Спосіб одержання інсулінотропних пептидів












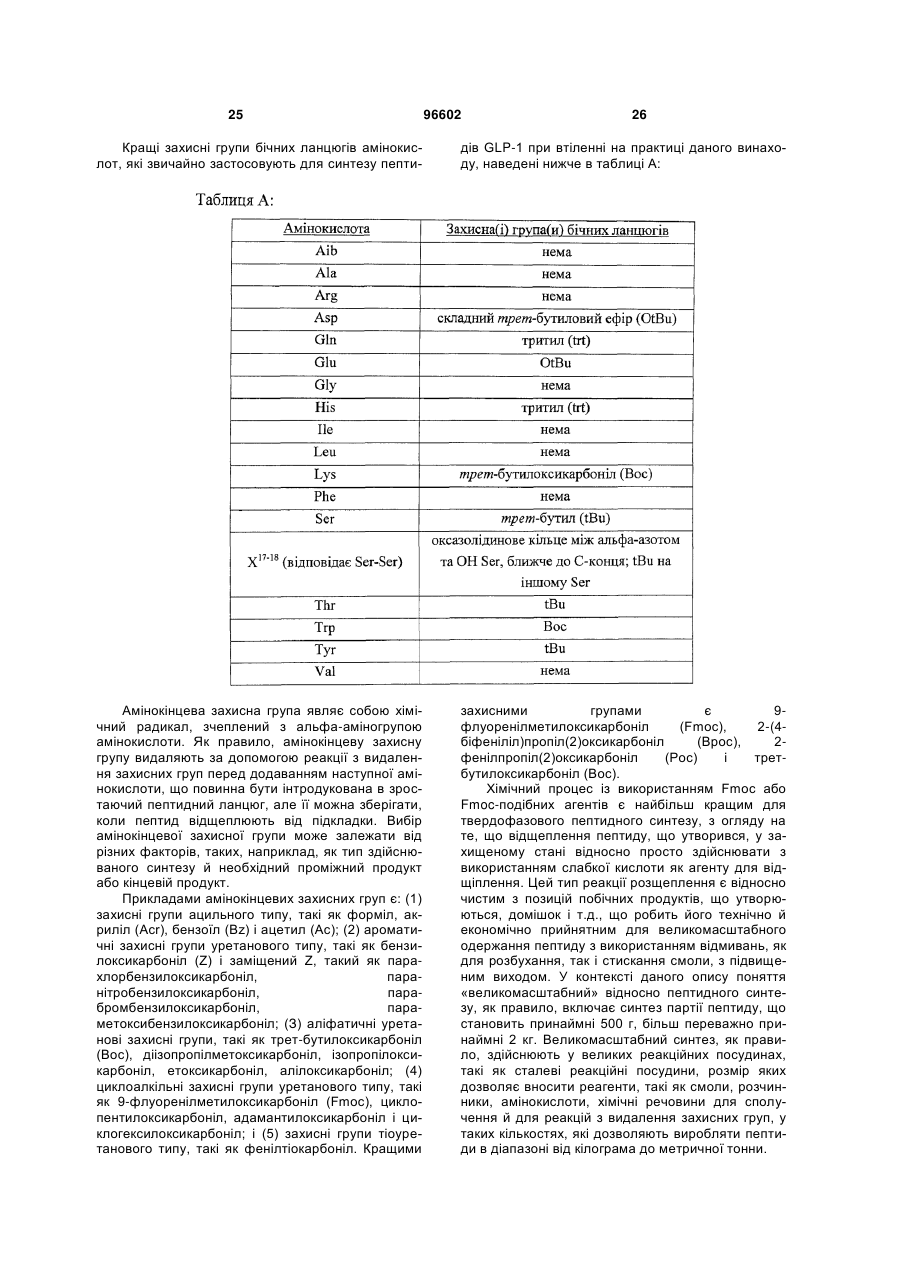


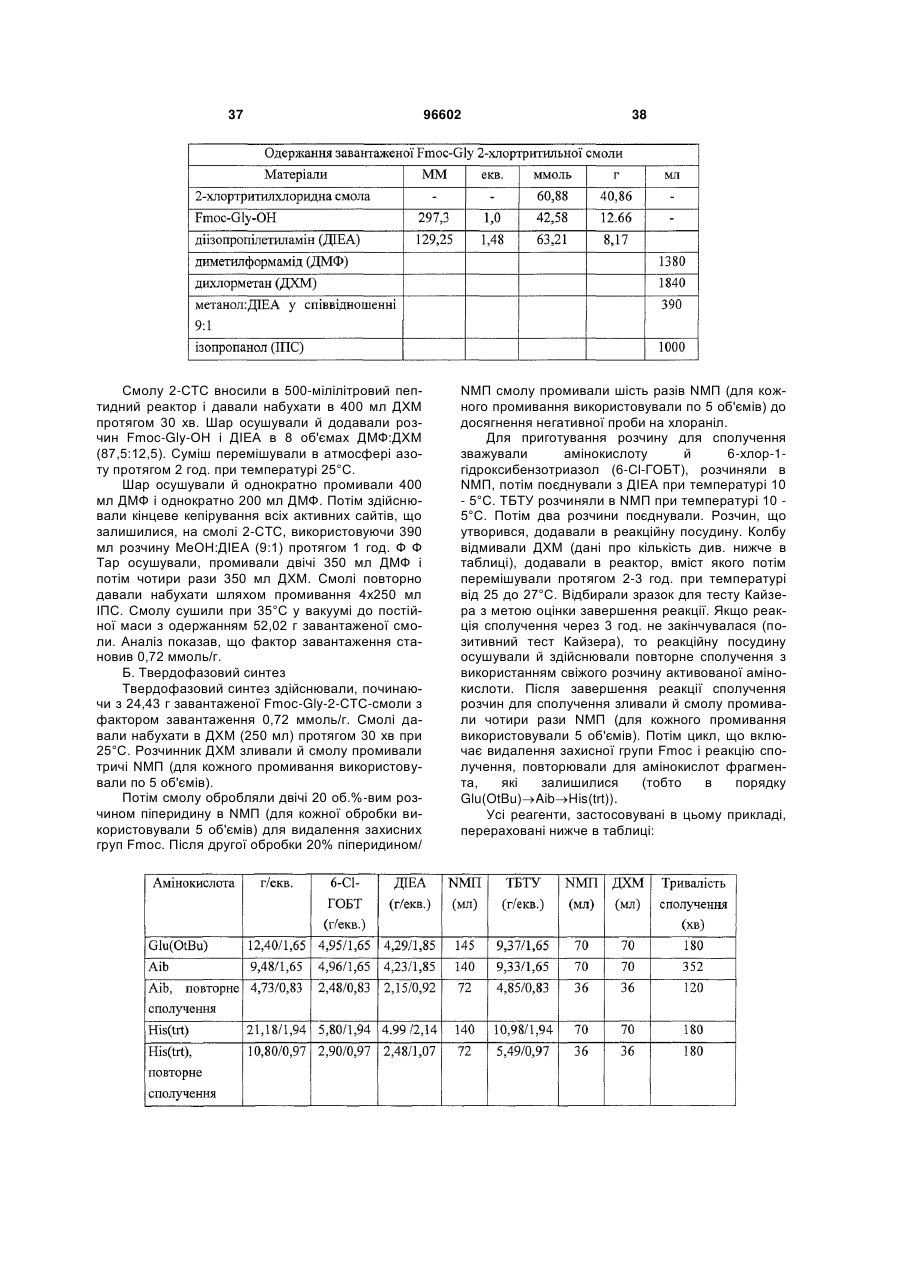
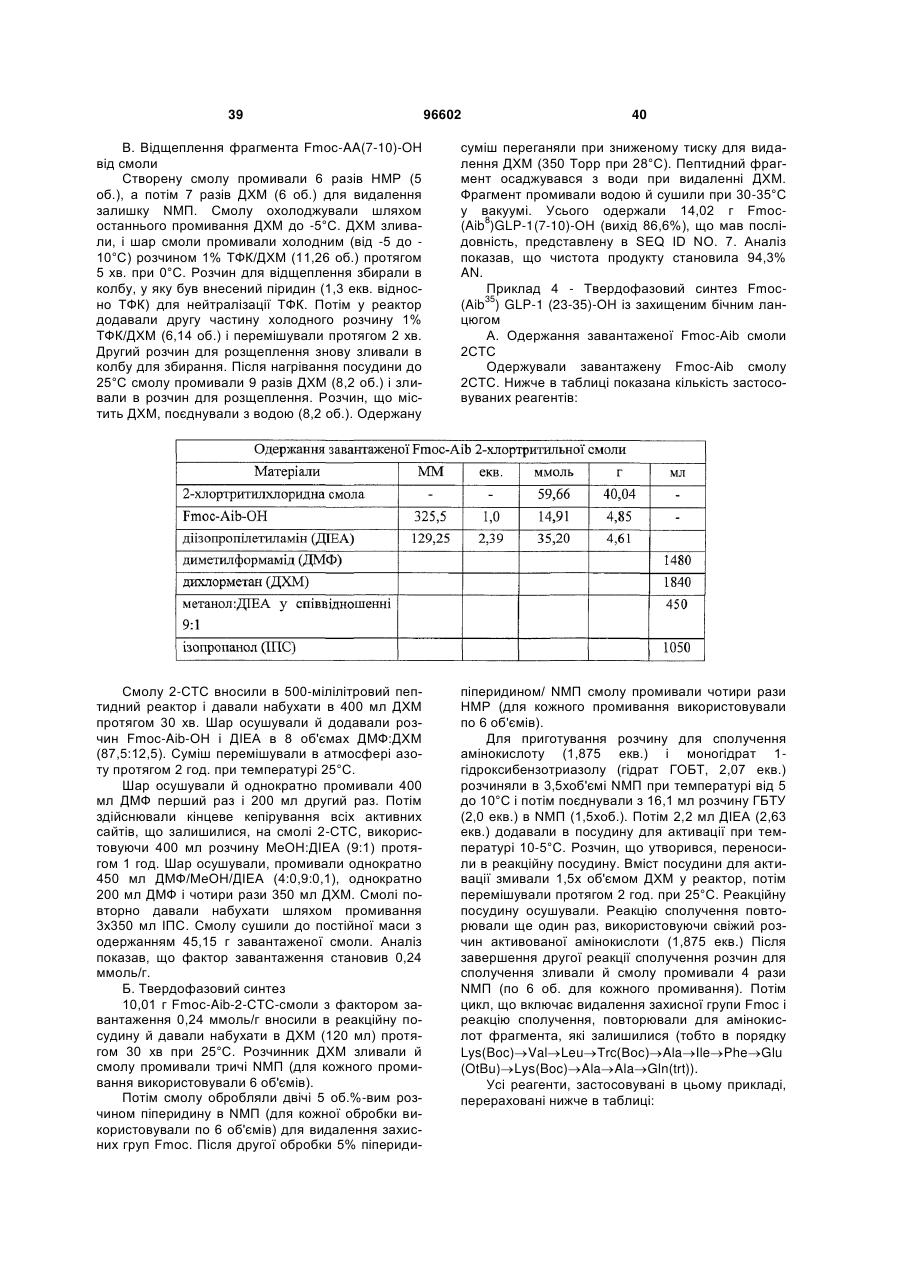
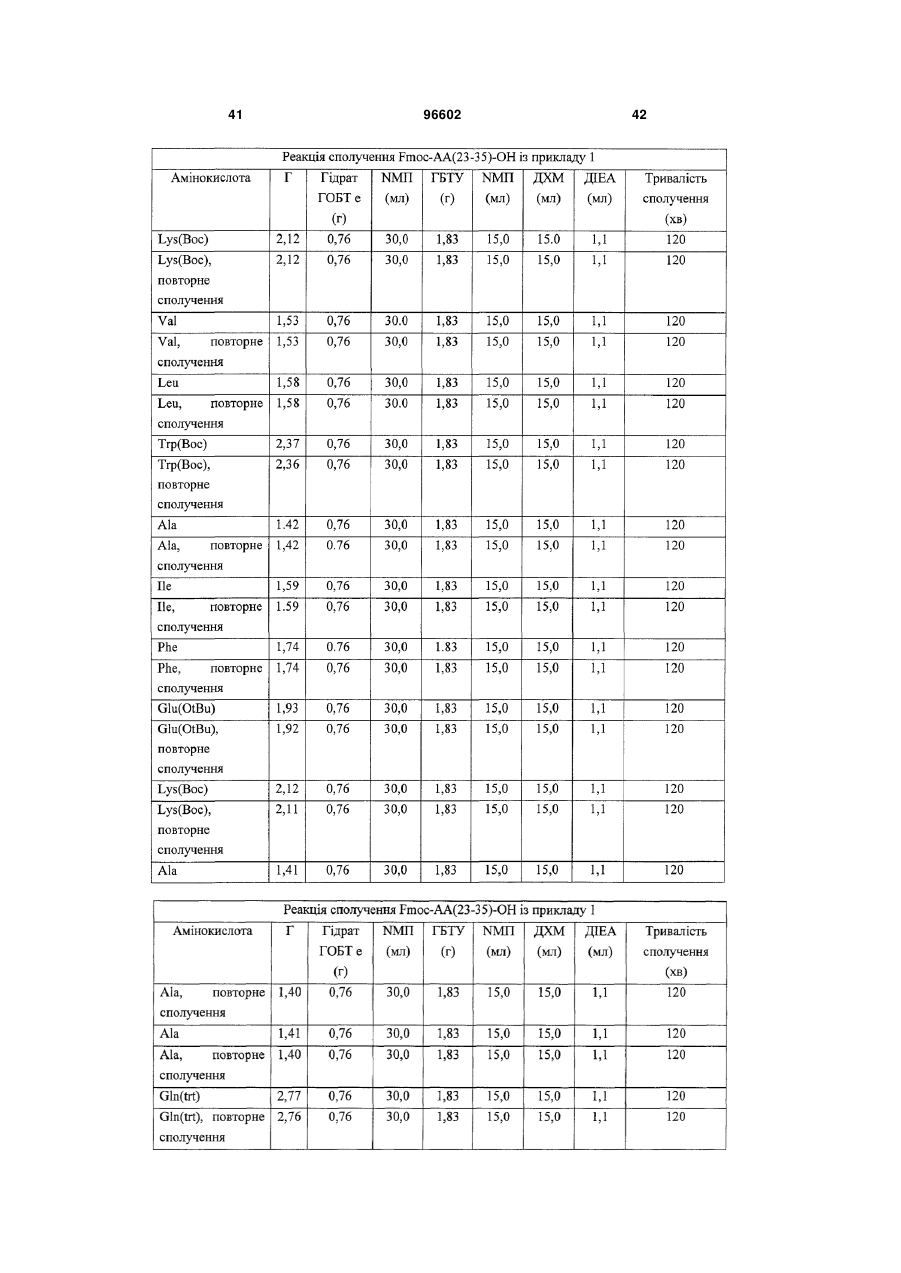
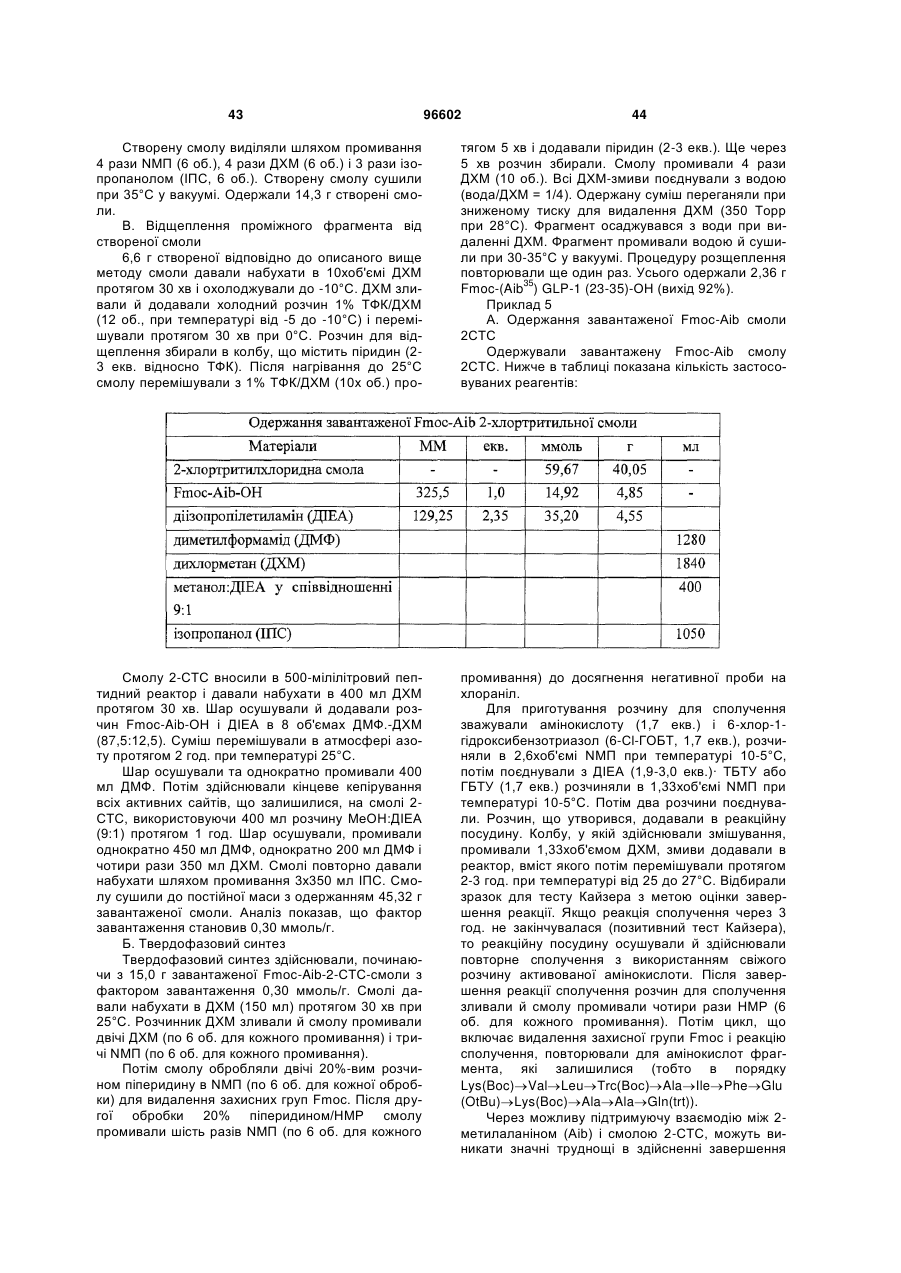









Формула / Реферат
1. Спосіб одержання інсулінотропного пептиду, який включає наступні стадії а) – е), на яких:
а) одержують перший пептидний фрагмент, який включає амінокислотну послідовність HX8EX10 (SEQ ID NO: 6), у якій X8 і X10 кожний означає залишки ахіральної амінокислоти, H і E кожний необов'язково несе захисну групу бічного ланцюга;
б) одержують другий пептидний фрагмент, який включає амінокислотну послідовність TFTSDVX17-18YLEG (SEQ ID NO: 8), у якій залишок, позначений символом X17-18, являє собою дипептидний залишок псевдопроліну, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга;
в) здійснюють сполучення першого фрагмента із другим фрагментом з одержанням третього пептидного фрагмента, який включає амінокислотну послідовність HX8EX10 TFTSDVX17-18YLEG (SEQ ID NO: 11), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга;
г) одержують четвертий пептидний фрагмент, який включає амінокислотну послідовність QAAKEFIAWLVKX35 (SEQ ID NO: 9), у якій X35 означає залишок ахіральної амінокислотної кислоти, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга;
д) здійснюють сполучення четвертого пептидного фрагмента з аргініном з одержанням п'ятого пептидного фрагмента, який включає амінокислотну послідовність QAAKEFIAWLVK X35R (SEQ ID NO: 12), де зазначені залишки послідовності необов'язково несуть захисну групу бічного ланцюга; і
е) здійснюють сполучення п'ятого фрагмента із третім фрагментом з одержанням інсулінотропного пептиду, який включає амінокислотну послідовність HX8EX10TFTSDVX17-18YLEGQAAKEFIAWLVK X35R (SEQ ID NO: 13), де зазначені залишки послідовності необов'язково несуть захисну групу бічного ланцюга.
2. Спосіб за п. 1, який полягає у тому, що додатково включає стадію, на якій:
ж) видаляють захисні групи бічних ланцюгів з одержанням інсулінотропного пептиду, який включає амінокислотну послідовність HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R (SEQ ID NO: 5), і його аналогів, де кожний із символів Х у положеннях 8, 10 і 35 незалежно один від одного означає ахіральний амінокислотний залишок, що необов’язково має стеричну перешкоду.
