Сполуки діазенійдіолату, спосіб їх одержання і фармацевтична композиція, яка їх містить
Номер патенту: 97035
Опубліковано: 26.12.2011
Автори: Аберкорн Лор, Вербюрен Тоні, Сімоне Серж, Курше Крістін, Корді Алексіс
















Формула / Реферат
1. Сполука формули (І):
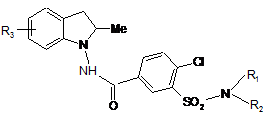 , (I)
, (I)
в якій:
R1 являє собою атом водню або -COOR групу, в якій R являє собою лінійну або розгалужену (С1-С6)алкільну групу або арил-(С1-С6)алкільну групу, в якій алкільна частина може бути лінійною або розгалуженою;
R2 являє собою групу G або лінійну або розгалужену (С1-С6)алкільну групу, заміщену групою G, в якій G являє собою (CH2)n-A-(CH2)m-B-(CR4R5)p-(CH2)o-R6 групу, в якій:
n являє собою 0, 1, 2 або 3,
m являє собою 0, 1, 2 або 3,
р являє собою 0 або 1,
о являє собою 0, 1 або 2,
R4 і R5, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу,
в якій одна з груп -СН2- або -CR4R5- ланцюгу G може бути в рівній мірі заміщена, якщо бажано, феніленом, -PhC(O)- або -PhC(O)O- групою (де Ph означає феніл),
А і В, які можуть бути однаковими або різними, кожний являє собою зв'язок, атом азоту або групу:
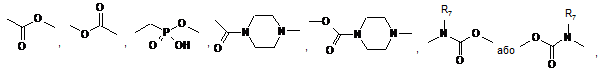
де R7 являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу,
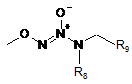 або
або 
і R6 являє собою групу,
в якій R8, R9, R10, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, незаміщену або заміщену аміногрупою, або R8 і R9 разом утворюють лінійний або розгалужений (С1-С6)алкіленовий ланцюг,
R3 являє собою атом водню, лінійну або розгалужену (С1-С6)алкільну групу або NO2 групу,
її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
2. Сполука формули (І) за п. 1, в якій R1 являє собою атом водню, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
3. Сполука формули (І) за п. 1, в якій R3 являє собою атом водню, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
4. Сполука формули (І) за п. 1, в якій R3 являє собою NO2 групу, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
5. Сполука формули (І) за п. 1, в якій R2 являє собою групу:
 ,
,
в якій R6 є таким же, як визначено в п. 1, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
6. Сполука формули (І) за п. 1, в якій R6 являє собою групу -O-N=N(O)-NR8R9, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
7. Сполука формули (І) за п. 1, в якій R8 і R9 являють собою лінійну (С1-С6)алкільну групу, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
8. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5-[(2-метил-5-нітро-2,3-дигідро-1Н-індол-1-іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
9. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 4-{[({2-хлор-5-[(2-метил-2,3-дигідро-1Н-індол-1-іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
10. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 4-({[(2-xлop-5-{[(2R)-2-мeтил-2,3-дигiдpo-1H-індол-1-іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензоат, її адитивні солі з фармацевтично прийнятною кислотою або основою.
11. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5-[(2-метил-2,3-дигідро-1Н-індол-1-іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою.
12. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5-[((2R)-2-метил-2,3-дигідро-1Н-індол-1-іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат, її адитивні солі з фармацевтично прийнятною кислотою або основою.
13. Спосіб одержання сполук формули (І) за п. 1, який відрізняється тим, що як вихідний матеріал використовують сполуку формули (II):
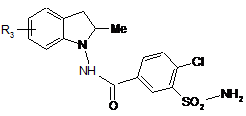 , (II)
, (II)
де R3 є таким же, як визначено для формули (І),
яку конденсують зі сполукою формули (III):
![]() , (III)
, (III)
де R є таким же, як визначено для формули (І),
з одержанням сполуки формули (IV):
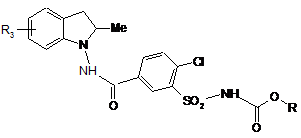 , (IV)
, (IV)
де R3 i R є такими ж, як визначено тут вище,
яку конденсують, у присутності основи, зі сполукою формули (V):
![]() , (V)
, (V)
в якій R2 є таким же, як визначено для формули (І), і X являє собою атом галогену,
з одержанням сполуки формули (І/а), окремого випадку сполук формули (І):
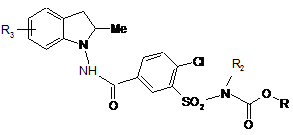 , (I/a)
, (I/a)
в якій R, R2 і R3 є такими ж, як визначено для формули (І), сполуки формули (І/а) можуть також бути одержані шляхом конденсації сполуки формули Cl-(CH2)o-R6, в якій о і R6 є такими ж, як визначено для формули (І), з карбоновою або фосфорамідною функцією, присутньою в групі G,
сполуки формули (І/а) необов'язково нагрівають в кислотному середовищі з одержанням сполуки формули (І/b), окремого випадку сполук формули (І):
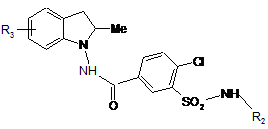 , (I/b)
, (I/b)
в якій R2 і R3 є такими ж, як визначено для формули (І),
сполуки формул (І/а) і (І/b), сукупність яких складає сполуки формули (І), очищують, коли прийнятно, відповідно до звичайної методики очищення, необов'язково розділяють на ізомери відповідно до звичайної методики розділення і перетворюють, коли бажано, в адитивні солі з фармацевтично прийнятною кислотою або основою.
14. Фармацевтична композиція, яка містить як активний інгредієнт сполуку за будь-яким з пп. 1-12 у поєднанні з одним або більше фармацевтично прийнятними, інертними, нетоксичними наповнювачами або носіями.
15. Фармацевтична композиція за п. 14, яка містить принаймні один активний інгредієнт за будь-яким з пп. 1-12 для застосування як лікарського засобу у лікуванні гіпертензії та серцево-судинних патологій і їх ускладнень, таких як ретинопатія, інсульти, недоумство, гіпертрофія лівого шлуночка, серцева недостатність, стенокардія, інфаркт міокарда і нефропатія; у лікуванні серцево-судинних патологій, пов'язаних з атеротромбозом, таких як інсульти і коронарні ураження, артеріїт і васкулопатії; у лікуванні судинних ускладнень численних розладів, таких як діабет, ожиріння, метаболічний синдром, рак, фіброз печінки, і у лікуванні гіпертензії легеневого, очного походження або портальної гіпертензії.
Текст
1. Сполука формули (І): UA (21) a201008615 (22) 10.12.2008 (24) 26.12.2011 (86) PCT/FR2008/001716, 10.12.2008 (31) 07.08604 (32) 11.12.2007 (33) FR (46) 26.12.2011, Бюл.№ 24, 2011 р. (72) КОРДІ АЛЕКСІС, FR, АБЕРКОРН ЛОР, FR, ВЕРБЮРЕН ТОНІ, FR, КУРШЕ КРІСТІН, FR, СІМОНЕ СЕРЖ, FR (73) ЛЕ ЛАБОРАТУАР СЕРВЬЄ, FR (56) JP07118231 A 09.05.1995 JP58124766 A 25.07.1983 FR2003311 A 07.11.1969 LIU D-G ET AL: "Acylsulfonamide-containing PTP1B inhibitors designed to mimic an enzyme-bound water of hydration" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 13, no. 18, 2003, pages 3005-3007, XP002337095 ISSN: 0960-894X WO9819996 A 14.05.1998 KONTER, JOERG ET AL: "Synthesis of diazen-1ium-1,2-diolates monitored by the "NOtizer" apparatus: relationship between formation rates, molecular structure and the release of nitric oxide" EUROPEAN JOURNAL OF ORGANIC CHEMISTRY , (4), 616 -624 CODEN: EJOCFK; ISSN: 1434-193X, 2007, XP002473169 SAAVEDRA, JOSEPH E. ET AL: "Piperazine as a linker for incorporating the nitric oxide-releasing diazeniumdiolate group into other biomedically relevant functional molecules" JOURNAL OF ORGANIC CHEMISTRY , 64(14), 5124 -5131 3 97035 де R7 являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, O O + N N N R8 -O + N R9 або N O R10 , і R6 являє собою групу, в якій R8, R9, R10, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, незаміщену або заміщену аміногрупою, або R8 і R9 разом утворюють лінійний або розгалужений (С1С6)алкіленовий ланцюг, R3 являє собою атом водню, лінійну або розгалужену (С1-С6)алкільну групу або NO2 групу, 4 її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 2. Сполука формули (І) за п. 1, в якій R1 являє собою атом водню, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 3. Сполука формули (І) за п. 1, в якій R3 являє собою атом водню, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 4. Сполука формули (І) за п. 1, в якій R3 являє собою NO2 групу, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 5. Сполука формули (І) за п. 1, в якій R2 являє собою групу: O O CH2 CH2 O R6 CH2 O CH2 , CH2 R6 , O O CH2 R6 або CH2 R6 CH2 в якій R6 є таким же, як визначено в п. 1, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 6. Сполука формули (І) за п. 1, в якій R6 являє собою групу -O-N=N(O)-NR8R9, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 7. Сполука формули (І) за п. 1, в якій R8 і R9 являють собою лінійну (С1-С6)алкільну групу, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 8. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5[(2-метил-5-нітро-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 9. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 4-{[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 10. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 4-({[(2-xлop-5{[(2R)-2-мeтил-2,3-дигiдpo-1H-індол-1іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензоа , т, її адитивні солі з фармацевтично прийнятною кислотою або основою. 11. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат, її енантіомери і діастереоізомери, а також її адитивні солі з фармацевтично прийнятною кислотою або основою. 12. Сполука формули (І) за п. 1, яка являє собою ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5[((2R)-2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат, її адитивні солі з фармацевтично прийнятною кислотою або основою. 