Кристалічні піперидиніл-n-алкілкарбоксилати (варіанти), спосіб їх одержання (варіанти), фармацевтична композиція та проміжні продукти
Номер патенту: 52577
Опубліковано: 15.01.2003
Автори: УОРД Джеффрі Алан, ПРЕЙЕТЕР Дуглас Едвард, Уернер Джон Арнольд, Франк Скотт Алан




Формула / Реферат
1. Кристаллический пиперидинил-N-карбоксилат формулы (2)
где R является С1-С6 алкилом,
Z- является группой, выбранной из гидрохлорида, гидробромида, сукцината и (+)- дибензоилтартрата, который может быть использован как промежуточный продукт.
2. Кристаллический пиперидинил-N-карбоксилат по п. 1, отличающийся тем, что в нем Z- является гидрохлоридом, а R является метилом.
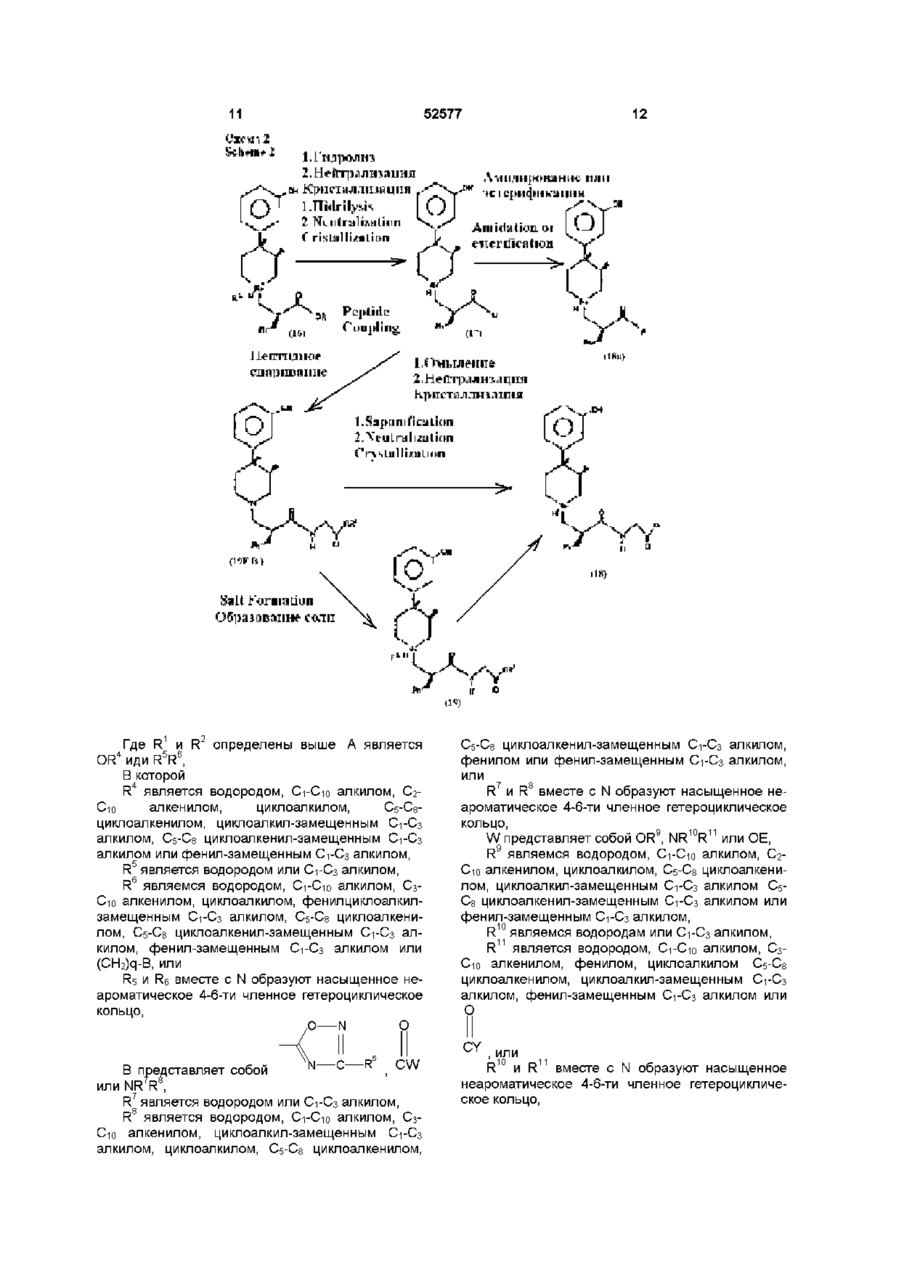
3. Кристаллический пиперидинил-N-карбоксилат формулы (4)
где R1 является C1-С6 алкилом, и соединение представляет собой соль, такую, как гидрохлорид ацетон моносольват, малат (1:1) и полуторамалат (3:2), который может быть использован как опиоидный антагонист.
4. Кристаллический пиперидинил-N-карбоксилат по п. 3, отличающийся тем, что в нем соединение формулы (4) представляет собой 2-метилпропиловый эфир (2S,3R,4R)[[2-[[4-(3-гидроксифенил)-3,4-диметил-1-пиперидинил]метил]-1-оксо-3-фенилпропил]-амино]уксусной кислоты.
5. Кристаллический пиперидинил-N-карбоксилат по пп. 3 или 4, отличающийся тем, что в нем соль представляет собой гидрохлорид ацетон моносольват.
6. Кристаллический пиперидинил-N-карбоксилат по пп. 3 или 4, отличающийся тем, что в нем соль представляет собой полуторомалат.
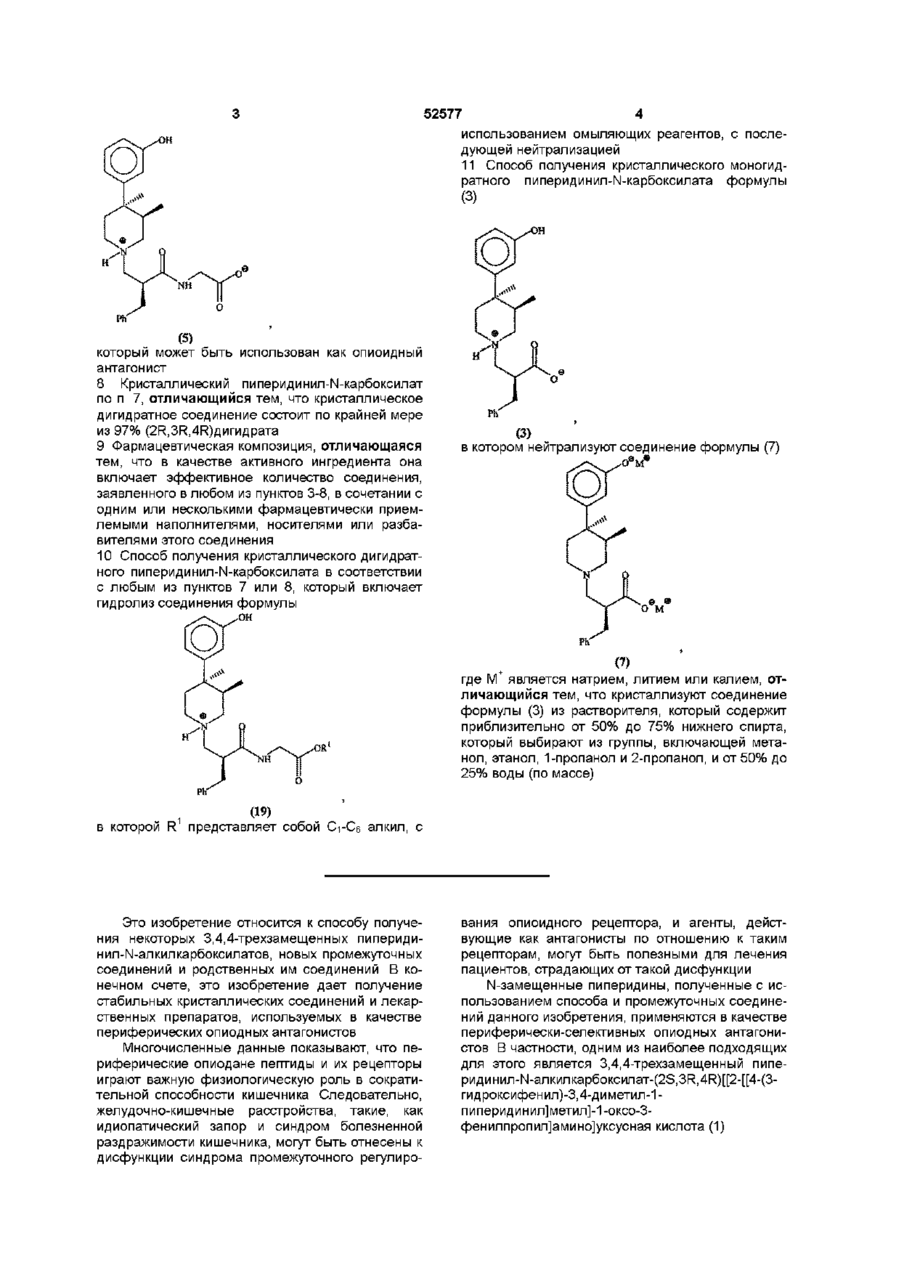
7. Кристаллический дигидратный пиперидинил-N-карбоксилат формулы (5)
который может быть использован как опиоидный антагонист.
8. Кристаллический пиперидинил-N-карбоксилат по п. 7, отличающийся тем, что кристаллическое дигидратное соединение состоит по крайней мере из 97% (2R,3R,4R)дигидрата.
9. Фармацевтическая композиция, отличающаяся тем, что в качестве активного ингредиента она включает эффективное количество соединения, заявленного в любом из пунктов 3-8, в сочетании с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями этого соединения.
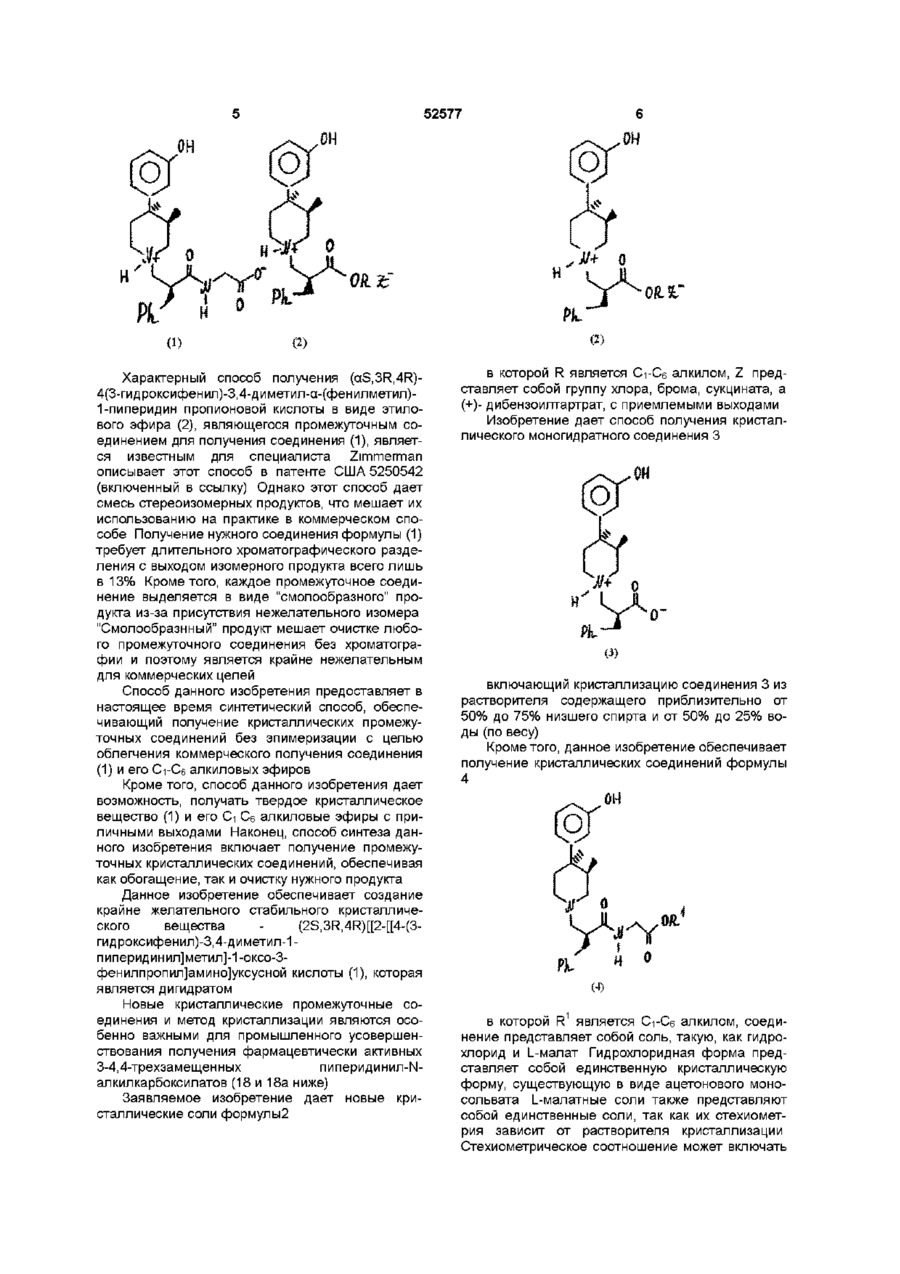
10. Способ получения кристаллического дигидратного пиперидинил-N-карбоксилата в соответствии с любым из пунктов 7 или 8, который включает гидролиз соединения формулы
в которой R1 представляет собой С1-С6 алкил, с использованием омыляющих реагентов, с последующей нейтрализацией.
