Ненуклеозидні інгібітори зворотної транскриптази та фармацевтична композиція на їх основі

















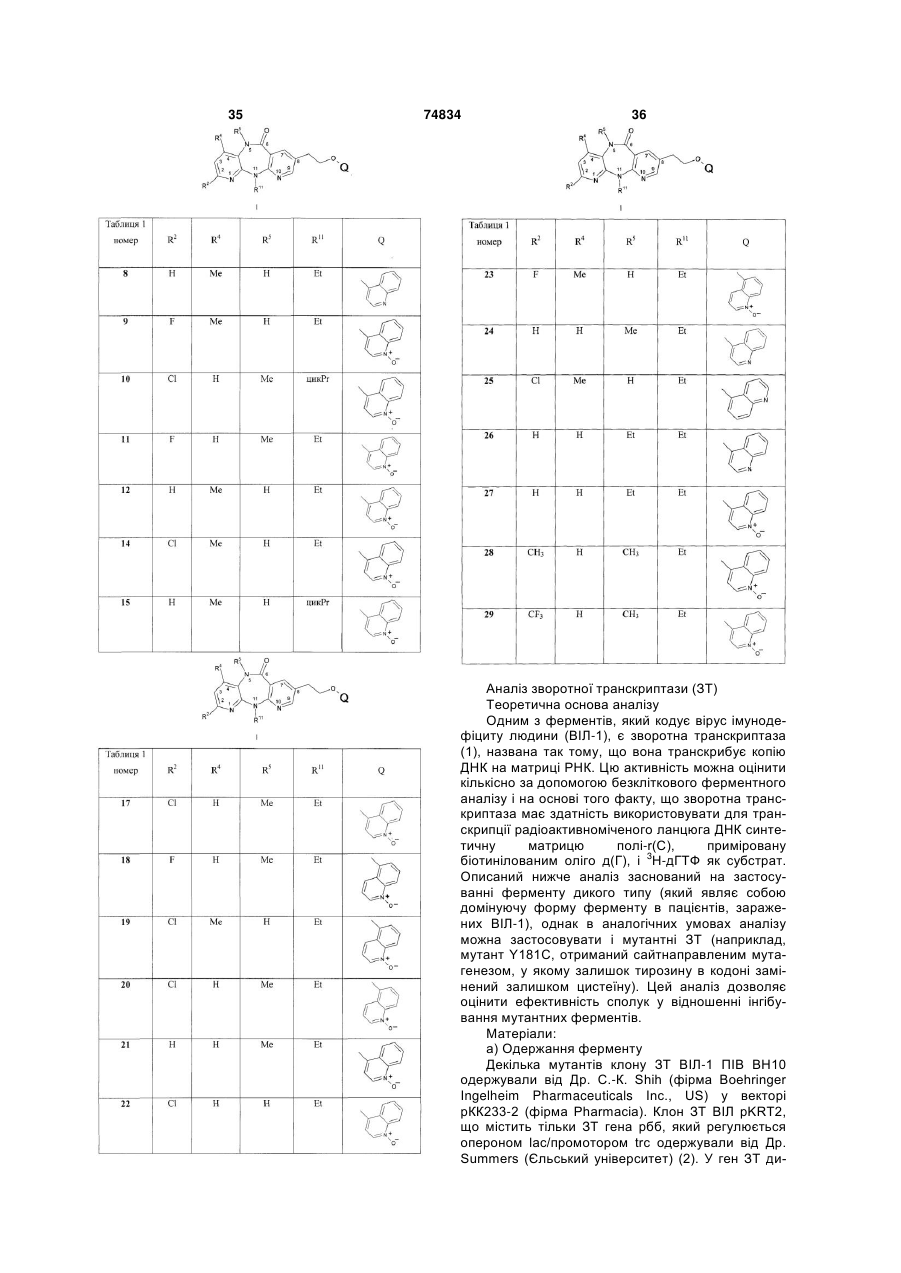

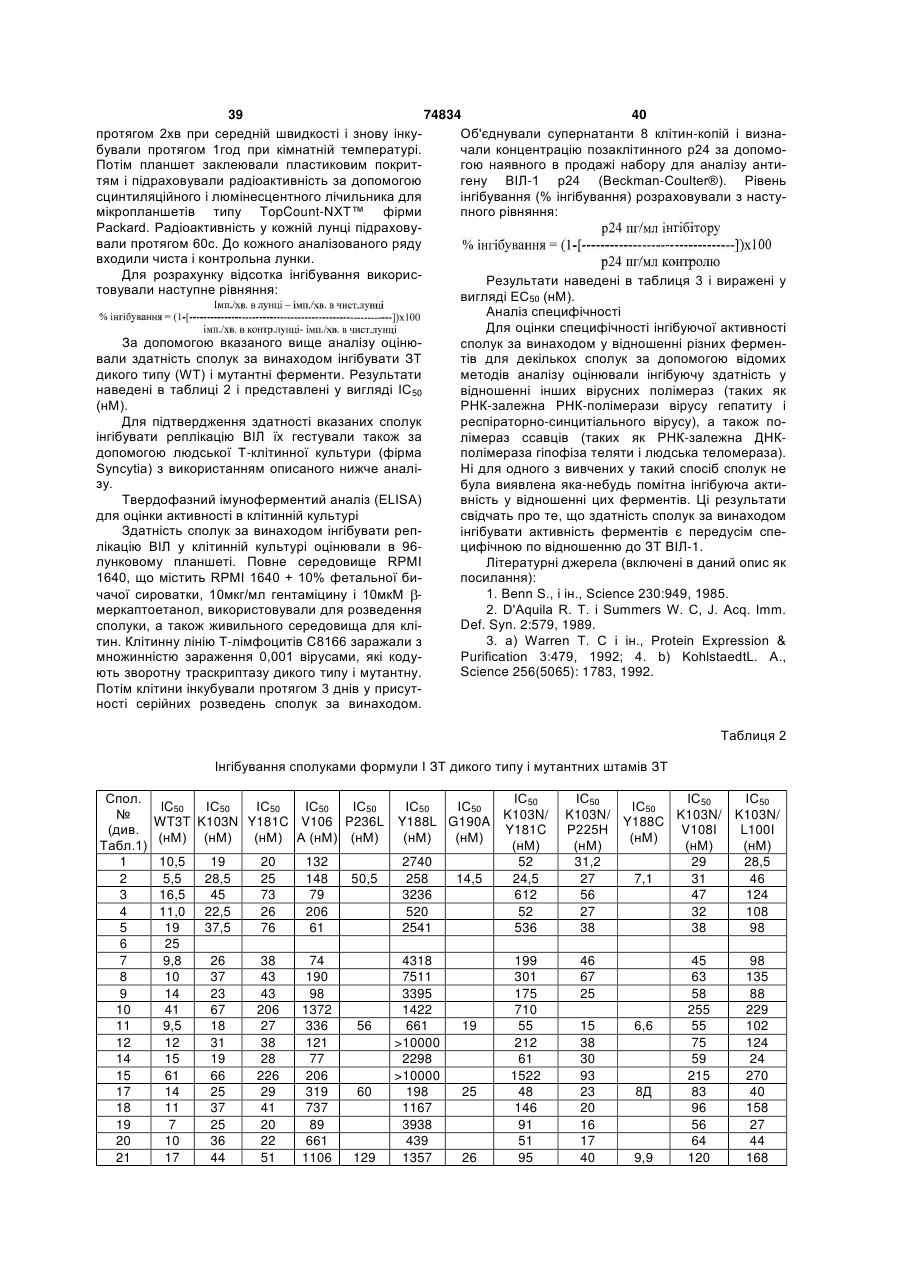
Формула / Реферат
1. Сполука загальної формули I:
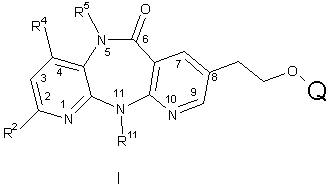 ,
,
де
R2 вибирають із групи, яка включає H, F, Cl, C1-С4алкіл, C3-С4циклоалкіл і CF3;
R4 означає H або Me;
R5 означає H, Me або Et за умови, що R4 і R5 обидва не означають Me, і, якщо R4 означає Me, то R5 не означає Et;
R11 означає Me, Et, циклопропіл, пропіл, ізопропіл або циклобутил; і
Q вибирають із групи, яка включає:
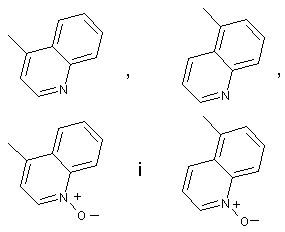 ;
;
або її фармацевтично прийнятна сіль.
