Заміщені піридини, фармацевтична композиція на їх основі та спосіб лікування опосередкованих циклооксигеназою захворювань
Номер патенту: 62935
Опубліковано: 15.01.2004
Автори: Вонг Заойін, Фортін Ріджен, Фресен Річард, Готьє Жак Ів, Дюб Деніел





















Формула / Реферат
1. Заміщені піридини формули I
або їх фармацевтично прийнятна сіль, де:
R1 вибраний з групи, що включає
(a) CH3,
(b) NH2,
(c) NHC(О)CF3, і
(d) NНСН3;
Ar являє собою моно-, ди- або три-заміщений феніл або піридиніл (або його N-оксид), де замісники вибрані з групи, що включає
(a) водень,
(b) галоген,
(c) C1-6алкокси,
(d) C1-6алкілтіо,
(e) CN,
(f) C1-6алкіл,
(g) C1-6фторалкіл,
(h) N3,
(i) - CO2R3,
(j) гідрокси,
(k) -С(R4)(R5)-OH,
(l) -C1-6алкіл-CO2-R6, і
(m) C1-6фторалкокси;
R2 вибраний з групи, що включає
(a) галоген,
(b) C1-6алкокси,
(c) C1-6алкілтіо,
(d) CN,
(e) C1-6алкіл,
(f) C1-6фторалкіл,
(g) N3,
(h) -CO2R7,
(i) гідрокси,
(j) -С(R8)(R9)-OH,
(k) -C1-6алкіл-CO2-R10,
(l) C1-6фторалкокси,
(m) NO2,
(n) NR11R12, і
(o) NHCOR13,
R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13 кожний, незалежно, вибраний з групи, що включає
(a) водень і
(b) C1-6алкіл,
або R4 і R5, R8 і R9 або R11 і R12 разом з атомом, до якого вони приєднані, утворюють насичене моноциклічне кільце з 3, 4, 5, 6 або 7 атомів, за умови, що, коли Ar являє собою фторфеніл, R2 вибраний з (a), (b), (с) або (е) - (о) з визначення R2, даного вище.
2. Сполука за п.1, де Ar являє собою моно-, ди- або тризаміщений 2-піридиніл.
3. Сполука за п.1, де Ar являє собою моно-, ди- або тризаміщений 3-піридиніл.
4. Сполука за п.1, де R1 являє собою CH3 або NH2.
5. Сполука за п.1, де Ar являє собою моно-, ди- або тризаміщений 2-піридиніл або 3-піридиніл, і замісники вибрані з групи, що включає
(a) водень,
(b) галоген,
(c) C1-3алкокси,
(d) C1-3алкілтіо,
(e) C1-3алкіл,
(f) CF3, і
(g) CN.
6. Сполука за п.1, де
R1 являє собою CH3 або NH2; і
Ar являє собою моно-, ди- або тризаміщений 2-піридиніл або 3-піридиніл, і замісники вибрані з групи, що включає
(a) водень,
(b) галоген,
(c) C1-3алкокси,
(d) C1-3алкілтіо,
(e) C1-3алкіл,
(f) CF3 і
(g) CN.
7. Сполука за п.1, де R2 являє собою
(a) галоген,
(b) C1-4алкокси,
(c) CN,
(d) C1-3алкіл,
(e) C1-3фторалкіл,
(f) - CO2H,
(g) - C1-3алкіл-CO2H,
(h) C1-3фторалкокси або
(i) NO2.
8. Сполука за п.1, де R2 являє собою галоген, CH3 або CF3.
9. Сполука за п.1, де:
R1 являє собою CH3 або NH2;
R2 являє собою галоген, CH3 або CF3; і
Ar являє собою моно-, ди- або тризаміщений 2-піридиніл або 3-піридиніл, і замісники вибрані з групи, що включає
(a) водень,
(b) галоген,
(c) C1-3алкокси,
(d) C1-3алкілтіо,
(e) C1-3алкіл,
(f) CF3 і
(g) CN.
10. Сполука за п.1, де:
R1 являє собою CH3 або NH2;
R2 являє собою галоген, CH3 або CF3; і
Ar являє собою моно-, ди- або тризаміщений 3-піридиніл, і замісники вибрані з групи, що включає
(a) водень,
(b) галоген,
(c) C1-3алкокси,
(d) C1-3алкілтіо,
(e) C1-3алкіл,
(f) CF3 і
(g) CN.
11. Сполука за п.1, де:
R1 являє собою CH3 або NH2;
R2 являє собою галоген, CH3 або CF3; і
Ar являє собою моно-, ди- або тризаміщений феніл, і замісники вибрані з групи, що включає
(a) водень,
(b) галоген,
(c) C1-3алкокси,
(d) C1-3алкілтіо,
(e) C1-3алкіл,
(f) CF3 і
(g) CN.
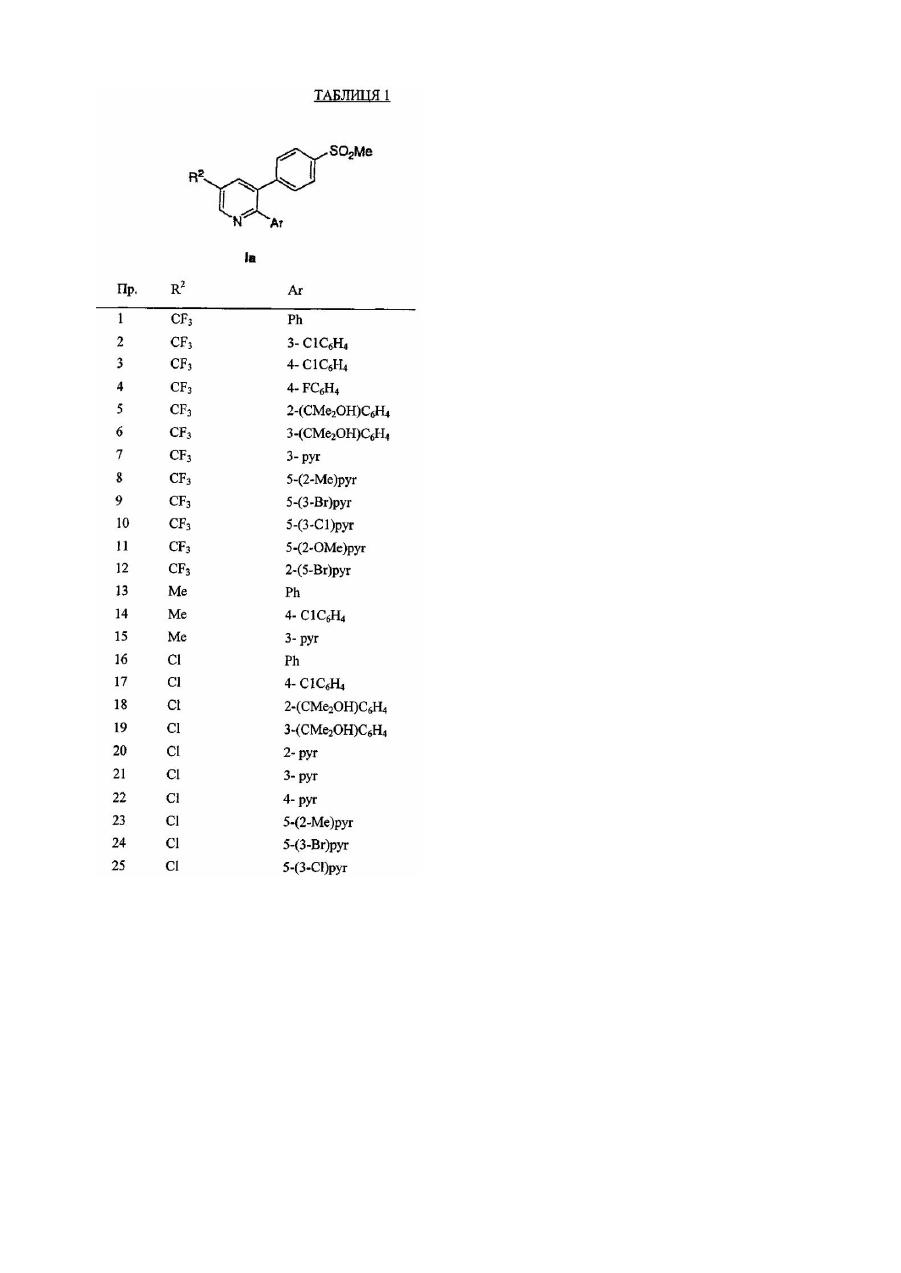
12. Сполука за п.1 формули Ia
або її фармацевтично прийнятна сіль, де R2 і Ar разом вибраніз наступних:
R2
Ar
(1)
CF3
Ph
(2)
CF3
3-ClC6H4
(3)
CF3
4-ClC6H4
(4)
CF3
4-FC6H4
(5)
CF3
2-(CMe2OH)C6H4
(6)
CF3
3-(CMe2OH)C6H4
(7)
CF3
3-pyr
(8)
CF3
5-(2-Me)pyr
(9)
CF3
5-(3-Br)pyr
(10)
CF3
5-(3-Cl)pyr
(11)
CF3
5-(2-OMe)pyr
(12)
CF3
2-(5-Br)pyr
(13)
Me
Ph
(14)
Me
4-ClC6H4
(15)
Me
3-pyr
(16)
Cl
Ph
(17)
Cl
4-ClC6H4
(18)
Cl
2-(CMe2OH)C6H4
(19)
Cl
3-(CMe2OH)C6H4
(20)
Cl
2-pyr
(21)
Cl
3-pyr
(22)
Cl
4-pyr
(23)
Cl
5-(2-Me)pyr
(24)
Cl
5-(3-Br)pyr
(25)
Cl
5-(3-Cl)pyr
(26)
Cl
5-(2-OMe)pyr
(27)
Cl
2-(5-Br)pyr
(28)
F
Ph
(29)
F
3-pyr
(30)
F
5-(2-Me)pyr
(31)
Br
Ph
(32)
Br
3-pyr
(33)
Br
5-(2-Me)pyr
(34)
NO2
Ph
(35)
NO2
3-pyr
(36)
NO2
5-(2-Me)pyr
(37)
OMe
Ph
(38)
OMe
3-pyr
(39)
OMe
5-(2-Me)pyr
(40)
NHCOMe
Ph
(41)
NHCOMe
3-pyr
(42)
MHCOMe
5-(2-Me)pyr
(43)
CO2Me
4-ClC6H4
(44)
CO2H
4-ClC6H4
(45)
CN
4-ClC6H4
13. Сполука за п.12, де фармацевтично прийнятна сіль являє собою сіль лимонної, бромистоводневої, хлористоводневої, малеїнової, метансульфонової, фосфорної, сірчаної або винної кислоти.
14. Сполука за п.13, де фармацевтично прийнятна сіль являє собою сіль хлористоводневої або метансульфонової кислоти.
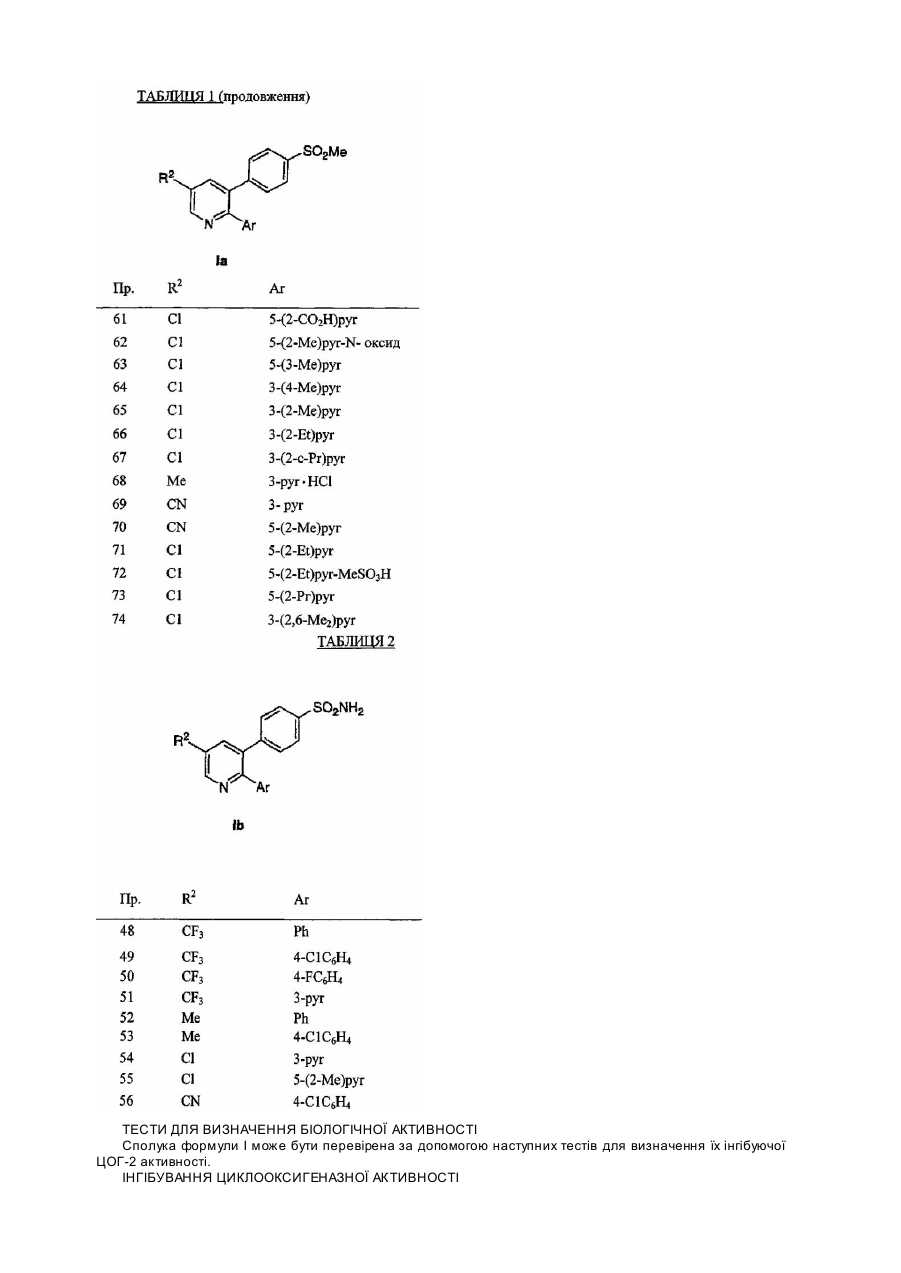
15. Сполука за п.1 формули Ib
або її фармацевтично прийнятна сіль, де R2 і Ar разом вибрані з наступних:
R2
Ar
(48)
CF3
Ph
(49)
CF3
4-ClC6H4
(50)
CF3
4-FC6H4
(51)
CF3
3-pyr
(52)
Me
Ph
(53)
Me
4-ClC6H4
(54)
Cl
3-pyr
(55)
Cl
5-(2-Me)pyr
(56)
CN
4-ClC6H4
(71)
Cl
5-(2-етил)pyr
(72)
Cl
5-(2-Et)pyr-MeSO3H
(73)
Cl
5-(2-c-Pr)pyr
(74)
Cl
3-(2,6-Me2)pyr
16. Сполука за п.15, де фармацевтично прийнятна сіль являє собою сіль лимонної, бромистоводневої, хлористоводневої, малеїнової, метансульфонової, фосфорної, сірчаної або винної кислоти.
17. Сполука за п.16, де фармацевтично прийнятна сіль являє собою сіль хлористоводневої або метансульфонової кислоти.
18. Сполука за п.1, де:
R2 вибраний з групи, що включає
(а) галоген,
(b) C1-6алкокси,
(c) C1-6алкілтіо,
(d) С1-6алкіл,
(e) N3,
(f) - СО2Н,
(g) гідрокси,
(h) С1-6фторалкокси,
(i) NO2,
(j) NR11R12,
(k) NHCOR13,
R3, R4, R5, R6, R11, R12, R13 кожний, незалежно, вибраний з групи, що включає
(a) водень і
(b) C1-6алкіл,
або R4 і R5 або R11 і R12 разом з атомом, до якого вони приєднані, утворюють насичене моноциклічне кільце з 3, 4, 5, 6 або 7 атомів.
19. Сполука за п.18 формули Ic:
,
де:
R1 вибраний з групи, що включає
(a) CH3,
(b) NH2,
R2 вибраний з групи, що включає
(a) хлор і
(b) метил,
і де можуть бути одна, дві або три групи X, незалежно вибрані з групи, що включає
(a) водень,
(b) галоген,
(c) C1-4алкокси,
(d) C1-4алкілтіо,
(e) CN,
(f) C1-4алкіл і
(g) CF3.
20. Сполука за п.19, де:
R2 являє собою хлор, де є одна група X, незалежно вибрана з групи, що включає
(a) водень,
(b) F або Cl,
(c) метил і
(d) етил.
21. Сполука за п. 20, де є одна група X, незалежно вибрана з групи, що включає
(a) водень,
(b) F або Cl і
(c) метил.