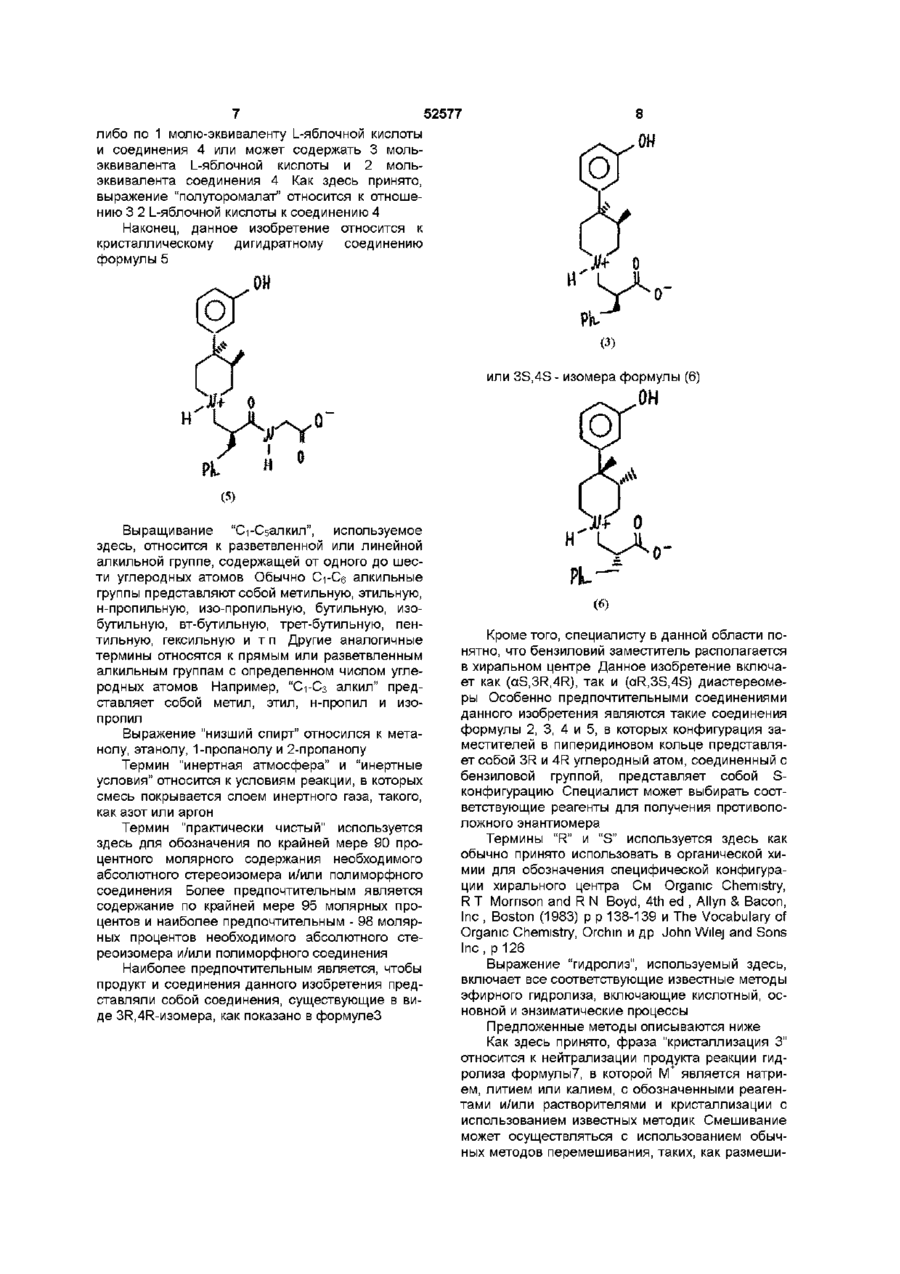
11. Способ получения кристаллического моногидратного пиперидинил-N-карбоксилата формулы (3)
в котором нейтрализуют соединение формулы (7)
где М+ является натрием, литием или калием, отличающийся тем, что кристаллизуют соединение формулы (3) из растворителя, который содержит приблизительно от 50% до 75% нижнего спирта, который выбирают из группы, включающей метанол, этанол, 1-пропанол и 2-пропанол, и от 50% до 25% воды (по массе).
Текст
1 Кристаллический пиперидинил-Nкарбоксилат формулы (2) .-он п где R является С-і-Сб алкилом, Z является группой, выбранной из гидрохлорида, гидробромида, сукцината и (+)- дибензоилтартрата, который может быть использован как промежуточный продукт 2 Кристаллический пиперидинил-Ы-карбоксилат по п 1, отличающийся тем, что в нем Z является гидрохлоридом, a R является метилом 3 Кристаллический пиперидинил-Ы-карбоксилат формулы (4) (4) где R1 является С-і-Сб алкилом, и соединение представляет собой соль, такую, как гидрохлорид ацетон моносольват, малат (1 1) и полуторамалат (3 2), который может быть использован как опиоидный антагонист 4 Кристаллический пиперидинил-Ы-карбоксилат по п 3, отличающийся тем, что в нем соединение формулы (4) представляет собой 2метилпропиловый эфир (2S,3R,4R)[[2-[[4-(3гидроксифенил)-3,4-диметил-1 пиперидинил]метил]-1-оксо-3-фенилпропил]амино]уксусной кислоты 5 Кристаллический пиперидинил-Ы-карбоксилат по пп 3 или 4, отличающийся тем, что в нем соль представляет собой гидрохлорид ацетон моносольват 6 Кристаллический пиперидинил-Ы-карбоксилат по пп 3 или 4, отличающийся тем, что в нем соль представляет собой полуторомалат 7 Кристаллический дигидратный пиперидинил-Ккарбоксилат формулы (5) О 1 ю ю 52577 использованием омыляющих реагентов, с последующей нейтрализацией 11 Способ получения кристаллического моногидратного пиперидинил-Ы-карбоксилата формулы (3) (5) который может быть использован как опиоидный антагонист 8 Кристаллический пиперидинил-Ы-карбоксилат по п 7, отличающийся тем, что кристаллическое дигидратное соединение состоит по крайней мере из 97% (2R,3R,4R)flHrHflpaTa 9 Фармацевтическая композиция, отличающаяся тем, что в качестве активного ингредиента она включает эффективное количество соединения, заявленного в любом из пунктов 3-8, в сочетании с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями этого соединения 10 Способ получения кристаллического дигидратного пиперидинил-Ы-карбоксилата в соответствии с любым из пунктов 7 или 8, который включает гидролиз соединения формулы ,он в котором нейтрализуют соединение формулы (7) в о м где М является натрием, литием или калием, отличающийся тем, что кристаллизуют соединение формулы (3) из растворителя, который содержит приблизительно от 50% до 75% нижнего спирта, который выбирают из группы, включающей метанол, этанол, 1-пропанол и 2-пропанол, и от 50% до 25% воды (по массе) (19) в которой R представляет собой С-і-Сє алкил, с Это изобретение относится к способу получения некоторых 3,4,4-трехзамещенных пиперидинил-ІЧ-алкилкарбоксилатов, новых промежуточных соединений и родственных им соединений В конечном счете, это изобретение дает получение стабильных кристаллических соединений и лекарственных препаратов, используемых в качестве периферических опиодных антагонистов Многочисленные данные показывают, что периферические опиодане пептиды и их рецепторы играют важную физиологическую роль в сократительной способности кишечника Следовательно, желудочно-кишечные расстройства, такие, как идиопатический запор и синдром болезненной раздражимости кишечника, могут быть отнесены к дисфункции синдрома промежуточного регулиро вания опиоидного рецептора, и агенты, действующие как антагонисты по отношению к таким рецепторам, могут быть полезными для лечения пациентов, страдающих от такой дисфункции N-замещенные пиперидины, полученные с использованием способа и промежуточных соединений данного изобретения, применяются в качестве периферически-селективных опиодных антагонистов В частности, одним из наиболее подходящих для этого является 3,4,4-трехзамещенный пиперидинил-N-an кил карбоксил aT-(2S,3R,4R)[[2-[[4-(3гидроксифенил)-3,4-диметил-1 пиперидинил]метил]-1-оксо-3фенилпропил]амино]уксусная кислота (1) 52577 (1) ^* ~ () 2 оа v Характерный способ получения (aS,3R,4R)4(3-гидроксифенил)-3,4-диметил-а-(фенилметил)1-пиперидин пропионовой кислоты в виде этилового эфира (2), являющегося промежуточным соединением для получения соединения (1), является известным для специалиста Zimmerman описывает этот способ в патенте США 5250542 (включенный в ссылку) Однако этот способ дает смесь стереоизомерных продуктов, что мешает их использованию на практике в коммерческом способе Получение нужного соединения формулы (1) требует длительного хром ато граф и чес ко го разделения с выходом изомерного продукта всего лишь в 13% Кроме того, каждое промежуточное соединение выделяется в виде "смолообразного" продукта из-за присутствия нежелательного изомера "Смолообразнный" продукт мешает очистке любого промежуточного соединения без хроматографии и поэтому является крайне нежелательным для коммерческих целей в которой R является СгСб алкилом, Z представляет собой группу хлора, брома, сукцината, а (+)- дибензоилтартрат, с приемлемыми выходами Изобретение дает способ получения кристаллического моногидратного соединения 3 Способ данного изобретения предоставляет в настоящее время синтетический способ, обеспечивающий получение кристаллических промежуточных соединений без эпимеризации с целью облегчения коммерческого получения соединения (1) и его СгСб алкиловых эфиров Кроме того, способ данного изобретения дает возможность, получать твердое кристаллическое вещество (1) и его Сі Сб алкиловые эфиры с приличными выходами Наконец, способ синтеза данного изобретения включает получение промежуточных кристаллических соединений, обеспечивая как обогащение, так и очистку нужного продукта Данное изобретение обеспечивает создание крайне желательного стабильного кристаллического вещества (2S,3R,4R)[[2-[[4-(3гидроксифенил)-3,4-диметил-1 пиперидинил]метил]-1-оксо-3фенилпропил]амино]уксусной кислоты (1), которая является дигидратом Новые кристаллические промежуточные соединения и метод кристаллизации являются особенно важными для промышленного усовершенствования получения фармацевтически активных 3-4,4-трехзамещенных пиперидинил-Nалкилкарбоксилатов (18 и 18а ниже) Заявляемое изобретение дает новые кристаллические соли формулы2 включающий кристаллизацию соединения 3 из растворителя содержащего приблизительно от 50% до 75% низшего спирта и от 50% до 25% воды (по весу) Кроме того, данное изобретение обеспечивает получение кристаллических соединений формулы 4 ОН (4) в которой R1 является С-і-Сб алкилом, соединение представляет собой соль, такую, как гидрохлорид и L-малат Гидрохлоридная форма представляет собой единственную кристаллическую форму, существующую в виде ацетонового моносольвата L-малатные соли также представляют собой единственные соли, так как их стехиометрия зависит от растворителя кристаллизации Стехиометрическое соотношение может включать 52577 либо по 1 молю-эквиваленту L-яблочной кислоты и соединения 4 или может содержать 3 мольэквивалента L-яблочной кислоты и 2 мольэквивалента соединения 4 Как здесь принято, выражение " п ол утором ал ат" относится к отношению 3 2 L-яблочной кислоты к соединению 4 Наконец, данное изобретение относится к кристаллическому дигид ратному соединению формулы 5 или 3S.4S - изомера формулы (6) Выращивание "СгСбалкил", используемое здесь, относится к разветвленной или линейной алкильной группе, содержащей от одного до шести углеродных атомов Обычно С-і-Сє алкильные группы представляют собой метильную, этильную, н-пропильную, изо-пропильную, бутильную, изобутильную, вт-бутильную, трет-бутильную, пентильную, гексильную и т п Другие аналогичные термины относятся к прямым или разветвленным алкильным группам с определенном числом углеродных атомов Например, "С-і-Сз алкил" представляет собой метил, этил, н-пропил и изопропил Выражение "низший спирт" относился к метанолу, этанолу, 1-пропанолу и 2-пропанолу Термин "инертная атмосфера" и "инертные условия" относится к условиям реакции, в которых смесь покрывается слоем инертного газа, такого, как азот или аргон Термин "практически чистый" используется здесь для обозначения по крайней мере 90 процентного молярного содержания необходимого абсолютного стереоизомера и/или полиморфного соединения Более предпочтительным является содержание по крайней мере 95 молярных процентов и наиболее предпочтительным - 98 молярных процентов необходимого абсолютного стереоизомера и/или полиморфного соединения Наиболее предпочтительным является, чтобы продукт и соединения данного изобретения представляли собой соединения, существующие в виде 3R,4R-H3OMepa, как показано в формулеЗ (6) Кроме того, специалисту в данной области понятно, что бензиловий заместитель располагается в хиральном центре Данное изобретение включает как (aS,3R,4R), так и (aR,3S,4S) диастереомеры Особенно предпочтительными соединениями данного изобретения являются такие соединения формулы 2, 3, 4 и 5, в которых конфигурация заместителей в пиперидиновом кольце представляет собой 3R и 4R углеродный атом, соединенный с бензиловой группой, представляет собой Sконфигурацию Специалист может выбирать соответствующие реагенты для получения противоположного энантиомера Термины "R" и "S" используется здесь как обычно принято использовать в органической химии для обозначения специфической конфигурации хирального центра См Organic Chemistry, R T Morrison and R N Boyd, 4th ed , Allyn & Bacon, Inc , Boston (1983) p p 138-139 и The Vocabulary of Organic Chemistry, Orchm и др John Wilej and Sons Inc , p 126 Выражение "гидролиз", используемый здесь, включает все соответствующие известные методы эфирного гидролиза, включающие кислотный, основной и энзиматические процессы Предложенные методы описываются ниже Как здесь принято, фраза "кристаллизация 3" относится к нейтрализации продукта реакции гидролиза формулы7, в которой М + является натрием, литием или калием, с обозначенными реагентами и/или растворителями и кристаллизации с