Вибірково діючі інгібітори тромбіну та фармацевтична композиція на іх основі
Номер патенту: 63887
Опубліковано: 16.02.2004
Автори: Йеонг Йі На, Лі Йонг Хі, Юн Май Кюнг, Шин Ю Сеунг, Хонг Сеонг Вон, Оу Йенг Со, Кім Сенг Со, Хванг Сеонг Рюл, Хванг Сенг Йеул, Лі Ку








Формула / Реферат
1. Сполука формули (І)
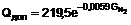 , (I)
, (I)
де R1 являє собою ацетил, заміщений арилом або арилокси, або сульфоніл, заміщений заміщеним або незаміщеним арилом або гетероциклічною групою, що містить N,
Х вибрано з групи:
![]() ,
,
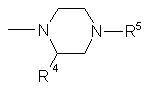 ,
,
R2 і R3 незалежно один від одного являють собою водень, циклоалкіл, заміщений або незаміщений карбоксилом або алкоксикарбонілом, арилалкілокси, гідрокси або нижчий алкіл, заміщений або незаміщений карбоксилом, алкоксикарбонілом або гідрокси, або
R2 і R3 разом з атомом азоту, до якого вони прикріплені, можуть формувати піперидинову групу, заміщену карбоксилом або алкоксикарбонілом,
R4 являє собою водень, нижчий алкіл або нижчий алкокси,
R5 являє собою алкансульфоніл, алкоксикарбоніл, алкілкарбоніл, форміл, нижчий алкіл, арил, заміщений або незаміщений алкокси або галоалкілом, або гідроксизаміщений нижчий алкіл,
R6 являє собою метил або аміно,
R7 являє собою водень,
або її фармацевтичнo прийнятна сіль, гідрат, сольват та ізомер.
2. Сполука формули (І) за п. 1, яка відрізняється тим, що R1 являє собою ацетил, заміщений нафтилом або нафтилокси, або сульфоніл, заміщений нафтилом або фенілом, які можуть бути заміщеними або незаміщеними від одного до чотирьох замісниками, вибраними з групи, що містить нижчий алкіл, нижчий алкокси і діалкіламіно,
Х вибрано з групи:
![]() ,
,
,
R2 і R3 незалежно один від одного являють собою С3-6циклоалкіл, заміщений або незаміщений карбоксилом або метоксикарбонілом, бензилокси, нижчий алкіл, заміщений або незаміщений карбоксилом, метоксикарбонілом або гідрокси, або гідрокси, або
R2 і R3 разом з атомом азоту, до якого вони прикріплені, можуть формувати піперидинову групу, заміщену карбоксилом або метоксикарбонілом,
R4 являє собою водень,
R5 являє собою метансульфоніл, етоксикарбоніл, форміл, етил, феніл, метилкарбоніл, гідроксіетил або феніл, який може бути заміщеним або незаміщеним трифторметилом або етокси.
3. Сполука формули (І) згідно з п. 2, яка відрізняється тим, що зазначену сполуку вибирають з групи, що містить:
(S)-N-циклопентил-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-бутил-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-пропіл-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-(2-бензилоксіетил)-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-бутил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-етил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-метил-3-[4-(метиламідино)феніл]-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-метил-3-(4-амідразонофеніл)-2-[(4-метокси-2,3,6-триметилбензол)сульфоніламіно]пропіонамід,
(S)-N-циклопентил-N-гідрокси-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-(2-гідроксіетил)-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопентил-N-метил-3-[4-(метиламідино)феніл]-2-[(4-метокси-2,3,6-триметилбензол)сульфоніламіно]пропіонамід,
(S)-N,N-диметил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N,N-диметил3-[4-(1-метиламідино)феніл]-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклогексил-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-N-циклопропіл-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід,
(S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(2–нафталін-1-іл-ацетиламіно)пропіонамід,
(S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(5-диметиламінонафталін-1-сульфоніламіно)пропіонамід,
(S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(5-метоксинафталін-1-сульфоніламіно)пропіонамід,
(S)-2-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(6,7-диметоксинафталін-2-сульфоніламіно)пропіонамід,
(S)-3-[4-(метиламідино)-феніл]-N-циклопентил-N-метил-2-(5-диметиламінонафталін-1-сульфоніламіно)пропіонамід,
(S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(нафталін-1-сульфоніламіно)пропіонамід,
(S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-[2-(нафталін-1-іл-оксі)ацетиламіно]пропіонамід,
(S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-[2-(нафталін-2-іл-оксі)ацетиламіно]пропіонамід,
метиловий ефір {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламінo}оцтової кислоти,
{[-3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}оцтова кислота,
метиловий ефір (S)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}пропіонової кислоти,
(S)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}пропіонова кислота,
метиловий ефір (R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл}метиламіно}пропіонової кислоти,
(R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}пропіонова кислота,
метиловий ефір (R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталiн-2-сульфоніламіно)пропіоніл]метиламіно}-3-метилмасляної кислоти,
(R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}-3-метилмасляна кислота,
метиловий ефір 3-{[-3-(4-амідразонофеніл)-(S)-2-(нафталiн-2-сульфоніламіно)пропіоніл]метиламіно}пропіонової кислоти,
3-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламінo}пропіонова кислота,
метиловий ефір 4-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}масляної кислоти,
4-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}масляна кислота,
метиловий ефір {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]циклопропіламіно}оцтової кислоти,
{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]циклопропіламіно}оцтова кислота,
метиловий ефір {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]бутиламіно}оцтової кислоти,
{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]бутиламіно}оцтова кислота,
метиловий ефір {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]циклопентиламіно}оцтової кислоти,
{[3-(4-(амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]циклопентиламіно}оцтова кислота,
метиловий ефір 1-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}циклопентанкарбонової кислоти,
1-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}циклопентанкарбонова кислота,
етиловий ефір 2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}циклопентанкарбонової кислоти,
2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}циклопентанкарбонова кислота,
метиловий ефір (S)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]метиламіно}-3-метилмасляної кислоти,
метиловий ефір 1-[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]піперидин-(R)-2-карбонової кислоти,
1-[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)пропіоніл]піперидин-(R)-2-карбонова кислота,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-(4-метилсульфонілпіперазиніл)-2-оксоетил]амід,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-(4-етоксикрбонілпіперазиніл)етил]амід,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-(4-формілпіперазиніл)-2-оксоетил]амід,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-(4-етилпіперазиніл)-2-оксоетил]амід,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-(4-фенілпіперазиніл)етил]амід,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-(4-(3-трифторметилфеніл)піперазиніл)етил]амід,
(S)-нафталін-2-сульфокислота[2-(4-ацетилпіперазиніл)-1-(4-амідразоно)бензил-2-оксоетил]амід,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-[4-(2-гідроксіетил)піперазиніл]етил]амід,
(S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-[4-(2-етоксифеніл)піперазиніл]етил]амід.
4. Фармацевтична композиція для інгібування тромбіну, яка відрізняється тим, що вона містить сполуку за будь-яким з пп. 1-3 як активний інгредієнт разом з фармацевтично придатним носієм.
5. Фармацевтична композиція за п. 4, яка відрізняється тим, що виготовлена як препарат для орального введення.
Текст
Цей винахід належить до нового вибірково діючого інгібітора тромбіну, який має формулу (І): у якій R1 становить собою ацетил, заміщений арилом або арилокси, або становить собою сульфоніл, заміщений заміщеним або незаміщеним арилом або гетероциклічною групою, що містить N, X становить собою групу формули R2 і R3 незалежно один від одного становлять собою водень; циклоалкіл, заміщений або незаміщений карбоксилом або алкоксикарбонілом; арилалкілокси; гідрокси; або нижчий алкіл, заміщений або незаміщений карбоксилом, алкоксикарбонілом або гідрокси, або R2 і R3 разом з атомом азоту, до якого вони прикріплені, можуть формувати піперидинову гр упу, заміщену карбоксилом або алкоксикарбонілом, R4 становить собою водень, нижчий алкіл або нижчий алкокси, R5 становить собою алкансульфоніл; алкоксикарбоніл; алкілкарбоніл; форміл; нижчий алкіл; арил, заміщений або незаміщений алкокси або галоалкілом; або гідрокси-заміщений нижчий алкіл, і R6 і R7 незалежно один від одного становлять собою водень, нижчий алкіл або аміно. Деякі сполуки фомули (І) можуть показувати ефективну інгібіторну активність відносно тромбіну навіть при оральному введенні і тому є дуже корисні. Цей винахід також стосується способу виготовлення сполуки формули (І) і фармацевтичної композиції, яка призначена для інгібування тромбіну і яка містить сполуку формули (І) як активний інгредієнт. У цілому відомо, що способ, призначений для коагуляції крові, включає декілька складних ензимних реакцій, які на кінцевому етапі включають реакцію конвертування прототромбіну у тромбін. Тромбін, одержаний на кінцевому етапі процесу коагуляції крові, активує тромбоцити і конвертує фібриноген у фібрин, який далі конвертується у ви щу молекулярну речовину завдяки полімеризуванню і зшиванню за рахунок дії активованого тромбіном фактора крові XIII з метою коагуляції нерозчиненої крові. Відповідним чином, тромбін грає важливу роль у процесі коагуляції крові. Тромбін також активує фактори крові V і VIII, які у свою чергу, прискорюють коагуляцію крові завдяки механізму зворотного зв'язку. Таким чином, оскільки інгібітори тромбіну діють як ефективні коагулянти і у той же самий час можуть інгібувати активність тромбоциту і одержання та стабілізацію фібрину протягом довгого строку, багато уваги приділяли знаходженню способу запобігання коагуляції крові і лікування різних тромбозів з використовуванням нової сполуки, яка може інгібувати активність тромбіну. Однак, сполука, яка здатна інгібувати тільки активність тромбіну, має обмеження у використанні як ефективний антикоагулянт і тромболітичний агент. Причиною цього є те, що, оскільки тромбін є одним із серин-протеаз, а численні серин-протеази аналогічні трипсину, типово плазміну, присутні у тілі людини, особливо у крові, ефективний інгібітор тромбіну звичайно також має високу інгібіторну активність проти зазначених серин-протеаз. Завдяки такої характерної особливості тромбіну при розробці інгібіторів тромбіну дуже важливим є те, щоб інгібіторна сполука мала меншу інгібіторну активність відносно серин-протеази прототипу, наприклад трипсину, ніж відносно тромбіну. У таких умовах були проведені численні дослідження щодо створення вибірково діючого інгібітора тромбіну, який може ефективно інгібувати тромбін і у той же час мати невелику інгібіторну активність до трипсину. У результаті був створений препарат аргатробан (Argatroban), який має нижченаведену формулу (А), як сполука на основі арилсульфоніларгініну (див. Патенти США № 4258192 і 4201863). Аргатробан показує високу інгібіторну активність до тромбіну, яка у 250 разів вища ніж активність до трипсину (див., Biochemistry, 1984, 23, p.85-90). Однак він може бути одержаний тільки завдяки складним процедурам синтезування. Він з'явився на ринку Японії у 1990р. Крім того, був розроблений препарат NAPAP, що має нижченаведену формулу (В), на основі бензоамідіну. Ця сполука може бути легко синтезована і має ефективну інгібіторну активність до тромбіну. Однак, вона має той недолік, що інгібіторна активність до тромбіну тільки у 50 разів вище ніж активність до трипсину (див., J. Biol. Chem. 1991, 266, р.20085-20093). Крім того, описано препарат Ro 46-6240, що має формулу (C), як сполука, що має більш високу селективність до тромбіну ніж до трипсину. Ця сполука показує можливість створення як внутрішньовенної ін'єкційної композиції завдяки її короткому строку зберігання у крові, але не показує будь-яких можливостей для орального введення (див., J. Med. Chem. 1994, 37, 3889-3901). Крім того, було повідомлено, що нещодавно створена сполука на основі піперазиду має якоюсь мірою можливість для орального введення пацюкам але має низьку селективність до тромбіну (див. Патент WO 94/18185). Таким чином, зазначені сполуки не придатні для використання у цій галузі. Внаслідок цього, автори цього винаходу здійснили широке дослідження можливості одержання певної сполуки, яка може бути легко синтезована, показує ефективну інгібіторну активність до тромбіну з високою селективністю до тромбіну відносно трипсину і може також вводитися орально. У результаті було встановлено, що інгібітор тромбіну формули (1) згідно з цим винаходом може досягнути таку ціль. Таким чином, ціллю цього винаходу є одержання нового інгібітора тромбіну формули (1), як це визначено вище, який може бути введений орально і який має високу селективність до тромбіну. Іншою ціллю цього винаходу є створення способу одержання інгібітора тромбіну формули (1). Крім того, ще однією ціллю цього винаходу є створення фармацевтичної композиції для запобігання коагуляції крові і для лікування різних тромбозів, яка містить інгібітор тромбіну формули (1) як активний інгредієнт. У одному аспекті цей винахід належить до нової сполуки, яка має нижченаведену формулу (1): її фармацевтично придатної солі, гідрату, сольвату і ізомеру, у якій R1 становить собою ацетил, заміщений арилом або арилокси, або становить собою сульфоніл, заміщений заміщеним або незаміщеним арилом або гетероциклічною групою, що містить N, X становить собою групу формули R2 і R3 незалежно один від одного становлять собою водень; циклоалкіл, заміщений або незаміщений карбоксилом або алкоксикарбонілом; арилалкілокси; гідрокси; або нижчий алкіл, заміщений або незаміщений карбоксилом, алкоксикарбонілом або гідрокси, або R2 і R3 разом з атомом азоту, до якого вони прикріплені, можуть формувати піперидинову гр упу, заміщену карбоксилом або алкоксикарбонілом, R4 становить собою водень, нижчий алкіл або нижчий алкокси, R5 становить собою алкансульфоніл; алкоксикарбоніл; алкілкарбоніл; форміл; нижчий алкіл; арил, заміщений або незаміщений алкокси або галоалкілом; або гідрокси-заміщений нижчий алкіл, і R6 і R7 незалежно один від одного становлять собою водень, нижчий алкіл або аміно. Стосовно до кожного замісника сполуки формули (1) згідно до цього винаходу термін "нижчий алкіл" означає насичений, розгалужений або нерозгалужений вуглеводневий радікал, що має від 1 до 4 атомів вуглецю, наприклад, метил, етил, ізопропіл, t - бутил та ін.; термін "аралкілокси" означає