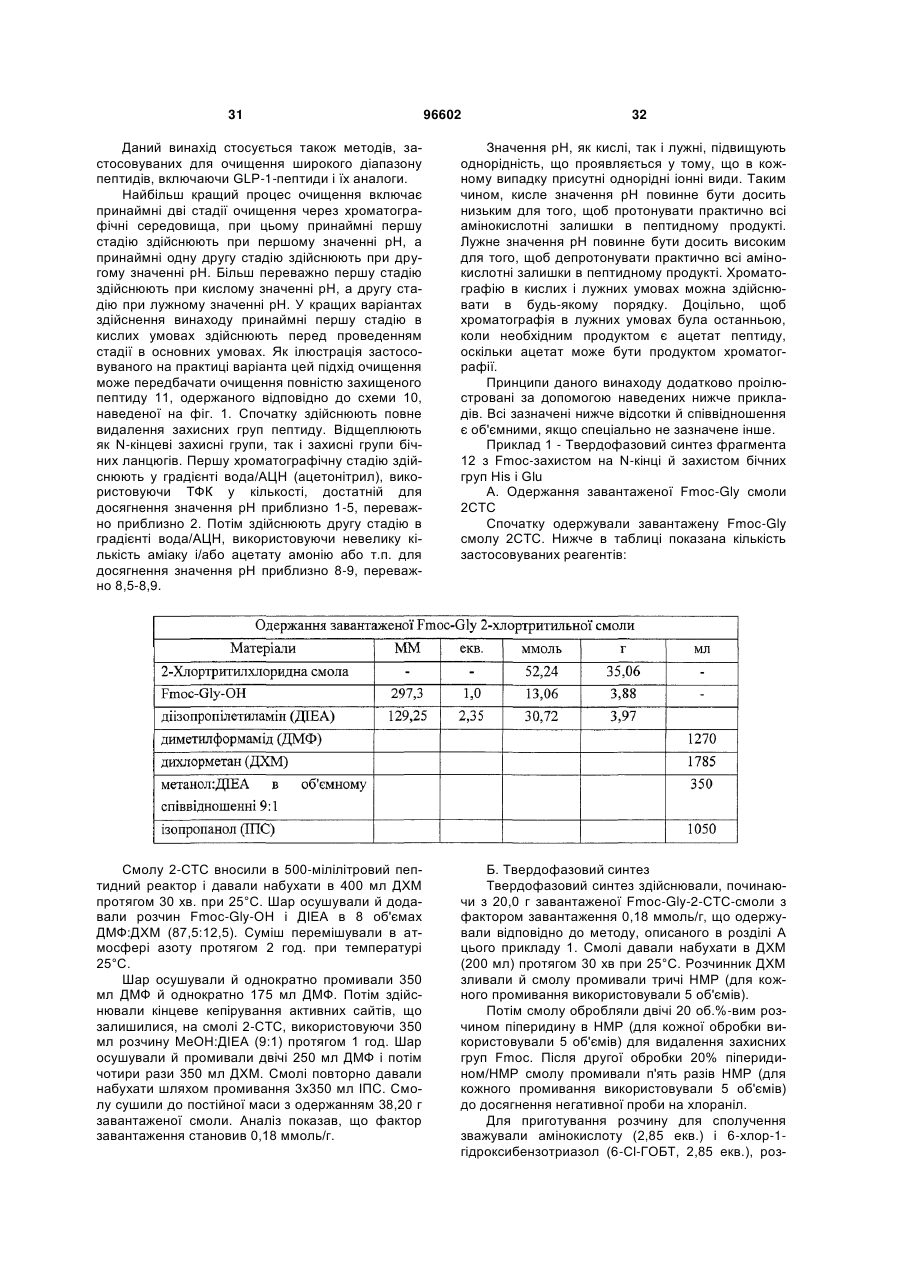
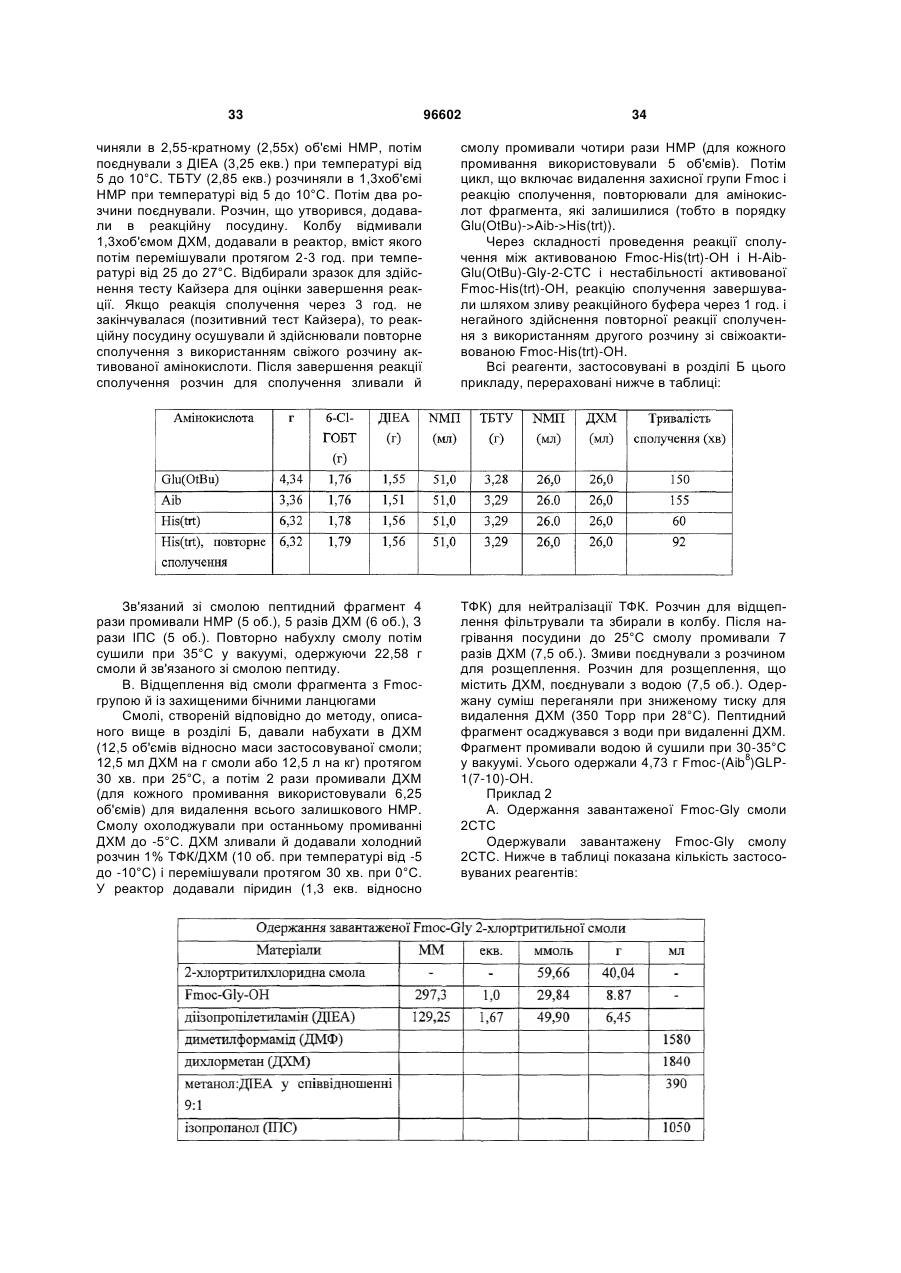
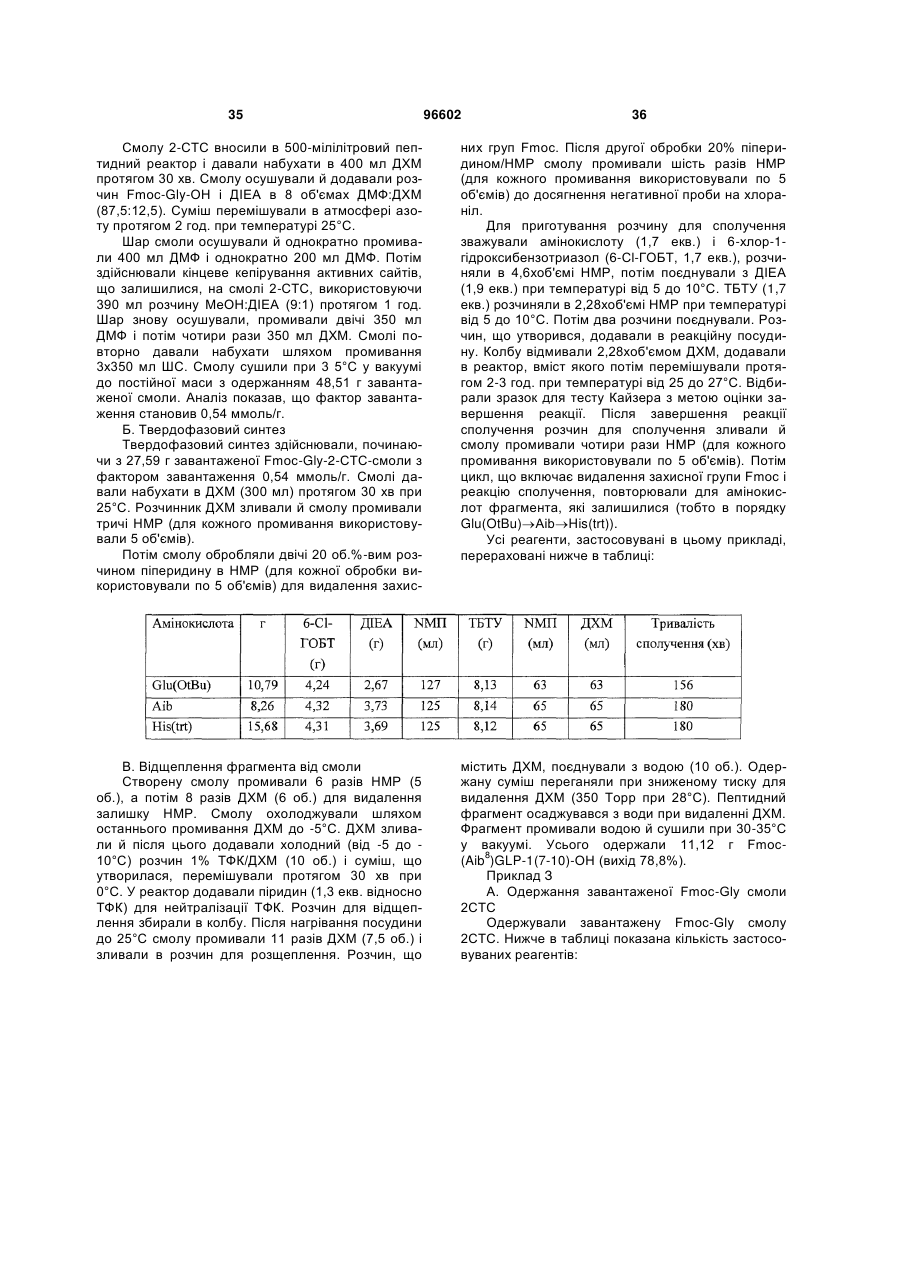
3. Спосіб одержання інсулінотропного пептиду за п. 1, який включає стадії, на яких:
а) одержують перший пептидний фрагмент, який включає амінокислотну послідовність HX8EX10 (SEQ ID NO: 6), у якій X8 означає амінокислотний залишок, що відповідає Aib, а X10 означає амінокислотний залишок, що відповідає гліцину, H і E кожний необов'язково несе захисну групу бічного ланцюга;
б) одержують другий пептидний фрагмент, який включає амінокислотну послідовність TFTSDVX17-18YLEG (SEQ ID NO: 8), у якій залишок, позначений символом X17-18, являє собою дипептидний залишок псевдопроліну, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга;
в) здійснюють сполучення першого фрагмента із другим фрагментом з одержанням третього пептидного фрагмента, який включає амінокислотну послідовність HX8EX10 TFTSDV X17-18YLEG (SEQ ID NO: 11), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга;
г) одержують четвертий пептидний фрагмент, який включає амінокислотну послідовність QAAKEFIAWLVKX35 (SEQ ID NO: 9), у якій X35 означає амінокислотний залишок, що відповідає Aib, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга;
д) здійснюють сполучення четвертого пептидного фрагмента з аргініном з одержанням п'ятого пептидного фрагмента, який включає амінокислотну послідовність QAAKEFIAWLVK X35R (SEQ ID NO: 12), де зазначені залишки послідовності необов'язково несуть захисну групу бічного ланцюга; і
е) здійснюють сполучення п'ятого фрагмента із третім фрагментом з наступним видаленням захисних груп бічних ланцюгів з одержанням інсулінотропного пептиду формули HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R (SEQ ID NО: 5) і його аналогів, де X8 і X35 означають амінокислотні залишки, що відповідають Aib, а X10 означає амінокислотний залишок, що відповідає гліцину.
4. Спосіб за п. 1 або п. 3, у якому X8 означає амінокислотний залишок, що відповідає метилаланіну.
5. Спосіб за п. 1 або п. 3, у якому X10 означає амінокислотний залишок, що відповідає гліцину.
6. Пептидний фрагмент, який має амінокислотну послідовність HX8EX10 (SEQ ID NO: 6), у якій X8 і X10 кожний означає залишки ахіральної амінокислоти, де H, E, X8 і X10 кожний необов'язково несе захисну групу бічного ланцюга.
7. Пептидний фрагмент за п. 6, у якому X8 означає амінокислотний залишок, що відповідає Aib, а X10 означає амінокислотний залишок, що відповідає гліцину.
8. Спосіб за п. 1, у якому інсулінотропний пептид включає амінокислотну послідовність (SEQ ID NО: 5)
HX8EX10TFTSDVSSYLEGQAAKEFIAWLVKX35R
і його аналоги, де кожний із символів X у положеннях 8, 10 і 35 незалежно один від одного означає ахіральний амінокислотний залишок, що необов’язково має стеричну перешкоду; і де один або декілька амінокислотних залишків необов'язково несуть захисну групу бічного ланцюга.
9. Спосіб за п. 8, у якому принаймні один із символів X8 і X35 означає залишок Aib.
10. Спосіб за п. 8, у якому X10 означає залишок гліцину.
11. Спосіб за п. 1, у якому X17-18 має формулу
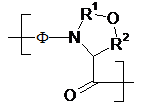 ,
,
у якій Φ означає залишок будь-якої амінокислоти, яка необов’язково несе захисну групу бічного ланцюга, та R1 і R2 кожний незалежно один від одного означає прийнятний двовалентний сполучний радикал.
12. Спосіб за п. 11, у якому Φ означає залишок Ser, який необов’язково несе захисну групу бічного ланцюга.
13. Спосіб за п. 11, у якому R2 означає –CH2-.
14. Спосіб за п. 11, у якому R1 означає
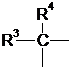 ,
,
де R3 і R4 кожний незалежно один від одного означає одновалентний радикал, вибраний з H або (низч.)алкілу; або R3 і R4 можуть також бути членами кільцевої структури.
15. Спосіб за п. 14, у якому R3 і R4 кожний означає метил.
16. Пептид або його аналог, які включають амінокислотну послідовність TFTSDVX17-18YLEG (SEQ ID NO: 8), у якій залишок, позначений символом X17-18, являє собою дипептидний залишок псевдопроліну; де зазначені амінокислотні залишки необов'язково несуть захисну групу бічного ланцюга.
17. Спосіб за п. 1, у якому X35 означає амінокислотний залишок метилаланіну.
18. Спосіб за пп. 1-2, у якому інсулінотропний пептид являє собою пептид, який має амінокислотну послідовність (SEQ ID NО: 4)
HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR,
або його аналог.
19. Спосіб за п. 18, у якому інсулінотропний пептид являє собою пептид, який має амінокислотну послідовність (SEQ ID NО: 4)
HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR,
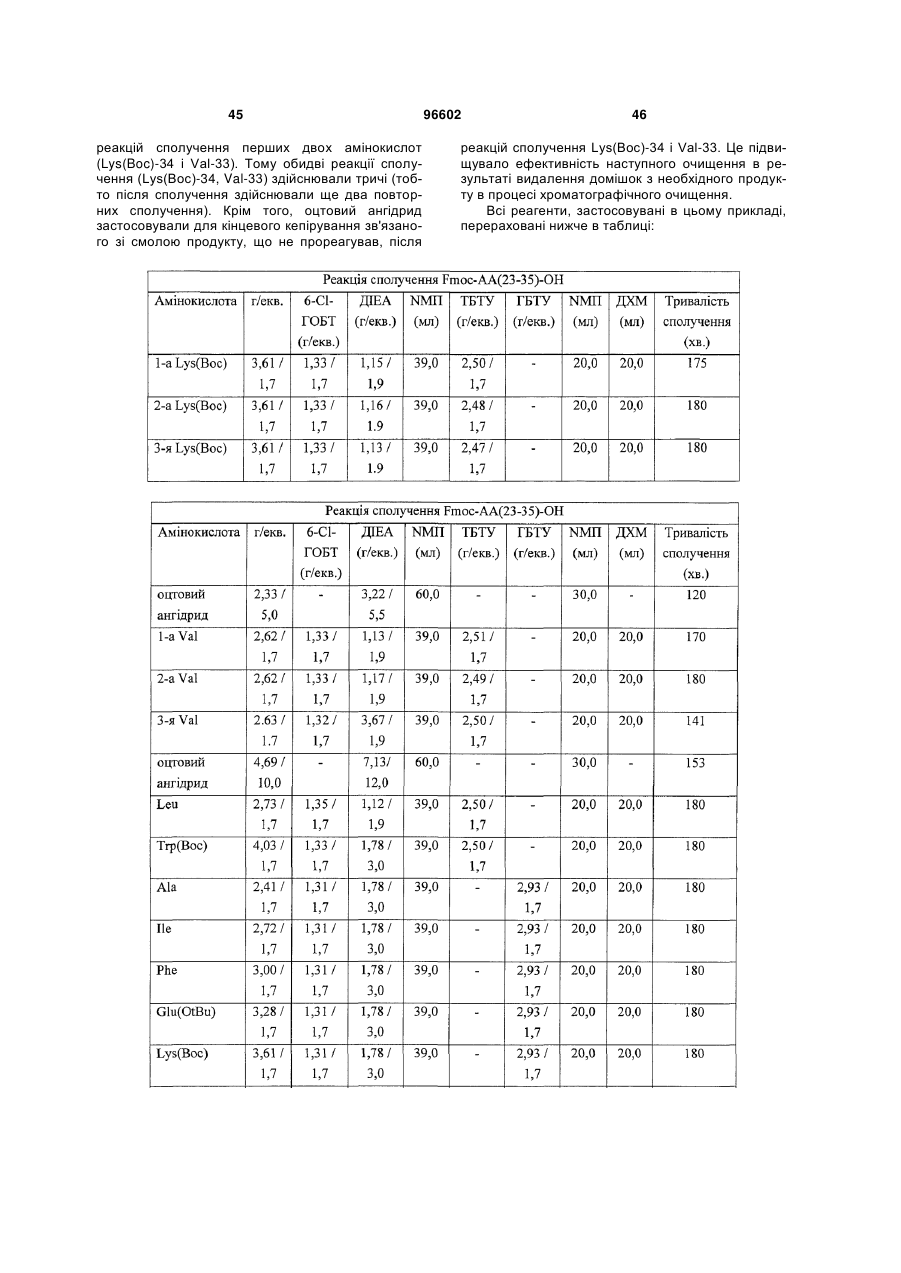
або його аналог, амідований на C-кінці.