13. Спосіб одержання сполук формули (І) за п. 1, який відрізняється тим, що як вихідний матеріал використовують сполуку формули (II): R3 Me N Cl NH O SO2 NH2 , (II) де R3 є таким же, як визначено для формули (І), яку конденсують зі сполукою формули (III): O O O R R O O , (III) де R є таким же, як визначено для формули (І), 5 97035 6 з одержанням сполуки формули (IV): R3 R3 Me N Cl NH O SO2 NH O O R , (IV) де R3 i R є такими ж, як визначено тут вище, яку конденсують, у присутності основи, зі сполукою формули (V): X R2 , (V) в якій R2 є таким же, як визначено для формули (І), і X являє собою атом галогену, з одержанням сполуки формули (І/а), окремого випадку сполук формули (І): Me N Cl NH O SO2 Cl NH O R3 Me N R2 N O R O , (I/a) в якій R, R2 і R3 є такими ж, як визначено для формули (І), сполуки формули (І/а) можуть також бути одержані шляхом конденсації сполуки формули Cl(CH2)o-R6, в якій о і R6 є такими ж, як визначено для формули (І), з карбоновою або фосфорамідною функцією, присутньою в групі G, сполуки формули (І/а) необов'язково нагрівають в кислотному середовищі з одержанням сполуки формули (І/b), окремого випадку сполук формули (І): Даний винахід стосується нових сполук діазенійдіолату, способу їх одержання і фармацевтичних композицій, які їх містять. Ці сполуки мають нову структуру і можуть бути застосовані в галузі лікування гіпертензії і серцево-судинних захворювань. Гіпертензія приводить до збільшеного ризику гострих судинних розладів, особливо на мозковому і коронарному рівнях. Її все більш і більше часто пов'язують з іншими патологіями, такими як атеросклероз, або з метаболічними розладами, такими як ожиріння, діабет або ниркова недостатність, яка відчутно збільшує ризик спазмів і тромбозів. Діуретики представляють клас дуже ефективних антигіпертензивних лікарських засобів. Їх основною дією є зниження загального периферичного опору судин шляхом збільшення виведення натрію і збільшення обсягу плазми. Діуретики зменшують кількість інсультів, а також є ефективними при серцевій недостатності. Їх перевагою є також те, що вони не мають значних протипоказань і добре переносяться. Їх можна поєднувати з інши SO2 NH R2 , (I/b) в якій R2 і R3 є такими ж, як визначено для формули (І), сполуки формул (І/а) і (І/b), сукупність яких складає сполуки формули (І), очищують, коли прийнятно, відповідно до звичайної методики очищення, необов'язково розділяють на ізомери відповідно до звичайної методики розділення і перетворюють, коли бажано, в адитивні солі з фармацевтично прийнятною кислотою або основою. 14. Фармацевтична композиція, яка містить як активний інгредієнт сполуку за будь-яким з пп. 1-12 у поєднанні з одним або більше фармацевтично прийнятними, інертними, нетоксичними наповнювачами або носіями. 15. Фармацевтична композиція за п. 14, яка містить принаймні один активний інгредієнт за будьяким з пп. 1-12 для застосування як лікарського засобу у лікуванні гіпертензії та серцево-судинних патологій і їх ускладнень, таких як ретинопатія, інсульти, недоумство, гіпертрофія лівого шлуночка, серцева недостатність, стенокардія, інфаркт міокарда і нефропатія; у лікуванні серцевосудинних патологій, пов'язаних з атеротромбозом, таких як інсульти і коронарні ураження, артеріїт і васкулопатії; у лікуванні судинних ускладнень численних розладів, таких як діабет, ожиріння, метаболічний синдром, рак, фіброз печінки, і у лікуванні гіпертензії легеневого, очного походження або портальної гіпертензії. ми класами антигіпертензивних засобів і їх систематично застосовують за необхідності у подвійній або потрійній терапії. З моменту відкриття його серцево-судинної дії в 1980 році, монооксид азоту (NO) було визнано як судинорозширювальну і судинозахисну молекулу, здатну запобігати вазоспазмам, атеросклерозу і тромбозу, що, будучи ендогенним медіатором, таким чином надає захист проти серцевосудинного захворювання. NO по суті продукується ендотеліальними клітинами і, при серцевосудинних патологіях, дисфункція ендотелію викликає нестачу ендогенного NO. Нітровазодилататорні сполуки, такі як нітрогліцерин, застосовувались протягом тривалого часу для лікування стенокардії і серцевої недостатності. Сприятлива дія цих продуктів пов'язана з їх здатністю утворювати NO (мимовільно або метаболічно). Їх застосування також призвело до спостереження, що, у пацієнта з підвищеним артеріальним тиском, ці NO донори викликають суттєве зниження систолічного артеріального тиску. Неконтрольований систолічний артеріальний тиск 7 97035 являє собою значний фактор ризику інсульту та інфаркту міокарда і часто є стійким до антигіпертензивного лікування. Дійсно, незважаючи на продемонстровані антигіпертензивні і судинозахисні дії діуретиків та інших класів антигіпертензивних продуктів, артеріальний тиск, особливо систолічний артеріальний тиск, залишається важко контролювати, і ступінь захворюваності і смертності залишається високою. Додавання властивості NO-донора до діуретичних продуктів вдосконалить їх антигіпертензивні, кардіозахисні і судинозахисні властивості, і це забезпечить безпосередню антитромботичну дію, NO, що має дію проти агрегації тромбоцитів і антитромботичну дію (Walford et al., 2003, J. Thromb. Haemost, 1, 2112-2118). В доповнення до нової структури, сполуки за даним винаходом проявляють таку подвійну фармакологічну активність, що наділяє їх цілком несподіваними і цінними властивостями в галузі лікування гіпертензії і серцево-судинних патологій. Більш конкретно, даний винахід стосується сполук формули (І): 8 в якій: R1 являє собою атом водню або -COOR групу, в якій R являє собою лінійну або розгалужену (С1С6)алкільну групу або арил-(С1-С6)алкільну групу, в якій алкільна частина може бути лінійною або розгалуженою; R2 являє собою групу G або лінійну або розгалужену (С1-С6)алкільну групу, заміщену групою G, в якій G являє собою (CH2)n-A-(CH2)m-B-(CR4R5)p(CH2)0-R6 групу, в якій: - n являє собою 0, 1, 2 або 3, - m являє собою 0, 1, 2 або 3, - p являє собою 0 або 1, - о являє собою 0, 1 або 2, - R4 і R5, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, в якій одна з груп -СН2- або -CR4R5- ланцюгу G може бути в рівній мірі заміщена, якщо бажано, феніленом, -PhC(O)- або -PhC(O)O- групою (де Ph означає феніл), - А і В, які можуть бути однаковими або різними, кожний являє собою зв'язок, -NH- або групу: , де R7 являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, - і R6 являє собою групу або , в якій R8, R9, R10, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену (С1С6)алкільну групу, незаміщену або заміщену аміногрупою, або R8 і R9 разом утворюють лінійний або розгалужений (С1-С6)алкіленовий ланцюг, R3 являє собою атом водню, лінійну або розгалужену (С1-С6)алкільну групу або NO2 групу, їх енантіомерів і діастереоізомерів, а також їх адитивних солей з фармацевтично прийнятною кислотою або основою. Серед фармацевтично прийнятних кислот можуть бути згадані, без будь-якого обмеження, наприклад, хлористоводнева кислота, бромистоводнева кислота, сірчана кислота, фосфонова кислота, оцтова кислота, трифтороцтова кислота, молочна кислота, піровиноградна кислота, малонова кислота, янтарна кислота, глутарова кислота, фумарова кислота, винна кислота, малеїнова кислота, лимонна кислота, аскорбінова кислота, метансульфонова кислота, камфорна кислота і т.д. Серед фармацевтично прийнятних основ можуть бути згадані, без будь-якого обмеження, гідроксид натрію, гідроксид калію, триетиламін, третбутиламін і т.д. Більш конкретно, даний винахід стосується сполуки формули (І), в якій R1 являє собою атом водню. Переважно, даний винахід стосується сполуки формули (І), в якій R3 являє собою атом водню або NО2 групу. R2 переважно являє собою групу: , 9 97035 10 , , або , в якій R6 є таким же, як визначено вище. Переважна R6 група відповідно до даного винаходу являє собою групу -О-N=N(O)-NR8R9. Зв'язок -N=N- групи R6 переважно має конфігурацію Z. R8 і R9 переважно являють собою лінійну (С1С6)алкільну групу, таку як, наприклад, етильна група. Навіть більш конкретно, даний винахід стосується наступних сполук формули (І): ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5[(2-метил-5-нітро-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат, ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат, - трет-бутил ({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1-іл)карбамоїл]-феніл}сульфоніл)[({[(1Z)2,2-діетил-1-оксидогідразоно]аміно}окси)метил]карбамат, - ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил Ν-(трет-бутоксикарбоніл)-N-({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1-іл)карбамоїл]феніл}сульфоніл)гліцинат, ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил N-({2-хлор-5-[(2метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)гліцинат, ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 4-{[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат, - ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 4-({[(2-xnop-5-{[(2R)-2-метил-2,3-дигідро1Н-індол-1-іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензоат, ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат, - ({[(1Z)-2,2-діетил-1-оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5-[((2R)-2-метил-2,3дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат. Винахід стосується також способу одержання сполук формули (І), який відрізняється тим, що як вихідний матеріал використовують сполуку формули (II): в якій R3 є таким же, як визначено для формули (І), яку конденсують зі сполукою формули (III): в якій R є таким же, як визначено для формули (І), для одержання сполуки формули (IV): , ще, в якій R3 і R є такими ж, як визначено тут ви яку конденсують, у присутності основи, зі сполукою формули (V): , в якій R2 є таким же, як визначено для формули (І), і X являє собою атом галогену, для одержання сполуки формули (І/а), окремого випадку сполук формули (І): , в якій R, R2 і R3 є такими ж, як визначено для формули (І), сполуки формули (І/а) можуть