Проміжна сполука для одержання агомелатину (варіанти) і спосіб її одержання (варіанти)
Номер патенту: 104684
Опубліковано: 25.02.2014
Автори: Жанг Пенг, Йу Ксіонг, Хуанг Йу, Йуан Жедонг, Шан Ханбін

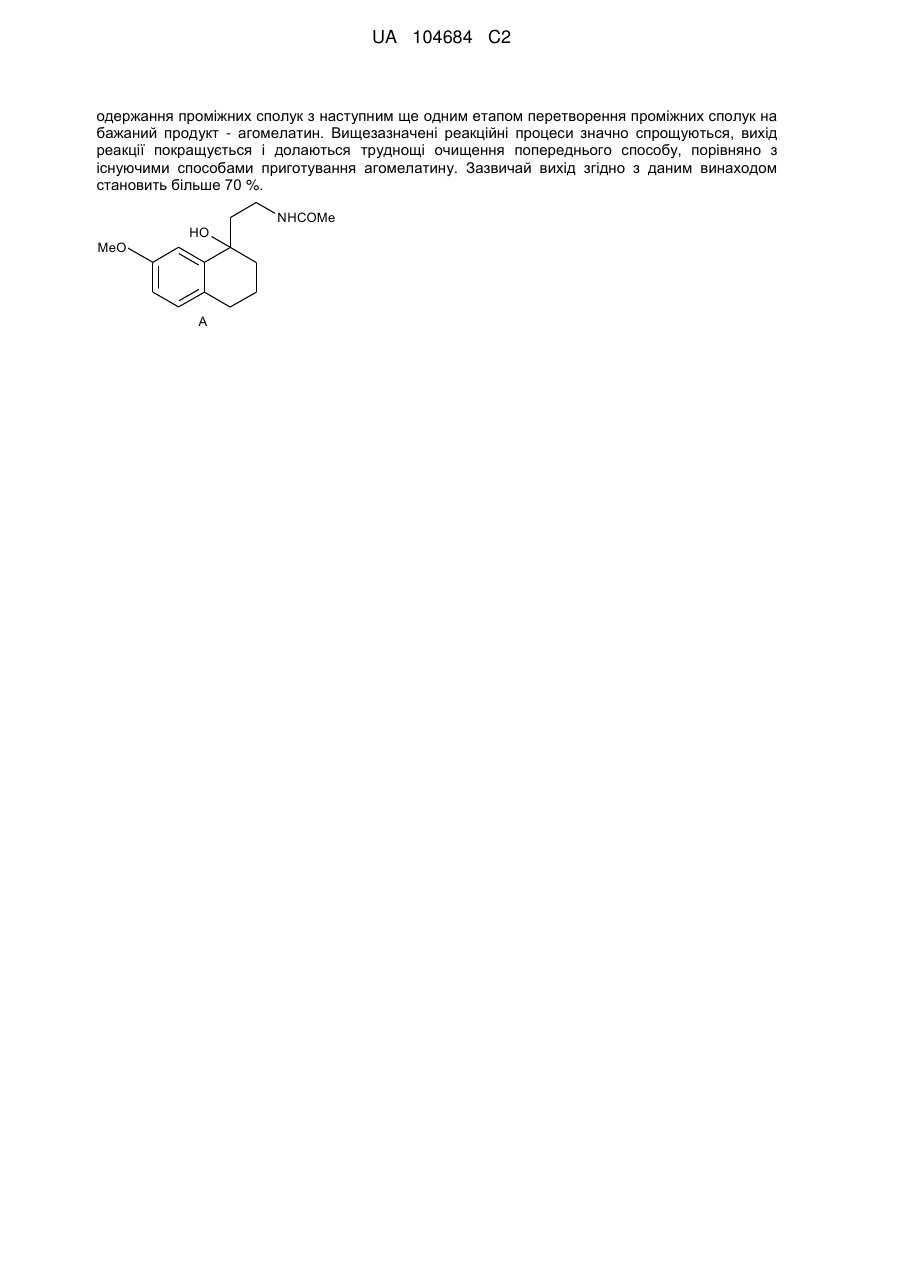








Формула / Реферат
1. Сполука А наступної формули:
 .
.
2. Сполука В наступної формули:
 .
.
3. Спосіб одержання сполуки А за п. 1, який відрізняється тим, що здійснюють відновлювальне ацилювання сполуки С за умови каталітичної гідрогенізації і у присутності оцтового ангідриду
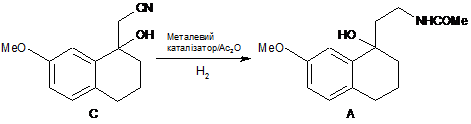 .
.
4. Спосіб за п. 3, який відрізняється тим, що вищезгаданий металевий каталізатор являє собою Ni-Ренея, кількість якого становить 0,1-0,3 частки від кількості сполуки С за масою.
5. Спосіб за п. 3, який відрізняється тим, що кількість вищезгаданого оцтового ангідриду становить 1-1,3 частки від молярної кількості сполуки С.
6. Спосіб одержання агомелатину з використанням сполуки А, за яким здійснюють обезводнення і ароматизацію сполуки А з одержанням бажаного продукту формули І:
 .
.
7. Спосіб одержання агомелатину з використанням сполуки А за п. 6, який відрізняється тим, що дегідрогенізуюча речовина, яку використовують у вищезгаданій ароматизації, являє собою дихлородиціанобензохінон.
8. Спосіб одержання агомелатину з використанням сполуки А за п. 6, який відрізняється тим, що кількість дегідрогенізуючої речовини становить 1-3 частки від молярної кількості сполуки А.
