Пептидні аргінали та спосіб лікування розсіяної внутрішньосудинної коагуляції















Формула / Реферат
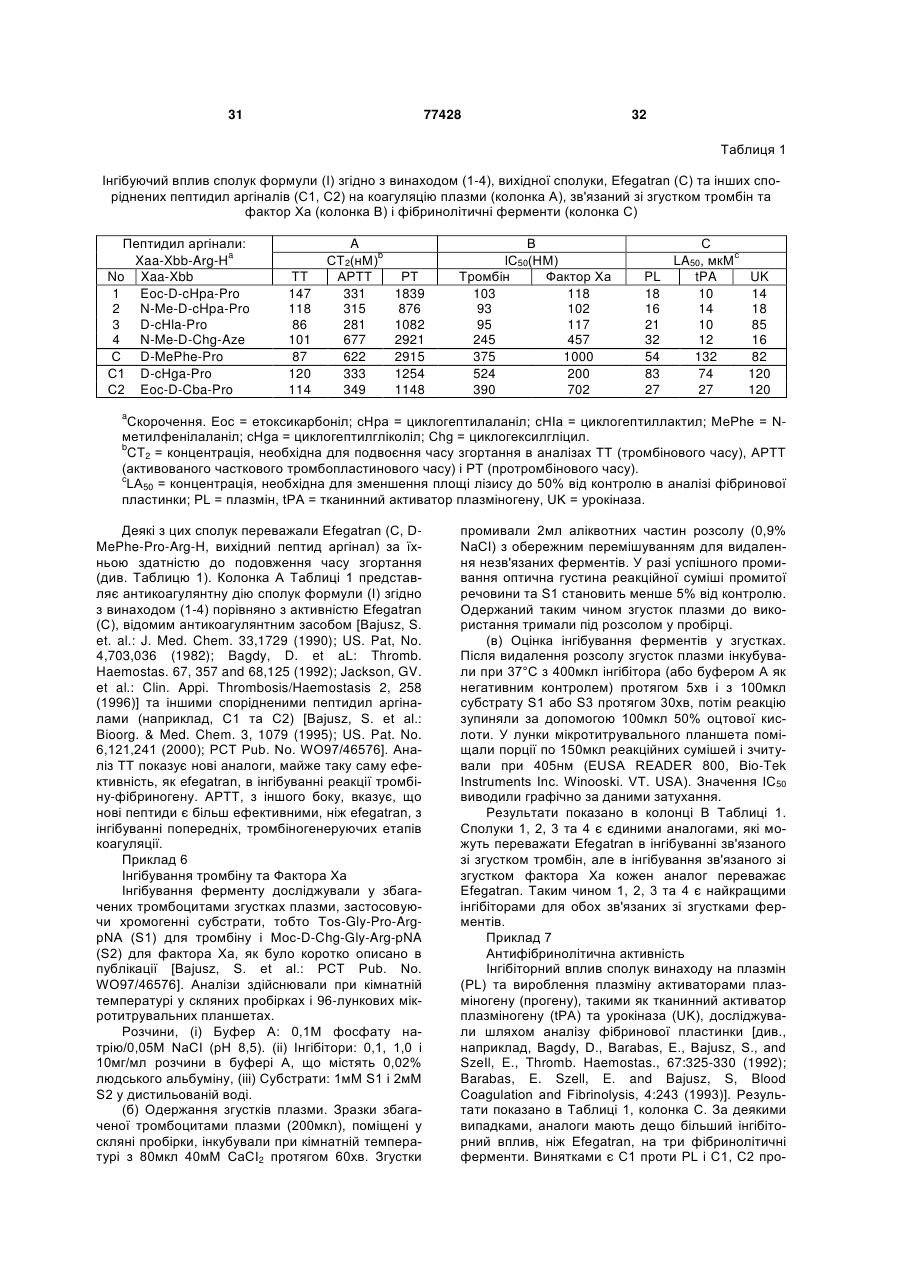
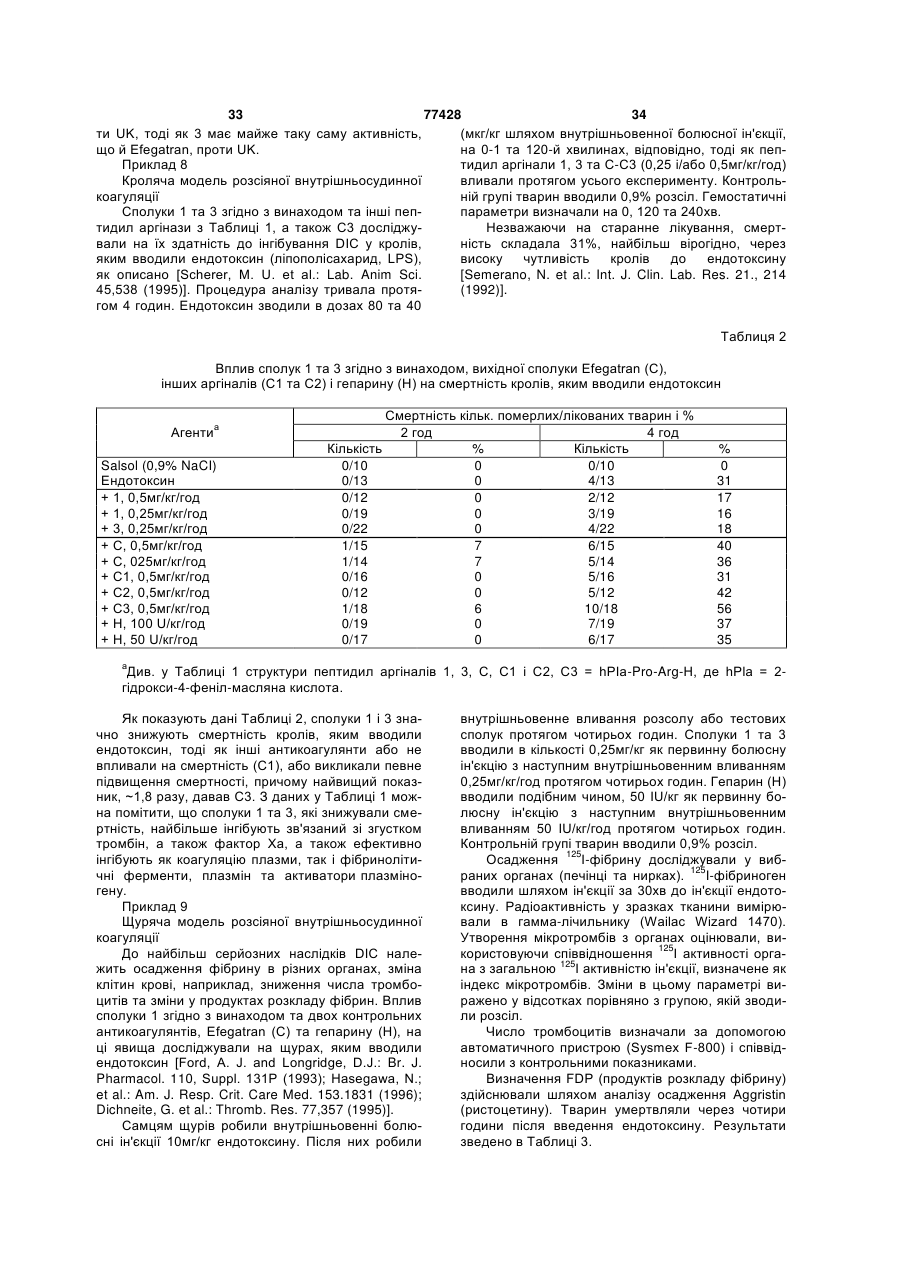
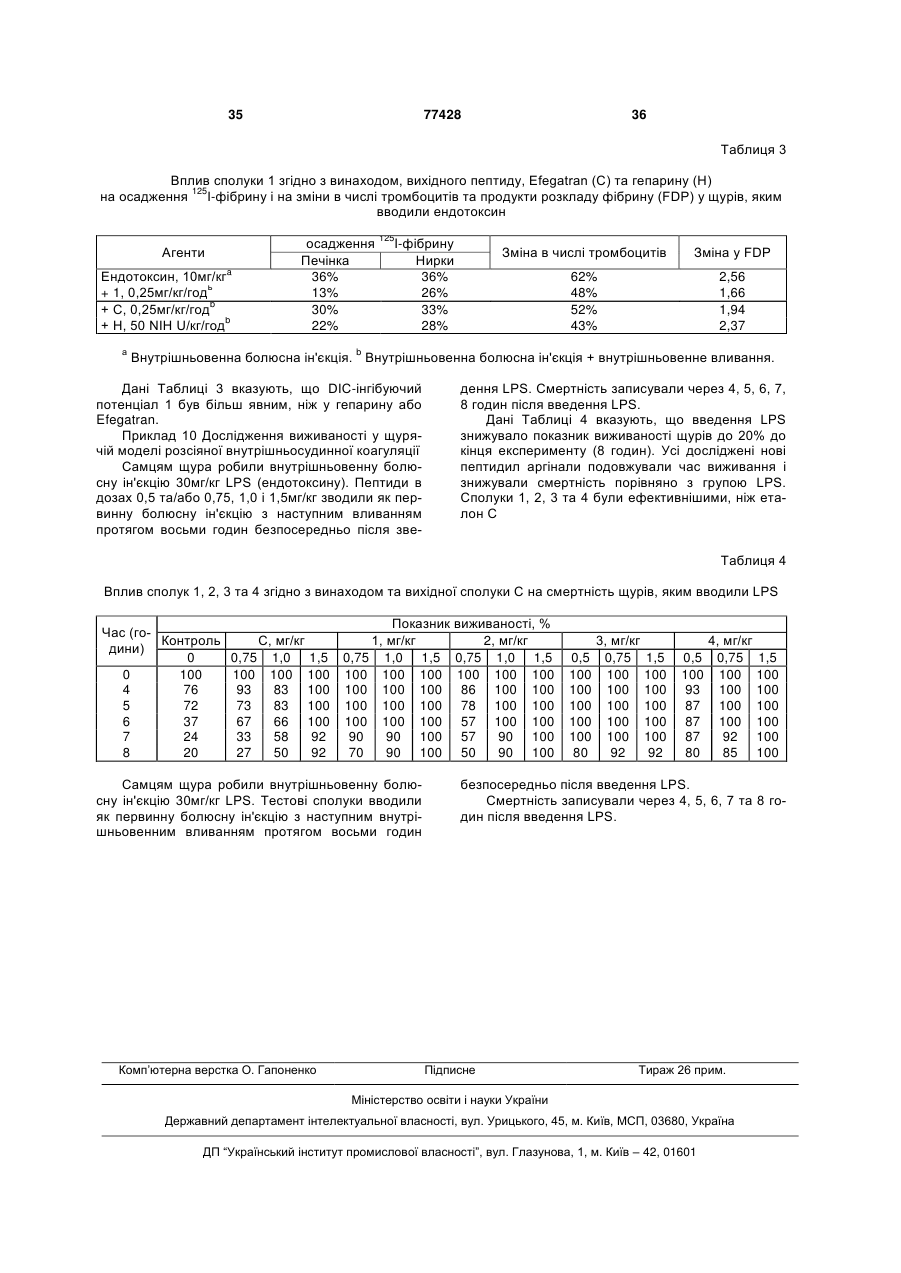
1. Сполука, яка має формулу (І)
Xaa-Xbb-Arg-H , (I)
де Хаа представляє альфа-заміщений карбоновокислотний залишок формули (II)
Q-CH(R)-CO , (II)
де Q представляє алкілоксикарбоніламіногрупу з 1-3 атомами вуглецю, метиламіногрупу або гідроксильну групу, і R представляє циклоалкілметильну групу з 7-9 атомами вуглецю, 1-адамантилметильну групу або циклоалкільну групу з 5-7 атомами вуглецю, і Хbb представляє L-пролін або L-азетидин-2-карбоновокислотний залишок, та її кислотно адитивні солі, утворені з органічною або неорганічною кислотою.
2. Сполука, яка має структуру 1:
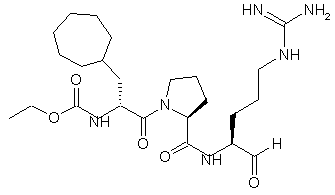 , 1
, 1
та її кислотно адитивні солі.
3. Сполука, яка має структуру 2:
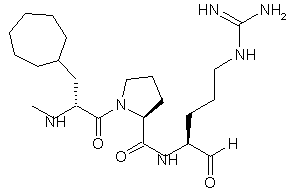 , 2
, 2
та її кислотно адитивні солі.
4. Сполука, яка має структуру 3:
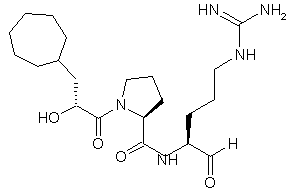 , 3
, 3
та її кислотно адитивні солі.
5. Сполука, яка має структуру 4:
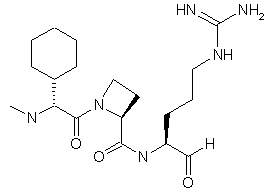 , 4
, 4
та її кислотно адитивні солі.
6. Фармацевтична композиція, яка містить сполуку, вказану в п. 1.
7. Фармацевтична композиція, яка містить сполуку, вказану в п. 2.
8. Фармацевтична композиція, яка містить сполуку, вказану в п. 3.
9. Фармацевтична композиція, яка містить сполуку, вказану в п. 4.
10. Фармацевтична композиція, яка містить сполуку, вказану в п. 5.
11. Сполука етоксикарбоніл-D-циклогептилаланілпроліл-NG-бензилоксикарбоніл-L-аргінін альдегід.
12. Сполука тетрагідропіраніл-D-циклогептиллактил-L-проліл-NG-бензилоксикарбоніл-L-аргінін альдегід.
13. Сполука бензилоксикарбоніл-N-метил-D-циклогептилаланіл-L-проліл-NG-бензилоксикарбоніл-L-аргінін альдегід.
14. Сполука N-метил-D-циклогексилгліцил-L-азетидин-2-карбоніл-L-аргінін альдегід.
15. Фармацевтична композиція за будь-яким з пп. 6-10, яка відрізняється тим, що вищезгадана композиція може бути у формі таблетки, капсули, порошку, пілюлі, драже, гранули, розчину, інфузії, супозиторія, пластиру або мазі.
16. Спосіб лікування пацієнта з розсіяною внутрішньосудинною коагуляцією, при якому пацієнту вводять пептидил аргінал, який має інгібуючу дію на зв'язаний зі згустком тромбін, фактор Ха, плазмін та активатори плазміногена.
17. Спосіб лікування пацієнта з розсіяною внутрішньосудинною коагуляцією, при якому пацієнту вводять пептидил аргінал, який має формулу (І)
Хаа-Xbb-Arg-H , (I)
де Хаа представляє альфа-заміщений карбоновокислотний залишок формули (II)
Q-CH(R)-CO , (II)
де Q представляє алкілоксикарбоніламіногрупу з 1-3 атомами вуглецю, метиламіногрупу або гідроксильну групу, і R представляє циклоалкілметильну групу з 6-9 атомами вуглецю, 1-адамантилметильну групу або циклоалкільну групу з 5-7 атомами вуглецю, і Xbb представляє L-пролін або L-азетидин-2-карбоновокислотний залишок, або його фармацевтично прийнятні кислотно адитивні солі.
18. Спосіб лікування пацієнта з розсіяною внутрішньосудинною коагуляцією, при якому пацієнту вводять пептидил аргінал, який має структуру 1
, 1
або його фармацевтично прийнятні кислотно адитивні солі.