алкокси - групу, заміщену ароматичним кільцем, наприклад, бензилокси та ін.; і термін "циклоалкіл" означає циклічну алкільну груп у, що має від 3 до 8 атомів вуглецю, наприклад, циклопентил. Серед вищенаведених сполук формули (1) найкращими є сполуки, у яких: R1 становить собою ацетил, заміщений нафтилом або нафтилокси, або становить собою сульфоніл, заміщений нафтилом або фенілом, які можуть бути заміщеними або незаміщеними від одного до чотирьох замісниками, вибраними з групи, що містить нижчий алкіл, нижчий алкокси і діалкіламіно, X становить собою групу формули R2 і R3 незалежно один від одного становлять собою С 3-6 циклоалкіл, заміщений або незаміщений карбоксилом або метоксикарбонілом; бензилокси; нижчий алкіл, заміщений або незаміщений карбоксилом, метоксикарбонілом або гідрокси; або гідрокси, або R2 і R3 разом з атомом азоту, до якого вони прикріплені, можуть створювати піперидинову групу, заміщену карбоксилом або метоксикарбонілом, R4 становить собою водень, R5 становить собою метансульфоніл, етоксикарбоніл, форміл, феніл, метилкарбоніл, гідроксиетил, або феніл, який може бути заміщеним або незаміщеним трифторметилом або етокси, і R6 і R7 незалежно один від одного становлять собою водень, метил або аміно. Типовими прикладами сполуки формули (1) згідно з цим винаходом є такі сполуки: (S)-N-циклопентил-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсуль фоніламіно)пропіонамід, (S)-N-бутил-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклопентил-N-пропіл-3-(4-амідразонофеніл)-2-(2нафтилсуль фоніламіно)пропіонамід, (S)-N-циклопентил-N-(2-бензилоксиетил)-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклопентил-N-бутил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклопентил-N-етил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклопентил-N-метил-3-[4-(метиламідино)феніл]-2-(2-нафтилсуль фоніламіно)пропіонамід, (S)-N-циклопентил-N-метил-3-[4-(1,1-диметиламідино)-феніл]-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклопентил-N-метил-3-(4-амідразонофеніл)-2-[(4-метокси-2,3,6-триметилбензол)сульфоніламіно] пропіонамід, (S)-N-циклопентил-N-гідрокси-3-(4-амідразонфеніл)-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклопентил-N-(2-гідроксиетил)-3-(4-амідразонфеніл)-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклопентил-N-метил-3-[4-(метиламідино)феніл]-2-[(4-метокси-2,3,6-триметилбензол) сульфоніламіно]пропіонамід, (S)-N,N-диметил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N,N-диметил3-[4-(1-метиламідино)феніл]-2-(2-нафтилсульфоніламіно)пропіонамід, (S)-N-циклогексил-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсуль фоніламіно)пропіонамід, (S)-N-циклопропіл-N-метил-3-(4-амідразонофеніл)-2-(2-нафтилсуль фоніламіно)пропіонамід, (S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(2-нафталін-1-іл-ацетиламіно)пропіонамід, (S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(5-диметиламіно-нафталін-1-сульфоніламіно) пропіонамід, (S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(5-метоксинафталін-1-сульфоніламіно)пропіонамід, (S)-2-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(6,7-диметокси-нафталін-2-сульфоніламіно) пропіонамід, (S)-3-[4-(метиламідино)-феніл]-N-циклопентил-N-метил-2-(5-диметиламіно-нафталін-1-сульфоніламіно) пропіонамід, (S)-3-[4-(амідразоно)-феніл]-N-циклопентил-N-метил-2-(нафталін-1-сульфоніламіно)пропіонамід, (S)-3-[4-(амідразоно)-феніл]-N-циклопентил-N-метил-2-[2-(нафталін-1-іл-окси)ацетиламіно]-пропіонамід, (S)-3-[4-(амідразоно)-феніл]-N-циклопентил-N-метил-2-[2-(нафталін-2-іл-окси)ацетиламіно]пропіонамід, метиловий ефір {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}оцтової кислоти, {[-3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}оцтова кислота, метиловий ефір(S)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} пропіонової кислоти, (S)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}пропіонова кислота, метиловий ефір(R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл} -метиламіно} пропіонової кислоти, (R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}пропіонова кислота, метиловий ефір(R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}-3метил масляної кислоти, (R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}-3-метил масляна кислота, метиловий ефір3-{[-3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} пропіонової кислоти, 3-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]метиламіно}пропіонова кислота, метиловий ефір4-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} масляної кислоти, 4-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}масляна кислота, метиловий ефір{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-циклопропіламіно} оцтової кислоти, {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-циклопропіламіно}оцтова кислота, метиловий ефір{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-бутиламіно}оцтової кислоти, {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-бутиламіно}оцтова кислота, метиловий ефір{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-циклопентиламіно} оцтової кислоти, {[3-(4-(амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-циклопентиламіно}оцтова кислота, метиловий ефір1-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} циклопентан карбонової кислоти, 1-{[3-(4-амідразонофеніл)-(S)-2-(нафталіїн-2-сульфоніламіно)-пропіоніл]-метиламіно}-циклопентан карбонова кислота, етиловий ефір2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}циклопентан карбонової кислоти, 2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}циклопентан карбонова кислота, метиловий ефір(S)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}-3метил-масляної кислоти, метиловий ефір1-[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-піперидин-(R)-2карбонової кислоти, 1-[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-піперидин-(R)-2-карбоновакислота, (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-(4-метилсульфоніл-піперазиніл)-2-оксоетил]амід, (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-(4-етоксикрбоніл-піперазиніл)етил]амід, (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-(4-форміл-піперазиніл)-2-оксоетил]амід, (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-(4-етил-піперазиніл)-2-оксоетил]амід, (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-(4-феніл-піперазиніл)-етил]амід, (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-(4-(3-трифторметилфеніл)-піперазиніл) етил] амід, (S)-нафталін-2-сульфокислота[2-(4-ацетил-піперазиніл)-1-(4-амідразоно)бензил-2-оксоетил]амід, (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-[4-(2-гідроксиетил)-піперазиніл]-етил]амід, і (S)-нафталін-2-сульфокислота[1-(4-амідразоно)бензил-2-оксо-2-[4-(-2-етоксифеніл)-піперазиніл]-етил]амід. Сполука формули (1) згідно з цим винаходом також може утворювати фармацевтично придатну сіль. Відповідні фармацевтично придатні солі для сполуки формули (1) можуть включати кислу додаткову сіль, створену за допомогою кислот, які можуть утворювати нетоксичну кислу додаткову сіль, яка містить фармацевтично придатний аніон, наприклад неорганічні кислоти, такі як хлористоводнева кислота, сірчана кислота, азотна кислота, бромистоводнева кислота, фосфорна кислота, йодистоводнева кислота та ін., органічні вугільні кислоти, такі як винна кислота, мурашина кислота, лимонна кислота, оцтова кислота, трихлороцтова кислота, глюконова кислота, бензойна кислота, молочна кислота, фумарова кислота, малеїнова кислота і т.д., сульфонові кислоти, такі як метансульфокислота, бензолсульфокислота, р толуолсульфокислота, нафталінсульфокислота та ін., та подібні кислоти. Крім того, оскільки сполука формули (1) згідно з цим винаходом може включати у своїй структурі асиметричний атом вуглецю, вона може існувати у формі рацемату, діастереометричній суміші та індивідуального діастереомеру. Усі зазначені ізомери включені у об'єм цього винаходу. У іншому аспекті, цей винахід також стосується способа одержання сполуки формули (1), як це визначено вище. Згідно з цим винаходом сполука формули (І) може бути одержана шляхом вступу в реакцію сполуки формули (II) із сполукою формули (III), як це показано у нищенаведеній схемі 1 реакції. Схема 1 реакції : У вищезазначеній схемі реакції X, R1, R6 і R7 визначені так, як це описано раніше. Як зображено на вищенаведеній схемі 1 реакції , сполука формули (1) згідно з цим винаходом може бути одержана шляхом вступу в реакцію метилмеркапто-сполуки формули (II) з амінопохідною формули (III) як нейклеофільної речовини. Зазначена реакція найкращим чином може бути здійснена у присутності розчинника. Хоч у цій реакції може бути застосований будь-який органічний розчинник, що не має шкідливого впливу на реакцію, у загальному випадку для цієї цілі найкраще використовувати спиртовий розчинник, наприклад метанол, етанол, пропанол та інші. У вищенаведеній реакції умови проведення реакції, включаючи кількість реактивів, температуру реакції, час реакції та інші, можна визначати залежно від типу конкретного реактиву, який застосований спеціалістом, який має звичайні знання у цій галузі. У загальному випадку, хоч температура реакції може змінюватись у значному діапазоні, особливо найкраще виконували реакцію при температурі, що знаходиться у межах від 0°С до 50°С. Крім того, у загальному випадку час реакції становить від 0,5 до 5 годин, найкраще - від 1 до 2 годин. Після завершення зазначеної реакції продукт реакції може бути виділений і очищений згідно із звичайним розробленим способом, наприклад, хроматографією, рекристалізацією та ін. Метилмеркапто-сполука формули (II), яку використовують як проміжну сполуку для одержання сполуки формули (І) в схемі реакції 1, може бути отримана згідно із схемами 2 або 3 реакції, які зображені нижче. Схема 2 реакції: У вищенаведених схемах реакції X, R1, R 6 і R7 визначені аналогічно описаним раніше, а р становить собою амінозахисну гр уп у. Схеми реакцій 2 і 3 детально пояснюються нижче. У схемі 2 реакції спочатку С - кінець сполуки [1] зв'язують з аміногрупою X для одержання сполуки [2], із якої видаляють амінозахисну гр уп у на N - кінці з метою виготовлення сполуки [3]. Далі до незахищенного N кінця сполуки [3] з метою виготовлення сполуки [4] вводять R1 . У протилежному випадку, згідно з схемою 3 реакції спочатку групу R1 вводять до N - кінця сполуки [7] і далі С -кінець зв'язують з аміногрупою X для одержання сполуки [4]. У даному випадку, згідно з схемою 2 реакції, сполуку формули [1] зв'язують з аміносполукою, яка відповідає заміснику X, з метою одержання сполуки формули [2], із якої видаляють амінозахисну гр упу на N кінці для одержання сполуки формули [3]. Потім групу R 1 вводять до N - кінця сполуки формули [3] з метою одержання нітрилової сполуки формули [4], яка далі насичується сірководнем у присутності піридину і триетиламіну для виготовлення тіоамідної сполуки формули [5]. Зазначену тіоамідну сполуку потім метилують за допомогою метилуючого агента, наприклад, йодометану, диметилсульфату, метилтрифлату та ін. для одержання бажаної метилмеркапто-сполуки формули [II]. Згідно з схемою 3 реакції спочатку груп у R1 вводять до N - кінця сполуки формули [7] шля хом реакції сполуки формули [7] з сполукою формули [6], а далі аміногрупу вводять до С - кінця одержаної сполуки формули [8] за допомогою зв'язування сполуки формули [8] з аміносполукою, що відповідає заміснику X, для одержання сполуки формули [4], яку потім піддають тим самим операціям, як і у схемі реакції 2 з метою одержання бажаної метилмеркапто-сполуки формули [II]. Зв'язувальний агент, що можна використовувати у операції зв'язування у схемах реакцій 2 і 3, включає одну або більше речовин, вибраних з групи, що містить дициклогексилкарбодіімід (ДЦК), 3 - етил - 3' (диметиламіно)пропілкарбодіімід (ЕДК), хлорід біс-(2-оксо-3-оксазолідиніл)фосфорної кислоти (БОФ - СІ) і дифенілфосфорилазід (ДФФА), але не обмежуючись зазначеними речовинами. Хоч сполуки карбоксильної кислоти [1] і [7], що застосовують у схемах реакцій 2 і 3, можна використовува ти у вигляді їх вільних кислот, найкраще їх можна використати у вигляді їх реакційних похідних, наприклад, похідної галоідангідриду або похідних інших активованих ефірів для полегшення ходу реакції. Зокрема, похідна активованого ефіру карбонової кислоти потрібна для здійснення реакції зв'язування із аміносполукою для формування амінозв'язку, або для реакції зв'язування із спиртом для формування ефірного зв'язку. Такі реакційні похідні включають звичайні похідні, які можна одержувати згідно з способом, який широко використовують у цій галузі техніки. Наприклад, як похідну галоідангідриду використовують хлорангідрид; а похідна активованого ефіру включає ангідрид карбоксильної кислоти, виділений з алкоксикарбонілового галоідангідриду, наприклад, метоксикарбонілхлорід, ізобутилоксикарбонілхлорід та ін., і зв'язувальний агент, N - гідроксифталімід - виділений ефір, N -гідроксисукцинімід - виділений ефір, N - гідрокси - 5 - норбонин -2', 3' -дікарбоксиімід - виділений ефір, 2, 4, 5 - трихлорфенол - виділений ефір та ін., але не обмежуючись вищезазначеним. Інгібіторна дія відносно тромбіну сполуки формули (І) згідно з цим винаходом може бути ідентифікована шляхом визначення константи дисоціації Кі, яку розраховують, ви ходячи із нижченаведеного рівнення згідно з відомим способом, який описан у літературі (див. Methods in enzymology V. 80, p341 - 361; Biochemistry 27, p2144 - 2151 (1988)). де * [E] - концентрація вільного ензиму, ** [I] - концентрація незв'язаного інгібітора, *** [El] - концентрація комплексу ензим - інгібітор. Константа дисоціації Кі означає ступінь дисоціації комплексу ензим -інгібітор тромбіну. Згідно з цим мала константа дисоціації відповідає високій ступені зв'язування інгібітора тромбіну з ензимом і, таким чином, указує на те, що інгібітор тромбіну має високу інгібіторну активність до тромбіну. Зазначена константа дисоціації може бути визначена шляхом реакції тромбіну з певним субстратом, який змінює колір, коли його гідролізують завдяки дії тромбіну, та подальшого вимірювання за допомогою спектрофотометрії ступені зміни кольору як функції часу. У цьому винаході Chromozym ТН (Gly - Pro - Arg - 4 - нітро - анілід ацетат) використовують як субстратну речовину для тромбіну, яка змінює колір під впливом дії тромбіну. Chromozym ТН гідролізують тромбіном для одержання пара -нітроаніліну жовтого кольору. Відповідним чином, кількість жовтого паранітроаніліну, одержаного вищенаведеним способом, можна вимірити як зміну спектральної поглинальної здатності протягом часу для визначення інгібіторної активності стосовно тромбіну сполуки згідно з цим винаходом. Тобто, ензимну активність можна визначити із швидкості зміни спектральної поглинальної здатності, а далі її можна безпосередньо зв'язати із здатністю інгібітора тромбіну інгібувати ензимну активність (див. Methods in Enzymology, V. 80, р341 - 361, Biochemistry 27, р2144-2151, 1988). Для ідентифікації селективності сполуки згідно з цим винаходом до тромбіну відносно трипсину, інгібіторну активність сполуки формули (І) до трипсину виміряють як величину Кі згідно із способом, який є аналогічним раніше наведеному способу для визначення інгібіторної активності до тромбіну, а далі розраховують співвідношення активності до трипсину і активності до тромбіну. У цьому випадку операція по визначенню інгібіторної активності до трипсину істотно ідентична операції по визначенню інгібіторної активності до тромбіну за винятком того, що як субстрат застосовують хлористоводневий N - бензоїл - Val - Gly - Arg пара нітроанілід . У результаті визначення інгібіторної активності сполуки формули (І) згідно з цим винаходом до тромбіну і трипсину, можна стверджувати, що сполука згідно з цим винаходом показує відмінну інгібіторну активність до тромбіну і, крім того, має високу селективність до тромбіну відносно трипсину. Зокрема, селективність сполук, що наведені у прикладах 1 і 7, до тромбіну відносно трипсину приблизно у 2900 разів і у 26304 разів відповідно більше, у той час коли аналогічні показники у відомих інгібіторів тромбіну, агротробану (А) і NAPAP (В), становлять тільки 250 і 50 разів відповідно. Таким чином, можна бачити, що сполука формули (І) згідно з цим винаходом має значну перевагу у селективності до тромбіну відносно трипсину. Як наведено вище, оскільки нова сполука формули (І) згідно з цим винаходом є інгібітором тромбіну, який показує потенційну інгібіторну активність до тромбіну навіть при оральному введенні, а також має високу селективність до тромбіну відносно трипсину, вона придатна для профілактики коагуляції крові та лікування різних тромбозів. Відповідним чином, третя ціль цього винаходу полягає в створенні фармацевтичної композиції для профілактики коагуляції крові та лікування тромбозів, яка містить сполуку формули (І) або її фармацевтично придатну сіль як активний інгредієнт. Коли сполуку формули (І) згідно з цим винаходом вводять у клінічних цілях пацієнту, щоденна доза сполуки (І) може змінюватись найкраще у діапазоні від 0,001мг до 10мг на 1кг ваги тіла. Однак, у залежності від конкретного пацієнта може бути призначено специфічне дозування, яке виходить за межі вищезазначеного діапазону доз залежно від специфічної сполуки, що застосовують, ваги, статі, стану здоров'я конкретного пацієнта, дієти, часу та способу введення, ступеня секреції сполуки, іншого активного компонента , що комбінується, і ступеня захворювання , що належить до лікування. Сполука згідно з цим винаходом може вводитися у вигляді ін'єкційноздатного препарату або орального препарату згідно з бажаною ціллю. Ін'єкційноздатний препарат, наприклад, стерилізовані ін'єкційноздатні водні або масляні суспензії, може бути вигото влений з використовуванням придатного диспергуючого агенту, зволожника або суспендуючого агенту згідно з відомим способом, що звичайно використовують у галузі виготовлення ін'єкцій. Як водний розчинник, придатний для зазначеної цілі, може бути використана вода, розчин Рінгера або ізотонічний розчин NaCI. Крім того, хоч стерилізоване нелетюче масло може використовуватися як розчинник або суспендуюче середовище, для цієї цілі можна використовувати будь - яке неподразливе нелетюче масло, включаючи моно або дигліцериди. Крім того, до ін'єкційного препарату може бути додана жирна кислота, наприклад, олеїнова кислота. Твердий препарат для орального введення може бути виготовлений у вигляді капсул, таблеток, пілюль, порошків і гранул, причому особливо придатними є капсульні та таблеткові рецептури. Таблетки і пілюлі можуть бути виго товлені переважно у вигляді препарату, покритого оболонкою. Як твердий препарат активна сполука формули (1) згідно з цим винаходом може бути скомбінована з фармацевтично придатним носієм. Наприклад, з одним або більше інертним розчинником(ами), такими як сукроза, лактоза, крохмаль та ін., змазуючими речовинами, такими як кремнекислий магній, диспергаторами, зв'язувальними речовинами та ін. Однією з характерних ознак сполуки формули (1) згідно з цим винаходом є те, що сполука формули (1) показує гарний фармакологічний ефект, навіть коли вона виготовлена у вигляді орального препарату, а потім вводиться оральним способом. Це може бути продемонстровано результатами фармакокінетичних експериментів на пацюках та собаках, які виступають у ролі експериментальних тварин. Тобто у цих експериментах було продемонстровано, що активна сполука згідно з цим винаходом зберігається у крові протягом більш тривалого часу, коли вона вводиться орально. Відповідно з цим сполука формули (1) згідно з цим винаходом є більш придатна з точки зору того факту, що її можна більш ефективно застосовувати у вигляді орального препарату. Крім того, з фармакокінетичних експериментів стало зрозуміло, що активна сполука формули (1) згідно з цим винаходом може забезпечити бажану ціль без сильної токсичності у ссавців, включаючи пацюків та собак. Коли інгібітор тромбіну згідно з цим винаходом вводять з метою досягнення антикоагуляційного та тромболітичного ефекту, його можна вводити у комбінації з однією або більшеречовиною(ами), які вибирають із групи, що містить тромболітичні агенти та агенти для інгібування тромбоцитової активності. Як тромболітичний агент, що може бути використаний з цією метою, можна згадати t - PA, урокіназу, стрептокіназу та ін.; як агент для інгібування тромбоцитової активності можна використовувати аспірин, тіклопідин, клопідрогель, моноклональне антитіло 7ЕЗ та ін. Однак, необхідно зрозуміти, що препарат, який містить активну сполуку згідно з цим винаходом для лікування та профілактики тромбозів, не обмежується вищенаведеними і до них може бути включений будьякий препарат, який придатний для тієї самої цілі. Нижче наведені приклади, за допомогою яких більш детально пояснюється цей винахід. Однак, треба зрозуміти, що зазначені приклади призначені тільки для пояснення цього винаходу і ніякою мірою не обмежують його об'єм. Препарат 1 Синтез циклопентил-метиламіну До розчину циклопентанону ( 10мл, 113мМ) у метанолі (50мл) і воді (50мл) додавали хлористоводневий метиламін (7,6г, 113ммол) і ціаноборогідрид натрію (NaBH3CN) (7,1г, 113ммол). Суміш нагрівали до стану флегми протягом 12 годин при pH 6. Метанол випарювали при зменшеному тиску та осад охолоджували до температури 0°С, регулюючи pH до величини, що дорівнює 2, використовуючи 3N соляну кислоту, а потім промивали тричі диетиловим ефіром. Водний шар знову о холоджували до температури 0°С, а потім регулювали pH до величини, що дорівнювала 11, використовуючи розчин їдкого натру (6N). До цієї суміші додавали ангідрид t-бутилоксикарбонілу (24,5г, 113ммол) у діоксані (50мл). Розчин перемішували протягом 3 годин при кімнатній температурі і концентрували при зменшеному тиску до одержання об'єму, що дорівнював приблизно 30мл. Осад екстрагували етилацетатом і промивали за допомогою водної 0,5N соляної кислоти та насичували розчинами бікарбонату натрію. Органічний шар висушували над безводним сульфатом магнію, фільтрували та концентрували при зменшеному тиску з метою одержання твердого тіла білого кольору, яке потім очищали за допомогою капілярної хроматографії (розчин становив собою етил ацетат : гексан у об'ємному співвідношенні 7:3). Після очищення одержаний твердий продукт розчинювали у розчині 4N НСІ діоксан (60мл) та одержаний розчин перемішували протягом 30 хвилин при кімнатній температурі. Розчинник випарювали у вакуумі з метою одержання вищеназваної сполуки (13,7г вихідного продукту : 90,5 %). 1 Н ЯМР*О 3OD, чнм**) : 3,50 (м, 1Н), 2,68 (s, 3Н), 2,10(м, 2Н), 1,86 - 1,50 (м, 6Н) - * Ядерний магнітний резонанс - ** Частин на мільйон Препарат 2 Синтез (S) - N - циклопентил - N - метил - 3 - (4 - ціанофеніл) - 2 - (бут -ілоксикарбоніламіно)пропіонаміду До розчину (S) - 3 - (4 - ціанофеніл) - 2 (бутилоксикарбоніламіно)пропіонової кислоти (0,7г, 2,41ммол) у диметилформаміді (ДМФ, 6мл) додавали хлористоводневий 1 - (3 -диметиламінопропіл) - 3 - етилкарбодіімід гідрохлорид (ЕДХ, 0,7г) і 1-гідроксибензотриазолгідрат (ГОБТ, 0,4г) при температурі 0°С. Суміш перемішували, доки речовини повністю не розчинювались у зазначеному розчині. До цієї реакційної суміші додавали сполуку (0,4г, 2,96ммол), одержану як Препарат 1, та N - метилморфолін (1,0мл), а потім температуру реакції повільно збільшували до кімнатної температури. Реакційний розчин перемішували протягом 3,5 годин. Після закінчення реакції реакційний розчин концентрували при зменшеному тиску для видалення летючих речовин, а осад, що залишився, розчинювали за допомогою етилацетату, послідовно промивали водним насиченим розчином кислої солі карбонату натрію, розчиняли соляною кислотою і сольовим розчином, висушували над безводним сульфатом натрію, фільтрували, а потім концентрували. Осад очищали за допомогою колонкової хроматографії (при об'ємному співвідношенні етилацетат : гексан, що дорівнював 7 : 3) для одержання очищеної вищеназваної сполуки (0,65г , вихід : 73 %). 1 НЯМР(СDСІ3, чнм) d : 7.61 (m, 2H) , 7.32 (m, 2Н) , 5.48, 5.01-4.86, 4.12(3m, 3Н), 2.75, 2.62(2s, 3Н), 2.901.20(m, 17Н) Маса (ФСА, m/е* ) : 372(M++1) * відношення маси до заряду Препарат 3 Синтез (S) - N -циклопентил - N - метил -3 - (4 - ціанофеніл) –2-(2-нафтилсульфоніламіно)пропіонаміду Сполуку (0,65г, 1,75ммол), виготовлену як Препарат 2, розчинювали у дихлорметані (3мл), а потім охолоджували до температури -10°С та додавали до неї трифтороцтову кислоту (ТФК, 1мл). Реакційну суміш перемішували протягом 5 хвилин, повільно нагрівали до кімнатної температури, перемішували протягом 30 хвилин, а далі концентрували при зменшеному тиску з метою видалення летючих речовин. Осад сушили за допомогою вакуумного насосу, а далі додавали до неї 6мл ДМК. Зазначену суміш охолоджували до 0°С та до неї додавали 1мл N, N - діізопропілетіламіну. Реакційну суміш нагрівали до кімнатної температури і перемішували протягом 5 хвилин. Після додавання 2 -нафталінсульфонілхлориду (0,47г, 2,07ммол) реакційну суміш перемішували протягом 1 години до закінчення реакції, а потім концентрували при зменшеному тиску з метою видалення летючих речовин. Осад розчинювали за допомогою етилацетату, промивали у насиченому розчині кислої солі карбонату натрію і соляному розчині, сушили над безводним сульфатом магнію, фільтрували і концентрували. Осад очищали за допомогою колонкової хроматографії (при об'ємному співвідношенні етилацетату : гексану, що дорівнював 1:1) для одержання вищеназваної сполуки (0,65г виходного продукту, 80,2%). 1 Н ЯМР (CDCL3, чнм) d : 8.28 (m, 1H) , 7.87 (m, 3Н), 7.73(m, 3Н) , 7.49(m, 2Н) , 5.92(m, 1Н), 4.50, 4.32, 3.76(m, m, m, 2Н), 2 . 95 (m, 2Н). 2.36, 2.22(s, s, 3Н), 1.60-1.20 (m, 6Н), 0.98, 0.80, 0.47(m, m, m, 2Н) Маса (ФСА, m/е) : 462 (М+ + 1) Приклад 1 Синтез (S) - N - циклопентил - N - метил-3 - (4 - амідразонофеніл)-2-(2-нафтилсуль фоніламіно) пропіонаміду Сполуку (0,65г, 1,41ммол), виготовлену як Препарат 3, розчинювали у піридині (10мл) і одержаний розчин розміщували у обладнану відведеннями колбу, у яку додавали триетиламін (0,45мл). Реакційна колба була обладнана таким чином, щоб сірководень (H2S) був здатний повільно утікати крізь одне з відведень колби, а витікати крізь друге відведення. Реакційний розчин насичували сірководнем, постійно перемішуючи протягом приблизно 10 хвилин, при цьому безкольоровий розчин ставав спочатку зеленого кольору , а далі поступово переходив до темнокоричневого кольору. Колбу закривали за допомогою гумової пробки і зберігали ЇЇ протягом 3 діб при кімнатній температурі до закінчення реакції. Потім реакційний розчин дистилювали при зменшеному тиску для видалення летючих речовин і висушували за допомогою вакуумного насосу. Для одержання твердого тіла жовтого кольору додавали разом ацетон (15мл) і йодометан (СН3І, 0,65мл) і одержану суміш нагрівали протягом 30 хвилин до одержання флегми. Зазначену реакційну суміш знову дистилювали при зменшеному тиску для видалення летючих речовин та висушували за допомогою вакуумного насосу. Осад розчинювали у чистому метанолі (8мл) і потім перемішували. До цієї суміші тричі порційно додавали 80 % - ний гідрат гідразину (H2NNH2 H2O, 0,12мл, 1,98ммол) через кожні 10 хвилин. Після закінчення реакції реакційний розчин концентрували, а потім очищали за допомогою рідинної хроматографії високого тиску (РХВТ) для одержання названої сполуки (0,63г, вихід : 73%). Умови РХВТ : - розчинник - метанол : вода (об'ємне співвідношення 75:25), кожен з яких містить CF3COOH з концентрацією 0,1% - довжина хвилі - 215нм - швидкість розчинення - 20мл/хв - колона Delta РАК С 18 100 ангстрем (30х300мм) 1 Н ЯМР (CD3OD, чнм) d : 8.28(d, 1Н) , 7.92 (m, 3Н) , 7.70-7.50(m, 5Н) , 7.35(dd, 2H) , 4.60, 4.45(t, t, 1H) , 4.12, 3.99(m, m, 1Н), 3.00, 2.87(m, m, 2Н) , 2.49, 2.26(s, s, 3Н), 1.60-1.00, 0.73-0.52(m, m, 8Н) Маса (ФСА, м/е) : 494 (М++1) Препарат 4 Синтез солі бутил-метиламінтрифтороцтової кислоти N - бутилоксикарбонілбутиламін (140мг, 0,80ммол) розчинювали у ДМФ (8мл) і туди додавали їдкий натр (NaH, 20мг, 1 еквівалентна вага) та йодометан (0,10мл, 2 еквівалентні ваги). Реакційну суміш перемішували протягом 30 хвилин при кімнатній температурі і фільтрували крізь шар целіту і концентрували при зменшеному тиску для видалення розчинника. Осад розчинювали за допомогою етилацетату, промивали у водному розчині соляної кислоти (0,5N), сушили над безводним сульфатом магнію і фільтрували. Органічний шар концентрували при зменшеному тиску і сушили за допомогою вакуумного насосу до одержання твердого тіла білого кольору, яке далі розчинювали у дихлорметані і охолоджували до температури 0°С. До цієї суміші додавали 1мл трифтороцтової кислоти (ТФК). Реакційний розчин перемішували протягом 30 хвилин при кімнатній температурі, концентрували при зменшеному тиску, а далі сушили за допомогою вакуумного насосу для одержання вищеназваної сполуки (0,16г) у кількісному виході. 1 Н ЯМР (CDCL3, чнм) d : 1.02(t, 3Н), 1.30-1.80(m, 4H), 3.04(s, 3Н), 3.52(t, 2Н) , 8.20(s, 2Н) Приклад 2 Синтез (S) - N - бутил - N - метил - 3 -(4 - амідразонофеніл) - 2 - (2 -нафтилсульфоніламіно)пропіонаміду Сполуку, вигото влену як Препарат 4, піддавали реакції згідно з тими ж самими операціями, що і препарати 2 і 3 з метою одержання проміжної сполуки (S) - N - бутил - N - метил - 3 - (4 - ціанофеніл) - 2 - (2 нафтилсуль фоніламіно)пропіонаміду (0,23г), яку використовували як вихідний матеріал. Зазначений вихідний матеріал оброблювали згідно з тими же операціями, що і у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,1г, вихід: 40,0%). 