Текст
1. Спосіб одержання інсулінотропного пептиду, який включає наступні стадії а) – е), на яких: а) одержують перший пептидний фрагмент, який 8 10 включає амінокислотну послідовність HX EX 8 10 (SEQ ID NO: 6), у якій X і X кожний означає за 2 (19) 1 3 3. Спосіб одержання інсулінотропного пептиду за п. 1, який включає стадії, на яких: а) одержують перший пептидний фрагмент, який 8 10 включає амінокислотну послідовність HX EX 8 (SEQ ID NO: 6), у якій X означає амінокислотний 10 залишок, що відповідає Aib, а X означає амінокислотний залишок, що відповідає гліцину, H і E кожний необов'язково несе захисну групу бічного ланцюга; б) одержують другий пептидний фрагмент, який 17включає амінокислотну послідовність TFTSDVX 18 YLEG (SEQ ID NO: 8), у якій залишок, позначений 17-18 символом X , являє собою дипептидний залишок псевдопроліну, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; в) здійснюють сполучення першого фрагмента із другим фрагментом з одержанням третього пептидного фрагмента, який включає амінокислотну 8 10 17-18 послідовність HX EX TFTSDV X YLEG (SEQ ID NO: 11), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; г) одержують четвертий пептидний фрагмент, який включає амінокислотну послідовність 35 35 QAAKEFIAWLVKX (SEQ ID NO: 9), у якій X означає амінокислотний залишок, що відповідає Aib, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; д) здійснюють сполучення четвертого пептидного фрагмента з аргініном з одержанням п'ятого пептидного фрагмента, який включає амінокислотну 35 послідовність QAAKEFIAWLVK X R (SEQ ID NO: 12), де зазначені залишки послідовності необов'язково несуть захисну групу бічного ланцюга; і е) здійснюють сполучення п'ятого фрагмента із третім фрагментом з наступним видаленням захисних груп бічних ланцюгів з одержанням інсулінотропного пептиду формули 8 10 35 HX EX TFTSDVSSYLEGQAAKEFIAWLVKX R 8 35 (SEQ ID NО: 5) і його аналогів, де X і X означають амінокислотні залишки, що відповідають Aib, а 10 X означає амінокислотний залишок, що відповідає гліцину. 8 4. Спосіб за п. 1 або п. 3, у якому X означає амінокислотний залишок, що відповідає метилаланіну. 10 5. Спосіб за п. 1 або п. 3, у якому X означає амінокислотний залишок, що відповідає гліцину. 6. Пептидний фрагмент, який має амінокислотну 8 10 8 послідовність HX EX (SEQ ID NO: 6), у якій X і 10 X кожний означає залишки ахіральної амінокис8 10 лоти, де H, E, X і X кожний необов'язково несе захисну групу бічного ланцюга. 8 7. Пептидний фрагмент за п. 6, у якому X означає 10 амінокислотний залишок, що відповідає Aib, а X означає амінокислотний залишок, що відповідає гліцину. 8. Спосіб за п. 1, у якому інсулінотропний пептид включає амінокислотну послідовність (SEQ ID NО: 5) 8 10 35 HX EX TFTSDVSSYLEGQAAKEFIAWLVKX R 96602 4 і його аналоги, де кожний із символів X у положеннях 8, 10 і 35 незалежно один від одного означає ахіральний амінокислотний залишок, що необов’язково має стеричну перешкоду; і де один або декілька амінокислотних залишків необов'язково несуть захисну групу бічного ланцюга. 9. Спосіб за п. 8, у якому принаймні один із симво8 35 лів X і X означає залишок Aib. 10 10. Спосіб за п. 8, у якому X означає залишок гліцину. 17-18 11. Спосіб за п. 1, у якому X має формулу 1 R O N 2 R O , у якій Φ означає залишок будь-якої амінокислоти, яка необов’язково несе захисну групу бічного лан1 2 цюга, та R і R кожний незалежно один від одного означає прийнятний двовалентний сполучний радикал. 12. Спосіб за п. 11, у якому Φ означає залишок Ser, який необов’язково несе захисну групу бічного ланцюга. 2 13. Спосіб за п. 11, у якому R означає –CH2-. 1 14. Спосіб за п. 11, у якому R означає R R 3 4 C , 3 4 де R і R кожний незалежно один від одного означає одновалентний радикал, вибраний з H або 3 4 (низч.)алкілу; або R і R можуть також бути членами кільцевої структури. 3 4 15. Спосіб за п. 14, у якому R і R кожний означає метил. 16. Пептид або його аналог, які включають аміно17-18 кислотну послідовність TFTSDVX YLEG (SEQ ID NO: 8), у якій залишок, позначений символом 17-18 X , являє собою дипептидний залишок псевдопроліну; де зазначені амінокислотні залишки необов'язково несуть захисну групу бічного ланцюга. 35 17. Спосіб за п. 1, у якому X означає амінокислотний залишок метилаланіну. 18. Спосіб за пп. 1-2, у якому інсулінотропний пептид являє собою пептид, який має амінокислотну послідовність (SEQ ID NО: 4) HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR, або його аналог. 19. Спосіб за п. 18, у якому інсулінотропний пептид являє собою пептид, який має амінокислотну послідовність (SEQ ID NО: 4) HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR, або його аналог, амідований на C-кінці. 5 Даний винахід стосується способів одержання інсулінотропних пептидів, насамперед глюкагонподібного пептиду-1 (GLP-1) і його аналогів, за допомогою методів твердофазового та рідиннофазового синтезу. Даний винахід стосується також пептидних фрагментів, які являють собою проміжні продукти, які можна застосовувати в зазначених способах. У літературі описані численні методи пептидного синтезу (див., наприклад, US 6015881; Mergler і ін., Tetrahedron Letters 29, 1988, ее. 40054008; Mergler і ін., Tetrahedron Letters 29, 1988, ее. 4009-4012; Peptides, Chemistry and Biology, під ред. Kamber і ін., вид-во ESCOM, Leiden, 1992, ее. 525-526; Riniker і ін., Tetrahedron Letters 49, 1993, ее. 9307-9320; Lloyd-Williams і ін., Tetrahedron Letters 49, 1993, cc. 11065-11133; і Andersson і ін., Biopolymers 55, 2000, cc. 227-250). Різні методи синтезу відрізняються фізичним станом фази, у якій протікає синтез, а саме тим, чи являє вона собою рідку фазу або тверду фазу. При твердофазовому пептидному синтезі (SPPS) амінокислоту або пептидну групу зв'язують із твердою підкладкою, що являє собою смолу. Потім до зв'язаного з підкладкою пептиду приєднують наступні амінокислоти або пептидні групи до одержання пептидного продукту, який являє інтерес. Після цього зв'язаний з підкладкою пептид, як правило, відщеплюють від підкладки й піддають додатковому процесуванню і/або очищенню. У деяких випадках твердофазовий синтез дозволяє одержувати зрілий пептидний продукт; в інших випадках пептид, відщеплений від підкладки (тобто пептидний фрагмент, що являє собою проміжний продукт («пептидний проміжний фрагмент»)) застосовують для одержання більшого зрілого пептидного продукту. Пептидні проміжні фрагменти, одержані у результаті твердофазових процесів, можна з'єднувати один з одним методом твердофазового синтезу або синтезу в розчині (далі в контексті даного опису позначений як «рідиннофазовий синтез»). Рідиннофазовий синтез найбільш доцільно застосовувати у випадках, коли синтез необхідного зрілого пептиду на твердій фазі або неможливий, або його важко здійснити на практиці. Наприклад, при твердофазовому синтезі більш довгі пептиди можуть випадково приймати неправильну конформацію, залишаючись при цьому зв'язаними із твердою підкладкою, що утруднює приєднання додаткових амінокислот або пептидного продукту до ланцюга, що подовжується. В міру подовження пептидого ланцюга на смолі, що являє собою підкладку, ефективність стадій процесу, таких як сполучення й видалення захисних груп, може знижуватися. Це у свою чергу може приводити крім зростаючої втрати вихідних продуктів, таких як здатні до активації амінокислоти, кореагенти та розчинники, до подовження часу процесування для компенсації зазначених проблем. Зазначені проблеми можуть збільшуватися в міру подовження пептиду. 96602 6 Таким чином, досить важко знайти зрілі пептиди, що складаються більш ніж з 30 амінокислот, синтезовані у вигляді одного фрагмента з використанням тільки методу твердофазового синтезу. Замість цього на твердій фазі можна синтезувати окремо індивідуальні фрагменти, а потім здійснювати їх сполучення за допомогою твердофазового і/або рідиннофазового синтезу з одержанням необхідного пептидного продукту. При використанні цього підходу потрібен ретельний вибір фрагментів-кандидатів. Хоча при виборі фрагментів можна користуватися деякими загальними принципами, досить часто потрібна емпірична оцінка фрагментів-кандидатів. Стратегії вибору фрагментів, які працюють в одних умовах, можуть не працювати в інші. Навіть коли виявлені прийнятні фрагментикандидати, можуть ще знадобитися інновації процесу для вибору стратегії синтезу, що може працювати в прийнятних з позицій вартості умовах. Таким чином, пептидний синтез, заснований на використанні гібридних схем, часто викликає питання й у багатьох випадках важко передбачити, які проблеми пов'язані з конкретною схемою синтезу, до фактичного здійснення синтезу. При сполученні в рідкій фазі два пептидних проміжних фрагменти або пептидний проміжний фрагмент і реактивну амінокислоту зв'язують у відповідному розчиннику, як правило, у присутності додаткових реагентів, які підвищують ефективність і якість реакції сполучення. Пептидні проміжні фрагменти в процесі реакції розташовуються таким чином, що N-кінець одного фрагмента виявляється зв'язаним з С-кінцем іншого фрагмента або навпаки. Крім того, захисні групи бічних ланцюгів, які присутні при твердофазовому синтезі, як правило, зберігають на фрагментах при сполученні в рідкій фазі для гарантії специфічної реактивності кінців фрагментів. Ці захисні групи бічних ланцюгів, як правило, не видаляють до одержання зрілого пептиду. Невеликі вдосконалення однієї або декількох стадій у загальній схемі синтезу можуть приводити до істотних покращень одержання зрілого пептиду. Такі покращення можуть приводити до істотного загального зниження часу одержання й споживання реагентів, а також можуть істотно підвищувати чистоту й вихід кінцевого продукту. Хоча обговорення важливості вдосконалень гібридного синтезу можна проводити для будьякого типу пептиду, одержуваного за допомогою зазначених процедур, це особливо важливо в контексті пептидів, які знаходять терапевтичне застосування і які виробляються в кількості, придатній для комерційного застосування в медицині. Синтез більших біомолекулярних фармацевтичних агентів, таких як терапевтичні пептиди, може бути дуже дорогим. Внаслідок значної вартості реагентів, тривалості синтезу, наявності великої кількості стадій синтезу, а також інших факторів, дуже незначні вдосконалення процесу синтезу цих великих бімолекулярних фармацевтичних агентів можуть суттєво вплинути навіть на те, чи буде економічно можливим виготовляти такий фарма 7 цевтичний засіб. Зазначені вдосконалення необхідні через високу вартість виробництва більших біомолекулярних фармацевтичних агентів, потреба у які підтверджується тим фактом, що в багатьох випадках існує небагато, якщо вони взагалі існують, прийнятних терапевтичних альтернатив зазначеним типам більших біомолекулярних фармацевтичних агентів. Це, мабуть, має місце у випадку глюкагонподібного пептиду-1 (GLP-1) і його аналогів. Зазначені пептиди, запропоновані як можливі терапевтичні агенти для лікування інсуліннезалежного цукрового діабету типу 2, а також родинних метаболічних порушень, таких як ожиріння (Gutniak M.K. і ін., Diabetes Care, 17, 1994, ее. 1039-1044). Lopez зі співавторами встановили, що нативний GLP-1 складається з 37 амінокислотних залишків (Lopez L. С. і ін., Proc. Natl. Acad. Sci. USA., 80, 1983, сс. 5485-5489). Ці дані були підтверджені дослідженнями Uttenthal L.O. і ін., J. Clin. Endocrinal. Metabol., 61, 1985, сс. 472-479. Нативний GLP-1 можна позначати як GLP-1 (1-37). Це позначення вказує на те, що до пептиду входять всі амінокислоти, починаючи з 1 (N-кінець) до 37 (С-кінець). Нативний GLP-1 має амінокислотну послідовність, представлену в SEQ ID NO. I: HDEFERHAEGTFTSDVSSYLEGQAAKEFIAWLV KGRG Установлено, що нативний GLP-1(1-37), як правило, не може опоередковувати біосинтез інсуліну, але біологічно важливі фрагменти цього пептиду мають інсулінотропні властивості. Наприклад, нативний пептид, що складається з 31 амінокислоти, GLP-1(7-37), послідовність якого представлена в SEQ ID NO. 2: HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRG, є інсулінотропним і складається з амінокислот, починаючи з положення 7 (N-кінець) до положення 37 (С-кінець) нативного GLP-1. GLP-1(7-37) має кінцевій залишок гліцину. Коли цей залишок гліцину відсутній, то пептид, що утворився, усе ще має інсулінотропну активність, і його позначають як GLP-1(7-36), його послідовність представлена в SEQ ID NO. 3: HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR. В GLP-1 (7-3 6) часто С-кінцевій залишок аргініну знаходиться в амідованій формі, і цю форму можна позначати як GLP-1(7-36)-NH2. GLP-1(1-37), як правило, перетворюється in vivo на свій аналог, що має інсулінотропну активність. Наприклад, GLP-1(l-37) у природних умовах перетворюється на GLP-1(7-37) in vivo. Цей пептид у свою чергу може також піддаватися додатковому процесингу в результаті протеолітичного видалення С-кінцевого залишку гліцину з утворенням GLP1(7-36), що часто існує в амідованій формі, тобто GLP-1(7-36)-NH2. Таким чином, терапевтичні застосування можуть включати введення GLP-1(137) або його аналога з розрахунком на те, що похідне, яке має інсулінотропну активність, утвориться in vivo. Однак більш часто в терапевтичних застосуваннях, які проходять дослідження, використовують введення самих фрагментів GLP-1, що мають інсулінотропну властивість. 96602 8 Згідно US 6887849 інсулінотропна активність GLP-1(7-37), GLP-1(7-36) і GLP-1(7-36)-NН2, ймовірно, є специфічною у відношенні панкреатичних бета клітин, у яких ці пептиди, очевидно, індукують біосинтез інсуліну. Це робить зазначені пептиди і їх фармацевтично прийнятні аналоги придатними для вивчення патогенезу цукрового діабету, що починається в дорослому віці, тобто стану, що характеризується гіперглікемією, при якій динаміка секреції інсуліну є аномальною. Крім того, зазначені глюкагонподібні пептиди можна застосовувати для терапії та лікування цього захворювання й для терапії та лікування гіперглікемії. Згідно ЕР 1137667В1 зазначені пептиди або їх фармацевтично прийнятні аналоги можна застосовувати для лікування інших типів діабету, ожиріння, глюкагоном, секреторних порушень дихальних шляхів, метаболічного порушення, артриту, остеопорозу, захворювання центральної нервової системи, рестенозу, нейродегенеративного захворювання, ниркової недостатності, застійної серцевої недостатності, нефротичного синдрому, цирозу, набряку легенів, гіпертензії і/або порушень, при яких бажане зниження споживання їжі. Нативний GLP-1(1-37) і його нативні аналоги, що мають інсулінотропну властивість, послідовності яких представлені в SEQ ID NO. 1-3, є метаболічно нестабільними, мають час напівжиття в плазмі, що становить тільки 1-2 хв in vivo. Екзогенно введений GLP-1 також швидко розщеплюється. Ця метаболічна нестабільність обмежує терапевтичний потенціал нативного GLP-1 і його нативних фрагментів. Створено синтетичні аналоги GLP-1-пептидів з поліпшеною стабільністю. Наприклад, в ЕР 1137667 В1 описаний пептид, послідовність якого представлена в SEQIDNO. 4: HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR. Цей пептид є аналогом нативного GLP-1(7-36), за винятком того, що ахіральний залишок альфааміноізомасляної кислоти (скорочено позначений на схемі як Aib) присутня у положеннях 8 і 35 замість відповідних амінокислот, що зустрічаються в природних умовах, у зазначених положеннях. Ахіральна альфа-аміноізомасляна кислота відома також під назвою метилаланін. Зазначений пептид 8,35 можна позначати формулою (Aib ) GLP-1 (7-3 6)NH2. В ЕР 1137667 зазначено, що пептид, який має послідовність, представлену в SEQ ID NO. 4, і його аналоги можна одержувати у вигляді індивідуального фрагмента за допомогою твердофазового синтезу. Запропонований в ЕР 1137667 підхід, що передбачає синтез індивідуального фрагмента, є проблематичним. З одного боку, цей підхід може приводити до високих рівнів епімеризації при сполученні кінцевої амінокислоти, наприклад, гістиди8 35 ну, у випадку, наприклад, (Aib ' ) GLP-1 (7-36). Крім того, може виявитися складним видаляти домішки в процесі хроматографічного очищення, і вихід може мати тенденцію до значного зниження. Отже, існує потреба в удосконалених стратегіях синтезу пептидів, послідовність яких представлена в SEQ ID NO. 4, з метою одержання можливості здійснювати виробництво цього пептиду і його 9 аналогів із прийнятними з комерційної точки зору виходом, чистотою та у прийнятній кількості. Крім цих міркувань великий вплив на можливість застосування конкретної схеми синтезу пептидів можуть здійснювати показники, пов'язані з виходом продукту й чистотою продукту при великомасштабному виробництві пептидів, а також з обробкою реагентів, зберіганням і розподілом. Таким чином, продовжує зберігатися потреба в методах пептидного синтезу, що забезпечують ефективне одержання пептидних продуктів, що являють комерційний інтерес, у вигляді великих партій з підвищеним виходом. Даний винахід стосується одержання інсулінотропних пептидів, які синтезують із використанням підходу, заснованого на застосуванні твердофазового й рідиннофазового («гібридного») синтезу. У цілому, зазначений підхід передбачає синтез трьох різних пептидних проміжних фрагментів за допомогою твердофазової хімії. Потім використовують рідиннофазову хімію для додавання додаткового амінокислотного продукту до одного із фрагментів. Після цього здійснюють сполучення фрагментів один з одним з використанням твердої та рідкої фаз. Застосування псевдопроліну в одному із фрагментів полегшує твердофазовий синтез цього фрагмента й полегшує також наступне сполучення цього фрагмента з іншими фрагментами в рідкій фазі. Даний винахід можна застосовувати, насамперед, для одержання інсулінотропних пептидів, таких як GLP-1, GLP-1(7-36) і їх аналогів, які зустрічаються в природних умовах і які не зустрічаються в природних умовах, зокрема GLP-1(7-36) і його аналогів, які зустрічаються в природних умовах і які не зустрічаються в природних умовах. Одним з об'єктів даного винаходу є спосіб одержання інсулінотропного пептиду, який полягає у тому, що: а) одержують пептидний фрагмент, який 8 10 включає амінокислотну послідовність НХ ЕХ 8 10 (SEQ ID NO. 6), у якій X і X кожний означає залишки ахіральної амінокислоти, або фрагмент, що є його аналогом, що включає залишки X і X , де Н, 8 10 Е, X і X кожний необов'язково несе захисну групу бічного ланцюга; і б) вбудовують пептидний фрагмент в інсулінотропний пептид. 