22. Сполука за п. 21, де R1 являє собою метил.
23. Сполука за п.1 формули Ic:
,
де:
R1 являє собою CH3 або NH2;
R2 являє собою
(a) галоген,
(b) C1-6алкокси,
(c) C1-6алкілтіо,
(d) C1-6алкіл,
(e) N3,
(f) - CO2H,
(g) гідрокси,
(h) C1-6фторалкокси,
(i) NO2,
(j) NR11R12 або
(k) NHCOR13, і
X являє собою водень.
24. Сполука за п.1, яка являє собою 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин або його фармацевтично прийнятну сіль.
25. Сполука за п.1, яка являє собою 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридил)піридин гідрометансульфонат.
26. Сполука за п.1, яка являє собою 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридил)піридин гідрохлорид.
27. Сполука за п.1, яка являє собою 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)пірідин або її фармацевтично прийнятну сіль.
28. Сполука за п.1, яка являє собою хлор-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)піридин гідрометансульфонат.
29. Сполука за п.1 формули Ic:
,
де:
R1 являє собою CH3 або NH2,
R2 являє собою
(a) галоген,
(b) C1-3алкокси,
(c) C1-3алкілтіо,
(d) C1-3алкіл,
(e) N3,
(f) CO2H,
(g) гідрокси,
(h) C1-3фторалкокси,
(i) NO2,
(j) NR11R12 або
(k) NHCOR13, і
X являє собою метил, етил, н-пропіл, ізопропіл або циклопропіл.
30. Сполука за п. 29, де X являє собою метил.
31. Сполука за п. 29, яка являє собою 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридин або його фармацевтично прийнятну сіль.
32. Сполука за п. 29, яка являє собою гідрохлорид 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридин.
33. Сполука за п.1, яка вибрана з групи, що включає:
3-(4-метилсульфоніл)феніл-2-феніл-5-трифторметилпіридин;
2-(3-хлорфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин;
2-(4-хлорфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин;
2-(4-фторфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин;
5-метил-3-(4-метилсульфоніл)феніл-2-фенілпіридин;
2-(4-хлорфеніл)-5-метил-3-(4-метилсульфоніл)фенілпіридин;
5-хлор-2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридин;
метиловий ефір 2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5-карбонової кислоти;
2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5-карбонову кислоту і
5-ціано-2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридин.
34. Сполука за п.1, яка вибрана з групи, що включає:
3-(4-метилсульфоніл)феніл-2-(3-піридиніл)-5-трифторметилпіридин;
5-метил-3-(4-метилсульфоніл)феніл-2-фенілпіридин;
5-хлор-3-(4-метилсульфоніл)феніл-2-(2-піридиніл)піридин;
5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин;
5-хлор-3-(4-метилсульфоніл)феніл-2-(4-піридиніл)піридин;
5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридин;
гідрометансульфонат 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридил)піридину;
гідрохлорид 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридил)піридину;
гідрохлорид 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридину;
5-хлор-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)піридин; і
гідрометансульфонат 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)піридину.
35. Сполука формули (I) за п.1 або її фармацевтично прийнятна сіль, де:
R1 являє собою CH3;
R2 вибраний з C1-6алкілу, галогену, СН3 або CN; і
Ar являє собою монозаміщений феніл або незаміщений або монозаміщений піридиніл, в якому монозамісник вибраний з C1-6алкілу, галогену або C1-6циклоалкілу.
36. Сполука за п. 35, де Ar являє собою вказаний монозаміщений феніл.
37. Сполука за п. 35, де Ar являє собою вказаний незаміщений або монозаміщений піридиніл.
38. Сполука за будь-яким з пп.1, 24, 25, 26, 27, 28, 31, 32, 33, 34, 35, 36 і 37 для приготування лікарського засобу для лікування запального захворювання.
39. Сполука за будь-яким з пунктів 1-33 або її фармацевтично прийнятна сіль для застосування як вибірного інгібітора ЦОГ-2 відносно ЦОГ-1.
40. Фармацевтична композиція для лікування опосередкованих циклооксигеназою-2 захворювань, що успішно виліковуються активним агентом, який вибірно інгібує ЦОГ-2 відносно ЦОГ-1, яка містить нетоксичну терапевтично ефективну кількість сполуки за п.1 і фармацевтично прийнятний носій.
41. Фармацевтична композиція за п. 40, де захворюванням є запальне захворювання, чутливе до лікування нестероїдним протизапальним агентом.
42. Фармацевтична композиція за п. 40, де сполукою є сполука за будь-яким з пп.1, 24, 25, 26, 27, 28, 31, 32, 33, 34, 35, 36 і 37.
43. Фармацевтична композиція за п. 40 або п. 41, де сполукою є сполука за будь-яким з пп.1-16 і 18-37.
44. Протизапальна фармацевтична композиція за п. 41, де сполукою є сполука формули (I) за будь-яким з пп.1-11, 18-21 і 29-32, або її фармацевтично прийнятна сіль.
45. Спосіб лікування опосередокованих циклооксигеназою захворювань, що успішно виліковуються активним агентом, який вибірно інгібує ЦОГ-2 відносно ЦОГ-1, в якому передбачають введення пацієнту, що потребує такого лікування, нетоксичної терапевтично ефективної кількості сполуки за п.1.
46. Спосіб лікування за п. 45, де захворюванням є запальне захворювання, чутливе до лікування нестероїдним протизапальним агентом.
Текст
Даний винахід відноситься до способів лікування опосередкованих циклооксигеназою захворювань і до деяких фармацевтичних композицій. Нестероїдні протизапальні лікарські засоби надають велику частину їх протизапальної, анальгезуючої і жарознижуючої активності і інгібірують скорочення матки, що викликаються гормоном, і деякі типи ракового зростання через інгібіювання простагландин G/H-синтази, відомої також як циклооксигеназа. Спочатку була відома тільки одна форма циклооксигенази, відповідна циклооксигеназі-1 (ЦОГ-1), або конститутивному ферменту, виявленому спочатку в бичачих насіннєвих пухирцях. Зовсім недавно ген для другої форми циклооксигенази, що індукується, циклооксигенази-2 (ЦОГ-2), був клонований, секвенований і охарактеризований спочатку з курки, миші і людини. Цей фермент відрізняється від ЦОГ-1, яка була клонована, секвенована і охарактеризована з різних джерел, в тому числі, з вівці, миші і людини. Друга форма циклооксигенази, ЦОГ-2, швидко і легко індукується рядом агентів, в тому числі, мітогенами, ендотоксином, гормонами, цитокінами і факторами рост. Оскільки простагландини грають як фізіологічну, так і патологічну роль, автори прийшли до висновку, що конститутивний фермент, ЦОГ-1, відповідальний, в основному, за ендогенне базальне вивільнення простагландинів і, отже, має важливе значення для їх фізіологічних функцій, таких як підтримка шлунково-кишкової цілісності і нирковового кровотоку. Протилежно до цього автори прийшли до висновку, що форма, яка індукується, ЦОГ-2, відповідальна, в основному, за патологічні дії простагландинів, коли індукція цього ферменту має місце у відповідь на такі агенти, як запальні агенти, гормони, фактори росту і цитокини. Таким чином, вибірний інгібітор ЦОГ-2 буде мати схожі протизапальні, жарознижуючі і анальгезуючі властивості в порівнянні із загальноприйнятим нестероїдним протизапальним лікарським засобом і, крім того, буде інгібувати скорочення матки, що індукуються гормонами, і мати потенційні протиракові ефекти, але буде володіти зниженою здатністю індукувати деякі побічні ефекти, що відносяться до механізму дії. Зокрема, таке сполука буде володіти зниженим потенціалом відносно шлунково-кишкової токсичності, зниженим потенціалом відносно ниркових побічних ефектів, зменшеною дією на час кровотечі і, можливо, ослабленою здатністю індукувати приступи астми у чутливих до аспірину суб'єктів. Крім того, така сполука буде також інгібувати скорочення гладких м'язів, що індукується простаноїдами, шляхом запобігання синтезу контрактильних простаноїдів і, отже, може бути використане при лікуванні дисменореї, передчасних родів, астми і пов'язаних з еозинофілами захворювань. Воно буде також застосовне при лікуванні хвороби Альцгеймера, для зменшення рарефікації кісток, зокрема, у жінок в постклімактеричному періоді (тобто для лікування остеопорозу) і для лікування глаукоми. Можливі області застосування вибірних інгібіторів циклооксигенази-2 обговорюються в наступних стаття х: 1. John Vane, "Towards a better aspirin" in Nature, Vol. 367, pp. 215-216, 1994. 2. Bruno Battistini, Regina Botting and Y.S. Bakhle, "COX-1 and COX-2: Toward the Development of More Selective NSAIDs", in Drug News and Perspectives, Vol. 7, pp. 501-512,1994. 3. David B. Reitz and Karen Seibert, "Selective Cyclooxygenase Inhibitors" in Annual Reports in Medicinal Chemistry, James A. Bristol, Editor, Vol. 30, pp. 179-188,1995. 4. Don E. Griswold and Jerry L. Adams, "Constituative Cyclooxygenase (COX-1) and Inducible Cyclooxygenase (COX-2): Rationale for Selective Inhibition and Progress to Date", in Medicinal Research Reviews. Vol. 16, pp. 181-206,1996. У WO 96/10012 (DuPont Merck, April 4, 1996) описані сполуки, представлені формулою А, як такі, що використовуються при лікуванні опосредкованих ЦОГ-2 захворювань завдяки їх виборчому інгібуванню ЦОГ-2 відносно ЦОГ-1. У наш час автори виявили, що підгрупа сполук, представлених А, в яких -J-K-Lозначає -NCHCH-, X означає зв'язок, R1 означає ароматичний радикала і R3 і R4 обидва не є воднем, виявляє несподівано перевершуючу вибірність у відношенні до інгібування ЦОГ-2 відносно ЦОГ-1 і/або перевершуючу активність в порівнянні з найбільш близькими сполуками, описаними в 96/10012. Ця підгрупа сполук є об'єктом даного винаходу і представлена формулою І: З більш ніж 175 характерних сполук, описаних в WO 96/10012, тільки 4 є піридинами, і жоден з цих піридинів не містить замісник (R3 або R4 в А) у піридиновому кільці. У WO 96/16934 (Searle, June 6, 1996) описані сполуки, представлені структурою В, як використовуваних для лікування запалення і пов'язаних захворювань. Хімічно ці сполука відрізняються від сполук даного винаходу тим, що центральне з трьох ароматичних кілець є переважніше бензолом, ніж піридином. Даний винахід відноситься до нової сполуки формули І, а також до способу лікування опосредкованих ЦОГ-2 захворювань, що передбачає введення пацієнту, що потребує такого лікування, нетоксичної терапевтично ефективної кількості сполуки формули І: Даний винахід відноситься також до деяких фармацевтичних композицій для лікування опосередкованих ЦОГ-2 захворювань, що містять сполуки формули І. Даний винахід відноситься до нової сполуки формули І, а також до способу лікування опосередкованих циклооксигеназою-2 захворювань шляхом введення пацієнту, що потребує такого лікування, нетоксичної терапевтично ефективної кількості сполук формули І: де: R1 вибраний з групи, що включає (a) СН3, (b) NH2, (з) NHC(O)CF3; (d) NHCH3; Аr означає моно-, ди- або тризаміщений феніл або піридиніл (або їх N-оксид), де замісники вибрані з групи, що включає (a) водень, (b) галоген, (c) Сl1-6-алкокси, (d) Сl1-6-алкілтіо, (є) CN, (f) Сl1-6-алкіл, (g) Сl1-6-фторалкіл, (h) N3, (і) - CO2R3, (j) гідрокси, (k) - С(R4)(R5)-OH, (l) - С1-6-алкіл-CO2-R6, (m) С1-6-фторалкокси; R2 вибраний з групи, що включає (a) галоген, (b) С1-6-алкокси, (c) С1-6-алкілтіо, (d) CN, (є) С1-6-алкіл, (f) С1-6-фторалкіл, (g) N3, (h) - CO2R7, (і) гідрокси, (j) - С(R8)(R9)-OH, (k) - С1-6-алкіл-CO2-R10, (1) С1-6-фторалкокси, (m) NO2, (n) NRHR12i (o) NHCOR13, R3, R4, R5, R 6, R7 , R8, R9, R10, R11 , R12, R13, вибрані, кожний незалежно, з групи, що включає (a) водень і (b) С1-6-алкіл, або R і R5, R8 і R9 або R11 і R12, разом з атомом, до якого вони приєднані, утворять насичене моноциклічне кільце з 3,4, 5, 6 або 7 атомів. Відповідні алкільні групи включають групи метил, етил, н-пропіл, ізопропіл і бутил, пентил і гексил. Переважною алкільною групою є метальна група. Як правило, R4 і R5, R8 і R9 і R11 і R12 не є залишками вищезгаданих моноциклічних кілець. Коли "алкіл" є частиною складового терміну, такого як алкокси, алкілтіо, фторалкіл, фторалкокси, значення алкіл відноситься також до складового терміну. Переважною підгрупою формули І є така, де Ar є моно- або дизаміщеним піридинілом. У цій підгрупі особливо переважні 3-піридинальні ізомери, такі як ізомери формули Іс. Коли Аr являє собою дизаміщений феніл, особливо переважно те, щоб один або обидва замісники були воднем. Іншою переважною підгрупою формули І є така, де Аr є моно- або дизаміщеним фенілом. Коли Аr являє собою дизаміщений феніл, особливо переважно те, щоб один із замісників був воднем або фтором, а другий був воднем, фтором, хлором, метилом, метокси або трифторметилом. Іншою переважною підгрупою формули І є підгрупа, в якій R1 означає СН3 або NH2. Загалом, СН3 переважний для специфічності ЦОГ-2, a NH 2 переважний для активності. Іншою переважною підгрупою формули І є підгр упа, в якої R 2 означає галоген, СН3 або CF3. Іншою переважною підгрупою формули І є підгр упа, в якій замісники Ar вибрані з групи, що включає (a) водень, (b) галоген, (c) С1-4-алкокси, (d) С1-4-алкілтіо, (є) С1-4-алкіл, (f) CF3 i (g) CN. У одному аспекті даний винахід відноситься до сполук формули І: де: R1 вибраний з групи, що складається з (a) СН3, (b) NH2, (c) NHC (О) CF3, (d) NHCH3; Аг означає моно- ди- або тризаміщений піридиніл (або його N-оксид), де замісники вибрані з групи, що включає (a) водень, (b) галоген, (c) С1-6-алкокси, (d) С1-6-алкілтіо, (є) CN, (f) С1-6-алкіл, (g) С1-6-фторалкіл, (h) N3, (і) -CO2R3, G) гідрокси, (к) -С(R4)(R5)-ОН, (l) С1-6-алкіл -CO2-R6, (m) С1-6-фторалкокси; R2 вибраний з групи, що включає (a) галоген, (b) С1-6-алкокси5 (c) С1-6-алкілтіо, (d) С1-6-алісіл, (є) N3, (f) -СО2Н, (g) гідрокси, (h) С1-6-фторалкокси, (і) NO2, (j) NR11R12 i (k) NHCOR13; R3, R4, R5, R 6, R11, R 12, R13 , кожний, незалежно, вибраний з групи, що включає (a) водень і (b) С1-6-алкіл, або R4 і R5 або R11 і R12 разом з атомом, до якого вони приєднані, утворять насичене моноциклічне кільце з 3,4, 5, 6 або 7 атомів. У цьому аспекті запропонована група сполук формули Іс: R1 вибраний з групи, що включає (a) СНз, (b) ΝH2; R2 вибраний з групи, що включає (a) хлор, (b) метил, і де можуть бути одна, дві або три групи X, незалежно вибрані з групи, що включає (a) водень, (b) галоген, (c) С1-4-алкокси, (d) С1-4-алкілтіо, (є) CN, (f) С1-4-алкіл, (д) CF3. У цій групі сполук формули Іс: є підгрупа, де: R1 вибраний з групи, що включає (a) СНз, (b) NH2; R2 означає хлор, де є одна група X, незалежно вибрана з групи, що включає (a) водень, (b) F або СІ, (c) метил, (d) етил. У цій групі сполук формули Іс : є підгрупа, де: R1 вибраний з групи, що включає (a) СНз, (b) NH2; R2 означає хлор, де є одна група X, незалежно вибрана з групи, що включає (a) водень, (b) F або СІ, (c) метил. Переважні сполуки формули І і Іс включають сполуки, де R2 означає галоген, зокрема, хлор. Переважні сполуки формули І і Іс включають сполуки, де Аr являє собою 3-піридиніл і X являє собою водень або