9. Спосіб одержання агомелатину з використанням сполуки А за п. 6, який відрізняється тим, що розчинник, який використовують в реакції, являє собою суміш толуолу і льодяної оцтової кислоти або суміш ацетонітрилу і льодяної оцтової кислоти, або льодяну оцтову кислоту.
10. Спосіб одержання сполуки В за п. 2, який відрізняється тим, що здійснюють обезводнення сполуки А в кислих умовах:
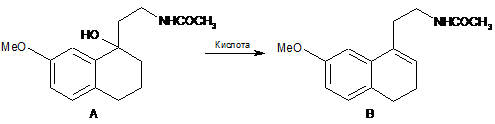 .
.
11. Спосіб одержання агомелатину з використанням сполуки В, за яким здійснюють взаємодію сполуки В з дегідрогенізуючою речовиною з одержанням бажаного продукту формули І:
 .
.
12. Спосіб одержання агомелатину з використанням сполуки В за п. 11, який відрізняється тим, що вищезгадана дегідрогенізуюча речовина являє собою дихлородиціанобензохінон.
13. Спосіб одержання агомелатину з використанням сполуки В за п. 11, який відрізняється тим, що кількість вищезгаданої дегідрогенізуючої речовини становить 1-3 частки від молярної кількості сполуки В.
14. Спосіб одержання агомелатину з використанням сполуки В за п. 11, який відрізняється тим, що органічний розчинник, який використовують в реакції, являє собою дихлорметан або толуол.
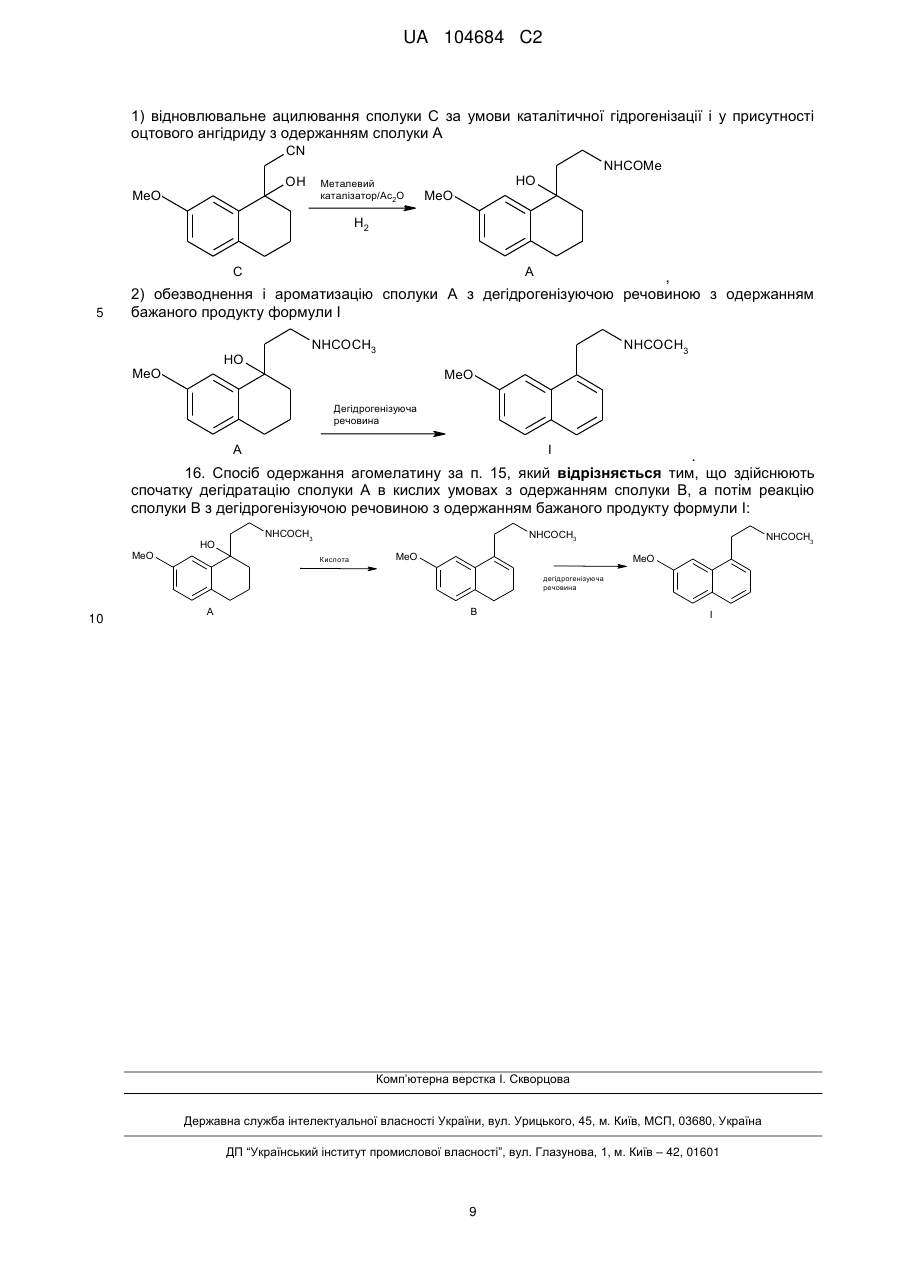
15. Спосіб одержання агомелатину, за яким здійснюють наступні етапи:
1) відновлювальне ацилювання сполуки С за умови каталітичної гідрогенізації і у присутності оцтового ангідриду з одержанням сполуки А
,
2) обезводнення і ароматизацію сполуки А з дегідрогенізуючою речовиною з одержанням бажаного продукту формули І
.