19. Спосіб лікування пацієнта з розсіяною внутрішньо судинною коагуляцією, при якому пацієнту вводять пептидил аргінал, який має структуру 2
, 2
або його фармацевтично прийнятні кислотно адитивні солі.
20. Спосіб лікування пацієнта з розсіяною внутрішньосудинною коагуляцією, при якому пацієнту вводять пептидил аргінал, який має структуру 3
, 3
або його фармацевтично прийнятні кислотно адитивні солі.
21. Спосіб лікування пацієнта з розсіяною внутрішньосудинною коагуляцією, при якому пацієнту вводять пептидил аргінал, який має структуру 4
, 4
або його фармацевтично прийнятні кислотно адитивні солі.
22. Спосіб за будь-яким із пп. 16-21, який відрізняється тим, що пацієнту вводять від приблизно 0,1 мг до приблизно 50 мг/кг пептидил аргіналу, залежно від маси тіла пацієнта.
23. Спосіб за будь-яким із пп. 16-21, який відрізняється тим, що пацієнту вводять пептидил аргінал у дозі, достатній для досягнення рівня в крові пептидил аргіналів від приблизно 6 мкм до приблизно 100 мкм.
24. Спосіб за будь-яким із пп. 16-23, який відрізняється тим, що пептидил аргінал вводять одночасно або послідовно.
Текст
1. Сполука, яка має формулу (І) Xaa-Xbb-Arg-H , (I) де Хаа представляє альфа-заміщений карбоновокислотний залишок формули (II) Q-CH(R)-CO , (II) де Q представляє алкілоксикарбоніламіногрупу з 1-3 атомами вуглецю, метиламіногрупу або гідроксильну групу, і R представляє циклоалкілметильну групу з 7-9 атомами вуглецю, 1адамантилметильну групу або циклоалкільну групу з 5-7 атомами вуглецю, і Хbb представляє L-пролін або L-азетидин-2-карбоновокислотний залишок, та її кислотно адитивні солі, утворені з органічною або неорганічною кислотою. 2. Сполука, яка має структуру 1: 3 77428 4 9. Фармацевтична композиція, яка містить сполуку, або його фармацевтично прийнятні кислотно адивказану в п. 4. тивні солі. 10. Фармацевтична композиція, яка містить сполу19. Спосіб лікування пацієнта з розсіяною внутріку, вказану в п. 5. шньо судинною коагуляцією, при якому пацієнту 11. Сполука етоксикарбоніл-Dвводять пептидил аргінал, який має структуру 2 G HN NH2 циклогептилаланілпроліл-N -бензилоксикарбонілL-аргінін альдегід. NH 12. Сполука тетрагідропіраніл-DG циклогептиллактил-L-проліл-N ,2 N NH бензилоксикарбоніл-L-аргінін альдегід. H O 13. Сполука бензилоксикарбоніл-N-метил-DO NH циклогептилаланіл-L-проліл-NGO бензилоксикарбоніл-L-аргінін альдегід. або його фармацевтично прийнятні кислотно ади14. Сполука N-метил-D-циклогексилгліцил-Lтивні солі. азетидин-2-карбоніл-L-аргінін альдегід. 20. Спосіб лікування пацієнта з розсіяною внутрі15. Фармацевтична композиція за будь-яким з пп. шньосудинною коагуляцією, при якому пацієнту 6-10, яка відрізняється тим, що вищезгадана вводять пептидил аргінал, який має структуру 3 композиція може бути у формі таблетки, капсули, HN NH2 порошку, пілюлі, драже, гранули, розчину, інфузії, NH супозиторія, пластиру або мазі. 16. Спосіб лікування пацієнта з розсіяною внутрі,3 N шньосудинною коагуляцією, при якому пацієнту HO вводять пептидил аргінал, який має інгібуючу дію H O O на зв'язаний зі згустком тромбін, фактор Ха, плазNH O мін та активатори плазміногена. 17. Спосіб лікування пацієнта з розсіяною внутріабо його фармацевтично прийнятні кислотно адишньосудинною коагуляцією, при якому пацієнту тивні солі. вводять пептидил аргінал, який має формулу (І) 21. Спосіб лікування пацієнта з розсіяною внутріХаа-Xbb-Arg-H , (I) шньосудинною коагуляцією, при якому пацієнту де Хаа представляє альфа-заміщений карбонововводять пептидил аргінал, який має структуру 4 HN NH2 кислотний залишок формули (II) Q-CH(R)-CO , (II) NH де Q представляє алкілоксикарбоніламіногрупу з 1-3 атомами вуглецю, метиламіногрупу або гідрок,4 сильну групу, і R представляє циклоалкілметильну N N групу з 6-9 атомами вуглецю, 1H O адамантилметильну групу або циклоалкільну групу O N з 5-7 атомами вуглецю, і Xbb представляє L-пролін O або L-азетидин-2-карбоновокислотний залишок, або його фармацевтично прийнятні кислотно адиабо його фармацевтично прийнятні кислотно адитивні солі. тивні солі. 22. Спосіб за будь-яким із пп. 16-21, який відріз18. Спосіб лікування пацієнта з розсіяною внутріняється тим, що пацієнту вводять від приблизно шньосудинною коагуляцією, при якому пацієнту 0,1 мг до приблизно 50 мг/кг пептидил аргіналу, вводять пептидил аргінал, який має структуру 1 HN NH2 залежно від маси тіла пацієнта. 23. Спосіб за будь-яким із пп. 16-21, який відрізNH няється тим, що пацієнту вводять пептидил аргіO нал у дозі, достатній для досягнення рівня в крові ,1 N пептидил аргіналів від приблизно 6 мкм до прибO NH лизно 100 мкм. H O O NH 24. Спосіб за будь-яким із пп. 16-23, який відрізO няється тим, що пептидил аргінал вводять одночасно або послідовно. Винахід стосується розсіяної внутрішньосудинної коагуляції. Точніше, винахід стосується медичного втручання у випадках розсіяної внутрішньосудинної коагуляції. Розсіяна внутрішньосудинна коагуляція (DIC) є вторинним захворюванням і може бути наслідком будь-якого з багатьох первинних захворювань. [Див. Віск, Disseminated Intravascular Coagulation and Related Syndromes. CRC Press, Boca Raton, (1983)]. До його характеристик належить систематична активація коагуляції крові, в результаті якої виробляється й осаджується фібрин, що призводить до мікросудинних тромбів у різних органах і сприяє розвиткові поліорганної недостатності. У роботі [Віск et al., Clin. АррІ Thrombosis Hemostasis і: 3-23 (1995)] вказується, що ще однією характе 5 77428 6 ристикою DIC є системно циркулюючий плазмін, де Q представляє алкілоксикарбоніламіногрузагальний протеолітичний фермент, який може пу з 1-3 атомами вуглецю, метиламіногрупу або біологічно розкладати різні білки плазми (фактори, гідроксильну групу, і R представляє циклоалкілмегормони і т. ін.) і може розщеплювати фібринотильну групу з 7-9 атомами вуглецю або циклоалген/фібрин, забезпечуючи продукти розкладу фібкільну групу з 5-7 атомами вуглецю, або 1риногену/фібрину. Ці продукти порушують гемосадамантилметильну групу, і Xbb представляє Lтаз і призводять до крововиливу. пролін або L-азетидин-2-карбоновокислотний заНайбільш серйозна клінічна форма DIC хараклишок, та її кислотно адитивні солі, утворені з ортеризується широкою витратою білків коагуляції, ганічною або неорганічною кислотою. значним осадженням фібрину та кровотечею. В оптимальних варіантах втілення такі сполуки Пацієнти з травмами піддаються підвищеному можуть мати такі структури: 1 (етоксикарбоніл-Dризикові DIC, особливо, якщо уражено велику циклогептилаланіл-L-проліл-L-аргінін альдегід, площу тканини (зокрема, мозку), в разі сепсису та Eoc-D-cHpa-Pro-Arg-H), або 2 (N-метил-Dполіорганної недостатності. Травма голови є осоциклогептилаланіл-L-проліл-L-аргінін альдегід, Nбливо поширеним випадком DIC у немовлят та Me-D-cHpa-Pro-Arg-H), або 3 (Dдітей через високий вміст тромбопластину в мозку циклогептиллактил-L-проліл-L-аргінін альдегід, Dта пропорційно збільшене співвідношення площі cHpl-Pro-Arg-H), або 4 (N-метил-Оповерхні голови з загальною площею поверхні циклогексилгліцил-L-азетидин-2-карбоніл-L-аргінін тіла. альдегід, N-Me-D-Chg-Aze-Arg-H), які відповідають Сепсис може траплятися у приблизно 40% від формулі (І), де Хаа представляє альфа-заміщені загальної кількості пацієнтів з травмами і є суттєалкільні карбоновокислотні залишки формули (II), вою первинною причиною DIC у всіх пацієнтів. де R представляє циклогептилметильну та циклоКлінічний стан погіршується вторинним фібринолігексильну групу, відповідно, Q представляє етокзом, що призводить до утворення FDP (продуктів сикарбоніламіно-, метиламіно- та гідроксильну розкладу фібриногену/фібрину) або "D-димерів", групу, відповідно, і Xbb представляє L-пролін та Lякі заважають нормальному утворенню фібрину та азетидиніл-2-карбоновокислотний залишок, відпофункції тромбоцитів. відно. Осадження фібрину при DIC може призводити до дисфункції інших органів. DIC є головною причиною гострої ниркової недостатності, а також сприяє поліорганній системній недостатності. І навпаки, пошкоджені органи сприяють DIC. На даний час єдиний визнаний спосіб лікування DIC обмежується спробами послабити первинне порушення. Без контролю DIC триває, незважаючи на форми терапії, спрямовані на виправлення кровотечі або тромболітичної проблеми. У деяких випадках, коли трапляється значна кровотеча, може допомогти замісна терапія з застосуванням свіжозамороженої плазми, компонентів плазми (наприклад, антитромбіну III) кріопреципітату, та/або концентратів тромбоцитів, доки контролюється основна проблема, але ці способи терапії є надзвичайно дорогими. Використання гепарину при DIC є дуже суперечливим і не має загального застосування для пацієнтів з проблемами, в основі яких лежать травми. Таким чином, існує потреба в нових поліпшених сполуках та способах з лікування від DIC [Див. також, наприклад, de Jonge et al, Drugs 55:767-777 (1998) and Levi et al, Thrombosis and Haemostasis 82: 695 (1999)]. Винахід забезпечує нові поліпшені сполуки та спосіб лікування від DIC. Несподівано було виявлено, що антикоагулянтні сполуки, які мають інгібуючу дію як на вільний, так і на зв'язаний зі згустком тромбін та фактор Ха, а також інгібують активатори плазміну та плазміногену, можуть бути корисними для лікування від DIC. У першому аспекті винахід забезпечує композицію речовини, яка включає пептидил аргінал формули (І) Xaa-Xbb-Arg-H (I) де Хаа представляє альфа-заміщений карбоновокислотний залишок формули (II) Q-CH(R)-CO (II) 7 У другому аспекті винахід забезпечує фармацевтичну композицію, яка включає антикоагулянтний пептидил аргінал або його фармацевтично прийнятну сіль згідно з першим аспектом винаходу та фармацевтично прийнятний носій, наповнювач або розріджувач. У третьому аспекті винахід забезпечує спосіб лікування розсіяної внутрішньосудинної коагуляції, який включає введення пацієнтові з розсіяною внутрішньосудинною коагуляцією антикоагулянтного пептидил аргіналу, який відповідає формулі (І) Xaa-Xbb-Arg-H (I) де Хаа представляє альфа-заміщений карбоновокислотний залишок формули (II) Q-CH(R)-OO (II) де Q представляє алкілоксикарбоніламіногрупу з 1-3 атомами вуглецю, метиламіногрупу або гідроксильну групу, і R представляє циклоалкілметильну групу з 7-9 атомами вуглецю, або 1адамантилметильну групу або циклоалкільну групу з 5-7 атомами вуглецю, і Xbb представляє L-пролін або азетидин-2-карбоновокислотний залишок, або його фармацевтично прийнятної кислотно адитивної солі. В оптимальних варіантах втілення такі сполуки можуть мати такі структури: 1 (етоксикарбоніл-D-циклогептилаланіл-L-проліл-L-аргінін альдегід, Eoc-D-cHpa-Pro-Arg-H), або 2 (N-метил-Dциклогептилаланіл-L-проліл-L-аргінін альдегід, NMe-D-cHpa-Pro-Arg-H), або 3 (Dциклогептиллактил-L-проліл-L-аргінін альдегід, DcHpl-Pro-Arg-H), або 4 (N-метил-Dциклогексилгліцил-L-азетидин-L-карбоніл-Lаргінін-альдегід, N-Me-D-Chg-Aze-Arg-H), які відповідають формулі (І), де Хаа представляє альфазаміщені алкільні карбоновокислотні залишки формули (II), де R представляє циклогептилметильну та циклогексильну групу, відповідно, Q представляє етоксикарбоніламіно-, метиламіно- та гідроксильну групу, відповідно, і Xbb представляє L пролін та і-азетидиніл-2-карбоновокислотний залишок, відповідно. 