С1-3-алкіл, зокрема, водень, п-метил і п-етил. Наступні сполуки ілюструють даний винахід: 3-(4-Метилсульфоніл)феніл-2-феніл-5-трифторметилпіридин; 2-(3-Хлорфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин; 2-(4-Хлорфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин; 2-(4-Фторфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин; 3-(4-Метилсульфоніл)феніл-2-(3-піридиніл)-5-трифторметилпіридин; 5-метил-3-(4-метилсульфоніл)феніл-2-фенілпіридин; 2-(4-Хлорфеніл)-5-3-(4-метилсульфоніл)фенілпіридин; 5-метил-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)пірвдин; 5-Хлор-2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридин; 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-піридиніл)піридин; 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин; 5-хлор-3-(4-метилсульфоніл)феніл-2-(4-піридиніл)піридин; 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл) піридин; Складний метиловий ефір 2-(4-Хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5-карбонової кислоти; 2-(4-Хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5-карбонова кислота; 5-Ціано-2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридин; Гідрометансульфонат 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридил)піридин; Гідрохлорид 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридиніл) піридин; Гідрохлорид 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл) піридин; 5-Хлор-3-(4-метилсульфоніл) феніл-2-(2-етил-5-піридиніл)піридин і Гідрометансульфонат 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)піридину. Переважні сполуки формули І і формули Іс включають сполуки, де R1 являє собою метил або NH2, зокрема, метил. У іншому аспекті даний винахід відноситься до фармацевтичної композиції для лікування запального захворювання, чутливого до лікування нестероїдним протизапальним агентом, яка має нетоксичну терапевтично ефективну кількість сполуки формули І і фармацевтично прийнятний носій. У іншому аспекті даний винахід відноситься також до фармацевтичної композиції для лікування опосередкованих циклооксигеназою захворювань, що успішно піддаються лікуванню активним агентом, який вибірково інгібує ЦОГ-2 в порівнянні з ЦОГ-1, що містить нетоксичну терапевтично ефективну кількість сполуки формули І і фармацевтично прийнятного носія. У іншому аспекті даний винахід відноситься також до способу лікування запального захворювання, чутливого до лікування нестероїдним протизапальним агентом, який полягає у введенні пацієнту, що потребує такого лікування, нетоксичної терапевтично ефективної кількості сполуки формули І і фармацевтично прийнятного носія. У іншому аспекті даний винахід відноситься також до способу лікування опосередкованих циклооксигеназою захворювань, що успішно виліковуються активним агентом, який вибірково інгібує ЦОГ-2 в порівнянні з ЦОГ-1, що полягає у введенні пацієнту, що потребує такого лікування, нетоксичної терапевтично ефективної кількості сполуки формули І. У іншому аспекті даний винахід відноситься також до застосування сполуки формули І або фармацевтичної композиції при отриманні лікарського засобу для лікування запального замінорювання, чутливого до лікування нестероїдним протизапальним агентом. Даний винахід ілюструється Прикладами 1-56. Наступні абревіатури мають вказані значення: АА = арахідонова кислота Ас = ацетил AIBN = 2,2-азобісізобутиронітрил ВНТ = бутильований гідрокситолуол Bn = бензил CSA = камфорсульфонова кислота (рацемічна) dba = дибензиліденацетон DMAP = 4-(диметиламіно)піридин DMF = Ν,N-диметилформамід DMSO = диметилсульфоксид EDTA = етилендіамінтетраоцтова кислота ESA = етансульфонова кислота Et3N = триетиламін HBSS = збалансований сольовий розчин Хенкса HEPES = N-[2-Гідроксіетил]піперазин-N'-[2-етансульфонова кислота] HWB = цільна кров людини KHMDS = калій-гексаметилдисилазан LDA = діізопропіламід літію LPS = ліпополісахарид тСРВА = м-хлорпербензойна кислота ММРР = монопероксифталат магнію Ms = метансульфоніл = мезил MsO = метансульфонат = мезилат NBS = N-бромсукцинімід NCS = N-хлорсукцинімід NIS = N-йодсукцинімід NMO = N-метилморфолін-N-оксид NMP = N-метилпірролідон NSAID = нестероїдний протизапальний лікарський засіб (НСПЗЛЗ)oxoneâ = 2KHSO5·KHSO4·K 2SO4 РСС = хлорхромат піридинію PDC = дихромат піридинію PEG = поліетиленгліколь Ph = феніл руr = піридиніл r.t. = кімнатна температура rас. = рацемічний Tf = трифторметансульфоніл = трифліл ТfO = трифторметансульфонат = трифлат THF = тетрагідрофуран TLC = тонкошарова хроматографія Ts = п-толуолсуль фоніл = тозил TsO = п-толуолсульфонат = тозилат Tz = 1Н(або 2Н)-тетразол-5-іл SO2 Me = метилсульфон (також С 2СН3) SO2NH2 = сульфонамід Абревіатури алкільних гр уп Me = метил Et = етил n-Рr = н-пропіл і-Рr = ізопропіл n-Bu = н-бутил і-Bu = ізобутил s-Bu = втор-бутил t-Bu = трет-бутил с-Рr = циклопропіл с-Вu = циклобутил с-Реn = циклопентил с-Нех = циклогексил Абревіатури доз bid = bis in die = два рази в день qid = quarter in die = чотири рази в день tid = ter in die = три рази в день Для цілей даного опису "алкіл" означає лінійні, розгалужені і циклічні структури з вказаним числом атомів Byraew.. Приклади алкільних груп включають метил, етил, пропіл, втор- і трет-бутил, бутил, пентил, гексил, 1,1-диметилетил, циклопропіл, циклобутил, циклогексилметил і т.п. Подібним чином алкокси і алкілтіо означають лінійні, розгалужені і циклічні структури з вказаним числом атомів вуглецю. Для цілей даного опису "фторалкіл" означає алкільні групи з вказаним числом атомів вуглецю, в яких один водень замінений фтором. Прикладами є -CF3, -CH2CH2F, -CH2CF3, c-Pr-F5, c-Hex-F 11 і т.п. Подібним чином фторалкокси означає лінійні, розгалужені і циклічні структури з вказаним числом атомів вуглецю. Для цілей даного опису в ситуаціях, коли термін зустрічається два чи більше разів, визначення цього терміну в кожному випадку не залежить від визначення в кожному іншому випадку. Для цілей даного опису галоген означає F, СІ, Вr або І. У іншому варіанті даний винахід відноситься до фармацевтичних композицій для інгібування ЦОГ-2 і для лікування опосередкованих ЦОГ-2 захворювань, описаних тут, що містить фармацевтично прийнятний носій і нетоксичну терапевтично ефективну кількість сполуки формули І, описаної вище. Ще в одному варіанті даний винахід відноситься до способу інгібування циклооксигенази і лікування опосередкованих циклооксигеназою захворювань, що успішно виліковуються активним агентом, який вибірно інгібує ЦОГ-2 в порівнянні з ЦОГ-1, як описано тут, який полягає у введенні пацієнту, що потребує такого лікування, нетоксичної терапевтично ефективної кількості сполуки формули І, як описано тут. Оптичні ізомери - Діастереомери - Геометричні ізомери Деякі з описаних тут сполук містять один або декілька асиметричних центрів і, отже, можуть давати діастереомери і оптичні ізомери. Даний винахід включає в себе такі можливі діастереомери, а також їх рацемічні і розділені, енантіомерно чисті форми і їх фармацевтично прийнятні солі. Деякі з описаних тут сполук містять етиленові подвійні зв'язки і, якщо немає інших вказівок, включають як Е, так і Ζ геометричні ізомери. Солі Фармацевтичні композиції даного винаходу містять сполуки формули І як активні інгредієнти або його фармацевтично прийнятну сіль і можуть також містити фармацевтично прийнятний носій і, необов'язково, інші терапевтичні інгредієнти. Термін "фармацевтично прийнятні солі" відноситься до солей, отриманих з фармацевтично прийнятних нетоксичних основ, в тому числі неорганічних основ і органічних основ. Солі, отримані з неорганічних основ, включають солі алюмінію, амонію, кальцію, міді, тривалентного заліза, двовалентного заліза, літію, магнію, солі тривалентного марганцю, солі двовалентного марганцю, калію, натрію, цинку і т.п. Особливо переважні солі амонію, кальцію, магнію, калію і натрію. Солі, отримані з фармацевтично прийнятних органічних нетоксичних основ, включають солі первинних, вторинних і третинних амінів, заміщених амінів, в тому числі циклічних амінів, що зустрічаються в природі заміщених амінів, і основних іонообмінних смол, таких як аргінін, бетаїн, кофеїн, холін, Ν,Ν-дибензилетилендіамін, діетиламін, 2-діетиламіноетанол, 2-диметиламіноетанол, етаноламін, етилендіамін, Ν-етилморфолін, Nетилпіперидин, N-метилглюкамін, глюкамін, глюкозамін, гістидин, гідрабамін, N-(2-гідроксиетил)піперидин, N-(2-гідрокси-етил)пірролідин, ізопропіламін, лізин, метилглюкамін, морфолін, піперазин, піперидин, поліамінові смоли, прокаїн, пурини, теобромін, триетиламін, триметиламін, трипропіламін, трометамін і т.п. Коли сполука даного винаходу є основною, солі можуть бути отримані з фармацевтично прийнятних нетоксичних кислот, в тому числі неорганічних і органічних кислот. Такі кислоти включають оцтову, адипінову, аспарагінову, 1,5-нафталіндисульфонову, бензолсульфонову, бензойну, камфорсульфонову, лимонну, 1,2-етандисульфонову, етансульфонову, етилендіамінтетраоцтову, фумарову, глюкогептонову, глюконову, глутамінову, йодоводневу, бромоводневу, хлороводневу, ізетіонову, молочну, малеїнову, яблучну, мигдальну, метансульфонову, муцинову, 2-нафталінсульфонову, азотну, щавлеву, памову, пантотенову, фосфорну, пивалінову, пропіонову, саліцилову, стеаринову, янтарну, сірчану, винну, птолуолсульфонову, ундеканову, 10-ундеценову кислоту і т.п. Особливо переважні лимонна, бромоводнева, хлороводнева, малеїнова, метансульфонова, фосфорна, сірчана і винна кислоти. Повинно бути зрозумілим, що при обговоренні способів лікування, яке представлене далі, посилання на сполуки формули І включають також фармацевтично прийнятні солі. Корисність Сполука формули І може використовуватися для ослаблення болю, жару і запалення при різних станах, що включають ревматичну атаку, симптоми, пов'язані з грипом або іншими вірусними інфекціями, простуду, біль в нижній частині спини і шиї, дисменорею,, головний біль, зубний біль, розтягнення, міозит, невралгію, синовит, артрит, в тому числі ревматоїдний артрит, дегенеративні захворювання суглобів (остеоартрит), подагру і анкілозуючий спондилоартрит, бурсит, опіки, пошкодження, супроводжуючі хірургічні і зубні процедури. Крім того, така сполука може інгібувати клітинні неопластичні перетворення і метастатичний ріст пухлини і, отже, може використовуватися при лікуванні раку. Сполука І може бути також використана при лікуванні і/або запобіганні опосередкованим циклооксигеназою проліферативним порушенням, таким, які можуть мати місце у випадку діабетичної ретинопатії і пухлинного ангіогенезу. Сполука І буде також інгібувати скорочення гладких м'язів, що індукується простаноїдом, шляхом запобігання синтезу простаноїїдів, що скорочуються і, отже, може бути використане при лікуванні дисменореї, передчасних родів, астми і пов'язаних з еозинофілами порушень. Воно також може використовува тися при лікуванні хвороби Альцгеймера, для зниження втрати кісткової тканини у жінок в постклімактеричному періоді (тобто при лікуванні остеопорозу) і при лікуванні глаукоми. Завдяки його високій інгібуючій ЦОГ-2 активності і/або його специфічності у відношенні до інгібування ЦОГ-2 в порівнянні з ЦОГ-1, сполука І буде корисною як альтернатива для загальноприйнятих НСПЗЛЗ (не стероїдних протизапальних лікарських засобів) (NSAID), зокрема, коли такі нестероїдні протизапальні лікарські засоби можуть бути протипоказані, наприклад, для пацієнтів з пептичними виразками, гастритом, регіонарним ентеритом, виразковим колітом, дивертикулітом або з рецидивуючим анамнезом шлунковокишкових пошкоджень; у випадку ШК (шлунково-кишкової) кровотечі, порушень згортання крові, в тому числі анемії, такої як гіпопротромбінемія, гемофілія, або інших пов'язаних з кровотечею проблем; у разі захворювання нирок; перед хірургічним втручанням або при прийомі антикоагулянтів. Фармацевтичні композиції Для лікування будь-якого з цих опосередкованих циклооксигеназою захворювань сполука І може бути введена перорально, місцево, парентерально, інгаляцією або ректально у вигляді стандартних лікарських препаратів, що містять загальноприйняті нетоксичні фармацевтично прийнятні носії, домішки і розріджувачі. Термін "парентеральний" в застосуванні тут включає підшкірні ін'єкції, способи внутрішньовенної, внутрішньом'язової, вн утрішньогрудинної ін'єкції або інфузії. Крім лікування таких теплокровних тварин, як миші, пацюка, коня, великої рогаті" худоби, вівці, собаки, кішки і т.д., сполука даного винаходу ефективна при лікуванні людини. Сполука даного винаходу, зокрема, є добре відповідною для коней. Як вказано вище, фармацевтичні композиції для лікування опосередкованих ЦОГ-2 захворювань, описаних вище, можуть необов'язково містити один або декілька інгредієнтів, перерахованих ви ще. Фармацевтичні композиції, що містять активний інгредієнт, можуть бути в формі, придатній для перорального застосування, наприклад, у вигляді таблеток, пастилок, коржиків, водних або масляних суспензій, диспергованих порошків або гранул, емульсій, твердих або м'яких капсул або сиропів або еліксирів. Композиції, призначені для перорального застосування, можуть бути приготовані згідно з будьяким способом, відомим в даній області для виробництва фармацевтичних композицій, і такі композиції можуть містити один або декілька агентів, вибраних з групи, що включає підсолоджувачі, ароматизатори, барвники і консерванти, для забезпечення фармацевтично прийнятних і приємних на смак препаратів. Таблетки містять активний інгредієнт в суміші з нетоксичними фармацевтично прийнятними наповнювачами, які використовуються для отримання таблеток. Ці наповнювачі можуть являти собою, наприклад, інертні розріджувачі, такі як карбонат кальцію, карбонат натрію, лактозу, фосфат кальцію або фосфа т натрію; гранулюючі або розпушуючі агенти, наприклад, кукурудзяний крохмаль або альгінову кислоту; зв'язуючі агенти, наприклад, крохмаль, желатин або аравійську камедь; і змочувальні агенти, наприклад, стеарат магнію, стеаринову кислоту або тальк. Таблетки можуть бути непокритими або можуть бути покриті відомими способами для сповільнення руйнування і всмоктування в шлунково-кишковому тракті і, отже, забезпечення уповільненої дії протягом більш тривалого періоду часу. Наприклад, може бути використаний такий матеріал для пролонгування дії, як гліцерилмоностеарат або гліцерилдистеарат. Вони можуть бути також покриті за способом, описаному в патентах США 4256108; 4166452 і 4265874, для отримання осмотичних терапевтичних таблеток для регульованого вивільнення. Препарати для перорального використання можуть бути також представлені у вигляді твердих желатинових капсул, в яких активний інгредієнт змішаний з інертним твердим розріджувачем, наприклад, карбонатом кальцію, фосфатом кальцію або каоліном, або м'яких желатинових капсул, в яких