Фенілвмісні n-ацильні похідні амінів і амінокислот, спосіб їх одержання, фармацевтична композиція і їх застосування
Номер патенту: 88688
Опубліковано: 10.11.2009
Автори: Кромова Татьяна Алєксандровна, Ковальова Віолєтта Лєонідовна, Нєбольсін Владімір Євгєньєвіч, Желтухіна Галіна Алєксандровна








Формула / Реферат
1. Фенілвмісні N-ацильні похідні біогенних амінів загальної формули:
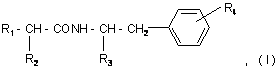
де R1 представляє
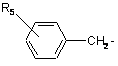
або
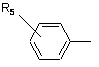 ,
,
де R5 представляє водень або гідроксильну групу;
R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6,
де R6 представляє С1-С6алкіл або
 ,
,
де R7 представляє водень або гідроксильну групу,
R4 представляє водень, гідроксильну групу;
за умови, що сполука загальної формули І не є фенілацетилтираміном,
3-(п-гідроксифеніл)пропіонілфенілетиламіном,
3-(п-гідроксифеніл)пропіонілтираміном,
3-фенілпропіонілфенілетиламіном,
3-фенілпропіонілтираміном,
3-(п-гідроксифеніл)пропіонілфенілаланіну метиловим ефіром,
3-(п-гідроксифеніл)пропіонілтирозином,
3-фенілпропіонілтирозином, фенілацетилтирозином,
3-(п-гідроксифеніл)пропіонілфенілаланіном,
3-фенілпропіонілфенілаланіном, фенілацетилфенілаланіном,
3-(п-гідроксифеніл)пропіонілтирозину метиловим ефіром,
3-фенілпропіонілтирозину метиловим ефіром, фенілацетилтирозину метиловим ефіром, фенілацетилфенілетиламіном,
3-фенілпропіонілфенілаланіну метиловим ефіром, фенілацетилфенілаланіну метиловим ефіром,
3-(п-гідроксифеніл)пропіонілтирозину бензиловим ефіром;
або їх фармацевтично прийнятні солі.
2. Сполука за п. 1, у якій R3 представляє -СООН, -СООСН3.
3. Сполука за п. 1, вибрана з
n-гідроксифенілацетилтирозину,
n-гідроксифенілацетилфенілаланіну,
n-гідроксифенілацетилтирозину метилового ефіру,
n-гідроксифенілацетилфенілаланіну метилового ефіру,
3-фенілпропіонілтирозину бензилового ефіру,
n-гідроксифенілацетилтирозину бензилового ефіру,
n-гідроксифенілацетилфенілаланіну бензилового ефіру,
N-ацетилтирозилфенілетиламіну,
N-ацетилтирозилтираміну,
n-гідроксифенілацетилтираміну,
n-гідроксифенілацетилфенілетиламіну,
або їх фармацевтично прийнятних солей.
4. Сполука за кожним з пп. 1-3, яка має інгібуючу циклооксигеназу активність.
5. Сполука за п. 4, яка має анальгетичну, протизапальну, спазмолітичну, антигіпоксичну, антидепресантну і протипаркінсонічну дію.
6. Сполуки за п. 4, здатні потенціювати дію інших анальгетиків, зокрема трамалу й анальгіну.
7. Спосіб одержання сполук загальної формули І
де R1 представляє
або
,
де R5 представляє водень або гідроксильну групу;
R2 представляє водень або аміногрупу, необовязково заміщену групою СН3(СН2)mСО-, де m=0-4;
R3 представляє водень, -СООН, -COOR6,
де R6 представляє С1-С6алкіл або
,
де R7 представляє водень або гідроксильну групу,
R4 представляє водень, гідроксильну групу;
в якому здійснюють активацію карбоксильної групи сполуки загальної формули
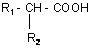 ,
,
взаємодією з дифенілфосфорилазидом і триетиламіном в органічному розчиннику, при охолодженні з наступною взаємодією з аміносполукою загальної формули
 ,
,
де R1-R4 приймають значення, визначені для сполук загальної формули І.
8. Спосіб за п. 6, у якому використовують 1-1,2 еквіваленти дифенілфосфорилазиду й триетиламіну.
9. Спосіб за п. 6 або 7, у якому як амінопохідні використовують ефіри тирозину або фенілаланіну.
10. Спосіб за будь-яким з пп. 6-9, у якому як органічний розчинник використовують N,N-диметилформамід або етилацетат.
11. Спосіб за будь-яким з пп. 6-9, який здійснюють в інтервалі температур від -25 °С до 0 °С.
12. Спосіб одержання сполук за п. 1 або їх фармацевтично прийнятних солей, в якому здійснюють перетворення п-гідроксифенілоцтової кислоти, фенілоцтової кислоти або N-заміщеного тирозину загальної формули
на активований N-оксисукцинімідний ефір загальної формули
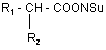
N,N'-дициклогексилкарбодіімідним методом, з наступною взаємодією активованого N-оксисукцинімідного ефіру з амінопохідним загальної формули
,
де R1-R4 приймають значення, визначені для сполук загальної формули І у п. 1.
13. Спосіб за п. 12, у якому як амінопохідні використовують ефіри тирозину або фенілаланіну.
14. Фармацевтична композиція, яка містить як активний агент ефективну кількість сполуки загальної формули (І)
де R1 представляє
або
,
де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6,
де R6 представляє С1-С6алкіл або
,
а R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; або її фармацевтично прийнятної солі і фармацевтично прийнятний носій.
15. Фармацевтична композиція за п. 14, яка має здатність інгібувати циклооксигеназу, анальгетичні, протизапальні, спазмолітичні, антигіпоксичні, антидепресантні й протипаркінсонічні властивості, а також здатність потенціювати дію інших анальгетиків.
16. Засіб, що має анальгетичні, протизапальні, спазмолітичні, антигіпоксичні, антидепресантні й протипаркінсонічні властивості, а також здатність потенціювати дію інших анальгетиків, який містить сполуку загальної формули (І)
де R1 представляє
або
,
де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6,
де R6 представляє С1-С6алкіл або
,
де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу;
або її фармацевтично прийнятні солі.
17. Застосування сполук загальної формули (І)
де R1 представляє
або
,
де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6,
де R6 представляє С1-С6алкіл або
,
де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; або їх фармацевтично прийнятних солей, для одержання лікарського засобу, що має здатність інгібувати циклооксигеназу.
18. Застосування сполук за п. 16 як анальгетичних, протизапальних, спазмолітичних, антигіпоксичних, антидепресантних і протипаркінсонічних засобів, а також засобів, що мають здатність потенціювати дію інших анальгетиків.
19. Спосіб лікування больових синдромів різного генезу, запальних і запально-дегенеративних захворювань суглобів і сполучної тканини, а також кістково-м'язової системи,інших захворювань, що супроводжуються запаленням, спазмами, депресією, гіпоксією, а також явищами Паркінсонізму, який включає введення ссавцеві ефективної кількості сполуки загальної формули (І)
де R1 представляє
або
,
де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6,
де R6 представляє С1-С6алкіл або
,
де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу;
або її фармацевтично прийнятної солі.
20. Спосіб за п. 19 лікування післяопераційного болю, посттравматичного болю, а також больових синдромів гінекологічної, неврологічної, онкологічної, стоматологічної природи, ревматоїдного артриту, артропатії, хвороби Бехтерєва, неспецифічних спондилоартритів, подагричного артриту, остеоартрозу, позасуглобового ревматизму й тромбофлебіту, а також емоційно-стресових станів і порушень, викликуваних спазмами, гіпоксією й супровідною хворобою Паркінсона.
21. Спосіб за п. 19 або 20, у якому сполуку загальної формули (І) вводять у сполученні з іншими анальгетиками.