77428 8 Винахід стосується розсіяної внутрішньосудинної коагуляції. Точніше, винахід стосується медичного втручання у випадках розсіяної внутрішньосудинної коагуляції. Винахід забезпечує нові й поліпшені сполуки та способи лікування DIC. Сполуки згідно з винаходом мають інгібіторну дію як на вільний, так і на зв'язаний зі згустком тромбін та фактор Ха, а також на активатори плазміну та плазміногену. У наведених автором патентах та публікаціях відображено відомості існуючого рівня техніки і, таким чином, вони включаються шляхом посилання в їх повному обсязі. Будь-яка невідповідність між цими патентами та публікаціями і даним описом має тлумачитись на користь даного опису. У першому аспекті винахід забезпечує композицію речовини, яка включає пептидил аргінал, який має формулу (І) Xaa-Xbb-Arg-H (I) де Хаа представляє альфа-заміщений карбоновокислотний залишок формули (II) Q-CH(R)-CO (II) де Q представляє алкілоксикарбоніламіногрупу з 1-3 атомами вуглецю, метилам і ногрупу або гідроксильну групу, і R представляє циклоалкілметильну групу з 7-9 атомами вуглецю, 1адамантилметильну групу або циклоалкільну групу з 5-7 атомами вуглецю, і Xbb представляє L-пролін або L-азетидиніл-2-карбоновокислотний залишок, та його кислотно адитивні солі, утворені з органічною або неорганічною кислотою. Варіант втілення згідно з цим аспектом винаходу, якому віддають особливу перевагу, відповідає структурі 1 (етоксикарбоніл-D 9 77428 10 циклогептилаланіл-Ьпроліл-Ьаргінін альдегід, Eocбензилоксикарбонільною групою, і відновлення D-cHpa-Pro-Arg-H): одержаного трипептид лактаму до захищеного трипептид альдегіду, з видаленням захисної групи з гуанідино-групи аргініну, і, в разі, якщо Q = метиламіно або гідроксил у формулі (II) - також з кінцевої метиламіно- або гідроксильної групи, і виділення пептидної похідної формули (І) як її адиційної солі, утвореної з органічною або неорганічною кислотою. Сполуки, представлені формулою (І), одержують і застосовують у формі кислотно адитивних солей через більшу стійкість у формі солі. У кислотно адитивних солях сполуки формули (І) активність міститься в основі, і кислота має менше знаІнший варіант втілення згідно з цим аспектом чення, хоча для терапевтичних цілей перевагу винаходу, якому віддають особливу перевагу, відвіддають застосуванню фармацевтично прийнятповідає структурі 2 (N-метил-D-циклогептилаланілних кислотно адитивних солей. Прикладами таких L-проліл-L-apriHiH альдегід, N-Me-D-cHpa-Pro-Argпридатних кислот є (а) мінеральні кислоти: хлориH): стоводнева, бромистоводнева, фосфорна, метафосфорна та сірчана кислоти, (б) органічні кислоти: винна, оцтова, лимонна, яблучна, молочна, фумарова, бензойна, гліколева, глюконова, бурштинова, памова та арилсульфонові кислоти, наприклад, р-толуолсульфонова кислота. Оптимальною кислотно адитивною сіллю є сульфат, особливо, гемісульфат. Кислотно адитивні солі одержують традиційним способом, наприклад, шляхом нейтралізації вільноосновної форми сполуки формули (І) кислотою. Двозалишковий кислотний компонент може Ще один варіант втілення згідно з цим аспекбути показаний як D-Xaa-Xbb, де Хаа представляє том винаходу, якому віддають особливу перевагу, -заміщений карбоновокислотний залишок форвідповідає структурі 3 (D-циклогептиллактил-Lмули Q-CH(R)-CO, де Q означає алкоксикарбоніпроліл-L-аргінін альдегід, D-Hpl-Pro-Arg-H): ламіно-групу з 1-3 атомами вуглецю, R є таким, як визначено вище, і Xbb представляє L-пролін або Lазетидин-2-карбоновокислотний залишок. Якщо Хаа, -заміщена алкільна кислота, є -метиламіно або -гідроксикислотою, тобто Q представляє метиламіно або гідроксильну групу, двозалишковий кислотний компонент може бути показаний як P-DXaa-Xbb, де Ρ представляє N-захисну групу, таку як бензилоксикарбонільна (Ζ) або третбутоксикарбонільна (Вос) група або 0-захисна група, в оптимальному варіанті - тетрагідропіранільна (ТНР) група. Ще один варіант втілення згідно з цим аспекАцил дипептид, який використовують як вихідтом винаходу, якому віддають особливу перевагу, ний матеріал для сполук, що містять -аміно або відповідає структурі 4 (N-метил-D-метиламінокислотний залишок, одержують шляциклогексилгліцил-L-азетидин-2-карбоніл-L-аргінін хом ацилювання -амінокислоти відповідним есальдегід, N-Me-D-Chg-Aze-Arg-H): тером хлоромурашиної кислоти для одержання алкоксикарбоніламінокислоти з 1-3 атомами вуглецю та бензилоксикарбоніламінокислоти, які після цього з'єднують з L-проліном або L-азетидин-2карбоновою кислотою для одержання D-Xaa-Xbb та Z-аміноацилу Xbb, який N-метилують для одержання потрібного P-D-Xaa-Xbb. D-Xaa, що вимагається для з'єднання з Xbb, в оптимальному варіанті одержують шляхом ацетилювання рацемічної DL-Xaa сполуки, перетворення DL-ацетиламінокислоти на її метиловий естер і ферментативного розкладу ацетил-DL-Xaa-OMe Сполуки згідно з формулою 1, 2, 3 та 4 одеррацемічного естеру. Одержаний таким чином ацежують, наприклад, шляхом конденсації N або Отил-D-Хаа-Ome після цього омилюють і деацетизаміщеного 2-залишкового кислотного компонента люють, а потім перетворюють на потрібну NL-аргінін лактамом, захищеним на гуанідино-групі захищену D-амінокислоту. 11 77428 12 від приблизно 0,1мг до приблизно 50 мг пептидил Потрібну D- -гідроксикислоту в оптимальному аргіналу на кг маси тіла на день. Може бути бажаваріанті одержують із відповідної D- ним одночасне або послідовне введення суб'єктові амінокислоти. Потім її перетворюють на її Отерапевтично ефективної кількості однієї або кільзахищену форму і з'єднують з Xbb для одержання кох терапевтичних композицій винаходу як одинипотрібного P-D-Xaa-Xbb. чний лікувальний захід. У другому аспекті винахід забезпечує фармаНижченаведені приклади представлені для цевтичну композицію, яка включає пептидил аргідетальнішого пояснення деяких оптимальних ванал згідно з першим аспектом винаходу та фармаріантів втілення винаходу і не мають на меті обцевтично прийнятний носій, наповнювач або меження обсягу винаходу. Якщо спеціально не розріджувач. вказано іншого, для представлених нижче експеФармацевтичні композиції включають ефектириментів людський тромбін (3,000 NIH U/мг), людвну кількість сполуки загальної формули (І) або її ський альбумін та людський фібриноген отримуфармацевтично прийнятної солі та відомі фармавали від Sigma Aldrich Kft. (Будапешт, Угорщина), цевтично прийнятні носії, наповнювачі, розріджуа людський фактор Ха (8мкг/U) - від Enzyme вачі та/або інші фармацевтичні наповнювачі. Research Laboratories (Суонсі, Великобританія). Вищезгадані носії, розріджувачі або наповнюРеагент АРТТ отримували від REANAL (Будапешт, вачі можуть бути водою, спиртами, желатином, Угорщина), а реагент PT, Simplastin D, купували в лактозою, сахарозою, крохмалем, пектином, стеаORGANON, TEKNIKA (Еппельгайм, Німеччина). ратом магнію, стеариновою кислотою, тальком, Скорочення для амінокислот, пептидів, замісрізними оліями тваринного або рослинного похоників та реагентів вжито згідно з конвенціями ІЮдження, а також гліколями, наприклад, пропіленгПАК-ІЮБ. В цій заявці зустрічаються такі скороліколем або поліетиленгліколем. чення: Arg = L-аргінін, Воc = трет-бутокси-карбоніл, Фармацевтичні наповнювачі можуть бути конBzl = бензил, Chg = L-циклогексилгліцин, DCHA = сервантами, різними природними або синтетичнидициклогексиламін, DHP = дигідропіран, Еос = ми емульгаторами, диспергаторами або зволожуетоксикарбоніл, Gly = гліцин, Me = метил, MePhe = ючими агентами, барвниками, ароматизаторами, N-метил-L-фенілаланін, Моc = метоксикарбоніл, буферами, матеріалами, які сприяють розщепленPro = L-пролін, pNA = р-нітроаніліно, TFA - триню, та іншими матеріалами, які поліпшують біодофторооцтова кислота, ТНР = тетрагідропіраніл, ступність активного інгредієнта. Tos = р-толуолсульфоніл, Ζ = бензилоксикарбоніл, Фармацевтичні композиції винаходу приготовRT = кімнатна температура. Скорочення рідкісних ляють як звичайні композиції, такі як пероральні кислот, вжиті в цій заявці, є такими: Ada - адаманкомпозиції (які вводять через рот, такі як таблетки, тил-L-аланін, Aze = L-азетидин-2-карбонова кислокапсули, порошки, пілюлі, драже або гранули), а та, N-Me-D-cHpa = N-метил-D-циклогептилаланін, також парентеральні композиції (ліки, які вводять, D-cHpa = D-циклогептилаланін або (R)-2-аміно-3обминаючи шлунково-кишкову систему, такі як циклогептилпропіонова кислота, D-HIa = D-циклоін'єкції, інфузії, супозиторії, пластирі або мазі). гептилмолочна кислота або (R)-2-гідрокси-3У третьому аспекті винахід забезпечує спосіб циклогептилпропіонова кислота. лікування пацієнта з розсіяною внутрішньосудинЗначення Rf, вказані у прикладах, визначали ною коагуляцією, включаючи введення пацієнтові з шляхом тонкошарової хроматографії, застосовуюрозсіяною внутрішньосудинною коагуляцією пепчи силікагель як адсорбент (DC-Alufolien Kieselgel тидил аргіналу, який відповідає формулі Xaa-Xbb60 F254, Merck, Дармштадт), у представлених ниArg-H (І), де Хаа та Xbb є такими, як визначено жче проявних системах. Номери застосованих вище, або його фармацевтично прийнятної кислосистем подано в дужках після скорочення Rf. тно адитивної солі. Пептидил аргінали також нази1. Етилацетат вають похідними пептидиларгінін альдегідів. 2. Етилацетат - n-гексан (1:4) Згідно з цим аспектом винаходу винахід за3. Етилацетат - n-гексан (1:1) безпечує спосіб лікування розсіяної внутрішньосу4. Етилацетат - циклогексан (15:85) динної коагуляції, який включає введення тварині, 5. Хлороформ - ацетон (95:5) включаючи людину, пептидил аргіналів згідно з 6. Етилацетат - піридин - оцтова кислота - вовинаходом. У способі згідно з цим аспектом винада (960:20:6:11) ходу терапевтично ефективну кількість пептидил 7. Етилацетат - піридин - оцтова кислота - воаргіналу згідно з винаходом вводять протягом теда (480:20:6:11) рапевтично ефективного періоду часу тварині, 8. Етилацетат - піридин - оцтова кислота - вовключаючи людину, яка має у своєму організмі да (240:20:6:11) розсіяну судинну коагуляцію. В оптимальному ва9. Етилацетат - піридин - оцтова кислота - воріанті таке введення краще здійснювати внутрішда (120:20:6:11) ньовенно або підшкірно, у найкращому варіанті 10. Етилацетат - піридин - оцтова кислота внутрішньовенно. Введення терапевтичних комповода (90:20:6:11) зицій здійснюють, застосовуючи відомі процедури, 11. Етилацетат - піридин - оцтова кислота в дозах і протягом періодів часу, ефективних для вода (60:20:6:11) послаблення симптомів або рівноцінних ознак DIC. 12. Етилацетат - піридин - оцтова кислота При системному введенні терапевтичну композивода (45:20:6:11) цію в оптимальному варіанті вводять у дозі, доста13. Етилацетат - піридин - оцтова кислота тній для досягнення рівня в крові пептидил аргінавода (30:20:6:11) Коефіцієнти навантаження (k'), лів від приблизно 6мкМ до приблизно 100мкМ. В вказані у прикладах, визначали за допомогою приоптимальному варіанті загальна доза становить 13 77428 14 строю "Pharmacia LKB Analytical HPLC System. (20,1мМ) ізобутилового хлороформату. Після 10 Two" таким чином: хвилин перемішування додавали вищезгаданий Колонка: LiChrospher RP-18:12мкм 240x4мМ розчин у диметилформаміді N GТемпература колонки: навколишня бензилоксикарбоніл-L-аргінін лактаму, потім триеЕлюенти: Розчинник А, 0,1% TFA/вода, Розтиламін у кількості, достатній для доведення рН чинник В, 0,1% TFA/ацетонітрил реакційної суміші до 8 (приблизно 2,8мл). Реакційну суміш перемішували при -10°С протягом 30 Градієнтний профіль: 0 15хв. 30 60% В, похвилин, потім при 0°С протягом однієї години. Пістім ізократичний 60% В. ля цього солі відфільтровували і фільтрат розвоШвидкість потоку розчинника: 1мл/хв. дили 100мл етилацетату. Одержаний у результаті Детектор: УФ-монітор LKB 2141; довжина хвирозчин промивали 3x25мл води, 10мл 1М KHSO4 і лі: 214нм. 3x10мл води, висушували над безводним Na2SO4 і Інжектор: Rheodyne 7125. Петля: 100мкл. випарювали при 2,0-2,5кПа. Одержаний продукт Насоси: тип 2 LKB 2148. Система регулюванпіддавали хроматографії на колонці з силікагелем, ня: LKB HPLC Manager. використовуючи 200г Kieselgel 60 (0,040-0,063мМ) Концентрація зразка: 1мг/мл у Розчиннику А, як адсорбент та етилацетат як елюент. Фракції, які об'єм нагнітання 25мкл. містили лише чистий продукт [(Rf (1) = 0,60], об'єдЧас аналізу 40хв. нували й випарювали при 2,0-2,5кПа. Залишок Ацил-аргінін альдегіди присутні у врівноважепісля випарювання кристалізували з діізопропілоних структурах, тобто у формі альдегіду, альдегідвого етеру. гідрату та двох формах аміноциклолу. Під час Вихід 10,84г (86,1%), Rf (1) - 0,55-0,65. HPLC-аналізу альдегідгідрат та одна або обидві FAB мас-спектр (627 [М+Н]+) підтверджував форми аміноциклолу виявляються як два або три передбачувану структуру. окремі піки. Ацил-аргінін альдегіди, описані у прикЕтап 2: Етоксикарбоніл-D-циклогептилаланілладах, характеризуються двома або трьома покаL-проліл-NG-бензилоксикарбоніл-L-аргінін альдегід зниками k'. 8,02г (12,8мМ) етоксикарбоніл-DMac-спектрометрія. Вимірювання FAB позитициклогептилаланіл-L-проліл-NGвної іонізації здійснювали у пристрої Finnigan MAT бензилоксикарбоніл-L-аргінін лактаму (Приклад 1, 8430. Зразки розчиняли у m-нітробензиловому Етап 1) розчиняли у 15мл тетрагідрофурану, а спирті (NBA) і вводили безпосередньо у джерело потім при перемішуванні і при температурі, яка не іонів. У спектрі пептидил-аргінін альдегідів виявперевищувала -50°С, додавали розчин 3,6мМ ляли додатковий молекулярний іон додаткової LiAIH4, розчиненого в тетрагідрофурані. Процес сполуки, утвореної з NBA: [М+Н]+ і [M+H+NBA]*. У відновлення контролювали шляхом тонкошарової прикладах дані про FAB спектри вказували відпохроматографії (розчинник 7 як проявний розчинвідним чином. Вимірювання ESI позитивної іонізаник) і, в разі необхідності, додавали ще одну порції здійснювали у пристрої VG Quattro (Fisons). цію LіАІН4. До цієї реакційної суміші по краплях Зразки розчиняли в суміші ацетонітрил-вода (1:1), додавали 0,5Μ KHSO4 з постійним перемішуваняка містила 1% (об'єм/об'єм) мурашиної кислоти і ням і охолодженням до досягнення рН 3, потім вводили з петлею зразка 10мл у джерело іонів зі 35мл води. Одержаний у результаті розчин екстрашвидкістю потоку 15-25мл/хв. гували 2x15мл гексану, потім 3x20мл дихлоромеПриклад 1 тану. Екстракти в дихлорометані об'єднували, Синтез гемісульфатуетоксикарбоніл-Dпромивали 3x15мл води, 15мл холодного 5% розциклогептилаланіл-L-проліл-L-аргінін альдегіду чину гідрокарбонату натрію і знову 15мл води, виЕтап 1: Етоксикарбоніл-D-циклогептилаланілсушували над безводним Na2SO4 і випарювали L-проліл-NG-бензилоксикарбоніл-L-аргінін лактам при 2,0-2,5кПа. Залишок після випарювання обро7,85г (20,1мМ) трет-бутилоксикарбоніл-Dбляли діізопропіловим етером, фільтрували й вибензилоксикарбоніл-L-аргінін лактаму [(Bajusz et сушували в умовах зниженого тиску. aI., J. Med. Chem. 33,1729 (1990)] суспендували у Вихід 7,08г (88%), Rf (8) = 0,40-0,50. 