Спосіб одержання платифіліну гідротартрату
Номер патенту: 32355
Опубліковано: 15.12.2000
Автори: Георгієвський Віктор Петрович, Шеїн Анатолій Тихонович, Акімов Володимир Андрійович


















Текст
МПК5 A 61 К 35/78 СПОСІБ ОДЕРЖАННЯ ПЛАТИФІЛІНУ ГІДРОТАРТРАТУ Винахід відноситься до медицини та хіміко-фармацевтичної промисловості, зокрема, до способів одержання алкалоїду платифіліну з рослинної сировини. Відомий спосіб одержання суми алкалоїдів з рослинної сировини, який здійснюють таким чином. Надземну частину барвінку малого з вмістом алкалоїдів 0,24% екстрагують водним розчином сірчаної кислоти. Одержаний сірчанокислий екстракт пропускають через батарею з чотирьох адсорберів, заповнених олеофільним катіонітом КО-40. Після закінчення адсорбції катіоніт промивають холодною водою, сушать до залишкової вологи 40-50% і проводять десорбцію 5% розчином хлористого натрію в 70% етиловому спирті. З одержаного елюату відганяють спирт під вакуумом. До одержаного водного розчину алкалоїдів додають 25% розчин аміаку до рН 9-9,5, і суму основ алкалоїдів, що випала, залишають для кристалізації при 4°С протягом 2 год, після чого суму основ відфільтровують і сушать протягом 11-12 год при 30-40°С. Одержану суму основ алкалоїдів розчиняють в етиловому спирті, насиченому хлористим воднем (рН 3,5). Спирт відганяють і одержують хлористоводневі солі алкалоїдів з вмістом суми алкалоїдів 95,0%. Катіоніт після проведення десорбції піддають регенерації (1). Відомий спосіб одержання алкалоїду скополаміну, який здійснюють таким чином. Подрібнена рослина дурман інденський (Dutara innoxia) з вмістом скополаміну 0,16%, екстрагують водою при ЮО^С та співвідношенні сировина : екстрагент 1:(10-12). Екстракцію проводять в батареї з чотирьох екстракторів по принципу протито 2 ку. Одержаний екстракт, що містить скополаміну основу, пропускають черев адсорбер, заповнений знебарвлювальним іонітом ИА-Ір. Очищений екстракт подають на адсорбційну батарею, що складається з п'яти адсорберів, заповнених сухим катіонітом КУ-2х8. Сорбцію скополаміну проводять у дінамічних умовах. Десорбцію ведуть в тих же адсорберах 1,5% розчином хлористого натрію в 65% спирті, після чого спирт відганяють під вакуумом, водний розчин підлужують 25% розчином аміаку до рН 9,0 і п'ятиразово здобувають скополамін порціями дихлоретану протягом 10 хв кожного разу. Дихлоретанові здобування сушать безводним сульфатом натрію, фільтрують і концентрують при нагріванні під вакуумом. Після відгонки дихлоретану одержують сиропоподібний залишок жовтого кольору, що є сумою алкалоїдів основ, до якого додають рівну кількість по об'єму спирту, охолоджують до 5°С і при перемішуванні додають 65% розчин бромистоводневої кислоти до рН 4,0. Масу залишають в кристалізаторі на 6 год, потім фільтрують і промивають невеликою кількістю холодного спирту, одержуючи технічний скополаміну гідробромід, який містить скополаміну основу. Технічний скополаміну гідробромід розчиняють при перемішуванні в триразовій кількості спирту з додаванням активованого вугілля. Потім вугілля відфільтровують, а спиртовий розчин залишають для кристалізації, після чого осадок відфільтровують, промивають два рази охолодженим спиртом і сушать при 35°С. Одержують скополаміну гідробромід, що відповідає фармакопейним вимогам (2). Відомий засіб одержання ергоалкалоїдів з рослинної сировини , який здійснюють таким чином. Подрібнені ріжки спориння екстрагують три рази до повного здобування 50% водним розчином ацетону, що містить сірчану кислоту у співвідношенні 1:1000 для утворення кислого середовища рН 4,5, при кімнатній температурі та постійному перемішуванні в екстракторі. Термін кожної екстракції складає з одну годину. Співвідношення сировини та екстрагенту 1:8. Одержані екстракти об'єднують і упарюють при температурі 40-50°С та залишковому тиску мінус 78,4-88,2 кїїа. Одержаний водний залишок доводять до рН 9-10, використовуючи 25% ровчин аміаку, і екстрагують 4 рази хлороформом. При цьому ергоалкалоїди у вигляді солей переходять у вільну основу. Об'єднані хлороформні екстракти фільтрують черев шар окису алюмінію, фільтр промивають хлороформом, упарюють на вакуум-випарному циркуляційному апараті при 30-4СГС та залишковому тиску 78,4-83,2 кПа до сухого залишку. До залишку додають етилацетат (1:5), розчиняють при слабкому нагріванні при 25-30°С і кристалізують, додаючи сірчаний ефір та затравку, при 5°С протягом 8 год. Після фільтрації одержують 12-15 г кристалічної фракції готового продукту з ступенем чистоти 93%. Вихід кристалічної субстанції ергоалкалоїдів складає 70%. (3). Відомий спосіб одержання ергоалкалоїдів, : який полягає у тому, що подрібнені ріжки спориння екстрагують дихлоретаном в лужному середовищ, обробляють екстракт розчином ортофосфорної кислоти з подальшою обробкою фосфорно-кислотного екстракту хлороформом. Хлороформне здобування упарюють, а потім осаджують петролейним ефіром. Одержану суму алкалоїдів фільтрують, промивають петролейним ефіром, вивітрюють під тягою в вакуум-сушильній шафі, одержуючи технічну суму алкалоїдів, яку розчиняють