обладающих противовирусной Д р и м е р 2. 1-(4-Амино-2-окактивностью. со-1Н-пиримидин-1-ил)-2-дрэокси~2,2-дифторорибоза. Цель изобретения - получение н о - 25 Б атмосфере а з о т а к 5,0 г 3 , 5 вых нуклеозидов, обладающих большей -бис(трет-бугилдиметилсилокси)-1-меактивностью, чем известный структуртане ульфонилокси-2-деэокси-2, 2-диный аналог - видарабин (Ара-А). фторорибозы в 1 мл сухого 1,2-дихлорП р и м е р 1. 1~(5-Метилг2,4этана добавляют 4,68 г бис-триметил30 силилацетилцитозина и затем 3,96 г -диоксо-1Н,ЗН-пиримидин-1-ил)-1, 2~дезокси~2,2-дифторорибоза, трифторметансульфонилоксиметилсилаВ атмосфере азота к 2,59 г 3,5— на. Раствор кипятят с обратным хо-бис(трет-бутилдиметилсилокси)-1-'мелодильником в интервале 3-15 ч . Р е тансульфонилокси-2-~дезокси-2,2-диакционную смесь охлаждают до комнатфторорибозы добавляют 1,60 г 5-ме— 45 ной температуры, вводят 2 мл метатил-2,4-бис-(триметилоксилилокси)пинола и полученную суспензию перемеримидина и 45 мл сухого 1,2-дихлор, шивают в течение 30 мин. Остаток э т а н а . Затем к этой смеси добавляют ' фильтруют, а фильтрат сгущают в в а 1,45 г трифторметансульфонилоксимет кууме до полного обезвоживания, Остйтилсилана и перемешивают чистый раст '40 ток растворяют в метиленхлориде, навор при кипячении с обратным холосыщают безводным НВг и перемешивают дильником, в течение 2-3 ч . Затем при комнатной температуре в течение продукты реакции охлаждают до комнат 45.мин. Полученную смесь сгущают до ной температуры,, добавляют 1,35 мл сухого состояния в вакууме, поглощаметанола и перемешивают полученную 45 ют метанольным аммонием и перемешисуспензию в течение 30 мин» Осадок вают в течение 15 мин при комнатной фильтруют, а фильтрат восстанавливатемпературе. Затем раствор сгущают ют до половины своего объема под в а до полного обезвоживания в вакууме, куумом и разбавляют равным образом трижды разбавляют водой и водную дихлорметана. Затем раствор промыва- 50 растворимую порцию вводят в колонку ют водным раствором бикарбоната натна силикагеле с обратной фазой, полурия и после этого насыщенным водным чая 100 мг требуемого продукта при раствором хлорида натрия и высушива,элюировании водой. ют над безводным сульфатом натрия; Раствор фильтруют, а фильтрат насыща J NMR(CD3OD,90 мГц), d1 : 3,7-4,65 55 ют безводным бромводородом. РеакциІ (серии m, 4H)', 4,83 (bS, 4H)', 5,97. онную смесь перемешивают в течение (d, J « 8 Гц, 1Н); 6,24 ( t , J - 8 Гц,' о 30 мин и затем сгущают под вакуумом. 1Н); 7,88 (d, J = 8 Гі\, 1Н); массОстаток растворяют в метаноле и раст спектр: m/e-263-исходныГг. '1442076 ( t , J = 8 Гц, 1Н); 7,94 (d, J = 7 Гц, " П р и м е р 3 . 