16. Спосіб одержання агомелатину за п. 15, який відрізняється тим, що здійснюють спочатку дегідратацію сполуки А в кислих умовах з одержанням сполуки В, а потім реакцію сполуки В з дегідрогенізуючою речовиною з одержанням бажаного продукту формули І:
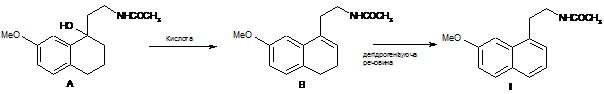 .
.
Текст
Реферат: Даний винахід стосується проміжних сполук для приготування агомелатину, а також способів їх приготування. Проміжною сполукою за даним винаходом для приготування агомелатину є сполука А, яка показана у нижченаведеній формулі. Також надані дві нові проміжні сполуки. Під час використання цих нових проміжних сполук для приготування агомелатину ними просто маніпулювати, ці сполуки є добре контрольованими, з високим ступенем чистоти, без складних операцій, таких як ректифікація і розділення за допомогою колонкової хроматографії, а також вони є придатними для промислового виробництва. Між тим, сам спосіб приготування двох нових проміжних сполук є простим і дає високий вихід, використовуючи тільки найчастіше використовуваний 7метокситетралон як первинний початковий матеріал, і проходить один етап реакції для UA 104684 C2 (12) UA 104684 C2 одержання проміжних сполук з наступним ще одним етапом перетворення проміжних сполук на бажаний продукт - агомелатин. Вищезазначені реакційні процеси значно спрощуються, вихід реакції покращується і долаються труднощі очищення попереднього способу, порівняно з існуючими способами приготування агомелатину. Зазвичай вихід згідно з даним винаходом становить більше 70 %. MeO HO A NHCOMe UA 104684 C2 5 10 15 20 25 30 35 40 Винахід стосується проміжних сполук для приготування агомелатину, а також способів їх приготування. Рівень техніки Агомелатин має хімічну структуру, продемонстровану Формулою (I), з хімічною назвою N-[2(7-метокси-1-нафтил)етил]ацетамід і торговою назвою Вальдоксан. Він має подвійні фармакологічні властивості і є не тільки агоністом рецепторів мелатонінергічної системи, але і антагоністом рецептора 5HT2C. Зазначені властивості забезпечують активність у центральній нервовій системі, зокрема, у лікуванні значної депресії, сезонних афективних розладів, порушень сну, серцево-судинних захворювань, захворювань системи травлення, безсоння і втоми, викликаних зміною часових поясів, порушень апетиту і ожиріння. Це перша антидепресивна речовина мелатонінового типу, яка може ефективно лікувати депресивні розлади, покращувати параметри сну і підтримувати статеві функції. Вона була схвалена ЄС 24 лютого 2009 року під торговою назвою Вальдоксан®/Тиманакс®. Беручи до уваги фармацевтичне значення цієї сполуки, дуже важливо одержувати цю сполуку за допомогою ефективного промислового синтетичного способу, який може бути легко конвертованим у великомасштабне виробництво у галузі, і одержати агомелатин з достатнім виходом і високим ступенем чистоти. Було зареєстровано багато способів синтезу агомелатину, які умовно можна розділити на чотири типи, в яких всі початкові матеріали є сполуками формули (II). Європейський опис винаходу до патенту EP0447285 повідомив про спосіб одержання агомелатину (I): взаємодія 7метокси-тетралону (II) з етил бромацетатом в реакції Реформатського, а потім дегідроароматизація із сіркою для одержання етилового ефіру (7-метокси-1-нафтил) оцтової кислоти (IV) з наступним гідролізом, ациловим хлоруванням, амоніфікацією, обезводненням і відновленням з виходом сполуки (VIII), яку наприкінці ацетилюють, щоб одержати агомелатин (I), як показано нижче: Однак, у наведеному вище способі є деякі недоліки, які включають: (1) для синтезу 2-(7-метокси-1-нафтил)етиламіну необхідно вісім етапів, що надає середній вихід менше 30 %; (2) під час конвертування вищезгаданого способу у промисловий масштаб стає важко проводити реакції в основному через погану відтворюваність першого етапу; перший етап включає взаємодію 7-метокси-тетралону (II) з етил бромацетатом відповідно до реакції Реформатського для виробництва етил (7-метокси-3,4-дигідронафталін-1(2Н)-іліден)ацетату, який як розчинник потребує бензол; враховуючи фактори навколишнього середовища, вищезгаданий етап не відповідає вимогам великомасштабного виробництва; і (3) наступний етап ароматизації етил (7-метокси-3,4-дигідронафталін-1(2Н)-іліден)ацетату звичайно є неповним і після омилення часто призводить до одержання суміші, з якої важко 1 UA 104684 C2 5 відокремити чистий продукт (IV). Китайський опис винаходу до патенту CN1680284 повідомив про інший спосіб синтезу агомелатину: взаємодія 7-метокси-тетралону (II) з ціано-оцтовою кислотою надає проміжну сполуку (IX), проміжну сполуку (IX) піддають дегідрогенізації у присутності каталізатора гідрогенізації Pd-C з алілметакрилатом як дегідрогенізуючою речовиною з наступним відновленням для одержання сполуки (VIII), і, наприкінці, сполуку (VIII) перетворюють на агомелатин (I) за допомогою ацетилювання. Загальний вихід становить приблизно 72 %, як продемонстровано нижче: 10 15 20 25 30 35 У наведеному вище способі є деякі недоліки: (1) у реакційному