2. Сполука за п. 1, де R11 означає Et або циклопропіл, або її фармацевтично прийнятна сіль.
3. Сполука за п. 1, де Q вибирають із групи, яка включає:
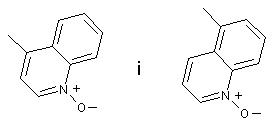 ;
;
або її фармацевтично прийнятна сіль.
4. Сполука за п. 1, де Q означає:
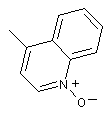 ;
;
або її фармацевтично прийнятна сіль.
5. Сполука за п. 1, де R5 означає Me,
або її фармацевтично прийнятна сіль.
6. Сполука за п. 1, де R11 означає Et, або її фармацевтично прийнятна сіль.
7. Сполука за п. 1, представлена формулою:
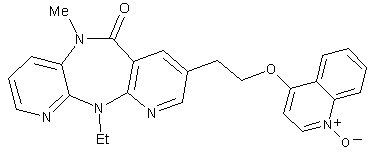 ;
;
або її фармацевтично прийнятна сіль.
8. Сполука за п. 1, представлена формулою:
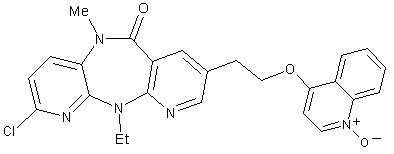 ;
;
або її фармацевтично прийнятна сіль.
9. Сполука за п. 1, представлена формулою:
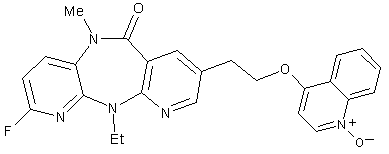 ;
;
або її фармацевтично прийнятна сіль.
10. Фармацевтична композиція, призначена для лікування або попередження ВІЛ-інфекції, яка містить сполуку формули І або її фармацевтично прийнятну сіль і фармацевтично прийнятний носій.
Текст
1. Сполука загальної формули I: _ ; або її фармацевтично прийнятна сіль. 5. Сполука за п. 1, де R5 означає Me, або її фармацевтично прийнятна сіль. 3 74834 4 6. Сполука за п. 1, де R11 означає Et, або її фарабо її фармацевтично прийнятна сіль. мацевтично прийнятна сіль. 9. Сполука за п. 1, представлена формулою: O Me 7. Сполука за п. 1, представлена формулою: O Me N N O O N N N F N N+ Et O _ ; або її фармацевтично прийнятна сіль.8. Сполука за п. 1, представлена формулою: O Me N N Et N N+ _ O ; або її фармацевтично прийнятна сіль. 10. Фармацевтична композиція, призначена для лікування або попередження ВІЛ-інфекції, яка містить сполуку формули І або її фармацевтично прийнятну сіль і фармацевтично прийнятний носій. O N Cl N Et N N+ O _ ; Винахід стосується нових сполук та їх фармацевтично прийнятних солей, їх застосування або індивідуально, або в сполученні з іншими терапевтичними агентами для лікування або профілактики ВІЛ-інфекції і фармацевтичних композиціям, які містять ці сполуки. Хвороба, відома як синдром набутого імунодефіциту (СНІД), викликається вірусом імунодефіциту людини (ВІЛ), насамперед штамом, відомим як ВІЛ-1. Для реплікації ВІЛ у клітині-хазяїні, інформація про вірусний геном повинна бути інтегрована в ДНК клітину-хазяїна. Однак ВІЛ являє собою ретровірус, тобто вірус, у якого генетична інформація зчитується з РНК. Внаслідок цього в циклі реплікації ВІЛ повинна бути присутня стадія транскрипції вірусного геному (РНК) з утворенням ДНК, яка є зворотною по відношенню до звичайного процесу. Транскрипція вірусної РНК з утворенням ДНК відбувається за допомогою ферменту, який тому називається зворотною транскриптазою (ЗТ). Віріон ВІЛ поряд з вірусною РНК включає копію ЗТ. Для зворотної транскриптази характерні три відомі ферментативні функції; вона діє як РНКзалежна ДНК-полімераза, як рибонуклеаза і як ДНК-залежна ДНК-полімераза. Діючи як РНКзалежна ДНК-полімераза, ЗТ транскрибує синтез копії одноланцюгової ДНК вірусної РНК. Діючи як рибонуклеаза, ЗТ розщеплює вихідну вірусну РНК і вивільняє ДНК, безпосередньо отриману на матриці вихідної РНК. І нарешті, діючи як ДНКзалежна ДНК-полімераза, ЗТ забезпечує одержання другого комплементарного ланцюга ДНК, використовуючи перший ланцюг ДНК як матрицю. Два ланцюги утворюють дволанцюгову ДНК, яка інтегрується в геном клітини-хазяїна за допомогою іншого ферменту, який називається інтегразою. Сполуки, що інгібують ферментативні активності зворотної транскриптази ВІЛ-1, можуть інгібувати реплікацію ВІЛ-1 у заражених клітинах. Такі сполуки можна застосовувати для попередження або лікування інфекції в людей, яка викликається ВІЛ-1, що продемонстровано при використанні відомих інгібіторів ЗТ, таких як 3'-азидо-3'дезокситимідин (AZT), 2',3'-дидезоксіінозин (ddl), 2',3'-дидезоксицитидин (ddC), d4T, ЗТС, невірапін, делавірдин, ефавіренз і абакавір, основних лікарських засобів, які в даний час дозволені для застосування при лікуванні СНІДу. Як і у випадку будь-якої антивірусної терапії, застосування інгібіторів ЗТ для лікування СНІДу зрештою приводить до зниження чутливості вірусу до даного лікарського засобу. Стійкість (знижена чутливість) до вказаних лікарських засобів є результатом мутацій, що торкаються сегменту зворотної транскриптази гена рої. До даного часу охарактеризовано декілька мутантних штамів ВІЛ, для яких встановлено, що стійкість до відомих терапевтичних агентів є результатом мутацій у гені ЗТ. Прикладами деяких з найбільш розповсюджених виявлених, пов'язаних з лікуванням мутацій є: мутація Y181C, що включає заміну тирозину (Y) у кодоні 181 на залишок цистеїну (С), і K103N, що включає заміну лізину (К) у положенні 103 на аспарагін (N). Інші мутанти, частота яких зростає в процесі лікування з використанням відомих антивірусних засобів, являють собою мутанти з однією мутацією V106А, G190A, Y188C і P236L; і мутанти з двома мутаціями K103N/Y181C, K103N/P225H, K103N/V108I і K103N/L100I. Оскільки лікування і попередження ВІЛінфекції з використанням антивірусних засобів продовжується, слід очікувати збільшення числа нових стійких штамів. Звідси зрозуміло, що існує потреба в розробці нових інгібіторів ЗТ, які відрізняються різним механізмом дії у відношенні різних мутантів. Сполуки, що мають трициклічні структури, які є інгібіторами ВІЛ-1, описані в патенті US No. 5366972. Інші інгібітори зворотної транскриптази ВІЛ-1 описані в Hargrave і ін., J. Med Chem., 34, 2231 (1991). 5 74834 6 У патенті US No. 5705499 як інгібітори ЗТ загає в тому, що пацієнту вводять інгібуючу ВІЛ кільпропоновані 8-арилалкіл- і 8-арилгетероалкіл-5,11кість сполуки формули І або її фармацевтично дигідро-6Н-дипіридо[3,2-В:2',3'-Е] [1,4] діазепіни. прийнятної солі. Встановлено, що наведені як приклад сполуки П'ятим об'єктом винаходу є фармацевтична мають певну активність у відношенні ЗТ ВІЛ-1 дикомпозиція, призначена для лікування або попекого типу і мутантної ЗТ, насамперед Y181C, але редження ВІЛ-інфекції, що містить сполуку форпри цьому вони менш ефективні у відношенні інмули І або її фармацевтично прийнятну сіль і фаших мутантів, які несуть одну мутацію, таких як рмацевтично прийнятний носій. K103N. Шостим об'єктом винаходу є спосіб одержання Встановлено, що сполуки за даним винаходом сполуки формули І або її фармацевтично прийнятє ефективними інгібіторами мутацій Y181C і ної солі. K103N, а також широкого спектру інших мутантів з Визначення однією