Похідні нуклеозиду як інгібітори рнк-залежної рнк вірусної полімерази
Номер патенту: 73843
Опубліковано: 15.09.2005
Автори: Пракаш Тхазха П., Бхат Балкрішен, Бхат Неєліма, Сонг Кванлай, Керролл Стівен С., Прхавк Маріджа, Кук Філліп Ден, Елдруп Енн Б., Олсен Девід Б., Маккосс Малкольм







Формула / Реферат
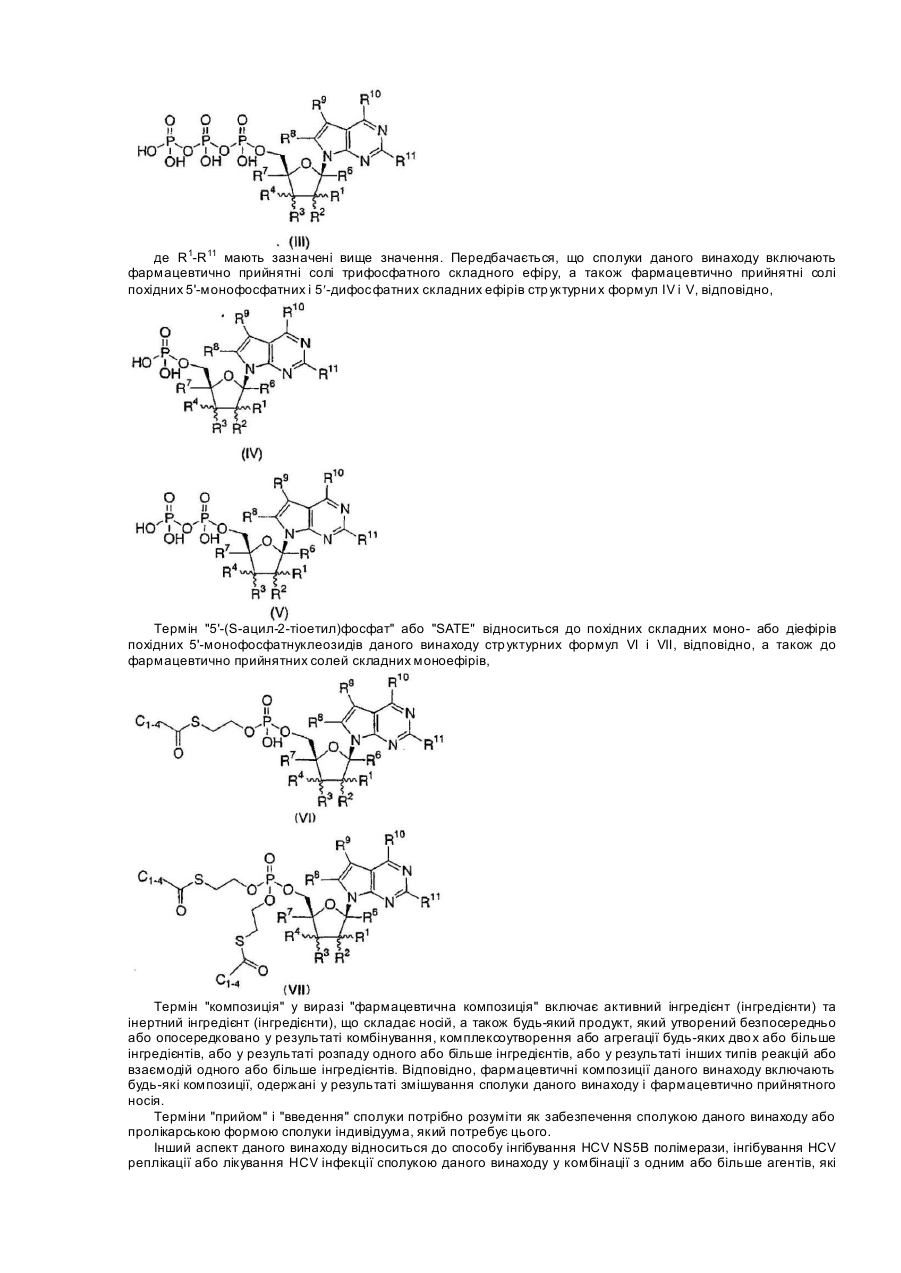
1. Сполука структурної формули I:
або її фармацевтично прийнятна сіль; де
R1 являє собою С2-4-алкеніл, С2-4-алкініл або С1-4-алкіл, де алкіл є незаміщеним або заміщений гідрокси, аміно, C1-4-алкокси, C1-4-алкілтіо або одним-трьома атомами фтору;
R2 являє собою водень, фтор, гідрокси, меркапто, C1-4-алкокси або С1-4-алкіл; або R1 і R2 разом з атомом вуглецю, до якого вони приєднані, утворюють 3-6-членну насичену моноциклічну кільцеву систему, яка необов'язково містить гетероатом, вибраний з О, S і NC0-4-алкілу;
R3 і R4 кожний незалежно вибирають з групи, яка складається з водню, ціано, азидо, галогену, гідрокси, меркапто, аміно, C1-4-алкокси, С2-4-алкенілу, С2-4-алкінілу і C1-4-алкілу, де алкіл є незаміщеним або заміщений гідрокси, аміно, C1-4-алкокси, C1-4-алкілтіо або одним-трьома атомами фтору;
R5 являє собою водень, C1-10-алкілкарбоніл, Р3О9Н4, Р2О6Н3 або P(О)R13R14;
R6 і R7 кожний незалежно являє собою водень, метил, гідроксиметил або фторметил;
R8 являє собою водень, C1-4-алкіл, С2-4-алкініл, галоген, ціано, карбокси, C1-4-алкілоксикарбоніл, азидо, аміно, C1-4-алкіламіно, ді(С1-4-алкіл)аміно, гідрокси, C1-6-алкокси, C1-6-алкілтіо, C1-6-алкілсульфоніл, (C1-4-алкіл)0-2-амінометил або С4-6-циклогетероалкіл, незаміщений або заміщений однією-двома групами, незалежно вибраними з галогену, гідрокси, аміно, C1-4-алкілу і С1-4-алкокси;
R9 являє собою водень, ціано, нітро, C1-3-алкіл, NHCONH2, CONR12R12, CSNR12R12, COOR12, C(=NH)NH2, гідрокси, C1-3-алкокси, аміно, С1-4-алкіламіно, ді(C1-4-алкіл)аміно, галоген, (1,3-оксазол-2-іл), (1,3-тіазол-2-іл) або (імідазол-2-іл); де алкіл є незаміщеним або заміщений однією-трьома групами, незалежно вибраними з галогену, аміно, гідрокси, карбокси і C1-3-алкокси;
R10 і R11 кожний незалежно являє собою водень, гідрокси, галоген, C1-4-алкокси, аміно, C1-4-алкіламіно, ді(С1-4-алкіл)аміно, С3-6-циклоалкіламіно, ди(С3-6-циклоалкіл)аміно або С4-6-циклогетероалкіл, незаміщений або заміщений однією-двома групами, незалежно вибраними з галогену, гідрокси, аміно, C1-4-алкілу і C1-4-алкокси;
кожний R12 незалежно являє собою водень або С1-6-алкіл; і
R13 і R14 кожний незалежно являє собою гідрокси, OCH2CH2SC(=О)С1-4-алкіл, ОСН2О(С=О)ОС1-4-алкіл, NHCHMeСО2Me, ОСН(C1-4-алкіл)О(С=О)С1-4-алкіл,
або
за умови, що якщо R1 являє собою β-метил і R4 являє собою водень, або R4 являє собою β-метил і R1 являє собою водень, R2 і R3 являють собою α-гідрокси, R10 являє собою аміно, і R5, R6, R7, R8 і R11 являють собою водень, тоді R9 не являє собою ціано або CONH2.
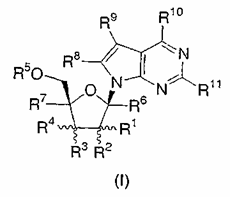
2. Сполука за п.1 структурної формули II:
або її фармацевтично прийнятна сіль; де
R1 являє собою C1-3-алкіл, де алкіл необов'язково заміщений гідрокси, аміно, C1-3-алкокси, C1-3-алкілтіо або одним-трьома атомами фтору;
R2 являє собою гідрокси, фтор або C1-4-алкокси;
R3 являє собою водень, галоген, гідрокси, аміно або C1-4-алкокси;
R5 являє собою водень, Р3О9Н4, Р2О6Н3 або РО3Н2;
R8 являє собою водень, аміно або C1-4-алкіламіно;
R9 являє собою водень, ціано, метил, галоген або CONH2; і
R10 і R11 кожний незалежно являє собою водень, галоген, гідрокси, аміно, С1-4-алкіламіно, ді(C1-4-алкіл)аміно або С3-6-циклоалкіламіно;
за умови, що якщо R1 являє собою β-метил, R2 і R3 являють собою α-гідрокси, R10 являє собою аміно, і R5, R8 і R11 являють собою водень, тоді R9 не являє собою ціано або CONH2.
3. Сполука за п. 2, де:
R1 являє собою метил, фторметил, гідроксиметил, дифторметил, трифторметил або амінометил;
R2 являє собою гідрокси, фтор або метокси;
R3 являє собою водень, фтор, гідрокси, аміно або метокси;
R5 являє собою водень або Р3O9Н4;
R8 являє собою водень або аміно;
R9 являє собою водень, ціано, метил, галоген або CONH2; і
R10 і R11 кожний незалежно являє собою водень, фтор, гідрокси або аміно;
за умови, що якщо R1 являє собою β-метил, R2 і R3 являють собою α-гідрокси, R10 являє собою аміно, і R5, R8 і R11 являють собою водень, тоді R9 не являє собою ціано або CONH2.
4. Сполука за п.1, вибрана з групи, яка складається з:
4-аміно-7-(2-С-метил-β-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-метиламіно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-диметиламіно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-циклопропіламіно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С-вініл-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С-гідроксиметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С-фторметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-5-метил-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-5-карбонової кислоти,
4-аміно-5-бром-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-5-хлор-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-5-фтор-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
2,4-діаміно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
2-аміно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
2-аміно-4-циклопропіламіно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
2-аміно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3H)-ону,
4-аміно-7-(2-С-етил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С,2-О-диметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3H)-ону,
2-аміно-5-метил-7-(2-C,2-O-диметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3H)-ону,
4-аміно-7-(3-дезокси-2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(3-дезокси-2-С-метил-β-D-арабiнофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-2-фтор-7-(2-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(3-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(3-C-метил-β-D-ксилофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2,4-ди-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину і
4-аміно-7-(3-дезокси-3-фтор-2-C-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
і відповідних їм 5'-трифосфатів;
або їх фармацевтично прийнятних солей.
5. Сполука за п.4, вибрана з групи, яка складається з:
4-аміно-7-(2-С-метил-β-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-7-(2-С-фторметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-5-метил-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-5-бром-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-5-хлор-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
4-аміно-5-фтор-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину і
4-аміно-7-(2-С,2-О-диметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину,
і відповідних їм 5'-трифосфатів;
або їх фармацевтично прийнятних солей.
6. Сполука за п. 5, яка є 4-аміно-7-(2-С-метил-β-D-арабiнофуранозил)-7Н-піроло[2,3-d]піримідином або його фармацевтично прийнятною сіллю.
7. Сполука за п. 5, яка є 4-аміно-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідином або його фармацевтично прийнятною сіллю.
8. Сполука за п. 5, яка є 4-аміно-7-(2-С-фторметил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідином або його фармацевтично прийнятною сіллю.
9. Сполука за п. 5, яка є 4-аміно-5-хлор-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідином або його фармацевтично прийнятною сіллю.
10. Сполука за п. 5, яка є 4-аміно-5-бром-7-(2-С-метил-β-D-рибофуранозил)-7Н-піроло[2,3-d]піримідином або його фармацевтично прийнятною сіллю.
11. Сполука за п. 1 для інгібування РНК-залежної РНК вірусної полімерази або інгібування РНК-залежної РНК вірусної реплікації у ссавців.
12. Сполука за п. 1 як активний агент для лікування РНК-залежної РНК вірусної інфекції у ссавців.
13. Сполука за п. 12, де зазначеною РНК-залежною РНК вірусною інфекцією є інфекція гепатиту С.
14. Сполука за п. 1 для виготовлення ліків для інгібування РНК-залежної РНК вірусної полімерази або для інгібування РНК-залежної РНК вірусної реплікації у ссавців.
15. Сполука за п. 1 для виготовлення ліків для інгібування РНК-залежної РНК вірусної інфекції у ссавців.
16. Сполука за п. 15, де зазначеною РНК-залежною РНК вірусною інфекцією є інфекція гепатиту С.
17. Фармацевтична композиція, що містить сполуку за п. 1 і фармацевтично прийнятний носій.
18. Фармацевтична композиція за п. 17, призначена для інгібування РНК-залежної РНК вірусної полімерази, інгібування РНК-залежної РНК реплікації і/або для лікування РНК-залежної РНК вірусної інфекції.
19. Фармацевтична композиція за п. 18, де зазначена РНК-залежна РНК вірусна полімераза є HCV NS5B полімеразою, зазначена РНК-залежна РНК вірусна реплікація є HCV реплікацією і зазначена РНК-залежна РНК вірусна інфекція є HCV інфекцією.
20. Спосіб інгібування РНК-залежної РНК вірусної полімерази і/або інгібування РНК-залежної РНК вірусної реплікації, при якому вводять ссавцю, який потребує такого інгібування, ефективну кількість сполуки за п. 1.
21. Спосіб за п. 20, де зазначена РНК-залежна РНК вірусна полімераза є HCV NS5B полімеразою і зазначена РНК-залежна РНК вірусна реплікація є HCV вірусною реплікацією.
22. Спосіб лікування РНК-залежної РНК вірусної інфекції, при якому вводять ссавцю, який потребує такого лікування, ефективну кількість сполуки за п. 1.
23. Спосіб за п. 22, де зазначена РНК-залежна РНК вірусна інфекція є HCV інфекцією.
24. Спосіб за п. 23, при якому вводять сполуку за п. 1 у комбінації з терапевтично ефективною кількістю іншого агента, активного проти HCV.
25. Спосіб за п. 24, де зазначений агент, активний проти HCV, є рибавірином, левовірином, тимозином альфа-1; інгібітором NS3 серинпротеази; інгібітором інозин монофосфатдегідрогенази; інтерфероном-α або пегільованим інтерфероном-α, одним або у комбінації з рибавірином або левовірином.
26. Спосіб за п. 25, де зазначеним агентом, активним проти HCV, є інтерферон-α або пегільований інтерферон-α, один або у комбінації з рибавірином.