при кімнатній температурі в хлороформі. Одержаний розчин пропускають під вакуумом через хроматографічну колонку, заповнену комбінованим сорбентом з окису алюмінію та активованого вугілля у співвідношенні 1:1. Алкалоїди обробляють хлороформом для негативної реакції на алкалоїди у пробі з петролейним ефіром. Елюат упарюють і осаджують очищену фракцію ергоалкаоїдів петролейним ефіром. Ємкість з осадком витримують 5 год, після чого осадок відфільтровують на фільтрі Еюхнера, промивають петролейним ефіром, вивітрюють під тягою протягом 5 год і потім сушать в вакуум-сушильній шафі. Вихід готового продукту складзє 59,61% від вмісту продукту в сироЕИНІ (4). Відомий спосіб одержання платифіліну гідротартрату, який здійснюють таким чином. До апарату ємкістю 50 л з нержавіючої сталі з оболонкою, удаваним дном, холодильником, мішалкою та барботером для відгонки дихлоретану поміщають 2 кг подрібненої трави жовтозілля, 1,6 л 12% водного розчину соди, 0,06 кг маїсового цукру та 20 л дихлоретану. Суміш кип'ятять при перемішуванні протягом 2 год. Потім вміст апарату охолоджують і зливають перший екстракт. Подальше дворазове вимивання алкалоїдів з трави проводять при кімнатній температурі. Після зливу третього екстракту до апарату подають через барботер гостру пару і відганяють дихлоретан, який залишився в траві. S першого та другого екстрактів послідовно здобувають алкалоїди 10% сірчаною кислотою. Незначну кількість алкалоїдів, які містять третій екстракт, використовують для здобування свіжої трави. З кислого розчину осаджують суму алкалоїдів 25% аміаком при температурі 5-10°С. Технічну суму алкалоїдів відсмоктують, промивають водою та висушують при темпаратурі Б0-60°С. Аміачний маточник здобувають дихлоретаном, який приєднують до третього екстракту. Одержану суму алкалоїдів кип'ятять зі спиртом для відокремлення основної частини сенецифіліну. Спиртовий розчин, який містить переважно платифілін, застосовують для одержання технічного бітартрату. Осадок, до не розчинився (сенецифілін), використовують як сировину для одержання диплацину. Технічний бітартрат перекристалізовують з 87% спирту (5). Найбільш близьким до заявляемого є спосіб одержання платифіліну гідротартрату, який здійснюють таким чином. ТраЕу жовтозілля плосколиотного подрібнюють до розміру частинок £-5 мм. Екстракцію проводять в екстракторах з оболонкою для обігріву або охолоджен 5 ня, зкомунікованою зі зворотним холодильником, з мішалкою, що робить 60 об/хв, удаваним днищем, заправленим оірошинельнш сукном. Першу екстракцію проводять киплячим дихлоретаном в присутності глюкози та 12,5% водного розчину соди протягом 2 год. Після зливу третього екстракту дихлоретанові екстракти передають на наступну стадію технологічного процесу. До оболонки екстрактора подають пару і при перемішуванні ведуть відгін дихлоретану від шроту, потім до оболонки екстрактора подають холодну воду для охолодження апарата, після чого шрот відвантажують і направляють у відЕал , а екстрактор підготовлюють для завантаження свіжої партДЇ сировини. З кожного дихлоретанового здобування алкалоїди екстрагують 10% водним розчином сірчаної кислоти протитоком, одержані сірчанокислотні здобування об'єднують і завантажують до реактору з оболонкою для охолодження при перемішуванні 60 об/хв. Охолоджують роз-' сольним розчином до 3-4°С і при перемішуванні додають тонким струмінцем 25% ЕОДНИЙ розчин аміаку до рН 9, внаслідок чого сума алкалоїдів випадає в осадок. Масу в реакторі витримують при 3°С протягом години для більш повного випадання суми алкалоїдів в осадок, після чого суспензію при перемішуванні спускають в нутч-фільтр, осадок на фільтрі промивають £-3 рази водою до зникнення аміаку, а потім сушать на дерев'яних лотках в камерній сушарці при 60-70°С протягом 6 год при періодичному перемішуванні. Висохлий осадок розтирають на порошок, після чого завантажують до реактору в оболонкою для обігріву та мішалкою 120 об/хв і змішують з 96% етиловим спиртом. Одержану суміш кип'ятять протягом ЗО хв, після чого суспензію технічного сенецифіліну в спиртовому ' розчині платифіліну фільтрують на нутч-фільтрі. Осадок технічного сенецифіліну промивають з 'виносом гарячим спиртом, відфільтровують, добре віджимають і висушують в вакуум-сушарці при 50-60°С і залишковому тиску 50-75 мм рт.ст. 6 Для одержання технічного платифіліну гідротартрату спиртовий розчин шіатифіліну-основи повертають до того ж реактора, до нього вносять ровраховану кількість виннокам'яної кислоти, вміст реактору нагрівають до 80°С при перемішуванні. Кристали платифіліну гідротартрату випадають вже при кип'ятінні або при охолодженні розчину до кімнатної температури. При цій температурі масу витримують протягом 12 год. Потім кристали технічного платифіліну гідротзртрату, що випали, відфільтровують, одержаний осадок промивають охолодженим спиртом і висушують в вакуум-сушильній шафі при 5О60°С і залишковому тиску 50-75 мм рт.