Інгібітори металопротеїнази




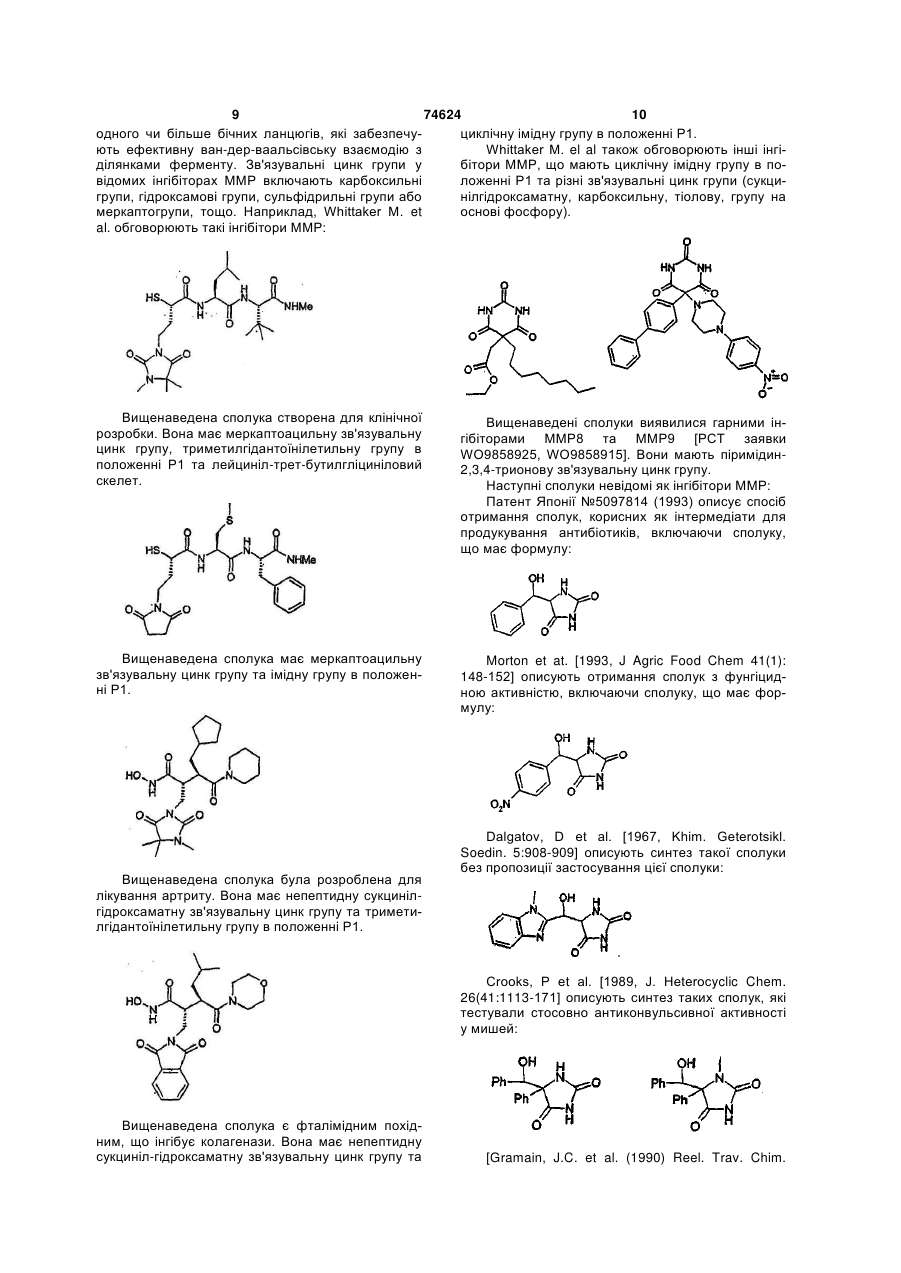











Формула / Реферат
1. Сполука формули I або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер
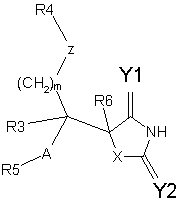 (І),
(І),
де
Х вибрано з групи: NR1, О, S;
Y1 та Y2 незалежно вибрані з групи: О, S;
Z вибрано з групи: NR2, О, S;
m дорівнює 0 чи 1;
А вибрано з групи: безпосередній зв'язок, (С1-6)алкіл, (С1-6)алкеніл, (С1-6 )галогеналкіл, або (С1-6)гетероалкіл, що містить гетерогрупу, вибрану з груп: N, О, S, SO, SO2, або містить дві гетерогрупи, вибрані з груп: N, О, S, SO, SO2 та розділені щонайменше двома атомами карбону;
R1 вибрано з групи: Н, алкіл, галогеналкіл;
R2 вибрано з групи: Н, алкіл, галогеналкіл;
R3 та R6 незалежно вибрані з групи: Н, галоген (переважно F), алкіл, галогеналкіл, алкоксіалкіл, гетероалкіл, циклоалкіл, арил, алкіларил, гетероалкіларил, гетероарил, алкілгетероарил, гетероалкілгетероарил, арилалкіл, арилгетероалкіл, гетероарилалкіл, гетероарилгетероалкіл, бісарил, арилгетероарил, гетероариларил, бісгетероарил, циклоалкіл або гетероциклоалкіл, що містять 3-7 кільцевих атомів, де радикали алкілу, гетероалкілу, арилу, гетероарилу, циклоалкілу або гетероциклоалкілу можуть бути, як варіант, заміщені однією або більше групами, незалежно вибраними з груп: гідроксил, алкіл, гетероалкіл, циклоалкіл, арил, гетероарил, галоген, галогеналкіл, гідроксіалкіл, алкоксил, алкоксіалкіл, галогеналкоксил, галогеналкоксіалкіл, карбоксил, карбоксіалкіл, алкілкарбоксил, аміногрупа,
N-алкіламіногрупа, N,N-діалкіламіногрупа, алкіламіногрупа, алкіл(N-алкіл)аміногрупа, алкіл(N,N-діалкіл)аміногрупа, амідогрупа, N-алкіламідогрупа, N,N-дiaлкiлaмiдoгрyпa, алкіламідогрупа, алкіл(N-алкіл)амідогрупа, алкіл(N,N-діалкіл)амідогрупа, тіол, сульфон, сульфонаміногрупа, алкілсульфонаміногрупа, арилсульфонаміногрупа, сульфонамідогрупа, галогеналкілсульфон, алкілтіогрупа, арилтіогрупа, алкілсульфон, арилсульфон, аміносульфон, N-алкіламіносульфон, N,N-діалкіламіносульфон, алкіламіносульфон, ариламіносульфон, ціаногрупа, алкілціаногрупа, гуанідиногрупа, N-ціаногуанідиногрупа, тіогуанідиногрупа, амідиногрупа, N-аміносульфонамідиногрупа, нітрогрупа, алкілнітрогрупа, 2-нітро-етен-1,1-діамін;
R4 вибрано з групи: Н, алкіл, гідроксіалкіл, галогеналкіл, алкоксіалкіл, галогеналкоксил, аміноалкіл, амідоалкіл, тіоалкіл;
R5 представляє моноциклічну групу, що містить 3-7 кільцевих атомів, незалежно вибраних з груп: циклоалкіл, арил, гетероциклоалкіл або гетероарил, як варіант, заміщений одним чи більше замісниками, незалежно вибраними з групи: галоген, гідроксил, галогеналкоксил, аміногрупа, N-алкіламіногрупа, N,N-діалкіламіногрупа, ціаногрупа, нітрогрупа, алкіл, алкоксил, алкілсульфон, галогеналкілсульфон, карбоніл, карбоксил, де будь-який алкіл у будь-якому заміснику може бути сам, як варіант, заміщеним одною чи більше групами, вибраними з груп: галоген, гідроксил, аміногрупа, N-алкіламіногрупа, N,N-діалкіламіногрупа, алкілсульфонаміногрупа, алкілкарбоксіаміногрупа, ціаногрупа, нітрогрупа, тіол, алкілтіол, алкілсульфоногрупа, алкіламіносульфоногрупа, алкілкарбоксилат, амідогрупа, N-алкіламідогрупа, N,N-діалкіламідогрупа, алкоксил, галогеналкоксил, карбоніл, карбоксил;
за умови, що:
коли Х представляє NR1, R1 представляє Н, Y1 представляє О, Y2 представляє О, Z представляє О, m дорівнює 0, А є безпосереднім зв'язком, R3 представляє Н, R4 представляє Н та R6 представляє Н, тоді R5 не представляє феніл, нітрофеніл, гідроксифеніл, алкоксифеніл або піридин;
коли Х представляє NR1, R1 представляє Н або метил, Y1 представляє О, Y2 представляє О, Z представляє О, m дорівнює 0, А є безпосереднім зв'язком, R3 представляє Н, R4 представляє Н та R6 представляє феніл, тоді R5 не представляє феніл;
коли Х представляє NR1, R1 представляє Н, Y1 представляє О, Y2 представляє О, Z представляє О, m дорівнює 0, А є безпосереднім зв'язком, R3 представляє феніл, R4 представляє Н та R6 представляє Н, тоді R5 не представляє феніл;
коли Х представляє S, щонайменше один з Y1 та Y2 представляє О, m дорівнює 0, А є безпосереднім зв'язком, R3 представляє Н або метил, R6 представляє Н або метил, тоді R5 не представляє феніл, піридин, пірол, тіофен або фуран;
коли Х представляє О, Y1 представляє О, Y2 представляє О, Z представляє О, m дорівнює 0, А є безпосереднім зв'язком, R3 представляє метилхлорид, R4 представляє Н та R6 представляє Н, тоді R5 не представляє феніл.
2. Сполука формули І за п. 1 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де Х представляє NR1, R1 представляє Н або (С1-3) алкіл, щонайменше один з Y1 та Y2 представляє О, Z представляє О, m дорівнює 0, та А є безпосереднім зв'язком.
3. Сполука за п. 1 або п. 2 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де R3 представляє Н, алкіл або галогеналкіл, R4 представляє Н, алкіл або галогеналкіл.
4. Сполука за будь-яким з попередніх пунктів або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де R5 представляє, як варіант, заміщене 5- або 6-членне кільце, незалежно вибране з групи: циклоалкіл, арил, гетероциклоалкіл або гетероарил.
5. Сполука за будь-яким з попередніх пунктів або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де R6 представляє Н, алкіл, гідроксіалкіл, аміноалкіл, циклоалкілалкіл, алкілциклоалкіл, арилалкіл, алкіларил, гетероалкіл, гетероциклоалкілалкіл, алкілгетероциклоалкіл, гетероарилалкіл або гетероалкіларил.
