Спосіб одержання периндоприлу або його солей (варіанти), проміжна сполука, спосіб її одержання (варіанти) та її застосування





Формула / Реферат
1. Спосіб одержання периндоприлу формули І або його фармацевтично прийнятних солей
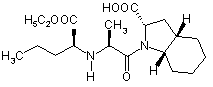 , I
, I
який відрізняється тим, що хлорангідрид формули III або його сіль
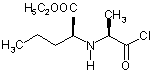 III
III
піддають реакції з (2S,3аS,7аS)-октагідроіндол-2-карбоновою кислотою або з її похідною формули IV
 , IV
, IV
де R позначає захисну групу, таку як бензилова, трет-бутилова, триметилсилілова група, або водень або катіон, такий як калій, літій або натрій, а R1 позначає водень або захисну групу, таку як триметилсилілова група,
з утворенням сполуки формули V
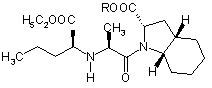 , V
, V
де R має значення, зазначене вище,
яке, у випадку, якщо R не є воднем, перетворюють у периндоприл формули І шляхом гідрогенолізу або гідролізу.
2. Спосіб за п. 1, який відрізняється тим, що хлорангідрид формули III
III
одержують шляхом перетворення з N-((S)-1-карбетоксибутил)-L-аланіну формули II
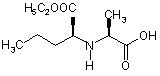 . II
. II
3. Спосіб за п. 1, який відрізняється тим, що в ньому використовують сіль хлорангідриду формули III.
4. Спосіб за п. 3, який відрізняється тим, що в ньому використовують гідрохлорид хлорангідриду III.
5. Спосіб за пп. 1-4, який відрізняється тим, що реакцію утворення пептидного зв'язку проводять у присутності основи в органічних розчинниках, таких як ацетон, ацетонітрил, діоксан, хлороформ, метиленхлорид, тетрагідрофуран, або їхньої комбінації з водою.
6. Спосіб за пп. 1-5, який відрізняється тим, що реакцію утворення пептидного зв'язку проводять при температурі від -20 °С до +20 °С, переважно при температурі від 0 °С до -20 °С.
7. Сполука формули III у формі гідрохлориду
. III
8. Спосіб одержання сполуки за п. 7, який відрізняється тим, що в ньому використовують сполуку формули II
ІІ
і реагенти для одержання хлоридів, такі як пентахлорид фосфору, трихлорид фосфору, фосфорилхлорид і тіонілхлорид.
9. Спосіб одержання сполуки за п. 7, який відрізняється тим, що в ньому використовують сполуку формули ІІ
ІІ
у формі гідрохлориду.
10. Спосіб за п. 8, який відрізняється тим, що реакцію проводять в інертному розчиннику, переважно в метиленхлориді.
11. Спосіб за п. 8, який відрізняється тим, що реакцію проводять при температурі від -30 °С до +30 °С, переважно від 0 °С до +10 °С.
12. Спосіб одержання периндоприлу або його фармацевтично прийнятних солей, який відрізняється тим, що в ньому використовують сполуку за п. 7.
13. Застосування сполуки за п. 7 для одержання периндоприлу або його фармацевтично прийнятних солей.