ст. В реакційний апарат завантажують технічний платифіліну гідротартрат, 90%. етиловий спирт, до оболонки апарата подається пара. Вміст апарата кип'ятять при температурі 80-83°С протягом 15 хв до ПОЕНОГО розчинення осадку, потім до розчину вносять порошкоподібне активоване вугілля, масу нагрівають 10-15 хв і фільтрують на нутч-фільтрі. Осадок вугілля на фільтрі промивають -гарячим спиртом. З нутч-фільтра ровчин зливають до крісталізатора, який охолоджують розсолом до температури 3-5°С, і витримують протягом 15 год. Кристалічний осадок кінцевого продукту, що виділився, відфільтровують, осадок на фільтрі два рази промивають охолодженим 95% етиловим спиртом, добре віджимають, помішають в скляні кристалізатори і висушують в вакуум-сушильній шафі при 50-60°С і залишковому тиску 50-100 мм рт.ст. (6). До недоліків прототипу слід віднести складність і нестабільність технологічного процесу, високу залежність результатів від виду та якості «вихідної сировини, невисокий вихід і недостатня чистота ЦІЛЬОЕОГО продукту. В основу винаходу поставлено завдання створення способу одержання платифіліну гідротартрату шляхом підбору технологічних операцій у такій послідовності та взаємозв'язку і з такими режи мами та параметрами, які б забезпечили спрощення способу за рахунок скорочення кількості стадій, універсальність способу за рахунок можливості переробки всіх частин рослини, підвищення ЕИКОДУ та якості цільового продукту. Поставлене завдання вирішується тим, що в заявляемому способі, який включає екстракцію подрібненого жовтозілля плосколистно-го органічним розчинником, а потім 10% сірчаною кислотою, нейтралізацію сірчанокислотного екстракту £5% розчином аміаку, кристалізацію, фільтрацію та сушіння одержаних кристалів, розчинення їх в киплячому 96% спирті етиловому, фільтрацію одержаної суспензії, змішування з виннокам'яною кислотою при нагріванні, охолодження та перекристалізацію з подальшою фільтрацією та сушінням готового продукту, у відповідності з винаходом екстракцію проводять 60-70% спиртом етиловим при співвідношенні сировина : екстрагент 1:(3-б), одержаний екстракт упарюють, підкислений водний концентрат витримують при перемішуванні, додають цинк, фільтрують, ДОЕОДЯТЬ £5% розчином аміаку до рН 9,0-9,3, екстрагують хлористим метиленом, а після екстракції хлористометиленового екстракту 10% сірчаною кислотою проводять очистку сірчанокислотного екстракту активованим Еугіллям, причому як сировину використовують надземну частину та/'або кореневище жовтозілля плосколистного, а водний концентрат під?;ислюють однаковими кількостями 20% соляної кислоти та £0% сірчаної кислоти. Технічний результат, якого досягають внаслідок здійснення винаходу, полягає у спрощенні способу одержання платифіліну гідротартрату *за рахунок скорочення кількості стадій, досягненні ' універсальності способу за рахунок можливості переробки всіх частин рослини, у підвищенні виходу та якості цільового продукту. Заявляємий спосіб здійснюють таким чином. Підготовлену сировину (надземна частина або кореневище жов 8 тозілля плосколистного) помішають до екстрактора з фільтруючим елементом і екстрагують 60-70°С спиртом етиловим при співвідношенні сировина : екстрагент 1 : (3-6). Одержаний екстракт упарюють під вакуумом, доводять до рН 3-4 20% соляною кислотою та 20% сірчаною кислотою, які беруть у однакових кількостях, і витримують при безперервному перемішуванні протягом 8-24 год. До підкисленого концентрату для відновлення N-окисів алкалоїдів вводять цинк і перемішують протягом 4 год під азотом, після чого фільтрують для відокремлення крупних конкрецій та згустків смол і установлюють рН 9,0-9,3 додаванням 25% ровчину аміаку. Нейтралізований концентрат екстрагують хлористим метиленом, а одержаний хлористометиленовий екстракт знову екстрагують 10%, розчином сірчаної кислоти. Одержаний сірчанокислотний екстракт очищають активованим вугіллям від ліпофільної та поліфенольної фракцій, охолоджують до 4°С, установлюють рН 9,0-9,3 додаванням 25% розчину аміаку і витримують в кристалізаторі протягом 1-5 год. Одержану кристалічну масу фільтрують для відокремлення рідкої фази, і кристали суми алкалоїдів сушать при 55-80°С протягом 10-24 год. Висушені кристали змішують зі спиртом етиловим (1:6) і кип'ятять протягом 30-45 хв. Одержану суспензію фільтрують і витримують при 4°С протягом 4-24 год, після чого знову фільтрують. Одержаний фільтрат змішують з виннокам'яною кислотою (2:1), нагрівають до розчинення кислоти і витримують в кристалізаторі при 4°С протягом 8-16 год. Одержану кристалічну масу фільтрують, осадок висушують на повітрі протягом 24 год, після чого готовий продукт фасують в герметичну упаковку. Лікарський васіб платифіліну гідротартрат, який одержують по заявляемому способу, належить до похідних, геліотридану (1-метил-пірралізидин). Платифіліну гідротартрат виявляє холінолітичну дію і по впливу на периферічні холінореактивні системи близький э до атропіну. На ЦНО, особливо на судинорухові центри, виявляє заспокійливу дію, має спазмолітичні властивості. Призначають платифіліну гідротартрат при спазмах гладких м'-явів органів черевної порожнини, виразці шлунка та дванадцятипалої кишки, бронхіальній астмі. Препарат вменшує опавми кровоносних судин, спазми судин головного мозку, для купірування гострого виразкового болю, а також кишкової, печінкової колік. В офтальмологічній практиці платифіліну гідрстартрат застосовують для розширення зіниці. Наводимо конкретні приклади здійснення винаходу. Приклад 1. 