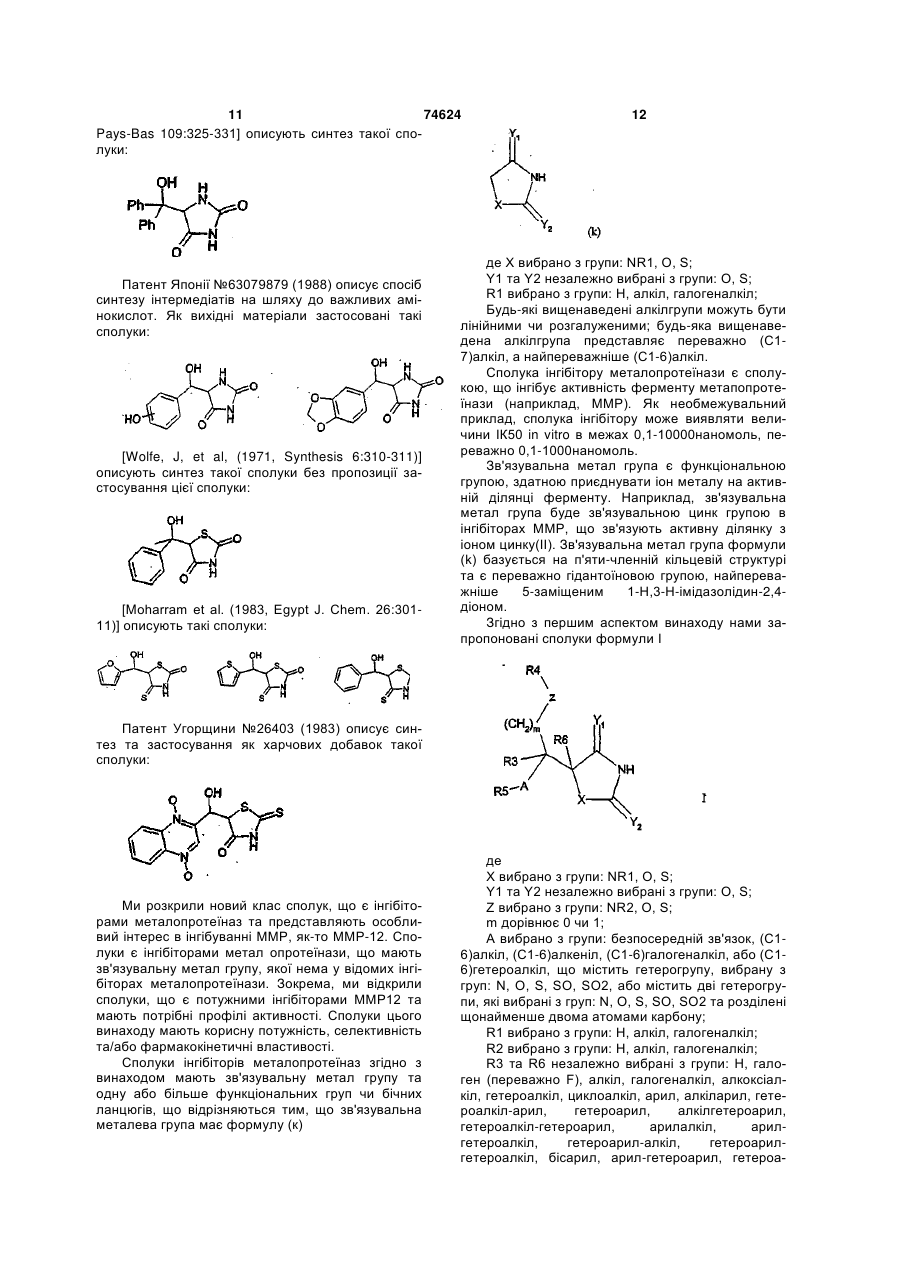
6. Сполука формули II або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер
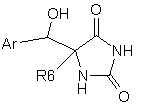 (ІІ),
(ІІ),
де
Ar є 5- або 6-членним арилом або гетероарилом, як варіант, заміщеним одним або двома замісниками, вибраними з групи: галоген, аміногрупа, нітрогрупа, (С1-6)алкіл, (С1-6)алкоксил або (С1-6)галогеналкоксил;
R6 вибрано з групи: Н, арил або (С1-6)алкіл та R6 є, як варіант, заміщеним групою, вибраною з групи: гідроксил, тіоалкіл, феніл, галогенфеніл, піридил або карбамат.
7. Сполука формули II за п. 6 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де Ar представляє феніл або заміщений феніл, або Ar представляє 5- або 6-членне гетероарильне кільце, що містить два гетероатоми, незалежно вибрані з групи: О та N.
8. Сполука формули II за п. 6 або п. 7 або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер, де R6 представляє феніл, феніл, заміщений галогеном, метиленпіридином, або (С1-3)алкіл, як варіант, заміщений гідроксилом, тіометилом або бензилкарбаматом.
9. Фармацевтична композиція, яка містить сполуку формули І за п. 1 або її фармацевтично прийнятну сіль чи здатний до гідролізу in vivo естер та фармацевтично прийнятний носій.
10. Фармацевтична композиція, яка містить сполуку формули II за п. 6 або її фармацевтично прийнятну сіль чи здатний до гідролізу in vivo естер та фармацевтично прийнятний носій.
11. Спосіб лікування опосередкованої металопротеїназою хвороби або стану, який полягає в уведенні теплокровній тварині терапевтично ефективної кількості сполуки формули І або формули II або її фармацевтично прийнятної солі чи здатного до гідролізу in vivo естеру.
12. Застосування сполуки формули І або формули II або її фармацевтично прийнятної солі чи здатного до гідролізу in vivo попередника у виробництві медикаменту для застосування у лікуванні хвороби або стану, опосередкованих одним чи більше металопротеїназними ферментами.
Текст
1. Сполука формули I або її фармацевтично прийнятна сіль чи здатний до гідролізу in vivo естер 2 (19) 1 3 74624 4 R5 представляє моноциклічну групу, що містить 3ране з групи: циклоалкіл, арил, гетероциклоалкіл 7 кільцевих атомів, незалежно вибраних з груп: або гетероарил. циклоалкіл, арил, гетероциклоалкіл або гетероа5. Сполука за будь-яким з попередніх пунктів або її рил, як варіант, заміщений одним чи більше замісфармацевтично прийнятна сіль чи здатний до гідниками, незалежно вибраними з групи: галоген, ролізу in vivo естер, де R6 представляє Н, алкіл, гідроксил, галогеналкоксил, аміногрупа, Nгідроксіалкіл, аміноалкіл, циклоалкілалкіл, алкілциалкіламіногрупа, N,N-діалкіламіногрупа, ціаногруклоалкіл, арилалкіл, алкіларил, гетероалкіл, гетепа, нітрогрупа, алкіл, алкоксил, алкілсульфон, гароциклоалкілалкіл, алкілгетероциклоалкіл, гетерологеналкілсульфон, карбоніл, карбоксил, де будьарилалкіл або гетероалкіларил. який алкіл у будь-якому заміснику може бути сам, 6. Сполука формули II або її фармацевтично прияк варіант, заміщеним одною чи більше групами, йнятна сіль чи здатний до гідролізу in vivo естер вибраними з груп: галоген, гідроксил, аміногрупа, OH O N-алкіламіногрупа, N,N-діалкіламіногрупа, алкілсульфонаміногрупа, алкілкарбоксіаміногрупа, ціаAr NH ногрупа, нітрогрупа, тіол, алкілтіол, алкілсульфоII R6 N ногрупа, алкіламіносульфоногрупа, H алкілкарбоксилат, амідогрупа, N-алкіламідогрупа, O (ІІ), N,N-діалкіламідогрупа, алкоксил, галогеналкоксил, де карбоніл, карбоксил; Ar є 5- або 6-членним арилом або гетероарилом, за умови, що: як варіант, заміщеним одним або двома замісниколи Х представляє NR1, R1 представляє Н, Y1 ками, вибраними з групи: галоген, аміногрупа, нітпредставляє О, Y2 представляє О, Z представляє рогрупа, (С1-6)алкіл, (С1-6)алкоксил або (С1О, m дорівнює 0, А є безпосереднім зв'язком, R3 6)галогеналкоксил; представляє Н, R4 представляє Н та R6 предстаR6 вибрано з групи: Н, арил або (С1-6)алкіл та R6 вляє Н, тоді R5 не представляє феніл, нітрофеніл, є, як варіант, заміщеним групою, вибраною з гругідроксифеніл, алкоксифеніл або піридин; пи: гідроксил, тіоалкіл, феніл, галогенфеніл, піриколи Х представляє NR1, R1 представляє Н або дил або карбамат. метил, Y1 представляє О, Y2 представляє О, Z 7. Сполука формули II за п. 6 або її фармацевтичпредставляє О, m дорівнює 0, А є безпосереднім но прийнятна сіль чи здатний до гідролізу in vivo зв'язком, R3 представляє Н, R4 представляє Н та естер, де Ar представляє феніл або заміщений R6 представляє феніл, тоді R5 не представляє феніл, або Ar представляє 5- або 6-членне гетефеніл; роарильне кільце, що містить два гетероатоми, коли Х представляє NR1, R1 представляє Н, Y1 незалежно вибрані з групи: О та N. представляє О, Y2 представляє О, Z представляє 8. Сполука формули II за п. 6 або п. 7 або її фарО, m дорівнює 0, А є безпосереднім зв'язком, R3 мацевтично прийнятна сіль чи здатний до гідролізу представляє феніл, R4 представляє Н та R6 предin vivo естер, де R6 представляє феніл, феніл, ставляє Н, тоді R5 не представляє феніл; заміщений галогеном, метиленпіридином, або (С1коли Х представляє S, щонайменше один з Y1 та 3)алкіл, як варіант, заміщений гідроксилом, тіомеY2 представляє О, m дорівнює 0, А є безпосередтилом або бензилкарбаматом. нім зв'язком, R3 представляє Н або метил, R6 9. Фармацевтична композиція, яка містить сполуку представляє Н або метил, тоді R5 не представляє формули І за п. 1 або її фармацевтично прийнятну феніл, піридин, пірол, тіофен або фуран; сіль чи здатний до гідролізу in vivo естер та фарколи Х представляє О, Y1 представляє О, Y2 мацевтично прийнятний носій. представляє О, Z представляє О, m дорівнює 0, А 10. Фармацевтична композиція, яка містить сполує безпосереднім зв'язком, R3 представляє метилку формули II за п. 6 або її фармацевтично прийнхлорид, R4 представляє Н та R6 представляє Н, ятну сіль чи здатний до гідролізу in vivo естер та тоді R5 не представляє феніл. фармацевтично прийнятний носій. 2. Сполука формули І за п. 1 або її фармацевтично 11. Спосіб лікування опосередкованої металопроприйнятна сіль чи здатний до гідролізу in vivo естеїназою хвороби або стану, який полягає в уветер, де Х представляє NR1, R1 представляє Н або денні теплокровній тварині терапевтично ефекти(С1-3) алкіл, щонайменше один з Y1 та Y2 предвної кількості сполуки формули І або формули II ставляє О, Z представляє О, m дорівнює 0, та А є або її фармацевтично прийнятної солі чи здатного безпосереднім зв'язком. до гідролізу in vivo естеру. 3. Сполука за п. 1 або п. 2 або її фармацевтично 12. Застосування сполуки формули І або формули прийнятна сіль чи здатний до гідролізу in vivo есII або її фармацевтично прийнятної солі чи здатнотер, де R3 представляє Н, алкіл або галогеналкіл, го до гідролізу in vivo попередника у виробництві R4 представляє Н, алкіл або галогеналкіл. медикаменту для застосування у лікуванні хворо4. Сполука за будь-яким з попередніх пунктів або її би або стану, опосередкованих одним чи більше фармацевтично прийнятна сіль чи здатний до гідметалопротеїназними ферментами. ролізу in vivo естер, де R5 представляє, як варіант, заміщене 5- або 6-членне кільце, незалежно виб 5 Даний винахід стосується сполук, корисних при інгібуванні металопротеїназ і, зокрема, фармацевтичних композицій, що їх містять, а також їх застосування. Сполуки цього винаходу є інгібіторами одного чи більше металопротеїназних ферментів. Металопротеїнази є надродиною протеїназ (ферментів), число яких в останні роки різко зросло. На основі структурних та функціональних досліджень ці ферменти було розподілені на родини та підродини, як описано в N.M. Hooper (1994) FEBS Letters 354:1-6. Приклади металопротеїназ включають матричні металопротеїнази (ММР), як-то колагенази (ММР1, ММР8, ММР13), желатинази (ММР2, ММР9), стромелізини (MMPS, ММР10, ММР11), матрилізин (ММР7), металоеластаза (ММР12), енамелізин (ММР19), МТ-ММР (ММР14, ММР15, ММР16, ММР17); репролізин або адамалізин, або MDC родину, яка включає секретази та шедази, як-то перетворюючі TNF ферменти (ADAM 10 та ТАСЕ); астацинову родину, що включає ферменти, ' як-то перетворююча проколаген протеїназа (РСР); та інші металопротеїнази, як-то агреканаза, родина перетворюючих ендотелій ферментів та родина перетворюючих ангіотензин ферментів. Металопротеїнази, можна вважати важливими при гіперволемії фізіологічних хворобливих процесів, що включають корекцію тканини, як-то розвиток ембріону, утворення кісток та корекцію матки при менструації. Це базується на здатності металопротеїназ розщеплювати багато матричних субстратів, як-то колаген, протеоглікан та фібронектин. Металопротеїнази, можна також вважати важливими при перетворенні або секреції біологічно важливих клітинних посередників, як-то фактор некрозу пухлин (TNF); та посттрансляційне протеолізне перетворення, або втрата, біологічно важливих мембранних білків, як-то рецептор CD23 з низькою спорідненістю до ІgЕ [для повнішого огляду див. N.М. Hooper et ah, (1997) BiochemJ. 