Текст
Даний винахід відноситься до нуклеозидних сполук і деяких їх похідних, до їх одержання та до їх використання як інгібіторів РНК-залежної РНК вірусної полімерази. Сполуки даного винаходу є інгібіторами РНК-залежної РНК вірусної реплікації, і можуть бути використані для лікування РНК-залежної РНК вірусної інфекції. Вони особливо корисні як інгібітори (HCV) NS5B полімерази вірусу гепатиту С, як інгібітори реплікації HCV, і для лікування інфекції гепатиту С. Інфекція вірусу гепатиту С (HCV) є основною проблемою для здоров'я, яка приводить до хронічних захворювань печінки, таких як цироз і гепатоклітинна карцинома, у значного числа інфікованих індивідуумів, число яких оцінюється як 2-15% населення усього світу. Тільки у Сполучених Штатах діагностовано 4,5 мільйони інфікованих людей, за даними Центру Охорони Здоров'я США. Відповідно до даних Всесвітньої Організації Здоров'я у всьому світі інфіковано більше 200 мільйонів чоловік, причому, щонайменше, від 3 до 4 мільйонів людей інфікується щорічно. Після інфікування близько 20% людей позбуваються вірусу, але інші є носіями вірусу HCV до кінця свого життя. У десяти-двадцяти відсотків хронічно інфікованих індивідуумів, вкінці кінців, розвивається цироз, що руйнує печінку, або рак печінки. Вірусне захворювання переноситься парентерально через заражену кров і продукти, які включають кров, забруднені голки або статевим шляхом і безпосередньо від інфікованих матерів або вагітних матерів їх потомству. Способи лікування HCV інфекції, що існують у наш час, які обмежуються імунотерапією з використанням одного рекомбінантного інтерферону-α або у комбінації з нуклеозидним аналогом рибавірину, дають обмежені позитивні клінічні результати. Більш того, для HCV не існує визнаної вакцини. Відповідно, існує нагальна потреба у вдосконалених терапевтичних агентах, які ефективно боролися б з хронічною HCV інфекцією. Стан справ, що існує відносно лікування HCV інфекції приведений в оглядах, приводяться посилання на наступні публікації: В. Dymock, et al., "Novel approaches to the treatment of hepatitis С virus infection", Antiviral Chemistry & Chemotherapy, 11: 79-96 (2000); H. Rosen, et al., "Hepatitis С virus: current understanding and prospects for future therapies", Molecular Medicine Today, 5: 393-399 (1999); D. Moradpour, et al., "Current and evolving therapies for hepatitis C", European J. Gastroenterol. Hepatol., 11:1189-1202 (1999); R. Bartenschlager, "Candidate Targets for Hepatitis С Virus-Specific Anti viral Therapy", Intervirology, 40: 378-393 (1997); G.M. Lauer and B.D. Walker, "Hepatitis С Virus Infection", N. Engl. J. Med., 345: 41-52 (2001); B.W. D ymock, "Emerging therapies for hepatitis С virus infection", Emerging Drugs, 6: 13-42 (2001); і С. Crabb, "Hard-Won Advances Spark Excitement about Hepatitis C", Science: 506-507 (2001); зміст яких включений як посилання. Були здійснені різні підходи до лікування HCV, які включають інгібування вірусної серинпротеїнази (NS3 протеаза), гелікази та РНК-залежної РНК полімерази (NS5B), і розробку вакцини. HCV віріон є оболонковим РНК вірусом з позитивним ланцюгом, з єдиною олігонуклеотидною геномною послідовністю приблизно з 9600 основ, яка кодує поліпротеїн, що складається з приблизно 3010 амінокислот. Білкові продукти HCV гена складаються із структурних білків С, E1 і Е2 та нестр уктурних білків NS2, NS3, NS4A і NS4B та NS5A і NS5B. Вважають, що неструктурні білки (NS) забезпечують каталітичний механізм вірусної реплікації. NS3 протеаза виділяє NS5B, РНК-залежну РНК полімеразу з поліпротеїнового ланцюга. HCV NS5B полімераза необхідна для синтезу дволанцюгової РНК з одноланцюгової вірусної РНК, що служить матрицею у циклі реплікації HCV. Тому NS5B полімеразу вважають істотним компонентом у HCV комплексі реплікації [див. К. Ishi, et al., "Expression of Hepatitis С Virus NS5B Protein: Characterization of Its RNA Polymerase Acti vity and RNA Binding", Hepatology, 29: 1227-1235 (1999), і V. Lohmann, et al., "Biochemical and Kinetic Analyses of NS5B RNA-Dependent RNA Polymerase of the Hepatitis С Virus", Virology, 249: 108-118 (1998)]. Інгібування HCV NS5B полімерази запобігає утворенню дволанцюгової HCV РНК і тому є привабливим підходом до розробки HCV-специфічних противірусних способів лікування. У наш час було виявлено, що нуклеозидні сполуки даного винаходу та деякі їх похідні є е фективними інгібіторами РНК-залежної РНК вірусної реплікації, і зокрема, HCV реплікації. 5'-трифосфатні похідні цих нуклеозидних сполук є інгібіторами РНК-залежної РНК вірусної полімерази, і зокрема, HCV NS5B полімерази. Розглянуті нуклеозидні сполуки є їх похідними і можуть бути корисними при лікуванні РНК-залежної РНК вірусної інфекції, і зокрема, HCV інфекції. Тому задачею даного винаходу є створення нуклеозидних сполук і деяких їх похідних, які могли б бути корисними як інгібітори РНК-залежної РНК вірусної полімерази, і зокрема, як інгібітори HCV NS5B полімерази. Іншою задачею даного винаходу є створення нуклеозидних сполук і деяких їх похідних, які могли б бути корисними як інгібітори реплікації РНК-залежного РНК вірусу, і зокрема, як інгібітори реплікації вірусу гепатиту С. Іншою задачею даного винаходу є створення нуклеозидних сполук і деяких їх похідних, які були б корисними при лікуванні РНК-залежної РНК вірусної інфекції, і зокрема, при лікуванні HCV інфекції. Іншою задачею даного винаходу є створення фармацевтичних композицій, що включають нуклеозидні сполуки разом з фармацевтично прийнятним носієм. Іншою задачею даного винаходу є створення фармацевтичних композицій, що включають нуклеозидні сполуки та їх похідні для використання як інгібіторів РНК-залежної РНК вірусної полімерази, і зокрема, як інгібіторів HCV NS5B полімерази. Іншою задачею даного винаходу є створення фармацевтичних композицій, що включають нуклеозидні сполуки та їх похідні для використання як інгібіторів РНК-залежної РНК вірусної реплікації, і зокрема, як інгібіторів HCV реплікації. Іншою задачею даного винаходу є створення фармацевтичних композицій, що включають нуклеозидні сполуки та їх похідні для використання при лікуванні РНК-залежної РНК вірусної інфекції, і зокрема, при лікуванні HCV інфекції. Іншою задачею даного винаходу є створення фармацевтичних композицій, що включають нуклеозидні сполуки та їх похідні у комбінації з іншими агентами, активними проти РНК-залежного РНК вірусу, і зокрема, проти HCV. Іншою задачею даного винаходу є створення способів інгібування РНК-залежної РНК вірусної полімерази, і зокрема, інгібування HCV NS5B полімерази. Іншою задачею даного винаходу є створення способів інгібування РНК-залежної РНК вірусної реплікації, і зокрема, інгібування HCV реплікації. Іншою задачею даного винаходу є створення способів лікування РНК-залежної РНК вірусної інфекції, і зокрема, лікування HCV інфекції. Іншою задачею даного винаходу є створення способів лікування РНК-залежної РНК вірусної інфекції у комбінації з іншими агентами, активними проти РНК-залежного РНК вірусу, і зокрема, лікування HCV інфекції у комбінації з іншими агентами, активними проти HCV. Іншою задачею даного винаходу є створення нуклеозидних сполук і деяких їх похідних та їх фармацевтичних композицій для використання як ліків для інгібування РНК-залежної РНК вірусної реплікації і/або для лікування РНК-залежної РНК вірусної інфекції, і зокрема, для інгібування HCV реплікації і/або лікування HCV інфекції. Іншою задачею даного винаходу є створення для використання нуклеозидних сполук і деяких їх похідних та їх фармацевтичних композицій для одержання ліків для інгібування РНК-залежної РНК вірусної реплікації і/або лікування РНК-залежної РНК вірусної інфекції, і зокрема, для інгібування HCV реплікації і/або лікування HCV інфекції. Ці та інші задачі стануть очевидними з наступного докладного опису. Даний винахід відноситься до сполук структурної формули І зазначеної стереохімічної конфігурації: або їх фармацевтично прийнятних солей; де R1 являє собою С2-4-алкеніл, С2-4-алкініл або С 1-4-алкіл, де алкіл є незаміщеним або заміщений гідрокси, аміно, С1-4-алкокси, С1-4-алкілтіо або одним-трьома атомами фтору; R2 являє собою водень, фтор, гідрокси, меркапто, С1-4-алкокси або С1-4-алкіл; або R1 і R2 разом з атомом вуглецю, до якого вони приєднані, утворюють 3-6-членну насичену моноциклічну кільцеву систему, яка необов'язково містить гетероатом, вибраний з О, S і NC0-4-алкілу; R3 і R4 кожний незалежно вибирають з групи, яка складається з водню, ціано, азидо, галогену, гідрокси, меркапто, аміно, С1-4-алкокси, С2-4-алкенілу, С2-4-алкінілу і С 1-4-алкілу, де алкіл є незаміщеним або заміщений гідрокси, аміно, С1-4-алкокси, С1-4-алкілтіо або одним-трьома атомами фтору; R5 являє собою водень, С1-10-алкілкарбоніл, Р3О9Н4, Р2О 6Н3 або P(O)R13R14 ; R6 і R7 кожний незалежно являє собою водень, метил, гідроксиметил або фторметил; R8 являє собою водень, С1-4-алкіл, С2-4-алкініл, галоген, ціано, карбокси, С1-4-алкілоксикарбоніл, азидо, аміно, С1-4-алкіламіно, ди(С 1-4-алкіл)аміно, гідрокси, С1-6-алкокси, С1-6-алкілтіо, С1-6-алкілсульфоніл, (С1-4алкіл)0-2-амінометил або С 4-6-циклогетероалкіл, незаміщений або заміщений однією-двома групами, незалежно вибраними з галогену, гідрокси, аміно, С1-4-алкілу і С 1-4-алкокси; R9 являє собою водень, ціано, нітро, С1-3-алкіл, NHCONH2, CONR12R12, CSNR12R12, COOR 12 , C(=NH)NH2, гідрокси, С1-3-алкокси, аміно, С1-4-алкіламіно, ди(С1-4-алкіл)аміно, галоген, (1,3-оксазол-2-іл), (1,3-тіазол-2-іл) або (імідазол-2-іл); де алкіл є незаміщеним або заміщений однією-трьома групами, незалежно вибраними з галогену, аміно, гідрокси, карбокси і С 1-3-алкокси; R10 і R11 кожний незалежно являє собою водень, гідрокси, галоген, С1-4-алкокси, аміно, С1-4-алкіламіно, ди(С1-4-алкіл)аміно, С3-6-циклоалкіламіно, ди(С 3-6-циклоалкіл)аміно або С 4-6-циклогетероалкіл, незаміщений або заміщений однією-двома групами, незалежно вибраними з галогену, гідрокси, аміно, С1-4-алкілу і С 1-4алкокси; кожний R12 незалежно являє собою водень або С1-6-алкіл; і R13 і R14 кожний незалежно являє собою гідрокси, ОСН2СН2SС(=О)С1-4-алкіл, ОСН2О(С= О)О1- 4-алкіл, NHCHMeCO2Me, ОСН(С1-4-алкіл)О(С=О)С 1-4-алкіл, за умови, що якщо R1 являє собою b-метил і R4 являє собою водень, або R4 являє собою b-метил і R1 являє собою водень, R2 і R3 являють собою a-гідрокси, R10 являє собою аміно і R5, R6, R7, R8 і R11 являють собою водень, тоді R9 не являє собою ціано або CONH2. Сполуки формули І можна використовувати як інгібітори РНК-залежної РНК вірусної полімерази, і зокрема, HCV NS5B полімерази. Вони також є інгібіторами РНК-залежної РНК вірусної реплікації, і зокрема, HCV реплікації, і їх можна використовувати для лікування РНК-залежної РНК вірусної інфекції, і зокрема, для лікування HCV інфекції. Крім того, в обсяг даного винаходу включені фармацевтичні композиції, які містять зазначені сполуки, взяті як окремо, так і у комбінації з іншими агентами, активними проти РНК-залежних РНК вірусів, і зокрема, проти HCV, а також способи інгібування РНК-залежної РНК вірусної реплікації і для лікування РНК-залежної РНК вірусної інфекції. Даний винахід відноситься до сполук структурної формули І зазначеної стереохімічної конфігурації: або їх фармацевтично прийнятних солей; де R1 являє собою С2-4-алкеніл, С2-4-алкініл або С 1-4-алкіл, де алкіл є незаміщеним або заміщений гідрокси, аміно, С1-4-алкокси, С1-4-алкілтіо або одним-трьома атомами фтору; R2 являє собою водень, фтор, гідрокси, меркапто, С1-4-алкокси або С1-4-алкіл; або R1 і R2 разом з атомом вуглецю, до якого вони приєднані, утворюють 3-6-членну насичену моноциклічну кільцеву систему, яка необов'язково містить гетероатом, вибраний з О, S і NC0-4-алкілу; R3 і R4 кожний незалежно вибирають з групи, яка складається з водню, ціано, азидо, галогену, гідрокси, меркапто, аміно, С1-4-алкокси, С2-4-алкенілу, С2-4-алкінілу і С 1-4-алкілу, де алкіл є незаміщеним або заміщений гідрокси, аміно, С1-4-алкокси, С1-4-алкілтіо або одним-трьома атомами фтору; R5 являє собою водень, С1-10-алкілкарбоніл, Р3О9Н4, Р2О 6Н3 або P(O)R13R14 ; R6 і R7 кожний незалежно являє собою водень, метил, гідроксиметил або фторметил; R8 являє собою водень, С1-4-алкіл, С2-4-алкініл, галоген, ціано, карбокси, С1-4-алкілоксикарбоніл, азидо, аміно, С1-4-алкіламіно, ди(С 1-4-алкіл)аміно, гідрокси, С1-6-алкокси, С1-6-алкілтіо, С1-6-алкілсульфоніл, (С1-4алкіл)0-2-амінометил або С 4-6-циклогетероалкіл, незаміщений або заміщений однією-двома групами, незалежно вибраними з галогену, гідрокси, аміно, С1-4-алкілу і С 1-4-алкокси; R9 являє собою водень, ціано, нітро, С1-3-алкіл, NHCONH2, CONR12R12, CSNR12R12, COOR 12 , C(=NH)NH2, гідрокси, С1-3-алкокси, аміно, С1-4-алкіламіно, ди(С1-4-алкіл)аміно, галоген, (1,3-оксазол-2-іл), (1,3-тіазол-2-іл) або (імідазол-2-іл); де алкіл є незаміщеним або заміщений однією-трьома групами, незалежно вибраними з галогену, аміно, гідрокси, карбокси і С 1-3-алкокси; R10 і R11 кожний незалежно являє собою водень, гідрокси, галоген, С1-4-алкокси, аміно, С1-4-алкіламіно, ди(С1-4-алкіл)аміно, С3-6-циклоалкіламіно, ди(С 3-6-циклоалкіл)аміно або С 4-6-циклогетероалкіл, незаміщений або заміщений однією-двома групами, незалежно вибраними з галогену, гідрокси, аміно, С1-4-алкілу і С 1-4алкокси; кожний R12 незалежно являє собою водень або С1-6-алкіл; і R13 і R14 кожний незалежно являє собою гідрокси, ОСН2СН2SС(=О)С1-4-алкіл, ОСН2О(С= О)ОС 1-4алкіл,NНCМеСО2Ме, ОСН(С1-4-алкіл)О(С=О)С 1-4-алкіл, за умови, що якщо R1 являє собою b-метил і R4 являє собою водень або R4 являє собою b-метил і R1 являє собою водень, R2 і R3 являють собою a-гідрокси, R10 являє собою аміно і R5, R6, R7, R8 і R11 являють собою водень, тоді R9 не являє собою ціано або CONH2. Сполуки формули І можна використовувати як інгібітори РНК-залежної РНК вірусної полімерази. Вони також є інгібіторами РНК-залежної РНК вірусної реплікації і їх