1 Н ЯМР (CD3OD, чнм) d : 8.30(d, 1Н), 7.98(m, 3Н), 7.81-7.30(m, 7Н), 4.50(m, 1Н), 3.25-2.55(m, 4Н) , 2.78, 2.45(2s, 3Н), 1.40-0.50(m, 7H) Маса (ФСА, м/е) : 482(М+ + 1) Приклад 3 Синтез (S)-N- циклопентил-N-пропил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно)пропіонаміду Для одержання хлористоводневого циклопентил-пропіламіну виконували такі ж самі операції, що і для одержання Препарату 1, за винятком того, що замість метиламіну застосовували пропиламін. Далі зазначений хлористоводневий циклопентил-пропиламін оброблювали тим же самим способом, що і Препарати 2 і 3 для одержання проміжної сполуки -(S)-N-циклопентил-N-пропіл-3-(4-ціанофеніл)-2-(2- нафтилсульфоніламіно) пропіонаміду (0,15г). Зазначену проміжну сполуку використовували як вихідний матеріал та оброблювали згідно з тим ж самим способом, що і у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,089г, вихід : 55,1 %). 1 Н ЯМР (CD3OD, чнм) d : 8.35-7.35(m, 11Н), 4.62-4.32(m, m, 1H) , 3.90, 3.70(m, m, 1H) , 3.10-2.50(m, 4H), 1.70-0.50(m, 13H) Маса (ФСА, m/e) : 522(M++1) Приклад 4 Синтез (S)-N-циклопентил-N-(2 -бензилоксиетил)-3-(4 –амідразонофеніл)-2-(2-нафтилсуль фоніламіно) пропіонаміду Для одержання хлористоводневого циклопентил -(2 бензилоксиетил)аміну виконували такі ж самі операції, що і для одержання Препарату 1, за винятком того, що замість метиламіну застосовували пропіламін. Далі одержану сполуку оброблювали згідно з тим же самим способом, що і Препарати 2 і 3, для одержання проміжної сполуки (S)-N-циклопентил-N-(2-бензилоксиетил)-3-(4-ціанофеніл)-2-(2-нафтилсульфоніламіно) пропіонамід (0,15г). Зазначену проміжну сполуку використовували як вихідний матеріал та оброблювали згідно з тим ж самим способом, що і у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,15г, вихід : 93,8%). 1 Н ЯМР (CD3OD, чнм) d : 8. 30-7.15 (m, 16Н), 4.64 (m, 1Н), 4.48, 4.39(s, s, 1Н), 4.14, 4.00, 3.75, 3.45(m, m, m, m, 3Н), 3.10-2.70(m, 5Н), 1.62-1.00(m, 8H) Маса(ФСА, м/е) : 614(М++1) Приклад 5 Синтез (S)-N-циклопентил-N-бутил-3-(4-амідразонофеніл)-2-(2-нафтилсуль фоніламіно)пропіонаміду Для одержання хлористоводневого бутил-циклопентиламіну виконували такі ж самі операції, що і для одержання Препарату 1, за винятком того, що замість метиламіну застосовували бутиламін. Далі зазначену сполуку оброблювали тим же самим способом, що і Препарати 2 і 3 для одержання проміжної сполуки (S)-Nциклопентил-N-бутил-3-(4-ціанофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід (0,28г). Зазначену проміжну сполуку використовували як вихідний матеріал та оброблювали згідно з тим же самим способом, що і у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,18г, вихід : 60%). 1 Н ЯМР (CD3 OD, чнм) 6 : 8.32(d, 1H), 7.96(m, 3Н), 7.78-7.55(m, 5Н), 7.42(dd, 2H), 4.62, 4.30(m, m, 1Н), 4.02, 3.90(m, m, 1Н), 3.10-2.75, 2.55 (m, m, 4Н), 1.65-0.80(m, 12Н), 0.65(t, 3Н) Маса (ФСА, м/e) : 536(М++ 1) Приклад 6 Cсинтез (S)-N-циклопентил-N-етил-3-(4-амідразонофеніл)-2-(2- нафтилсульфоніламіно)пропіонаміду Для одержання хлористоводневого циклопентил-етиламіну виконували такі ж самі операції, що і для одержання Препарату 1, за винятком того, що замість метиламіну застосовували етиламін. Далі зазначену сполуку оброблювали тим же самим способом, що і Препарати 2 і 3 для одержання проміжної сполуки (S)-Nциклопентил -N-етил-3-(4-ціанофеніл)-2-(2-нафтилсульфоніламіно)пропіонамід (0,21г). Зазначену проміжну сполуку використовували як вихідний матеріал та оброблювали згідно з тим ж самим способом, що і у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,11г, вихід : 50%). 1 Н ЯМР (CD3OD, чнм) d : 8.32 (d, 1Н), 7.97 (m, 3Н), 7.75-7.55(m, 5Н), 7.42(m, 2Н), 4.60, 4.38(m, m, 1Н), 3.98, 3.87(m, m, 1Н), 3.20-2.70(m, 4Н), 1.65-1.00(m, 8Н), 0.95, 0.58(t, t, 3Н) Маса (ФСА, m/е) : 508(М++1) Приклад 7 Синтез (S)-N-циклопентил-N-метил-3-[4-(метиламіндіно)-феніл]-2-(2-нафтилсульфоніламіно) пропіонаміду Проміжну сполуку, виготовлену як Препарат 3, використовували як вихідний матеріал і оброблювали згідно з тим же способом, як і у Прикладі 1, за винятком того, що замість гідразину тричі додавали метиламін з інтервалами в 1 годину для одержання очищеної вищеназваної сполуки (0,064г, вихід : 8%). 1 Н ЯМР (CD3OD, чнм) d : 0.50-1.60 (m, 8Н), 2.00-2.49 (2s, 3Н), 2.84(m, 1Н), 2.96(m, 1Н), 3.00(s, 3Н), 4.05(m, 1Н), 4.50(m, 1Н), 7.20-8.30(m, 11Н) Маса(ФСА, м/е) : 493(М++1) Приклад 8 Синтез (S)-N-циклопентил-N-метил-3-[4-(1,1-диметиламідіно)-феніл]-2-(2-нафтилсуль фоніламіно) пропіонаміду Проміжну сполуку, виготовлену як Препарат 3, використовували як вихідний матеріал і оброблювали згідно з тим же способом, як і у Прикладі 1, за винятком того, що замість гідразину тричі додавали диметиламін з інтервалами в 1 годину для одержання очищеної вищеназваної сполуки (0,18г, вихід : 22%). 1 Н ЯМР (CD3 OD, чнм) d : 0.60-1.60(m, 8Н), 2.29, 2.54(2s, 3Н), 2.95(m, 1Н), 3.06(m, 1Н), 3.09(s, 3Н), 3.31(s, 3Н) , 4.16(m, 1Н) , 4.60(m, 1Н), 7.20-8.30(m, 11Н) Маса(ФСА, т/е) : 507(М+ + 1) Приклад 9 Сиатез (S)-N-циклопентил-N-метил-3-(4-амідразонофеніл)-2-[(4-метокси-2,3,6-триметилбензол) сульфоніламіно] пропіонаміду Сполуку, виготовлену як Препарат 2, оброблювали згідно з тим же самим способом, як і Препарат 3, за винятком того, що замість 2-нафталінсульфохлориду використовували 4-метокси-2,3,6триметилбензолсульфохлорид для одержання проміжної сполуки (S)-N-циклопентил-N-метил-3-(4ціанофеніл)-2-(4-метокси-2,3,6-триметилбензолсульфоніламіно)пропіонамід (0,27г). Цю проміжну сполуку використовували як вихідний матеріал і оброблювали згідно з тим же способом, як і у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,16г, вихід : 57,1%). 1 Н ЯМР (CD3OD, чнм ) d : 7.62(d, 2Н), 7.40(dd, 2Н), 6.70(d, 1Н), 4.35, 3.90(m, m, 2H), 3.83(d, 3Н), 3.00(m, 2Н), 2.50(m, 9Н), 2.1(s, 3Н), 1.75-0.90(m, 8Н) Маса(ФСА, м/е) : 516(М++1) Препарат 5 Синтез циклопентил-гідроксиламіну Хлористоводневий гідроксиламін (H2NOH HCI, 5,0г, 71,95ммол) розчинювали у воді (14мл) і до нього додавали метанол (30мл) і циклопентанон (5,1мл, 57,66ммол). Суміш перемішували, охолоджували до температури 0°С, а далі регулювали pH до величини, що дорівнювала 8, шляхом додавання 6N водного розчину гідроксиду натрію. Потім до зазначеної суміші додавали ціаноборогідрид натрію (NaBH3CN, 1,9г, 30,24ммол). Суміш нагрівали до кімнатної температури, а потім перемішували. Підтримували величину pH реакційної суміші, що дорівнювала 4,0, за допомогою порційного додавання перемішаного розчину 6N НСІ (20мл) і метанолу (30мл) у процесі реакції. Після 5 годин pH реакційної суміші регулювали до величини, що дорівнювала 7,0, та дистилювали при зменшеному тиску з метою видалення метанолу. Залишковий реакційний розчин охолоджували до температури 0°С, знову регулювали pH до величини, що дорівнювала 11, насичували хлорідом натрію, а далі чотири рази екстрагували за допомогою хлороформу. Одержаний екстракт сушили над безводним сульфатом магнію, фільтрували і концентрували для одержання вищезазначеної сполуки (3,4г, вихід : 58,3 %). 1 Н ЯМР(СDСL3, чнм) d : 7.20-5.00(bs, 1H), 3.56 (m, 1H), 1.90-1.45 (m, 9H) Приклад 10 Синтез (S)-N-циклопентил-N-гідрокси-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно) пропіонаміду Для одержання проміжної сполуки (S)-N-циклопентил-N-гідрокси-3-(4-ціанофеніл)-2-(2нафтилсуль фоніламіно) пропіонамід (0,1г) сполуку, виготовлену як Препарат 5, оброблювали згідно з тим же самим способом, що і Препарати 2 і 3. Зазначену проміжну сполуку використовували як вихідний матеріал та оброблювали згідно з тим ж самим способом, що і у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,07г, вихід : 63,6 %). 1 Н ЯМР(СD3ОО, чнм) d : 8.25-7.30(m, 11Н), 4.75(m, 1Н), 4.30(m, 1Н), 3.10, 2.75(m, m, 2Н), 1.70-1.00(m, 8Н) Маса(ФСА, m/е) : 496(М++1) Приклад 11 Синтез (S)-N-циклопентил-N-(2-гідроксиетил)-3-(4-амідразонофеніл)-2(2-нафтилсуль фоніламіно) пропіонаміду Сполуку (0,15г, 0,24ммол), виготовлену так, як це описано у Прикладі 4, розчинювали у метанолі (10мл), а потім до неї додавали гідрооксид паладію (0,02г). До реакційної судини прикріплювали балон з воднем. Після перемішування протягом 2 днів реакційну суміш фільтрували крізь шар целіту і концентрували. Осад очищали за допомогою РХВТ для одержання вищеназваної сполуки (0,08г, вихід : 63,7%). Умови проведення РХВТ були ті ж самі, що і у Прикладі 1. 1 Н ЯМР(СD3OD, чнм) d : 7.80-7.00(m, 11Н), 4.47(m, 1Н), 4.00(m, 1Н), 3.60(m, 2Н), 3.10-2.70(m, 4Н), 1.701.10(m, 8Н) Маса (ФСА, m/е) : 524(М++1) Приклад 12 Синтез (S)-N-циклопентил-N-метил-3-[4-(метиламідіно)-феніл]-2-(4-метокси-2,3,6триметилбензолсульфоніламіно) пропіонаміду Для одержання очищеної вищеназваної сполуки (0,08г, вихід : 28,3%) використовували той же самий спосіб, що і у Прикладі 9, за винятком того, що замість гідразину тричі додавали метиламін з інтервалом в 1 годину. 1 Н ЯМР(СD3OD, чнм) d : 7.65(d, 2Н), 7.40(dd, 2Н), 6.70(d, 1Н), 4.40, 3.90(m, m, 2Н), 3.85(d, 3Н), 3.20-2.90(m, 5Н), 2.55(m, 9Н), 2.10(s, 3Н), 1.75-0.90(m, 8Н) Маса(ФСА, m/е) : 515(М++1) Приклад 13 Синтез (S)-N,N-диметил-3-(4-амідразонофеніл)-2-(2-нафтилсульфоніламіно) пропіонаміду Такі ж самі операції, що і при виготовленні Препаратів 2 і 3, за винятком того, що використовували диметиламін, виконували з метою одержання проміжної сполуки (S) -N,N-диметил-3 -(4- ціанофеніл)-2 -(2нафтилсуль фоніламіно) пропіонаміду (0,11г). Зазначену проміжну сполуку використовували як вихідний матеріал і оброблювали згідно з тими ж самими операціями, що указані у Прикладі 1 для одержання очищеної вищеназваної сполуки (0, 080г, вихід : 53%) 1 Н ЯМР(СD3OD, чнм) d : 2.19(s, 3Н), 2.62(s, 3Н) , 2.70-2.95(m, 2Н), 4.41(m, 1H), 7.20-8.30(m, 11Н) Маса (ФСА, m/е) : 440 (М+ + 1) Приклад 14 Синтез (S) - N,N -диметил - 3 - [4 - (метиламідіно)феніл] –2-(2-нафтилсульфоніламіно) пропіонаміду Для одержання очищеної вищеназваної сполуки (0,10г, вихід : 48%) використовували той же самий спосіб, що і у Прикладі 1, за винятком того, що замість гідразину тричі додавали метиламін з інтервалом в 1 годину. 1 Н ЯМР (CD3OD, чнм) d : 2.31(s, 3Н), 2.75(s, 3Н), 2.80-3.05(m, 2Н), 3.07(s, 3Н), 4.54 (m, 1Н), 7.30-8.40(m, 11Н) Маса (ФСА, м/е) : 439(М++1) Приклад 15 Синтез (S)-N-циклогексил-N-метил-3-(4-амідразоно феніл)-2(2-нафтилсульфоніламіно) пропіонаміду Такі ж самі операції, що і при виготовленні препарату 1, за винятком того , що використовували циклогексанон замість циклопентанону, виконували з метою одержання хлористоводневого циклогексил метиламіну, який оброблювали згідно з тими ж самими операціями, що і при виготовленні препаратів 2 і 3 для одержання проміжної сполуки (S)-N-циклогексил-N-метил-3-(4-ціанофеніл)-2-(2-нафтилсульфоніламіно) пропіонаміду (0,21г). Зазначену проміжну сполуку використовували як вихідний матеріал і оброблювали згідно з тими ж самими операціями, що указані у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,17г, вихід : 73,9%) 1 Н ЯМР (CD3OD, чнм) d : 8.32(d, 1Н), 7.95 (m, 3Н), 7.75-7.53(m, 5Н), 7.40(dd, 2H), 4.50, 4.21, 3.62(m, m, m, 2Н), 3.05(m, 1Н), 2.90(m, 1Н), 2.55, 2.40(s, s, 3Н), 1.80-0.62(m, 10Н) Маса(ФСА, м/е) : 508(М++1) Приклад 16 Синтез (S)-N-циклопропіл-N-метил-3-(4-амідразонофеніл)-2(2-нафтилсуль фоніламіно)пропіонаміду Такі ж самі операції, що і при виготовленні Препарату 1, за винятком того , що використовували циклопропанон замість циклопентанону, виконували з метою одержання хлористоводневого циклопропил метиламіну, який оброблювали згідно з тими ж самими операціями, що і при виготовленні препаратів 2 і 3, для одержання проміжної сполуки (S)-N-циклопропил-N-метил-3-(4-ціанофеніл)-2-(2-нафтилсульфоніламіно) пропіонаміду (0,21г). Зазначену проміжну сполуку використовували як вихідний матеріал і "оброблювали згідно з тими ж самими операціями, що указані у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,06г, ви хід : 12%) 1 Н ЯМР (CD3 OD, чнм) d : 0.40-0.90(m, 4Н), 2.40(s, 3Н), 2.75(m, 1Н), 2.95(m, 1Н), 7.20-8.30(m, 11Н) Маса (ФСА, м/е) : 466 (М++ 1) Препарат 5 Синтез(S)-3-(4-ціанофеніл)-N-циклопентил-N-метил-2-(2-нафталін-1-іл-ацетиламіно)пропіонаміду Сполуку (0,51г, 1,34ммол), виготовлену як Препарат 2, розчинювали у 3мл дихлорметану і охолоджували до -10°C, а далі у одержаний розчин додавали трифтороцтову кислоту (ТФК, 3мл). Реакційну суміш перемішували протягом 5 хвилин, повільно нагрівали до кімнатної температури, знову перемішували протягом ЗО хвилин, а потім дистилювали при зменшеному тиску з метою видалення летючої речовини. Осад сушили за допомогою вакуумного насосу, а далі до нього додавали 10мл ДМК. Зазначений розчин охолоджували до 10°С і додавали до нього хлористоводневий 1-(3-диметиламінопропіл)-3-етилкарбодіімід (ЕДХ, 0,4г) і 1гідроксибензотриазол (ГОБТ, 0,2г), а далі перемішували до повного завершення розчинення. До одержаного розчину додавали 1-нафталін оцетову кислоту (0,26г, 1,4ммол) і N,N -діізопропілетиламін (1,2мл). Реакційну суміш повільно нагрівали до кімнатної температури і перемішували протягом 3 годин. Після завершення реакції реакційний розчин дистилювали при зменшеному тиску з метою видалення летючої речовини. Осад розчинювали за допомогою етилацетату, послідовно промивали насиченим водним розчином кислої солі карбонату натрію (NaHCO3), розчином соляної кислоти і сольовим розчином, сушили над безводним сульфатом магнію і далі фільтрували. Одержаний фільтрат концентрували і осад очищали за допомогою колонкової хроматографії (об'ємне співвідношення метанол : хлороформ дорівнювало 1 : 99) для одержання вищеназваної сполуки (0,42г, вихід : 68%). 1 Н ЯМР (CDCL3, чнм) d : 8.0-6.8(m, 11H), 6.5 (m, 1Н), 5.1-5.3(m, 1Н), 4.8-4.2(m, 1Н), 4.1-3.8(m, 2Н), 3.02.6(m, 5Н), 1.8-1.3(m, 8Н) Приклад 17 Синтез (S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(2-нафталін-1- іл ацетиламіно)пропіонаміду Сполуку (0,21г, 0.48ммол), виготовлену як Препарат 6, розчинювали у піридині (3мл) і одержаний розчин розміщували у обладнану відведеннями колбу, у яку додавали триетиламін (0,2мл). Реакційна колба була обладнана таким чином, щоб сірководень (H2S) був здатний повільно утікати крізь одне з відведень колби, а витікати крізь друге відведення. Реакційний розчин насичували сірководнем, постійно перемішуючи протягом приблизно 10 хвилин, при цьому безкольоровий розчин ставав спочатку зеленого кольору , а далі поступово переходив до темнокоричневого кольору. Колбу закривали за допомогою гумової пробки і зберігали її протягом 3 діб при кімнатній температурі до закінчення реакції. Потім реакційний розчин дистилювали при зменшеному тиску для видалення летючих речовин і висушували за допомогою вакуумного насосу. До одержаного твердого тіла жовтого кольору додавали разом ацетон (10мл) і йодометан (СН3І, 0,3мл) і одержану суміш нагрівали до одержання флегми протягом 30 хвилин. Зазначену реакційну суміш знову дистилювали при зменшеному тиску для видалення летючих речовин та висушували за допомогою вакуумного насосу. Осад дистилювали у чистому метанолі (5мл) і потім перемішували. До зазначеної суміші тричі порційно з інтервалом 10 хвилин додавали 80 - процентний гідрат гідразину (H2NNH2 × Н2О, 0,04мл, 0,72ммол). Після завершення реакції реакційний розчин концентрували, а потім очищали за допомогою РХВТ для одержання вищеназваної сполуки (0,16г, вихід : 70%). 