Спосіб одержання похідних 4-оксохіноліну або хромону







Формула / Реферат
1. Порошкообразная смесь химических веществ, предназначенная для получения огнеупорной композиции, включающая наполнитель из огнеупорных частиц, частицы, содержащие пероксид металла из группы: Са, Mg, Ba, Sr и металлические частицы, отличающаяся тем, что частицы, содержащие пероксид металла, содержат пероксида кальция не более 75 мас.%, предпочтительно не более 65 мас.%, пероксида магния не более 30 мас.% , пероксида бария не более 92 мас.% и/или пероксида стронция не более 90 мас.% при следующем соотношении компонентов, мас.%:
наполнитель из огнеупорных частиц
20-85
указанные частицы, содержащие пероксид
12-24
металлические частицы
остальное.
2. Смесь по п. 1, отличающаяся тем, что она содержит металлические частицы, образованные главным образом кремнием, и частицы, содержащие пероксид кальция.
3. Смесь по п. 1, отличающаяся тем, что она включает металлические частицы, образованные главным образом алюминием, и частицы, содержащие пероксид магния.
4. Смесь по п. 1, отличающаяся тем, что она включает металлические частицы, образованные главным образом алюминием, и частицы, содержащие пероксид бария.
5. Смесь по п. 1, отличающаяся тем, что она включает металлические частицы, образованные главным образом магнием, и частицы, содержащие пероксид стронция.
6. Смесь по любому из пп. 1- 5, отличающаяся тем, что она содержит частицы по крайней мере одного из следующих металлов: Al, Si, Mg, Fe, Сr, Са, Ba, Sr, Zr, Ті, Be в различной форме или в виде сплавов этих металлов.
7. Смесь по любому из пп. 1- 6, отличающаяся тем, что огнеупорный наполнитель включает по крайней мере один из оксидов, карбидов и/или нитридов следующих металлов: Si, Al, Zr, Ca, Mg, Ті, Сr, в частности, в виде любой из их минералогических структур и/или в смешанных формах, таких, как оксинитриды, оксикарбиды, карбонитриды.
8. Смесь по любому из пп. 1- 7, отличающаяся тем, что она содержит по крайней мере одно соединение металла, которое способно путем разложения и/или окисления, образовывать огнеупорный оксид.
9. Смесь по любому из пп. 1-8, отличающаяся тем, что содержание огнеупорного наполнителя составляет предпочтительно 50-85 мас. %, в расчете на общую массу смеси.
10. Смесь по любому из пп. l- 9, отличающаяся тем, что частицы наполнителя имеют средний диаметр 200-800 микрон, причем максимальный диаметр составляет 1 мм.
11. Смесь по любому из пп. 1-10, отличающаяся тем, что гранулометрический состав наполнителя близок к составу, предусматриваемому законом Андреазена.
12. Смесь по любому из пл. 1-11, отличающаяся тем, что вышеуказанные металлические частицы имеют гранулометрию 10-30 микрон, тогда как гранулометрия частиц, содержащих пероксид, составляет 5-30 микрон.
13. Смесь по любому из пп. 1-12, отличающаяся тем, что соотношение различных компонентов выбирают таким образом, чтобы получить огнеупорную композицию, содержащую наполнитель из огнеупорных частиц, находящийся в связующей фазе, имеющей температуру плавления ниже температуры плавления этого наполнителя и содержащий по крайней мере 20 %, предпочтительно более 50 %, по крайней мере одного смешанного огнеупорного оксида по крайней мере из двух различных металлов.
14. Способ получения огнеупорной композиции путем реакции окисления и/или разложения исходных химических веществ порошкообразной смеси, содержащей наполнитель из огнеупорных частиц, частицы, содержащие пероксид металла из группы Са, Mg, Ba, Sr, и металлические частицы для получения огнеупорных оксидов различных металлов в таких соотношениях, чтобы между собой прореагировало по крайней мере 50 % образовавшихся огнеупорных оксидов с получением смешанного огнеупорного оксида, термодинамически устойчивого в условиях этой реакции взаимодействия, причем природу и количество реагирующих химических соединений выбирают таким образом, чтобы вышеуказанные реакции были экзотермическими и приводили к образованию смешанного оксида в расплавленном состоянии, отличающаяся тем, что используют частицы, содержащие пероксид металла, содержащие не более 75 мас.%, предпочтительно не более 65 мас.% пероксида кальция, не более 30 мас.% пероксида магния, не более 92 мас.% пероксида бария и не более 90 мас.% пероксида стронция при следующем соотношении компонентов порошкообразной смеси, мас.% :
наполнитель из огнеупорных частиц
20-85
указанные частицы, содержащие пероксид
12-24
металлические частицы
остальное
15. Способ по п. 14, отличающийся тем, что при получении огнеупорных оксидов дополнительно вводят одно или несколько металлических соединений в жидком и/или газообразном виде.
16. Способ по п. 14 или 15, отличающийся тем, что регулируют количество металлов, пероксидов металлов и/или металлических соединений, вводимых в реакцию для получения огнеупорных оксидов таким образом, чтобы массовое соотношение образующихся оксидов составляло величину 0,5-2,0 от стехиометрического соотношения оксидов, образующих смешанный оксид.
17. Способ по любому из пп. 14-16, отличающийся тем, что во время образования вышеуказанных огнеупорных оксидов поддерживают температуру на 50-200° выше температуры плавления образующихся смешанных оксидов.
18. Способ по любому из пп. 14-17, отличающийся тем, что во время образования огнеупорных оксидов температуру реакции регулируют путем выбора соответствующего количества и гранулометрии огнеупорного оксида или огнеупорных оксидов, вводимых в качестве наполнителя.
19. Способ по любому из пп. 14-18, отличающийся тем, что реакции для получения огнеупорных оксидов возбуждают с помощью внешнего источника тепла и регулируют температуру этих реакций с учетом начальной температуры системы.
20. Способ по любому из пп. 14-19, отличающийся тем, что регулируют количество и природу металлов, пероксидов и/или металлических соединений, вводимых в реакцию таким образом, чтобы в образующейся композиции смешанный оксид находился в форме диоксида, монтичеллита и/или мервинита.
21. Способ по п. 20, отличающийся тем, что регулируют количество и природу металлов, пероксидов и/или металлических соединений, вводимых в реакцию таким образом, чтобы в образующейся композиции смешанный оксид находился в форме 12СаО×Аl2О3, СаО×Аl2О3 и/или СаО×2Аl2O3.
22. Способ по п. 21, отличающийся тем, что регулируют количество и природу металлов, пероксидов и/или металлических соединений, вводимых в реакцию таким образом, чтобы в получаемой композиции смешанный оксид находился в форме ВаО×SiO2, и/или ВаО×Аl2О3.
