Тетрагідро-4н-піридо[1,2-а]піримідини та фармацевтична композиція, що застосовуються як інгібітори віл-інтегрази
Номер патенту: 80737
Опубліковано: 25.10.2007
Автори: Муралья Естер, Крешенци Бенедетта, Пескаторе Джованна, Сумма Вінченцо, Кінцель Олаф, Орвьєто Федеріка, Роулі Майкл








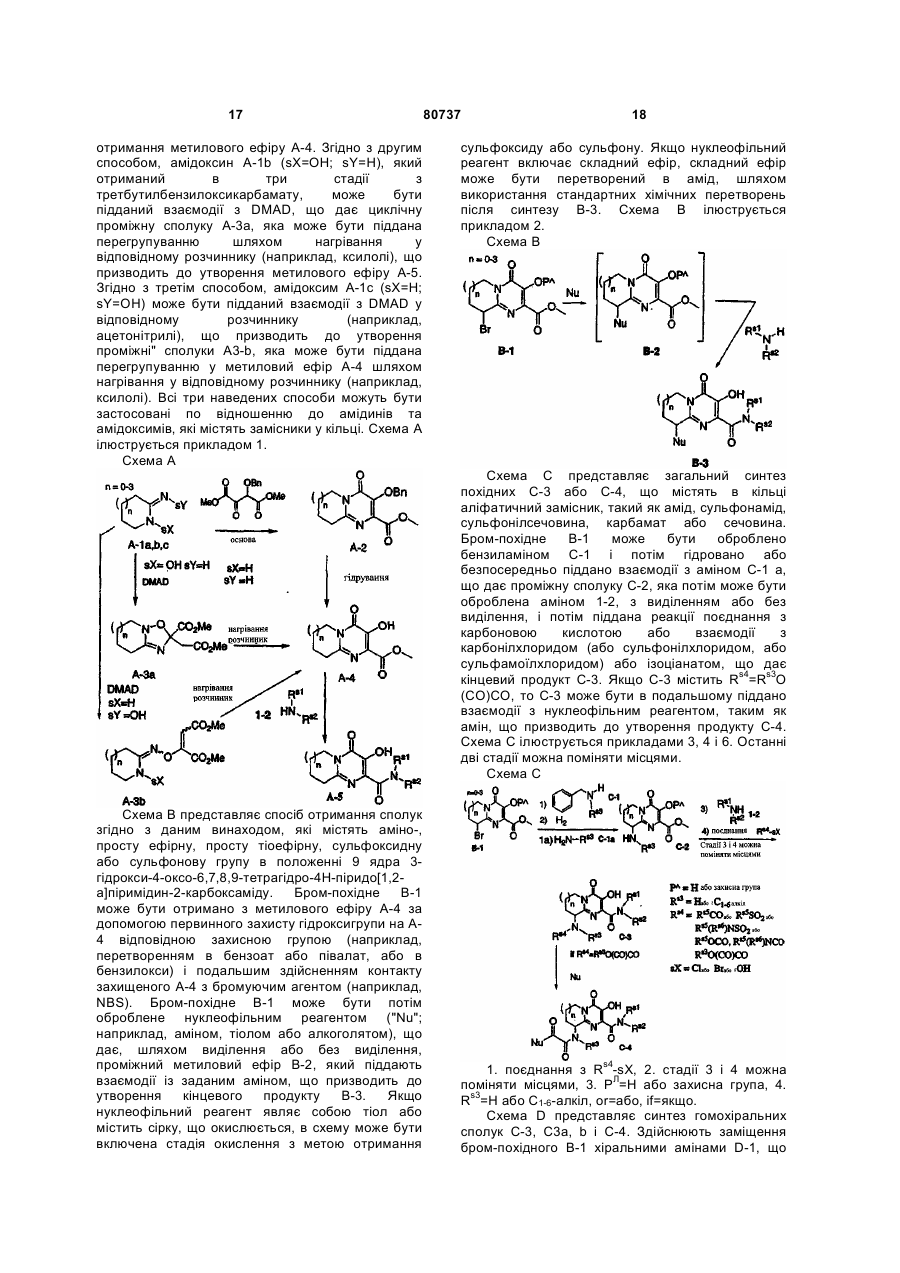











Формула / Реферат
1. Сполука формули (І) або її фармацевтично прийнятна сіль:
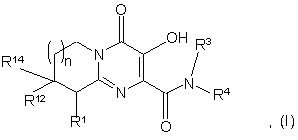
де R1 означає Н або NR2R5;
R2 означає СН3;
R5 означає:
1) C(O)CH2SO2CH3,
2) C(O)C(O)N(CH3)2,
3) SO2N(CH3)2 aбo
4) SO2R20, де R20 означає:
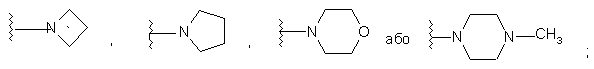
або альтернативно, R2 і R5 разом з атомом азоту, до якого приєднані, утворюють
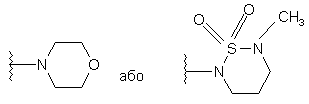 ;
;
R3 означає водень;
R4 означає:
1) п-фторбензил,
2) 4-фтор-3-метилбензил,
3) 3-хлорбензил або
4) 3-хлор-4-метилбензил;
R12 та R14 обидва означають Н, за винятком, коли R5 означає C(O)C(O)N(CH3)2 і R4 означає п-фторбензил, і n дорівнює 1, то R12 і R14 або обидва означають Н, або обидва означають СН3; та
n означає ціле число, яке дорівнює нулю, 1 або 2.
2. Сполука за п.1 або її фармацевтично прийнятна сіль, де R1 означає NR2R5 і n дорівнює 1 або 2.
3. Сполука за п.1 або її фармацевтично прийнятна сіль, де
R1 означає NR2R5;
R2 означає СН3;
R5 означає C(O)C(O)N(CH3)2 або SO2R20, де R20 означає
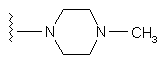 ;
;
R3 означає водень;
R4 означає п-фторбензил або 4-фтор-3-метилбензил;
R12 і R14 обидва означають Н, за винятком, коли R5 означає C(O)C(O)N(CH3)2 і R4 означає п-фторбензил, і n дорівнює 1, то R12 і R14 або обидва означають Н, або обидва означають СН3; і n означає ціле число, що дорівнює 1 або 2.
4. Сполука формули II або її фармацевтично прийнятна сіль:
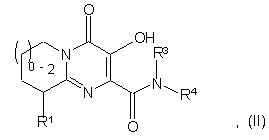
де R1 означає водень, NR2R5, OR2, SR2, SOR2, SO2R2, SO2NR2R5 або OC(O)NR2R5;
R3 означає водень;
R4 означає
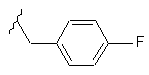 ;
;
R2 означає
1) водень,
2) CH3 або
3)
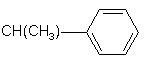 ;
;
і
R5 означає
1) С(О)СН3,
2) C(O)CH2SO2CH3,
3) СН3,
4) C(O)C(O)N(CH3)2,
5) SO2CH3,
6) SO2N(CH3)2,
7) C(O)CH2N(CH3)2,
8) SO2CH2SO2CH3,
9) C(O)CF3,
10)
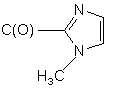 ,
,
11)
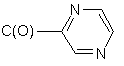
або
12)
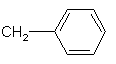 ;
;
або R2 і R5, разом з атомом азоту, до якого вони приєднані, утворюють гетероциклічний цикл, що вибирається з групи, яка включає
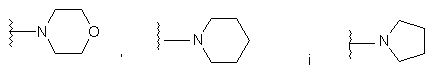 .
.
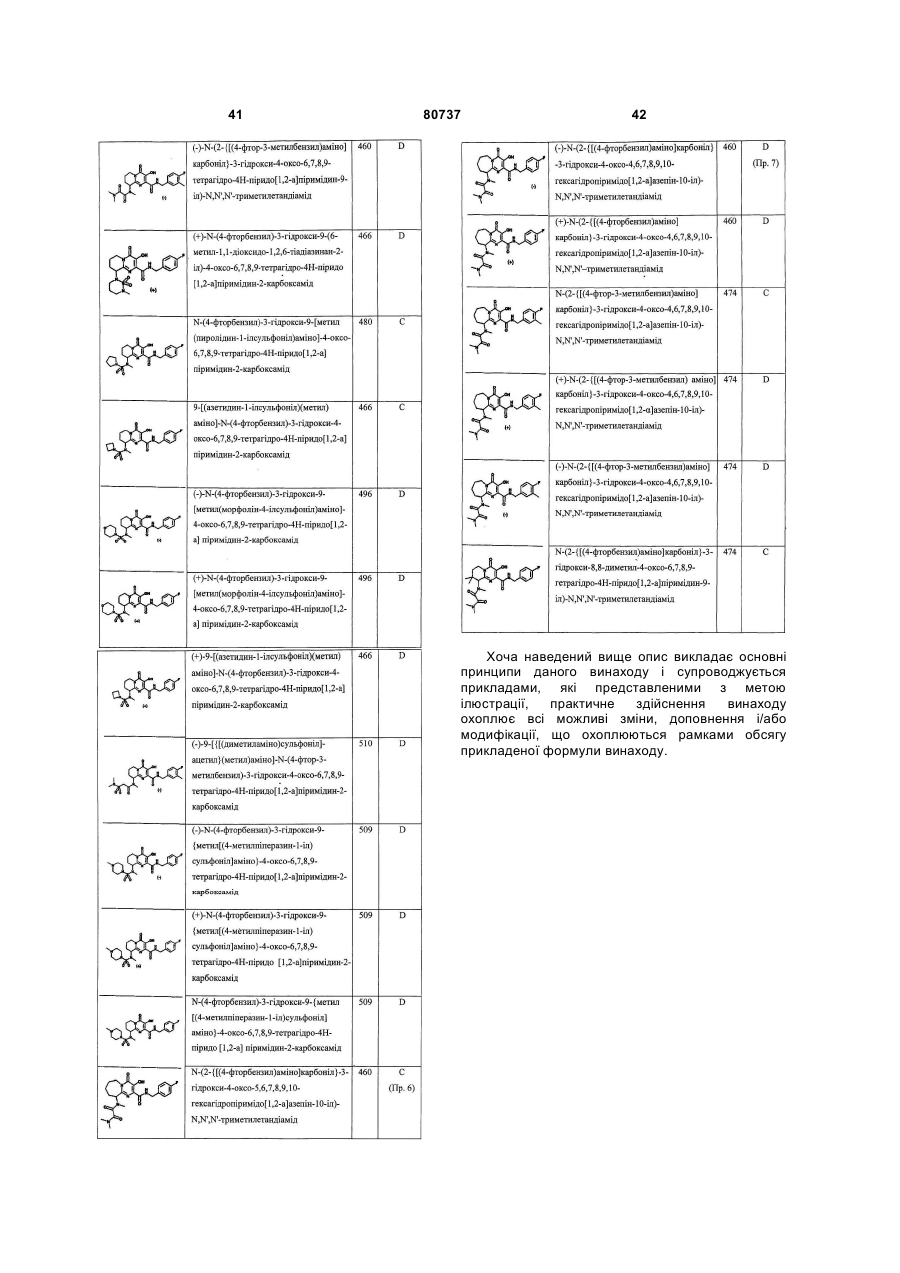
5. Сполука за п.1 або її фармацевтично прийнятна сіль, що вибирається з групи, що включає:
N-(4-фторбензил)-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N-(4-фторбензил)-3-гідрокси-9-морфолін-4-іл-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
9-[[(диметиламіно)сульфоніл](метил)аміно]-N-(4-фторбензил)-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N1-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідрокси-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N1,N2,N2-триметилетандіамід;
(+)-N1-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N1,N2,N2-триметилетандіамід;
(-)-N-(4-фторбензил)-3-гідрокси-9-{метил[(метилсульфоніл)ацетил]аміно}-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N,N',N'-триметилетандіамід;
N-(2-{[(3-хлор-4-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N,N',N'-триметилетандіамід;
N-(2-{[(3-хлорбензил)аміно]карбоніл}-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N,N',N'-триметилетандіамід;
N-(4-фторбензил)-3-гідрокси-9-(6-метил-1,1-діоксидо-1,2,6-тіадіазинан-2-іл)-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(-)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N,N',N'-триметилетандіамід;
(+)-N-(4-фторбензил)-3-гідрокси-9-(6-метил-1,1-діоксидо-1,2,6-тіадіазинан-2-іл)-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N-(4-фторбензил)-3-гідрокси-9-[метил(піролідин-1-ілсульфоніл)аміно]-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
9-[(азетидин-1-ілсульфоніл)(метил)аміно]-N-(4-фторбензил)-3-гідрокси-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(-)-N-(4-фторбензил)-3-гідрокси-9-[метил(морфолін-4-ілсульфоніл)аміно]-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(+)-N-(4-фторбензил)-3-гідрокси-9-[метил(морфолін-4-ілсульфоніл)аміно]-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(+)-9-[(азетидин-1-ілсульфоніл)(метил)аміно]-N-(4-фторбензил)-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(-)-9-[{[(диметиламіно)сульфоніл]ацетил}(метил)аміно]-N-(4-фтор-3-метилбензил)-3-гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(-)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4-метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(+)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4-метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N-(4-фторбензил)-3-гідрокси-9-{метил[(4-метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N-(2-{(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(+)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(+)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-a]aзепін-10-іл)-N,N',N'-триметилетандіамід;
(-)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[ 1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідрокси-8,8-диметил-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N,N',N'-триметилетандіамід.
6. Сполука за п. 5 або її фармацевтично прийнятна сіль, яка вибирається з групи, що включає:
N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(+)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(-)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(-)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4-метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
(+)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4-метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N-(4-фторбензил)-3-гідрокси-9-{метил[(4-метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2-карбоксамід;
N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідрокси-8,8-диметил-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9-іл)-N,N',N'-триметилетандіамід;
N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(+)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N'N'-триметилетандіамід.
7. Сполука за п. 6 або її фармацевтично прийнятна сіль, вибрана з групи, яка включає:
N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід;
(+)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід.
8. Сполука за пунктом 7, яка являє собою (-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід або його фармацевтично прийнятну сіль.
9. Фармацевтична композиція, що містить терапевтично ефективну кількість сполуки за будь-яким з пп.1-8 або її фармацевтично прийнятної солі і фармацевтично прийнятний носій.
10. Сполука за будь-яким з пп. 1-8, або її фармацевтично прийнятна сіль, для застосування в отриманні лікарського засобу для інгібування ВІЛ-інтегрази, для профілактики або лікування ВІЛ-інфекції або профілактики, лікування або затримки вияву СНІДу у пацієнта, який цього потребує.