использованием известных методик Смешивание может осуществляться с использованием обычных методов перемешивания, таких, как размеши 52577 вание, встряхивание и т п Кроме того, специалисту понятно, что процессы кристаллизации могут включать затравку, резкое охлаждение, "скребление" стекла реакционного сосуда и другие, обычно используемые для этого способы Исходные материалы для данного изобретения могут быть получены множеством разнообразных методов, хорошо известных специалистам данной отрасли 3-замещенные-4-метил-4-(3гидрокси- или алканоилоксифенил)пиперидиновые производные, используемые в качестве исходных веществ в способе данного изобретения, могут быть получены с применением основного метода, предложенного Zimmerman в патенте США №4115400 (1978) и Zimmerman и др в патенте США №4891379 (1990) Патенты США №№4115400 и 4891379 объединены здесь ссылкой Исходное вещество для синтеза соединений данного изобретения (3R,4R)-4-(3гидроксифенил)-3,4-диметил пиперидин может быть получено по методу Zimmerman патент США 5250542, включенное здесь в ссылку Специалист должен обратить особое внимание на примері ссылки Zimmerman 5250542 Исходное вещество 14, полученное как описано в методике, может быть использовано в способе по схеме 1 (ниже) в которой R1 определен выше R2 являемся хлоридом, бромидом, (+)-дабензоилтартратом или сукцинатом Как проиллюстрировано в схемеї, соединение 14 контактирует с алкилакрилатом (R1 акрилат) с 10 1 образованием соединения 15 R определен выше Подходящими растворителями являются метанол, тетрагидрофуран, этанол и другие Наиболее подходящими растворителями являются метанол и тетрагидрофуран Соединение 15 подвергается депротонированию и контактированию с бензилбромидом Депротонирование может быть осуществлено с использованием соответствующего основания Примерами подходящих основных реагентов являются литий диизопропиламид или литий гексаметилдисилазид Предпочтительными растворителями для реакции с основанием являются тетрагидрофуран и 1,2-диметоксиэтан Специалисту понятно, что могут быть подходящими и другие растворители При использовании в качестве основания литий диизопропиламида (ЛДА) наиболее предпочтительным является присутствие 2 эквивалентов бензилбромида Продукт алкилирования представляет собой смесь 1 1 (aS,3R,4R)изомера и (aR,3R,4R)-H3OMepa Кристаллические соединения формулы 16 являются новыми уникальными Только четыре конкретные соли формулы 16 являлись как стабильными кристаллическими солями, так и характеризовались желательным диастереомерным обогащением Были исследованы каждая из следующих кислот при использовании четырех различных систем растворителей HCI, НВг, (+)дибензоил винная, янтарная, (-)-ди-п-толуил винная, (+)-ди-п-толуил винная, (-)-дибензоил винная, (Ш,33)-(+)-камфарная, гиппуровая, бензойная, Lяблочная, D-яблочная, малоновая, Dаспаркановая, (-)-винная (+)-винная, (-)миндальная, (+)-миндальная, L-аскорбиновая, малеиновая, серная, уксусная, фосфорная, лимонная, молочная, п-толуолсульфоновая, Dарабаскорбиновая и L-аспартиковая Таким образом, среди исследованных 110 кристаллических соединений только четыре стабильные кристаллические соли характеризовались как обогащенные Обогащение и выход четырех стабильных кристаллических солей иллюстрируется в таблице1 Таблицаї Кристаллические соли эфира Соль Выход Растворитель кристаллизации Гидрохлорид Гидробромид (+)-ДБТАа сукцинат а Диастер отношение 88/12 79/21 71/29 83/17 39% 42% 25% 25% метанол метанол этилацетат ацетон (1 1) этилацетат ацетон (1 1) (+)-дибензоилтартрат Как иллюстрируется Схемой2 (ниже), соединение 16 подвергается гидролизу с образованием соединения 17 Специалисту понятно, что соединение 17 может быть использовано для получения других полезных соединений, как это иллюстрируется соединением 18а Соединения 18а описаны в основном в патенте США 5250542 как полезные в качестве опиодных антагонистов Прежде всего, возможно получение абсолютно чистых стереохимических изомеров (18 и 18а) без утомительного хроматографического разделения, используя новые промежуточные соединения данного изобретения 11 12 52577 Пептидное СПІЩ не а шіе К]* иста л .ти Ї Л ШІЯ I S L 5 V V v " M) B ^ Где R1 и R2 определены выше А является OR иди R5R6, В которой 4 R является водородом, Сі-Сю алкилом, С-АСю алкенилом, циклоал килом, Cs-Csциклоалкенилом, циклоалкил-замещенным СгСз алкилом, Сб-Св циклоалкенил-замещенным СгСз алкилом или фенил-замещенным С-і-Сз алкилом, R5 является водородом или СгСз алкилом, 6 R являемся водородом, Сі-Сю алкилом, СзСю алкенилом, циклоалкилом, фенилциклоалкилзамещенным С-і-Сз алкилом, Cs-Cs циклоалкенилом, Сб-Св циклоалкенил-замещенным СгСз алкилом, фенил-замещенным С-і-Сз алкилом или (CH2)q-B, или Rs и R6 вместе с N образуют насыщенное неароматическое 4-6-ти членное гетероциклическое кольцо, ,0—N О 4 Сб-Св циклоалкенил-замещенным С-і-Сз алкилом, фенилом или фенил-замещенным СгСз алкилом, или 7 8 R и R вместе с N образуют насыщенное неароматическое 4-6-ти членное гетероциклическое кольцо, Q C Y N R C W В представляет собой ^ , или NR?R , R7 является водородом или С-і-Сз алкилом, R8 является водородом, Сі-Сю алкилом, СзСю алкенилом, циклоал кил-замещенным СгСз алкилом, циклоалкилом, Cs-Cs циклоалкенилом, 1П 11 W представляет собой OR , NR R или ОЕ, R9 являемся водородом, Сі-Сю алкилом, С2Сю алкенилом, циклоалкилом, Cs-Cs циклоалкенилом, циклоалкил-замещенным С-і-Сз алкилом CsCs циклоалкенил-замещенным СгСз алкилом или фенил-замещенным СгСз алкилом, R10 являемся водородам или С-і-Сз алкилом, R11 является водородом, Сі-Сю алкилом, СзСю алкенилом, фенилом, циклоалкилом Cs-Cs циклоал кенилом, циклоалкил-замещенным С-і-Сз алкилом, фенил-замещенным СгСз алкилом или О , или R и R вместе с N образуют насыщенное неароматическое 4-6-ти членное гетероциклическое кольцо, является 14 52577 13 (СН2)Г -D О ,13 1 -R,12 -OCR или является С-і-Сз алкилзамещенным мети R леном R1 является Сі-Сю алкилом, D представляет собой OR или NR R , R14 является водородом, С-і-Сю алкилом, С2Сю алкенилом, циклоалкилом, Cs-Cs циклоалкенилом, циклоалкил-замещенным С-і-Сз алкилом или Сб-Св-циклоалкенил-замещенным Сі-Сз алкилом или фенил-замещенным Сі-Сз алкилом, R15 является водородом, Сі-Сю алкилом СзСю алкенилом, фенилом, фенил-замещенным СіСз алкилом, циклоалкилом, Cs-Cs циклоалкенилом циклоалкил-замещенным Сі-Сз алкилом или Сб-Св циклоалкенил-замещенным Сі-Сз алкилом и R1 является водородом или С-і-Сз алкилом, или R15 и R16 вместе с N образуют насыщенное неароматическое 4-6-ти членное тетрациклическое кольцо, У является OR17 или NR18R19, R17 является водородом, Сі-Сю алкилом С2Сю алкенилом, циклоалкилом, Cs-Cs циклоалкенилом циклоалкил-замещенным Сі-Сз алкилом, CsCs циклоалкенил-замещенным Сі-Сз алкилом или фенил-замещенным С-і-Сз алкилом, R18 являемся водородом ила Сі-Сз алкилом и R19 является водородом, С-і-Сю алкилом, СзСю алкенилом, фенилом, циклоалкилом, Cs-Cs циклоалкенилом, циклоалкил-замещенным С-і-Сз алкилом, Сб-Св циклоалкенил-замещенным Сі-Сз алкилом или фенил-замещенным С-і-Сз алкилом, или R и R вместе с N образуют насыщенное неароматическое 4-6-ти членное гетероциклическое кольцо, q равняется 1-4, m равняется 1-4 Заместитель "А" описывается в патенте США 5250542 Реакция гидролиза может быть завершена при использовании известных методик кислотного гидролиза Примером одного из таких методов кислотного гидролиза является действие водного раствора кислоты в диоксане в условиях дефлегмирования Более предпочтительным является проведение реакции гидролиза с использованием условий омыления с тем, чтобы избежать эпимеризации Примерами омыляющих реагентов являются гидроокись лития, гидроокись натрия, гидроокись калия и т п Продукт реакции гидролиза (карбоксплатная соль) подвергают регулированию до изоэлектрической точки аминокислоты, используя водную кислоту для получения амфотерного соединения 17 Кристаллизация моногидрата 17 должна бить завершена с использованием 50-75% низшего спирта и 50-25% воды Таблицаї иллюстрирует критическую зависимость кристаллизации от приблизительного состава растворителя 1 1 для низшего спирта и води Выражение "смоляной ком" относится к коагуляции липкого полутвердого продукта в аморфную массу Таблицаї № % сорастворителя Концентрация кислоты 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 нет нет нет нет нет нет нет нет 6 СНзОН 12СН 3 ОН 25 СНзОН 25 СНзОН 50 СНзОН 50 СНзОН 50 СНзОН 50 СНзОН 50 і-Рг 50ACN 12N НСІ 12N НСІ 12N НСІ 1N НСІ 6N НСІ 1М Н3Р04 6М Н3Р04 1М АсОН 12N НСІ 12N НСІ 12N НСІ 12N НСІ 12N НСІ 12N НСІ 12N НСІ 12N НСІ 12N НСІ 12N НСІ ACN относится к ацетонитрилу, i-Pr - относится к изопропиловому спирту * Относится к реакционной концентраций ЮмЛ растворителя/г соединения 16 Выход Примечания 86% 88% 93% 90% 91% 93% 97% 99% 88% 87% 82% 90% 58%* 82% 90%** 95 9**% 73% 23% Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком Смоляной ком кристаллы кристаллы кристаллы кристаллы кристаллы Повышение выхода связано с испарением метанола дистилляцией после осуществления кристаллизации 16 15 52577 Как это проиллюстрировано в схеме2, продукт растворе Конечно, специалист ожидает, что от17 может быть непосредственно использован в ношение составит 1 1 Кроме того, кристаллизаэтапе амидирования/эстерификации Когда требуция полуторомалатной соли в "не образующем ется амидирование должна, быть выбрана аминоп ол утором ал ата" растворителе или системах раскислота с целью образования необходимых сотворителей дает только кристаллическая мономаединений формул 18 и 18а Аминокислота латная соль с выходом, близким к количественноподвергается действию глицинового эфира в расму, при этом избыток L-яблочной кислоты творителе, таком, как диметилформамид или тетостается в маточной жидкости рагидрофуран Кристаллизация полуторомалатной соли дает продукт фармацевтически приемлемой чистоты В качестве сопряженного реагента использупри высоком выходе, и этот продукт состоит из ется дициклогексилкарбодиимид В качестве кристаллов с веской равномерностью кристалливспомогательного нуклеофильного вещества доческой формой и величиной кристаллов Гидробавляется N-гидроксибензотриазол Реакция сохлоридные и полуторомалатные соли могут быть пряжения может осуществляться в