20,0 кг підготовленої сировини (трава жовтозілля плосколистного) поміщають до екстрактора з фільтруючим елементом і екстрагують 60 л 50% спирту ЄТИЛОЕОГО (1:3). Одержаний екстракт упарюють під вакуумом, доводять до рН 3,0 однаковими кількостями £0% соляної кислоти та £0% сірчаної кислоти (усього 6,Е5 л) і витримують при безперервному перемішуванні протягом 10 год. До підкисленого водного концентрату для відновлення N-окисів алкалоїдів вводять 2,2 кг цинку і перемішують протягом 4 год під азотом, після чого фільтрують для відокремлення крупних конкрецій та згустків смол і установлюють рН 9,0 додаванням £5% розчину аміаку. Нейтралізований концентрат екстрагують ЗО л хлористого метилену , а потім одержаний хлористометиленовий екстракт знову екстрагують 3 л 10% розчину сірчаної кислоти. Одержаний сірчанокислотний екстракт очищають активованим вугіллям від ліпофільної та поліфенольної фракцій, охолоджують до 4°С, установлюють рН 9,0 додаванням 25Z розчину аміаку і витримують в кристалізаторі протягом 1 год. Одержану кристалічну масу фільтрують для відокремлен-' ня рідкої фази, і кристали суми алкалоїдів сушать при 80°С в слабкому тоці псшітря протягом 10 год. Висушені кристали вмішують зі спиртом етиловим (1:6) і кип'ятять протягом ЗО ХЕ. Одержану суспензію фільтрують і витримують при 4°С протягом 4 год, 10 після чого внову фільтрують. Одержаний фільтрат змішують з виннокам'яною кислотою (2:1), нагрівають до розчинення кислоти і вит-'** римують в кристалізаторі при 4°С протягом 8 год. Одержану кристалічну масу фільтрують, осадок висушують на повітрі протягом 24 год. Вихід платифіліну гідротартрату складає 1,15 кг на одну тону сировини в вмістом алкалоїдів 0,4%. Приклад 2. 20,0 кг підготовленої сировини (трава жовтозілля плосколистного) поміщають до екстрактора з фільтруючим елементом і екстрагують 100 л 70% спирту етилового (1:5). Одержаний екстракт упарюють під вакуумом, доводять до рН 4,0 однаковими кількостями 20% соляної кислоти та £0% сірчаної кислоти (усього £,25 л) і витримують при безперервному перемішуванні протягом 8 год. До підкисленого водного концентрату для відновлення-N-окисів алкалоїдів вводять 0,8 кг цинку і перемішують протягом 4 год під азотом, після чого фільтрують для відокремлення крупних конкрецій та згустків смол і установлюють рН 9,2 додаванням 25% розчину аміаку. Нейтралізований концентрат екстрагують 25 л хлористого метилену, а потім одержаний хлорнетометиленовий екстракт знову екстрагують 5 л 10% розчину сірчаної кислоти. Одержаний сірчанокислотний екстракт очишають активованим вугіллям від ліпофільної та поліфенольної фракцій, охолоджують до 4°С/ установлюють рН 9,2 додаванням 25% розчину аміаку і витримують в кристалізаторі протягом 4 год. Одержану кристалічну масу фільтрують для відокремлення рідкої фази, і кристали суми алкалоїдів сушать при 55°С в слабкому тоці повітря протягом 24 год. Висушені кристали змішують щзі спиртом етиловим (1:6) і кип'ятять протягом 45 хв. Одержану ' суспензію фільтрують і витримують при 4°С протягам 16 год, після чого знову фільтрують. Одержаний фільтрат змішують з виннокам'яною кислотою (2:1), нагрівають до розчинення кислоти і витримують в кристалізаторі при 4°С протягом 16 год. Одержану кристаліч ну масу фільтрують, осадок висушують НЕ повітрі протягам 24 год. Вихід платифіліну гідротартрату складає 1,35 кг на одну тону сировини з вмістом алкалоїдів 0,4%. Приклад 3. 20,0 кг підготовленої сировини (кореневиїда жовтозілля плосколистного) помішають до екстрактора з фільтруючим елементом і екстрагують 80 л 70% спирту етилового (1:4). Одержаний екстракт упарюють під вакуумом, доводять до рН 3,5 однаковими кількостями 20% соляної кислоти та 20% сірчаної кислоти (усього 3,75 л) і витримують при безперервному перемішуванні протягом 24 год. До підкисленого водного концентрату для ВІДНОЕЛЄННЯ N-окисів алкалоїдів вводять 1,25 кг цинку і перемішують протягом 4 год під азотом, після чого фільтрують для відокремлення крупних конкрецій та згустків смол і установлюють рН 9,2 додаванням 25% розчину аміаку. Нейтралізований концентрат екстрагують 20 л хлористого метилену, а потім одержаний хлористометиленовий екстракт знову екстрагують 10 л 10% розчину сірчаної кислоти. Одержаний сірчанокислотний екстракт очищають активованим вугіллям від ліпофільної та поліфенольної фракцій, охолоджують до 4°С, установлюють рН 9,2 додаванням 25% розчину аміаку і витримують в кристзлізаторі протягом 6 год. Одержану кристалічну масу фільтрують для відокремлення рідкої фази, і кристали суми алкалоїдів сушать при 60°С в слабкому тоці повітря протягом 24 год. Висушені кристали (540 г) змішують з 3,24 л спирту етилового (1:6) і кип'ятять протягом 45 хв. Одержану суспензію фільтрують і витримують при 4°С протягом 24 год, після чого знову фільтрують. Одержаний фільтрат змішують з 270 г виннокам'яної кислоти (2:1), нагрівають до розчинення чсислоти і витримують в кристалізаторі при 4°С протягом 16 год. Одержану кристалічну масу фільтрують, осадок висушують на повітрі протягом 24 год. Вихід платифіліну гідротартрату складає 12,95 кг на одну тону сировини з вмістом алкалоїдів 3%. 