також бути одержання шляхом конденсації сполуки формули CI(CH2)0-R6, в якій о і R6 є такими ж, як визначено для формули (І), з карбоновою або фосфорамідною функцією, присутньою в групі G, сполуки формули (І/а) необов'язково нагрівають в кислотному середовищі для одержання сполуки формули (І/b), окремого випадку сполук формули (І): 11 97035 , в якій R2 і R3 є такими ж, як визначено для формули (І), сполуки формул (І/а) і (І/b), сукупність яких складає сполуки формули (І), очищують, коли прийнятно, відповідно до звичайної методики очищення, необов'язково розділяють на ізомери відповідно до звичайної методики розділення і перетворюють, коли бажано, в адитивні солі з фармацевтично прийнятною кислотою або основою. Сполуки формули (II), як визначено тут вище, одержують за допомогою звичайних реакцій органічної хімії, таких як, наприклад, спосіб, розкритий в патенті FR 2003311. Беручи до уваги їх фармакологічні властивості, сполуки за даним винаходом є корисними при лікуванні гіпертензії та серцево-судинних патологій і їх ускладнень, таких як ретинопатія, інсульти, недоумство, гіпертрофія лівого шлуночку, серцева недостатність, стенокардія, інфаркт міокарду і нефропатія. Сполуки за даним винаходом є такою корисними при серцево-судинних патологіях, пов'язаних з атеротромбозом, таких як інсульти і коронарні ураження, артеріїт і васкулопатії, а також при судинних ускладненнях численних розладів, таких як діабет, ожиріння, метаболічний синдром, рак, фіброз печінки і т.д. Сполуки є також корисними при гіпертензії легеневого, очного походження або портальній гіпертензії. Даний винахід стосується також фармацевтичних композицій, які містять як активний інгредієнт щонайменшу одну сполуку формули (І), її оптичний ізомер або її адитивну сіль з фармацевтично прийнятною кислотою або основою, одну або в поєднанні з одним або більше фармацевтично прийнятним, інертним, нетоксичним наповнювачем або носієм. Серед фармацевтичних композицій за даним винаходом можуть бути згадані більш конкретно ті, які прийнятні для орального, парентерального (внутрішнього, внутрішньом'язового або підшкірного), перкутанного або черезшкірного, внутрішньовагінального, ректального, назального, під'язикового, буккального, очного або респіраторного введення. Фармацевтичні композиції за даним винаходом для парентеральних ін'єкцій включають, зокрема, стерильні водні і неводні розчини, дисперсії, суспензії і емульсії, а також стерильні порошки для розчинення розчинів або дисперсій для ін'єкцій. Фармацевтичні композиції відповідно до даного винаходу для орального введення у твердій формі включають, зокрема, таблетки або драже, під'язикові таблетки, саше, капсули і гранули, а для рідкого орального, назального або очного введення вони включають, зокрема, емульсії, розчини, суспензії, краплі, сиропи і аерозолі. 12 Фармацевтичні композиції для ректального або вагінального введення переважно являють собою супозиторії, а ті, які призначені для перкутанного або черезшкірного введення, включають, зокрема, порошки, аерозолі, креми, мазі, гелі і пластирі. Фармацевтичні композиції, згадані вище, демонструють даний винахід, але не обмежують його жодним чином. Серед фармацевтично прийнятних, інертних, нетоксичних наповнювачів або носіїв можуть бути згадані, з метою прикладу і без будь-якого обмеження, розріджувачі, розчинники, консерванти, змочувальні агенти, емульсифікатори, дисперсанти, зв'язувальні речовини, агенти, які викликають набухання, розпушувачі, уповільнювачі, абсорбенти, суспендуючі агенти, барвники, віддушки і т.д. Використовуване дозування змінюється в залежності від віку і ваги пацієнта, шляху введення, залученої фармацевтичної композиції, природи і тяжкості захворювання і призначення будь-яких пов'язаних лікувань. Дозування знаходиться в діапазоні від 0,1 мг до 1 г за одне або більше введень в день. Наступні приклади демонструють даний винахід, але жодним чином не обмежують його. Залучені вихідні матеріали являють собою відомі продукти або продукти, які одержують відповідно до відомих методик. Структури описаних у Прикладах сполук визначають відповідно до звичайних спектрофотометричних методик (інфрачервоний, ЯМР, масспектрометрія...). Наступні Приготування дають проміжні сполуки синтезу для застосування у приготуванні сполук за даним винаходом. Проміжна сполука 1: 5-(Бензолокси)-5оксопентанова кислота -2 Глутаровий ангідрид (8,76x10 моль) розчиняють в дихлорметані (300 мл) і піддають перемішуванню. Додають 4-диметиламінопіридин -2 -2 (7,88x10 моль) і бензиловий спирт (7,88x10 моль) і потім залишають реакційну суміш при температурі навколишнього середовища. Через 4 години 30 хвилин суміш гідролізують 5% водним розчином карбонату натрію (200 мл). Дві фази розділяють декантуванням. Потім водну фазу ацилюють водним 1М розчином хлористоводневої кислоти і пізніше екстрагують етилацетатом. Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі твердої речовини білого кольору, яку використовують без подальшого очищення. Проміжна сполука 2: Бензил хлорметил пентандіоат -2 Проміжну сполуку 1 (2,24x10 моль) розчиняють в дихлорметані (75 мл), додають воду (75 мл) і потім охолоджують до 0°С при енергійному перемішуванні. Пізніше, додають гідрокарбонат натрію -2 (8,91x10 моль) і сульфат тетрабутиламонію -3 (2,24x10 моль). Через 15 хвилин краплями дода-2 ють розчин хлорметил хлорсульфату (2,70x10 моль) в дихлорметані (20 мл). Реакційну суміш 13 енергійно перемішують при 0°С протягом 1 години і потім дають повернутись до температури навколишнього середовища. Через 1 годину 30 хвилин дві фази розділяють декантуванням. Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і потім випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі безбарвної олії, яку використовують без подальшого очищення. Проміжна сполука 3: Бензил йодметил пентандіоат -2 Йодид натрію (2,70x10 моль) розчиняють в ацетоні (100 мл). Піддають перемішуванню і потім -2 додають розчин проміжної сполуки 2 (2,25x10 моль) в ацетоні (40 мл). Реакційну суміш нагрівають при 45°С. Через 48 годин реакційну суміш фільтрують. Твердий залишок промивають ацетоном і потім фільтрат випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі в'язкого залишку коричневого кольору, який використовують без подальшого очищення. Проміжна сполука 4: 4-(Бензилокси)-4оксобутанова кислота Янтарний ангідрид (0,10 моль) розчиняють в дихлорметані (300 мл) і піддають перемішуванню. -2 Додають 4-диметиламінопіридин (8,3x10 моль) і -2 бензиловий спирт (8,3x10 моль) і потім реакційну суміш залишають при температурі навколишнього середовища. Через 1 годину 30 хвилин суміш гідролізують 5% водним розчином карбонату натрію (200 мл). Дві фази розділяють декантуванням. Потім водну фазу ацилюють водним 1М розчином хлористоводневої кислоти і пізніше екстрагують етилацетатом. Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі твердої речовини білого кольору, яку використовують без подальшого очищення. Проміжна сполука 5: Бензил хлорметил бутандіоат -3 Проміжну сполуку 4 (4,80x10 моль) розчиняють в дихлорметані (15 мл), додають воду (15 мл) і потім охолоджують до 0°С при енергійному перемішуванні. Пізніше, додають гідрокарбонат натрію -2 (1,90x10 моль) і сульфат тетрабутиламонію -4 (4,80x10 моль). Через 15 хвилин краплями дода-3 ють розчин хлорметил хлорсульфату (5,76x10 моль) в дихлорметані (4 мл). Реакційну суміш енергійно перемішують при 0°С протягом 1 години і потім дають повернутись до температури навколишнього середовища. Через 5 годин 30 хвилин дві фази розділяють декантуванням. Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і потім випаровують досуха під зниженим тиском. Безбарвний маслянистий сирий продукт хроматографують на кварцевій колонці, використовуючи як елюент суміш 80/20 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі безбарвної олії. Проміжна сполука 6: Бензил йодметил бутандіоат 97035 14 -3 Йодид натрію (4,67x10 моль) розчиняють в ацетоні (18 мл). Піддають перемішуванню і потім -3 додають розчин проміжної сполуки 5 (3,89x10 моль) в ацетоні (7,5 мл). Реакційну суміш нагрівають при 45°С. Через 6 годин реакційну суміш фільтрують. Твердий залишок промивають ацетоном і потім фільтрат випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі в'язкого залишку коричневого кольору, який використовують без подальшого очищення. Проміжна сполука 7: Метил 2(бромметил)бензоат -2 Метил 2-метилбензоат (3,33x10 моль) розчиняють в хлороформі (30 мл). Додають N-2 бромсукцинімід (3,50x10 моль), 2,2'-азобіс(2-4 метилпропіонітрил) (3,65x10 моль) і нагрівають поступово до 65°С, щоб ініціювати вільнорадикальну реакцію. Пізніше, реакційну суміш нагрівають зі зворотним холодильником протягом 5 годин. Суміш потім охолоджують до температури навколишнього середовища. Осад, який утворився, відфільтровують. Фільтрат випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі олії оранжевого кольору, яку використовують без подальшого очищення. Проміжна сполука 8: 4-Хлор-N-[(2R)-2-метил2,3-дигідро-1Н-індол-1-іл]-3-сульфамоїл бензамід Вказаний у заголовку продукт одержують шляхом розділення 3-(аміносульфоніл)-4-хлор-N-(2метил-2,3-дигідро-1Н-індол-1-іл)бензаміду, яке проводять за допомогою препаративної хіральної хроматографії. Проміжна сполука 9: Метил 4-(2брометил)бензоат -2 4-(2-Брометил)бензойну кислоту (1,31x10 моль) суспендують в метанолі (40 мл). Суміш піддають перемішуванню, додають сірчану кислоту (1 мл) і нагрівають при 60°С. Через приблизно 10 хвилин суміш стає відмінно прозорою. Після нагрівання протягом 2 годин реакцію зупиняють. Температурі суміші дають повернутись до температури навколишнього середовища, а потім виливають її у воду (100 мл). Екстрагують етилацетатом (2x100 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і потім випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі безбарвної олії, яку використовують без подальшого очищення. Проміжна сполука 10: Метил 4-(1брометил)бензоат -3 4-(1-Брометил)бензойну кислоту (2,18x10 моль) розчиняють в метанолі (12 мл). Суміш піддають перемішуванню і потім повільно додають сірчану кислоту (0,5 мл). Після перемішування протягом ночі при температурі навколишнього реакційну суміш виливають у воду (50 мл). Екстрагують дихлорметаном (3x30 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і потім випаровують досуха під зниженим тиском. 