инертных усиспользованы в качестве про-лекарства, так как ловиях Более предпочтительным является изобутиловый эфир легко расщепляется in vitro использованием в качестве растворителя тетрагидрофурана в реакции пептидного сопряжения Кислотами, которые не образуют кристаллиСпециалисту понятно, что могут быть эффективно ческих солей, стабильных при 25°С, являются НВг, использованы также и другие способы пептидного H2SO4, гиппуровая, d-винная, І-винная, малоновая, сопряжения янтарная, уксусная, арабаскорбиновая, аскорбиновая, лимонная, бензойная, молочная, (S)-(+)Или же, кристаллическая соль соединения 19 миндальная и (Р)-(-)-миндальная кислоты СО (свободное основание) может быть получена, как это иллюстрируется в схеме2 выше Были Продукт реакции амидировапроведены исследования по кристаллизации с ния/эстерификации или его солевые формы могут использованием 17 различных кислот с тремя быть гидролизованы с использованием стандартрастворителями этилацетат, ацетон и этанол Из ных методик Предпочтительно используются меэтих 51 экспериментов только L-яблочная и хлотоды основного гидролиза Предпочтительными ристоводородная кислоты дали кристаллические реагентами омыления являются гидроокись насоли, стабильные при 25°С трия, гидроокись калия, гидроокись лития и т п Наиболее предпочтительным является осуществКристаллическая хлористоводородная соль ление этапа омыления с использованием гидрополучается контактированием 19 СО с безводной окиси натрия и растворителя Особенно предпочHCI в ацетоне Капиллярный газохроматографитительными растворителями (1 1) являются ческий анализ показал, что соль образуется в виметанол вода и (2 1) этанол вода Реакция преде моносольвата в ацетоне Эта единственная кращается при использовании кислоты, такой, как моносольватная кристаллическая форма позвохлористоводородная кислота После нейтрализаляет выделить соединение 19 в существенно чисции (рН=6), твердый кристаллический дигидраттом виде Контактирование соединения 19 СО с ный продукт 18 выделяется непосредственно безводным HCI в других растворителях дает фильтрацией Выделенный продукт 18 обладает аморфное твердое вещество без существенной фармацевтически приемлемой чистотой без поочистки следующих этапов очистки Заявители открыли, что соль L-яблочной кислоты может быть получена в виде стабильного Особенно ценным является дигидрат 18, так кристаллического твердого вещества, имеющего как это соединение является стабильным кридва отношения 19 СО по отношению к L-яблочной сталлическим твердым веществом, с равномерной кислоте в зависимости от растворителя кристалкристаллической формой и размером частиц, что лизации В том случае, когда кристаллизация обеспечивает воспроизводимые скорости раствоосуществляется в растворителях, таких, как метирения, оно характеризуется фармацевтически лэтил кетон, ацетон, ацетонл'-бутил метиловий желаемыми качествами эфир или ацетон/гептан была установлена предСоединения формул 5 и 4, приведенных выполагаемая стехиометрия 1 1 в 19 СО по отношеше, используются для блокирования периферичению к L-яблочной кислоте Однако, когда кристалских опиодных рецепторов и для предотвращения лизация проводится в системе растворителей периферических индуцированных препаратами ацетон/этил ацетат, ацетонл'олуол или этаопия побочных эффектов Эти побочные эффекты Honfronyon, кристаллическая соль дает единстиндуцируются приемом млекопитающими препавенное стехиометрическое отношение 3 2 Lрата опия, такого как морфин Индуцированные яблочной кислоты по отношению к 19 СО (полупрепаратом опия побочные эффекты могут вклюторный малат) Такой результат является отчасти чать запор, тошноту и рвоту Таким образом, сонеожиданным, когда полуторный малат получаетединения данного изобретения используются для ся даже при отношении 1 1 яблочной кислоты и 19 лечения одного или нескольких индуцированных СО в определенных растворителях Действительпрепаратами опия побочных эффектов Эти соно, когда соединяются эквимолярные количества единения могут использоваться также для лечеL-яблочной кислоты и 19 СО в растворителе или ния синдрома болезненной раздражимости кисистеме растворителей, образующих полуторный шечника, неязвенной дисперсии и малат, этот полуторный малат является единстидиопатического запора Эти соединения незнавенной солью, образованной с выходом около чительно преодолевают гематоэнцефалический 67% в массовом равновесии 19 СО в маточном барьер и поэтому не снижают опиодного эффекта 18 17 52577 на центральные (мозговых и позвоночных тяжей) может быть определена сравнением ADso для опиодные рецепторы Следовательно, эти харакмыши, подвергшейся испытанию на сокращение, с теристики показывают, что соединения свободны ED50 для мыши, подвергшейся испытанию на пов основном от других центральных промежуточнос При более высоком отношении наблюдается ных эффектов более высокий относительный антагонизм периферических опиодных рецепторов, обусловленДля того, чтобы определить in vivo антагонизм ный конкретным соединением опиодного рецептора, бала использована мышь, находящаяся под воздействием анальгезиса ИсЗначения AD50 для соединений данного изопытуемые соединения оценивались по их способбретения составляют свыше 8 мг/кг, в то время как ности к блокированию индуцированной морфолизначения ED50 составляют ниже 1 ном анальгезии Кроме того, было установлено, что соединеПять мышей мужской особи CF-1 (Chrles River, ния формул 5 и 4, приведенные выше, показываPortage, Ml), весом приблизительно в 20г, после ют прекрасную активность в анализе связывания голодания в течение ночи подвергались одновреопиодного рецептора, которая оценивается сродменно наблюдениям с точки зрения реакции на ством соединений к связыванию Мю рецепторов сокращение Ответ на сокращение определялся Этот анализ проводился в соответствии со слекак сокращение брюшной мускулатуры, за котодующей методикой рым следует вытяжение задних конечностей, и Мужские особи крыс Spraque Dawley были индуцировался внутрибрюшным приемом 0,6% умерщвлены обезглавливанием и их мозг бил изуксусной кислоты в объеме 1мл/100г веса тела влечен Мозговая ткань с выделенным мозжечком Период наблюдения длился 10 минут, начиная с гомогенизировалась в тефлоне и в стеклянном 5-ти минут после инъекции уксусной кислоты гомогенизаторе для тканей Всплывшая I, шарикоПроцент торможения сокращения высчитывался, вая IV фракция бала заморожена в азотном замоисходя из среднего числа сокращений в контрольраживателе при концентрации 1 ЗЗг/мЛ и храниной (не подвергнутой лекарственному воздейстлась до использования не более пяти недель вию) группе ED50 определялась как доза вещестУвеличивающиеся концентрации эксперименва-агониста, имеющего ингибирующее значение тального соединения (от 0,1 до ЮООнаномол на сокращение 50% ADso определялась как доза (нМ)), буфер Krebs-Hepes pH7,4 и тритиевый наантагониста, которая, снижает ингибирование солоксон (0 5нМ) (3Н лиганд) помещались в поликращения, создаваемое дозой сульфата морфина стирольные пробирки при комнатной температуре 1 25мг/кг, до 50% Каждая мышь подвергалась Реакция инициировалась добавлением ресуспениспытанию только одни раз Все лекарственные зированной ткани, которая была предварительно препараты вводились подкожно (1мл/100г веса) за термостатирована при 37°С в течение 20 минут 20 минут до инъекции уксусной кислоты Реакционная смесь термостатировалась при 37°С на водяной бане в течение 20 минут Реакция заПроводились определения периферической канчивалась быстрой фильтрацией (Brandel cell опиодной активности Мыши выдерживались как harvester) через стеклянные фильтры Whatman минимум в течение 10 дней на 0 01М сахариновой GF/B, которые были предварительно пропитаны воде, содержащей 1 г/Л сульфата морфина при буфером Krebs-Hepes с рН7,4 Затем фильтры среднем потреблении 3,0г воды на мышь в день в промывалась 2 раза 5мЛ ледяного буфера Krebsтечение по крайней мере 3 дней Морфиновая Hepes с рН7,4 Промытые фильтры помещались в вода удалялась за 45 минут до инъекции предпосцинциляционные ампулы, было добавлено ЮмЛ лагаемого опиоидного антагониста (Brande) и образцы подвергались обсчету на бета После приема опиодного антагониста мыши счетчике Searle D-300 Время термостатировання помещались в пластмассовые цилиндры, дно кореакционной смеси составляло 20 минут при 37°С торых было выстлано белыми бумажными полоЗначения К, и Ко рассчитывались с использованитенцами ем стандартных методик Мыши подвергались визуальному наблюдению в течение 30 минут после инъекции - на налиСоединения данного изобретения выявляют чие подергивания и поноса Подергивание расцевысокие значения желаемой активности Значение нивалось как позитивное, если происходило по процента замещения испытуемыми соединениями крайней мере одно подергивание в 30 минут Попри концентрации ЮнМ было выше 75% и свыше нос расценивался положительно, когда эскремен80% при ЮОнМ Это особенно желательно в свете ты были достаточно влажными и оставляли пятна значений AD50 и ED50, приведенных выше Резульна белой бумаге, выстилающей дно цилиндра таты показывают, что соединения данного изобреЧерез 30 минут испытаний мыши возвращались в тения характеризуются благоприятной активносвои первоначальные клетки, снова сажались на стью для использования их в лечении синдрома морфиновую воду и не подвергались повторным раздражимости кишечника и условий, относящихиспытаниям в течение 48 часов Были испытаны ся к связыванию мю рецепторов более низкие доза соединений антагонистов, пока Хотя и возможен прием соединения данного не были определены пороговые дозы поноса Поизобретения непосредственно, без каких-либо нос представляет собой периферически усредсоставов, предпочтительно использование таких ненное значение стремительного, вызванного соединений в виде фармацевтических композипрепаратом опия воздержания ций, включающих фармацевтически приемлемый наполнитель и по крайней мере одно из соединеСтепень воздействия на периферическую акний изобретения Эффективный интервал дозиротивность представленных соединений по сравневок соединений данного изобретения достаточно нию с воздействием на центральную активность 19 52577 широк Так, эти композиции содержат приблизительно от 0,1% по весу до 90,0% по весу