Текст
1. Спосіб одержання периндоприлу формули І або його фармацевтично прийнятних солей HOOC H H5C2OOC CH 3 2 3 H5 C2 OOC H 3C 83139 H5 C2 OOC CH 3 Cl N H O . III 8. Спосіб одержання сполуки за п. 7, який відрізняється тим, що в ньому використовують сполуку формули II H5 C2 OOC CH 3 H 3C 4 OH N H O ІІ і реагенти для одержання хлоридів, такі як пентахлорид фосфору, три хлорид фосфору, фосфорилхлорид і тіонілхлорид. 9. Спосіб одержання сполуки за п. 7, який відрізняється тим, що в ньому використовують сполуку формули ІІ Даний винахід відноситься до області органічної хімії, зокрема, до способу одержання периндоприла і до солі, яку використовують як проміжний продукт при синтезі периндоприлу. Периндоприл, переважно його mpemбутиламіновая сіль, є сполукою, що має здатність інгібувати ангіотензин-перетворюючий фермент, (АПФ). Існує потреба в поліпшеному способі синтезу периндоприлу для використання в промислових масштабах, за допомогою якого можна досягти високої чистоти і високого виходу кінцевого продукту без утворення небажаних побічних продуктів. Периндоприл, хімічне найменування якого (2S,3aS,7aS)-1-((2S)-2-(((1 S)-I-(етоксикарбоніл)бутил)аміно)-1-оксопропіл)октагідро-1Н-індол-2карбонова кислота, розкрите [у EP 49 658]. Описаний у цьому джерелі синтез є багатостадійним процесом і включає поділ ізомерів колончатою хроматографією. [EP 308 341] описує поліпшений спосіб великомасштабного синтезу периндоприлу у формі трет-бутиламінової солі. /7-толуолсульфонову сіль бензилового ефіру (28,За,7а)-октагідроіндол-2карбонової кислоти і N-((S)-1-карбетоксібутил)-і_аланін піддають взаємодії в присутності триетиламіну, N1N-дициклогексилкарбодиіміду і 1гідроксибензотріазолу. Після завершення реакції одержують бензиловий ефір периндоприлу, який зжижають, піддають ліофільному сушінню і перетворюють у сіль із mpem-бутиламіном у етилацетаті. У [WO 01/58868] розкрите використання більш низького молярного співвідношення між птолуолсульфоновою сіллю бензилового ефіру (2S,3a,7a)-октагідроіндол-2-карбонової кислоти, і N((S)-1-карбетоксибутил)-L-аланіном, триетиламіном, N,N-дициклогексилкарбодиімідом і 1- гідро H 3C CH 3 OH N H O ІІ у формі гідрохлориду. 10. Спосіб за п. 8, який відрізняється тим, що реакцію проводять в інертному розчиннику, переважно в метиленхлориді. 11. Спосіб за п. 8, який відрізняється тим, що реакцію проводять при температурі від -30°С до +30°С, переважно від 0°С до +10°С. 12. Спосіб одержання периндоприлу або його фармацевтично прийнятних солей, який відрізняється тим, що в ньому використовують сполуку за п. 7. 13. Застосування сполуки за п. 7 для одержання периндоприлу або його фармацевтично прийнятних солей. ксибензотріазолом з метою зменшити утворення побічних продуктів реакції між бензиловим ефіром периндоприлу і ^^діциклогексилкарбодиімідом. Незважаючи на низьке співвідношення, побічні продукти все ж таки присутні. У [WO 03/064388] розкритий синтез периндоприлу без побічних продуктів реакції з N.Nдициклогексилкарбодиімідом. Реакцію проводять між (2S,3a,7a)-октагідроіндол-2-карбоновою кислотою і хлоридом-N-((S)-1-карбетоксибутил)-і_аланіну, захи щеного N-ацилом. Хлорангідрид одержують з використанням тіоніл хлориду і попередньо захищеного N-((S)-1-карбетоксибутил)-Lаланіну. Недоліком зазначеного способу є низький вихід, одержуваний після конденсації і видалення захисної групи, а саме усього лише від 35 до 55%. Було несподівано