Текст
1. Фенілвмісні N-ацильні похідні біогенних амінів загальної формули: R4 R1 - CH - CONH - CH - CH2 R2 R3 , (I) де R1 представляє R5 CH2 або R5 , де R5 представляє водень або гідроксильну групу; 4 R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6, де R6 представляє С1-С6алкіл або R7 - CH2 , де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; за умови, що сполука загальної формули І не є фенілацетилтираміном, 3-(п-гідроксифеніл)пропіонілфенілетиламіном, 3-(п-гідроксифеніл)пропіонілтираміном, 3-фенілпропіонілфенілетиламіном, 3-фенілпропіонілтираміном, 3-(п-гідроксифеніл)пропіонілфенілаланіну метиловим ефіром, 3-(п-гідроксифеніл)пропіонілтирозином, 3-фенілпропіонілтирозином, фенілацетилтирозином, 3-(п-гідроксифеніл)пропіонілфенілаланіном, 3-фенілпропіонілфенілаланіном, фенілацетилфенілаланіном, 3-(п-гідроксифеніл)пропіонілтирозину метиловим ефіром, 3-фенілпропіонілтирозину метиловим ефіром, фенілацетилтирозину метиловим ефіром, фенілацетилфенілетиламіном, 3-фенілпропіонілфенілаланіну метиловим ефіром, фенілацетилфенілаланіну метиловим ефіром, 3-(п-гідроксифеніл)пропіонілтирозину бензиловим ефіром; або їх фармацевтично прийнятні солі. 2. Сполука за п. 1, у якій R3 представляє -СООН, СООСН3. 3. Сполука за п. 1, вибрана з n-гідроксифенілацетилтирозину, n-гідроксифенілацетилфенілаланіну, n-гідроксифенілацетилтирозину метилового ефіру, n-гідроксифенілацетилфенілаланіну метилового ефіру, 3-фенілпропіонілтирозину бензилового ефіру, n-гідроксифенілацетилтирозину бензилового ефіру, n-гідроксифенілацетилфенілаланіну бензилового ефіру, N-ацетилтирозилфенілетиламіну, N-ацетилтирозилтираміну, n-гідроксифенілацетилтираміну, n-гідроксифенілацетилфенілетиламіну, або їх фармацевтично прийнятних солей. 4. Сполука за кожним з пп. 1-3, яка має інгібуючу циклооксигеназу активність. 5. Сполука за п. 4, яка має анальгетичну, протизапальну, спазмолітичну, антигіпоксичну, антидепресантну і протипаркінсонічну дію. 6. Сполуки за п. 4, здатні потенціювати дію інших анальгетиків, зокрема трамалу й анальгіну. 7. Спосіб одержання сполук загальної формули І R4 R1 - CH - CONH - CH - CH2 R2 R3 , (I) 5 де R1 представляє R5 CH2 або R5 88688 6 N,N'-дициклогексилкарбодіімідним методом, з наступною взаємодією активованого Nоксисукцинімідного ефіру з амінопохідним загальної формули R4 NH2 - CH - CH2 R3 , де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необовязково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6, де R6 представляє С1-С6алкіл або R7 , де R1-R4 приймають значення, визначені для сполук загальної формули І у п. 1. 13. Спосіб за п. 12, у якому як амінопохідні використовують ефіри тирозину або фенілаланіну. 14. Фармацевтична композиція, яка містить як активний агент ефективну кількість сполуки загальної формули (І) R4 R1 - CH - CONH - CH - CH2 - CH2 , де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; в якому здійснюють активацію карбоксильної групи сполуки загальної формули R1 - CH - COOH R2 , взаємодією з дифенілфосфорилазидом і триетиламіном в органічному розчиннику, при охолодженні з наступною взаємодією з аміносполукою загальної формули R4 NH2 - CH - CH2 R3 , де R1-R4 приймають значення, визначені для сполук загальної формули І. 8. Спосіб за п. 6, у якому використовують 1-1,2 еквіваленти дифенілфосфорилазиду й триетиламіну. 9. Спосіб за п. 6 або 7, у якому як амінопохідні використовують ефіри тирозину або фенілаланіну. 10. Спосіб за будь-яким з пп. 6-9, у якому як органічний розчинник використовують N,Nдиметилформамід або етилацетат. 11. Спосіб за будь-яким з пп. 6-9, який здійснюють в інтервалі температур від -25 °С до 0 °С. 12. Спосіб одержання сполук за п. 1 або їх фармацевтично прийнятних солей, в якому здійснюють перетворення п-гідроксифенілоцтової кислоти, фенілоцтової кислоти або N-заміщеного тирозину загальної формули R1 - CH - COOH R2 на активований N-оксисукцинімідний ефір загальної формули R1 - CH - COONSu R2 R2 R3 , (I) де R1 представляє R5 CH2 або R5 , де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6, де R6 представляє С1-С6алкіл або R7 - CH2 , а R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; або її фармацевтично прийнятної солі і фармацевтично прийнятний носій. 15. Фармацевтична композиція за п. 14, яка має здатність інгібувати циклооксигеназу, анальгетичні, протизапальні, спазмолітичні, антигіпоксичні, антидепресантні й протипаркінсонічні властивості, а також здатність потенціювати дію інших анальгетиків. 16. Засіб, що має анальгетичні, протизапальні, спазмолітичні, антигіпоксичні, антидепресантні й протипаркінсонічні властивості, а також здатність потенціювати дію інших анальгетиків, який містить сполуку загальної формули (І) R4 R1 - CH - CONH - CH - CH2 R2 R3 де R1 представляє , (I) 7 88688 R5 CH2 або R5 , де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6, де R6 представляє С1-С6алкіл або R7 - CH2 , де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; або її фармацевтично прийнятні солі. 17. Застосування сполук загальної формули (І) R4 R1 - CH - CONH - CH - CH2 R2 R3 8 18. Застосування сполук за п. 16 як анальгетичних, протизапальних, спазмолітичних, антигіпоксичних, антидепресантних і протипаркінсонічних засобів, а також засобів, що мають здатність потенціювати дію інших анальгетиків. 19. Спосіб лікування больових синдромів різного генезу, запальних і запально-дегенеративних захворювань суглобів і сполучної тканини, а також кістково-м'язової системи, інших захворювань, що супроводжуються запаленням, спазмами, депресією, гіпоксією, а також явищами Паркінсонізму, який включає введення ссавцеві ефективної кількості сполуки загальної формули (І) R4 R1 - CH - CONH - CH - CH2 R2 R3 , (I) де R1 представляє R5 CH2 або R5 , (I) де R1 представляє R5 CH2 або R5 , де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6, де R6 представляє С1-С6алкіл або R7 - CH2 , де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; або їх фармацевтично прийнятних солей, для одержання лікарського засобу, що має здатність інгібувати циклооксигеназу. Даний винахід належить до галузі біоорганічної хімії й стосується нових сполук - фенілвмісних N-ацильних похідних біогенних амінів, а також способу синтезу нових і відомих сполук, їхнього застосування в медицині як потенційних анальге , де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -COOR6, де R6 представляє С1-С6алкіл або R7 - CH2 , де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; або її фармацевтично прийнятної солі. 20. Спосіб за п. 19 лікування післяопераційного болю, посттравматичного болю, а також больових синдромів гінекологічної, неврологічної, онкологічної, стоматологічної природи, ревматоїдного артриту, артропатії, хвороби Бехтерєва, неспецифічних спондилоартритів, подагричного артриту, остеоартрозу, позасуглобового ревматизму й тромбофлебіту, а також емоційно-стресових станів і порушень, викликуваних спазмами, гіпоксією й супровідною хворобою Паркінсона. 21. Спосіб за п. 19 або 20, у якому сполуку загальної формули (І) вводять у сполученні з іншими анальгетиками. тичних, протизапальних, спазмолітичних і антигіпоксичних засобів, а також засобів, що мають антидепресантну, протипаркінсонічну дію й здатність потенціювати дію інших анальгетиків. 9 У публікації міжнародної заявки WO 97/23202 розкриті фенілвмісні N-ацильні похідні амінів загальної формули (XV) яка, серед інших, включає 3-(пгідроксифеніл)пропіоніл феніл етил амін, 3-(пгідроксифеніл)пропіонілтирамін і 3фенілпропіонілфенілетиламін (сполуки IX, X, XI даного винаходу, відповідно), як проміжні сполуки, а також синтез сполук, загальної формули (XV) і їхнє застосування як селективних лігандів підтипів NMDA рецепторів, використовуваних для лікування хронічного болю, мігреневого головного болю, а також анестетиків. Однак, у зазначеній публікації не описані й не охарактеризовані конкретні структури, що відповідають сполукам X і XI даного винаходу й відсутні які-небудь дані, що підтверджують заявлений вид активності, а сполука IX як проміжна сполука і її синтез розкриті лише в способі одержання інших похідних амінів. Сполуки IX, X і XI даного винаходу також описані в більш ранніх публікаціях, що стали загальнодоступними до дати пріоритету вищевказаної міжнародної заявки WO 97/23202, для використання за іншим призначенням. 3-(п-Гідроксифеніл)пропіонілфенілетиламін (IX) розкритий в Jacobson K.A., Kirk K.L. New highperformance liguid chromatographic procedure for the detection and quantification of β-phenyletylamine. // J. Chromatography. 1987. V.415. P.124-128); 3-(пгідроксифеніл)пропіонілтирамін (X) - в R.B. Herbert, A.E.Kattah. The biosynthesis of Sceletium alkaloids in Sceletium subvelutinum L. Bolus. // Tetrahedron. 1990. V.46. №20. P.7105-7118 і (3фенілпропіонілфенілетиламін (XI) - в Maldonado Ε., Hernandez Ε., Ortega A. Amides, coumarine and other constituents from simsia cronquistii. II Phytochem. 