заявленных здесь соединений Само по себе, представленное изобретение представляет также фармацевтические композиции, включающие соединение данного изобретения с фармацевтически приемлемым наполнителем При приготовлении композиций данного изобретения активный ингредиент обычно смешиваемся с наполнителем, который может быть носителем или разбавителем, или может быть разбавлен носителем, или включен в носитель, который может представлять собой капсулу, пакетный, бумажный или другой контейнер В том случае, когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким веществом, служащим связующим, наполнителем или средой для активного ингредиента Таким образом, композиции могут иметь вид таблеток, пилюль, порошков, лепешек, пакетиков, облаток, элексиров, эмульсий, растворов, сиропов, суспензий, аэрозолей (как в твердой, так и в жидкой среде), суппозиториев, мягких и твердых желатиновых капсул Соединения данного изобретения могут вводиться трансдермально, если это желательно Специалисту хорошо известны трансдермальные усилители проницаемости и доставляющие системы, включающие аппликации и т п Примерами соответствующих носителей, наполнителей и разбавителей являются лактоза, декстроза, сахароза, сорбит, маннит, крахмалы, аравийская камедь, фосфат кальция, альгинаты, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, целлюлоза, трагакант, желатин, сироп, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния, вода и минеральное масло Композиции могут включать также увлажняющие агенты, эмульгирующие и суспендирующие агенты, консервирующие агенты, подслащивающие вещества или облагораживающие агенты Композиции изобретения могут быть приготовлены как для быстрого, так и отложенного или замедленного выделения активного компонента после приема его пациентом - применением хорошо известных специалисту приемов Композиции данного изобретения могут вводиться трансдермально с использованием известных трансдермальных поставляющих систем и наполнителей Более предпочтительным является смешение соединения данного изобретения с усилителями проницаемости, включающими, но не ограниченными пропиленгликоль, полиэтиленгликоль, монолаурат и азациклоалкан-2-оны и с включенными в аппликацию аналогичными поставляющими системами Дополнительными наполнителями являются желатинирующие агенты, эмульгаторы и буферные вещества, которые могут быть добавлены в случае необходимости в трансдермальную композицию Для орального приема соединения данного изобретения могут быть в идеальном случае смешаны с носителями и разбавителями и сплавлены в таблетки или помещены в желатиновые капсулы Соединения данного изобретения могут быть приготовлены в виде микрочастиц или микроша 20 риков Микрочастицы могут быть приготовлены с использованием пол игл и кол и да, полилактида или других полимеров для облегчения непрерывного выделения активного соединения или пролекарства Композиции готовятся предпочтительно в единичной дозированной форме, при этом каждая дозировка содержит приблизительно от 1 до 500мг, более предпочтительно от 5 до 300мг активного ингредиента Другой предпочтительный интервал составляет приблизительно от 0,5мг до 60мг активного ингредиента на единичную дозированную форму Термин "единичная дозированная форма" относится к физически дискретным единицам, подходящим в качестве стандартных дозированных форм для человека и других млекопитающих, при этом каждая единица содержит предварительно определенное количество активного вещества, рассчитанная для обеспечения желаемого терапевтического эффекта в сочетании с соответствующим фармацевтическим носителем Специалисту понятно, что соединения данного изобретения могут быть составлены с применением других известных лекарственных препаратов Такие составляющие могут сообщать синергический терапевтический эффект Например, антацид может быть соединен с соединениями данного изобретения для создания желаемого желудочнокишечного эффекта Для более полной иллюстрации действия, данного изобретения ниже приводятся примеры составов Примеры только иллюстрируют, но не ограничивают объем изобретения Композиции могут использовать в качестве активных соединений любые соединения денного изобретения Композицияі Твердые желатиновые капсулы готовятся с использованием следующих ингредиентов ., Количество на капсулу (2S,3R,4R)[[2-[[4-(3гидроксифенил)-3,4диметил-1пиперидинил] метил]-1-оксо-3фенилпропил] амино] уксусная кислота - метиловый эфир, гидрохлорид Высушенный крахмал Стеарат магния Концентрач ^ ция по весу (процент) 250мг 55 0 200мг 10мг 460мг 43 0 20 100 0 Перечисленные выше ингредиенты смешиваются и помещаются в твердую желатиновую капсулу в количестве 460мг Композиция2 Капсулы с содержанием каждая в 20мг лекарственного препарата готовятся следующим образом 52577 21 ., Количество на капсулу (2S,3R,4R)[[2-[[4-(3гидроксифенил)-3,4диметил-1-пиперидинил]метил]-1оксо-3фенилпропил] амино] уксусная кислота, этиловый эфир, гидрохлорид моногидрат Крахмал Микрокристаллическая целлюлоза Стеарат магния Концентрач ^ ция по весу (процент) 20мг 89мг 10 44 5 89мг 2мг 200мг 44 5 1 0 100 0 Активный ингредиент, целлюлоза, крахмал и стеарат магния смешиваются, пропускаются через сита США с размером N45 меш и помещаются в твердую желатиновую капсулу КомпозицияЗ Капсулы, каждая из которых содержит 100мг лекарственного препарата, готовятся следующим образом ., Количество на капсулу (2S,3R,4R)[[2-[[4(3- гидроксифенил)-3,4-диметил1-пиперидинил]метил]-1оксо-3фенилпропил] амино] уксусная кислота, дигидрат Полиоксиэтиленсорбит, мо-ноолеат Порошок крахмала Концентрация ч ^ ч по весу (процент) кислота, этиловый эфир, полуто ром ал ат Крахмал Микрокристаллическая целлюлоза Поливинилпирролидон (в виде 10%-го раствора в воде) Натрий карбоксиметил крахмал Стеарат маг-ния Тальк 45мг 45 0 35мг 35 0 4мг 40 4 5мг 45 0 5мг 05 1мг 10 100мг 100 0 Активный ингредиент, крахмал и целлюлоза пропускаются через сито США с размером отверстий 45 меш и тщательно перемешиваются С образующимся порошком смешивается раствор поливинилпирролидона после чего смесь пропускается через сито США с размером отверстий 14 меш Полученные таким образом гранулы сушатся при 50-60°С и пропускаются через сита США в 18 меш Натрий карбоксиметил крахмал, стеарат магния и тальк, предварительно пропущенные через сито США с отверстиями в 60 меш, затем добавляются к гранулам, которые после смешения прессуются на таблетирующей машине с получением таблеток весом 100мг Композиция5 Могут быть приготовлены таблетки следующего состава ., Количество с на таблетку 3 100мг 30 0 50мг 0 02 250мг 350мг 69 98 100 0 Перечисленные выше ингредиенты тщательно перемешиваются и помещаются в пустую желатиновую капсулу Композиция4 Таблетки, содержащие 10мг активного ингредиента, готовятся следующим образом Количество на таблетку (2S,3R,4R)[[2-[[4(3- гидроксифенил)-3,4-диметил1-пиперидинил]метил]-1оксо-3фенилпропил] амино] уксусная 22 10мг Концентрация по весу (процент) 100 (2S,3R,4R)[[2-[[4(3- гидроксифенил)-3,4-диметил1-пиперидинил]метил]-1оксо-3фенилпропил] амино] уксусная кислота, дигидрат Микрокристаллическая целлюлоза Двуокись кремния дымящая Стеариновая кислота Концентрация ч ^ ч по весу , ' -. (процент) 250мг 38 0 400мг 60 0 10мг 1 5 5мг 665мг 05 100 0 Компоненты смешиваются и прессуются в виде таблеток весом 665мг Композицияб Твердые желатиновые капсулы могут быть приготовлены с использование следующих ингредиентов Количество на капсулу Концентрация по весу 23 52577 (процент) (2S,3R,4R)[[2-[[4(3- гидроксифенил)-3,4-диметил1-пиперидинил]метил]-1оксо-3фенилпропил] амино] уксусная кислота, дигидрат Полиэтиленгликоль 66мг 18 300мг 366мг 82 100 Все твердые ингредиенты пропускают через сито Полиэтиленгликоль расплавляют и поддерживают в расплавленном состоянии Лекарственный препарат помещают в расплавленное связующее Сплавленная гомогенная суспензия помещается в твердую желатиновую капсулу соответствующего веса и объема с использованием подходящего для наполнения маслообразной пасты оборудования Капсулы, содержащие 6мг активного вещества, могут быть приготовлены точно, как описано выше, однако количество дигидратного соединения должно быть снижено до 6,6мг на капсулу Капсулы, содержащие 0,6мг активного вещества, могут быть приготовлены, как описано выше, однако количество дигидрата должно быть снижено до 0,66мг с 200мг полиэтиленгликоля на капсулу Промежуточные соединения и способы, соответствующие данному изобретению, могут использоваться для получения соединений, характеризующихся успешной периферической опиоидной антагонистической активностью, в объеме данного изобретения предлагаются определенные соединения и условия Следующие далее условия, осуществления изобретения и характеристики соединений, приведенные в виде таблицы, независимо могут комбинироваться для создания множества предлагаемых соединений и условий способа Следующий далее перечень осуществлений данного изобретения не имеет в виду каким-либо образом ограничить объем данного изобретения A) Кристаллическое соединение 2 представляет собой метиленовый сложный эфир B) Кристаллическое соединение 2 представляет собой (аЗ,ЗР,4Р)-3-[[4-(3-гидроксифенил)-3,4диметил-а-(фе-нилметил)-1-пиперидин пропионовую кислоту, гидрохлорид C) Кристаллическое соединение 2 представляет собой этиловый сложный эфир D) Кристаллическое соединение 2 представляет собой НВг-соль E) Низший спирт является метанолом F) Соотношения низшего спирта и воды составляют Низшего спирта 50-60% и 50-40% воды G) R1 представляет собой С1-С4 алкил Н) Кристаллические соединения формулы 4 представляют собой полуторомалатную соль 24 I) Кристаллические соединения формулы 4 представляют собой гидрохлорид ацетоновый моносольват J) Практически чистый дигидрат формулы 5 представляет собой 97% или более 2S.3R.4R дигидрата К) Фармацевтическая композиция включает дигидратное соединение формулы 5 и один или несколько фармацевтически приемлемых наполнителя L) Фармацевтическая композиция включает полуторомалатную соль соединения формулы 4 М) Способ использования соединения формулы 5 состоит в лечении синдрома раздражимости кишечника N) Способ использования одного или нескольких соединений формулы 4 состоит в лечении синдрома раздражимости