20мл хлороформу, потім 20мл етилацетату, насиFAB мас-спектр (629 [М+Н]+, 782 [M+H+NBA]+) ченого газом НСІ (0,11-0,15г/мл), додавали при підтверджував передбачувану структуру. перемішуванні та охолодженні льодом. РозщепЕтап 3: Гемісульфат етоксикарбоніл-Dлення Вос групи контролювали шляхом тонкошациклогептилаланіл-L-проліл-L-аргінін альдегіду рової хроматографії [Rf (11) = 0,5 (вільна сполука); 6,91г (11,0мМ) етоксикарбоніл-D1.0 (Вос-сполука)]. До завершення реакції суспенциклогептилаланіл-L-проліл-Lзію розводили 40мл діетилового етеру, утворену бензилоксикарбоніл-L-аргінін альдегіду (Приклад кристалічну масу фільтрували, промивали 10мл 1, Етап 2) розчиняли у 85мл етанолу й додавали ацетону та 10мл діетилового етеру й висушували 11,25мл 0,5Μ сірчаної кислоти, потім 0,7г Pd-C в умовах зниженого тиску над КОН. Одержаний у каталізатора, суспендованого у 14мл води і суміш результаті NG-бензилоксикарбоніл-L-аргінін лактам гідрогенізували при приблизно 10°С. Процес реакгідрохлорид розчиняли у 20мл диметилформаміції контролювали шляхом тонкошарової хроматогду, охолоджували до -20°С і додавали до ангідрирафії. Після завершення реакції (приблизно 15 ду з подальшим перемішуванням. хвилин) каталізатор фільтрували і фільтрат конце7,12г (20,1мМ) етоксикарбоніл-Dнтрували до приблизно 7-9мл при 2,0-2,5кПа. Зациклогептилаланіл-L-проліну (Приклад 1, Етап J) лишок розводили 80 мл води, екстрагували розчиняли у 20мл диметилформаміду, охолоджу4x15мл дихлорометану і водний розчин залишали вали до -15°С, потім при перемішуванні додавали стояти при 20-22°С протягом 24 годин. Розчин 2,23мл (20,1мМ) N-метил-морфоліну та 2,65мл 15 77428 16 знову екстрагували 3x15мл дихлорометану і рН 302,38мМ), Rf (14) = 0,55-0,65), який додавали до доводили до 3,5 іонообмінною смолою Dowex AG суміші у 450мл метиленхлориду та 450мл води, 1-X8 (НО-), потім розчин піддавали ліофілізації. що містила 47,7г (450мМ) Na2СО3. Реакційну суміш перемішували до наступного дня. Дві фази відоВихід 4,90г (82%). Rf (11) = 0,35-0,45. [ ]D20 = кремлювали, органічну фазу промивали метилен77,6° (с=1,018; вода). хлоридом (2x100мл). Комбіновані органічні шари HPLC: k' = 1,695 і 2,328. промивали водою, висушували над безводним FAB мас-спектр (495 [M+H]+, 648 [M+H+NBA]+) Na2SO4, потім випарювали при 2,0-2,5кПа. Одерпідтверджував передбачувану структуру. жаний у результаті маслянистий продукт (36,97г, Синтез вихідних матеріалів: 263,64мМ) був чистим 1-циклогептилЕтоксикарбоніл-D-циклогептилаланіл-L-пролін ацетальдегідом, який використовували безпосеЕтап А; 1-циклогептилацетил-2,5редньо для наступної реакції. Rf (2) = 0,7-0,8. диметилпіразол Етап С: 5-циклогептилметилгідантоїн 58,6г (375мМ) циклогептилоцтової кислоти До розчину альдегіду (Приклад 1, Етап В) у [Protiva et al, Collect. Czech., Chem., Commun., 55: 50% водному етанолі (857мл; 3,25мл/мМ) при 501278-1289 (1990)] розчиняли у 375мл тетрагідро55°С при перемішуванні додавали 15,5г (290мМ; фурану, охолоджували до - 15°С, потім при пере1,1еквів.) хлориду амонію, 27,87г (659мМ; мішуванні додавали 41,3мл (375мМ) N2,5еквів.) карбонату амонію і 18,9г (290мМ; метилморфоліну, 49,50мл (375мМ) ізобутилхло1,1еквів.) ціаніду калію, і перемішування продовроформату і після 10 хвилин перемішування при жували при 50°С протягом 48 годин. Осаджений 10°С додавали розчин 37,85г (393,75мМ) 3,5матеріал відфільтровували, промивали 50% етадиметилпіразолу у 300мл тетрагідрофурану при нолом (100мл) і висушували у вакуум-ексикаторі. У 10°С. Перемішування продовжували при -10°С першій партії одержували 4226г (206,97мМ) матепротягом 30хв, при 0°С протягом години і при кімріалу. З маточного розчину відганяли етанол, занатній температурі протягом 3 годин. Після цього лишок екстрагували етилацетатом (1x250мл і солі відфільтровували, фільтрат випарювали в 2x100мл), органічні розчини комбінували, промиумовах зниженого тиску й залишок розчиняли у вали водою (3x50мл), висушували над безводним 900 мл етилацетату. Одержаний у результаті розNa2SO4, потім випарювали при 2,0-2,5кПа. Заличин послідовно промивали 3x80мл 1М NaOH, 89мл шок розтирали з діізопропіловим етером, фільтруводи, 3x80мл 1М НСІ і водою до нейтральності. вали й висушували. У другій партії одержували (Основні промиї комбінували й підкислювали для 3,6г (17,12мМ) матеріалу; загальний вихід складав одержання ~5,8г, 37,1мМ, циклогептилоцтової ки45,86г (218мМ, 82,5%) 5слоти). Розчин етилацетату висушували над безциклогептилметилгідантоїну. Т. пл.: 240,9°С. Rf (6) водним Na2SO4, потім випарювали при 2,0-2,5кПа. = 0,73-0,78. Одержаний у результаті маслянистий продукт розАналіз для C11H18N2O2 (210,28). Розраховано: глядали як 340мМ 1-циклогептилацетил-2,5С% = 62,83; Η % = 8,63; Ν% = 13,32. Виявлено: С% диметилпіразолу й використовували для наступно= 63,09; Η % = 8,67; N % = 13,25. го етапу без подальшого очищення. Rf (2) = 0,8-0,9 Етап D: DL-циклогептилаланін гідрохлорид (кислота: 0,3-0,4). 87г (1,55Μ) КОН розчиняли у 435мл nЕтап В: 1-циклогептилацетальдегід бутанолу при перемішуванні й нагріванні і додаваДо 350мл тетрагідрофурану, охолодженого до ли 45,71г (217,4мМ) 5-циклогептилметилгідантоїну -30°С додавали 12,83г (338мМ) LiAIH4. Після цього (Приклад 1, Етап С). Одержаний таким чином розпри перемішуванні по краплях додавали розчин чин піддавали дефлегмації протягом 72 годин, маслянистого продукту (Приклад 1, Етап А) у потім розводили водою й випарювали в умовах 250мл холодного тетрагідрофурану при -25°С. За зниженого тиску. Залишок розчиняли у 220мл вореакцією стежили шляхом TLC. У разі необхідності ди, підкислювали до рН 2 з використанням сс НСІ додавали 30-40мл розчину LіАІН4 у тетрагідрофу(~130мл) і тримали в холодильнику до наступного рані (3г/100мл). Після завершення відновлення дня. Осаджений матеріал відфільтровували, прохолодну суміш підкислювали 6Μ НСІ і розводили мивали 50мл води і висушували у вакууметилацетатом (500мл). Водну фазу екстрагували ексикаторі. Одержаний таким чином продукт етилацетатом, органічні розчини комбінували, (106,5г) розглядали як 217мМ DLпромивали водою до нейтральності, висушували циклогептилаланіну і використовували для поданад безводним Na2SO4, а потім випарювали при льших реакцій. Rf (11) = 0,2-0,3. 2,0-2,5кПа. Одержаний у результаті маслянистий Етап Е: гідрохлорид DL-циклогептилаланін мепродукт був необробленим 1тилового естеру циклогептилацетальдегідом, забрудненим 2,523,65мл (325,25мМ) SOCI2 при перемішуванні диметилпіразолом (DMP). Rf (2) = 0,7-0,8 (DT. ПЛ.: по краплях додавали у 217мл метанолу при тем0,25-0,35). пературі від 0°С до -5°С. Після цього додавали DLДо одержаного в результаті маслянистого циклогептилаланін (217мМ з Прикладу 1, Етап D) і продукту (62,6г), розчиненого у 350мл метанолу перемішували протягом 24 годин. Після перетвопри перемішуванні додавали розчин 36,4г NaHSO3 рення здійснювали TLC Rf (11) = 0,75-0,85 (естер), у 70мл води. Реакційну суміш до наступного дня (0,3-0,4 (кислота). Якщо виявляли непрореаговану тримали в холодильнику. Осаджений матеріал амінокислоту, реакційну суміш охолоджували до відфільтровували, промивали холодною сумішшю 5°С, по краплях додавали 11,8мл SOCI2 і реакцій35мл метанолу та 7мл води, потім діетиловим ну суміш перемішували ще 24 години. Після цього етером, і висушували. Твердий матеріал являє нерозчинені солі відфільтровували, промивали собою адукт гідросульфіт натрію (73,87г, 17 77428 18 метанолом (2x50мл) і комбіновані розчини мета7,24г (30мМ) ацетил-D-естеру (Приклад 1, нолу випарювали. Залишок повторно розчиняли в Етап G) суспендували у 120мл 6М НСІ і піддавали метанолі і знову випарювали. І нарешті, залишок дефлегмації протягом 3 годин. Вільну амінокислорозтирали з діізопропіловим етером, відфільтроту відокремлювали у вигляді кристалів. Реакційну вували, промивали діізопропіловим етером і висусуміш охолоджували, тримали в холодильнику до шували у вакуум-ексикаторі над КОН та Р2О5. наступного дня, фільтрували, промивали холодОдержували 33,68г (142,85мМ) гідрохлориду DLною водою та етером, потім висушували у вакуумциклогептилаланін метилового естеру. Rf (11) = ексикаторі. Одержували 5,9г (26,69мМ, 89%) D0,5-0,6. Т. пл. 95,4-96,5°С. циклогептилаланін гідрохлориду. Rf (12) = 0,10Етап F: Ацетил-DL-циклогептилаланін метило0,15. [ ]D20 = -11° (c=0,4; 1M HCI). вий естер Аналіз для C10H19NO2.HCI (221,728). РозрахоДо розчину 33,68г (142,85мМ) гідрохлориду вано: С % = 54,17; Η % = 9,09; Ν % = 6,32; СІ % = DL-циклогептилаланін метилового естеру (Прик15,99. Виявлено: С % = 54,27; Η % = 9,27; Ν % = лад 1, Етап Е) у 140мл піридину протягом години 6,30; СІ % = 16,2. по краплях додавали оцтовий ангідрид (162мл, Етап І: Етоксикарбоніл-D-циклогептилаланін 171,43мМ) при перемішуванні й охолодженні у До розчину 4,43г (20мМ) D-циклогептилаланін ванні з крижаною водою. Реакційну суміш перемігідрохлориду (Приклад 1, Етап Н) у 20мл диметишували протягом 24 годин за навколишньої темлформаміду додавали 5,6мл (40мМ) триетиламіну ператури, потім випарювали в умовах зниженого та 3,95г (21мМ) (N-гідроксисукцинімідил)-етил картиску. Залишок розчиняли у 300мл етилацетату, бонату.* Після перемішування при кімнатній темпромивали 1М KHSO4 (3x50мл) та водою (3x50мл), пературі протягом 3 годин реакційну суміш випависушували над безводним Na2SO4, a потім випарювали, залишок, розчинений у 40мл етилацетату, рювали при 2,0-2,5кПа. Одержаний у результаті промивали 2x30мл 1М KHSO4 та водою до нейтмаслянистий продукт розтирали з діізопропіловим ральності. Після цього органічний шар висушували етером до твердого матеріалу, який відфільтровунад безводним Na2SO4, потім випарювали при 2,0вали, промивали діізопропіловим етером, потім 2,5кПа. Одержаний у результаті маслянистий проводою, і висушували в ексикаторі. Одержували дукт (4,47г, ~17мМ) був етоксикарбоніл-D24,36г (100,94мМ, 70,4%) метил ацетил-DLциклогептилаланіном [Rf (7) = 0,85-0,90], який вициклогептилаланінату (ацетил-DL-естер). Rf (1) = користовували безпосередньо на наступному ета0,55-0,65. Т. пл.: 69-71°С. пі. [ ]D20 = +6,3° (с=1, метанол). Аналіз для C13H23NO3 (241,332). Розраховано: *Одержання (N-гідроксисукцинімідил)-етилС % = 64,70; Η % = 9,61; Ν% = 5,80. Виявлено: С % карбонату = 64,75; Η % = 9,76; Ν % = 5,85. 11,5г (100мМ) N-гідроксисукциніміду розчиняЕтап G: Ацетил-D-циклогептилаланін метилоли у 100мл тетрагідрофурану, охолоджували до вий естер (ферментативне розчинення ацетил-DL10°С, потім при перемішуванні додавали 15,4мл циклогептилаланін метилового естеру) (110мМ) триетиламіну та 10,45мл (110мМ) етилДо розчину 8,69г (36мМ) ацетил-DLхлорокарбонату. Після 2 годин перемішування при циклогептилаланін метилового естеру, ацетил-DLкімнатній температурі суміш фільтрували і фільтестерy (Приклад 1, Етап F), у 36мл толуолу, додарат випарювали в умовах зниженого тиску. Масвали 72мл води і 36мг Subtilisin Carlsberg (Протеалянистий залишок після охолодження кристалізуза, тип VIII, Sigma). Ферментативний гідроліз Lвався. Кристалічний матеріал суспендували в енантіомеру, ацетил-L-естерy, що відбувався при петролейному етері, відфільтровували і висушурН 7,0, підтримували за допомогою автотитратора, вали у вакуум-ексикаторі. Вихід 12,78г (68,3%). Т. заповненого 3М NaOH. Після припинення витрати пл.: 39,4-39,7°С. NaOH (5,8мл, 17,4мМ) реакційну суміш розводили Аналіз для C7H9NO5 (187,15). Розраховано: С, 36мл толуолу і два шари відокремлювали. Водну 44,92; Н, 4,85; N, 7,48. фазу промивали 2x30мл толуолу. Комбіновані роВиявлено: С % = 44,67; Н% = 4,81; N% = 7,27. зчини в толуолі містили ацетил-D-естер, а комбіЕтап J: Етоксикарбоніл-D-циклогептилаланілновані водні розчини містили натрієву сіль ацетилL-пролін L-кислоти. Розчин етоксикарбоніл-D-циклогептилаланіну Після висушування над безводним Na2SO4 (~17мМ, Приклад 1, Етап І) у 17мл THF комбінувакомбіновані розчини в толуолі випарювали в умоли з 374г (20мМ) N-гідроксисукциніміду, охолоджувах зниженого тиску для одержання 3,8г (15,75мМ) вали до 10°С і комбінували з розчином 4,12г ацетил-D-естерy, Rf (1) = 0,55-0,65, який викорис(20моль) 1,3-дициклогексилкарбодиіміду у приблитовували безпосередньо на наступному етапі. зно 20мл THF. Суміш перемішували протягом приКомбіновані водні розчини підкислювали й екблизно 5год при 22°С, після чого утворення активстрагували 3x30мл етилацетатом. Комбіновані ного естеру вважали завершеним згідно з даними розчини в етилацетаті промивали водою, висушуTLC. вали над безводним Na2SO4, і випарювали в умоL-пролін (1,95г, 17мМ) додавали до перемішувах зниженого тиску для одержання 4,0г (17,6мМ) ваної реакційної суміші з наступним додаванням ацетил-L-кислоти. Rf (7) = 0,38-0,42. 