23. Способ получения огнеупорного покрытия путем нанесения на поверхность порошкообразной смеси химических веществ, содержащей наполнитель из огнеупорных частиц, частицы, содержащие пероксид металла из группы Са, Mg, Ba, Sr и металлические частицы, и нагрева нанесенной смеси химических веществ и образующейся из неё огнеупорной композиции до температуры выше температуры плавления смешанного оксида, но ниже температуры плавления огнеупорного наполнителя, отличающийся тем, что используют частицы, содержащие пероксид металла, содержащие не более 75 мас.%, предпочтительно не более 65 мас.% пероксида кальция, не более 30 мас.% пероксида магния, не более 92 мас.% пероксида бария и/или не более 90 мас.% пероксида стронция при следующем соотношении компонентов порошкообразной смеси, мас.%:
наполнитель из огнеупорных частиц
20-85
указанные частицы, содержащие пероксид
12-24
металлические частицы
остальное
24. Способ по п. 23, отличающийся тем, что для нанесения указанной смеси используют газ-носитель, содержащий по крайней мере 21 % кислорода, предпочтительно, по крайней мере 50 % кислорода.
25. Способ по п. 23 или п. 24, отличающийся тем, что по крайней мере часть тепла, необходимого для доведения этой смеси до вышеуказанной температуры, подводят к ней до того, как смесь достигнет поверхности, образуя in situ огнеупорные оксиды, которые должны образовать смешанный оксид в вышеуказанной композиции.
26. Способ по любому из пп. 23-25, отличающийся тем, что получают смешанный оксид in situ во время нанесения на вышеуказанную поверхность.
Текст
35545 Настоящее изобретение относится к получению гетероциклических соединений и, в частности, к получению новых соединений, которые могут быть полезны в качестве антагонистов специфических 5гидрокситриптаминовых (5-НТ) рецепторов. В ряде описаний известных патентов раскрываются 5-НТз антагонисты различных структур, например, в ЕР-А-0200444, GB-A-2153821, GB-A-2125398 и ЕР-А-323077. Новые соединения, получаемые по способу настоящего изобретения, представляют соединения общей формулы: , (I) а также их фармацевтически приемлемые кислотно-аддитивные соли или их N-оксиды. В указанной формуле R1 и R1¢, независимо друг от друга, водород, С1-С4-алкил, С1-С4-алкоксигруппа, галоген, трифторметил или R1 и R1¢ вместе образуют метилендиоксигруппу, Х представляет собой -О- или -NR2-, где R2 представляет собой С1-С4алкил, С1-С4алкенил, С3С6циклоалкил, С3-С6циклоалкил С1-С4алкил, бензил, фенил, необязательно замещенный атомом галогена, группу формулы -(CH2)r-Y'-R8 (где r представляет собой целое число в интервале от 1 до 4, Y¢ представляет собой О и R8 представляет собой С1-С4алкил) или группу формулы -Z-, которая связана с положением 8 ароматического кольца с образованием гетероциклического кольца из 5-7 кольцевых элементов, в котором кольцевые элементы, обозначенные индексом Z, представляют собой две или более метиленових групп, необязательного замещенных одной или более С1-С4алкилами, Y представляет собой NR3, где R3 представляет собой атом водорода или С1-С4алкил, В представляет собой насыщенное азабициклическое кольцо, причем насыщенное азабициклическое кольцо отвечает формуле: , в которой m равно 2 или 3, а R4 представляет собой водород или С1-С4алкил, или . Термин "алкил", используемый в тексте, относится к радикалу, содержащему до 6 углеродных атомов. Предпочтительно, такой радикал содержит до 4 углеродных атомов. Так, например, низшая алкильная группа может иметь нормальное или разветвленное строение и может представлять собой: метил, этил, пропил или бутил. Предпочтительным примером низшего алкенила является алкил. Низшая алкокси группа может представлять собой, например, метокси, этокси, пропокси или бутокси. Цикло (низшая) алкильная группа может представлять собой, например, циклопропил, циклобутил, циклопентил или циглогексил. Галоид (низший) алкил, предпочтительно, представляет собой трифторметил. Предпочтительным примером цикло(низший)алкил(низшего) алкила является циклопропилметил, Соединения настоящего изобретения могут содержать один или более асимметричных углеродных атомов, в связи с чем они могут существовать в различных стереоизомерных формах. Так, например, такие соединения могут существовать в виде рацематов или в оптически активных формах. Оптически активные формы могут быть получены расщеплением рацематов или путем использования оптически активной формы исходного вещества, Кроме этого, радикалы формулы Y могут иметь различные конфигурации, соответствующие эндо-конфигурации, как в случае тропина, и экзо-конфигурации, как в случае псевдотропина. Эндо-конфигурация является предпочтительной. Способ настоящего изобретения осуществляется с помощью ацилирования амина формулы (II): HNR3В, где R3 и В имеют указанные выше значения, кислотой формулы (III): (II) 35545 (III) где R1, R1¢ и Х имеют указанные выше значения, или ее ацилирующим производным и выделяют в свободном виде или в виде фармацевтически приемлемой соли, или осуществляют, в случае необходимости, окисление соединений формулы (I), с получением его М-оксида, или превращают соль соединения формулы (І) в свободное основание, или свободное основание превращают в его фармацевтически приемлемую соль. Примерами ацилирующих производных могут служить галоидангидриды (например, хлорангидриды), азиды, ангидриды, имидазолиды (например, полученные из карбонилдиимидазола), активированные сложные эфиры или 0-ацилмочевины, полученные из такого карбодиимида, как диалкилкарбодиимид, особенно дициклогексилкарбодиимид. Предпочтительно, амин