321:265-279]. Металопротеїнази було пов'язано з багатьма хворобами або станами. Інгібування активності одної чи більше металопротеїназ може бути дуже корисним при цих хворобах або умови, наприклад, при різних запальних та алергічних хворобах, якто, запалення суглобів (особливо ревматоїдний артрит, остеоартрит та подагра), запалення шлунково-кишкового тракту (особливо запальна хвороба кишечнику, виразковий коліт та гастрит), запалення шкіри (особливо псоріаз, екзема, дерматит); при метастазах або інвазії пухлин; при хворобі, пов'язаній з нерегульованою деградацією екстрацелюлярної матриці, як-то остеоартрит; при резорбтивній хворобі кісток (як-то остеопороз та хвороба Педжета); при хворобах, пов'язаних з порушеним ангіогенезом; пов'язана з діабетом посилена корекція колагену, хвороба зубів (як-то гінгівіт), покриття виразками роговиці, покриття виразками шкіри, постопераційні стани (як-то анастомоз товстої кишки) та загоєння поранень шкіри; демієлінізуючі хвороби центральної та периферійної нервових систем (як-то розсіяний склероз); хвороба Альцгеймера; корекція екстрацелюлярної 74624 6 матриці, яку спостерігають при серцево-судинних хворобах, як-то рестеноз та атеросклероз; астма, риніт; та хронічні обструктивні хвороби легенів (COPD). ММР12, відома також як еластаза або металоеластаза макрофагів, була спочатку клонована у мишах за допомоги Shapiro et. al [1992, J. Biol. Chem. 267: 4664] та у людини за допомоги тієї ж групи у 1995. ММР-12 преференційно експресуеться в активованих макрофагах, і було виявлено, що вона секретується з альвеолярних макрофагів курців [Shapiro etal., 1993, J. Biol. Chem., 268: 23824], а також у пінних клітинах в атеросклеротичних ураженнях [Matsumoto et al, 1998, Am J Pathol 153: 109]. Мишача модель COPD базується на контрольному зараженні мишей сигаретним димом протягом 6 місяців, двома сигаретами 6 діб на тиждень Дикі миші формують емфізему легенів після цієї обробки. Коли уражених ММР12 мишей тестували у цій моделі, вони не формували значної емфіземи, чітко вказуючи, що ММР-12 є ключовим ферментом у патогенезі COPD. Роль ММР, як-то ММР12 у COPD (емфізема та бронхіт) було обговорено [Anderson та Shinagawa, 1999, Current Opinion in Anti-inflammatory, та Immunomodulatory Investigational Drugs 1(1): 29-38]. Зараз було виявлено, що паління збільшує інфільтрацію макрофагів та похідну від макрофагів експресію ММР-12 у бляшках Кангаварі сонної артерії людини [Matetzky S, Fishbein MC et al., Circulation 102:(181. 36-39 Suppl. S. Oct 31, 2000]. MMP13, або колагеназа 3, була спочатку клонована з похідної пухлин молочної залози бібліотеки кДНК [J. M. P. Freije et al. (1994) J. Biol. Chem. 269(24): 16766-16773]. ПЛР-РНК-аналіз РНК з великого числа тканин показав, що експресія ММР13 була обмежена карциномами молочної залози, оскільки її не була виявлено у фіброаденомах молочної залози, нормальних або спочиваючих грудних залозах, плаценті, печінці, яєчнику, матці, простаті або завушній залозі або у лініях клітин раку молочної залози (T47-D, MCF-7 та ZR75-1). На додаток до цього спостереження, ММР13 було виявлено у трансформованих епідермальних кератиноцитах [N. Johansson et al., (1997) Cell Growth Differ. 8j2): 243-250], сквамозно-клітинній карциномі [Ν. Johansson et al., (1997) Am. J. Pathol. 151(2):499-508] та епідермальних пухлинах [К. Airola et al., (1997) J. Invest. Dermatol. 109(2): 225231]. Ці результати підтверджують, що ММР13 секретується у трансформованих епітеліальних клітинах та може бути включена у деградацію екстрацелюлярної матриці та клітинно-матричну взаємодію, пов'язану з метастазом, як зокрема спостерігали при ураженнях раком молочної залози та при злоякісному рості епітелію при карциногенезі шкіри. Нещодавно опубліковані дані свідчать, що ММР13 грає важливу роль в обороті інших сполучних тканин. Наприклад, сумісна з ММР13субстратною специфічністю та перевагою стосовно деградації колагену типу II [P.G. Mitchell et al., (1996) J. Clin. Invest 97(3): 761-76S: V. Knauper et al., (1996) The Biochemical Journal 271: 1544-1550]. MMP13, як припущено, грає роль під час первин 7 74624 8 ного формування кісток та корекції скелета [М. надосадкової рідинах від нелікованих астматиків Stahle-Backdahl et al., (1997) Lab. Invest. 76(5):717порівняно з астматиками з інших популяцій [Am. J. 728: N. Johansson et al., (1997) Dev. Dyn. 208(3): Resp. Cell & Моl. Biol., (Nov 1997) 17(5): 583-591]. 387-397], при деструктивних хворобах суглобів, якТакож, збільшену експресію ММР9 виявлено у то ревматоїдний та остео-артрит [D. Wernicke et деяких інших патологічних станах, що свідчить про al., (1996) J. Rheumatol. 23: 590-595; P.G. Mitchell et залучення ММР9 у хворобливі процеси, як-то al., (1996) J. Clin. Invest 97(3): 761-768; O. Lindy et COPD, артрит, метастази пухлин, хвороба Альцal., (1997) Artritis Rheum 40(8): 1391-1399]; та при геймера, розсіяний склероз, та руйнування тромасептичному ослабленні заміни стегна [S. Imai et боцитів при атеросклерозі, що призводить до гостal., (1998) J. Bone Joint Surg. Br, 80(4): 701-710]. рих коронарних станів, як-то інфаркт міокарду. MMP13 також було залучено у хронічний пародонММР-8 (колагеназа-2, нейтрофільна колагенатоз у дорослих, оскільки вона локалізована в епіза) є ферментом розміром 53кДа родини матричтелії хронічно запаленої слизової тканини ясен них металопротеїназ, що преференційно експрелюдини [V.J. Uitto et al., (1998) Am. J. Pathol. сується у нейтрофілах. Останні. дослідження 152(6): 1489-1499] та при корекції колагенної матсвідчать, що ММР-8 експресується також в інших риці при хронічних пораненнях [М. Vaalamo etal, клітинах, як-то остеоартритні хондроцити. [Shlopov (1997) J. Invest Dermatol. 109(1 ): 96-101]. et al., (1997) Artritis Rheum, 40:2065]. Вироблена MMP9 (Желатиназа В; 92кДа Колагеназа типу нейтрофілами ММР може викликати корекцію ткаIV; 92кДа Желатиназа) є секретованим білком, нини, а тому блокування ММР-8 повинно мати поякий спочатку очищали, потім клонували та секвезитивний вплив у фіброзних хворобах, наприклад, нсували, у 1989 [S.M. Wilhelm et al., (1989) J. Biol легенів, та при дегратативних хворобах типу емфіChem. 264 (29'): 17213-17221; опублікована помиземи легенів. ММР-8 була також виявлена як звелка у J. Biol Chem. (1990) 265 (36): 22570]. Нещорхрегульована при остеоартриті, свідчачи тим, що давній огляд ММР9 пропонує чудове джерело деблокування ММР-8 може також бути корисним при тальної інформації та посилань на цю протеазу: цій хворобі. Т.Н. Vu & Ζ. Werb (1998) [In: Matrix ММР-3 (стромелізин-1) є ферментом розміром Metalloproteinases. 1998. Edited by W.C. Parks & 53кДа родини матричних металопротеїназ. АктивR.P. Mecham. pp.115-148. Academic Press. ISBN 0ність ММР-3 продемонстрована у фібробластах, 12-545090-7]. Наступні відомості взяті з цього виділених із запалених ясен [Uitto V. J. et al., огляду Т.Н. Vu & Ζ. Werb (1998). (1981) J. Periodontal Res., 16:417-424], та рівні феЕкспресія ММР9 звичайно обмежена кількома рменту скорельовані з суворістю хвороби ясен типами клітин, включаючи трофобласти, остеокла[Overall С.Μ. et al., (1987) J. Periodontal Res., 22:81сти, нейтрофіли та макрофаги. Однак, її експресію 88]. ММР-3 продукуються також у базальних кераможна індукувати у тих же самих клітинах та у інтиноцитах у багатьох хронічних виразках ших типах клітин кількома посередниками, вклю[Saarialho-Kere U.К. et al, (1994) J. Clin. Invest, чаючи обробку клітин факторами росту або цито94:79-88]. MMP-3 mPHK та білок було виявлено у кінами. Вони є посередниками, часто залученими у базальних кератиноцитах, суміжних з, але на відспочаткову запальну реакцію. Як інші секретовані тані від краю поранення, в якому можливо предММР, ММР9 вивільняється як неактивний профеставлено ділянки проліферуючого епідермісу. рмент, який далі розщеплюється з утворенням ММР-3 може тим самим заважати загоєнню епідеактивного ферменту. Потрібні для цієї активації рмісу. Кілька дослідників продемонстрували стійке протеази in vivo невідомі. Баланс активної ММР9 підвищення ММР-3 у синовіальних з рідинах від відносно неактивного ферменту далі регулюється пацієнтів з ревматоїдним та остеоартритом порівin vivo взаємодією з ТІМР-1 (Інгібітор тканинної няно з контрольною групою [Walakovits L.A. et al., металопротеїнази-1), природно існуючим білком. (1992) Artritis Rheum., 35:35-42; Zafarullah Μ. et al., ТІМР-1 приєднується до С-термінального регіону (1993) J. Rheumatol., 20:693-697]. Ці дослідження ММР9, призводячи до інгібування каталітичного дають основу для думки, що інгібітор ММР-3 лікудомену ММР9. Баланс індукованої експресії ватиме хвороби, при яких залучено руйнування РrоММР9, розщеплення Pro- в активну ММР9 та екстрацелюлярної матриці, що призводить до наявність ТІМР-1 комбінують для визначення кільобумовленого інфільтрацією лімфоцитів запаленкості каталітично активної ММР9, яка є присутня або втрати необхідної для функції органу струкньою на локальній ділянці. Протеолітично активна турної цілісності. ММР9 атакує субстрати, що включають желатин, Ряд інгібіторів металопротеїнази відомі [див., еластин, та природні колагени типу IV та типу V; наприклад, огляд інгібіторів ММР Beckett R.P. та вона не має активності проти природного колагену Whittaker Μ., 1998, Exp. Opin. Then Патенти, типу І, протеогліканів або ламінінів. 