1 Н ЯМР (CD3OD, чнм) d : 8.0-7.3(m, 11Н), 5.3-5.1(m, 1Н), 4.8-4.2(m, 1Н), 3.9(s, 2H). 3.2-2.9(m, 2Н), 2.78, 2.72 (2s, 3Н), 1.8-1.3(m, 8Н) Маса(ФСА, м/е) : 472(М++1) Приклад 18 Синтез (S)-3-[4-(амідразоно)феніл]-N-циклопентил-N-метил-2-(5-диметиламінонафталін-1сульфоніламіно) пропіонаміду Такі ж самі операції, що і при виготовленні Препарату 3, за винятком того , що замість 2 нафтилсуль фонілхлоріду використовували (N,N-диметиламіно)-1-нафталінсульфоніл хлорід, виконували з метою одержання проміжно/ сполуки (S)-3-(4-ціаноонофеніл)-N-циклопентил-N-метил-2-(5-диметиламінонафталін-1-сульфоніламіно) пропіонаміду (0,55г, 1,09ммол). Зазначену проміжну сполуку далі оброблювали згідно з тими ж самими операціями, що указані у Прикладі 1 для одержання очищеної вищеназваної сполуки (0,35г вихідного продукта : 60%) 1 Н ЯMP(CD3OD, чнм) d : 8.6-7.3(m, 10Н), 4.6-3.9(m, 2Н), 3.11(s, 3Н), 3.0(s, 3Н), 3.05-2.8(m, 2Н), 2.5, 2.4(2s, 3Н), 1.7-0.9(m, 10Н) Маса(ФСА, m/е) : 537(М++1) Приклад 19 Синтез (S)-3-[4-амідразоно)-пентил]-N-циклопентил-N-метил-2-(5-диметокси-нафталін-1- сульфоніламіно) пропіонаміду Такі ж самі операції, що і при виготовленні препарату 3, за винятком того, що замість 2нафталінсульфохлориду використовували 5-метокси-1-нафтилсульфо хлоріду, виконували з метою одержання проміжної сполуки (S)-3-(4-ціанофеніл)-N-циклопентил-N-метил-2-(5-метокси-нафталін-1-сульфоніламіно) пропіонаміду (0,18г, 0,37ммол).3азначену проміжну сполуку далі оброблювали згідно з тими ж самими операціями, що указані у Прикладі 1 для одержання очищеної вищеназваної сполуки (0,12г вихідного продукта : 65%) 1 Н ЯМР (CD3OD, чнм) d : 8.4-7.3 (m, 9Н), 4.7-4.0 (m, 2Н), 4.2-4.0 (2s, 6H), 3.2-2.8 (m, 2H), 2.6-2.2(2s, 3Н), 1.71.0(m, 8Н) Маса (ФСА, m/е) : 523 (М++1) Приклад 20 Синтез (S)-3-[4-амідразоно)-феніл]-N-циклопентил-N-метил-2-(6,7-диметокси-нафталін-2сульфоніламіно)пропіонаміду Такі ж самі операції, що і при виготовленні препарату 3, за винятком того , що замість 2 нафталінсульфохлоріду використовували 6,7-диметокси-2-нафталінсульфохлорід, виконували з метою одержання проміжної сполуки (S)-3-(4-ціанофеніл)-N-циклопентил-N-метил-2-(6,7-диметокси-нафталін-2сульфоніламіно) пропіонаміду (2,2г, 2,2ммол). Потім зазначену проміжну сполуку оброблювали згідно з тими ж самими операціями, що описані у Прикладі 1, для одержання очищеної вищеназваної сполуки (1,55г, вихід : 67%). 1 Н ЯМР (CD3OD, чнм) d : 8.5-7.0 (m, 10Н), 4.6-4.0 (m, 2Н), 4.2(s, 3Н), 3.0-2.8(m, 2Н), 2.48-2.45(2s, 3Н), 1.71.0(m, 8Н) Маса (ФСА, м/е) : 554(М++1) Приклад 21 Синтез (S)-3-[4-метиламідіно)-феніл]-N-циклопентил-N-метил-2-(5-диметиламіно-нафталін-1сульфоніламіно)пропіонаміду Такі ж самі операції, що і при виготовленні препарату 3, за винятком того, що замість 2нафталінсульфохлоріду використовували 5-(N,N-диметиламіно)-1-нафталінсульфо хлорід, виконували з метою одержання проміжної сполуки (S)-3-(4-ціанофеніл)-N-циклопентил-N-метил-2-(5-диметиламінонафталін-1-сульфоніламіно) пропіонаміду (0,3г, 0,6ммол). Потім зазначену проміжну сполуку оброблювали згідно з тими ж самими операціями, що описані у Прикладі 1, за винятком того, що для одержання очищеної вищеназваної сполуки (0,08г, вихід : 25%) замість 80% гідрату гідразину використовували метиламін. 1 Н ЯМР (CD3OD, чнм) d : 8.52-7.25(m, 10H), 4.55-4.42(m, 2Н), 4.23-3.98(m, 2Н), 3.09(s, 3Н), 3.06-2.80(m, 2Н), 2.90(m, 6Н), 2.45(s, 3Н), 2.38(s, 3Н), 1.76-0.90(m, 8Н) Маса(ФСА, м/е) : 536(М++1) Приклад 22 Синтез (S)-3-[4-амідразоно)-феніл]-N-циклопентил-N-метил-2-(нафталін-1-сульфоніламіно)-пропіонаміду Такі ж самі операції, що і при виготовленні Препарату 3, за винятком того , що замість 2нафталінсульфохлориду використовували 1-нафталінсульфо хлорид, виконували з метою одержання проміжної сполуки (S)-3-(4-ціанофеніл)-N-циклопентил-N-метил-2-(нафталін-1-сульфоніламіно) пропіонаміду (0,5г, 1ммол). Зазначену проміжну сполуку оброблювали згідно з тими ж самими операціями, що описані у Прикладі 1, для одержання очищеної вищеназваної сполуки (0,2г, вихід : 40%). 1 Н ЯМР (CD3OD, чнм) d : 8.61-7.28(m, 11Н), 4.55-4.41(m, 2Н), 4.23-4.00(m, 2Н), 3.00-2.84 (m, 2Н), 2.49, 2.41(2s, 3H), 1.70-1.00(m, 8H) Маса(ФСА, м/е) : 494 (М++ 1) Приклад 23 Синтез (S)-3-[4-амідразоно)-феніл]-N-циклопентил-N-метил-2-[2-(нафталін-1-іл-окси)ацеталаміно]пропіонаміду Такі ж самі операції, що і при виготовленні препарату 6, за винятком того , що замість 1 - нафталін оцтової кислоти використовували (1-нафтокси) оцтову кислоту, виконували з метою одержання проміжної сполуки (S)3-(4-ціанофеніл)-N-циклопентил-N-метил-2-[2-(нафталін-1-ілокси)-ацетиламіно]-пропіонамід (0,59г, 1,3ммол). Зазначену проміжну сполуку потім оброблювали згідно з тими ж самими операціями, що описані у Прикладі 17, для одержання очищеної вищеназваної сполуки (0,38г, вихід : 60%). 1 Н ЯМР (CD3OD, чнм) d : 8.25-6.8(m, 11Н), 5.38-5.22 (m, 2Н), 4.77-4.39(m, 2Н), 4.70(s, 2H), 3.21-3.05(m, 2Н), 2.88-2.77 (2s, 3Н), 1.92-1.40(m, 8Н) Маса (ФСА, м/е) : 488 (М+ + 1) Приклад 24 Синтез (S)-3-[4-амідразоно)-феніл]-N-циклопентил-N-метил-2-[2-(нафталін-2-іл-окси)ацетиламіно]пропіонаміду Такі ж самі операції, що і при виготовленні препарату 3, за винятком того , що замість 1 - нафталін оцтової кислоти використовували (2-нафтокси) оцтову кислоту, виконували з метою одержання проміжної сполуки (S)3-[4-аміно-гідразонометил)-феніл)-N-циклопентил-N-метил-2-[2-(нафталін-1-іл-окси)ацетиламіно]пропіонаміду (0,7г, 1,54ммол). Зазначену проміжну сполуку потім оброблювали згідно з тими ж самими операціями, що описані у Прикладі 17 для одержання очищеної вищеназваної сполуки (0, 59г, ви хід : 63%). 1 Н ЯМР (CD3 OD, чнм) d : 7.80-7.14 (m, 11H), 5.35-5.18 (m, 1Н) , 4.76-4.35(m, 1Н), 4.61(s, 2Н), 3.18-3.05(m, 2Н), 2.85-2.75(2s, 3Н), 1.85-1.25(m, 8Н) Маса (ФСА, м/е) : 488(М++1) Препарат 7 Синтез метилового ефіру N-t-бутоксикарбоніл-N-метиламінооцтової кислоти Хлористоводневий метиловий ефір гліцину (1,0г, 8,2ммол) розчинювали у воді (12мл) і 1N водному розчині гідроксиду натрію (8,2мл), а далі додавали 1,4-діоксан (20мл). До цієї суміші додавали ді-t бутилдикарбонат (2,2г, 9,8ммол) при 0°С і суміш нагрівали до кімнатної температури та перемішували протягом 2 годин. Летючу речовину видаляли з реакційної суміші при зменшеному тиску і осад розчинювали за допомогою етилацетату, послідовно промивали за допомогою водного насиченого розчину кислої солі карбонату натрію, соляною кислотою і насиченим сольовим розчином, сушили над безводним сульфатом натрію, фільтрували, а потім концентрували. Одержаний продукт розчинювали у диметилформаміді (ДМФ, 10мл). До цього розчину повільно додавали 60%-ний гідрид натрію (NaH, 0,25г, 6,4ммол) при 0°С, а далі додавали по краплям йодометан (СН3І, 1,1мл). Суміш повільно нагрівали до кімнатної температури і перемішували протягом 3 годин при тієї же самої температурі. Суміш фільтрували крізь шар целіту і концентрували при зменшеному тиску. Осад розчинювали за допомогою етилацетату, послідовно промивали за допомогою вводного насиченого розчину кислої солі карбонату натрію, розчином соляної кислоти і сольовим розчином, сушили над безводним сульфатом натрію, фільтрували, а далі концентрували. Осад очищали за допомогою колонкової хроматографії, використовуючи як розчинник етилацетат/гексан (об'ємне співвідношення 6 : 4) для одержання очищеної вищеназваної сполуки (1,0г, вихід : 65%). 1 H ЯMP(CD3CD, чнм) d : 1.45(d, 9Н), 2.95(s, 3Н), 3.78(s, 3Н), 3.92 (s, 1Н), 4.00 (s, 1Н) Маса (ФСА, м/е) : 204 (М + 1) Препарат 8 Синтез метилового ефіру {[(S)-2-(t-бутоксикарбоніламіно)-3-(4-ціанофеніл)-пропіоніл]-метиламіно} оцтової кислоти (S)-2-(t-бутоксикарбоніламіно)-3-(4-ціанофеніл)-пропіонову кислоту (0,5г, 1,72ммол) розчинювали у диметилформаміді (ДМФ). Одержаний розчин охолоджували до 0°С, а далі до нього додавали хлористоводневий 1-(3-диметиламінопропіл)-3-метилкарбодіімід (ЕДХ, 0,39г) і 1- гідробензотриазол (ГОБТ, 0,28г) і перемішували до повного розчинювання. Окремо сполуку (0,35 г, 1,72 ммол), виготовлену як Препарат 7, розчинювали у ди хлорметані (2мл) і охолоджували до -10°С. До зазначеної сполуки додавали трифтороцтову кислоту (2мл) і суміш перемішували протягом 5 хвилин, повільно нагрівали до кімнатної температури, знову перемішували протягом 30 хвилин, а потім дистилювали при зменшеному тиску з метою видалення летючої речовини. Одержану таким способом сполуку і N-метилморфолін (1мл) додавали до вищенаведеного розчину, а потім реакційний розчин повільно нагрівали до кімнатної температури і перемішували протягом 3,5 годин. Після завершення реакції реакційний розчин дистилювали при зменшеному тиску з метою видалення летючої речовини. Осад розчинювали за допомогою етилацетату, послідовно промивали за допомогою водного насиченого розчину кислої солі карбонату натрію, розчином соляної кислоти і сольовим розчином, сушили над безводним сульфатом натрію, фільтрували, а далі концентрували. Осад очищали за допомогою колонкової хроматографії, використовуючи як розчинник етилацетат/гексан (об'ємне співвідношення 3 : 7) для одержання очищеної вищеназваної сполуки (0,58г, вихід : 90%). 1 Н ЯМР (CD3CD,4HM) d : 1.40(m, 9Н), 3.08 (s, 3Н), 2.95-3.25(m, 2Н), 3.78(s, 3Н), 3.89-4.35(m, 2Н), 4.95(m, 1Н), 5.52(d, 1Н), 7.35(m, 2Н), 7.60(m, 2Н) Маса (ФСА, м/е) : 376 (М + 1) Препарат 9 Синтез метилового ефіру 1-{[3-(4-ціанофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} оцтової кислоти Сполуку (0,57г, 1,52ммол), виготовлену як Препарат 8, розчинювали у дихлорметані (2мл) і охолоджували до -10°С, а далі до розчину додавали трифтороцтову кислоту (ТФК, 2мл). Реакційну суміш перемішували протягом 5 хвилин, повільно нагрівали до кімнатної температури, знову перемішували протягом 30 хвилин, а далі дистилювали при зменшеному тиску з метою видалення летючої речовини. Осад сушили за допомогою вакуумного насосу, а потім до нього додавали ДМФ (10мл). Зазначений розчин охолоджували до температури -10°С і до нього додавали N,N - діізопропілетиламін (1мл). Зазначений реакційний розчин нагрівали до кімнатної температури і перемішували протягом приблизно 5 хвилин, а далі до нього додавали 2 нафталінсульфохлориду (0,41г, 1,82ммол). Реакційну суміш перемішували протягом 1 години до завершення реакції і дистилювали при зменшеному тиску для видалення летючої речовини. Осад розчинювали етилацетатом, двічі промивали водою і сушили над безводним сульфатом магнію, а далі фільтрували. Фільтрат концентрували і осад очищали за допомогою колонкової хроматографії, використовуючи як розчинник етилацетат/гексан (у об'ємному співвідношенні 1 : 1) для одержання очищеної вищеназваної сполуки (0,55г, вихід : 78%). 1 Н ЯМР(СD3СD, чнм) d : 2.88(s, 3Н), 2.80-3.20(m, 2H), 3.80(d, 3Н), 4.12 (d, 2H), 4.58(m, 1Н), 6.40(d, 1H), 7.20-8.40(m, 11Н) Маса (ФСА, м/е) : 466 (М + 1) Приклад 25 Синтез метилового ефіру {[3-(4-іімідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-проліоніл]метиламіно} оцтової кислоти Сполуку (0,55г, 1,18ммол), виготовлену як Препарат 9, розчинювали у піридині (10мл) і одержаний розчин розміщували у обладнану відведеннями колбу, у яку додавали триетиламін (0,45мл). Реакційна колба була обладнана таким чином, щоб сірководень (H2S) був здатний повільно утікати крізь одне з відведень колби, а витікати крізь друге відведення. Реакційний розчин насичували сірководнем, постійно перемішуючи протягом приблизно 10 хвилин, при цьому безкольоровий розчин ставав спочатку зеленого кольору, а далі поступово переходив до темнокоричневого кольору. Колбу закривали за допомогою гумової пробки і зберігали її протягом 3 діб при кімнатній температурі. Після закінчення реакції реакційний розчин дистилювали при зменшеному тиску для видалення летючих речовин і висушували за допомогою вакуумного насосу. До одержаного твердого тіла жовтого кольору додавали разом ацетон (10мл) і йодометан (СН3І, 0,55мл) і одержану суміш нагрівали до одержання флегми протягом 30 хвилин. Зазначену реакційну суміш знову дистилювали при зменшеному тиску для видалення летючих речовин та висушували за допомогою вакуумного насосу. Осад розчинювали у чистому метанолі (5мл) і потім перемішували. До цієї суміші тричі порційно додавали 80% - ний гідрат гідразину (H2NNH2 H2O, 0,1мл 1,77ммол) через кожні 10 хвилин. Після завершення реакції реакційний розчин концентрували, а далі очищали за допомогою РХВТ з метою одержання вищеназваної сполуки (0,25г, вихід : 43%). 1 Н ЯМР(СD3СD, чнм) d : 2.95(s, 3Н), 2.70-3.20(m, 2H), 3.54(s, 3Н), 3.80(d, 2H), 4.55(m, 1Н), 7.20-8.30(m, 11Н) Маса (ФСА, m/е) : 498 (М + 1) Приклад 26 Синтез {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} оцтової кислоти Сполуку (160мг, 0,32ммол), виготовлену згідно з операціями, описаними у Прикладі 25, розчинювали у розчиннику, що становить собою суміш метанолу (4мл) і води ( у співвідношенні 3:1). До цього розчину повільно додавали гідрат гідроокиду літію (LiOH. Н2О, 0,016г, 0,38ммол) при 0°С і суміш перемішували протягом 2 годин при кімнатній температурі. Після завершення реакції реакційний розчин концентрували і очищали за допомогою РХВТ для одержання вищеназваної сполуки (50мг, вихід : 32%). 1 Н ЯМР (CD3CD, чнм) d : 2.20-2 .60 (m, 2Н), 2.48(s, 3Н), 2.78(s, 3Н), 2.32(m, 2Н), 4.12 (m, 1Н), 6.80-7.80(m, 11Н) Маса (ФСА, м/е) : 484 (М + 1) Приклад 27 Синтез метилового ефіру (S)-2-{[3-(4-амідразонофеніл)-(S)-2- (нафталін-2-сульфоніламіно)-пропіоніл]метиламіно} пропіонової кислоти Для одержання метилового ефіру (L)-(N-t-бутоксикарбоніл-N-метил)-аланіну виконували такі ж самі операції, що і для одержання Препарату 7, за винятком того, що замість метилового ефіру гліцину застосовували метиловий ефір (L)-аніліну, який далі оброблювали згідно з тим же самим способом, що і Препарати 8 і 9, для одержання проміжної сполуки метилового ефіру (S)-2-{[3-(4-ціанофеніл)-(S)-2-(нафталін 2-сульфоніламіно)-пропіоніл]-метиламіно} пропіонової кислоти (1,43г). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,64г, вихід : 48%). 1 H ЯMP(CD3CD, чнм) d : 0.69, 0.88(d, d, 3Н), 2.79, 2.95(s, s, 3H), 2.80, 3.06(m, m, 2H), 3.48, 3.57(s, s, 3H), 4.29(m, 1H), 4.55 (m, 1H), 7.30-8.30(m, 11H) Маса (ФСА, m/e) : 512(M + 1) Приклад 28 Синтез (S) - 2 - {[3 - (4 - амідразонофеніл) - (S) - 2 - (нафталін - 9 - сульфоніламіно) - пропіоніл] – метиламіно} пропіонової кислоти Сполуку, виготовлену згідно з операціями, описаними у Прикладі 27, оброблювали згідно з операціями, описаними у Прикладі 26, для одержання очищеної вищеназваної сполуки (0,06г, вихід : 41%). 1 Н ЯМР (CD3CD, чнм) d : 0.64, 0.95 (d, d, 3Н), 2.76, 2.92(s, s, 3Н), 2.83, 3.09(m, m, 2H), 4.37 (m, 1H), 4.54 (m, 1Н) 7.30-8.40(m, 11H) Маса (ФСА, m/e) :498(M + 1) Приклад 29 Синтез метилового ефіру (R) - 2 - {[3 - (4 - амідразонофеніл) - (S) - 2 - (нафталін - 2 - сульфоніламіно) пропіоніл] - метиламіно} пропіонової кислоти Для одержання метилового ефіру (D) - (N - t - бутоксикарбоніл - N -метил) - аланіну виконували такі ж самі операції, що і для одержання Препарату 7, за винятком того, що замість метилового ефіру гліцину застосовували метиловий ефір (D) - аніліну, який далі оброблювали згідно з тим же самим способом, що і препарати 8 і 9, для одержання проміжної сполуки метилового ефіру (R) - 2 - {[3 - (4 - ціанофеніл) - (S) - 2 (нафталін - 2 - сульфоніламіно) - пропіоніл] - метиламіно} пропіонової кислоти (0,78г). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,58г, вихід : 70%). 1 Н ЯМР(СD3СD, чнм) d : 0.89, 1.21(d, d, 3Н), 2.46, 2.94(s, s, 3Н), 2.80, 3.08 (m, m, 2H), 3.49, 3.78(s, s, 3Н), 4.29(m, 1H), 4.59 (m, 1H), 7.30-8.40(m, 11H) Маса (ФСА, m/e) : 512 (M +1) Приклад 30 Синтез (R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} пропіонової кислоти Сполуку (0,03г, 0,059ммол), виготовлену згідно з операціями, описаними у Прикладі 29, оброблювали згідно з операціями, описаними у Прикладі 26, для одержання очищеної вищеназваної сполуки (0,01г, вихід : 33%). 1 Н ЯМР (CD3CD, чнм) d : 0.90, 1.18(d, d, 3Н), 2.44, 2.92(s, s, 3Н), 2.82, 3.08(m, m, 2H), 4.33 (m, 1H), 4.62 (m, 1H), 7.30-8.40 (m, 11H) Маса (ФСА, m/e) : 498 (M + 1) Приклад 31 Cинтез метилового ефіру (R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]метиламіно}-3-метилмасляної кислоти Для одержання метилового ефіру (D)-(N-t-бутоксикарбоніл-N-метил)-валіну виконували такі ж самі операції, що і для одержання Препарату 7, за винятком того, що замість метилового ефіру гліцину застосовували метиловий ефір (D)-(N-t-бутоксикарбоніл-N-метил)-валіну, який далі оброблювали згідно з тим же самим способом, що і препарати 8 і 9, для одержання проміжної сполуки метилового ефіру (R)-2-{[3-(4ціанофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}-3-метилмасляної кислоти (0,19г). Зазначену проміжну сполуку оброблювали згідно з тим же самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,11г, вихід : 55%). 1 Н ЯМР(СD3СD, чнм) d : 0.59, 0.70(d, d, 3Н), 0.89, 0.98(d, d, 3Н), 2.09, 2.21(m, m, 1H), 2.75, 3.06(s, s, 3H), 3.40, 3.68(s, s, 3H), 4.34, 4.38(d, d, 1H), 4.63, 4.70(m, m, 1H), 7.20-8.40(m, 11H) Маса(ФСА, м/e) : 540(M++1) Приклад 32 Синтез (R)-2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}3-метилмасляної кислоти Сполуку, виготовлену згідно з операціями, описаними у Прикладі 31, оброблювали згідно з операціями, описаними у Прикладі 26, для одержання очищеної вищеназваної сполуки (0,04г, вихід : 40%). 1 Н ЯМР(СD3СD, чнм) d : 0.57, 0.63(d, d, 3Н), 0.92, 0.99(d, d, 3Н), 2.09, 2.18(m, m, 1H), 2.74, 3.08(s, s, 3H), 4.18, 4.36(d, d, 1H), 4.64, 4.70(m, m, 1H), 7.20-8.40(m, 11H) Маса (ФСА, m/e) : 526 (M + 1) Приклад 33 Синтез метилового ефіру 3-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]метиламіно} пропіонової кислоти Для одержання метилового ефіру 3-(N-t-бутоксикарбоніл-N-метил)-амінопропіонової кислоти виконували такі ж самі операції, що і для одержання Препарату 7, за винятком того, що замість метилового ефіру гліцину застосовували метиловий ефір 3-амінопропіонової кислоти, який далі оброблювали згідно з тим же самим способом, що і Препарати 8 і 9, для одержання проміжної сполуки метилового ефіру 3-{[3-(4-ціанофеніл)-(S)-2 -(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} пропіонової кислоти (0,69г). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,55г, вихід : 74%). 1 Н ЯМР (CD3CD, чнм) d : 8.31, 7.97, 7.68, 7.48(d, m, m, m, 11Н), 4.62, 4.51(m, m, 1H), 3.62, 3.55(s, s, 3H), 3.05, 2.85(m, m, 4H), 2.80, 2.45(s, s, 3H), 2.38, 1.91(m, m, 2H) Маса (ФСА, m/e) : 512(M++1) Приклад 34 Синтез 3-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]- метиламіно} пропіонової кислоти Сполуку, виготовлену згідно з операціями, описаними у Прикладі 33, оброблювали згідно з операціями, описаними у Прикладі 26, для одержання очищеної вищеназваної сполуки (0,17г, вихід : 32%). 1 Н ЯМР (СD3СD, чнм) d : 8.31, 7.98, 7.78-7.37(d, m, m, 11Н), 4.65, 4.52(m, m, 1H), 3.20-2.85(m, 4H), 2.80, 2.45(s, s, 3H) , 2.38, 1.91(m, m, 2H) Маса (ФСА, m/e) : 498 (М+ + 1) Приклад 35 Синтез метилового ефіру 4-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]метиламіно} масляної кислоти Для одержання метилового ефіру 4-(N-t-бутоксикарбоніл-N-метил)-аміно-пропіонової кислоти виконували такі ж самі операції, що і для одержання Препарату 7, за винятком того, що замість метилового ефіру гліцину застосовували метиловий ефір 4-аміномасляної кислоти, який далі оброблювали згідно з тим же самим способом, що і Препарати 8 і 9, для одержання проміжної сполуки метилового ефіру 4-{[3-(4- іанофеніл)-(S)-2(нафталін-2-сульфоніламіно)-пропіоніл] метиламіно} масляної кислоти (0,51г). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,40г, вихід : 74%). 1 НЯМР(СD3СD, чнм) d : 8.32(s, 1Н), 7.98 (m, 3Н), 7.78-7.36(m, 7Н), 4.55(m, 1Н), 3.72, 3.60(s, s, 3Н), 3.10, 2.81(m, m, 4Н), 2.79, 2.55(s, s, 3Н), 2.22(m, 1Н), 1.89(m, 1Н), 1.63, 1.42(m, m, 1Н), 1.18(m, 1Н) Маса (ФСА, м/е) : 526(М++1) Приклад 36 Синтез 4-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно} масляної кислоти Сполуку, що виготовляли, як це описано у Прикладі 35, оброблювали згідно з операціями, наведеними у Прикладі 26 для одержання очищеної вищеназваної сполуки (0,12г, вихід 32%). 1 Н ЯМР(СD3СD, чнм) d : 8.32 (m, 1Н), 7.98 (m, 3Н), 7.78-7.35(m, 7Н), 4.55(m, 1Н), 3.05, 2.81(m, m, 4Н), 2.79, 2.50(s, s, 3Н), 2.18(m, 1Н), 1.89(m, 1Н), 1.35(m, m, 1Н), 1.16(m, 1Н) Маса (ФСА, м/е) : 512(М++1) Препарат 10 Синтез метилового ефіру N-t-бутоксикарбоніл-N-циклопропіл-оцтової кислоти Циклопропіламін (1,34г, 23, 49ммол) змішували з ДМФ (15мл) і триетиламіном (3мл) і зазначену суміш вводили до реакційної судини. Метилбромацетат (2,2мл, 23,49ммол) і ДМФ вводили у краплинну лійку. Реакційну судину о холоджували до 0°С, а потім розчин, що містився у краплинній лійці по краплям додавали до реакційної судини. Після закінчення додавання реакційну суміш нагрівали до кімнатної температури і здійснювали реакцію протягом 3,5 годин. Після завершення реакції до зазначеної суміші додавали воду (10мл) і гідроксид натрію (3N) (8мл). Дo реакційної суміші додавали 1,4-діоксану (10мл), потім додавали ангідридбутилкарбоніл (6,1г, 27,95ммол). Реакцію зазначеної суміші здійснювали протягом 3 годин при кімнатній температурі і дистилювали при зменшеному тиску з метою видалення летючої речовини. Осад розчинювали за допомогою метилацетату і послідовно промивали насиченим кислим розчином карбоната натрію, розчином соляної кислоти та сольовим розчином. Відділяли органічний шар і сушили його над безводним сульфатом магнію та фільтрували. Розчинник видаляли із фільтру при зменшеному тиску. Осад очищали за допомогою колонкової хроматографії (розчинник етилацетат/гексан (при об'ємному співвідношенні 6 : 4)) для одержання очищеної вищеназваної сполуки (2,3 г, ви хід : 43%). 1 Н ЯМР (СD3СD, чнм) d: 3.95(m, 2H), 3.72 (m, 3Н), 2.75, 2.52(bs, bs, 1H), 1.45, 1.47(s, s, 9H), 0.80-0.45(m, 4H) Маса (ФСА, м/e) : 230(M++1) Приклад 37 Синтез метилового ефіру {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]циклопропіламіно} оцтової кислоти Для одержання проміжної сполуки метилового ефіру {[3-(4-ціанофеніл)-(S)-2-(нафталін-2сульфоніламіно)-пропіоніл]-циклопропіламіно} оцтової кислоти (0,30г) сполуку, одержану як Препарат 10, оброблювали згідно з тими же самими операціями, що і Препарати 8 і 9. Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,25г, вихід : 77%). 1 Н ЯМР(СD3СD, чнм) d: 8.24 (s, 1Н), 7.93 (m, ЗН), 7.65 (m, 3Н), 7.42(d, 2Н), 7.35(d, 2H), 5.02(m, 1Н), 3.92(d, 1Н), 3.64(d, 1Н), 3.60(s, 3Н), 3.19(dd, 1H), 2.80(m, 2Н), 0.95, 0.85, 0.61 (m, m, m, 4Н) Маса (ФСА, м/е) : 524(М++1) Синтез {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-циклопропіламіно}оцтової кислоти Сполуку, що виготовляли, як це описано у Прикладі 37, оброблювали згідно з операціями, наведеними у Прикладі 26 для одержання очищеної вищеназваної сполуки (0,07г, вихід : 29%). 1 Н ЯMP(СD3СD, чнм) d: 8.24(s, 1Н), 7.93 (m, ЗН), 7.42 (d, 2Н), 7.35(d, 2Н), 5.02(m, 1Н), 3.95(d, 1Н), 3.54(d, 1Н), 3.20(dd, 1Н), 2.80(m, 2Н), 0.95, 0.85, 0.61(m, m, m, 4Н) Маса (ФСА, м/е) : 510(М++1) Приклад 39 Синтез метилового ефіру {[3-(4-амідразонофеніл)-(S)-2-нафталін-2-сульфоніламіно)-пропіоніл]бутиламіно} оцтової кислоти Для одержання метилового ефіру N-t-бутоксикарбоніл-N-бутиламіно оцтової кислоти здійснювали ті ж самі операції, що і при виготовленні Препарату 10, за винятком того, що замість циклопропіламіну використовували бутиламін. Зазначену сполуку оброблювали згідно з тими же самими операціями, що і при виго товленні Препаратів 8 і 9, для одержання проміжної сполуки метилового ефіру {[З -(4- ціанофеніл)- (S)-2 - (нафталін- 2сульфоніламіно)-пропіоніл]-бутиламіно} оцтової кислоти (0,31г). Зазначену проміжну сполуку оброблювали згідно з тими ж самими операціями, що і у Прикладі 25 для одержання очищеної вищеназваної сполуки (0,19г, вихід : 58%). 1 Н ЯМР(СD3СD, чнм) d: 8.30-7.32(m, 11Н), 4 .32-4.09(m, 3Н), 3.55(s, 3Н), 3.57-2.50(m, 4Н), 1.26-0.50(m, 7Н) Маса (ФСА, м/е) : 540(М++1) Приклад 40 Синтез {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2- сульфоніламіно)-пропіоніл]-бутиламіно} оцтової кислоти Сполуку, що виготовляли, як це описано у Прикладі 39, оброблювали згідно з операціями, наведеними у Прикладі 26 для одержання очищеної вищеназваної сполуки (0,12г, вихід : 67%). 1 Н ЯМР (СD3СD, чнм) d: 8.30-7.10(m, 11Н), 4.31-4.10(m, 3Н), 3.52-2.55(m, 4Н), 1.25-0.50(m, 7Н) Маса (ФСА, м/е) : 526 (М+1) Приклад 41 Синтез метилового ефіру {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]циклопентиламіно} оцтової кислоти Для одержання метилового ефіру N-t-бутоксикарбоніл-N-циклопентиламіно оцтової кислоти здійснювали ті ж самі операції, що і при виготовленні Препарату 10, за винятком того, що замість циклопропіламіну використовували циклопентиламін. Зазначену сполуку оброблювали згідно з тими же самими операціями, що і при виготовленні Препаратів 8 і 9, для одержання проміжної сполуки метилового ефіру {[3-(4-ціанофеніл)-(S)2-(нафталін-2-сульфоніламіно)-пропіоніл]-циклопентиламіно} оцтової кислоти (0,23г). Зазначену проміжну сполуку оброблювали згідно з тими ж самими операціями, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,12г, вихід : 50%). 1 Н ЯМР (СD3СD, чнм) d : 8.35-7.35(m, 11Н), 4.66, 4.33(m, m, 1H), 4.15 (m, 1H) , 3.75, 3.53(m, m, 1H), 3.61(s, 3H), 3.40-2.80(m, 3H), 1.90-0.60(m, 8H) Маса (ФСА, м/е) : 552 (М++ 1) Приклад 42 Синтез {[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-циклопентиламіно} оцтової кислоти Сполуку, що виготовляли, як це описано у Прикладі 41, оброблювали згідно з операціями, наведеними у Прикладі 26, для одержання очищеної вищеназваної сполуки (0,4г, ви хід : 33%). 1 Н ЯМР (СD3СD, чнм) d: 8.35-7.35(m, 1Н), 4.65, 4.32(m, m, 1H), 4.15(m, 1H), 3.75, 3.52(m, m, 1H), 3.41, 3.283.10, 2.80(m, m, m, 3H), 1.90-0.60(m, 8H) Маса (ФСА, м/e) : 538 (M+ + 1) Препарат 11 Синтез метилового ефіру 1-(N-t-бутоксикарбоніл-N-метиламіно) циклопентан карбонової кислоти Циклолейцин (3г, 23,2ммол) розчинювали у 1N водного розчину гідроксиду натрію (23,2мл) і дистильованої воді (7мл), а потім додавали 1,4-діоксан (30мл). До зазначеної суміші додавали ди-tбутилдикарбонат (6,1г, 27,8ммол) при температурі 0° С і наведену суміш нагрівали до кімнатної температури, а потім перемішували протягом 2 годин. Летючу речовину видаляли при зменшеному тиску і осад розчинювали за допомогою етилацетату, послідовно промивали водним насиченим розчином карбонату натрію, розчином соляної кислоти і сольовим розчином, сушили над безводним сульфатом натрію, фільтрували, а потім концентрували. Одержаний твердий продукт білого кольору розчиняли у диметилформаміді (ДМФ, 30мл). До зазначеного розчину додавали карбонат калію (4,8г, 34,8ммол), а далі по краплям додавали йодометан (СН3І, 14,4мл, 232ммол). Реакційну суміш перемішували протягом 2 годин при кімнатній температурі протягом 2 годин і дистилювали при зменшеному тиску для видалення летючої речовини. Залишковий розчин розчинювали за допомогою етилацетату, послідовно промивали водним насиченим розчином кислої солі карбонату натрію, розчином соляної кислоти і насиченим сольовим розчином, сушили над безводним сульфатом натрію, фільтрували, а потім концентрували. Одержаний продукт розчинювали у диметилформаміді (ДМФ, 20мл). До цього розчину повільно додавали 60%-ний гідрид натрію (NaH, 0,46г, 11,4ммол) при температурі 0°С, а потім по краплям додавали йодометан (СН3І, 1,8мл, 28,4ммол). Зазначену суміш повільно нагрівали до кімнатної температури і перемішували протягом 3 годин при вищенаведеній температурі. До реакційної суміші з метою видалення залишкового гідриду натрію додавали воду. Суміш фільтрували і концентрували при зменшеному тиску. Осад розчинювали за допомогою етилацетату, послідовно промивали водним насиченим розчином кислої солі карбоната натрію, розчином соляної кислоти і насиченим сольовим розчином, сушили над безводним сульфатом натрію, фільтрували, а потім концентрували. Осад очищали за допомогою колонкової хроматографії, використовуючи як розчинник етилацетат/гексан у об'ємному співвідношенні 3 : 7 для одержання очищеної вищеназваної сполуки (2,0г, вихід : 34%). 1 Н ЯМР(СD3СD, чнм) d: 1.35(s, 9H), 1.62 (m, 4H) , 1.78 (m, 2Н), 2.18(m, 2Н), 2.90(s, 3Н), 3.62(8, 3Н) Маса (ФСА, м/е) : 258 (М + 1) Приклад 43 Синтез метилового ефіру 1-{[3-(4-амідразонофеніл)-(S)-2 –(нафталін-2-сульфоніламіно)-пропіоніл]метиламіно}циклопентан карбонової кислоти Для одержання проміжної сполуки метилового ефіру 1-{[3-(4-аміногідразонометил)-феніл-(S)-2- (нафталін - 2-сульфоніламіно)-пропіоніл]-метиламіно} циклопентан карбонової кислоти (0,28г) сполуку, одержану як Препарат 11, оброблювали згідно з тими же самими операціями, що і Препарати 8 і 9. Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,56г, вихід : 53%). 1 Н ЯМР (СD3СD, чнм) d : 0.52 (m, 1Н), 0.89(m, 1Н), 1.29(m, 2Н), 1.52 (m, 1Н), 1.76(m, 2Н), 2.05 (m, 1Н), 2.75, 3.00 (m, m, 2Н), 2.88(s, ЗН), 3.50(s, ЗН), 4.48(m, 1Н), 6.38(m, 1Н), 7.30-8.40(m, 11Н) Маса (ФСА, м/е) : 552 (М + 1) Приклад 44 Синтез 1-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}- циклопентан карбонової кислоти Сполуку, що виготовляли, як це описано у Прикладі 43, оброблювали згідно з операціями, наведеними у Прикладі 26 для одержання очищеної вищеназваної сполуки (0,01г, вихід : 17%). 