Текст
1. Сполука формули (І) або її фармацевтично прийнятна сіль: O R14 КОМПОЗИЦІЯ, 3 80737 O 0 -2 2 OH R3 N N N R4 , (II) O де R1 означає водень, NR2R5, OR2, SR2, SOR2, SO2R2, SO2NR2R5 або OC(O)NR2R5; R3 означає водень; R4 означає R1 F ; R2 означає 1) водень, 2) CH3 або 3) CH(CH3) ; і R5 означає 1) С(О)СН3, 2) C(O)CH2SO2CH3, 3) СН3, 4) C(O)C(O)N(CH3)2, 5) SO2CH3, 6) SO2N(CH3)2, 7) C(O)CH2N(CH3)2, 8) SO2CH2SO2CH3, 9) C(O)CF3, 10) N C(O) N H3 C , 11) N C(O) N або 12) CH2 ; або R 2 і R 5 , разом з атомом азоту, до якого вони приєднані, утворюють гетероциклічний цикл, що вибирається з групи, яка включає N O , N N i . 5. Сполука за п.1 або її фармацевтично прийнятна сіль, що вибирається з групи, що включає: 4 N-(4-фторбензил)-3-гідроксі-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; N-(4-фторбензил)-3-гідрокси-9-морфолін-4-іл-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; 9-[[(диметиламіно)сульфоніл](метил)аміно]-N-(4фторбензил)-3-гідроксі-4-оксо-6,7,8,9-тетрагідро4Н-піридо[1,2-а]піримідин-2-карбоксамід; N1-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідрокси-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9іл)-N1,N2,N2-триметилетандіамід; (+)-N1-(2-{[(4-фторбензил)аміно]карбоніл}-3гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-іл)-N1,N2,N2-триметилетандіамід; (-)-N-(4-фторбензил)-3-гідрокси-9{метил[(метилсульфоніл)ацетил]аміно}-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-іл)-N,N',N'-триметилетандіамід; N-(2-{[(3-хлор-4-метилбензил)аміно]карбоніл}-3гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-іл)-N,N',N'-триметилетандіамід; N-(2-{[(3-хлорбензил)аміно]карбоніл}-3-гідроксі-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9іл)-N,N',N'-триметилетандіамід; N-(4-фторбензил)-3-гідрокси-9-(6-метил-1,1діоксидо-1,2,6-тіадіазинан-2-іл)-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; (-)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3гідроксі-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-іл)-N,N',N'-триметилетандіамід; (+)-N-(4-фторбензил)-3-гідрокси-9-(6-метил-1,1діоксидо-1,2,6-тіадіазинан-2-іл)-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; N-(4-фторбензил)-3-гідрокси-9-[метил(піролідин-1ілсульфоніл)аміно]-4-оксо-6,7,8,9-тетрагідро-4Нпіридо[1,2-а]піримідин-2-карбоксамід; 9-[(азетидин-1-ілсульфоніл)(метил)аміно]-N-(4фторбензил)-3-гідрокси-4-оксо-6,7,8,9-тетрагідро4Н-піридо[1,2-а]піримідин-2-карбоксамід; (-)-N-(4-фторбензил)-3-гідрокси-9-[метил(морфолін4-ілсульфоніл)аміно]-4-оксо-6,7,8,9-тетрагідро-4Нпіридо[1,2-а]піримідин-2-карбоксамід; (+)-N-(4-фторбензил)-3-гідрокси-9[метил(морфолін-4-ілсульфоніл)аміно]-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; (+)-9-[(азетидин-1-ілсульфоніл)(метил)аміно]-N-(4фторбензил)-3-гідроксі-4-оксо-6,7,8,9-тетрагідро4Н-піридо[1,2-а]піримідин-2-карбоксамід; (-)-9[{[(диметиламіно)сульфоніл]ацетил}(метил)аміно]N-(4-фтор-3-метилбензил)-3-гідроксі-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; (-)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; 5 (+)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; N-(4-фторбензил)-3-гідрокси-9-{метил[(4метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; N-(2-{(4-фторбензил)аміно]карбоніл}-3-гідроксі-4оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10іл)-N,N',N'-триметилетандіамід; (-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2а]азепін-10-іл)-N,N',N'-триметилетандіамід; (+)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін10-іл)-N,N',N'-триметилетандіамід; N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2а]азепін-10-іл)-N,N',N'-триметилетандіамід; (+)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2a]aзепін-10-іл)-N,N',N'-триметилетандіамід; (-)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}3-гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[ 1,2-а]азепін-10-іл)-N,N',N'-триметилетандіамід; N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідрокси8,8-диметил-4-оксо-6,7,8,9-тетрагідро-4Нпіридо[1,2-а]піримідин-9-іл)-N,N',N'триметилетандіамід. 6. Сполука за п. 5 або її фармацевтично прийнятна сіль, яка вибирається з групи, що включає: N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}-3гідроксі-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2а]азепін-10-іл)-N,N',N'-триметилетандіамід; (+)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}3-гідроксі-4-оксо-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'триметилетандіамід; (-)-N-(2-{[(4-фтор-3-метилбензил)аміно]карбоніл}3-гідроксі-4-оксо-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-10-іл)-N,N',N'триметилетандіамід; (-)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; (+)-N-(4-фторбензил)-3-гідрокси-9-{метил[(4метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо Даний винахід відноситься до тетрагідро-4Нпіридо[1,2-а]піримідинів, споріднених сполук і їх фармацевтично прийнятних солей, синтезу і застосуванню цих сполук як інгібіторів фермент ВІЛ-інтегрази. Сполуки і їх фармацевтично прийнятні солі згідно з даним винаходом корисні для профілактики або лікування ВІЛ-інфекції і для лікування або затримки вияву СНІДу. Ретровірус, який називається вірусом імунодефіциту людини (ВІЛ), є етіологічним 80737 6 6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; N-(4-фторбензил)-3-гідрокси-9-{метил[(4метилпіперазин-1-іл)сульфоніл]аміно}-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід; N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідрокси8,8-диметил-4-оксо-6,7,8,9-тетрагідро-4Нпіридо[1,2-а]піримідин-9-іл)-N,N',N'триметилетандіамід; N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10іл)-N,N',N'-триметилетандіамід; (-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10іл)-N,N',N'-триметилетандіамід; (+)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін10-іл)-N,N'N'-триметилетандіамід. 7. Сполука за п. 6 або її фармацевтично прийнятна сіль, вибрана з групи, яка включає: N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10іл)-N,N',N'-триметилетандіамід; (-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10іл)-N,N',N'-триметилетандіамід; (+)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4оксо-4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10іл)-N,N',N'-триметилетандіамід. 8. Сполука за пунктом 7, яка являє собою (-)-N-(2{[(4-фторбензил)аміно]карбоніл}-3-гідроксі-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)N,N',N'-триметилетандіамід або його фармацевтично прийнятну сіль. 9. Фармацевтична композиція, що містить терапевтично ефективну кількість сполуки за будь-яким з пп.1-8 або її фармацевтично прийнятної солі і фармацевтично прийнятний носій. 10. Сполука за будь-яким з пп. 1-8, або її фармацевтично прийнятна сіль, для застосування в отриманні лікарського засобу для інгібування ВІЛ-інтегрази, для профілактики або лікування ВІЛ-інфекції або профілактики, лікування або затримки вияву СНІДу у пацієнта, який цього потребує. фактором комплексного захворювання, що включає прогресуюче руйнування імунної системи (синдром набутого імунодефіциту; СНІД) і дегенерацію центральної і периферичної нервової системи. Цей вірус раніше був відомий як LAV, HTLV-III або ARV. Загальною ознакою реплікації ретровірусів є вставка ДНК провірусу, що кодує вірусну інтегразу в геном клітки-хазяїна, обов'язкова стадія в реплікації ВІЛ в Т-лімфоцитах і моноцитарних клітинах людини. Вважається, що 7 80737 інтеграція під дією інтегрази відбувається в три стадії: збирання стабільного нуклерпротеїдного комплексу, з послідовностями вірусної ДНК; відщеплення двох нуклеотидів від 3' кінців лінійної ДНК провірусу; ковалентне зв'язування звільнених 3' ОН-кінців ДНК провірусу з ДНК клітини-хазяїна на ступінчасто розташованих сайтах розщеплення. Четверта стадія процесу, репаративний синтез двохниткового розриву, що утворився, може бути виконана за допомогою клітинних ферментів. Секвенування нуклеїнової кислоти ВІЛ свідчить про наявність pol-гену в одній відкритій рамці зчитування [Ratner, L. et al., Nature, 313, 277(1985)]. Гомологія послідовностей амінокислот свідчить про те, що pol-послідовність кодує зворотну транскриптазу, інтегразу і ВІЛ-протеазу [Toh, Η. et al., EMBO J. 4, 1267 (1985); Power, M.D. et al., Science, 231, 1567 (1986); Pearl, L.H. et al., Nature, 329, 351 (1987)]. Встановлено, що всі три ферменти необхідні для реплікації ВІЛ. Відомо, що деякі противірусні сполуки, що діють як інгібітори реплікації ВІЛ, є ефективними засобами в лікуванні СНІДу і схожих захворювань, включаючи інгібітори зворотної транскриптази, такі як азидотимін (AZT) і ефавіренз, і інгібітори протеази, такі як індинавір і нелфінавір. Сполуки згідно з даним винаходом є інгібіторами ВІЛінтегрази і інгібіторами реплікації ВІЛ. Інгібування інтегрази in vitro і реплікації ВІЛ в клітинах є прямим наслідком інгібування реакції перенесення ланцюга, що каталізується рекомбінантною інтегразою in vitro в клітинах, заражених ВІЛ. Особлива перевага даного винаходу полягає у високо специфічному інгібуванні ВІЛ-інтегрази і реплікації ВІЛ. Даний винахід стосується нових піридопіримідинових похідних і споріднених сполук. Ці сполуки корисні в інгібуванні ВІЛінтегрази, профілактиці ВІЛ-інфекції і в профілактиці, лікуванні або затримці вияву СНІДу і/або СНІД-асоційованого комплексу (ARC), або у вигляді їх сполук, або у їх вигляді фармацевтично прийнятних солей або гідратів (коли це доцільне), або як інгредієнтів фармацевтичні композиції, в комбінації з іншими ВІЛ/СНІД-противірусними засобами, протиінфекційними засобами, імуномодуляторами, антибіотиками або вакцинами, або за відсутності вказаних засобів. Більш конкретно, даний винахід включає сполуки формули І і їх фармацевтично прийнятні солі: (I) R1 означає Η або NR2R5; R2 означає СН3; R5 означає 1) C(O)CH2SO2CH3, 2) C(O)C(O)N(CH3)2, 3) SO2N(CH3)2 або 4) SO2R20, де R20 означає: 8 або альтернативно, R2 і R5 разом з атомом азоту, до якого приєднані, утворюють Помилка! Об'єкти не можуть бути розкриті на основі кодів поля редагування, або R3 означає водень; R4 означає: 1) п-фторбензил, 2) 4-фтор-3-метилбензил, 3) 3-хлорбензил або 4) 3-хлор-4-метилбензил; R12 і R14 - обидва означають Н, за винятком, що коли R5 означає C(O)C(O)N(CH3)2 і R4 означає п-фторбензил, і n дорівнює 1, то R12 і R14 або обидва означають Н, або обидва означають СН3; і n означає ціле число, що дорівнює нулю, 1 або 2. Даний винахід також включає фармацевтичні композиції, що містять сполуки згідно з даним винаходом, і способи отримання таких фармацевтичних композицій. Крім того, даний винахід включає способи лікування СНІДу, способи затримки вияву СНІДу, способи профілактики СНІДу, способи профілактики ВІЛінфекції і способи лікування ВІЛ-інфекції. Інші варіанти здійснення, аспекти і відмітні ознаки даного винаходу або зазначені нижче в приведеному описі, або стають очевидними з подальшого опису, прикладів і формули винаходу. Даний винахід включає сполуки приведеної вище формули І і їх фармацевтично прийнятні солі. Ці сполуки і відповідні фармацевтично прийнятні солі є інгібіторами ВІЛ-інтегрази. Варіантом здійснення даного винаходу є сполука формули І або її фармацевтично прийнятна сіль, де R1 означає NR2R5; n означає ціле число, що дорівнює 1 або 2; а всі інші змінні приймають попередньо визначені значення. Іншим варіантом здійснення даного винаходу є сполука формули І або її фармацевтично прийнятна сіль, де R1 означає NR2R5; R2 означає СН3; R5 означає 1) C(O)C(O)N(CH3)2 або 2) SO2R20, де R20 означає R3 означає водень; R4 означає п-фторбензил або 4-фтор-3-метилбензил; R12 і R14 - обидва означають Н, за винятком, що коли R5 означає C(O)C(O)N(CH3)2 і R4 означає п-фторбензил, і n дорівнює 1, то R12 і R14 або обидва означають Н, або обидва означають СН3; і n означає ціле число, яке дорівнює 1 або 2. Даний винахід також включає сполуку формули II або її фармацевтично прийнятну сіль: (II) 9 де R1 означає водень, NR2R5; OR2, SR2, SOR2, SO2R2, SO2NR2R5 або OC(O)NR2 R5; R3 означає водень; R4 означає R2 означає 1) водень, 2) CH3 або 3) і R5 означає 1) С(О)СН3, 2) C(O)CH2SO2CH3, 3) СН3, 4) C(O)C(O)N(CH3)2, 5) SO2CH3, 6) SO2N(CH3)2, 7) C(O)CH2N (CH3)2, 8) SO2CH2SO2CH3, 9) C(O)CF3, 10) 11) або 12) або R2 і R5, разом з атомом азоту, до якого приєднані, утворюють гетероциклічне кільце, що вибирається з групи, яка включає Іншим варіантом здійснення даного винаходу є сполука або її фармацевтично прийнятна сіль, яка вибирається з групи, що включає сполуки, які перераховані в таблиці І, що наводиться нижче. Додаткові варіанти здійснення даного винаходу включають наступне: (a) Фармацевтичну композицію, що містить сполуку формули (І) та фармацевтично прийнятний носій. (b) Фармацевтичну композицію, що містить продукт, який отриманий комбінуванням (наприклад, змішуванням) ефективної кількості сполуки формули (І) і фармацевтично прийнятного носія. (з) Фармацевтичну композицію згідно з п.