виявлено, що за допомогою даного винаходу, спрямованого на розробку методу синтезу периндоприлу з використанням незахи щеного-N-((S)-1-карбетоксибутил)-L-аланіну у формі його хлорангідриду або солі хлорангідриду можна уникнути вищезгаданих недоліків. До основних переваг даного винаходу відноситься одержання периндоприлу високої якості і чистоти з високим виходом без побічних продуктів. Винахід відноситься, насамперед, до способу одержання периндоприлу формули І або його фармацевтично прийнятних солей, HOOC H H5C2OOC CH 3 H 3C N N H O H ,I що характеризується тим, що хлорангідрид формули III або його сіль 5 H5 C2 OOC H 3C 83139 CH 3 Cl N H O III піддають реакції з (28,3а,7а)-октагідроіндол-2карбоновою кислотою формули IV, H COOR H N R1 , IV де R позначає захисну гр упу, таку як бензилова, mpem-бутилова, триметилсилілова група, або водень або катіон, такий як калій, літій або натрій, a R1 позначає водень або захисну гр упу, таку як триметилсилілова група, с утворенням сполуки формули V, ROOC H5 C2OOC H CH3 H 3C N N H O H ,V де R має значення, зазначене вище, яке, у випадку якщо R не є воднем, перетворюють у периндоприл формули І шляхом гідрогенолізу або гідролізу. Після цієї реакції периндорпил можуть перетворювати в бажану сіль. Переважно, щоб у конкретному випадку реакції хлорангідриду формули III з (28,За,7а)-октагідроіндол-2-карбоновою кислотою формули IV R не був бензилом. Також переважно, щоб хлорангідрид формули III H5 C2 OOC CH 3 H 3C Cl N H O . III був о триманий шляхом перетворення з N-((S)1-Kap6eTOKcn6yTnn)-L-аланіну формули Il H5 C2 OOC CH 3 H 3C OH N H O ІІ Також було показано, у способі за винаходом переважно використовувати сіль хлорангідриду формули III, переважно, гідрохлорид. Ці солі легше очищати, ніж сполуку III, завдяки чому можна використовува ти високоочищену вихідн у речовину. Це, у свою чергу, дозволяє одержати більш чистий кінцевий продукт. Реакцію конденсації, тобто утворення пептидного зв'язку між сполукам формули III і сполуками формули IV, переважно проводити в органічних розчинниках, таких як ацетон, ацетонітрил, діоксан, хлороформ, метиленхлорид, тетрагідрофуран або, можливо, у їхню суміші з водою, переважно, у присутності основи, при температурі від -20°C до 6 +20°C, переважно, при температурі від 0°C до 20°C. Сполука формули І і його фармацевтично прийнятні солі виділяють відомими або звичайними способами. Сполуку можна виділити у формі трет-бутиламінової солі, що відома з [EP 308 341] і може існувати у вигляді різних поліморфних форм, таких як, наприклад, розкриті в [WO 01/87835] (поліморфна форма альфа), [WO 01/87836] (поліморфна форма бета) і [WO 01/83439] (поліморфна форма гама). Сполука І може бути також виділена у вигляді інших фармацевтично прийнятних солей, таких як, наприклад, аргінінова сіль, розкрита в [WO 03/087050]. Винахід відноситься також до сполуки формули III H5 C2 OOC CH 3 H 3C Cl N H O . III у формі його солі. Ця сіль переважно являє собою гідрохлорид. Сіль за винаходом можна одержати відповідним перетворенням сполуки III. Однак, цю сіль також можна одержати безпосередньо із сполуки формули Il у формі солі, переважно у формі гідрохлориду. Гідрохлорид сполуки формули Il описаний у [Tetrahedron Lett. 1982, 23 (16) 1677-1680, Drug Design and Discovery 1992, 9 (1) 11-28, і в EP 1 403 278]. Переважно на першій стадії здійснювати перетворення сполуки II, наприклад, з використанням HCI у