можна використовувати для лікування РНКзалежної РНК вірусної інфекції. В одному з варіантів сполуки структурної формули І є сполуками структурної формули II: або їх фармацевтично прийнятною сіллю; де R1 являє собою С 1-3-алкіл, де алкіл необов'язково заміщений гідрокси, аміно, С1-3-алкокси, С1-3-алкілтіо або одним-трьома атомами фтору; R2 являє собою гідрокси, фтор або С1-3-алкокси; R3 являє собою водень, галоген, гідрокси, аміно або С 1-3-алкокси; R5 являє собою водень, Р3О6Н4, Р 2О6Н 3 або РО3Н2 ; R8 являє собою водень, аміно або С 1-4-алкіламіно; R9 являє собою водень, ціано, метил, галоген або CONH 2; і R10 і R11 кожний незалежно являє собою водень, галоген, гідрокси, аміно, С1-4-алкіламіно, ди(С1-4алкіл)аміно або С3-6-циклоалкіламіно; за умови, що якщо R1 являє собою b-метил, R2 і R3 являють собою a-гідрокси, R10 являє собою аміно і R5, R8 і R11 являють собою водень, тоді R9 не являє собою ціано або CONH2. У другому варіанті сполуки структурної формули І являють собою сполуки структурної формули II, де: R1 являє собою метил, фторметил, гідроксиметил, дифторметил, трифторметил або амінометил; R2 являє собою гідрокси, фтор або метокси; R3 являє собою водень, фтор, гідрокси, аміно або метокси; R5 являє собою водень або Р3О9Н4; R8 являє собою водень або аміно; R9 являє собою водень, ціано, метил, галоген або CONH 2; і R1 і R11 кожний незалежно являє собою водень, фтор, гідрокси або аміно; за умови, що якщо R1 являє собою b-метил, R2 і R3 являють собою a-гідрокси, R10 являє собою аміно і R5, 8 R і R11 являють собою водень, тоді R9 не являє собою ціано або CONH2. Ілюстративними, але не обмежувальними прикладами сполук даного винаходу стр уктурної формули І, які можна використовувати як інгібітори РНК-залежної РНК вірусної полімерази, є наступні: 4-аміно-7-(2-С-метил-b-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-метиламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-диметиламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-циклопропіламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(2-С-вініл-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(2-С-гідроксиметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(2-С-фторметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-5-метил-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-5-карбонова кислота, 4-аміно-5-бром-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-5-хлор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-5-фтор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 2,4-діаміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 2-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 2-аміно-4-циклопропіламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 2-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он, 4-аміно-7-(2-С-етил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(2-С,2-О-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он, 2-аміно-5-метил-7-(2-С,2-O-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он, 4-аміно-7-(3-дезокси-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(3-дезокси-2-С-метил-b-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-2-фтор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(3-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(3-С-метил-b-D-ксилофуранозил)-7Н-піроло[2,3-d]піримідин, 4-аміно-7-(2,4-ди-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин і 4-аміно-7-(3-дезокси-3-фтор-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин та відповідні 5'-трифосфати; або їх фармацевтично прийнятні солі. Додатковими ілюстраціями даного винаходу є сполуки, вибрані з групи, яка складається з: 4-аміно-7-(2-С-метил-b-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідину, 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину, 4-аміно-7-(2-С-фторметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину, 4-аміно-5-метил-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину, 4-аміно-5-бром-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину, 4-аміно-5-хлор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину, 4-аміно-5-фтор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину і 4-аміно-7-(2-С,2-O-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину та відповідних 5'трифосфаті в; або їх фармацевтично прийнятних солей. В одному втіленні даного винаходу н уклеозидні сполуки можна використовувати як інгібітори позитивносмислової одноланцюгової РНК-залежної РНК вірусної полімерази, інгібітори позитивносмислової одноланцюгової РНК-залежної РНК вірусної реплікації і/або для лікування позитивносмислової одноланцюгової РНК-залежної РНК вірусної інфекції. У класі цього втілення позитивносмисловий одноланцюговий РНК-залежний РНК вірус є Flavi viridae вірусом або Picornaviridae вірусом. У підкласі цього класу Picornaviridae вірус є риновірусом, поліовірусом або вірусом гепатиту А. У др угому підкласі цього класу Flaviviridae вірус вибирають з групи, яка складається з вірусу гепатиту С, вірусу жовтої лихоманки, вірусу Денге, вірусу За хідного Нілу, вірусу японського енцефаліту, вірусу Банзі та вірусу коров'ячої вірусної діареї (BVD V). У підгр упі цього підкласу Flavi viridae вірус є вірусом гепатиту С. Інший аспект даного винаходу відноситься до способу інгібування РНК-залежної РНК вірусної полімерази, до способу інгібування РНК-залежної РНК вірусної реплікації і/або до способу лікування РНК-залежної РНК вірусної інфекції у ссавців, які потребують цього, що включає введення ссавцю терапевтично ефективної кількості сполуки структурної формули І. В одному втіленні цього аспекту даного винаходу РНК-залежна РНК вірусна полімераза є позитивносмисловою одноланцюговою РНК-залежною РНК вірусною полімеразою. У класі цього втілення позитивносмислова одноланцюгова РНК-залежна РНК вірусна полімераза є Flaviviridae вірусною полімеразою або Picornaviridae вірусною полімеразою. У підкласі цього класу Picornaviridae вірусна полімераза є риновірусною полімеразою, поліовірусною полімеразою або полімеразою вірусу гепатиту А. У др угому підкласі цього класу Flavi viridae вірусну полімеразу вибирають з групи, яка складається з полімерази вірусу гепатиту С, полімерази вірусу жовтої лихоманки, полімерази вірусу Денге, полімерази вірусу За хідного Нілу, полімерази вірусу японського енцефаліту, полімерази вірусу Банзі та полімерази вірусу коров'ячої вірусної діареї (BVDV). У підгрупі цього підкласу Flavi viridae вірусна полімераза є полімеразою вірусу гепатиту С У другому втіленні цього аспекту даного винаходу РНК-залежна РНК вірусна реплікація є позитивносмисловою одноланцюговою РНК-залежною РНК вірусною реплікацією. У класі цього втілення позитивносмислова одноланцюгова РНК-залежна РНК вірусна реплікація є Flaviviridae вірусною реплікацією або Picornaviridae вірусною реплікацією. У підкласі цього класу Picornaviridae вірусна реплікація є риновірусною реплікацією, поліовірусною реплікацією або реплікацією вірусу гепатиту А. У др угому підкласі цього класу Flavi viridae вірусну реплікацію вибирають з групи, яка складається з реплікації вірусу гепатиту С, реплікації вірусу жовтої лихоманки, реплікації вірусу Денге, реплікації вірусу За хідного Нілу, реплікації вірусу японського енцефаліту, реплікації вірусу Банзі та реплікації вірусу коров'ячої вірусної діареї. У підгрупі цього підкласу Flavi viridae вірусна реплікація є реплікацією вірусу гепатиту С. У третьому втіленні цього аспекту даного винаходу РНК-залежна РНК вірусна інфекція є позитивносмисловою одноланцюговою РНК-залежною РНК вірусною інфекцією. У класі цього втілення позитивносмислова одноланцюгова РНК-залежна РНК вірусна інфекція є Flaviviridae вірусною інфекцією або Picornaviridae вірусною інфекцією. У підкласі цього класу Picornaviridae вірусна інфекція є риновірусною інфекцією, поліовірусною інфекцією або інфекцією вірусу гепатиту А. У другому підкласі цього класу Flaviviridae вірусну інфекцію вибирають з групи, яка складається з інфекції вірусу гепатиту С, інфекції вірусу жовтої лихоманки, інфекції вірусу Денге, інфекції вірусу За хідного Нілу, інфекції вірусу японського енцефаліту, інфекції вірусу Банзі та інфекції вірусу коров'ячої вірусної діареї. У підгрупі цього підкласу Flaviviridae вірусна інфекція є інфекцією вірусу гепатиту С. У тексті заявки, що розглядається, наступні терміни мають зазначені далі значення: Вказані вище алкільні групи включають алкільні групи зазначеної довжини у лінійній або розгалуженій конфігурації. Прикладами таких алкільних гр уп є метил, етил, пропіл, ізопропіл, бутил, фтор-бутил, трет-б утил, пентил, ізопентил, гексил, ізогексил і т.п. Термін "алкеніл" означає нерозгалужений або розгалужений алкеновий ланцюг, що містить від дво х до шести атомів вуглецю або будь-яке число у цьому інтервалі (наприклад, етеніл, пропеніл, бутеніл, пентеніл і т.д.). Термін "алкініл" означає нерозгалужений або розгалужений алкіновий ланцюг, що містить від двох до шести атомів вуглецю або будь-яке число у цьому інтервалі (наприклад, етиніл, пропініл, бутиніл, пентиніл і т.д.). Термін "циклоалкіл" означає циклічні алканові кільця, які містять від трьох до восьми атомів вуглецю або будь-яке число у цьому інтервалі (тобто циклопропіл, циклобутил, циклопентил, циклогексил, циюіогептил або циклооктил). Термін "циклогетероалкіл" включає неароматичні гетероцикли, які містять один або два гетероатоми, вибрані з атомів азоту, кисню і сірки. Приклади 4-6-членних циклогетероалкілів включають азетидиніл, піролідиніл, піперидиніл, морфолініл, тіаморфолініл, імідазолідиніл, тетрагідрофураніл, тетрагідропіраніл, тетрагідротіофеніл, піперазиніл і т.п. Термін "алкокси" відноситься до нерозгалуженого або розгалуженого ланцюга алкоксидів, що містить зазначену кількість атомів вуглецю (наприклад, С1-4-алкокси) або будь-яке число у зазначеному інтервалі [тобто метокси (МеО-), етокси, ізопропокси і т.д.]. Термін "алкїлтіо" відноситься до нерозгалуженого або розгалуженого ланцюга алкілсульфідів, що містить зазначене число атомів вуглецю (наприклад, С1-4-алкілтіо) або будь-яке число у зазначеному інтервалі [тобто метилтіо (MeS-), етилтіо, ізопропілтіо і т.д.]. Термін "алкіламіно" відноситься до нерозгалужених або розгалужених алкіламінів, які містять зазначене число атомів вуглецю (наприклад, С1-4-алкіламіно) або будь-яке число у зазначеному інтервалі [тобто метиламіно, етиламіно, ізопропіламіно, трет-бутиламіно і т.д.]. Термін "алкілсульфоніл" відноситься до нерозгалужених або розгалужених алкілсульфонів, які містять зазначене число атомів вуглецю (наприклад, С1-6-алкілсульфоніл) або будь-яке число у зазначеному інтервалі [тобто метилсульфоніл (MeSO2-), етил сул ьфоніл, ізопропілсульфоніл і т.д.]. Термін "алкілоксикарбоніл" відноситься до складних ефірів з нерозгалуженим або розгалуженим ланцюгом похідного карбонової кислоти даного винаходу, що містить зазначене число атомів вуглецю (наприклад, С1-4алкілоксикарбоніл) або будь-яке число у зазначеному інтервалі [тобто метилоксикарбоніл (МеОСО-), етилоксикарбоніл або бутилоксикарбоніл]. Термін "арил" включає як феніл, нафтил, так і піридил. Арильна група необов'язково заміщена однієютрьома групами, незалежно вибраними з С 1-4-алкілу, галогену, ціано, нітро, трифторметилу, С1-4-алкокси і С 1-4алкілтіо. Термін "галоген" включає атоми галогенів: фтору, хлору, брому та йоду. Термін "заміщений" включає множинний ступінь заміщення зазначеними замісниками. Коли розкрита або заявлена велика кількість груп, які заміщають, заміщена сполука може бути незалежно заміщена одним або більше з розкритих або заявлених груп замісників, по одному або декілька. Термін "5'-трифосфат" відноситься до похідного 5'-гідроксильної групи складного ефіру трифосфорної кислоти групи нуклеозидних сполук даного винаходу, які мають наступну загальну стр уктурн у формулу III: де R1-R11 мають зазначені вище значення. Передбачається, що сполуки даного винаходу включають фармацевтично прийнятні солі трифосфатного складного ефіру, а також фармацевтично прийнятні солі похідних 5'-монофосфатних і 5¢-дифосфатних складних ефірів стр уктурни х формул IV і V, відповідно, Термін "5'-(S-ацил-2-тіоетил)фосфат" або "SATE" відноситься до похідних складних моно- або діефірів похідних 5'-монофосфатнуклеозидів даного винаходу стр уктурних формул VI і VII, відповідно, а також до фармацевтично прийнятних солей складних моноефірів, Термін "композиція" у виразі "фармацевтична композиція" включає активний інгредієнт (інгредієнти) та інертний інгредієнт (інгредієнти), що складає носій, а також будь-який продукт, який утворений безпосередньо або опосередковано у результаті комбінування, комплексоутворення або агрегації будь-яких дво х або більше інгредієнтів, або у результаті розпаду одного або більше інгредієнтів, або у результаті інших типів реакцій або взаємодій одного або більше інгредієнтів. Відповідно, фармацевтичні композиції даного винаходу включають будь-які композиції, одержані у результаті змішування сполуки даного винаходу і фармацевтично прийнятного носія. Терміни "прийом" і "введення" сполуки потрібно розуміти як забезпечення сполукою даного винаходу або пролікарською формою сполуки індивідуума, який потребує цього. Інший аспект даного винаходу відноситься до способу інгібування HCV NS5B полімерази, інгібування HCV реплікації або лікування HCV інфекції сполукою даного винаходу у комбінації з одним або більше агентів, які можна використовувати для лікування HCV інфекції. Такі агенти, активні проти HCV, включають (але цим не обмежуються) рибавірин, левовірин, вірамідин, тимозин альфа-1, інтерферон-α, пегільований інтерферон-α (пегінтерферон-α), комбінацію інтерферону-α і рибавірину, комбінацію пегінтерферону-α і рибавірину, комбінацію інтерферону-α і левовірину та комбінацію пегінтерферону-α і левовірину. Інтерферон-a включає (але цим не обмежується) рекомбінантний інтерферон-a2а (такий