шляху використовуються деякі канцерогенні речовини, наприклад, високотоксична каталітична система бензиламін/гептанова кислота використовується у перетворенні формули (II) на формулу (IX); (2) під час перетворення формули (IX) на формулу (VII) як дегідрогенізуючу речовину використовують пропіл метакрилат, в результаті чого відбувається сильне забруднення навколишнього середовища, крім того, виявилось, що цей етап реакції насправді дає низький вихід і є важким для відтворювання; і (3) у процесі гідрогенізації під час перетворення формули (VII) на формулу (VIII) утворюється побічний продукт, що має формулу (XII); оскільки побічний продукт за характером схожий на бажаний продукт, і цей етап є передостаннім етапом, бажаний продукт важко очистити і втрати виходу після перекристалізації є великими. Враховуючи лікувальну цінність і хороші ринкові перспективи агомелатину, важливо синтезувати сполуку з формулою (I) ефективним для індустріалізації чином. Опис винаходу Однією метою цього винаходу є надання двох нових проміжних сполук для приготування агомелатину. Під час використання цих нових сполук для приготування агомелатину ними просто маніпулювати і легко обробляти (без складних операцій, таких як ректифікація і розділення за допомогою колоночної хроматографії), ці сполуки є добре контрольованими, з високим ступенем чистоти і виходу і є придатними для промислового виробництва. Іншою метою цього винаходу є створення способів приготування двох вищезазначених проміжних сполук і їх застосування. З цією метою в даному винаході використовуються наступні технічні розчини. Використовується сполука з формулою (A): 2 UA 104684 C2 . Спосіб приготування сполуки з формулою (A) являє собою відновлювальне ацилювання сполуки з формулою (C) за умови каталітичної гідрогенізації у присутності оцтового ангідриду. 5 10 15 Каталізатор, який використовують у перетворенні сполуки з формулою (С) на сполуку з формулою (A), є звичайним металевим каталізатором, таким як активований кобальт, активований нікель (Ni), переважно - Ренея-Ni; кількість каталізатора може становити 0,1-0,3 частки від кількості сполуки C за масою; кількість оцтового ангідриду становить 1-3 частки від молярної кількості сполуки C, переважно - 1-1,3 частки. Органічний розчинник, який використовується у цій реакції, є звичайно використовуваним органічним розчинником, таким як діоксан, ТГФ, ацетонітрил або оцтовий ангідрид, переважно - ТГФ. Оптимальна температура реакції становить 10-50 °C, переважно - 20-30 °C. Час реакції залежить від виявленої повної витрати реагентів, зазвичай становить 6-12 годин. Після завершення реакції процедура обробки реакційної суміші може бути виконана відповідно до загальноприйнятих у даній галузі техніки способів. Спосіб приготування агомелатину з використанням сполуки А, наданий в даному винаході, включає обезводнення і ароматизацію сполуки А для одержання бажаного продукту з формулою I: 20 25 30 35 . У перетворенні сполуки A на сполуку I шляхом ароматизації, як продемонстровано вище, дегідрогенізуюча речовина переважно являє собою дихлоро-диціанобензохінон (ДДХ), кількість вищезгаданої дегідрогенізуючої речовини переважно становить 1-3 частки від молярної кількості сполуки A, більш переважно - 1-1,3 частки. Органічний розчинник, який використовується в цій реакції, є звичайно використовуваним органічним розчинником, наприклад, одним із толуолу, діоксану, ТГФ, ацетонітрилу або крижаної оцтової кислоти, або будь-якої їх суміші, переважно - суміші толуолу і крижаної оцтової кислоти, суміші ацетонітрилу і крижаної оцтової кислоти, або крижаною оцтовою кислотою. Кількість вищезгаданого органічного розчинника звичайно становить 10-50 мл/г сполуки A. Температура реакції переважно становить 30-150 °C, більш переважно - 50-100 °C. Час реакції залежить від виявленої повної витрати реагентів, зазвичай становить від 30 хвилин до 12 годин. Після завершення реакції процедура обробки реакційної суміші може бути виконана відповідно до загальноприйнятих у даній галузі техніки способів. Використовується сполука з формулою (B): 3 UA 104684 C2 . Способом приготування сполуки з формулою (B) є обезводнення сполуки A в кислих умовах: 5 10 15 20 25 30 . Кислота, яка використовується у перетворенні сполуки A на сполуку B, є звичайно використовуваною кислотою, такою як галогеноводнева кислота, сірчана кислота, оцтова кислота тощо. Органічний розчинник, який використовується, є звичайно використовуваним органічним розчинником, таким як спирти, діоксан, ТГФ або ацетонітрил, переважно - спиртовим розчинником, наприклад, етилацетатом, ацетоном тощо. Кількість вищезгаданого органічного розчинника звичайно становить 10-50 мл/г сполуки A. Температура реакції зазвичай становить 20-40 °C, переважно - 0-30 °C. Час реакції залежить від виявленої повної витрати реагентів, зазвичай становить 1-3 години. Після завершення реакції процедура обробки реакційної суміші може бути виконана відповідно до загальноприйнятих у даній галузі техніки способів. Спосіб