(а) або (b), що додатково містить терапевтично ефективну кількість засобу для лікування ВІЛінфекції/СНІДу, який вибирається з групи, що включає ВІЛ/СНІД-противірусні засоби, імуномодулятори та протиінфекційні засоби. (d) Фармацевтичну композицію згідно з п.(с), де засіб для лікування ВІЛ-інфекції/СНІДу являє собою противірусний засіб, який вибирається з групи, що включає: інгібітори ВІЛ-протеази, ненуклеозидні інгібітори зворотної транскриптази ВІЛ і нуклеозидні інгібітори зворотної транскриптази ВІЛ. (e) Комбінацію, яка є корисною для інгібування ВІЛ-інтегрази, лікування або профілактики ВІЛінфекції, або профілактики, лікування або 80737 10 затримки виявлення СНІДу, що являє собою терапевтично ефективну кількість сполуки формули (І) і терапевтично ефективну кількість засобу для лікування ВІЛ-інфекції/СНІДу, який вибирається з групи, що включає: ВІЛ/СНІДпротивірусні засоби, імуномодулятори та протиінфекційні засоби. (f) Комбінацію згідно з п.(e), де засіб для лікування інфекції ВІЛ/СНІДу являє собою противірусний засіб, який вибирається з групи, що включає: інгібітори ВІЛ-протеази, ненуклеозидні інгібітори зворотної транскриптази ВІЛ і нуклеозидні інгібітори зворотної транскриптази ВІЛ. (g) Спосіб інгібування ВІЛ-інтегрази у пацієнта, який цього потребує, що полягає у введенні пацієнту терапевтично ефективної кількості сполуки формули (І) (h) Спосіб профілактики або лікування ВІЛінфекції у пацієнта, який цього потребує, що полягає у введенні пацієнту терапевтично ефективної кількості сполуки формули (І). (і) Спосіб згідно з п.(h), де сполуку формули (І) вводять в комбінації з терапевтично ефективною кількістю, щонайменше, одного противірусного засобу, який вибирається з групи, що включає: інгібітори ВІЛ-протеази, ненуклеозидні інгібітори зворотної транскриптази ВІЛ і нуклеозидні інгібітори зворотної транскриптази ВІЛ. (j) Спосіб профілактики, лікування або затримки прояву СНІДу у пацієнта, який цього потребує, що включає введення пацієнту терапевтично ефективної кількості сполуки формули (І). (k) Спосіб згідно з п.(j), де сполуку вводять в комбінації з терапевтично ефективною кількістю, щонайменше, одного противірусного засобу, який вибирається з групи, що включає: інгібітори ВІЛпротеази, ненуклеозидні інгібітори зворотної транскриптази ВІЛ і нуклеозидні інгібітори зворотної транскриптази ВІЛ. (1) Спосіб інгібування ВІЛ-інтегрази у пацієнта, який цього потребує, що включає введення пацієнту фармацевтичної композиції згідно з п.(а), (b), (с) або (d), або комбінації згідно з п.(e) або (f). (m) Спосіб профілактики або лікування ВІЛінфекції у пацієнта, який цього потребує, що включає введення пацієнту фармацевтичної композиції згідно з п. (а), (Ь), (с) або (d), або комбінації згідно з п.(e) або (f). (n) Спосіб профілактики, лікування або затримки вияву СНІДу у пацієнта, який цього потребує, що включає введення пацієнту фармацевтичної композиції згідно з п.(а), (b), (с) або (d), або комбінації згідно з п.(e) або (f). Даний винахід також включає сполуку (і) для застосування з метою, (іі) для застосування як лікарський засіб з метою, або (ііі) для застосування з метою отримання лікарського засобу, що використовується з метою: (а) інгібування ВІЛінтегрази, (b) профілактики або лікування ВІЛінфекції або (с) профілактики, лікування або затримки вияву СНІДу. При застосуванні у наведених цілях сполуки згідно з даним винаходом, необов'язково, можуть бути 11 використані в комбінації з одним або більше засобами для лікування ВІЛ/СНІДу, які вибираються з групи, що включає: ВІЛ/СНІДпротивірусні засоби, протиінфекційні засоби та імуномодулятори. Сполуки згідно з даним винаходом можуть мати асиметричні центри і можуть існувати, за винятком випадків, які зазначаються окремо, у вигляді сумішей стереоізомерів або у вигляді індивідуальних діастереомерів або енантіомерів, при цьому всі ізомерні форми охоплені даним винаходом. N-заміщені гідроксипіримідинонові сполуки згідно з даним винаходом можуть також існувати у вигляді таутомерів, таких, як наведений нижче таутомер сполуки формули І: Зрозуміло, що даний винахід включає всі таутомери гідроксипіримідинонових сполук формули І (або II), як окремо, так і в сумішах. Сполуки згідно з даним винаходом корисні в інгібуванні ВІЛ-інтегрази, для профілактики або лікування інфекції вірусу імунодефіциту людини (ВІЛ) і профілактики, лікування або затримки прояву подальших патологічних станів, таких як СНІД. Профілактика СНІДу, лікування СНІДу, затримка у прояві СНІДу, або профілактика або лікування ВІЛ-інфекції визначаються, як такі, що включають, але не в порядку обмеження, лікування широкого ряду станів при ВІЛ-інфекції: СНІДу, ARC (СНІД-асоційований комплекс), як симптоматичних, так і асимптоматичних, та існуючого або потенційного контакту з ВІЛінфекцією. Наприклад, сполуки згідно з даним винаходом корисні в лікуванні ВІЛ-інфекції після припустимого контакту з ВІЛ, що відбувся шляхом переливання крові, заміни рідини організму, укусу, випадкового уколу зараженою голкою або контакту з кров'ю пацієнта під час хірургічної операції. Характерні сполуки згідно з даним винаходом були перевірені на інгібування шляхом дослідження активності інтегрази у реакції перенесення ланцюгів. Випробування проводять способом, [який описаний у WO 02/30930. Випробування також відповідає наведеному у Wolfe, A.L. et al., J. Virol. 1996, 70: 1424-1432 для рекомбінантної інтегрази, за винятком того, що: (і) для аналізу використовують заздалегідь зібрані комплекси перенесення інтеграза-ланцюг; (іі) реакцію перенесення ланцюга здійснюють в присутності інгібітору в 2,5 мМ MgCl2, використовуючи від 0,5 до 5 нМ 3'-ФІТЦ-міченого субстрату ДНК-мішені, і (ііі) продукти реакції перенесення ланцюга розпізнають, використовуючи анти-ФІТЦ, антитіло, яке кон'юговане з лужною фосфатазою, та хемілюмінесцентний субстрат для лужної фосфатази. Характерні сполуки згідно з даним 80737 12 винаходом, та відповідно до даного випробування, здійснюють інгібуючу дію на активність в реакції перенесення ланцюга. Наприклад, сполуки, які представлені в таблиці 1, що наведена нижче, досліджені згідно з методом аналізу активності інтегрази і дають значення ІС50 порядку 5мкмоль/л або нижче. Додатково опис проведення випробування з використанням заздалегідь зібраних комплексів може бути знайдений [у Hazuda et al., J. Virol. 1997, 71. 7005-7011; Hazuda et al., Drug Design and Discovery 1997,15: 17-24 та Hazuda et al., Science 2000,287: 646-650]. Деякі характерні сполуки згідно з даним винаходом були також вивчені під час випробування на інгібування гострої ВІЛ-інфекції Т-лімфоцитів, яка здійснюється згідно [з Vacca, J.P. et al., Proc. Natl. Acad. Sci. USA 1994, 91: 4096]. Ці сполуки, які включають сполуки, наведені нижче в таблиці 1, дають значення ІС95 порядку 20мкмоль/л або нижче. Сполуки згідно з даним винаходом можуть також діяти як інгібітори ВІЛ-рибонуклеази Η (РНКази Н). Зворотна транскриптаза (RT) вірусу імунодефіциту людини тину 1 (ВІЛ-1) каталізує конверсію геномної РНК в дволанцюгову ДНК провірусу після проникнення в клітину, за допомогою використання РНК- і ДНК-залежних полімеразних активностей і активності РНКази Н. RT ВІЛ-1 є асиметричним димером, що складається з полипептидів р66 і р51. Каталітичні активності RT керуються окремими сайтами в субодиниці р66; тобто, N-кінець рбб відповідає за РНК- та ДНК-залежну ДНК-полімеразну активність, і домен р15 на С-кінці відповідає за активність РНКази Н. РНКаза Η потрібна для розщеплення РНК-ланцюга РНК:ДНК гетеродуплексних проміжних продуктів в зворотній транскрипції. Сполуки згідно з даним винаходом можуть селективно зв'язуватися з доменом РНКази Η RT ВІЛ-1 та інгібувати активність вказаного домену. Інгібуюча активність сполук по відношенню до РНКазе Η може бути виміряна в ході проведення відповідних випробувань, що відомі з рівня техніки, таких як випробування, що описане [у Shaw-Reid et al., J. Biol. Chem. 2003, 278 (5): 2777-2780]. Таким чином, даний винахід включає спосіб інгібування ВІЛ-РНКази Η у пацієнта, який цього потребує, що полягає у введенні пацієнту ефективної кількості сполуки згідно з винаходом. Даний винахід включає також сполуки (і) для застосування в цілях, (іі) для застосування у вигляді лікарського засобу в цілях, або (ііі) для застосування з метою отримання лікарського засобу, що використовується в цілях інгібування ВІЛ-РНКази Н. Сполуки згідно з даним винаходом можуть бути введені в формі фармацевтично прийнятних солей. Термін "фармацевтично прийнятна сіль" означає сіль, що володіє ефективністю початкової сполуки, яка не є біологічно або іншим чином неприйнятною (наприклад, не є ні токсичною, ні іншим чином шкідливою по відношенню до реципієнта). Відповідні солі включають кислотноадитивні солі, які можуть, наприклад, бути отримані шляхом змішування розчину сполуки 13 згідно з даним винаходом з розчином фармацевтично прийнятної кислоти, такої як хлористоводнева кислота, сірчана кислота, оцтова кислота, трифтороцтова кислота або бензойна кислота. Коли сполуки згідно з винаходом містять кислотну складову, відповідні фармацевтично прийнятні солі можуть включати солі лужних металів (наприклад, солі натрію або калію), солі лужноземельних металів (наприклад, солі кальцію або магнію) і солі, які утворені з відповідними органічними лігандами, такі як четвертинно амонієві солі. Також, у разі наявності кислотної (СООН) або спиртової групи, фармацевтично прийнятні складні ефіри можуть бути використані для зміни розчинності або гідролізних характеристик сполуки. З метою інгібування ВІЛ-інтегрази або ВІЛРНКази Н, профілактики або лікування ВІЛінфекції або профілактики, лікування або затримки у проявленні СНІДу, сполуки згідно з даним винаходом можуть бути введені перорально, парентерально (підшкірні ін'єкції, внутрішньовенні, внутрішньом'язові, підложкові ін'єкції або інфузії включно), за допомогою аерозольної інгаляції або ректально, в формі стандартної дози фармацевтичної композиції, що містить терапевтично ефективну кількість сполуки і загальноприйняті нетоксичні фармацевтично прийнятні носії, допоміжні речовини і розчинники. Термін "введення" і варіанти цього терміну (наприклад, "прийом" сполуки) по відношенню до сполуки згідно з винаходом означають, що індивідууму, який потребує лікування, вводять сполуки або пролікові сполуки. Коли сполуки згідно з винаходом або проліки вводять в комбінації з одним або більше іншими активними засобами (наприклад, противірусними засобами, які є корисними для лікування інфікування вірусом ВІЛ або СНІДу), зрозуміло, що термін "введення" і кожний з варіантів зазначеного терміну включають одночасне і послідовне введення сполуки або проліків та інших засобів. Мається на увазі, що термін "композиція", як він використовується тут, стосується продукту, що включає зазначені інгредієнти у вказаних кількостях, а також будь-якого продукту, що утворюється прямо або непрямо, шляхом комбінування вказаних інгредієнтів у зазначених кількостях. "Фармацевтично прийнятні" означає, що інгредієнти фармацевтичної композиції повинні бути сумісні один з одним і не завдавати шкоди реципієнту. Термін "суб'єкт" (який альтернативно називається тут "пацієнт"), як він використовується тут, відноситься до тварини, переважно до ссавця, більш переважно, до людини, що є об'єктом лікування, спостереження або дослідження. Термін "терапевтично ефективна кількість", як він використовується тут, означає таку кількість активної сполуки або фармацевтичного засобу, який викликає біологічний або фармакологічний відгук в тканині, системі, організмі тварини або людини, якої домагався дослідник, ветеринар, лікар або інший клініцист, що включає ослаблення 80737 14 або профілактику симптомів, які лікуються або попереджаються, хвороби або стану. Термін також включає кількість активної сполуки, яка достатня для інгібування ВІЛ-інтегрази і/або РНКази Н, і тим самим, що викликає очікуваний відгук. Коли активну сполуку (тобто активний інгредієнт) вводять у вигляді солі, зазначена кількість активного інгредієнта відноситься до сполуки у формі вільної кислоти або вільної основи. Фармацевтичні композиції можуть бути в формі суспензій, що вводяться перорально, або таблеток, або капсул, назальних спреїв, стерильних препаратів для ін'єкції, наприклад, у вигляді стерильних водних або олійних суспензій для ін'єкції, або супозиторіїв. Ці композиції можуть бути отримані добре відомими з рівня техніки способами і містять загальноприйняті наповнювачі. Придатні способи та інгредієнти [описані в Remington's Pharmaceutical Sciences, 18 edition, edited by A. R. Gennaro, Mack Publishing Co., 1990], який повністю включений тут як посилання. Сполуки згідно з даним