1-(4-Амин£>-5-иод1Н); масс-спектр: m/e ~ 282-исходный. -2-оксо-1Н-пиримидин-1-ил)-2-дезокси-2,2-дифторорибоза, П р и м е р 5 , 1-(5-Метил-2,4-диокси-1Н,ЗН-пиримидин-1-ил)-2-дезВ атмосфере азота к 1,99 г 3 , 5 5 окси-2,2-дифторорибоза. -бис(трет-бутшщиметнлснлокси)-1-ме~ тансульфонилокси-2-дезокси~2,2-дифтор5,4' г 3,5-бис(трет-бутиддиметил— орибозы в 1,35 мл сухого 1,2-дихлорсилилокси)-1-метансульфонилокси-2-дез этане добавляют 2,08 г трис-триметилокси-2,2-дифторорибозы и 45,4 г 5-месилнл-5-йодцитозина и 1,11 г трифтор-10 тил-2,4-бис(триметилсилилокси)пиримиметансульфонилоксиметнлсилана. Растдина соединяют и нагревают в атмосфевор кипятят с обратным холодильником ре азота ііри перемешивании, при 100°С в интервале 3-15 ч . Реакционную смесь в течение 1 ч и при 150°С в течение охлаждают до комнатной температуры, 1 ч . После этого смесь охлаждают до добавляют 5,0 мл метанола, полученную 15 комнатной температуры, разбавляют суспензию перемешивают приблизитель25 мл воды и 10 мл метанола. Суспенно в течение 30 мин. Остаток фильтрузию фильтруют через фильтрат со с л о ют и фильтрат сгущают в вакууме до ем кизельгура и слой промывают ацетон сухости. Остаток растворяют в метиленном. Соединенные фильтраты выпаривахлориде, насыщают безводным НВг и пе-20 ют под вакуумом и получают 5,3 г масремешивают при комнатной температуре лянистого о с т а т к а . Остаток растворяв течение 45 мин. Смесь сгущают до ют в 10 мл ацетона и вводят в 4,5 см „fcyxoro состояния в вакууме. Остаток колонку, набитую 80 г силикагеля. • трижды насыщают водой, нейтрализуют , Проводят алюирование смесью дихлорме— NaHCO3 и разделяют на колонне с с и - . 25 тан:метанол:триэтиламин 1 5 : 1 : 1 . ликагелем с обратной фазой (Н^О: Первые 100 мл элюента отбрасывают, МеОН(9:1) с получением 26 мл требуеа следующие 300 мл выпаривают под мого продукта. вакуумом и получают 4,1 г сиропообразного сырого продукта, который раствор NMR(CD30D, 90 МГц), 4: 3;64-4,73 (серии m, 4Н); 4,9 (bS, 4H); '6,25 30 ряют в 40 мл ацетона. Через раствор в течение 1 ч барботируют хлористый ( t , J = 8 Гц, Ш ) ; 8,44 (S, 1Н),массводород, а затем в течение еще 1 ч спектр: т / е = 389 = исходный. , барботируют бромистый водород. После : П р и м е р 4 , 1-(2,4-Диоксо-5этого раствор выпаривают при 62° и -фтор-1Н,ЗН-пиримидин-1-ил)-2-дезокси-2,2-дифторорибоза. , 35 получают 4,4 г маслянистого темного продукта. В атмосфере азота к 1,1 г 3,5-бисУказанный продукт растворяют в (тр|ет-бутилдиметилсилокси)-1-метан10 мл теплой смеси дихлорметан:уксуссульфонилокси-2-дезокси-2,2-дифторная кислота 3:1 и вводят в колонку орибозы в 20 мл сухого дихлорэтана добавляют 2,88 г бис-триметилсилил-5- 40 4,5 см, набитую 45 г силикагеля. Для первых 1000 мл в качестве элюента -фторурацила и 0,66 г трифторметаниспользуют смесь дихлорметан:уксусная сульфонилокситриметилсилана. Раствор кислота 3 : 1 , после чего в качестве охлаждают до комнатной температуры, элюента используют только уксусную добавляют 1,0 мл метанола'и полученную суспензию перемешивают примерно 45 кислоту. Большая часть целевого продукта находится в фракциях между в течение 30 мин. Остаток фильтруют 1000 и 1400 мл, выходящих из колонки» и фильтрат сгущают в вакууме до сухокак показывает тонкослойная