мутацією і деяких відомих мутантів з поУ контексті даного опису поняття «С1-С4алкіл» двійною мутацією, таких як К103N/Y181C і означає алкільні радикали з прямим або розгалуK103N/P225H. женим ланцюгом, що містять від 1 до 4 атомів вугДаний винахід дозволяє подолати складності і лецю, і вони являють собою метил, етил, пропіл, недоліки прототипів за допомогою нових сполук, ізопропіл, бутил, втор-бутил і трет-бутил. які є ефективними інгібіторами штамів ВІЛ-1, що У контексті даного опису поняття «С3несуть одну і дві мутації ЗТ. С4циклоалкіл» означає насичені циклічні вуглевоПершим об'єктом винаходу є сполука загальдневі радикали, що містять 3-4 атоми вуглецю, і ної формули І: вони являють собою циклопропіл і циклобутил. Докладний опис переважних варіантів здійснення Переважним варіантом здійснення винаходу є сполуки формули І, у яких R2 переважно означає СІ, F або Н. Більш переважно R2 означає СІ або Н. Найбільш переважно R2 означає Н. Переважним варіантом здійснення винаходу є сполуки формули І, у яких R4 переважно означає Н. Іншим варіантом здійснення винаходу є сполуки формули І, у яких переважно R5 означає Me. Переважними сполуками за винаходом є споде 2 луки формули І, у яких R11 означає Et або циклопR вибирають із групи, яка включає Н, F, СІ, ропіл. Більш переважно R11 означає Et. С1-С4алкіл, С3-С4циклоалкіл і CF3; Переважним варіантом здійснення винаходу є R4 означає Η або Me; сполуки формули І, у яких Q переважно вибирають R5 означає Н, Me або Et за умови, що R4 і R5 із групи, яка включає: обидва не означають Me, і, якщо R4 означає Me, то R5 не означає Et; R11 означає Me, Et, циклопропіл, пропіл, ізопропіл або циклобутил; і Q вибирають із групи, яка включає: Більш переважно Q означає: або її фармацевтично прийнятна сіль. Другим об'єктом винаходу є інгібітор реплікації ВІЛ загальної формули І або його фармацевтично прийнятна сіль. Третім об'єктом винаходу є інгібітор зворотної транскриптази ВІЛ загальної формули І або його фармацевтично прийнятна сіль. Четвертим об'єктом винаходу є спосіб лікування або попередження ВІЛ-інфекції, який поля Іншими переважними варіантами здійснення винаходу є сполуки, вибрані з групи, яка включає: 7 74834 8 дами таких носіїв є вода, желатин, тальк, крохмаль, стеарат магнію, гуміарабік, рослинні олії, поліалкіленгліколі, вазелін і т.п. Фармацевтичні композиції можна готувати загальноприйнятим методом і отримані в результаті дозовані форми можуть являти собою тверді дозовані форми, наприклад, таблетки, драже, капсули і т.п., або рідкі дозовані форми, наприклад розчини, суспензії, емульсії і т.п. Фармацевтичні препарати можна піддавати загальноприйнятим фармацевтичним обробкам, таким як стерилізація. Крім того, фармацевтичні препарати можуть місСполуки за винаходом є ефективними інгібітотити звичайні ад'юванти, такі як консерванти, старами зворотної транскриптази дикого типу, вони білізатори, емульгатори, коригенти. змочувальні також мають здатність інгібувати, ферменти, наагенти, буфери, солі для зміни осмотичного тиску і приклад, які несуть одну мутацію, такі як Y181C, т.п. Тверді носії, який можна використовувати, явK103N, V106A, G190A, Y188C і P236L. Сполуки ляють собою, наприклад, крохмаль, лактозу, маніт, інгібують також ферменти, які несуть дві мутації, метилцелюлозу, мікрокристалічну целюлозу, такі як K103N/Y181C, K103N/P225H, K103N/V108I і тальк, діоксид кремнію, вторинний кислий фосфат K103N/L100I. кальцію і високомолекулярні полімери (такі як поСполуки формули І мають інгібуючу активність ліетиленгліколь). у відношенні зворотної транскриптази ВІЛ-1. При При парентеральному застосуванні сполуку введенні у вигляді прийнятних дозованих форм їх формули І можна вводити у водному або неводможна застосовувати для лікування СНІДу, СНІДному розчині, суспензії або емульсії у фармацевасоційованого комплексу (ARC) і аналогічних, потично прийнятному маслі або суміші рідин, які мов'язаних із зараженням ВІЛ-1 захворювань. Таким жуть містити бактеріостатичні агенти, чином, ще одним об'єктом винаходу є спосіб лікуантіоксиданти, консерванти, буфери або інші інгвання інфекції, яка викликається ВІЛ-1, що полягає редієнти, які надають розчину ізотонічність із кроу введенні інфікованій ВІЛ-1 людині терапевтично в'ю, загусники, суспендувальні агенти або інші фаефективної кількості описаної вище нової сполуки рмацевтично прийнятні добавки. Добавки формули І. Незалежно від того, рекомендовані вказаного типу включають, наприклад, тартратний, вони для лікування або профілактики, сполуки цитратний і ацетатний буфери, етанол, пропіленгможна застосовувати також для попередження ліколь, поліетиленгліколь, комплексоутворювачі перинатальної передачі ВІЛ-1 від матері до дити(такі як ЕДТА), антіоксиданти (такі як бісульфіт ни, шляхом їх введення матері до народження натрію, метабісульфіт натрію й аскорбінова кислодитини. та), високомолекулярні полімери (такі як рідкі поСполуки формули І можна вводити у вигляді ліетиленоксиди) для регуляції в'язкості і поліетиоднократної або розділених доз пероральним або ленові похідні ангідридів сорбіту. При необхідності парентеральним шляхом. Прийнятна пероральна можна також додавати консерванти, такі як бендоза сполуки формули І становить приблизно від зойна кислота, метил- або пропіл парабен, бенза0,5мг до 1г на день. Переважна пероральна доза лконійхлорид і інші четвертинні амонійні похідні. сполуки формули І становить приблизно 100Сполуки за винаходом можна також вводити у 800мг на день для пацієнта вагою 70кг. У композивигляді назальних розчинів, і вони можуть містити ціях для парентерального введення прийнятна у водному носії крім сполук за винаходом прийнястандартна доза може містити від 0,1 до 250мг тні буфери, регулятори тонічности, консерванти, вказаних сполук, переважно від 1 до 200мг. Однак які перешкоджають мікробному зараженню, антіоповинно бути очевидно, що доза, яка вводиться, ксиданти й агенти, які підвищують в'язкість. Прикповинна змінюватися від пацієнта до пацієнта і ладами агентів, які застосовують для підвищення доза, призначена для введення будь-якому конкв'язкості, є полівініловий спирт, похідні целюлози, ретному пацієнту, повинна залежати від рекоменполівінілпіролідон, полісорбати або гліцерин. Задацій лікуючого лікаря, який як критерії для призстосовувані консерванти, які перешкоджають мікначення конкретної відповідної дози повинен робному зараженню, можуть являти собою бензавикористовувати габарити і стан пацієнта, а також лконійхлорид, тимеросал, хлорбутанол або реакцію пацієнта на лікарський засіб. фенілетиловий спирт. Коли сполуки за даним винаходом підлягають Сполуки за винаходом можна вводити також введенню пероральним шляхом, їх можна вводити за допомогою супозиторію. як лікарські засоби у формі фармацевтичних преСполуки за винаходом можна одержувати за паратів, які містять їх у сполученні із сумісним фадопомогою відомих методів органічного синтезу. рмацевтичним носієм. Такий носій може являти Приклади реакційних схем наведені на схемах 1-6. собою інертний органічний або неорганічний носій, придатний для перорального введення. Прикла 9 Послідовність здійснення реакцій, представлених на схемі 2, аналогічні до послідовності, описаної в J.M. Klunder / ін.