винаходом можуть бути введені перорально в межах доз від 0,001 до 1000мг/кг маси тіла ссавця (наприклад, людини) на день у вигляді єдиної дози або декількох доз. Найкращий інтервал доз складає від 0,01 до 500мг/кг маси тіла на день, перорально, у вигляді єдиної дози або декількох доз. Інший найкращий інтервал доз складає від 0,1 до 100мг/кг маси тіла на день, перорально, у вигляді єдиної дози або декількох доз. Для перорального прийому композиції можуть бути представлені в формі таблеток або капсул, що містять 1,0-500 міліграм активного інгредієнта, зокрема 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400 і 500 міліграм активного інгредієнта для симптоматичного підбору дозування пацієнту, який проходить лікування. Конкретний рівень доз і частота введення дози для кожного окремого пацієнта можуть варіюватися в залежності від факторів, що включають активність конкретної сполуки, яка використовується, метаболічну стійкість та тривалість дії цієї сполуки, вік, масу тіла, загальний стан здоров'я, стать, дієту, спосіб і час введення, швидкість виведення з організму, лікарську комбінацію, тяжкість конкретного стану і реципієнта, що проходить терапію. Як зазначено вище, даний винахід також стосується використання інгібуючих ВІЛ-інтегразу сполук згідно з даним винаходом з одним або більше засобами, які є корисними у терапії ВІЛінфекції або СНІДу. Наприклад, сполуки згідно з даним винаходом, можуть бути ефективно застосовані, незалежно, шляхом попереднього впливу і/або подальшого впливу, в комбінації з ефективною кількістю одного або більше ВІЛ/СНІД-противірусних засобів, імуномодуляторів, протиінфекційних засобів або вакцин, що є корисними для терапії ВІЛ-інфекції або СНІДу, таких як засоби, що наведені [в таблиці 1 у WO 01/38332 або в таблиці у WO 02/30930], обидва документи повністю включені тут як посилання. Очевидно, що кількість комбінацій сполук згідно з даним винаходом з ВІЛ/СНІД 15 противірусними засобами, імуномодуляторами, протиінфекційними засобами або вакцинами, не обмежується списком, що наведений у вищезгаданих [таблицях у WO 01/38332 і WO 02/30930], і включають, в принципі, будь-яку комбінацію з будь-якою фармацевтичною композицією, яка є корисною для лікування СНІДу. ВІЛ/СНІД-противірусні засоби і інші засоби звичайно використовують в цих комбінаціях на рівні загальноприйнятих доз і згідно з відомими з рівня техніки схемами прийому, включно з, наприклад, дозуванням, що вказане [у Physicians' Desk Reference, 57th edition, Thomson PDR, 2003]. Діапазон доз сполуки згідно з даним винаходом у таких комбінаціях відповідає наведеному вище. Скорочення, що містяться в даному описі, зокрема, на схемах і в прикладах, включають наступні: АIDS=синдром набутого імунодефіциту, ARC=CHIД-асоційований комплекс, Вn=бензил, CBZ (або Сbz)=бензилоксикарбоніл, DBU=1,8діазабіцикло[5.4.0]ундец-7-ен, DМАD=диметилацетилендикарбоксилат, ДМФА=N,N-диметилформамід, ДМСОиметилсульфоксид , ЕtOАстилацетат , = д = е FІА-МS=мас-спектрометрія з аналізом за методом вприскування в потік, h=тодина(и), ВІЛ=вірус імунодефіциту людини, ВЕРХвисокоефективна = рідинна хроматографія, і= ІРА зопропанол , літійдіізопропіламід = LDА , метил = Ме , = метанол, МеОН NMP=N-метилпіролідинон, ядерний = ЯМР магнітний резонанс , Рd/C=паладієвий каталізатор на вугіллі, ОФВЕРХ ВЕРХ = із зверненою фазою , трифтороцтова ТРА кислота , ТНF=тетрагідрофуран. Сполуки згідно з даним винаходом легко можуть бути отримані відповідно до наступних реакційних схем і прикладів або модифікацією зазначених прийомів, з використанням легко доступних вихідних матеріалів і реагентів. У наведених нижче реакційних схемах можливе також використання варіантів, які самі по собі відомі фахівцеві у даній галузі, але не наведені детально. Крім цього, інші способи отримання сполук згідно з винаходом стануть очевидними для фахівця в даній галузі в світлі схем і прикладів, що наведені нижче. Якщо це не обумовлено окремо, змінні, які перераховані в схемах 1, А, В, С і D, мають наступні значення: Р^ означає водень або захисну групу, наприклад, складний ефір, такий як, але не в порядку обмеження, бензоат та півалат, або простий ефір, такий як, але не в порядку обмеження, бензиловий ефір, який звичайно видаляють в умовах, що використовуються для перетворення складного метилового ефіру в амід або видаляють на іншій стадії. Захисну групу зазвичай використовують для синтезу і/або очищення. Р^ означає водень або С1-6-алкіл. Υ означає водень або NRsaRsb. Rsa означає С1-6-алкіл, C(O)Rsc, sc sd sc C(O)C(O)NR R , SO2R , SO2NRscRsd, sc sc sd C(O)CH2SO2R , C(O)CH2NR R , SO2CH2SO2Rsc або CH(CH3)Rsc або Rsa і Rsb, разом з атомом 80737 16 азоту, до якого приєднані, утворюють гетероциклічне кільце, що містить 1 або 2 гетероатоми. Rsb означає водень, С1-6-алкіл або C(O)CF3, або Rsa і Rsb, разом з атомом азоту, до якого приєднані, утворюють гетероциклічне кільце, що містить 1 або 2 гетероатоми. Rsc означає С1-6-алкіл, арил, 5- або 6-членне гетероарильне кільце, яке не заміщене або заміщене С1-6-алкілом, С(О)СН2SО2С1-6-алкілом або (СН2)1-6-арилом. Rsd означає С1-6-алкіл. Rs5 означає Rsc, C(O)NRscRsd, CH2SO2Rsc, CH2NRscRsd, NRscRsd або CH2SO2Rsc. Rs1 означає водень. Rs2 означає CH2Rse, де Rse означає незаміщений арил або арил, що заміщений галогеном. Сполуки згідно з даним винаходом можуть бути отримані поєднанням відповідних амінів з відповідним заміщеним алкіл-3-гідрокси-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксилатом (або карбоновими кислотами або галогенідами) або алкіл-3-гідрокси-4-оксо-4,6,7,8тетрагідропіроло[1,2-а]піримідин-2-карбоксилатом (або карбоновими кислотами або галогенідами), або алкіл-3-гідрокси-4-оксо-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-2-карбоксилатом (або карбоновими кислотами або галогенідами), або алкіл-3-гідрокси-4-оксо-6,7,8,9,10,11гексагідро-4Н-піримідо[1,2-а]азепін-2карбоксилатом (або карбоновими кислотами або галогенідами), як представлено на схемі 1. Схема 1 Способи поєднання похідних карбонових кислот з амінами з утворенням карбоксамідів добре відомі з рівня техніки. Відповідні способи описані, наприклад, [у Jerry March, Advanced Organic Chemistry,_3rd edition, John Wiley & Sons, 1985, pp.370-376]. Аміни формули 1-2 можуть бути отримані з використанням способів, що описані [у Richard Larock, Comprehensive Organic Transformations, VCH Publishers Inc, 1989, pp.385438], або стандартних варіантів вказаних способів. Схема А наводить загальний синтез карбоксамідів А-5. Метиловий ефір А-4 може бути підданий взаємодії з аміном 1-2 в розчинниках, таких як ДМФА, метанол, етанол, толуол, NMP, при відповідній температурі (наприклад, від 20 до 150°С), що дає кінцеву сполуку А-5. Метиловий ефір А-4 може бути синтезований одним з трьох синтетичних способів. Згідно з першим способом, амідингідрохлорид Α-1a (sX=H; sY=H) може бути підданий взаємодії з диметил-2-(бензилокси)-3оксосукцинатом в присутності основи, що дає проміжний захищений метиловий ефір А-2, захист якого легко може бути знятий, що призводить до 17 отримання метилового ефіру А-4. Згідно з другим способом, амідоксин A-1b (sX=OH; sY=H), який отриманий в три стадії з третбутилбензилоксикарбамату, може бути підданий взаємодії з DMAD, що дає циклічну проміжну сполуку А-3а, яка може бути піддана перегрупуванню шляхом нагрівання у відповідному розчиннику (наприклад, ксилолі), що призводить до утворення метилового ефіру А-5. Згідно з третім способом, амідоксим A-1c (sX=H; sY=OH) може бути підданий взаємодії з DMAD у відповідному розчиннику (наприклад, ацетонітрилі), що призводить до утворення проміжні" сполуки А3-b, яка може бути піддана перегрупуванню у метиловий ефір А-4 шляхом нагрівання у відповідному розчиннику (наприклад, ксилолі). Всі три наведених способи можуть бути застосовані по відношенню до амідинів та амідоксимів, які містять замісники у кільці. Схема А ілюструється прикладом 1. Схема А 80737 18 сульфоксиду або сульфону. Якщо нуклеофільний реагент включає складний ефір, складний ефір може бути перетворений в амід, шляхом використання стандартних хімічних перетворень після синтезу В-3. Схема В ілюструється прикладом 2. Схема В Схема С представляє загальний синтез похідних С-3 або С-4, що містять в кільці аліфатичний замісник, такий як амід, сульфонамід, сульфонілсечовина, карбамат або сечовина. Бром-похідне В-1 може бути оброблено бензиламіном С-1 і потім гідровано або безпосередньо піддано взаємодії з аміном С-1 а, що дає проміжну сполуку С-2, яка потім може бути оброблена аміном 1-2, з виділенням або без виділення, і потім піддана реакції поєднання з карбоновою кислотою або взаємодії з карбонілхлоридом (або сульфонілхлоридом, або сульфамоїлхлоридом) або ізоціанатом, що дає кінцевий продукт С-3. Якщо С-3 містить Rs4=Rs3O (СО)СО, то С-3 може бути в подальшому піддано взаємодії з нуклеофільним реагентом, таким як амін, що призводить до утворення продукту С-4. Схема С ілюструється прикладами 3, 4 і 6. Останні дві стадії можна поміняти місцями. Схема С Схема В представляє спосіб отримання сполук згідно з даним винаходом, які містять аміно-, просту ефірну, просту тіоефірну, сульфоксидну або сульфонову групу в положенні 9 ядра 3гідрокси-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-2-карбоксаміду. Бром-похідне В-1 може бути отримано з метилового ефіру А-4 за допомогою первинного захисту гідроксигрупи на А4 відповідною захисною групою (наприклад, перетворенням в бензоат або півалат, або в бензилокси) і подальшим здійсненням контакту захищеного А-4 з бромуючим агентом (наприклад, NBS). Бром-похідне В-1 може бути потім оброблене нуклеофільним реагентом ("Nu"; наприклад, аміном, тіолом або алкоголятом), що дає, шляхом виділення або без виділення, проміжний метиловий ефір В-2, який піддають взаємодії із заданим аміном, що призводить до утворення кінцевого продукту В-3. Якщо нуклеофільний реагент являє собою тіол або містить сірку, що окислюється, в схему може бути включена стадія окислення з метою отримання 1. поєднання з Rs4-sX, 2. стадії 3 і 4 можна поміняти місцями, 3. РЛ=Н або захисна група, 4. Rs3=Н або С1-6-алкіл, оr=або, іf=якщо. Схема D представляє синтез гомохіральних сполук С-3, С3а, b і С-4. Здійснюють заміщення бром-похідного В-1 хіральними амінами D-1, що 19 призводить до суміші діастереоізомерів, з подальшим або одночасним видаленням захисної групи. Проводять відновлюване алкілування аміногрупи в положенні 9 за допомогою альдегідів або кетонів D-2, що дає проміжну сполуку D-3. Суміш діастереоізомерів може бути розділена шляхом кристалізації або хроматографії, що призводить до окремих діастереоізомерів D-3a, b. Rs6 може бути видалений шляхом гідрування, що дає гомохіральну проміжну сполуку С-2а, b. Подальша взаємодія з аміном 1-2 та поєднання з карбоновою кислотою або обробка карбоніл хлоридом (або сульфонілхлоридом або сульфамоїлхлоридом) або ізоцинатом призводить до кінцевого гомохірального продукту С-3 а, b. Як і у випадку рацемічних сполук С-3 на схемі С, може бути проведена додаткова стадія з метою отримання гомохіральних сполук С-4. Схема D ілюструється прикладами 5 і 7. Схема D Р^=Н або захисна група Rs6=xipanbHHU алкільний залишок (наприклад, (S)-а-метилбензил) Rsp, Rsq=Η або С1-6-алкіл Rs4=Rs5CO або Rs5SO2 або Rs5(Rs6)NSO2 або s5 R OCO,, Rs3O(CO)CORs5(Rs6)NCO sX=Cl або Вr або ОН Наступні приклади служать виключно з метою ілюстрації винаходу і його практичного здійснення. Приклади не треба розглядати як такі, що обмежують об'єм або суть винаходу. Приклад 1 М-(4-Фторбензил)-3-гідрокси-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід Стадія 1а: Третбутилбензилокси(4ціанобутил)карбамат [Bergeron, R. J., McManis, J. S., Tetrahedron 45 (16), 4939-4944 (1989)]. До розчину третбутилбензилоксикарбамату в безводному диметилформаміді додають 5моль% йодиду натрію і порціями 1,36екв. гідриду натрію (60% дисперсія в мінеральному маслі). Суміш перемішують при кімнатній температурі протягом 15хв., після чого додають 1,05екв. 4хлорвалеронітрилу. Суміш нагрівають до 85°С і перемішують протягом 3,5год. Після охолодження до кімнатної температури суміш гасять водою і екстрагують діетиловим ефіром. Об'єднані органічні фази концентрують і промивають напівнасиченим водним тіосульфатом натрію, водою і насиченим розчином солі. Органічну фазу 80737 20 сушать над сульфатом натрію, фільтрують і концентрують шляхом зниженого тиску. Маслянистий залишок промивають петролейним ефіром і сушать при високому вакуумі, отримуючи третбутилбензилокси(4-ціанобутил)карбамат у вигляді світло-жовтого масла. 