ацилируют кислотой в присутствии такого агента сочетания, как дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, изобутилхлорформиат или дифенидфосфинилхлорид. Кислоты формулы (III) являются известными соединениями или могут быть получены известными методами. Так, например, кислота, в которой Х представляет собой -NR2- может быть получена, согласно следующей реакционной схеме 35545 Полученные, согласно способу изобретения, соединения могут быть переведены в N-оксиды с помощью окисления. Если, согласно способу изобретения, соединение получают в виде кислотно-аддитивной соли, то свободное основание может быть получено воздействием на раствор соли основанием. Если продукт реакции представляет собой свободное основание, то кислотно-аддитивную соль» особенно фармацевтически приемлемую соль, можно получить растворением свободного основания в подходящем органическом растворителе и обработкой раствора кислотой, в соответствии с традиционными методиками получения кислотно-ад-дитивных солей из оснований. Примерами солей могут служить соли, полученные из таких неорганических и органических кислот, как серная, хлористоводородная, бромистоводородная, фосфорная, винная, фумаровая, малеиновая, лимонная, уксусная, муравьиная, метансульфокислота, п-толуолсульфокислота, щавелевая и янтарная кислота. Соединения настоящего изобретения обладают фармакологической активностью. Как правило, они являются антагонистами специфических 5-гидрокситриптаминовых рецепторов теплокровных животных. Такие соединения обладают 5-НТ3 антагонистической активностью и, следовательно, являются ценными веществами в тех случаях, когда желателен антагонизм 5-НТ3 рецепторов. 5-НТ3-антагонисты обозначаются также, как "антагонисты" "нейронных" 5-гидрокситриптаминовых рецепторов" и "серотонин (5гидрокситриптамин) М-рецепторные антагонисты". Соединения настоящего изобретения испытывали на 5-НТ3 антагонистическую активность в блуждающем нерве крыс, в соответствии со следующей методикой: Такой метод аналогичен, описанному авторами Ireland and Tyers, в Br. J. Farmac.,1987, 90, 229-238, и зависит от способности 5-НТ деполяризовать блуждающий нерв ин витро. Сегменты блуждающего нерва крыс разновидности Sprague Dawley помещали в перспексовую камеру к заливали растворов Кребса. Электроды, помещенные на каждом конце сегмента нерва, использовали для записи разности потенциалов, которая возникала в ходе добавления различных концентраций 5-НТ к одному из концов сегмента нерва. Этим методом получали зависимости концентрация - реакция на действие 5-НТ до и после уравновешивания сегмента нерва раствором Кребса, содержащим испытуемое вещество. На основе этих результатов осуществляли анализ Шилда с целью получения меры мощности антагониста, выраженной величиной рА2. Результаты испытаний приведены в нижеследующей таблице. Таблица 5-HT3 - антагонистическая активность Соединение Пример 1 2 3 4 5 6 7 8 9 11 12 13 14 15 16 17 18 20(a) 20(b) 20(e) 20(f) 20(h) 20(e) pA 8.4 9.2* 6.9 7.65 7.76 8.9* 8.3 9.7* 9.1* 6.9* 9.6 9,6* 8.6 8.2 7.9 8.4 8.2 8.8 8.8 8.5* 8.9 8.2 9.8 Как отмечалось, продукты способа настоящего изобретения полезны в качестве антагонистов 5-НТ3 рецепторов млекопитающих. 5-НТ3 антагонисты могут использоваться при лечении нейро-психиатрических расстройств, таких как беспокойство, психических расстройств (например, шизофрении), зависимости от лекарств или других веществ при злоупотреблении ими, нарушения сознания, при лечении таких желудочно-кишечных нарушений, как рвота и тошнота, и при лечении мигрени. В случае некоторых из указанных выше состояний, совершенно ясно, что соединения изобретения могут использоваться профилактически, а также для облегчения острых симптомов. Следует иметь в виду, что используемый в тексте термин "лечение" или аналогичный термин включают как профилактику, так и лечение острых состояний. Следующие ниже примеры иллюстрируют настоящее изобретение. 35545 Пример 1. (Эндо)-N'-(8-метил-8 -азабицикло[3.2.1]октан-3-ил) -1,4-дигидро-1-метил-4-оксохинолин-3-карбо-ксамид. Суспензию 1,4-дигидро-1-метил-4-оксохино-лин-3-карбоновой кислоты (1,02 г, 5 ммоля) и карбонилдиимидазола (0,85 г, 5 ммоля) в диметилформамиде (15 мл) перемешивали и нагревали в течение 0,5 часа при 80°С с получением прозрачного раствора. В систему добавляли (эндо)-3-аминотропан дигидрохлорид (1,06 г, 5 ммоля), после чего добавляли триэтиламин (13 г) в реакционную смесь, перемешивали при 80°С еще в течение 2 часов. Затем смесь охлаждали, разбавляли водой (25 мл), и осажденный продукт (1,2 г) собирали и перекристаллизовывали из воды (150 мл) с получением 0,7 г целевого основания. Данное основание растворяли в этаноле (7 мл) и подкисляли эфирным раствором хлористоводородной кислоты с осаждением целевого соединения в виде гидрохлорида (0,55 г), т.пл. > 300°С. Пример 2. (Эндо)-N-(8- метил-8-азабицикло[3.2.1]октан-3-ил)- 1, 4 -дигидро-1-бутил-4-оксохинолин-3-карбо-ксамид. Смесь 1-бутил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (0,93 г, 4 ммоля), карбонилдимидазола (0,7 г, 4.4. ммоля) и диметилформамида (12 мл) перемешивали в течение 1,5 часа при 80°С. Затем добавляли (эндо)-3-аминотропан (0,56 г, 4 ммоля) и перемешивание продолжали еще в течение 1,5 часов при той же температуре. Растворитель удаляли, и остаток разбавляли водой со льдом (15 г). Осажденное твердое вещество собирали, промывали охлажденной льдом водой и сушили на воздухе. Основание растворяли в горячей смеси воды (5 мл) и этанола (3 мл), затем охлаждали льдом и подщелачивали до рН II путем добавления концентрированного водного раствора аммиака с целью освоения кристаллического продукта, который собирали и промывали холодным разбавленным раствором аммиака. Затем очищенное основание (0,82 г) растворяли в этаноле (8 мл), подкисляли этанольным раствором НСl и разбавляли эфиром (3 л). В результате охлаждения льдом получали продукт в виде гидрохлорида (0,49 г) т.