15 Вказаний у заголовку продукт одержують у формі безбарвної олії, яку використовують без подальшого очищення. Приклад 1: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5[(2-метил-5-нітро-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат Стадія А: трет-Бутил [(2-хлор)-5-{[(2-метил-2,3дигідро-1Н-індол-1іл)аміно]карбоніл}феніл)сульфоніл]карбамат 3-(Аміносульфоніл)-4-хлор-N-(2-метил-2,3-2 дигідро-1H-1-іл)бензамід (1,37x10 моль) суспендують у дихлорметані (125 мл) і піддають енергійному перемішуванню. Додають триетиламін -2 -3 (1,50x10 моль), 4-диметиламінопіридин (1,36x10 моль) і потім розчин ди-трет-бутил дикарбонату -2 (1,57x10 моль) в дихлорметані (50 мл). Спостерігають виділення газу і швидко реакційна суміш стає відмінно прозорою. Через 1 годину 30 хвилин реакційну суміш концентрують досуха, використовуючи роторний випарник. Одержаний залишок додають в етилацетат (50 мл) і потім промивають водою (200 мл). Водну фазу ацилюють 1Ν розчином хлористоводневої кислоти у воді і потім екстрагують дихлорметаном. Органічну фазу промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском для одержання вказаного у заголовку продукту у формі твердої речовини лимонно-жовтого кольору, яку використовують без подальшого очищення. Стадія В: Метил 3-({(трет-бутоксикарбоніл)[(2хлор-5-{[(2-метил-2,3-дигідро-1Н-індол-1іл)аміно]карбоніл}феніл)сульфоніл]аміно}метил)бе нзоат -3 Сполуку, одержану на Стадії А, (1,29x10 моль) розчиняють в ацетонітрилі (6 мл), додають -3 діізопропілетиламін (1,54x10 моль) і піддають перемішуванню. Додають метил-3-3 бромметилбензоат (1,41x10 моль) і нагрівають реакційну суміш при 80°С. Після нагрівання протягом 3 годин суміш випаровують досуха, використовуючи роторний випарник. Одержаний сирий продукт хроматографують на кварцевій колонці, використовуючи як елюент суміш 90/10, потім 75/25 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору. Стадія С: 3-({(трет-Бутоксикарбоніл)[(2-хлор-5{[(2-метил-2,3-дигідро-1Н-індол-1іл)аміно]карбоніл}феніл)сульфоніл]аміно}метил)бе нзойна кислота -3 Сполуку, одержану на Стадії В, (1,14x10 моль) розчиняють в суміші ацетонітрил/вода (40 -2 мл/8 мл). Додають гідроксид літію (1,14x10 моль) і нагрівають при 50°С з перемішуванням. Після нагрівання протягом 2 годин реакційну суміш виливають у воду (100 мл). Екстрагують 3 рази етилацетатом (50 мл). Органічну фазу потім промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Після сушіння під вакуумною установкою вказаний у заголовку продукт одержують у формі тве 97035 16 рдої речовини коричневого кольору і використовують її без подальшого очищення. Стадія D: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 3-({(третбутоксикарбоніо)[(2-хлор-5-{[(2-метил-5-нітро-2,3дигідро-1Н-індол-1іл)аміно]карбоніл}феніл)сульфоніл]аміно}метил}бе нзоат -4 Сполуку, одержану на Стадії С, (8,41x10 моль) розчиняють в диметилформаміді (5 мл). Піддають перемішуванню під азотом, а потім додають розчин N-[(Z)-(хлорметокси)-NNО-азокси]-N-4 етилетанаміну (9,25x10 моль) в диметилформаміді (4 мл). Додають карбонат цезію одразу -4 (8,41x10 моль) і залишають реакційну суміш при температурі навколишнього середовища з перемішуванням. Через 2 години суміш виливають у воду (50 мл). Екстрагують 3 рази з етилацетатом (20 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і концентрують під зниженим тиском. Одержаний маслянистий сирий продукт оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 65/35, потім 50/50 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору. Стадія Е: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5[(2-метил-5-нітро-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат -4 Сполуку, одержану на Стадії D, (4,38x10 моль) розчиняють в діоксані (25 мл). Піддають перемішуванню і потім додають 37% хлористоводневу кислоту (3,5 мл) і нагрівають при 70°С. Через 2 години реакційну суміш виливають у воду (50 мл). Екстрагують 3 рази етилацетатом (20 мл). Органічну фазу промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і концентрують, використовуючи роторний випарник. Одержаний маслянистий сирий продукт оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 60/40 n-гептан/етилацетат. Після сушіння під вакуумною установкою при 60°С вказаний у заголовку продукт одержують у формі твердої речовини злегка жовтого кольору. Точка плавлення: 85-86°С Елементний мікроаналіз: С Η Ν S % теоретично: 50,47 4,67 14,21 4,65 % експериментально: 51,14 5,04 13,19 4,55 Приклад 2: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат Стадія А: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 3-({(третбутоксикарбоніл)[(2-хлор-5-{[(2-метил-2,3-дигідро1Н-індол-1іл)аміно]карбамоїл}феніл)сульфоніл]аміно}метил) бензоат 17 Продукт, одержаний на Стадії С Прикладу 1, піддають реакції з N-[(Z)-(хлорметокси)-NNOазокси]-N-етилетанаміну відповідно до умов способу, розкритих на Стадії D Прикладу 1. Вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору після хроматографії на кварцевій колонці, використовуючи як елюент суміш 65/35 n-гептан/етилацетат. Стадія В: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат Методика є ідентичною тій, що розкрита на Стадії Ε Прикладу 1, починаючи зі сполуки, одержаної на Стадії А. Вказаний у заголовку продукт одержують у формі твердої речовини білуватого кольору після хроматографії на кварцевій колонці, використовуючи як елюент суміш 60/40 nгептан/етилацетат. Пізніше її висушують під вакуумною установкою при 60°С протягом 24 годин. Точка плавлення: 66-67°С Елементний мікроаналіз: С Η Ν S % теоретично: 53,99 5,16 13,03 4,97 % експериментально: 54,76 5,50 12,20 4,98 Приклад 3: трет-Бутил ({2-хлор-5-[(2-метил2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)[({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил]карбамат Продукт, одержаний на Стадії А Прикладу 1, -3 (1,00x10 моль) розчиняють в ацетонітрилі (5 мл). Розчин піддають перемішуванню. Додають діізоп-3 ропілетиламін (1,20x10 моль) і потім розчин Ν[(Ζ)-хлорметокси)-ΝΝΟ-азокси]-N-етилетанаміну -3 (1,10x10 моль) в ацетонітрилі (2 мл). Реакційну суміш нагрівають при 70°С протягом 2 годин 30 хвилин. Виливають реакційну суміш у воду (20 мл) і екстрагують 3 рази етилацетатом (15 мл). Органічну фазу промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і концентрують досуха, використовуючи роторний випарник. Одержаний маслянистий сирий продукт оранжевого кольору хроматографують на колонці силікагелю, використовуючи як елюент суміш 80/20 n-гептан/етилацетат. Тверду речовину жовтуватого кольору відновлюють. Вказаний у заголовку продукт одержують у формі твердої речовини оранжевого кольору після гель-фітльтрації через Sephadex LH-20, використовуючи як елюент суміш 1/1 ацетон/дихлорметан. Точка плавлення: 80-81°С Елементний мікроаналіз: С Η Ν S % теоретично: 51,10 5,77 13,75 5,25 % експериментально: 51,36 5,90 13,16 5,34 Приклад 4: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил N-(третбутоксикарбоніл)-N-({2-хлор-5-[(2-метил-2,3дигідро-1H-індол-1-іл)карбамоїл]феніл}сульфоніл)гліцинат Стадія А: Бензил N-(трет-бутоксикарбоніл)-N[(2-хлор-5-{[(2-метил-2,3-дигідро-1H-індол-1іл)аміно]карбоніл}феніл)сульфоніл]гліцинат 97035 18 Сполуку, одержану на Стадії А Прикладу 1, піддають реакції з бензил бромацетатом відповідно до умов способу, розкритого на Стадії В Прикладу 1. Одержаний сирий продукт жовтуватого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 80/20 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини злегка жовтого кольору. Стадія В: N-(трет-Бутоксикарбоніл)-N-[(2-хлор5-{[(2-метил-2,3-дигідро-1H-індол-1іл)аміно]карбоніл}феніл)сульфоніл]гліцин -3 Сполуку, одержану на Стадії А, (1,34x10 моль) розчиняють в етилацетаті (50 мл). Додають каталізатор паладій-на-вуглеці (10%, 82 мг) і потім вміщують реакційну суміш під водень при температурі навколишнього середовища і атмосферному тиску. Через 24 години реакційну суміш фільтрують через целіт і потім фільтрат випаровують досуха під зниженим тиском. Одержаний маслянистий залишок оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент дихлорметан і потім суміш 98/2, 95/5 і 90/10 дихлорметан/метанол. Вказаний у заголовку продукт одержують у формі жовтуватої твердої речовини. Стадія С: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил N-(третбутоксикарбоніл)-N-({2-хлор-5-[(2-метил-2,3дигідро-1H-індол-1іл)карбамоїл]феніл}сульфоніл)гліцинат -3 Продукт, одержаний на Стадії В, (1,01x10 моль) розчиняють в диметилформаміді (5 мл). Піддають перемішуванню і додають розчин Ν-[(Ζ)(хлорметокси)-NNO-азокси]-N-етилетанаміну -3 (2,23x10 моль) в диметилформаміді (3 мл). Пізніше додають карбонат цезію за один раз -3 (1,01x10 моль). Через 2 години реакційну суміш виливають у воду (30 мл). Екстрагують 3 рази етилацетатом (20 мл). Органічну фазу промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і концентрують під зниженим тиском. Одержаний залишок хроматографують на кварцевій колонці, використовуючи