2,3мл (17мМ) триетиламіну. Реакційну суміш пеПодібний спосіб одержання, починаючи з 9,65г ремішували при 22°С протягом приблизно 15год, (40мМ) ацетил-DL-естеру (Приклад 1, Етап F) дапісля чого витрату активного естеру вважали завав на виході 4,6г (19,06мМ) ацетил-D-естеру та вершеною згідно з даними TLC. Реакційну суміш 4,44г (19,55мМ) ацетил-L-кислоти. фільтрували, осад на фільтрі промивали 10мл Етап Н: D-циклогептилаланін гідрохлорид THF і фільтрат випарювали. Залишок розчиняли у 1977428 20 30мл етилацетату та 30мл води. Водну фазу проFAB мас-спектр (703 [М+Н]+) підтверджував мивали 2х20мл етилацетату, підкислювали 20мл передбачувану структуру. 1М KHSO4 і екстрагували 3х20мл етилацетату. Етап 2: Бензилоксикарбоніл-N-метил-DКомбіновані розчини в етилацетаті промивали циклогептилаланіл-L-проліл-NGводою до нейтральності, висушували над безводбензилоксикарбоніл-L-аргінін альдегід ним Na2SO4, потім випарювали при 2,0-2,5кПа. 5,62г (8мМ) бензилоксикарбоніл-N-метил-DПісля розтирання з діізопропіловим етером залициклогептилаланіл-L-проліл-NGшок кристалізувався. Цю суспензію кристалів охобензилоксикарбоніл-L-аргінін лактаму (Приклад 2, лоджували в холодильнику, фільтрували з петроЕтап 1) розчиняли у 10мл тетрагідрофурану, а лейним етером і висушували у вакуум-ексикаторі. потім при перемішуванні і при температурі, яка не Одержували 4,2г (11,85мМ, 70%) етоксикарбонілперевищувала -50°С, додавали розчин 2,25мМ D-циклогептилаланіл-L-проліну. Rf (7) = 0,45-0,55. LіАІН4, розчиненого в тетрагідрофурані. Процес Аналіз для C18H30N2O4 (354,45). Розраховано: відновлення контролювали шляхом тонкошарової С, 60,99; Н, 8,53; N, 7,90. Виявлено: С % = 60,14; Η хроматографії (розчинник 7 як проявний розчин% = 8,55; Ν% = 7,38. ник) і, в разі необхідності, додавали ще одну порFAB мас-спектр (355 [M+H]+) підтверджував цію LіАІН4. До цієї реакційної суміші по краплях передбачувану структуру. додавали 0,5Μ KHSO4 з постійним перемішуванПриклад 2 ням і охолодженням до досягнення рН 3, потім Синтез N-метил-D-циклогептилаланіл-L25мл води. Одержаний у результаті розчин екстрапроліл-L-аргінін альдегід сульфату гували 2x10мл гексану, потім 3x15мл дихлоромеЕтап 1: Бензилоксикарбоніл-N-метил-Dтану. Екстракти з дихлорометані об'єднували, циклогептилалант-L-пролт-NG-бензилпромивали 3x15мл води, 15мл холодного 5% розоксикарбоніл-L-аргінін лактам чину NaHCO3 і знову 15мл води, висушували над 3,93г (10мМ) трет-бутилоксикарбоніл-NGбезводним Na2SO4 і випарювали при 2,0-2,5кПа. бензилоксикарбоніл-L-аргінін лактаму [(Bajusz et Залишок після випарювання обробляли діізопроal, J. Med. Chem. 33, 1729 (1990)] суспендували у піловим етером, фільтрували і висушували в умо10мл хлороформу, потім при перемішуванні й оховах зниженого тиску. лодженні льодом додавали 10мл етилацетату, Вихід 4,95г (88%), Rf (1) = 0,20-0,25. насиченого газом НСІ (0,11-0,15г/мл). РозщепленFAB мас-спектр (705 [М+Н]+, 858 [M+H+NBA]4) ня Вос групи контролювали шляхом тонкошарової підтверджував передбачувану структуру. хроматографії [Rf (11) = 0,5 (вільна сполука); 1,0 Етап 3: N-метил-D-циклогептилаланіл-L(Вос-сполука)]. До завершення реакції суспензію проліл-L-аргінін альдегід сульфат розводили 20мл діетилового етеру, утворену крис4,6г (6,5мМ) бензилоксикарбоніл-N-метил-Dталічну масу фільтрували, промивали 5мл ацетону циклогептилаланіл-L-проліл-NGта 5мл діетилового етеру й висушували в умовах бензилоксикарбоніл-L-аргінін альдегіду (Приклад зниженого тиску над КОН. Одержаний у результаті 2, Етап 2) розчиняли у 65мл етанолу й додавали гідрохлорид NG-бензилоксикарбоніл-Ьаргінін лак13,5мл 0,5Μ сірчаної кислоти, потім 0,4г Pd-C катаму розчиняли у 10мл диметилформаміді, охолоталізатора, суспендованого у 10мл води, і суміш джували до -20°С і додавали до ангідриду з подагідрогенізували при приблизно 10°С Процес реакльшим перемішуванням. ції контролювали шляхом тонкошарової хроматог4,32г (10мМ) бензилоксикарбоніл-N-метил-Dрафії. Після завершення реакції (приблизно 15 циклогептилаланіл-L-проліну (Приклад 2, Етап С) хвилин) каталізатор фільтрували і фільтрат концерозчиняли у 10мл диметилформаміду, охолоджунтрували до приблизно 5-7мл при 2,0-2,5кПа. Завали до -15°С, потім при перемішуванні додавали лишок розводили 50мл води, екстрагували 4x10мл 1,12мл (10,1мМ) N-метил-морфоліну та 1,33мл дихлорометану і водний розчин залишали стояти (10,1мМ) ізобутил хлороформату. Після 10 хвилин при 20-22°С протягом 24 годин. Розчин знову експеремішування додавали вищезгаданий розчин у трагували 3x10мл дихлорометану і рН доводили диметилформаміді NG-бензилоксикарбоніл-Lдо 3,5 іонообмінною смолою Dowex AG 1-X8 (НО), аргінін лактаму, потім триетиламін у кількості, доспотім розчин піддавали ліофілізації. татній для доведення рН реакційної суміші до 8 Вихід 2,85г (82%). Rf (9) = 0,35-0,45. [ ]D20 = (вимагалося приблизно 1,4мл). Реакційну суміш 79,6° (с=1; вода). перемішували при -10°С протягом 30 хвилин, поFAB мас-спектр (437 [M+H]+', 590 [M+H+NBA]+) тім при 0°С протягом однієї години. Після цього підтверджував передбачувану структуру. солі відфільтровували і фільтрат розводили 100мл Синтез вихідних матеріалів: етилацетату. Одержаний у результаті розчин проБензилоксикарбоніл-N-метил-Dмивали 3x15мл води, 6мл 1М KHSO4 і 3x6мл води, циклогептилаланіл-L-пролін висушували над безводним Na2SO4 і випарювали Етап А: Синтез N-бензилоксикарбоніл-Dпри 2,0-2,5кПа. Одержаний продукт піддавали циклогептилаланіну хроматографії на колонці з силікагелем, викорисD-Циклогептилаланін гідрохлорид (Приклад 1, товуючи 100г Kieselgel 60 (0,040-0,063мМ) як адЕтап Н) (11,09г, 50мМ) комбінували з 40мл THF та сорбент і етилацетат як елюент. Фракції, які місти40мл води при 0°С. Перемішувану суміш доводили ли лише чистий продукт [(Rf (1) = 0,70], до приблизно рН 10 додаванням 5М розчину об'єднували й випарювали при 2,0-2,5кПа. ЗалиNaOH. До реакційної суміші додавали бензил хлошок після випарювання кристалізували з діізопророформат (8,22мл, 55,4мМ), одночасно підтримупілового етеру. ючи температуру приблизно 3°С і рН приблизно Вихід 6,0г (85%), Rf (1) = 0,65-0,75. 10, додаючи 5М NaOH, якщо потрібно. Після заве 21 77428 22 ршення додавання бензил хлороформату реакційводи до реакційної суміші, одночасно підтримуючи ну суміш перемішували протягом 1год при 0°С і температуру нижче 13°С і контролюючи спінюванпідтримували рН приблизно 10. Перемішування ня. Гашену реакційну суміш перемішували протяпродовжували до наступного дня при RT і завергом приблизно 20хв при 22°С, а потім концентрушення реакції перевіряли за допомогою TLC (7). У вали до приблизно 40мл в умовах зниженого тиску разі необхідності до реакційної суміші додавали при температурі нижче 30°С. До залишку додаваще певну кількість бензил хлороформату (1-2 x ли воду (70мл) з наступним додаванням 30мл t075мл, 5мМ), одночасно підтримуючи температуру бутилметилового етеру. Фази відокремлювали і приблизно 3°С і рН приблизно 10, додаючи 5М водну фазу знову промивали 30мл tNaOH. Додавали t-бутилметиловий етер (25мл) і бутилметилового етеру. Водну фазу комбінували з перемішувану суміш нагрівали до 22°С. Водну фа40мл етилацетату й доводили до рН 2,2 за допозу відокремлювали і промивали другою 25мл пормогою 3М розчину сірчаної кислоти. Фази відокрецією t-бутилметилового етеру. Вміст органічної млювали і водну фазу піддавали зворотній екстрафази перевіряли за допомогою TLC і, в разі необкції 40мл етилацетату. Комбіновану органічну хідності, органічну фазу піддавали зворотній екстфазу промивали 70мл 5% розчину тіосульфату ракції 25мл води. Водні фази та 25мл етилацетату натрію. Фази відокремлювали й органічну фазу комбінували й доводили до рН 2 концентрованою концентрували до малого об'єму в умовах вакууму НСІ. Фази відокремлювали й водну фазу екстрагу(33-45кПа), одночасно підтримуючи температуру вали другою 25мл порцією етилацетату. Комбінонижче 50°С. Залишок комбінували з 18,6мл THF та вану органічну фазу промивали 20мл 1М KHSO4 і 100мл води й доводили до рН 85 циклогексиламі2x30мл води, висушували над безводним Na2SO4 і ном. Одержану в результаті гідросуміш концентрувипарювали при 2,0-2,5кПа. Залишок після випавали до 100мл в умовах зниженого тиску (9-33кПа) рювання розчиняли у 45мл THF і цей розчин бенпри 25-55°С, доводили до 25°С, розводили 71,5мл зилоксикарбоніл-D-циклогептилаланіну тримали води й перемішували приблизно 10,5год. Гідросудля застосування на наступному етапі без подаміш фільтрували, промивали водою й висушували льшого очищення. на повітрі при 45°С для одержання 16,72г солі беЕтап В: Бензилоксикарбоніл-Dнзилоксикарбоніл-N-метил-D-циклогептилаланіл-Lциклогептилаланіл-L-пролін Розчин у TFA бензипролін циклогексиламіну (63% вихід із Dлоксикарбоніл-D-циклогептилаланіну, одержаного циклогептилаланіну). Rf (8) = 0,55-0,65. у Прикладі 2, Етап А, комбінували з 5,9г (5124мМ) Приклад 3 N-гідроксисукциніміду, охолоджували до 10°С і Синтез гемісульфату D-циклогептиллактил-Lкомбінували з розчином 11г (53,3мМ) 1,3проліл-L-аргінін альдегіду дициклогексилкарбодиіміду у приблизно 25мл Етап 1: Тетрагідропіраніл-DTHF. Суміш перемішували протягом приблизно циклогептиллактил-L-проліл-NG4,5год при 22°С, після чого утворення активного бензилоксикарбоніл-L-аргінін лактам естеру вважали завершеним згідно з даними TLC. 5,08г (13мМ) трет-бутилоксикарбоніл-NGL-пролін (5,9г, 51,24мМ) додавали до перемібензилоксикарбоніл-L-аргінін лактаму [(Bajusz et шуваної реакційної суміші з наступним додаванal, J. Med Chem. 33, 1729 (1990)] суспендували у ням 7,2мл (51,24мМ) триетиламіну. Реакційну су13мл хлороформу, потім при перемішуванні та міш перемішували при 22°С протягом приблизно охолодженні льодом додавали 13мл етилацетату, 15год, після чого витрату активного естеру вважанасиченого газом НСІ (0,11-0,15г/мл). Розщепленли завершеною згідно з даними TLC. Реакційну ня Вос групи контролювали шляхом тонкошарової суміш фільтрували, осад на фільтрі промивали хроматографії [Rf (11) = 0,5 (вільна сполука); 1,0 25мл THF і фільтрат випарювали. Залишок розчи(Вос-сполука)]. До кінця реакції суспензію розвоняли у 50мл етилацетату та 50мл води. Водну фадили 25мл діетилового етеру, утворену кристалічзу промивали 2x20 мл етилацетату, підкислювали ну масу фільтрували, промивали 7мл ацетону та 20мл 1Μ KHSO4 і екстрагували 3x40мл етилацета7мл діетилового етеру й висушували в умовах ту. Комбіновані розчини в етилацетаті промивали зниженого тиску над гідроксидом калію. Одержаводою до нейтральності, висушували над безводний у результаті NG-бензилоксикарбоніл-Ьаргінін ним Na2SO4, потім випарювали при 2,0-2,5кПа. лактам гідрохлорид розчиняли у 13мл диметилЗалишок, бензилоксикарбоніл-Dформаміду, охолоджували до -20°С і додавали до циклогептилаланіл-L-пролін, розчиняли у 60мл ангідриду з подальшим перемішуванням. THF і використовували на наступному етапі без Розчин солі тетрагідропіраніл-Dподальшого очищення. циклогептиллактил-L-пролін триетил-амонію, одеЕтап С: Бензилоксикарбоніл-N-метил-Dржаного на Етапі 1 Прикладу 3 (12мМ) охолоджуциклогептилаланіл-L-пролін вали до -20°С, потім при перемішуванні додавали Йодометан (17,95мл, 288мМ) додавали до ро1,6мл (12мМ) ізобутил хлороформату. Після 10 зчину у THF бензилоксикарбоніл-Dхвилин перемішування додавали вищезгаданий циклогептилаланіл-L-проліну з Прикладу 2, Етап В. розчин у диметилформаміді NGЦей розчин охолоджували до 8°С і переносили бензилоксикарбоніл-L-аргінін лактаму, потім триеразом з 20мл THF для промивання до перемішутиламін у кількості, достатній для доведення рН ваної гідросуміші 4,75г (119мМ) гідриду натрію реакційної суміші до 8 (вимагалося приблизно 60% у 35мл THF, одночасно підтримуючи темпе1,8мл). Реакційну суміш перемішували при -10°С ратуру нижче 13°С. Реакційну суміш перемішували протягом 30 хвилин, потім при 0°С протягом однієї при 11°С протягом 24год. Надлишок гідриду нагодини. Після цього солі відфільтровували і фільттрію розкладали обережним додаванням 1,6мл рат розводили 65мл етилацетату. Одержаний у 23 77428 24 результаті розчин промивали 3x25мл води, 7мл підтверджував передбачувану структуру. 