приготування агомелатину з використанням сполуки B, наданий в даному винаході, включає реакцію сполуки B з дегідрогенізуючою речовиною для одержання бажаного продукту з формулою I: . У перетворенні сполуки B на сполуку I дегідрогенізуюча речовина переважно являє собою дихлоро-диціанобензохінон (ДДХ), кількість вищезгаданої дегідрогенізуючої речовини переважно становить 1-3 частки від молярної кількості сполуки B, більш переважно - 1-1,3 частки. Органічний розчинник, який використовується в цій реакції, є звичайно використовуваним органічним розчинником, таким як дихлорметан, діоксан, ТГФ, ацетонітрил, крижана оцтова кислота тощо, переважно - дихлорметан або толуол. Кількість вищезгаданого органічного розчинника звичайно становить 10-50 мл/г сполуки B. Температура реакції переважно становить 0-50 °C, більш переважно 10-30 °C. Час реакції залежить від виявленої повної витрати реагентів, зазвичай становить від 30 хвилин до 6 годин. Після завершення реакції процедура обробки реакційної суміші може бути виконана відповідно до загальноприйнятих у даній галузі техніки способів. Спосіб приготування агомелатину включає наступні етапи: a. відновлювальне ацилювання сполуки C за умови каталітичної гідрогенізації і у присутності оцтового ангідриду для одержання сполуки A ; 4 UA 104684 C2 b. обезводнення і ароматизація сполуки A з дегідрогенізуючою речовиною для одержання бажаного продукту з формулою I 5 10 15 20 25 30 35 40 . У способі приготування агомелатину, згідно із даним винаходом, ми також можемо використовувати наступний шлях, який включає спочатку дегідратацію сполуки А в кислих умовах з одержанням сполуки B, а потім реакцію сполуки B з дегідрогенізуючою речовиною для одержання бажаного продукту з формулою I . Проміжна сполука формули C може бути зроблена шляхом конденсації формули II і ацетонітрилу у присутності каталізатора . Каталізатором, що використовується у перетворенні сполуки з формулою II на сполуку з формулою (С), є бутиллітій. І кількість каталізатора, і кількість ацетонітрилу становить 1-3 частки від молярної кількості сполуки II, переважно 1-1,3 частки. Органічний розчинник, який використовується в цій реакції, являє собою безводний органічний розчинник, такий як діоксан, ТГФ тощо, який потребує обробки для обезводнення або може бути придбаним безпосередньо у комерційних постачальників. Кількість вищезгаданого органічного розчинника звичайно становить 5-20 мл/г сполуки II. Оптимальна температура реакції становить від -80 °C до -50 °C, переважно - від -70 °C до -60 °C. Час реакції залежить від виявленої повної витрати реагентів, зазвичай становить від 1 хвилини до 3 годин. Після завершення реакції процедура обробки реакційної суміші може бути виконана відповідно до загальноприйнятих у даній галузі техніки способів. Сполуку C також можна одержати згідно зі способами, описаними у відповідній літературі, такій як Journal of Medicinal Chemistry, 1976, 19(6), 803. Реагенти і початкові матеріали, що використовуються у даному винаході, якщо не зазначене інше, є комерційно доступними. Перевагами даного винаходу є: винахід надає дві нові проміжні сполуки; коли ми використовуємо ці нові сполуки для приготування агомелатину, ними просто маніпулювати і легко обробляти, без складних операцій, таких як ректифікація і розділення за допомогою колонкової хроматографії, ці сполуки є добре контрольованими, з високим ступенем чистоти і є придатними для промислового виробництва. Між тим, сам спосіб приготування двох нових проміжних сполук є простим і дає високий вихід, використовуючи тільки найчастіше використовуваний 7-метокси-тетралон (II) як первинний початковий матеріал, і проходить один етап реакції для одержання проміжних сполук з наступним ще одним етапом перетворення проміжних сполук на бажаний продукт агомелатин. Вищезазначені реакційні процеси значно спрощуються, вихід реакції покращується, і долаються труднощі попереднього способу в очищенні, порівняно з існуючими способами приготування агомелатину. Зазвичай вихід, згідно з даним винаходом, становить більше 70 %. Приклади Наступні приклади використовуються для подальшої ілюстрації даного винаходу, але вони не призначені для обмеження обсягу винаходу будь-яким чином. 5 UA 104684 C2 Приклад 1: 1) Синтез 2-(1-гідроксил-7-метокси-1,2,3,4-тетрагідро-нафталін-1-іл)- ацетонітрилу (сполука C) 5 10 15 20 25 30 35 40 45 50 55 60 У реакційну посудину додають ацетонітрил (19,0 мл) і безводний ТГФ (50 мл), охолоджують до -70 °C сухим льодом/етанолом, потім повільно краплями додають розчин n-бутиллітію в nгексані (2,5 М, 142,0 мл). Після перемішування протягом півгодини за цієї температури розчин сполуки II (44,6 г) у безводному ТГФ (300 мл) додають повільно краплями і перемішують протягом 1 год. за тієї ж температури. Реакцію гасять додаванням насиченого водного розчину хлориду амонію (700 мл), екстрагують етилацетатом (350 мл x 3). Органічні шари об'єднують, промивають насиченим водним розчином NaCl (350 мл), сушать над безводним сульфатом натрію