хромаго состояния. Остаток растворяют в то гр афия на сштика геле с использоваметиленхлориде, насыщают безводным НВг и перемешивают при комнатной тем- 50 нием смеси дихлорметан:метанол 1 5 : 1 . Эти фракции объединяют и выпаривают пературе приблизительно в течение под вакуумом, а остаток растворяют в 45 мин. Смесь сгущают до сухого с о с 15 мл холодного ацетона и фильтруют. тояния в вакууме. Остаток трижды н а Фильтрат выпаривают под^ вакуумом и сыщают Н^О, нейтрализуют NaHCO3 и получают масло, которое растворяют разделяют на колонке с силикагелем 5с в 5 мл ацетона и хроматографирует с обратной фазой при элюировлнии в о на 20 г силикагеля с использованием дой с получением требуемого продукта. смеси дихлорметан:метанол 1 5 : 1 . СоNMR(CD3OD, 90 МГц), J ; 3,5-4,5 держащие продукт фракции объединяют, (серии m, 4H); 4,65. (bS-, 3H) і 5,89 1442076 выпаривают под вакуумом и получают использованием смеси воДа/метахюл 300 мг полужидкого соединения. Этот (объем/объем, 9:1) в качестве элюенпродукт растворяют в 5 мл ацетона и та, получают 2,21 г 1-(5-метил-2,4~ фильтруют, фильтрат выпаривают под -диоксо-1Н,ЗН-пиримидин-1-ил)-2-деэ5 вакуумом и получают 230 мг светлоокси-2,2-дифтороркбозы. Я Р (CD3GD, М коричневого полутвердого соединения, 90 МГц),
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparing a nucleoside a pharmaceutically acceptable salt thereof
Автори англійськоюLarry Wayne Hertel
Назва патенту російськоюСпособ получения нуклеозида или его фармацевтически приемлемых солей
Автори російськоюЛарри Вейн Хертел
МПК / Мітки
МПК: A61K 31/708, C07D 307/33, C07H 19/04, C07H 11/00, A61K 31/7072, A61K 31/7068, C07H 19/16, A61K 31/7042, A61K 31/70, C07D 317/30, C07H 19/06, C07H 5/00, A61P 31/12, A61K 31/7076, A61K 31/7064, A61K 31/7052
Мітки: фармацевтично, придатних, нуклеозиду, солей, спосіб, отримання
Код посилання
<a href="https://ua.patents.su/10-5955-sposib-otrimannya-nukleozidu-abo-jjogo-farmacevtichno-pridatnikh-solejj.html" target="_blank" rel="follow" title="База патентів України">Спосіб отримання нуклеозіду або його фармацевтично придатних солей</a>
Спосіб отримання циклоалкомедів (8-beta)-1алкіл-6-(заміщеного) ерголіну або їх фармацевтично придатних кислотно-адитивних солей

Номер патенту: 6022
Опубліковано: 29.12.1994
Автори: Марк Мортенсен Форман, Кетлін Роуз Віттен, Віллям Лі Гарбрехт, Джіффорд Пурнелл Марзоні
МПК: C07D 457/00
Мітки: придатних, отримання, спосіб, циклоалкомедів, ерголіну, солей, фармацевтично, 8-beta)-1алкіл-6-(заміщеного, кислотно-адітивних
Формула / Реферат:
Способ получения циклоалкиламидов (8-b)-І-алкил-6-(замещенного) эрголина общей формулыгде R1 - С1-С4-алкил; R2 - С1-С4-алкил с прямой цепью; R3 - водород или С1-С4-алкил с прямой цепью; R4 -водород, С1-С4-алкил, оксигруппа или С1-С4-алкоксигруппа; m=0,1 или 2,при условии, что когда R1 и R2каждый оначает метил, а R3 и R4каждый оначает водород, m не равно 0; или их фармацевтически...