кишечника О) Способ связывания мю рецептора состоит в приеме эффективного количества соединения формулы 5 Р) Способ связывания мю рецептора, состоит в приеме эффективного количества одного или нескольких соединений формулы 4 Предложенные осуществления данного изобретения представлены пунктами А-Р Следующие далее примеры даются с целью иллюстрации и не могут быть рассмотрены в качестве ограничивающих объем притязаний изобретения Концентрации реагентов не являются критическими для осуществления изобретения Специалист в данной области может изменить концентрации реагентов с целью достижения желаемой скорости реакции и выхода продукта Продолжительность времени осуществления описанных процессов не является критической Как во всех случаях химических процессов, скорость реакции зависит от множества факторов, таких, как температура и точные соединения, которые должны быть приготовлены За ходом реакции можно следить, используя такие методы, как тонкослойная хроматография (ТСХ), высокоэффективная жидкостная хроматография (ВЭЖХ), газовая хроматография (ГХ) и ядерная магнитная резонансная спектроскопия (ЯМР) - с целью определения степени завершения реакции Исполнитель может получить максимальные выходы, увеличивая время реакции Или же, исполнитель может пожелать получить максимальную производительность, прекращая реакцию в точке, в которой достигается максимальное завершение с экономической точки зрения Как принято в дальнейших примерах, следующие выражения имеют такие значения "ГОБТ" относится к 1-гидроксибензотриазол гидрату "ТГФ" означает тетрагидрофуран "ДМФ" относится к даметилформамиду "ТЭА" означает триэтиламин "ДЦК" означает дициклогексилкарбодиимид Препараті (ЗР,4Р)-4-(3-гидроксифенил)-3,4-диметил-1пиперидин-пропионовая кислота, метиловый эфир Круглодонная колба загружалась ТГФ (ЮООмЛ) и (+)-3-(3,4-диметил-4пиперидинил)фенолом (70,46г, 0,343мол) Суспен 25 Остаток (гомогенное масло) растворялся в этилацетате (450мЛ) пока был еще теплым и раствор переносился в 3-х горлую круглодонную колбу, снабженную механической мешалкой и конденсатором дефлегматорного типа К раствору при комнатной температуре при перемешивании добавлялся гексан (450мЛ) Пастообразная смесь затем нагревалась с обратным холодильником для повторного растворения твердого вещества, и раствору давали медленно охлаждаться при перемешивании Затем раствор затравливали при 38°С для инициирования кристаллизации После охлаждения до комнатой температуры смесь охлаждали до 5°С и перемешивали еще час Продукт выделяли фильтрацией через воронку со стеклянным фильтром, сушили на воздухе в течение 1/2 часа и затем сушили в течение ночи в вакуумной печи (40°С, 5мм рт ст) Общий выход белого кристаллического вещества составлял 89,1г (97,9%) Тпл 77 2-79 6°С 26 рКа (67% водню ДМФ) = 7 68 ИК (СНСІз) 3300-2600, 3018, 2970, 1752, 1213, 1125, 1033, 1011см 1 1 Н-ЯМР (300МГц, CDCI3) 5 0,82 (д, 6Н, I = 6,9), 1 79 (септ , 1Н, I = 6,8), 2 33 (с, ЗН), 3,66 (уш р, с 2Н), 3 78 (д, 2Н, I = 6,6), 7 10 (д 2Н, I = 8,1), 7,72 (д, 2Н, I = 8,2), 8,03 (уш с , ЗН) ІЗ С-ЯМР (75 4МГц, CDCI3) 5 18,9, 21,3, 27,4, 40,3, 72,0, 126,1, 128,9, 140,3, 141,4, 167,5 52577 зия нагревалась до 40-45°С и в течение 3 минут добавлялся метилакрилат (46,4мЛ 0,515мол, 1,5экв ) Изменения температуры не наблюдалось Реакционная смесь перемешивалась при 45°С и развитие ее наблюдали, используя метод ВЭЖХ Реакционная смесь оставалась мутной Через 4 часа реакционную смесь охлаждали до комнатной температуры и фильтровали через диатомовую землю Растворитель и избыток метилакрилата удаляли концентрированием раствора посредством ротационного испарения при 40°С до общего веса 120г Неочищенный продукт подвергали повторному растворению в ДМФ (180г) с получением раствора в ЗЗ.Звес % для использования его в способе примера2 Количественный выход по ВЭЖХ [a]\S(20,D) 75,3° (С 1 01, МеОН), [а]20зб5 245 6° (с 1,01 МеОН) ИКС (СНС1_3) 3600,3600-3100,1732,1440 см 1 1 Н-ЯМР (CDCL3) 5 0,72 (д, ЗН, I =7,0Гц), 1,30 (с, ЗН), 1,59 (уш д , 1Н), 1,90-2,03 (м, 1Н), 2,25-2,50 (м, 2Н), 2,5—2,90 (м, 7Н), 3,66 (с, ЗН), 6,63 (дд, 1Н, 1 7,8, 2,0Гц), 6 73 (уш с , 1Н), 6 81 (д, 1Н, 1 7 8Гц), = = 7,15 (т, 1Н, І=8,0Гц) 1 Х-ЯМР (CDCL3) 5 16,1, 27,4, 30,8, 32,0, 38,4, 38,9, 49,9, 51,7, 53,9, 55,7, 55,8, 112,5, 112,6, 113,0, 113,2, 117,6 117,7, 129,2, 151,6 156,1, 173,4, УФ (ЕЮН) Атах 274нм, є 2028, 202нм, є 17350 МС (FAB) m/z 292 (100%, М+1), 292 (18%, М+), 218(65%) Препарат2 Изобутилглицин, соль п-толуолсульфоновой кислоты Круглодонная колба наполнялась толуолом (бООмЛ), глицином (22,53г, О.ЗОмол), моногидратом п-толуолсульфоновой кислоты (62,76г, О.ЗЗмол, 1,1экв) и изобутиловым спиртом (бОмЛ, 0,65мол, 2,17экв) Гетерогенная реакционная смесь перемешивалась и нагревалась при дефлегмировании с наружным обогревом для азеотропного удаления воды по мере ее образования Через два часа реакционная смесь была гомогенной Спустя еще 1,5 часа реакционная смесь охлаждалась до 50°С и концентрировалась с применением ротационного испарения при 60°С, до конечного веса 135г Анализ C13H21NO5S Рассчитано С 51,47, Н 6,98, N4,62, S 10,57 Установлено С 51,74, Н 6,77, N 4,76, S 10,73 ПримерІ (23,ЗР,4Р)[[2-[[4-(3-гидроксифенил)-3,4диметил-1-пиперидинилметил-і-оксо-Зфенилпропил]-амино]-уксусная кислота, 2мегилпропиловый эфир Круглодонная колба наполнялась (aS,3R,4R)4-(3-гидроксифенил)-3,4-диметил а- (фенилметил1-пиперидин пропионовой кислотой (20,11г, 0 0522мол, 1экв), соединением препарата 2 (17,60г, 0,058мол, 1,11 экв), гидроксибензотриазол моногидратом (7,83г, 0,058мол, 1 11 экв) и сухим тетрагидрофураном (144мЛ) К смеси добавлялся триэтиламин (8,08мЛ, 0,058мол, 1,11 экв), затем следовало добавление дициклогексилкарбодиимида (11,97г, 0,058мол, 1,11 экв), растворенного в тетрагидрофуране (бОмЛ) Смесь перемешивалась в течение двух дней при 25°С в атмосфере азота Завершение реакции определяли с использованием ВЭЖХ Пастообразный осадок охлаждали при 0°С в течение двух часов и затем фильтровали Затем фильтрат выпаривали до состояния, близкого к сухому, при пониженном давлении (10 Торр, 1333,22Па) при 40°С Масло растворяли в 250мЛ этилацетата Органический слой промывался 250мЛ 0,5М буферным раствором СОз 2 /НСО3 с рН9,8 рН доводили до 3,5-9,8 Органический раствор промывался 250мЛ насыщенного раствора соли Органический слой сушили над Na2SO4, охлаждали при перемешивании до -20°С и оставляли стоять без перемешивания при -20°С на ночь (16 часов) Осажденный DCU удалялся фильтрованием Этилацетат испаряли при пониженном давлении (10 Торр, 1333,22Па), получая 25,0г (95%) аморфного твердого вещества ИКС (СНзСІз) 2897, 1740, 1659 см \ 1 Н ЯМР (300МГц, CDCI3) 5 8,94 (дд, 1Н, I = 2,0Гц), 8,40 (уш с, 1Н), 7,20-6,93 (м 4Н) 6,60-6,50 (м, ЗН), 4,04, 3,95 (м, 2Н), 3,80-3,65 (м, 2Н), 3,16 (дд, 1Н, I = 13,8Гц, I = 4,4Гц), 2,69 (уш д , 1Н), I = 10,2Гц, 2,63-2,41 (м 4Н) 2,40-2,15 (м 4Н), 1 841 71 (м 2Н), 1,42 (ушд, 1Н, I = 12,4Гц), 1,10 (с ЗН), 0 77 (д 6Н, I = 6,9Гц), 0,57 (д, ЗН, 1 6,9Гц), = 1 Н ЯМР (300 МГц, ДМСО-de) 5 9,17 (уш с, 1Н), 840 (ушт, 1Н, I = 2,0Гц), 7,26-7,14 (м, 4Н) 7,04 (т, 1Н, I = 7,8Гц) 6,63 (м, 2Н), 6,52 (д, 1Н, I = 8,1 Гц) 3,81-3,79 (м, 4Н) 2,90-2,43 (м, 6Н), 2,37 (д, 1Н, I = 12,4Гц) 2,33-2,03 (м, 2Н), 1,95-1,65 (м, 2н), 1,43 (д, 1Н, I = 12,4Гц), 1,17 (с, ЗН), 0,85 (д, 6Н, I = 6,7Гц), 0,65 (д, ЗН, I = 6,8Гц), I3 C ЯМР (75,4МГц, ДМСО-de) 5 174,03, 169,78, 157,05, 151,71, 140,08, 128,80, 128,71, 125,77, 115,93, 112,36, 112,06, 69,96, 59,73, 54,95, 49,87, 27 52577 28 44,24, 40,59, 38,03, 37,83, 35,61, 29,93, 27,19, Чистота 96,2% (aS,3R,4R)-4-(327,08, 18,72, 15,79, гидроксифенил)-3,4-диметил-а-(фенилметил)-1пиперидинпропионовая кислота, метиловый эфир MC (FD) m/z 481 (M+), гидрохлорид, 2,9% (aR,3R,4R)-4-(3[a]25589 + 57,23°, [а]25зб5 + 177° (MeOH, c=1,01), УФ (MeOH) 274,4нм (є= 2093), 202,8нм гидроксифенил)-3,4-диметил-а-(фенилметил)-1пиперидинпропионовая кислота, метиловый эфир Анализ C29H40N2O4 монохлорид и 0,7% (aS,3R,4R)-4-(3Рассчитано С 72,47, Н 8,39, N 5,83 гидроксифенил)-3,4-диметил-а-(фенилметил)-1Найдено С 72,49, Н 8,59, N 5,63 пиперидинпропионовая кислота моногидрат Пример2 (ВЭЖХ поверх %) (аЗ,ЗР,4Р)-4-(3-Гидроксифенил)-3,4-диметила-(фенилметил)-1-пиперидинпропионовая кислот пл 230-232°С (разл ), та, метиловый эфир гидрохлорид ИКС (КВг) 3174, 1732, 1620, 1586, 1276, 785, 749, 706 см 1 Круглодонная колба продувалась азотом и за1 гружалась ТГФ (1 ООмЛ) и 2М раствором LDA Н-ЯМР (ДМСО-de) [0,78 (д, 0,85 х ЗН, I = (17,6мЛ, 35,18ммол, 2,05экв) Раствор охлаждался 7,2Гц) и 1,02 (д, 0,15 х ЗН, I = 7,2Гц), диастереодо -30°С и к нему добавлялся раствор соединения мерные соли], [1,28 (с, 0,15 х ЗН), 1,34 (с, 0,85 х препарата 1 (15,24г, 17,6ммол, 1,0экв, 32,8вес % в ЗН), диастереоизомерные соли], 1,76 (уш д , 1Н), ТГФ) - в течение 20 минут при поддержании тем2,10-2,43 (м, 2Н), 2,75-3,65 (м, 12Н), 6,60-6,90 (м, пературы между -26° и -28°С ЗН), 7,11 (т, 1Н, I = 7,8Гц), 7,15-7,35 (м, 5Н), 9,43 (ушс, 1Н), 9,75 (уш с, 1Н) После перемешивания втечение 15 минут при -25°С медленно добавлялся бензилбромид (5,81 г, MC (FD) m/z 381 (100%, M-HCI) 34,32ммол, 2,0экв) при поддержании температуАнализ C24H32CINO3 ры между -17° и -20°С Реакционная смесь переРассчитано С 68,97, Н 7,72, N 3,35, CI 8,48 мешивалась в течение трех часов при температуНайдено С 68,27, Н 7,84, N 3,42, CI 8,38 ре от -15° до -20°С Отношение ((aS,3R,4R)-4-(3ПримерЗ гидроксифенил)-3,4-диметил-а-(фенилметил)-1(+)-(аЗ,ЗК4[?)