1992. P.1413-1414. У публікації міжнародної заявки WO 97/23202 зазначена можливість використання сполук загальної формули (XV) для запобігання деяким специфічним видам болю, таким як мігреневий головний біль, хронічний біль, а також застосування їх для анестезії, обумовлена здатністю даних сполук проявляти дію селективних лігандів підтипів NMDA рецепторів. Однак, в WO 97/23202 немає якихнебудь даних, що підтверджують заявлену активність зазначеної групи сполук і, отже, можливість застосування даних сполук за зазначеним призначенням, на конкретних моделях на тваринах in vivo, і, таким чином, висновки про можливі фармакологічні ефекти засновані винятково на твердженні про те, що всі розкриті в зазначеній міжнародній заявці сполуки є селективними лігандами підтипів NMDA-рецепторів. У публікації міжнародної заявки WO 97/23202 описаний спосіб синтезу 3-(пгідроксифеніл)пропіонілфенілетиламіну (IX) з використанням 1-гідроксибензотриазолу в присутності Ν,Ν'-дициклогексилкарбодііміду (DCC). Не описаний спосіб виділення й очищення даної сполуки, 88688 10 з фізико-хімічних констант наведена температура плавлення й дані 1H-ЯМР-спектроскопії. У статті Jacobson K.A., Kirk K.L. New highperformance liguid chromatographic procedure for the detection and quantification of β-phenyletylamine. // J. Chromatography. 1987. V.415.P.124-128 розкритий синтез 3-(пгідроксифеніл)пропіонілфенілетиламіну (IX) із застосуванням модифікованого Nоксисукцинімідного ефіру 3-(пгідроксифеніл)пропіонової кислоти. Реакцію проводять у суміші метанол- 1М Na2HPO4, pH 8 (1:1), використовуючи сульфосукцинімідил-3-(пгідроксифеніл)пропіонат (сульфатований реагент Bolton-Hunte). Одержаний продукт охарактеризований тільки температурою плавлення. Відповідно до даної статті, одержаний 3-(пгідроксифеніл)пропіонілфенілетиламін використовують як внутрішній стандарт в електрохімічному детекторі при кількісному визначенні рівня ендогенного фенілетиламіну в біологічних рідинах методом ВЕРХ. У статті Herbert R.B., Kattah A.E. The biosynthesis of Sceletium alkaloids in Sceletium subvelutinum L. Bolus. // Tetrahedron. 1990. V.46. №20. P.7105-7118 описане застосування 3-(пгідроксифеніл)пропіонілтираміну (X) як проміжного продукту у синтезі алкалоїдів Sceletium subvelutinum, а також спосіб його синтезу методом DCC. Недоліком даного способу є необхідність застосування для очищення цільового продукту колонкової хроматографії, його порівняно невисокий вихід - близько 48%. В статті Maldonado Ε., Hernandez E., Ortega A. Amides, coumarine and other constituents from simsia cronquistii. // Phytochem. 1992. P.1413-1414 описане виділення 3фенілпропіонілфенілетиламіну (XI) з наземної частини рослин Simsia cronquistii і представлені дані мас-спектрометрії, 1Н-ЯМР-спектроскопії, а також температура плавлення. Даних з біологічної активності не наведено. Синтез сполуки XI із застосуванням конденсувального агента 4-(4,6-диметокси-1,3,5-триазин-2іл)-4-метилморфоліну хлориду (DMT-MM) описаний в Kunishima Μ., Kawachi C, Hioki К. et al. Formation of carboxamides by direct condensation of carboxylic acids and amines in alcohols using a new alcohol- and water-soluble condensing agent: DMTMM. // Tetrahedron. 2001. V.57. №8. P.1551-1558. Недоліком даного способу синтезу є утворення побічного продукту й необхідність застосування препаративної тонкошарової хроматографії для очищення цільового продукту, що ускладнює процес і повинно неминучо приводити до зниження виходу. Незважаючи на це, вказується високий вихід продукту (XI), що становить 98%. Сполука XI була синтезована з метою вивчення застосовності нового конденсувального агента DMT-MM. Синтез похідних амінокислот тирозину й фенілаланіну 3-(п-гідроксифеніл)пропіонілтирозину, фенілпропіонілтирозину, фенілацетилтирозину, фенілпропіонілфенілаланіну й фенілпропіонілтирозину метилового ефіру (сполуки XIV, XV, XVI XVIII і XXI даного винаходу, відповідно) і вивчення 11 їх інгібувальної дії на нейрон TAN, ідентифікований у ганглії равлика Achatina fulica ferussac, описані в статтях Takeuchi Η., Ariyoshi Y., Effects of Nbeta-phenylpropionyl-L-tyrosine and its derivatives on the excitability of an identifiable giant neuron of Achatina fulica ferussac. // Comparative biochemistry and physiology. C: Comparative pharmacology. 1982. V.72. №2. P.225-229 і Y. Ariyoshi. H. Takeuchi. Structure-activity relationships of Ν-βphenylpropionyl-L-tyrosine and its derivatives on the inhibition of an identifiable giant neurone of an identifiable giant neurone of an African giant snail. // Br.J. Pharmacol. 1982. V.77. P.631-639. У статті Y.Ariyoshi. Η. Takeuchi. Structure-activity relationships of Ν-β-phenylpropionyl-L-tyrosine and its derivatives on the inhibition of an identifiable giant neurone of an identifiable giant neurone of an African giant snail. // Br.J. Pharmacol. 1982. V.77. P.631-639. Описано типову методику синтезу сполук XIV, XV, XVI, XVIII, XXI методом активованих Nоксисукцинімідних ефірів з використанням як амінопохідного метилового ефіру тирозину, з наступним його омиленням (для сполук XIV, XV, XVI, XVIII), але фізико-хімічні константи й виходи для зазначенихсполук не наведені. Крім того, синтез фенілацетилтирозину (XV) з високим виходом (94%) з використанням 1-гідроксибензотриазолу й етил-3(3-диметиламіно)пропілкарбодііміду, з використанням як вихідних сполук етилового ефіру тирозину й фенілпропіонової кислоти, з наступним омиленням етилового ефіру описаний в Tangpasuthadol V., Pendharkar S.M., Kohn J. Hydrolytic degradation of tyrosine-derived polycarbonates, a class of new biomaterials. Part I: Study of model compounds. // Biomaterials. 2000. V.21. №23. P.2371-2378. Наведені дані 1Н-ЯМРспектроскопії й температура плавлення. Синтез фенілпропіонілфенілаланіну (XVIII) хлорангідридним методом у присутності КОН розкритий в Lustig N., Spiegelstein-Klarfeld H., Schneider Ε., Lichtenstein N. Phenylacetyl and phenylpropionyl amino acids. Their inhibitory effect on glutamine synthetase and their resistance to acylase. I. // Israel Journal of Chemistry. 1974. V.12. №3. P.757-763. Наведено температуру плавлення й елементний аналіз. Синтез був проведений для вивчення ступеня інгібування сполукою XVIII глутамінсинтетази. Фенілпропіонілтирозину метиловий ефір (XXI) згадується в патенті Японії JP 57193437 (приклад 4), де його синтез здійснений методом активованих N-оксисукцинімідних ефірів. Синтез фенілацетилфенілаланіну (XIX), подібний до синтезу сполуки XVIII, з використанням хлорангідриду фенілоцтової кислоти розкритий в Chen H.M., Hsu M.S., Huang L.J., et al. Effect of Nphenylacetyl L-amino acids on the differentiation of HL-60 cells // Chinese Pharmaceutical Journal. 2001. V.53. №3. P.157-167. Наведено фізико-хімічні характеристики цільової сполуки: температуру плавлення, дані 1Н-ЯМР- і ІЧ-спектроскопії, масспектрометрії. Було встановлено, що фенілацетилфенілаланін (XIX) є індуктором диференціювання клітин. 88688 12 3-(п-Гідроксифеніл)пропіонілтирозину метиловий ефір (XX) згадується в публікації міжнародної заявки WO 99/52962, однак методика синтезу й фізико-хімічні характеристики не наведені. Сполука (XX) синтезована з метою її використання як мономера для одержання біорозкладаних полімерів, сумісних із тканинами. Природна сполука, виділена із симбіотичної бактерії Xenorhabdus nematophilus, фенілацетилфенілетиламін (XXIII) була синтезована хлорангідридним методом і охарактеризована фізикохімічними даними 1Н-ЯМР-, 13С-ЯМР- і ІЧспектроскопії, мас-спектрометрії, температурою плавлення в публікації міжнародної заявки WO 01/49656. Досліджена протипухлинна активність сполуки XXIII in vitro. Загальна формула сполук, розкритих у публікації міжнародної заявки WO 01/49656, охоплює також і інші сполуки даного винаходу: пгідроксифенілацетилтирамін, пгідроксифенілацетилфенілетиламін і фенілацетилтирамін (сполуки VII, VIII і VI даного винаходу, відповідно). Однак, дана публікація не розкриває ні конкретних структурних формул зазначених сполук, ні методик їхнього синтезу, ні фізикохімічних констант, ні даних з біологічної активності. Фенілпропіонілтирамін (XII) згадується в статті Takeuchi Hiroshi; Tamura Hiroko. The effects of aromatic amino acid derivatives on the excitability of an identifiable giant neuron of the African giant snail (Achatina fulica Ferassac). // British Journal of Pharmacology. 1980. V.69. №1. P.29-34, але без опису його синтезу, фізико-хімічних характеристик і призначення. У статті Garrett С.Е., Jiang X., Prasad К., Repic О. New observations on peptide bond formation using CDMT. // Tetrahedron Letters. 