\ J. Med. Chem., 41, 296071, 1998, і в C.L. Cywin / iн.;J. Med. Chem., 41, 2972-84, 1998. 74834 10 Як вказано вище, сполуки за винаходом інгібують ферментативну активність ЗТ ВІЛ-1. Грунтуючись на описаній нижче оцінці біологічної активності, встановлено, що вказані сполуки інгібують такий вид активності ЗТ ВІЛ-1, як активність РНКзалежна ДНК-полімерази ВІЛ-1. Встановлено (дані не представлені), що вони інгібують також такий вид активності ЗТ ВІЛ-1, як активність ДНКзалежної ДНК-полімерази. Встановлено, що за допомогою описаного нижче аналізу зворотної транскриптази (ЗТ), можна тестувати здатність сполук інгібувати активність ЗТ ВІЛ-1 як РНКзалежна ДНК-полімерази. За допомогою цього була вивчена активність деяких конкретних сполук, наведених нижче в прикладах. Результати дослідження представлені в таблиці 2 у вигляді значень ІС50 (нМ) і в таблиці 3 у вигляді значень ЕС50 (нМ). Приклади Даний винахід додатково проілюстрований за допомогою прикладів, які не обмежують його обсяг. Усі реакції здійснювали в атмосфері азоту або аргону. Температура наведена в градусах Цельсія. Якщо не вказане інше, то процентний вміст або співвідношення компонентів у розчині виражене в об.%. Нижче наведені скорочення або символи, які застосовуються в даному описі: ДЕАД: діетилазодикарбоксилат; ДІАД: діізопропілазодикарбоксилат; ДІЕА: діізопропілетиламін; ДМАП: 4-(диметиламіно)піридин; ДМСО: диметилсульфоксид; ДМФ: диметилформамід; MC ES: мас-спектрометрія з використанням електронного пучка; Et: етил; ЕtOАс: етилацетат; Et2O: простий діетиловий ефір; РХВР: високоефективна рідинна хроматогра 11 74834 12 ли Н2О, а потім гексаном. Після сушіння протягом iРr: ізопропіл; ночі при зниженому тиску одержували вказану в Me: метил; заголовку сполуку у вигляді твердої речовини чорМеОН: метанол; ного кольору вказану в заголовку сполуку (54,9г, MeCN: ацетонітрил; вихід 69%). NБC: N-бромсукцинімід; г) 8-Бром-5,11-дигідро-11-етил-6HPh: феніл; дипіридо[3,2-b:2',3'-е][1,4]діазепін-6-он ТБЕ: трис-борат-ЕДТА; До розчину 2-хлор-N-{2-(етиламіно)-3ТБТУ: тетрафторборат 2-(1Н-бензотриазол-1піридиніл}-5-бром-3-піридинкарбоксаміду (54,9г, іл)-N,N,N',N'-тетраметилуронію; 154,4ммоля) у піридині (308мл) при 50°С додавали ТФК: трифтороцтова кислота; по краплях 1М розчин NaГМДС (гексаметилдисиТГФ: тетрагідрофуран; лазид натрію) у ТГФ (355мл, 355ммолів). Через MC (ES): мас-спектрометрія з використанням 10хв реакційній суміші давали охолонути до кімнаелектронного пучка; тної температури, а потім зливали на льодяну воMC (FAB) або FAB/MC: мас-спектрометрія з ду (2л). Утворений твердий продукт фільтрували, використанням бомбардування швидкими атомапромивали водою, а потім гексаном. Твердий проми; дукт сушили при зниженому тиску, одержуючи вкаВРМС: мас-спектрометрія високого розділензану в заголовку сполуку (36г, вихід 75%) у вигляді ня; твердої речовини темно-зеленого кольору. БУЕ: бляшкоутворюючі одиниці; д) 8-Бром-5,11-дигідро-11-етил-5-метил-6HДЕПК: діетилпірокарбонат; дипіридо[3,2-b:2',3' e] [1,4] діазепін-6-он ДМСО: диметилсульфоксид; До розчину 8-бром-5,11-дигідро-11-етил-6НДТТ: дитіотреїтол; дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону (36,7г, ЕДТА: етилендіамінтетраацетат; 115ммолів) у ДМФ (380мл) додавали NaH (3,5г, УМФ: уридин-5'-монофосфат; 138ммолів) і суміш нагрівали до 50°С протягом УТФ: уридин-5'-трифосфат. 30хв. Реакційну суміш охолоджували до кімнатної Синтез температури й обробляли МеІ (14,3мл, У наведених нижче прикладах проілюстровані 230ммолів). Через 1,5год реакційну суміш зливали методи одержання сполук за винаходом. на льодяну воду. Твердий продукт фільтрували, Приклад І (номер 24, таблиця 1) промивали водою, а потім гексаном, одержуючи 5,11-Дигідро-11-етил-5-метил-8-{2-(4після сушіння вказану в заголовку сполуку (37,9г, вихід 99%) у вигляді твердої речовини темнохінолінілокси)етил}-6H-дипіридо[3,2-b:2 ,3'-е] зеленого кольору. [1,4]діазепін-6-он а) 2-(Етиламіно)-3-нітропіридин e) 5,11-Дигідро-11-етил-5-метил-8-(2До розчину 2-хлор-3-нітропіридину (51г, пропеніл)-6H-дипіридо[3,2-b:2',3' e] [1,4]діазепін-6325ммолів) у ТГФ (650мл) додавали 2М розчин он етиламіну ТГФ (365мл, 731ммоль). Реакційну суАлілтрибутилолово (30,7мл, 99,0ммолів) і міш перемішували при кімнатній температурі проРа(Рh3Р)4 (5,20г, 4,50ммоля) додавали до дегазотягом ночі. Реакційну суміш зливали на воду ваного (отриманого шляхом пропускання через ( 1,5л) і утворений твердий продукт фільтрували і розчин N2 протягом 30хв) розчину 8-бром-5,11сушили при зниженому тиску, одержуючи вказану дигідро-11-етил-5-метил-6H-дипіридо[3,2-b:2',3'в заголовку сполуку (52г). е][1,4]діазепін-6-ону (30,0г, 90,0ммолів) у ДМФ б) Аміно-2-(етиламіно)піридин (450мл) при кімнатній температурі. Суміш переміРозчин 2-(етиламіно)-3-нітропіридину (52г) у шували при 90°С протягом 1,5год, потім охолоМеОН (600мл) перемішували протягом ночі при джували до кімнатної температури і концентрувакімнатній температурі в атмосфері водню (1атм.) у ли при зниженому тиску. Залишок очищали присутності 20% Pd(OH)2/C (10,4г). Каталізатор експрес-хроматографією (гексан: ЕtOАс, від 8:2 до видаляли фільтрацією через діатомову землю. 