1 Н-ЯМР(400МГц, CDCI3) 5: 7.38(м, 5Н), 4.84(с, 2Н), 3.45(т, J=6.4Гц, 2Н), 2.34(т, J=6.8Гц, 2Н), 1.70(м, 4Н), 1.52(с, 9Н). MC m/z: 271(M+H)+. Стадія 2а: 1-(Бензилокси)піперідин-2імінгідрохлорид Третбутилбензилокси-(4-ціанобутил)карбамат розчиняють в розчині 4Μ НСI в 1,4-діоксані і суміш перемішують протягом 18год. при кімнатній температурі. Розчинник видаляють при зниженому тиску і залишок обробляють етилацетатом і діетиловим ефіром. Отриману тверду речовину, що промита діетиловим ефіром, фільтрують і сушать при високому вакуумі, що дає 1(бензилокси)піперідин-2-імінгідрохлорид у вигляді блідо-жовтої твердої речовини. 1 Н-ЯМР(400МГц, ДМСО-d6) δ:9.53(с, 1Н), 8.97(с, 1Н), 7.57-7.41(м, 5Н), 5.05(с, 2Н), 3.67(т, J=6.0Гц, 2Н), 2.64(т, J=6.4Гц, 2Н), 1.90-1.84(м, 2Н), 1.69-1.63(м, 2Н). МС m/z: 205(M+H)+. Стадія 3а; 2-Імінопіперідин-1-ол гідрохлорид Розчин 1-(бензилокси)піперідин-2-імін гідрохлориду у метанолі, що містить паладій на активованому вугіллі (10%, мас/мас.) перемішують в середовищі водня при атмосферному тиску протягом 3год. Каталізатор відділяють шляхом фільтрування і розчин концентрують досуха при зниженому тиску. Залишок розтирають з діетиловим ефіром, фільтрують і сушать при високому вакуумі, отримуючи 2-імінопіперідин-1-ол гідрохлорид у вигляді блідо-жовтої твердої речовини. 1 Н-ЯМР (400МГц, ДМСО-d6) δ: 11.76(с, 1Н), 8.82(с, 1Н), 8.49(с, 1Н), 3.63(т, J=6.0Гц, 2Н), 2.63(т, J=6.0Гц, 2Н), 1.87(м, 2Н), 1.66 (м, 2Н). 13С-ЯМР (150МГц, ДМСО-d6) 5:159.06, 50.92, 25.76, 22.01, 17.22. MCm/z: 115(М+Н)+. Стадія 4а: Метил-2-(2-метокси-2-оксоетил)5,6,7,8-тетрагідро-2Н-[1,2,4]оксадіазоло[2,3а]пірідин-2-карбоксилат До розчину 2-імінопіперідин-1-ол гідрохлориду у хлороформі додають триетиламін. Суміш перемішують протягом 5 хв. при кімнатній температурі, потім охолоджують до 0°С і додають по краплях шляхом перемішування 1,2екв. диметилацетилендікарбоксилату. Охолоджуючу баню прибирають і суміш перемішують при кімнатній температурі протягом однієї години. Розчинник видаляють при зниженому тиску і розчин розподіляють між етилацетатом і напівнасиченим водним хлоридом амонію. Водну фазу додатково екстрагують етилацетатом. Об'єднані органічні фази сушать над сульфатом натрію і фільтрують через силікагель. Розчинник видаляють у вакуумі, отримуючи метил-2-(2метокси-2-оксоетил)-5,6,7,8-тетрагідро-2Н 21 [1,2,4]оксадіазоло[2,3-а]піридин-2-карбоксилат у вигляді світло-жовтого масла. 1 Н-ЯМР(400МГц, CDCI3) δ: 3.82 (с, 3Н), 3.70 (с, 3Н), 3.51 (м, 1Н), 3.36 (м, 1Н) 3.31 (д, J=16.6Гц, 1Н), 2.98 (д, J=16.6Гц, 1Н), 2.53 (м, 2Н), 1.94 (м, 2Н), 1.74 (м, 2Н). 13С-ЯМР (100МГц, CDCI3) 6:169.15, 168.88, 164.97, 103.27, 55.71, 52.97, 51.84, 42.26, 26.06, 23.49,22.83. МС m/z: 257 (M+H)+. Стадія 5а: Метил-3-гідрокси-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а] піримідин-2карбоксилат Розчин метил-2-(2-метокси-2-оксоетил)5,6,7,8-тетрагідро-2Н-[1,2,4]оксадіазоло[2,3а]піридин-2-карбоксилату у безводному о-ксилолі вміщують у двугорлу круглодону колбу. Колбу забезпечують термометром і закривають мембраною. Суміш нагрівають до 148-150°С протягом 5год. Нагріваючу баню прибирають і суміш залишають стояти при кімнатній температурі протягом 16 год. До суміші, що містить осад, додають діетиловий ефір. Через 5хв. осад відділяють фільтруванням, промивають діетиловим ефіром і сушать у вакуумі. Продукт, метил-3-гідрокси-4-оксо-6,7,8,9-тетрагідро-4Нпіридо[1,2-а]піримідин-2-карбоксилат, отримують у вигляді блідо-коричневої твердої речовини. 1 Н-ЯМР(400МГц, ДМСО-d6) δ: 10.03 (с, 1Н), 3.86 (т, J=6.0Гц, 2Н), 3.80 (с, 3Н), 2.75 (т, J=6.8Гц, 2Н), 1.90-1.70 (м, 4Н). 13С-ЯМР (150МГц, ДМСО-d6) 6:165.81, 158.65, 148.60, 143.10, 127.07, 51.98, 42.87, 30.32, 20.91, 18.40. МС m/z: 115(М+Н)+. Наступна методика являє собою альтернативний спосіб синтезу метил-3-гідрокси-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин2-карбоксилату. Стадія 1b: Диметил-2-(бензилокси)-3оксосукцинат Розчин метил(бензилокси)ацетату (1екв.) та диметилоксалату (1,2екв.) в сухому ТГФ охолоджують до -78°С і додають по краплях LDA (2M в суміші ТГΦ-гептан, 1,2екв.). Після перемішування протягом однієї години охолоджуючу баню прибирають і перемішування продовжують ще одну годину. Реакцію гасять при 0°С, виливаючи в охолоджену 1н водну НСI, і водну фазу екстрагують ЕtOАс; органічний шар промивають насиченим розчином солі, сушать і концентрують, що дає сирий продукт, який використовують без додаткового очищення. Стадія 2b: Метил-3-(бензилокси)-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а] піримідин-2карбоксилат 2-імінопіперидингідрохлорид (1,5екв.), що випускається промислово, додають при кімнатній температурі до розчину оксосукцинату, отриманого на стадії lb, (1екв.) в МеОН. Після додання по краплях чистого DBU (4,5екв.) реакційну суміш перемішують протягом 2 днів. Випаровування розчинника дає залишок, який поглинають ЕtOАс і промивають 1н НСI та насиченим розчином солі; органічний шар сушать над Na2SO4 і розчинник видаляють. Сирий продукт використовують без додаткового очищення. 80737 22 Аналітичний зразок цього продукту очищають флеш хроматографією (суміш петролійний ефір/ЕtOАс 1:2-1:5), отримані наступні спектроскопічні дані: 1 Н-ЯМР(400МГц, CDCl3) δ: 7.49-7.30 (м, 5Н), 5.25(с, 2Н), 4.00 (т, J=6.2Гц, 2Н), 3.86 (с, 3Н), 2.94 (т, J=6.6Гц, 2Н), 2.01-1.95 (м, 2Н), 1.92-1.87 (м, 2Н). 13 С-ЯМР (75МГц, CDCI3) δ:164.1, 159.3, 154.1, 141.1, 140.6, 136.0, 128.1, 127.8, 127.7, 73.8, 52.2, 42.7, 30.9, 21.0, 18.4. MC m/z: 315(M+H)+. Стадія 3b: Метил-3-гідрокси-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а] піримідин-2карбоксилат Проміжний метил-3-(бензилокси)-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а] піримідин-2карбоксилат, отриманий на стадії 2Ь, розчиняють в МеОН і додають при кімнатній температурі каталітичний 10% Pd/C. Суміш перемішують в атмосфері Н2 протягом 3,5 годин. Фільтрування каталізатора і випаровування метанолу дає залишок, до якого додають діетиловий ефір; фільтрування призводить до отримання метил-3гідрокси-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-2-карбоксилату у вигляді блідокоричневої твердої речовини зі спектроскопічними характеристиками, що ідентичні характеристикам сполуки, яка описана на стадії 5а. Стадія 6: М-(4-Фторбензил)-3-гідрокси-4-оксо6,7,859-тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксамід Розчин метил-3-гідрокси-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксилату, отриманого на стадії 3b або стадії 5а, та 4-фтор-бензиламіну (2екв.) у метанолі перемішують і нагрівають до 65°С протягом 22год. Розчинник видаляють при зниженому тиску-і зазначений у заголовку продукт отримують за допомогою препаративної ОФ-ВЕРХ, використовуючи воду (0,1% TFA) і ацетонітрил (0,1% TFA) як елюенти (колонка: С18). Об'єднані фракції, які містять продукт, ліофілізують, отримуючи сполуку, що зазначена у заголовку, у вигляді рихлого білого матеріалу. 1 Н-ЯМР(400МГц, ДМСО-d6) δ: 12,12 (с, 1Н), 9.35(м, 1Н) 7,36 (м, 2Н), 7.15 (м, 2Н), 4.44 (м, 2Н), 3.84 (т, J=6.4Гц, 2Н), 2.80 (т, J=6.8Гц, 2Н), 1.901.73 (м, 4Н). MC m/z: 318 (M+H)+. Приклад 2 N-(4-Фторбензил)-3-гідрокси-9-морфолін-4-іл4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-2-карбоксамід гідрохлорид Стадія 1: Метил-3-(бензоїлокси)-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксилат До розчину метил-3-гідрокси-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а] піримідин-2карбоксилату (отриманого згідно з прикладом 1) у піридині додають бензойний ангідрид (1,55екв.). Суміш перемішують при кімнатній температурі протягом 16год. Розчинник видаляють при зниженому тиску і залишок розподіляють між етилацетатом і 0,5Μ водним НСI. Водну фазу екстрагують етилацетатом і об'єднані органічні 23 фази промивають 0,5Μ водним НСI, водою і насиченим розчином солі. Органічну фазу сушать над сульфатом натрію, фільтрують і концентрують досуха у вакуумі. Сполуку, що зазначена у заголовку, отримують після флеш хроматографії (елюент - суміш петролійний ефір/етилацетат, 1:2) у вигляді незабарвленої твердої речовини. 1 H-ЯМР (400МГц, ДМСО-d6) δ: 8.07 (м, 2Н), 7.78 (м, 1Н), 7.62 (м, 2Н), 3.86 (т, J=6.0Гц, 2H), 3.74 (с, 3Н), 2.92 (т, J=6.4Гц, 2Н), 1.93-1.81(м, 4Н). МС m/z: 329 (M+H)+. Стадія 2: Метил-3-(бензоїлокси)-9-бром-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин2-карбоксилат Суміш метил-3-(бензоїлокси)-4-оксо-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-2карбоксилату, N-бромсукциніміду (1,2екв.) та дибензоїлпероксиду (70%, 0,13екв.) у чотирьоххлористому вуглеці перемішують шляхом нагрівання до температури кипіння із зворотним холодильником протягом однієї години. Суміш охолоджують до кімнатної температури, сукцинімід відділяють фільтруванням і розчинник видаляють при зниженому тиску. Метил-3-(бензоїлокси)-9бром-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-2-карбоксилат отримують після флеш хроматографії (елюент суміш петролійний ефір/етилацетат, 1:1) у вигляді блідо-жовтого масла. 1 Н-ЯМР (400МГц, ДМСО-d6) δ:8.08 (м, 2Н), 7.79 (м, 1Н), 7.63 (м, 2Н), 5.58 (м, 1Н), 4.24 (м, 1Н), 3.77 (с, 3Н), 3,72 (м, 1Н), 2.43-2.35 (м, 1Н), 2.302.05 (м, 3Н). МС m/z: 409/407 (M+H)+. Стадія 3: N-(4-Фторбензил)-3-гідрокси-9морфолін-4-іл-4-оксо-6,7,8,9-тетрагідро-4Нпіридо[1,2-а]піримідин-2-карбоксамід. До розчину метил-3-(бензоїлокси)-9-бром-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин2-карбоксилату в ДМФА додають морфолін (3екв.) і суміш перемішують при кімнатній температурі протягом 1год. Розчинник видаляють при зниженому тиску і залишок розтирають з діетиловим ефіром. Сирий продукт розчиняють у метанол, додають 4-фторбензиламін (3екв.) і суміш перемішують протягом 1,5год. при 65°С. Розчинник видаляють при зниженому тиску і продукт очищають за допомогою препаративної ОФ-ВЕРХ, використовуючи воду (0,1% TFA) та ацетонітрил (0,1% TFA) як елюенти (колонка: С18). Об'єднані фракції, які містять продукт, ліофілізують і знову розчиняють в 1н НСI. Розчинник видаляють при зниженому тиску і залишок ліофілізують з суміші вода/ацетонітрил, отримуючи гідрохлорид N-(4-фторбензил)-3гідрокси-9-морфолін-4-іл-4-оксо-6,7,8,9-тетрагідро4Н-піридо[1,2-а]піримідин-2-карбоксаміду у вигляді трохи рожевого рихлого матеріалу. 1 Н-ЯМР (400МГц, ДМСО-d6) δ: 12.34 (с, 1Н), 10.99 (с, 1Н), 10.47 (с, 1Н), 7.44 (м, 2Н), 7.16 (м, 2Н), 4.85 (м, 1Н), 4.60-4.40 (м, 3Н), 4.10-3.85 (м, 4Н), 3.60-3.05 (м, 5Н замаскований сигналом води), 2.35-2.15 (м, 2Н), 2.03-1.80 (м, 2Н). МС m/z: 403(M+H)+. Приклад 3 80737 24 (+/-)-9[[(Диметиламіно)сульфоніл](метил)аміно]-N-(4фторбензил)-3-гідрокси-4-оксо-6,7,8,9-тетрагідро4Н-піридо[1,2-а]піримідин-2-карбоксамід С-3 Стадія 1: Метил-9-[бензил(метил)аміно]-3гідрокси-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-2-карбоксилат гідрохлорид До розчину бром-похідного, що перемішується, метил-3-(бензоїлокси)-9-бром-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин2-карбоксилату (який отриманий згідно з прикладом 2, стадія 2), в безводному диметилформаміді додають N-бензил-Nметиламін (3екв.). Суміш перемішують протягом 1,5год. при кімнатній температурі, після чого додають діетиловий ефір і 2М НСI в діетиловому ефірі. Суміш перемішують протягом 5хв., осад, що утворився, відділяють фільтруванням і промивають діетиловим ефіром. Осад розчиняють в безводному метанолі і розчин концентрують досуха при зниженому тиску. Зазначений у заголовку сирий продукт, отриманий у вигляді блідо-жовтого масла, що містить надлишок Nбензил-N-метиламінгідрохлориду, використовують без додаткового очищення. МС m/z: 344 (М+Н)+. Стадія 2: Метил-3-гідрокси-9-(метиламіно)-4оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин2-карбоксилат Отриманий на стадії 1 розчин сирого продукту у метанолі, що містить паладій на активованому вугіллі (10% мас/мас.) перемішують у середовищі водню при атмосферному тиску протягом 3год. Каталізатор відділяють фільтруванням і розчин концентрують досуха при зниженому тиску. Залишок розтирають з діетиловим ефіром, фільтрують і сушать при високому вакуумі, отримуючи сирий продукт у вигляді жовтої твердої речовини, який використовують на наступній стадії без додаткового очищення. МС m/z: 254 (М+Н)+. Стадія 3: N-(4-Фторбензил)-3-гідрокси-9(метиламіно)-4-оксо-6,7,8,9-тетрагідро-4Н-тридо[ 1,2-а]піримідин-2-карбоксамід До розчину сирого продукту, отриманого на стадії 2, в сухому метанолі додають надлишок триетиламіну. Розчинник видаляють при зниженому тиску і потім при високому вакуумі. Оліїстий залишок розчиняють в безводному метанолі і додають 4-фторбензиламін (3,1екв., теор.). Суміш перемішують і нагрівають до 60°С протягом ночі. Після охолоджування до кімнатної температури розчинник видаляють при зниженому тиску. Залишок розтирають з діетиловим ефіром і залишають при високому вакуумі на 15 хв. Зазначений у заголовку сирий продукт отримують у вигляді жовтої твердої речовини, яка містить надлишок 4-фторбензиламіну (близько 3,5екв.), і використовують без додаткового очищення. МС m/z: 347 (М+Н)+. Стадія 4: (+/-)-9[[(Диметиламіно)сульфоніл](метил)аміно]-N-(4фторбензил)-3-гідрокси-4-оксо-6,7,8,9-тетрагідро4Н-піридо[1,2-а]піримідин-2-карбоксамід 25 До розчину неочищеної сполуки, отриманого на стадії 3, в суміші 2:1 тетрагідрофурану і 2Μ водного гідроксиду натрію додають Ν,Νдиметилсульфамоїлхлорид (4.6екв.). Суміш перемішують при кімнатній температурі протягом 16год. Суміш концентрують при зниженому тиску і продукт виділяють препаративною ОФ-ВЕРХ, з використанням води (0,1% TFA) і ацетонітрилу (0,1% TFA) як елюентів (колонка: С18). Об'єднані фракції, що містять продукт, ліофілізують, отримуючи сполуку, що зазначена у заголовку, у вигляді рихлої, трохи рожевої речовини. 