2002. V.43. №23. P.4161-4165 розкритий фенілпропіонілфенілаланіну метиловий ефір (XXIV) і спосіб його синтезу із застосуванням нового конденсувального агента 2хлор-4,6-диметокси-1,3,5-триазину (CDMT) у присутності N-метилморфоліну. Однак, не наведено ні фізико-хімічних характеристик вказаної сполуки, ні даних з її активності, лише повідомляється, що даний спосіб синтезу має переваги: синтез в одну стадію й виділення продукту шляхом висаджування водою приводять до хроматографічно чистого продукту з високим виходом 90%. У статті Peric M., Vercek В., Petric A. ωDiazoacetophenones as reagents for a mild and selective protection of an amino group. // Acta Chimica Slovenica. 1996. V.43. №2. P.163-173 описаний синтез фенілацетилтирозину метилового ефіру (XXII), проміжної сполуки для синтезу пептидів, конденсацією фенілоцтової кислоти з метиловим ефіром тирозину через утворення діазокетону. Для очищення сполуки XXII обов'язково використовували колонкову хроматографію. Наведено температуру плавлення, дані !Н-ЯМР спектроскопії й елементного аналізу. Фенілацетилфенілаланіну метиловий ефір (XXV), відповідно до Votano J. R., Altaian J., Wilchek M., Potential use of biaromatic Lphenylalanyl derivatives as therapeutic agents in the treatment of sickle cell disease. // Proceedings of the 13 National Academy of Sciences of the United States of America. 1984. V.81. №10. P.3190-3194, був синтезований методом активованих Nоксисукцинімідних ефірів, з наступним очищенням колонковою хроматографією. Фізико-хімічних констант для даної сполуки не наведено. У зазначеній статті сполука XXV є проміжною у синтезі сполуки XIX, яка досліджується як потенційний засіб для лікування серпоподібноклітинного захворювання. Крім того, відомий ферментативний спосіб синтезу сполуки XXV [Didziapetris R., Drabnig В., Schellenberger V., Jakubke H.D., Svedas V. Penicillin acylase-catalyzed protection and deprotection of amino groups as a promising approach in enzymic peptide synthesis. // FEBS Letters. 1991. V.287. №12. P.31-33]. У патенті Bok S., Lee S., Jeong Т., Phenolic acid derivatives and composition for preventing or treating blood lipid level-related diseases comprising the same. US 2003199566 описаний синтез 3-(пгідроксифеніл)пропіонілфенілаланіну (XVII) і 3-(пгідроксифеніл)пропіоніл фенілаланіну метилового ефіру (ХІІІ) з використанням 1гідроксибензотриазолу й 1-[3(диметиламіно)пропіл]-3-етилкарбодііміду гідрохлориду в присутності триетиламіну. Для одержання 3-(п-гідроксифеніл)пропіонілфенілаланіну (XVII) далі проводили омилення сполуки (XIII) з виходом цільового продукту 78%. Для обох сполук наведені дані 1Н-ЯМР- і 13С-ЯМР-спектроскопії. Сполуки XVII і XIII пропонується використовувати для попередження й лікування захворювань, пов'язаних з рівнем ліпідів у крові. У публікації міжнародної заявки WO 9952962 описаний 3-(п-гідроксифеніл)пропіонілтирозину бензиловий ефір (XXXIV). Наведено температуру плавлення, дані 1Н-ЯМР- і 13С-ЯМР-спектроскопії. Відомо, що анальгетична дія може здійснюватися відповідно до різних механізмів, зокрема, шляхом інгібування ферменту циклооксигенази в каскаді арахідонової кислоти [Машковский М.Д. Лекарственные средства. // Москва. Новая волна. 2005. С.163-164]. Найбільш вираженим знеболювальним ефектом серед препаратів, що знижують синтез альгогенів, мають ненаркотичні анальгетики й нестероїдні протизапальні засоби. Ненаркотичні анальгетики представлені саліцилатами (аспірин), похідними піразолону (амідопірин, анальгін) і параамінофенолу (парацетамол). До нестероїдних протизапальних засобів належать похідні саліцилової, оцтової, пропіонової і антранілової кислот. Ненаркотичні анальгетики й нестероїдні протизапальні засоби, поряд з болезаспокійливим ефектом, мають протизапальну й жарознижувальну дію [Кукушкин Μ.Л., Хитров Н.К. Общая патология боли. / Москва. Медицина. 2004. 142с.]. Основним побічним ефектом нестероїдних протизапальних засобів є ульцерогенність. У анальгетиків з іншим механізмом дії часто спостерігається проспазматичний побічний ефект [Машковский М.Д. Лекарственные средства. // Москва. Новая волна. 2005. С.154.]. Відомі протипаркінсонічні властивості нестероїдних протизапальних препаратів саліцилату на 88688 14 трію, індометацину й піроксикаму [Кадиева М.Г., Оганесян Э.Т., Мацуева С.Х.. Нейротоксины и средства для лечения болезни Паркинсона. III. Средства, опосредованно влияющие на дофаминэргическую систему. Хим.-фарм. Журнал. 2005. Т.39. №11. С.3-11]. Припускають, що така активність, зокрема, реалізовується опосередковано через простагландини, що впливають на дофамінову систему. Відомо також, що антисеротонінові препарати впливають на дофамінову систему при хворобі Паркінсона, сприяючи зв'язуванню рецепторів з дофаміновими антагоністами [Кадиева М.Г., Оганесян Э.Т., Мацуева С.Х. Нейротоксины и средства для лечения болезни Паркинсона. III. Средства, опосредованно влияющие на дофаминэргическую систему. Хим.-фарм. Журнал. 2005. Т.39. №11. С.3-11], існують і інші механізми дії протипаркінсонічних препаратів [Машковский М.Д. Лекарственные средства. // Москва. Новая волна. 2005. С.138.]. Залежно від механізму дії антидепресанти поділяються на кілька груп, зокрема інгібітори моноамінооксидази, трициклічні антиоксиданти, блокатори гістамінових, серотонінових, холецистокінінових, α-адренорецепторів [Машковский М.Д. Лекарственные средства. // Москва. Новая волна. 2005. С.109.]. Оскільки застосування відомих антидепресантів і структурно родинних сполук супроводжується численними серйозними побічними ефектами, пошук нових більш безпечних і ефективних препаратів такої дії є актуальним. Застосування сполук даного винаходу для купірування депресивних станів не було відоме. Гіпоксія спостерігається при численних патологічних станах, у тому числі порушеннях функцій мозку. Антигіпоксанти поліпшують утилізацію організмом циркулюючого кисню, підвищуючи стійкість організму до кисневої недостатності. Препарати такої дії нечисленні [Машковский М.Д. Лекарственные средства. // Москва. Новая волна. 2005. С.729.]. Багато препаратів, у тому числі регулюючі діяльність ЦНС, мають додатково антигіпоксичні властивості, що підвищують ефективність їхньої дії. Для групи сполук даного винаходу, антигіпоксичний ефект раніше описаний не був. Метою даного винаходу є синтез і застосування нових і відомих фенілвмісних N-ацильних похідних біогенних амінів і амінокислот як нетоксичних, більш ефективних анальгетиків і протизапальних засобів, без побічних ефектів, зокрема ульцерогенності й проспазматичної дії, які мають також антигіпоксичну, антидепресантну і протипаркінсонічну дію, а також здатність потенціювати дію інших анальгетиків. Даний винахід належить до нових фенілвмісних N-ацильних похідних амінів загальної формули І: 15 де R1 представляє 88688 16 або , де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, СООН, -СООR6, де R6 представляє С1-С6алкіл або , де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; за умови, що сполука загальної формули І не є фенілацетилтираміном, 3-(п-гідроксифеніл)пропіонілфенілетиламіном, 3-(п-гідроксифеніл)пропіонілтираміном, 3-фенілпропіонілфенілетиламіном, 3-фенілпропіонілтираміном, З-(п-гідроксифеніл)пропіонілфенілаланіну метиловим ефіром, 3-(п-гідроксифеніл)пропіонілтирозином, 3-фенілпропіонілтирозином, фенілацетилтирозином, 3-(п-гідроксифеніл)пропіонілфенілаланіном, 3-фенілпропіонілфенілаланіном, фенілацетилфенілаланіном, 3-(п-гідроксифеніл)пропіонілтирозину метиловим ефіром, 3-фенілпропіонілтирозину метиловим ефіром, фенілацетилтирозину метиловим ефіром, фенілацетилфенілетиламіном, 3-фенілпропіонілфенілаланіну метиловим ефіром, фенілацетилфенілаланіну метиловим ефіром, 3-(п-гідроксифеніл)пропіонілтирозину бензиловим ефіром; або їх фармацевтично прийнятних солей, що мають інгібуючу циклооксигеназу активність, протизапальну і анальгетичну дію, спазмолітичну, антигіпоксичну, антипаркінсонічну і антидепресивну дію, а також здатність потенціювати дію інших анальгетиків. Даний винахід також належить до застосування сполук загальної формули І: де R1 або представляє , де R5 представляє водень або гідроксильну групу; R2 представляє водень або аміногрупу, необов'язково заміщену групою СН3(СН2)mСО-, де m=0-4; R3 представляє водень, -СООН, -СOOR6, де R6 представляє С1С6алкіл або , де R7 представляє водень або гідроксильну групу, R4 представляє водень, гідроксильну групу; або їх фармацевтично прийнятних солей як інгібіторів циклооксигенази, анальгетичних і протизапальних, спазмолітичних, антигіпоксичних, антипаркінсонічних і антидепресантних засобів, а також засобів здатних потенціювати дію інших анальгетиків. Далі, даний винахід належить до фармацевтичної композиції або засобу, що має інгібуючу циклооксигеназу активність, протизапальну і анальгетичну дію, а також антидепресантну, спазмолітичну, антигіпоксичну і протипаркінсонічну дію, які містять ефективну кількість сполуки загальної формули І або її фармацевтично прийнятної солі і необов'язково фармацевтично прийнятний носій. Ще одним об'єктом винаходу є спосіб лікування больових синдромів різного генезу, а також захворювань, що супроводжуються запаленням, спазмами, гіпоксією, депресією і явищами Паркінсонізму, який включає введення ефективної кількості сполуки загальної формули І або її фармацевтично прийнятної солі, необов'язково в сполученні з іншими анальгетиками. Даний винахід також належить до нових способів одержання сполук загальної формули І. Переважними сполуками формули І є сполуки, в яких R3 представляє -СООН, -СООСН3. Нові переважні сполуки загальної формули І представлені в Таблиці 1. 