8 X означає амінокислотний залишок, що пере10 важно відповідає метилаланіну (Aib). X означає амінокислотний залишок, що переважно відповідає гліцину. Наступним об'єктом винаходу є пептидний фрагмент, який має амінокислотну послідовність 8 10 8 10 НХ ЕХ (SEQ ID NO. 6), у якій X і X кожний 8 означає залишок ахіральної амінокислоти, Н, Е, X 10 і X кожний необов'язково несе захисну групу біч8 ного ланцюга. Переважно X означає амінокислот10 ний залишок, що відповідає Aib, a X означає амінокислотний залишок, що відповідає гліцину. Наступним об'єктом даного винаходу є спосіб одержання інсулінотропного пептиду, який полягає у тому, що: а) одержують пептидний фрагмент або його аналог, які включають амінокислотну послідовність 96602 10 17-18 TFTSDVX YLEG (SEQ. ID No. 8), у якій залишок, 17-18 позначений символом X , являє собою залишок псевдопроліну; і б) вбудовують пептидний фрагмент в інсулінотропний пептид. Ще одним об'єктом даного винаходу є пептид або його аналог, які включають амінокислотну по17-18 слідовність TFTSDVX YLEG (SEQ. ID No. 8), у 17-18 якій залишок, позначений символом X , являє собою залишок псевдопроліну; де зазначені амінокислотні залишки необов'язково несуть захисну групу бічного ланцюга. Наступним об'єктом даного винаходу є спосіб одержання інсулінотропного пептиду за п. 1 формули винаходу, який полягає у тому, що: а) здійснюють сполучення першого пептидного фрагмента, який включає амінокислотну послідов8 10 8 10 ність НХ ЕХ (SEQ ID NO. 6), у якій X і X кожний означає залишки ахіральної амінокислоти, З і Е кожний необов'язково несе захисну групу бічного ланцюга, із другим пептидним фрагментом, який 17включає амінокислотну послідовність TFTSDVX 18 YLEG (SEQ ID NO. 8), у якій залишок, позначений 17 18 символом X ' , являє собою дипептидний залишок псевдопроліну, де зазначені залишки амінокислотної послідовності необов'язково несуть захисну групу бічного ланцюга, з одержанням третього пептидного фрагмента, який включає амінокисло8 10 17-18 тну послідовність НХ ЕХ TFTSDVX YLEG (SEQ ID NO. 11), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; і б) вбудовують пептидний фрагмент в інсулінотропний пептид. Ще одним об'єктом даного винаходу є спосіб одержання інсулінотропного пептиду, який полягає у тому, що: а) одержують пептидний фрагмент або його аналог, які включають амінокислотну послідовність 35 35 QAAKEFIAWLVKX (SEQ ID NO. 9), у якій X означає залишок ахіральної амінокислоти, де зазначені залишки амінокислотної послідовності необов'язково несуть захисну групу бічного ланцюга; і б) вбудовують пептидний фрагмент в інсулінотропний пептид. Ще одним об'єктом даного винаходу є спосіб одержання інсулінотропного пептиду, який полягає у тому, що здійснюють одну або декілька стадій, на яких: а) одержують перший пептидний фрагмент, який включає амінокислотну послідовність 8 10 8 10 НХ ЕХ (SEQ ID NO. 6), у якій X і X кожний означає залишки ахіральної амінокислоти, З і Е кожний необов'язково несе захисну групу бічного ланцюга; б) одержують другий пептидний фрагмент, який включає амінокислотну послідовність 17-18 TFTSDVX YLEG (SEQ ID NO. 8), у якій залишок, 17-18 позначений символом X , являє собою дипептидний залишок псевдопроліну, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; в) здійснюють сполучення першого фрагмента й другого фрагмента з одержанням третього пеп 11 тидного фрагмента, який включає амінокислотну 8 10 17-18 послідовність HX EX TFTSDVX YLEG (SEQ ID NO. 11), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; г) одержують четвертий пептидний фрагмент, який включає амінокислотну послідовність 35 35 QAAKEFIAWLVKX (SEQ ID NO. 9), у якій X означає залишок ахіральної амінокислоти, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; д) здійснюють сполучення четвертого пептидного фрагмента з аргініном з одержанням п'ятого пептидного фрагмента, який включає амінокисло35 тну послідовність QAAKEFIAWLVK X R (SEQ ID NO. 12), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; і є) здійснюють сполучення п'ятого фрагмента із третім фрагментом з одержанням інсулінотропного пептиду, який включає амінокислотну послідов8 10 17-18 ність HX EX TFTSDVX YLEGQAAKEFIAWLVK 35 X R (SEQ ID NO. 13), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга. Переважно даний винахід стосується способу одержання інсулінотропного пептиду, який полягає у тому, що: а) одержують перший пептидний фрагмент, який включає амінокислотну послідовність 8 10 8 10 НХ ЕХ (SEQ ID NO. 6), у якій X і X кожний означає залишки ахіральної амінокислоти, З і Е кожний необов'язково несе захисну групу бічного ланцюга; б) одержують другий пептидний фрагмент, який включає амінокислотну послідовність 17-18 TFTSDVX YLEG (SEQ ID NO. 8), у якій залишок, 17-18 позначений символом X , являє собою дипептидний залишок псевдопроліну, зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; в) здійснюють сполучення першого фрагмента й другого фрагмента з одержанням третього пептидного фрагмента, який включає амінокислотну 8 10 17-18 послідовність НХ ЕХ TFTSDVX YLEG (SEQ ID NO. 11), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; г) одержують четвертий пептидний фрагмент, який включає амінокислотну послідовність 35 35 QAAKEFIAWLVKX (SEQ ID NO. 9), у якій X означає залишок ахіральної амінокислоти, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; д) здійснюють сполучення четвертого пептидного фрагмента з аргініном з одержанням п'ятого пептидного фрагмента, який включає амінокисло35 тну послідовність QAAKEFIAWLVK X R (SEQ ID NO. 12), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; і є) здійснюють сполучення п'ятого фрагмента із третім фрагментом з одержанням інсулінотропного пептиду, який включає амінокислотну послідов8 10 17-18 ність HX EX TFTSDVX YLEGQAAKEFIAWLVK 96602 12 35 X R (SEQ ID NO. 13), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга. Наступним об'єктом даного винаходу є описаний вище спосіб, який полягає у тому, що здійснюють наступну додаткову стадію, на якій: є) видаляють захисні групи бічних ланцюгів з одержанням інсулінотропного пептиду, який включає амінокислотну послідовність 8 10 35 HX EX TFTSDVSSYLEGQAAKEFIAWLVKX R (SEQ ID NO. 5), і його аналогів, у яких кожний із символів X у положеннях 8, 10 і 35 незалежно один від одного означає ахіральний амінокислотний залишок, що необов'язково має стеричну перешкоду. Видалення захисних груп бічних ланцюгів переважно здійснюють за допомогою розчину для видалення захисних групу, що містить принаймні один ацидолітичний агент і принаймні один акцептор катіонів. Ацидолітичні реагенти для повного видалення захисних груп переважно вибирають із групи, що включає трифтороцтову кислоту (ТФК), НСl, кислоти Льюїса, такі як BF3Et2O або Me3SiBr, рідку плавикову кислоту (HP), бромистий водень (НВr), трифторметансульфонову кислоту і їх комбінації. Прийнятні акцептори катіонів переважно вибирають із динітротреїтолу (ДТТ), анізолу, паракрезолу, етандитіолу та диметилсульфіду. Розчин для видалення захисних груп може також включати воду. Наступним об'єктом даного винаходу є спосіб одержання інсулінотропного пептиду, який полягає у тому, що: а) одержують перший пептидний фрагмент, який включає амінокислотну послідовність 8 10 8 10 НХ ЕХ (SEQ ID NO. 6), у якій X і X кожний означає залишки ахіральної амінокислоти, З і Е кожний необов'язково несе захисну групу бічного ланцюга; б) одержують другий пептидний фрагмент, який включає амінокислотну послідовність 17-18 TFTSDVX YLEG (SEQ ID NO. 8), у якій залишок, 17-18 позначений символом X , являє собою дипептидний залишок псевдопроліну, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; в) здійснюють сполучення першого фрагмента й другого фрагмента з одержанням третього пептидного фрагмента, який включає амінокислотну 8 10 17-18 послідовність НХ ЕХ TFTSDVX YLEG (SEQ ID NO. 11), де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; г) одержують четвертий пептидний фрагмент, який включає амінокислотну послідовність 35 35 QAAKEFIAWLVKX (SEQ ID NO. 9), у якій X означає залишок ахіральної амінокислоти, де зазначені амінокислотні залишки послідовності необов'язково несуть захисну групу бічного ланцюга; д) здійснюють сполучення четвертого пептидного фрагмента з аргініном з одержанням п'ятого пептидного фрагмента, який включає амінокисло35 тну послідовність QAAKEFIAWLVK X R (SEQ ID NO. 12), де зазначені амінокислотні залишки пос 13 лідовності необов'язково несуть захисну групу бічного ланцюга; і є) здійснюють сполучення п'ятого фрагмента із третім фрагментом з одержанням інсулінотропного пептиду формули (SEQ. ID No. 5) 8 10 35 HX EX TFTSDVSSYLEGQAAKEFIAWLVKX R і його аналогів, у яких кожний із символів X у положеннях 8, 10 і 35 незалежно один від одного означає ахіральний амінокислотний залишок, що необов'язково має стеричну перешкоду; і в яких один або декілька амінокислотних залишків необов'язково несуть захисну групу бічного ланцюга. На фіг. 1 показана схематична діаграма схеми синтезу, запропонованої в даному винаході. Фрагмент 12 являє собою пептидний фрагмент, який включає амінокислотну послідовність 8 10 НХ ЕХ (SEQ ID NO. 6). Фрагмент 14 являє собою пептидний фрагмент, який включає амінокислотну 11 15 17-18 20 послідовність T FTSD VX YL EG (SEQ ID NO. 8). Фрагмент 16 являє собою пептидний фрагмент, який включає амінокислотну послідовність 23 25 30 35 Q AA KEFIA WLVKX (SEQ ID NO. 9). Проміжний фрагмент 18 являє собою пептидний фрагмент, який включає амінокислотну послідовність 7 8 10 15 17-18 20 H X EX TFTSD VX YL EG (SEQ ID NO. 11). Проміжний фрагмент 20 являє собою пептидний фрагмент, який включає амінокислотну послідов23 25 30 35 ність Q AA KEFIA WLVKX R (SEQ ID NO. 12). Продукт 11 являє собою необхідний захищений 7 8 10 15 17-18 20 пептид H X EX TFTSD VX YL EG 25 30 35 QAA KEFIA WLVKX R (SEQ ID NO. 13), у якому Ser-Ser у положеннях 17 і 18 ще знаходяться у формі захищеного псевдопроліну. Нижче діаграма буде описана більш докладно. Наведені нижче варіанти здійснення даного винаходу у вигляді конкретних форм, докладно описаних нижче, не є вичерпними й не спрямовані на обмеження винаходу. Скоріше, варіанти здійснення винаходи вибрані й описані таким чином, щоб інші спеціалісти в даній галузі змогли оцінити й зрозуміти принципи та варіанти втілення на практиці даного винаходу. Даний винахід стосується способів синтезу для одержання пептидів, таких як глюкагонподібний пептид-1 (GLP-1) і його аналогів, які зустрічаються в природних умовах і які не зустрічаються в природних умовах, що мають інсулінотропну властивість, з використанням методів твердофазового і/або рідиннофазового синтезу. Пептидні молекули, пропоновані у винаході, можуть бути захищеними, незахищеними або частково захищеними. Захист може являти собою N-кінцевій захист, захист бічного ланцюга і/або С-кінцевій захист. Хоча винахід стосується насамперед синтезу глюкагонподібних пептидів, їх аналогів, фрагментів і їх аналогів, і злитих продуктів і їх аналогів, запропоновані у винаході способи можна застосовувати також для синтезу інших пептидів, зокрема, пептидів, які синтезують із використанням комбінації твердофазових і рідиннофазових підходів. Даний винахід стосується також синтезу пептидних проміжних фрагментів, асоційованих з домішками, насамперед піроглутаматними домішками. Кращі молекули GLP-1, які можна застосовувати при втіленні на практиці даного винаходу, являють собою GLP 96602 14 1(7-36) і їх аналоги, які зустрічаються в природних умовах і які не зустрічаються в природних умовах. У контексті даного опису поняття «включає амінокислотну послідовність» переважно означає «має амінокислотну послідовність». У контексті даного опису поняття «аналог» стосується аналогів, похідних, злитих сполук, солей або т.п. пептиду, які зустрічається в природних умовах і які не зустрічається в природних умовах. У контексті даного опису поняття «пептидний аналог», як правило, стосується пептиду, що має модифіковану амінокислотну послідовність, наприклад, у результаті однієї або декількох амінокислотних замін, делецій, інверсій і/або додавань відносно іншого пептиду або пептидного аналога. Заміни можуть включати одну або декілька амінокислот, які зустрічаються в природних умовах і які не зустрічаються в природних умовах. Заміни переважно можуть бути консервативними або висококонсервативними. Поняття «консервативна заміна» стосується заміни амінокислоти на іншу амінокислоту, що має в цілому такий же чистий електронний заряд і в цілому такий же розмір і форму. Наприклад, амінокислоти з аліфатичними бічними ланцюгами або амінокислоти із заміщеними аліфатичними бічними ланцюгами мають приблизно однаковий розмір, коли загальна кількість атомів вуглецю й гетероатомів у їх бічних ланцюгах відрізняється не більш ніж приблизно на чотири. Вони мають приблизно однакову форму, коли кількість гілок у їх бічних ланцюгах відрізняється не більш ніж приблизно на одну або дві. Вважається, що амінокислоти з фенільними або заміщеними фенільними групами в їх бічних ланцюгах мають приблизно однаковий розмір і форму. Нижче перераховано п'ять груп амінокислот. Заміна амінокислоти в сполуці на іншу амінокислоту із цієї ж групи, як правило, приводить до консервативної заміни. Група І: гліцин, аланін, валін, лейцин, ізолейцин, серин, треонін, цистеїн, метіонін і амінокислоти, що не зустрічаються в природних умовах, з С1С4аліфатичними або із заміщеними гідроксилом С1-С4аліфатичними бічними ланцюгами (прямоланцюжкові або однорозгалужені). Група II: глутамінова кислота, аспарагінова кислота та амінокислоти, що не зустрічаються в природних умовах, із заміщеними карбоновою кислотою С1-С 4аліфатичними бічними ланцюгами (які є нерозгалуженими або мають одну точку розгалуження). Група III: лізин, орнітин, аргінін і амінокислоти, що не зустрічаються в природних умовах, із заміщеними аміном або гуанідиногрупою С 1С4аліфатичними бічними ланцюгами (які є нерозгалуженими або мають одну точку розгалуження). Група IV: глутамін, аспаргін і амінокислоти, що не зустрічаються в природних умовах, із заміщеними амідом С1-С4аліфатичними бічними ланцюгами (які є нерозгалуженими або мають одну точку розгалуження). Група V: фенілаланін, фенілгліцин, тирозин і триптофан. 15 У контексті даного опису поняття «аналог» більш переважно стосується солей пептидів або їх похідних, амідованих на С-кінці. «Висококонсервативна заміна» являє собою заміну амінокислоти іншою амінокислотою, що має таку ж функціональну групу в бічному ланцюзі й приблизно такий же розмір і форму. Амінокислоти з аліфатичними або амінокислоти із заміщеними аліфатичними бічними ланцюгами мають приблизно однаковий розмір, коли загальна кількість атомів вуглецю й гетероатомів у їх бічних ланцюгах відрізняється не більш ніж приблизно на два. Вони мають приблизно однакову форму, коли вони мають однакову кількість гілок у їх бічних ланцюгах. Прикладами висококонсервативних замін є заміни валіну на лейцин, треоніну на серин, аспарагінової кислоти на глутамінову кислоту й фенілгліцину на фенілаланін. Поняття «пептидне похідне», як правило, стосується пептиду, пептидного аналогу або іншої пептидної копії, які мають хімічну модифікацію в одній або декількох їх бічних групах, альфаатомах вуглецю, кінцевій аміногрупі і/або кінцевій карбоксильній групі. Наприклад, хімічна модифікація включає (але, не обмежуючись тільки ними) додавання хімічних залишків, створення нових зв'язків і/або видалення хімічних залишків. Модифікації бічних груп амінокислот включають (але, не обмежуючись тільки ними) ацилювання аміногруп лізину, N-алкілювання аргініну, гістидину або лізину, алкілювання карбоксильних груп глутамінової або аспарагінової кислоти й деамідування глутаміну або аспарагіну. Модифікації кінцевих аміногруп включають (але, не обмежуючись тільки ними) модифікації дез-аміно-типу, з використанням N-(низч.)алкілу, N-ди(низч.)алкілу й N-ацилу (наприклад, -СО-(низч.)алкілу). Модифікації кінцевої карбоксильної групи включають (але, не обмежуючись тільки ними) модифікації з використанням аміду, (низч.)алкіламіду, діалкіламіду й (низч.)алкілового складного ефіру. Кращими похідними є похідні, амідовані на кінцевій карбоксильній групі, наприклад, амід, (низч.)алкіламід або діалкіламід пептиду. Таким чином, частково або повністю захищені пептиди належать до пептидних похідних. При втіленні даного винаходу на практиці вважається, що сполука має «інсулінотропну» активність, якщо вона може стимулювати або сприяти стимуляції синтезу або експресії гормону інсуліну. У кращих варіантах втілення на практиці інсулінотропну активність можна продемонструвати за допомогою аналізів, описаних в US 6887849 і 6703365. Кращими варіантами здійснення даного вина8 10 ходу є шляхи синтезу синтетичних (X , X , 35 Х )GLР-1(7-36)-пептидів, що мають наступну формулу (SEQ. ID NO. 5): 8 10 35 HX EX TFTSDVSSYLEGQAAKEFIAWLVKX R і їх аналогів, у яких кожний із символів X у положеннях 8, 10 і 35 незалежно один від одного означає ахіральний амінокислотний залишок, що необов'язково має стеричну перешкоду. Кожний із 8 10 35 залишків X , X і/або X необов'язково може нести захисну(і) групу(и) бічних ланцюгів. Пептиди, що мають зазначену формулу, відрізняються від 96602 16 нативного GLP-1(7-36) принаймні тим, що нативні амінокислотні залишки в положеннях 8 і 35 замі8 35 нені на хіральні амінокислотні залишки X і X , що необов'язково мають стеричну перешкоду. Зали10 шок X може бути похідним нативної ахіральної амінокислоти гліцину або інший ахіральної амінокислоти. Застосування ахіральних амінокислот у 8 10 35 положеннях X , X і X не тільки сприяє стабілізації пептиду, що утворився, але також, як установлено при створенні даного винаходу, застосування зазначених амінокислот як конструктивних елементів полегшує пропонований у даному винаході шлях синтезу, що представлений на фіг. 1 і додатково описаний нижче. s 10 35 Найбільш кращим варіантом (X , X , X ) GLP11(7-36)-пептиду, який можна синтезувати відповідно до принципів, пропонованих у даному винаході, є пептид формули (SEQ ID NO. 4): HAibEGTFTSDVSSYLEGQAAKEFIAWLVKAibR і його аналоги, переважно амідовані на С-кінці. У цьому пептиді застосовують ахіральний залишок альфа-аміноізомасляної кислоти (або метилаланін, скорочено позначений на схемі як Aib) як X , так і X , він переважно містить амід на С-кінці, залишок нативного G у положенні 10, і він може бути 8,35 позначений формулою (Aib )GLP-1(7-36)-NH2. Це умовне зображення означає, що амінокислотний залишок, який відповідає амінокислоті «Aib», присутній у положеннях 8 і 35 замість нативного аланіну. Ахіральну альфа-аміноізомасляну кислоту називають також метилаланіном. Пептид, який має послідовність, представлену в SEQ ID NO. 4, описаний в ЕР 1137667 В1. Присутність залишків Aib у положеннях 8 і 35 сповільнює метаболічне розщеплення в організмі, роблячи цей пептид більш стабільним в організмі в порівнянні з нативним пептидом GLP-1(7-36). Даний винахід стосується вдосконаленої методології одержання пептидів GLP-1(7-36), таких 8,35 як (Aib )GLP-1(7-36)-NH2. Наприклад, на фіг. 1 схематично проілюстрована одна схема 10 синтезу пептидів GLP-1(7-36) і їх аналогів. Схему 10, представлену на фіг. 1, очевидно, найбільш доцільно застосовувати для великомасштабного синтезу пептидів GLP-1(7-36). Великомасштабні процеси, як правило, здійснюють для одержання пептиду в кількості, яку можна застосовувати для продажу. Наприклад, кількість пептиду при великомасштабному процесі може становити 500 г або 1 кг на партію й більш часто від десятків до сотень кілограмів на партію або більше. У кращих варіантах здійснення винаходу способи, пропоновані у винаході, можуть забезпечувати вдосконалення, які приводять до зниження часу процесингу (синтезу), підвищенню виходу продуктів, поліпшення чистоти продукту і/або зниженню кількості необхідних реагентів і вихідних продуктів. Схема синтезу 10, представлена на фіг. 1, заснована на сполученні твердофазових і рідиннофазових методів для одержання пептидного продукту 11. Як видно з фіг. 1, відповідно до схеми 10 синтезують пептидні проміжні фрагменти 12, 14 і 16 на твердій фазі. Фрагмент 12 являє собою пептид 17 ний фрагмент, який включає амінокислотні залишки, представлені в SEQ ID NO. 6: 8 10 НХ ЕХ , 8 10 де X і X мають зазначені вище значення, або являє собою його аналог, що включає залиш8 10 ки X і X . Один або декілька амінокислотних залишків можуть нести загальноприйняті захисні групи бічних ланцюгів. У деяких варіантах здійснення винаходу пептидний фрагмент 12 може бути зв'язаний зі смолою через С-кінець. Цей фрагмент необов'язково може нести захисні групи на N-кінці і/або на С-кінці. Установлено, що Fmoc є найбільш кращою захисною групою для N-кінця для твердофазового синтезу пептидного фрагмента. Фрагмент 12 включає 4 амінокислотних залишки, що відповідають амінокислотам у положеннях 7-10 нативного пептиду GLP-1(7-36), і тому його 8 10 можна позначати як (X , X )GLP-1(7-10). У кращих 8 варіантах здійснення винаходу X означає Aib, а 10 X означає гліцин згідно SEQ ID N0. 7: 7 10 H AibEG або означає його аналог, що містить залишок Aib у положенні 10. Пептидний фрагмент, представлений в SEQ ID NO. 7, може бути позначений 8 як (Aib )GLP-1(7-10), у цьому позначенні проілюстрована заміна на Aib нативного аланіну в положенні 8 нативного GLP-1(7-10). Твердофазовий синтез, як правило, здійснюють у напрямку від С-кінця до Текінця фрагмента 10 12. Так, амінокислота X , що присутня в Скінцевій ділянці фрагмента, являє собою перший амінокислотний залишок, що сполучать зі смолоюпідкладкою для твердофазового синтезу. Потім здійснюють твердофазовий синтез шляхом послідовного додавання амінокислотних залишків у порядку, що відповідає необхідній послідовності. Синтез пептидного проміжного фрагмента завершують після досягнення N-кінцевого залишку (наприклад, N-кінцевій залишок гістидину (Н) додавали до пептидного ланцюга, що утворюється). Вибір і застосування пептидного фрагмента, що має послідовність, представлену в SEQ ID NO. 6 і 7, забезпечує значну перевагу для синтезу, представленого на схемі 10. По-перше,Н може являти собою амінокислотний залишок, який важко включати в зростаючий пептидний ланцюг, принаймні частково через епімеризацію. Однак фрагмент 12 є в достатньому ступені невеликим, що значно полегшує ці проблеми. При цьому фрагмент 12 є досить довгим, що дозволяє мати два хіральні центри. У результаті фрагмент можна очищати за допомогою простої кристалізації. Якщо фрагмент 12 закінчується Aib, то фрагмент буде мати тільки один хіральний центр і в результаті його буде складніше очищати рацемічно. Позиціювання ахірального G на С-кінці дозволяє уникати також пов'язаних з рацемізацією проблем, які можуть виникати, якщо фрагмент 12 має на С-кінці хіральний Е. Коротше кажучи, вибір фрагмента 12 як конструктивного елемента пептиду полегшує створення фрагмента, його очищення й сполучення з іншим пептидним продуктом. Вибір фрагмента обумовлений також низькою рацемізацією Н. При створенні винаходу несподівано було встановлено, що додавання З до зазначеного фрагмента 96602 18 приводить до дуже незначного збільшення ступеня епімеризації, наприклад, приблизно на 3 мас.