як елюент суміш 80/20, а потім 70/30 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини злегка жовтого кольору. Точка плавлення: 76-77°С Елементний мікроаналіз: С Η Ν S % теоретично: 50,26 5,57 12,56 4,79 % експериментально: 50,34 5,74 12,36 4,65 Приклад 5: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил N-({2-хлор-5-[(2метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)гліцинат Сполуку, одержану на Стадії С Прикладу 4, -4 (4,06x10 моль) розчиняють у дихлорметані (12 мл). Розчин охолоджують до 0°С і піддають перемішуванню. Додають трифтороцтову кислоту -3 (4,06x10 моль). Через 1 годину реакційну суміш вміщують в умови температури навколишнього середовища. Через 3 дні суміш концентрують досуха, використовуючи роторний випарник. Віднов 19 лений маслянистий сирий продукт хроматографують на кварцевій колонці, використовуючи як елюент суміш 60/40, а потім 50/50 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору після гель-фільтрації через Sephadex LH-20, використовуючи як елюент суміш 1/1 ацетон/дихлорметан. Точка плавлення: 68-69°С Елементний мікроаналіз: С Η Ν S % теоретично: 48,55 5,14 14,77 5,64 % експериментально: 48,16 5,24 14,17 5,46 Приклад 6: [(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1H-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил ({[(1Z)2,2-діетил-1-оксидогідразоно]аміно}окси)метил пентандіоат Стадія А: Бензил [(трет-Бутоксикарбоніл)({2хлор-5-[(2-метил-2,3-дигідро-1H-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил пентандіоат Сполуку, одержану на Стадії А Прикладу 1, -3 (2,15x10 моль) розчиняють в ацетонітрилі (10 мл) і розчин піддають перемішуванню. Додають діізоп-3 ропілетиламін (2,57x10 моль) і потім розчин про-3 міжної сполуки 3 (2,36x10 моль) в ацетонітрилі (10 мл). Протягом додавання реакційна суміш стає червонуватого кольору і потім швидко стає безбарвною. Суміш нагрівають при 60°С. Через 1 годину 30 хвилин реакційну суміш випаровують досуха під зниженим тиском. Одержаний твердий залишок жовтого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 80/20, а потім 70/30 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору. Стадія В: 5-{[(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метокси}-5оксопентанова кислота -3 Сполуку, одержану на Стадії А, (1,45x10 моль) розчиняють в етилацетаті (100 мл). Додають каталізатор паладій-на-вуглеці (10%, 100 мг) і потім при температурі навколишнього середовища реакційну суміш вміщують під водень при атмосферному тиску. Через 36 годин реакційну суміш фільтрують через целіт і знову додають новий каталізатор паладій-на-вуглеці (10%, 100 мг). Суміш знову вміщують під водень, в тих же умовах, що і описані вище, на додаткові 24 години. Реакцію потім зупиняють, суміш фільтрують через целіт і фільтрат випаровують досуха під зниженим тиском. Одержаний залишок хроматографують на кварцевій колонці, використовуючи як елюент суміш 98/2, а потім 97/3 дихлорметан/метанол. Вказаний у заголовку продукт одержують у формі твердої речовини кремового кольору. Стадія С: [(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1H-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил ({[(1Z)2,2-діетил-1-оксидогідразоно]аміно}окси)метил пентандіоат -4 Сполуку, одержану на Стадії В, (9,23x10 моль) розчиняють в диметилформаміді (10 мл). 97035 20 Піддають перемішуванню під азотом, і додають розчин N-[(Z)-(хлорметокси)-NNO-азокси]-N-3 етилетанаміну (1,11x10 моль) в диметилформаміді (3 мл). Пізніше додають карбонат цезію за -4 один раз (9,23x10 моль). Через 1 годину 30 хвилин реакційну суміш виливають у воду (100 мл). Екстрагують 3 рази етилацетатом (75 мл). Органічну фазу промивають водою і потім розсолом. Пізніше її висушують над сульфатом магнію, фільтрують і потім випаровують досуха під зниженим тиском. Одержаний маслянистий залишок оранжевого кольору очищують за допомогою хроматографії на кварцевій колонці, використовуючи як елюент суміш 70/30, а потім 60/40 nгептан/етилацетат. Є необхідною друга хроматографія, обернена фаза, на колонці Lichroprep RP18. Умови елюювання є наступними: (елюент А: 1000 мл Н2О/25мл CH3CN; елюент В: 25 мл Н2О/1000 мл CH3CN) 10% В протягом 15 хвилин, від 10 до 75% В за 60 хвилин, від 75 до 100% В за 20 хвилин. Вказаний у заголовку продукт одержують у формі меренги злегка жовтуватого кольору. Елементний мікроаналіз: С Η Ν S % теоретично: 50,89 5,74 11,13 4,25 % експериментально: 50,83 5,74 11,16 3,99 Приклад 7: [(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1H-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил ({[(1Z)2,2-діетил-1-оксидогідразоно]аміно}окси)метил бутандіоат Стадія А: Бензил [(трет-Бутоксикарбоніл)({2хлор-5-[(2-метил-2,3-дигідро-1H-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил бутандіоат Сполуку, одержану на Стадії А Прикладу 1, -3 (1,50x10 моль) розчиняють в ацетонітрилі (7 мл) і розчин піддають перемішуванню. Додають діізоп-3 ропілетиламін (1,80x10 моль) і потім розчин про-3 міжної сполуки 6 (1,65x10 моль) в ацетонітрилі (10 мл). Протягом додавання реакційна суміш стає червонуватого кольору і потім швидко стає безбарвною. Суміш нагрівають при 55°С. Через 1 годину 30 хвилин реакційну суміш випаровують досуха під зниженим тиском. Одержаний твердий залишок лимонно-жовтого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 80/20, 70/30, а потім 60/40 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги жовтого кольору. Стадія В: 4-{[(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метокси}-4оксобутанова кислота -3 Сполуку, одержану на Стадії А, (1,08x10 моль) розчиняють в етилацетаті (75 мл). Додають каталізатор паладій-на-вуглеці (10%, 75 мг) і потім при температурі навколишнього середовища реакційну суміш вміщують під водень при атмосферному тиску. Через 36 годин реакційну суміш фільтрують через целіт і знову додають новий каталізатор паладій-на-вуглеці (10%, 75 мг). Суміш знову вміщують під водень, в тих же умовах, що і 21 описані вище, на додаткові 24 години. Реакцію потім зупиняють, суміш фільтрують через целіт і фільтрат випаровують досуха під зниженим тиском. Одержаний залишок хроматографують на кварцевій колонці, використовуючи як елюент суміш 98/2 дихлорметан/метанол. Вказаний у заголовку продукт одержують у формі твердої речовини кремового кольору. Стадія С: [(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1H-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил ({[(1Z)2,2-діетил-1-оксидогідразоно]аміно}окси)метил бутандіоат -4 Сполуку, одержану на Стадії В, (8,30x10 моль) розчиняють в диметилформаміді (9 мл). Піддають перемішуванню під азотом, і додають розчин N-[(7)-(хлорметокси)-NNO-азокси]-N-4 етилетанаміну (9,96x10 моль) в диметилформаміді (3 мл). Пізніше додають карбонат цезію за -4 один раз (8,30x10 моль). Через 1 годину реакційну суміш виливають у воду (100 мл). Екстрагують 3 рази етилацетатом (75 мл). Органічну фазу промивають водою і потім розсолом. Пізніше її висушують над сульфатом магнію, фільтрують і потім випаровують досуха під зниженим тиском. Одержаний маслянистий залишок оранжевого кольору очищують за допомогою хроматографії на кварцевій колонці, використовуючи як елюент суміш 70/30, а потім 60/40 n-гептан/етилацетат. Є необхідною друга хроматографія, обернена фаза, на колонці Lichroprep RP-18 (60x400 мм). Умови елюювання є наступними: (елюент А: 1000 мл Н2О/25мл CH3CN; елюент В: 25 мл Н2О/1000 мл CH3CN) А/В: 40/60, швидкість потоку: 12 мл/хв. Вказаний у заголовку продукт одержують у формі меренги оранжевого кольору. Елементний мікроаналіз: С Η Ν S % теоретично: 50,23 5,58 11,34 4,33 % експериментально: 50,51 5,67 10,91 4,48 Приклад 8: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат Стадія А: Метил 4-{[(трет-бутоксикарбоніл)({2хлор-5-[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат Сполуку, одержану на Стадії А Прикладу 1, -3 (1,72x10 моль) розчиняють в ацетонітрилі (8 мл) і розчин піддають перемішуванню. Додають діізоп-3 ропілетиламін (2,06x10 моль) і потім, після перемішування протягом 10 хвилин, метил 4-3 бромметилбензоат (2,57x10 моль). Суміш нагрівають при 60°С. Через 2 години 30 хвилин реакційну суміш виливають у воду (30 мл). Екстрагують 3 рази етилацетатом (20 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Одержаний маслянистий залишок оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 90/10 до 75/25 n-гептан/етилацетат. 97035 22 Вказаний у заголовку продукт одержують у формі меренги жовтого кольору. Стадія В: 4-{[(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензой на кислота -3 Сполуку, одержану на Стадії А, (1,30x10 моль) розчиняють у суміші ацетонітрилу (45 мл) і -2 води (9 мл). Додають гідроксид літію (1,30x10 моль) і нагрівають при 50°С при перемішуванні. Після нагрівання протягом 5 годин темнокоричневу суміш виливають у воду (100 мл). Екстрагують 3 рази етилацетатом (50 мл). Органічну фазу потім промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Після висушування під вакуумною установкою вказаний у заголовку продукт одержують у формі меренги коричневого кольору, яку використовують без подальшого очищення. Стадія С: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат -4 Сполуку, одержану на Стадії В, (8,96x10 моль) розчиняють в диметилформаміді (8 мл). Піддають перемішуванню під азотом, і потім додають розчин N-[(Z)-(хлорметокси)-NNO-азокси]-N-3 етилетанаміну (1,17x10 моль) в диметилформаміді (1 мл). Додають карбонат цезію за один раз -4 (9,41x10 моль) і реакційну суміш залишають при температурі навколишнього середовища при перемішуванні. Через 2 години реакційну суміш виливають у воду (50 мл). Екстрагують 3 рази етилацетатом (20 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і концентрують під зниженим тиском. Одержаний маслянистий сирий продукт оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 65/35 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору. Стадія D: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат -4 Сполуку, одержану на Стадії С, (4,74x10 моль) розчиняють в 1,4-діоксані (5 мл). Розчин піддають перемішуванню при температурі навколишнього середовища і потім додають 10 мл 4Ν розчину НСІ в 1,4-діоксані. Спочатку жовтувата реакційна суміш стає темніше і темніше доти, доки вона не стане практично чорною. Після перемішування протягом 2 днів при температурі навколишнього середовища реакційну суміш випаровують досуха під зниженим тиском. Одержаний залишок чорного кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 50/50 nгептан/етилацетат. 