12 Послідовність та взаємозв'язок технологічних операцій заявляемого способу, підбір режимів та параметрів забезпечують вико* нання поставленого у винаході завдання. Екстракція рослинної сировини 60-70% спиртом етиловим забез печує найбільш повне здобування суми алкалоїдів з одночасним вик люченням повторних екстракцій в критичних для рослинної сировини умовах. При використанні в прототипі кип'ятиння сировини процес ВІДНОЕЛЄННЯ N-окисів алкалоїдів відбувається НЄПОЕНІСТЮ І , окрім реакції відновлення N-окисів, має місце процес їх гідролізу в вод но- лудному середовищі, що призводить до значного забруднення про дукту, а отже до необхідності застосування додаткових стадій очистки, ' " * ' При концентрації спирту етилового менш заявляємих значень (60%) не забезпечується повнота здобування суми алкалоїдів. При концентрації спирту етилового більше гаявляємих значень (7'0%) з'являється небажаний ефект здобування більшої кількості баластних речовин - ліпофільних, поліфенольних білків, ферментів та інше. Співвідношення сировина : екстрагент 1г(3-6) визначено експериментально і є оптимальним для заявляемого способу. При кількості екстрагенту меншезаявляємих значень не забезпечується повнота здійснення екстракції суми алкалоїдів. Використання його в кількостях більше заявляємих недоцільно, тому що призводить до перевитрати цінної сировини без підвищення виходу цільового продукту. Введення стадії упарювання екстракту має перевагу у порівнянні з кип'ятінням через щадящий вплив на продукти екстракції коли під , дією високих температурних режимів можуть виникнути структурно механічні зміни речовин (прототип). Окрім того стадія , концентрації (упарювання) спиртового екстракту необхідна для підготовки гарантованого проведення кристалізації суми алкалоїдів: в 13 дуже розведеному розчині кристалізація моде значно затриматися або не відбутися. Підкислення водного концентрату, одержаного після екстракції та упарювання, 20% соляною кислотою та 20% сірчаною кислотою з подальшим витримуванням при перемішуванні передбачене для переходу солей алкалоїдів в розчин а саме, сполучення однакових кіль, костей цих кислот дає оптимальний результат Кількість підкислю. вального агента залежить від кількості одержаного водного кон центрату. Введення цинку необхідно для відновлення N-окисів алкалоїдів, кількість його визначено експериментально і залежить від кількості підкислювального агента. Підлужування охолодженого кислого розчину £5% розчином аміаку до рН 9,0-9,3 необхідно для осадження суми алкалоїдів, яку потім очищають хлористим метіленом від баластних речовин, що раніше не розчинилися в органічних розчинниках Причому саме хлористий . метилен має найбільшу перевагу перед іншими екстрагентами у по рівнянні, наприклад, з хлороформом, використання якого в цьому випадку призводить до значного подорожчання кінцевого продукту : окрім того, що хлороформ приблизно в 15 разів дорожче хлористого метилену, его "летюча" сутність призводить до великих втрат реагенту і до необхідності застосування особливих заходів для дотримання техніки безпеки виробництва. Обробка хлористометиленового екстракту 10% сірчаною кислотою є додатковою стадією очистки суми алкалоїдів, при якій видобуваються ліпофільні та поліфенольні фракції, які потім вилучаються за допомогою активованого вугілля. Вихід платифіліну гідротартрату при здійсненні заявляемого способу складає 1850 г на одну тону при ЕМІСТІ алкалоїдів 0,4£. В прототипі відповідна - 650 г. 1 4 кзводятьсл далі для порівняльного аналізу заявлявшего способу та способу-прототипу. Спосіб-прототип і. Подрібнення сировини - трави жовтозілля плосколіїстного до розміру частинок 2-5 мм. 2. Перша екстракція подрібненої сировини киплячим дихлорета ном Е присутності глюкози та 12,5% водного розчину соди протягом 2 год. 3. Друга екстракція сировини при кімнатній температурі про тягом 2 гол. 4. Третя екстракція сировини при кімнатній температурі про тягом 2 год. 5. Протитокова екстракція кожного з лихлоретакоЕих здобу вань алкалоїдів 10% водним розчином сірчаної кислоти. 6. Охолодження об'єднаних сірчанокислотних здобувань алкало їдів до 3-4°С при перемішуванні. 7. Дсдавання 25%. водного розчину аміаку (рН 9) до випадання суми алкалоїдів в осадок. 8. Витримування маси протягом і год при 3°С до повного випа дання суми алкалоїдів в осадок у вигляді суспензії. 9. Фільтрація одержаної суспензії на нутч-фільтрі. 10. Промивка осадку водою до псвногс зникнення запаху аміаку. 11. Сушіння осадку суми алкалоїдів при 60-70иС протягом 6 год при періодичному перемішуванні. 12. Подрібнення висушеного озадку на порошок* 13. Розчинення порошку в киплячому 9Є% етанолі протягом 3D хв до розчинення платифіліну. 14. Фільтрапія на нутч-фільтрі суспензії сенецифіліну в спир товому розчині платифіліну. 