7:3), одержуючи вказану в заголовку сполуку Фільтрат концентрували при зниженому тиску, (22,19г, вихід 84%). одержуючи вказану в заголовку сполуку у вигляді ж) 5,11-Дигідро-11-етил-8-(2-гідроксіетил)-5твердої речовини чорного кольору (39г, загальний метил-6H-дипіридо[3,2-b:2',3'е][1,4]діазепін-6-он вихід після стадій а) і б): 88%). Струменем озонованого кисню протягом в) 2-Хлор-N-{2-(етиламіно)-3-піридиніл}-52,5год барботували холодний (-78°С) розчин 5,11бром-3-піридинкарбоксамід дигідро-11-етил-5-метил-8-(2-пропеніл)-6HДо охолодженого розчину 3-аміно-2дипіридо[3,2-b:2',3'-е][1,4]діазепін-6-ону (22,19г, (етиламіно)піридину (30,6г, 223ммоля) у MeCN 75,4ммоля) у СН2Сl2 (150мл) і МеОН (150мл). По(740мл) додавали твердий NаНСО3 (56,3г, тім розчин протягом 15хв барботували N2, після 669ммолів). Через 5хв додавали неочищений 5цього до розчину додавали твердий NaBH4 (4,99г, бром-2-хлор-3-піридинкарбонілхлорид (1екв. 132ммоля). Реакційній суміші давали нагрітися до 223ммоля) (отриманий з 5-бром-2-гідрокси-3кімнатної температури. Через 1 год додавали водпіридинкарбонової кислоти і SOCl2 [відповідно до ний насичений розчин NH4CI (200мл) і суміш пеметоду, описаному в Т. W. Gero і ін. у: Synth. ремішували при кімнатній температурі протягом Commun., 19, 553-559, 1989 (публікація включена 2год. Органічні розчинники видаляли при знижев даний опис як посилання), але за винятком того, ному тиску. До залишку додавали воду (300мл) і що не проводили обробку водою]. Через 2год реаСНСl3 (300мл). Фази розділяли і водний шар ексткційну суміш зливали на суміш лід /Н2О (1,5л) і рагували СНСl3 (3 300мл). Об'єднані органічні утворену тверду речовину фільтрували, промивафія; 13 74834 14 шари сушили (MgSO4), фільтрували і концентру(180мл), що містить твердий NaHCO3 (14,2г, вали при зниженому тиску. Залишок очищали екс169ммоля). Суміш перемішували при кімнатній прес-хроматографією (ЕtOАс:СНСl3, 4:1), одержутемпературі протягом 1год. Додавали воду ючи вказану в заголовку сполуку (16,1г, вихід 72%) (200мл) і суміш перемішували протягом 10хв. у вигляді твердої речовини білого кольору. Утворену суспензію фільтрували. Твердий продукт з) 5,11-Дигідро-11-етил-5-метил-8-{2-(4промивали Еt2О (50мл) і концентрували з розчину хінолінілокси)етил}-6H-дипіридо[3,2b:2',3'-e] піридину (3 50мл), одержуючи вказану в заголовку [1,4]діазепін-6-он сполуку (28,4г, вихід 75%). Діетилазодикарбоксилат (ДЕАД) (12,8мл, г) 8-Бром-2-хлор-5,11-дигідро-11-етил-6H81,0ммоль) додавали при кімнатній температурі по дипіридо[3,2-b:2',3'-е] [1,4] діазепін-6-он краплях до розчину, що містить 5,11-дигідро-111М розчин NаГМДС у ТГФ (167,5мл, етил-8-(2-гідроксіетил)-5-метил-6H-дипіридо[3,2167,5моля) повільно додавали до розчину 5-бромb:2',3'-е] [1,4]діазепін-6-он (16,1г, 54,0ммоля), 42-хлор-N-{2-(етиламіно)-6-хлор-3-піридиніл}-3гідроксихінолін (11,6г, 81,0ммоль) і Рh3Р (213г, піридинкарбоксаміду (28,4г, 72,8ммоля) у піридині 81,0ммоль) у ΤΓΦ (270мл). Суміш перемішували (146мл), нагрітому до 50°С. Реакційну суміш перепри кімнатній температурі протягом 1год, потім мішували при 50°С протягом 1,5год. Потім суміш концентрували при зниженому тиску. Залишок зливали на суміш вода/лід (1л) і через 1год утвоочищали експрес-хроматографією (EtOAc:MeOH; рену суспензію фільтрували. Твердий продукт 95:5), одержуючи вказану в заголовку сполуку промивали водою і сушили при зниженому тиску, (17,7г, вихід 77% ) у вигляді твердої речовини біодержуючи вказану в заголовку сполуку (23,4г, лого кольору: MC (ESI) m/z 426 (МН)+. вихід 91%). Приклад II (номер 2, таблиця 1) д) 8-Бром-2-хлор-5,11-дигідро-11-етил-52-Хлор-5,11-дигідро-11-етил-5-метил-8-{2-(4метил-6H-дитридо[3,2-b:2',3'-е] [1,4] діазепін-6-он хінолінілокси)етил}-6H-дипіридо[3,2-b:2',3'-е] Твердий NaH (60%-на масляна дисперсія, [1,4]діазепін-6-он 3,46г, 86,1ммоля) додавали протягом 30хв до роза) 6-Хлор-2-(етиламіно)-3-нітропіридин чину 8-бром-2-хлор-5,11-дигідро-11-етил-6HОхолоджений на льоду розчин EtNH2 (49,8г, дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону (23,4г, 1,10моля) у толуолі (200мл) додавали протягом 66,3ммоля) у ДМФ (220мл) при 50°С. Суміш пере15хв до охолодженого на льоду розчину 2,6мішували при 50°С протягом 1год, потім давали дихлор-3-нітропіридину (100,0г, 0,52моля) у толуохолонути до кімнатної температури. Суміш злиолі (225мл). Суміш перемішували при 0°С протявали на воду (1л) і утворену суспензію фільтрувагом 45хв. Додавали воду (500мл) і ЕtOАс (500мл) і ли. Твердий продукт послідовно промивали водою розділяли фази. Органічний шар послідовно проі гексаном, потім сушили при зниженому тиску, мивали водою (200мл) і соляним розчином одержуючи вказану в заголовку сполуку (23,0г, (200мл), сушили (MgSO4), фільтрували і концентвихід 94%). рували при зниженому тиску. Твердий продукт, що e) 2-Хлор-5,11-дигідро-11-етил-5-метил-8-(2залишився, перекристалізовували з МеОН, одерпропеніл)-6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6жуючи вказану в заголовку сполуку (83,7г, вихід он 80%) у вигляді голчастих кристалів жовтого кольоАлілтрибутилолово (21,3мл, 68,7ммоля) і ру. Pd(Ph3P)4 (3,61г, 3,12ммоля) додавали до дегазоб) 3-Аміно-6-хлор-2-(етиламіно)піридин ваного (пропускання N2 через розчин протягом Розчин, що містить SnCl2 H2O (616,3г, 30хв) розчину 8-бром-2-хлор-5,11-дигідро-11-етил2,73моля) у водному 12н. розчині НСl (500мл), 5-метил-6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6швидко додавали при кімнатній температурі до ону (23,0г, 62,5ммоля) у ДМФ (312мл). Суміш нарозчину 6-хлор-2-(етиламіно)-3-нітропіридину грівали до 90°С протягом 2год. Суміш концентру(169,5г, 0,84моля) у АсОН (1,7л). Через 20хв суміш вали при зниженому тиску. Залишок очищали ексохолоджували до 0°С і додавали воду (250мл). прес-хроматографією (гексан:ЕtOАс, 7:3), Потім невеликими порціями додавали NaOH одержуючи вказану в заголовку сполуку (13,4г, (240г). Утворену суспензію фільтрували, видаляювихід 65%). чи солі олова. Фільтрат розбавляли водою (3,5л), ж) 2-Хлор-5,11-дигідро-11-етил-8-(2розчин підлуговували, додаючи водний 10н. NaOH, гідроксіетил)-5-метил-6H-дипіридо[3,2-b:2',3'-e] [1,4]діазепін-6-он а потім екстрагували ЕtOАс (3 1,7л). Об'єднані Озонований кисень вносили в холодний (органічні шари промивали соляним розчином (1л), 78°С) розчин 2-хлор-5,11-дигідро-11-етил-5-метилсушили (MgSO4), фільтрували і концентрували при 8-(2-пропеніл)-6H-дипіридо[3,2-b:2',3'-е] зниженому тиску. Залишок очищали експрес[1,4]діазепін-6-ону (13,4г, 40,7ммоля) у МеОН хроматографією (гексан:EtOAc, 3:2), одержуючи (102мл) і СН2Сl2 (102мл) до повного зникнення вказану в заголовку сполуку (89,9г, вихід 62%) у алкену. Розчин барботували азотом, видаляючи вигляді твердого коричневого кольору: MC (ESI) надлишок О3. Потім невеликими порціями додаваm/z 172/174 (МН)+. ли твердий NaBH4 (2,69г, 71,1ммоля) і суміші дав) 5-Бром-2-хлор-N-{2-(етиламіно)-6-хлор-3вали нагрітися до кімнатної температури. Через 1 піридиніл}-3 піридинкарбоксамід год додавали водний насичений розчин NH4Cl Розчин 5-бром-2-хлор-3(150мл) і суміш перемішували протягом 