і концентрують, щоб одержати жовтуватий названий у заголовку продукт (54,3 г). Вихід: 98,3 %. Приклад 2: 2) Синтез N-[2-(1-гідроксил-7-метокси-1,2,3,4-тетрагідро-нафталін-1-іл)- етил]ацетаміду (сполука A) Сполуку C (54,3 г) розчиняють в ТГФ (500 мл) і потім додають оцтовий ангідрид (33,1 г) і Ренея-Ni (10 г). Реакційну суміш гідрогенізують під тиском водню, який підтримується на рівні 1,1 Мпа, за температури 30 °C до завершення реакції. Одержану суміш охолоджують до кімнатної температури, фільтрують і концентрують для видалення ТГФ. Залишок розбавляють етилацетатом (500 мл), промивають насиченим водним розчином NaHCO3 (150 мл), водою (150 мл) і насиченим водним розчином NaCl (150 мл). Одержану органічну фазу висушують над безводним сульфатом натрію, фільтрують і концентрують, щоб одержати жовтуватий продукт A (56,0 г). Вихід: 85 % 1HЯМР(CDCl3) δ: 1,77-1,98 (м, 4H), 1,92 (с, 3H), 2,01-2,11 (м, 2H), 2,28 (с, 0H), 2,67-2,77 (м, 2H), 3,28-3,50 (м, 2H), 3,80 (с, 3H), 6,32 (с, NH), 6,74-7,27 (м, 3H). ЕСІ-МС (m/z): 286,1 (M+Na). Т. пл.: 106-109 °C. Приклад 3: 3) Синтез N-[2-(7-метокси-1-нафтил)етил]ацетаміду (сполука I) Сполуку A (56,0 г) розчиняють в толуолі (500 мл) та оцтовій кислоті (50 мл), додають ДДХ (53,2 г) і суміш нагрівають за температури 40 °C протягом приблизно 5 год. Після завершення реакції суміш фільтрують і фільтрат промивають насиченим водним розчином NaHCO3 (250 мл x 2), водою (250 мл) і насиченим водним розчином NaCl (250 мл). Одержану органічну фазу висушують над безводним сульфатом натрію, фільтрують і розчинник випарюють. Залишок перекристалізовують із суміші етанол-вода (1:1), висушують в сушильній шафі, щоб одержати названий у заголовку продукт у вигляді білого порошку (43,8 г). Вихід: 85 %. 1HЯМР(CDCl3) δ: 1,922 (с, 3H), 3,21-3,24 (т, 2H), 3,56-3,61(к, 2H), 3,96 (с, 3H), 5,97 (с, 1H), 7,14-7,16 (к, 1H), 7,22-7,26 (м, 2H), 7,46-7,47 (м, 1H), 7,64-7,67 (м, 1H), 7,72-7,74 (д, 1H). ЕСІ -МС(m/z): 244,14 (M+H). Приклад 4: 1) Синтез 2-(1-гідроксил-7-метокси-1,2,3,4-тетрагідро-нафталін-1-іл)- ацетонітрилу (сполука C) У реакційну посудину додають ацетонітрил (9,5 мл) і безводний ТГФ (25 мл), охолоджують до -70 °C сухим льодом/етанолом, потім повільно краплями додають розчин n-бутиллітію в nгексані (2,5 M, 71,0 мл). Після перемішування протягом півгодини за цієї температури додають повільно краплями розчин сполуки II (22,3 г) у безводному ТГФ (150 мл) і перемішують протягом 1 год. за тієї ж температури. Реакцію гасять додаванням насиченого водного розчину хлориду амонію (350 мл), екстрагують етилацетатом (200 мл x 3). Органічні шари об'єднують, промивають насиченим водним розчином NaCl (200 мл), сушать над безводним сульфатом натрію і концентрують, щоб одержати жовтуватий названий у заголовку продукт (27,2 г). Вихід: 98,4 %. Приклад 5: 2) Синтез N-[2-(1-гідроксил-7-метокси-1,2,3,4-тетрагідро-нафталін-1-іл)- етил]ацетаміду (сполука A) Сполуку C (27,2 г) розчиняють в ТГФ (250 мл) і потім додають оцтовий ангідрид (15,6 г) і Ренея-Ni (4 г). Реакційну суміш гідрогенізують під тиском водню, який підтримується на рівні 1,1 Мпа, за температури 30 °C до завершення реакції. Одержану суміш охолоджують до кімнатної температури, фільтрують і концентрують для видалення ТГФ. Залишок розбавляють етилацетатом (250 мл), промивають насиченим водним розчином NaHCO3 (100 мл), водою (100 мл) і насиченим водним розчином NaCl (100 мл). Одержану органічну фазу висушують над 6 UA 104684 C2 5 10 15 20 25 30 безводним сульфатом натрію, фільтрують і концентрують, щоб одержати жовтуватий названий у заголовку продукт (28,0 г). Вихід: 85 %. 1HЯМР(CDCl3) δ: 1,77-1,98 (м, 4H), 1,92 (с, 3H), 2,01-2,11 (м, 2H), 2,28 (с, 0H), 2,67-2,77 (м, 2H), 3,28-3,50 (м, 2H), 3,80 (с, 3H), 6,32 (с, NH), 6,74-7,27 (м, 3H). ЕСІ-МС (m/z): 286,1 (M+Na). Т.пл.: 106-109 °C. Приклад 6: 3) Синтез N-[2-(7-метокси-3,4-дигідро-нафталін-1-іл)етил]ацетаміду (сполука B) Сполуку A (28,0 г) розчиняють в етилацетаті (300мл) з утворенням суспензії, до якої потім додають краплями концентрований розчин HCl (12 M, 13,3 мл) за кімнатної температури (КТ). Суспензія поступово стає прозорою. Реакційний розчин далі перемішують протягом 2 год. і виливають у воду (150 мл). Після розділення шарів органічну фазу промивають насиченим водним розчином NaHCO3 (150 мл x 2) і насиченим водним розчином NaCl (150 мл), висушують над безводним сульфатом натрію, фільтрують і концентрують, щоб одержати названий у заголовку продукт у вигляді масла (25,5 г). Вихід: 97,8 %. 1HЯМР(CDCl3) δ: 1,944 (с, 3H), 2,21-2,27 (м, 2H), 2,61-2,69 (м, 4H), 3,40-3,45 (м, 2H), 3,80 (с, 3H), 5,59 (с, NH), 5,90-5,93 (м, 1H), 6,68-7,05 (м, 3H). ЕСІ-МС (m/z): 268,3 (M+Na). Приклад 7: 4) Синтез N-[2-(7-метокси-1-нафтил)етил]ацетаміду (сполука I) Сполуку B (25,5 г) розчиняють в дихлорметані (250 мл), порціями додають ДДХ (26,1 г) і суміш перемішують протягом ночі за КТ. Після завершення реакції суміш фільтрують і фільтрат промивають насиченим водним розчином NaHCO3 (150 мл x 2), водою (150 мл) і насиченим водним розчином NaCl (150 мл). Одержану органічну фазу висушують над безводним сульфатом натрію, фільтрують і розчинник випарюють. Залишок перекристалізовують із суміші етанол-вода (1:1), висушують в сушильній шафі, щоб одержати білий порошок (46,4 г). Вихід: 91,8 %. 