15. Промивка осадку сенецифіліну гарячіш спиртом на фільтрі. -1 СГ 16. Віджиманняf висушування під вакуумом при 50-60°С та за лишковому тиску 50-75 мм рт.ст. 17. Змішування спиртового розчину платифіліну гідротартрату э розрахованою кількістю виннокам'янсї кислоти при кагріванні до 80°0 до розчинення виннокам'яної кислоти. 18. Охолодження одержаної маси де кімнатко: температури. 19. Витримування маси протягом 12 год до повного випадання осадку технічного платифіліну гідротартрату. 20. Фільтрація осадку, що випав. 21. Промивка осадку платифіліну гідротартрату охолодженим спиртом. 22. Сушіння під вакуумом при 50-60°С та залишковому тиску 5075 мм рт.ст. 23. Змішування технічного платифіліну гідротартрату з 96% ети ловим спиртом з подальшим кип'ятінням одержаної суміші при 80-83°0 протягом 15 ХЕ ДО ПОЕНОГО розчинення осадку. 24. Счистка розчину шляхом змішування його з активованим ву гіллям при нагріванні протягом 10-15 ХЕ. 2-з. Фільтрація сдержано і суміші. 26. Еитримування осадку в кристалізаторі при 2-5JC протягом 15 год. 27. Фільтрація кристалічного осадку. 28. Дворазова промивка фільтрату охолодженим 95% етиловим спиртом з подальшим віддиманням. 29. Сушка платифіліну гідротартрату під вакуумом при 50-60°С та залишковому тиску 50-100 мм рт.ст. 30. Фасовка готового продукту в герметичну упаковку. Вихід платифіліну гідретартрату на одну тону сировини при вмісті алкалоїдів иЛ% - 550 г. Чистота цільового продукту - 99%. 16 Заявлявшій спосіб 1. Подрібнення сировини (надземної частини та/або кореневищ жовтозілля плосколиотного) до насипної щільності маси 0,55 кг/дм3. 2. Екстракція подрібнено: сировини 60-70% спиртом етиловим на фільтрі під вакуумом при кімнатнії! температурі при співвідношенні сировина : екстрагент 1:(3-б). 3. Уларквзннл екстракту в плівковому випарнику. 4. Підкислювання одержаного концентрату до рН 3-4 однаковими кількостями £0* соляно": кислоти та 2С% сірчаної кислоти. 5. Витримування сдержано! суміші протягом 3-24 год при Єезперервному перемішуванні. 5. Додавання до суміші цинку та перемішування протягом 4 год під азотом. 7. Фільтрація суміші, 3. Нейтралізація Фільтрату 2Б% розчином аміаку ло рН 9,0-9,3. 9. Протитокова екстракція хлористим метиленом. 10. Екстракція хлсристометіїленового екстракту 10% розчином сір чаної кислоти. 11. Очистка сірчанокислотного екстракту активованій: вугіллям. 12. Охолодження очищеного екстракту до 4°С. 13. Нейтралізація екстракту 25% розчином аміаку. 14. Витримування екстракту в кристалізаторі до відокремлення кристалів суми алкалоїдів протягом 1-5 год. 15. Фільтрація кристалів від рідкої фази. 16. Сушіння кристалів при 55-80°С протягом 10-24 год. 17. Змішування сухої суми алкалоїдів з 96% спиртом етиловим (1:5) і кип'ятіння протягом 30-45 ХЕ. 17 18. Фільтрація одержаної суміші, 19. Охолодження суспензії до 4lJC та Ектрішузання протягом 4-24 год. £0. Фільтрація суспензій й1. Змішування фільтрату з сухою винникам*яною кислотою та нагрівання до розчинення випискам-'яно: кислоти. 22. Витримування в кристалізаторі при 4°С протягом 8-16 год. йо. УДІЛЛІ рад І зі і\рЯиТсиичНиі MdUi'i Td СушлННЯ ^ьЗДгЛ nd ьидира протягом 24 год. 24. Фасовка готового продукту в герметичну упаковку. Вихід платифіліну пдротартрату на одну тону сировини при вмісті алкалоїдів 0,4% - 1850 г. Чистота цільового продукту - не менше 99,53%. Як відомо, головним алкалоїдом, що супроводжує платифілін в жсвтозіллі є , оенецифілін, Інші побічні алкалоїди присутні в ньому у незначній кількості, Окрім того з технічній суміші алкалоїдів яку ., 5 виділяють з жовтозілля міститься велика кількість смолистих , речовин, барвників, хлорофілів. Наведені дані свідчать, шо зачвляБМИЙ спосіб дозволяє досягти високого ступеня чистоти цільового продукту - не менше 99,53%. Слід мзгл на увазі, ще сенецифілін ЕІДкеситься до високотоксичних алкалоїдів, таким як лазіокарпін, гелїосупін. Для нього в характерною виражена здатність діяти на певні тканини, Е основному, на печінку, нирки, легені. Окрім гепатотоксичнеге ефекту, вік здатний при достатньо тривалому зведенні в органігм викликати розвиток пухлин в печінці тварин В зв'язку з цим , неприпустима присутність сенецифіліну Б субстанції платифіліну гідротартраті більше 0Д%. Таким чином, заявляємий спосіб одержання платифілліну гідротартрату дозволяє виділити цільовий продукт з більшим виходом та кращої якості, ніж в прототипі та аналогах, спростити та уніфікувати технологію за рахунок скорочення кількості стадій та можливості переробки сировини різних видів та якості. ЛІТЕРАТУРА: 1. А. с. N 618112, кл. А 61 К 35/78. Опубл. БИ, 1978, N 29. 2. А. с. N 1681429, кл. А 61 К 35/78. Опубл. БИ, 1991, N 37. 3. Патент РФ N 2058150, кл. А 61 К 35/78. Опубл. Бй, 1996, N 11. 4. Промышленный регламент N 64-1800, ВИЛР, 1987. 5. Данилова А.В. и др. Новый метод получения платифиллина из крестовника широколистного /Мед. пром. СССР, 1960, т. 14, N 4, с. 28-39. 6. Захаров В.П., Либизов Н.И., Асланов Х.А. Лекарственные вещества из растений и способы их производства. Ташкент, 1980. С. 72-74 (прототип).