% у деяких варіантах здійснення винаходу на практиці. Фрагмент 14 являє собою пептидний фрагмент, який включає амінокислотні залишки, представлені в SEQ ID NO. 8: 11 15 17-18 20 T FTSD VX YL EG, 17-18 де залишок, позначений символом X , являє собою дипептидний залишок псевдопроліну, що додатково описаний нижче, або його аналог, 17-18 що містить Х у положеннях 17 і 18. Фрагмент 14 включає амінокислотні залишки, які в цілому відповідають амінокислотним залишкам у положеннях 11-22 нативного пептиду GLP-1(7-36), за винятком того, що дипептидний залишок псевдоп17-18 роліну X застосовують замість залишків SS (Ser-Ser), які знаходяться у відповідних положеннях 17 і 18 нативного GLP-1(7-36). Один або декілька амінокислотних залишків фрагмента 14 можуть нести загальноприйняті захисні групи. У деяких варіантах здійснення винаходу пептидний фрагмент 14 може зв'язуватися зі смолою через С-кінець. Цей фрагмент необов'язково може нести захисні групи на N-кінці і/або Скінці. Установлено, що Fmoc є найбільш кращою для твердофазового синтезу пептидного фрагмента захисною групою для N-кінця. Пептидний фрагмент, який має послідовність, представлену в SEQ 17-18 ID NO. 8, можна умовно позначати як (X )GLP1(11-22), у цьому позначенні проілюстрована замі17 18 на ч " на залишок псевдопроліну залишку SerSer у положеннях 17 і 18. При втіленні даного винаходу на практиці поняття «псевдопролін» стосується дипептиду, який включає залишок, що містить функціональний гідроксил амінокислоти, такий як Ser або Thr, у якому функціональний гідроксил бічного ланцюга захищений з утворенням нестійкого в присутності ТФК оксазолідинового кільця, що нагадує пролін, між альфа-аміногрупою і гідроксилом бічного ланцюга. Через наявність оксазолідинового кільця дипептид функціонує як зворотній міметик проліну. У цілому, типовий залишок псевдопроліну, включений у пептид, може бути представлений формулою , у якій Ф означає залишок будь-якої амінокис1 2 лоти, a R і R кожний незалежно один від одного означає прийнятний двовалентний сполучний ра1 дикал. Часто R означає двовалентний радикал формули , 3 4 у якій R і R кожний незалежно один від одного означає одновалентний радикал, такий як З або 19 96602 3 4 (низч.)алкіл, наприклад, метил. R і R можуть разом бути членами кільцевої структури. Переважно 3 4 R і R кожний означає метил. У випадку захище2 ного оксазолідиніновим кільцем Ser, R означає 2 двовалентний радикал СН2, а у випадку Thr, R означає двовалентний радикал (СН3)СН. Поняття «(низч.)алкіл» стосується розгалуженого або прямоланцюжкового одновалентного алкільного радикалу, що складається з 1-6 атомів вуглецю, переважно 1-4 атомів вуглецю. Додатковими прикладами радикалів, які підпадають під зазначене поняття, є метил, етил, н-пропіл, ізопропіл, н-бутил, ізобутил, трет-бутил, н-пентил, 3метилбутил, н-гексил, 2-етилбутил і т.п. Кращими (низч.)алкільними радикалами є метил і етил, особливо переважно метил. У процесі видалення захисної групи радикал 1 R відщеплюється з утворенням дипептидиого залишку наступної формули: , 2 у якій Ф і R мають зазначені вище значення. У фрагменті 14 залишок псевдопроліну переважно відповідає залишку Ser-Ser, у якому Ser, найбільш близько розташований відносно С-кінця, захищають оксазолідиновим кільцем, і він має наступну структуру: . Бічний ланцюг Ser, що несе гідроксил і є найближчим до N-кінця, захищають, наприклад, за допомогою захисної групи трет-Вu. Коли захисну оксазолідинову структуру й трет-Вu відщеплюють, то утворюється залишок Ser-Ser. Застосування зазначеного пролінового міметика як конструктивного елемента при синтезі фрагмента 14 забезпечує істотні переваги в контексті даного винаходу. По-перше, значно полегшується твердофазовий синтез фрагмента 14. Коли не застосовують псевдопролін у процесі твердофазового синтезу фрагмента 14, то можуть виникати істотні проблеми з видаленням Fmocгрупи із залишків 13-11. Ймовірно, що ці труднощі можуть бути наслідком бета-складчастої конформації. Застосування псевдопроліну дуже спрощує видалення зазначених Fmoc-груп, ймовірно, у результаті зниження рівня бета-складчастої конформації. По-друге, значно полегшується описане нижче наступне твердофазове сполучення фрагмента 14 із фрагментом 12 з утворенням фрагмента 18. У відсутності залишку псевдопроліну розчинність фрагмента 18 у типових розчинниках, застосовуваних для рідиннофазового сполучення, є дуже поганою. Псевдопролін підвищує 20 характеристики розчинності фрагмента 18, полегшуючи рідиннофазове сполучення цього фрагмента із фрагментом 20 (див. нижче обговорення фіг. 1). Твердофазовий синтез, як правило, здійснюють у напрямку від С-кінця до N-кінця фрагмента 14. Так, амінокислота G, що присутня у С-кінцевій ділянці фрагмента, являє собою перший амінокислотний залишок, що сполучають зі смолою, використовуваною як підкладка для твердофазового синтезу. Потім здійснюють твердофазовий синтез шляхом послідовного додавання амінокислотних залишків у порядку, що відповідає необхідній пос17-18 лідовності. Однак як X додають дипептид псевдопролін до зростаючого ланцюга в положенні 17 і 18 GLP-1(7-36) замість послідовного додавання пари нативних залишків Ser у положеннях 17 і 18. Синтез пептидного проміжного фрагмента завершують після N-кінцевого залишку ((наприклад, Nкінцевій залишок треоніну (Т) додавали до пептидного ланцюга, що утворювався). Фрагмент 16 являє собою пептидний фрагмент або його аналог, що включає амінокислотні залишки, представлені в SEQ ID NO. 9: 23 25 30 35 Q AA KEFIA WLVKX , 35 у якій X має зазначені вище значення, або 35 означає його аналог, що включає залишок X . Один або декілька амінокислотних залишків можуть нести загальноприйняті захисні групи. Фрагмент 16 включає амінокислотні залишки, що відповідають амінокислотам у положеннях 23-35 нативного пептиду GLP-1(7-36) за винятком того, 35 що Х у положенні 35 заміняє нативну амінокислоту в цьому положенні. Фрагмент 16 можна умов35 но позначати як (X )GLP-1(23-35). У деяких варіантах здійснення винаходу пептидний фрагмент 16 можна зв'язувати зі смолою через С-кінець. Цей фрагмент необов'язково може нести захисні групи бічного ланцюга, N-кінця і/або С-кінця. Установлено, що Fmoc є найбільш кращою захисною групою для N-кінця для твердофазового синтезу пептидного фрагмента. 35 У кращих варіантах здійснення винаходу X означає Aib у послідовності, представленій в SEQ ID NO. 10: 23 25 30 35 Q AA KEFIA WLVKAib або її аналогу, що включає Aib у положенні 35. Пептидний фрагмент, послідовність якого представлена в SEQ ID NO. 10, можна позначати як 35 (Aib )GLP-1(23-35), у цьому позначенні проілюстрована заміна на Aib нативної амінокислоти в положенні 35 нативного GLP-1(7-36). Слід зазначити, що фрагмент 16, послідовність якого представлена в SEQ ID NO. 9 і 10, ще не включає залишок аргініну R (arg) у положенні 36 на С-кінці. Потім здійснюють сполучення Arg з С-кінцем фрагмента 16 у рідкій фазі, переважно використовуючи Arg без захисту бічного ланцюга. Ця стратегія забезпечує значні переваги при використанні схеми 10, представленої на фіг. 1, оскільки дозволяє уникати небажаних реакцій за участю бічного ланцюга, які мають тенденцію відбуватися при використанні захищеного Arg. Наприклад, після видалення захисної групи захищеного Arg побічні продукти, що виникають при видаленні захис 21 ної групи, можуть мати тенденцію до взаємодії з іншими компонентами пептиду, наприклад, із триптофаном. Це знижує кількість необхідного пептиду, доступну в неочищеному призначеному для очищення продукті. Твердофазовий синтез, як правило, здійснюють у напрямку від С-кінця до N-кінця фрагмента 35 16. Так, амінокислота X , що присутня в Скінцевій ділянці фрагмента, являє собою перший амінокислотний залишок, що сполучають зі смолою, використовуваною як підкладка для твердофазового синтезу. Потім здійснюють твердофазовий синтез шляхом послідовного додавання амінокислотних залишків у порядку, що відповідає необхідній послідовності. Синтез пептидного проміжного фрагмента завершують після досягнення N-кінцевого залишку (наприклад, N-кінцевій залишок глутаміну (Q) додавали до пептидного ланцюга, що утворюється). Будь-яка амінокислота, застосовувана для синтезу фрагмента 16, може нести загальноприйняті захисні групи бічних ланцюгів. Через стеричну перешкоду, найближчу до за35 лишку X , який здійснює зв'язок зі смолоюпідкладкою, сполучення лізину (34) й валіну (33) зі зростаючим пептидним ланцюгом може виявитися проблематичним. Навіть при надлишку амінокислоти важко довести ці реакції сполучення до їх завершення. Вибір розчинника і/або кінцевого кепірування може сприяти рішенню цієї проблеми. Було встановлено, що природа застосовуваного для сполучення розчинника може впливати на рівень сполучення, що досягає при завершенні реакції. Наприклад, в одній серії експериментів реакції сполучення здійснювали в суміші 3:1 NМП/ДХМ, 1:1 NMP/ДХМ, 1:1 ДМФ/ДХМ і 3:1 ДМФ/ДХМ. Співвідношення зазначених комбінацій розчинників дані у вигляді об'ємних співвідношень. NMП означає N-метилпіролідон, ДХМ означає дихлорметан, а ДМФ означає диметилформамід. Було встановлено, що реакції сполучення більшою мірою досягають завершення при використанні 1:1 ДМФ/ДХМ. Кінцеве кепірування після кожного сполучення лізину й валіну можна використовувати також для попередження участі в додаткових реакціях сполучення смоли, яка являє собою матеріал підкладки та не прореагувала. Матеріал з кінцевим кепіруванням легше видаляти при необхідності в процесі очищення. Можна використовувати загальноприйняті методи кінцевого кепірування. Продовжуючи посилання на схему, наведену на фіг. 1, фрагменти 12, 14 і 16, а також Arg, складають для завершення складання необхідного пептиду 11. Для цього фрагмент 12 додають до фрагмента 14 на твердій фазі з одержанням більшого проміжного фрагмента 18, що включає амінокислотні залишки, представлені в SEQ ID NО.11: 7 8 10 15 17-18 20 H x EX TFTSD VX YL EG, 8 10 17-18 де X , X і X мають зазначені вище зна8 чення. У кращому варіанті здійснення винаходу X 10 17-18 означає Aib, X означає нативний G, а X означає дипептидний залишок псевдопроліну, описаний вище. Цей проміжний пептидний фрагмент 8 10 17-18 можна позначати як (X , X , X ) GLP-1(7-22). 96602 22 На фіг. 1 показано також, що Arg додають до С-кінця фрагмента 16 у рідкій фазі з одержанням більшого проміжного пептидного фрагмента 20, що включає амінокислотні залишки, представлені в SEQ ID NO. 12: 23 25 30 35 Q AA KEFIA WLVKX R, 35 у якій X має зазначені вище значення й переважно означає Aib. Переважно Arg, доданий у такий спосіб до пептидному фрагмента, не має захисних груп бічного ланцюга. Цей проміжний пептидний фрагмент 20 може бути позначений як 35 (X )GLP-1(23-36). Потім здійснюють сполучення пептидних фрагментів 18 і 20 у рідкій фазі з одержанням необхідного захищеного пептиду 11, послідовність якого представлена в SEQ ID NO. 13, де Ser-Ser у положеннях 17 і 18 усе ще являють собою псевдопролін у захищеній формі: 7 8 10 15 1718 20 25 30 H X EX TFTSD VX YL EGQAA KEFIA W 35 LVKX R, 8 10 1748 Пептид 11 можна позначити як (X , X , X , 35 X )GLP-1(7-36). У процесі здійснення цієї стадії бажано зберігати наявний ступінь захисту бічних ланцюгів інших амінокислот. При здійсненні реакцій, представлених на реакційній схемі, на фіг. 1, твердофазовий і рідиннофазовий синтез можна проводити за допомогою стандартних методів, відомих у промисловості. У репрезентативних варіантах здійснення винаходу на практиці пептиди синтезують на твердій фазі з використанні хімічних методів, згідно яким амінокислоти додають, починаючи з С-кінця в напрямку до N-кінця. Таким чином, амінокислота або пептидна група, найближча до С-кінця конкретного фрагмента, являє собою першу групу, яку слід зв'язувати зі смолою. Це здійснюють шляхом взаємодії С-кінцевої функціональної групи амінокислоти або пептидної групи з комплементарною функціональною групою смоли-підкладки. N-кінцеву ділянку амінокислоти або пептидної групи маскують із метою попередження небажаних побічних реакцій. Бажано також, щоб бічні групи амінокислоти або пептидної групи були захищені. Потім до зв'язаного з підкладкою пептидного продукту приєднують наступні амінокислоти або пептидні групи до утворення пептиду, який являє інтерес. Більшість із них має також захищені бічні ланцюги відповідно до загальноприйнятої практики. При кожному наступному сполученні маскувальну групу на N-кінці зв'язаного зі смолою пептидного продукту видаляють. Потім її піддають взаємодії з С-кінцем наступної амінокислоти, N-кінець якої замаскований. Таким чином, продукт твердофазового синтезу являє собою пептид, зв'язаний зі смолоюпідкладкою. Можна використовувати будь-який тип підкладки, що застосовують на практиці при здійсненні твердофазового синтезу пептидів. У кращих варіантах здійснення винаходу підкладка містить смолу, що може складатися з одного або декількох полімерів, співполімерів або комбінацій полімерів, таких як поліамід, полісульфамід, заміщені поліетилени, поліетиленгліколь, фенольні смоли, полісахариди або полістирол. Полімерна підкладка може також мати будь-яку твердість, достатню для того, щоб бути нерозчинною й інертною відно 23 сно розчинників, застосовуваних у пептидному синтезі. Тверда підкладка, як правило, включає сполучний радикал, з яким у процесі синтезу сполучать зростаючий пептид і який можна розщеплювати в необхідних умовах для вивільнення пептиду з підкладки. Прийнятні тверді підкладки можуть містити лінкери, які можуть розщеплюватися під дією світла, розщеплюватися в присутності ТФК, розщеплюватися в присутності HF, розщеплюватися в присутності іона фтору, розщеплюватися в присутності відновника; розщеплюватися в присутності Pd(O); розщеплюватися в присутності нуклеофілу; або розщеплюватися в присутності радикалів. Кращі сполучні радикали можуть розщеплювати в умовах, при яких бічні групи пептиду, який відщеплюють, усе ще в цілому залишаються захищеними. В одному із кращих способів синтезу пептидні проміжні фрагменти синтезують на чутливій до кислоти твердій підкладці, що містить тритильні групи, і більш переважно на смолі, що включає тритильні групи з «підвішеними» атомами хлору, наприклад 2-хлортритилхлоридній (2-СТС) смолі (Barlos і ін., Tetrahedron Letters 30(30), 1989, ее. 3943-3946). Прикладами є також тритилхлоридна смола, 4-метилтритилхлоридна смола, 4метокситритилхлоридна смола, 4-амінобутан-1-ол2-хлортритильна смола, 4-амінометилбензоїл-2хлортритильна смола, З-амінопропан-1-ол-2хлортритильна смола, бромоцтова кислота-2хлортритильна смола, ціаноцтова кислота-2хлортритильна смола, 4-ціанбензойна кислота-2хлортритильна смола, гліцинол-2-хлортритильна смола, пропіонова-2-хлортритильна смола, етиленгліколь-2-хлортритильна смола, N-Fmocгідроксиламін-2-хлортритильна смола, гідразин-2хлортритильна смола. Деякі кращі тверді підкладки включають полістирол, який можна піддавати співполімеризації з дивінілбензолом з одержанням матеріалу підкладки, у який можна «заякорювати» реактивні групи. Інші смоли, які можна використовувати у твердофазовому синтезі, включають смоли «Ванга», які містять співполімер стиролу й дивінілбензолу з 4-гідроксиметилфенілоксиметильними заякорювальними групами (Wang S.S., J. Am. Chem. Soc, 1973), і смолу на основі 4-гідроксиметил-Зметоксифеноксимасляної кислоти (Richter і ін., Tetrahedron Letters 35(27), 1994, ее. 4705-4706). Смолу Ванга, 2-хлортритилхлоридну смолу та смолу на основі 4-гідроксиметил-Зметоксифеноксимасляної кислоти можна купувати, наприклад, у фірми Calbiochem-Novabiochem Corp., Сан-Дієго, шт. Каліфорнія. Для підготовки смоли для твердофазового синтезу смолу можна попередньо відмивати в прийнятному (их) розчиннику (ах). Наприклад, використовувану як тверда фаза смолу, таку як смола 2СТС, додають у камеру для синтезу пептидів і попередньо відмивають прийнятним розчинником. Розчинник для попереднього відмивання можна вибирати з розчинників різних типів (або суміші розчинників), які застосовують у реакції сполучення або навпаки. Придатні для відмивання, а також для наступної реакції сполучення розчинники яв 96602 24 ляють собою дихлорметан (ДХМ), дихлоретан (ДХЕ), диметилформамід (ДМФ) і т.п. а також суміші зазначених реагентів. Інші прийнятні розчинники являють собою ДМСО, піридин, хлороформ, діоксан, тетрагідрофуран, етилацетат, Nметилпіролідон і їх суміші. У деяких випадках сполучення можна проводити в бінарній системі розчинників, такій як суміш ДМФ і ДХМ в об'ємному співвідношенні від 9:1 до 1:9, більш переважно від 4:1 до 1:4. Синтез, пропонований у даному винаході, переважно здійснюють у присутності відповідних захисних груп, якщо не зазначене інше. Природа захисних груп і їх застосування широко відомі. Як правило, прийнятною захисною групою є група будь-якого типу, що може сприяти попередженню участі атома або радикала, до якого вона приєднана, наприклад, кисню або азоту, у небажаних реакціях під час процесування й синтезу. Захисні групи включають захисні групи бічних ланцюгів і аміно- або N-кінцеві захисні групи. Захисні групи можуть попереджати також взаємодію або зв'язування карбонових кислот, тіолів і т.п. Поняття «захисна група бічного ланцюга» стосується хімічного радикала, зчепленого з бічним ланцюгом (тобто R-група в загальній формулі амінокислот H2N-C(R)(H)-COOH) амінокислоти, що сприяє попередженню взаємодії частини бічного ланцюга з хімічними речовинами, застосовуваними на стадіях пептидного синтезу, процесингу й т.д. Вибір захисної групи бічного ланцюга може залежати від різних факторів, наприклад, типу здійснюваного синтезу, процесингу, якому повинен піддаватися пептид, і необхідного проміжного продукту або кінцевого продукту. Природа захисної групи бічного ланцюга залежить також на природи самої амінокислоти. Як правило, захисну групу бічного ланцюга вибирають так, щоб у процесі видалення захисних груп вона зберігалася на баміногрупі при твердофазовому синтезі. Тому захисна група -аміногрупи й бічного ланцюга, як правило, не є однаковими. У деяких випадках і залежно від типу реагентів, застосовуваних для твердофазового синтезу й іншого процесингу пептидів, для амінокислоти не потрібна присутність захисної групи бічного ланцюга. Такі аміногрупи, як правило, не містять реактивний кисень, азот або інший реактивний радикал у бічному ланцюзі. Прикладами захисних груп бічних ланцюгів є ацетил (Ас), бензоїл (Bz), трет-бутил, трифенілметил (тритил), тетрагідропіраніл, простий бензиловий ефір (Bzl) і 2,6-дихлорбензил (ДХБ), wpemбутоксикарбоніл (Вос), нітрогрупа, паратолуолсульфоніл (Tos), адамантилоксикарбоніл, ксантил (Хаn), бензил, 2,6-дихлорбензил, складний метиловий, етиловий і трет-бутиловий ефір, бензилоксикарбоніл (Z), 2хлорбензилоксикарбоніл (2-Cl-Z), третамілоксикарбоніл (Аос) і ароматичні або аліфатичні захисні групи уретанового типу, фотолабільні групи, такі як нітровератрилоксикарбоніл (NVOC); і нестійкі в присутності фтористих сполук групи, такі як триметилсилілоксикарбоніл (ТЕОС). 25 96602 26 Кращі захисні групи бічних ланцюгів амінокислот, які звичайно застосовують для синтезу пепти дів GLP-1 при втіленні на практиці даного винаходу, наведені нижче в таблиці А: Амінокінцева захисна група являє собою хімічний радикал, зчеплений з альфа-аміногрупою амінокислоти. Як правило, амінокінцеву захисну групу видаляють за допомогою реакції з видалення захисних груп перед додаванням наступної амінокислоти, що повинна бути інтродукована в зростаючий пептидний ланцюг, але її можна зберігати, коли пептид відщеплюють від підкладки. Вибір амінокінцевої захисної групи може залежати від різних факторів, таких, наприклад, як тип здійснюваного синтезу й необхідний проміжний продукт або кінцевій продукт. Прикладами амінокінцевих захисних груп є: (1) захисні групи ацильного типу, такі як форміл, акриліл (Асr), бензоїл (Bz) і ацетил (Ас); (2) ароматичні захисні групи уретанового типу, такі як бензилоксикарбоніл (Z) і заміщений Z, такий як парахлорбензилоксикарбоніл, паранітробензилоксикарбоніл, парабромбензилоксикарбоніл, параметоксибензилоксикарбоніл; (З) аліфатичні уретанові захисні групи, такі як трет-бутилоксикарбоніл (Вос), діізопропілметоксикарбоніл, ізопропілоксикарбоніл, етоксикарбоніл, алілоксикарбоніл; (4) циклоалкільні захисні групи уретанового типу, такі як 9-флуоренілметилоксикарбоніл (Fmoc), циклопентилоксикарбоніл, адамантилоксикарбоніл і циклогексилоксикарбоніл; і (5) захисні групи тіоуретанового типу, такі як фенілтіокарбоніл. Кращими захисними групами є 9флуоренілметилоксикарбоніл (Fmoc), 2-(4біфеніліл)пропіл(2)оксикарбоніл (Врос), 2фенілпропіл(2)оксикарбоніл (Рос) і третбутилоксикарбоніл (Вос). Хімічний процес із використанням Fmoc або Fmoc-подібних агентів є найбільш кращим для твердофазового пептидного синтезу, з огляду на те, що відщеплення пептиду, що утворився, у захищеному стані відносно просто здійснювати з використанням слабкої кислоти як агенту для відщіплення. Цей тип реакції розщеплення є відносно чистим з позицій побічних продуктів, що утворюються, домішок і т.д., що робить його технічно й економічно прийнятним для великомасштабного одержання пептиду з використанням відмивань, як для розбухання, так і стискання смоли, з підвищеним виходом. У контексті даного опису поняття «великомасштабний» відносно пептидного синтезу, як правило, включає синтез партії пептиду, що становить принаймні 500 г, більш переважно принаймні 2 кг. Великомасштабний синтез, як правило, здійснюють у великих реакційних посудинах, такі як сталеві реакційні посудини, розмір яких дозволяє вносити реагенти, такі як смоли, розчинники, амінокислоти, хімічні речовини для сполучення й для реакцій з видалення захисних груп, у таких кількостях, які дозволяють виробляти пептиди в діапазоні від кілограма до метричної тонни. 