дихлорметані, у його сіль, а потім перетворювати цю сіль у сполук у III за винаходом. Сполуку формули III можна одержати з N-((8)1-карбетоксібутил)-L-аланіну, тобто, сполуку II, використовуючи реагенти для одержання хлоридів, такі як пентахлорид фосфору, три хлорид фосфору, хлористий фосфорил або хлористий тіоніл. Для одержання хлориду можна використовувати надлишок реагенту від 10 до 50%, причому реакцію звичайно проводять в інертному розчиннику, такому як метиленхлорид, при температурі від 30°C to +30°C, переважно від 0°C до +10°C. Сполуку формули III у формі солі, переважно у формі гідрохлориду, можна виділити частковим випарюванням розчинника й осадженням за допомогою осаджуючого реагенту, такого як різні прості ефіри або вуглеводні. Крім того, винахід також відноситься до способу одержання периндоприлу або його фармацевтично прийнятних солей, при якому використовують сіль за винаходом. Нарешті, винахід відноситься також до використання солі за винаходом для одержання периндоприлу або його фармацевтично прийнятних солей. Публікації, що відносяться до реагентів, які використовуються у ви щезгаданих способах, приведені нижче. 7 83139 Похідна L-аланіну формули Il розкрита в [EP 308 340, EP 308 341, EP 309 324, EP 1 362 845, EP 1 400 431, EP 1 400 531, WO 01/56353 і WO 01/56972]. Октагідроіндол-2-карбонова кислота формули IV добре відома з [EP 37231, EP 308339, EP 308341, EP 1323729, US 5258525 і EP 1338591]. Даний винахід проілюстрований, але не обмежений наступними прикладами. Приклади Приклад 1: Одержання гідрохлориду N-((S)-1карбетоксібутил )-L-аланіл хлориду. До суспензії М-((8)-1-карбетоксібутил)-і.аланіну (13,2г) у ди хлорметані (80 мл) додавали HCI при перемішуванні при температурі від 20 до 25°C до одержання прозорого розчину. Прозорий розчин прохолоджували до температури від -5°C до 0°C, додавали PCIs (12,9г) і продовжували перемішування при тій же температурі протягом наступних п'яти годин. Приблизно половину дихлорметану випарювали із суспензії, додавали диізопропіловий ефір (180ml) і продовжували перемішування при температурі від 10 до 25°C протягом наступної години. Кристали, що випали в осад, відфільтровували і промивали диізопропіловим ефіром (90мл). Одержували гідрохлорид N-((S)-Ікарбетоксібутил)-L-аланілхлориду (15,1г). Температурний інтервал плавлення = 89-98°C (розкладання), IK (см -1): 2972, 1793, 1742, 1470 і 1206. 1 Н ЯМР (300 МГц, ДМСО-d6) d 0.90 (3Н, t, J=7.15 Гц), 1.24 (3H, t J=7.14 Гц), 1.40 (2H, m), 1.51 (3H, d, J = 7.14 Гц) , 1.86 (2H, m) , 4.07 (2Н, m) , 4.21 (2Н, m, J = 7.14 Гц), і 9.71 (2Н, b). Приклад 2: Одержання периндоприлу ербуміну (2-метилпропан-2-амінової солі ((2S,3a,7a)-1 ((2S)-2-(((1 S)-1 -(етоксикарбоніл)бутил)аміно)-1 оксопропіл)октагідро-1 Н-індол-2-карбонової кислоти). Триметилхлорсилан (2,86мол) і триетиламін (3,08мол) додали до (28,3а,7а)-октагідроіндол-2карбонової кислоти (3,72г) у ди хлорметані (60мл) при 20-25°C і перемішували при 20-25°C протягом двох годин. Через дві години туди додавали триетиламін (2,8мл), і прохолоджували суспензію до 15°C. Далі туди додавали суспензію гідрохлориду М-((Б)-1-карбетоксибутил)- L-аланілхлориду (5,5г) у ди хлорметані (60 мл), попередньо охолоджену до -15°C, і продовжували перемішування при 15°C протягом двох годин. Реакційний розчин нагрівали до 0°C, додавали 25мл розчину NaOH (0,8г) і доводили рН до 4,2 за допомогою 20% розчину NaOH. Органічну фазу