1HЯМР(CDCl3) δ: 1,922 (с, 3H), 3,21-3,24 (т, 2H), 3,56-3,61 (к, 2H), 3,96 (с, 3H), 5,97 (с, 1H), 7,14-7,16 (к, 1H), 7,22-7,26 (м, 2H), 7,46-7,47 (м, 1H), 7,64-7,67 (м, 1H), 7,72-7,74 (д, 1H). ЕСІ-МС(m/z): 244,14 (M+H). ФОРМУЛА ВИНАХОДУ 1. Сполука А наступної формули: MeO HO NHCOMe A 35 . 2. Сполука В наступної формули: NHCOCH3 MeO B . 3. Спосіб одержання сполуки А за п. 1, який відрізняється тим, що здійснюють відновлювальне ацилювання сполуки С за умови каталітичної гідрогенізації і у присутності оцтового ангідриду 7 UA 104684 C2 CN OH MeO NHCOMe Металевий каталізатор/Ас2О HO MeO H2 A C 5 . 4. Спосіб за п. 3, який відрізняється тим, що вищезгаданий металевий каталізатор являє собою Ni-Ренея, кількість якого становить 0,1-0,3 частки від кількості сполуки С за масою. 5. Спосіб за п. 3, який відрізняється тим, що кількість вищезгаданого оцтового ангідриду становить 1-1,3 частки від молярної кількості сполуки С. 6. Спосіб одержання агомелатину з використанням сполуки А, за яким здійснюють обезводнення і ароматизацію сполуки А з одержанням бажаного продукту формули І: MeO HO NHCOCH3 NHCOCH3 MeO Дегідрогенізуюча речовина A 10 15 I . 7. Спосіб одержання агомелатину з використанням сполуки А за п. 6, який відрізняється тим, що дегідрогенізуюча речовина, яку використовують у вищезгаданій ароматизації, являє собою дихлородиціанобензохінон. 8. Спосіб одержання агомелатину з використанням сполуки А за п. 6, який відрізняється тим, що кількість дегідрогенізуючої речовини становить 1-3 частки від молярної кількості сполуки А. 9. Спосіб одержання агомелатину з використанням сполуки А за п. 6, який відрізняється тим, що розчинник, який використовують в реакції, являє собою суміш толуолу і льодяної оцтової кислоти або суміш ацетонітрилу і льодяної оцтової кислоти, або льодяну оцтову кислоту. 10. Спосіб одержання сполуки В за п. 2, який відрізняється тим, що здійснюють обезводнення сполуки А в кислих умовах: MeO HO NHCOCH3 Кислота NHCOCH3 MeO A 20 В . 11. Спосіб одержання агомелатину з використанням сполуки В, за яким здійснюють взаємодію сполуки В з дегідрогенізуючою речовиною з одержанням бажаного продукту формули І: NHCOCH3 MeO NHCOCH3 MeO Дегідрогенізуюча речовина В 25 30 I . 12. Спосіб одержання агомелатину з використанням сполуки В за п. 11, який відрізняється тим, що вищезгадана дегідрогенізуюча речовина являє собою дихлородиціанобензохінон. 13. Спосіб одержання агомелатину з використанням сполуки В за п. 11, який відрізняється тим, що кількість вищезгаданої дегідрогенізуючої речовини становить 1-3 частки від молярної кількості сполуки В. 14. Спосіб одержання агомелатину з використанням сполуки В за п. 11, який відрізняється тим, що органічний розчинник, який використовують в реакції, являє собою дихлорметан або толуол. 15. Спосіб одержання агомелатину, за яким здійснюють наступні етапи: 8 UA 104684 C2 1) відновлювальне ацилювання сполуки С за умови каталітичної гідрогенізації і у присутності оцтового ангідриду з одержанням сполуки А CN NHCOMe OH MeO Металевий каталізатор/Ас2О HO MeO H2 A C 5 , 2) обезводнення і ароматизацію сполуки А з дегідрогенізуючою речовиною з одержанням бажаного продукту формули І HO MeO NHCOCH3 NHCOCH3 MeO Дегідрогенізуюча речовина A I . 16. Спосіб одержання агомелатину за п. 15, який відрізняється тим, що здійснюють спочатку дегідратацію сполуки А в кислих умовах з одержанням сполуки В, а потім реакцію сполуки В з дегідрогенізуючою речовиною з одержанням бажаного продукту формули І: MeO HO NHCOCH3 NHCOCH3 Кислота MeO NHCOCH3 MeO дегідрогенізуюча речовина 10 A ВI Комп’ютерна верстка І. Скворцова Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601 9
ДивитисяДодаткова інформація
Назва патенту англійськоюAgomelatine intermediates and preparation method thereof
Автори англійськоюZhang, Peng, Huang, Yu, Yuan, Zhedong, Shan, Hanbin, Yu, Xiong
Автори російськоюЖанг Пенг, Хуанг Йу, Йуан Жедонг, Шан Ханбин, Йу Ксионг
МПК / Мітки
МПК: C07C 231/12, A61P 25/06, C07C 233/18, A61P 25/24, A61P 25/22, C07C 231/06, A61K 31/165, A61P 25/20
Мітки: варіанти, проміжна, одержання, агомелатину, спосіб, сполука
Код посилання
<a href="https://ua.patents.su/11-104684-promizhna-spoluka-dlya-oderzhannya-agomelatinu-varianti-i-sposib-oderzhannya-varianti.html" target="_blank" rel="follow" title="База патентів України">Проміжна сполука для одержання агомелатину (варіанти) і спосіб її одержання (варіанти)</a>
Водорозчинні проліки блокованих спиртів чи фенолів, фармацевтична композиція на їх основі, спосіб їх одержання, проміжна сполука (варіанти) та спосіб її одержання (варіанти)

Номер патенту: 73479
Опубліковано: 15.08.2005
Автори: Сафаді Мухаммед С., Стелла Валентіно Дж., Георг Інгрід Гунда, Зігмунт Джан Дж.