1М гідросульфату калію та 3x7мл води, висушуваСинтез вихідних матеріалів: ли над безводним Na2SO4 і випарювали при 2,0Триетиламонієва сільтетрагідропіраніл-D2,5кПа. Одержаний продукт піддавали хроматогциклогептиллактил-L-проліну рафії на колонці з силікагелем, використовуючи Етап А: Хлороацетил-DL-циклогептилаланін 130г Kieselgel 60 (0,040-0,063мМ) як адсорбент та метиловий естер етилацетат як елюент. Фракції, які містили лише До розчину 15,2г (65мМ) гідрохлориду DLчистий продукт [(Rf (1) = 0,60], об'єднували і випациклогептилаланін метилового естеру (Приклад 1, рювали при 2,0-2,5кПа. Залишок після випарюванЕтап Е) у 65мл дихлорометану додавали 9,1мл ня кристалізували з діізопропілового етеру. (65мМ) триетиламіну та 14,9г (78мМ) (NВихід 5,0г (7,8 мМ, 64%), Rf (1) - 0,6 гідроксисукцинімідил)-хлорацетату*. Після переFAB мас-спектр (640 (М+Н+) підтверджував мішування при кімнатній температурі протягом 3 передбачувану структуру. годин реакційну суміш розводили 65мл дихлороЕтап 2: Тетрагідропіраніл-Dметану й послідовно промивали 3x30мл води, 1М циклогептиллактил-L-проліл-NGKHSO4, водою, 5% NaHCO3, і, нарешті, водою до бензилоксикарбоніл-L-аргінін-альдегід нейтральності. Після цього органічний шар вису4,8г (7,51мМ) тетрагідропіраніл-Dшували над безводним Na2SO4, потім випарювали циклогептиллактил-L-проліл-NGпри 2,0-2,5кПа. Одержаний у результаті маслянисбензилоксикарбоніл-Ьаргінін лактаму (Приклад 3, тий продукт розтирали з петролейним етером. Етап 1) розчиняли у 15мл тетрагідрофурану, потім Твердий матеріал відфільтровували, промивали при перемішуванні при температурі, яка не перепетролейним етером і висушували у вакуумвищувала -50°С, додавали розчин 3,6мМ алюмогіексикаторі. Одержували 17,54г (63,6мМ, ~98%) дриду літію, розчиненого в тетрагідрофурані. Прохлороацетил-DL-циклогептилаланін метилового цес відновлення контролювали шляхом естеру, який використовували безпосередньо для тонкошарової хроматографії з проявним розчиннинаступної реакції. Rf (7) = 0,73-0,83. Т. пл.: 78-80°С. ком №8 і, в разі необхідності, додавали ще одну Аналіз для C13H22NO3CI (275,777). Розраховапорцію алюмогідриду літію. До цієї реакційної суно С % = 56,62; Η % = 8,04; Ν % = 5,08; СІ % = міші по краплях додавали 0,5Μ сірчаної кислоти з 12,86. Виявлено: С % = 55,65; Η % = 7,93; Ν % = постійним перемішуванням та охолодженням до 5,06; СІ % = 12,72. досягнення рН 3, потім додавали 35мл води. Оде*Одержання (N-гідроксисукцинімідил) хлораржаний у результаті розчин екстрагували 2x15мл цетату гексану, потім 3x20мл дихлорометану. Екстракти в 32мл (450мМ) хлороацетил хлориду додавали дихлорометані об'єднували, промивали 3x15мл до 23г (200мМ) Ν-гідроксисукциніміду і суміш підводи, 15мл холодного 5% розчину гідрокарбонату давали дефлегмації протягом 10 хвилин, потім натрію і знову 15мл води, висушували над безводвиливали на подрібнений лід, фільтрували, проним сульфатом натрію і випарювали при 2,0мивали холодною водою і висушували у вакуум2,5кПа. Залишок після випарювання обробляли ексикаторі. діізопропіловим етером, фільтрували й висушуваВихід 20,23г (105,9мМ, 53%). Т. пл.: 113,3ли в умовах зниженого тиску. 113,7°С. Аналіз для C6H6NO4CI C7H9NO5 (191.55). РозВихід 3,9г (6,07мМ, 81%). Rf (8) = 0,40. [ ]D20 = раховано: С% = 37,62; Н% = 3,16; N% = 7,31; СІ% +16,0°(с=1, тетрагідрофуран). = 18,51. Виявлено: С % = 37,37; Н% = 3,16; N % = FAB мас-спектр (642 [М+Н+, 795 [M+H+NBA]+) 7,23; СІ% = 18,35. підтверджував передбачувану структуру. Етап В: Хлороацетил-D-циклогептилаланін Етап 3: Гемісульфат D-циклогептиллактил-Lметиловий естер (ферментативне розчинення проліл-L-аргінін альдегіду хлороацетил-DL-циклогептилаланін метилового 3,21г (5мМ) тетрагідропіраніл-Dестеру) циклогептиллактил-L-проліл-NGДо розчину 8,71г (31,6моль) хлороацетил-DLбензилоксикарбоніл-L-аргінін альдегіду (Приклад циклогептилаланін метилового естеру, DL-естер, 3, Етап 2) розчиняли у 40мл етанолу і додавали (Приклад 3, Етап А) у 30мл толуолу додавали 5мл 0,5Μ сірчаної кислоти, потім 0,3г Pd-C каталі70мл води та 50мг Subtilisin Carlsberg (Протеаза, затора, суспендованого у 6мл води, і суміш гідротип VIM, Sigma). Ферментативний гідроліз Lгенізували при приблизно 10°С. Процес реакції енантіомеру, L-естер, що відбувався при рН 7,0, контролювали шляхом тонкошарової хроматограпідтримували за допомогою автотитратора, заповфії. Після завершення реакції (приблизно 15 хвиненого 3Μ NaOH. Після припинення витрати NaOH лин) каталізатор відфільтровували і фільтрат кон(5,522мл, 16,57мМ) реакційну суміш розводили центрували до приблизно 4-6мл при 2,0-2,5кПа. 30мл толуолу і два шари відокремлювали. Водну Залишок розводили 40мл води, екстрагували фазу промивали 2x20мл толуолу. Комбіновані ро4x7мл дихлорометану і водний розчин залишали зчини в толуолі містили D-естер, а комбіновані стояти при 20-22°С протягом 24 годин. Розчин водні розчини містили натрієву сіль L-кислоти. знову екстрагували 3x15мл дихлорометану і рН Після висушування над безводним Na2SO4 доводили до 3,5 іонообмінною смолою Dowex AG комбіновані розчини в толуолі випарювали в умо1-Х8 (НО-), потім розчин піддавали ліофілізації. вах зниженого тиску для одержання 4,18г Вихід 1,65г (3,5мМ, 70%) [ ]D20 = -94,7° (с=1, (15,16мМ) D-естеру. Rf (7) = 0,73-0,83, який виковода) ристовували безпосередньо для одержання DHPLC: k' = 2,702 і 3,010. циклогептилаланіну. FAB мас-спектр (424 [M+H]+, 577 [M+H+NBA]+) 25 77428 26 Комбіновані водні розчини підкислювали й екАналіз для С10Н18О3 (186,52). Розраховано: С страгували 3x30мл етилацетату. Комбіновані роз% = 64;48; Η % = 9,74. Виявлено: С % = 64,54; Н% чини в етилацетаті промивали водою, висушували = 9,86. над безводним Na2SO4 і випарювали в умовах Етап Е: Бензиловий естер Dзниженого тиску для одержання 3,46г (13,22мМ) Lциклогептилмолочної кислоти кислоти [Rf (7) = 0,45-0,50], яку використовували До розчину 11,21г (30,5мМ) дициклогексилабезпосередньо для одержання Lмонієвої солі D-циклогептилмолочної кислоти циклогептилаланіну. (Приклад 3, Етап D) у 30мл диметилформаміду Подібний спосіб одержання, починаючи з 8,25г додавали 3,57мл (30мМ) бензилброміду. Суміш (30мМ) DL-естеру (Приклад 2, Етап А) давав на перемішували при кімнатній температурі протягом виході 4,08г (14,79мМ) D-естеру і 3,23г (12,34мМ) 24 годин, потім фільтрували і випарювали при 2,0L-кислоти. 2,5кПа. Залишок розчиняли у 20мл 0,5Μ гідрокарЕтап С: D-циклогептилаланін гідрохлорид бонату калію та 60мл діетилового етеру. Органічну 8,27г (30мМ) D-естеру (Приклад 3, Етап В) суфазу послідовно промивали 20мл води, 0,5Μ спендували у 120мл 6М НІ і піддавали дефлегмаKHSO4 та водою, потім висушували над безводції протягом 3 годин. Вільну амінокислоту відокреним сульфатом натрію і випарювали в умовах млювали у вигляді кристалів. Реакційну суміш зниженого тиску. Маслянистий залишок являв соохолоджували, тримали в холодильнику до настубою 8,3г (~30мМ) бензилового естеру Dпного дня, фільтрували, промивали холодною воциклогептилмолочної кислоти [Rf (3) = 0,2-0,3], дою та етером, потім висушували у вакуумякий використовували безпосередньо на Етапі F. ексикаторі. Одержували 5,9г (26,69мМ, 89%) DЕтап F: Бензиловий естер тетрагідропіраніл-Оциклогептилаланін гідрохлориду. Rf (12) = 0,10циклогептилмолочної кислоти До розчину 8,29г (30мМ) бензилового естеру 0,15. [ ]D20 = -11° (с=0,4; 1 МНСІ). D-циклогептилмолочної кислоти (Приклад 3, Етап Аналіз для C10H19NO2.HCI (221,728). РозрахоЕ) у 30мл дихлорометану додавали 3,01мл (33мМ) вано: С % = 54,17; Η % = 9,09; Ν % = 6,32; СІ % = 3,4-дигідро-2Н-пірану і 0,3мл ~3Μ НСІ в етилаце15,99. Виявлено: С % = 54,27; Η % = 9,27; Ν % таті і суміш залишали стояти при кімнатній темпе6,30; Cl% = 16,2. ратурі протягом 16 годин. Після цього реакційну Етап D: Дициклогексиламонієва сіль Dсуміш розводили 40мл дихлорометану, промивали циклогептилмолочної кислоти 3x20мл води, висушували над безводним сульфа5,78г (26,15мМ) D-циклогептилаланін гідрохтом натрію і випарювали при 2,0-2,5кПа. Залишок лориду (Приклад 3, Етап С) розчиняли у 26мл вопіддавали хроматографії на колонці з силікагелем, ди, розводили 105мл води та 52,5 л крижаної оцвикористовуючи 200г Kieselgel 60 (0,040-0,063мМ) тової кислоти й охолоджували до 5°С. До цієї як адсорбент та 85:15 суміш циклогексану та етисуміші по краплях додавали розчин 18,0г (261мМ) лацетату як елюент. Фракції, які містили лише чисNaNO2 у 30мл води при перемішуванні й охолотий продукт [Rf (4) = 0,60-0,70], об'єднували і випадженні. Перемішування продовжували при 5°С рювали при 2,0-2,5кПа. Маслянистий залишок протягом години і при кімнатній температурі до являв собою 7,9г (21,9мМ, 73%) бензилового еснаступного дня. Наступного дня реакційну суміш теру тетрагідропіраніл-D-циклогептилмолочної підкислювали 25мл сс НСІ при перемішуванні. кислоти, який використовували безпосередньо на Після цього суміш випарювали до сухого стану при наступному етапі. 50°С в умовах зниженого тиску. Залишок розчиняЕтап G: Триетиламонієва сіль тетрагідропірали у 100мл води і так само випарювали, розтирали ніл-О-циклогептилмолочної кислоти з толуолом і випарювали знову. Кінцевий залишок 7,21г (20мМ) бензилового естеру тетрагідропірозчиняли 50мл етилацетату та 50мл води. Водну раніл-D-циклогептилмолочної кислоти (Приклад 3, фазу промивали етилацетатом і комбіновані розЕтап F) розчиняли у 20мл диметилформаміду, чини в етилацетаті промивали водою до нейтрадодавали 2,8мл (20мМ) триетиламіну й гідрогенільності, висушували над безводним Na2SO4 і визували у присутності 0,1г каталізатора Pd/C. Пропарювали в умовах зниженого тиску. Одержану в цес реакції контролювали шляхом тонкошарової результаті тверду речовину розчиняли у 20мл дііхроматографії [Rf (1) = 0,30 (естер), 0,00 (кислозопропілового етеру. До цього розчину додавали та)]. Після завершення реакції каталізатор відфі5мл (25мМ) дициклогексиламіну, після чого крисльтровували і промивали 2x5мл диметилформаталічну сіль відокремлювали. Після охолодження міду. Фільтрат та промиї комбінували і кристали відфільтровували, промивали холодним використовували на наступному етапі як розчин етером і висушували у вакуум-ексикаторі для оде20мМ триетиламонієвої солі тетрагідропіраніл-Dржання 5,5г (14,96мМ, 57,2%) дициклогексиламіциклогептилмолочної кислоти. нової солі D-циклогептилмолочної кислоти, DЕтап Н: Бензиловий естер тетрагідропіраніл-DcHla.DCHA. Rf (7) = 0,53-0,60. [ ]D20 = +19,5° (с=1; циклогептиллактил-L-проліну метанол.)· Т. пл.: 147-150°С. Розчин триетиламонієвої солі тетрагідропіраАналіз для С10Н18О3 (C22H41NO3). Розраховано: ніл-D-циклогептилмолочної кислоти, одержаної на С % = 71,89; Η % = 11,24; Ν % = 3,81. Етапі G Прикладу 3 (20мМ), охолоджували до +5°С Виявлено: С% = 71,57; Η % = 11,34; Ν % = і при перемішуванні додавали 2,7г (20мМ) 13,83. гідроксибензотриазолу, 4,83г (20мМ) гідрохлориду Перетворення 0,35г (1мМ) D-cHla.DCHA на віL-пролін бензилового естеру та 4,12г (20мМ) дицильну -гідроксикислоту давало на виході 0,15г 20 клогексилкарбодиіміду. Реакційну суміш тримали (0,8мМ) D-cHIa, [ ]D = +10,1° (с=1; метанол.); т. при кімнатній температурі до наступного дня, потім пл.: 125-127°С. 27 77428 28 фільтрували і випарювали при 2,0-2,5кПа. Залитягом однієї години. Після цього солі відфільтрошок розчиняли у 50мл етилацетату і промивали вували і фільтрат розводили 50мл етилацетату. 20мл води та 5% гідрокарбонату натрію, висушуОдержаний у результаті розчин промивали 3x7мл вали над безводним Na2SO4 і випарювали в умоводи, 3мл 1М KHSO4 і 3x3мл води, висушували вах зниженого тиску. Залишок піддавали хроматонад безводним Na2SO4 і випарювали при 2,0графії на колонці з силікагелем, використовуючи 2,5кПа. Одержаний продукт піддавали хроматог200г Kieselgel 60 (0,040-0,063мМ) як адсорбент і рафії на колонці з силікагелем, використовуючи 1:1 суміш n-гексану та етилацетату як елюент. 