23 Після перекристалізації з n-гептану вказаний у заголовку продукт одержують у формі твердої речовини білого кольору. Точка плавлення: 96-97°С Елементний мікроаналіз: С Η Ν S % теоретично: 53,99 5,16 13,03 4,97 % експериментально: 54,24 5,27 12,65 4,60 Приклад 9: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 2-{[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат Стадія А: Метил 2-{[(трет-бутоксикарбоніл)({2хлор-5-[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат Сполуку, одержану на Стадії А Прикладу 1, -3 (1,72x10 моль) розчиняють в ацетонітрилі (8 мл) і розчин піддають перемішуванню. Додають діізоп-3 ропілетиламін (2,06x10 моль) і потім, після перемішування протягом 10 хвилин, проміжну сполуку -3 7 (2,62x10 моль). Суміш нагрівають при 60°С. Через 5 годин реакційну суміш виливають у воду (30 мл). Екстрагують 3 рази етилацетатом (20 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Одержаний маслянистий залишок оранжево-коричневого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 90/10 до 65/35 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги жовтого кольору. Стадія В: 2-{[(трет-Бутоксикарбоніл)({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензой на кислота -3 Сполуку, одержану на Стадії А, (1,26x10 моль) розчиняють у суміші ацетонітрилу (45 мл) і -2 води (9 мл). Додають гідроксид літію (1,26x10 моль) і нагрівають при 50°С при перемішуванні. Після нагрівання протягом 5 годин 30 хвилин темно-коричневу реакційну суміш виливають у воду (100 мл). Екстрагують 3 рази етилацетатом (50 мл). Органічну фазу потім промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Після висушування під вакуумною установкою вказаний у заголовку продукт одержують у формі меренги коричневого кольору, яку використовують без подальшого очищення. Стадія С: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 2-{[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}бензоат -4 Сполуку, одержану на Стадії В, (8,25x10 моль) розчиняють в диметилформаміді (8 мл). Піддають перемішуванню під азотом, і потім додають розчин N-[(7)-(хлорметокси)-NNО-азокси]-N-3 етилетанаміну (1,07x10 моль) в диметилформа 97035 24 міді (2 мл). Додають карбонат цезію за один раз -4 (8,66x10 моль) і реакційну суміш залишають при температурі навколишнього середовища при перемішуванні. Через 3 години реакційну суміш виливають у воду (50 мл). Екстрагують 3 рази етилацетатом (20 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і концентрують під зниженим тиском. Одержаний маслянистий сирий продукт коричневого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 70/30 nгептан/етилацетат. Є необхідною друга хроматографія, обернена фаза, на колонці Lichroprep RP18. Умови елюювання є наступними: (елюент А: 1000 мл Н2О/25мл CH3CN; елюент В: 25 мл Н2О/1000 мл CH3CN) 20% В протягом 15 хвилин, від 20 до 83% В за 60 хвилин, від 83 до 100% В за 15 хвилин. Після перекристалізації з суміші nгептан/етилацетат вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору. Точка плавлення: 79-81°C Елементний мікроаналіз: С Η Ν S % теоретично: 54,80 5,54 11,28 4,30 % експериментально: 54,57 5,65 10,82 3,88 Приклад 10: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-({[(2-хлор-5{[(2R)-2-метил-2,3-дигідро-1Н-індол-1іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензоат Стадія А: трет-Бутил [(2-хлор-5-{[(2R)-2-метил2,3-дигідро-1Н-індол-1іл]карбамоїл}феніл)сульфоніл]карбамат -3 Проміжну сполуку 8 (6,0x10 моль) суспендують в дихлорметані (55 мл) і піддають енергійному -3 перемішуванню. Додають триетиламін (6,60x10 -4 моль), 4-диметиламінопіридин (6,0x10 моль) і -3 потім розчин ди-трет-бутил дикарбонату (6,90x10 моль) в дихлорметані (20 мл). Спостерігають виділення газу, і реакційна суміш швидко стає відмінно прозорою. Через одну годину 30 хвилин реакційну суміш виливають у воду (100 мл). Екстрагують 3 рази дихлорметаном (50 мл). Органічну фазу потім промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі твердої речовини лимонно-жовтого кольору, яку використовують без подальшого очищення. Стадія В: Метил 4-({(трет-бутоксикарбоніл)[(2хлор-5-{[(2R)-2-метил-2,3-дигідро-1Н-індол-1іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензоат -3 Сполуку, одержану на Стадії А, (7,57x10 моль) розчиняють в ацетонітрилі (35 мл) і розчин піддають перемішуванню. Додають діізопропіле-3 тиламін (9,09x10 моль) і потім, після перемішування протягом 10 хвилин, метил 4-4 бромметилбензоат (1,15x10 моль). Суміш нагрівають при 60°С. Через 1 ніч реакційну суміш виливають у воду (100 мл). Екстрагують 3 рази етилацетатом (50 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фі 25 льтрують і випаровують досуха під зниженим тиском. Одержаний залишок порошкують в суміші nгептан/етилацетат, доводячи до осаджування твердої речовини, яку відтворюють шляхом фільтрування. Вказаний у заголовку продукт одержують у формі твердої речовини білого кольору. Стадія С: 4-({(трет-Бутоксикарбоніл)[(2-хлор-5{[(2R)-2-метил-2,3-дигідро-1Н-індол-1іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензой на кислота -3 Сполуку, одержану на Стадії В, (5,03x10 моль) розчиняють у суміші ацетонітрилу (175 мл) і -2 води (35 мл). Додають гідроксид літію (5,03x10 моль) і нагрівають при 50°С при перемішуванні. Після нагрівання протягом 3 годин темнокоричневу реакційну суміш виливають у воду (200 мл). Екстрагують 3 рази етилацетатом (150 мл). Органічну фазу потім промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Одержаний маслянистий залишок порошкують в суміші n-гептан/етилацетат, доводячи до осаджування твердої речовини, яку відновлюють шляхом фільтрування. Вказаний у заголовку продукт одержують у формі твердої речовини кремового кольору. Стадія D: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-({(третбутоксикарбоніл)[(2-хлор-5-{[(2R)-2-метил-2,3дигідро-1Н-індол-1іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензоат -3 Сполуку, одержану на Стадії С, (2,90x10 моль) розчиняють в диметилформаміді (30 мл). Піддають перемішуванню під азотом і потім додають розчин N-[(Z)-(хлорметокси)-NNO-азокси]-N-3 етилетанаміну (4,88x10 моль) в диметилформаміді (3 мл). Додають карбонат цезію за один раз -3 (3,05x10 моль) і реакційну суміш залишають при температурі навколишнього середовища при перемішуванні. Через 3 години 30 хвилин реакційну суміш виливають у воду (200 мл). Екстрагують 3 рази етилацетатом (150 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і концентрують під зниженим тиском. Одержаний маслянистий сирий продукт оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 65/35 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини жовтого кольору. Стадія Е: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-({[(2-хлор-5{[(2R)-2-метил-2,3-дигідро-1Н-індол-1іл]карбамоїл}феніл)сульфоніл]аміно}метил)бензоат -3 Сполуку, одержану на Стадії D, (1,47x10 моль) розчиняють в 20 мл 4Ν розчину НСІ в 1,4діоксані і піддають перемішуванню. Спочатку жовтувата реакційна суміш стає темніше і темніше доти, доки вона не стане практично чорною. Після перемішування протягом 18 годин при температурі навколишнього середовища реакційну суміш випаровують досуха під зниженим тиском. Одержа 97035 26 ний залишок чорного кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 50/50 n-гептан/етилацетат. Після перекристалізації з n-гептану вказаний у заголовку продукт одержують у формі твердої речовини білого кольору. Точка плавлення: 78-79°С Елементний мікроаналіз: С Η Ν S % теоретично: 53,99 5,16 13,03 4,97 % експериментально: 53,77 4,94 12,90 5,06 Приклад 11: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил водень ({2хлор-5-[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)амідофосфат Стадія А: Діетил ({2-хлор-5-[(2-метил-2,3дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)амідофосфат У тригирлій колбі з двома додатковими ампулами розчиняють 3-(аміносульфоніл)-4-хлор-N-(2метил-2,3-дигідро-1H-індол-1-іл)бензамід -2 (1,37x10 моль) у водному 1Ν розчині гідроксиду натрію (25 мл). Одержаний розчин лимонножовтого кольору піддають перемішуванню при температурі навколишнього середовища і під азотом. Краплями додають розчин діетилхлорфосфа-2 ту (9,56x10 моль) в тетрагідрофурані (50 мл) і одночасно водний розчин 3Ν гідроксиду натрію (45 мл) таким чином, щоб підтримувати реакційну суміш відмінно гомогенною. Після закінчення додавання реакційну суміш залишають при температурі навколишнього середовища на 2 години. Реакційну суміш виливають у воду (200 мл) і екстрагують 3 рази етилацетатом. Водну фазу ацилюють до рН 4 і потім знову екстрагують етилацетатом. Органічні фази об'єднують, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі олії оранжевого кольору, яку використовують без подальшого очищення. Стадія В: ({2-Хлор-5-[(2-метил-2,3-дигідро-1Ніндол-1-іл)карбамоїл]феніл}сульфоніл)фосфорамідна кислота -2 Сполуку, одержану на Стадії А, (1,22x10 моль) розчиняють в дихлорметані (130 мл). Розчин піддають перемішуванню при 0°С і під азотом. Пізніше краплями додають триметилсиліл йодид -2 (5,41x10 моль). Після завершення додавання все залишають до повернення до температури навколишнього середовища, при перемішуванні. Через 18 годин реакційну суміш випаровують досуха під зниженим тиском. Залишок коричневого кольору додають у суміш 25мл/1 мл ацетон/вода, перемішують протягом 15 хвилин і потім випаровують знову досуха. Одержаний залишок коричневого кольору у формі меренги очищують хроматографією з оберненою фазою на колонці Lichroprep RP18. Умови елюювання є наступними: (елюент А: 1000 мл Н2О/25 мл CH3CN; елюент В: 25 мл Н2О/1000 мл CH3CN) 100% А; 95/5, а потім 90/10 А/В. Фракції, які містять очікуваний продукт, ліофілізують. 