МПК: C07F 9/6515, C07C 323/12, C07K 7/64, A61K 31/7042, C07D 311/72, A61P 25/00, A61K 31/665, A61K 31/7048, C07F 9/09, A61K 31/661, C07F 9/6561, C07C 319/00, C07F 9/655, C07H 17/04, A61K 38/00, A61K 31/437, A61K 31/675, A61K 31/355, A61P 35/00, C07D 491/052, A61K 31/10, A61K 31/352
Мітки: варіанти, спосіб, проміжна, фармацевтична, одержання, фенолів, композиція, сполука, проліки, блокованих, водорозчинні, основі, спиртів
Формула / Реферат:
1. Сполука відповідно до формули І:, IдеR-O- - залишок спиртвмісної або фенолвмісної фармацевтичної сполуки, крім таксолу і його похідних,R1 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота,R2 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота, іn являє...
Спосіб одержання 1н-індол-3-гліоксамідів, заміщених в положенні 4 (варіанти), проміжна сполука та спосіб її одержання (варіанти)
Номер патенту: 56245
Опубліковано: 15.05.2003
Автори: Кхау Вьєн Ван, Мартінеллі Майкл Джон, Полак Джозеф Метью
МПК: C07D 209/22, C07D 209/14, C07D 209/08
Мітки: положенні, варіанти, проміжна, сполука, 1н-індол-3-гліоксамідів, спосіб, одержання, заміщених
Формула / Реферат:
1. Спосіб одержання сполуки формули І або фармацевтично прийнятної солі чи проліків сполуки формули І:, (І)деR1 вибрано із групи, яка складається з:С7-С20 алкілу,, та,деR10 вибрано із групи, яка складається з галоїду, С1-С10 алкілу, С1-С10 алкоксигрупи, -S-(С1-С10 алкілу) та галоїдного (С1-С10) алкілу, a t - ціле число від 0 до 5 включно;R2 вибрано із групи, яка складається...
Спосіб одержання периндоприлу або його солей (варіанти), проміжна сполука, спосіб її одержання (варіанти) та її застосування

Номер патенту: 83139
Опубліковано: 10.06.2008
Автори: Мерславік Марйо, Смід Янья, Томсік Зденка
МПК: C07D 209/42, A61P 9/12
Мітки: варіанти, сполука, спосіб, одержання, солей, периндоприлу, проміжна, застосування
Формула / Реферат:
1. Спосіб одержання периндоприлу формули І або його фармацевтично прийнятних солей, Iякий відрізняється тим, що хлорангідрид формули III або його сіль IIIпіддають реакції з (2S,3аS,7аS)-октагідроіндол-2-карбоновою кислотою або з її похідною формули IV
Похідні 2,3-(1н, 4н)-хіноксаліндіону, фармацевтична комозиція, спосіб лікування, проміжна сполука (варіанти), спосіб одержання сполуки (варіанти)

Номер патенту: 58534
Опубліковано: 15.08.2003
Автори: Вейт Дейвід Чарльз, Готьє Елізабет Колетт Луїз, Крук Роберт Джеймс, Стоубі Алан
МПК: A61P 25/28, A61P 25/10, A61P 25/30, A61K 31/498, A61P 25/16, A61P 27/16, A61P 9/10, A61P 25/00, A61P 43/00, A61P 25/02, C07D 401/14, A61P 25/06
Мітки: 2,3-(1н, варіанти, сполуки, похідні, сполука, проміжна, 4н)-хіноксаліндіону, фармацевтична, спосіб, комозиція, лікування, одержання
Формула / Реферат:
1. Похідні 2, 3 – (1Н, 4Н) – хіноксаліндіону загальної формули (І)або її фармацевтично прийнятна сіль або сольват.2. Сполука за п. 1, яка відрізняється тим, що її чистота становить щонайменше 90 % (за масою).3. Сполука за п. 2, яка відрізняється тим, що її чистота становить щонайменше 95 % (за масою).4. Сполука за п. 3, яка відрізняється тим, що її чистота становить щонайменше 98 % (за масою).5....
Спосіб одержання невірапіну, проміжна сполука (варіанти)

Номер патенту: 80140
Опубліковано: 27.08.2007
Автори: Ло Юонг Сек, Бозуелл Роберт Фредерік Джр., Гаптон Бернард Франклін
МПК: C07D 213/80, C07D 213/85, C07D 471/10, C07D 213/82, C07D 213/803, C07D 471/14, C07D 213/78
Мітки: сполука, невірапіну, спосіб, варіанти, одержання, проміжна
Формула / Реферат:
1. Спосіб одержання невірапіну, який полягає в тому, що:(а) піддають взаємодії 2-гало-3-піридинкарбонітрил формули,де X означає атом фтору, хлору, брому або йоду, переважно хлору або брому, із циклопропіламіном з одержанням 2-(циклопропіламіно)-3-піридинкарбонітрилу;(б) гідролізують 2-(циклопропіламіно)-3-піридинкарбонітрил з одержанням...
Попередній патент: Спосіб одержання шаруватого метал-інтерметалідного композиційного матеріалу
Наступний патент: Спосіб виготовлення спіненого матеріалу з використанням розплавленого шлаку
Випадковий патент: Ківш драглайна