як Roferon інтерферон, доступний від Hoffmann-LaRoche, Nutley, NJ), пегільований інтерферон-a2а (Pegasys™), інтерферон-a2b (такий як Intron-A інтерферон, доступний від Schering Corp., Kenilworth, NJ), пегільований інтерферон-a2b (Peglntron™), рекомбінантний консенсусний інтерферон (такий, як інтерферон alphacon-1) і очищений продукт інтерферонуα. Рекомбінантний консенсусний інтерферон Amgen має торгову марку Infergen®. Левовірин являє собою Lенантіомер рибавірину, який демонструє імуномодуляторну активність, аналогічну рибавірину. Вірамідин являє собою аналог рибавірину, розкритий у WO01/60379 (правонаступник ICN Pharmaceuticals). Відповідно до способу даного винаходу окремі компоненти комбінації можна вводити окремо у різні моменти часу протягом курсу лікування або одночасно у розділеній або єдиній комбінованій формі. Тому даний винахід потрібно розуміти як такий, що включає всі такі схеми одночасного лікування, або лікування, що чергується, і термін "введення" потрібно інтерпретувати відповідним чином. Необхідно враховува ти, що обсяг комбінацій сполук даного винаходу з іншими агентами, які можна використовувати для HCV інфекції, включає в принципі будь-яку комбінацію з будь-якою фармацевтичною композицією для лікування HCV інфекції. Коли сполуку даного винаходу або її фармацевтично прийнятну сіль використовують у комбінації з другим терапевтичним агентом, активним проти HCV, доза кожної із сполук може бути однаковою або відрізнятися від дози у тому випадку, коли цю сполуку використовують окремо. Для лікування HCV інфекції сполуки даного винаходу можна також вводити у комбінації з агентом, який є інгібітором HCV NS3 серинпротеази. HCV NS3 серинпротеаза є основним вірусним ферментом, і було показано, що вона є чудовою мішенню для інгібування HCV реплікації. Інгібітори HCV NS3 протеази, як на субстраті, так і без субстрату, розкриті в WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929 і GB-2337262. HCV NS3 протеаза, як мішень для розробки інгібіторів HCV реплікації і для лікування HCV інфекції, обговорена у B.W. D ymock, "Emerging therapies for hepatitis С virus infection", Emerging Drugs, 6: 13-42 (2001). Рибавірин, левовірин та вірамідин можуть виявляти свою анти-HCV дію, модулюючи внутрішньоклітинний об'єм гуаніннуклеотидів за рахунок інгібування внутрішньоклітинного ферменту інозинмонофосфатдегідрогенази (IMPDH). IMPDH є ферментом, який обмежує швидкість у біосинтетичній схемі біосинтезу гуаніннуклеотиду de novo. Рибавірин легко фосфорилюється внутрішньоклітинно, і монофосфатне похідне є інгібітором IMPDH. Таким чином, інгібування IMPDH являє собою ще одну придатну мішень для виявлення інгібіторів HCV реплікації. Тому сполуки даного винаходу можна також вводити у комбінації з інгібітором IMPDH, таким як VX-497, що розкритий у WO 97/41211 і WO 01/00622 (правонаступник Verte x); ще одним IMPDH інгібітором, таким як той, що розкритий у WO 00/25780 (правонаступник Bristol-Myers Squibb); або з мікофенолатмофетилом (mycophenolate mofetil) [див. A.C. Allison and E.M. Eugui, Agents Action. 44 (Suppl.): 165 (1993)]. Для лікування HCV інфекції сполуки даного винаходу можна також вводити у комбінації з противірусним агентом амантадином (1-аміноадамантан) [докладний опис цього агента див.: J. Kirschbaum, Anal. Profiles Drug Subs. 12: 1-36 (1983)]. Термін "фармацевтично прийнятний" має на увазі, що носій, розріджувач або ексципієнт повинен бути сумісним з іншими інгредієнтами композиції та не повинен здійснювати шкідливого впливу на реципієнта. В обсяг даного винаходу включені також фармацевтичні композиції, які включають нуклеозидні сполуки та їх похідні разом з фармацевтично прийнятним носієм. Іншим прикладом даного винаходу є фармацевтична композиція, одержана у результаті комбінації будь-якої з описаних вище сполук і фармацевтично прийнятного носія. Іншою ілюстрацією винаходу є спосіб одержання фармацевтичної композиції, що включає комбінування будь-якої з описаних вище сполук і фармацевтично прийнятного носія. В обсяг даного винаходу включені також фармацевтичні композиції, придатні для інгібування РНКзалежної РНК вірусної полімерази, зокрема, HCV NS5B полімерази, які містять ефективну кількість сполуки даного винаходу та фармацевтично прийнятний носій. Фармацевтичні композиції, придатні для лікування РНКзалежної РНК вірусної інфекції, зокрема, HCV інфекції, також включені в обсяг даного винаходу, також як і спосіб інгібування РНК-залежної РНК вірусної полімерази, зокрема, HCV NS5B полімерази, і спосіб інгібування РНК-залежної вірусної реплікації, і зокрема, HCV реплікації. Крім того, даний винахід відноситься до фармацевтичної композиції, яка включає терапевтично ефективну кількість сполуки даного винаходу у комбінації з терапевтично ефективною кількістю іншого агента, активного проти РНК-залежного РНК вірусу, і зокрема, проти HCV. Агенти, активні проти HCV, включають (але цим не обмежуються) рибавірин, левовірин, вірамідин, тимозин альфа-1, інгібітор HCV NS3 серинпротеази, інтерферон-α, пегільований інтерферон-α (пегінтерферон-a), комбінацію інтерферону-α і рибавірину, комбінацію пегінтерферону-α і рибавірину, комбінацію інтерферону-α і левовірину і комбінацію пегінтерферону-a і левовірину. Інтерферон-а включає (але цим не обмежується) рекомбінантний інтерферон-a2а (такий як Roferon інтерферон, доступний від HoffmannLaRoche, Nutley, NJ), інтерферон-a2b (такий як Intron-A інтерферон, доступний від Schering Corp., Kenilworth, NJ), консенсусний інтерферон і очищений продукт інтерферону-a. Обговорення рибавірину та його активності проти HCV див.: J.O. Saunders and S.A. Raybuck, "Inosine Monophosphate Dehydrogenase: Consideration of Structure, Kinetics, and Therapeutic Potential", Ann. Rep. Med. Chem., 35: 201-210 (2000). В іншому аспекті даного винаходу запропоноване використання нуклеозидних сполук і їх по хідних та їх фармацевтичних композицій для виготовлення ліків для інгібування РНК-залежної РНК вірусної реплікації, зокрема, HCV реплікації, і/або для лікування РНК-залежної РНК вірусної інфекції, зокрема, HCV інфекції. Ще в одному аспекті даного винаходу нуклеозидні сполуки, їх похідні та їх фармацевтичні композиції запропоновані як ліки для інгібування РНК-залежної РНК вірусної реплікації, зокрема, HCV реплікації, і/або для лікування РНК-залежної РНК вірусної інфекції, зокрема, HCV інфекції. Фармацевтичні композиції даного винаходу включають сполуку стр уктурної формули І як активний інгредієнт або її фармацевтично прийнятну сіль і можуть також містити фармацевтично прийнятний носій і необов'язково інші терапевтичні інгредієнти. Композиції винаходу включають композиції для перорального, ректального введення, зовнішнього застосування, парентерального (включаючи підшкірне, внутрішньом'язове і внутрішньовенне), окулярного (очного), легеневого (через ніс або за допомогою інгаляції) або назального введення, хоча найбільш придатний у даному випадку спосіб буде залежати від характеру і тяжкості стану, який підлягає лікуванню, та від природи активного інгредієнта. Вони звичайно можуть бути представлені в одиничній (стандартній) дозованій формі і приготовані будь-яким з відомих фа хівцям-фармацевтам способів. На практиці сполуки структурної формули І можна комбінувати як активний інгредієнт в однорідній суміші з фармацевтичним носієм, використовуючи звичайну фармацевтичну методику змішування. Носій може приймати велику кількість різних форм в залежності від потрібної для введення форми препарату, наприклад, перорального або парентерального (включаючи внутрішньовенне). При готуванні композицій для пероральних дозованих форм можна використовувати будь-яке звичайне фармацевтичне середовище, таке як, наприклад, вода, гліколі, олії, спирти, смакові агенти, консерванти, барвники і т.п. у випадку рідких препаратів для перорального введення, наприклад, суспензій, еліксирів і розчинів; або носії, такі як крохмалі, цукри, мікрокристалічна целюлоза, розріджувачі, гранулюючі агенти, змащувальні агенти, в'яжучі речовини, розпушувачі і т.п. у випадку твердих препаратів для перорального введення, таких як, наприклад, порошки, тверді і м'які капсули і таблетки, причому тверді препарати для перорального введення є більш переважними, ніж рідкі препарати. Завдяки простоті введення, таблетки і капсули є найбільш зручними дозованими стандартними одиничними формами для перорального введення, причому у цьому випадку звичайно використовують тверді фармацевтичні носії. За бажанням на таблетки може бути нанесене покриття з використанням стандартної водної або безводної методики. Такі композиції і препарати повинні містити, щонайменше, 0,1% активної сполуки. Природно, відсоток активної сполуки у цих композиціях може варіюватися і звичайно може бути в інтервалі від близько 2 до близько 60мас. % на одиницю. Кількість активної сполуки у таких терапевтично придатних композиціях повинна бути такою, щоб досягалася величина ефективної дози. Активні сполуки можна також вводити інтраназально, наприклад, у формі рідких крапель або спреїв. Таблетки, пілюлі, капсули і т.п. можуть також містити в'яжучу речовину, таку як трагакантова камедь, акація, кукурудзяний крохмаль або желатин; такі ексципієнти, як дикальційфосфат; розпушувальний агент, такий як кукурудзяний крохмаль, картопляний крохмаль, альгінова кислота; змащувальний агент, такий як стеарат магнію; і агент, що підсолоджує, такий як сахароза, лактоза або сахарин. Коли одинична доза приготована у формі капсули, вона може містити, крім матеріалів зазначеного вище типу, рідкий носій, такий як жирна олія. Різні інші матеріали можуть бути присутнім у вигляді покриттів або для модифікації фізичної форми дозованої одиниці. Наприклад, таблетки можуть бути покриті шелаком, цукром або тим і іншим разом. Сироп або еліксир можуть містити на додаток до активного інгредієнта сахарозу як агент, що підсолоджує, метил- і пропілпарабени як консерванти, барвники і смакові агенти, такі як вишневий або апельсиновий аромат. Сполуки структурної формули І можна також вводити парентерально. Розчини або суспензії цих активних сполук можна приготувати у воді відповідним чином змішаній з поверхнево-активною речовиною, такою як гідроксипропілцелюлоза. Дисперсії можна також приготувати у гліцерині, рідкому поліетиленгліколі та в їх сумішах в оліях. У звичайних умовах збереження і використання ці препарати містять консервант для запобігання росту мікроорганізмів. Фармацевтичні форми, придатні для використання у вигляді ін'єкцій, включають стерильні водні розчини або дисперсії і стерильні порошки для готування стерильних розчинів або дисперсій для ін'єкцій безпосередньо перед використанням. В усіх випадках форма повинна бути стерильною і повинна бути рідкою, щоб її можна було легко вводити через шприц. Вона повинна бути стабільною в умовах готування та збереження і повинна бути захищена від забруднень, які викликаються діями мікроорганізмів, таких як бактерії і грибки. Носієм може бути розчинник або дисперсійне середовище, які містять, наприклад, воду, етанол, поліол (наприклад, гліцерин, пропіленгліколь та рідкий поліетиленгліколь), їх придатні суміші та рослинні олії. Будь-який придатний спосіб введення можна використовувати для забезпечення ссавця, особливо людини, ефективною дозою сполуки даного винаходу. Наприклад, можна використовувати пероральний, ректальний, зовнішній, парентеральний, окулярний, легеневий, назальний і т.п. способи. Дозовані форми включають таблетки, пастилки, дисперсії, суспензії, розчини, капсули, креми, мазі, аерозолі і т.п. Переважно сполуки структурної формули І вводити перорально. Для перорального введення людям інтервал доз складає від 0,01 до 1000мг/кг маси тіла у роздільних дозах. В одному втіленні інтервал доз складає від 0,1 до 100мг/кг маси тіла у роздільних дозах. В іншому втіленні інтервал доз складає від 0,5 до 20мг/кг маси тіла у роздільних дозах. Для перорального введення композиції, переважно, готують у формі таблеток або капсул, що містять від 1,0 до 1000мг активного інгредієнта, зокрема, 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900 і 1000мг активного інгредієнта для симптоматичного підбору дози для пацієнта, який підлягає лікуванню. Використовувана ефективна доза активного інгредієнта може змінюватися в залежності від конкретно використовуваної сполуки, способу введення, стану, що підлягає лікуванню, і тяжкості стану, що підлягає лікуванню. Такі дози легко може визначити фа хівець у даній області. Таку схему прийому доз можна підібрати, щоб забезпечити оптимальну терапевтичну реакцію. Сполуки даного винаходу містять один або більше асиметричних центрів, і тому можуть існувати у вигляді рацематів або рацемічних сумішей, окремих енантіомерів, сумішей діастереоізомерів і окремих діастереоізомерів. Мається на увазі, що даний винахід включає нуклеозидні сполуки, які мають b-Dстереохімічну конфігурацію п'ятичленного фуранозного кільця, як представлено структурною формулою далі, тобто нуклеозидні сполуки, в яких замісники у С-1 і С-4 п'ятичленного фуранозного кільця мають bстереохімічну конфігурацію (орієнтація "вгору", як позначено жирною лінією). Деякі з розкритих тут сполук містять олефінові подвійні зв'язки, і якщо немає інших вказівок, мається на увазі, що вони включають як Е-, так і Z-геометричні ізомери. Деякі з розкритих тут сполук можуть існувати у та утомерній формі, такій як кетоенольні таутомери. Окремі таутомери, а також їх суміші, включені у сполуки структурної формули І. Приклад кетоенольних таутомерів, які включені у сполуки даного винаходу, проілюстрований далі: Сполуки структурної формули І можна розділити на окремі діастереоізомери, наприклад, за допомогою фракційної кристалізації з придатного розчинника, наприклад, метанолу або етилацетату, або їх суміші, або за допомогою хіральної хроматографії, використовуючи оптично активну нерухому фаз