27 Вказаний у заголовку продукт одержують у формі пластівчастої твердої речовини жовтого кольору. Стадія С: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил водень ({2хлор-5-[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}-сульфоніл)амідофосфат -3 Сполуку, одержану на Стадії В, (3,81x10 моль) розчиняють у Ν,Ν'-диметил-N,N'полісечовині (25 мл). Додають триетиламін -3 (8,88x10 моль) і потім перемішують при температурі навколишнього середовища протягом 10 хвилин. Пізніше додають N-[(Z)-(хлорметокси)-NNО-3 азокси]-N-етилетанамiн (15,25x10 моль), йодид -3 натрію (7,62x10 моль) і реакційну суміш нагрівають при 50°С. Через одну ніч реакційну суміш виливають в діетиловий ефір (200 мл). Нерозчинний матеріал, поглинений в ацетонітрилі, відфільтровують. Тверду речовину, що залишається у суспензії, відфільтровують і фільтрат випаровують досуха під зниженим тиском. Одержаний залишок очищують шляхом трьох послідовних хроматографій з оберненою фазою на колонці Lichroprep RP-18. Умови елюювання є наступними: (елюент А: 1000 мл Н2О/25 мл CH3CN; елюент В: 25 мл Н2О/1000 мл CH3CN) 5% В протягом 10 хвилин, від 5 до 75% В за 60 хвилин, від 75% до 100% за 10 хвилин. Фракції, які містять очікуваний продукт, ліофілізують. Вказаний у заголовку продукт одержують у формі пластівчастої твердої речовини жовтого кольору. Елементний мікроаналіз: С Η Ν S СІ % теоретично: 42,68 4,78 14,22 5,43 6,00 % експериментально: 42,58 5,24 13,20 5,13 5,62 Приклад 12: (({[(1Z)-2,2-Діетил-1-оксидогідразоно]аміно}окси)метил 4-{[({2-хлор-5-[(2-метил-2,3дигідро-1Н-індол-1-іл)карба-моїл]феніл}сульфоніл)аміно]метил}феніл)ацетат Стадія А: (4-{[(трет-Бутоксикарбоніл)({2-хлор5-[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}феніл) оцтова кислота Сполуку, одержану на Стадії А Прикладу 1, -3 (3,22x10 моль) розчиняють в ацетонітрилі (15 мл) і розчин піддають перемішуванню. Додають діізоп-3 ропілетиламін (3,86x10 моль) і потім 4-3 бромметилфенілоцтову кислоту (7,08x10 моль). Суміш нагрівають при 65°С. Через 3 години реакційну суміш випаровують досуха під зниженим тиском. Одержаний маслянистий залишок оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 60/40, а потім 50/50 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі твердої речовини білуватого кольору. Стадія В: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил (4-{[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}феніл) ацетат 97035 28 -3 Сполуку, одержану на Стадії А, (2,03x10 моль) розчиняють в диметилформаміді (10 мл). Піддають перемішуванню під азотом і потім додають розчин N-[(Z)-(хлорметокси)-NNO-азокси]-N-3 етилетанаміну (2,64x10 моль) в диметилформаміді (2 мл). Додають карбонат цезію за один раз -3 (2,13x10 моль) і реакційну суміш залишають при температурі навколишнього середовища при перемішуванні. Через 2 години реакційну суміш виливають у воду (60 мл). Екстрагують 3 рази етилацетатом (30 мл). Органічну фазу промивають розсолом, висушують над сульфатом магнію, фільтрують і концентрують під зниженим тиском. Одержаний маслянистий сирий продукт оранжевого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 65/35 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги жовтого кольору. Стадія С: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}феніл)ацетат -4 Сполуку, одержану на Стадії В, (8,86x10 моль) розчиняють в 1,4-діоксані (5 мл). Розчин піддають перемішуванню при температурі навколишнього середовища і потім додають 10 мл 4Ν розчину НСІ в 1,4-діоксані. Спочатку жовтувата реакційна суміш стає темніше і темніше доти, доки вона не стане практично чорною. Після перемішування протягом 36 годин при температурі навколишнього середовища реакційну суміш випаровують досуха під зниженим тиском. Одержаний залишок чорного кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 60/40 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги злегка жовтого кольору. С Η Ν S % теоретично: 54,66 5,35 12,75 4,86 % експериментально: 54,60 5,43 12,30 4,96 Приклад 13: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)-аміно]етил}бензоат Стадія А: Метил 4-{2-[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат Сполуку, одержану на Стадії А Прикладу 1, -2 (1,4x10 моль) розчиняють в ацетонітрилі (20 мл) і розчин піддають перемішуванню. Додають діізоп-2 ропілетиламін (1,24x10 моль) і потім, після перемішування протягом 10 хвилин, проміжну сполуку -2 9 (2,20x10 моль), розчинену в ацетонітрилі (20 мл). Суміш нагрівають при 60°С. Через 5 днів реакційну суміш випаровують досуха під зниженим тиском. Одержаний залишок хроматографують на кварцевій колонці, використовуючи як елюент суміш 75/25 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги блідо-жовтого кольору. Стадія В: 4-{2-[(трет-Бутоксикарбоніл)({2-хлор5-[(2-метил-2,3-дигідро-1Н-індол-1 29 іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензойн а кислота -4 Сполуку, одержану на Стадії А, (5,10x10 моль) розчиняють в суміші ацетонітрилу (18 мл) і -3 води (3 мл). Додають гідроксид літію (5,10x10 моль) і нагрівають при 55°С при перемішуванні. Після нагрівання протягом 4 годин реакційну суміш темно-коричневого кольору виливають у воду (100 мл). Екстрагують 3 рази етилацетатом (75 мл). Органічну фазу потім промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі меренги коричневого кольору, яку використовують без подальшого очищення. Стадія С: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{2-[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат -4 Сполуку, одержану на Стадії В, (4,0x10 моль) розчиняють в диметилформаміді (3 мл). Піддають перемішуванню під азотом і потім додають розчин N-[(7)-(хлорметокси)-NNO-азокси]-N-етилетанаміну -4 (5,21x10 моль) в диметилформаміді (2 мл). Пізніше додають карбонат цезію за один раз (4,20x10 4 моль). Через 1 годину реакційну суміш виливають у воду (50 мл). Екстрагують 3 рази етилацетатом (50 мл). Органічну фазу промивають водою, а потім розсолом. Пізніше її висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Одержаний маслянистий залишок коричневого кольору очищують за допомогою хроматографії на кварцевій колонці, використовуючи як елюент суміш 65/35 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги оранжевого кольору. Стадія D: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат -4 Сполуку, одержану на Стадії С, (2,17x10 моль) розчиняють в 1,4-діоксані (2 мл). Розчин піддають перемішуванню при температурі навколишнього середовища і потім додають 4 мл 4Ν розчину НСІ в 1,4-діоксані. Спочатку жовтувата реакційна суміш стає темніше і темніше доти, доки вона не стане практично чорною. Після перемішування протягом 40 годин при температурі навколишнього середовища реакційну суміш випаровують досуха під зниженим тиском. Одержаний залишок чорного кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 55/45 n-гептан/етилацетат. Є необхідною друга хроматографія, обернена фаза, на колонці Lichroprep RP-18. Умови елюювання є наступними: (елюент А: 1000 мл Н2О / 25мл CH3CN; елюент В: 25 мл Н2О/1000 мл CH3CN) 10% В протягом 20 хвилин, від 10 до 80% В за 70 хвилин, від 80 до 100% В за 15 хвилин. Вказаний у заголовку продукт одержують у формі меренги злегка жовтого кольору. С Η Ν S % теоретично: 54,66 5,35 12,75 4,86 97035 30 % експериментально: 54,34 5,43 12,46 4,72 Приклад 14: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{2-[({2-хлор-5[((2R)-2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат Починаючи з проміжної сполуки 8, вказаний у заголовку продукт одержують відповідно до методики, розкритої на Стадіях A-D Прикладу 13. Приклад 15: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{1-[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат Стадія А: Метил 4-{1-[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат Сполуку, одержану на Стадії А Прикладу 1, -3 (2,42x10 моль) розчиняють в ацетонітрилі (8 мл) і розчин піддають перемішуванню. Додають діізоп-3 ропілетиламін (2,90x10 моль) і потім, після перемішування протягом 10 хвилин, проміжну сполуку -3 10 (3,38x10 моль), розчинену в ацетонітрилі (2 мл). Суміш нагрівають при 60°С. Через 3 дні реакційну суміш випаровують досуха під зниженим тиском. Одержаний маслянистий залишок жовтого кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 75/25 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги блідо-жовтого кольору. Стадія В: 4-{1-[(трет-Бутоксикарбоніл)({2-хлор5-[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензойн а кислота -3 Сполуку, одержану на Стадії А, (1,58x10 моль) розчиняють в суміші ацетонітрилу (60 мл) і -2 води (10 мл). Додають гідроксид літію (1,58x10 моль) і нагрівають при 55°С при перемішуванні. Після нагрівання протягом 4 годин реакційну суміш темно-коричневого кольору виливають у воду (200 мл). Екстрагують 3 рази етилацетатом (100 мл). Органічну фазу потім промивають водою і потім розсолом, висушують над сульфатом магнію, фільтрують і випаровують досуха під зниженим тиском. Вказаний у заголовку продукт одержують у формі меренги коричневого кольору, яку використовують без подальшого очищення. Стадія С: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{1-[(третбутоксикарбоніл)({2-хлор-5-[(2-метил-2,3-дигідро1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат -3 Сполуку, одержану на Стадії В, (1,07x10 моль) розчиняють в диметилформаміді (6 мл). Піддають перемішуванню під азотом і потім додають розчин N-[(Z)-(хлорметокси)-NNO-азокси]-N-3 етилетанаміну (1,40x10 моль) в диметилформаміді (2 мл). Пізніше додають карбонат цезію за -3 один раз (1,13x10 моль). Через 1 годину реакційну суміш виливають у воду (50 мл). Екстрагують 3 рази етилацетатом (50 мл). Органічну фазу промивають водою, а потім розсолом. Пізніше її висушують над сульфатом магнію, фільтрують і потім випаровують досуха під зниженим тиском. 