27 Крім того, захисну групу Fmoc можна вибірково видаляти з пептиду поза залежністю від захисних груп бічного ланцюга, так що захист бічних ланцюгів зберігається, коли відщеплюють Fmoc. Такий тип вибіркової дії є важливим при сполученні амінокислот, мінімізуючи реакції за участю бічних ланцюгів. Крім того, захисні групи бічних ланцюгів можна вибірково відщеплювати для видалення їх поза залежністю від Fmoc, залишаючи Fmoc на місці. Остання зазначена вибіркова дія забезпечує дуже велику перевагу при схемах очищення, які додатково описані нижче. Твердофазову реакцію сполучення можна здійснювати в присутності одного або декількох сполук, які підсилюють або підвищують ефективність реакції сполучення. Сполуки, які можуть підвищувати швидкість реакції й знижують швидкість побічних реакцій, являють собою солі фосфонію й уронію, які можуть у присутності третинної основи, наприклад, діізопропілетиламіну (ДІЕА) і триетиламіну (TEA), перетворювати захищені амінокислоти на активовані види (наприклад, ВОР, РуВОРО, ГБТУ й ТБТУ, які всі утворюють складні ГОБТефіри). Інші реагенти сприяють попередженню рацемізації, пов'язаної із захисним реагентом. Ці реагенти включають карбодііміди (наприклад, ДЦК або ВРКДІ) з доданим допоміжним нуклеофілом (наприклад, 1-гідроксибензотриазол (ГОБТ), 1гідроксіазабензотриазол (ГОАТ) або HOSu). Поряд з азидним методом можна застосовувати також метод, заснований на застосуванні змішаних ангідридів, з використанням ізобутилхлорформіату з доданим допоміжним нуклеофілом або без нього завдяки низькій рацемізації, пов'язаній з ним. Ці типи сполук також можуть підвищувати швидкість опосередковуваних карбодіімідом сполучень, а також попереджувати дегідратацію залишків Asn і Gin. Після визначення того, що сполучення завершене, реакційну суміш для сполучення відмивають за допомогою розчинника й цикл сполучення повторюють для кожного наступного амінокислотного залишку пептидного продукту. Для сполучення наступної амінокислоти видалення N-кінцевої захисної групи (наприклад, Fmoc-групи) зі зв'язаного зі смолою продукту, як правило, здійснюють обробкою реагентом, що включає 20-50 мас.% піперидину як розчинник, наприклад, N-метилпіролідону (НМР) або диметилформаміду (ДМФ). Після видалення захисної Fmoc-групи, як правило, здійснюють декілька відмивань для видалення піперидину, що залишився, й побічних продуктів Fmoc (таких як дибензофулвен і його піперидиновий аддукт). Наступні амінокислоти можна використовувати в стехіометричному надлишку амінокислот відносно фактора завантаження пептидного продукту на підкладці зі смоли. Як правило, кількість амінокислот, застосовуваних на стадії сполучення, принаймні еквівалентна фактору завантаження першої амінокислоти в смолі (1 еквівалент або більше). Краща кількість амінокислот, застосовуваних на стадії сполучення, становить принаймні 1,3 еквівалента (надлишок 0,3) або більше й найбільш переважно принаймні приблизно 1,5 еквівалента (надлишок 0,5) або більше. У деяких випад 96602 28 ках на стадії сполучення використовують кількість еквівалентів амінокислот у діапазоні від 1 до 3. Після останнього циклу сполучення смолу відмивають розчинником, таким як НМР, а потім відмивають інертним другим розчинником, таким як ДХМ. Для виділення синтезованого пептидного продукту зі смоли здійснюють розщеплення в такому режимі, при якому відщеплений пептидний продукт усе ще мав достатню кількість захисних груп бічних ланцюгів і кінцевих захисних груп. Збереження захисних груп сприяє попередженню небажаного сполучення або інших небажаних реакцій пептидних фрагментів у процесі або після відщеплення від смоли. У випадку застосування Fmoc або аналогічної хімії для синтезу пептиду захист від розщеплення можна здійснювати будьяким необхідним чином, наприклад, шляхом використання відносно слабкого кислотного реагенту, такого як оцтова кислота або розведена ТФК у розчиннику, такому як ДХМ. Як правило, застосовують від 0,5 до 10 мас.%, переважно від 1 до 3 мас.% ТФК у ДХМ (див., наприклад, US 6281335). Стадії відщеплення пептидного проміжного фрагмента від смоли, що являє собою тверду фазу, можна здійснювати за допомогою процесу, наведеного нижче як приклад. Однак можна застосовувати будь-який придатний процес, при використанні якого відбувається ефективне відщеплення пептидного проміжного фрагмента від смоли. Наприклад, у посудину, що містить зв'язаний зі смолою пептидний продукт, додають приблизно 5-20, переважно приблизно 10 об'ємів розчинника, який містить кислий розщеплюючий агент. Потім у реагент занурюють смолу, як правило, у формі гранул. Реакція розщеплення відбувається при перемішуванні рідкого вмісту при прийнятній температурі протягом необхідного періоду часу. Перемішування дозволяє попереджати грудкування гранул. Необхідний час і температурні умови повинні залежати від таких факторів, як застосовуваний кислотний реагент, природа пептиду, природа смоли й т.п. У цілому, можна здійснювати перемішування при температурі від приблизно -15°С до приблизно 5°С, переважно від приблизно -10°С до приблизно 0°С протягом проміжку часу, що становить від 5 хв. до 2 год., переважно від приблизно 25 хв. до приблизно 45 хв. Тривалість розщеплення може становити від приблизно 10 хв. до приблизно 2 год. або навіть до одного дня. Розщеплення бажано здійснювати при охолодженні, при такому діапазоні температур, щоб компенсувати екзотермічну реакцію, яка, як правило, може мати місце. Крім того, низька температура реакції розщеплення попереджає видалення на цій стадії чутливих захисних груп бічних ланцюгів, таких як тритильні групи. Після розщеплення реакцію припиняють. Для цієї мети можна застосовувати, наприклад, комбінацію розщеплюючого реагенту, із прийнятною основою, такою як піридин або т.п., і продовжувати збовтування та перемішування протягом додаткового періоду часу, наприклад, додатково протягом 5 хв. - 2 год., переважно від приблизно 20 хв. до приблизно 40 хв. Додавання основи та продовження перемішування приводить до підвищення 29 температури в посудині. Наприкінці перемішування вміст посудини може мати температуру, що становить від приблизно 0°С до приблизно 15°С, переважно від приблизно 5°С до приблизно 10°С. Для підвищення виходу пептиду в загальний процес синтезу, необов'язково можна включати такі фактори, як розбухання й стискування смоли. Ці методи, описані, наприклад, в опублікованій заявці на патент США 2005/0164912 А1. Відповідно до деяких об'єктів винаходу, відщиплені пептидні фрагменти можна підготовляти для сполучення в рідкій фазі з іншими пептидними фрагментами і/або амінокислотами. Відомості про реакції пептидного сполучення в рідкій фазі узагальнені, наприклад, в: New Trends in Peptide Coupling Reagents; Albericio Fernando; Chinchilla Rafeal; Dodsworth David J.; і Najera Armen; Organic Preparations and Procedures International, 33(3), 2003, cc. 203-303. Сполучення пептидних проміжних фрагментів з іншими фрагментами або амінокислотою(ами) у рідкій фазі можна здійснювати in situ з використанням реагентів для сполучення, таких, наприклад, як гексафторфосфат 2-(1Н-бензотриазол-1іл)трис(диметиламіно)фосфонію (БОП), гексафторфосфат орто-(бензотриазол-і-іл)-N,N,N',N'тетраметилуронію (ГБТУ), гексафторфосфат орто(7-азобензотриазол-1-іл)-1,1,3,3-тетраметилуронію (ГАТУ), дициклокарбодіімід (ДЦК), водорозчинний карбодіімід (ВРКДІ) або тетрафторборат орто(бензотриазол-1-іл)-N,N,N’,N'-тетраметилуронію (ТБТУ). В інших методах сполучення застосовують попередньо одержані активні складні ефіри, такі як гідроксисукцинімідні (HOSu) і пара-нітрофенолові (HONp) складні ефіри; попередньо одержані симетричні ангідриди; несиметричні ангідриди, такі як N-карбоксіангідриди (NCA); або галогенангідриди, такі як фтористий ацил, а також хлористий ацил. При здійсненні реакції сполучення в рідкій фазі можна використовувати прийнятний розчинник для сполучення. Очевидно, що застосовуваний(і) для сполучення розчинник(и) може(уть) впливати на ступінь рацемізації утвореного пептидного зв'язку; розчинність пептиду і/або пептидних фрагментів; і швидкість реакції сполучення. У деяких варіантах здійснення винаходу розчинник для сполучення включає один або декілька реагентів, що змішуються з водою. Прикладами реагентів, що змішуються з водою, є, наприклад, ДМСО, піридин, хлороформ, діоксан, тетрагідрофуран, етилацетат, Nметилпіролідон, диметилформамід або їх суміші. В інших варіантах здійснення винаходу в реакції сполучення можна застосовувати один або декілька реагентів, що не змішуються з водою. Прикладом розчинника, що не змішується з водою, є метиленхлорид. У цих варіантах здійснення винаходу розчинник, що не змішується з водою, переважно сумісний з реакцією з видалення захисних груп; наприклад, якщо переважно застосовують розчинник, що не змішується з водою, то він не здійснює шкідливого впливу на реакцію з видалення захисних груп. Після одержання пептиду 11 цей продукт можна піддавати при необхідності видаленню захис 96602 30 них груп, очищенню, ліофілізації, додатковому процесингу (наприклад, взаємодії з іншим пептидом з одержанням злитого білка); їх комбінаціям і/або т.п. Наприклад, відповідно до винаходу захисні групи бічних ланцюгів, як правило, зберігаються в пептидних проміжних фрагментах у процесі твердофазового синтезу, а також при здійсненні реакцій сполучення в процесі рідиннофазового синтезу. Як правило, після завершення стадії сполучення в рідкій фазі можна здійснювати одну або декілька стадій з видалення захисних груп для видалення однієї або декількох захисних груп у пептиду. Для видалення захисних груп бічних ланцюгів у процесі повного видалення захисних груп, як правило, використовують розчин для видалення захисних груп, що включає ацидолітичний агент для відщеплення захисних груп бічних ланцюгів. Загальноприйняті ацидолітичні реагенти для повного видалення захисних груп включають нерозбавлену трифтороцтову кислоту (ТФК), НСl, кислоти Льюїса, такі як BF3Et2O або Me3SiBr, рідку плавикову кислоту (HF), бромистий водень (НВr), трифторметансульфонову кислоту і їх комбінації. Розчин для видалення захисних груп може включати також один або декілька придатних акцепторів катіонів, наприклад, динітротреїтол (ДТТ), анізол, пара-крезол, етандитіол або диметилсульфід. Розчин для видалення захисних груп може також включати воду. У контексті даного опису кількість реагентів, що присутня у композиції для видалення захисних груп, як правило, виражають у вигляді співвідношення, у якому кількість індивідуального компонента виражають у чисельнику в «частинах», таких як «масові частини» або «об'ємні частини», а знаменник означає загальну кількість частин у композиції. Наприклад, розчин для видалення захисних груп, що містить ТФК:Н2О:ДТТ у співвідношенні 90:5:5 (мас./мас./мас), містить 90/100 мас.част. ТФК, 5/100 мас.част. Н2О, 5/100 мас.част. ДТТ. Відповідно до деяких варіантів здійснення винаходу можна здійснювати реакцію з видалення захисних груп з використанням такої кількості ацидолітичного агента, переважно ТФК, у композиції для видалення захисних груп на частку якого припадає більше 90/100 мас.част. Переважно використовувати також кількість 93/100 мас.част. або більше, або в діапазоні, що становить від 93/100 мас.част. до 95/100 мас.част. Осадження, як правило, здійснюють за допомогою простого ефіру, наприклад, діетилового ефіру або МТБЕ (метил-трет-бутиловий ефір). Після осадження пептид бажано виділяти й сушити, а потім поєднувати з іншими інгредієнтами, ліофілізувати, упаковувати, зберігати, додатково процесувати і/або іншим способом обробляти. Це можна здійснювати будь-яким прийнятним шляхом. Відповідно до одного прийнятного підходу пептид збирають фільтрацією, відмивають, використовуючи достатню кількість відмивань МТБЕ для зниження кінцевого вмісту солей до прийнятного рівня, і потім сушать. 31 96602 32 Даний винахід стосується також методів, застосовуваних для очищення широкого діапазону пептидів, включаючи GLP-1-пептиди і їх аналоги. Найбільш кращий процес очищення включає принаймні дві стадії очищення через хроматографічні середовища, при цьому принаймні першу стадію здійснюють при першому значенні рН, а принаймні одну другу стадію здійснюють при другому значенні рН. Більш переважно першу стадію здійснюють при кислому значенні рН, а другу стадію при лужному значенні рН. У кращих варіантах здійснення винаходу принаймні першу стадію в кислих умовах здійснюють перед проведенням стадії в основних умовах. Як ілюстрація застосовуваного на практиці варіанта цей підхід очищення може передбачати очищення повністю захищеного пептиду 11, одержаного відповідно до схеми 10, наведеної на фіг. 1. Спочатку здійснюють повне видалення захисних груп пептиду. Відщеплюють як N-кінцеві захисні групи, так і захисні групи бічних ланцюгів. Першу хроматографічну стадію здійснюють у градієнті вода/АЦН (ацетонітрил), використовуючи ТФК у кількості, достатній для досягнення значення рН приблизно 1-5, переважно приблизно 2. Потім здійснюють другу стадію в градієнті вода/АЦН, використовуючи невелику кількість аміаку і/або ацетату амонію або т.п. для досягнення значення рН приблизно 8-9, переважно 8,5-8,9. Значення рН, як кислі, так і лужні, підвищують однорідність, що проявляється у тому, що в кожному випадку присутні однорідні іонні види. Таким чином, кисле значення рН повинне бути досить низьким для того, щоб протонувати практично всі амінокислотні залишки в пептидному продукті. Лужне значення рН повинне бути досить високим для того, щоб депротонувати практично всі амінокислотні залишки в пептидному продукті. Хроматографію в кислих і лужних умовах можна здійснювати в будь-якому порядку. Доцільно, щоб хроматографія в лужних умовах була останньою, коли необхідним продуктом є ацетат пептиду, оскільки ацетат може бути продуктом хроматографії. Принципи даного винаходу додатково проілюстровані за допомогою наведених нижче прикладів. Всі зазначені нижче відсотки й співвідношення є об'ємними, якщо спеціально не зазначене інше. Приклад 1 - Твердофазовий синтез фрагмента 12 з Fmoc-захистом на N-кінці й захистом бічних груп His і Glu А. Одержання завантаженої Fmoc-Gly смоли 2СТС Спочатку одержували завантажену Fmoc-Gly смолу 2СТС. Нижче в таблиці показана кількість застосовуваних реагентів: Смолу 2-СТС вносили в 500-мілілітровий пептидний реактор і давали набухати в 400 мл ДХМ протягом 30 хв. при 25°С. Шар осушували й додавали розчин Fmoc-Gly-OH і ДІЕА в 8 об'ємах ДМФ:ДХМ (87,5:12,5). Суміш перемішували в атмосфері азоту протягом 2 год. при температурі 25°С. Шар осушували й однократно промивали 350 мл ДМФ й однократно 175 мл ДМФ. Потім здійснювали кінцеве кепірування активних сайтів, що залишилися, на смолі 2-СТС, використовуючи 350 мл розчину МеОН:ДІЕА (9:1) протягом 1 год. Шар осушували й промивали двічі 250 мл ДМФ і потім чотири рази 350 мл ДХМ. Смолі повторно давали набухати шляхом промивання 3x350 мл ІПС. Смолу сушили до постійної маси з одержанням 38,20 г завантаженої смоли. Аналіз показав, що фактор завантаження становив 0,18 ммоль/г. Б. Твердофазовий синтез Твердофазовий синтез здійснювали, починаючи з 20,0 г завантаженої Fmoc-Gly-2-СТС-смоли з фактором завантаження 0,18 ммоль/г, що одержували відповідно до методу, описаного в розділі А цього прикладу 1. Смолі давали набухати в ДХМ (200 мл) протягом 30хв при 25°С. Розчинник ДХМ зливали й смолу промивали тричі НМР (для кожного промивання використовували 5 об'ємів). Потім смолу обробляли двічі 20 об.%-вим розчином піперидину в НМР (для кожної обробки використовували 5 об'ємів) для видалення захисних груп Fmoc. Після другої обробки 20% піперидином/НМР смолу промивали п'ять разів НМР (для кожного промивання використовували 5 об'ємів) до досягнення негативної проби на хлораніл. Для приготування розчину для сполучення зважували амінокислоту (2,85 екв.) і 6-хлор-1гідроксибензотриазол (6-Сl-ГОБТ, 2,85 екв.), роз 33 96602 34 чиняли в 2,55-кратному (2,55х) об'ємі НМР, потім поєднували з ДІЕА (3,25 екв.) при температурі від 5 до 10°С. ТБТУ (2,85 екв.) розчиняли в 1,3хоб'ємі НМР при температурі від 5 до 10°С. Потім два розчини поєднували. Розчин, що утворився, додавали в реакційну посудину. Колбу відмивали 1,3хоб'ємом ДХМ, додавали в реактор, вміст якого потім перемішували протягом 2-3 год. при температурі від 25 до 27°С. Відбирали зразок для здійснення тесту Кайзера для оцінки завершення реакції. Якщо реакція сполучення через 3 год. не закінчувалася (позитивний тест Кайзера), то реакційну посудину осушували й здійснювали повторне сполучення з використанням свіжого розчину активованої амінокислоти. Після завершення реакції сполучення розчин для сполучення зливали й смолу промивали чотири рази НМР (для кожного промивання використовували 5 об'ємів). Потім цикл, що включає видалення захисної групи Fmoc і реакцію сполучення, повторювали для амінокислот фрагмента, які залишилися (тобто в порядку Glu(OtBu)->Aib->His(trt)). Через складності проведення реакції сполучення між активованою Fmoc-His(trt)-OH і H-AibGlu(OtBu)-Gly-2-CTC і нестабільності активованої Fmoc-His(trt)-ОН, реакцію сполучення завершували шляхом зливу реакційного буфера через 1 год. і негайного здійснення повторної реакції сполучення з використанням другого розчину зі свіжоактивованою Fmoc-His(trt)-OH. Всі реагенти, застосовувані в розділі Б цього прикладу, перераховані нижче в таблиці: Зв'язаний зі смолою пептидний фрагмент 4 рази промивали НМР (5 об.), 5 разів ДХМ (6 об.), З рази ІПС (5 об.). Повторно набухлу смолу потім сушили при 35°С у вакуумі, одержуючи 22,58 г смоли й зв'язаного зі смолою пептиду. В. Відщеплення від смоли фрагмента з Fmocгрупою й із захищеними бічними ланцюгами Смолі, створеній відповідно до методу, описаного вище в розділі Б, давали набухати в ДХМ (12,5 об'ємів відносно маси застосовуваної смоли; 12,5 мл ДХМ на г смоли або 12,5 л на кг) протягом 30 хв. при 25°С, а потім 2 рази промивали ДХМ (для кожного промивання використовували 6,25 об'ємів) для видалення всього залишкового НМР. Смолу охолоджували при останньому промиванні ДХМ до -5°С. ДХМ зливали й додавали холодний розчин 1% ТФК/ДХМ (10 об. при температурі від -5 до -10°С) і перемішували протягом 30 хв. при 0°С. У реактор додавали піридин (1,3 екв. відносно ТФК) для нейтралізації ТФК. Розчин для відщеплення фільтрували та збирали в колбу. Після нагрівання посудини до 25°С смолу промивали 7 разів ДХМ (7,5 об.). Змиви поєднували з розчином для розщеплення. Розчин для розщеплення, що містить ДХМ, поєднували з водою (7,5 об.). Одержану суміш переганяли при зниженому тиску для видалення ДХМ (350 Торр при 28°С). Пептидний фрагмент осаджувався з води при видаленні ДХМ. Фрагмент промивали водою й сушили при 30-35°С 8 у вакуумі. Усього одержали 4,73 г Fmoc-(Aib )GLP1(7-10)-OH. Приклад 2 А. Одержання завантаженої Fmoc-Gly смоли 2СТС Одержували завантажену Fmoc-Gly смолу 2СТС. Нижче в таблиці показана кількість застосовуваних реагентів: 35 96602 36 Смолу 2-СТС вносили в 500-мілілітровий пептидний реактор і давали набухати в 400 мл ДХМ протягом 30 хв. Смолу осушували й додавали розчин Fmoc-Gly-OH і ДІЕА в 8 об'ємах ДМФ:ДХМ (87,5:12,5). Суміш перемішували в атмосфері азоту протягом 2 год. при температурі 25°С. Шар смоли осушували й однократно промивали 400 мл ДМФ і однократно 200 мл ДМФ. Потім здійснювали кінцеве кепірування активних сайтів, що залишилися, на смолі 2-СТС, використовуючи 390 мл розчину МеОН:ДІЕА (9:1) протягом 1 год. Шар знову осушували, промивали двічі 350 мл ДМФ і потім чотири рази 350 мл ДХМ. Смолі повторно давали набухати шляхом промивання 3x350 мл ШС. Смолу сушили при 3 5°С у вакуумі до постійної маси з одержанням 48,51 г завантаженої смоли. Аналіз показав, що фактор завантаження становив 0,54 ммоль/г. Б. Твердофазовий синтез Твердофазовий синтез здійснювали, починаючи з 27,59 г завантаженої Fmoc-Gly-2-СТС-смоли з фактором завантаження 0,54 ммоль/г. Смолі давали набухати в ДХМ (300 мл) протягом 30 хв при 25°С. Розчинник ДХМ зливали й смолу промивали тричі НМР (для кожного промивання використовували 5 об'ємів). Потім смолу обробляли двічі 20 об.