пл. 267-268°С. Пример 3. (Эндо)- N -(8-метил-8-азабицикло[3.2.1]-октан-3-ил)- 1 -бензил- 1,4-дигидро-4-оксохинолин-3-кар-боксид. 1-Бензил - 1,4-дигидро-4-оксохинолин-3-карбо-новую кислоту (1,96 г, 7.03 ммоля) в сухом ДМФ (20 мл) обрабатывали карбонилдиимидазолом (1,14 г, 7.04 ммоля) при комнатной температуре, и смесь в течение 3 часов нагревали при 80°С. Добавляли (эндо)-3-аминотропан (0,39 г, 7.0 ммоля), и нагревание продолжали в течение ночи (19 часов) с получением суспензии. Полученную смесь разбавляли водой (40 мл) и рН устанавливали равным 9-10 путем добавления концентрированного водного раствора карбоната калия. Твердое вещество собирали, промывали водой, сушили и перекристаллизовывали из этанола (20 мл) и воды (20 мл) с получением свободного основания (1,72 г). Данное вещество растворяли в кипящем этаноле (15 мл), и раствор подкисляли этанольным раствором хлористого водорода. Полученный в результате осадок собирали, промывали этанолом и сушили при 80°С в вакууме с получением целевого соединения в виде гидрохлорида, гидрата, 1,3 этанолята (1,91 г) т.пл. 296-297°С. Пример 4. (Эндо)-N-(8 – метил-8-азабицикло[3.2.1]-октан-3-ил)хромон-3-карбоксаиид. (а) Смесь хромон-3-карбоновой кислоты (1,25 г) и тионилхлорида (6 мл) нагревали с обратным холодильником в течение 5 минут. Затем реакционную смесь разбавляла циклогексаном (15 мл) и охлаждали льдом. Кристаллический осадок собирали фильтрованием, промывали циклогексаном и сушили в вакууме с получением хромон-3-карбонилхлорида (1,2 г). (в) Раствор хромон-3-карбонилхлорида (1,04 г, 5 ммоля) в СН2СI2 (20 мл) прикапывали в течение 5 минут к охлажденной льдом перемешиваемой смеси (эндо)-3-аминотропана (0.7 г, 5 ммоля), без-водного К2СО3 (3 г) и CH2CI2 (20 мл). После завершения добавления перемешивание продолжали еще в течение 0,5 часа, и смесь разбавляли водой (50 мл). Органическую фазу отделяли, сушили (Nа2SO4) и выпаривали с образованием твердого вещества (1,7 г). Данное основание растворяли в этаноле (15 мл) и подкисляли этанольным раствором НСl с осаждением сырого гидрохлорида (0,9 г). В результате трехкратной перекристаллизации из этанола получали целевое соединение в виде чистого гидрохлорида (0,3 г), т.пл. > 300°С. Пример 5. (Эндо)-N-(3-метил-9-азабицикло[3.3.1]нонан-3-ил)- 1, 4 -дигидро-1-метил-4-оксохинолин-3-карбо-ксамид. Суспензию 1,4-дигидро-1-метил-4-оксохино-лин-3-карбоновой кислоты (1,02 г, 5 ммоля) и карбонилдиимидазола (0,85 г, 5 ммолей) в ДМФ (15 мл) перемешивали и нагревали в течение 3 часов при 85°С с получением прозрачного раствора. Добавляли (эндо)- 3 -аминогомотропан дигид рохлорид (1,13 г, 5 ммолей) и диизопропилэтиламин (1,29 г, 10 ммолей), и нагревание продолжали в течение ночи (19 часов). Раствор охлаждали до комнатной температуры и разбавляли водой (25 мл). Величину рН устанавливали равной 10-11 путем добавления небольшого количества водного раствора гидроксида калия. Осажденное твердое вещество собирали, промывали водой и сушили с 35545 получением целевого основания (1,25 г), которое трижды перекристаллизовывали из смесей вода/этанол. Основание растворяла в горячем этаноле (12 мл) и подкисляли этанольным раствором хлористого водорода с получением целевого соединения в виде гидрохлорида, 1,25 гидрата (0,92 г), т.пл. 278-81°С (разл.) Пример 6. (Эндо)-N-(8- метил-8-азабицикло[3.2.1]октан-3-ил)- 1,8-этил-1,4-дигидро-4-оксохинолин-карбокса-мид. Суспензию 1,8-этил-1,4-дигидро-4-оксохино-лин-3-карбоновой кислоты (1,08 г, 5 ммолей) и карбонилдиимидазола (0,39 г, 5,5 ммолей) в диметилформамиде (15 мл) перемешивали и нагревала в течение 1,25 часа при 80°С с получением прозрачного раствора. В систему добавляли (эндо)-3-аминотропан (0,7 г, 5 ммолей) в виде одной порции, и реакционную смесь перемешивали в течение 2 часов при 80°С. Реакционную смесь охлаждали льдом и разбавляли водой (25 мл), и осажденный продукт собирали и дважды перекристаллизовывали из смеси этанол:вода (2:1) с получением 0,7 г целевого основания. Основание растворяли в горячем этаноле (15 мл) и подкисляли этанольным раствором хлористого водорода с получением целевого соединения в виде гидрохлорида (0,55 г), т.пл. > 300°С. Пример 7. (Эндо)-N-(8-метил-8-азабицикло [3.2,1]октан-3-ил)- 1, 4 -дигдро-1,8-пропанохинолин-4-он-3-карбо-ксамид. Целевое соединение получала по методике примера 6, заменяя 1,8-этано-1,4-дигидро-4-оксо-хинолин-3карбоновую кислоту 1,8-пропано-1,4-ди-гидро-4-оксохинолин-3-карбоновой кислотой. Продукт реакции получали в виде гидрохлорида, т. пл. > 300°С. Пример 8. (Эндо)-N-(8-метил-8-азабицикло[3.2.1]октан- 3-ил)-1,4-дигидро - 4 -оксо-1-н-пропилхинолин-3-карбоксамид. 