8(3):25 9-282]. Відмінні класи сполук можуть мати З'являється багато даних стосовно ролі ММР9 відмінні ступені потужності та селективності інгібуу різних фізіологічних та патологічних процесах. вання різних металопротеїназ. Фізіологічні ролі включають інвазію ембріональних [Whittaker Μ. et al., (1999) Chemical Reviews трофобластів через епітелій матки на ранніх ета99(9):2735-2776] розглядають багато відомих спопах ембріональної імплантації; деяку роль у рості лук інгібіторів ММР. Вони констатують, що ефекта розвитку кісток; та міграцію запальних клітин з тивний інгібітор ММР потребує зв'язувальної цинк судинної системи у тканини. групи або ZBG (функціональної групи, здатної хеВивільнення ММР-9, виміряне з використанлатувати активну ділянку з іоном цинку(ІІ)), щоням ферментного імунодослідження, було значно найменше одної функціональної групи, яка забезпідвищеним у тканинних рідинах та в АМпечує водневий зв'язок з основою ферменту, та 9 74624 10 одного чи більше бічних ланцюгів, які забезпечуциклічну імідну групу в положенні Р1. ють ефективну ван-дер-ваальсівську взаємодію з Whittaker Μ. el al також обговорюють інші інгіділянками ферменту. Зв'язувальні цинк групи у бітори ММР, що мають циклічну імідну групу в повідомих інгібіторах ММР включають карбоксильні ложенні Р1 та різні зв'язувальні цинк групи (сукцигрупи, гідроксамові групи, сульфідрильні групи або нілгідроксаматну, карбоксильну, тіолову, групу на меркаптогрупи, тощо. Наприклад, Whittaker Μ. et основі фосфору). al. обговорюють такі інгібітори ММР: Вищенаведена сполука створена для клінічної розробки. Вона має меркаптоацильну зв'язувальну цинк групу, триметилгідантоїнілетильну групу в положенні Р1 та лейциніл-трет-бутилгліциніловий скелет. Вищенаведені сполуки виявилися гарними інгібіторами ММР8 та ММР9 [РСТ заявки WO9858925, WO9858915]. Вони мають піримідин2,3,4-трионову зв'язувальну цинк групу. Наступні сполуки невідомі як інгібітори ММР: Патент Японії №5097814 (1993) описує спосіб отримання сполук, корисних як інтермедіати для продукування антибіотиків, включаючи сполуку, що має формулу: Вищенаведена сполука має меркаптоацильну зв'язувальну цинк групу та імідну групу в положенні Р1. Morton et at. [1993, J Agric Food Chem 41(1): 148-152] описують отримання сполук з фунгіцидною активністю, включаючи сполуку, що має формулу: Вищенаведена сполука була розроблена для лікування артриту. Вона має непептидну сукцинілгідроксаматну зв'язувальну цинк групу та триметилгідантоїнілетильну групу в положенні Р1. Dalgatov, D еt аl. [1967, Khim. Geterotsikl. Soedin. 5:908-909] описують синтез такої сполуки без пропозиції застосування цієї сполуки: Crooks, Ρ еt аl. [1989, J. Heterocyclic Chem. 26(41:1113-171] описують синтез таких сполук, які тестували стосовно антиконвульсивної активності у мишей: Вищенаведена сполука є фталімідним похідним, що інгібує колагенази. Вона має непептидну сукциніл-гідроксаматну зв'язувальну цинк групу та [Gramain, J.C. et al. (1990) Reel. Trav. Chim. 11 74624 Pays-Bas 109:325-331] описують синтез такої сполуки: Патент Японії №63079879 (1988) описуєспосіб синтезу інтермедіатів на шляху до важливих амінокислот. Як вихідні матеріали застосовані такі сполуки: [Wolfe, J, et al, (1971, Synthesis 6:310-311)] описують синтез такої сполуки без пропозиції застосування цієї сполуки: [Moharram еt аl. (1983, Egypt J. Chem. 26:30111)] описують такі сполуки: 12 де X вибрано з групи: NR1, О, S; Y1 та Y2 незалежно вибрані з групи: О, S; R1 вибрано з групи: Н, алкіл, галогеналкіл; Будь-які вищенаведені алкілгрупи можуть бути лінійними чи розгалуженими; будь-яка вищенаведена алкілгрупа представляє переважно (С17)алкіл, а найпереважніше (С1-6)алкіл. Сполука інгібітору металопротеїнази є сполукою, що інгібує активність ферменту метапопротеїнази (наприклад, ММР). Як необмежувальний приклад, сполука інгібітору може виявляти величини ІК50 in vitro в межах 0,1-10000наномоль, переважно 0,1-1000наномоль. Зв'язувальна метал група є функціональною групою, здатною приєднувати іон металу на активній ділянці ферменту. Наприклад, зв'язувальна метал група буде зв'язувальною цинк групою в інгібіторах ММР, що зв'язують активну ділянку з іоном цинку(ІІ). Зв'язувальна метал група формули (k) базується на п'яти-членній кільцевій структурі та є переважно гідантоїновою групою, найпереважніше 5-заміщеним 1-Н,3-Н-імідазолідин-2,4діоном. Згідно з першим аспектом винаходу нами запропоновані сполуки формули І Патент Угорщини №26403 (1983) описує синтез та застосування як харчових добавок такої сполуки: Ми розкрили новий клас сполук, що є інгібіторами металопротеїназ та представляють особливий інтерес в інгібуванні ММР, як-то ММР-12. Сполуки є інгібіторами метал опротеїнази, що мають зв'язувальну метал групу, якої нема у відомих інгібіторах металопротеїнази. Зокрема, ми відкрили сполуки, що є потужними інгібіторами ММР12 та мають потрібні профілі активності. Сполуки цього винаходу мають корисну потужність, селективність та/або фармакокінетичні властивості. Сполуки інгібіторів металопротеїназ згідно з винаходом мають зв'язувальну метал групу та одну або більше функціональних груп чи бічних ланцюгів, що відрізняються тим, що зв'язувальна металева група має формулу (к) де X вибрано з групи: NR1, О, S; Y1 та Y2 незалежно вибрані з групи: О, S; Ζ вибрано з групи: NR2, О, S; m дорівнює 0 чи 1; А вибрано з групи: безпосередній зв'язок, (С16)алкіл, (С1-6)алкеніл, (С1-6)галогеналкіл, або (С16)гетероалкіл, що містить гетерогрупу, вибрану з груп: N, О, S, SO, SO2, або містить дві гетерогрупи, які вибрані з груп: N, О, S, SO, SO2 та розділені щонайменше двома атомами карбону; R1 вибрано з групи: Н, алкіл, галогеналкіл; R2 вибрано з групи: Н, алкіл, галогеналкіл; R3 та R6 незалежно вибрані з групи: Н, галоген (переважно F), алкіл, галогеналкіл, алкоксіалкіл, гетероалкіл, циклоалкіл, арил, алкіларил, гетероалкіл-арил, гетероарил, алкілгетероарил, гетероалкіл-гетероарил, арилалкіл, арилгетероалкіл, гетероарил-алкіл, гетероарилгетероалкіл, бісарил, арил-гетероарил, гетероа 13 74624 14 рил-арил, бісгетероарил, циклоалкіл або гетероR3 представляє Н, R4 представляє Η та R6 предциклоалкіл, що містять 3-7 кільцевих атомів, де ставляє Н, тоді R5 не представляє феніл, нітрорадикали алкілу, гетероалкілу, арилу, гетероарифеніл, пдроксифеніл, алкоксифеніл або піридин; лу, циклоалкілу або гетероцикпоалкілу можуть коли X представляє NR1, R1 представляє Η бути, як варіант, заміщеними одною чи більше або метил, Υ1 представляє О, Υ2 представляє О, групами, незалежно вибраними з груп: гідроксил, Ζ представляє О, m дорівнює 0, А є безпосереднім алкіл, гетероалкіл, циклоалкіл, арил, гетероарил, зв'язком, R3 представляє Н, R4 представляє Η та галоген, галогеналкіл, гідроксіалкіл, алкоксил, алR6 представляє феніл, тоді R5 не представляє коксіалкіл, галогеналкоксил, галогеналкоксіалкіл, феніл; карбоксил, карбоксіалкіл, алкілкарбоксил, аміногколи X представляє NR1, R1 представляє Η, рупа, N-алкіламіногрупа, Ν,Ν-діалкіламіногрупа, Υ1 представляє О, Υ2 представляє О, Ζ представалкіламіногрупа, алкіл(Ν-алкіл)аміногрупа, алляє О, m дорівнює 0, А є безпосереднім зв'язком, кіл(Ν,N-діалкіл)аміногрупа, амідогрупа, NR3 представляє феніл, R4 представляє Η та R6 алкіламідогрупа, Ν,Ν-діалкіламідогрупа, алкіламіпредставляє Н, тоді R5 не представляє феніл; догрупа, aлкiл(N-aлкiл)aмiдoгрyпa, алкіл(Ν,Νколи X представляє S, щонайменше один з Υ1 діалкіл)амідогрупа, тіол, сульфон, сульфонаміногта Υ2 представляє О, m дорівнює 0, А є безпосерупа, алкілсульфонаміногрупа, арилсульфонаміреднім зв'язком, R3 представляє Η або метил, R6 ногрупа, сульфонамідогрупа, галогеналкіл сульпредставляє Η або метил, тоді R5 не представляє фон, алкілтіогрупа, арилтіогрупа, алкілсульфон, феніл, піридин, пірол, тіофен або фуран; арилсульфон, аміносульфон, Nколи X представляє О, Υ1 представляє О, Υ2 алкіламіносульфон, Ν,Ν-діалкіламіносульфон, представляє О, Ζ представляє О, m дорівнює 0, А алкіламіносульфон, ариламіносульфон, ціаногрує безпосереднім зв'язком, R3 представляє метилпа, алкілціаногрупа, гуанідиногрупа, N-ціанохлорид, R4 представляє Η та R6 представляє Н, гуанідиногрупа, тіогуанідиногрупа, амідиногрупа, тоді R5 не представляє феніл. N-аміносульфон-амідиногрупа, нітрогрупа, алкілніБажаними сполуками формули І є сполуки, де трогрупа, 2-нітро-етен-1,1-діамін; використано один чи більше з наступного: R4 вибрано з групи: Н, алкіл, гідроксіалкіл, гаX представляє NR1; логеналкіл, алкоксіалкіл, галогеналкоксил, аміноаЩонайменше один з Υ1 та Υ2 представляє О; лкіл, амідоалкіл, тіоалкіл; зокрема, обидва Υ1 та Υ2 представляють О; R5 представляє моноциклічну групу, що місΖ представляє О; тять 3-7 кільцевих атомів, незалежно вибраних з m дорівнює 0; групи: циклоалкіл, арил, гетероциклоалкіл або геА є безпосереднім зв'язком; тероарил, як варіант, заміщений одним чи більше R1 представляє Н, (С1-3)алкіл або (С1замісниками, незалежно вибраними з групи: гало3)галогеналкіл; зокрема, R1 представляє Η або ген, гідроксил, галогеналкоксил, аміногрупа, N(С1-3)алкіл; найпереважніше R1 представляє Н; алкіламіногрупа, Ν,Ν-діалкіламіногрупа, ціаногруR3 представляє Н, алкіл або галогеналкіл; па, нітрогрупа, алкіл, алкоксил, алкілсульфон, гаособливо R3 представляє Н, (С1-6)алкіл або (С1логеналкіл сульфон, карбоніл, карбоксил, де будь6)галогеналкіл; найпереважніше R3 представляє який алкіл у будь-якому заміснику може сам бути, Н; як варіант, заміщеним одною чи більше групами, R4 представляє Н, алкіл або галогеналкіл; зовибраними з групи: галоген, гідроксил, аміногрупа, крема, R4 представляє Н, (С1-6)алкіл або (С1N-алкіламіногрупа, Ν,Ν-діалкіламіногрупа, алкіл6)галогеналкіл; найпереважніше R4 представляє сульфонаміногрупа, алкілкарбоксіаміногрупа, ціаН; ногрупа, нітрогрупа, тіол, алкілтіол, алкілсульфоR5 представляє, як варіант, заміщене 5 або 6 ногрупа, алкіламіносульфоногрупа, членне кільце, незалежно вибране з групи: циклоалкілкарбоксилат, амідогрупа, N-алкіламідогрупа, алкіл, арил, гетероциклоалкіл або гетероарил; Ν,Ν-діалкіламідогрупа, алкоксил, галогеналкоксил, зокрема, R5 представляє 5 або 6 членний арил карбоніл, карбоксил; або гетероарил; Будь-яка гетероалкілгрупа, визначена вище, є R6 представляє Η, алкіл, пдроксіалкіл, аміноазаміщеним гетероатомом алкілом, що містить одну лкіл, цикпоалкіл-алкіл, алкіл-циклоалюл, арилалчи більше гетерогруп, незалежно вибраних з грукіл, алкіларил, гетероалкіл, гетероциклоалкілпи: N, О, S, SO, SO2, (а гетерогрупою є гетероатом алкіл, алкіл-гетероциклоалкіл, гетероарил-алкіл або група атомів); або гетероалкіл-арил; зокрема, R6 представляє Будь-який гетероциклоалкіл або гетероарил, алкіл, аміноалкіл або гетероарил-алкіл. визначений вище, містить одну чи більше гетерогОсобливі сполуки даного винаходувключають руп, незалежно вибраних з групи: N, О, S, SO, сполуки формули II: SO2; Будь-який алкіл, алкеніл