50г Kieselgel 60 (0,040-0,063мМ) як адсорбент і Фракції, які містили лише чистий продукт [Rf (3) = етилацетат як елюент. Фракції, які містили лише 0,3-0,4], об'єднували і випарювали при 2,0-2,5кПа. чистий продукт [(Rf (1) = 0,70], об'єднували і випаМаслянистий залишок являв собою 5,95г (13мМ, рювали при 2,0-2,5кПа. Залишок після випарюван65%) бензилового естеру тетрагідропіраніл-Dня кристалізували з діізопропілового етеру. циклогептиллактил-L-проліну, який використовуВихід 2,35г (71%), Rf (1) = 0,45-0,55 вали безпосередньо на наступному етапі. FAB мас-спектр (661 [МН+Н]+) підтверджував Етап І: Триетиламонієва сіль тетрагідропірапередбачувану структуру. ніл-D-циклогептиллактил-L-проліну Етап 2: Бензилоксикарбоніл-N-метил-D5,5г (12мМ) бензилового естеру тетрагідропіциклогексилгліцил-L-азетидин-2-карбоніл-NGраніл-D-циклогептиллактил-L-проліну (Приклад 3, бензилоксикарбоніл-L-аргінін альдегід Етап Н) розчиняли у 12мл диметилформаміду, 2,15г (3,25мМ) бензилоксикарбоніл-N-метил-Dдодавали 1,68мл (12мМ) триетиламіну й гідрогеніциклогексилгліцил-L-азетидин-2-карбоніл-NGзували у присутності 0,1г каталізатора Рd/С. Пробензилоксикарбоніл-L-аргінін лактаму (Приклад 4, цес реакції контролювали шляхом тонкошарової Етап 1) розчиняли у 5мл тетрагідрофурану, а потім хроматографії [Rf (1) = 0,30 (естер), 0,00 (кислопри перемішуванні і при температурі, яка не перета)]. Після завершення реакції каталізатор відфівищувала -50°С, додавали розчин 2,25мМ LiAIH4, льтровували і промивали 2x2мл диметилформарозчиненого в тетрагідрофурані. Процес відновміду. Фільтрат та промиї комбінували і лення контролювали шляхом тонкошарової хровикористовували як розчин, що містив 12мМ триематографії (розчинник 7 як проявний розчинник) і, тиламонієвої солітетрагідропіраніл-Dв разі необхідності, додавали ще одну порцію циклогептиллактил-L-проліну. LiAIH4. До цієї реакційної суміші по краплях додаПриклад 4 вали 0,5Μ KHSO4 з постійним перемішуванням і Синтез гемісульфату N-метил-Dохолодженням до досягнення рН 3, потім 13мл циклогексилгліцил-L-азетидин-2-карбоніл-L-аргінін води. Одержаний у результаті розчин екстрагуваальдегіду ли 2x5мл гексану, потім 3x7мл дихлорометану. Етап 1: Бензилоксикарбоніл-N-метил-DЕкстракти в дихлорометані об'єднували, промивациклогексилгліцил-L-азетидин-2-карбоніл-NGли 3x7мл води, 7мл холодного 5% розчину бензилоксикарбоніл-L-аргінін лактам NаНСО3 і знову 7мл води, висушували над безво1,97г (5мМ) трет-бутилоксикарбоніл-NGдним Nа2SO4 і випарювали при 2,0-2,5кПа. Залибензилоксикарбоніл-L-аргінін лактаму [(Bajusz et шок після випарювання обробляли діізопропілоaI., J. Med. Chem. 33,1729 (1990)] суспендували у вим етером, фільтрували і висушували в умовах 5мл хлороформу, потім при перемішуванні та охозниженого тиску. лодженні льодом додавали 5мл етилацетату, наВихід 1,6г (74%), Rf (7) = 0,33-0,43 сиченого газом НСІ (0,11-0,15г/мл). Розщеплення FAB мас-спектр (663 [М+Н]+) підтверджував Вос групи контролювали шляхом тонкошарової передбачувану структуру. хроматографії [Rf (11) = 0,5 (вільна сполука); 1,0 Етап 3: N-метил-D-циклогексилгліцил-L(Вос-сполука)]. До завершення реакції суспензію азетидин-2-карбоніл-L-аргінін альдегід сульфат розводили 10мл діетилового етеру, утворену крис1,53г (2,3мМ) бензилоксикарбоніл-N-метил-Dталічну масу фільтрували, промивали 3мл ацетону циклогексилгліцил-азетидин-2-карбоніл-NGта 3мл діетилового етеру й висушували в умовах бензилоксикарбоніл-L-аргінін альдегіду (Приклад зниженого тиску над КОН. Одержаний у результаті 4, Етап 2) розчиняли у 23мл етанолу, додавали NG-бензилоксикарбоніл-L-аргінін лактам гідрохло4,8мл 0,5Μ сірчаної кислоти, потім 0,15г каталізарид розчиняли у 5мл диметилформаміду, охолотора Pd-C, суспендованого у 3,5мл води і суміш джували до -20°С і додавали до ангідриду з подагідрогенізували при приблизно 10°С. Процес реакльшим перемішуванням. ції контролювали шляхом тонкошарової хроматог1,95г (5мМ) бензилоксикарбоніл-N-метил-Dрафії. Після завершення реакції (приблизно 15 циклогексилгліцил-L-азетидин-2-карбонової кислохвилин) каталізатор фільтрували і фільтрат концети (Приклад 4, Етап В) розчиняли у 5мл диметилнтрували до приблизно 2-3мл при 2,0-2,5кПа. Заформаміду, охолоджували до -15°С, потім при лишок розводили 20мл води, екстрагували 4x4мл перемішуванні додавали 0,56мл (5,05мМ) Nдихлорометану і водний розчин залишали стояти метил-морфоліну та 0,665мл (5,05мМ) ізобутил при 20-22°С протягом 24 годин. Розчин знову ексхлороформату. Після 10 хвилин перемішування трагували 3x4мл дихлорометану і рН доводили до додавали вищезгаданий розчин у диметилформа3,5 іонообмінною смолою Dowex AG 1-X8 (НО-), міді NG-бензилоксикарбоніл-L-аргінін лактаму, попотім розчин піддавали ліофілізації. тім триетиламін у кількості, достатній для довеВихід 1,02г (90%). Rf (12) = 0,40. дення рН реакційної суміші до 8 (вимагалося FAB мас-спектр (395 [М+Н]+, 548 [M+H+NBA]+) приблизно 0,7мл). Реакційну суміш перемішували підтверджував передбачувану структуру. при -10°С протягом 30 хвилин, потім при 0°С проСинтез вихідних матеріалів: 29 77428 30 Бензилоксикарбоніл-N-метил-Dвисушували над безводним Na2SO4 і випарювали циклогексилгліцил-L-азетидин-2при 2,0-2,5кПа. карбоновокислотний Залишок після випарювання є бензилоксикарЕтап А: Синтез 2, 4, 5-трихлорофенілового есбоніл-D-циклогексилгліцил-L-азетидин-2теру бензилоксикарбоніл-D-циклогексилгліцину карбоновою кислотою, яку розчиняли у 10мл тетДо перемішуваної суспензії 5,42г (20мМ) трирагідрофурані і комбінували з 5,0мл (80мМ) йодофтороацетатної солі D-циклогексилгліцину та метану й охолоджували до 0°С. До цього розчину 5,6мл (40мМ) триетиламіну у 50мл диметилфордодавали 1,2г (30мМ) 60% гідриду натрію й реакмаміду додавали 8,8г (22мМ) бензилційну суміш перемішували при RT до наступного пентахлорофеніл карбонату [Anteunis et al: Bui. дня. Надлишок гідриду натрію розкладали обереSoc. Chim. Belg. 96, 775 (1987)] і 6,16мл (44мМ) жним додаванням 0,4мл води. Гашену реакційну триетиламіну. Після 3 годин перемішування реаксуміш концентрували до приблизно 10мл в умовах ційну суміш випарювали в умовах зниженого тиску, зниженого тиску при температурі нижче 30°С. Зазалишок розчиняли у 60мл діетилового етеру та лишок розводили 15мл води та 10мл t60 води. Фази відокремлювали, органічну фазу бутилметилового етеру. Фази відокремлювали і промивали водою і комбіновані водні фази промиводну фазу знову промивали 10мл tвали діетиловим етером, підкислювали 1М KHSO4 бутилметилового етеру. Водну фазу продукту до рН 3, потім екстрагували 3x30мл етилацетату. комбінували з 15мл етилацетату й доводили до рН Органічну фазу промивали водою до нейтрально22 за допомогою ЗМ розчину сірчаної кислоти. сті, висушували над безводним Na2SO4 і випарюФази відокремлювали і водну фазу піддавали звовали при 2,0-2,5кПа. ротній екстракції 10мл етилацетату. Комбіновану Залишок після випарювання був бензилоксиорганічну фазу промивали 15мл 5% розчину тіосукарбоніл-D-циклогексилгліцином, який розчиняли у льфату натрію. Фази відокремлювали й органічну 20мл тетрагідрофурану і комбінували з 4,54г фазу випарювали під тиском при температурі ниж(22мМ) 2,4,5-трихлоро-фенолу та 4,54г (22мМ) че 40°С. Маслянистий залишок являє собою 3,75г дициклогексилкарбодиіміду. Через три години реабензилоксикарбоніл-N-метил-D-циклогексилгліцилкційну суміш фільтрували, фільтрат та промиї L-азетидин-2-карбонової кислоти (9,65мМ, Rf (7) = комбінували і випарювали в умовах зниженого 0,85-0,95), яку розчиняли у 9,65мл тетрагідрофутиску. Твердий залишок очищали шляхом хромарану і тримали для застосування на Етапі 1 Приктографії на колонці з силікагелем, використовуючи ладу 4. 140г Kieselgel 60 (0,040-0,063мМ) як адсорбент і FAB мас-спектр (403 [М+Н]+) підтверджував 95:5 суміш хлороформу та ацетону як елюент. передбачувану структуру. Фракції, які містили лише чистий продукт [Rf (5) = Приклад 5 0,7-0,8], об'єднували і випарювали при 2,0-2,5кПа. Інгібування згортання пептидил аргіналами Маслянистий залишок розтирали діетиловим етеінгібіторний вплив на коагуляцію плазми оціром, фільтрували, промивали діетиловим етером і нювали шляхом аналізів тромбінового часу (ТТ), висушували. Вихід: 8,34г (88,5%) чистого 2,4,5активованого часткового тромбопластинового часу трихлорофенілового естеру бензилоксикарбоніл(АРТТ) та протромбінового часу (РТ) з викорисD-циклогексилгліцину. танням очищеної людської плазми як субстрату Етап В: Синтез бензилоксикарбоніл-D-метил[Bagdy, D. et al: Thromb. Haemostas. 67J325 D-циклогексилгліцил-L-азетидин-2-карбонової кис(1992)]. В аналізі ТТ вимірюють інгібування одного лоти етапу, коагуляцію фібриногену під дією екзогенноL-азетидин2-карбонову кислоту (1,01г, 10мМ) го тромбіну (кінцева концентрація 2,5 NIH U/мл). та триетиламін (1,4мл, 10мМ) додавали до розчиЗгортання плазми в аналізах АРТТ та РТ викликану 2,4,5-трихлорофенілового естеру бензилоксили рекальцифікацією. Вироблений ендогенний карбоніл-D-циклогексилгліцину (5,18г, 11мМ, з тромбін теоретично може бути присутній у кінцевій Прикладу 4, Етап А) у 10мл піридину. Після переконцентрації 50 NIH U/мл (АРТТ, РТ). Ці аналізи мішування до наступного дня реакційну суміш вивиявляють сумарну інгібіторну дію на утворення парювали в умовах зниженого тиску й залишок фібрину та вироблення тромбіну, яка включає кірозчиняли у 50мл 5% NaHCO3 та 50мл діетилового лька протеолітичних реакцій, опосередкованих етеру. Органічну фазу промивали водою і комбітромбіном або іншими ферментами, наприклад, новані водні фази промивали діетиловим етером, fXa. Антикоагулянтну активність виражено у СТ2, підкислювали 1М KHSO4 до рН 3, потім екстрагуяка є концентрацією (нМ), необхідною для подвовали 3x50мл етилацетату. Екстракти в етилацетаті єння часу згортання. комбінували, промивали водою до нейтральності, 31 77428 32 Таблиця 1 Інгібуючий вплив сполук формули (І) згідно з винаходом (1-4), вихідної сполуки, Efegatran (С) та інших споріднених пептидил аргіналів (С1, С2) на коагуляцію плазми (колонка А), зв'язаний зі згустком тромбін та фактор Ха (колонка В) і фібринолітичні ферменти (колонка С) Пептидил аргінали: Xaa-Xbb-Arg-Ha No Xaa-Xbb 1 Eoc-D-cHpa-Pro 2 N-Me-D-cHpa-Pro 3 D-cHla-Pro 4 N-Me-D-Chg-Aze С D-MePhe-Pro С1 D-cHga-Pro С2 Eoc-D-Cba-Pro TT 147 118 86 101 87 120 114 A СТ2(нМ)b АРТТ 331 315 281 677 622 333 349 РТ 1839 876 1082 2921 2915 1254 1148 В lС50(НМ) Тромбін Фактор Ха 103 118 93 102 95 117 245 457 375 1000 524 200 390 702 PL 18 16 21 32 54 83 27 С LA50, мкМс tPA 10 14 10 12 132 74 27 UK 14 18 85 16 82 120 120 a Скорочення. Еос = етоксикарбоніл; сНра = циклогептилаланіл; сНІа = циклогептиллактил; MePhe = Nметилфенілаланіл; cHga = циклогептилгліколіл; Chg = циклогексилгліцил. b CT2 = концентрація, необхідна для подвоєння часу згортання в аналізах ТТ (тромбінового часу), АРТТ (активованого часткового тромбопластинового часу) і РТ (протромбінового часу). с LА50 = концентрація, необхідна для зменшення площі лізису до 50% від контролю в аналізі фібринової пластинки; PL = плазмін, tPA = тканинний активатор плазміногену, UK = урокіназа. Деякі з цих сполук переважали Efegatran (С, DMePhe-Pro-Arg-H, вихідний пептид аргінал) за їхньою здатністю до подовження часу згортання (див. Таблицю 1). Колонка А Таблиці 1 представляє антикоагулянтну дію сполук формули (І) згідно з винаходом (1-4) порівняно з активністю Efegatran (C), відомим антикоагулянтним засобом [Bajusz, S. et. al.: J. Med. Chem. 33,1729 (1990); US. Pat, No. 4,703,036 (1982); Bagdy, D. et aL: Thromb. Haemostas. 67, 357 and 68,125 (1992); Jackson, GV. et al.: Clin. Appi. Thrombosis/Haemostasis 2, 258 (1996)] та іншими спорідненими пептидил аргіналами (наприклад, С1 та С2) [Bajusz, S. et al.: Bioorg. & Med. Chem. 3, 1079 (1995); US. Pat. No. 6,121,241 (2000); PCT Pub. No. WO97/46576]. Аналіз ТТ показує нові аналоги, майже таку саму ефективність, як efegatran, в інгібуванні реакції тромбіну-фібриногену. АРТТ, з іншого боку, вказує, що нові пептиди є більш ефективними, ніж efegatran, з інгібуванні попередніх, тромбіногенеруючих етапів коагуляції. Приклад 6 Інгібування тромбіну та Фактора Ха Інгібування ферменту досліджували у збагачених тромбоцитами згустках плазми, застосовуючи хромогенні субстрати, тобто Tos-Gly-Pro-ArgpNA (S1) для тромбіну і Moc-D-Chg-Gly-Arg-pNA (S2) для фактора Ха, як було коротко описано в публікації [Bajusz, S. et al.: PCT Pub. No. WO97/46576]. Аналізи здійснювали при кімнатній температурі у скляних пробірках і 96-лункових мікротитрувальних планшетах. Розчини, (і) Буфер А: 0,1Μ фосфату натрію/0,05Μ NaCI (pH 8,5). (іі) Інгібітори: 0,1, 1,0 і 10мг/мл розчини в буфері А, що містять 0,02% людського альбуміну, (ііі) Субстрати: 1мМ S1 і 2мМ S2 у дистильованій воді. (б) Одержання згустків плазми. Зразки збагаченої тромбоцитами плазми (200мкл), поміщені у скляні пробірки, інкубували при кімнатній температурі з 80мкл 40мМ СаСІ2 протягом 60хв. Згустки промивали 2мл аліквотних частин розсолу (0,9% NaCI) з обережним перемішуванням для видалення незв'язаних ферментів. У разі успішного промивання оптична густина реакційної суміші промитої речовини та S1 становить менше 5% від контролю. Одержаний таким чином згусток плазми до використання тримали під розсолом у пробірці. (в) Оцінка інгібування ферментів у згустках. Після видалення розсолу згусток плазми інкубували при 37°С з 400мкл інгібітора (або буфером А як негативним контролем) протягом 5хв і з 100мкл субстрату S1 або S3 протягом 30хв, потім реакцію зупиняли за допомогою 100мкл 50% оцтової кислоти. У лунки мікротитрувального планшета поміщали порції по 150мкл реакційних