активні інгредієнти змішані з водою або розчинниками, що змішуються, такими як пропiленгліколь, ПЕГ і етанол, або масляним середовищем, наприклад, арахісовою олією, рідким парафіном або оливковою олією. Водні суспензії містять активний матеріал в суміші з наповнювачами, придатними для отримання водних суспензій. Такими наповнювачами є суспендуючі агенти, наприклад, натрійкарбоксиметилцелюлоза, метилцелюлоза, гідроксипропілметил-целюлоза, альгінат натрію, полівінілпіролідон, трагакантова камедь і аравійська камедь; диспергуючі або змочувальні агенти можуть бути фосфатидом, що зустрічається в природі, наприклад, лецитином, або продуктами конденсації алкіленоксиду з жирними кислотами, наприклад, поліоксіетиленстеаратом, або продуктами конденсації етиленоксиду з аліфатичними спиртами з довгим ланцюжком, наприклад, гептадекаетиленоксицетанолом, або продуктами конденсації етиленоксиду з неповним складним ефіром, отриманим з жирних кислот і гекситу, такими як моноолеат поліоксіетиленсорбіту, або продуктами конденсації етиленоксиду з неповним складним ефіром, отриманим з жирних кислот і ангідридів гекситу, таким як моноолеат поліетиленсорбітану. Водні суспензії можуть також містити один або декілька консервантів, наприклад, етил- або н-пропіл-п-гідроксибензоат, бензиловий спирт, один або декілька барвників, один або декілька ароматизаторів і один або декілька підсолоджувачів, таких як сахарин або аспартам. Масляні суспензії можуть бути приготовані суспендуванням активного інгредієнта в рослинній олії, наприклад, арахісовій олії, оливковій олії, кунжутній олії або кокосовій олії, або мінеральному маслі, такому як рідкий парафін. Масляні суспензії можуть містити загусники, наприклад, бджолиний віск, твердий парафін або цетиловий спирт. Підсолоджувані, такі як описані вище, і ароматизатори можуть бути додані для отримання смачного перорального препарату. Ці композиції можуть зберігатися при додаванні антиоксиданту, такого як аскорбінова кислота. Дисперговані порошки і гранули, придатні для приготування водної суспензії шляхом додання води, містять активний інгредієнт в суміші з диспергуючим або зволожуючим агентом, суспендуючим агентом і одним або декількома консервантами. Придатні диспергуючі або зволожуючі агенти і суспендуючі агенти вже описані вище. Можуть також бути присутніми додаткові наповнювачі, наприклад, підсолоджувачі, ароматизатори і барвники. Фармацевтичні композиції даного винаходу можуть бути також у вигляді емульсій типу олія-в-воді. Масляною фазою може бути рослинна олія, наприклад, оливкова олія або арахісова олія, або мінеральне масло, наприклад, рідкий парафін, або їх суміші. Придатними емульгуючими агентами можуть бути фосфа тиди, що зустрічаються в природі, наприклад, лецитин сої, і складний ефір або неповний складний ефір, отриманий з жирних кислот і ангідридів гекситу, наприклад, моноолеат сорбітану, і продукти конденсації вказаного неповного складного ефіру з етиленоксидом, наприклад, моноолеат поліоксиетиленсорбітану. Емульсії можуть також містити підсолоджувачі і ароматизатори. Сиропи і еліксири можуть бути приготовані з підсолоджувачами, наприклад, гліцерином, пропіленгліколем, сорбітом або сахарозою. Такі препарати можуть також містити зменшуючий подразнення засіб, консервант і ароматизатори і барвники. Фармацевтичні композиції можуть бути у вигляді стерильної ін'єктованої водної або масляної суспензії. Така суспензія може бути приготована відповідно до відомих в даній області способів з використанням придатних диспергуючих або зволожуючих агентів і суспендуючи х агентів, які згадувалися вище. Стерильний ін'єкційний препарат може бути також стерильним ін'єкційним розчином або суспензією в нетоксичному парентерально прийнятному розріджувачі або розчиннику, наприклад, у вигляді розчину в 1,3-бутандіолі. Серед прийнятних носіїв і розчинників, які можуть бути використані, знаходяться вода, розчин Рінгера і ізотонічний розчин хлорида натрію. Можуть також використовува тися співрозчинники, такі як етанол, пропіленгліколь або поліетиленгліколі. Крім того, в якісті розчинника або суспендуюче середовище зручно використати нелеткі масла. Для цієї мети може використовува тися будь-яке м'яке нелетке масло, що включає синтетичні моно- або дигліцериди. Крім того, при приготуванні ін'єкційних розчинів можуть знайти застосування жирні кислоти, такі як олеїнова кислота. Сполука формули І може бути також введена у вигляді супозиторіїв для ректального введення лікарського засобу. Ці композиції можуть бути приготовані змішуванням лікарського засобу з відповідним неподразнюючим наповнювачем, який є твердим при звичайній температурі, але рідким при ректальной температурі і, отже, буде розплавлятися в прямій кишці з вивільненням лікарського засобу. Такими матеріалами є масло какао і поліетиленгліколі. Для зовнішнього застосування використовують креми, мазі, гелі, розчини або суспензії і т.д., що містять сполуку формули І. (Для цілей даної заявки місцеве застосування включає також використання рідини для полоскання рота і горла). Препарати для місцевого застосування звичайно можуть містити фармацевтичний носій, співрозчинник, емульгатор, підсилювач проникності, консервант і пом'якшувач. Діапазони доз При лікуванні вищезгаданих станів застосовні рівні доз близьковід біля 0,01мг до біля 140мг/кг маси тіла в день або, альтернативно, від біля 0,5мг до біля 7г на пацієнта в день. Наприклад, запалення можна ефективно лікувати введенням від біля 0,01 до 50мг сполуки на кг маси тіла в день або, альтернативно, від біля 0,5мг до біля 3,5г на пацієнта в день. Кількість активного інгредієнта, яка може бути об'єднана з матеріалами-носіями для отримання одиничної дозованої форми, буде змінюватися в залежності від підлягаючого лікуванню хазяїна і від конкретного способу введення. Наприклад, препарат, призначений для перорального введення людині, може містити від 0,5мг до 5г активного агента, об'єднаного з відповідною і зручною кількістю матеріалуносія, яка може змінюватися від біля 5 до біля 95% всієї композиції. Одиничні дозовані форми звичайно містять від біля 1мг до біля 500мг активного інгредієнта, звичайно 25мг, 50мг, 100мг, 200мг, 300мг, 400мг, 500мг, 600мг, 800мг або 1000мг. Однак, повинно бути зрозуміло, що рівень конкретної дози для будь-якого конкретного пацієнта буде залежати від безлічі чинників, в тому числі від віку, маси тіла, загального стану здоров'я, статі, харчового раціону, часу введення, способу введення, швидкості виведення, комбінації лікарських засобів і тяжкості конкретного захворювання, що підлягає терапії. Комбінації з іншими ліками Подібним чином сполука формули І буде застосовна як частковий або повний замінник загальноприйнятих НСПЗЛЗ в препаратах, в яких вони в цей час вводяться спільно з іншими агентами або інгредієнтами. Таким чином, в додаткових аспектах даний винахід включає фармацевтичні композиції для лікування опосередкованих ЦОГ-2 захворювань, таких як описані вище, утримуючу нетоксичну терапевтично ефективну кількість сполуки формули І, описані вище, і один або декілька інгредієнтів, таких як інший болезаспокійливий засіб, в тому числі ацетамінофен або фенацетин; потенціюючий засіб, що включає кофеїн; Нг-антагоніст, гідроксид алюмінію або магнію, симетикон, протизастійний засіб, в тому числі фенілефрин, фенілпропаноламін, псевдофедрин, оксиметазолін, епінефрин, нафазолін, ксилометазолін, пропілгекседрин або леводезоксиефедрин; противокашльовий засіб, в тому числі кодеїн, гідрокодон, карамифен, карбетапентан або декстраметорфан; простагландин, в тому числі мізопростол, енпростил, ріопростил, орнопростол або розапростол; сечогінний засіб; седативний або неседативний антігистамінний засіб. Крім того, даний винахід включає спосіб лікування опосередкованих циклооксигеназою захворювань, що передбачає введення пацієнту, що потребує такого лікування, нетоксичної терапевтично ефективної кількості сполуки формули І, що вводиться необов'язково з одним або декількома інгредієнтами, щойно перерахованими вище. Способи синтезу Сполуки формули І даного винаходу можуть бути отримані згідно з шляхами синтезу, представленими в схемах 1-2, і за допомогою описаних у винаході способів. СХЕМА 1 Піридини формули Іа і Іb можуть бути отримані шляхом багатостадійної послідовності реакцій з необхідного 2-амінопіридину II. Початкове бромування II бромом в оцтовій кислоті дає бромід III. Каталізоване паладієм зв'язування III з 4-(метилтіо)фенілбороновою кислотою в присутності відповідної основи, такої як карбонат натрію, дає сульфід IV, який може бути окислений за допомогою одного або декількох окислювачів, таких як ММРР, оксонâ, або OsO4/NMO до відповідного сульфона V. Амінопіридин V може бути перетворений в галогенід VI за допомогою одного з декількох способів. Наприклад, обробка V нітритом натрію в присутності НСl і брому дає бромід VI (Х=Вr). Альтернативно, обробка V нітритом натрію і НС1 з подальшою взаємодією з РОСl3 дає відповідний хлорид VI (Х=С l). Др уге катализоване паладієм зв'язування VI з відповідним чином заміщеною метальованою ароматичною сполукою, такою як арилборонова кислота або арилстанан, дає піридин формули Іа. Відповідна модифікація замісника R 2 в Іа дає додаткові приклади Іа. Наприклад, коли R2=Me, окислення таким окислювачем, як КМпО4, дає відповідну кислоту (R2 Іа = СО2Н), яка потім може бути перетворена в складний метиловий ефір (R2 Іа = СОгМе) із застосуванням такого реагенту, як діазометан. Альтернативно, обробка цієї кислоти хлорсуль фонілізоціанатом і ДМФ дає нітрил (R2 Іа = CN). Піридинметилсульфони Іа можуть бути перетворені у відповідні піридинсульфонаміди Іb із застосуванням процедур, описаних в літературі (Huang et al.. Tetrahedron Lett. 1994,39, 7201). СХЕМА 2 2-Галогенпіридини VI Схеми 1 можуть бути отримані багатостадійним процесом з відповідних 2гідроксипіридинів VII. Спочатку, обробка VII бромом в оцтовій кислоті дає бромід VIII. Подальша взаємодія VIII з бензилбромідом в присутності основи, такої як карбонат срібла, дає бензиловий ефір IX, який може бути перетворений в сульфон X через послідовність реакцій, подібних до описаних для перетворення броміду III в V в С хемі 1. Бензильна захисна група може бути видалена обробкою IX кислотою, такою як трифтороцтова кислота, з отриманням гідроксипіридину X. Нагрівання X з РОВr3 або РОСl3 дає відповідні 2галогенпіридини VI (X = Вr, СІ) Схеми 1. ХАРАКТЕРНІ СПОЛУКИ Таблиці 1 і 2 ілюструють сполуки формули Іа і Іb, які є характерними сполуками даного винаходу. ТЕСТИ ДЛЯ ВИЗНАЧЕННЯ БІОЛОГІЧНОЇ АКТИВНОСТІ Сполука формули І може бути перевірена за допомогою наступних тестів для визначення їх інгібуючої ЦОГ-2 активності. ІНГІБУВАННЯ ЦИКЛООКСИГЕНАЗНОЇ АКТИВНОСТІ Cполука тестують в якості інгібіторів циклооксигеназной активність в циклооксигеназних тестах на цілих клітинах. Обидва ці тести визначають синтез простагландина Ед у відповідь на АА (арахидонову кислоту) при допомозі радіоімуноаналізу. Клітками, що використовуються для цих тестів, є лінії клітин 143 остеосаркоми людини (які специфічно експресують ЦОГ-2) і лінії клітин U-937 людини (які специфічно експресують ЦОГ-1). У цих тестах 100% активністю вважають відмінність між синтезом простагландина Е2 у відсутність і в присутності арахiдоната. Тести на цілих клітинах Для циклооксигеназних тестів клітини остеосаркоми культивують в 1мл середи в 24-луночних мультипланшетах (Nunclon) до конфлюентности (1-2х105 клітин/лунку). Клітини U-937 вирощують в роллерних колбах і ресуспендують до кінцевої щільності 1,5 х106клітин/мл в 24-луночних мультипланшетах (Nunclon). Після промивання і ресуспендуовання клітин остеосаркоми і U-937 в 1мл HBSS додають 1мкл розчини в ДМСО тест-сполуки або ДМСО-носія і пробу ретельно змішують. Всі тести виконують, повторюючи 3 рази. Потім проби інкубують на протязі 5 або 15 хвилин при 37°С перед доданням АА. АА (не утримуючу пероксида. Cayman Chemical) готують у вигляді 10мМ початкового розчину в етанолі і потім розбавляють в 10 раз в HBSS. До клітин додають Аліквоту 10мкл цього розбавленого розчину з отриманням кінцевої концентрації АА 10мкМ. Контрольні проби інкубують з етанолом-носієм замість АА. Проби знову ретельно змішують і інкубують ще протягом 10 хвл. при 37°С. Для клітин остеосаркоми реакції зупиняють після цього доданням 100мкл 1 N НСl з перемішуванням і швидким видаленням розчину з монослоїв клітин. Для клітин U-937 реакції зупиняють доданням 100мкл 1N НСl з перемішуванням. Потім проби нейтралізували доданням 100мкл 1 N NaOH і рівні PGE2 вимірюють радіоімуноаналізом. Тести на цілих клітинах для ЦОГ-2 і ЦОГ-1 із застосуванням ліній трансфіційованих клітин ЯКХ Лінії клітин яєчника китайського хомячка (ЯКХ), які були стабільно трансфіційовані еукаріотичним експресуючим вектором pCDNAIII, що містить кДНК або ЦОГ-1, або ЦОГ-2 людини, використовують для цього тесту. Ці клітинні лінії називаються, відповідно, ЯКХ[хЦОГ-1] і ЯКХ[хЦОГ-2]. Для циклооксигеназних тестів клітини ЯКХ[хЦОГ-1] з суспензіонних культур і клітини ЯКХ[хЦОГ-2], о тримані трипсинізацією прикріплених культур, збирають центрифугуванням (300xg, 10хв.) і промивають один раз в HBSS, що містить 15мМ HEPES, рН 7,4, і ресуспендують в HBSS, 15мМ HEPES, рН 7,4, при концентрації клітин 1,5 х106клітин/мл. Лікарські засоби, що тестуються, розчиняють в ДМСО в 66,7 раз для найвищої концентрації лікарського засобу. Сполуку звичайно тестують при 8 концентраціях, повторюючи два рази, із застосуванням 3-кратних серійних розбавлень в ДМСО найвищої концентрації лікарського засобу. Клітини (0,3х106клітин в 200мкл) проінкубували з 3мкл лікарського засобу, що тестується, або ДМСО-носія протягом 15хв. при 37°С. Робочі розчини не утримуючої пероксида АА (5,5мкМ і 110мкМ АА для тестів ЯКХ[хЦОГ-1] і ЯКХ[хЦОГ-2], відповідно) отримують 10-кратним розведенням концентрованого розчину АА в е танолі в HBSS, що містить 15мМ HEPES, рН 7,4. Потім клітини стимулюють в присутності або у відсутності лікарського засобу розчином AA/HBSS з отриманням кінцевої концентрації 0,5мкМ АА в тесті ЯКХ[хЦОГ-1] і кінцевій концентрації 10мкМ в тесті ЯКХ[хЦОГ-2]. Реакцію зупиняють доданням 10мкл 1 N НСl з подальшою нейтралізацією 20мкл 0,5 N NaOH. Проби центрифугують при 300xg при 4°С протягом 10хв. і пробу освітленого супернатанта відповідним чином розбавляють для визначення рівнів PGE2 з використанням імуноферментного аналізу для PGE2 (Correlate PGE2 enzyme immunoassay kit. Assay Designs, Inc.). Циклооксигеназну активність у відсутність тесту-сполук визначають як різницю рівнів PGE2 клітин, стимульованих АА, і клітин, удаваностимульованих етанолом-носієм. Інгібування синтезу РСЕ2 тестомсполуками розраховують у вигляді проценту активності в присутності лікарського засобу відносно