або алкініл, визначені вище, можуть бути лінійними чи , розгалуженими; якщо не встановлено інше, будь-яка вищенаведена алкілгрупа представляє переважно (С1-7)алкіл, а найпереважніше (С1-6)алкіл; За умови, що: коли X представляє NR1, R1 представляє Н, Y1 представляє О, Y2 представляє О, Ζ представде ляє О, m дорівнює 0, А є безпосереднім зв'язком, Аr є 5 або 6 членним арилом або гетероари 15 74624 16 лом, як варіант, заміщеним одним або двома замісниками, вибраними з групи: галоген, аміногрупа, нітрогрупа, (С1-6)алкіл, (С1-6)алкоксил або (С1-6) галогеналкоксил; R6 вибрано з групи: Н, арил або (С1-6)алкіл та R6 є, як варіант, заміщений групою, вибраною з групи: гідроксил, тіоалкіл, феніл, галогенфеніл, піридил або карбамат. Бажаними сполуками формули II є ті, де викоR= Η, (С1-6)алкіл, ОН, СН3О, CF3, CF3O, F, ристано один чи більше з наступного: СІ, Br, I Аr представляє феніл або заміщений феніл, Х=О, S або N окрема, феніл, заміщений одним або двома галоПридатні значення для R6 у сполуках формугеновими замісниками; або Аr представляє 5 або ли І або формули II включають наступні: 6-членне гетероарильне кільце, що містить два гетероатоми, незалежно вибрані з групи: О та Ν; R6 представляє феніл, феніл, заміщений з галогеном, метиленпіридином, або (С1-3)алкіл, як варіант, заміщений з гідроксилом, тіометилом або Метил Етил Пропіл Бутил бензилкарбаматом. Придатні значення для R5 у сполуках формули І або для Аг у сполуках формули II включають: Треба розуміти, що певні замісники та ряд за місників у сполуках формули І або формули II ви 17 74624 18 бирають так, щоб попередити стерично небажані наступним обстеженням рідин з організму тесткомбінації. тварини. Придатні здатні до гідролізу in vivo естеКожна сполука-приклад представляє особлири для карбоксилу включають метоксиметил, а вий та незалежний аспект винаходу. для гідроксилу включають форміл та ацетил, зокКоли у сполуках формули І або формул II наярема ацетил. вні оптично активні центри, ми розкриваємо усі Для застосування сполуки інгібітору металопіндивідуальні оптично активні форми та їх комбіротеїнази винаходу (сполуки формули І або форнації як певні індивідуальні втілення винаходу, а мули II) або її фармацевтично прийнятної солі або також їх відповідні рацемати. Рацемати можна здатного до гідролізу in vivo естеру для терапевтирозділити на індивідуальні оптично активні форми чного лікування (включаючи профілактичне лікуз використанням відомих способів [cf. Advanced вання) ссавців, включаючи людину, її звичайно Organic Chemistry: 3rd Edition: author J March, формують згідно зі стандартною фармацевтичною ст.104-107], включаючи, наприклад, утворення практикою як фармацевтичну композицію. діастереомерних похідних, що мають звичайні Тому, згідно з наступним аспектом нами заоптично активні допоміжним, з їх наступним віддіпропоновано фармацевтичну композицію, яка місленням та розщепленням. тить сполуку винаходу (сполуку формули І або Треба розуміти, що сполуки згідно з винахоформули II) або її фармацевтично прийнятну сіль дом можуть містити один чи більше асиметрично або здатний до гідролізу in vivo естер та фармацезаміщених атомів карбону. Наявність одного чи втично прийнятний носій. більше цих асиметричних центрів (хіральні центри) Фармацевтичні композиції цього винаходу моу сполуці формули І або формул II може давати жна уводити стандартним чином при хворобі або стереоізомери, та у кожному випадку, як зрозумістані, що треба лікувати, наприклад пероральним, ло, поширюватися на всі такі стереоізомери, вклюлокальним, парентеральним, букальним, назальчаючи енантіомери та діастереомери, та суміші, ним, вагінальним або ректальним уведенням або включаючи їх рацемічні суміші. інгаляцією. Для цього, сполуки цього винаходу Коли у сполуках формули І або формули II наможна формувати відомими у рівні техніки засоявні таутомери, ми розкриваємо усі індивідуальні бами, наприклад, у таблетки, капсули, водні або таутомерні форми та їх комбінації як певні індивімасляні розчини, суспензії, емульсії, креми, мазі, дуальні втілення винаходу. гелі, назальні спреї, супозиторії, високодисперсні Як визначено вище, сполуки винаходу є інгібіпорошки або аерозолі для інгаляції, а для паренторами металопротеїнази, зокрема, вони є інгібітерального застосування (включаючи внутрішньоторами ММР12. Кожне з вищенаведених визнавенне, внутрішньом'язове або вливанням) стеричень для сполук формули І або формули II льні водні або масляні розчини або суспензії або представляє незалежне та особливе втілення вистерильні емульсії. находу. На додаток до сполук винаходу, фармацевтиПевні сполуки винаходу мають особливе викочна композиція цього винаходу може також містиристання як інгібітори ММР13 та/або ММР9 та/або ти, або може бути уведеною разом (одночасно або ММР8 та/або ММР3. Певні сполуки винаходу мапослідовно) з одним чи більше потрібними фармають особливе використання як інгібітори агреканакологічними засобами при лікуванні одної чи більзи, тобто інгібітори деградації агрекану. ше хвороб або станів, згаданих вище. Сполуки даного винаходу виявляють сприятФармацевтичні композиції цього винаходу ливий профіль селективності. Не вдаючись до звичайно вживатимуться людиною так, щоб, натеоретичних міркувань, можна вважати, що сполуприклад, отримувати добову дозу 0,5-75мг/кг маси ки винаходу виявляють селективне інгібування для тіла (та переважно 0,5-30мг/кг маси тіла). Цю добудь-якого одного з вищенаведених визначень бову дозу можна давати поділеними дозами, якщо стосовно будь-якої інгібіторної активності відносно необхідно, точна кількість отриманої сполуки та ММР1, як необмежувальний приклад, вони можуть шлях уведення залежить від маси, віку та статі виявляти 100-1000-кратно більшу селективність по пацієнта, якого лікують, та від певної хвороби або відношенню до інгібіторної активності відносно стану, якого лікують згідно з відомими у рівні техніММР1. ки принципами. Сполуки даного винаходу можна забезпечуваЗвичайно одиничні дозовані форми містять ти як фармацевтично прийнятні солі. Вони вклюприблизно 1мг-500мг сполуки цього винаходу. чають кислотно-адитивні солі, як-то гідрохлорид, Тому згідно з наступним аспектом, нами загідробромід, цитрат та малеат, та солі, утворені з пропоновано сполуку формули І або формул II або фосфатною та сульфатною кислотами. Згідно з її фармацевтично прийнятну сіль або здатний до наступним аспектом, придатними солями є солі гідролізу in vivo естер для застосування у способі основ, як-то сіль лужного металу, наприклад, натерапевтичного лікування людини або тварин, або трію або калію, сіль лужноземельного металу, надля застосування як терапевтичного засобу. Ми приклад, кальцію або магнію, або сіль органічного розкрили застосування при лікуванні хвороби або аміну, наприклад, триетиламіну. стану, опосередкованих одним чи більше металоїх можна також забезпечувати як здатні до гідпротеїназними ферментами, Зокрема, ми розкриролізу in vivo естери. Ними є фармацевтично прили застосування при лікуванні хвороби або стану, йнятні естери, що гідролізуються у тілі людини, опосередкованих ММР12 та/або ММР13 та/або утворюючи вихідну сполуку. Такі естери можна ММР9 та/або ММР8 та/або ММР3 та/або агреканаідентифікувати уведенням, наприклад внутрішньозою; особливо застосування при лікуванні хвороби венно тестованій тварині, досліджуваної сполуки, з або стану, опосередкованих ММР12 або ММР9; 19 74624 20 найкраще, застосування при лікуванні хвороби або стану, опосередкованих ММР12. Згідно з подальшим аспектом нами запропоновано спосіб лікування опосередкованих металопротеїназою хвороби або стану, що включає уведення теплокровній тварині терапевтично ефективної кількості сполуки формули І або формули II або її фармацевтично прийнятної солі або здатного до гідролізу in vivo естеру. Ми також розкрили застосування сполуки формули І або форАльдегіди або кетони формули ІІа та сполуки мули II або її фармацевтично прийнятної солі або формули ІІІа у придатному розчиннику обробляздатного до гідролізу in vivo попереднику у виробють основою, переважно у межах температури від ництві медикаменту для застосування при лікузовнішньої до температури кипіння під зворотним ванні хвороби або стану, опосередкованих одним холодильником. Кращі комбінації основачи більше металопротеїназними ферментами. розчинник включають аліфатичні аміни, як-то триОпосередковані металопротеїназою хвороби метиламін, піролідин або піперидин у розчинниках, або стани включають астму, риніт, хронічні обяк-то метанол, етанол, тетрагідрофуран, ацетонітструктивні хвороби легенів (COPD), артрит (як-то рил або диметилформамід, з додаванням води за ревматоїдний артрит та остеоартрит), атероскленеобхідності для розчинення реагентів [Phillips, АР роз та рестеноз, рак, інвазію та метастаз, хвороби, та Murphy, JG, 1951, J. Org. Chem. 16]; або гексапри яких залучено деструкцію тканин, послабленметилдисилазан літію у тетрагідрофурані [Міо, S et ня заміни суглобу стегна, хворобу зубів, фіброзну al., 1991, Тетраhеdrоn 47:2121-2132]; або октагідхворобу, інфаркт та хворобу серця, фіброз печінки рат гідроксиду барію у суміші ізопропанол-вода та нирок, ендометріоз, хвороби, пов'язані з ослаб[Ajinomoto KK, 1993, Патент Японії №05097814]. ленням екстрацелюлярної матриці, серцеву неПереважно, при отриманні сполуки формули І стачу, аневризми аорти, хвороби центральної нецим способом, R3, R5 або R6 не містять додаткорвової системи, як-то хвороба Альцгеймера та вих функціональних груп, альдегідних, кетонних, розсіяний склероз (MS), гематологічні розлади. галогенованих радикалів або будь-яких інших раОтримання сполук винаходу дикалів, що добре відомі фахівцям, які мають поЗгідно з наступним аспектом, даний винахід