20хв. Орпіридинкарбонілхлориду (30,0г, 97,0ммоля) у ганічні розчинники видаляли при зниженому тиску. MeCN (100мл) додавали при кімнатній температурі До водного розчину додавали воду (100мл). Розчерез канюлю до розчину 3-аміно-6-хлор-2чин екстрагували СНСІ3 (3 200мл). Об'єднані ор(етиламіно)піридину (16,6г, 97,0ммоля) у MeCN 15 74834 16 ганічні шари сушили (MgSO4, фільтрували і концедої речовини білого кольору: MC (ESI) m/z 472/474 нтрували при зниженому тиску. Залишок очищали (MH)+. експрес-хроматографією (ЕtOАс:СНСl3, 4:1), одерПриклад IV (номер 4, таблиця 1) жуючи вказану в заголовку сполуку (10,4г, вихід 5,11-Дигідро-11-етил-2-фтор-5-метил-8-{2-(477% ). хінолінілокси)етил}-6Н-дипіридо[3,2-b:2',3'-e] з) 2-Хлор-5,11-дигідро-11-етил-5-метил-8-{2-(4[1,4]діазепін-6-он хінолінілокси)етил}-6H-дипіридо[3,2-b:2',3'-е] а) 2,6-Дифтор-3-нітропіридин [1,4]діазепін-6-он До суміші концентрованої сірчаної кислоти Діетилазодикарбоксилат (ДЕАД) (4,26мл, (600мл) і димлячої азотної кислоти (90%, 400мл) у 27,0ммолів) додавали при кімнатній температурі льодяній бані (внутрішня температура підтримупо краплях до розчину, що містить 2-хлор-5,11валася на рівні 5-10°С) додавали по краплях 2,6дигідро-11-етил-8-(2-гідроксіетил)-5-метил-6Hдифторпіридин (200г, 1,74моля). Утворену суміш дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-он (6,00г, перемішували протягом ночі при кімнатній темпе18,0ммоля), 4-гідроксихінолін (3,92г, 27,0ммолів) і ратурі. Суміш повільно зливали на 3кг льоду й Ph3P (7,09г, 27,0ммолів) у ТГФ (90мл). Суміш пеекстрагували Et2O (2 2л). Об'єднані органічні шари ремішували при кімнатній температурі протягом промивали водним 1,5н. розчином NaOH (2x1л), 1год, потім концентрували при зниженому тиску. потім водним насиченим розчином NaHCO3 Залишок очищали експрес-хроматографією (400мл) до того часу, поки значення pH не досяга(EtOAc:MeOH, 95:5), одержуючи вказану в загололо приблизно 8-9. Органічні шари сушили над вку сполуку (6,22г, вихід 75%) у вигляді твердої MgSО4, фільтрували і концентрували при знижеречовини білого кольору: MC (ESI) m/z 460/462. ному тиску до постійної маси (до видалення не Приклад III (номер 6, таблиця 1) прореагованого 2,6-дифторпіридину: 10-12%). У 2-Хлор-11-циклопропіл-5,11-дигідро-5-метилрезультаті одержали вказану в заголовку сполуку 8-{2-(4-хінолінілокси)етил}-6H-дипіридо[3,2-b:2',3'у вигляді рідини жовтого кольору (207,3г, вихід e] [1,4]діазепін-6-он 74% ). а) 6-Хлор-2-(циклопропіламіно)-3-нітропіридин б) 2-(Етиламіно)-6-фтор-3-нітропіридин Розчин циклопропіламіну (1,25г, 22,0ммоля) у До розчину 2,6-дифтор-3-нітропіридину (45,7г, толуолі (11мл) додавали протягом 10хв до охоло285ммолів) у ТГФ (500мл) при -40°С додавали по дженого на льоду розчину 2,6-дихлор-3краплях розчин етиламіну (25,7г, 570ммолів) у ТГФ нітропіридину (2,00г, 10,4ммоля) у толуолі (10мл). (250мл). Через 30хв реакційну суміш концентруваСуміш перемішували при 0°С протягом 1год при ли при зниженому тиску і залишок розчиняли в кімнатній температурі протягом 2год. До суміші EtOAc. Органічну фазу промивали соляним розчидодавали воду (50мл) і розділяли фази. Органічном, сушили (MgSO4), фільтрували і концентруваний шар промивали соляним розчином (25мл), ли. Утворений продукт жовтого кольору очищали сушили (MgSO4), фільтрували і концентрували при експрес-хроматографією (15% ЕtOАс у гексані), зниженому тиску. Залишок очищали експресодержуючи вказану в заголовку сполуку (43,2г, хроматографією (гексан:ЕtOАс, 3:2), одержуючи вихід 82%) у вигляді твердої речовини жовтого вказану в заголовку сполуку (1,97г, вихід 89%) у кольору. вигляді твердої речовини жовтого кольору. в) 3-Аміно-2-(етиламіно)-6-фторпіридин б) 2-Хлор-11-циклопропіл-5,11-дигідро-8-(2Розчин 2-(етиламіно)-6-фтор-3-нітропіридину гідроксіетил)-5-метил-6H-дипіридо[3,2-b:2',3'-е] (43,2г, 230ммолів) у ТГФ (1л) перемішували протя[1,4]діазепін-6-он гом ночі при кімнатній температурі в атмосфері Вказану в заголовку сполуку одержували з 6водню (1атм.) у присутності 20% Рd(ОН)2/С (4,35г). хлор-2-(циклопропіламіно)-3-нітропіридину відпоКаталізатор видаляли фільтрацією через діатомовідно до методу, аналогічному до описаного у приву землю. Фільтрат концентрували при зниженому кладі II для одержання 11-етильного аналога. тиску, одержуючи вказану в заголовку сполуку в) 2-Хлор-11-циклопропіл-5,11-дигідро-5(36,3г, вихід 95%) у вигляді твердої речовини чорметил-8-{2-(4-хінолінілокси)етил}-6Н-дипіридо[3,2ного кольору. b:2',3'-e] [1,4]діазепін-6-он г) 2-Хлор-N-{2-(етиламіно)-6-фтор-3Діетилазодикарбоксилат (ДЕАД) (85,6мкл, піридиніл}-5-бром-3-піридинкарбоксамід 0,54ммоля) додавали при кімнатній температурі по До охолодженого розчину (4°С) 3-аміно-2краплях до розчину, що містить 2-хлор-11(етиламіно)-6-фторпіридину (31,0г, 200ммолів) у циклопропіл-5,11-дигідро-8-(2-гідроксіетил)-5MeCN (160мл) додавали твердий NaHCO3 (50,4г, метил-6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-он 600ммолів). Через 15хв додавали розчин 5-бром(125мг, 0,36ммоля), 4-гідроксихінолін (78,9мг, 2-хлор-3-піридинкарбонілхлориду (1екв., 0,54ммоля) і Ph3P (143мг, 0,54ммоля) у ТГФ 200ммолів) у MeCN (155мл). Після витримування (1,8мл). Суміш перемішували при кімнатній темпепротягом 60хв при кімнатній температурі реакційну ратурі протягом 3год і потім концентрували при суміш зливали на воду (1,2л) і перемішували прозниженому тиску. Залишок частково очищали екстягом 30хв. Утворену тверду речовину фільтрувапрес-хроматографією (ЕtOАс:МеОН; 95:5). Тверли, сушили протягом ночі при зниженому тиску при дий продукт додатково очищали за допомогою 50°С. У результаті одержали вказану в заголовку РХВР із оберненою фазою (колонка CombiPrep сполуку (73,7г, вихід 99% ) у вигляді твердої речоADS-AQ 50 20мм, 5мкм, 120 Å, 5-100% MeCN + вини чорного кольору. 0,10% ТФК / вода + 0,10% ТФК протягом 25хв), д) 8-Бром-5,11-дигідро-11-етил-2-фтор-6Hодержуючи вказану в заголовку сполуку у вигляді дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-он трифторацетату (17,7г. вихід 77% ) у вигляді тверДо розчину 2-хлор-N-{2-(етиламіно)-6-фтор-3 17 74834 18 піридиніл}-5-бром-3-піридинкарбоксаміду (73,5г, го кольору. 