1 Н-ЯМР (300МГц, CDCI3) δ: 11.95 (с, 1Н). 9.13 (м, 1Н), 7.38 (м, 2Н), 7.04 (м, 2Н), 4.98 (м, 1Н), 4.56 (м, 2Н), 4.36 (м, 1Н), 3.62 (м, 1Н), 2.84 (с, 6Н), 2.57 (с, 3Н), 2.38-1.85 (м, 4Н). 13С-ЯМР (100МГц, CDCI3) 5:167.53, 162.55, 160.11, 157.74, 145.76, 144.11, 132.70, 128.89, 128.81, 124.82, 114.60, 114.39, 58.06, 42.93, 41.53, 37.00, 29.03, 23.89, 20.09. МС m/z: 454 (M+H)+. Приклад 4 (+/-)-N1-(2-{[(4-Фторбензил)аміно]карбоніл}-3гідрокси-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-іл)-N1, Ν2, N2-триметилетандіамід Стадія 1: (+/-)-N1-(2-{[(4Фторбензил)аміно]карбоніл}-3-гідрокси-4-оксо6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9іл)N1,N2,N2-тріметилетандіамід До розчину сирого N-(4-фторбензил)-3гідрокси-9-(метиламіно)-4-оксо-6,7,8,9-тетрагідро4Н-піридо[1,2-а]піримідин-2-карбоксаміду (який синтезовано, як описано в прикладі 3, стадія 3), що перемішується в дихлорметані додають 6екв. триетиламіну і 6екв. метилхлороксоацетату. Суміш перемішують при кімнатній температурі протягом 2год., розчинник видаляють при зниженому тиску і залишок розчиняють в розчині диметиламіну (2М) у тетрагідрофурані. Суміш перемішують при 57°С протягом ночі. Після охолоджування до кімнатної температури розчинник видаляють при зниженому тиску і продукт виділяють препаративною ΟΦΒЕΡΧ, з використанням води (0,1% TFA) та ацетонітрилу (0,1% TFA) як елюентів (колонка: С18). Об'єднані фракції, що містять продукт, ліофілізують, отримуючи сполуку, що зазначена у заголовку, у вигляді рихлої, трохи рожевої речовини. Згідно з ЯМР продукт являє собою суміш ротамерів. 1 H-ЯМР (400МГц, ДМСО-d6) δ: 12.05 (с, 0,2 Η), 11.89 (с, 0,8 Η), 9.21 (м, 0,8 Η), 8.74 (м, 0,2 Η), 7.407.28 (м, 2Η), 7.20-7.10 (м, 2Н), 5.17 (м, 0,8 Н), 4.634.35 (м, 2,2 Н), 4.13-4.00 (м, 1Н), 3.65-3.53 (м, часткове накладення сигналу води), 2.95-2.75 (м, 9Н), 2.15-1.80 (м, 4Н). 13С-ЯМР (100МГц, ДМСО-d6) 5:167.87, 167.73, 165.92, 165.46, 164.51, 164.30, 162.42, 160.01, 157.50, 157.41, 146.27, 146.18, 145.76, 145.49, 134.44, 129.43, 129.35, 129.08, 129.00, 125.17, 125.05, 115.07, 114.85, 57.47, 53.60, 43.14, 41.37, 36.49, 35.95, 32.92, 32.64, 32.36, 28.19, 23.88,22.12, 19.67, 19.35. MC m/z: 115 (M+H)+. Приклад 5 (+)-N1-(2-{[(4-Фторбензил)аміно]карбоніл}-3гідрокси-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-іл)-N1,Ν2,N2-триметилетандіамід 80737 26 Стадія 1: (+)3-Гідрокси-2-(метоксикарбоніл)-Nметил-4-оксо-N-[(1S)-1 -фенілетил]-6,7,8, 9тетрагідро-4Н-піридо[ 1,2-а]піримідин-9-амоній трифторацетат До суміші 7:3 (об./об.) метанолу і води при 30°С, що містить (1S)-1-фенілетиламін (4,5екв.), додають бром-похідне, метил-3-(бензоїлокси)-9бром-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-2-карбоксилат (синтезований як указано в прикладі 2, стадія 2) (1,0екв.). Суміш енергійно перемішують протягом 1,5 год. при 30°С. Охолоджувальну баню прибирають і перемішування продовжують протягом 1 год. при кімнатній температурі. РН доводять приблизно до 5 за допомогою оцтової кислоти, після чого додають 37% водний формальдегід (11,5екв.) та ціаноборгідрид натрію (3,25екв.). Після перемішування при кімнатній температурі протягом 20хв. об'єм зменшують приблизно до 1/4 при зниженому тиску. Білий осад, що утворився, відділяють фільтруванням і фільтрат підкислюють до рН2-3 за допомогою трифтороцтової кислоти. Розчин наносять на картріджи з катіонообмінною смолою (Varian MEGA BOND ELUTE SCX), картріжди промивають метанолом і сирий продукт вимивають за допомогою 2Μ аміаку у метанолі. Об'єднані елюенти концентрують досуха при зниженому тиску, і маслянистий залишок розчиняють в метанолі і нейтралізують трифтороцтовою кислотою. Після видалення розчинника отримують маслянистий залишок. Отримані у співвідношенні 1:3 діастереоізомери розділяють шляхом очищення препаративною ОФВЕРХ (колонка: С18), елюенти: вода (0,1% TFA), ацетонітрил (0,1% TFA). Основний діастереоізомер виходить другим піком, і після ліофілізації отримують сполуку, що зазначена у заголовку, у вигляді трохи рожевої твердої речовини. 1 Н-ЯМР (500МГц, піридин-d5) δ:7.53 (м, 2Н), 7.39 (м, 2Н), 7.29 (м, 1Н), 4.45 (м, 1Н), 4.38 (м, 1Н), 4.14 (м, 1Н), 3.91 (с, 3Н), 3.80 (м, 1Н), 2.13 (с, 3Н), 1.95-1.82 (м, 3Н), 1.70-1.60 (м, 1Н), 1.49 (д, J=6.4Гц, 3Н). МС m/z: 358 (M+H)+. Стадія 2: (-)3-Гідрокси-2-(метоксикарбоніл)-Nметил-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-амонію трифторацетат Розчин (+)-3-гідрокси-2-(метоксикарбоніл)-Nметил-4-оксо-N[-(1S)-1-фенілетил]-6,7,8,9тетрагідро-4Н-піридо[1,2-а]піримідин-9-амонію трифторацетату в метанолі, що містить паладій на активованому вугіллі (10%, мас/мас.) перемішують у середовищі водню при атмосферному тиску протягом 1,5 год. Каталізатор відділяють шляхом фільтрування і розчин концентрують досуха при зниженому тиску, отримуючи сполуку, що зазначена у заголовку, у вигляді трохи рожевої олії. 1 Н-ЯМР (400МГц, CD3OD) δ: 4,41 (м, 1Н), 4,14 (м, 1н), 3,99 (с, 3Н), 3,91 (м, 1Н), 2,86 (с, 3Н), 2,50 (м, 1Н), 2,26 (м, 1Н), 2,08 (м, 1Н), 1,86 (м, 1Н). МС m/z: 254(М+Н)+. Стадія 3: (+)-N1-(2-{[(4Фторбензил}аміно]карбоніл}-3-гідрокси-4-оксо 27 6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-9іл)N1,N2,N2-триметилетандіамід Розчин (-)-3-гідрокси-2-(метоксикарбоніл)-Nметил-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2а]піримідин-9-амонію трифторацетату, пфторбензиламін (2,2екв.) і триетиламін (1,3екв.) у метанолі перемішують і нагрівають до 65°С протягом 3год. Розчинник видаляють у вакуумі і залишок розчиняють у безводному дихлорметані. Додають метилхлороксоацетат (5екв.) та триетиламін (5екв.), і суміш перемішують при кімнатній температурі протягом 50хв. Розчинник видаляють при зниженому тискуі залишок розчиняють у 2Μ розчині диметиламіну у тетрагідрофурані. Суміш перемішують при 57°С протягом ночі. Після охолоджування до кімнатної температури розчинник видаляють при зниженому тиску, і продукт виділяють препаративною ОФВЕРХ, з використанням води (0,1% TFA) і ацетонітрилу (0,1% TFA) як елюентів (колонка: С18). Об'єднані фракції, що містять продукт, ліофілізують отримуючи зазначений у заголовку продукт у вигляді рихлого білого матеріалу (енантіомерний надлишок якого 94,4%). Сполуку розчиняють у суміші етилацетат/гептан (суміш 3:2,5 (об./об.)) і залишають стояти при кімнатній температурі протягом чотирьох днів. Видаляють супернатант з осаду, що утворився, концентрують при зниженому тиску і залишок ліофілізують з суміші вода/ацетонітрил, отримуючи зазначений у заголовку енантіомірно чистий продукт, е.е. 100% (е.е. визначають з допомогою Chiral HPLC Chiralpak AS, рухома фаза суміш 0,2% TFA- нгекс./ІРА), спектроскопічні характеристики якого ідентичні відповідним характеристикам сполуки, яка описана у прикладі 4, стадія 1. [a]20D=+36,5+2,5° (С=0,63 в етанолі). Приклад 6 (+/-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3гідрокси-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2а]азепін-10-іл)-N,N',N'-триметилетандіамід Сполуку, що зазначена у заголовку, отримують згідно з синтетичною послідовністю, наведеною в Прикладі 4, з наступними змінами: Стадія 1: 1-(Бензилокси)азепан-2-імін Третбутил(бензилокси)-(5ціанопентил)карбамат (синтезований згідно з методикою, що описана в Прикладі 1 - стадія 1а, виходячи з 6-бромгексаннітрилу) розчиняють у насиченому розчині НСI в EtOH, і суміш перемішують протягом 45 хвилин. Через розчин барботують азот для видалення надлишку НСІ. Розчинник видаляють при зниженому тиску і залишок, який розчинений у 1,4-діоксані, обробляють триетиламіном, доводячи рН до 10. Етанол видаляють і сполуку, що зазначена у заголовку, яка містить надлишок три етиламонійхлориду і етил-6[(бензилокси)аміно]гексаноат використовують без додаткового очищення. Аналітичний зразок очищають препаративною ОФ-ВЕРХ, з використанням води (0,1% TFA) та ацетонітрилу (0,1% TFA) як елюентів (колонка: С18). Об'єднані фракції, які містять продукт, ліофілізують. 80737 28 1 H-ЯМР (400МГц, ДМСО-d6, 300К) δ: 9.39 (с, 1Н), 8.81 (с, 1Н), 7.61-7.52 (м, 2Н), 7.48-7.38 (м, 3Н), 5.03 (с, 2Н), 4.02-3.93 (м, 2Н), 2.70-2.59 (м, 2Н), 1.69-1.54 (м, 6Н). MC m/z: 219 (M+H)+. Стадія 2: (+/-)-N(2-{[(4Фторбензил)аміно]карбоніл}-3-гідрокси-4-оксо-46,7,8,9,10-гексагідропіримідо[1, 2-а]азепін-10-іл)N,N',N'-триметилетандіамід До розчину сирого N-(4-фторбензил)-3гідрокси-10-(метиламіно)-4-оксо-4,6,7,8,9,10гексагідропіримідо[1,2-]азепін-2-карбоксаміду (синтезованого виходячи з 1 -(бензилокси)азепан2-іміну згідно з методикою, що використовується в аналогічних 6-членних серіях (Приклад 3 - Стадія 3)), що перемішується у дихлорметані, додають 3екв. триетиламіну, 2екв. калій(диметиламіно)(оксо)ацетату, 2,2екв. 1-(3диметиламінопропіл)-3етилкарбодіімідгідрохлориду і 2,2екв. 1гідроксибензотриазолу. Суміш перемішують при кімнатній температурі протягом ночі. Розчинник видаляють при зниженому тиску і залишок розподіляють між етилацетатом і 1Μ водним НСІ. Водну фазу екстрагують етилацетатом і об'єднані органічні фази сушать над сульфатом натрію, фільтрують і концентрують досуха у вакуумі. Зазначений у заголовку продукт виділяють препаративною ОФ-ВЕРХ, з використанням води (0,1% TFA) і ацетонітрилу (0,1% TFA) як елюентів (колонка: С18). Об'єднані фракції, що містять продукт, ліофілізують отримуючи сполуку, що зазначена у заголовку, у вигляді рихлого, білого матеріалу. Згідно з 1Н-ЯМР продукт являє собою суміш ротамерів. 1 Н-ЯМР (400МГц, ДМСО-d6, 300К) δ: 12.29 (уш.с, 0.1Н), 11.95 (уш.с, 0.9Н), 9,30 (уш.с, 0,9Н), 8.45 (уш.т, 0.1Н), 7.38 (дд, J=8.33, 5.5Гц, 1.8Η), 7.33 (дд, J=8.33, 5.5Гц, 0.2Η), 7.15 (т, J=9.0Гц, 2Н), 5.45-5.25 (м, 0.9Н), 4.94 (дд, J=14, 5.7Гц, 1.0Η), 4.84-4.79 (м, 0.1Η), 4.57-4.43 (м, 2Η), 3.54 (дд, J=14, 11Гц, 0.9Η), 3.28-3.18 (м, 0.1Η), 3.05 (с, 0.3Н), 2.92 (с, 2.7Η), 2.90 (с, 5.4Η), 2.81 (с, 0.3Н), 2.76 (с, 0.3Н), 2.19-1.78 (м, 5Н), 1.41-1.27 (м, 1Н). МС m/z: 460 (M+H)+. Приклад 7 (-)-N-(2-{[(4-фторбензил)аміно]карбоніл}-3гідрокси-4-оксо-4,6,7,8,9,10-гексагідропіримідо[1,2а]азепін-10-іл)-N,N',N'-триметилетандіамід Стадия 1: Диметил-(2Е)-2-[(азепан-2іліденаміно)окси]бут-2-ендіоат і диметил-(2Z)-2[(азепан-2-іліденаміно)окси]бут-2-ендіоат До суспензії азепан-2-оноксиму в ацетонітрилі додають по краплях шляхом перемішування 1,1екв. диметилацетилендикарбоксилату. Суміш перемішують при кімнатній температурі протягом 1 години. Розчинник видаляють при зниженому тиску, отримуючи суміш 8/1 диметил-(2Е)-2[(азепан-2-іліденаміно)окси]бут-2-ендіоату і диметил-(22)-2-[(азепан-2-іліденаміно)окси]бут-2ендіоату у вигляді жовтого масла. Для кращої ідентифікації зазначених у заголовку сполук, невелику кількість сирого продукту очищають препаративною ОФ-ВЕРХ, з використанням води (0,1% TFA) і ацетонітрилу (0,1% TFA) як елюентів 29 (колонка: С18). Об'єднані фракції, що містять продукт, ліофілізують. Ізомер Е: 1 H-ЯМР (400МГц, ДМСО-d6, 300К) δ: 7.08 (уш.с, 1Н), 5,63 (с, 1Н), 3.77 (с, 3Н), 3.59 (с, 3Н), 3.19-3.11 (м, 2Н), 2.29-2.21 (м, 2Н), 1.66-1.42 (м, 6Н). 13 С-ЯМР (125ГМц, ДМСО-d6,- 300К) δ: 166.20, 162.81, 161.90, 161.61, 92.38, 52.42, 50.92, 42.28, 29.84, 29.32, 28.81, 25.40. MC m/z: 271 (M+H)+. Ізомер Z: 1Н-ЯМР (400МГц, ДМСО-d6, 300К) δ: 6.66 (уш.с, 1Н), 5,63 (с, 1Н), 3.74 (с, 3Н), 3.61 (с, 3Н), 3.24-3.16 (м, 2Н), 2.20-2.12 (м, 2Н), 1.65-1.44 (м, 6Н). 13 С-ЯМР (125МГц, ДМСО-d6, 300К) δ: 165.01, 163.01, 161.45, 154.11, 101.09, 52.32, 51,05, 42.24, 29.93, 29.31, 28.45, 25.14. MC m/z: 271 (M+H)+. Стадія 2: Метил-3-гідрокси-4-оксо-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-2-карбоксилат Суміш диметил-(2Е)-2-[(азепан-2іліденаміно)окси]бут-2-ендіоату і диметил-(2Z)-2[(азепан-2-іліденаміно)окси]бут-2-ендіоату у співвідношенні 8/1 розчиняють в о-ксилолі і нагрівають до температури кипіння із зворотним холодильником. Через 16 год. розчинник видаляють при зниженому тиску і залишок, який розчинений в етилацетаті, екстрагують насиченим розчином NaHCO3 у воді. РН водної фази доводять приблизно до 3 шляхом додання 6М водного НСI і розчин екстрагують дихлорметаном. Органічну фазу сушать над сульфатом натрію і концентрують. 1 H-ЯМР (400МГц, ДМСО-d6, 300К) δ: 10.12 (с, 1Н), 4.29-4.16 (м, 2Н), 3.80 (с, 3Н), 2.95-2.78 (м, 2Н), 1.79-1.41 (м, 6Н). 