активності в позитивних контрольних пробах. Тести активності ЦОГ-1 мікросом з клітин U-937 Клітини U-937 осаджують центрифугуванням при 500xg протягом 5хв і промивають один раз забуференим фосфатом сольовим розчином і повторно осаджують. Клітини ресуспендують в буфері для гомогенізації, що складається з 0,1Μ Трис-НСІ, рН 7,4, 10мМ ЕДТА, 2мкг/мл лейпептина, 2мкг/мл соєвого інгібітора трипсину, 2мкг/мл апротиніну і 1мМ фенілметилсульфонілфториду. Клітинну суспензію обробляють ультразвуком 4 рази протягом 10сек і центрифугують при 10000xg протягом 10хв при 4°С. Супернатант центрифугують при 100000xg протягом 1год. при 4°С. Осадок мікросом 100000xg ресуспендують в 0,1 Μ Трис-НСІ, рН 7,4, 10мМ ЕДТА до приблизно 7мг білки/мл і зберігають при -80°С. Препарати мікросом відтаюють безпосередньо перед використанням, піддають короткочасній обробці ультразвуком і потім розбавляють до концентрації білка 125мкг/мл в 0,1Μ Трис-НСІ-буфері, рН 7/4, що містить 10мМ ЕДТА, 0,5мМ фенол, 1мМ відновлений глутатион і 1мкМ гематин. Тести виконують, повторюючи два рази, в кінцевому об'ємі 250мкл. Спочатку 5мкл носія ДМСО або лікарського засобу в ДМСО додають до 20мкл 0,1Μ Трис-НСІ-буфера, рН 7,4, що містить 10мМ ЕДТА, в лунках 96-луночного поліпропиленового титраційного планшета з глибокими лунками. Потім додають 200мкл препарату мікросом і преінкубують протягом 15 хв. при кімнатній температурі перед доданням 25мкл 1 Μ арахідонової кислоти в 0,1Μ Трис-НСІ і 10мМ ЕДТА, рН 7,4. Проби інкубують протягом 40 хв. при кімнатній температурі і реакцію зупиняють доданням 25мкл 1 N НСl. Проби нейтралізували 25мкл 1 N NaOH перед визначенням кількості PGE2 при допомозі радіоімуноаналізу (тест-набори Dupont-NEN або Amersham). Циклооксигеназну активність визначають у вигляді різниці між рівнями PGE2 в пробах, інкубованих в присутності арахідонової кислоти і етанольного носія. Тест активності очищеної ЦОГ-2 людини Активність ферменту вимірюють із застосуванням хромогенного тесту, заснованого на окисленні Ν,Ν,Ν',N'-тетраметил-п-фенілендіаміну (TMPD) під час відновлення PGG2 до PGH2 ферментом ЦОГ-2 (Copeland et al. (1994) Proc. Natl. Acad. Sci. 91,11202-11206). Рекомбінантну ЦОГ-2 людини очищають з клітин Sf9, як описано раніше (Percival et al (1994) Arch. Biochem. Biophys. 15, 111-118). Тест-суміш (180мкл) містить 100мМ фосфат натрію, рН 6,5, 2мМ генапол Х100, 1мкМ гематин, 1 мг/мл желатин, 80-100 одиниць обчищеного ферменту (одна одиниця ферменту визначена як кількість ферменту, що потребується для отримання зміни OD 0,001/хв. при 610нм) і 4мкл тесту-сполуки в ДМСО. С уміш преінкубують при кімнатній температурі (22°С) протягом 15 хвилин перед ініціюванням ферментативної реакції шляхом додання 20мкл обробленого ультразвуком розчину 1мМ АА і 1мМ TMPD в тесті-буфері (без ферменту або гематину). Активність ферменту вимірюють оцінкою початкової швидкості окислення TMPD протягом перших 36 сек. реакції. Неспецифічну швидкість окислення спостерігають у відсутності ферменту (0,007-0,010 OD/хв.) і віднімають перед розрахунком % інгібування. Величини ІС50 визначають методом найменших квадратів для нелінійного регресійного аналізу з 4 параметрами графіку вираженої в логарифмічних одиницях концентрації проти % інгібування. АН АЛІЗ Ц ІЛЬНОЇ КРОВІ ЛЮДИНИ Обґрунтування Цільна кров людини забезпечує багате білком і клітинами середовище, придатне для дослідження біохімічної ефективності протизапальних сполук, таких як вибірні інгібітори ЦОГ-2. Дослідження показали, що кров здорової людини не містить ферменту ЦОГ-2. Це узгоджується із спостереженням, що інгібітори ЦОГ-2 не впливають на утворення PGE2 в нормальній крові. Ці інгібітори активні тільки після інкубування суцільної крові людини з LPS (ліпополісахаридом), яка індукує ЦОГ-2. Цей тест може бути використаний для оцінки інгібуючої дії вибірних інгібіторів ЦОГ-2 на утворення PGE2. Тромбоцити в цільній крові також містять велику кількість ферменту ЦОГ-1. Відразу ж після згортання крові тромбоцити активуються по опосредкованому тромбіном механізму. Ця реакція приводить до продуціювання тромбоксану Вr (ТхВr) через активацію ЦОГ-1. Отже, дія тесту-сполук на рівні ТхВг після згортання крові може бути визначена і використана як показник активності ЦОГ-1. Таким чином, міра вибірковості тестом-сполукою може бути визначена вимірюванням рівнів PGE2 після індукції LPS (ЦОГ-2) і ТхВ2 після згортання крові (ЦОГ-1) в одному і тому ж тесті. МЕТОД Стадія А: ЦОГ-2 (індуковане LPS продукування PGE2) Свіжу кров збирають в гепаринізовані пробірки венопункцією у волонтерів (чоловіків і жінок). Ці суб'єкти не мають видимих запальних станів і вони не приймали яких-небудь НСПЗЛЗ протягом щонайменше 7 днів перед взяттям крові. Відразу ж отримують плазму крові з аліквоти 2мл крові для застосування як сліпий контроль (фонових рівнів PGE2). Іншу кров інкубують з LPS (100мкг/мл, кінцева концентрація, Sigma Chem. #L-2630 з Е. coli; розбавлений в 0,1% БСА (в забуференому фосфатом сольовому розчині) протягом 5 хвилин при кімнатній температурі. Аліквоти 500мкл крові інкубують або з 2мкл носія (ДМСО), або з 2мкл тесту-сполуки при кінцевих концентраціях, що варіюють від 10нМ до 30мкМ, протягом 24 годин при 37°С. У кінці інкубування кров центрифугують при 12000xg протягом 5 хвилин для отримання плазми. Аліквоту 100мкл плазми змішують з 400мкл метанолу для осадження білка. Отримують супернатант і його аналізують на PGE2 за допомогою набору для радіоімуноаналізу (Airiersham, RPA#530) після перетворення PGE2 в його метилоксиматне похідне відповідно до процедури виготовлювача. Стадія В: ЦОГ-1 (індуковане згортанням продукування ТхВ 2) Свіжу кров збирають у вакутейнери, що не містять антикоагулянтів. Аліквоти 500мкл відразу ж переносять в силіконізовані мікроцентрифужні пробірки, заздалегідь завантажені (2мкл) або ДМСО, або тестом-сполукою при кінцевих концентраціях від 10нМ до 30мкМ. Пробірки перемішують вортексом і інкубують при 37°С протягом 1 години, даючи крові скипатися. У кінці інкубування сироватку отримують центрифугуванням (12000xg протягом 5хвл.). Аліквоту 100мкл сироватки змішують з 400мкл метанолу для осадження білка. Отримують супернатант і його аналізують на ТхВr із застосуванням набору для ферментного імуноаналізу (Cayman #519031) відповідно до інструкції виготівника. ТЕСТ НАБРЯКУ ЛАПКИ ПАЦЮКА Протокол Самців пацюків Sprague-Dawley (150-200 г) піддають голодуванню протягом ночі і вводять їм перорально або носій (1% метоцел або 5% Твій 80), або тест-сполуку. Через 1 годину проводять стійким маркером лінію на рівні вище голеностопного суглоба на одній задній лапці для визначення зони лапки, що підлягає спостереженню. Об'єм лапки (Vo) вимірюють при допомозі плетизмометру (Ugo-Basile, Italy), заснованого на принципі витіснення води. Потім тварин ін'єкують субплантарно (через підошву) 50мкл 1% розчину карагенану в сольовому розчині (FMC Corp. Maine) в лапку за допомогою інсулінового шприца з голкою 25G (тобто 500мкг карагенану на лапку). Через 3 години вимірюють об'єм лапки (V3) і розраховують збільшення в об'ємі лапки (V3-Vo). Тварин умертвляють асфіксією за допомогою СО2 і оцінюють відсутність або наявність пошкоджень шлунку. Дані порівнюють з величинами для контролю-носія і розраховують процентне інгібування. Всі групи обробки шифрують для виключення спотворення (яке викликається помилкою спостерігача). НСПЗЛЗ ГАСТРОПАТІЯ, ЩО ІНДУКУЄТЬСЯ У ПАЦЮКІВ Обґрунтування Основною побічною дією загальноприйнятих НСПЗЛЗ є їх здатність виробляти шлункові пошкодження у людей. Вважається, що ця дія зумовлена інгібуванням ЦОГ-1 в шлунково-кишковому тракті. Пацюки особливо чутливі до дій НСПЗЛЗ. Дійсно, щурячі моделі використали звичайно в минулому для оцінки шлунково-кишкових побічних ефектів існуючих загальноприйнятих НСПЗЛЗ. У даному тесті НСПЗЛЗ шлунково-кишкове пошкодження, що індукується, спостерігають шляхом вимірювання фекальної екскреції 51 Сr після системної ін'єкції 51Сr-мічених еритроцитів. Фекальна 51Сr-екскреция являє собою добре відпрацьований і чутливий спосіб для виявлення шлунково-кишкової цілісності у тварин і людей. Способи Самцям пацюків Sprague-Dawley (150-200 г) вводять перорально тест-сполуки або один раз (гостре дозування), або b.i.d. (два рази в день) протягом 5 днів (хронічне дозування). Відразу ж після введення останньої дози пацюків ін'єкують через хвостову вену 0,5мл 51Сr-мічених еритроцитів від пацюка-донора. Тварин вмішують індивідуально в клітки для дослідження метаболізму з кормом і водою ad libitum. Фекалії збирають протягом періоду 48 годин і 51Сrекскрецію розраховують у вигляді процента від загальної ін'єкованої дози. 51Сr-мічені еритроцити отримують за допомогою наступних процедур. 10мл крові збирають в гепаринизовані пробірки через vena cava (порожнисту вену) від пацюка-донора. Плазму видаляють центрифугуванням і знов заповнюють рівним об'ємом HBSS. Еритроцити інкубують з 400мкКи 51хромату натрію протягом 30хв. при 37°С. У кінці інкубування еритроцити промивають двічі 20мл HBSS для видалення вільного 51хромату натрію. Нарешті, еритроцити відтворюють (реконституюють) в 10мл HBSS і 0,5мл цього розчину (біля 20мкКи) ін'єкують на пацюка. ГАСТРОПАТІЯ З ВТРАТОЮ БІЛКА У БІЛЯЧИХ МАВП Обґрунтування Гастропатія з втратою білку (що виявляється у вигляді появи циркулюючих клітин і білків плазми в шлунково-кишковому тракті) є істотною і обмежуючою дозу реакцією у відповідь на стандартні нестероїдні протизапальні лікарські засоби (НСПЗЛЗ). Вона може бути кількісно оцінена внутрішньовенним введенням розчину 51СrСl3. Цей ізотопний іон може авідно зв'язуватися з клітиною і сироваточними глобінами і ендоплазматичним ретикулумом клітини. Вимірювання радіоактивності, що з'являється в фекаліях, які збираються протягом 24год. після введення ізотопу, забезпечує, отже, чутливий і кількісний показник гастропатії з втратою білку. Способи Групи самців білячих мавп (0,8-1,4кг) обробляють шляхом годування через шлунковий зонд або 1% метоцелом, або 5% Твіном 80 в Н2О-носіях, (3мл/кг b.i.d.), або тестом-сполуками при дозах від 1 до 100мг/кг b.i.d. протягом 5 днів. 51Сr (5мкКи/кг в 1мл/кг забуференого фосфатом сольового розчину (PBS)) вводять внутрішньовенно Через 1год. Після останньої дози лікарського засобу/носія і фекалії збирають протягом 24год. в клітці для дослідження метаболізму і оцінюють по гаммі-рахунку 51Сr. Венозну кров збирають через 1год. і 8год. після останньої дози лікарського засобу і концентрації лікарського засобу в плазмі вимірюють при допомозі ОФ-ВЕЖХ. LPS гіпертермзя, що індукується, ν пацюків, що знаходяться в свідомості. Самців пацюків Sprague-Dawley (150-200г) піддавали голодуванню протягом 16-18год. перед використанням. Приблизно в 9год. 30хв. ранку тварин вміщували тимчасово в фіксуючі камери з плексигласу і їх фонову ректальну температуру реєстр ували за допомогою гнучкого температурного зонда (YSI series 400), сполученого з цифровим термометром (Model 08502, Cole Farmer). Одні і ті ж зонди і термометр використали для всіх тварин для зменшення експериментальної помилки. Після вимірювань температури тварин повертали в їх клітини. При часі нуль, пацюків ін'єкували внутричеревно або сольовим розчином, або LPS (ліпополісахаридом) (2мг/кг, Sigma Chem.) і ректальну температуру повторно вимірювали при 5, 6 і 7год. після ін'єкції LPS. Після вимірювання при 5год., коли збільшення температури досягало плато, ін'єкованим LPS пацюкам давали перорально або носій (1% метоцел), або тест-сполуку для визначення, чи може ця сполука обертати гіпертермію. Процент обертання гіпертермії розраховували з використанням ректальної температури, отриманої при 7год. в контрольній (обробленої носієм) групі, як порівняльна (обертання нуль) точка. Повне обертання гіпертермії до величини фону перед LPS приймали за 100%. LPS гіпертермія, що індукується у білячих мавп, що знаходяться в свідомості Температурні зонди хірургічно імплантували під шкіру живота в гр упі білячих мавп (Salmlrl sciureusj (1,01,7 кг). Це дозволяє спостерігати температуру тіла у не підданих фіксації мавп, що знаходяться в свідомості, за допомогою телеметричної сприймаючої системи (Data Sciences International, Minnesota). Тварин піддавали голодуванню і вміщували в індивідуальні клітини для акліматизації за 13-14год. до використання. На бічній стороні клітин встановлювали електронні приймачі, які приймали сигнали від імплантованих температурних зондів. Приблизно в 9год. 00хв. ранку в день експеримент мавп тимчасово фіксували в стільцях для дресирування і давали їм болісну I.V. (вн утрішньовенну) ін'єкцію LPS (6мг/кг, розчинені в стерильному сольовому розчині). Тварин повертали в їх клітини і температур у тіла реєстрували безперервно кожні 5 хв. Через дві години після ін'єкції LPS, коли температура тіла збільшувалася на 1,52°С, мавпам давали перорально дозу або носій (1% метоцел), або тест-сполуку (3мг/кг). Через 100 хвилин визначали різницю між температурою тіла і фоновою величиною. Процентне інгібування розраховували, приймаючи величину в контрольній групі за 0% інгібування. Гостра запальна гіпералгезія, індукована карагенаном у пацюків Експерименти проводили, використовуючи самців пацюків Sprague-Dawley (90-110г). Гіпералгезію до механічного стиснення задньої лапки індукували інтраплантарно (в підошву) ін'єкцією карагенану (4,5мг в одну задню лапку) за 3год. до використання. Контрольні тварини отримували рівний об'єм сольового розчину (0,15мл в підошву). Тест-сполуку (0,3-30мг/кг, суспендування в 0,5% метоцелі в дистильованій воді) або носій (0,5% метоцел) вводили перорально (2мл/кг) через 2год. після карагенану. Голосову реакцію у відповідь на стиснення задньої лапки вимірювали через 1год. при допомозі алгезиметру Ugo Basile. Статистичний аналіз гіпералгезії, що індукується каррагенаном, виконували за допомогою однофакторного дисперсійного аналізу ANOVA (BMDP Statistical Software Inc.). Гіпералгезію визначали відніманням голосового порогу у ін'єкованих сольовим розчином пацюків з голосового порога, який отримували у тварин, іньецийованих каррагенаном. Оцінки гіпералгезії для пацюків виражали у вигляді процента цієї відповіді. Потім величини ID5o (дози, що дає 50% максимальної відповіді, що спостерігаються) розраховували за допомогою нелінійного регресіонного аналізу середніх величин із застосуванням методу найменших квадратів з використанням GraFit (Erithacus Software). Артрит, що індукується ад'ювантом у пацюків Сімдесят 6,5-7,5-тижневих самиць пацюків Lewis (вага тіла 146-170г) зважували, мітили (вушною міткою) і розподіляли на групи (група негативного контролю, в якій не індукували артрит, група контролю з носієм, група позитивного контролю, якій вводили індометацин з добовою дозою 1мг/кг, і чотири групи, яким