відокремлювали, а водяну фракцію повторно екстрагували дихлорметаном (20мл). Об'єднані дихлорметанові фракції випарювали, осад розчиняли у етилацетаті (100мл), частину, яка не розчинилась, відфільтровували, а до фільтрату додавали трет-бутиламін (2.2мл). Осаджені кристали розчиняли при температурі кипіння розчину, прозорий розчин прохолоджували до 10-20°C і продовжували перемішування протягом двох годин. Через дві години осаджені кристали відфільтровували, промивали етилаце 8 татом (12мл) і сушили при 35-40°C у повітряній сушарці. Одержували периндоприл ербумін (7,1г; 80%) у формі а, з чистотою більше 99%, із вмістом окремих домішок не більше 0,1% кожної. Приклад 3: Одержання периндоприлу ербуміну (2-метилпропан-2-амінової солі ((2S,3a,7a)-1 ((2S)-2-(((1S)-І-(етоксікарбоніл)бутил)аміно)-1 оксопропіл)октагідро-1Н-індол-2-карбонової кислоти) з використанням ізопропіл ацетату. Триметилхлорсілан (2,92мл) і триетиламін (3,2мл) додавали до (2S,3a,7a)-октагідроіндол-2карбонової кислоти (3,72г) у ди хлорметані (40мл) при 20-25°C і перемішували при 20-25°C протягом двох годин. Через дві години туди додавали триетиламін (2,77мл), суспензію прохолоджували до 15°C, туди ж додавали попередньо охолоджену до -15°C суспензію гідрохлориду N-((S)-1карбетоксібутил)-L-аланілхлориду (5,5г) у ди хлорметані (80мл) і продовжували перемішування при 15°C протягом двох годин. Реакційний розчин нагрівали до 0°C, осад гідрохлориду триетиламіну відфільтровували і промивали дихлорметаном (10мл), потім додавали до фільтрату 33мл води з розчиненим у ній 0,8г NaOH, і потім рН доводили до 4,2 20% розчином NaOH. Органічну фазу відокремлювали, а водяну фракцію двічі екстрагували дихлорметаном (2 х 20 ml). Об'єднані дихлорметанові фракції випарювали, осад розчиняли у ізопропілацетаті (125мл), частину, яка не розчинилась, відфільтровували і додавали до фільтрату mpem-бутиламіну (2,2мл). Кристали, що випали в осад, розчиняли при температурі кипіння розчину, прозорий розчин прохолоджували до 10-20°C і проводили перемішування протягом двох годин. Через дві години кристали, що випали, відфільтровували, промивали ізопропілацетатом (15мл) і сушили при 35-40°C у повітряній сушилці. Був отриманий периндоприл ербумін (7.65г; 86%) у формі а с чистотою більше 99% із вмістом окремих домішок не більше 0,1% кожної. Приклад 4: Одержання периндоприл ербуміну (2-метилпропан-2-амінової солі ((2S,3a,7a)-1 ((2S)-2-(((l)-1 -(етоксикарбоніл)бутил)аміно)-1оксопропіл)октагідро-1Н-/ндол-2-карбонової кислоти) з використанням N,N-диметилформаміду. Триметилхлорси лан (2,92мл) і триетиламін (3,2мл) додавали до (28,3а,7а)-октагідроіндол-2карбонової кислоти (3,77 г) у дихлорметані (40 мл) при 20-25°C і перемішували при 20-25°C протягом двох годин. Через дві години додавали триетиламін (2,77мл), суспензію прохолоджували до -15°C, туди ж додавали попередньо охолоджену до -15°C суспензію гідрохлориду N-((S)-I-карбетоксибутил)L-аланілхлориду (5,5г) у дихлорметані (40мл) і перемішували при -15°C протягом двох годин. Реакційний розчин нагрівали до 0°C, випавший в осад гідрохлоридтриетиламін відфільтровували і промивали дихлорметаном (10мл), потім додавали до фільтрату 33мл води (з розчиненим у ній 0,8г NaOH, а потім доводили рН до 4,2 20%-ним розчином NaOH. Органічну фазу відокремлювали, а водяну фракцію двічі екстрагували дихлорметаном (2х20мл). Об'єднані дихлорметанові фракції 9 83139 випарювали, залишок