1 Н ЯМР(СD3СD, чнм) d : 0.42 (m, 1Н), 0.74 (m, 1Н), 1.25 (m, 2Н), 1.50(m, 1Н), 1.78 (m, 2Н), 2.05(m, 1Н), 2.75, 3.08(m, m, 2Н), 2.98(s, ЗН), 4.58(m, 1Н), 7.40-8.40(m, 11Н) Маса (ФСА, м/е) : 538 (М + 1) Препарат 12 Синтез етилового ефіру 2-(N-t-бутоксикарбоніл-N-метил)-аміно-циклопентан карбонової кислоти Етил 2-оксициклопентан карбоксилат (10мл, 67,49ммол) вводили разом з етанолом (100мл) до реакційної судини. Потім для розчину реактивів у зазначену судину додавали хлористоводневий метиламін (4,69г, 68,13ммол) і воду (10мл). Далі до реакційної судини додавали ціаноборогідрид натрію (4,3г, 68,43ммол) і pH суміші регулювали до величини, що дорівнювала 6; а далі реакцію здійснювали протягом 12 або більше годин при температурі від 30°С до 40°С. Далі реакційну суміш концентрували при зменшеному тиску, охолоджували до 0°С, регулювали pH до величини, що дорівнювала 2, за допомогою 6N соляної кислоти і тричі промивали диетиловим ефіром. pH водяного шару знову регулювали до величини, що дорівнювала 10, і додавали ту ж саму кількість діоксану. До зазначеної суміші додавали одну еквівалентну вагу ангідриду бутилоксикарбонілу. Реакцію зазначеної реакційної суміші здійснювали протягом 3 годин при кімнатній температурі. Після завершення реакції реакційний розчин дистилювали при зменшеному тиску для видалення летючої речовини, розчинювали етилацетатом і послідовно промивали насиченим розчином кислої солі карбонату натрію, розчином соляної кислоти і насиченим сольовим розчином. Органічний шар сушили над безводним сульфатом магнію, фільтрували і дистилювали при зменшеному тиску з метою видалення розчинника. Осад очищали за допомогою колонкової хроматографії (розчинник становив собою етилацетат : гексан при об'ємному співвідношенні 1:1) для одержання вищеназваної сполуки (5,82г, вихід : 32%). 1 Н ЯМР (СD3СD, чнм) d : 4.55 (m, 1Н), 4.10(m, 2Н), 2.79(s, 3Н), 2.73(s, 1H), 2.00-1.40(m, 6Н), 1.45 (s, 9Н), 1.24(t, 3Н) Маса (ФСА, m/е) : 272(М++1) Приклад 45 Синтез етилового ефіру 2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]метиламіно} циклопентан карбонової кислоти Для одержання проміжної сполуки етилового ефіру 2-{[3-(4-ціанофеніл)-(S)-2-(нафталін-2сульфоніламіно)-пропіоніл]-метиламіно} циклопентан карбонової кислоти (0,48г) сполуку, одержану як Препарат 12, оброблювали згідно з тими же самими операціями, що і Препарати 8 і 9. Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки. (0,36г, вихід : 71%). 1 H ЯMP (СD3СD, чнм) d : 8.39-7.25(m, 11Н), 4.78-4.40(m, 2Н), 4.05 (m, 2Н), 3.05(m, 1Н), 2.90-2.65(m, ЗН), 2.50-2.40(m, 2Н), 2.05, 1.90-1.30, 0.85 (m, m, m, 6Н), 1.28-1.15(m, 3Н) Маса (ФСА, m/е) : 566 (М+ + 1) Приклад 46 Синтез 2-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}-циклопентан Сполуку, що виготовляли, як це описано у Прикладі 45, оброблювали згідно з операціями, наведеними у Прикладі 26 для одержання очищеної вищеназваної сполуки (0,086г, вихід : 25%). 1 Н ЯМР (СD3СD, чнм) d : 8.39-7.20 (m, 11H), 4.78-4.50 (m, m, 2Н), 3.05(m, 1Н), 2.90-2.40(m, 5Н), 2.10, 1.901.20, 0.75(m, m, m, 6Н) Маса (ФСА, m/e) : 538(М++1) Приклад 47 Синтез метилового ефіру (S)-2-{[3- (4-амідцазонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл}метиламіно} 3-метил масляної кислоти Для одержання метилового ефіру (L)-(N-t-бутоксикарбоніл-N-метил)-валіну виконували такі ж самі операції, що і для одержання Препарату 7, за винятком того, що замість метилового ефіру гліцину застосовували метиловий ефір (L)-(N-t-бутоксикарбоніл-N-метил)-валіну, який далі оброблювали згідно з тим же самим способом, що і 8 і 9, для одержання проміжної сполуки метилового ефіру (S)-2-{[3-(4-ціанофеніл) (S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]-метиламіно}-3-метил масляної кислоти (0,13г). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,09г, вихід : 69%). 1 Н ЯМР(СD3СD, чнм) d : 8.30, 7.95, 7.75-7.20(m, m, m, 11Н), 4.75-4.25(m, 2Н), 4.65, 3.39(m, m, 3Н), 3.152.65(m, 4Н), 2.22, 2.15(s, s, 1Н), 2.08, 1.90(m, m, 1Н), 1.00-0.55, 0.20(m, m, 6Н) Маса (ФСА, m/е) : 540(М++1) Приклад 48 Синтез метилового ефіру 1-{[3-(4-амідразонофеніл)-(S)-2-(нафталін-2-сульфоніламіно)-пропіоніл]піперидин-(R)-2-карбонової кислоти Для одержання проміжної сполуки метилового ефіру 1-{[3-(4-ціанофеніл)-(S)-2-(нафталін-2сульфоніламіно)-пропіоніл]-піперидін -(R) карбонової кислоти (0,18г) виконували такі ж самі операції, що і для одержання Препарату 7, за винятком того, що замість метилового ефіру гліцину застосовували метиловий ефір (D)-піпеколінової кислоти, який далі оброблювали згідно з тим же самим способом, що і Препарати 8 і 9. Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 25, для одержання очищеної вищеназваної сполуки (0,16г, вихід : 84%). 1 Н ЯМР(СD3СD, чнм) d : 8.32, 7.95, 7.78-7.35(m, m, m, 11H), 4.71, 4.52(m, m, 1Н), 3.97, 3.80(d, d, 1H), 3.73, 3.43(s, s, 3H), 3.10, 2.83, 2.39(m, m, m, 3H), 2.05(m, 1H), 1.65-1.00, 0.30(m, m, 6H) Маса (ФСА, m/e) : 538 (M+ + 1) Приклад 49 Синтез 1-[3-(4-амідразонофеніл)-(S)-2- (нафталін-2-сульфоніламіно)-пропіоніл]-піперидин(R)-2- карбонової кислоти Сполуку, що виготовляли, як це описано у Прикладі 48, оброблювали згідно з операціями, наведеними у Прикладі 26, для одержання очищеної вищеназваної сполуки (0,03г, вихід : 19%). 1 H ЯMP (СD3СD, чнм) d: 8.35, 8.00, 7.75-7.30(m, m, m, 11Н), 4.50, 4.20(m, m, 1Н), 3.89(m, 1Н), 3.10, 2.82, 2.45(m, m, m, 3Н), 2.10(m, 1Н), 1.65-1.00, 0.25(m, m, 6Н) Маса (ФСА, m/е) : 524 (М+ + 1) Препарат 13 Синтез (S)-4-[2-(бутилоксикарбоніл-аміно)-3-(4-метилсульфоніл-піперазиніл)-3-оксо пропіл]-бензонітрилу (S)-3-(4-ціанофеніл)-2-(бутилоксикарбоніламіно) пропіонову кислоту (0,5г, 1,7ммол) розчинювали у диметилформаміді (ДМФ, 20мл) і потім охолоджували до температури 0°С. Потім до цього розчину додавали хлористоводневий 1-(3-диметиламінопропіл)-3 - етилкарбодіімід (ЕДХ, 0,5г) і гідрат 1-гідроксибензотриазолу (ГОБТ, 0,3г ) і перемішували, доки вони повністю не розчинювались. До цієї реакційної суміші додавали 1метансульфоніл - піперазин (0,3г) і N-метилморфолін (0,2мл), а далі температуру повільно збільшували до кімнатної температури. Зазначений реакційний розчин перемішували протягом 3,5 годин. Після завершення реакції реакційний розчин дистилювали при зменшеному тиску для видалення летючої речовини, розчинювали етилацетатом і послідовно промивали насиченим водним розчином кислої солі карбонату натрію, розчином соляної кислоти і насиченим сольовим розчином, сушили над безводним сульфатом натрію, фільтрували і потім концентрували. Осад очищали за допомогою колонкової хроматографії, використовуючи як розчинник етилацетат/гексан (при об'ємному співвідношенні 6 : 4) для одержання вищеназваної сполуки ( 0,7г, ви хід : 93%). 1 Н ЯМР (СD3СD, чнм) d : 7.7-7.3 (m, 4Н), 5.3 (m, 1Н), 4.8(m, 1Н), 3.9-3.55(m, 2Н), 3.55-2.8(m, 8Н), 2.7(s, 3Н), 1.5(s, 9H) Препарат 14 Синтез(S)-нафталін-2-сульфокислота[1-(4-ціанобензил)-2-(4-метилсульфоніл-піперазиніл)-2-оксоетил] аміду Сполуку (0,7г, 1,6ммол), виготовлену як Препарат 13, розчинювали у дихлорметані (3мл), а потім охолоджували до -10°С і додавали до цього розчину трифтороцтову кислоту (ТФК, 3мл). Реакційну суміш перемішували протягом 5 хвилин, повільно нагрівали до кімнатної температури, перемішували протягом З хвилин, а потім дистилювали при зменшеному тиску для видалення летючої речовини. Залишковий розчин висушували за допомогою вакуумного насосу і далі додавали до нього 20мл ДМФ. Суміш охолоджували до 0°С і до неї додавали 2-нафталін-сульфохлорид (0,5г) та діізопропілетиламін (0,9мл). Зазначену суміш перемішували, доки реактиви не розчинювались повністю. Реакційну суміш повільно нагрівали до кімнатної температури і перемішували протягом 3 годин. Після завершення реакції реакційний розчин дистилювали при зменшеному тиску для видалення летючої речовини. Залишковий розчин розчинювали за допомогою хлороформу і послідовно промивали насиченим водним розчином кислої солі карбонату натрію, розчином соляної кислоти і сольовим розчином, сушили над безводним сульфатом натрію, фільтрували і потім концентрували. Осад очищали за допомогою колонкової хроматографії, використовуючи як розчинник хлороформ / метанол (при об'ємному співвідношенні 95 : 5) для одержання вищеназваної сполуки (0,8г, вихід: 95%). 1 Н ЯМР (СD3СD, чнм) d : 8.3-7.2(m, 11Н), 5.8(m, 1Н), 4.5 (m, 1Н), 3.5-3.2(m, 2Н), 2.45(s, 3Н), 3.1-2.3(m, 8Н) Приклад 50 Синтез (S)нафталін-2-сульфокислота – [1-(4-амідразоно)-бензил-2-(4-метилсульфоніл-піперазиніл)-2оксо-етил] аміду Сполуку (0,4г, 0,76ммол), виготовлену як Препарат 14, розчинювали у піридині (5мл) і одержаний розчин розміщували у обладнану відведеннями колбу, у яку додавали триетиламін (0,3мл). Реакційна колба була обладнана таким чином, щоб сірководень (H2S) був здатний повільно утікати крізь одне з відведень колби, а витікати крізь друге відведення. Реакційний розчин насичували сірководнем, постійно перемішуючи протягом приблизно 10 хвилин, при цьому безкольоровий розчин ставав спочатку зеленого кольору , а далі поступово переходив до темнокоричневого кольору. Колбу закривали за допомогою гумової пробки і зберігали ЇЇ протягом 3 діб при кімнатній температурі до закінчення реакції. Потім реакційний розчин дистилювали при зменшеному тиску для видалення летючих речовин і висушували за допомогою вакуумного насосу. До одержаного твердого тіла жовтого кольору додавали разом ацетон (10мл) і йодометан (СН3І, 0,5мл) і одержану суміш нагрівали до одержання флегми протягом 30 хвилин. Зазначену реакційну суміш знову дистилювали при зменшеному тиску для видалення летючих речовин та висушували за допомогою вакуумного насосу. Осад розчинювали у чистому метанолі (5мл) і потім перемішували. До цієї суміші тричі порційно додавали 80% - ний гідрат гідразину (H2NNH2 Н2О, 0,06мл) через кожні 10 хвилин. Після закінчення реакції реакційний розчин концентрували, а потім очищали за допомогою рідинної хроматографії високого тиску (РХВТ) для одержання названої сполуки (0,3г, вихід : 65%). 1 H ЯMP (СD3СD, чнм) d : 8.4-7.4(m, 11Н), 4.6 (m, 1Н), 3.5-3.2(m, 2Н), 2.55(s, 3Н), 3.1-2.2(m, 8Н) Маса (ФСА, m/е) : 559 (М++ 1) Приклад 51 Синтез (S)-нафталін-2-сульфокислота-[1-(4-амідразоно)-бензил-2-оксо-2-(4-етоксикарбоніл-піперазиніл)етил] аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1метансульфоніл-піперазину застосовували 1-піперазин етил карбоксилат, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S) нафталін-2-сульфокислота-[1-(4-ціанобензил)-2-оксо-2-(4-етоксикарбоніл-піперазиніл)-етил] аміду (1г, 1,9ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,6г, вихід : 56%). 1 Н ЯМР (СD3СD, чнм) d : 8.5-7.3 (m, 11Н), 4.55(m, 1Н), 4.05(m, 2Н), 3.2-2.4(m, 10Н), 1.2(m, 3Н) Маса (ФСА, m/е) : 553 (М+ + 1) Приклад 52 Синтез (S)-нафталін-2-сульфокислота-[1-(4-амідразоно)-бензил-2-(4-форміл-піперазиніл)-2-оксоетил]аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1метансульфоніл-піперазину застосовували 1-піперазинетилкарбоксалдексид, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S)нафталін-2-сульфокислота-[1-(4-ціанобензил)-2- (4-форміл-піперазиніл)-2-оксоетил] (7г, 1,6ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,5г, вихід : 62%). 1 Н ЯМР (СD3СD, чнм) d : 8.36-7.44(m, 11Н), 4.60 (m, 1Н), 3.46-2.80(m, 9Н), 2.55(m, 1Н) Маса (ФСА, m/е) : 509 (М+ + 1) Приклад 53 Синтез (S)-нафталін-2-сульфокислота-[1-(4-амідразоно)-бензил-2-(4-етил-піперазиніл)-2-оксоетил] аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1метансульфонілпіперазину використовували 1-етилпіперазин, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S)нафталін-2-сульфокислота-[1-(4-ціанобензил)-2-(4-етил-піперазиніл)-2-оксоетил]аміду (0,3г, 0,6ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,2г, вихід : 56%). 1 Н ЯМР (СD3СD, чнм) d: 8.5-7.3 (m, 11Н), 4.6 (m, 1Н), 3.5-2.7(m, 10Н), 2.2(s, 2H), 1.4-1.2(m, 3Н) Маса (ФСА, m/е) : 509(М++1) Приклад 54 Синтез (S)-нафталін-2-сульфокислота-[1-(4-амідразоно)-бензил-2-оксо-2-(4-фенілпіперазиніл)-етил] аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1 метансульфонілпіперазину використовували 1-фенілпіперазин, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S) нафталін-2-сульфокислота-[1-(4-ціанобензил)-2-оксо-2-(4-феніл-піперазиніл)-етил] аміду (0,5г, 0,97ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,3г, вихід : 57%). 1 Н ЯМР (СD3СD, чнм) d: 8.35-7.54(m, 11Н), 7.20(m, 2H), 6.86(m, 1Н), 6.67(m, 2H), 4.56(m, 1Н), 3.45(m, 1Н), 3.25-2.92 (m, 5Н), 3.72 (m, 2Н), 2.42 (m, 1Н), 2.05(m, 1Н) Маса (ФСА, m/е) : 557(М++1) Приклад 55 Синтез (S)-нафталін-2-сульфокислота-[1-(4-амідразоно)-бензил-2-оксо-2-(4-(3-трифторметил-феніл)піперазиніл] -етил] аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1 метансульфонілпіперазину використовували 1-(a, a, a,-трифтор-m-толил) піперазин, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S)-нафталін-2-сульфокислота-[1-(4-ціанобензил)-2-оксо-2-[4-трифторметил-феніл)-піперазиніл]- етил] аміду (0,5г, 0,8ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,3г, ви хід: 55%). 1 H ЯMP (СD3СD, чнм) d : 8.35-6.7 (m, 15Н), 3.47(m, 1H) , 3.3-3.0 (m, 5Н), 3.76(m, 2Н), 2.45 (m, 1Н), 2.03(m, 1Н) Маса (ФСА, m/е) : 625(М++1) Приклад 56 Синтез (S)-нафталін-2-сульфокислота-[2-(4-ацетил-піперазиніл)-1-(4-амідразоно)бинзил-2-оксоетил]аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1метансульфонілпіперазину використовували 1-ацетил-піперазин, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S) нафталін-2-сульфокислота-[2-(4-ацетил-піперазиніл)-1-(ціанобензил)-2-оксо-етил] аміду (1,25г, 2,55ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,7г, вихід : 53%). 1 Н ЯМР (СD3СD, чнм) d : 8.4-7.4(m, 11Н), 4.5(m, 1Н), 3.5-3.2(m, 2Н), 3.1-2.2(m, 8Н), 2.0(s, 3Н) Маса (ФСА, m/е) : 523 (М+ + 1) Приклад 57 Синтез (S)-нафталін-2-сульфокислота-[1-(4-амідразоно)-бензил-2-оксо-2-[4-(2-гідроксиетил)-піперазиніл]етил] аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1метансульфонілпіперазину використовували 1-піперазин етан, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S)нафталін-2-сульфокислота-[1-(4-ціанобензил)-2-оксо-2-[4-(2-гідроксиетил)-піперазиніл]-етил] аміду (0,07г, 0,14ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,05г, вихід : 68,5%). 1 Н ЯМР (СD3СD, чнм) d : 8.35-7.30(m, 11Н), 4.60(m, 1Н), 3.96-3.7(m, 4Н), 3.70-2.80(m, 10Н) Маса (ФСА, m/е) : 525(М++1) Приклад 58 Синтез (S)-нафталін-2-сульфокислота-[1-(4-амідразоно)-бензил-2-оксо-2-[4-(2-етоксифеніл)-піперазиніл]етил] аміду Виконували такі ж самі операції, що і для одержання Препарату 13, за винятком того, що замість 1метансульфонілпіперазину використовували 1-(2-ето-ксифеніл) піперазин, і одержаний при цьому продукт далі оброблювали згідно з тим же самим способом, що і Препарат 14, для одержання проміжної сполуки (S)нафталін-2-сульфокислота-[1-(4-ціанобензил)-2-оксо-2-[4-(2-етоксифеніл)-піперазиніл]-етил] аміду (0,4г, 0,7ммол). Зазначену проміжну сполуку оброблювали згідно з тим ж самим способом, що і у Прикладі 50, для одержання очищеної вищеназваної сполуки (0,26г, вихід : 62%). 