1,4-Дигидро-4-оксо - 1-н-пропилхинолин-3-кар-боновую кислоту (1,58 г, 6.82 ммоля) и триэтиламин (0.7 г, 7 ммоля) растворяли в дихлорметане (20 мл) в атмосфере аргона. Сразу же при перемешивании добавляли хлорид дифенилфосфина (1,6 г, 6.76 ммоля). Полученный раствор оставляли на 6 часов и затем добавляли (эндо)-3-амино-пропан (1,0 г, 7.14 ммоля) и триэтиламин (0.7 г, 7 ммолей). Полученный раствор оставляли (на 3 дня и затем выпаривали. Остаток растворяли в воде и подкисляли) концентрированной хлористоводородной кислотой. Осадок отфильтровывали, промывали водой и отбрасывали. Фильтрат подщелачивали карбонатом натрия и выпаривали. Остаток дважды обрабатывали этилацетатом, этилацетат выпаривали, и остаток обрабатывали водой (6 мл) и концентрированным аммиаком (1 мл) с получением белого твердого вещества (1,34 г). Данное вещество (3,68 ммоля) растворяли в горячем этаноле (15 мл) и добавляли горячий дигидрат щавелевой кислоты (0,47 г, 3.73 ммоля). Полученный в результате раствор хранили в течение ночи в холодильнике. Осадок собирали, промывали этанолом и сушили с получением целевого соединения в виде полугидрата оксалата (1,24 г), т.пл. 227-231°C. Примеры 9-14. Следуя методике примера 1, но, заменяя 1,4-дигидро-1-метил-4-оксохинолин-3-карбоновую кислоту следующими реагентами, получали следующие ниже продукты: Пример 9. Реагент: 1,4-дигидро-1-этил-4-оксохинолин-3-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицикло[3.2. 1]октан-3-ил)-1,4-днгидро-1-этил4-оксохинолин-3карбоксамид, гидрохлорид, полугидрат, т.пл. 298-302°С. Пример 10. Реагент: 1,4-дигидро-1-(2-метоксиэтил)-4-оксо-хинолин-3-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицик-ло[3.2. 1]октан-3-ил)- 1,4 -дигидро-1-(2-метоксиэтил)-4-оксохинолин-3-карбоксамид, 1:1 фумарат, т.пл. 243-245°С. Пример 11. Реагент: 9-фтор-6,7-дигидро-5-метил-1-оксо-IH,5Н-бензо[e, f]хинолизин-2-карбоновая кислота. Продукт: (эндо-N-(8-метил-8-азабицик-ло[3.2. 1]октан-5-ил)-9-фтор-6,7-дигидро- 5 -метил-1-оксо-ІН,5Нбензо[e,f]хинолизин-2-карбоксамид, гидрохлорид, 1/4 гидрат, т.пл. 320°C Пример 12. Реагент: 1-цикогексил-1,4-дигидро-4-оксохино-лин-3-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицик-ло[3.2. 1]октан-3-ил)- 1-циклогексил-1,4-дигидро-4-оксохи-нолин-3карбоксамид, 1:1 оксалат, 1,5 гидрат, т.пл. 217°С. Пример 13. Реагент: 1-(циклопропилметил)-1,4-дигидро-4-оксохинолин-3-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицикло[3.2. 1]октан-3-ил)-1-циклопропилметил-1,4-дигидро- 4 оксохинолин-3-карбоксамид, гидрохлорид, 0.75 ги-драт, т.пл. 164-166°С (разл.). Пример 14. Реагент: 1-(4-фторфенил)-1,4-дигидро-4-окса-хинолин-3-карбоновая кислота. Продукт: (эндо)-N-(8-метил-8-азабицик-ло[3.2. 1]октан-3-ил)-1-(4-фторфенил)-1,4-дигидро-4-оксо-хинолин3-карбоксамид, гидрохлорид, т.пл. 240°С (разл.). Пример 15. Следуя методике примера 1, но, заменяя (эндо)-3-аминотpoпaн 1-азабицикло[2.2.2]октан-3-амином (3аминохинукледином), получали N-(1-азабицикло[2.2.2]октан-3-ил)-1,4-дигидро-1-метил-4-оксохинолин-3карбоксамид, гидрохлорид, гидрат, т.пл. 179-181°С. Пример 16. (Эндо)-N-(3-метил-9-азабицикло[3.3.1]нонан-3-ил)-1, 4-дигидро-1-этил-4-оксохинолин-3-карбокса-мид. Суспензию 1,4-дигидро-1-этил-4-оксохинолин-3-карбоновой кислоты (1.52 г, 7 ммолей) и триэтиламина (0,7 г, 7 ммолей) в дихлорметане (20 л) перемешивали при комнатной температуре в атмосфере аргона в 35545 течение 1 часа. Добавляли изо-бутилхлорформиат (0.36 г, 7.03 ммоля), и смесь перемешивали в течение часа. Добавляли триэтиламин (1.4 г. 14 ммолей) и (эндо)-3-амино-9-ме- тил – 9 - азабицикло[3.3.1]нонан дигидрохлорид (1,58 г 6,96 ммоля). Через 3 дня реакционную смесь охлаждали метанолом и растворители выпаривали. Остаток обрабатывали водой (10 мл) и концентрированным аммиаком (2 мл), твердое вещество собирали, промывали концентрированным раствором аммиака и сушили. Данное вещество превращали в его соль фумаровой кислоты в соотношении 1:1 в системе ІРА:метанол (2: :1,15 мл) с получением целевого соединения в виде 1:1 фумарата, полугидрата (73.1%), т.пл. 184-185°С. Пример 17. (Эндо)-1-(9-метил- 9-азабицикло[3.3.1]нонан-3-ил)-1-бутил- 1, 4 -дигидро-4-оксохинолин-3-карбо-ксамид. 1,4-Дигидро-1-бутил-4-оксохинолин-3-карбоно-вая кислота подвергалась реакции с (эндо)-3-амино-9метил-9-азабицикло[3.3.1]нонаном, согла-сно методике примера 16, и целевое соединение получали в виде малеата 1:1, т.пл. 203-205°С. Пример 18. (Эндо)-N-(8 -метил-8-азабицикло[3.2.1]октан-3-ил-1-этил6-фтор-1,4-дигидро-4-оксохинолин-3-карбоксамид. Проводили реакцию между 1,4-дигидро-1-этил-6-фтор-4-оксохинолин-3-карбоновой кислотой и (эндо)-3аминотропаном, согласно методике примера 16, и целевое соединение получали в виде гидрохлорида, 0,75 гидрата, т.пл. 315-317°С. Пример 19. Следуя методикам приведенным выше, с использованием соответствующих реагентов получала: (эндо)N-(8-метил-8-азабицикло[3.2.1]октан-3-ил)-1,4-дигидро-1-циклопропил-4-оксохинолин-3-карбоксамид и соответствующие 1-циклобутил, 1-циклопентил, 1-трет.бутил и 1-(бут-3-енил) аналоги, (эндо)-N-(8-метил-8азабицикло[3.2.1]октан-3-ил)- 1,4-дигидро-1-этил-6,7-метилендиокси-4-оксо-хинолин-3-карбоксамид, (эндо)-N(8-метил-8-аза-бицикло[3.2.1]октан-3-ил) - 1,4 - дигидро-1-этил-7-фтор-4-оксохинолин-3-карбоксамид и аналоги, в которых 7-фтор заместитель заменен 7-три-фторметилом, 8-фтором, 6.7-дифтором и 6-хлор-8метилом. Пример 20. Используя способ Примера 16, получают следующие соединения: (а) (эндо) - N - (8-метил-8-азабицикло[3.2.1]ок-тан-3-ил)-1,4-дигидро – 1 -этил-8-фтор-4-оксохино-лин-3карбоксамид, гидрохлорид, четвертьгидрат, т.п. 296-300°С (разл.). (в) (эндо) – N - (8-метил-8-азабицикло[3.2.1]ок-тан-3-ил)-1-(3 - бутенил)- 1, 4-дигидро-4-оксохино-лин-3карбоксамид, фумарат, 0,75 гидрат, т.пл. 213-216°С (с) (R)-(-)-N-(1-азабицикло[2.2.2]октан-3-ил)-1-циклогексил- 1, 4-дигидро-4-оксохинолин-3-карбо-ксамид, 1:1 оксалат, 0,75 гидрат, т.п. 173-177°С (d) (S)-(+)-N-(1-азабицикло[2.2.2]октан-3-ил)-1-циклогексил-1, 4 -дигидро-4-оксохинолин-3-карбо-ксамид, 1:1 оксалат, 1,25 гидрат, т.