у. Альтернативно, будь-який стереоізомер сполуки структурної формули І можна одержати у результаті стереоспецифічного синтезу, використовуючи оптично чисті вихідні матеріали або реагенти відомої конфігурації. Стереохімію замісників у положеннях С-2 і С-3 фуранозного кільця сполук даного винаходу структурної формули І позначають хвилястими лініями, які означають, що замісники R1, R2, R3 і R4 можуть бути в а(замісник "вниз") або b-(замісник "вгору") конфігурації незалежно один від одного. Позначення стереохімії жирною лінією, як у С-1 і С-4 фуранозного кільця, означає, що замісник має b-конфігурацію (замісник "вгору"). Сполуки даного винаходу можна вводити у формі фармацевтично прийнятної солі. Термін "фармацевтично прийнятна сіль" відноситься до солей, одержаних з фармацевтично прийнятних нетоксичних основ або кислот, включаючи неорганічні або органічні основи та неорганічні або органічні кислоти. Солі основних сполук, що охоплюються терміном "фармацевтично прийнятна сіль", відносяться до нетоксичних солей сполук даного винаходу, які звичайно одержують, здійснюючи взаємодію вільної основи з відповідною органічною або неорганічною кислотою. Представницькі солі основних сполук даного винаходу включають (але цим не обмежуються) наступні: ацетат, бензолсульфонат, бензоат, бікарбонат, бісульфат, бітартрат, борат, бромід, камзилат (camsylate), карбонат, хлорид, клавуланат (clavulanate), цитрат, дигідрохлорид, едетат, едисилат, естолат, езилат, фумарат, глуцептат, глюконат, глутамат, гліколіларсанілат, гексилрезорцинат, гідрабамін, гідробромід, гідрохлорид, гідроксинафтоат, йодид, ізотіонат, лактат, лактобіонат, лаурат, малат, малеат, манделат, мезилат, метилбромід, метилнітрат, метилсульфат, мукат, напсилат, нітрат, амонієву сіль N-метилглюкаміну, олеат, оксалат, памоат (ембонат), пальмітат, пантотенат, фосфа т/ди фосфат, полігалактуронат, саліцилат, стеарат, сульфа т, субацетат, сукцинат, танат, тартрат, теоклат, тозилат, триетіодид і валерат. Крім того, якщо сполуки даного винаходу містять кислотну груп у, тоді їх фармацевтично прийнятні солі включають (але цим не обмежуються) солі, одержані з неорганічних основ, включаючи алюміній, амоній, кальцій, мідь, залізо(lll), залізо(ІІ), літій, магній, марганець(lll), марганець(ll), калій, натрій, цинк і т.п. Найбільш переважні солі амонію, кальцію, магнію, калію та натрію. Солі, одержані з фармацевтично прийнятних органічних нетоксичних основ, включають солі первинних, вторинних і третинних амінів, циклічних амінів і основних іонообмінних смол, таких як аргінін, бетаїн, кафеїн, холін, Ν,Νдибензилетилендіамін, діетиламін, 2-діетиламіноетанол, 2-диметиламіноетанол, етаноламін, етилендіамін, Nетилморфолін, N-етилпіперидин, глюкамін, глюкозамін, гістидин, гідрабамін, ізопропіламін, лізин, метилглюкамін, морфолін, піперазин, піперидин, поліамінові смоли, прокаїн, пурини, теобромін, триетиламін, триметиламін, трипропіламін, трометамін і т.п. Крім того, якщо у сполуках даного винаходу присутня група карбонової кислоти (-СООН) або спиртова група, можна використовувати фармацевтично прийнятні складні ефіри похідних карбонової кислоти, такі як метиловий, етиловий або півалоїлоксиметиловий, або ацильні похідні спиртів, такі як ацетат або малеат. Включені також ті складноефірні та ацильні групи, які відомі фахівцям у даній області для модифікації розчинності або характеристик гідролізу для використання як композицій пролонгованої дії або пролікарських форм. Одержання нуклеозидних сполук і похідних даного винаходу Нуклеозидні сполуки та їх похідні даного винаходу можна одержати відповідно до широко відомих способів синтезу, які використовують у хімії нуклеозидів і нуклеотидів. Посилання зроблене на наступний текст для опису способів синтезу, що використовуються при одержанні сполук даного винаходу: "Chemistry of Nucleosides and Nucleotides", L.B. Townsend, ed., Vols. 1-3, Plenum Press, 1988, який включений в опис як посилання у повному обсязі. Представницький загальний спосіб одержання сполук даного винаходу представлений далі на схемі 1. Ця схема ілюструє синтез сполук даного винаходу структурної формули 1-7, де фуранозне кільце має b-Dрибоконфігурацію. Ви хідним матеріалом служить 3,5-біс-О-захищений алкілфуранозид, такий як метилфуранозид структурної формули 1-1. С-2 гідроксильну груп у окиснюють, використовуючи придатний агент, що окиснює, такий як триокис хрому або реагент хромату, періодинан Dess-Martin, або використовуючи окиснення Сверна, одержуючи С-2 кетон структурної формули 1-2. Приєднання реагенту Гриньяра, такого як алкіл-, алкеніл- або алкінілмагнійгалогенід (наприклад, MeMgBr, EtMgBr, вініл-MgBr, аліл-MgBr і етиніл-MgBr) або алкіл-, алкеніл- або алкініллітій, такого як MeLi, по карбонільному подвійному зв'язку 1-2 у придатному органічному розчиннику, такому як тетрагідрофуран, діетиловий ефір і т.п., приводить до одержання С-2 третинного спирту структурної формули КЗ. Потім вводять придатну захисну груп у (таку як СІ, Вr та І) у С-1 (аномерному) положенні фуранозного цукрового похідного, використовуючи обробку фуранозиду формули 1-3 галогенводнем у придатному органічному розчиннику, такому як бромистий водень в оцтовій кислоті, одержуючи проміжний фуранозилгалогенід 1-4. С-1 сульфонат, такий як метансульфонат (MeSO 2O-), трифторметансульфонат (CF3SO2 O-) або п-толуолсульфонат (-OTs), також можуть служити придатними відщеплюваними групами у наступній реакції одержання глікозидного (нуклеозидного) зв'язку. Нуклеозидний зв'язок створюють, обробляючи проміжну сполуку структурної формули 1-4 сіллю металу (такого як літій, калій або натрій) відповідним чином заміщеного 1Н-піроло[2,3-d]піримідину 1-5, такого як відповідним чином заміщений 4-гало-1Н-піроло[2,3-d]піримідин, який можна одержати in situ у результаті обробки гідридом лужного металу (таким як гідрид натрію), гідроксидом лужного металу (таким як гідроксид калію), карбонатом лужного металу (таким як карбонат натрію) або гексаметилдисилазидом лужного металу (таким як NaHMDS) у придатному безводному органічному розчиннику, такому як ацетонітрил, тетрагідрофуран, 1-метил-2піролідинон або Ν,Ν-диметилформамід (ДМФ). Реакцію заміщення можна вести з каталізатором, використовуючи каталізатор фазового перенесення, такий як TDA-1 або триетилбензиламонійхлорид, у двофазовій системі (тверде-рідина або рідина-рідина). Необов'язкові захисні групи у захищеному нуклеозиді структурної формули 1-6 потім відщеплюють, використовуючи звичайні способи видалення захисних груп, такі як описані у T.W. Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis", 3rd ed., John Wiley & Sons, 1999. Необов'язкове введення аміногрупи у положення 4 піроло[2,3-гі]піримідинових ядер здійснюють, використовуючи обробку 4-галогенної проміжної сполуки 1-6 відповідним аміном, таким як спиртовий аміак або рідкий аміак, для одержання первинного аміну у положенні С-4 (-NH2) алкіламіном для одержання вторинного аміну (-NHR), або діалкіламіном для одержання третинного аміну (-NRR1). Сполуку 7Н-піроло[2,3-d]піримідин4(3H)-он можна одержати у результаті гідролізу 1-6 водною основою, такою як водний гідроксид натрію. У результаті алкоголізу (такого як метаноліз) 1-6 одержують С-4 алкоксид (-OR), тоді як у результаті обробки алкілмеркаптидом одержують С-4 алкілтіо (-SR) похідне. Наступні хімічні маніпуляції, добре відомі фахівцям в області органічної/медичної хімії, можуть знадобитися для одержання потрібних сполук даного винаходу. У прикладах, що приводяться далі, є посилання на літературні публікації, які містять докладні вказівки щодо способів одержання цільових сполук або проміжних сполук, що використовуються при одержанні цільових сполук даного винаходу. Н уклеозидні сполуки даного винаходу були одержані відповідно до способів, докладно описаних у наступних прикладах. Ці приклади жодним чином не обмежують обсяг даного винаходу, і їх не слід розглядати таким чином. Фахівцям в області синтезу нуклеозидів і нуклеотидів буде очевидно, що для одержання цих та інши х сполук даного винаходу можна використовувати відомі варіанти умов і процесів способів одержання, що приводяться, цих сполук. Усі температури вказані у градуса х Цельсію, якщо немає інших вказівок. Приклад 1 4-аміно-7-(2-С-метил-b-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідин До триокису хрому (1,57г, 1,57моль) у ди хлорметані (DCM) (10мл) при 0°С додавали оцтовий ангідрид (145мг, 1,41моль) і потім піридин (245мг, 3,10моль). Суміш перемішували протягом 15 хвилин, потім додавали розчин 7-[3,5-О-[1,1,3,3-тетракіс(1-метилетил)-1,3-дисилоксандііл]-b-D-рибофуранозил]-7Нпіроло[2,3d]піримідин-4-аміну [спосіб одержання див.: J. Am. Chem. Soc. 105: 4059 (1983)] (508мг, 1,00моль) у DCM (3мл). Одержаний розчин перемішували протягом 2 годин, потім виливали в етилацетат (10мл) і потім фільтрували через силікагель, використовуючи етилацетат як елюент. Об'єднані фільтрати випарювали у вакуумі, поміщали у суміш діетиловий ефір/ТГФ (1:1) (20мл), охолоджували до -78°С та по краплях додавали метилмагнійбромід (3М, у ТГФ) (3,30мл, 10моль). Суміш перемішували при -78°С протягом 10 хвилин, потім залишали охолоджуватися до кімнатної температури і гасили, додаючи насичений водний амонійхлорид (10мл), і екстрагували DCM (20мл). Органічну фазу випарювали у вакуумі і сирий продукт очищали на силікагелі, використовуючи 5% метанол у дихлорметані як елюент. Фракції, які містять продукт, об'єднували і випарювали у вакуумі. Одержану олію поміщали у ТГФ (5мл) і додавали тетрабутиламонійфторид (TBAF) на силікагелі (1,1моль/г силікагелю) (156мг). Суміш перемішували при кімнатній температурі протягом 30 хвилин, фільтрували та випарювали у вакуумі. Сирий продукт очищали на силікагелі, використовуючи 10% метанол у дихлорметані як елюент. Фракції, які містять продукт, об'єднували і випарювали у вакуумі, одержуючи потрібну сполуку (49мг) у вигляді безбарвної твердої речовини. 1 Н-ЯМР (ДМСО-d6): δ 1,08 (с, 3Н), 3,67 (м, 2Н), 3,74 (м, 1H), 3,83 (м, 1Н), 5,19 (м, 1H), 5,23 (м, 1Н), 5,48 (м, 1Н), 6,08 (1H, с), 6,50 (м, 1H), 6,93 (шир.с, 2Н), 7,33 (м, 1Н), 8, 02 (с, 1H). Приклад 2 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 3,5-біс-О-(2,4-дихлорфенілметил)-1-О-метил-a-D-рибофураноза Суміш 2-O-ацетил-3,5-біс-O-(2,4-дихлорфенілметил)-1-0-метил-a-D-рибо-фуранози [спосіб одержання див.: Helv. Chim. Acta 78: 486 (1995)] (52,4г, 0,10моль) у метанольному К2СО3 (500мл, насичений при кімнатній температурі) перемішували при кімнатній температурі протягом 45 хвилин і потім концентрували при зниженому тиску. Маслянистий залишок суспендували в СН 2СІ2 (500мл), промивали водою (300мл + 5x200мл) і сольовим розчином (200мл), сушили (Na2SO4), фільтр ували та концентрували, одержуючи зазначену у заголовку сполуку (49,0г) у вигляді безбарвної олії, яку використовували без додаткового очищення на стадії В далі. 1 H-ЯМР (ДМСО-d6): δ 3,28 (с, 3Н, ОСН3), 3,53 (д, 2Н, J5,4=4,5 Гц, Н-5а, H-5b), 3,72 (дд, 1H, J3, 4=3,6 Гц, J3,2=6,6 Гц, Н-3), 3,99 (ддд, 1H, J2,1=4,5 Гц, J2,OH-2=9,6 Гц, Н-2), 4,07 (м, 1H, Н-4), 4,50 (с, 2Н, CH2Ph), 4,52, 4,60 (2д, 2Н, Jgem=13,6 Гц, CH2Ph), 4,54 (д, 1H, ОН-2), 4,75 (d, 1H, Н-1), 7,32-7,45, 7,52-7,57 (2м, 10Н, 2Ph). 13 С-ЯМР (ДМСО-d6): δ 55,40, 69,05, 69,74, 71,29, 72,02, 78,41, 81,45, 103,44, 127,83, 127,95, 129,05, 129,28, 131,27, 131,30, 133,22, 133,26, 133,55, 133,67, 135,45, 135,92. Стадія В: 3,5-біс-О-(2,4-дихлорфенілметил-1-О-метил-a-D-еритропенто-фураноз-2-улоза До охолодженої льодом суспензії Dess-Martin періодинану (50,0г, 118моль) у безводному СН 2Сl2 (350мл) в атмосфері аргону (Ar) додавали по краплях розчин сполуки стадії А (36,2г, 75моль) у безводному СН 2Сl2 (200мл) протягом 0,5 години. Реакційну суміш перемішували при 0°С протягом 0,5 години і потім при кімнатній температурі протягом 3 діб. Суміш розбавляли безводним Et2O (600мл) та виливали в охолоджену льодом суміш Na2S2 O3×5H2O (180г) у насиченому водному NaHCO3 (1400мл). Шари розділяли і органічний шар промивали насиченим водним NaHCO3 (600мл), водою (800мл) і сольовим розчином (600мл), сушили (MgSO4), фільтрували та випарювали, одержуючи зазначену у заголовку сполуку (34,2г) у вигляді безбарвної олії, яку використовували без додаткового очищення на стадії С далі. 1 Н-ЯМР (CDCl3): δ 3,50 (с, 3Н, ОСН3), 3,79 (дд, 1H, J5a,5b=11,3 Гц, J5a, 4=3,5 Гц, Н-5а), 3,94 (дд, 1Н, J5b,4=2,3 Гц, Н-5b), 4,20 (дд, 1Н, J3,1=1,3 Гц, J 3,4=8,4 Гц, Н-3), 4,37 (ддд, 1H, Н-4), 4,58, 4,69 (2д, 2Н, Jgem=13,0 Гц, CH2Ph), 4,87 (д, 1H, Н-1), 4,78, 5,03 (2д, 2Н, Jgem=12,5 Гц, CH2Ph), 7,19-7,26, 7,31-7,42 (2м, 10Н, 2Ph). 13 С-ЯМР (ДМСО-d6): δ 55,72, 69,41, 69,81, 69,98, 77,49, 78,00, 98,54, 127,99, 128,06, 129,33, 129,38, 131,36, 131,72, 133,61, 133,63, 133,85, 133,97, 134,72, 135,32, 208,21. Стадія С: 3,5-біс-O-(2,4-дихлорфенілметил)-2-С-метил-1-O-метил-a-D-рибо-фураноза До розчину MeMgBr у безводному Et2 O (0,48Μ, 300мл) при -55°С додавали по краплях розчин сполуки стадії В (17,40г, 36,2моль) у безводному Et2O (12 мл). Реакційну суміш залишали нагріватися до -30°С і перемішували протягом 7 годин при температурі від -30°С до -15°С, потім виливали в охолоджену льодом воду (500мл) і суміш інтенсивно перемішували при кімнатній температурі протягом 0,5 години. Суміш фільтрували через шар целіту (10x5см), який ретельно промивали Et2O. Органічний шар сушили (MgSO4), фільтрували та концентрували. Залишок розчиняли у гексані (~30мл), вводили у колонку з силікагелем (10x7см, попередньо набиту у гексані) та елюювали гексаном і гексаном/EtOAc (9/1), одержуючи зазначену у заголовку сполуку (16,7г) у ви гляді безбарвного сиропу. 1 H-ЯМР (CDCl3): δ 1,36 (д, 3Н, JMe,OH=0,9 Гц, 2С-Ме), 3,33 (к, 1H, ОН), 3,41 (д, 1Н, J3,4=3,3 Гц), 3,46 (с, 3Н, ОСН3), 3,66 (д, 2Н, J5 ,4=3,7 Гц, Н-5а, Н-5b), 4,18 (уявний кв, 1H, Н-4), 4,52 (с, 1Н, Н-1), 4,60 (с, 2Н, CH2Ph), 4,63, 4,81 (2д, 2Н, Jgem=13,2 Гц, CH2Ph), 7,19-7,26, 7,34-7,43 (2м, 10Н, 2Ph). 