17 88688 18 19 Відомі переважні сполуки загальної формули І представлені в Таблиці 2. 88688 20 21 88688 22 23 88688 24 Сполуки загальної формули І одержують активацією карбоксильної групи пгідроксифенілоцтової кислоти або фенілоцтової кислоти взаємодією з дифенілфосфорилазидом (DPPA) і триетиламіном (TEA) в органічному розчиннику, переважно Ν,Ν-диметилформаміді, етилацетаті при охолодженні, переважно в інтервалі від -25° до +0° з наступним здійсненням взаємодії з амінопохідним. Переважно активацію карбоксильної групи здійснюють із використанням 1-1,2 еквівалентів DPPA і TEA. Як амінопохідне можуть бути використані ефіри тирозину й фенілаланініну. Для одержання сполук II і III як вихідне амінопохід не використовують бензилові ефіри тирозину й фенілаланіну, відповідно, з наступним видаленням бензильної групи шляхом каталітичного гідрогенолізу. На відміну від раніше використовуваних способів синтезу відомих сполук загальної формули І, застосування дифенілфосфорилазидного способу дозволило зменшити число стадій, а саме, виключити стадію виділення активованого похідного карбоксильного компонента, обмежитися екстракцією для виділення цільових речовин і підвищити виходи (≥90%). Загальна схема синтезу дифенілфосфорилазним методом представлена на Схемі 1. Нові сполуки II, III, IV, V, VII, VIII, у тому числі, які містять фенольні гідроксильні групи, можуть бути одержані також методом активованих Nоксисукцинімідних ефірів, перевагою якого є доступність реагентів, водорозчинність Nгідроксисукциніміду, що виділяється, швидкість протікання як реакції одержання Nоксисукцинімідних ефірів ацилуючих агентів, так і реакції утворення амідного зв'язку, і можливість досягнення високих виходів цільових продуктів (70-80%), незважаючи на наявність у них фенольного гідроксилу. Відповідно до пропонованого способу синтез N-оксисукцинімідних ефірів ацилуючих агентів здійснюється перетворенням пгідроксифенілоцтової кислоти або фенілоцтової кислоти на активований N-оксисукцинімідний ефір Ν,Ν'-дициклогексилкарбодіімідним методом (DCCметодом) з високим виходом (близько 90%), з наступним утворенням амідного зв'язку реакцією Νоксисукцинімідних ефірів з амінопохідним, також з високими виходами (70-80%), за короткий час (1-2 години) і без застосування хроматографічного очищення цільового продукту. Як амінопохідне можуть бути використані ефіри тирозину й фенілаланініну. Аналогічно можуть бути одержані відомі сполуки X, XI, XII, XIII, XV, XVII, XIX, XX, XXII, XXIII, XXIV, синтез яких методом активованих Nоксисукцинімідних ефірів не описаний у рівні техніки. Загальна схема синтезу сполук загальної формули І методом активованих N-оксисукцинімідних ефірів представлена на Схемі 2. Синтез гідроксифенілпропіонілтирозину (XIV) може бути здійснений також методом активованих N-оксисукцинімідних ефірів, причому, з метою зменшення числа стадій може бути використаний незахищений по С-кінцю тирозин. Крім того, це дозволяє уникнути тривалого впливу лугу, який був би необхідний для омилення метилового ефіру тирозину, що могло б несприятливо відбитися на оптичній чистоті одержуваної сполуки [Шредер Э., Любке К. // Пептиды. / Μ. Мир. 1967. 2т.; Гросс Э., Майенхофер И. // Пептиды. Основные методы образования пептидной связи / Москва. Мир. 1983г. с.422]. Проблема досить низької розчинності незахищеного тирозину як в органічних розчинниках, так і у воді вирішена шляхом його переве дення в розчинну Na-сіль у результаті додавання до суспензії тирозину в DMF 2-х еквівалентів 1N розчину NaOH, у результаті чого спостерігалося повне розчинення амінокислоти. Реакція одержаного в такий спосіб розчину амінопохідного з Nоксисукцинімідним ефіром 3-(nгідроксифеніл)пропіонової кислоти проходить практично повністю й швидко (за 2 години). Після виділення екстракцією без застосування хроматографічного очищення, вихід цільового (XIV) продукту склав близько 63%. Сполуки загальної формули І також можуть бути одержані у вигляді фармацевтично прийнятних адитивних солей з нетоксичними кислотами, такими як фумарова кислота, малеїнова кислота, 25 бурштинова кислота, оцтова кислота, лимонна кислота, винна кислота й подібні, і солей з основами, такими як гідроксид натрію, гідроксид калію, карбонат натрію й подібні. Сполуки загальної формули І мають інгібуючою циклооксигеназу активність й можуть бути використані для лікування больових синдромів різного генезу, запальних і запальнодегенеративних захворювань суглобів і сполучної тканини, а також кістково-м'язової системи, інших захворювань, що супроводжуються запаленням, спазмами, гіпоксією, для потенціювання інших анальгетиків, а також порушень, обумовлених депресією й хворобою Паркінсона. Зокрема, сполуки даного винаходу можуть бути використані для лікування післяопераційного болю, посттравматичного болю, а також больових синдромів гінекологічної, неврологічної, онкологічної, стоматологічної природи, ревматоїдного артриту, артропатії, хвороби Бехтерева, неспецифічних спондилоартритів, подагричного артриту, остеоартрозу, позасуглобового ревматизму й тромбофлебіту, інших захворювань, що супроводжуються запаленням, спазмами, гіпоксією, а також порушень, обумовлених хворобою Паркінсона, емоційно-стресовими станами. Сполуки даного винаходу вводяться в ефективній кількості, яка забезпечує бажаний терапевтичний результат. Сполуки формули (І) можуть бути введені перорально, місцево, парентерально, шляхом інгаляцій і ректально у вигляді стандартних лікарських форм, що містять нетоксичні фармацевтично прийнятні носії. Використовуваний у даному описі термін «парентеральне введення» означає підшкірні, внутрішньовенні, внутрішньом'язові або внутрішньогрудні ін'єкції або вливання. Сполуки даного винаходу можуть бути введені пацієнтові в дозах, що становлять від 0,1 до 10мг/кг ваги тіла в день, переважно в дозах від 0,5 до 5мг/кг один або більше разів на день. Прицьому слід зазначити, що конкретна доза для кожного конкретного пацієнта буде залежати від багатьох факторів, включаючи активність даної використовуваної сполуки, вік, вагу тіла, стать, загальний стан здоров'я і режим харчування пацієнта, час і спосіб введення лікарського засобу, швидкість його виведення з організму, конкретно використовувану комбінацію лікарських засобів, а також тяжкість захворювання, що піддається лікуванню. Фармацевтичні композиції за даним винаходом містять сполуку за даним винаходом в кількості, ефективній для досягнення бажаного результату й можуть бути введені у вигляді стандартних лікарських форм (наприклад, у твердій, напівтвердій або рідкій формах), що містять сполуки даного винаходу як активний інгредієнт у суміші з носієм або наповнювачем, придатним для внутрішньом'язового, внутрішньовенного, перорального, сублінгвального, інгаляційного й інтраректального введення. Активний інгредієнт може бути включений у композицію разом зі звичайно використовуваними нетоксичними фармацевтично прийнятними носіями, придатними для виготовлення розчинів, таб 88688 26 леток, пігулок, капсул, драже, супозиторіїв, емульсій, суспензій, мазей, гелів і будь-яких інших лікарських форм. Як наповнювачі можуть бути використані різні речовини, такі як сахариди, наприклад глюкоза, лактоза або сахароза, маніт або сорбіт, похідні целюлози й/або фосфати кальцію, наприклад, трикальцій фосфат або кислий фосфат кальцію, як зв'язуючого компонента можуть бути використані такі, як крохмальна паста, наприклад кукурудзяний, пшеничний, рисовий, картопляний крохмаль, желатин, трагакант, метилцелюлоза, гідроксипропілметилцелюлоза, натрій карбоксиметилцелюлоза й/або полівінілпіролідон. При необхідності можуть бути використані розпушувальні агенти, такі як вищезгадані крохмалі й карбоксиметилкрохмаль, що поперечнозшитий полівінілпіролідон, агар або альгінова кислота або її сіль, така як альгінат натрію. Можуть бути використані необов'язкові добавки, такі як агенти, що регулюють текучість, і мастильні агенти, такі як діоксид кремнію, тальк, стеаринова кислота і її солі, такі як стеарат магнію або стеарат кальцію й/або пропіл енгліколь. Ядро драже звичайно покривають шаром, який стійкий до дії шлункового соку. Для цієї мети можуть бути використані концентровані розчини сахаридів, які можуть необов'язково містити аравійську камедь, тальк, полівінілпіролідон, поліетиленгліколь і/або діоксид титану, і придатні органічні розчинники або їхні суміші. Як добавки можуть бути також використані стабілізатори, загусники, барвники й віддушки. Як мазева основа можуть