вводили тест-сполуку при загальних добових дозах 0,10-3,0мг/кг), так щоб маси тіл були рівними в кожній групі. Шість груп з 10 пацюків кожна отримували ін'єкції в задню лапку 0,5мг Mycobacterium butyrlcum в 0,1мл легкого мінерального масла (ад'юванту), а група негативного контролю з 10 пацюків не отримувала ін'єкцій ад'ювантом. Маси тіл, об'єми контралатеральних лапок (що визначаються плетизмографією з витісненням ртуті) і латеральні радіографи (рентгенограми) (отримані під анестезією із застосуванням Кетаміну і Ксилазіну) визначали до ін'єкції ад'юванту (день -1) і через 21 день після ін'єкції ад'юванту і первинні об'єми лапки визначали до ін'єкції ад'юванту (день -1) і в дні 4 і 21 після ін'єкції ад'юванту. Пацюків анестезували внутрішньом'язовою ін'єкцією 0,03-0,1мл комбінації Кетаміну (87мг/кг) і Ксилазіну (13мг/кг) для радіографів і ін'єкцій ад'юванту. Радіографи отримували для обох задніх лапок в дні 0 і 21 з використанням Faxitron (45 kvp, 30 секунд) і плівки Kodak X-OMAT TL і виявляли в автоматизованому процесорі. Радіографи (рентгенограми) оцінювалися на зміни в м'яких і твердих тканинах дослідником, який не знав експериментальних варіантів. Наступні радіографичні зміни розподілялися по ступенях у вигляді цифр відповідно до тяжкості; збільшений об'єм м'якої тканини (0-4), звуження або розширення порожнин суглобів (0-5), субхондральна ерозія (0-3), периостеальна реакція (0-4), остеоліз (0-4), підвивих (0-3) і дегенеративні зміни суглоба (0-3). Для встановлення цифрової оцінки тяжкості для кожної зміни на рентгенограмі використали специфічні критерії. Максимальна можлива оцінка на лапку була 26. Тест-сполука при загальних добових дозах 0,1, 0,3, 1 і 3мг/кг/день, індометацин при загальній добовій дозі 1мг/кг/день або носій (0,5% метоцел в стерильній воді) вводили перорально b.i.d. (два рази в день), починаючи після ін'єкції адъюванта і продовжуючи протягом 21 дня. Сполуку готували щотижня, тримали в холодильнику в темряві до використання і перемішували вортексом безпосередньо перед введенням. Двухфакторний ( "варіант обробки" і "час") дисперсійний аналіз з вимірюваннями, що повторяються у "часі" застосовували до % змін ваги тіла і об'ємів лапок (стоп) і до впорядкованих даних загальних оцінок радіографів. Проводили post hoe-тест Dunnett для порівняння дії варіантів обробок з носієм. Однофакторний дисперсійний аналіз застосовували для ваги тимуса і селезінки з подальшим тестом Durmett для порівняння дії обробок з носієм. Криві доза-відповідь для % інгібування об'ємів лапок в дні 4, 14 і 21 будували з використанням логистичної функції з 4 параметрами за допомогою нелінійної регресії методом найменших квадратів. ID50 визначали як дозу, відповідну 50% зменшенню від носія, і отримували інтерполяцією з отриманого рівняння з 4 параметрами. ФАРМАКОКІНЕТИКА В П АЦЮКАХ Пероральна фармакокінетика в пацюках Тварин утримують, годують і забезпечують доглядання відповідно до Правил Канадської Ради по догляду за тваринами. Самців пацюків Sprague-Dawley (325-375г) піддають голодуванню протягом ночі для кожного дослідження рівня в крові при пероральному введенні. Пацюків вміщують в фіксуючу камеру по одній і камеру міцно фіксують. Нульове значення проби крові отримують відсіканням невеликого (1мМ або менш) шматочка від кінчика хвоста. Потім хвіст гладять міцним, але м'яким рухом зверху вниз для "видоєння" крові. Приблизно 1мл крові збирають в гепаринізовану пробірку-вакутейнер. Сполуку готують, як годиться, в стандартному об'ємі для дозування 10мл/кг і вводять перорально проведенням голки для годування через зонд 16G, 3" в шлунок. Подальше взяття крові виконується так само, як взяття крові для нульового значення, за винятком того, що немає необхідності знову відсікати кінчик хвоста. Хвіст очищають шматочком марлі і "видоюють "/гладять, як описано вище, у відповідно помічені пробірки. Відразу ж після взяття крові кров центрифугують, відділяють, вміщують в чітко помічені флакони і зберігають в холодильнику до аналізу. Типові тимчасові точки для визначення рівнів в крові пацюка після РО-дозування: 0,15 хвилин, 30 хвилин, 1 година, 2 години, 4 години, 6 годин. Після 4-часової точки взяття крові пацюкам дають корм ad libitum. Воду надають весь час в процесі дослідження. Носії У визначеннях рівнів в крові пацюків при РО-дозуванні можуть бути використані наступні носії: PEG 200/300/400: обмеження до 2мл/кг Methocel 0,5% -1 %: 10мл/кг Твін 80: 10мл/кг Сполуки для РО-рівнів в крові можуть знаходитися в формі суспензій. Для кращого розчинення розчин може бути вміщений в прилад для обробки ультразвуком приблизно на 5 хвилин. Для аналізу аліквоти розбавляють рівним об'ємом ацетонітрилу і центрифугують для видалення осаду білку, Супернатант ін'єкують безпосередньо на колонку 3-18 ВЕЖХ з УФ-детектування. Кількісне визначення виконують відносно чистої проби крові, в яку введена відома кількість лікарського засобу. Біодоступність (F) оцінюють по порівняльній площі під кривою (AUC) i.v. проти р.о. AUCpo DOSEiv F= x x100% AUCiv DOSEpo Швидкості кліренсу розраховують з наступного відношення: DOSEiv(мг / кг ) CL = AUCiv Одиниці CL - мл/ч·кг (мілілітри в годину на кілограм). Внутрішньовенна фармакокінетика у пацюків Тварин утримують, годують і забезпечують догляд відповідно до Правил Канадської Ради по догляду за тваринами. Самців пацюків Sprague-Dawley (325-375г) вміщують в пластикові клітини з розміром коробок для туфель з підвішеним дном, кришкою клітини, поїлкою для води і кормом. Сполуку готують, як годиться, в стандартному об'ємі для дозування 1мл/кг. У пацюків беруть пробу крові для нульової величини і їх дозують при впливі СО2 як седативного засобу. Пацюків, по одному, вміщують в заповнену СО2 камеру і виймають, як тільки вони втрачають їхній рефлекс випрямлення. Потім пацюка вміщують на фіксуючий столик, носовий конус з подачею СО2 вміщують над мордою і пацюка закріпляють на столику еластичними бинтами. За допомогою пінцета і ножиць оголяють яремну вену і беруть нульову пробу, після чого відміряну дозу сполуки ін'єкують в яремну вену. Легко затискають пальцем місце ін'єкції і носовий конус видаляють. Помічають час. Цей час є часом нульової точки. 5-хвилинне взяття крові виконують відсіканням шматочка (1-2мМ) від кінчика хвоста. Потім гладять міцним, але м'яким рухом зверху вниз для "видоєння" крові. Приблизно 1мл крові збирають в гепаринізований флакон для збору крові. Подальше взяття крові виконується подібним чином, за винятком того, що немає необхідності знову відсікати кінчик хвоста. Хвіст очи щають шматочком марлі і беруть кров, як описано вище, у відповідні помічені пробірки. Типові тимчасові точки для визначення рівнів в крові після I.V.-дозування: 0,5 хвилини, 15 хвилин, 30 хвилин, 1 година, 2 години, 6 годин або 0,5 хвилини, 30 хвилин, 1 година, 2 години, 4 години, 6 годин. Носії: У визначеннях рівнів в крові пацюків при IV-дозуванні можуть бути використані наступні носії: Декстроза 1мл/кг Moleculosol 25% 1мл/кг обмеження до об'єму дози ДМСО: 0,1мл на тварину не більше за 60% в суміші з PEG 200 40% стерильної води - 1мл/кг У випадку декстрози, якщо розчин каламутний, може бути доданий бікарбонат натрію або карбонат натрію. Для аналізу аліквоти розбавляють рівним об'ємом ацетонітрилу і центрифугують для видалення осаду білку. Супернатант ін'єкують безпосередньо на колонку 3-18 ВЕЖХ з УФ-детектування. Кількісне визначення виконують відносно чистої проби крові, в яку введена відома кількість лікарського засобу. Біодоступність (F) оцінюють по порівняльній площі під кривою (AUC) i.v. проти р.о. AUCpo DOSEiv F= x x100% AUCiv DOSEpo Швидкості кліренсу розраховують з наступного відношення: DOSEiv(мг / кг ) CL = AUCiv Одиниці CL - мл/ч·кг (мілілітри в годину на кілограм). РЕПРЕЗЕНТАТИВНІ БІОЛОГІЧНІ ДАНІ Сполуки даного винаходу є інгібіторами ЦОГ-2 і тому застосовні для лікування опосередкованих ЦОГ-2 захворювань, перерахованих ви ще. Активність сполук проти циклооксигенази можна бачити в репрезентативних результатах, показаних нижче. У цьому тесті інгібування визначають вимірюванням кількості простагландина E2 (PGE2), синтезованої в присутності АА, ЦОГ-1 або ЦОГ-2 і передбачуваному інгібітора. Величини ІС50 представляють концентрацію передбачуваного інгібітора, що вимагається для зниження синтезу PGE2 до 50% синтезу, отриманого в порівнянні з неінгібованим контролем. Дані з трьох цих біологічних тестів дані для репрезентативних сполук разом з порівняльними даними для наступних дво х сполук з World Patent Application 96/10012: Як можна бачити з цих даних, сполуки даного винаходу виявляють більш високу вибірковість у відношенні ЦОГ-2 і активність, чим Аа і Аb. Крім того, основність піридинового кільця в цих прикладах дозволяє утворення кислотно-адитивних солей, що приводить до збільшеної водорозчинності і створює потенціал для перентерального введення у водних носіях. Далі даний винахід проілюстрований наступними необмежуючими прикладами, в яких, якщо немає інших вказівок: (і) всі операції проводили при кімнатній температурі або температурі навколишнього середовища, тобто при температурі в межах 18-25°С, і висушування органічних речовин виконували з використанням MgSO4; (іі) випаровування розчинника проводили з використанням роторного випарника при зниженому тиску (600-4000Па: 4,5-30мМ рт. ст.) з температурою бані до 60°С; (ііі) хід реакції контролювали за допомогою тонкослойної хроматографії (ТСХ), і час реакції указано лише для цілей ілюстрації; (iv) точки плавлення дані без поправок і "розкл." вказує розкладання; представлені точки плавлення є температурами плавлення, отриманими для речовин, приготованих, як описано; поліморфізм може приводити до виділення речовин з різними точками плавлення в деяких препаратах; (ν) стр уктур у і чистоту всіх кінцевих продуктів підтверджували щонайменше одним з наступних способів: ТСХ, мас-спектрометрія, спектрометрія ядерного магнітного резонансу (ЯМР) або мікроаналітичними даними; (vi) ви ходи дані тільки для ілюстрації; (vii) якщо дані ЯМР представлені, то вони дані в формі величин дельта (d) для основних діагностичних протонів, представлених в мільйонних частках (ррт), відносно тетраметилсилану (TMS) як внутрішній стандарт, визначених при 300мГЦ або 400мГЦ з використанням вказаного розчинника; загальноприйнятими абревіатурами, що використовуються для форми сигналу, є: s, синглет, d, дуплет, t, триплет, т, мультиплет, br, широкий; і т.д., крім того, "Аr" означає ароматичний сигнал; (viii) хімічні символи мають їх звичайні значення; використовувалися також наступні абревіатури: V (об'єм), W (вага), т. кип. (точка кипіння), т. пл. (точка плавлення), л (літр, літри), мл (мілілітри), г (грами), мг (міліграми), моль (молі), мМоль (чи міллімоллі ), екв. (еквіваленти), к.т. (кімнатна температура). ПРИКЛАД 1 3-(4-Метілсульфоніл)феніл-2-феніл-5-трифторметилпіридин Стадія 1: 2-Аміно-3-бром-5-трифторметилпіридин До розчину 2-аміно-5-трифторметюшіридину (9г) в оцтовій кислоті (75мл) повільно додавали бром (5,8мл). Через 1год. кислоту нейтралізували обережним доданням гідроксиду натрію (10N) при 0°С. Отриманий оранжевий залишок розчиняли в ефірі і промивали послідовно насиченим карбонатом калію, насиченим NA2SO 3 і насиченим розчином солі, сушили і концентрували. Тверду речовину, що залишилася інтенсивно перемішували в гексані протягом 1год. з отриманням, після фільтрування, сполуки заголовка у вигляді білої твердої речовини (10,2г). Стадія 2: 2-Аміно-3-(4-метилтіо)феніл-5-трифторметилпіридин Суміш броміду зі стадії 1,4-метилтіобензолборонової кислоти (Li, et al. J. Med. Chem. 1995, 38,4570) (8,5г), 2Μ водного карбонату натрію (60мл) і паладій-тетракис(трифенілфосфину) (490мг) в суміші етанол/бензол (100мл, 1:1) нагрівали із зворотним холодильником протягом 15год. Суміш охолоджували до кімнатної температури, розбавляли водою і екстрагували ефіром. Органічні речовини концентрували і залишок енергійно перемішували в суміші ефір/гексан протягом 1год. з отриманням, після фільтрування, сполуки заголовку (11,2г) у вигляді бежового твердого продукту. Стадія 3: 2-Аміно-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин Суміш 2-аміно-3-(4-метилтю)феніл-5-трифторметилпіридину (9,7 г), OsC4 (2мл 4% розчину у воді) і Ν ΜΟ (13г) в суміші ацетон/вода (60мл: 5мл) перемішували при кімнатній температурі протягом ночі. Потім додавали насичений водний Na2SO3 і отриману суміш перемішували протягом 30хв. Ацетон випаровували і отриману суміш екстрагували ефіром і етилацетатом. Об'єднані органічні екстракти промивали Na2SO3, водою, насиченим розчином солі і потім концентрували. Твердий залишок інтенсивно перемішували в гексані і ефірі протягом 1год. і потім фільтрували з отриманням сполуки заголовка у вигляді блідо-жовтої твердої речовини (9,9г). Стадія 4: 2-3-(4-метилсульфоніл)феніл-5-трифторметил-піридин До розчину 2-аміно-3-(4-метилсульфоніл)феніл-5-трифторметил-піридин (1,2г) в суміші вода/концентруючий НСl (9,5мл: 1мл) при 0°С додавали розчин нітриту натрію (262мг) в 5мл води. Суміш нагрівали до кімнатної температури і перемішували протягом ночі. Додавали ще 30мг нітриту натрію і після 3год. гетерогену суміш фільтрували. Частину твердої речовини (250мг) і РОСl3 (110мкл) в ДМФ (2мл) нагрівали при 70°С протягом 60год. Суміш охолоджували до кімнатної температури, розбавляли водою і екстрагували етилацетатом. Органічний шар промивали насиченим розчином солі, сушили і концентрували з отриманням сполуки заголовка у вигляді блідо-жовтої твердої речовини (270мг), яке використали відразу в наступній реакції. Стадія 5: 3-(4-Метілсульфоніл)феніл-2-феніл-5-трифторметил-піридин Суміш 2-хлор-3-(4-метилсульфоніл)феніл-5-трифторметил-піридин (260мг), бензолборонової кислоти (113мг), 2Μ водних карбонату натрію (2,1мл) і паладія-тетракис(трифенілфосфину) (30мг) в суміші етанол/бензол (8мл, 1:1) нагрівали із зворотним холодильником протягом 24год. Суміш охолоджували до кімнатної температури, розбавляли водою і екстрагували етилацетатом. Органічний шар концентрували і тверду речовину піддавали флеш-хроматографії (з елююванням сумішшю гексан/етилацетат, 4:1 об/об) з отриманням сполуки заголовка у вигляді білої твердої речовини, т. пл. 191-192°С (215мг). ПРИКЛАД 2 2-(3-Хлорфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин Стадія 1: 2-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин До розчину 2-аміно-3-(4-метилсульфоніл)феніл-5-трифторметил-піридин прикладу 1, стадії 3 (2г) в 48% НВr (25мл) при 0°С додавали порціями бром (3мл) і потім нітрит натрію (1,1г). Після 2год. розчин нейтралізували доданням гідроксиду натрію (10Ν) і потім екстрагували етилацетатом. Органічний шар промивали насиченим Na 2SO3 і насиченим розчином солі, сушили і концентрували. Флеш-хроматографія