1 Н ЯМР (СD3СD, чнм) d : 8.36-6.54 (m, 15Н), 4.6 (m, 1H), 4.01(m, 2Н), 3.48(m, 1Н), 3.28-3.08(m, 5Н), 2.69(m, 2Н), 2.38(m, 1Н), 2.08(m, 1Н), 1.35(m, 3Н) Маса (ФСА, m/е) : 601(М++1) Тест 1: Інгібіторна активність до тромбіну Здатність сполуки згідно з цим винаходом інгібувати активність тромбіну визначали отаким чином. У кювету з ємностю 1,5мл додавали 1160мкл 0,1М трометамінового (тріс) буферного розчину (pH 7,8), який містив 150мМ NaCI та 0,1% ПЕГ - 8000 (поліетиленгліколь, молекулярна вага приблизно 8000). Хромозим ТН розчинювали в диметилсульфооксиді (ДМСО), при концентрації, що дорівнювала 10мМ, і одержаний розчин розчинювали за допомогою вищенаведеного трометамінового буферного розчину до концентрації 0,1мМ, який потім використовували як основний розчин. До кювети додавали 225мкл одержаного таким шляхом 0,1мМ основного розчину. Розчин інгібітора готували шля хом розчинення інгібіторної відносно тромбіну сполуки згідно з цим винаходом у диметилсульфоксиді з концентрацією 10мг/мл, а потім розчинювали одержаний таким чином розчин вищенаведеним трометаміновим буферним розчином до концентрації 0,1мг/мл , 0,01мг/мл, 0,001мг/мл і 0,0001мг/мл. Одержаний інгібіторний розчин брали у кількості, що відповідала 0-10мкг інгібітора, а потім розчинювали трометаміновим буферним розчином до досягнення загального об'єму 100мкл та додавали до кювети. З метою ініціації реакції гідролізу ензиму до кювети додавали 15мкл розчину бичачого тромбіну, розчиненого у вищезазначеному трометаміновому буферному розчині у концентрації 0,1мг/мл. Кількість пара нітроаніліну, одержаного протягом 2 хвилин після додавання ензиму, контролювали за допомогою абсорбції при довжині хвилі 381нм. Зображали безперервний спектр абсорбції за час реакції. Такі ж самі експерименти проводили для різних концентрацій інгібітора для одержання безперервного спектра. У кожному спектрі початкову швидкість Vн визначали по нахилу кривої для перших 30 секунд часу реакції, а потім робили графік для зворотньої величини початкової швидкості (1/Vн ) відносно концентрації інгібітора. З цього графіку одержували перше рівнення, що задовольняє точкам на вищезазначеному графіку, а далі, використовуючи рівнення для реакції ензиму, по значенням X первинного рівнення розраховували величину Кі. Величина Kм, яку використовували для цього розрахування, дорівнювала 8,3мкМ і була одержана шляхом змінювання концентрації субстрату при постійній концентрації ензиму. Швидкісну константу Кш визначали, використовуючи той самий розчин при аналогічній концентрації, як і при визначенні величини Кі, але при застосуванні нижченаведеної експериментальної процедури. У цьому випадку до кювети, що мала ємкість 1,5мл, додавали 1160мкл буферного розчину та 15мкл розчину бичачого тромбіну при концентрації 0,1мг/мл і 100мкл розчину інгібітора. Суміш тримали при кімнатній температурі протягом 15 хвилин. Далі під час додавання 225мкл 0,1мМ основного розчину протягом 2 хвилин контролювали зміну абсорбції як функції часу. Із одержаного безперервного спектру визначали похил прямої ділянки та репрезентували його як значення Vш. Аналогічний експеримент здійснювали для різних концентрацій інгібітора для визначення величини Vш при кожній концентрації інгібітора, а потім робили графік залежності 1/Vш як функції концентрації інгібітора. Використовуючи цей графік, одержували первинне рівнення, яке задовольняє точкам на графіку, а потім, використовуючи рівнення реакції ензиму, по значенням X первинного рівнення розраховували величину Кі. Особливо, інгібіторну активність до трипсину сполуки згідно з цим винаходом визначали таким же чином, як і визначення інгібіторної активності сполуки до тромбіну, що описана вище. Як субстрат використовували 20мкМ розчину хлористоводневого N-бензоїл-Val-Gly-Arg-р-нітроаніліду, а інгібітор застосовували при різних концентраціях у діапазоні від 0 до 120мкг. Крім того, трипсин розчинювали у 0.1N НСІ, регулювали до концентрації 45мкг/мл за допомогою вищезазначеного тріс - буферного розчину безпосередньо перед експериментом, а далі використовували у кількості, що дорівнювала 40мкл. Як і у експерименті з тромбіном, загальний об'єм реакційного розчину становив 1,5 мл, а наступні операції проводили аналогічним способом. Величина Kм, яку використовували для розрахунку величини Кі і визначали згідно з тим же самим способом, як і у ви щенаведеному експерименті з тромбіном, дорівнювала 20,2мкМ. Інгібіторну активність сполуки згідно з цим винаходом відносно активності кожного ензиму, яку визначали згідно з вищенаведеним способом, репрезентували величинами Кі та Кш, а селективність до тромбіну репрезентували як співвідношення інгібіторної активності до трипсину і інгібіторної активності до тромбіну. Одержані таким чином результати показані у нижченаведеній Таблиці 1. Таблиця 1 Інгібіторна активність сполуки згідно з цим в инаходом для тромбіну та трипсину Сполука Інгібіторна № актив ність до (Експ.) тромбіну Кі= 0.0038 mM 1 Ks= 0.0011 mM 2 Кі= 0.041 mM 3 Кі= 0.093 mM 4 Кі= 0.345 mM 5 Кі = 0.231 mM 6 Кі= 0.00435 mM 7 Кі= 0.0138 mM 9 Кі= 0.227 mM 10 Кі= 0.367 mM 13 Кі= 0.216 mM 15 Кі= 0.152 mM 16 КІ= 0.247 mM 17 КІ= 0.016 mM Кі= 0.011 mM 18 Ks= 0.002 mM 19 КІ= 0.0053 mM 20 Кі= 0.0217 mM Кі= 0.025 mM 21 Ks= 0.009 mM 22 Кі= 0.017 mM 23 Кі= 2.59 mM 24 Кі= 20.1 mM 25 Кі= 0.259 mM 27 Кі= 0.065 mM 29 Кі= 0.089 mM 31 Кі= 0.165mM 33 КІ= 0.035 mM 35 Кі= 0.612 mM 39 Кі= 0.165 mM 45 Кі= 0.346 mM 46 Кі= 0.880 mM 47 Кі= 0.665 mM 48 Кі- 0.013 mM 49 Кі= 0.406 mM Кі= 0.011 mM 50 Ks= 0.006 mM 51 Кі= 3.3 mM 52 Кі= 0.05 mM 53 Кі= 0.530 mM 54 Кі= 5.380 mM 55 Кі = 10.000 mM 56 Кі= 0.280 mM 57 Кі = 0.088 mM Інгібіторна Селектив ність актив ність до (трипсин/тромбін) трипсину Кі= 3.19 mM 2900 Кі= 363 mM 26304 Кі= 3.98 mM 2000 Кі = 5.25 mM Кі = 2.3 mM Кі > 100 mM 990 106 >10000 Кі = 21.7 mM 8 Кі = 5.79 mM 965 58 Кі = 9.130 mM Тест 2- Фармакокінетичний тест Спосіб виконання тесту Самців пацюків та собак годували протягом 24 годин та використовували як експериментальних тварин. Використовуючи фізіологічний розчин, готували 1%-ний розчин (10мг/мл) сполуки, одержаний, як це описано у Прикладі 1, а потім вводили його до експериментальних тварин внутрішньовенно і орально. Через визначені інтервали часу у тварин забирали кров і одразу змішували її з метанолом і сульфатом цинку. Потім проводили кількісний аналіз поверхневого шару суміші, використовуючи ультрофіолетове випромінювання при довжині хвилі, що дорівнювала 231нм, з метою вимірювання концентрації ліків у крові за допомогою РВХТ. Результати тесту: Концентрація ліків фармакокінетичні параметри сполуки, що наведені у Прикладі 1, після внутрішньовенного та орального введення показані у Таблицях 2-7. При внутрішньовенній ін'єкції сполуки, що наведена у Прикладі 1, вона швидко поширювалась, а далі повільно зникала як у пацюків, так і у собак, причому період напіврозпаду у собак був удвічі або більше разів довше, ніж у пацюків. Крім того, період напіврозпаду сполуки, що наведена у Прикладі 1, у собак була удвічі або більше разів довше, ніж період полурозпаду комерційно придатного Аргатробану (40 хвилин) у людини (див., Osamu et al., Pharmacology and Therapy, vol. 14, suppl. 5, 1986). Проміж цим, було показано, що біопридатність сполуки, що наведена у Прикладі 1, становить 15% у пацюків та 61% у собак при її оральному введенні. Однак було показано, що Аргатробан не абсорбується у тварин і людини при його оральному введенні. З ви щенаведених результатів зрозуміло, що сполука, наведена у Прикладі 1, показує кращі фармакокінетичні характеристики ніж Аргатробан при оральному введенні та періоду напіврозпаду. Таблиця 2 Концентрація у кров і сполуки, що нав едена у Прикладі 1, після її в нутрішньов енної ін'єкції (10мг/кг) до пацюків Час Концентрація кров і (нг/мл) Середня (хв ил.) Пацюк -1 Пацюк-2 Пацюк-3 (+помилка) (нг/мл) 31503 23000 1 31964 28822 (2914) 8105 6773 3 7884 7587(412) 3976 3066 5 4223 3755(352) 1907 1828 10 1928 1888(31) 918 898 20 883 900(10) 631 600 30 591 607(12) 355 408 45 398 387(16) 270 242 60 289 267(14) 168 166 90 184 173(6) 76 97 120 119 97(12) 87 73 180 78 79(4) Таблиця 3 Концентрація у кров і сполуки, що нав едена у Прикладі 1, після її орального вв едення (15 мг/кг) до пацюків Час Концентрація кров і (нг/мл) Середня (хв ил.) Пацюк -1 Пацюк-2 Пацюк-3 (+помилка) (нг/мл) 5 1610 822 570 1001(313) 10 1478 686 384 850(326) 20 569 166 97 255(147) 30 232 82 113 143(46) 45 145 107 112 121(12) 60 143 123 110 125(9) 90 66 186 119 124(35) 120 34 71 53(19) 180 24 238 64 109(66) Таблиця 4 Фармакокінетичні параметри сполуки, що нав едена у Прикладі 1, у пацюків Параметр Середня +(помилка) Період напів розпаду 47 (1) (хв ил.) Біопридатність (%) 15 (2) Таблиця 5 Концентрація у кров і сполуки, що нав едена у Прикладі 1, після її в нутрішньов енної ін'єкції (10мг/кг) до собак Час (хв ил.) 2 5 10 20 30 45 60 90 120 240 300 360 Концентрація кров і (нг/мл) Собака-1 Собака-2 29121 36648 9082 5228 5935 3282 3610 1801 2433 1335 1937 859 1471 690 1269 501 1090 377 544 129 315 107 317 92 Середня (+помилка) (нг/мл) 32885(3764) 7155(1927) 4608(1327) 2706(904) 1884(549) 1398(539) 1081(391)і 885(384) 733(357) і 337(207) 211(104) 204(112) Таблиця 6 Концентрація у кров і сполуки, що нав едена у Прикладі 1, після її орального вв едення (10мг/кг) до собак Час (хв ил.) 10 20 30 45 60 90 120 180 240 300 360 Концентрація кров і (нг/мл) Собака-1 Собака-2 1389 15 2076 17 1942 105 1891 558 2215 1312 1273 935 774 730 663 616 812 318 51 222 118 153 Середня (+помилка) (нг/мл) 702(687) 1047(1029) 1023 (918) 1225(667) 1763 (451) 1104 (169) 752(22) 640(24) 565(247) 136(86) 135(18) Таблиця 7 Фармакокінетичні параметри сполуки, що нав едена у Прикладі 1, у собак Параметр Період напів розпаду (хв ил.) Біопридатність (%) Середня +(помилка) 98 (15) 61 (15) Хоч цей винахід описаний на прикладі його найкращого варіанта виконання з певною мірою особливості, спеціалістам у цій галузі зрозуміло, що цей опис найкращого варіанта зроблений тільки як приклад і можуть бути передбачені численні зміни у деталях складання, комбінування та у структурі частин винаходу без відхилення від суті та об'єму цього винаходу.
ДивитисяДодаткова інформація
Назва патенту англійськоюSelective thrombin inhibitors and a pharmaceutical composition based thereon
Автори англійськоюLEE GU
Назва патенту російськоюВыборочно действующие ингибиторы тромбина и фармацевтическая композиция на их основе
Автори російськоюЛи Ку
МПК / Мітки
МПК: A61P 9/00, C07D 295/185, C07D 241/04, C07C 257/00, A61K 31/155
Мітки: тромбіну, діючі, інгібітори, фармацевтична, вибіркової, композиція, основі
Код посилання
<a href="https://ua.patents.su/24-63887-vibirkovo-diyuchi-ingibitori-trombinu-ta-farmacevtichna-kompoziciya-na-ikh-osnovi.html" target="_blank" rel="follow" title="База патентів України">Вибірково діючі інгібітори тромбіну та фармацевтична композиція на іх основі</a>
Інгібітори тромбіну, спосіб їх одержання, фармацевтична композиція на їх основі та спосіб лікування

Номер патенту: 61057
Опубліковано: 17.11.2003
Автори: Нюстрьом Ян-Ерік, Густафссон Давід
МПК: C07D 405/06, A61P 7/02, C07D 205/00, A61K 31/40, A61K 31/395, A61K 31/445, A61K 31/443, C07D 211/60, C07D 207/16, A61K 31/4025, A61K 31/397
Мітки: основі, спосіб, одержання, фармацевтична, тромбіну, лікування, композиція, інгібітори
Формула / Реферат:
1. Сполука формули І, Iде р та q незалежно дорівнюють 0, 1, 2, 3 або 4;R1 - Н, 2,3-епоксипропіл, C1-6алкіл (в якому остання група необов‘язково заміщена або закінчена одною чи більше гідроксильними групами), структурний фрагмент формули Іа, Iaв якій А1 - одинарний зв'язок або С1-4алкілен, a Rx - Н або С1-4алкіл при умові, що у ланцюгу Rx-C-C-A1 не більше шести атомів карбону, або, коли р дорівнює 0, разом з...
Піразинонові інгібітори тромбіну, фармацевтична композиція, спосіб інгібування утворення тромбів у крові, спосіб лікування станів, пов’язаних із тромбоутворенням

Номер патенту: 58636
Опубліковано: 15.08.2003
Автори: КОБЕРН Крейг А., САНДЕРСОН Філіп Е., Бергі Крістофер С., ЛАЙЛ Террі А., УІЛЛЬЯМС Пітер Д., РОБІНСОН Кайл А.
МПК: A61P 43/00, C07D 401/12, A61K 31/495, A61P 7/02, A61P 9/10, A61K 31/497, A61P 9/00, C07D 401/14
Мітки: композиція, інгібування, фармацевтична, лікування, піразинонові, станів, інгібітори, пов'язаних, крові, спосіб, тромбів, тромбоутворенням, тромбіну, утворення
Формула / Реферат:
1. Сполука формули:,де W вибирають із групи, що складається з1) водню,2) 5-7-членного моно- або 9-10-членного конденсованого біциклічного гетероциклічного кільця, що містить кільцеві атоми вуглецю і кільцеві гетероатоми, яке може бути насиченим або ненасиченим, причому вказане кільце міститьa) від одного до чотирьох гетероатомів, вибраних із групи, що складається з N, О і S, і де кільце є незаміщеним,...
Сульфонамідні похідні, спосіб їх одержання та фармацевтична композиція на їх основі

Номер патенту: 63990
Опубліковано: 16.02.2004
Автори: Гадек Томас, Кнолле Йохен, Гурвест Жан-Франсуа, Катбертсон Роберт Ендрю, Бодарі Сара Кетрін, Пейман Ануширван, Вілл Девід Вільям, Карніато Дені, Шойнеманн Карлхайнц, Макдауелл Роберт
МПК: A61K 31/197, C07C 311/19, A61P 13/12, A61P 9/10, A61P 27/02, A61P 43/00, C07D 401/12, A61P 29/00, C07D 409/12, A61K 31/505, A61P 9/00, A61K 31/506, A61P 19/10, A61P 35/00, C07D 239/16
Мітки: композиція, одержання, фармацевтична, основі, сульфонамідні, похідні, спосіб
Формула / Реферат:
1. Сульфонамідні похідні формули І:, (I)в якійR1 і R2, незалежно один від одного, означають водень чи (С1-С6)-алкіл, незаміщений чи заміщений радикалом R3,чи в якій радикали R1 і R2, спільно, являють собою насичений чи ненасичений двовалентний (С2-С9)-алкіленовий радикал, що є незаміщеним чи заміщеним однією чи більше групами з числа наступних:...
Інгібітори матричних металопротеаз та фармацевтична композиція на їх основі

Номер патенту: 50707
Опубліковано: 15.11.2002
Автори: КАСТЕЛЯНО Арліндо Л., ЛАЙЕК Тенг Дж., ЮАН Женгьо, ХОРН Стівен, КРАНТЦ Александер
МПК: A61K 31/40, C07F 9/6561, A61P 35/00, A61P 43/00, C07D 498/08, A61P 1/02, C07F 9/645, C07D 487/04, C07D 487/08, A61P 29/00
Мітки: фармацевтична, основі, металопротеаз, матричних, інгібітори, композиція
Формула / Реферат:
1. Соединение формулы (I):где штриховые линии обозначают необязательные двойные связи; и,если n равно 1, 2 или 3; m равно 3 или 4; тоА обозначает –СН2-;R1 обозначаетa) -СН2-R4, где R4 обозначает меркапто, ацетилтио, карбокси, гидроксиаминокарбонил, N-гидроксиформамидометил, алкоксикарбонил, арилоксикарбонил, аралкоксикарбонил, бензилоксиаминокарбонил илигде R6 обозначает необязательно...
1н-індол-3-гліоксиламіди як інгібітори spla2 та фармацевтична композиція на їх основі

Номер патенту: 47387
Опубліковано: 15.07.2002
Автори: Бах Ніколас Джеймс, Діллард Роберт Ділейн, Драхейм Сюзан Елізабет
МПК: C07D 209/22, A61K 31/405, C07D 403/12, C07D 209/12
Мітки: 1н-індол-3-гліоксиламіди, основі, інгібітори, фармацевтична, композиція, spla2
Формула / Реферат:
1. 1Н-индол-3-глиоксиламид, представленный формулой (I), или его фармацевтически приемлемая соль, сольват или пролекарство:в которойкаждый Χ независимо является кислородом или серой;R1 выбирают из групп (а), (b) или (с), где:(a) - С7-С20 алкил, С7-С20 алкенил, С7-С20 алкинил, карбоциклические радикалы или гетероциклические радикалы;(b) - член группы (а), замещенный одним или несколькими...
Попередній патент: Пристрій для посадки насіння
Випадковий патент: Двоканальний адаптивний пристрій контролю вологості