тенціал впливу на реакцію утворення зв'язку, констосується способів отримання сполуки формули І курування з нею або її інгібування. або її фармацевтично прийнятної солі або здатноСлід зазначити, що багато потрібних вихідних го до гідролізу in vivo естеру, як описано в (а) до матеріалів є комерційно або інакше доступними, (д) нижче (X, Y1, Y2, Z, m, А та R1-R6 визначені або їх можна синтезувати відомими способами чи раніше для сполуки формули І). знайти у науковій літературі. (a) Сполуку формули І можна перетворити у Для отримання сполук винаходу загальної фосіль, зокрема, у фармацевтично прийнятну сіль, рмули Illa (R6 описано вище), сполуки формули або навпаки, відомими способами; у сіль, зокрема, ІІІа, в яких R6 представляє Н, можуть реагувати з фармацевтично прийнятну сіль, сполуку формули прийнятним альдегідом або кетоном, з наступною І можна перетворити у відмінну сіль, зокрема, фадегідратацією та наступним відновленням утворермацевтично прийнятну сіль, відомими способами. ного подвійного зв'язку способами, що добре відо(b) Сполуки формули І, в якій Ζ=Ο, a R4=H, мі фахівцям. можна отримати реакцією сполуки формули На зі (с) Сполуки формули І, в якій Ζ=О, R4=Η та сполукою формули ІІІа або з придатно захищеною Х=N або NR1, зокрема, їх певні стереоізомери, формою сполуки формули ІІІа (як показано на можна також отримати, як описано для двох з чосхемі 1), та, як варіант, після цього, утворенням її тирьох можливих стереоізомерів на схемах 2 та 3 фармацевтично прийнятної солі або здатного до нижче. гідролізу in vivo естеру: 21 Починаючи з пропеноатних похідних формули IV, через діоли Vla або Vlb асиметричним епоксидуванням, з наступним регіоселективним розкриттям водою, або асиметричним дигідроксилуванням. Залежно від хірального допоміжнику при епоксидуванні або дигідроксилуванні, можна отримати показані стереоізомери або їх енантіомери діолів формул Vla або Vlb. [Наприклад, Ogino, Y. et al., 1991, Тетраhеdrоn Lett. 32 (41):5761-5764; Jacobsen, E.N. et al., 1994, Тетраhеdrоn, 50(15):4323-4334; Song, С.Ε. et al., 1997, Тетраhеdrоn Asymmetry, 8 (6):841-844]. Обробка органічною основою та тіонілхлоридом та наступне каталізоване окиснення тетраоксидом рутенію дає циклічні сульфати Vlla та Vllb. Циклічні сульфати формули Vlla та Vllb перетворюють у гідроксіазиди (Схема 3) формули Vllla та Vlllb обробкою азидом натрію у диметилформаміді з наступним обережним гідролізом напівсульфатних інтермедіатів перед обробкою водою. [Gao, Sharpless, 1988, J. Am. Chem. Soc., 110:7538: Kim, Sharpless, 1989, Тетраhеdrоn Lett., 30:655]. Гідроксіазиди формули Vllla та Vlllb гідролізують та відновлюють до -гідрокси- -амінокислоти (не показано на схемі 3), переважно гідроліз LiOH у ТГФ, з наступним відновленням гідросульфідом, магнієм у метанолі або органічними фосфінами способом Стаудингера. -гідрокси- -амінокислоти у свою чергу дають сполуки формули Іа при обробці ціанатом та кислотою у водному середовищі. (d) Сполуки формули І, в якій Ζ=О, a R4 не представляє Н, зокрема їх певні стереоізомери, можна також отримати, як описано для двох з чотирьох можливих стереоізомерів на схемах 2 та 3. Сполуки можна отримати реакцією епоксидів формули V на схемі 2 зі спиртом формули R4-OH, отримавши спирти Via. Наступне перетворення в азиди фосфоазидатом [Thompson, A. S. et al., 1993, J. Org. Chem. 58(22):5 8 86-5 8 88] дає етерні 74624 22 аналоги азидоестерів VІlla на схемі 3, які можна довести до кінцевих продуктів, як описано у способі (с). Радикал R4 у спиртах R4-ОH та радикали R3, R5 та R6 можна придатно захищати. Захисні групи можна видаляти на останньому етапі після перетворення у гідантоїни формули І. (е) Сполуки формули І, в якій Ζ представляє S або NR2 та Υ1 та/або Υ2 представляє О, зокрема їх певні стереоізомери, можна також отримати, як описано для двох з чотирьох можливих стереоізомерів на схемах 2 та 3. Сполуки можна синтезувати розкриттям епоксидів формули V (Схема 2) тіолами R4-SH або амінами R4-NH2 та після цього піддавати аналогічним перетворенням, як описано для спиртів VІlla та Vlllb на схемі 3. Коли аміни R4NH2 використовують, можна виявитися необхідним N-захист проміжних аміноспиртів, зокрема, коли радикал R4 представляє н-алкілгруп. (f) Сполуки формули І, в якій X представляє S та Y1 та/або Y2 представляє О, зокрема їх певні стереоізомери, можна також отримати, як описано для двох з чотирьох можливих стереоізомерів на схемах 2 та 3. Сполуки можна отримати реакцією циклічних сульфатів формули VІla або Vllb, або гідроксилових естерів формули VІa через їх сульфонатні естери, з тіосечовиною та кислотою [1997, Патент Японії №09025273]. Пропеноатні похідні формули IV легко досяжні, наприклад, з альдегідів та фосфонієвих або фосфонатних похідних оцтової кислоти реакціями Віттига або Хорнера-Еммонса [наприклад, van Heerden, P.S. et al., 1997, J. Chem. Soc., Perkin Trans. 1(8):141-1(146)]. (g) Сполуки формули І, в якій X=NR1 та R1=H можна отримати реакцією прийнятного заміщеного альдегіду або кетону формули lІd з карбонатом амонію та ціанідом калію у водних спиртах при 50100°С у герметичній посудині протягом 4-24 годин. 23 74624 24 аналізу частково очищеного, виділеного ферменту, який отримано з мембран ТНР-1, як описано Κ.Μ. Mohler et al., (1994) Nature 370:218-220. Активність очищеного ферменту та його інгібування визначають інкубуванням частково очищеного ферменту при наявності чи відсутності тест-сполук з використанням субстрату 4',5'диметоксифлуорецеїніл Ser.Pro.Leu.Ala.Gln.Ala.Val.Arg.Ser.Ser.Ser.Arg.Cys( 4-(3-сукцінімід-1-іл)-флуорецеїн)-NН2 у буфері для аналізу (50мМ Tris-HCI, pH 7,4, що містить 0,1% Сполуки даного винаходу можна оцінювати, (за масою/об'ємом) Triton Х-100 та 2мМ СаСІ2), наприклад, у таких аналізах: при 26°С протягом 18 годин. Ступінь інгібування Аналізи виділених ферментів визначають як для ММР13 за винятком того, що Родина матричних металопротеїназ, включавикористовували екс 490нм та ем 530нм. Субючи наприклад ММР12, ММР13. страт синтезували таким чином. Пептидну частину Рекомбінантний каталітичний домен ММР12 субстрату сажали на Fmoc-N-H-Rink-MBHAлюдини можна експресувати та очищати, як опиполістирольну смолу вручну або на автоматичносано Parkar A.A. et al., (2000), Protein Expression му синтезаторі пептидів стандартними способами, and Purification, 20:152. Очищений фермент можна в яких залучено використання Fmoc-амінокислоти використовувати для контролю активності інгібітота гексафлуорфосфату О-бензотриазол-і-іл-Nрів таким чином: ММР12 (50нг/мл кінцева концент,N,N',N'-тетраметилуронію (HBTU) як засобу спорація) інкубують протягом 30 хвилин при кімнатній лучення з щонайменше 4- або 5-кратним надлиштемпературі у буфері для аналізу (0,1Μ Tris-HCI, ком Fmoc-амінокислоти та HBTU. Ser1 та Pro2 були pH 7,3, що містить 0,1Μ NaCI, 20мМ СаСІ2, подвійно сполучені. Застосовували наступну стра0,040мМ ZnCІ2 та 0,05% (за масою/об'ємом) Brij тегію захисту бічного ланцюга; Ser1(But), 35) з використанням синтетичного субстрату Mac5 8,12 9,10,11 Ghi (Trityl), Arg (Pmc або Pbf), Ser (Trityl), Pro-Cha-GІy-Nva-His-Ala-Dpa-NH2 за наявності чи Cys13(Trityl). Після приєднання N-термінальну відсутності інгібіторів. Активність визначають виміFmoc-захисну групу видаляли обробкою Fmocрюванням флуоресценції при екс 328нм та ем пептидильної смоли у ДМФ. Отриману так аміно393нм. Процент інгібування розраховують таким пептидильну смолу активували обробкою протячином: % Інгібування дорівнює [Флуоресценціяз гом 1,5-2 годин при 70°С з 1,5-2 еквавалентами інгібітором - Флуоресценціяфонова] поділені на [Флуо4',5'-диметокси-флуорецеїн-4(5)-карбонової кислоресценціябез інгібітору - Флуоресценціяфонова]. ти [Khanna & Ullman, (1980) Anal Biochem. 108:156Рекомбінантний рrоММР13 людини можна 161), яка попередньо активована діізопропілкарекспресувати та очищати, як описано Knauper et бодіімідом та 1-гідроксибензотриазолом у ДМФ]. al. [V. Knauper et al, (1996) The Biochemical Journal Диметоксифлуорецеїніл-пептид далі одночасно 271:1544-1550 (1996)]. Очищений фермент можна позбавляли захисту та відщеплювали від смоли використовувати для контролю активності інгібітообробкою трифлуороцтовою кислотою, що містить рів таким чином: очищений рrоММР13 активують з по 5% кожного з води та триетилсилану. Диметоквикористанням І мМ амінофенілртутної кислоти сифлуорецеїніл-пептид виділяли випарюванням, (АРМА), 20 годин при 21°С; активований ММР13 розтиранням з діетиловим етером та фільтруван(11,25нг на аналіз) інкубують протягом 4-5 годин ням. Виділений пептид реагував з 4-(Nпри 35°С у буфері для аналізу (0,1Μ Tris-HCI, pH малеїнімідо)-флуорецеїном у ДМФ, що містить 7,5, що містить 0,1Μ NaCI, 20мМ СаСІ2, 0,02мМ діізопропілетиламін, продукт очищали за допомоZnCI2 та 0,05% (за масою/об'ємом) Brij 35) з викогою ОФ-ВЕРХ та під кінець виділяли сублімацією з ристанням синтетичного субстрату (7водної оцтової кислоти. Продукт характеризували метоксикумарин-4-іл)ацетил.Рrо.Lеu.Glу.Lеu.N-3за допомогою МС MALDI-TOF та амінокислотного (2,4-динітрофеніл)-1-2,3аналізу. діамінопропіоніл.АІа.Аrg.NН2 за наявності чи відсуПриродні субстрати тності інгібіторів. Активність визначають вимірюАктивність сполук винаходу як інгібіторів деванням флуоресценції при екс 328нм та ем градації агрекану можна аналізувати з викорис393нм. Процент інгібування розраховують таким танням способів, основаних, наприклад, на відкчином: % Інгібування дорівнює [Флуоресценціяз ритті [Е.С. Arner et al., (1998) Osteoarthritis and інгібітором - Флуоресценціяфонова] поділені на [ФлуоCartilage 6:214-228; (1999) J. Biol. Chem., 274 (10) ресценціябез інгібітору - Флуоресценціяфонова]. 