13 С-ЯМР (CDCl3): δ 24,88, 55,45, 69,95, 70,24, 70,88, 77,06, 82,18, 83,01, 107,63, 127,32, 129,36, 130,01, 130,32, 133,68, 133,78, 134,13, 134,18, 134,45, 134,58. Стадія D: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-метил-b-D-рибо-фуранозил)-7Н-піроло[2,3d]піримідин До розчину сполуки стадії С (9,42г, 19моль) у безводному дихлорметані (285мл) при 0°С по краплях додавали НВr (5,7Μ в оцтовій кислоті, 20мл, 114моль). Одержаний розчин перемішували при 0°С протягом 1 години і потім при кімнатній температурі протягом 3 годин, випарювали у вакуумі, і випарювали разом з безводним толуолом (3x40мл). Маслянистий залишок розчиняли у безводному ацетонітрилі (50мл) і додавали до розчину натрієвої солі 4-хлор-1Н-піроло[2,3-d]піримідину в ацетонітрилі [одержували in situ з 4-хлор-1Нпіроло[2,3-d]піримідину [спосіб одержання див.: J. Chem. Soc: 131 (1960)] (8,76г, 57моль) у безводному ацетонітрилі (1000мл) і NaH (60% у мінеральному маслі, 2,28г, 57моль) після 4 годин інтенсивного перемішування при кімнатній температурі]. Об'єднану суміш перемішували при кімнатній температурі протягом 24 годин і потім випарювали досуха. Залишок суспендували у воді (250мл) та екстрагували EtOAc (2x500мл). Об'єднані екстракти промивали сольовим розчином (300мл), сушили над Na2SO4, фільтрували та випарювали. Сирий продукт очищали на колонці з силікагелем (10см ´ 10см), використовуючи етилацетат/гексан (1:3 і 1:2) як елюент. Фракції, які містять продукт, об'єднували і випарювали у вакуумі, одержуючи потрібний продукт (5,05г) у вигляді безбарвної піни. 1 H-ЯМР (CDCl3): δ 0,93 (с, 3Н, СН3), 3,09 (с, 1Н, ОН), 3,78 (дд, 1Н, 1 5¢,5² =10,9Гц, J5¢,4=2,5 Гц, Н-5¢), 3,99 (дд, 1Н, J5² ,4=2,2 Гц, Н-5"), 4,23-4,34 (м, 2Н, Н-3', Н-4¢), 4,63, 4,70 (2д, 2Н, Jgem=12,7 Гц, CH2Ph), 4,71, 4,80 (2д, 2Н, Jgem=12,1 Гц, CH2Ph), 6,54 (д, 1Н, J5,6=3,8 Гц, Н-5), 7,23-7,44 (м, 10Н, 2Ph). 13 С-ЯМР (CDCl3): 5 21,31, 69,10, 70,41, 70,77, 79,56, 80,41, 81,05, 91,11, 100,57, 118,21, 127,04, 127,46, 127,57, 129,73, 129,77, 130,57, 130,99, 133,51, 133,99, 134,33, 134,38, 134,74, 135,21, 151,07, 151,15, 152,47. Стадія Е: 4-хлор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До розчину сполуки стадії D (5,42г, 8,8моль) у дихлорметані (175мл) при температурі -78°С по краплях додавали трихлористий бор (1Μ у ди хлорметані, 88мл, 88моль). Суміш перемішували при -78°С протягом 2,5 години, потім при температурі від -30°С до -20°С протягом 3 годин. Реакцію гасили, додаючи метанол/дихлорметан (1:1) (90мл) і одержану суміш перемішували при -15°С протягом 30 хвилин, потім нейтралізували водяним аміаком при 0°С та перемішували при кімнатній температурі протягом 15 хвилин. Тверду частин у фільтрували і промивали СН 2Сl2/МеOН (1/1, 250мл). Об'єднаний фільтрат випарювали і залишок очищали, використовуючи фле ш-хроматографію на силікагелі, використовуючи градієнтне елюювання СН 2СІ2 і СН 2Сl2:МеОН (99:1, 98:2, 95:5 і 90:10), одержуючи потрібну сполуку (1,73г) у вигляді безбарвної піни, яка перетворювалася в аморфну тверду речовину після обробки MeCN. 1 Н-ЯМР (ДМСО-d 6): δ 0,64 (с, 3Н, СН3), 3,61-3,71 (м, 1H, Н-5'), 3,79-3,88 (м, 1H, Н-5"), 3,89-4,01 (м, 2Н, Н-3¢, Н-4¢), 5,15-5,23 (м, 3Н, 2'-ОН, 3'-ОН, 5'-ОН), 6,24 (с, 1Н, Н-1¢), 6,72 (д, 1Н, J5,6=3,8 Гц, Н-5), 8,13 (д, 1H, Н-6), 8,65 (с, 1Н, Н-2). 13 С-ЯМР (ДМСО-d6): δ 20,20, 59,95, 72,29, 79,37, 83,16, 91,53, 100,17, 117,63, 128,86,151,13,151,19, 151,45. Стадія F: 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До сполуки стадії Ε (1,54г, 5,1моль) додавали метанольний аміак (насичений при 0°С; 150мл). Суміш нагрівали в автоклаві з нержавіючої сталі при 85°С протягом 14 годин, потім охолоджували та випарювали у вакуумі. Сиру суміш очищали на колонці з силікагелем, використовуючи СН2СІ2/МеOН (9/1) як елюент і одержуючи зазначену у заголовку сполуку у вигляді безбарвної піни (0,8г), яку виділяли у вигляді аморфної твердої речовини після обробки MeCN. Аморфну тверду речовину перекристалізовували з метанолу/ацетонітрилу; т.пл. 222°С. 1 H-ЯМР (ДМСО-d 6): δ 0,62 (с, 3Н, СН3), 3,57-3,67 (м, 1H, Н-5¢), 3,75-3,97 (м, 3Н, Н-5", Н-4¢, Н-3¢), 5,00 (с, 1Н, 2'-ОН), 5,04 (д, 1Н, J3¢OH,3' = 6,8 Гц, 3'-ОН), 5,06 (т, 1Н, J5¢OH,5',5² =5,1 Гц, 5¢-OH), 6,11 (с, 1H, Н-1¢), 6,54 (д, 1Н, J5,6=3,6 Гц, Н-5), 6,97 (шир.с, 2Н, NH2), 7,44 (д, 1Н, Н-6), 8,02 (с, 1Н, Н-2). 13 С-ЯМР (ДМСО-d6): δ 20,26, 60,42, 72,72, 79,30, 82,75, 91,20, 100,13, 103,08, 121,96, 150,37, 152,33, 158,15. Рідинна хроматографія-мас-спектроскопія (РХ-МС): знайдено: 279,10 (М-Н+); розраховано для C12H16N4 O4+H+: 279,11. Приклад 3 4-аміно-7-(2-С-етил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 3,5-біс-О-[2,4-дихлор фенілметил)-2-С-етил-1-О-метил-a-b-рибо-фураноза До діетилового ефіру (300мл) при -78°С повільно додавали EtMgBr (3,0Μ, 16,6мл) і потім по краплях сполуку стадії В прикладу 2 (4,80г, 10,0моль) у безводному Et2O (100мл). Реакційну суміш перемішували при 78°С протягом 15 хвилин, залишали нагріватися до -15°С та перемішували протягом ще 2 годин, потім виливали у суміш, що перемішується, води (300мл) і Et2O (600мл). Органічну фазу виділяли, сушили (MgSO4) і випарювали у вакуумі. Сирий продукт очищали на силікагелі, використовуючи етилацетат/гексан (1:2) як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (3,87г) у вигляді безбарвної олії. Стадія В: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-етил-b-D-рибо-фуранозил]-7Н-піроло[2,3d]піримідин До розчину сполуки стадії А (1,02мг, 2,0моль) у дихлорметані (40мл) по краплях додавали НВr (5,7Μ в оцтовій кислоті) (1,75мл, 10,0моль) при 0°С. Одержаний розчин перемішували при кімнатній температурі протягом 2 годин, випарювали у вакуумі і двічі випарювали разом з толуолом (10мл). Маслянистий залишок розчиняли в ацетонітрилі (10мл) і додавали до суміші, що інтенсивно перемішується, 4-хлор-1H-піроло[2,3d]піримідину (307мг, 2,0моль), гідроксиду калію (337мг, 6,0моль) і трис[2-(2-метоксіетокси)етил]амiну (130мг, 0,4моль) в ацетонітрилі (10мл). Одержану суміш перемішували при кімнатній температурі протягом ночі і потім виливали у суміш, що перемішується, насиченого амонійхлориду (100мл) та етилацетату (100мл). Органічний шар виділяли, промивали сольовим розчином (100мл), сушили над MgSO4, фільтрували та випарювали у вакуумі. Сирий продукт очищали на силікагелі, використовуючи е тилацетат/гексан (1:2) як елюент і одержуючи потрібний продукт (307мг) у вигляді безбарвної піни. Стадія С: 4-хлор-7-(2-С-етил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До розчину сполуки стадії В (307мг, 0,45моль) у ди хлорметані (8мл) додавали трихлористий бор (1Μ у дихлорметані) (4,50мл, 4,50моль) при -78°С. Суміш перемішували при -78°С протягом 1 години, потім при 10°С протягом 3 годин. Реакцію гасили, додаючи метанол/дихлорметан (1:1) (10мл), перемішували при -15°С протягом 30 хвилин і нейтралізували, додаючи водний гідроксид амонію. Суміш випарювали при зниженому тиску і одержану олію очищали на силікагелі, використовуючи метанол/дихлорметан (1:9) як елюент. Фракції, які містять продукт, об'єднували і випарювали у вакуумі, одержуючи потрібний продукт (112мг) у вигляді безбарвної піни. Стадія D: 4-аміно-7-(2-С-етил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До сполуки стадії С (50мг, 0,16моль) додавали насичений аміак у метанолі (4мл). Суміш перемішували при 75°С протягом 72 годин у закритому контейнері, охолоджували і випарювали у вакуумі. Сиру суміш очи щали на силікагелі, використовуючи метанол/дихлорметан (1:9) як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (29мг) у ви гляді безбарвного порошку. 1 H-ЯМР (200 МГц, ДМСО-d6): δ 0,52 (т, 3Н), 1,02 (м, 2Н), 4,01-3,24 (м, 6Н), 5,06 (м, 1Н), 6,01 (с, 1Н), 6,51 (д, 1Н), 6,95 (шир.с, 2Н), 6,70 (д, 1Н), 7,99 (с, 1Н). РХ-МС: знайдено: 295,2 (М+Н+); розраховано для С13Η18Ν4Ο4+Η+: 295,14. Приклад 4 2-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3H)-он Стадія А: 2-аміно-4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-метил-b-D-рибофуранозил]-7Нпіроло[2,3-d]піримідин До охолодженого льодом розчину продукту стадії С прикладу 2 (1,27г, 2,57моль) у СН2Сl2 (30мл) по краплях додавали НВr (5,7Μ в оцтовій кислоті; 3мл). Реакційну суміш перемішували при кімнатній температурі протягом 2 годин, концентрували при зниженому тиску і випарювали разом з толуолом (2x15мл). Одержану олію розчиняли в ацетонітрилі (MeCN) (15мл) і додавали по краплях до суміші, яка добре перемішується, 2аміно-4-хлор-7Н-піроло[2,3-d]піримідину [спосіб одержання див.: Heterocycles 35: 825 (1993)] (433мг, 2,57моль), КОН (85%, порошок) (0,51г, 7,7моль), трис[2-(2-метоксіетокси)етил]аміну (165мкл, 0,51моль) в ацетонітрилі (30мл). Одержану суміш перемішували при кімнатній температурі протягом 1 години, фільтрували та випарювали. Залишок очищали на колонці з силікагелем, використовуючи гексан/ЕtOАс, 5/1, 3/1 і 2/1, як елюент і одержуючи зазначену у заголовку сполуку у вигляді безбарвної піни (0,65г). Стадія В: 2-аміно-4-хлор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До розчину продукту стадії А (630мг, 1,0моль) у СН2Сl2 (20мл) при -78°С додавали трихлористий бор (1Μ у СН2СІ2) (10мл, 10моль). Суміш перемішували при -78°С протягом 2 годин, потім при -20°С протягом 2,5 години. Реакцію гасили, використовуючи СН 2Сl2/МеOН (1:1) (10мл), перемішували при -20°С протягом 0,5 години і нейтралізували при 0°С водним аміаком. Тверду частину фільтрували, промивали СН2Сl2/МеOН (1:1) і об'єднаний фільтрат випарювали у вакуумі. Залишок очищали на колонці з силікагелем, використовуючи СН2Сl2/МеOН, 50/1 і 20/1, як елюент, одержуючи зазначену у заголовку сполуку у вигляді безбарвної піни (250мг). Стадія С: 2-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3H)-он Суміш продукту стадії В (90мг, 0,3моль) у водному NaOH (2н, 9мл) нагрівали при температурі кипіння зі зворотним холодильником протягом 5 годин, потім нейтралізували при 0°С 2н водною НСl та випарювали досуха. У результаті очищення на колонці з силікагелем з використанням СН 2Сl2/МеOН, 5/1, як елюенту, одержували зазначену у заголовку сполук у у вигляді твердої речовини білого кольору (70мг). 1 H-ЯМР (200 МГц, CD3OD): δ 0,86 (с, 3Н), 3,79 (м, 1Н), 3,90-4,05 (м, 3Н), 6,06 (с, 1H), 6,42 (д, J=3,7 Гц, 1Н), 7,05 (д, J=3,7 Гц, 1H). Приклад 5 2-аміно-4-циклопропіламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Розчин 2-аміно-4-хлор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину (приклад 4, стадія В) (21мг, 0,07моль) у циклопропіламіні (0,5мл) нагрівали при 70°С протягом 2 діб, потім випарювали до маслянистого залишку та очи щали на колонці з силікагелем, використовуючи СН2Сl2/МеOН, 20/1, як елюент, одержуючи зазначену у заголовку сполуку у вигляді твердої речовини білого кольору (17мг). 1 Н-ЯМР (200 МГц, CD3CN): δ 0,61 (м, 2Н), 0,81 (м, 2Н), 0,85 (с, 3Н), 2,83 (м, 1Н), 3,74-3,86 (м, 1H), 3,93-4,03 (м, 2Н), 4,11 (д, J=8,9 Гц, 1H), 6,02 (с, 1Н), 6,49 (д, J=3,7 Гц, 1H), 7,00 (д, J=3,7 Гц, 1Н). Приклад 6 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-5-карбонітрил Дану сполуку одержували за способом, описаним Y. Murai et al. у Heterocycles 33:391-404(1992). Приклад 7 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-5-карбоксамід Дану сполуку одержували за способом, описаним Y. Murai et al. у Heterocycles 33:391-404(1992). Приклад 8 Загальний спосіб одержання SATE пролікарського фрагмента S-ацил-2-тіоетил (SATE) пронуклеотиди обговорені у C.R. Wagner, V.V. Iyer, and E.J. Mclntee, "Pronucleotides: Toward the In Vivo Delivery of Antiviral and Anticancer Nucleotides", Med. Res. Rev., 20: 1-35 (2000), причому зміст включений в опис як посилання у повному обсязі. SATE похідні нуклеозидів розкриті також у патентах США №№5770725, 5849905 і 6020482, зміст яких також включений в опис як посилання у повному обсязі. Біс(S-ацетил-2-тіоетил)-N,N-діізопропілфосфорамідит 2-меркаптоетанол (5г, 64моль) розчиняли у СН 2Сl2 (50мл). До цього розчину додавали триетиламін (7,67мл, 57,6моль) і реакційну суміш охолоджували у бані з льодом до 0°С. За 10 хвилин по краплях додавали оцтовий ангідрид (4,54мл, 48моль) і реакційну суміш перемішували протягом 1 години при 0°С. Потім реакційній суміші давали нагрітися до кімнатної температури протягом 2 годин. Реакційну суміш розбавляли СН2СІ2 (50мл), промивали водою (75мл), 5% водним NaHCO3 (75мл) та сольовим розчином (75мл). Органічну фазу сушили над безводним Na2SO4 і концентрували у вакуумі, одержуючи олію. Потім дану олію розчиняли у безводному ТГФ (40мл) і додавали безводний триетиламін (7,76мл). До цієї суміші додавали активовані молекулярні сита (4Å) і витримували при кімнатній температурі протягом 10 хвилин. Реакційну суміш охолоджували у бані з льодом до 0°С та додавали діізопропілфосфорамідний дихлорид (6,47г, 32,03моль). Реакційну суміш перемішували при 0°С протягом 2 годин в інертній атмосфері. До реакційної суміші додавали гексан (40мл) і осад, що утворювався, відфільтровували. Одержаний фільтрат концентрували до чверті об'єму, очищали хроматографічно на колонці з силікагелем та елюювали гексаном, який містить 3% триетиламіну і зростаючу кількість етилацетату (0-7%), одержуючи зазначену у заголовку сполуку у вигляді олії (2,36г). 1 H-ЯМР (CDCl3): δ 1,17 (с, 6Н), 1,21 (с, 6Н), 2,36 (с, 6Н), 3,14 (т, J=6,44 Гц), 3,51-3,84 (м, 6Н). 13 С-ЯМР (CDCl3): δ 24,47, 24,61, 30,48, 42,85,43,1, 61,88, 62,23, 195,26. 