-4-(3-гидроксифенил)-3,4пропионовая кислота, метиловый диметил-а-(фенилметил)-1эфир/(аР,ЗР,4Р)-4-(3-гидроксилфенил)-3,4пиперидинпропионовая кислота, моногидрат диметил-1-пиперидин пропионовая кислота, метиВ круглодонную колбу помещалась дистиллиловый эфир составляло 97/3 рованная вода (230мЛ) вместе с 50вес % раствора гидроокиси натрия (20,02г, 250ммол, 4,2экв) В Реакционная смесь резко охлаждалась 1N HCI колбу добавлялась одна порция продукта приме(22мЛ, 22ммол) рН регулировалось от 10,6 до 9,5 ра2 (25,Ог, бОммол, 1экв) Смесь перемешивалась с помощью 12N HCI (2,ЗмЛ), после чего низкотемпри комнатой температуре и фильтровалась пературная баня удалялась Добавлялся гептан Фильтровальная бумага промывалась ЗЗмЛ 1N (50мЛ) и слои разделялись К органическому слою раствора гидроокиси натрия Раствор переносилдобавлялся метанол (25мЛ) и раствор охлаждался в круглодонную колбу, пригодную для вакуумся К нему добавлялась безводная HCI (1,3) при ной перегонки К раствору добавлялся метанол поддержании температуры ниже 5°С - до тех пор, (240мЛ) рН раствора доводили до 6,0, используя пока смесь не становилась кислой В процессе концентрированную хлористоводородную кислоту добавления осаждалась соль хлористоводород(32,14г) Метанол удаляли при пониженном давной кислоты Смесь подвергалась концентрировалении (100-20мм ртст) и температуре (45-50°С) нию до конечного веса 32,58г Затем к масляному Метанол испаряли до тех пор, пока вес конценконцентрату добавлялся метанол (ЗбмЛ) и через трата не достиг приблизительно 31 Зг Пастообнесколько минут образовался осадок Смесь перазный осадок перемешивался втечение четырех ремешивали в течение ночи при комнатной темчасов рН раствора регулировали до 6,0, и затем пературе осадок охлаждали в течение 1,5 часов при 0-5°С После охлаждения до 0°С втечение 1,25 часа Нужный продукт отфильтровывали и три раза осадок фильтровали, колбу промывали ЮмЛ промывали 50мЛ дистиллированной воды Затем фильтрата а отжатый осадок промывали холодпродукт сушили Нужный моногидратный продукт ным метанолом (ЮмЛ) Твердое вещество высувыделяли в виде белого гранулированного твершивали с получением 2,93г (выход 40,9%) белого дого вещества весом 21,Зге выходом 92% порошка Анализ, проведенный с применением ВЭЖХ, т п л 178-180°С (разлож) 1 показал, что продукт представлял собой смесь Н-ЯМР (300МГц, ДМСО) 5 0,64 (д, ЗН, I 86 14 стереоизомеров =6,9Гц), 1,19 (с, ЗН), 1,51 (д 1Н, I = 13,1 Гц), 1,972,00 (м, 1Н), 2,11 (тд, 1Н, 1 3,6Гц, 12,7Гц), 2,34= Неочищенная хлористоводородная соль 2,95 (м, 9Н), 6,54 (д, 1Н, I = 8,1 Гц), 6,66 (м, 2Н), (2,75г) помещалась в метанол (13,75мЛ) и полу7,06 (т, 1Н, I = 7,9Гц), 7,14-7,28 (м, 5Н), 9,22 ченный пастообразный продукт нагревался при (ушс,1Н), дефлегмировании в течение двух часов Смесь 13 охлаждалась до температуры, приблизительно С ЯМР (75,5МГц, ДМСО) 5 15,5, 26,9, 29,5, 0°С Осадок фильтровался, колба промывалась 35,2, 37,5, 37,7, 42,7, 49,7, 53,7, 58,8, 112,2, 112,3, фильтратом, а отжатый осадок промывали холод115,9, 126,0, 128,2, 128,7, 128,9, 139,4, 151,2, 157, ным метанолом (1,5мЛ) Продукт сушили с полу175,1 чением 2,32г белого твердого продукта (выход УФ (МеОН) Атах 203, є 17 860, 275, є 2356 84,4%) Общий выход 34,5% (алкилирование и MC (FD) m/z 368 выделение осадка) 29 52577 ИКС (КВг) 3360, 3272, 2967, 1622, 1585, 1363, 1 844 см 20 [а] зб5 304° (с 1,01, МеОН), KF=4,07% (Подсчитано для моногидрата 4,70%) Анализ C23H31NO4 Рассчитано С 71,66, Н 8,10, N 3,63 Найдено С 72,29, Н 8,10, N 3,71 Пример4 (23,ЗР,4Р)[[2-[[4-(3-гидроксифенил)-3,4диметил-1-пиперидинил]метил]-1-оксо-3фенилпропил]-амино] уксусная кислота, 2метилпропиловый эфир п ол утором ал атная соль (1 15) Соединение примераї (2,5г, 5,2ммол 1экв) растворялось в 50мЛ этилацетата К смеси добавлялась L-яблочная кислота (1,03г, 7,8ммол, 1,5экв) После расиворения L-яблочной кислотой при перемешивании раствор нагревали до 70°С и добавляли 4мЛ ацетона Раствор кристаллизовался Продукт выделяли фильтрацией Фильтрат в виде спекшегося осадка промывали этилацетатом Соль сушили до остаточного уровня этилацетата 1% Указанное в заглавии соединение выделяли в виде белого кристаллического вещества Образец анализировали с применением метода дифракции рентгеновских лучей Т пл 94-95°С ИКС (КВг) 3346,92, 2972,68, 1741,94, 1601,12 см \ 1 НЯМР(300МГц, flCMO-d6)5 9,70 (уш с, 1Н), 8,47 (т, 1Н, I = 1,9Гц), 7,27-7,13 (м, 4Н), 7,06 (т, 1Н, I = 7,9Гц), 6,67 (д, 1Н, I = 8,0Гц), 6,63 (с, 1Н), 6,53 (дд, 1Н, 8Гц, I = 1,7Гц) 4,18 (т, 1,5Н, I = 5,8Гц), 3,82-3,78 (м, ЗН), 3,33-1,8 (м, 16Н), 1,48 (уш д, 1Н, 1 13,0 Гц), 1,18 (с, ЗН), 0,85 (д, 6Н, I = 6,7Гц), 0,64 = (д, ЗН, I = 6,9Гц) 13 С ЯМР (75,4 МГц, ДМСО - d6) 5 175,63, 175,42, 171,44, 158,66, 138,63, 138,60, 130,50, 130,23, 129,66, 128,02, 114,07 114,05, 114,01, 113,94, МС (FD) m/z 481 (М+), УФ (МеОН) 272,8нм (є = 1797), 202,4нм (є = 20576), Анализ C70H98N4O23 Рассчитано С 61,65, Н 7,38, N4,10, 0 26,98 Найдено С 61,40, Н 7,23, N 4,1, О 26,66 Пример5 (2S, 3R, 4Р)[[2-[[4-(3-гидроксифенил)-3,4диметил-1-пиперидил]метил]-1-оксо-2фенилпропил]амино] уксусная кислота, дигидрат Раствор соединения примераї (12,5г, 0,026мол, 1,0экв) в 315мЛ ЗА этанола загружался в круглодонную колбу К смеси добавлялась вода (74,0мЛ) По каплям в течение 10-15 минут добавлялся водный раствор гидроокиси натрия (1,0М), (0,77мол, З.Оэкв) при температуре 25-30°С Раствор перемешивался и затем фильтровался рН раствора доводилось с 12,5 до 6,0 добавлением концентрированной хлористоводородной кислоты Раствор затравливали и в течение 10-15 минут начинала осаждаться (2S,3R,4R)[[2-[[4-(3гидроксифенил)-3,4-диметил-1-пиперидил]метил]1-оксо-2-фенилпропил]амино] уксусная кислота Кристаллизация осуществлялась при перемешивания в течение двух часов при 25°С и затем (23^^)-[[2-[[4-(3-гидроксифенил)-3,4-диметил 30 1-пиперидил]метил]-1-оксо-2-фенилпропил]амино] уксусная кислота, фильтровалась при применении легкого отсасывания к влажному осадку Кристаллы разбавлялись бОмЛ воды и отфильтровывалить при отсасывании до образования твердого осадка Кристаллы высушивались в течение ночи (16 часов) на воздухе при 25°С и относительной влажности 33% при прокачивании воздуха через продукт на фильтровальной воронке и легком отсасывании Озаглавленный продукт выделялся с 85%-ным выходом (10,2г) из (aS,3R,4R)-4-(3гидроксифенил)-3,4-диметил-а-(фенилметил)-1пиперидин пропионовой кислоты моногидрата в виде белого кристаллического вещества с четкой точкой плавления при 208°С Образец анализировался с применением метода дифракции рентгеновских лучей ИКС (КВг) 3419, 3204, 3028, 1684, 1591см \ 1 Н ЯМР (300МГц, de) 5 9,18 (уш с, 1Н), 8,34 (т, 1Н, I = 5,5Гц), 7,26-7,12 (м, 6Н), 7,05 (т, 1Н, I = 7,9Гц) 6,67 (д, 1Н, I = 8,0Гц), 6,63 (с 1Н), 6,52 (дд, 1Н, I = 8 ОГц, I = 1,8Гц), 3,65 (д, 2Н, I = 5,7Гц), 2,892,10 (м, 14Н), 1,91 (ушд, 1Н, I = 6,7Гц), 1,18 (с, ЗН), 0,64 (д, ЗН, I = 6,9Гц), I3 C ЯМР (75,4МГц, ДМСО-de) 5 173,54 71,30, 157,05, 151,28, 139,83, 148,83, 128,73, 128,05, 125,82, 115,97, 112,14, 59,62, 54,59, 49,92, 43,75, 41,12, 39,95, 39,67, 39,39, 39,12, 38,84, 37,80, 37,73,35,42,29,68,27,04 15,54, МС (FD) m/z 425 М+ - 2Н2О) УФ (МеОН) 275,0 (є = 2246), 202,6 (є = 22709,4), [а]25зб5= -1,18 (МеОН, с=1,0) Анализ C25H36N2O6 Рассчитано С 65,20, Н 7,88, N 6,08, О 20,84 Найдено С 64,96, Н 7,74, N 6,10, О 20,82 Примерб (2S,3R,4R)[[2-[[4-(3-rHflpoKCH4>eHiin)-3,4диметил-1-пиперидинил]-метил]-1-оксо-3фенилпропил]амино] уксусная кислота, дигидрат Этанол (2400мЛ, ЗА) и соединение примера 4 (146г с 5% ЕЮ Ас, 138,7г чистого вещества (0,203мол, 1,0экв, 0,085мольн) помещалась в круглодонную колбу Добавлялся 1,0М водный раствор гидроокиси натрия (1200мл, 1,2мол, 5,9экв) - по каплям в течение 20 минут при 2530°С Раствор перемешивался, затем фильтровался рН раствора доводилось с 12,96 до 6,00 добавлением концентрированной хлористоводородной кислоты Раствор затравляли и через 1015 минут начинала осаждаться (2S,3R,4R)[[2-[[4(3-гидроксифенил)-3,4-диметил-1-пиперидинил]метил]-1-оксо-3-фенилпропил]амино] уксусная кислота Кристаллизация происходила при перемешивании в течение двух часов Пастообразный осадок охлаждался до 0°С и перемешивался Продукт в виде (2S,3R,4R)[[2-[[4-(3-rnflpoKCHфенил)-3,4-диметил-1-пиперидинил]-метил]-1оксо-3-фенилпропил]амино] уксусной кислоты, отфильтровывался при легком отсасывании влажного осадка Кристаллы соединялись с 500мЛ воды при 25°С при перемешивании, затем следовало легкое отсасывание, промывание 500мЛ воды и фильтрование с отсасыванием до твердого осадка Кристаллы сушились до дигидрата на воздухе при 35%-ой относительное влажности при 25°С и продувании воздуха через продукт на 32 31 52577 фильтровальной воронке при слабом отсасыва112,9, 112,3, 53,9, 57,1, 70,2, 48,1, 46,4, 40,8, 37,3, нии Озаглавленное соединение выделялось с 36,9, 27,3, 27,0, 26,5, 18,8, 15,1, выходом > 93% (88г) в виде белах кристаллов с УФ (МеОН) 274, (є = 2738), 202,2 (є =28413), + четкой точкой плавления при 208°С МС (FD) 481 (М -НСІ-Н2О) Анализ C25H36N2O6 Рассчитано С 65,20, Н 7,88, N 6,08, О 20,84 Найдено С 65,38, Н 7,87, N 6,25, О 20,90 Пример7 (23,ЗР,4Р)[[2-[[4-(3-гидроксифенил)-3,4диметил-1-пиперидинил]метил]-1-оксо-3фенилпропил]-амино] уксусная кислота, 2метилпропиловый эфир гидрохлорид ацетон моносольват 6г образца соединения примераї растворяли в 60,0мЛ сухого ацетона Порция в 0,45г (0,98экв) газообразного хлористого водорода, растворенная в ЗОмЛ сухого ацетона, по каплям при 25°С добавлялась к первому раствору Газообразный НСІ в ацетоне добавлялся по каплям, пока рН не достигало 3 Когда рН раствора достигло 3,0, добавлялась вторая аликвотная часть 1,0мЛ соединения примераї при такой же концентрации, как в исходном растворе Образовался осадок Реакционная смесь перемешивалась при 25°С в течение часа, затем охлаждалась до 0°С Затем перемешивалась при 0°С еще два часа Желаемый продукт (23,ЗР,4Р)[[2-[[4-(3-гидроксифенил)-3,4диметил-1-пиперидинил]метил]-1-оксо-3фенилпропил]-амино] уксусная кислота, 2метилпропиловый эфир -гидрохлоридная соль отфильтровывался фильтрованием под давлением с использованием азота (2S,3R,4R)[[2-[[4-(3гидроксифенил)-3,4-диметил-1 пиперидинил]метил]-1-оксо-3-фенилпропил]амино] уксусная кислота, 2-метилпропиловый эфир - гидрохлоридная соль -сушилась потоком азота над фильтратом с образованием моносольвата ацетона Ацетоновый моносольват характеризовался с помощью капиллярного газохроматографического анализа, который выявил 9,3-9,97% (по весу) ацетона (теоретическое значение 10 процентов) Продукт характеризовался удалением молекулы ацетона из сольвата Гидрохлорид ацетонового моносольвата сушился далее в вакуумной печи в течение 2-8 дней при 50°С Образование гидрохлорид моногидрата было затронуто распространением кристаллов на большую поверхность при 25°С при 40%-ой относительной влажности в течение 2 дней Выход составлял > 85% с чистотой около 99,3% по оценке ВЭЖХ с обращенными фазами Тпл 70-75°С, ИКС (КВг) 3217,7, 3063,4, 2965,0, 1749,7, 1671,5 1 Н ЯМР (300МГц, ДМСО-de) 5 9,45 (уш с, 1Н), 9,37 (с, 1Н), 8,94 (т, 0,85Н, І = 1,5Гц) 8,92 (т, 0,15Н, I = 1,5Гц), 7,28-7,20 (м, Н), 7,09 (т, 1Н, I = 7,8Гц), 6,67-6,56 (м, ЗН), 3,83-3,76 (м, 4Н), 3,47-3,10 (м, 5Н), 2,83 (д кв , 2Н, I = 18,0гц, I = 5,5гц, I = 2,0Гц), 2,7-2,0 (м, 5Н), 1,82 (септ, 1Н, I = 6,7Гц), 1,70 (д, 1Н, 1= 12,0Гц), 1,29 (с, 0,85Н), 1,24 (с, 0,15Н), 0,99 (д, 0,45Н, I = 7,4Гц), 0,85 (д, 6Н, I = 6,6Гц), 0,71 (д, 2,55Н, І = 7,ЗГц), ІЗ С ЯМР (75,4Гц, ДМСО-de) 5 172,7, 169,8 157,4, 149,4, 129,3, 128,3, 121,6, 118,6, 115,7, Анализ C29H41N2O4-H2O Рассчитано С 65,09, Н 8,10, N 5,23, О 14,95, СІ 6,63 Найдено С 66,06, Н 7,92, N 5,27, О 15,19 СІ 6,92 Пример8 (аЗ,ЗР,4Р)-4-(3-гидроксифенил)-3,4-диметила-(фенилметил)-1-пиперидинпропионовая кислота, этиловый эфир гидрохлорид Образец (+)-3-(3,4-диметил-4пиперидинил)фенола (50,Ог, 243,5ммол, 1 эквивалент) помещалсяв круглодонную колбу Добавлялись тетрагидрофуран (1Л) и этилацетет (ЗЗ.