розчиняли в N, Nдиметилформаміде (90мл). Частину, що нерозчинилася, відфільтровували і додавали до фільтрату mpem-бутиламін (2,2мл). Кристали, що випали в осад, розчиняли при 80°C, прозорий розчин прохолоджували до 10-20°C і перемішували протягом двох годин. Через дві години кристали, що випали Комп’ютерна в ерстка В. Клюкін 10 в осад, відфільтровували, промивали N,Nдиметилформамідом (9мл) і висушили при 3540°C у повітряній сушил ці. Був отриманий периндоприл ербумін (6,9г; 78%) у формі а з чистотою більше 99% із вмістом окремих домішок не більше 0.1% кожної. Підписне Тираж 26 прим. Міністерство осв іт и і науки України Держав ний департамент інтелектуальної в ласності, вул. Урицького, 45, м. Київ , МСП, 03680, Україна ДП “Український інститут промислов ої в ласності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of perindopril and salts thereof (variants), intermediate, process for the preparation (variants) and use thereof
Автори англійськоюMerslavic Marjo, Smid Janja, Tomsic Zdenka
Назва патенту російськоюСпособ получения периндоприла или его солей (варианты), промежуточное соединение, способ его получения (варианты) и его применение
Автори російськоюМерславик Марйо, Смид Янья, Томсик Зденка
МПК / Мітки
МПК: A61P 9/12, C07D 209/42
Мітки: периндоприлу, варіанти, проміжна, одержання, застосування, спосіб, сполука, солей
Код посилання
<a href="https://ua.patents.su/5-83139-sposib-oderzhannya-perindoprilu-abo-jjogo-solejj-varianti-promizhna-spoluka-sposib-oderzhannya-varianti-ta-zastosuvannya.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання периндоприлу або його солей (варіанти), проміжна сполука, спосіб її одержання (варіанти) та її застосування</a>
Способи одержання комбретастатину та його солей, проміжна сполука

Номер патенту: 76079
Опубліковано: 15.06.2006
Автори: Казімір Жан-Поль, Лавінь Мішель, Мютті Стефан, Малєжонок Іріна
МПК: C07C 213/00, C07D 263/06
Мітки: проміжна, способи, солей, комбретастатину, одержання, сполука
Формула / Реферат:
1. Спосіб одержання комбретастатину наступної загальної формули (І):, (I)в якій А означає аміногрупу, який відрізняється тим, що після конденсації за методом Віттіга в присутності броміду або хлориду (3,4,5-триметоксибензил)-трифенілфосфонію і 3-нітро-4-метоксибензальдегіду або 3,4,5-триметоксибензальдегіду з сіллю (4-метокси-3-нітробензил)-трифенілфосфонію...
Бензамід 3- або 4-заміщеного 4-(амінометил)піперидину, спосіб його одержання (варіанти), фармацевтична композиція на його основі, спосіб її одержання, проміжна сполука та спосіб її одержання

Номер патенту: 67745
Опубліковано: 15.07.2004
Автори: Суркін Мішель, де Клеін Мішель Анна Жозеф, Босманс Жан-Поль Рене Марі Андре
МПК: A61P 43/00, A61P 1/10, A61P 1/00, A61K 31/4427, C07D 405/12, A61K 31/4525, A61K 31/4545, A61K 31/497, A61K 31/502, C07D 211/42, A61K 31/501, C07D 401/06, C07D 405/06, C07D 405/14, A61K 31/453, A61K 31/443, A61K 31/454, C07D 211/44, A61K 31/445
Мітки: основі, фармацевтична, спосіб, проміжна, сполука, варіанти, композиція, 4-(заміщеного, 4-(амінометил)піперидину, бензамід, одержання
Формула / Реферат:
1. Сполука бензаміду 3- або 4-заміщеного 4-(амінометил)піперидину формули (I), (I)її стереоізомерні форми, N-оксиди або фармацевтично прийнятні солі з кислотами або лугами, деR1 і R2, взяті разом, утворюють бівалентний радикал формули-O-CH2-O- (a-1),-O-CH2-CH2- (a-2),-O-CH2-CH2-O- (a-3),-O-CH2-CH2-CH2-...