%-вим розчином піперидину в НМР (для кожної обробки використовували по 5 об'ємів) для видалення захис них груп Fmoc. Після другої обробки 20% піперидином/НМР смолу промивали шість разів НМР (для кожного промивання використовували по 5 об'ємів) до досягнення негативної проби на хлораніл. Для приготування розчину для сполучення зважували амінокислоту (1,7 екв.) і 6-хлор-1гідроксибензотриазол (6-Сl-ГОБТ, 1,7 екв.), розчиняли в 4,6xоб'ємі НМР, потім поєднували з ДІЕА (1,9 екв.) при температурі від 5 до 10°С. ТБТУ (1,7 екв.) розчиняли в 2,28xоб'ємі НМР при температурі від 5 до 10°С. Потім два розчини поєднували. Розчин, що утворився, додавали в реакційну посудину. Колбу відмивали 2,28xоб'ємом ДХМ, додавали в реактор, вміст якого потім перемішували протягом 2-3 год. при температурі від 25 до 27°С. Відбирали зразок для тесту Кайзера з метою оцінки завершення реакції. Після завершення реакції сполучення розчин для сполучення зливали й смолу промивали чотири рази НМР (для кожного промивання використовували по 5 об'ємів). Потім цикл, що включає видалення захисної групи Fmoc і реакцію сполучення, повторювали для амінокислот фрагмента, які залишилися (тобто в порядку Glu(OtBu)АіbHis(trt)). Усі реагенти, застосовувані в цьому прикладі, перераховані нижче в таблиці: В. Відщеплення фрагмента від смоли Створену смолу промивали 6 разів НМР (5 об.), а потім 8 разів ДХМ (6 об.) для видалення залишку НМР. Смолу охолоджували шляхом останнього промивання ДХМ до -5°С. ДХМ зливали й після цього додавали холодний (від -5 до 10°С) розчин 1% ТФК/ДХМ (10 об.) і суміш, що утворилася, перемішували протягом 30 хв при 0°С. У реактор додавали піридин (1,3 екв. відносно ТФК) для нейтралізації ТФК. Розчин для відщеплення збирали в колбу. Після нагрівання посудини до 25°С смолу промивали 11 разів ДХМ (7,5 об.) і зливали в розчин для розщеплення. Розчин, що містить ДХМ, поєднували з водою (10 об.). Одержану суміш переганяли при зниженому тиску для видалення ДХМ (350 Торр при 28°С). Пептидний фрагмент осаджувався з води при видаленні ДХМ. Фрагмент промивали водою й сушили при 30-35°С у вакуумі. Усього одержали 11,12 г Fmoc8 (Aib )GLP-1(7-10)-OH (вихід 78,8%). Приклад З А. Одержання завантаженої Fmoc-Gly смоли 2СТС Одержували завантажену Fmoc-Gly смолу 2СТС. Нижче в таблиці показана кількість застосовуваних реагентів: 37 Смолу 2-СТС вносили в 500-мілілітровий пептидний реактор і давали набухати в 400 мл ДХМ протягом 30 хв. Шар осушували й додавали розчин Fmoc-Gly-OH і ДІЕА в 8 об'ємах ДМФ:ДХМ (87,5:12,5). Суміш перемішували в атмосфері азоту протягом 2 год. при температурі 25°С. Шар осушували й однократно промивали 400 мл ДМФ і однократно 200 мл ДМФ. Потім здійснювали кінцеве кепірування всіх активних сайтів, що залишилися, на смолі 2-СТС, використовуючи 390 мл розчину МеОН:ДІЕА (9:1) протягом 1 год. Ф Ф Тар осушували, промивали двічі 350 мл ДМФ і потім чотири рази 350 мл ДХМ. Смолі повторно давали набухати шляхом промивання 4x250 мл ІПС. Смолу сушили при 35°С у вакуумі до постійної маси з одержанням 52,02 г завантаженої смоли. Аналіз показав, що фактор завантаження становив 0,72 ммоль/г. Б. Твердофазовий синтез Твердофазовий синтез здійснювали, починаючи з 24,43 г завантаженої Fmoc-Gly-2-СТС-смоли з фактором завантаження 0,72 ммоль/г. Смолі давали набухати в ДХМ (250 мл) протягом 30 хв при 25°С. Розчинник ДХМ зливали й смолу промивали тричі NМП (для кожного промивання використовували по 5 об'ємів). Потім смолу обробляли двічі 20 об.%-вим розчином піперидину в NМП (для кожної обробки використовували 5 об'ємів) для видалення захисних груп Fmoc. Після другої обробки 20% піперидином/ 96602 38 NМП смолу промивали шість разів NМП (для кожного промивання використовували по 5 об'ємів) до досягнення негативної проби на хлораніл. Для приготування розчину для сполучення зважували амінокислоту й 6-хлор-1гідроксибензотриазол (6-Сl-ГОБТ), розчиняли в NМП, потім поєднували з ДІЕА при температурі 10 - 5°С. ТБТУ розчиняли в NМП при температурі 10 5°С. Потім два розчини поєднували. Розчин, що утворився, додавали в реакційну посудину. Колбу відмивали ДХМ (дані про кількість див. нижче в таблиці), додавали в реактор, вміст якого потім перемішували протягом 2-3 год. при температурі від 25 до 27°С. Відбирали зразок для тесту Кайзера з метою оцінки завершення реакції. Якщо реакція сполучення через 3 год. не закінчувалася (позитивний тест Кайзера), то реакційну посудину осушували й здійснювали повторне сполучення з використанням свіжого розчину активованої амінокислоти. Після завершення реакції сполучення розчин для сполучення зливали й смолу промивали чотири рази NМП (для кожного промивання використовували 5 об'ємів). Потім цикл, що включає видалення захисної групи Fmoc і реакцію сполучення, повторювали для амінокислот фрагмента, які залишилися (тобто в порядку Glu(OtBu)AibHis(trt)). Усі реагенти, застосовувані в цьому прикладі, перераховані нижче в таблиці: 39 96602 40 В. Відщеплення фрагмента Fmoc-AA(7-10)-OH від смоли Створену смолу промивали 6 разів НМР (5 об.), а потім 7 разів ДХМ (6 об.) для видалення залишку NМП. Смолу охолоджували шляхом останнього промивання ДХМ до -5°С. ДХМ зливали, і шар смоли промивали холодним (від -5 до 10°С) розчином 1% ТФК/ДХМ (11,26 об.) протягом 5 хв. при 0°С. Розчин для відщеплення збирали в колбу, у яку був внесений піридин (1,3 екв. відносно ТФК) для нейтралізації ТФК. Потім у реактор додавали другу частину холодного розчину 1% ТФК/ДХМ (6,14 об.) і перемішували протягом 2 хв. Другий розчин для розщеплення знову зливали в колбу для збирання. Після нагрівання посудини до 25°С смолу промивали 9 разів ДХМ (8,2 об.) і зливали в розчин для розщеплення. Розчин, що містить ДХМ, поєднували з водою (8,2 об.). Одержану суміш переганяли при зниженому тиску для видалення ДХМ (350 Торр при 28°С). Пептидний фрагмент осаджувався з води при видаленні ДХМ. Фрагмент промивали водою й сушили при 30-35°С у вакуумі. Усього одержали 14,02 г Fmoc8 (Aib )GLP-1(7-10)-OH (вихід 86,6%), що мав послідовність, представлену в SEQ ID NO. 7. Аналіз показав, що чистота продукту становила 94,3% AN. Приклад 4 - Твердофазовий синтез Fmoc35 (Aib ) GLP-1 (23-35)-ОН із захищеним бічним ланцюгом А. Одержання завантаженої Fmoc-Aib смоли 2СТС Одержували завантажену Fmoc-Aib смолу 2СТС. Нижче в таблиці показана кількість застосовуваних реагентів: Смолу 2-СТС вносили в 500-мілілітровий пептидний реактор і давали набухати в 400 мл ДХМ протягом 30 хв. Шар осушували й додавали розчин Fmoc-Aib-OH і ДІЕА в 8 об'ємах ДМФ:ДХМ (87,5:12,5). Суміш перемішували в атмосфері азоту протягом 2 год. при температурі 25°С. Шар осушували й однократно промивали 400 мл ДМФ перший раз і 200 мл другий раз. Потім здійснювали кінцеве кепірування всіх активних сайтів, що залишилися, на смолі 2-СТС, використовуючи 400 мл розчину МеОН:ДІЕА (9:1) протягом 1 год. Шар осушували, промивали однократно 450 мл ДМФ/МеОН/ДІЕА (4:0,9:0,1), однократно 200 мл ДМФ і чотири рази 350 мл ДХМ. Смолі повторно давали набухати шляхом промивання 3x350 мл ІПС. Смолу сушили до постійної маси з одержанням 45,15 г завантаженої смоли. Аналіз показав, що фактор завантаження становив 0,24 ммоль/г. Б. Твердофазовий синтез 10,01 г Fmoc-Aib-2-СТС-смоли з фактором завантаження 0,24 ммоль/г вносили в реакційну посудину й давали набухати в ДХМ (120 мл) протягом 30 хв при 25°С. Розчинник ДХМ зливали й смолу промивали тричі NМП (для кожного промивання використовували 6 об'ємів). Потім смолу обробляли двічі 5 об.%-вим розчином піперидину в NМП (для кожної обробки використовували по 6 об'ємів) для видалення захисних груп Fmoc. Після другої обробки 5% піпериди піперидином/ NМП смолу промивали чотири рази НМР (для кожного промивання використовували по 6 об'ємів). Для приготування розчину для сполучення амінокислоту (1,875 екв.) і моногідрат 1гідроксибензотриазолу (гідрат ГОБТ, 2,07 екв.) розчиняли в 3,5xоб'ємі NМП при температурі від 5 до 10°С і потім поєднували з 16,1 мл розчину ГБТУ (2,0 екв.) в NМП (1,5хоб.). Потім 2,2 мл ДІЕА (2,63 екв.) додавали в посудину для активації при температурі 10-5°С. Розчин, що утворився, переносили в реакційну посудину. Вміст посудини для активації змивали 1,5х об'ємом ДХМ у реактор, потім перемішували протягом 2 год. при 25°С. Реакційну посудину осушували. Реакцію сполучення повторювали ще один раз, використовуючи свіжий розчин активованої амінокислоти (1,875 екв.) Після завершення другої реакції сполучення розчин для сполучення зливали й смолу промивали 4 рази NМП (по 6 об. для кожного промивання). Потім цикл, що включає видалення захисної групи Fmoc і реакцію сполучення, повторювали для амінокислот фрагмента, які залишилися (тобто в порядку Lys(Boc)ValLeuTrс(Вoc)AlaIlePheGlu (OtBu)Lys(Boc)AlaAlaGln(trt)). Усі реагенти, застосовувані в цьому прикладі, перераховані нижче в таблиці: 41 96602 42 43 96602 44 Створену смолу виділяли шляхом промивання 4 рази NМП (6 об.), 4 рази ДХМ (6 об.) і 3 рази ізопропанолом (ІПС, 6 об.). Створену смолу сушили при 35°С у вакуумі. Одержали 14,3 г створені смоли. В. Відщеплення проміжного фрагмента від створеної смоли 6,6 г створеної відповідно до описаного вище методу смоли давали набухати в 10хоб'ємі ДХМ протягом 30 хв і охолоджували до -10°С. ДХМ зливали й додавали холодний розчин 1% ТФК/ДХМ (12 об., при температурі від -5 до -10°С) і перемішували протягом 30 хв при 0°С. Розчин для відщеплення збирали в колбу, що містить піридин (23 екв. відносно ТФК). Після нагрівання до 25°С смолу перемішували з 1% ТФК/ДХМ (10х об.) про тягом 5 хв і додавали піридин (2-3 екв.). Ще через 5 хв розчин збирали. Смолу промивали 4 рази ДХМ (10 об.). Всі ДХМ-змиви поєднували з водою (вода/ДХМ = 1/4). Одержану суміш переганяли при зниженому тиску для видалення ДХМ (350 Торр при 28°С). Фрагмент осаджувався з води при видаленні ДХМ. Фрагмент промивали водою й сушили при 30-35°С у вакуумі. Процедуру розщеплення повторювали ще один раз. Усього одержали 2,36 г 35 Fmoc-(Aib ) GLP-1 (23-35)-OH (вихід 92%). Приклад 5 А. Одержання завантаженої Fmoc-Aib смоли 2СТС Одержували завантажену Fmoc-Aib смолу 2СТС. Нижче в таблиці показана кількість застосовуваних реагентів: Смолу 2-СТС вносили в 500-мілілітровий пептидний реактор і давали набухати в 400 мл ДХМ протягом 30 хв. Шар осушували й додавали розчин Fmoc-Aib-OH і ДІЕА в 8 об'ємах ДМФ.-ДХМ (87,5:12,5). Суміш перемішували в атмосфері азоту протягом 2 год. при температурі 25°С. Шар осушували та однократно промивали 400 мл ДМФ. Потім здійснювали кінцеве кепірування всіх активних сайтів, що залишилися, на смолі 2СТС, використовуючи 400 мл розчину МеОН:ДІЕА (9:1) протягом 1 год. Шар осушували, промивали однократно 450 мл ДМФ, однократно 200 мл ДМФ і чотири рази 350 мл ДХМ. Смолі повторно давали набухати шляхом промивання 3x350 мл ІПС. Смолу сушили до постійної маси з одержанням 45,32 г завантаженої смоли. Аналіз показав, що фактор завантаження становив 0,30 ммоль/г. Б. Твердофазовий синтез Твердофазовий синтез здійснювали, починаючи з 15,0 г завантаженої Fmoc-Aib-2-СТС-смоли з фактором завантаження 0,30 ммоль/г. Смолі давали набухати в ДХМ (150 мл) протягом 30 хв при 25°С. Розчинник ДХМ зливали й смолу промивали двічі ДХМ (по 6 об. для кожного промивання) і тричі NМП (по 6 об. для кожного промивання). Потім смолу обробляли двічі 20%-вим розчином піперидину в NМП (по 6 об. для кожної обробки) для видалення захисних груп Fmoc. Після другої обробки 20% піперидином/НМР смолу промивали шість разів NМП (по 6 об. для кожного промивання) до досягнення негативної проби на хлораніл. Для приготування розчину для сполучення зважували амінокислоту (1,7 екв.) і 6-хлор-1гідроксибензотриазол (6-Сl-ГОБТ, 1,7 екв.), розчиняли в 2,6xоб'ємі NМП при температурі 10-5°С, потім поєднували з ДІЕА (1,9-3,0 екв.)· ТБТУ або ГБТУ (1,7 екв.) розчиняли в 1,33хоб'ємі NМП при температурі 10-5°С. Потім два розчини поєднували. Розчин, що утворився, додавали в реакційну посудину. Колбу, у якій здійснювали змішування, промивали 1,33xоб'ємом ДХМ, змиви додавали в реактор, вміст якого потім перемішували протягом 2-3 год. при температурі від 25 до 27°С. Відбирали зразок для тесту Кайзера з метою оцінки завершення реакції. Якщо реакція сполучення через 3 год. не закінчувалася (позитивний тест Кайзера), то реакційну посудину осушували й здійснювали повторне сполучення з використанням свіжого розчину активованої амінокислоти. Після завершення реакції сполучення розчин для сполучення зливали й смолу промивали чотири рази НМР (6 об. для кожного промивання). Потім цикл, що включає видалення захисної групи Fmoc і реакцію сполучення, повторювали для амінокислот фрагмента, які залишилися (тобто в порядку Lys(Boc)ValLeuTrс(Вoc)AlaIlePheGlu (OtBu)Lys(Boc)AlaAlaGln(trt)). Через можливу підтримуючу взаємодію між 2метилаланіном (Aib) і смолою 2-СТС, можуть виникати значні труднощі в здійсненні завершення 45 реакцій сполучення перших двох амінокислот (Lys(Boc)-34 і Val-33). Тому обидві реакції сполучення (Lys(Boc)-34, Val-33) здійснювали тричі (тобто після сполучення здійснювали ще два повторних сполучення). Крім того, оцтовий ангідрид застосовували для кінцевого кепірування зв'язаного зі смолою продукту, що не прореагував, після 96602 46 реакцій сполучення Lys(Boc)-34 і Val-33. Це підвищувало ефективність наступного очищення в результаті видалення домішок з необхідного продукту в процесі хроматографічного очищення. Всі реагенти, застосовувані в цьому прикладі, перераховані нижче в таблиці: 47 В. Відщеплення фрагмента від створеної смо 96602 48 Створену відповідно до описаного вище методу смолу 7 разів промивали ДХМ (по 6 об. для кожного промивання) для видалення залишкових кількостей NМП і смолу охолоджували за допомогою останнього промивання ДХМ до -5°С. ДХМ зливали й додавали холодний розчин 1% ТФК/ДХМ (12 об., при температурі від -5 до -10°С) і перемішували протягом 30 хв. при 0°С. Розчин для відщеплення збирали в колбу, що містить піридин (1,3 екв. відносно ТФК). Після нагрівання посудини до 25°С смолу промивали 9 разів ДХМ (10 об.) і зливали в розчин для розщеплення. Розчин ДХМ поєднували з водою (6 об.). Одержану суміш переганяли при зниженому тиску для видалення ДХМ (350 Торр при 28°С). Фрагмент осаджувався з води при видаленні ДХМ. Фрагмент промивали й сушили при 30-35°С у вакуумі. У цьому прикладі процедуру розщеплення повторювали ще один раз. 35 Усього одержали 6,78 г Fmoc-(Aib ) GLP-1 (23-35)ОН (вихід 68,1%) із чистотою 87,3% AN. Приклад 6 А. Одержання завантаженої Fmoc-Aib смоли 2СТС Одержували завантажену Fmoc-Aib смолу 2СТС. Нижче в таблиці показані кількості застосовуваних реагентів: Смолу 2-СТС вносили в 500-мілілітровий пептидний реактор і давали набухати в 400 мл ДХМ протягом 30 хв. Шар осушували й додавали розчин Fmoc-Aib-OH і ДІЕА в 8 об'ємах ДМФ:ДХМ (87,5:12,5). Суміш перемішували в атмосфері азоту протягом 2 год. при температурі 25°С. Шар осушували та промивали 400 мл ДМФ. Потім здійснювали кінцеве кепірування всіх активних сайтів, що залишилися, на смолі 2-СТС, використовуючи 400 мл розчину МеОН:ДІЕА (9:1) протягом 1 год. Шар осушували, промивали однократно 400 мл ДМФ, промивали однократно 200 мл ДМФ і промивали чотири рази 350 мл ДХМ. Смолі повторно давали набухати шляхом промивання 3x350 мл ІПС. Смолу сушили до постійної маси з одержанням 47,56 г завантаженої смоли. Аналіз показав, що фактор завантаження становив 0,37 ммоль/г. Б. Твердофазовий синтез Твердофазовий синтез здійснювали, починаючи з 25,0 г завантаженої Fmoc-Aib-2-СТС-смоли з фактором завантаження 0,37 ммоль/г. Смолі давали набухати в ДХМ (250 мл) протягом 30 хв при 25°С. Розчинник ДХМ зливали й смолу промивали двічі ДХМ (по 6 об. для кожного промивання) і тричі NМП (по 6 об. для кожного промивання). Потім смолу обробляли двічі 20 об.%-вим розчином піперидину в NМП (6 об. для кожної обробки) для видалення захисних груп Fmoc. Після другої обробки 20% піперидином/ NМП смолу промивали шість разів NМП (6 об. для кожного промивання) до досягнення негативної проби на хлораніл. Для приготування розчину для сполучення зважували амінокислоту й 6-хлор-1гідроксибензотриазол (6-Сl-ГОБТ), розчиняли в 3,2xНМР (або ДМФ для Lys-34, Val-33 і Gln-23), потім поєднували з ДІЕА при температурі від 10 до 5°С. ТБТУ розчиняли в 1,6xоб'ємі НМР (або ДМФ для Lys-34, Val-33 і Gln-23) при температурі від 10 до 5°С. Потім два розчини поєднували. Розчин, що утворився, додавали у реакційну посудину й колбу ли 49 промивали 1,6 x об'ємом ДХМ, змиви вносили в реактор, перемішували зі смолою протягом 2-3 год. при температурі від 25 до 27°С. Відбирали зразок для тесту Кайзера з метою оцінки завершення реакції. Якщо реакція сполучення через 3 год. не закінчувалася (позитивний тест Кайзера), то реакційну посудину осушували й здійснювали повторне сполучення з використанням свіжого розчину активованої амінокислоти. Після завершення реакції сполучення розчин для сполучення зливали й смолу промивали чотири рази NМП (по 6 об. для кожного промивання). Потім цикл, що включає видалення захисної групи Fmoc і реакцію сполучення, повторювали для амінокислот фрагмента, які залишилися (тобто в порядку Lys(Boc)ValLeuTrс(Вoc)AlaIlePheGlu (OtBu)Lys(Boc)AlaAlaGln(trt)). Через можливу підтримуючу взаємодію між 2метилаланіном (Aib) і смолою 2-СТС, можуть ви 96602 50 никати значні труднощі в здійсненні завершення реакцій сполучення перших двох амінокислот (Lys(Boc)-34 і Val-ЗЗ). Умови сполучення Lys(Boc)34, Val-33 і Gln(trt)-23 модифікували, підвищуючи застосовувані кількості, як амінокислот, так і 6-СlГОБТ із 1,7 екв. до 2,5 екв., а ДІЕА з 1,9 екв. до 3,0 екв. Розчинник для реакції сполучення також змінювали, використовуючи замість NМП ДМФ, для завершення реакції сполучення. Крім того, у цьому прикладі застосовували оцтовий ангідрид для кінцевого кепірування зв'язаного зі смолою продукту, що не прореагував, після реакцій сполучення Lys(Boc)-34 і Val-33. Це підвищувало ефективність наступного очищення в результаті видалення домішок з необхідного продукту в процесі хроматографічного очищення. Всі реагенти, застосовувані в цьому прикладі, перераховані нижче в таблиці: 51 В. Відщеплення фрагмента від створеної смоли Створену відповідно до описаного вище методу смолу 6 разів промивали ДХМ (по 6 об. для кожного промивання) для видалення NМП і смолу охолоджували за допомогою останнього промивання ДХМ до -5°С. ДХМ зливали й додавали холодний розчин 1% ТФК/ДХМ (10 об., при температурі від -5 до -10°С) і перемішували протягом 30 хв. при 0°С. Розчин для відщеплення збирали в колбу, що містить піридин (1,3 екв. відносно ТФК). Після нагрівання посудини до 25°С смолу промивали 7 разів ДХМ (6 об.) і зливали в розчин для розщеплення. Розчин ДХМ поєднували з водою (10 об.). Одержану суміш переганяли при зниженому тиску для видалення ДХМ (350 Торр при 28°С). Фрагмент осаджувався з води при видаленні ДХМ. Фрагмент промивали й сушили при 3035°С у вакуумі. У цьому прикладі процедуру розщеплення повторювали ще один раз. Усього оде35 ржали 12,36 г Fmoc-(Aib ) GLP-1 (23-35)-OH (вихід 59,35%) із чистотою 84,3% AN. Приклад 7 1. Промивання смоли Процес починали з попередньо завантаженої Fmoc-Gly-O-2-СТС-смоли (завантаження від 0,18 до 0,65 ммоль/г) або Fmoc-Aib-O-2-CTC (завантаження від 0,25 до 0,65 ммоль/г), застосовуючи стандартну Fmoc-хімію. Смолі спочатку давати набухати в 10xоб'ємі ДХМ протягом 30-60 хв. Потім ДХМ зливали й смолу промивали 10xоб’ємом NМП протягом 3-5 хв (по 5 хв. щоразу). 