216молів) у піридині (435мл) при 50°С додавали по и) 5,11-Дигідро-11-етил-2-фтор-5-метил-8-{2краплях 1М розчин NaГМДС у ТГФ (520мл, (4-хінолінілокси)етил}-6H-дипіридо[3,2-b:2',3'-e] 520ммолів). Через 10хв реакційній суміші давали [1,4]діазепін-6-он охолонути до кімнатної температури, потім зливаДіетилазодикарбоксилат (ДЕАД) (14,3мл, ли на льодяну воду (2л). Утворений твердий про91,0ммоль) додавали при кімнатній температурі по дукт фільтрували, промивали водою і потім гексакраплях до розчину, що містить 5,11-дигідро-11ном. Твердий продукт сушили при зниженому етил-2-фтор-8-(2-гідроксіетил)-5-метил-6Hтиску, одержуючи вказану в заголовку сполуку дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-он (19,2г, (50,6г, вихід 69%) у вигляді твердої речовини тем60,7ммоля), 4-гідроксихінолін (13,2г, 91,0ммоль) і но-зеленого кольору. Ph3P (23,9г, 91,0ммоль) у ТГФ (300мл). Суміш пеe) 8-Бром-5,11-дигідро-11-етил-2-фтор-5ремішували при кімнатній температурі протягом метил-6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-он 1год, потім концентрували при зниженому тиску. До розчину 8-бром-5,11-дигідро-11-етил-2Залишок очищали експрес-хроматографією фтор-6H-дипіридо[3,2-b:2',3'-е][1,4]діазепін-6-ону (ЕtOАс:МеОН; 95:5), одержуючи вказану в заголо(44г, 130,5ммоля) у ДМФ (520мл) додавали NaH вку сполуку (17,9г, вихід 67%) у вигляді твердого (4,28г, 178ммолів) і суміш нагрівали до 50°С пропродукту білого кольору: MC (ESI) m/z 444 (МН)+. тягом 30хв. Реакційну суміш охолоджували до кімПриклад V (номер 1, таблиця 1) натної температури й обробляли МеІ (24,4мл, 2-Хлор-5,11-дигідро-11-етил-4-метил-8-{2-(4522ммоля). хінолінілокси)етил}-6H-дипіридо[3,2-b:2',3'-е] Через 1,5год реакційну суміш зливали на льо[1,4]діазепін-6-он дяну воду. Твердий продукт фільтрували, промиа) 2-Хлор-N-(2,6-дихлор-4-метил-3-піридиніл)вали водою і потім гексаном, сушили при зниже3-піридинкарбоксамід ному тиску, одержуючи вказану в заголовку NaHCO3 (21,72г, 258,6ммоля) додавали до росполуку (43,2г, вихід 94%) у вигляді твердої речозчину 3-аміно-2,6-дихлор-4-метилпіридину (41,61г, вини темно-сірого кольору. 235,1ммоля; отриманому відповідно до методу, ж) 5,11-Дигідро-11-етил-2-фтор-5-метил-8-(2описаному в К. G. Grozinger / ін. J. Heterocyclic пропеніл)-6H-дипіридо[3,2-b:2'.3'-е] [1,4] діазепін-6Chem. 32, 259-263, 1995) у MeCN (350мл) і утвоон рену суспензію перемішували протягом 15хв. ПоАлілтрибутилолово (32,0мл, 103,4ммоля) і тім протягом 30хв вводили розчин 2Pd(Ph3P)4 (5,43г, 4,70ммоля) додавали до дегазохлорнікотинілхлориду (43,44г, 246,8ммоля) у ваного (через розчин протягом 45хв пропускали N2 MeCN (500мл). Утворену суспензію перемішували ) розчину 8-бром-5,11-дигідро-11-етил-2-фтор-5при кімнатній температурі. Через 24год було встаметил-6H-дипіридо[3,2-b:2',3'-е] [1,4]діазепін-6-ону новлено, що розчин був кислим і тому в нього вво(33,0г, 94,0ммоля) у ДМФ (470мл). Для завершендили додаткову кількість NаНСО3 (3,00г, ня реакції вводили додаткову кількість Pd(Ph3)4 35,7ммоля). Потім суспензію перемішували при (1,09г, 0,94ммоля вносили в реакційну суміш через кімнатній температурі протягом ще 2 днів. Потім 1, 2, 3, 4, 5год). Суміш нагрівали до 90°С протягом суміш зливали на суміш води (2л) і льоду (200г) і 6год. Суміш концентрували при зниженому тиску. перемішували протягом 20хв. Твердий продукт Залишок очищали експрес-хроматографією (гекфільтрували і промивали водою (500мл) і потім сан:EtOAc, від 8:2 до 7:3), одержуючи вказану в утворений твердий продукт сушили над Р2О5 при заголовку сполуку (22,4 г, вихід 76%). зниженому тиску, одержуючи вказану в заголовку з) 5,11-Дигідро-11-етил-2-фтор-8-(2сполуку (62,55г, вихід 84%) у вигляді порошку білогідроксіетил)-5-метил-6H-дипіридо[3,2-b:2',3'го кольору. е][1,4]діазепін-6-он б) N-2,6-дихлор-4-метил-3-піридиніл)-2Струменем озонованого кисню протягом 3год (етиламіно)-3-піридинкарбоксамід барботували холодний (-78°С) розчин 5,11Розчин 2-хлор-N-2,6-дихлор-4-метил-3дигідро-11-етил-2-фтор-5-метил-8-(2-пропеніл)-6Hпіридиніл)-3-піридинкарбоксаміду (63,17г, 194,0ммоля) і етиламіну (28,0г, 583ммоля) у ксидипіридо[3,2-b:2 ,3 -е][1,4]діазепін-6-ону (22,38г, лолі (250мл) перемішували при температурі 12071,6ммоля) у СН2Сl2 (100мл) і МеОН (100мл). По125°С у сталевому автоклаві протягом 7год. Утвотім розчин барботували струменем N2 протягом рену суспензію зливали на воду (1л), перемішува15хв і потім до розчину додавали твердий NaBH4 ли протягом 15хв і фільтрували. Залишок розчи(5,05г, 133ммоля). Реакційній суміші давали нагріняли в ЕЮ Ас і промивали водою (тричі), соляним тися до кімнатної температури. Через 1год у реакрозчином і сушили над MgSO4. Фільтрат екстрагуційну суміш вносили додаткову порцію NaBH4 вали EtOAc і об'єднані органічні екстракти проми(1,62г, 43,0моля). Після витримування протягом вали водою і соляним розчином і сушили над ще 1год додавали насичений розчин NH4Cl MgSO4. Потім дві фракції об'єднували і надлишок (150мл) і суміш перемішували при кімнатній темксилолу видаляли спільною перегонкою з бензопературі протягом 30хв. Органічні розчинники вилом, одержуючи вказану в заголовку сполуку даляли при зниженому тиску. Додавали воду (200 (61,98г, тверда речовина рожевого кольору) у вимл) і суміш екстрагували СНСІ3 (3 300мл). Об'єдгляді основного компонента суміші сполук. Неочинані органічні шари сушили (MgSO4), фільтрували і щений продукт використовували безпосередньо в концентрували при зниженому тиску. Залишок наступній реакції. очищали експрес-хроматографією (ЕtOАс:СНСl3, в) 5-Бром-N-(2,6-дихлор-4-метил-3-піридиніл)4:1), одержуючи вказану в заголовку сполуку 2-(етиламіно)-3-піридинкарбоксамід (19,7г, вихід 72%) у вигляді твердої речовини біло 19 74834 20 Твердий КОАс (22,2г, 229ммолів) додавали до нагрітися до кімнатної температури. Через 30хв неочищеного розчину N-(2,6-дихлор-4-метил-3вносили додаткову кількість NaBH4 (500мг). Через піридиніл)-2-(етиламіно)-3-піридинкарбоксаміду 1год додавали водний насичений розчин NH4Cl і (61,98г, 10000 212 15 19 28 77 2298 61 61 66 226 206 >10000 1522 14 25 29 319 60 198 25 48 11 37 41 737 1167 146 7 25 20 89 3938 91 10 36 22 661 439 51 17 44 51 1106 129 1357 26 95 ІС50 ІС50 ІС50 ІС50 K103N/ K103N/ K103N/ Y188C Р225Н V108I L100I (нМ) (нМ) (нМ) (нМ) 31,2 29 28,5 27 7,1 31 46 56 47 124 27 32 108 38 38 98 46 67 25 15 38 30 93 23 20 16 17 40 6,6 8Д 9,9 45 63 58 255 55 75 59 215 83 96 56 64 120 98 135 88 229 102 124 24 270 40 158 27 44 168 41 22 23 24 25 26 27 28 29 45 6,1 5,6 8 8,9 38 19 35 19 40 17 51 74834 2990 5614 627 25 2537 5204 5335 1190 25 578 2493 105 344 84 1745 2271 605 893 42 336 247 70 64 200 299 141 84 38 147 25 9,0 169 28 443 35 23 85 28 Таблиця 3 Інгібування сполуками формули І штамів дикого типу і мутантних штамів ВІЛ у клітинних культурах 1 2 3 4 5 6 7 8 9 10 11 12 14 15 17 18 19 20 21 22 23 24 25 26 28 29 ЕС50 Y181C (нМ) ЕС50 V106A (нМ) ЕС50 Y188L (нМ) ЕС50 K103N/ Y181C (нМ) 5 ЕС50 ЕС50 Спол. № (див. WT3T K103N таблицю 1) (нМ) (нМ) 118 7,6 ЕС50 ЕС50 K103N/P ЕС50 K103N/ K103N/L 225Н (нМ) V108I (нМ) 1001 (нМ) 2,6 2,2 1,3 0,98 0,83 1,1 0,93 0,92 0,34 0,26 0,67 2 2,8 4,1 2,7 28 12,4 52 7,5 2,0 1,5 3,9 5,0 3,2 2,9 0,34 0,34 0,37 0,40 0,53 1,5 2,8 24 58 1,2 2,7 1,2 3,5 4,8 65 67 2,3 95 22 11 4,3 2,9 5,0 5,9 2,3 2,6 42 83 70 65 7,5 3,5 2,2 2,2 1,3 1,6 Комп’ютерна верстка Л. Купенко Підписне Тираж 26 прим. Міністерство освіти і науки України Державний департамент інтелектуальної власності, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюNon-nucleoside inhibitors of reverse transcriptase and a pharmaceutical composition based thereon