бути використані вуглеводневі мазеві основи, такі як вазелін білий й жовтий (Vaselinum album, Vaselinum flavum), вазелінове масло (Oleum Vaselini), мазь біла й рідка (Unguentum album, Unguentum flavum), а як добавки для надання більш щільної консистенції, такі як твердий парафін і віск; абсорбтивні мазеві основи, такі як гідрофільний вазелін (Vaselinum hydrophylicum), ланолін (Lanolinum), кольдкрем (Unguentum leniens); мазеві основи, що змиваються водою, такі як гідрофільна мазь (Unguentum hydrophylum); водорозчинні мазеві основи, такі як поліетиленгліколева мазь (Unguentum Glycolis Polyaethyleni), бентонітові основи й інші. Як основа для гелів можуть бути використані метилцелюлоза, натрієва сіль карбоксиметилцелюлози, оксипропілцелюлоза, поліетиленгліколь або поліетиленоксид, карбопол. Як основа для супозиторія можуть бути використані основи, не розчинні у воді, такі як масло какао; основи, розчинні у воді або змішувані із водою, такі як желатиногліцеринові або поліетиленоксидні; комбіновані основи - мильно-гліцеринові. При приготуванні стандартної лікарської форми кількість активного інгредієнта, використовуваного в комбінації з носієм, може варіюватися залежно від реципієнта, що піддається лікуванню, від конкретного способу введення лікарського засобу. Так, наприклад, при використанні сполук даного винаходу у вигляді розчинів для ін'єкцій, вміст активного агента в них становить 0,01-5%. Як роз 27 ріджувачі можуть бути використані 0,9% розчин хлориду натрію, дистильована вода, розчин новокаїну для ін'єкцій, розчин Рінгера, розчин глюкози, специфічні добавки для розчинення. При введенні в організм сполук даного винаходу у вигляді таблеток і супозиторіїв, їхня кількість становить 5,0500 мг на стандартну лікарську форму. Лікарські форми даного винаходу одержують за стандартними методиками, такими як, наприклад, процеси змішування, гранулювання, формування драже, розчинення й ліофілізація. Слід зазначити, що сполуки даного винаходу проявляють біологічну активність у дозах на дватри порядки нижче в порівнянні з відомими препаратами, використаними для порівняння, при практично однаковій ефективності, і для них не виявлено негативних побічних дій і не виявлено протипоказань до застосування. При цьому, при дослідженні токсичності сполук даного винаходу в дозі 1000мкг/кг, перорально, не зареєстрували загибелі експериментальних тварин. Детальний опис сполук даного винаходу, їх одержання й дослідження фармакологічної активності представлено в нижченаведених прикладах, призначених для ілюстрації переважних варіантів винаходу, і не обмежуючих його об'єм. Приклади синтезу сполук даного винаходу Індивідуальність одержаних сполук перевіряли методом ТШХ на пластинках "Kieselgel 60 F254" ("Merck", Німеччина) у системі розчинників: хлороформ-метанол 9:1 (1), хлороформ-метанолетилацетат 6:1:3 (2), хлороформ-метанол-аміак 6:3:0,5 (3). Хроматограми проявляли хлортолідиновим реактивом, нінгідрином, йодом і по світінню в УФсвітлі. 1 H-ЯМР реєстрували на приладі "АМХ-400 Bruker" (Німеччина). ІЧ-Фур'є спектри знімали в таблетках КВr на приладі "Magna 750" ("Nicolet" США). Температуру плавлення визначали на приладі "Boetius" (Німеччина). Мас-спектри високого розрізнення одержували на часопрольотному мас-спектрометрі методом матриксної лазернодесорбційної іонізації, з використанням як матриці 2,5-дигідроксибензойної кислоти, на приладах і REFLEX™ III (Bruker, Німеччина). Аналітичну обернено-фазову ВЕРХ проводили на приладах: - хроматограф "Breeze", детектор "Waters" (США), детекція при 214нм, швидкість елюювання 1мл/хв., в умовах (1): колонка Symmetry 300 C18, 3,9×150мм, 5мкм, елюція 0,1%-ний водної TFA із градієнтом ацетонітрилу від 0% до 60% за 18хв.; - хроматографи "System Gold" ("Beckman", США), швидкість елюювання 0,25мл/хв., детекція при 220нм, в умовах (2): колонка "Phenomenex" (США) C18, 2×250мм, 5мкм, елюція 0,1%-ний TFA із градієнтом 0,08% TFA в 100% MeCN від 0% до 100% за 50хв. - хроматограф "Breeze", детектор "Waters" (США), детекція при 214нм, швидкість елюювання 1мл/хв., в умовах (3): колонка Symmetry 300 С18, 4,6×250мм, 20мкм, елюція 0,1%-ний TFA із градіє 88688 28 нтом 0,09% TFA у суміші 60:40 ацетонітрил-вода від 0% до 100% за 15хв. Приклад 1 п-Гідроксифенілацетилтирамін (VII) Методика А При перемішуванні до розчину 0,40г (2,63ммоль) п-гідроксифенілоцтової кислоти в 3,5мл DMF додавали 0,35г (2,63ммоль) тираміну. Розчин охолоджували до -10°С і додавали 0,68мл (3,16ммоль) дифенілфосфорилазиду й 0,44мл (3,16ммоль) триетиламіну. Перемішували 2год. при -10°С і залишали при 20°С на 15год. До реакційної маси додавали 35мл води, екстрагували 20мл етилацетату. Етилацетатний шар промивали 10мл 5% розчину Na2CO3, водою до рН 7, 10мл 5% розчину НСl, водою до рН 7. Етилацетатний шар сушили над Na2SO4, відфільтровували Na2SO4, етилацетат видаляли у вакуумі. Маслоподібний залишок розтирали із сумішшю ефір-гексан (1:1). Білий осад, що утворюється, відфільтровували й сушили у вакуумі над СаСl2. Вихід 0,68г (95%). Rf 0,7 (1). Тпл=147-149°. [М]+ 271,6. 1 Н-ЯМР, CD3OD, δ, м.ч.: 2,65 (т, J=7Гц, 2H, αСН2-ТА), 3,29-3,32 (м, 4Н, β-СН2-ТА, CH2-(OHPhAc)), 6,63-6,75 (м, 4Н, о-СН-аром.), 6,90-7,06 (м, 4Н, м-СН-аром.). ІЧ-Фур'є, см-1: 3276 (вал. ОН); 3108 (вал., =СН, аром.); 1612 (амід І); 1591 (амід II); 1515 (аром. С-С-); 1226 (вал., -С-О, фенольний). Знайдено, %: С 70,57; Η 6,43; Ν 5,50 C16H17NO3. Обчислено, %: С 70,83; Η 6,32; Ν 5,16. ВЕРХ в умовах (1): індивідуальний пік, час утримання 8,71хв. Методика Б До розчину 0,70г (4,60ммоль) пгідроксифенілоцтової кислоти в 17мл етилацетату при перемішуванні додавали 0,53г (4,60ммоль) Νгідроксисукциніміду, розчин охолоджували до 0°С і додавали 0,95г (4,60ммоль) Ν,Ν'дициклогексилкарбодіімід (DCC). Перемішували 2 години при 0°С і залишали на 20 годин при 4°С. Осад Ν,Ν'-дициклогексилсечовини (DCU) відфільтрували. Розчинник видалили у вакуумі. Маслоподібний залишок розтирали з гексаном. Білий твердий осад, що утворився, відфільтровували, промивали гексаном і сушили у вакуумі над СаСl2. Одержали 1,08 г (94,6%). Rf 0,58 (1). При перемішуванні до розчину 0,30г (1,2ммоль) N-оксисукцинімідного ефіру nгідроксифенілоцтової кислоти в 8мл Ν,Νдиметилформаміду (DMF) додавали 0,16г (1,2ммоль) тираміну. Реакційну суміш перемішували 2 години при 20°С, залишали при 4°С на 20 годин. DMF видаляли у вакуумі. Маслоподібний залишок розтирали з водою. Білий осад, що утворюється, відфільтровували, промивали водою. Вихід 0,26г (80%). Rf 0,68 (1). Тпл=446-148°. [М+Н]+ 272,3. Знайдено, %: С 71,05; Η 6,10; Ν 5,25 С16Н17NO3. Обчислено, %: С 70,83; Η 6,32; N 5,16. 29 Приклад 2 п-Гідроксифенілацетилфенілетиламін (VIII) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,57г (90,5%). Rf 0,82 (1). Тпл=69-70°. [М]+ 255,5. 1 Н-ЯМР DMSO-d6, δ, м.ч.: 2,68 (т, J=8Гц, 2Н, βСН2-РЕА), 3,22-3,26 (м, α-СН2-РЕА), 3,36 (с, 2Н, CH2-(OH-PhAc)), 6,66 (д, J=4Гц, 2Н, м-СН-аром. OH-PhAc), 7,00 (д, J=4Гц, 2Н, м-СН-аром. OHPhAc), 7,14-7,28 (м, 5Н, аром. -СН-РЕА), 8,0 (уш. с., 1H, NH-PEA), 9,20 (с, 1H, OH-(OH-PhAc)). ІЧ-Фур'є, см-1: 3332 (вал. ОН); 3087 (вал., =СН, аром.); 1626 (амід І); 1558 (амід II); 1515 (аром. С-С-); 1249 (вал., -С-О, фенольний). Знайдено, %: С 75,57; Η 6,80; Ν 5,77 C16H17NO2. Обчислено, %: С 75,27; Η 6,71; Ν 5,49. ВЕРХ в умовах (1): індивідуальний пік, час утримання 11,17хв. Синтез проводили відповідно до методики Б, наведеної для сполуки VII. Вихід 0,50г (79,4%). Rf 0,85(1). Тпл=68-70°. [М]+ 255,7. Знайдено, %: С 75,17; Η 6,87; Ν 5,75 C16H17NO2. Обчислено, %: С 75,27; Η 6,71; Ν 5,49. Приклад 3 3-(п-гідроксифеніл)пропіонілтирамін (X) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,41г (95%). Rf 0,38 (1). Тпл=174-176°. 1 Н-ЯМР, DMSO-d6, δ, м. ч.: 2,26 (т, J=8Гц, 2H, α-CH2-(HO-PhPr)), 2,53 (т, J=6Гц, 2Н, β-СН2-Туrа), 2,67 (т, J=8Гц, 2Н, β-CH2-(HO-PhPr)), 3,16 (т, J=6Гц, 2Н, α-СН2-Туrа), 6,62 (д, J=1Гц, 2Н, м-CHBzl-Tyra), 6,65 (д, J=1Гц, 2Н, м-СН-Вzl-(НО-PhPr)), 6,92-6,96 (м, 4Н, o-CH-Bzl-Tyra і o-CH-Bzl-(HOPhPr)), 7,79 (с, 1H, NH-Tyra), 9,09 (уш. с, 2Н, OHTyra i OH-(HO-PhPr)). ІЧ-Фур'є, см-1: 3249 (вал. ОН), 1621 (амід І), 1515 (аром.), 1541 (амід II). Знайдено %: С 71,56; Η 6,78; Ν 4,97. Обчислено %: С 71,56; Η 6,71; Ν 4,91, C17H19NO3. ВЕРХ в умовах (2): індивідуальний пік, час утримання 25,62хв. Синтез проводили відповідно до методики Б, наведеної для сполуки VII. Вихід 0,37г (85%). Rf 0,35(1). Тпл=172-174°. [М]+ 285,3. Приклад 4 3-Фенілпропіонілфенілетиламін (XI) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,26г (97%). Rf 0,78(1). Тпл=94-96°. 1 Н-ЯМР, DMSO-d6, δ, м.