п. 179-183°С. (e) (эндо) – N -(8-метил-8-азабицикло[3.2.1]ок-тан-3-ил)-циклопропил-І,4-дигидро-4-оксохинолин-3карбоксамид, 1:1 фумарат, 0,75 гидрат, т.п. 201-3°С (f) (эндо)-N-(8-метил-8-азабицикло[3.2.і]октан-3-ил)1 -циклобутил-1,4-дигидро-4-оксохинолин-3карбоксаид, фумарат 1.25 гидрат, т.п. 232°С (разл.) (g) (эндо)-N-(8-метил-8-азабицпкло[3.2.1]ок-тан-3-ил)- 1,4-дигидро-1-этил-4-оксо-7-трифторме-тилхинолин3-карбоксамид, фумарат, 0,25 гидрат, т.п. 225-227°С (h) (эндо)- N -(8-метил-8-азабицикло[3.2.1]ок-тан-3-ил)-1-(1,1-диметилэтил) -1,4-дигидро-4-оксо-хинолин-3карбоксамид, 1:1 малеат, т.п. 226-229°С (разл.) (i) (эндо)-N-(8-метил-8-азабицикло[3.2.1]октан-3-ил)1,4 -дигидро-1-этил-6,7-метилендиокси-4-оксохинолин-3-карбоксамид, 1:1 малеат, т.п. 240°С (разл.) (j) (эндо)-N-(8-метил-8-азабицкло[3.2.1]октан-3-ил)-1-циклопентил-1,4-дигидро-4-оксохинолин-3карбоксамид, 1:1 малеат, т.п. 181-184°С. Пример 21. Эндо- N -(8 - этил-8-азабицикло[3.2.1]октан-3-ил)-1-циклогексил- 1, 4 -дигидро-4-оксохинолин-3карбоксамид. N-метилморфолин (0.4 мл, 3,64 ммоль) доба-вляли к раствору 1 - цилогексил -1, 4 - дигидро4-оксохинолин 3 карбоновой кислоты 2.95 ммоль) в безводном ТГФ (25 мл) в атмосфере аргона. Раствор охлаждался до -15°С в течение (0,80 г, 0,25 ч, прежде чем был добавлен хлороформат изобутила (0.4 мл, 3,08 ммоль). К полученному раствору добавляли раствор дигидрохлорида эндо-N-(8-этил-8-азабицикло[3.2. 1]октан-3ил)амина (0,61 г, 2,69 ммоль), N-метил-морфолин (0,4 мл, 3,64 ммоль), безводный ДМФ (5 мл) и безводный ТГФ (10 мл) при -10°С. После перемешивания в течение 20 ч добавляли раствор триэтиламина (1.0 мл; 7,2 моль) в хлороформе (10 мл), и смесь перевешивалась при комнатной температуре в течение 25 часов. К реакционной смеси добавлялся хлороформ и разбавленный раствор NaOH. Органический слой удаляли, и водный слой промывали хлороформом. Объединенные органические экстракты промывали рассолом, сушили (безводным Na2SO4) и выпаривали в вакууме до получения прозрачной жидкости, из которой выделили сое-динение, указанное в заголовке, в виде основания (0.74 г). Основание растворяли в этанольном растворе хлористого водорода и в осадок выпадало целевое соединение в виде гидрохлорида, 1 1/4 гидрата, т.п. > 250°C (разл. свыше 202°C). Пример 22. 35545 (Эндо) - N - (8-азабицикло[3.2.1]октан-3-ил)-1-циклогексил - 1,4 -дигидро-4-оксохинолин-3-карбо-ксамид. (Эндо)- N-(8-метил-8-азабицикло[3.2.1]октан-3-ил)-1-циклогексил - 1,4 - дигидро-4-оксохинолин-3карбоксамид (1,0 г, 2.54 ммоль) высушивали и суспендировали в 1,2-дихлорэтане (30 мл), в атмосфере аргона, и охлаждали льдом. Добавляли 1-хлорэтил хлорформат (0.28 мл, 0.37 г, 2.59 ммоль) и оставляли в течение 1 часа подогреваться до комнатной температуры. Затем раствор кипятили с обратным холодильником в течение 1 часа, и растворитель выпаривали. Остаток растворяли в метаноле и кипятили с обратным холодильником в течение суток (24 часа). Реакционную смесь выпаривали, и остаток хроматографировали на основной окиси алюминия (активность 11-111), элюировали смесью хлороформ: :метанол (10:0.1®0.4). Весь выделенный продукт (1,54 г, 4,06 ммоль) растворяли в горячем этаноле (10 мл) и добавляли фумаровую кислоту (0.45 г, 3,88 ммоль). Раствор фильтровали, охлаждали и перекристаллизовывали из этанола (20 мл) и небольшого количества воды, получая (эндо)-N-(8азабицикло[3.2.1]октан-3-ил)-1-циклогексил-1,4-ди-гидро-4-оксохинолин-3-карбоксамида, 1:1 фумарат, полугидрат в виде белого твердого вещества (0,77 г), т.п. 237-239°С. Пример 23. (Эндо, анти)-и (эндо, син)-N-(8-метил-8-азаби-цикло[3.2.1]октан-3-ил)-1-циклогексил-1.4-дигидро-4оксохинолин-3-карбоксамид-N-оксид. (Эндо)-N-(8-метил-8-азабицикло[3.2.1]октан-3-ил)-1-циклогексил-1,4-дигидро – 4 - оксохинолин-3карбоксамид (3.93 г, 10 ммолей) в метаноле (10 мл) обрабатывали 27.5 вес/вес% водной перекисью водорода (3.2 г). Раствор разбавляли водой (30 мл) и метанол выпаривали. Добавляли суспензию платина/катализатора углерод с водой, и смесь фильтровали. Фильтрат выпаривали досуха, и остаток выпаривали со смесью толуол: этанол 4:1, получая твердый остаток (2.48 г), содержащий смесь анти и син N-оксидов. Твердый остаток растворяли в горячем этилацетате (25 мл) и метаноле (1,5 мл) и кипятили до тех пор, пока не начиналось осаждение, затем охлаждали. Твердое вещество собирали, промывали этилацетатом и высушивали. Получали (эндо, анти)-N-(8-метил-8азабицикло[3.2.1]октан-3-ил)-1-циклогексил- 1,4 -дигидро-4-оксохинолин-3-карбоксамид N-оксид, дигидрат в виде белого твердого вещества (0,91 г), т.п. 174-177°С. Порцию (250 мг) вторично собранного вещества, содержащую смесь син и анти N-оксидов, разделяли с помощью хроматографии на вращающейся пластине на силикагеле (толщ. 2 мм) и элюировали смесью хлороформ:метанол: 0,888 аммиак (10:1:0.1). Син-N-оксид получался в виде стекла, которое подвергали кристаллизации с холодным эфиром, а затем горячим этилацетатом. Твердое вещество растиранием собирали и сушили в вакууме при 75°С в течение ночи. Получали (эндо, син)-N-(8-метил-8азабицикло[3.2.1]октан-3-ил)-1-циклогексил - 1,4 - дигидро-4-оксохинолин-3-карбоксамид N-оксид, 0,9 гидрохлорид, в виде белого твердого вещества (50 мг), т.п. 223-232°С. Пример 24. Превращение оксалата примера 12 в свободное основание. Перемешиваемый раствор оксалата примера 12 (2,5 г) в теплой воде (100 мл) подщелачивали до рН 9 добавлением насыщенного водного раствора карбоната калия. Затем смесь охлаждали льдом, и осажденный продукт собирали с помощью фильтрования и промывали водой, в результате получали 1,7 г продукта с температурой плавления 190-193°С.