ДивитисяДодаткова інформація
Назва патенту англійськоюMethod for manufacturing alkaloid platyphyllin hydrotartrate
Автори англійськоюAkimov Volodymyr Andriiovych, Heorhiievskyi Viktor Petrovych, Chein Anatolii Tykhonovych
Назва патенту російськоюСпособ получения алкалоида платифиллина гидротартрата
Автори російськоюАкимов Владимир Андреевич, Георгиевский Виктор Петрович, Шеин Анатолий Тихонович
МПК / Мітки
МПК: A61K 36/24
Мітки: одержання, гідротартрату, платифіліну, спосіб
Код посилання
<a href="https://ua.patents.su/18-32355-sposib-oderzhannya-platifilinu-gidrotartratu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання платифіліну гідротартрату</a>
Спосіб отримання гідротартрату платифіліну

Номер патенту: 24931
Опубліковано: 26.02.1999
Автори: Ємельянов Віктор Іванович, Зубченко Тамара Миколаївна, Заболотний Вадим Олександрович, Савченко Віра Ашмуханівна, Чернобай Володимир Тимофійович, Комісаренко Олена Пантелеївна, Лещенко Валентина Олексіївна, Супрун Ольга Всеволодівна
МПК: A61K 135/00, A61K 31/00, A61K 36/28
Мітки: платифіліну, гідротартрату, спосіб, отримання
Формула / Реферат:
Способ получения гидротартрата платифиллина, включающий измельчение травы крестовника плосколистного, экстракцию из сырья суммы алкалоидов, восстановление алкалоидов, получение сернокислых солей алкалоидов, очистку, и получение гидротартрата платифиллина, отличающийся тем, что измельчение травы крестовника плосколистного осуществляют вальцеванием, экстракцию суммы алкалоидов осуществляют 40 - 70% спирто-водным раствором, после чего...
Спосіб одержання пектинатів та пектину

Номер патенту: 30439
Опубліковано: 15.11.2000
Автори: Радуль Любов Павлівна, Сокол Евгенія Іванівна, Ровенський Володимир Терентійович, Казаров Альберт Артемович, Демченко Павло Іванович, Сторожук Нінель Борисівна
МПК: C08B 37/06, A23L 1/0524
Мітки: пектинатів, пектину, одержання, спосіб
Формула / Реферат:
1. Спосіб одержання пектинатів та пектину, який включає кислотній гідроліз пектинвміщуючої рослинної сировини, відокремлення рідкої фази, її нейтралізація, добавка осаджувача, відокремлення коагуляту у вигляді пектинатів і регенерацію до пектину, який відрізняється тим, що нейтралізацію гідролізату проводять до рН=3,0-5,2, в ролі осаджувача використовують сполуки металів в кількості 10-25% від маси сировини, а регенерацію здійснюють...
Спосіб одержання харчового білка з рослинної сировини

Номер патенту: 29197
Опубліковано: 16.10.2000
Автори: Карнаушенко Лідія Іванівна, Лукіна Галина Дмитріївна, Золотарьова Людмила Анатоліївна, Іоргачова Катерина Георгіївна
МПК: A23J 1/14
Мітки: спосіб, харчового, білка, одержання, рослинної, сировини
Формула / Реферат:
Спосіб одержання харчевого білка з рослинної сировини, передбачаючий подрібнення рослинної сировини, екстракцію білка лугом, відділення екстракта від нерозчинного твердого залишка, осадження білка кислотою, його відділення, промивку та сушку, який відрізняється тим, що в якості рослинної сировини використовують шрот амаранта, а екстракцію ведуть 0,2%-ним розчином гідроксиду натрію при гідромодулі 10, t=20-25°С протягом 55-60 хвилин, причому...