Спосіб отримання похідних імідазола або його фармацевтично прийнятих солей

Номер патенту: 2697
Опубліковано: 26.12.1994
Автори: Джон Джонес Вітотес Данкіе, Дейвід Джон Керіні
Мітки: імідазола, похідних, отримання, солей, прийнятих, фармацевтично, спосіб
Текст:
...5 Гц). пластинках для ТСХ. Масс-спектр 325. ЯМР (200 мГц, Е. 1~(4~Аминобензил)~5-гидроксиме3 э 8,00-6,80 (м, 8Н), тил-2-(2*-метоксиэтил~-4-«зшоримидазол. 5,15 ( с , 2Н>, 4,45 ( с , 2Н), 3,60 ( т , Соединение СИНТеЗИРУЮТ П Примеру О 2Н, 5 Г ц ) , 3,15 ( с , ЗН), 2,75 54, Е из 5-тидроксимет'ил—2~(2-меток~ ( т , 2Н, 5 Г ц ) 0 П р и м е р ы сиэгил)— 1 — (4—нитроб ензил) — 4—хлорими— 57—71„ Соединения, синтезированные по дазола ( 2 , 2 г, 6,75...
Спосіб одержання похідних 2-тієнілоксіуксусної кислоти чи їх фармацевтично прийнятних солей

Номер патенту: 4227
Опубліковано: 27.12.1994
Автори: Дітер Біндер, Хуберт Петер Фербер, Франц Ровенсцкі
МПК: C07D 333/32, A61K 31/381, A61P 7/02, A61P 9/00, A61K 31/38, A61P 11/00, A61P 43/00, A61P 29/00
Мітки: фармацевтично, солей, кислоти, спосіб, похідних, 2-тієнілоксіуксусної, прийнятних, одержання
Формула / Реферат:
1. Способ получения производных 2-тиенил-оксиуксусной кислоты общей формулы где R - незамещенный или замещенный галогеном фенил, или их фармацевтически применимых солей, отличающийся тем, что соединение общей формулыгде R имеет указанные значения, подвергают окислению окисью серебра в водно-щелочной среде с последующим выделением целевого продукта в свободном виде или в виде фармацевтически применимой если....
Засіб отримання похідних гуаніну або їх кислотно-адітивних фармацевтично припущених солей

Номер патенту: 2668
Опубліковано: 26.12.1994
Автори: Томас Ентоні Креницкі, Ліліа Мері Бьючемп
Мітки: припущених, похідних, фармацевтично, засіб, кислотно-адітивних, гуаніну, солей, отримання
Формула / Реферат:
1. Способ получения производных гуанина общей формулы Iгде R - группа -СН(СН3)2 или группа-СН(СН3)СН2СН3,или их кислотно-аддитивных фармацевтически приемлемых солей, отличающийся тем, что 9-(2-гид-роксиэтоксиметил)гуанинподвергают взаимодействию с N-защищенным...
Спосіб отримання (+)-4-ді-(н-пропіл)-аміно-6-карбамоіл-1,3,4,5-тетрагідробенз (с,д) індола або його фармацевтично придатної солі

Номер патенту: 5954
Опубліковано: 29.12.1994
Автор: Майкл Едвард Флауф
МПК: A61K 31/403, A61P 25/30, A61P 25/26, A61P 3/04, C07D 209/90, A61K 31/40, A61P 25/24, A61P 25/28
Мітки: фармацевтично, придатної, отримання, +)-4-ді-(н-пропіл)-аміно-6-карбамоіл-1,3,4,5-тетрагідробенз, індола, спосіб, с,д, солі
Формула / Реферат:
Способ получения (± )-4-ди(н-пропил)амино-6-карбамоил-1, 3, 4, 5-тетрагидробенз [с, d] формулы Іили его фармацевтически приемлемой соли, отличающийся тем, что (±)-4-ди(н-пропил)амино-6-циано-1, 3, 4, 5-тетрагидробенз [с, d] индол формулыповергают гидролизу гидроокисью калия в среде трет-бутанола в атмосфере азота в присутствии диметилсульфоксида с выделением целевого продукта хроматографическим разделением и при...
Попередній патент: Вставка теплообмінної труби радіатору водяного охолодження
Наступний патент: Засіб для захисту зернових культур від фітотоксичної дії гербіцидів
Випадковий патент: Ущільнювач субстрату