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for the preparation of 4-oxoquinoline or chromone derivatives
Автори англійськоюWord Terens James, White Jennet Christin
Назва патенту російськоюСпособ получения производных 4-оксохинолина или хромона
Автори російськоюУорд Теренс Джеймс, Уайт Дженет Кристин
МПК / Мітки
МПК: A61K 31/35, C07D 405/12, C07D 403/12, A61K 31/47
Мітки: похідних, 4-оксохіноліну, хромону, спосіб, одержання
Код посилання
<a href="https://ua.patents.su/7-35545-sposib-oderzhannya-pokhidnikh-4-oksokhinolinu-abo-khromonu.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання похідних 4-оксохіноліну або хромону</a>
Спосіб одержання похідних бензімідазолу у вигляді суміші ізомерів або індивідуальних ізомерів у вільному вигляді або у вигляді їх фізіологічно прийнятих солей

Номер патенту: 8022
Опубліковано: 26.12.1995
Автори: Антоніо Жакетті, Артуро Донетті, Розамарія Мікелетті, Марко Турконі, Аннамарія Уберті, Массімо Нікола, Ернесто Монтанья
МПК: C07D 235/26, C07D 487/18, A61K 31/4164
Мітки: ізомерів, суміші, індивідуальних, вигляді, фізіологічно, вільному, прийнятих, одержання, бензимідазолу, солей, похідних, спосіб
Формула / Реферат:
Способ получения производных бензимидазола общей формулыгде R1 - водород, С1-С6-алкил;Y - кислород или группа NH;А - радикал общей формулыгде R2 - С1-С6-алкил или группагде R3 - водород или аминогруппа;n = 2 или 3,в виде смеси изомеров или индивидуальных изомеров в свободном виде или в виде их физиологически приемлемых солей, отличающийся тем, что соединение общей...
Спосіб одержання похідних хінолінкарбонової кислоти або її фармацевтично придатних солей та проміжні сполуки для їх одержання
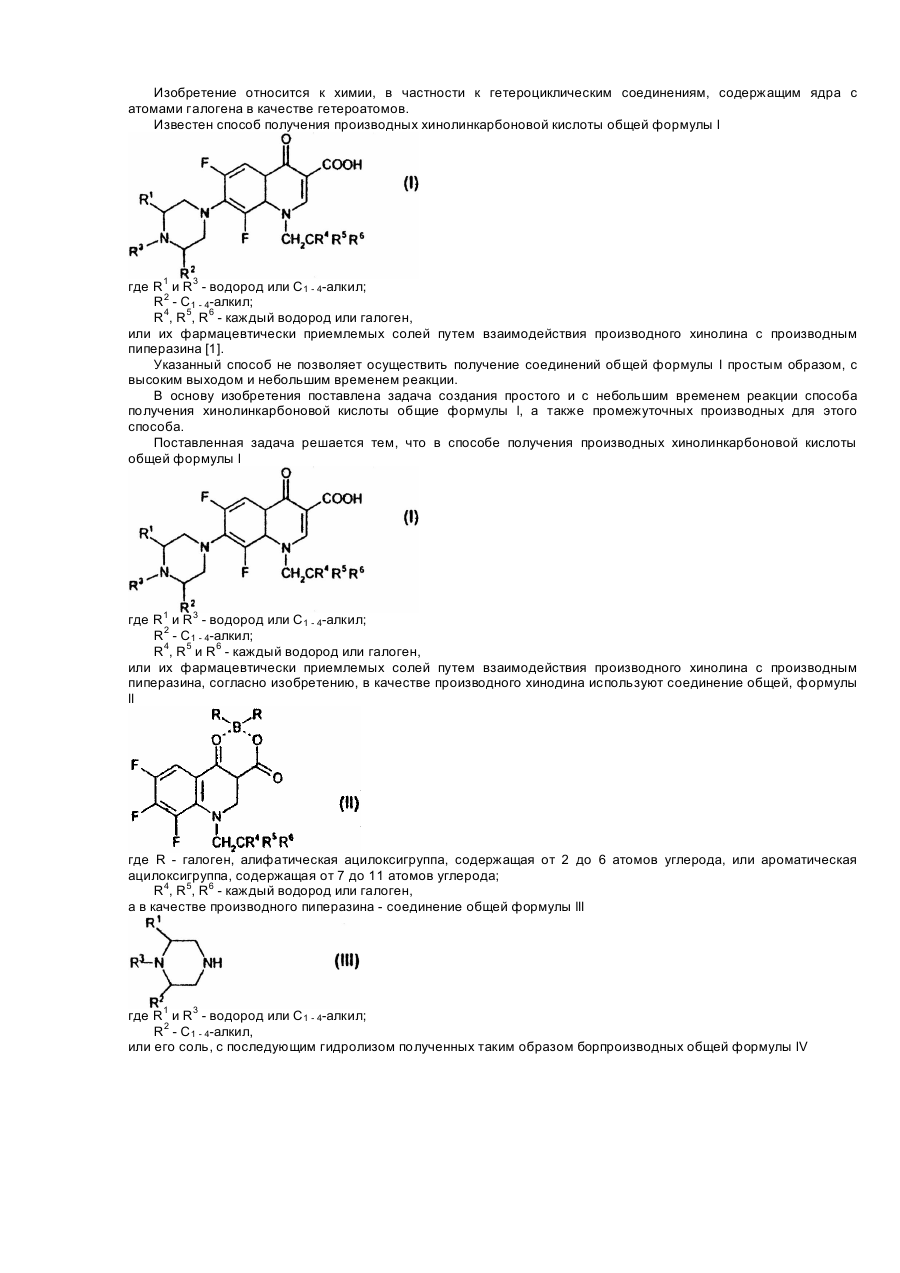
Номер патенту: 26568
Опубліковано: 11.10.1999
Автори: Пайор Аніко, Вашварі Лелле, Керестурі Геза, Рітлі Петер, Хермец Іштван, Хорват Агнеш, Балог Марія, Шіпош Юдіт
МПК: C07D 215/56, A61K 31/495, A61P 31/04, C07D 401/04, C07F 5/00
Мітки: проміжні, похідних, сполуки, хінолінкарбонової, солей, фармацевтично, спосіб, одержання, кислоти, придатних
Формула / Реферат:
1. Способ получения производных хинолинкарбоновой кислоты общей формулы lгде R1 и R3 - водород или C1 - 4-алкил;R2 - C1 - 4-алкил;R4, R5 и R6 - каждый - водород или галоген,или ее фармацевтически приемлемых солей, взаимодействием производного хинолина с производным пиперазина, отличающийся тем, что в качестве производного хинолина используют соединение общей формулы llгде R - галоген,...