Спосіб одержання активного червоного сш моноазобарвника
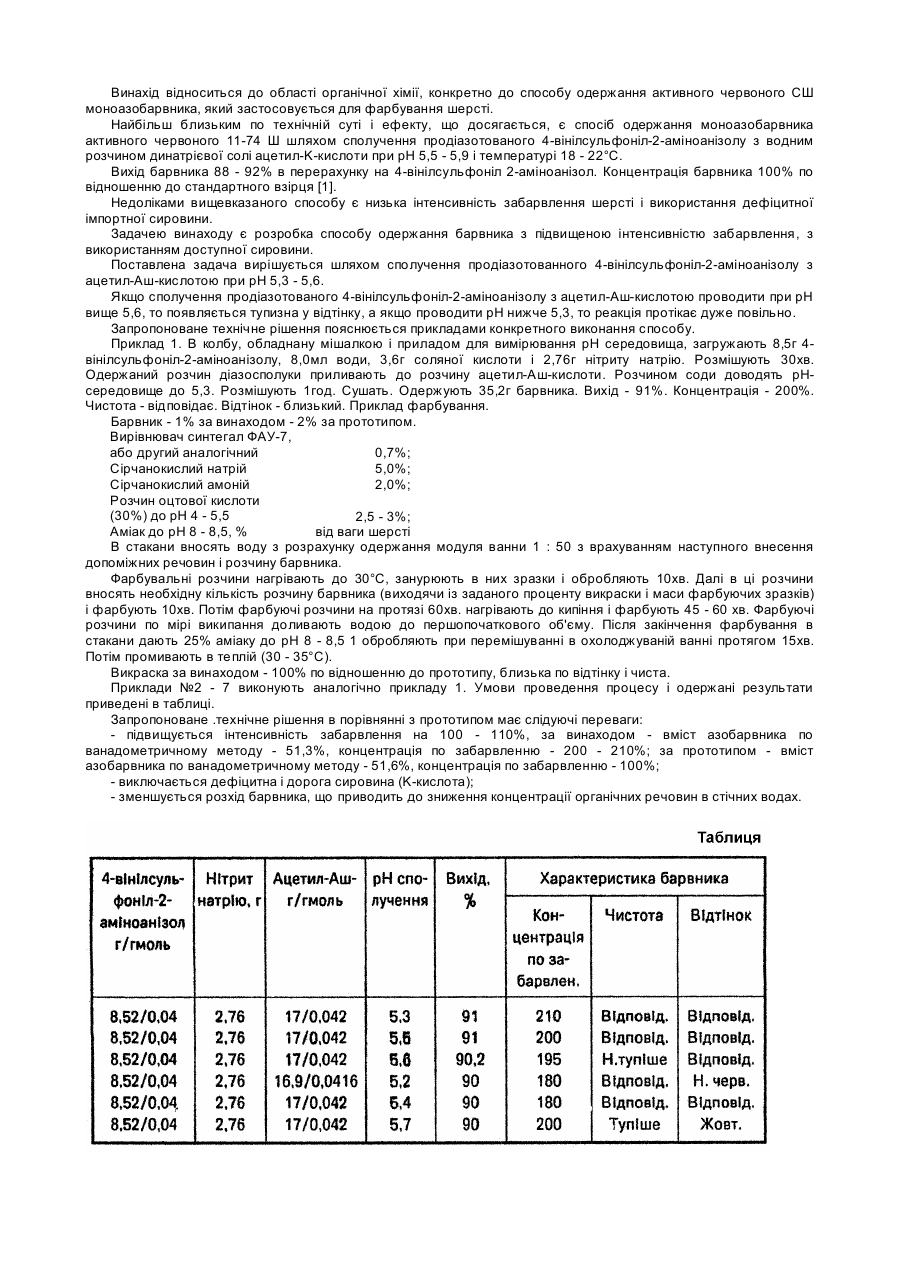
Номер патенту: 20450
Опубліковано: 15.07.1997
Автори: Литвин Борис Львович, Василишин Євген Володимирович, Лучкевич Євген Романович, Великорода Ольга Григорівна, Хомут Віра Миронівна, Клюба Оксана Іванівна
МПК: C09B 29/00
Мітки: одержання, моноазобарвника, червоного, активного, спосіб
Формула / Реферат:
Спосіб одержання активного червоного СШ моноазобарвника шляхом сполучення продіазото-ваного 4-вінілсульфоніл-2-аміноанізолу з азо-складовою, який відрізняється тим, що в якості азоскладової використовують ацетил-Аш кислоту і сполучення проводять при рН 5,3-5,6.
Спосіб одержання фітозасобу з протигрибковою дією

Номер патенту: 31288
Опубліковано: 15.12.2000
Автори: Бензель Леонід Васильович, Виноград Іван Андрійович, Зузук Богдан Михайлович, Грицик Андрій Романович, Виноград Наталія Олексіївна, Федущак Надія Казимирівна
МПК: A61K 36/70
Мітки: фітозасобу, протигрибковою, дією, одержання, спосіб
Текст:
...альпійського (Rhizoma cum rad. Rumex alp.) попередньо висушені та подрібнені до розміру частинок 0,5-2 мм ектрагувалн 30-70 % етиловим спиртом протягом 5 діб у співвідношенні сировина-екстрагент 1:5 -1:10 двічі. Екстракти об'єднували, відстоювали протягом 1 доби, фільтрували і доводили екстрагентом до початкового об"єму. Отриманий фітозасіб являє собою спиртовий екстракт світло-коричневого кольору специфічного запаху і гіркого смаку. В...
Попередній патент: Спосіб реабілітації жінок, які перенесли перитоніт після кесаревого розтину
Наступний патент: Електростимулятор
Випадковий патент: Фармацевтична композиція, що містить тіазолідиндіон і метформін (варіанти), та спосіб лікування цукрового діабету