Назва патенту російськоюНенуклеозидные ингибиторы обратной транскриптазы и фармацевтическая композиция на их основании
МПК / Мітки
МПК: A61P 31/18, A61K 31/551, A61P 43/00, C07D 471/14
Мітки: ненуклеозидні, композиція, фармацевтична, зворотної, основі, інгібітори, транскриптази
Код посилання
<a href="https://ua.patents.su/21-74834-nenukleozidni-ingibitori-zvorotno-transkriptazi-ta-farmacevtichna-kompoziciya-na-kh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Ненуклеозидні інгібітори зворотної транскриптази та фармацевтична композиція на їх основі</a>
Спірогетероциклічні нітрили як інгібітори зворотної дії цистеїнових протеаз, фармацевтична композиція та спосіб лікування захворювань

Номер патенту: 73378
Опубліковано: 15.07.2005
Автори: Сан Санхінг, Ворд Янси Девід, Сперо Деніс Мері, Томсон Девід С., Патель Уша Р., Янг Ерік Річард Роуш, Хікей Еужен Річард, Беккалі Юнес, Ліу Веймін
МПК: A61K 31/40, A61K 31/5377, C07D 413/12, C07D 417/12, A61P 21/04, A61P 29/00, A61K 31/5365, A61K 31/538, A61P 31/00, C07D 405/12, C07D 211/66, A61P 25/28, A61P 37/00, A61K 31/55, A61P 3/10, A61K 31/496, A61K 31/541, A61K 31/517, A61P 9/10, A61K 31/4468, C07D 487/08, C07D 413/14, A61P 37/06, A61K 31/506, C07D 207/16, A61P 1/04, C07D 455/00, A61P 11/06, C07D 401/12, C07D 211/60, A61P 19/10, A61K 31/454
Мітки: фармацевтична, зворотної, захворювань, протеаз, інгібітори, цистеїнових, нітрили, лікування, композиція, дії, спосіб, спірогетероциклічні
Формула / Реферат:
1. Сполука формули (Ia): , (Ia)у якій її фрагменти , і , позначені як A, B і C...
Вибірково діючі інгібітори тромбіну та фармацевтична композиція на іх основі

Номер патенту: 63887
Опубліковано: 16.02.2004
Автори: Хванг Сенг Йеул, Лі Ку, Йеонг Йі На, Шин Ю Сеунг, Юн Май Кюнг, Кім Сенг Со, Лі Йонг Хі, Хванг Сеонг Рюл, Оу Йенг Со, Хонг Сеонг Вон
МПК: C07C 257/00, C07D 241/04, A61P 9/00, C07D 295/185, A61K 31/155
Мітки: композиція, фармацевтична, вибіркової, основі, інгібітори, діючі, тромбіну
Формула / Реферат:
1. Сполука формули (І), (I)де R1 являє собою ацетил, заміщений арилом або арилокси, або сульфоніл, заміщений заміщеним або незаміщеним арилом або гетероциклічною групою, що містить N,Х вибрано з групи:,
Інгібітори серинових протеаз, фармацевтична композиція на їх основі

Номер патенту: 74372
Опубліковано: 15.12.2005
Автори: Уайлі Уільм Александер, Шіхан Скотт Мартін, Уотсон Брайан Морган, Джоунз Стьюарт Доналд, Лібешюц Джон Уолтер, Маррі Крістофер Уільям, Мейер Майкл Джон, Мастерз Джон Джозеф, Камп Ніколас Пол, Енджел Дейвід Біренбаум, Гуццо Пітер Роберт, Янг Стівен Клінтон, Уайлі Майкл Роберт
МПК: C07D 309/14, C07D 295/16, C07D 213/38, C07D 403/12, C07D 213/36, A61P 17/02, A61K 31/495, C07D 335/00, A61P 11/06, A61K 31/501, C07D 207/14, C07D 409/12, C07D 211/26, C07D 207/12, A61K 31/496, A61P 11/00, C07D 401/12, A61P 19/02, A61P 1/16, C07D 211/46, C07D 521/00, C07D 417/12, A61P 9/00, C07D 213/56, C07D 295/185, C07D 309/34, C07D 417/14, A61P 43/00, A61P 9/10, A61P 29/00, A61P 7/02, C07D 401/14, A61K 31/497, A61P 35/00
Мітки: фармацевтична, інгібітори, композиція, серинових, протеаз, основі
Формула / Реферат:
1. Інгібітор серинових протеаз – сполука формули (I),деR2 є:(i) феніл, факультативно заміщений у положенні 3 та/або 4 галогеном, нітрогрупою, тіолом, галогеналкоксигрупою, гідразидогрупою, алкілгідразидогрупою, аміногрупою, ціаном, галогеналкілом, алкілтіогрупою, алкенілом, алкінілом, ациламіногрупою, три- або дифторметоксигрупою, карбоксилом, ацилоксигрупою, MeSO2- або R1, та факультативно заміщений у...
Інгібітори тирозинкінази та фармацевтична композиція на їх основі

Номер патенту: 74560
Опубліковано: 16.01.2006
Автори: Кім Юнтае, Хартман Джордж Д., Білодо Марк Т., Аррінгтон Кеннет Л., Фрелі Марк Е., Хангейт Рендалл В., Хоффман Вілльям Ф.
МПК: A61P 17/06, A61P 27/02, A61K 31/5377, A61P 17/02, A61K 31/506, A61K 45/06, A61P 19/08, C07D 409/14, A61K 31/541, A61P 19/02, A61P 43/00, C07D 405/14, A61N 5/10, A61P 35/00, A61K 31/495, C07D 401/04, C07D 413/14, A61K 45/00, A61K 31/551, A61K 31/496, A61P 29/00, C07D 417/14, A61P 9/10, C07D 401/14, A61K 31/4709
Мітки: тирозинкінази, основі, фармацевтична, композиція, інгібітори
Формула / Реферат:
1. Сполука формули IІабо її фармацевтично прийнятна сіль або стереоізомер, деZ представляє формулу,а дорівнює 0 або 1;b дорівнює 0 або 1;m дорівнює 0, 1 або 2;s дорівнює 1 або 2;t дорівнює 1, 2 або 3;R1 і R5 незалежно вибрані...
Заміщені 8-арилхінолінові інгібітори фосфодіестерази-4, фармацевтична композиція на їх основі (варіанти)

Номер патенту: 74815
Опубліковано: 15.02.2006
Автори: Галлан Мішель, Дюб Деніел, Дешен Дені, Перьє Елєн, Жирар Ів, Макдональд Дуайт, Лякомб Патрік, Мастраччо Ентоні
МПК: A61P 37/08, A61P 17/06, C07D 417/14, C07D 215/12, A61K 31/4709, A61P 13/12, A61P 11/00, A61P 1/04, A61P 17/00, C07D 401/10, C07D 401/14, A61P 19/10, A61P 7/12, C07D 417/12, A61P 19/02, C07D 409/10, A61K 31/47, C07D 413/10, A61P 17/04, A61P 9/00, A61P 11/06, C07D 405/12, A61K 45/00, A61P 11/16, C07D 413/14, C07D 417/10, C07D 215/14
Мітки: варіанти, фармацевтична, заміщені, композиція, 8-арилхінолінові, інгібітори, основі, фосфодіестерази-4
Формула / Реферат:
1. Сполука, представлена формулою (І):, (I)або її фармацевтично прийнятна сіль, деS1, S2 і S3 незалежно являють собою Н, -ОН, галоген, -С1-С6алкіл, -NO2, -CN або –С1-С6алкокси, де алкільна і алкоксигрупи необов'язково заміщені 1-5 замісниками; де кожний замісник незалежно являє собою галоген або ОН;R1 являє собою Н, ОН, галоген, карбоніл,...
Попередній патент: Лічильний пристрій для контролю витрати енергоносіїв в автоматизованій системі
Наступний патент: Спосіб фторування питної води
Випадковий патент: Пилогазомазутний пальник із примусовою подачею повітря