ОмЛ, 304,4ммол, 1,25 эквивалентов) и гетерогенная реакционная смесь перемешивалась в течение нескольких дней при комнатной температуре Реакционная смесь фильтровалась через диатомовую землю и прозрачный раствор выделял вязкое янтарное масло весом 75,Ог Порция аминоэфира (1,16г, 3,80ммол, 1,0 эквивалент) растворялась в ЮмЛ тетрагидрофурана (ТГФ) и добавлялась при -75°С к раствору литий диизопропиламида (3,90мЛ, 7,80ммол, 2,05 эквивалентов) в ТГФ (20мЛ) Дополнительно выдерживали смесь приблизительно пять минут Затем, пастообразный осадок перемешивался при -70°С в течение 15 минут и добавлялся бензилбромид (0,47мЛ, 3,99ммол, 1,05 эквивалентов) Реакционной смеси дали нагреться до -25 - -30°С и перемешивали ее в течение 3 часов Затем, реакционная смесь быстро заливается ЮмЛ насыщенного раствора хлорида аммония, ЮмЛ НгО и 20мЛ этилацетата Водный слой отделялся Органический слой промывали насыщенным солевым раствором Затем органический слой сушили над MgSO4 Смесь фильтровали, и образующийся раствор подвергали выпариванию на ротационном испарителе с получением желтого масла весом 1,80г Смесь продукта и исходного вещества затем подвергали флеш-хроматографированию смесью этилацетата и гексана с выделением 1,07г (71%) этилового эфира Приведенный выше этиловый эфир (14,8г, 37,4ммол) растворяли затем в 150мЛ этанола В раствор пропускали безводный хлористый водород, а этанол удаляли испарением на ротационном испарителе Затем твердое вещество растирали в порошок с 50мЛ этилацетата и фильтровали Твердое вещество сушили в течение ночи при 30°С с выделением 12,25г гидрохлорида (76%, точка плавления 179-181 °С) Соотношение диастереомеров составляло 49% aS,3R,4R (нужный диастереомер) и 5 1 % aR,3R,4R (ненужный диастереомер) Гидрохлоридная соль (1,02г) смешивалась с 5мЛ этанола и нагревалась с обратным холодильником в течение 3 часов Смеси давали охлаждаться до комнатной температуры и перемешивали Затем смесь перемешивали в течение часа при 0°С и отфильтровывали Соль сушили в течение ночи при 40°С Выделяли белое твердое ве 33 52577 щество весом 0,42г (47%) Соотношения диастереомеров было 76% aS,3R,4R и 24% aR,3R,4R Гидрохлоридная соль (0,42г) смешивалась с 6мл этанола и нагревалась с обратным холодильником в течение 2 часов, после чего ее охлаждали до комнатной температуры и перемешивали Пастообразный осадок охлаждали до 0°С в течение часа и фильтровали Твердое вещество сушили с получением палочки соли весом 0,31г (74%) Соотношение диастереомеров составляло 92% aS,3R,4R и 8% aR,3R,4R 34 Часть соли (0,24г) смешивали в третий раз с 2,5мЛ этанола Смесь нагревали в течение трех часов с обратным холодильником, затем давали ей охладиться до комнатной температуры и перемешивали Пастообразный осадок охлаждали до 0°С в течение 1 часа и затем фильтровали Твердое вещество сушили Диастереомерно чистая (98% aS,3R,4R) гидрохлоридная соль этилового эфира имела вес 0,23г (96%) ТОВ "Міжнародний науковий комітет" вул Артема, 77, м Київ, 04050, Україна (044)236-47-24
ДивитисяДодаткова інформація
Назва патенту англійськоюCrystalline piperidinyl-n-alkylcarboxylates (variants), a process for production thereof (variants) the pharmaceutical composition and intermediate products
Автори англійськоюFrank Scott Alan
Назва патенту російськоюКристаллические пиперидинил-n-алкилкарбоксилаты (варианты), способ их получения (варианты), фармацевтическая композиция и промежуточные продукты
Автори російськоюФранк Скотт Алан
МПК / Мітки
МПК: A61P 1/00, A61K 31/451, C07D 211/22, A61K 31/445
Мітки: фармацевтична, піперидиніл-n-алкілкарбоксилати, композиція, проміжні, продукти, варіанти, кристалічні, спосіб, одержання
Код посилання
<a href="https://ua.patents.su/17-52577-kristalichni-piperidinil-n-alkilkarboksilati-varianti-sposib-kh-oderzhannya-varianti-farmacevtichna-kompoziciya-ta-promizhni-produkti.html" target="_blank" rel="follow" title="База патентів України">Кристалічні піперидиніл-n-алкілкарбоксилати (варіанти), спосіб їх одержання (варіанти), фармацевтична композиція та проміжні продукти</a>
Індолінові похідні, а також їх фармацевтично прийнятні солі, що є антагоністами вазопресинових v1-рецепторів, спосіб їх одержання, проміжні продукти і фармацевтична композиція

Номер патенту: 27238
Опубліковано: 15.08.2000
Автори: Нісато Діно, Тоннер Бернар, Плузан Клод, Серадей-Легаль Клодін, Ваньон Жан
МПК: A61P 9/10, A61K 31/4427, A61K 31/55, A61K 31/40, C07D 417/06, A61K 31/445, C07C 317/30, A61P 15/00, A61K 31/425, A61P 25/04, C07D 403/06, C07D 401/12, A61P 9/12, C07D 209/42, C07C 311/08, C07D 401/06, A61P 7/02, A61K 31/404, A61P 9/08, A61P 25/18, A61K 31/495, A61K 31/403, A61P 9/00, A61P 1/04, A61K 31/535, A61K 31/54, A61P 13/02
Мітки: продукти, індолінові, одержання, проміжні, вазопресинових, прийнятні, v1-рецепторів, похідні, також, антагоністами, фармацевтично, солі, композиція, фармацевтична, спосіб
Текст:
...пл. ° С) (или их точкой кипения Т кип ) и/или их ЯМР спектром, снятым при 200 МГц в ДМСО и/или показателем вращения плоскости Бполяризации (альфаО), измеренной при 25 °С (если нет других указаний). Измеренное значение вращения плоскости поляризации зависит от количества остаточного растворителя, присутствующего в приготовленном продукте. За исключением особо указанных случаев обозначение "цисизомер" или "трансизомер" означает, что выделенное...
Похідні бензотіофену, спосіб їх одержання (варіанти), проміжні сполуки, фармацевтична композиція

Номер патенту: 44710
Опубліковано: 15.03.2002
Автори: Трашер Кеннет Джефф, Палковіч Алан Девід
МПК: A61P 5/00, A61K 31/4535, A61P 19/10, A61K 31/4025, A61K 31/565, A61K 31/381, A61P 43/00, A61P 13/02, C07D 333/62, A61K 31/55, A61P 15/00, A61K 31/38, A61K 31/57, C07D 333/64
Мітки: фармацевтична, проміжні, сполуки, одержання, похідні, спосіб, композиція, варіанти, бензотіофену
Формула / Реферат:
1. Бензотиофен формулы I: гдеR1 представляет -Н, -ОН, -О(С1-С4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), или –OSO2(С2-С6алкил);R2 представляет -Н, -ОН, -O(С1-С4алкил), -ОСОС6Н5, -ОСО(С1-С4алкил), -OSO2(С2-С6алкил) или галоид;R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино, диизопропиламино или...
Похідні n-алкіленпіперидинів як антагоністи рецептора нейрокініну, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 26894
Опубліковано: 29.12.1999
Автори: Пруаєтто Венсанзо, Мартінез Серж, ван Броєкк Дідьє, Емонд-Альт Ксавьє
МПК: C07D 401/12, A61K 31/4465, A61K 31/4427, A61P 43/00, C07D 401/06, C07D 409/12, C07D 211/54, C07D 211/58, A61K 31/445, A61K 31/454, A61K 31/4433, C07D 403/12, A61K 31/44, C07D 211/46, A61K 31/4545, A61K 31/4468
Мітки: композиція, спосіб, n-алкіленпіперидинів, сполуки, фармацевтична, рецептора, похідні, нейрокініну, антагоністи, проміжні, одержання
Текст:
...где Alk является (С,-Сэ)алкильной группой, R представляет собой атом водорода 40 или С^-С^алкил, Z представляет собой фенил, незамещенный или моно- или дизамещенный галогеном или (С^-Сзіалкоксигруппой, наф 26894 7 тил, замещенный галогеном, фенилметильной группой, замещенной на фениле (С,С4}алкоксигруппой, * обозначает хиральный центр, в виде рацемата или оптически чистых изомеров, или их соли с минеральными или органическими кислотами,...
Похідні 2,3-бензодіазепіну, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 45358
Опубліковано: 15.04.2002
Автори: Лінг Іштван, Абрахам Гізелла, Берженьї Пал, Андраші Ференц, Хорват Каталін, Гал Мелінда, Сьолльоші Марта, Тарнава Іштван, Чузді Емеше, Моравчік Імре, Хаморі Тамаш, Шойом Шандор
МПК: A61P 43/00, A61K 31/551, A61P 25/00, C07D 243/02, A61P 25/28, A61P 25/08, A61K 31/55
Мітки: одержання, композиція, проміжні, 2,3-бензодіазепіну, похідні, сполуки, фармацевтична, спосіб
Формула / Реферат:
1. Производные 2,3-бензодиазепина формулы (I):(I),гдеR1 и R2 означают, независимо друг от друга, водород, галоген, C1-4 алкильную группу, C1-4 алкоксигруппу, нитрогруппу, трифторметильную группу или группу формулы -NR8R9, где R8 и R9 означают, независимо друг от друга, водород, С1-4 алкильную группу или группу формулы -COR10, гдеR10 означает водород, C1-6 алкильную группу, которая может быть замещена, С6-10...
Сполуки,які містять біциклічне конденсоване ядро, спосіб їх одержання, проміжні сполуки, спосіб їх одержання (варіанти) та фармацевтична композиція для інгібування перетворення ферменту ангіотензину та нейтраль

Номер патенту: 39924
Опубліковано: 16.07.2001
Автори: Робл Джефрі А., Годфрей Джоллі Д., Кронентал Девід Р.
МПК: A61P 9/04, A61P 9/12, A61P 43/00, C07K 5/078, C07C 323/59, C07C 327/00, A61K 31/535, A61K 31/553, A61K 31/55, C07C 317/50, A61K 31/542, C07C 319/00, A61K 38/00, A61K 31/554, A61K 31/54, C07D 513/04, C07D 498/04
Мітки: інгібування, сполуки, композиція, ядро, проміжні, спосіб, ферменту, ангіотензину, біциклічне, варіанти, сполуки,які, містять, перетворення, нейтраль, одержання, фармацевтична, конденсоване
Формула / Реферат:
1. Соединения, содержащие конденсированное бициклическое ядро формулы,или их фармацевтически приемлемая соль, где А представляетили,Χ представляет О или S,r1 и R12 независимо...
Попередній патент: Система захисту від корозії
Наступний патент: Пристрій для обробки повітря в приміщеннях
Випадковий патент: Перекривний клапан для контейнера-цистерни