продукту, що залишився (з елююванням сумішшю гексан/етилацетат, 7:3-3:7 об/об), давала сполуку заголовку у вигляді білої твердої речовини (435 мг). Стадія 2: 2-(3-Хлорфеніл)-3-(4-метилсульфоніл)феніл-5-трифтор-метилпіридин Суміш 2-бром-3-(4-метилсульфоніл)феніл-5-трифторметил-піридин (178мг), 3-хлорфенілбороновой кислоти (110мг), фосфату калію (225мг) і паладію-тетракис(трифенілфосфина) (20мг) в діоксані (10мл) нагрівали із зворотним холодильником протягом 24год. Після охолоджування при кімнатній температурі суміш розбавляли водою і екстрагували ефіром. Органічний екстракт сушили і концентрували і продукт, що залишився, піддавали флеш-хроматографії (з елююванням сумішшю гексан/етилацетат, 7:3 об/об). Отриману тверду речовину енергійно перемішували в суміші гексан/ефір протягом 1год. з отриманням сполуки заголовку у вигляді блідо-жовтої твердої речовини, т. пл. 136-137°С (115мг). ПРИКЛАД 3 2-(4-Хлорфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин Згідно з прийомами, описаними в прикладі 1 стадії 5, але із заміною бензолборонової кислоти 4хлорфенілбороновою кислотою сполуку заголовку отримували у вигляді білої твердої речовини, т. пл. 192193°С (155 мг). ПРИКЛАД 4 2-(4-Фторфеніл)-3-(4-метилсульфоніл)феніл-5-трифторметилпіридин Згідно з прийомами, описаними в прикладі 1 стадії 5, але із заміною бензолборонової кислоти 4фтор фенілбороновою кислотою сполуку заголовку отримували у вигляді білої твердої речовини, т. пл. 163164°C (152мг). ПРИКЛАД 7 3-(4-Метилсульфоніл)феніл-2-(3-піридиніл)-5-трифторметилпіридин Суміш 2-бром-3-(4-метилсульфоніл)феніл-5-трифторметил-піридин (600мг) (приклад 2, стадія 2), діетил-3-піридинілборану (255мг), карбонату натрію (2М, 2,2мл) і диброміду біс(трифенілфосфін)паладію (25мг) в суміші бензол/етанол (1:1/ 32мл) нагрівали із зворотним холодильником протягом 24год. Після охолоджування до кімнатної температури суміш концентрували, розбавляли водою і екстрагували етилацетатом. Органічний екстракт концентрували і продукт, що залишився, розчиняли в суміші 10% НСІ/ефір. Органічну фазу видаляли і водну фазу доводили до рН ~10 шля хом додання насиченого бікарбонату натрію. Суміш екстрагували етилацетатом і об'єднані органічні екстракти концентрували і піддавали флеш-хроматографії (з елююванням етилацетатом) з отриманням сполуки заголовка в вигляді білої твердої речовини, т. пл. 171-172° (180мг). ПРИКЛАД 13 5-3-(4-метилсульфоніл)феніл-2-фенілпіридин Стадія 1: 2-Аміно-3-бром-5-метилпіридин До розчину 2-аміно-5-піколину (5 г) в оцтовій кислоті (40мл) при кімнатній температурі повільно додавали бром (2,6мл/ Через 1год. кислоту нейтралізували обережним доданням гідроксиду натрію (10N) при 0°С. Отриманий оранжевий залишок розчиняли в ефірі і промивали послідовно насиченим карбонатом калію, насиченим Na2S2O3 і насиченим розчином солі, сушили і концентрували. Флеш-хроматографія (з елююванням сумішшю гексан/етилацетат, 3:2 об/об) твердої речовини, що залишилася давала сполуку заголовка у вигляді блідо-жовтої твердої речовини (7,1г). Стадія 2: 2-Аміно-5-метил-3-(4-метилсульфоніл)фенілпіридин Згідно з прийомами, описаними в прикладі 1, стадії 2 і 3, але із заміною 2-аміно-3-бром-5трифторметилпіридину 2-аміно-3-бром-5-метилпіридином (7,1 г) зі стадії 1, сполуку заголовка отримували у вигляді блідо-жовтої твердої речовини (3,7 г). Стадія 3: 2-Бром-5-метил-3-(4-метилсульфоніл)фенілпіридин Згідно з прийомами, описаними в прикладі 2, стадія 1, але із заміною 2-аміно-3-(4 метилсульфоніл)феніл-5-трифторметилпіридину 2-аміно-5-метил-3-(4-метилсульфоніл)фенілпіридином (3г) зі стадії 2, сполуки заголовку отримували в вигляді білої твердої речовини (2,7г). Стадія 4: 5-3-(4-метилсульфоніл)феніл-2-фенілпіридин Згідно з прийомами, описаними в прикладі 1, стадія 5, але із заміною 2-хлор-3-(4метилсульфоніл)феніл-5-трифторметилпіридину 2-бром-5-метил-3-(4-метилсульфоніл)фенілпіридином (300мг) зі стадії 3 сполуки заголовку отримували у вигляді блідо-жовтої твердої речовини (270 мг). 1Н-ЯМР (300 MHz, CDCl3) d 2,42 (s, 3Н), 3,03 (s, 3Н), 7,19-7,28 (m, 5H), 7,35 (d, 2H), 7,51 (d, 1H), 7,81 (d,2H), 8,56 (d, 1H). ПРИКЛАД 14 2-(4-Хлорфеніл)-5-3-(4-метилсульфоніл)фенілпіридин Згідно з прийомами, описаними в прикладі 3, але із заміною 2-хлор-3-(4-метилсульфоніл)феніл-5трифторметилпіридину 2-бром-5-метил-3-(4-метилсульфоніл)фенілпіридином (300мг) зі стадії 3 прикладу 14 сполуку заголовка отримували у вигляді білої твердої речовини, т. пл. 155-156°С (125мг). ПРИКЛАД 15 5-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин Стадія 1: Три-н-пропокси-3 -піридинілборонат літію До розчину 3-бромпіридину (39,5г) в ефірі (800мл) при -90°С (внутрішня температура) додавали H-BuLi (100мл, 2,5М) при такій швидкості, щоб внутрішня температура не перевищувала -78°С. Отриману суміш перемішували протягом 1год. при -78°С і потім додавали триізопропоксиборат (59мл) і отриману суміш нагрівали до 0°С. Додавали метанол і суміш випаровували три рази з метанолу і потім два рази з нпропанолу. Залишок відкачували при високому вакуумі протягом 3 днів і отриману піну (76г суміші 1:1 сполуки заголовка/н-пропанол) використали як таку на наступній стадії. Стадія 2: 5-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин Згідно з прийомами, описаними в прикладі 14, але із заміною 4-хлорфенілборонової кислоти три-нпропокси-3-піридинілборонатом літію зі стадії 1 отримували сполуку заголовка у вигляді білої твердої речовини, т. пл. 166-167°С (2,1г). ПРИКЛАД 17 5-Хлор-2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридин Стадія 1: 2-Аміно-3-бром-5-хлорпіридин До розчину 2-аміно-5-хлорпіридину (10г) в оцтовій кислоті (75мл) при кімнатній температурі повільно додавали бром (2,6мл). Через 30хв. кислоту нейтралізували обережним доданням гідроксиду натрію (10N) при 0°С. Отриманий оранжевий залишок розчиняли в етилацетаті і промивали послідовно насиченим карбонатом калію, насиченим Na2S2O3 і насиченим розчином солі, сушили і концентрували. Флешхроматографія (з елююванням сумішшю гексан/етилацетат, 3:1 об/об) твердої речовини, що залишилася, давала сполука заголовка у ви гляді блідо-жовтої твердої речовини (14,8г). Стадія 2: 2-Аміно-5-хлор-3-(4-метилсульфотл)фенілпіридин Згідно з прийомами, описаними в прикладі 1, стадії 2 і 3, але із заміною 2-аміно-3-бром-5трифторметилпіридину 2-аміно-3-бром-5-хлорпіридином (5г) зі стадії 1 сполуку заголовка отримували у вигляді білої твердої речовини (5г) Стадія 3: 2-Бром-5-хлор-3-(4-метилсульфоніл)фенілпіридин До охолодженого (баня з льодом) розчину 2-аміно-5-хлор-3-(4-метилсульфоніл)фенілпіридину (5г) зі стадії 2 в суміші вода/концентруючий НСl (50мл/10мл) додавали нітрит натрію (1,5г) у воді (10мл). Отриману суміш перемішували при кімнатній температурі протягом 15год. і отриманий осадок видаляли фільтруванням і промивали послідовно водою і СС14. Після сушки на повітрі білу тверду речовину (4,8г) і РОВr3 (10,5г) в ДМФ (40мл) нагрівали при 100°С протягом 2 днів. Отриману суміш виливали в суміш води з льодом і екстрагували етилацетатом. Водну фазу підлужували 1N NaOH і екстрагували етилацетатом. Об'єднані органічні екстракти сушили і концентрували. Флеш-хроматографія (з елююванням сумішшю гексан/етилацетат, 1:1 об/об) залишку давала сполука заголовку у вигляді білої твердої речовини (2,7г). Стадія 4: 5-Хлор-2-(4-хлорфеніл)-3-(4-метилсульфоніл)феніл-піридин Згідно з прийомами, описаними в прикладі 3, але із заміною 2-хлор-3-(4-метилсульфоніл)феніл-5трифторметилпіридин 2-бром-5-хлор-3-(4-метилсульфо-нил)фенілпіридином (300 мг) зі стадії 3 отримували сполуку заголовку у вигляді білої твердої речовини, т. пл. 187-188°С (200мг). ПРИКЛАД 20 5-3-(4-метилсульфоніл)феніл-2-(2-піридиніл)піридин Стадія 1: 3-Бром-5-Хлор-2-гідроксипіридин Суміш 5-хлор-2-гідроксипіридину (100г) і брому (40,1мл) в оптовій кислоті (400мл) перемішували при кімнатній температурі протягом 1год. Суміш виливали в 3 л води і перемішували протягом 30хв. і потім фільтрували. Тверду речовину, що залишилася, промивали 2л холодної води, сушили на повітрі і потім випаровували разом з толуолом три рази і з бензолом два рази. Отриману таким чином тверду речовину (81г) використали в наступній реакції. Стадія 2: 2-Бензілокси-3-бром-5-хлорпіридин Суміш 3-бром-5-хлор-2-гідроксипіридину (81г), бензилброміду (52мл) і карбонату срібла (97г) в бензолі (1л) нагрівали при 70°С протягом 1год. Суміш охолоджували до кімнатної температури і потім фільтрували через шар целіту. Фільтрат концентрували і тверду речовину нестандартного білого кольору, що залишилася перекристалізовували з гексану з отриманням сполуки заголовка у вигляді білої твердої речовини (102г). Стадія 3: 2-Бензилокси-5-хлор-3-(4-метилсульфоніл)фенілпіридин Згідно з прийомами, описаними в прикладі 1, стадії 2 і 3, але із заміною 2-аміно-3-бром-5трифторметилпіридину 2-бензилокси-3-бром-5-хлорпіридином (81г) зі стадії 2 отримували сполуку заголовку у вигляді білої твердої речовини (72г). Стадія 4: 5-Хлор-2-гідрокси-3-(4-метилсульфоніл)фенілпіридин Розчин 2-бензилокси-5-хлор-3-(4-метилсульфоніл)фенілпіридин (72г) в трифтороцтовій кислоті (250мл) перемішували при 40°С протягом 15хв. і потім виливали в суміш води з льодом (~1л). Після перемішування протягом 10хв. білу тверду речовину фільтрували, промивали двічі ще 1л води і потім сушили на повітрі з отриманням сполуки заголовка. Стадія 5: 2,5-Дихлор-3-(4-метилсульфоніл)фенілпіридин Неочищений 5-Хлор-2-гідрокси-3-(4-метилсульфоніл)фенілпіридин зі стадії 4 нагрівали в запаяній посудині при 150°С з РОСІ3 (400мл) протягом 15год. Після охолоджування до кімнатної температури надлишок РОС13 видаляли відгонкою під вакуумом. Залишок розбавляли етилацетатом і водою і потім нейтралізували гідроксидом натрію (10N) до рН ~7. Органічний екстракт видаляли, промивали насиченим розчином солі і концентрували. Тверду речовину, що залишилася перекристалізовували з ефіру з отриманням сполуки заголовка у вигляді білої твердої речовини (61г). Стадія 6: Три-н-пропокси-2-піридилборонат літію Згідно з прийомами, описаними в прикладі 15, стадія 1, але із заміною 3-бромпіридину 2-бромпіридином отримували сполуку заголовка у вигляді твердої речовини нестандартного білого кольору (4,1г). Стадія 7: 5-3-(4-метилсульфоніл)феніл-2-(2-піридиніл)піридин Суміш 2,5-дихлор-3-(4-метилсульфоніл)фенілпіридину (1г), три-н-пропокси-2-піридилборонату літію (1,22г), карбонату натрію (5мл, 2М) і диброміда біс (трифенілфосфин) паладію 520мг) в толуолі (100мл), ізопропанолі (10мл) і воді (25мл) нагрівали із зворотним холодильником протягом 7год. Суміш охолоджували до кімнатної температури, розбавляли етилацетатом і фільтрували через шар целіту. Фільтрат екстрагували 6 N НСl і водний шар промивали етилацетатом. Водну фазу підлужували до рН ~10 за допомогою 10N гідроксиду натрію і потім екстрагували етилацетатом. Органічний екстракт промивали насиченим розчином солі, сушили і концентрували. Флеш-хроматографія (з елююванням сумішшю гексан/етилацетат, 1:1об/об) залишку давала сполуку заголовку у вигляді білої твердої речовини, т. пл. 134135°C (350мг). ПРИКЛАД ,21 5-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин Згідно з прийомами, описаними в прикладі 20, стадія 7, але із заміною три-н-пропокси-2піридинілборонату літію три-н-пропокси-3 -піридинілборонатом літію за прикладом 15, стадія 1, отримували сполуку заголовку у вигляді білої твердої речовини, т. пл. 166-169°С ПРИКЛАД 22 5-3-(4-метилсульфоніл)феніл-2-(4-піридиніл)піридин Стадія 1: Триметокси-4-піридинілборонат літію Згідно з прийомами, описаними в прикладі 15, стадія 1, але із заміною 3-бромпіридину 4-бромпіридином отримували сполуку заголовку. Неочищений продукт використали перед випаровуванням з н-пропанолу. Стадія 2: 5-3-(4-метилсульфоніл)феніл-2-(4-піридиніл)піридин Згідно з прийомами, описаними в прикладі 20, стадія 7, але із заміною три-н-пропокси-2піридинілборонату літію три-н-метокси-4-піридинілборонатом літію зі стадії 1 отримували сполуку заголовку у вигляді білої твердої речовини, т. пл. 187-188°С ПРИКЛАД 23 5-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридин Стадія 1: 2-метилпіридин-5-іловий складний ефір трифторметан-сульфонової кислоти До суміші 5-гідрокси-2-метилпіридину (2г) і піридин (1,9мл) в дихлорметані (100мл) при 0°С додавали ангідрид трифторметансульфонової кислоти (3,4мл). Суміш перемішували при цій температурі протягом 15хв. і потім при кімнатній температурі протягом 45 хв. Додавали ацетат амоній (25%) і органічні речовини видаляли і промивали 1 N НСl, сушили і концентрували. Сполуку заголовку отримували у вигляді бежової рідини (4г), яку використали як таку. Стадія 2: 2-Метил-5-триметилстанілпіридин Суміш 2-метилпіридин-5-илового складного ефіру трифторметан-сульфонової кислоти (2,1г), гексаметилдиолову (2,85г), хлориду лі тію (1,1г) і паладію-тетракис(трифенілфосфина) (190 мг) нагрівали із зворотним холодильником протягом 180хв. і потім охолоджували до кімнатної температури. Суміш фільтрували через шар целіту, промивали етилацетатом. Фільтрат промивали двічі. 5% фторидом калію, сушили і концентрували. Флеш-хроматографія (з елююванням сумішшю гексан/етилацетат, 6:1 про/про) залишку давала сполука заголовку у вигляді блідо-жовтого масла (1,3г). Стадія 3: 5-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридин Суміш 2,5-дихлор-3-(4-метилсульфоніл) фенілпіридину за прикладом 20, стадія 5 (750мг), 2-метил-5триметилстанілпіридину (1,3г) і паладій-тетракис(трифенілфосфина) (290мг)в NМР (10мл) нагрівали при 100°С протягом 15год. Суміш охолоджували до кімнатної температури, розбавляли етилацетатом і фільтрували через шар целіту. Фільтрат промивали водою, двічі 5% фторидом калію і потім екстрагували 1 N НСl. Водну фазу нейтралізували 10 N гідроксидом натрію і потім екстрагували етилацетатом. Органічний шар концентрували і залишок піддавали флеш-хроматографії (з елююванням етилацетатом) з отриманням сполуки заголовку у вигляді твердої речовини, т. пл. 127-128°С. ПРИКЛАД 43 Складний метиловий ефір 2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5-карбонової кислоти До розчину 2-(4-хлорфеніл)-5-3-(4-метилсульфоніл) фенілпіридину (приклад 14, 1,9г) в суміші третбутанол/вода (1:2, 60мл) при 90°С додавали порціями твердий перманганат калію (2,5г) протягом 2год. Отриману суміш перемішували при 90°3 протягом 15год. і охолоджували до кімнатної температури. Суміш фільтрували через шар целіту і фільтрат підкисляли до рН ~2 з допомогою 6 N НСl. Білий осадок фільтрували і потім частину цього продукту поглинали в суміші ТГФ/дихлорметан. До цього розчину додавали диазометан в ефірі до припинення виділення пухирців при доданні. Отриману суміш концентрували і піддавали флеш-хроматографії (з елююванням сумішшю гексан/етилацетат, 1:1 об/об). Сполуку заголовку отримували у вигляді білої твердої речовини, т. пл. 216-218°С. ПРИКЛАД 44 2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5-карбонова кислота До розчину складного метилового ефіру 2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5карбонової кислоти (140мг) в суміші етанол/вода (1:1, 10мл) додавали гідроксид літію (0,35мл, 3 N) і отриману суміш перемішували при кімнатній температурі протягом 45хв. Додавали насичений бікарбонат натрію і водний шар екстрагували етилацетатом. Водну фазу обробляли 3N НСд до встановлення рН ~2 і потім екстрагували хлороформом. Органічний шар концентрували з отриманням сполуки заголовку у вигляді білої твердої речовини, т. пл. >300°С (60мг). ПРИКЛАД 45 5-Ціано-2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридин До 2-(4-хлорфеніл)-3-(4-метилсульфоніл)фенілпіридиніл-5-карбонової кислоти (300мг) в дихлорметані (10мл) при нагріванні із зворотним холодильником додавали хлорсульфоніл-ізоцианат (0,4мл). Через 90хв. при нагріванні із зворотним холодильником суміш охолоджували до кімнатної температури і потім повільно додавали ДМФ (1,5мл). Через15хв. додавали воду і суміш екстрагували етилацетатом. Органічну фазу промивали водою, насиченим розчином солі, сушили і концентрували. Флеш-хроматографія (з елююванням сумішшю етилацетат/гексан, 2:1 об/об) залишку давала сполуку заголовку у вигляді білої твердої речовини (100мг). 