13 Р-Я МР (CDCl 3): δ 146,96. Приклад 9 5-трифосфатні похідні Нуклеозидні 5'-трифосфати даного винаходу одержували за способами, описаними у Chem. Rev. 100: 2047 (2000). Приклад 10 Очищення і аналіз ступеня чистоти 5'-трифосфатних по хідних Трифосфатні похідні очищали за допомогою аніонообмінної (АХ) хроматографії, використовуючи колонку 30x100мм Mono Q (Pharmacia) з буферною системою 50мм Tris, pH 8. Градієнти елюювання звичайно складали від 40 мм NaCl до 0,8 Μ NaCl у двох об'ємах сто впчика при швидкості 6,5мл/хв. Відповідні фракції, одержані за допомогою аніонообмінної хроматографії, збирали та знесолювали за допомогою хроматографії з оберненою фазою (RP), використовуючи колонку Luna C18 250x21мм (Phenomenex) при швидкості потоку 10мл/хв. Градієнти елюювання звичайно складали від 1% до 95% метанолу через 14 хвилин при постійній концентрації 5мМ триетиламонійацетату (ТЕАА). Мас-спектри очищених трифосфа тів одержували, використовуючи прилад, де скомбіновані послідовно ВЕРХ і мас-спектрометр Hewlett-Packard (Palo Alto, CA) MSD 1100. Для ВЕРХ з оберненою фазою використовували Phenomenex Luna (С 18(2)), 150x2мм, плюс 30x2мм guard колонку з розміром частинок 3мкм. Здійснювали градієнтне елюювання з лінійним градієнтом від 0 до 50% (15 хвилин) ацетонітрилу в 20мм ТЕАА (триетиламонійацетат) pH 7 з підключеним послідовно мас-спектральним детектором у режимі негативної іонізації. Для створення потоку іонів використовували газоподібний азот і пневматичний розпилювач. Одержували маси в інтервалі 150-900. Молекулярні маси визначали, використовуючи аналітичні програми HP Chemstation analysis package. Ступінь чистоти очищених трифосфатів визначали за допомогою аналітичної ВЕРХ з оберненою фазою та аніонообмінної ВЕРХ. ВЕРХ з оберненою фазою з колонкою Phenomonex Luna або Jupiter (250x4,6мм), з розміром частинок 5-мкм звичайно здійснювали з градієнтом 2-70% ацетонітрилу у 100мМ ТЕАА, pH 7, через 15 хвилин. Аніонообмінну ВЕРХ здійснювали на колонці 1,6x5мм Mono Q (Pharmacia). Трифосфати елюювали з градієнтом від 0 до 0,4Μ NaCl при постійній концентрації 50мМ Tris, pH 8. Ступінь чистоти трифосфатів звичайно складає більше 80%. Приклад 11 5'-монофосфатні похідні Нуклеозидні 5¢-монофосфати даного винаходу одержували за способом, описаним у Tetrahedron Lett. 50: 5065 (1967). Приклад 12 Mac-спектральні характеристики 5'-трифосфатних по хідних Мас-спектри 5'-трифосфатів сполук даного винаходу одержували за способом прикладу 10. У наступній таблиці перераховані розрахункові та експериментальні маси для представницьких 5'-трифосфатів, одержаних за способом прикладу 9. Номера прикладів відповідають вихідній сполуці 5'-трифосфату. Приклад 1 2 3 4 Розраховано 520,0 520,0 534,0 536,0 Знайдено 519,9 520,0 534,0 536,0 Приклад 13 [4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин]-5'-монофосфат До сполуки стадії F прикладу 2 (14мг, 0,05моль) (висушеної у результаті спільного випарювання з піридином і декілька разів з толуолом) додавали триметилфосфат (0,5мл). Суміш перемішували протягом ночі у запаяному контейнері. Потім її охолоджували до 0°С та через шприц додавали хлорокис фосфору (0,0070мл, 0,075моль).Суміш перемішували протягом 3 годин при 0°С, потім реакцію гасили, додаючи бікарбонат тетраетиламонію (ТЕАВ) (1 М) (0,5мл) і воду (5мл). Реакційну суміш очи щали і аналізували за способом прикладу 10. Мас-спектр з розсіюванням електронів (ES-MS): знайдено: 359,2 (М-Н+), розраховано для С12Н17N4О 7Р-Н+: 359,1. Приклад 14 [4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин]-5'дифосфат До сполуки стадії F прикладу 2 (56мг, 0,20моль) (висушеної спільним випарюванням з піридином і декілька разів з толуолом) додавали триметилфосфат (який зберігався над ситами) (1,0мл). Суміш перемішували протягом ночі у запаяному контейнері. Потім її охолоджували до 0°С та через шприц додавали хлорокис фосфор у (0,023мл, 0,25моль). Суміш перемішували протягом 2 годин при 0°С, потім додавали трибутиламін (0,238мл, 1,00моль) і трибутиламонійфосфат (одержаний з фосфорної кислоти і трибутиламіну у піридині з наступною азеотропною перегонкою з піридином і ацетонітрилом) (1,0моль у 3,30мл ацетонітрилу). Суміш перемішували протягом додаткових 30 хвилин при 0°С, потім відкривали запаяну ампулу і реакцію гасили, додаючи ТЕАВ (1 М) (1,0мл) і воду (5мл). Реакційну суміш очи щали та аналізували за способом прикладу 10. ES-MS: знайдено: 439,0 (М-Н+), розраховано для C12H18N4O 10P 2-H+: 439,04. Приклад 15 [4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин]-5'-трифосфат До сполуки стадії F прикладу 2 (20мг, 0,07моль) (висушеної спільним випарюванням з піридином і декілька разів з толуолом) додавали триметилфосфат (який зберігався над ситами) (0,4мл). Суміш перемішували протягом ночі у запаяному контейнері. Потім її охолоджували до 0°С та через шприц додавали хлорокис фосфор у (0,0070мл, 0,075моль). Суміш перемішували протягом 3 годин при 0°С, потім додавали трибутиламін (0,083мл, 0,35моль), трибутиламонійфосфат (127мг, 0,35моль) і ацетонітрил (який зберігався над ситами) (0,25мл). Суміш перемішували протягом додаткових 30 хвилин при 0°С, потім відкривали запаяну ампулу, і реакцію гасили, додаючи ТЕАВ (1М) (0,5мл) і воду (5мл). Реакційну суміш очи щали та аналізували за способом прикладу 10. ES-MS: знайдено: 519,0 (М-Н+), розраховано для C12Η19N4Ο 13P 3-Η+: 519,01. Приклад 16 7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он До сполуки стадії Ε прикладу 2 (59мг, 0,18моль) додавали водний гідроксид натрію (1М). Суміш нагрівали при кипінні зі зворотним холодильником протягом 1 години, охолоджували, нейтралізували водною НСl (2М) і випарювали у вакуумі. Залишок очищали на силікагелі, використовуючи ди хлорметан/метанол (4:1) як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (53мг) у вигляді безбарвної олії. 1 H-ЯМР (CD3CN): δ 0,70 (с, 3Н), 3,34-4,15 (такий, що перекривається м, 7Н), 6,16 (с, 1H), 6,57 (д, 3,6 Гц, 1H), 7,37 (д, 3,6 Гц, 1H), 8,83 (с, 1H). Приклад 17 4-аміно-5-хлор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До попередньо охолодженого (0°С) розчину сполуки стадії F прикладу 2 (140мг, 0,50моль) у ДМФ (2,5мл) по краплях додавали N-хлорсукцинімід (0,075г, 0,55моль) у ДМФ (0,5мл). Розчин перемішували при кімнатній температурі протягом 1 години, реакцію гасили, додаючи метанол (4мл), і випарювали у вакуумі. Сирий продукт очищали на силікагелі, використовуючи метанол/дихлорметан (1:9) як елюент. Фракції, які містять продукт, об'єднували і випарювали у вакуумі, одержуючи потрібний продукт (55мг) у вигляді безбарвної твердої речовини. 1 H-ЯМР (CD3CN): δ 0,80 (с, 3Н), 3,65-4,14 (такий, що перекривається м, 7Н), 5,97 (шир.с, 2Н), 6,17 (с, 1H), 7,51 (с, 1Н), 8,16 (с, 1Н). ES-MS: знайдено: 315,0 (М+Н+), розраховано для С12Н15СlN4O4+H+: 315,09. Приклад 18 4-аміно-5-бром-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До попередньо охолодженого (0°С) розчину сполуки стадії F прикладу 2 (28мг, 0,10моль) у ДМФ (0,5мл) по краплях додавали N-бромсукцинімід (0,018г, 0,10моль) у ДМФ (0,5мл). Розчин перемішували при 0°С протягом 20 хвилин, потім при кімнатній температурі протягом 10 хвилин. Реакційний розчин гасили, додаючи метанол (4мл), і випарювали у вакуумі. Сирий продукт очищали на силікагелі, використовуючи метанол/дихлорметан (1:9) як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (13,0мг) у вигляді безбарвної твердої речовини. 1 Н-ЯМР (CD3CN): δ 0,69 (с, 3Н), 3,46-4,00 (такий, що перекривається м, 7Н), 5,83 (шир.с, 2Н), 6,06 (с, 1H), 7,45 (с, 1Н), 8,05 (с, 1H). ES-MS: знайдено: 359,1 (М+Н+), розраховано для C12H15BrN4O4+H+: 359,04. Приклад 19 2-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Суміш 2-аміно-4-хлор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідину (приклад 4, стадія В) (20мг, 0,07моль) у EtOH (1,0мл), піридину (0,1мл) і 10% Pd/C (6мг) в атмосфері Н 2 (тиск атмосферний) перемішували протягом ночі при кімнатній температурі. Суміш фільтрували через шар целіту, який ретельно промивали EtOH. Об'єднаний фільтрат випарювали та очищали на колонці з силікагелем, використовуючи СН2Сl2/МеОН, 20/1 і 10/1, як елюент, одержуючи зазначену у заголовку сполуку у вигляді твердої речовини білого кольору (16мг). 1 Н-ЯМР (200 МГц, CD3OD): δ 0,86 (с, 3Н, 2'С-Ме), 3,82 (дд, J 5¢4¢.=3,6 Гц, J5¢,5² =12,7 Гц, 1Н, Н-5¢), 3,94-4,03 (м, 2Н, Н-5¢, Н-4'), 4,10 (д, J3¢ 4¢=8,8 Гц, 1Н, Н-3¢), 6,02 (с, 1Н, Н-1¢), 6,41 (д, J 5,6=3,8 Гц, 1H, Н-5), 7,39 (д, 1H, Н-6), 8,43 (с, 1H, Н-4). ES-MS: 281,4 (МН+). Приклад 20 2-аміно-5-метил-7-(2-С,2-О-диметил-b-D-рибофуранозил)-7Н-піроло[2,3d]піримідин-4(3H)-он Стадія А: 2-аміно-4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-метил-b-D-рибофуранозил1]-5-метил7Н-піроло[2,3-d]піримідин До охолодженого льодом розчину продукту стадії С прикладу 2 (1,57г, 3,16моль) у СН2Сl2 (50мл) по краплях додавали НВr (5,7Μ в оцтовій кислоті; 3,3мл). Реакційну суміш перемішували при 0°С протягом 1 години і потім при кімнатній температурі протягом 2 годин, концентрували у вакуумі та випарювали разом з толуолом (2x20мл). Одержану олію розчиняли у MeCN (20мл) і додавали по краплях до розчину натрієвої солі 2-аміно-4-хлор-5-метил-1H-піроло[2,3-d]піримідину в ацетонітрилі [одержаний in situ з 2-аміно-4-хлор-5-метил1H-піроло[2,3-d]піримідину [спосіб одержання див.: Liebigs Ann. Chem. 1984: 708-721] (1,13г, 6,2моль) у безводному ацетонітрилі (150мл) і NaH (60% у мінеральному маслі, 248мг, 6,2моль), після 2 годин перемішування при кімнатній температурі]. Об'єднану суміш перемішували при кімнатній температурі протягом 24 годин і потім випарювали досуха. Залишок суспендували у воді (100мл) та екстрагували ЕtOАс (300+150мл). Об'єднані екстракти промивали сольовим розчином (100мл), сушили над (Na2SO 4), фільтрували і випарювали. Сирий продукт очищали на колонці з силікагелем (5x7см), використовуючи етилацетат/гексан (від 0 до 30% ЕtOАс; 5% ступінчастий градієнт) як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (0,96г) у вигляді безбарвної піни. Стадія В: 2-аміно-4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С,2-О-диметил-b-D-рибофуранозил]-5метил-7Н-піроло[2,3-d]піримідин До охолодженої льодом суміші продукту стадії А (475мг, 0,7моль) у ТГФ (7мл) додавали NaH (60% у мінеральному маслі, 29мг) та перемішували при 0°С протягом 0,5 години. Потім додавали МеІ (48мкл) і реакційну суміш перемішували при кімнатній температурі протягом 24 годин. Реакцію гасили МеОН і суміш випарювали. Сирий продукт очищали на колонці з силікагелем (5x3,5см), використовуючи гексан/етилацетат (9/1, 7/1, 5/1 і 3/1) як елюент. Фракції, які містять продукт, об'єднували та випарювали, одержуючи потрібну сполуку (200мг) у ви гляді безбарвної піни. Стадія С: 2-аміно-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С,2-О-диметил-b-D-рибофуранозил]-5-метил-7Нпіроло[2,3-а]піримідин-4(3H)-он Суміш продукту стадії В (200мг, 0,3моль) у 1,4-діоксані (15мл) і водного NaOH (2н, 15мл) в автоклаві нагрівали протягом ночі при 135°С. Потім суміш охолоджували до 0°С, нейтралізували 2н водною НСl та випарювали досуха. Сирий продукт суспендували в МеОН, фільтрували і тверду частину ретельно промивали МеОН. Об'єднаний фільтрат концентрували, і залишок очищали на колонці з силікагелем (5x5см), використовуючи СН 2Сl2/МеOН (40/1, 30/1 і 20/1) як елюент, одержуючи потрібну сполуку (150мг) у вигляді безбарвної піни. Стадія D: 2-аміно-5-метил-7-(2-С,2-О-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3H)-он Суміш продукту стадії С (64мг, 0,1моль) у МеОН (5мл), Et3N (0,2мл) і 10% Pd/C (24мг) гідрували в апараті для гідрування Пара при тиску 50 псі при кімнатній температурі протягом 1,5 доби, потім фільтрували через шар целіту, який ретельно промивали МеОН. Об'єднаний фільтрат випарювали і залишок очищали на колонці з силікагелем (3x4см), використовуючи СН 2Сl2/МеOН (30/1, 20/1) як елюент, одержуючи 2-аміно-5-метил-7-(5O-бензил-2-С,2-O-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-4(3Н)-он. Дану сполуку гідрували далі в EtOH (2мл), використовуючи 10% Pd/C при атмосферному тиску водню. Після перемішування протягом 2 днів при кімнатній температурі реакційну суміш фільтрували через целіт, фільтрат випарювали і сирий продукт очищали на колонці з силікагелем (1x7см), використовуючи СН2Сl2/МеOН (30/1, 20/1 і 10/1) як елюент, одержуючи після сушіння виморожуванням зазначену у заголовку сполуку (12мг). 1 H-ЯМР (200 МГц, CD3OD): δ 0,81 (с, 3Н, 2'С-Ме), 2,16 (д, JH-6,C5-Me=1,3 Гц, 3Н, С5-Ме), 3,41 (с, 3Н, 2'-ОМе), 3,67 (дд, J5¢,4¢-=3,4 Гц, J5¢,5² =126 Гц, 1Н, Н-5'), 3,81-3,91 (м, 3Н, Н-5", Н-4', Н-3'), 6,10 (с, 1H, Н-1¢), 6,66 (д, 1H, Н6). ES-MS: 323,3 (М-Н)+. Приклад 21 4-аміно-5-метил-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-метил-b-D-рибофуранозил]-5-метил-7Нпіроло[2,3-d]піримідин До охолодженого льодом розчину продукту стадії С прикладу 2 (1,06г, 2,1моль) у СН2Сl2 (30мл) по краплях додавали НВr (5,7Μ в оцтовій кислоті; 2,2мл). Реакційну суміш перемішували при 0°С протягом 1 години і потім при кімнатній температурі протягом 2 годин, концентрували у вакуумі та випарювали разом з толуолом (2x15мл). Одержану олію розчиняли MeCN (10мл) і додавали по краплях до розчину натрієвої солі 4-хлор-5метил-1H-піроло[2,3-d]піримідину в ацетонітрилі [одержаний in situ з 4-хлор-5-метил-1H-піроло[2,3d]піримідину [спосіб одержання див.: J. Med. Chem. 33: 1984 (1990)] (0,62г, 3,7моль) у безводному ацетонітрилі (70мл) і NaH (60% у мінеральному маслі, 148мг, 3,7моль), після 2 годин інтенсивного перемішування при кімнатній температурі]. Об'єднану суміш перемішували при кімнатній температурі протягом 24 годин, а потім випарювали досуха. Залишок суспендували у воді (100мл) та екстрагували ЕtOАс (250+100мл). Об'єднані екстракти промивали сольовим розчином (50мл), сушили над Na2SO4, фільтрували і випарювали. Сирий продукт очищали на колонці з силікагелем (5x5см), використовуючи градієнт гексан/етилацетат (9/1, 5/1, 3/1) як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (0,87г) у вигляді безбарвної піни. Стадія В: 4-хлор-5-метил-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До розчину сполуки стадії А (0,87г, 0,9моль) у дихлорметані (30мл) при -78°С по краплях додавали трихлористий бор (1Μ у дихлорметані, 9,0мл, 9,0моль). Суміш перемішували при -78°С протягом 2,5 години, потім при температурі від -30°С до -20°С протягом 3 годин. Реакцію гасили, додаючи метанол/дихлорметан (1:1) (9мл), і одержану суміш перемішували при -15°С протягом 30 хвилин, потім нейтралізували водним аміаком при 0°С та перемішували при кімнатній температурі протягом 15 хвилин. Тверду частину фільтрували і промивали СН 2Сl2/МеOН (1/1, 50мл). Об'єднаний фільтрат випарювали і залишок очищали на колонці з силікагелем (5x5см), використовуючи СН 2Сl2 і градієнт СН2Сl2/МеOН (40/1 і 30/1) як елюент до одержання потрібної сполуки (0,22г) у вигляді безбарвної піни. Стадія С: 4-аміно-5-метил-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До сполуки стадії В (0,2г, 0,64моль) додавали метанольний аміак (насичений при 0°С; 40мл). Суміш нагрівали в автоклаві з нержавіючої сталі при 100°С протягом 14 годин, потім охолоджували та випарювали у вакуумі. Сиру суміш очищали на колонці з силікагелем (5x5см), використовуючи градієнт СН 2Сl2/МеOН (50/1, 30/1, 20/1) як елюент, одержуючи зазначену у заголовку сполуку у вигляді твердої речовини білого кольору (0,12г). 