Спосіб одержання похідних хіноліна або їх солей

Номер патенту: 4875
Опубліковано: 28.12.1994
Автори: Цутому Ірікура, Хіросі Кога, Сатосі Мураяма
МПК: C07D 401/04, C07D 215/233, C07D 215/18, C07D 215/14
Мітки: похідних, спосіб, хіноліна, одержання, солей
Формула / Реферат:
Способ получения производных хинолинагде R- 4-метил-1-пиперазинильная группа, или их солей, отличающийся тем, что соединение формулывводят в реакцию с N -метилпиперазином в инертном растворителе, например пиридине, при кипячении реакционной массы с выделением целевого продукта в свободном виде или в виде соли.
Спосіб одержання похідних 6-фтор-1, дігідро-4-оксо-7-заміщений піперазінілхінолін-3-карбонової кислоти або їх фармацевтично прийнятних солей
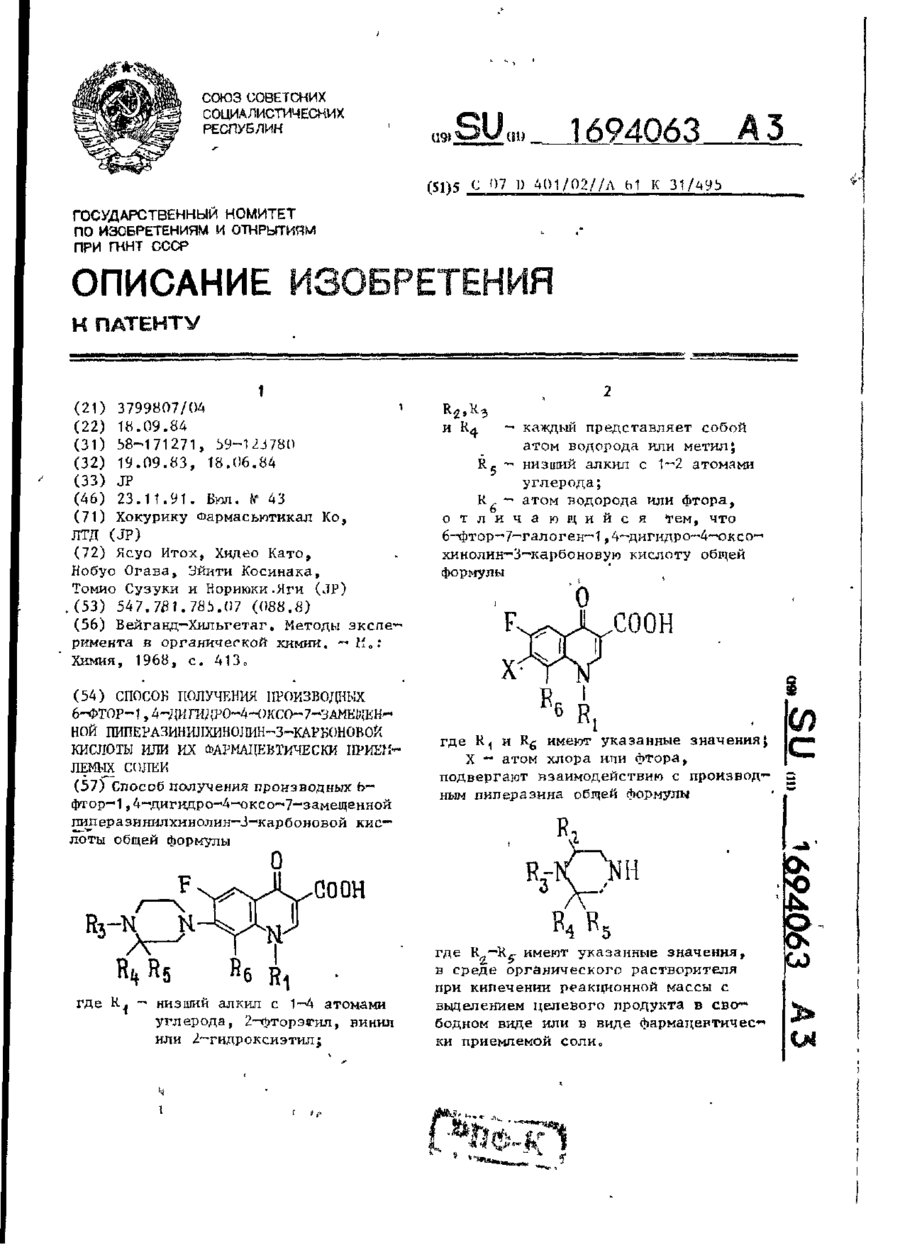
Номер патенту: 6324
Опубліковано: 29.12.1994
Автори: Ейіті Косінака, Хідео Като, Норіюкі Ягі, Томіо Сузукі, Нобуо Огава, Ясуо Ітох
МПК: C07D 401/04, C07D 241/00, A61P 31/04, C07D 487/00, C07D 215/00, A61K 31/47, A61K 31/495, C07D 241/04, C07D 215/56
Мітки: похідних, піперазінілхінолін-3-карбонової, 6-фтор-1, фармацевтично, солей, прийнятних, одержання, спосіб, кислоти, дігідро-4-оксо-7-заміщений
Формула / Реферат:
Способ получения производных 6-фтор-1, 4-дигидро-4-оксо-7- замещенной пиперазинилхинолин-3-карбоновой кислоты общей формулы 1где R1 - низший алкил с 1-4 атомами углерода, 2-фторэтил, винил или 2-гидроксиэтил; R2, R3 и R4 - каждый представляет собой атом водорода или метил;R5 - низший алкил с 1-2 атомами углерода; R6 -атом водорода или фтора, отличающийся тем, что...
Спосіб одержання похідних азепіна або їх солей, або енантіомерів

Номер патенту: 8041
Опубліковано: 26.12.1995
Автори: Армін Фюгнер, Карл-Гейнц Вебер, Клаус Шнейдер, Герхард Вальтер
МПК: C07D 487/04, A61P 7/02, A61K 31/55, C07D 498/04, A61P 43/00, A61P 37/08, C07D 223/00, C07D 267/00, C07D 513/04, C07D 281/00
Мітки: солей, одержання, спосіб, енантіомерів, похідних, азепіна
Формула / Реферат:
Способ получения производных азепина общей формулыгде R1 = Н; 7-Сl, R2 = Н при Х = СН2, О, S;R1 = Н, 6-Сl, 6-СН3, R2 = Н, 12-Сl при Х = О;R1 = H, R2 =12-Cl при X = O, S;R1 = 6 - ОСН3, R2 = Н при Х = S,или их солей, отличающийся тем, что соединение общей формулыгде R1, R2, Х имеют указанные значения, подвергают взаимодействию с бромцианом в среде тетрагидрофурана с последующим...
Попередній патент: Колієукладальна машина, яка безперервно переміщується, для ущільнення щебеневого баластного шару колії і спосіб безперервного опускання колії в задане положення
Наступний патент: Гетероциклічні похідні або їх солі, що мають антагоністичну активність по відношенню до фат-ацетеру
Випадковий патент: Шліцьовий вал та спосіб відновлення профілю його шліців