13 С-ЯМР (100МГц, ДМСО-d6, 300К) δ: 165.79, 158.54, 153.41, 143.55, 126.61, 52.04, 43.02, 35.75, 28.76, 26.94, 24.58. МС m/z: 239 (М-Н)+. Стадія 3: Метил-3-(бензоїлокси)-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а] азепін-2карбоксилат До розчину метил-3-гідрокси-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а] азепін-2карбоксилату в піридині додають бензойний ангідрид (1,1екв.) і каталітичну кількість диметиламінопіридину. Суміш перемішують при кімнатній температурі протягом 3 год. Розчинник видаляють при зниженому тиску і залишок розподіляють між дихлорметаном і 1Μ водн. НСI. Водну фазу екстрагують дихлорметаном і об'єднані органічні фази промивають 1 Μ водним НСI і насиченим розчином солі. Органічну фазу сушать над сульфатом натрію, фільтрують і концентрують досуха у вакуумі. Сполуку, що зазначена у заголовку, отримують після флеш хроматографії (елюенти петролійний ефір/етилацетат, суміш 6:4) у вигляді коричневої твердої речовини. 1 Н-ЯМР (400МГц, ДМСО-d6, 300К) δ: 8.07 (дд, J=8.6, 1.3Гц, 2Н), 7.78 (т, J=7.5Гц, 1Н), 7.62 (т, J=7.9Гц, 2Н), 4.31-4.29 (м, 2Н), 3.74 (с, 3Н), 3.063.04 (м, 2Н), 1.82-1.65 (м,6Н). 80737 30 13 С-ЯМР (75МГц, ДМСО-d6, 300К) δ: 162.78, 162.65, 162.43, 157.01, 140.29, 135.23, 134.18, 129.63, 128.86, 127.59, 52.46, 43.13, 36.07, 28.47, 26.12, 23.68. МС m/z: 343 (M+H)+. Стадія 4: Метил-3 -(бензоїлокси)-10-бром-4оксо-4,6,7,8,9,10-гексагідропіримідо[ 1,2-а]азепін-2карбоксилат Суміш метил-3-(бензоїлокси)-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а] азепін-2карбоксилату, N-бромсукциніміду (2екв.) і а,а'азоізобутиронітрилу (0,45екв.) у чотирьоххлористому вуглеці перемішують шляхом нагрівання до температури кипіння із зворотним холодильником протягом 14 годин. Суміш охолоджують до кімнатної температури, сукцинімід відділяють фільтруванням і розчинник видаляють при зниженому тиску. Метил-3(бензоїлокси)-10-бром-4-оксо-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-2-карбоксилат отримують після флеш хроматографії (елюенти петролійний ефір/етилацетат, суміш 8:2) у вигляді блідо-жовтої твердої речовини. 1 Н-ЯМР (400МГц, ДМСО-d6, 300К) δ: 8.07, (дд, J=8.3, 0.9Гц, 2Н), 7.79 (т, J=7.5Гц, 1Н), (7.63(t/=7.9Гц, 2Н), 5.63 (дд, J=5.9,2.2Гц, 1Н), 4.98 (дд, J=14.3), 6.1Гц, 1Н), 3.97 (дд, J=14.3, 11.0Гц, 1Н), 3.76 (с, 3Н), 2.31-2.13 (м, 2Н), 2.10-1.79 (м, 3Н), 1.-1.61-1.48 (м,1Н). 13 С-ЯМР (75МГц, ДМСО-d6, 300К) δ: 162.65, 162.14, 157.22, 157.10, 139.45, 136.59, 134.37, 129.72, 128.94, 127.36, 53.56, 52.67, 42.37, 31.52, 25.78, 24.40. MCm/z: 423/421(М+Н)+. Стадія 5: Метил-3-гідрокси-4-оксо-10-{[(1R)-1фенілетил]аміно}-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-2-карбоксилат Метил-3-(бензоїлокси)-10-бром-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-2карбоксилат (1,0екв.) додають до розчину (1R)-1фенілетиламіну (2,2екв.) та триетиламіну (1екв.), які розчинені у Ν,Ν-диметилформаміді. Суміш енергійно перемішують протягом 2 годин при кімнатній температурі і потім при 50°С протягом 30 хвилин. Розчинник видаляють при зниженому тиску, і сирий продукт (суміш діастереоізомерів 1:1), якій зазначено у заголовку, придатний для застосування без додаткового очищення. Згідно з альтернативною методикою (стадія 5А), твердий метил-3-(бензоїлокси)-10-бром-4-оксо-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-2-карбоксилат (1,0екв.) додають до розчину (1R)-1фенілетиламіну (4,5екв.), якій розчинений у суміші 7:3 метанол/вода при -30°С. Взаємодію здійснюють протягом ночі, потім температуру підіймають до кімнатної температури і розчинник концентрують, отримуючи білу тверду речовину, яку відділяють фільтруванням і відкидають. Сполуку, що зазначена у заголовку (у вигляді суміші діастереоізомерів 7:3) екстрагують дихлорметаном з маточного розчину з метою застосування на наступній стадії без додаткового очищення. МС m/z: 358 (М+Н)+. 31 Стадія 6: N-(4-фторбензил)-3-гідрокси-4-оксо10-{[(1R)-1-фенілетил]аміно}-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-2-карбоксамід п-Фторбензиламін (3екв.) додають до метил-3гідрокси-4-оксо-10-{[(1R)-1-феніл етил]аміно}4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-2карбоксилату (отриманий як описано на стадії 5 або стадії 5А), що розчинений в метанолі. Суміш перемішують при 70°С протягом ночі, потім охолоджують до кімнатної температури для безпосереднього застосування на стадії 7. Згідно з альтернативною методикою (стадія 6А), розчинник видаляють і сирий продукт декілька разів кристалізують з ацетонітрилу, що призводить до отримання окремого діастереоізомеру сполуки, яка зазначена у заголовку, у вигляді 4фторбензиламонієвої солі з d.e.>95%. Стадія 7: (+)N-(4-фторбензил)-3 -гідрокси-10{метил-[(1R)-1-фенілетил]аміно}-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-2карбоксамід N-(4-фторбензил)-3-гідрокси-4-оксо-10-{[(1R)1-фенілетил]аміно}-4,6,7,8,9,10гексагідропіримідо[1,2-а]азепін-2-карбоксамід, отриманий на стадії 6, розчиняють в метанолі і рН доводять приблизно до 5 за допомогою оцтової кислоти, після чого додають 37% водний формальдегід (6екв.) та ціаноборгідрид натрію (6,25екв.). Суміш перемішують при кімнатній температурі протягом ночі. Розчинник видаляють при зниженому тиску і залишок, якій розчинений в мінімальній кількості метанолу, підкислюють до рН2-3 за допомогою трифтороцтової кислоти. Розчин наносять на картріджи з катіонообмінною смолою (Varian MEGA BOND ELUTE SCX), картріджи промивають метанолом і сирий продукт вимивають за допомогою 2 Μ аміаку в метанолі. Об'єднані елюенти концентрують досуха при зниженому тиску, отримуючи маслянистий залишок. Продукт, суміш діастереоізомерів, розділяють шляхом очищення препаративної ОФВЕРХ (колонка: С18), елюенти: вода (0,1% TFA), ацетонітрил (0,1% TFA). Зазначений у заголовку діастереоізомер виходить першим піком, і після ліофілізації сполуку, що зазначена у заголовку, отримують у вигляді білої твердої речовини (TFAсіль). Згідно з альтернативною методикою (стадія 7А), продукт стадії 6А піддають взаємодії тим же способом, що і продукт зі стадії 6 при отриманні окремого діастереомеру без очищення методом ВЕРХ. 1 Н-ЯМР (300МГц, ДМСО-d6-TFA, 300К) δ: 9.42 (т, J=6.2Гц, 1Н), 9.20 (уш.с, 1Н), 7.60 (уш.д, J=7.3Гц, 2Н), 7.51-7.29 (м, 5Н), 7.21 (т, J=8.9Гц, 2Н), 4.98-4.75 (м, 3Н), 4.69 (дд, J=15.5, 6.9Гц, 1Н), 4.47 (дд, J=15.5, 5.5Гц, 1Н), 3.66 (т, J=12.8Гц, 1Н), 2.94-2.81 (м, 3Н), 1.97-1.81 (уш.м, 2Н), 1.79-1.33 (м, 7Н). МС m/z: 465 (M+H)+. [a]20D=+62±2 (C=0.1 у хлороформі). Стадія 8: (-)2-{[(4-Фторбензил)аміно]карбоніл}3-гідрокси-N-метил-4-оксо-4,6,7,8,9,10гексагідроиіримідо[1,2-а]азепін-10амонійтрифторацетат 80737 32 Розчин TFA-солі продукту зі стадії 7 (або.7А) у метанолі, що містить паладій на активованому вугіллі (10%, мас./мас), перемішують у середовищі водню при атмосферному тиску протягом 4 год. Каталізатор відділяють фільтруванням і розчин концентрують досуха при зниженому тиску, отримуючи сполуку, що зазначена у заголовку. 1 Н-ЯМР (300МГц, ДМСО-d6-TFA, 300К) δ: 9.88 (уш.с, 1Н), 9.56 (уш.с, 1Н), 9.14 (уш.с, 1Н), 7.41 (дд, J=8.6, 5.7Гц, 2Н), 7.17 (т, J=8.8Гц, 2Н), 4.92 (дд, J=14.6, 4.6Гц, 1Н), 4.72 (уш.м, 1Н), 4.58-4.44 (м, 2Н), 3.51 (дд, J=13.9, 11.7Гц, 1Н), 2.66 (т, J=4.9Гц, 3Н), 2.29 (д, J=13.3Гц, 1Н), 2.02-1.57 (м, 4Н), 1.451.27 (м, 1Н). MC m/z: 361 (M+H)+. [a]20D=-4±2 (C=0.4 у метанолі). Стадия 9: (-)-N-(2-{[(4Фторбензил)аміно]карбоніл}-3-гідрокси-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10іл)N,N',N'-триметилетандіамід Метилхлороксоацетат (2-6екв.) і Nетилдіізопропіламін (4екв.) додають до розчину амонійтрифторацетатної сполуки зі стадії 8 у хлороформі. Суміш перемішують при кімнатній температурі протягом 1 години. Розчинник видаляють при зниженому тиску і залишок, що розчинений у 2Μ розчині диметиламіну в метанолі, перемішують при кімнатній температурі протягом 6 годин. Розчинник видаляють при зниженому тиску і залишок, що розчинений в дихлорметані, промивають 1Μ НСI у воді. Органічну фазу сушать над безводним Na2SO4 і розчинник видаляють при зниженому тиску. Продукт, що зазначений у заголовку, виділяють препаративною ОФ-ВЕРХ, з використанням води (0,1% TFA) і ацетонітрилу (0,1% TFA) як елюентів (колонка: С18). Об'єднані фракції, що містять продукт, ліиофілізують отримуючи продукт, що зазначений у заголовку, з енантіомірним лишком 99,5% (е.е. визначають за допомогою Chiral HPLC Chiralpak AD, рухома фаза суміш 0,2% TFA- н-гекс/0,2% TFA- етанол з 3% метанолу). Аморфну калієву сіль сполуки, що зазначена у заголовку, отримують обробкою сполуки, яка розчинена у ацетонітрилі, водним КОН і подальшою ліофільною сушіння. Спектр 1Н-ЯМР ідентичний відповідному спектру сполуки, що описана у прикладі 6. 13 С-ЯМР (100МГц, ДМСО-d6, 300К) δ: 168.01, 165.80, 165.03, 161.30 (д, JC-F=243Гц), 157.68, 149.67, 145.94, 134.59, 129.56 (д, JC-F=8.5Гц), 124.72, 115.10 (д, JC-F=21Гц), 55.88, 42.42, 41.56, 36.24, 32.79, 32.34, 28.83, 27.10, 26.15. МС m/z: 460 (M+H)+. [a]20D=-72±2 (C=0.1 у хлороформі). Приклад 8 Рацемічний N-(2-{[(4фторбензил)аміно]карбоніл}-3-гідрокси-4-оксо4,6,7,8,9,10-гексагідропіримідо[1,2-а]азепін-10-іл)N,N',N'-триметилетандіамід Стадія 1: Отримання w-гідрокси-Nметиламінонітрилу 3 33 До 5% водного розчину H2SO4 додають 3,4дигідро-2Н-піран (DHP; 21,1г), при 20-35°С. Отриманий розчин витримують при 20-35°С протягом 1год. Реакційну суміш охолоджують до 05°С і нейтралізують до рН=6-7 за допомогою 40% водного метиламіну. До реакційної суміші додають, у зазначеному порядку, метиламінгідрохлорид і ціанід натрію. Розчин, що утворився, витримують при кімнатній температурі протягом 36 год. Реакційну суміш екстрагують IP Ac (6´150мл). Об'єднані органічні шари концентрують до загального об'єму порядку 150мл (вихід в експерименті орієнтовно 91%) і використовують на наступній стадії. 1 Н-ЯМР(СDСІ3, 400МГц) δ: 3.81 (м, 1Н), 3.45 (м, 2Н), 2.47 (с, 3Н), 1.90-1.40 (м, 6Н). Стадія 2: Отримання w-гідрокси-N-метил-NВос-амінонітрилу 4 До розчину w-гідрокси-N-метиламінонітрилу 3 (0,2106моль) в IPAc (зі стадії 1) додають (Вос)2О (48,3г) при кімнатній температурі. Отриманий розчин витримують при 30-35°С протягом 2год. (100% конверсія згідно з 1Н-ЯМР). Реакційну суміш охолоджують до 0-5°С і додають суміш 5% NH2OH/10% NH4Cl (35мл). Суміш, що утворилася, витримують при 10-20°С протягом 3год. Після розділення фаз водний шар екстрагують ІРАс (80мл), об'єднані органічні шари промивають насиченим розчином солі (50мл) та потім концентрують і розчинник замінюють на ІРА (загальний об'єм 230мл), який використовують на наступній стадії. 1 Н-ЯМР (CDCI3, 400МГц) δ: 5.18 (м, 1Н), 3.64 (кв, J=5.7Гц, 2Н), 2.88 (с, 3Н), 1.88-1.75 (м, 3Н), 1.65-1.61 (м, 2Н), 1.49-1.46 (м, 1Н), 1.18 (с, 9Н). Стадія 3: Отримання гідроксіамідину 5 80737 34 До розчину N-Boc-амінонітрилу 4 (0,2106моль) в ІРА (загальний об'єм 230мл) додають 50% гідроксиламін (16,2мл) при температурі навколишнього середовища. Отриманий розчин витримують при 60°С протягом 3год. Реакційну суміш потім концентрують і розчинник замінюють, отримуючи метанольний розчин (загальний об'єм 230мл), який використовують на наступній стадії. 1 H-ЯМР (CDCI3, 400МГц) δ: 7.53 (уш.с, 1Н), 4.84 (уш.с, 2Н), 4.84 (т, J=7.1Гц, 1Н), 3.71-3.62 (м, 2Н), 2.72 (с, 3Н), 2.00 (уш.с, 1Н), 1.92-1.82 (м, 1Н), 1.76 (1.55(м, 3Н), 1.49 (с, 9Н), 1.42-1.23 (м, 2Н). Умови ВЕРХ: колонка: Zorbax, Rx С8 250´4,6мм; температура: 30°С; детектування при 210нм; рухома фаза: суміш 0,1% води. Н3РО4 (A)/MeCN (В); градієнт: від 90:10 суміші (А)/(В) до 10:90 за 15хв., 10:90 підтримують протягом 5хв., від 10:90 до 90:10 суміші (А)/(В) за 10 секунд; швидкість потоку: 1мл/хв. Час втримання: амідоксим - 6,152 хвилини і 6,256 хвилини (два ізомери). Стадія 4: Отримання О-алкенамідоксиму 6 До розчину гідроксіамідину 5 (близько 0,2106моль) у метанолі (загальний об'єм 230мл) додають диметилацетилендикарбоксилат (27,10мл) при кімнатній температурі. Отриманий розчин витримують при кімнатній температурі протягом 16год. Реакційну суміш концентрують і розчинник замінюють на кумол при 40-60°С (загальний об'єм 430мл). Розчин використовують на наступній стадії. 1 Н-ЯМР (CDCI3, 400МГц) δ: 5.82 (с, 0.28Η), 5.73 (с, 0.72Η), 5.44 (уш.с, 1.77Η), 5.25 (уш.