ч.: 2,34 (т, J=8Гц, 2Н, α-CH2-PhPro), 2,66 (т, J=6Гц, 2H, β-ΟΗ2-ΡΕΑ), 2,79 88688 30 (т, J=8Гц, 2H, β-CH2-PhPro), 3,24 (т, J=6Гц, 2Н, αCH2-PEA), 7,25-7,30 (м, 10Н, СН-аром.), 7,89 (уш. с, 1Н, ΝΗ-ΡΕΑ). ІЧ-Фур'є, см-1: 1637 (амід І), 1546 (амід ІІ). Знайдено %: С 80,24; Η 7,61; Ν 5,54. Обчислено %: С 80,60; Η 7,56; Ν 5,53. C17H19NO3. ВЕРХ в умовах(2): індивідуальний пік, час утримання 37,86хв. Синтез проводили відповідно до методики Б, наведеної для сполуки VII. Вихід 0,20г (77%). Rf 0,80 (1). Знайдено %: С 80,39; Η 7,53; Ν 5,30. Обчислено %: С 80,60; Η 7,56; Ν 5,53. C17H19NO3. Приклад 5 3-(п-Гідроксифеніл)пропіонілфенілетиламін (IX) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,20г (90%). Rf (11) 0,4. Тпл=102-104°. Літ. [84] 102-104°. [М]+ 269,6. 1 Н-ЯМР, CDCІ3, δ, м.ч.: 2,39 (т, J=7Гц, 2H, αCH2-(HO-PhPr)), 2,73 (м, 2Н, β-СН2-РЕА), 2,86 (т, J=1Гц, 2Н, β-CH2-(HO-PhPr)), 3,48 (м, 2Н, α-СН2РЕА), 6,75 (м, 2Н, м-СН-аром. HO-PhPr), 7,03 (м, 2Н, о-СН-аром. HO-PhPr), 7,09 (м, 2Н, о-СН-аром. PEA), 7,3 (м, 3Н, м,n-СН-аром. PEA). ІЧ-Фур'є, см-1: 3263 (вал. ОН); 1618 (амід І); 1537 (амід ll). Знайдено %: С 75,57; Η 6,93; Ν 5,09. C17H19NO2. Обчислено %: С 75,81; Η 7,11; Ν 5,20. ВЕРХ в умовах (3): індивідуальний пік, час утримання 14,77хв. Приклад 6 п-Гідроксифенілацетилтирозину метиловий ефір (IV) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,17г (39%). Rf 0,56(2). [М]+ 329,85. [α]D25+12,22° (С 0,36; MeOH). 1 Н-ЯМР, DMSO-d6, δ, м.ч.: 2,78 (дд, 1H, CH2Tyr), 2,9 (дд, 1H, СН2-Туr), 3,25-3,45 (м, 2Н, CH2HOPhAc), 4,3-4,4 (м, 1Η, α-CH-Tyr), 3,6 (с, 3Н, ОСН3 Туr), 6,55-7,1 (м, 8Н, аром. Н), 8,25 (д, 1Н, NH-Tyr). ІЧ-Фур'є, δ, см-1: 1649 (амід І); 1515 (амід II); 1263 (амід III). Знайдено, %: С 65,75; Η 5,75; Ν 4,23. Обчислено, %: С 65,64; Η 5,81; Ν 4,25. ВЕРХ в умовах (3), індивідуальний пік, час утримання 7,25хв. Приклад 7 п-Гідроксифенілацетилфенілаланіну метиловий ефір (V) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,40г (39%), масло. Rf 0,70(2). [М]+ 313,83. [α]D20+35,05° (С 0,19, етилацетат). 31 1 Н-ЯМР, DMSO-d6, δ, м.ч.: 2,9 (дд, 1H, CH2Phe), 3,05 (дд, 1H, CH2-Phe), 3,25-3,4 (м, 2Н, CH2HOPhAc), 3,6 (с, 3Н, ОСН3 Phe), 4,4-4,5 (м, 1H, αCH-Phe), 6,55-6,95 (м, 4Н, аром. Η HOPhAc), 7,17,3 (м, 5Н, аром. Η Phe), 8,3 (д, 1H, NH-Phe), 9,2 (с, 1H, OH-Ar HOPhAc). ІЧ-Фур'є, δ, см-1: 1663 (амід І); 1515 (амід II); 1263 (амід III). Знайдено, %: С 69,08; Η 6,05; Ν 4,45. Обчислено, %: С 68,99; Η 6,11; Ν 4,47. ВЕРХ в умовах (3), індивідуальний пік, час утримання 8,57хв. Приклад 8 Фенілацетилтирамін (VI) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,35г (37,6%). Rf 0,85 (2). Tпл=105-108°. [M+1]+ 256,2. 1 Н-ЯМР, DMSO-d6, δ, м.ч.: 2,6 (т, 2Н, α-CH2TA), 3,2 (кв, 2Н, β-СН2-ТА), 3,4 (с, 2Н, CH2-PhAc), 6,6-7,0 (м, 4Н, аром. Η ТА), 7,15-7,3 (м, 5Н, аром. Η PhAc), 8,0 (т, 1Н, NH-TA), 9,1 (с, 1Н, ОН-ТА). ІЧ-Фур'є, δ, см-1: 1646 (амід І); 1516 (амід II); 1264 (амід III). Знайдено, %: С 75,37; Η 6,69; Ν 5,45. Обчислено, %: С 75,27; Η 6,71; Ν 5,49. ВЕРХ в умовах (3), індивідуальний пік, час утримання 8,06хв. Приклад 9 3-(п-Гідроксифеніл)пропіонілфенілаланіну метиловий ефір (XIII) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,37г (38%), масло. Rf 0,73 (2). [М+1]+ 328,21. [α]D25-6,95° (С 0,46; МеОН). 1 Н-ЯМР, DMSO-d6, δ, м.ч.: 2,3 (т, 2Н, 1-СН2 HOPhPr), 2,6 (т, 2Н, 2-СН2 HOPhPr), 2,85 (дд, 1Н, CH2-Phe), 3,0 (дд, 1Н, CH2-Phe), 3,6 (с, 3Н, ОСН3 Phe), 4,4-4,5 (м, 1Н, α-CH-Phe), 6,6-6,95 (м, 4Н, аром. Η HOPhPr), 7,15-7,3 (м, 5Н, аром. Η Phe), 8,22 (д, 1Н, NH-Phe), 9,1 (с, 1Н, ОН-Аr HOPhAc). ІЧ-Фур'є, δ, см-1: 1651 (амід І); 1516 (амід II); 1266 (амід III). Знайдено, %: С 69,61; Η 6,49; Ν 4,29. Обчислено, %: С 69,71; Η 6,47; Ν 4,28. ВЕРХ в умовах (3), індивідуальний пік, час утримання 8,9хв. Приклад 10 Бензиловий ефір пгідроксифенілацетилтирозину (XXVI) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,59г (55.7%). Rf 0,57 (2). [M+1]+ 406,0. [α]D20-9,18° (С 0,20, МеОН). ІЧ-Фур'є, δ, см-1: 1649 (амід І); 1515 (амід II); 1737 (вал С=О склади, еф.). Знайдено, %: С 71,05; Η 5,70; Ν 3,43. Обчислено, %: С 71,10; Η 5,72; Ν 3,45. 88688 32 Приклад 11 п-Гідроксифенілацетилтирозин (II) До розчину 0,59г (1,47ммоль) бензилового ефіру п-гідроксифенілацетилтирозину в 10мл метанолу додавали 0,20г 10%-го паладію-на-вугіллі й при інтенсивному перемішуванні гідрували в струмені водню протягом 1,5год. Каталізатор відфільтровували. Розчинник з фільтрату видаляли у вакуумі. Маслоподібний залишок розтирали із сумішшю ефір-гексан (1:1). Білий осад, що утворюється, відфільтровували й сушили у вакуумі над СаСl2 і Р2О5. Одержували 0,32г (68%). Вихід 37%. Rf 0,28 (3). [М+1]+ 316,07. [α]D20+28,03° (С 0,31, МеОН). 1 Н-ЯМР, DMSO-d6, δ, м.ч.: 2,75 (дд, 1Н, СН2Туr), 2,9 (дд, 1H, СН2-Туr), 3,2-3,4 (м, 2Н, CH2HOPhAc), 4,3-4,4 (м, 1H, α-CH-Tyr), 6,55-7,1 (м, 8Н, аром. Н), 8,05 (д, 1Н, NH-Tyr). ІЧ-Фур'є, δ, см4: 1614 (амід І); 1516 (амід II); 1254 (амід III). Знайдено, %: С 64,65; Η 5,41; Ν 4,37. C17H17NO5. Обчислено, %: С 64,75; Η 5,43; Ν 4,44. ВЕРХ в умовах (1), індивідуальний пік, час утримання 6,33хв. Приклад 12 Бензиловий ефір пгідроксифенілацетилфенілаланіну (XXVII) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,76г (74%). Rf 0,87(2). [M+1]+ 390,1. [α]D20 -19,47° (С 0,19, МеОН). ІЧ-Фур'є, δ, см4: 1649 (амід І); 1515 (амід II); 1740 (вал С=О склади, еф.). Знайдено, %: С 74,12; Η 5,92; Ν 3,57. Обчислено, %: С 74,02; Η 5,95; Ν 3,60. Приклад 13 п-Гідроксифенілацетилфенілаланін (III) До розчину 0,65г (1,67ммоль) бензилового ефіру п-гідроксифенілацетилфенілаланіну в 10мл метанолу додавали 0,30г 10%-го паладію-навугіллі й при інтенсивному перемішуванні гідрували в струмені водню протягом 1,5год. Каталізатор відфільтровували. Розчинник з фільтрату видаляли у вакуумі. Маслоподібний залишок розтирали з гексаном. Білий осад, що утворюється, відфільтровували й сушили у вакуумі над СаСl2 і P2O5. Одержували 0,27г (53%). Вихід 39,2%. Rf 0,42 (3). [M+1]+ 300,09. [α]D25+18,57° (С 0,44; MeOH). 1 H-ЯМР, DMSO-d6, δ, м.ч.: 2,85 (дд, 1H, CH2Phe), 3,1 (дд, 1Н, CH2-Phe), 3,2-3,35 (м, 2Н, CH2HOPhAc), 4,4-4,5 (м, 1Н, α-CH-Phe), 6,55-6,95 (м, 4Н, аром. Н HOPhAc), 7,1-7,3 (м, 5H, аром. Η Phe), 8,15 (д, 1Н, NH-Phe). ІЧ-Фур'є, δ, см-1: 1611 (амід І); 1512 (амід II). Знайдено, %: С 68,30; Η 5,68; Ν 4,65. Обчислено, %: С 68,21; Η 5,72; Ν 4,68. ВЕРХ в умовах (3), індивідуальний пік, час утримання 7,59хв. 33 88688 Приклад 14 Бензиловий ефір 3-фенілпропіонілтирозину (XXX) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,94г (70%). Rf 0,72(1). [М]+403,5. [α]D20-11,93° (С 0,18, MeOH). Знайдено, %: С 74,22; Η 6,92; Ν 3,57. C24H23NO4. Обчислено, %: С 74,42; Η 6,25; Ν 3,47. Приклад 15 Ацетилтирозилфенілетиламін (XXVIII) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,36г (50%). Rf 0,57(1). [М]+ 326,9. [α]D20+9,06° (С 0,30, MeOH). ІЧ-Фур'є, δ, см-1: 1651 (амід І); 1616 (амід II). Знайдено, %: С 69,22; Η 6,52; Ν 8,27. C24H23NO4. Обчислено, %: С 69,92; Η 6,79; Ν 8,58. Приклад 16 Ацетилтирозилтирамін (XXIX) Синтез проводили відповідно до методики А, наведеної для сполуки VII. Вихід 0,77г (65%). Rf 0,41(1). [M]+ 342,7. Знайдено, %: С 66,25; Η 6,32; Ν 8,25. C24H23NO4. Обчислено, %: С 66,65; Η 6,48; Ν 8,18. Тести на біологічну активність Приклад 17 Дослідження впливу сполук загальної формули І на метаболізм [14С]арахідонової кислоти в 34 безклітинному гомогенаті легеневої тканини миші in vitro Дослідження метаболізму арахідонової кислоти проводили на мишах-самках лінії СВА, що знаходилися на стандартному раціоні віварію. Тварин (мишей) забивали, витягали легені, гомогенізували в скляному гомогенізаторі фірми "Wheaton" (США) при 4°С у 10 об'ємах 0,05Μ трис-НСІ буфера. Аліквоти (0,5мл) супернатанту інкубували з 0,5мкКю [1-С14]-арахідонової кислоти ([С14]-АА), "Amersham", Англія; питома активність 5060мКю/ммоль) при 37°С протягом 30хв. Екстракцію неметаболізованої [С14]-АК і продуктів її метаболізму здійснювали в 20 об'ємах суміші хлороформу й метанолу (1:1), при ефективності екстракції не менше 90%, оціненої за допомогою [С14]-ПГF2α. Поділ і ідентифікацію [С14]-АА і її метаболітів здійснювали за допомогою ТШХ (пластини Kieselgel 60 фірми "Merck", Німеччина), з використанням як органічної фази системи розчинників (етилацетат, ізооктан, оцтова кислота, вода - 110:50:20:100) і мічених стандартів. Авторадіохроматограми, одержані на рентгенівській плівці X-Omat AR ("Kodak", США) і HS 11 ("ORWO", Німеччина), денситометрували на денсискані KS 3 ("Кірр and Zonnen", Голландія). Кількісний аналіз окремих ейкозаноїдів проведений за допомогою радіометрії фракцій, одержаних високоефективною рідинною хроматографією (ВЕРХ-система фірми "'Gilson", Франція; колонка ZORBAX С8 фірми "Du Pont", США) і елююванням плям на ТШХ-пластинках. Тестовані сполуки вводили в концентрації 10-4М. Одержані дані представлені в Таблиці 3. Таблиця 3 Вплив сполук загальної формули І (у концентрації 10-4Μ) на метаболізм [14С]арахідонової кислоти в безклітинному гомогенаті легеневої тканини миші in vitro № спол. IX Χ XIV XII VII VIII 6-кето-ПГТ1α -30 -9 -24 -42 -45 -45 ПГF2α -27 -15 -24 ТХВ2 -40 -42 -49 ПГЕ2 -38 -38 -54 -47 -32 -33 АК +47 +27 +84 +42 +22 +40 Простаноїди -33 -22 -35 -44 -40 -40 ПГ - простагландини ТХ - тромбоксан АК - арахідонова кислота Одержані дані по профілю ейкозаноїдів демонструють здатність сполук загальної формули І інгібувати циклооксигеназу на 22÷44% і свідчать про їхню перспективність як потенційних анальгетичних і протизапальних засобів. Приклад 18 Анальгетична й протизапальна активність сполук загальної формули (І) Дослідження анальгетичної активності на моделі "оцтові корчі" Тести проводили на білих безпородних мишах-самцях масою 22-24г. Специфічну больову реакцію ("корчі") викликали внутрішньочеревинним введенням мишам 0,75% розчину оцтової кислоти. Враховували кількість судорожних скорочень черевних м'язів, що супроводжуються витягуванням задніх кінцівок і прогинанням спини. Анальгетичну дію оцінювали по зменшенню числа корчів у тварин в % до контролю протягом 15 хв. після введення оцтової кислоти. Методика проведення тестів описана в Koster R., Anderson M., de Beer Ε. // Fed. Proc. 1959. V.18. P.412. Досліджувані сполуки вводили внутрішньошлунково (за допомогою зонда) у дозі 10мг/кг за 60хв. до ін'єкції кислоти. Як препарат порівняння 35 88688 використовували диклофенак (10мг/кг). Анальгетичний ефект розраховували за формулою: Ck − Co × 100, (%) Co 36 де Сk - кількість корчів у контрольній групі, Со кількість корчів у досліджуваній групі. Одержані дані представлені в Таблиці 4. Таблиця 4 Анальгетична активність досліджуваних сполук загальної формули І у дозі 10мг/кг у тесті "оцтові корчі" (число корчів за 15хв.) Сполука Число мишей С±m С, % до контролю II III контроль 1 IV V контроль 2 VIII контроль 3 IX контроль 4 X диклофенак 10мг/кг контроль 5 XI XII контроль 6 VI XIII Вольтарен 8 мг/кг контроль 7 XXVI XXVII контроль 8 XXX контроль 9 XXVIII XXIX контроль 10 10 8 10 10 10 10 8 8 8 8 8 8 8 10 10 9 8 8 8 8 8 9 9 8 8 9 10 9 24,2±1,9* 19,4±3,3* 32,2±1,6 20,8±1,9* 16,2±2,6* 26,7±0,79 16,0±4,5 36,8±3,5 11,8±2,9 36,8±3,5 11,0±2,4* 12,9±3,13* 25,4±2,4 21,2±2,5** 20,1±2,1** 34,3±3,0 21,1±1,8* 14,6±1,8** 15,8±2,6* 28,4±2,5 22,4±2,0* 20,1±1,7* 29,8±2,3 11,9±1,7** 18,6±1,4 15,9±2,4* 15,7±1,9* 27,4±2,6 75,2 60,2 100 77,9 60,7 100 43,5 100 32 100 46,0 50,8 100 61,8 58,6 100 74,5 51,6 55,5 100 73 67,4 100 63,9 100 57,9 57,1 100 Анальгетичний ефект (%) 24,8 39,9 22,1 39,3 56,5 68 54 49,2 0 38,2 41,4 25,5 48,4 44,5 27 32,6 36,1 42,1 42,9 * Ρ
ДивитисяДодаткова інформація
Назва патенту англійськоюPhenyl-containing n-acyl amine and aminoacid derivatives, methods for the production thereof, a pharmaceutical composition and the use thereof
Автори англійськоюNiebolsin Vladimir Yevhenievich, Kromova Tatiana Aleksandrovna, Zheltukhina Galina Aleksandrovna, Kovaliova Violietta Leonidovna
Назва патенту російськоюФенилсодержащие n-ацилные производные аминов и аминокислот, способ их получения, фармацевтическая композиция и их применение
Автори російськоюНебольсин Владимир Евгеньевич, Кромова Татьяна Александровна, Желтухина Галина Александровна, Ковалёва Виолетта Леонидовна
МПК / Мітки
МПК: A61K 31/165, C07C 237/22, A61P 29/00, C07C 233/51, C07K 5/065, A61P 25/16, C07C 237/20, A61K 38/05
Мітки: амінів, n-ацильні, застосування, похідні, спосіб, амінокислот, композиція, фармацевтична, фенілвмісні, одержання
Код посилання
<a href="https://ua.patents.su/24-88688-fenilvmisni-n-acilni-pokhidni-aminiv-i-aminokislot-sposib-kh-oderzhannya-farmacevtichna-kompoziciya-i-kh-zastosuvannya.html" target="_blank" rel="follow" title="База патентів України">Фенілвмісні n-ацильні похідні амінів і амінокислот, спосіб їх одержання, фармацевтична композиція і їх застосування</a>
Похідні індолу, фармацевтична композиція та спосіб одержання сполук (варіанти)

Номер патенту: 68388
Опубліковано: 16.08.2004
Автори: Хьонке Хрістоф, Бєхтєль Вольф-Дітріх, Вайзер Томас, Паллук Райнер, Пшорн Уве, Грауерт Маттіас, Картер Адріан
МПК: C07D 401/04, C07D 209/04, A61P 25/28, A61P 9/10, A61P 9/06, A61K 31/404, C07D 209/60, A61P 23/02
Мітки: похідні, фармацевтична, сполук, спосіб, індолу, композиція, варіанти, одержання
Формула / Реферат:
1. Похідні індолу загальної формули 1, 1деХ означає простий зв'язок, -О-, С1-С4алкіл, С1-С3алкоксигрупу, -О-СН2-СН2-О- чи -O-CH2-CH2-NH-,R1 означає водень, метил, етил чи феніл, R2 означає водень або метил,R3 означає водень, F, Сl, Br, гідрокси- чи метоксигрупу, R4 означає водень, метил чи етил, R5 означає водень, метил чи етил, R6 означає водень, метил чи етил, R7 означає...
Похідні амінокислот, спосіб їх одержання і фармацевтична композиція, що їх містить

Номер патенту: 77763
Опубліковано: 15.01.2007
Автори: Верберен Тоні, Рюпен Ален, ГЛОАНЕК Філіп, Де Нантей Гійом
МПК: A61P 7/04, C07D 231/56, A61K 31/4409, C07D 213/68, A61K 31/27, A61K 31/5375, C07D 221/04, C07K 5/065, C07D 295/22, C07C 257/00, C07D 213/73, C07C 303/00, A61K 31/223, C07C 311/10, A61K 31/18, C07D 295/26, A61K 31/44, C07D 261/20, C07K 5/06, A61P 43/00, C07C 259/00, C07C 311/19, A61K 31/416, A61K 31/423, A61K 31/198, C07D 295/08, A61K 31/435, A61K 31/277, C07C 311/13, A61K 31/165, C07D 213/34
Мітки: одержання, амінокислот, спосіб, містить, фармацевтична, похідні, композиція
Формула / Реферат:
1. Сполука формули (І): , (I)деR1 являє собою арильну групу, гетероарильну групу або лінійну чи розгалужену (С1-С6)алкільну групу, необов'язково заміщену однією або більшою кількістю однакових або різних груп, вибраних з арилу і гетероарилу, або R1 являє собою групу формули -(CO)-CR6R7NR8R9, де:- R6 являє собою атом водню або групу, вибрану з...
Гетероцикло-циклічні похідні амінів або їх фармацевтично прийнятні солі, проміжні сполуки, фармацевтична композиція, спосіб інгібування холінестерази, спосіб одержання гетероцикло-циклічних похідних амінів
Номер патенту: 45944
Опубліковано: 15.05.2002
Автори: Віллалобос Анабелла, Чен Юпанг Ліанг, Нейджел Артур Адам
МПК: C07D 211/32, C07D 239/74, A61K 31/415, C07D 417/06, C07D 211/60, A61K 31/496, C07D 231/56, C07D 211/24, C07D 413/14, A61K 31/472, A61K 31/505, A61K 31/4427, C07D 413/12, C07D 217/14, A61K 31/41, C07D 211/30, C07D 498/04, C07D 413/06, A61K 31/517, A61K 31/47, C07D 491/056, A61K 31/425, C07D 513/04, A61P 25/28, C07D 211/38, A61K 31/445, C07D 253/00, C07D 261/20, C07D 401/06, A61P 25/02, A61K 31/451, A61P 43/00, C12N 9/99, C07D 241/42
Мітки: сполуки, похідні, фармацевтично, похідних, фармацевтична, холінестерази, одержання, амінів, гетероцикло-циклічні, проміжні, композиція, прийнятні, гетероцикло-циклічних, спосіб, інгібування, солі
Формула / Реферат:
1. Гетероцикло-циклические производные аминов общей формулы (I): (I)где R1 и R2 независимо выбирают из водорода, (С1-С6)алкокси, бензилокси, фенокси, гидрокси, фенила, бензила, галогена, нитро, циано, COR5, COOR5, CONHR5, NR5R6, -NR5OR6, OCON5R6, -NHCOOR5, (С1-С6)алкила, необязательно замещенного 1-3 атомами фтора, SOpCH2-фенила или SOр(С1-С6)алкила, где р = 0, 1 или 2, пиридилметилокси, тиенилметилокси, 2-оксазолила,...
Похідні ехінокандину, спосіб їх одержання, їх застосування, проміжні сполуки та фармацевтична композиція

Номер патенту: 72200
Опубліковано: 15.02.2005
Автори: Шио Лоран, Мішель Жан-Марк, Мелон Мангер Домінік, Куртен Олів'є, Фово Патрік, Маркус Астрид
МПК: C07K 7/56, A61P 31/10, A61K 38/04
Мітки: ехінокандину, застосування, композиція, проміжні, фармацевтична, похідні, сполуки, одержання, спосіб
Формула / Реферат:
1. Похідні ехінокандину формули (І):(І),їх можливі ізомерні форми або їх суміші, в яких:або R1 і R2, однакові або відмінні один від одного, означають атом водню, гідроксил; лінійний, розгалужений або циклічний алкільний, алкенільний або алкінільний радикал, що містить до 8 атомів вуглецю, що можливо переривається атомом кисню, можливо заміщений атомом...
Похідні пептидів, їх застосування, спосіб одержання, спосіб лікування або профілактики, фармацевтична композиція та косметичний засіб

Номер патенту: 70301
Опубліковано: 15.10.2004
Автори: Небольсин Володимир Євгенійович, Євстигнєєва Рима Порфіріївна, Желтухіна Галина Олександрівна
МПК: C07D 207/09, C07D 213/40, C07D 333/20, C07D 209/16, C07K 5/037, C07D 233/54, C07D 401/12, C07D 521/00
Мітки: профілактики, спосіб, пептидів, похідні, одержання, композиція, лікування, застосування, фармацевтична, косметичний, засіб
Формула / Реферат:
1. Похідні пептидів загальної формули (І) (I)або їх фармацевтично прийнятні солі, де R1 означає атом водню або С1-С3-вуглеводневий радикал, заміщений функціональною групою, вибраною з аміно-, С1-С5-амідо-, С1-С7-уретано- або карбоксильної груп, причому карбоксильна група може бути етерифікована, а аміногрупа може бути заміщена ацильним замісником або ефіром вугільної кислоти; або С1-С3-вуглеводневий радикал, одночасно заміщений...
Попередній патент: Бурова вишка та гідравлічний механізм подачі
Наступний патент: Спосіб одержання фторапатиту кальцію у вигляді плівок або порошку
Випадковий патент: Пристрій для нанесення покриття на внутрішню поверхню виробів