6594-6601 та описаних там антитілах]. Потужність Подібний протокол можна застосовувати для сполук як інгібіторів проти колагенази можна виінших експресованих та очищених рrоММР з викозначити, як описано [Т. Cawston та A. Barrett.O979) ристанням субстратів та буферів, оптимальних Anal. Biochem. 99:340-345]. для певних ММР, наприклад, як описано [С. Інгібування активності металопротеїнази в акGraham Knight et al., (1992) FEES Lett. 296(3):263тивності на базі клітин/тканин 266]. Тест як засіб інгібування мембранних шедаз, Адамалізинова родина, включаючи наприклад як-то TNF-конвертази TNF-конвертазу Здатність сполук цього винаходу інгібувати Здатність сполук інгібувати фермент proTNF клітинне перетворення продукування TNF можна конвертази можна визначити з використанням визначити у клітинах ТНР-1 з використанням 25 74624 26 ELISA для детектування вивільненого TNF, як сою/об'ємом PEG400) та у наступний момент часу описано по суті [K.М. Mohler et al., (1994) Nature (наприклад, 5, 15, 30, 60, 120, 240, 480, 720, 1220 370:218-220]. Подібним чином перетворення або хвилин) зразки крові переносять з прийнятної повтрату інших мембранних молекул, як-то описаних судини у 10U гепарину. Фракції плазми отримують, у [N.М. Hooper et al., (1997) Biochem. J. 321:265центрифугують та білки плазми осаджують ацето279] можна тестувати, застосовуючи прийнятні нітрилом (80% за масою/об'ємом кінцева ι конценлінії клітин та з придатними антитілами для визнатрація). Через 30 хвилин при -20°С білки плазми чення відкинутого білку. осаджують центрифугуванням та надосадкову Тест як засіб інгібування інвазії на базі клітин фракцію випарюють до суха з використанням апаЗдатність сполуки цього винаходу інгібувати рату Savant speed vac. Осад відтворюють у буфері міграцію клітин у аналізі інвазії можна визначити, для аналізу досліджуваної сполуки, а далі аналіяк описано [A. Albini et al., (1987) Cancer Research зують на вміст сполуки аналізом з синтетичним 47:3239-3245]. субстратом. Коротше, для оцінюваних сполук буТест як засіб інгібування шедазної активності дують криву концентрація сполуки - реакція. СеTNF суцільної крові рійні розбавлення екстрактів відтвореної плазми Здатність сполук цього винаходу інгібувати аналізують на активність, та кількість сполуки, представленої у вихідному зразку плазми, розрапродукування TNF визначають у аналізі суцільної ховують з використанням кривої концентрація крові людини, де для стимуляції вивільнення TNF сполуки - реакція, зважаючи на фактор розбаввикористовують LPS. Гепаризовану лення загальної плазми. (10одиниць/мл) кров людини, отриману від волонДослідження in vivo Тест як засобу проти TNF терів, розбавляють 1:5 середовищем (RPMI1640 + Здатність сполук цього винаходу як ex vivo гідрокарбонат, пеніцилін, стрептоміцин та глютаTNF інгібіторів визначають на щурах. Коротше, мін) та інкубують (160мкл) з 20мкл тест-сполуки групи самців щурів Wistar Alderley Park (АР) (180(при потроєнні), у ДМСО або прийнятному носії, 210г) дозують сполукою (6 щурів) або носієм ліків протягом 30 хвилин при 37°С у зволоженому (10 щурів) прийнятним шляхом наприклад, перо(5%СО2/95%повітря) інкубаторі, перед додаванням ральним, інтраперитональним, підшкірним. Через 20мкл LPS (E. coli. 0111:B4; кінцева концентрація 90 хвилин щурів вбивали з використанням збіль10мкг/мл). Кожний аналіз включає контролі розбашеної концентрації СО2 та позбавляли крові через вленої крові, інкубованої із одним середовищем сідничну вену у 5 одиниць натрій-гепарину/мл кро(6комірок/планшет) або з відомим інгібітором TNF ві. Зразки крові негайно поміщають на лід та як стандартом. Планшети далі інкубують протягом центрифугують при 2000об./хвил. протягом 10 6 годин при 37°С (зволожений інкубатор), центрихвилин при 4°С та зібрані плазми заморожують фугують (2000об/хвил протягом 10 хвилин; 4°С), при -20°С для наступного аналізу їх дії на продукуплазму збирають (50-100мкл) та зберігають у 96вання TNF стимульованою LPS кров'ю людини. коміркових планшетах при -70°С до наступного Зразки плазми щурів розтоплюють та 175мкл кожаналізу концентрації TNF за допомогою ELISA. ного зразку додають у 96-комірковий планшет. Тест як засіб інгібування in vitro деградації П'ятдесят мкл гепаризованої крові людини далі хряща додають до кожної комірки, змішують та планшет Здатність сполук цього винаходу інгібувати деінкубують протягом 30 хвилин при 37°С (зволожеградацію агреканового або колагенового компонений інкубатор). LPS (25мкл; кінцева концентрація нтів хряща можна визначити, як описано по суті 10мкг/мл) додають до комірок та інкубування про[K.М. Bottomley et al., (1997) Biochem J. 323:483довжують ще 5,5 годин. Контрольні комірки інку488]. бують з 25мкл одного середовища. Планшети далі Фармакодинамічний тест центрифугують протягом 10 хвилин при Для оцінки здатності до виведення та біозас2000об./хвил. та 200мкл надосадкової рідини певоюваності сполук цього винаходу ex vivo застосореносять у комірковий планшет та заморожують вують фармакодинамічний тест, який використопри -20°С для наступного аналізу концентрації вує вищенаведені аналізи з синтетичним TNF за допомогою ELISA. субстратом або альтернативно ВЕРХ або масРезультати аналізу розраховують за допомоспектрометричний аналіз. Це є загальним тестом, гою програмного забезпечення для кожної сполуякий можна використовувати для оцінки швидкості ки/дози: виведення сполук через ряд видів. Тварин (наприклад, щурів, мавп) дозують внутрішньовенно або перорально розчинною композицією сполуки (як-то 20% за масою/об'ємом ДМСО, 60% за ма % інгібування TNF Значення TNF Контролі Значення TNF оброблені Значення TNF Контролі Тест як засобу проти артриту Активність сполуки як засобу проти артриту тестують у індукованому колагеном артриті (СІА) як визначено [D.Е. Trentham et al., (1977) J. Exp. Med. 146,:857]. У цій моделі кислотний розчинний природний колаген типу II викликає поліартрит у 100 щурів при застосуванні у неповному ад'юванті Фрейнда. Подібні умови можна використовувати для виклику артриту у мишей та приматів. Тест як засобу проти раку Активність сполуки як засобу проти раку можна визначити, як описано по суті [І.J. Fidler (1978) 27 74624 28 Methods in Cancer Research .15:399-439], застосоal. (1997) Science, 277:2002]. вуючи, наприклад, лінію клітин В16 [описану В. Винахід далі ілюстровано, але без обмеження Hibner et al., Abstract 283 p75 10th NCI-EORTC наступними прикладами: Symposium, Amsterdam June 16-19 1998]. Отримання вихідних матеріалів Тест як засобу проти емфіземи Згідно зі схемою 4 нижче, гідантоїни 5 отримуАктивність сполуки як засобу проти емфіземи вали двома етапами із загальних амінокислот 3 з можна визначити, як описано по суті [Hautamaki et виділенням інтермедіатів 4. В таблиці 1 наведено деякі з вихідних матеріалів, 5, що були синтезовані. Загальний спосіб отримання був таким. Кашку амінокислоти 3 (25ммоль) та ціанату калію (5,1г, 63ммоль) у воді (75мл) гріли при 80°С протягом приблизно 1 годин. Прозорий розчин охолоджували до 0°С та підкислювали до рН приблизно 1 концентрованою гідрохлоридною кислотою (водн). Утворений білий осад 4 нагрівали при температурі кипіння під зворотним холодильником для 0,5-1 годин, а тоді охолоджували на льоді. У деяких випадках повного перетворення не було досягнуто через 1 годину нагрівання. У цих випадках сирий матеріал обробляли за тим же протоколом знову. Білий твердий продукт фільтрували, промивали водою, сушили та аналізували за допомогою HNMP та LCMS. Таблиця 1 Вихідні матеріали Сполуки 5 на схемі 4 5-(4-Хлор-бензил)-імідазолідин-2,4-діон [3-(2,5-Діоксо-імідазолідин-4-іл)-пропіл]-карбамової кислоти бензиловий естер 5-ізобутил-імідазолідин-2,4-діон 5-Метилсульфанілметил-імідазолідин-2,4-діон 5-втор-бутил-імідазолідин-2,4-діон 5-(2-Гідроксіетил)-імідазолідин-2,4-діон Приклад 1 5-[Гідрокси-(4-йод-феніл)-метил]-5-метилімідазолідин-2,4-діон 4-Йод-бензальдегід (9,280г, 40,0ммоль), 5метил-гідантоїн (4,564г, 40,0ммоль) та 45% водного триметиламіну (6,40мл, 40,0ммоль) нагрівали при температурі кипіння під зворотним холодильником в етанолі (60мл) та воді (40мл) протягом 20 годин під азотом. Утворився білий осад. Після охолодження при кімнатній температурі протягом приблизно 15 хвилин осад. збирали фільтруванням, промивали послідовно етанолом (50%, 50мл), водою (50мл) та діетиловим етером (50мл). Сушіння відсмоктуванням повітря дала потрібну сполуку (7,968г, 23,0моль) з 57,5% виходом як білий твердий продукт з у формі чистого сііастереоізомера. 1 Н ЯМР (300МГц, ДMCO-d6): 10,19 (1Η, s); Вихід (%) 87 АРСІ MS m/z: [МН+] 224,9 50 292,0 85 45 52 36 157,0 161,0 157,0 8,08 (1H, s); 7,64 (2H, d, J=8,6Гц,); 7,07 (2H, d, J=8,4Гц); 5,98 (1H, d, J=4,5Гц,); 4,57 (1H, d, J=4,3Гц); 1,40 (3H, s), APCI-MS m/z: 346,9 [MH+]. Хроматографічне розділення: Частину з 0,158г діастереомерно чистого 5(гідрокси-(4-йодфеніл)-метил)-5-метилімідазолідин-2,4-діону розчиняли у 205мл суміші абсолютний етанол/ізо-гексан (50:50) та фільтрували через 0,45мкм нейлоновий фільтр. Об'єми 5,0мл повторювально уводили у хіральну колонку (Chiralpak AD-H (2см ID х 25см л)), з'єднану з УФдетектором (254нм) та колектором фракцій. Розділення проводили з суміші абсолютний етанол/ізогексан (50:50) як елюент при 6,0мл/хвилину швидкості потоку та чисті енантіомери елюювали. Фракції, що містять один енантіомер, поєднували, концентрували та аналізували на оптичну чистоту хіральною хроматографією (див. нижче). Енантіомер А ("ранішні" фракції) Вихід: 0,068г білого твердого продукту Хіральна хроматографія (Chiralpak AD-H (0,45см І.D х 25см л) при 0,43мл/хвилину суміш абсолютний етанол/ізо-гексан (50:50)) Час утримання: 10,5 хвилин Оптична чистота: 99,9% е.