с, 0.56Η), 4.61 (м, 1Н), 3.89 (с, 0.84Η), 3.84 (с, 2.16Η), 3.72 (с, 2.16Η), 3.68 (с, 0.84Η), 3.65-3.58 (м, 2Н), 2.73 (с, 0.84Н), 2.71 (с, 2.16Н), 1.90-1.52 (м, 4H), 1.47 (с, 9Н), 1.43-1.30 (м, 2Н). Умови ВЕРХ: колонка: Zorbax, Rx C8 250´4,6мм; температура: 30°С; детектування при 210нм; рухома фаза: суміш 0,1% води. Н3РО4 (A)/MeCN (В); градієнт: від 90:10 суміші (А)/(В) до 10:90 за 15хв., 10:90 підтримують протягом 5хв., від 10:90 до 90:10 суміші (А)/(В) за 10 секунд; швидкість потоку: 1мл/хв. Час втримування: амідоксим 6-12,051, 12,315 хвилин, співвідношення близько 3,6:1. Стадія 5: Отримання піримідину 7 35 Розчин О-алкенамідоксиму 6 (близько 0,2106моль) у кумолі (загальний об'єм 430мл) нагрівають до 120°С (внутрішня температура) протягом 12год. Реакційну суміш потім охолоджують приблизно до 60°С, концентрують до загального об'єму 250мл, потім розбавляють ЕtOАс (250мл) і охолоджують до 25-35°С. Потім повільно додають 5% бікарбонат натрію (330мл, біля 1еквів.), і отриманий розчин витримують при 25-35°С протягом 0,5год. Після розділення фаз органічний шар знову екстрагують 5% бікарбонатом натрію (180мл). Об'єднані водні екстракти підкислюють за допомогою 5н НСI до рН=2-3 і екстрагують за допомогою ЕtOАс (3´250мл). Об'єднані органічні шари промивають насиченим розчином солі (150мл). Органічний розчин концентрують і розчинник замінюють на ТГФ (орієнтовно 30-40% загальний вихід, KF орієнтовно 100-150ч/млн). 1 Н-ЯМР (CDCI3, 400МГц) δ: 10.66 (уш.с, 2Н), 4.77 (м, 1Н), 4.01 (с, 3Н), 3.72-3.67 (м, 2Н), 2.77 (с, 3Н), 2.20-1.55 (м, 5Н), 1.48 (с, 9Н), 1.43-1.35 (м, 1Н). Умови ВЕРХ: колонка: Zorbax, Rx С8 250´4,6мм; температура: 30°С; детектування при 210нм; рухома фаза: суміш 0,1% води. Н3РO4 (A)/MeCN (В); градієнт: від 90:10 суміші (А)/(В) до 10:90 за 15хв., 10:90 підтримують протягом 5хв., від 10:90 до 90:10 суміші (А)/(В) за 10 секунд; швидкість потоку: 1мл/хв. Час утримування: піримідин 7 - 9,905 хвилин. Стадія 6: Отримання бісмезилпіримідину 8 До розчину піримідину 7 (43,5г, близько 80% чистоти, 0,09029моль) у ТГФ (275мл) повільно додають TEA (37,8мл) і MsCl (21,0мл), одночасно, при 0-5°С за 1год. Отриманий розчин витримують при тій же температурі протягом 4год. Тверду речовину відділяють фільтруванням, промивають 80737 36 ТГФ (3´100мл). Об'єднані фільтрати концентрують і розчинник замінюють на метанол (загальний об'єм 200мл). До розчину тримезилпіримідину в метанолі додають карбонат калію (12,5г, 0,09029моль) при 10-20°С. Отриманий розчин витримують при тій же температурі протягом 6-10 год. (моніторинг методом ВЕРХ). Реакційну суміш нейтралізують до рН=6-7 за допомогою 5н НСI і концентрують до загального об'єму орієнтовно 100 мл. Додають 16% розчин солі (100мл) і отриманий розчин екстрагують ЕtOАс (3´100мл). Об'єднані органічні шари промивають насиченим розчином солі (50мл), концентрують і розчинник замінюють на ДМФА. Побічний продукт (MeSO3Me), що утворюється в кількості 1еквів. в ході селективного гідролізу тримезилпіримідину, видаляють азеотропною перегонкою з ДМФА при 60-65°С (моніторинг методом 1Н-ЯМР до 97А%). 1 Н-ЯМР (CDCI3, 400МГц) δ: 11.85 (уш.с, 1Н), 7.84 (уш.с, 0.5 Η), 7.68 (уш.с, 0.5 Η), 7.31 (м, 2Н), 7.04 (м, 2Η), 5.40-4.90 (м, 2Н), 4.53 (м, 2Н), 3.38 (м, 1Н), 2.87 (с, 3Н), 2.20-2.15 (м, 3Н), 1.90-1.40 (м, 3Н), 1.37 (с, 9Н). Умови ВЕРХ: колонка: Zorbax, Rx C8 250´4,6мм; температура: 30°С; детектування при 210нм; рухома фаза: суміш 0,1% водн. Н3РО4 (A)/MeCN (В); градієнт: від 90:10 суміші (А)/(В) до 10:90 за 15хв., 10:90 підтримують протягом 5хв., від 10:90 до 90:10 суміші (А)/(В) за 10 секунд; швидкість потоку: 1мл/хв. Час утримування: піримідин з семичленним циклом 10-15,467 хвилин. Стадія 9: Отримання гідрохлориду піримідинаміду з семичленним 11 Через розчин етилацетату (3,5мл) барботують газоподібний НСI (0,5389г), при температурі від -30 до -20°С. В НСІ-ЕtOАс-розчин вносять при температурі від -30 до -20°С піримідинамід з NВос-семичленним циклом 10 (кристалічна тверда речовина). Отриманий розчин повільно нагрівають до кімнатної температури за 2,5год. і витримують при кімнатній температурі протягом 0,5год. (100% конверсія згідно з ВЕРХ). Реакційну суміш розбавляють EtOAc (7мл). Отриману суспензію витримують при 0-5°С протягом 1год. Кристалічну тверду речовину відділяють фільтруванням, промивають EtOAc, гексаном, сушать у вакуумі шляхом продування азотом і отримують необхідний продукт 11 (98% ізольований вихід, >97А% чистота). 1 Н-ЯМР (CDCI3, 400МГц) δ: 12.35 (с, 1Н), 9.96 (т, J=6.3Гц, 1Н), 9.51 (уш.с, 1Н), 9.19 (уш.с, 1Н), 7.42 (дд, J=8.5, 5.6Гц, 2Н), 7.19 (т, J=8.5Гц, 2Н), 4.92 (дд, J=14.5, 5.1Гц, 1Н), 4.71 (м, 1Н), 4.57-4.45 (м, 2Н), 3.52 (т, J=14.5 Гц), 2.65 (т, J=5.0Гц, 3Н), 2.30 (уш.д, J=12.6Гц, 1Н), 1.99-1.92 (м, 1Н), 1.901,75 (м, 2Н), 1.68-1.60 (м, 1Н), 1.41-1.33 (м, 1Н). Умови ВЕРХ: колонка: Zorbax, Rx C8 250´4,6мм; температура: 30°С; детектування при 210нм; рухома фаза: суміш 0,1% водн. Н3РО4 (A)/MeCN (В); градієнт: від 90:10 суміші (А)/(В) до 10:90 за 15хв., 10:90 підтримують протягом 5хв., від 10:90 до 90:10 суміші (А)/(В) за 10 секунд; швидкість потоку: 1 мл/хв. Час утримування: гідрохлорид піримідинаміду з семичленним циклом - 8,118 хвилин. 39 Стадія 10: Отримання рацемічного N-(2-{[(4фторбензил)аміно]карбоніл}-3-гідрокси-4-оксо4,6,7,8,9,10-гексагідро-піримідо[1,2-а]азепін-10-іл)N,N',N'-триметил-етандіаміду 14 До розчину кислоти 12 (122мг) у ТГФ (3мл) додають етилхлорформіат (92мкл) при 0-5°С. Потім до реакційної суміші повільно додають при 0-5°С 4-NMM (106мкл). Реакційну суміш витримують при тій же температурі протягом 2год. До розчину змішаного ангідриду додають у вигляді твердої речовини при 0-5°С гідрохлорид піримідину 11 (79,4мг) і витримують при тій же температурі протягом 5год. і потім при 5-10°С протягом ще 2год. (100% конверсія згідно з ВЕРХ). До реакційної суміші додають водний диметиламін (40%, 158мкл), і суміш витримують при 10-15°С протягом 1 год., після закінчення якого здійснюють моніторинг реакції методом ВЕРХ для підтвердження повноти перетворення. Реакційну суміш підкислюють 2н НСI до рН=3-4 при 5-15°С. Додають, у зазначеному порядку, ЕtOАс (6мл) і насичений розчин солі (2мл). Після розділення фаз органічний шар промивають 1н НСI (2мл), насиченим розчином солі (2´2мл). Органічний шар концентрують до сумарного об'єму 1мл. Повільно, за 0,5год., додають гексан (5мл). Отриману суспензію витримують при 0-5°С протягом 1год. Кристалічну тверду речовину відділяють фільтруванням, промивають сумішшю гексан/ЕtOАс (5:1), МТВЕ, сушать у вакуумі шляхом продуття азотом і отримують зазначену у заголовку сполуку 14 (15,6мг, 82%). 1 Н-ЯМР (CDCI3, 400МГц) δ: 12.13 (с, 1Н), 9.41 (уш.с, 1Н), 7.38 (дд, J=8.5, 5.4Гц, 2Н), 7.00 (т, J=8.5Гц, 2Н), 5.40 (уш.с, 1Н), 5.29 (дд, J=14.5, 6.0Гц, 1Н), 4.60 (дд, J=14.5, 6.6Гц, 1Н), 4.52 (дд, J=14.5, 6.3Гц, 1Н), 3.35 (дд, J=14.5, 11.6Гц, 1Н), 3.04 (с, 3Н), 3.01 (с, 3Н), 2.98 (с, 3Н), 2.23-2.12 (м, 3Н), 1.95-1.81 (м, 2Н), 1.58-1.49 (м, 1Н). Умови ВЕРХ: колонка: Zorbax, Rx С8 250´4,6мм; температура: 30°С; детектування при 210нм; рухома фаза: суміш 0,1% водн. Н3РО4 80737 40 (A)/MeCN (В); градієнт: від 90:10 суміші (А)/(В) до 10:90 за 15хв., 10:90 підтримують протягом 5хв., від 10:90 до 90:10 суміші (А)/(В) за 10 секунд; швидкість потоку: 1мл/хв. Час утримування: зазначена у заголовку сполука 14 - 12,191 хвилин. У таблиці 1, що наведена нижче, наведений список отриманих сполук згідно з даним винаходом. У таблиці представлені структура і назва кожної сполуки, маса молекулярного іону плюс 1 (М+), що встановлені за допомогою FIA-MS, і синтетична схема, яка використовується для отримання кожної сполуки. 41 80737 42 Хоча наведений вище опис викладає основні принципи даного винаходу і супроводжується прикладами, які представленими з метою ілюстрації, практичне здійснення винаходу охоплює всі можливі зміни, доповнення і/або модифікації, що охоплюються рамками обсягу прикладеної формули винаходу.
ДивитисяДодаткова інформація
Назва патенту англійськоюTetrahydro-4h-pyrido[1,2-a]pyrimidines and related compounds useful as hiv integrase inhibitors
Автори англійськоюCrescenzi Benedetta, Kinzel Olaf, Muraglia Ester, Orvieto Federica, Pescatore Giovanna, Rowley Michael, Summa Vincenzo
Назва патенту російськоюТетрагидро-4н-пиридо[1,2-а]пиримидины и фармацевтическая композиция, которые применяются как ингибиторы вич-интегразы
Автори російськоюКрешенци Бенедетта, Кинцель Олаф, Муралья Эстер, Орвьето Федерика, Пескаторе Джованна, Роули Майкл, Сумма Винченцо
МПК / Мітки
МПК: C07D 487/04, A61P 31/18, C07D 471/04
Мітки: тетрагідро-4н-піридо[1,2-а]піримідини, застосовуються, інгібітори, фармацевтична, композиція, віл-інтегрази
Код посилання
<a href="https://ua.patents.su/21-80737-tetragidro-4n-pirido12-apirimidini-ta-farmacevtichna-kompoziciya-shho-zastosovuyutsya-yak-ingibitori-vil-integrazi.html" target="_blank" rel="follow" title="База патентів України">Тетрагідро-4н-піридо[1,2-а]піримідини та фармацевтична композиція, що застосовуються як інгібітори віл-інтегрази</a>
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Зембровскі Уільям Джеймс, Міларі Банавара Лакшман, Маррі Джеррі Ентоні, Чу-Моєр Маргарет Юхуа
МПК: C07D 471/04, C07D 403/12, A61K 31/53, C07D 513/10, C07D 413/12, C07D 403/04, C07D 401/12, C07D 409/12, A61P 43/00, C07D 451/06, C07D 405/14, C07D 409/14, C07D 239/42, C07D 491/048, C07D 498/04, C07D 521/00, A61P 3/10, A61K 31/5377, A61K 31/517, A61P 9/10, C07D 491/04, C07D 471/08, C07D 401/04, C07D 487/08, C07D 405/12, C07D 417/14, A61K 31/506, C07D 451/02, C07D 417/12, C07D 403/14, C07D 491/10, C07D 491/20, C07D 401/14, C07D 487/04
Мітки: піримідини, сполуки, спосіб, проміжні, одержання, проміжної, фармацевтична, сорбітдегідрогенази, композиція, інгібітори, містить
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...
Заміщені 2-арил-3-(гетероарил)-імідазо[1,2-a]піримідини, спосіб їх одержання (варіанти) та фармацевтична композиція на їх основі

Номер патенту: 70350
Опубліковано: 15.10.2004
Автори: Генрі Джеймс Р., Додд Джон Х., Руперт Кеннет С.
МПК: A61P 37/08, A61P 1/02, A61P 3/10, A61P 31/12, A61P 1/16, A61P 31/04, A61P 37/06, A61P 25/28, A61P 17/06, A61P 1/04, A61K 31/519, C07D 487/04, A61P 19/02, A61P 17/14, A61P 1/18, A61P 9/00, A61P 9/10, A61P 29/00, A61P 31/18, A61P 19/10, A61P 11/00, A61P 25/04
Мітки: заміщені, варіанти, композиція, спосіб, фармацевтична, основі, одержання, 2-арил-3-(гетероарил)-імідазо[1,2-a]піримідини
Формула / Реферат:
1. Заміщений 2-арил-3-(гетероарил)-імідазо[1,2-a]піримідин формули І, (I)або його фармацевтично прийнятна сіль, де(а) R1 вибрано із NH2, C1-5алкіламіно, діC1-5алкіламіно, гідрокси, С1-5алкокси, фенілметиламіно, гетероциклілметил, С1-5алкілкарбоніламіно та заміщеного фенілкарбоніламіно, дезазначені фенілметиламіно та гетероциклілметил можуть бути заміщені по своїй фенольній складовій одним або більшою кількістю...
Спосіб одержання 3-{2-[4-(6-фторбензо[d]ізоксазол-3-іл)піперидин-1-іл]етил}-2-метил-6,7,8,9-тетрагідро-4н-піридо[1,2-а]піримідин-4-ону та проміжні сполуки цього способу

Номер патенту: 80114
Опубліковано: 27.08.2007
Автори: Кампс Франсеск Кс., Фогет Рафаель, Раментоль Хорхе, Петсчен Інес, Гордо Естер, Сальярес Хуан, Фернандес-Кано Дьєго, Пасто Мірейя, Арменголь Мігель П.
МПК: C07D 471/04, C07D 413/14, A61K 31/505
Мітки: цього, проміжні, способу, одержання, сполуки, 3-{2-[4-(6-фторбензо[d]ізоксазол-3-іл)піперидин-1-іл]етил}-2-метил-6,7,8,9-тетрагідро-4н-піридо[1,2-а]піримідин-4-ону, спосіб
Формула / Реферат:
1. Спосіб одержання 3-{2-[4-(6-фторбензо[d]ізоксазол-3-іл)піперидин-1-іл]етил}-2-метил-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-4-ону формули I:, Iякий включає конденсацію (2-метил-4-оксо-6,7,8,9-тетрагідро-4Н-піридо[1,2-а]піримідин-3-іл)ацетальдегіду формули II:,...
Антихолінергічні засоби, які застосовуються як медикаменти, спосіб їх одержання та фармацевтична композиція

Номер патенту: 73804
Опубліковано: 15.09.2005
Автори: Поль Геральд, Спек Георг, Райхль Річард, Піпер Міхаель Пауль, Моршойзер Герд, БАНХОЛЬЦЕР Рольф, Майсснер Гельмут
МПК: A61P 9/06, A61K 45/00, A61P 15/08, A61P 11/06, A61K 31/439, A61P 43/00, C07D 451/10, C07B 55/00, A61P 13/02, A61P 1/06
Мітки: медикаменти, спосіб, композиція, застосовуються, засоби, фармацевтична, одержання, антихолінергічні
Формула / Реферат:
1. Сполуки загальної формули 1 , 1у якійA означає залишок із двома зв'язками, вибраний із групи, яка включає, X- означає однозарядний аніон, R1 і R2 мають ідентичні або різні значення і являють собою залишок, вибраний із групи, яка включає метил, етил, н-пропіл...
Похідні 1н-піридо[3,4-b]індол-4-карбоксаміду та фармацевтична композиція

Номер патенту: 57037
Опубліковано: 16.06.2003
Автори: Севрен Мірей, Малуазель Крістіан, Жорж Паскаль, Легаллудек Одетт, Еванно Яннік
МПК: C07D 471/04, A61P 25/22, A61P 25/00, A61P 25/20, A61K 31/437, A61P 25/08
Мітки: композиція, фармацевтична, 1н-піридо[3,4-b]індол-4-карбоксаміду, похідні
Формула / Реферат:
1. Похідні 1Н-піридо[3,4-b]індол-4-карбоксаміду, як варіант, у формі чистого оптичного ізомеру або суміші таких ізомерів, загальної формули (I), (I)деX - атом гідрогену або галогену або (С1-С3)алкіл, (С1-С3)алкоксил, трифлуорметил або фенілметоксил,R1 - атом гідрогену або (С1-С3)алкіл, циклопропіл або фенілметил,R2 - (С1-С3)алкільна група, як варіант, заміщена метоксильною групою, або феніл(С1-С3)алкільна...