2. Загальний цикл синтезу: Використовуючи смолу, промиту відповідно до методу, описаного в розділі А прикладу 7, для видалення Fmoc застосовували дві обробки 10хрозчином 20% піперидину в NМП (об./об.). Кожну обробку здійснювали протягом 15-30 хв. Після кожної обробки розчин піперидину /НМР зливали. Потім смолу промивали NМП 4-5 разів (10хоб'єм, по 5 хв. щоразу). Для приготування розчину для сполучення зважували захищену за допомогою Fmoc амінокислоту (АА) і ГОБТ (з розрахунку 1,5-2,0 екв.), розчиняли в 4хоб'ємі NМП при 10°С і поєднували з ДІЕА (1,5-2,0 екв.). ГБТУ (1,5-2,0 екв.) розчиняли в 3хоб'ємі NМП при 10°С. Потім два розчини поєднували й змішували з 3хоб'ємом ДХМ протягом 1-2 хв. Розчин, що утворився, додавали в реакційну посудину й змішували зі смолою при перемішуванні протягом 1,5-5 год. Відбирали зразок для тесту Кайзера з метою оцінки завершення реакції. Продукти незавершеної реакції сполучення піддавали повторному сполученню. Після завершення реакції сполучення розчин для сполучення злива 96602 52 ли й смолу промивали 5 разів NМП (10хоб'єм, по 5 хв. щоразу). A. Після здійснення загальної процедури синтезу фрагмент Fmoc-GLP-1(11-22)-2-СТС, що має нативну послідовність, створювали за допомогою постадійного процесу, використовуючи таку ж процедуру синтезу, фрагмент, послідовність якого представлена в SEQ ID NO. 7, сполучили із фрагментом, що утворився, Fmoc-GLP-1(11-22)-2-СТС, одержаним відповідно до описаного в цьому розділі А методом, використовуючи загальну процедуру синтезу. Б. Після здійснення загальної процедури синтезу фрагмент Fmoc-GLP-1(11-22)-2-СТС створювали за допомогою постадійного процесу. Однак умови, у яких здійснювали видалення Fmoc, заміняли, використовуючи розчин, що містить 10% піперидину й 2% ДБУ в НМР (об./об.) замість 20% піперидину в NМП (об./об.). Крім того, час обробки в положенні АА14 [Fmoc-Ser(tBu)-] і в положенні АА13 [Fmoc-Thr(tBu)-] подовжували до 1,5 год. із метою завершення реакції. Використовуючи таку ж загальну процедуру синтезу фрагмент, послідовність якого представлена в SEQ ID NO. 7, сполучили з одержаним фрагментом Fmoc-GLP- 1(1122)-2-СТС за допомогою загальної процедури синтезу. B. Дотримуючись загальної процедури синтезу з використанням розчину для видалення Fmoc, що містить 10% піперидину й 2% ДБУ в НМР (об./об.), 17-18 створювали фрагмент Fmoc-(X ) GLP-1(11-22)2-CTC за допомогою постадійного процесу. Потім фрагмент, послідовність якого представлена в SEQ ID NO. 7, сполучили з одержаним фрагмен17-18 том Fmoc-(X ) GLP-1(U-22)-2-CTC за допомогою загальної процедури синтезу. Г. Дотримуючись загальної процедури синтезу, 17-18 створювали фрагмент Fmoc-(X ) GLP-1(11-22)2-CTC за допомогою постадійного процесу. Потім фрагмент, послідовність якого представлена в SEQ ID NO. 7, сполучили з одержаним фрагмен17-18 том Fmoc-(X ) GLP-1(11-22)-2-CTC за допомогою загальної процедури синтезу. Д. Дотримуючись загальної процедури синтезу, викладеної вище в розділі Г, і за винятком розходжень, які зазначені в наведеній нижче таблиці, 17-18 створювали фрагмент Fmoc-(X ) GLP-1(11-22)2-CTC за допомогою постадійного процесу. Потім фрагмент, послідовність якого представлена в SEQ ID NO. 7, сполучили з одержаним фрагмен17-18 том Fmoc-(X ) GLP-1(11-22)-2-CTC за допомогою загальної процедури синтезу. Нижче в таблиці узагальнені деталі процедур, викладених у розділах А-Д даного прикладу: 53 Е. Відщеплення фрагмента: Для здійснення відщеплення кожного з пептидних фрагментів, синтезованих відповідно до процедури, описаної в цьому прикладі, смолі зі створеним фрагментом давали набухати в 10хоб'ємі ДХМ протягом 30 хв. і охолоджували до -10°С. ДХМ зливали й додавали розчин 1% ТФК/ДХМ (10хоб'єм) і перемішували протягом 30 хв. Розчин для розщеплення збирали в колбу, що містить піридин (2-3 екв. відносно ТФК). Після нагрівання до 25°С смолу обробляли 1% ТФК/ДХМ (10хоб'єм) протягом 5 хв, а потім додавали піридин (2-3 екв. відносно ТФК). Після перемішування протягом ще 5 хв розчин збирали. Потім смолу промивали 4 рази 10хоб'ємом ДХМ (щоразу 5 хв). Розчини, одержані після всіх промивань, і розчин для розщеплення поєднували й змішували з водою (об'ємне співвідношення вода/ДХМ ~1/4). Одержану суміш переганяли при зниженому тиску для видалення ДХМ (350 Торр/28°С). Пептидний фрагмент осаджувався з води при видаленні ДХМ, і його фільтрували. Пептидний фрагмент промивали водою й сушили при 30°С у вакуумі. Приклад 8 - Синтез у розчині: додавання Are 35 Фрагмент (Aib ) GLP-1 (23-35) (несучий захищений бічний ланцюг (див. таблицю А) і несучий захисну групу Fmoc HaN-кінці (9,11 г, 3,94 ммоль) розчиняли в ДМСО (90 мл). До цього розчину додавали ГОБТ (2,42 г, 4 екв.), ГБТУ (5,98 г, 4 екв.), ДІЕА (3,44 мл, 5 екв.) і H-Arg (2HC1)-NH2 (3,88 г, 4 екв.) в 10 мл ДМСО. Реакційну суміш перемішували й здійснювали моніторинг за допомогою ВЕРХ. Через 4 год. реакція не була завершена, тому додавали 2 мл ДІЕА. Реакцію проводили протягом ночі. Потім у реакційну суміш додавали піперидин (5 мл). Видалення Fmoc здійснювали протягом 2 год. Для припинення реакції додавали льодяну воду (800 мл) і перемішували протягом 40 хв. Твердий продукт, що утворився, білого кольору фільтрували, промивали водою (400 мл) і сушили про35 тягом ночі з одержанням фрагмента (Aib ) GLP-1 (23-36) (9,65 г, масовий вихід 109%). 96602 54 Приклад 9 8 17-18 Фрагмент (Aib , X ) GLP-1 (7-22) (7,73 г, 2,88 ммоль), що несе захисні групи бічних ланцюгів (див. таблицю А) і захисну групу Fmoc на N-кінці, розчиняли в ДМСО (65 мл). До цього розчину додавали ГОБТ (0,73 г, 4,77 ммоль), ГБТУ (1,46 г, 3,85 ммоль), ДІЕА (0,71 мл, 35 7,40 ммоль) і фрагмент (Aib ) GLP-1(23-36) (8,5 г, 3,45 ммоль) в 20 мл ДМСО. Реакційну суміш перемішували й здійснювали моніторинг за допомогою ВЕРХ. Через 3 год. реакція була завершена. Потім у реакційну суміш додавали піперидин (5 мл). Видалення Fmoc здійснювали протягом 2 год. Для припинення реакції додавали льодяну воду (800 мл) і перемішували протягом 30 хв. Твердий продукт, що утворився, білого кольору фільтрували, промивали МТБЕ (400 мл) і сушили протягом 8 17ночі з одержанням захищеного пептиду (Aib , X 18 35 , Aib ) GLP-1 (7-36). Приклад 10 - Повне видалення захисних груп Пептид, одержаний відповідно до методу, описаного в прикладі 9 (16,24 г), обробляли розчином ТФК/ДТТ/вода (100 мл/5 г/2,0 мл) протягом 2 год. і розчин, що утворився, зливали в МТБЕ (800 мл) у льодяній бані. Після перемішування протягом 30 хв тверду речовину, що утворилася, білого кольору фільтрували, промивали МТБЕ (400 мл) і сушили з одержанням неочищеного пептидного продукту (16,0 г, масовий вихід 138%). Пептид, що утворився, з вилученими захисними групами далі в контексті даного опису позначений як «неочищений» пептид. Приклад 11 - Очищення Очищення неочищеного пептиду здійснювали на колонці Kromasil® С4, 10 мкм, 2,0 ч 25 см з одержанням очищеного пептиду із чистотою не менше 98% (98+%) і виходом -60% у перерахунку на загальний вміст. Очищення передбачало 1-у стадію хроматографічного очищення при рН 2, з наступною 2-ою стадією при рН 9. Після такого очищення очищений пептид можна додатково обробляти або процесувати різними шляхами. Наприклад, одержаний очищений пул після другої стадії можна ліофілізувати або пропускати через 55 концентрувальну колонку, і виділяти осадженням. Після обох шляхів виділення одержують необхідний пептид (ацетат). При здійсненні обох стадій процесу неочищений пептид розчиняли з розрахунку 5 мг/мл (у перерахунку на загальний вміст) у суміші 10% ацетонітрил/вода (0,2М оцтова кислота). Ін'єкували з дублюванням по 1000 мг суміші (у перерахунку на загальний вміст), використовуючи градієнт ацетонітрил/ТГФ/вода/ТГФ, і поєднували фракції, що мають чистоту -95%. Також поєднували «рециркуляційну фракцію», що має чистоту -70%, на частку якої припадає -34% виходу. Рециркуляційну фракцію повторно ін'єкували, використовуючи такий же градієнт ацетонітрил/ТГФ/вода/ТГФ, і поєднували фракцію, що має чистоту -85%, з основним пулом. Після 1-ої стадії хроматографічного очищення вихід у перерахунку на загальний вміст становив 70%. Об'єднаний пул після 1-ої стадії (рН 2) потім очищали додатково на другій стадії на такій же колонці Kromasil® C4, але використовуючи градієнт ацетонітрил/ТГФ/вода/ацетат амонію (рН 8,8). Поєднували фракції, що мають чистоту 98 +%, на частку яких припадало -85% виходу після 2-ої стадії. Загальний вихід обох стадій очищення становив 60% у перерахунку на загальний вміст. Об'єднаний пул після 2-ої стадії (рН 8,8) потім концентрували за допомогою такої ж колонки Kromasil® C4, але використовуючи східчастий градієнт метанол/вода/ацетат амонію. Фракції поєднували, і вони були готові до виділення. А. Приготування розчину неочищеного пептиду: 8 г неочищеного пептиду розчиняли в 500 мл розчину, що містить 90% 0,2н. оцтової кислоти/10% ацетонітрилу. Розчин фільтрували спочатку через фільтр із розміром отворів 0,45 мкм типу Durapore (діаметром 47 мм) (фірма Milipore), а потім через дисковий мікронний патронний фільтр типу Supor EKV (0,65/діаметр 0,2 мм) (фірма Pall Filters). Неочищений ін'єкований розчин аналізували за допомогою ВЕРХ, і було виявлено, що він містить 5,02 мг (мас.%) пептиду, що входить до його складу, на мл (500 мл ч 5,02 мг/мл = 2512 мг пептиду, щовходить до складу). Б. Хроматографія - 1-а стадія при рН ~2: Ін'єкували в чотирьох повторюваннях неочищений розчин за наступних умов: Колонка: Виготовлена фірмою Kromasil, упакована С4, 10 мкм і має розміри 2,0 ч 25 см Детектор: Ультрафіолетовий детектор, встановлений на 280 нм (8 нм ширина смуги, 350/20 нм станд.) Температура колонки: температура навколишнього середовища Швидкість потоку: 13,0 мл/хв (-50 бар протитиск) Рухома фаза: А = суміш, що містить 0,1% трифтороцтової кислоти/15% ацетонітрилу/85% води Б = суміш, що містить 0,1% трифтороцтової кислоти/15% тетрагідрофурану/70% ацетонітрилу/15% води 96602 56 Градієнт: Градієнт починали з 100% рухомої фази й підтримували протягом 0,1 хв. Потім зразок завантажували вручну в колонку за допомогою помпи С (див. нижче опис помпи С). Після внесення зразка здійснювали погон з використанням лінійного градієнта від 100% А до 83% А протягом 1 хв. Потім підтримували рухому фазу, що містить 83% А, протягом наступних 11 хв. Потім здійснювали погон з використанням лінійного градієнта, що містить 73% А, протягом 10 хв. Потім підтримували рухому фазу, що містить 73% А, протягом наступних 15 хв. Потім погон завершували й здійснювали 10-хвилинний погон з використанням 100% Б з наступним повторним зрівноважуванням колонки за допомогою 100% А. Зразок вносили за допомогою окремої ізократичної помпи HP 1100 зі швидкістю 9,0 мл/хв (насос - С). Домішки, які елюювалися на початку процесу, відокремлювали від основного піка шляхом початкового 11-хвилинного ізократичного підтримування 83% А з наступним лінійним градієнтом до 73% А протягом наступних 10 хв. Потім елюювали основний пептидний пік протягом 15 хв., підтримуючи 73% А. Фракції збирали протягом 15 хв., підтримуючи 73% А. В. 1-а стадія рециклу при рН ~2: Пул для рециклу одержували із фракцій, для яких було встановлено, що їх чистота нижче тієї, з якої можна додавати фракції в об'єднаний пул. Ці фракції поєднували окремо й розводили рівним об'ємом води. Продукт для рециклу знову ін'єкували в колонку, з якої був одержаний цей конкретний об'єднаний пул. Використовували такі ж умови хроматографування й фракції, що мають прийнятну чистоту, поєднували, розводили рівним об'ємом води й додавали в об'єднаний пул 1-ої стадії. Ін'єкований об'єм продукту для рециклу істотно нижче в порівнянні з ін'єкцією неочищеного продукту через збільшену кількість домішок, які елююються поблизу піка пептиду. У середньому здійснювали 1 ін'єкцію продукту для рециклу на кожні 2 ін'єкції неочищеного продукту. Г. 2-а стадія препаративної хроматографії при рН 8,8: Кінцевій об'єднаний пул після 1-ої стадії додатково очищали за допомогою повторної хроматографії при рН 8,8. Застосування значення рН 8,8 для 2-ої стадії істотно змінювало рівень елюції домішок, що дозволяло одержувати більш очищений продукт із більш високим виходом. Для цієї 2ої хроматографічної стадії використовували наступні умови: Колонка: Виготовлений фірмою Kromasil, упакована С4, 10 мкм і має розміри 2,0 x 25 см Детектор: Ультрафіолетовий детектор, установлений на 280 нм (8 нм ширина смуги, 350/20 нм станд.) Температура колонки: температура навколишнього середовища Швидкість потоку: 13,0 мл/хв (~50 бар протитиск) Рухома фаза: А = суміш, що містить 15% ацетонітрилу / 85% води, що включає 2 г на 1 л ацетату амонію й 1 мл на 1 л концентрованого гідроксиду амонію 57 Б = суміш, що містить 15% тетрагідрофурану / 60% ацетонітрилу / 25% води, що включає 2 г/л ацетату амонію й 1 мл/л концентрованого гідроксиду амонію Градієнт: Градієнт починали з 100% рухомої фази й підтримували протягом 0,1 хв. Потім зразок завантажували вручну в колонку за допомогою помпи С Після внесення зразка здійснювали погон з використанням лінійного градієнта від 100% А до 673% А протягом 2 хв. Потім підтримували рухому фазу, що містить 67% А, протягом наступних 33 хв. Потім погон завершували й здійснювали 5хвилинний погон з використанням 100% Б с наступним повторним зрівноважуванням колонки за допомогою 100% А. Зразок вносили за допомогою окремої ізократичної помпи HP 1100 зі швидкістю 9,0 мл/хв (насос - С). Після 30-хвилинної стадії завантаження здійснювали короткочасний погон з використанням градієнта від 100% А до 67% А у момент часу від 30,2 до 32 хв. Основний пептидний пік елюювався після підтримки протягом 33 хв 67% А. Фракції збирали, починаючи через 20 хв після завантаження колонки, і закінчували збирання після завершення елюції основного піка. Фракції збирали протягом приблизно 15 хв. Прийнятні фракції поєднували й розводили рівним об'ємом води. На цій стадії очищення можливе використання рециклу, але відпрацьовані фракції, як правило, не містили достатню кількість пептиду для того, щоб здійснювати рециркуляційну ін'єкцію. Це може ставати доцільним, якщо здійснюють велику кількість ін'єкцій і відпрацьовані фракції поєднують для рециклу. Д. Концентрувальний погон: Кінцевій об'єднаний одержаний після 2-ої стадії пул вносили на ту ж колонку і швидко елюювали, використовуючи іншу рухому фазу для концентрування пептиду перед виділенням. На цій стадії використовували наступні умови: Колонка: Виготовлений фірмою Kromasil, упакована С4, 10 мкм і має розміри 2,0 х 25 см Детектор: Ультрафіолетовий детектор, установлений на 280 нм (8 нм ширина смуги, 350/20 нм станд.) Температура колонки: температура навколишнього середовища Швидкість потоку: 13,0 мл/хв (-50 бар протитиск) Рухома фаза: А = суміш, що містить 10% метанолу / 90% 20мМ ацетату амонію Б = суміш, що містить 90% метанолу /10% 20мМ ацетату амонію Градієнт: Градієнт починали з 100% рухомої фази й підтримували протягом 0,1 хв. Потім зразок завантажували вручну в колонку за допомогою помпи С Після завантаження зразка рухому фазу підтримували у вигляді 100% А протягом 5 хв. Потім рухому фазу негайно східчасто заміняли на 100% Б и підтримували протягом наступних 20 хв. Потім погон завершували й здійснювали повторне зрівноважування колонки протягом 15 хв із використанням 100% А. Зразок вносили за допомогою окремої ізократичної помпи HP 1100 зі швидкістю 9,0 мл/хв (на 96602 58 сос - С). Після 45-хвилинної стадії завантаження протягом нетривалого періоду часу (5 хв) підтримували 100% А, потім здійснювали погон з використанням східчастого градієнта до 100% Б протягом 20 хв для елюції пептиду. Основний пік починав елююватися через 2 хв. після східчастого градієнта до 100% Б. Наступні 12 хв. збирали фракції, що містять пептид, і поєднували для виділення. Приклад 12 - Ліофілізація Визначали масу 1-літрового широкогорлого флакона типу FLPE. У цей флакон додавали очи8 17-18 35 щений пул (210 мл) ацетату (Aib , X , Aib )GLP1(7-36) у водному ацетонітрилі /ацетаті амонію. Контейнер змивали деіонізованою водою 3 х 5 мл і змиви додавали до пула. Цей розчин інтенсивно перемішували до гомогенізації й потім закривали й посудину заморожували в рідкому азоті. Заморожений флакон відкривали й горло закривали подвійною безволоконною серветкою типу Kim Wipe, для утримання якої на місці використовували гумову смужку. Флакон поміщали у вакутайнері в ліофілізатор і ліофілізували. Температура конденсатора становила -89°С, тиск 19мкм. Через 24 год. вакутайнер відкривали для оцінки розвитку процесу; у ньому ще був присутній помітний товстий шматок льоду. Ліофілізацію починали знову. Ще через 18 год. флакон видаляли з ліофілізатора й зважували. Маса виявилося нестабільною, вона швидко зростала через гігроскопічну природу продукту. Липкий продукт швидко переносили у зважений сцинтиляційний флакон і зважували, установлено, що одержали 0,822 г пептиду. Приклад 13: Осадження очищеного пептиду У колбі розчин, що містить 100,5 мг чистого 8 17-18 35 ацетату (Aib , X , Aib )GLP-1(7-36) в 2 мл 20мМ ацетату амонію в суміші МеОН/вода (9:1, об./об.), розводили 1 мл 20мМ ацетату амонію в суміші МеОН/вода (9:1, об./об.). При струшуванні повільно вводили в колбу 20 мл ізопропанолу (ПТС) при температурі від 20 до 25°С. Суміш у посудині ставала мутною після додавання 15 мл ІПС. Перемішування продовжували протягом ночі при температурі від 20 до 25°С. Осілий продукт фільтрували й промивали 5 мл ІПС, а потім сушили при 25°С у вакуумі до постійної маси. Одержали 90,9 мг пептиду (вихід 90,42%). Приклад 14: Виділення очищеного пептиду У колбі розчин, що містить 497,7 мг чистого 8 17-18 35 ацетату (Aib , X , Aib )GLP-1(7-36) в 10 мл 20мМ ацетату амонію в суміші МеОН/вода (9:1), розводили 6 мл 20мМ ацетату амонію в суміші МеОН/вода (9:1). При струшуванні повільно вводили в колбу 40 мл ізопропанолу (ІПС) при температурі від 20 до 25°С протягом 35 хв. Суміш у посудині ставала мутною після додавання 2 мл ІПС. Перемішування продовжували протягом 1 год. при температурі від 20 до 25°С. Осілий продукт фільтрували й промивали 5 мл ІПС, а потім сушили при 25°С у вакуумі до постійної маси. Одержали 458,4 мг пептиду (вихід 92%). Приклад 15: Виділення очищеного пептиду У колбу повільно додавали при перемішуванні 900 мл ІПС у розчин, що містить -1000 мг очище8 17-18 35 ного ацетату (Aib , X , Aib )GLP-1(7-36) в 150 59 мл 20мМ ацетату амонію в суміші МеОН/вода (9:1), через концентрувальну колонку при температурі від 20 до 25°С. Це додавання завершували через 45 хв. Суміш у посудині ставала мутною після додавання 260 мл ІПС. Перемішування про 96602 60 довжували протягом 40 хв. при температурі від 20 до 25°С. Осілий продукт фільтрували й промивали 5 мл ІПС, а потім сушили при температурі від 20 до 25°С у вакуумі до постійної маси. Одержали 746 мг пептиду (вихід 74,6%).
ДивитисяДодаткова інформація
Назва патенту англійськоюInsulinotropic peptide synthesis
Автори англійськоюChen Lin, Han Yeun-Kwei, Roberts Christopher R.
Назва патенту російськоюСпособ получения инсулинотропных пептидов
Автори російськоюЧень Линь, Хань Ень-Гуй, Робертс Кристофер Р.
МПК / Мітки
МПК: A61K 38/26, C07K 14/605
Мітки: пептидів, інсулінотропних, спосіб, одержання
Код посилання
<a href="https://ua.patents.su/35-96602-sposib-oderzhannya-insulinotropnikh-peptidiv.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання інсулінотропних пептидів</a>
Спосіб одержання полімерзв’язаних циклічних пептидів

Номер патенту: 72460
Опубліковано: 15.03.2005
Автори: Менсел Джеймс Дж., Следескі Адам В.
МПК: C07K 7/06, C07K 14/635, C07K 1/04, C07K 5/062, C07K 7/04, C07K 7/54, C07K 5/113, C07K 17/00, C07K 7/08, C07K 5/09
Мітки: циклічних, пептидів, одержання, спосіб, полімерзв'язаних
Формула / Реферат:
1. Спосіб одержання, ІІв якій J, L і М представляють лінійні пептидні фрагменти;К1 відсутній або представляє циклічний пептидний фрагментіК2 представляє циклічний пептидний фрагмент,який включає:(1) одержання послідовним приєднанням придатним чином заміщених амінокислотних залишків до полімеру з одержанням:, ІІІде представляє придатний синтетичний полімер для пептиду, і М...
Фармацевтична гелева композиція пептидів, спосіб її одержання, набір для її одержання та її застосування

Номер патенту: 83996
Опубліковано: 10.09.2008
Автори: Ромеіз Петер, Роесслер Бертольд, Бауер Хорст, Райссманн Томас
МПК: A61P 15/00, A61K 47/02, A61K 9/10, A61K 38/04, A61P 5/24
Мітки: одержання, гелева, фармацевтична, застосування, набір, композиція, пептидів, спосіб
Формула / Реферат:
1. Фармацевтична гелева композиція, що включає суміш з:щонайменше однієї фармацевтично активної іонної пептидної сполуки, яка має довжину від 8 до 12 амінокислот, у ліофілізованій формі з концентрацією пептиду від 5 до 50 мг на мл композиції, та водного розчину солі неорганічної або оцтової кислоти з концентрацією від 0,01 до 0,9 % (мас./об. %), причому композиція придатна для введення або відразу після змішування вищевказаних...
Спосіб ферметативного одержання пептидів

Номер патенту: 1271
Опубліковано: 30.12.1993
Автори: Фред Відлер, Джек Таанінг Йохансен
МПК: C12P 21/02, C07K 5/06, C07K 5/083, C07K 5/078, C12P 21/04, C07K 5/072, C07K 1/02, C07K 5/103, C07K 5/087
Мітки: пептидів, одержання, ферметативного, спосіб
Формула / Реферат:
Формула изобретения1. Способ ферментативного получения пептидов общей формулы А-В, где A:N - концевой защищенный L-аминокислотный остаток или пептидный остаток состава 2-6 L-аминокислот, содержащий L-аминокислоту на его С-терминальном конце и необязательно защитную группу на N-терминальном конце, В:L - аминокислотный остаток, который может быть С-терминально защищен, путем взаимодействия аминокислотной и субстратной компонент в...
Спосіб одержання пептидів у вигляді кислотно-адітивних солей

Номер патенту: 3468
Опубліковано: 27.12.1994
Автори: Олга Ньєкі, Іштван Шон, Дьордь Хайош, Ласло Спорні, Юліа Ембер, Лайош Кішфалуді, Ласло Денеш, Бела Сенде
МПК: C07K 5/10, C07K 1/113, C07K 7/06, A61K 38/00, C07C 231/00, C07K 5/117, C07C 67/00, C07K 14/575, C07K 5/11, C07K 5/09, A61P 37/00, C07K 5/08, C07K 14/66, C07K 5/103, A61K 38/22
Мітки: одержання, спосіб, кислотно-адітивних, пептидів, солей, вигляді
Формула / Реферат:
Способ получения пептидов общей формулы Х-Аrg-Lуs-V, где X - водород, Glp; V-Аsр, Аsр-Vаl, Аsр-Vаl-NН2, Аsр-Vаl-ОМе, Аsn-Vаl, Аsр-Аlа, Аsр-Іlе, Аlа-Vаl, Glu-Vаl, Аsu-Vаl, в виде кислотно-аддитивных солей, отличающийся тем, что синтез осуществляют путем ступенчатого наращивания пептидной цепи, начиная с эфира С-концевой аминокислоты, методом активированных эфиров и методом смешанных ангидридов с последующим отщеплением от конечного...
Спосіб одержання суміші амінокислот і низькомолекулярних пептидів з біомаси дріжджів

Номер патенту: 24965
Опубліковано: 25.12.1998
Автор: Шахрай Олександр Олександрович
МПК: C12R 1/865, C07C 227/00, A23J 1/18, C12P 21/06
Мітки: одержання, низькомолекулярних, суміші, біомаси, амінокислот, дріжджів, спосіб, пептидів
Формула / Реферат:
Способ получения смеси аминокислот и низкомолекулярных пептидов из биомассы дрожжей, предусматривающий автолиз дрожжей при нагревании в тесном контакте с плазмолизирующим и стерилизующим агентом с последующим отделением от реакционной массы клеточных остатков, очисткой на ионообменных смолах, концентрированием раствора и удалением аммиака упариванием с получением готового продукта в распылительной сушилке, отличающийся тем, что перед...
Попередній патент: Спосіб валідації системи контролю/керування
Наступний патент: Спосіб одержання плющеної стрічки
Випадковий патент: 1-(1-метил-1н-піразол-4-іл)-n-((1r,5s,7s)-9-метил-3-окса-9-азабіцикло[3.3.1]-нонан-7-іл)-1н-індол-3-карбоксамід як антагоніст 5-ht3 рецептора