н (нема енантіомеру 29 В) Енантіомер В ("пізніші" фракції) Вихід: 0,071г білого твердого продукту Хіральна хроматографія (Chiralpak AD-H (0,45см I.D х 25см л) при 0,43мл/хвилину суміш абсолютний етанол/ізо-гексан (50:50)) Час утримання: 12,2 хвилин Оптична чистота: 99,6% е.н (представлено 0,24% енантіомеру В) Спектри ЯМР чистих енантіомерів співпадали з спектрами чистих діастереоізомерів. Наступні Приклади отримували наступним способом з прикладу 1. Якщо не встановлено інше, кінцеві сполуки представляють суміш чотирьох стереоізомерів. Колонкову хроматографію використовували для кінцевої очистки або для розділення сііастереоізомерів. Приклад 2 5-((4-Хлор-Феніл)-гідрокси-метил)Іімідазолідин-2,4-діон 74624 30 Приклад 5 5-((4-Трифлуорметил-феніл)-гідрокси-метил]імідазолідин-2,4-діон Приклад 6 5-((3-Трифлуорметил-феніл)-гідрокси-метил]імідазолідин-2,4-діон Приклад 7 5-((2-ТриФлуорметил-Феніл)-гідрокси-метил)імідазолідин-2,4-діон Діастереоізомер А 1 Н ЯМР (400МГц, ДMCO-d6): 10,32 (1Н, s); 8,07 (1Н, s); 7,37 (2Н, d, J=8,5Гц); 7,30 (2H,d, J=8,5Гц); 5,94 (1H, d, J=3,9Гц); 4,92 (1H, t, J=3,2Гц); 4,35 (1H, dd, J=3,1, 1,0гГц). 13 С ЯМР (400МГц, ДMCO-d6): 173,00; 157,36; 138,41; 131,98; 128,86; 127,52; 71,65; 63,88. APCI-MS m/z: 241 [МН+]. Діастереоізомер В 1 Н ЯМР (400МГц, ДMCO-d6): 10,53 (1Н, s); 7,54 (1Н, s); 7,42-7-37 (4Н, m); 5,83 (1Н, d, J=5,6Гц); 4,91 (1Н, dd, J=5,6, 2,6Гц); 4,23 (1Н, dd, J=2,6, 1,5Гц). 13 С ЯМР (400МГц, ДMCO-d6): 173,97; 158,04; 140,62; 131,67; 128,15; 127,89; 70,08; 63,93. APCI-MS m/z: 241 [ΜΗ4]. Приклад 3 5-((4-Хлор-феніл)-гідрокси-метил}1-5-фенілімідазолідин-2,4-діон Приклад 8 5-((4-Трифлуорметокси-феніл)-гідроксиметил]-імідазолідин-2,4-діон Приклад 9 5-((3-Хлор-феніл)-гідрокси-метил]імідазолідин-2,4-діон APCI-MS m/z: 317,1 [МН+]. Приклад 4 5-(4-Ціано-феніл)-гідрокси-метил)-5-ізобутилімідазолідин-2,4-діон Приклад 10 5-((2-Хлор-феніл)-гідрокси-метил)імідазолідин-2,4-діон 31 74624 32 J=8,3Гц); 5,91 (1H, d, J=3,9Гц); 4,87 (1H, t, J=2,7Гц); 4,34 (1H, d, J=2,5Гц). APCI-MS m/z: 333,1 [MH+]. Приклад 16 (3-{4-[Гідрокси-(4-йод-феніл)-метил]-2,5-діоксоімідазолідин-4-іл}-пропіл)-карбамової кислоти бензиловий естер Приклад 11 5-((4-Хлор-3-флуор-феніл)-гідрокси-метил]імідазолідин-2,4-діон Приклад 12 5-((4-Хлор-3-флуор-феніл)-гідрокси-метил)-5метил-імідазолідин-2,4-діон Приклад 13 5-((4-Хлор-3-флуор-Феніл)-гідрокси-метил)-5ізобутил-імідазолідин-2,4-діон APCI-MS m/z: 315.9 [MH+] Приклад 14 5-(1-Гідрокси-3-феніл-аліл)-5-метилімідазолідин-2,4-діон Приклад 17 5-[(4-Бром-феніл)-гідрокси-метил]-5-метилімідазолідин-2,4-діон Отримано альдольною конденсацією 4-бромбензальдегіду та 5-Метил-імідазолідин-2,4-діону. 1 Н ЯМР(400МГц, ДMCO-d6): 10,18 (1 Η, s); 8,08 (1H, s); 7,46 (2H, d, J=8,4Гц); 7,20 (2H, d, J=8,4Гц); 5,99 (1H, d, J=4,4Гц); 4,59 (1H, d, 3,81Гц); 1,39 (3H,s). APCI-MS m/z: 298,9 [MH+]. Приклад 18 5-[(3,5-Диметил-ізоксазол-4-іл)-гідроксиметил]-5-метил-мідазолідин-2,4-діон Отримано альдольною конденсацією 3,5диметил-ізоксазол-4-карбальдегіду та 5-Метилімідазолідин-2,4-діону. APCI-MS m/z: 240 [MH+] 5 Приклад 19 5-[(4-Бром-феніл)-гідрокси-метил]-5метилсульфанілметил-імідазолідин-2,4-діон Отримано альдольною конденсацією 4-бромбензальдегіду та 5-метилсульфанілметилімідазолідин-2,4-діону. 1 H ЯМР (400МГц, ДMCO-d6): 10,45 (1Η, s); 7,88 (1H, s); 7,38-7,22 (5H, m); 6,54 (1H, d, J=16,1Гц); 6,22 (1H, dd, J=7,3, 7,6Гц); 5,56 (1H, d, J=4,5Гц); 4,09 (1H, d, J=3,6, 4,5Гц); 1,27 (3H,s). APCI-MS m/z: 247,1 [МН+]. Приклад 15 5-[Гідрокси-(4-йод-феніл)-метил]-імідазолідин2,4-діон 1 H ЯМР (300МГц, ДМСО-d6): 10,32 (1Η, s); 8,06 (1H, s); 7,66 (2H, d, J=8,1Гц); 7,10 (2H, d, Приклад 20 5-[(4-Бром-феніл)-гідрокси-метил]-5-(2гідроксіетил)-імідазолідин-2,4-діон Отримано альдольною конденсацією 4-бромбензальдегіду та 5-(2-гідроксі-етил)-імідазолідин2,4-діону. 33 74624 34 Приклад 22 5-[(4-Бромфеніл)гідрокси-метил]-5-піридин-2ілметил-імідазолідин-2,4-діон Отримано альдольною конденсацією 4-бромбензальдегіду та 5-піридин-4-ілметилімідазолідин-2,4-діону. Приклад 21 5-[(4-Бром-феніл)-гідрокси-метил]-5-(4хлорбензил)-імідазолідин-2,4-діон Отримано альдольною конденсацією 4-бромбензальдегіду та 5-(4-хлор-бензил)-імідазолідин2,4-діону. Комп’ютерна верстка О. Гапоненко Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюMetalloproteinase inhibitors
Автори англійськоюLepistoe Matti
Назва патенту російськоюИнгибиторы металлопротеиназы
Автори російськоюЛеписте Матти
МПК / Мітки
МПК: C07D 401/10, C07D 409/06, C07D 409/12, A61P 43/00, A61P 1/04, A61P 15/00, A61P 19/02, C07D 277/20, A61P 17/00, C07D 405/06, A61P 1/02, A61P 19/08, A61K 31/4174, A61P 11/02, A61P 17/16, C07D 417/10, A61P 3/10, A61P 35/04, A61K 31/433, C07D 277/34, C07D 405/10, A61K 31/5377, A61P 25/28, A61P 9/00, C07D 235/02, A61P 37/08, C07D 403/06, A61P 1/16, A61P 27/02, A61P 7/00, C07D 403/10, A61P 17/02, A61K 31/496, C07D 403/12, C07D 401/12, A61P 11/06, A61P 17/06, A61P 9/10, C07D 233/78, A61P 25/00, A61P 35/00, C07D 409/14, A61P 11/00, A61K 31/497, A61K 31/4178, A61K 31/426, A61K 31/454, A61P 19/10, A61K 31/4439, A61P 29/00, A61P 19/00, C07D 401/06
Мітки: металопротеїнази, інгібітори
Код посилання
<a href="https://ua.patents.su/17-74624-ingibitori-metaloprotenazi.html" target="_blank" rel="follow" title="База патентів України">Інгібітори металопротеїнази</a>
Пептидні інгібітори вірусу гепатиту с, спосіб їх одержання та фармацевтична композиція

Номер патенту: 73480
Опубліковано: 15.08.2005
Автори: Ранкур Жан, Гудро Наталі, Гіро Еліз, Пупар Марк-Андре, Кемерон Дейл, Лінас-Брюне Монтсе, Бейлі Мьоррі Д., Тсантріцос Юла С.
МПК: C12N 15/09, A61K 31/13, A61P 1/16, A61P 43/00, A61P 31/14, A61K 45/00, C07K 5/10, C07K 5/107, A61P 31/12, C07K 7/06, A61K 38/21, C07K 1/10, C07K 14/81, A61K 38/55, A61K 38/00, A61K 31/7052
Мітки: інгібітори, вірусу, пептидні, композиція, спосіб, одержання, фармацевтична, гепатиту
Формула / Реферат:
1. Сполука формули (І), включаючи її рацемати, діастереоізомери і оптичні ізомери: (І),у якійа дорівнює 0 або 1,b дорівнює 0 або 1,Y означає Н або С1-С6алкіл,В означає Н, дансил або ацильне похідне формули R7-C(O)- ,R7 означає С1-С6алкіл або Het, який вибирають з групи, що містить: , , ,, , або,R6 означає С1-С6алкіл, заміщений карбоксилом, R5 означає...
Тієнопіримідини як інгібітори фосфодіестерази, спосіб їх одержання та фармацевтичний препарат на їх основі

Номер патенту: 72256
Опубліковано: 15.02.2005
Автори: Джонас Рохус, Шеллінг П'єр, Крістадлер Марія, Клюхен Франц-Вернер
МПК: A61P 9/04, A61P 9/00, C07D 495/04, A61P 15/10, A61K 31/519, A61P 43/00, A61P 15/00
Мітки: інгібітори, одержання, фармацевтичний, основі, спосіб, фосфодіестерази, тієнопіримідини, препарат
Формула / Реферат:
1. Сполука формули I, Ів якійR1, R2 у кожному випадку незалежно один від одного являють собою Н, А, ОН, ОА або Hal,Х являє собою R4, R5 або R6, монозаміщений R7,R4 являє собою нерозгалужений або розгалужений алкілен з 1-10 С-атомами, в якому одна або дві CH2-групи можуть бути заміщені групами -СН=СН-,R5 являє собою циклоалкіл або циклоалкілалкілен, що містить 5-12 С-атомів,R6 являє собою феніл...
Заміщені похідні пурину як інгібітори клітинної адгезії

Номер патенту: 72923
Опубліковано: 16.05.2005
Автори: Кнолле Йохен, Гурве Жан-Франсуа, Рюксер Жан-Марі, Гейдек Томас Р., Пейман Ануширван
МПК: A61P 29/00, A61P 3/14, A61P 19/00, A61K 31/52, C07D 473/34, A61P 19/10, A61P 43/00, A61P 1/02, A61P 5/24
Мітки: адгезії, інгібітори, пурину, клітинної, похідні, заміщені
Формула / Реферат:
l. Сполука формули І, Ів якійВ - (С1-С18)-алкіл, (С3-С14)-циклоалкіл, (С3-С14)-циклоалкіл-(С1-С8)-алкіл-, (С3-С14)-арил, (С5-С14)-арил-(С1-С8)-алкіл-, (С5-С14)-гетероарил, (С5-С14)-гетероарил-(С1-С8)-алкіл-, фтор, хлор, бром, гідрокси, ціано, трифторметил, нітро, гідроксикарбоніл-, (С1-С6)-алкокси, (С1-С6)-алкоксі-(С1-С6)-алкіл-, (С1-С6)-алкоксикарбоніл-, (С1-С6)-алкілкарбоніл-, (С5-С14)-арилкарбоніл-,...
Дициклічні гетероароматичні сполуки як інгібітори білкової тирозинкінази

Номер патенту: 66827
Опубліковано: 15.06.2004
Автори: Сміт Катрін Джейн, Лекі Керін Елізабет, Картер Малколм Клайв, Кокерілл Джордж Стюарт, антріп Стівен Беррі
МПК: C07D 417/04, A61K 31/519, C07D 405/04, C07D 417/14, C07D 239/94, A61K 31/517, A61P 35/00, A61P 43/00, C07D 405/14, A61P 29/00, C07D 471/04, A61P 17/06, A61K 31/4709
Мітки: дициклічні, гетероароматичні, білкової, інгібітори, тирозинкінази, сполуки
Формула / Реферат:
1. Дициклічні гетероароматичні сполуки формули (І): (І)або їх солі чи сольвати як інгібітори білкової тирозинкінази,деY - CR1, a V - N,або Y - CR1, a V - CR2,R1 - група CH3SO2CH2CH2NHCH2-Ar, причому Аr вибрано з групи, яка складається з фурану та тіазолу, кожний з яких заміщено, як варіант, одним чи двома галогенами,...
Інгібітори фарнезил білок трансферази із радіосенсибілізуючими in vivo властивостями

Номер патенту: 64796
Опубліковано: 15.03.2004
Автори: ван Гінкел Робер Франциск, Енд Девід Вілліам, Флорен Вім Джоанна, Вутерс Вальтер Будев'єн Леопольд
МПК: A61K 41/00
Мітки: радіосенсибілізуючими, трансферази, інгібітори, властивостями, фарнезил, білок
Формула / Реферат:
1. Використання принаймні інгібітора фарнезил білок трансферази для отримання фармацевтичної композиції, що має радіосенсибілізуючі властивості, для введення in vivo до, в процесі та після опромінення пухлини при лікуванні раку.2. Використання за п. 1, за яким вказаний інгібітор фарнезил білок трансферази є сполукою формули (І) або сполукою за формулами (II) або (III), які метаболізуються in vivo у сполуку формули (І), причому вказані...
Попередній патент: Роторно-імпульсний апарат
Наступний патент: Накопичувальна інформаційно-вимірювальна система силової установки літального апарата
Випадковий патент: Спосіб диференційної діагностики ступенів тяжкості перебігу хронічного обструктивного захворювання легень