сумішей і зчитували при 405нм (EUSA READER 800, Bio-Tek Instruments Inc. Winooski. VT. USA). Значення ІС50 виводили графічно за даними затухання. Результати показано в колонці В Таблиці 1. Сполуки 1, 2, 3 та 4 є єдиними аналогами, які можуть переважати Efegatran в інгібуванні зв'язаного зі згустком тромбін, але в інгібування зв'язаного зі згустком фактора Ха кожен аналог переважає Efegatran. Таким чином 1, 2, 3 та 4 є найкращими інгібіторами для обох зв'язаних зі згустками ферментів. Приклад 7 Антифібринолітична активність Інгібіторний вплив сполук винаходу на плазмін (PL) та вироблення плазміну активаторами плазміногену (прогену), такими як тканинний активатор плазміногену (tPA) та урокіназа (UK), досліджували шляхом аналізу фібринової пластинки [див., наприклад, Bagdy, D., Barabas, E., Bajusz, S., and SzeIl, E., Thromb. Haemostas., 67:325-330 (1992); Barabas, E. Szell, E. and Bajusz, S, Blood Coagulation and Fibrinolysis, 4:243 (1993)]. Результати показано в Таблиці 1, колонка С. За деякими випадками, аналоги мають дещо більший інгібіторний вплив, ніж Efegatran, на три фібринолітичні ферменти. Винятками є С1 проти PL і С1, С2 про 33 77428 34 ти UK, тоді як 3 має майже таку саму активність, (мкг/кг шляхом внутрішньовенної болюсної ін'єкції, що й Efegatran, проти UK. на 0-1 та 120-й хвилинах, відповідно, тоді як пепПриклад 8 тидил аргінали 1, 3 та С-С3 (0,25 і/або 0,5мг/кг/год) Кроляча модель розсіяної внутрішньосудинної вливали протягом усього експерименту. Контролькоагуляції ній групі тварин вводили 0,9% розсіл. Гемостатичні Сполуки 1 та 3 згідно з винаходом та інші пеппараметри визначали на 0, 120 та 240хв. тидил аргінази з Таблиці 1, а також С3 досліджуНезважаючи на стараннелікування, смертвали на їх здатність до інгібування DIC у кролів, ність складала 31%, найбільш вірогідно, через яким вводили ендотоксин (ліпополісахарид, LPS), високу чутливість кролів до ендотоксину як описано [Scherer, Μ. U. et al.: Lab. Anim Sci. [Semerano, N. et al.: Int. J. Clin. Lab. Res. 21., 214 45,538 (1995)]. Процедура аналізу тривала протя(1992)]. гом 4 годин. Ендотоксин зводили в дозах 80 та 40 Таблиця 2 Вплив сполук 1 та 3 згідно з винаходом, вихідної сполуки Efegatran (C), інших аргіналів (С1 та С2) і гепарину (Н) на смертність кролів, яким вводили ендотоксин Агентиа Salsol (0,9% NaCI) Ендотоксин + 1, 0,5мг/кг/год + 1, 0,25мг/кг/год + 3, 0,25мг/кг/год + С, 0,5мг/кг/год + С, 025мг/кг/год + С1, 0,5мг/кг/год + С2, 0,5мг/кг/год + С3, 0,5мг/кг/год + Н, 100 U/кг/год + Н, 50 U/кг/год Смертність кільк. померлих/лікованих тварин і % 2 год 4 год Кількість % Кількість 0/10 0 0/10 0/13 0 4/13 0/12 0 2/12 0/19 0 3/19 0/22 0 4/22 1/15 7 6/15 1/14 7 5/14 0/16 0 5/16 0/12 0 5/12 1/18 6 10/18 0/19 0 7/19 0/17 0 6/17 % 0 31 17 16 18 40 36 31 42 56 37 35 а Див. у Таблиці 1 структури пептидил аргіналів 1, 3, С, С1 і С2, С3 = hРІа-Pro-Arg-H, де hPla = 2гідрокси-4-феніл-масляна кислота. Як показують дані Таблиці 2, сполуки 1 і 3 значно знижують смертність кролів, яким вводили ендотоксин, тоді як інші антикоагулянти або не впливали на смертність (С1), або викликали певне підвищення смертності, причому найвищий показник, ~1,8 разу, давав С3. З даних у Таблиці 1 можна помітити, що сполуки 1 та 3, які знижували смертність, найбільше інгібують зв'язаний зі згустком тромбін, а також фактор Ха, а також ефективно інгібують як коагуляцію плазми, так і фібринолітичні ферменти, плазмін та активатори плазміногену. Приклад 9 Щуряча модель розсіяної внутрішньосудинної коагуляції До найбільш серйозних наслідків DIC належить осадження фібрину в різних органах, зміна клітин крові, наприклад, зниження числа тромбоцитів та зміни у продуктах розкладу фібрин. Вплив сполуки 1 згідно з винаходом та двох контрольних антикоагулянтів, Efegatran (С) та гепарину (Н), на ці явища досліджували на щурах, яким вводили ендотоксин [Ford, A. J. and Longridge, D.J.: Br. J. Pharmacol. 110, Suppl. 131Ρ (1993); Hasegawa, N.; et al.: Am. J. Resp. Crit. Care Med. 153.1831 (1996); Dichneite, G. et al.: Thromb. Res. 77,357 (1995)]. Самцям щурів робили внутрішньовенні болюсні ін'єкції 10мг/кг ендотоксину. Після них робили внутрішньовенне вливання розсолу або тестових сполук протягом чотирьох годин. Сполуки 1 та 3 вводили в кількості 0,25мг/кг як первинну болюсну ін'єкцію з наступним внутрішньовенним вливанням 0,25мг/кг/год протягом чотирьох годин. Гепарин (Н) вводили подібним чином, 50 IU/кг як первинну болюсну ін'єкцію з наступним внутрішньовенним вливанням 50 IU/кг/год протягом чотирьох годин. Контрольній групі тварин вводили 0,9% розсіл. Осадження 125І-фібрину досліджували у вибраних органах (печінці та нирках). 125I-фібриноген вводили шляхом ін'єкції за 30хв до ін'єкції ендотоксину. Радіоактивність у зразках тканини вимірювали в гамма-лічильнику (Wailac Wizard 1470). Утворення мікротромбів з органах оцінювали, використовуючи співвідношення 125І активності органа з загальною 125І активністю ін'єкції, визначене як індекс мікротромбів. Зміни в цьому параметрі виражено у відсотках порівняно з групою, якій зводили розсіл. Число тромбоцитів визначали за допомогою автоматичного пристрою (Sysmex F-800) і співвідносили з контрольними показниками. Визначення FDP (продуктів розкладу фібрину) здійснювали шляхом аналізу осадження Aggristin (ристоцетину). Тварин умертвляли через чотири години після введення ендотоксину. Результати зведено в Таблиці 3. 35 77428 36 Таблиця 3 Вплив сполуки 1 згідно з винаходом, вихідного пептиду, Efegatran (С) та гепарину (Н) на осадження 125І-фібрину і на зміни в числі тромбоцитів та продукти розкладу фібрину (FDP) у щурів, яким вводили ендотоксин Агенти a Ендотоксин, 10мг/кг + 1, 0,25мг/кг/годь + С, 0,25мг/кг/годb + Н, 50 NIH U/кг/годb а осадження 125I-фібрину Печінка Нирки 36% 36% 13% 26% 30% 33% 22% 28% Зміна в числі тромбоцитів Зміна у FDP 62% 48% 52% 43% 2,56 1,66 1,94 2,37 Внутрішньовенна болюсна ін'єкція. b Внутрішньовенна болюсна ін'єкція + внутрішньовенне вливання. Дані Таблиці 3 вказують, що DIC-інгібуючий потенціал 1 був більш явним, ніж у гепарину або Efegatran. Приклад 10 Дослідження виживаності у щурячій моделі розсіяної внутрішньосудинної коагуляції Самцям щура робили внутрішньовенну болюсну ін'єкцію 30мг/кг LPS (ендотоксину). Пептиди в дозах 0,5 та/або 0,75, 1,0 і 1,5мг/кг зводили як первинну болюсну ін'єкцію з наступним вливанням протягом восьми годин безпосередньо після зве дення LPS. Смертність записували через 4, 5, 6, 7, 8 годин після введення LPS. Дані Таблиці 4 вказують, що введення LPS знижувало показник виживаності щурів до 20% до кінця експерименту (8 годин). Усі досліджені нові пептидил аргінали подовжували час виживання і знижували смертність порівняно з групою LPS. Сполуки 1, 2, 3 та 4 були ефективнішими, ніж еталон С Таблиця 4 Вплив сполук 1, 2, 3 та 4 згідно з винаходом та вихідної сполуки С на смертність щурів, яким вводили LPS Показник виживаності, % Час (гоКонтроль С, мг/кг 1, мг/кг 2, мг/кг дини) 0 0,75 1,0 1,5 0,75 1,0 1,5 0,75 1,0 1,5 0 100 100 100 100 100 100 100 100 100 100 4 76 93 83 100 100 100 100 86 100 100 5 72 73 83 100 100 100 100 78 100 100 6 37 67 66 100 100 100 100 57 100 100 7 24 33 58 92 90 90 100 57 90 100 8 20 27 50 92 70 90 100 50 90 100 Самцям щура робили внутрішньовенну болюсну ін'єкцію 30мг/кг LPS. Тестові сполуки вводили як первинну болюсну ін'єкцію з наступним внутрішньовенним вливанням протягом восьми годин Комп’ютерна верстка О. Гапоненко 3, мг/кг 0,5 0,75 1,5 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 80 92 92 4, мг/кг 0,5 0,75 1,5 100 100 100 93 100 100 87 100 100 87 100 100 87 92 100 80 85 100 безпосередньо після введення LPS. Смертність записували через 4, 5, 6, 7 та 8 годин після введення LPS. Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюPeptides arginals and a method for the treatment of disseminated intravascular coagulation
Назва патенту російськоюПептидные аргиналы и способ лечения рассеянной внутрисосудистой коагуляции
МПК / Мітки
МПК: A61P 31/04, A61P 7/02, A61P 43/00, A61K 38/00, C07D 207/16, A61P 13/12, C07K 5/065, C07K 5/062, A61K 31/401
Мітки: аргінали, розсіяної, пептидні, спосіб, коагуляції, внутрішньосудинної, лікування
Код посилання
<a href="https://ua.patents.su/18-77428-peptidni-arginali-ta-sposib-likuvannya-rozsiyano-vnutrishnosudinno-koagulyaci.html" target="_blank" rel="follow" title="База патентів України">Пептидні аргінали та спосіб лікування розсіяної внутрішньосудинної коагуляції</a>
Фармацевтичний препарат для лікування хворих з порушеннями функції коагуляції, спосіб лікування таких хворих, спосіб одержання фармацевтичного препарату

Номер патенту: 35567
Опубліковано: 16.04.2001
Автори: НОРДФАНГ Оле, МОЛЛЕР Нілс Петер Хундахл, Хеднер Улла
МПК: A61K 39/395, A61P 7/04, A61K 38/55
Мітки: таких, одержання, препарат, спосіб, лікування, хворих, препарату, порушеннями, функції, фармацевтичного, фармацевтичний, коагуляції
Формула / Реферат:
1. Фармацевтический препарат для лечения больных с нарушениями функции коагуляции, содержащий активный компонент, который обладает воздействием на время коагуляции, отличающийся тем, что в качестве активного компонента указанный препарат содержит эффективное количество EPI-ингибитора.2. Фармацевтический препарат по п.1, отличающийся тем, что указанное нарушение функции коагуляции связано с гемофилией А или В.3. Фармацевтический...
Пептидні інгібітори вірусу гепатиту с, спосіб їх одержання та фармацевтична композиція

Номер патенту: 73480
Опубліковано: 15.08.2005
Автори: Тсантріцос Юла С., Кемерон Дейл, Лінас-Брюне Монтсе, Пупар Марк-Андре, Ранкур Жан, Бейлі Мьоррі Д., Гудро Наталі, Гіро Еліз
МПК: C07K 14/81, C07K 7/06, A61K 38/55, A61P 31/12, A61K 38/00, A61P 1/16, C07K 5/107, A61P 43/00, A61K 45/00, C12N 15/09, A61K 38/21, A61K 31/7052, A61K 31/13, A61P 31/14, C07K 1/10, C07K 5/10
Мітки: фармацевтична, одержання, пептидні, композиція, вірусу, гепатиту, інгібітори, спосіб
Формула / Реферат:
1. Сполука формули (І), включаючи її рацемати, діастереоізомери і оптичні ізомери: (І),у якійа дорівнює 0 або 1,b дорівнює 0 або 1,Y означає Н або С1-С6алкіл,В означає Н, дансил або ацильне похідне формули R7-C(O)- ,R7 означає С1-С6алкіл або Het, який вибирають з групи, що містить: , , ,, , або,R6 означає С1-С6алкіл, заміщений карбоксилом, R5 означає...
Пептидні аналоги паратиреоїдних гормонів

Номер патенту: 62967
Опубліковано: 15.01.2004
Автори: Моріз Ізабелль, Кондон Стефен М.
МПК: C07K 7/50, A61P 3/14, C07K 7/64, C07D 211/34, A61P 19/10, C12N 15/09, A61P 43/00, A61P 5/18, C07K 14/635, A61K 38/00
Мітки: паратиреоїдних, пептидні, гормонів, аналоги
Формула / Реферат:
1. Циклічна пептидна сполука формули:Х-А10-А11-А12-А13-А14-А15-A16-A17-А18-A19-A20-A21-A22-A23-A24-A25-A26-A27-Y або її фармацевтично прийнятна сіль чи пролікарський попередник,де Х вибирають з групи, що складається з:(a) R1а-А0-А1-А2-А3-А4-А5-А6-А7-А8-А9-,(b) R1а-А2-А3-А4-А5-А6-А7-А8-А9-,(c) R1b-А3-А4-А5-А6-А7-А8-А9-,(d) R1а-А4-А5-А6-А7-А8-А9-,(e) R1a-A5-A6-A7-A8-A9-,(f)...
Спосіб лазерної коагуляції кровоносних судин

Номер патенту: 73370
Опубліковано: 15.07.2005
Автор: Хомчєнко Владімір Валєнтіновіч
МПК: A61B 18/20
Мітки: коагуляції, судин, лазерної, кровоносних, спосіб
Формула / Реферат:
1. Спосіб лазерної коагуляції кровоносних судин шляхом впливу на судину імпульсним випромінюванням з довжиною хвилі 500 - 600 нм, щільністю енергії, не більшою ніж 10 Дж/см2, протягом не більше ніж 10 мс, який відрізняється тим, що одночасно на цю ж судину впливають додатковим випромінюванням принаймні з ще однією довжиною хвилі в діапазоні 800 - 1400 нм, з щільністю енергії, не більшою ніж 100 Дж/см2.2. Спосіб за п. 1, який...
Пептидні похідні

Номер патенту: 65518
Опубліковано: 15.04.2004
Автори: Бюлунд Рут Ельвю, Нільссон Нільс Олов Інгемар, Антонссон Карл Томас, Густафссон Нільс Давід
МПК: A61K 38/00, C07K 2/00, A61P 43/00, C07K 5/078, C07K 1/10, C07C 257/00, C12N 9/99, C07K 5/02, C07K 5/06, A61K 31/397, C07K 5/062, C07D 239/14, A61P 29/00, A61P 25/28, C07D 211/16, A61P 11/02, C07C 271/64, A61P 17/00, C07K 5/065, A61P 1/00, C07C 237/12, C07D 239/12, C07D 211/26, A61K 31/445, C07D 207/09, C07D 205/00, A61K 38/55, A61P 11/06, A61P 19/02, A61P 7/02, C07D 211/60
Формула / Реферат:
1. Соединение общей формулы:A1---A2---NH---(CH2)n---B---D ,IгдеА1 представляет структурный фрагмент формул IIа, IIb, IIc, IId или IIе: гдеk = 0, 1, 2, 3 или 4,m = 1, 2, 3 или 4,q = 0, 1, 2 или 3,R1 представляет Н,...
Попередній патент: Спосіб зниження частоти коливального процесу
Наступний патент: Упаковка для кріоконсервації спермодоз плідників сільськогосподарських тварин
Випадковий патент: Спосіб диференційної діагностики артропатичного псоріазу