31 Одержаний маслянистий залишок коричневого кольору очищують за допомогою хроматографії на кварцевій колонці, використовуючи як елюент суміш 65/35 n-гептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги оранжевого кольору. Стадія D: ({[(1Z)-2,2-Діетил-1оксидогідразоно]аміно}окси)метил 4-{1-[({2-хлор-5[(2-метил-2,3-дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]етил}бензоат -4 Сполуку, одержану на Стадії С, (7,07x10 моль) розчиняють в 1,4-діоксані (7 мл). Розчин піддають перемішуванню при температурі навколишнього середовища і потім додають 15 мл 4Ν розчину НСІ в 1,4-діоксані. Спочатку жовтувата реакційна суміш стає темніше і темніше доти, доки вона не стане практично чорною. Після перемішування протягом 2 днів при температурі навколишнього середовища реакційну суміш випаровують досуха під зниженим тиском. Одержаний залишок чорного кольору хроматографують на кварцевій колонці, використовуючи як елюент суміш 60/40 nгептан/етилацетат. Вказаний у заголовку продукт одержують у формі меренги блідо-жовтого кольору. С Η Ν S % теоретично: 54,66 5,35 12,75 4,86 % експериментально: 54,36 5,46 12,28 4,71 Фармакологічне дослідження продуктів за даним винаходом Приклад А: Сечогінна дія In vivo Сечогінну дію досліджують на свідомих щурах Wistar. Тваринам не дають їжу протягом 18 годин перед експериментом і дають тільки воду за 90 хвилин до початку експерименту. Досліджуваний продукт потім вводять орально шляхом насильницького годування, щура вміщують в камеру для дослідження метаболізму і обсяг сечі вимірюють через 6 годин. Обсяг виражають по відношенню до обсягу, виміряного у контрольної групи щурів. Результати: збільшення обсягу сечі, одержаного при використанні досліджуваних продуктів, складає більше або дорівнює 20%. З метою прикладу, при оральному дозуванні 10 мг/кг сполуки Прикладів 1 і 10 збільшують виділення сечі на 108 і 248%, відповідно. Комп’ютерна верстка О. Гапоненко 97035 32 Приклад В: NO-донорна активність In vitro Використовують кільця аорти без ендотелію. Після першого скорочення, індукованого 60 мМ KСІ, щоб характеризувати сенситивність кільця, і промивання, стабільне скорочення індукують норадреналіном (0,1-0,3 мкМ) у присутності або за відсутності ODQ. Застосовують серію кумулятивних концентрацій і активність досліджуваного продукту розраховують через IC50 (доза, яка інгібує максимальний ефект на 50%). Результати: сполуки за даним винаходом мають досить значний ефект релаксанту зі значенням IC50 менше ніж 1 мкМ. З метою прикладу, сполуки Прикладів 1 і 10 мають значення ІС50 на рівні 0,12 і 0,04 мкМ, відповідно. In vivo NO-донорний ефект сполуки оцінюють шляхом зменшення тиску, викликаного у SD щурів, анестезованих пентобарбіталом. Після стабілізації артеріального тиску досліджуваний продукт вводять внутрішньовенно наростаючими дозами. Результати: зменшення артеріального тиску принаймні на 50% спостерігають для сполук за даним винаходом при дозуванні, меншому або яке дорівнює 0,3 мг/кг. З метою прикладу, при внутрішньовенному дозуванні 100 мкг/кг сполуки Прикладів 1 і 10 зменшують артеріальний тиск на 56 і 55%, відповідно. Приклад С: Фармацевтична композиція Формула приготування 1000 таблеток, кожна з яких містить дозу 100 мг ({[(1Z)-2,2-діетил-1оксидогідразоно]аміно}окси)метил 3-{[({2-хлор-5-[(2-метил-5-нітро-2,3100 г дигідро-1Н-індол-1іл)карбамоїл]феніл}сульфоніл)аміно]метил}-бензоату (Приклад 1) Гідроксипропілцелюлоза 2г Пшеничний крохмаль 10 г Лактоза 100 г Стеарат магнію 3г Тальк 3г Підписне Тираж 23 прим. Державна служба інтелектуальної власності України, вул. Урицького, 45, м. Київ, МСП, 03680, Україна ДП “Український інститут промислової власності”, вул. Глазунова, 1, м. Київ – 42, 01601
ДивитисяДодаткова інформація
Назва патенту англійськоюDiazeniumdiolate derivatives, method for preparing same and pharmaceutical compositions containing same
Автори англійськоюCordi, Alexis, Haberkorn, Laure, Verbeuren, Tony, Courchay, Christine, Simonet, Serge
Назва патенту російськоюСоединения диазенийдиолата, способ их получения и фармацевтическая композиция, которая их содержит
Автори російськоюКорди Алексис, Аберкорн Лор, Вербюрен Тони, Курше Кристин, Симоне Серж
МПК / Мітки
МПК: A61K 31/404, C07D 209/08
Мітки: композиція, містить, сполуки, фармацевтична, яка, одержання, спосіб, діазенійдіолату
Код посилання
<a href="https://ua.patents.su/16-97035-spoluki-diazenijjdiolatu-sposib-kh-oderzhannya-i-farmacevtichna-kompoziciya-yaka-kh-mistit.html" target="_blank" rel="follow" title="База патентів України">Сполуки діазенійдіолату, спосіб їх одержання і фармацевтична композиція, яка їх містить</a>
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Маррі Джеррі Ентоні, Зембровскі Уільям Джеймс, Міларі Банавара Лакшман, Чу-Моєр Маргарет Юхуа
МПК: C07D 491/20, C07D 401/04, A61P 43/00, A61P 9/10, A61K 31/506, C07D 403/04, C07D 409/12, A61K 31/5377, C07D 521/00, C07D 413/12, C07D 487/04, C07D 409/14, C07D 491/10, A61P 3/10, C07D 471/08, A61K 31/53, C07D 487/08, C07D 403/12, C07D 417/12, C07D 239/42, C07D 405/14, C07D 451/06, A61K 31/517, C07D 405/12, C07D 498/04, C07D 401/12, C07D 417/14, C07D 491/04, C07D 471/04, C07D 401/14, C07D 451/02, C07D 403/14, C07D 513/10, C07D 491/048
Мітки: фармацевтична, проміжної, містить, одержання, проміжні, спосіб, інгібітори, сполуки, піримідини, сорбітдегідрогенази, композиція
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...
Сполуки фенілпіридилпіперазину, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 87284
Опубліковано: 10.07.2009
Автори: Дезо Патріс, Корді Алексіс, Лєстаж П'єр
МПК: C07D 401/02, C07D 213/72, A61K 31/44
Мітки: фармацевтична, композиція, фенілпіридилпіперазину, спосіб, містить, яка, сполуки, одержання
Формула / Реферат:
1. Сполуки формули (І):, (І)в якій:X являє собою С(O) або SO2-групу,R1 являє собою:арильну групу,або групу NR3R4, в якій R3 і R4, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену (С1-С6)алкільну групу, (С3-С8)циклоалкільну групу або (С3-С8)циклоалкіл(С1-С6)алкільну групу, в якій...
Сполуки індолу, спосіб їх одержання і фармацевтична композиція (варіанти), яка їх містить

Номер патенту: 91646
Опубліковано: 10.08.2010
Автори: П'єсар Сільві, Маршан Паскаль, Бутан Жан Альбер, Одіно Валері, Дюфло Мюріель, Делагранж Філіпп, Кеняр Даніель-Анрі, Бабоно Вінсан
МПК: A61K 31/404, A61P 35/00, C07D 209/42, A61P 25/00, A61P 15/00, A61P 3/04
Мітки: фармацевтична, спосіб, індолу, містить, яка, варіанти, композиція, одержання, сполуки
Формула / Реферат:
1. Сполука формули (І):, (I) в якій: R1 являє собою лінійну або розгалужену (С1-С6)алкільну групу, лінійну або розгалужену (С3-С8)циклоалкільну групу або (С3-С8)циклоалкіл(С1-С6)алкільну групу, в якій алкільна частина може бути лінійною або розгалуженою, R2 являє собою лінійну або розгалужену...
Заміщені сполуки [1,4]бензодіоксино[2,3-e]ізоіндолу, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 79997
Опубліковано: 10.08.2007
Автори: Пьєр Ален, Кеньяр Даніель-Енрі, Ренар Пьєр, Леонс Стефан, Лепіфр Франк, Рутьє Сільвен, Ікман Джон, Ейерб Наталі, Кудер Жерар
МПК: C07D 471/14, C07D 498/14, C07D 491/14, C07D 491/147, C07D 487/04, A61P 35/00, C07D 491/22
Мітки: спосіб, одержання, фармацевтична, яка, сполуки, 1,4]бензодіоксино[2,3-e]ізоіндолу, містить, заміщені, композиція
Формула / Реферат:
1. Сполука формули (І): (I),де:А, разом з атомами вуглецю, з якими він зв’язаний, являє собою групу формули (a) або (b): (a) (b),де:W1, разом з атомами вуглецю, з якими він зв'язаний, являє...
Сполуки піролідину і тіазолідину, спосіб їх одержання і фармацевтична композиція, яка їх містить

Номер патенту: 84553
Опубліковано: 10.11.2008
Автори: Бенуат Ален, Арлей Елізабет, Де Нантей Гійом, Комбетт Мюріель
МПК: C07D 207/16, A61P 43/00, A61K 31/426, C07D 311/96, A61K 31/427, A61P 3/10, C07D 295/18, C07D 417/12, C07D 207/06, C07D 277/06, C07D 401/12, A61K 31/4025, A61K 31/40, A61K 31/438, C07D 405/12, C07D 409/12
Мітки: спосіб, сполуки, тіазолідину, піролідину, яка, містить, одержання, фармацевтична, композиція
Формула / Реферат:
1. Сполука формули (І):, (I) де:Х1 являє собою атом або групу, вибрану з CR4aR4b, О, S(O)q1 і NR5,де R4а і R4b, які можуть бути однаковими або відрізнятись, кожний являє собою атом водню або лінійну або розгалужену С1-С6алкільну групу,або R4a і R4b разом утворюють, з атомом вуглецю, який несе їх, С3-С7циклоалкільну групу,q1 являє...
Попередній патент: Спосіб і пристрій обробки розплавлених металів або сплавів при безперервному розливанні
Наступний патент: Похідні циклопропіламіну
Випадковий патент: Безступенева коробка передач