1Н Я МР (300 MHz, CDCI3) d 3,06 (s, 3Н), 7,21-7,28 (m, 4H), 7,37 (d, 2H), 7,90 (d, 2H), 7,96 (d, 1Н), 8,94 (d, 1Н). ПРИКЛАД 46 Гідрометансульфонат-5-хлор-3-(4-метидсудьфоніл)феніл-2-(3-піридиніл) піридин Розчин 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин (5,31г, приклад 21) в етилацетаті (100мл) обробляли по краплях метансульфоновою кислотою (1мл) в етилацетаті (20мл). Отриманий осадок фільтрували і сушили у вакуумі з отриманням сполуки заголовку (6,4г) у вигляді білої твердої речовини. 1Н ЯМР(300МНr, CD3OD) d 2,68 (s, 3Н), 3,15 (s, 3Н), 7,60 (d, 2H), 7,96-8,00 (m, 3Н), 8,14 (d, 1H), 8,47 (dt, 1Н), 8,80 (d, 1H), 8,86 (m, 2H). ПРИКЛАД 47 Гідрохлорид 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридиніл) піридин Розчин 5-хлор-3-(4-метилсульфоніл)феніл-2-(3-піридиніл)піридин (2,05г, приклад 21) в гарячому етилацетаті (75мл) обробляли по краплях хлороводородной) кислотою (1,5мл, 4Μ в диоксані). Отриманий осадок фільтрували і сушили під вакуумом з отриманням сполуки заголовку (2,2г) у вигляді білої твердої речовини. 1Н ЯМР(300МHz, D MSO-d6)5 3,24 (s, 3Н), 7,59 (d, 2H), 7,80 (dd, 1H), 7,91 (d, 2Н), 8,15 (d, 1H), 8,22 (d, 1H), 8,69 (d, 1H), 8,77 (d, 1H), 8,90 (d, 1H). ПРИКЛАД 59 Гідрохлорид 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл) піридин Згідно з прийомами, описаними в прикладі 47, але із заміною 5-хлор-3-(4-метилсульфоніл)феніл-2-(3піридиніл)піридин 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-метил-5-піридиніл)піридин (за прикладом 23) отримували сполуку заголовку у вигляді білої твердої речовини. 1Н ЯМР (300 MHz, D MSO-d 6): d 2,68 (s, 3Н), 3,25 (s, 3Н), 7,61 (d, 2H), 7,70 (d, 1H), 7,92 (d, 2H), 8,07 (dd, 1H), 8,21 (d, 1H), 8,54 (d, 1H), 8,88 (d, 1H). ПРИКЛАД 71 5-3-(4-метилсульфонід)феніл-2-(2-етил-5-піридиніл)тридин Стадія 1: Три-н-пропоксі-2-етил-5-піридилборонат літію Згідно з прийомами, описаними в прикладі 15, стадія 1, але із заміною 3-бромпіридину 2-етил-5бромпіридином (Tilley and Zawoiski, J. Org. Chem. 1988, 53, 386) (4,6г) отримували сполуку заголовку у вигляді жовтої твердої речовини. Стадія 2: 5-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)піридин Суміш 2,5-дихлор-3-(4-метилсульфоніл)фенілпіридину (приклад 20, стадія 5) (5,6г), три-н-пропокси-2етил-5-піридилборонату літію (стадія 1) (4,0г), карбонату натрію (17мл, 2М) і диброміду біс (трифенілфосфин) паладію (420 мг) в толуолі (75мл), етанолі (75мл) і воді (15мл) нагрівали із зворотним холодильником протягом 7год. Суміш охолоджували до кімнатної температури, розбавляли етилацетатом і фільтрували через шар целіту. Фільтрат екстрагували 6 N НСl і водний шар промивали етилацетатом. Водну фазу підлужували до рН -10 за допомогою 10 N гідроксиду натрію і потім екстрагували етилацетатом. Органічний екстракт промивали насиченим розчином солі, сушили і концентрували. Флеш-хроматографія (з елююванням сумішшю гексан/етилацетат, 1:1 об/об) залишку давала сполуку заголовка у вигляді білої твердої речовини (4,0г). 1Н ЯМР (500MHz, ацетон-d6): d 1,21 (t, 3Н), 2,74 (q, 2Н), 3,14 (s, 3Н), 7,14 (d, 1H), 7,59 (m, 3Н), 7,93 (d, 2Н), 7,99 (d, 1Н), 8,44 (d, 1H), 8, 75 (d, 1H). ПРИКЛАД 71 - Альтернативний спосіб 5-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)піридин Стадія 1: 5-Бром-2-етилпіридин 280мл 5 N гідроксиду натрію (1,4мл, 2,09екв.) додавали до розчину 158г 2,5-дибромпіридину (0,67 моль, 1 екв.) в 1,4л ТГФ. До отриманого розчину додавали 700мл 1N триетилбору в ТГФ (0,70 моль, 1,04екв.), 195мг хлориду біс(ацетонітрил)паладію(П) (0,75мМоль, 0,0011екв.) і 414мг 1,1’біс(дифенілфосфино)фероцену (0,75мМоль, 0,0011екв.). Реакційну суміш повільно нагрівали до дуже слабкого кипіння із зворотним холодильником протягом 3 годин. Потім її охолоджували до 0°С і обробляли послідовно 140мл 5 N гідроксиду натрію (0,70 моль, 1,04екв.) і 53мл 30% пероксиду водні (0,70 моль, 1,05екв.) при такій швидкості, щоб температура не перевищувала 10°С. Суміш екстрагували е фіром і ефірні екстракти промивали гідроксидом натрію, водою, насиченим розчином солі і сушили над MgSO4. Потім ефірний розчин концентрували і коричневий залишок переганяли у вакуумі (40°С, 1 тор) з отриманням 112г сполуки заголовку у вигляді прозорого масла, злегка забрудненого початковим продуктом і 2,5диетилпіридином. Вихід -90%. 1Н ЯМР порівняємо з повідомленим J.W. Tilley and S. Zawoiski; J. Org. Chem. 1988, 53, 386-390. Цей продукт придатний для використання на наступній стадії без додаткового очищення. Стадія 2: Три-н-пропоксі-2-етил-5-піридилборонат літію Згідно з прийомами, описаними в прикладі 15, стадія 1, але із заміною 3-бромпіридину 5-бром-2етилпіридином зі стадії 1 отримували сполуку заголовку у вигляді жовтої твердої речовини. Стадія 3: 5-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)тридин Суміш 2,5-дихлор-3-(4-метилсульфоніл)фенілпіридину (приклад 20, стадія 5) (5,6г), три-н-пропокси-2етил-5-піридилборонату літію (стадія 2) (4,0г), карбонату натрію (17мл, 2М) і диброміду біс(трифенілфосфин)паладію (420мг) в толуолі (75мл), етанолі (75мл) і воді (15мл) нагрівали із зворотним холодильником протягом 7год. Суміш охолоджували до кімнатної температури, розбавляли етилацетатом і фільтрували через шар целіту. Фільтрат екстрагували 6 N НСl і водний шар промивали етилацетатом. Водну фазу підлужували до рН -10 за допомогою 10 N гідроксиду натрію і потім екстрагували етилацетатом. Органічний екстракт промивали насиченим розчином солі, сушили і концентрували. Флеш-хроматографія (з елююванням сумішшю гексан/етилацетат, 1:1 про/про) залишку давала сполуку заголовку у вигляді білої твердої речовини (4,0г). 1Н ЯМР (500MHz, ацетон-d6): d 1,21 (t, 3Н), 2,74 (q, 2Н), 3,14 (s, 3Н), 7,14 (d, 1Н), 7,59 (m, 3Н), 7,93 (d, 2H), 7,99 (d, 1H), 8,44 (d, 1H), 8, 75 (d, 1H). ПРИКЛАД 72 Гідрометансульфонат 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридинід) піридин Розчин 5-хлор-3-(4-метилсульфоніл)феніл-2-(2-етил-5-піридиніл)піридин (3,4 г, приклад 71) в етилацетаті (40мл) і ефірі (100мл) обробляли по краплях метансульфоновою кислотою (873мг) в ефірі (20мл). Отриманий осадок охолоджували до -20°С, фільтрували і сушили під вакуумом з отриманням сполуки заголовку (4,3г) у вигляді білої твердої речовини. 1Н Я МР (500MH z, CD3 OD) d 1,40 (t, 3Н), 2,68 (s, 3H), 3,07 (q, 2H), 3,15 (s, 3Н), 7,60 (d, 2H), 7,86 (d, 1H), 7,99 (d, 2H), 8,11 (d, 1H), 8,34 (m, 1H), 8,69 (d, 1H), 8,83 (d, 1H). ПРИКЛАД 73 5-3-(4-метилсульфоніл)феніл-2-(2-циклопропіл-5-піридиніл)піридин 1 Н ЯМР (ацетон-dб ) d 0,9 (m, 4Н), 2,0 (m, 1Н), 3,14 (s, 3Н), 7,15 (d, 1H), 7,50 (dd, 1H) 7,59 (m, 2H), 7,95 (m, 3H), 8,36 (d, 1H), 8,72 (d, 1H). ПРИКЛАД 74 5-3-(4-метилсульфоніл)феніл-2-(2,6-диметил-3-піридиніл)піридин Аналіз для: С Η Ν обчислено: 61,20 4,60 7,51 знайдено: 61,52 4,52 7,87
ДивитисяДодаткова інформація
Назва патенту англійськоюSubstituted pyridins as selective inhibitors of cyclogenase-2
Назва патенту російськоюЗамещенные пиридины в качестве выборочных ингибиторов циклооксигеназы-2
МПК / Мітки
МПК: A61P 19/02, A61K 31/4418, A61K 31/00, A61P 27/06, A61P 17/00, A61P 21/00, A61P 35/04, A61P 1/04, A61P 43/00, A61K 31/44, A61P 31/12, A61P 35/00, A61P 27/02, A61K 31/444, A61P 7/00, A61P 25/28, A61P 25/04, A61P 11/06, C07D 213/75, A61P 19/06, A61P 29/00, C07D 213/34, A61P 19/10, A61P 19/00, C07D 213/82, A61P 1/00, A61P 13/12, C07D 213/61, C07D 213/64, A61P 15/00, C07D 213/89, A61P 17/02, A61P 3/10, A61P 15/06, A61P 15/12, C07D 213/85, C07D 213/65, C07D 213/80
Мітки: композиція, піридини, фармацевтична, лікування, спосіб, заміщені, основі, захворювань, опосередкованих, циклооксигеназою
Код посилання
<a href="https://ua.patents.su/23-62935-zamishheni-piridini-farmacevtichna-kompoziciya-na-kh-osnovi-ta-sposib-likuvannya-oposeredkovanikh-ciklooksigenazoyu-zakhvoryuvan.html" target="_blank" rel="follow" title="База патентів України">Заміщені піридини, фармацевтична композиція на їх основі та спосіб лікування опосередкованих циклооксигеназою захворювань</a>
Похідні піридо[2,3-d] піримідину, фармацевтична композиція, спосіб лікування захворювань, опосередкованих клітинною проліферацією, та спосіб лікування раку, атеросклерозу, псоріазу або рестенозу

Номер патенту: 47427
Опубліковано: 15.07.2002
Автори: Доерті Еннет Меріен, Клачко Сільвестер, Пенек Роберт Лі, Босчеллі Дайяне Херріс, Бленклі Кліфтон Джон, Хембі Джеймс Маріно
МПК: A61K 31/505, A61P 43/00, A61P 35/00, A61P 9/10, C07D 471/04
Мітки: псоріазу, атеросклерозу, фармацевтична, рестенозу, захворювань, піридо[2,3-d, похідні, піримідину, спосіб, композиція, раку, проліферацією, клітинною, лікування, опосередкованих
Формула / Реферат:
1. Похідні піридо[2,3-d]піримідину загальної формули (І):, (I)де означає іміногрупу, N-ацил, кисень або сірку;R1 означає NR3R4, S(O)mR3, де m є 0, 1 або 2, або ОR3;R2, R3 та R4 незалежно означають водень, (CH2)nPh, де Ph є фенілом, незаміщеним або заміщеним 1-3 залишками, що вибирають з групи, що містить галоген, гідроксил, карбоксил, алкіл з 1-6 атомами вуглецю, незаміщений або заміщений гідроксилом,...
Спосіб інгібування активності impdh, похідні сечовини, фармацевтична композиція для лікування або профілактики impdн-опосередкованих захворювань (варіанти), спосіб лікування або профілактики impdн-опосередкован

Номер патенту: 61074
Опубліковано: 17.11.2003
Автори: Ронкін Стівен М., Саундерс Джеффрі О., Франк Катаріна А., Бадія Майкл.К, Армістед Девід М., Бетайл Ренді С., Новак Перрі М., Беміс Гай В.
МПК: A61P 35/00, A61P 29/00, A61K 31/18, C07D 413/12, C07D 263/32, C07C 275/42, A61P 43/00, A61K 31/404, A61P 31/12, A61K 31/422, A61K 31/17, A61P 9/14, A61K 31/421, A61P 37/06, A61K 31/42
Мітки: варіанти, спосіб, композиція, impdн-опосередкованих, impdh, лікування, активності, фармацевтична, інгібування, захворювань, impdн-опосередкован, похідні, сечовини, профілактики
Формула / Реферат:
1. Спосіб інгібування активності IMPDH у ссавця, при якому вводять зазначеному ссавцеві сполуку формули:,у якій:В означає насичену, ненасичену або частково насичену моноциклічну або біциклічну кільцеву систему, яка не обов'язково має до 4 гетероатомів, вибраних із N, O або S, яку вибирають із сполук, виражених формулами:,де кожний Х означає кількість атомів водню, необхідних для досягнення потрібної...
Заміщені похідні 4-бісфеніл-4-гідроксимасляної кислоти як інгібітори матричної металопротеази, спосіб їх одержання, фармацевтична композиція та спосіб лікування на їх основі

Номер патенту: 57047
Опубліковано: 16.06.2003
Автори: Клуендер Гарольд С.Є., Зад'юра Лайза Марія, Брубейкер Уільям Фредерік, Бйорге Сьюзен М.
МПК: A61P 1/02, A61K 31/4035, A61P 13/00, A61P 29/00, C07C 235/42, C07C 231/00, A61P 27/02, A61P 19/02, A61P 35/04, A61P 19/08, A61P 43/00, C07C 235/84, A61P 7/02, A61P 9/00, C07C 323/56, C07C 323/61, C07D 209/48, C07C 59/00, C07C 51/347, C07C 323/62
Мітки: матричної, інгібітори, одержання, композиція, заміщені, металопротеази, кислоти, фармацевтична, похідні, лікування, спосіб, 4-бісфеніл-4-гідроксимасляної, основі
Формула / Реферат:
1. Сполука, яка має інгібуючу активність у відношенні до матричної металопротеази загальної формули,(I)деΤ є фармацевтично придатною замінною групою;х дорівнює 0, 1 або 2;m дорівнює 0 або цілому числу від 1 до 4;n дорівнює 0 або 1; іабоА і G обидва є СН2,абоА є хімічним зв'язком і G є...
Трициклічні бензазепіни як судинозвужувальні антагоністи, спосіб їх одержання, фармацевтична композиція та спосіб лікування захворювань

Номер патенту: 54383
Опубліковано: 17.03.2003
Автори: Сум Фук-Вах, Дусза Джон Пол, Венкатесан Аранапакам Мудумбай, Олбрайт Джей Дональд
МПК: A61P 9/00, A61P 9/12, C07D 487/04, A61P 1/16, C07D 471/14, A61K 31/495, C07D 471/04, A61P 7/08, C07D 487/14, A61K 31/55
Мітки: композиція, трициклічні, захворювань, судинозвужувальні, одержання, бензазепіни, спосіб, фармацевтична, лікування, антагоністи
Формула / Реферат:
1. Трициклічні бензазепіни загальної формули (І): (I),де Y означає фрагмент, вибраний з: -(СН2)n-, де n означає ціле число від 0 до 2,>СН-нижчий алкіл (C1-С3) і -С(O)-;А-В означає фрагмент, вибраний з -(CH2)m-NR3- і -NR3-(CH2)m-,де m означає ціле число від 1 до 2 за умови, що, коли Y означає -(СН2)n-, і n означає 2, m може бути 0, і коли n означає 0, то m може також означати 3, за умови, що, коли Y...
Заміщені гідроксилом азетидинони, придатні як гіпохолестеринемічний засіб, фармацевтична композиція на їх основі та спосіб лікування або профілактики

Номер патенту: 41948
Опубліковано: 15.10.2001
Автори: Дугар Сандіп, Мекіттрік Брайєн А., Кладер Джон В., Розенблум Стюарт Б, Бьорнетт Дуане А.
МПК: A61K 31/397, A61P 9/10, A61K 31/395, C07D 205/00
Мітки: профілактики, придатні, гіпохолестеринемічний, гідроксилом, фармацевтична, спосіб, азетидинони, лікування, композиція, заміщені, засіб, основі
Формула / Реферат:
1. Замещенные гидроксилом азетидиноны общей формулыили их фармацевтически приемлемые соли, где:Аr1 и Аr2 независимо друг от друга выбраны из группы, включающей арили замещенный остатком R4 арил,Аr3 - арил или замещенный остатком R5 арил,X, Y и Z независимо друг от друга выбраны из группы, включающей-СН2-, -СН(низший алкил)-, -С(ди-низший алкил)-,R и R2 независимо друг от друга выбраны из...
Наступний патент: N6 гетероциклічні заміщені похідні аденозину
Випадковий патент: Інгібітори мітозу для інтенсифікації процесу апоптозу при терапії