Заміщені бензоїлом серин-аміди, їх застосування як гербіцидів, спосіб одержання (варіанти), проміжна сполука, засіб та спосіб одержання засобу, спосіб боротьби з небажаним ростом рослин

Номер патенту: 82454
Опубліковано: 10.04.2008
Автори: Хупе Айке, Цагар Сірілл, Штельцер Франк, Райнхард Роберт, Кюн Торальф, Зіверніх Бернд, Ерхардт Томас, Парра Рападо Ліліана, Вітшель Маттіас
МПК: C07D 333/28, C07D 213/30, C07D 307/56, C07D 333/16, C07D 207/44, C07D 209/12, A01N 43/50, C07D 249/08, A01N 43/38, A01N 43/56, A01N 43/40, C07D 213/89, C07D 231/12, C07D 233/54, C07D 213/61
Мітки: рослин, гербіцидів, небажаним, заміщені, ростом, сполука, серин-аміди, спосіб, проміжна, одержання, варіанти, бензоїлом, боротьби, застосування, засобу, засіб
Формула / Реферат:
1. Заміщені бензоїлом серин-аміди формули І ,Iу якій Het означає моно- або біциклічний гетероарил з від 5 до 10 кільцевими членами, який містить від 1 до 4 гетероатомів із групи азот, кисень і сірка, який частково або повністю може бути галогенованим і/або може мати від 1 до 3 залишків із групи ціано, нітро, C1-С6-алкіл, С1-С6-галогеналкіл, гідрокси,...
Водорозчинні проліки блокованих спиртів чи фенолів, фармацевтична композиція на їх основі, спосіб їх одержання, проміжна сполука (варіанти) та спосіб її одержання (варіанти)

Номер патенту: 73479
Опубліковано: 15.08.2005
Автори: Стелла Валентіно Дж., Сафаді Мухаммед С., Зігмунт Джан Дж., Георг Інгрід Гунда
МПК: C07D 311/72, C07F 9/09, C07F 9/6561, C07H 17/04, A61K 31/10, A61K 31/355, C07K 7/64, A61K 31/7042, C07D 491/052, C07C 319/00, A61P 35/00, C07C 323/12, A61K 31/661, C07F 9/655, A61P 25/00, A61K 38/00, A61K 31/352, A61K 31/437, A61K 31/7048, C07F 9/6515, A61K 31/665, A61K 31/675
Мітки: проліки, фармацевтична, водорозчинні, варіанти, спосіб, фенолів, проміжна, композиція, одержання, основі, сполука, спиртів, блокованих
Формула / Реферат:
1. Сполука відповідно до формули І:, IдеR-O- - залишок спиртвмісної або фенолвмісної фармацевтичної сполуки, крім таксолу і його похідних,R1 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота,R2 - атом водню або іон лужного металу, або протонований амін, або протонована амінокислота, іn являє...
Спосіб одержання невірапіну, проміжна сполука (варіанти)

Номер патенту: 80140
Опубліковано: 27.08.2007
Автори: Ло Юонг Сек, Гаптон Бернард Франклін, Бозуелл Роберт Фредерік Джр.
МПК: C07D 213/803, C07D 471/10, C07D 471/14, C07D 213/82, C07D 213/78, C07D 213/80, C07D 213/85
Мітки: одержання, варіанти, проміжна, сполука, спосіб, невірапіну
Формула / Реферат:
1. Спосіб одержання невірапіну, який полягає в тому, що:(а) піддають взаємодії 2-гало-3-піридинкарбонітрил формули,де X означає атом фтору, хлору, брому або йоду, переважно хлору або брому, із циклопропіламіном з одержанням 2-(циклопропіламіно)-3-піридинкарбонітрилу;(б) гідролізують 2-(циклопропіламіно)-3-піридинкарбонітрил з одержанням...
Попередній патент: Комбінація декоративної кришки і закупорювального ковпачка
Наступний патент: Маятниковий креномір-сигналізатор
Випадковий патент: Спосіб виготовлення прокату з вуглецевої сталі