1 H-ЯМР (ДМСО-d 6): δ 0,60 (с, 3Н, 2'С-Ме), 2,26 (с, 3Н, 5С-Ме), 3,52-3,61 (м, 1Н, Н-5¢), 3,70-3,88 (м, 3Н, Н-5", Н-4', Н-3¢), 5,00 (с, 1Н, 2'-ОН), 4,91-4,99 (м, 3Н, 2¢-ОН, 3'-ОН, 5'-ОН), 6,04 (с, 1Н, Н-1¢), 6,48 (шир.с, 2Н, ΝΗ2), 7,12 (с, 1H, Н-6), 7,94 (с, 1Н, Н-2). ES-MS: 295,2 (МН+). Приклад 22 4-аміно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин-5-карбонова кислота Сполуку прикладу 6 (0,035г, 0,11моль) розчиняли у суміші водного аміаку (4мл, 30мас. %) та насиченого метанольного аміаку (2мл) і додавали розчин Н2О2 у воді (2мл, 35мас. %). Реакційну суміш перемішували при кімнатній температурі протягом 18 годин. Розчинник видаляли при зниженому тиску і одержаний залишок очищали за допомогою ВЕРХ на колонці з оберненою фазою (Altech Altima С-18, 10x299мм, А=вода , В=ацетонітрил , 10-60% В протягом 50 хвилин, швидкість потоку 2мл/хв), одержуючи зазначену у заголовку сполуку (0,015г, 41%) у вигляді твердої речовини білого кольору. 1 H-ЯМР (CD3OD): δ 0,85 (с, 3Н, Me), 3,61 (м, 1H), 3,82 (м, 1H), 3,99-4,86 (м, 2Н), 6,26 (с, 1H), 8,10 (с, 2Н) 8,22 (с, 1Н). 13 С-ЯМР (CD3OD): δ 20,13, 61,37, 73,79, 80,42, 84,01, 93,00, 102,66, 112,07, 130,07, 151,40, 152,74, 159,12, 169,30. Мас-спектр високого розрізнення (HRMS) (з бомбардуванням швидкими атомами (FAB)): розраховано для C13H17N4 O6+: 325,1148, знайдено: 325,1143. Приклад 23 4-аміно-7-(2-С-вініл-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 3,5-біс-О-(2,4-дихлорфенілметил)-2-С-вініл-1-О-метил-a-D-рибофураноза Гептагідрат хлориду церію (50г, 134,2моль) тонко подрібнюють у попередньо нагрітій ступці і переносять у круглодонну колбу, оснащену механічною мішалкою. Колбу нагрівали при високому вакуумі протягом ночі при 160°С. Вакуум замінювали атмосферою аргону і колбу охолоджували до кімнатної температури. У колбу через канюлю вводили безводний ТГФ (300мл). Одержану суспензію перемішували при кімнатній температурі протягом 4 годин і потім охолоджували до -78°С. Додавали вінілмагнійбромід (1Μ у ТГФ, 120мл, 120моль) і перемішування продовжували при -78°С протягом 2 годин. До цієї суспензії по краплях додавали розчин 3,5біс-O-(2,4-дихлорфенілметил)-1-O-метил-a-D-еритропентофураноз-2-улози (14м, 30моль) [прикладу 2, стадії В] у безводному ТГФ (100мл) при постійному перемішуванні. Реакційну суміш перемішували при -78°С протягом 4 годин. Реакцію гасили насиченим розчином амонійхлориду та залишали нагріватися до кімнатної температури. Суміш фільтрували через целіт і залишок промивали Et2O (2x500мл). Органічний шар виділяли і водний шар екстрагували Et2O (2x200мл). Об'єднані органічні шари сушили над безводним Na 2SO4 і концентрували до в'язкої олії жовтого кольору. Дану олію очищали, використовуючи флеш-хроматографію (SiO2, 10% EtOAc у гексані). Зазначену у заголовку сполуку (6,1г, 13,2моль) одержували у вигляді олії блідожовтого кольору. Стадія В: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-вініл-b-D-рибо-фуранозил]-7Н-піроло[2,3d]піримідин До розчину сполуки стадії А (6,4г, 12,6моль) у безводному дихлорметані (150мл) при -20°С по краплях додавали НВr (30% розчин в АсОН, 20мл, 75,6моль). Одержаний розчин перемішували при температурі між 10°С і 0°С протягом 4 годин, випарювали у вакуумі та випарювали разом з безводним толуолом (3x40мл). Маслянистий залишок розчиняли у безводному ацетонітрилі (100мл) і додавали до розчину натрієвої солі 4хлор-1H-піроло[2,3-d]піримідину (5,8г, 37,8моль) в ацетонітрилі (одержаної in situ за способом прикладу 2) при -20°С. Одержану суміш залишали нагріватися до кімнатної температури і перемішували при кімнатній температурі протягом 24 годин. Потім суміш випарювали досуха, поміщали у воду та екстрагували ЕtOАс (2x300мл). Об'єднані екстракти сушили над Na2SO4, фільтрували і випарювали. Сиру суміш очищали, використовуючи флеш-хроматографію (SiO2, 10% ЕtOАс у гексані), і зазначену у заголовку сполуку (1,75г) виділяли у вигляді піни білого кольору. Стадія С: 4-аміно-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-вініл-b-D-рибофуранозил]-7Н-піроло[2,3d]піримідин Сполуку стадії В (80мг) розчиняли у мінімальній кількості 1,4-діоксану та поміщали у контейнер з нержавіючої сталі. Контейнер охолоджували до -78°С і додавали рідкий аміак. Контейнер запаювали і нагрівали при 90°С протягом 24 годин. Аміаку давали випаруватися і залишок концентрували до твердої речовини білого кольору, яку використовували на наступній стадії без додаткового очищення. Стадія D: 4-аміно-7-(2-С-вініл-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До розчину сполуки стадії С (60мг) у ди хлорметані при -78°С по краплях додавали трихлористий бор (1Μ у дихлорметані). Суміш перемішували при -78°С протягом 2,5 годин, потім при температурі від -30°С до -20°С протягом 3 годин. Реакцію гасили, додаючи метанол/дихлорметан (1:1), і одержану суміш перемішували при 15°С протягом 0,5 години, потім нейтралізували водним аміаком при 0°С і перемішували при кімнатній температурі протягом 15 хвилин. Тверду частин у фільтрували і промивали метанолом/дихлорметаном (1:1). Об'єднаний фільтрат випарювали і залишок очищали, використовуючи флеш-хроматографію (SiO2, 10% метанол в ЕtOАс, що містить 0,1% триетиламіну). Фракції, які містять продукт, випарювали, одержуючи зазначену у заголовку сполук у у вигляді твердої речовини білого кольору (10мг). 1 H-ЯМР (ДМСО-d6): δ 3,6 (м, 1H, Н-5¢), 3,8 (м, 1H, Н-5"), 3,9 (мд, 1H, Н-4¢), 4,3 (т, 1H, Н-3¢), 4,8-5,3 (м, 6Н, СН=СН2, 2'-ОН, 3'-ОН, 5'-ОН), 6,12 (с, 1H, Н-1¢), 6,59 (д, 1H, Н-5), 7,1 (шир.с, 1Н, ΝΗ2), 7,43 (д, 1H, Н-6), 8,01 (с, 1Н, Н-2). ES-MS: знайдено: 291,1 (М-Н-), розраховано для С13Н16N4O 4-Н-: 291,2. Приклад 24 4-аміно-7-(2-С-гідроксиметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-гідроксиметил-b-D-рибофуранозил]-7Нпіроло[2,3-в]піримідин До розчину сполуки прикладу 23, стадії В (300мг, 0,48моль) у 1,4-діоксані (5мл) додавали Тметилморфолін-N-оксид (300мг, 2,56моль) і чотириокис осмію (4% розчин у воді, 0,3мл). Суміш перемішували у темряві протягом 14 годин. Осад видаляли фільтруванням через шар целіту, розбавляли водою (3х) та екстрагували EtOAc. EtOAc шар сушили над Na2SO4 і концентрували у вакуумі. Маслянистий залишок поміщали у ди хлорметан (5мл) і перемішували над NaIO4 на силікагелі (3г, 10% NaIO4) протягом 12 годин. Силікагель видаляли фільтрацією, а залишок випарювали та поміщали в абсолютний етанол (5мл). Розчин охолоджували у бані з льодом і невеликими порціями додавали боргідрид натрію (300мг, 8моль). Одержану суміш перемішували при кімнатній температурі протягом 4 годин і потім розбавляли EtOAc. Органічний шар промивали водою (2x20мл), сольовим розчином (20мл) і сушили над Na2SO4. Розчинник випарювали і залишок очищали, використовуючи флеш-хроматографію (SiO2, 2:1 гексан/EtOAc), одержуючи зазначену у заголовку сполуку (160мг, 0,25моль) у вигляді пластівців білого кольору. Стадія В: 4-аміно-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-гідроксиметил-b-D-рибофуранозил]-7Нпіроло[2,3-d]піримідин Сполуку стадії А (150мг, 0,23моль) розчиняли у мінімальній кількості 1,4-діоксану (10мл) і поміщали у контейнер з нержавіючої сталі. Бомбу охолоджували до -78°С та додавали рідкий аміак. Контейнер запаювали і потім нагрівали при 90°С протягом 24 годин. Аміаку давали випаруватися і залишок концентрували до твердої речовини білого кольору, яку використовували на наступній стадії без додаткового очищення. Стадія С: 4-аміно-7-(2-С-гідроксиметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Сполуку стадії В (120мг, 0,2моль) розчиняли у 1:1 метанол/дихлорметан, додавали 10% Pd/C і одержану суспензію перемішували в атмосфері Н 2 протягом 12 годин. Каталізатор видаляли фільтруванням через шар целіту і промивали великими кількостями метанолу. Об'єднаний фільтрат випарювали у вакуумі і залишок очищали, використовуючи флеш-хроматографію (SiO2, 10% метанол у EtOAc, що містить 0,1% триетиламіну), одержуючи зазначену у заголовку сполуку (50мг) у вигляді порошку білого кольору. 1 H-ЯМР (CD3OD): δ 3,12 (д, 1H, СH2¢, 3,33 (д, 1Н, СН2"), 3,82 (м, 1H, Н-5'), 3,99-4,1 (м, 2Н, Н-4¢, Н-5"), 4,3 (д, 1Н, Н-3¢), 6,2 (с, 1H, Н-1¢), 6,58 (д, 1Н, Н-5), 7,45 (д, 1H, Н-6), 8,05 (с, 1H, Н-2). РХ-МС: знайдено: 297,2 (М+Н+), розраховано для C12Η16Ν4O5+Η+: 297,3. Приклад 25 4-аміно-7-(2-С-фторметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-фторметил-b-D-рибофуранозил]-7Н-піроло[2,3d]піримідин До розчину сполуки прикладу 24, стадії А (63мг, 0,1моль) у безводному дихлорметані (5мл) в атмосфері аргону додавали 4-диметиламінопіридин (DМАР) (2мг, 0,015моль) і триетиламін (62мкл, 0,45моль). Розчин охолоджували у бані з льодом і додавали п-толуолсульфонілхлорид (30мг, 0,15моль). Реакційну суміш перемішували при кімнатній температурі протягом ночі, промивали NaHCO3 (2x10мл), водою (10мл), сольовим розчином (10мл), сушили над Na2SO4 і концентрували у вакуумі до твердої речовини рожевого кольору. Тверду частину розчиняли у безводному ТГФ (5мл) і охолоджували у бані з льодом. Додавали тетрабутиламонійфторид (1Μ розчин у ТГФ, 1мл, 1моль) і суміш перемішували при кімнатній температурі протягом 4 годин. Розчинник видаляли у вакуумі, залишок поміщали у дихлорметан і промивали NaHCO3 (2x10мл), водою (10мл) і сольовим розчином (10мл). Шар дихлорметану сушили над безводним Na2SO4, концентрували у вакуумі та очищали, використовуючи фле ш-хроматографію (SiO2, 2:1 гексан/EtOAc), одержуючи зазначену у заголовку сполуку (20мг) у вигляді твердої речовини білого кольору. Стадія В: 4-аміно-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С-фторметил-b-D-рибофуранозил]-7Н-піроло[2,3dа]піримідин Сполуку стадії А (18мг, 0,03моль) розчиняли у мінімальній кількості 1,4-діоксану та поміщали у контейнер з нержавіючої сталі. Контейнер охолоджували до -78°С і додавали рідкий аміак. Контейнер запаювали і потім нагрівали при 90°С протягом 24 годин. Аміаку давали випаруватися і залишок концентрували до твердої речовини білого кольору, яку використовували на наступній стадії без додаткового очищення. Стадія С: 4-аміно-7-(2-С-фторметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Сполуку стадії В (16мг) розчиняли в 1:1 метанолі/дихлорметані, додавали 10% Pd/C і одержану суспензію перемішували в атмосфері Н 2 протягом 12 годин. Каталізатор видаляли фільтруванням через шар целіту і промивали великими кількостями метанолу. Об'єднаний фільтрат випарювали у вакуумі і залишок очищали, використовуючи фле ш-хроматографію (SiO2, 10% метанол у EtOAc, що містить 0,1% триетиламіну), одержуючи зазначену у заголовку сполуку (8мг) у вигляді порошку білого кольору. 1 H-ЯМР (ДМСО-d6): δ 3,6-3,7 (м, 1H, Н-5¢), 3,8-4,3 (м, 5Н, Н-5", Н-4¢, Н-3¢, СН2), 5,12 (т, 1Н, 5'-ОН), 5,35 (д, 1H, 3'-Н), 5,48 (с, 1H, 2'-ОН), 6,21 (с, 1H, Н-1¢), 6,52 (д, 1H, Н-5), 6,98 (шир.с, 2Н, NH2), 7,44 (д, 1Н, Н-6), 8,02 (с, 1H, Н-2). 19 F-Я МР (ДМСО-d 6): δ -230,2 (т). ES-MS: знайдено: 299,1 (М+Н+), розраховано для C12H15FN4 O4+H+: 299,27. Приклади 26 і 27 4-аміно-7-(3-дезокси-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин і 4-аміно-7-(3-дезокси-2-Сметил-b-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 7-[2,5-біс-О-(трет-бутилдиметилсиліл)-b-D-рибофуранозил]-7Н-піроло[2,3-d]піримідин і 7-[3,5-бісО-(трет-бутилдиметилсиліл)-b-D-рибофуранозил]-7Н-піроло[2,3-d]піримідин До розчину, що перемішується, туберцидину (5,0г, 18,7моль) у суміші піридину (7,5мл) і ДМФ (18,5мл) додавали нітрат срібла (6,36г, 38,8моль). Дану суміш перемішували при кімнатній температурі протягом 2 годин. Її о холоджували у бані з льодом, додавали ТГФ (37,4мл) і трет-бутилдиметилсилілхлорид (5,6г, 37моль) і суміш перемішували при кімнатній температурі протягом 2 годин. Потім суміш фільтрували через шар целіту і промивали ТГФ. Фільтрат і промивки розбавляли простим ефіром, який містить невелику кількість хлороформу. Органічний шар промивали послідовно бікарбонатом натрію і водою (3x50мл), сушили над безводним сульфатом натрію та концентрували. Піридин видаляли випаровуванням разом з толуолом, і залишок очищали, використовуючи флеш-хроматографію на силікагелі, використовуючи 5-7% МеОН у СН 2Сl2 як елюент; вихід 3,0г. Стадія В: 7-[2,5-біс-О-(трет-бутилдиметилсиліл)-b-D-рибофуранозил]-4-[ди(4метоксифеніл)фенілметил]аміно-7Н-піроло[2,3-d]піримідин і 7-[3,5-біс-О-(трет-бутилдиметилсиліл)-b-Dрибофуранозил]-4-[ди(4-метоксифеніл)фенілметил]аміно-7Н-піроло[2,3-d]піримідин До розчину суміші сполук стадії А (3,0г, 6,0моль) у безводному піридині (30мл) додавали 4,4'диметокситритилхлорид (2,8г, 8,2моль) і реакційну суміш перемішували при кімнатній температурі протягом ночі. Потім дану суміш ретельно розтирали з водним піридином і екстрагували ефіром. Органічний шар промивали водою, сушили над безводним сульфатом натрію та концентрували до піни жовтого кольору (5,6г). Залишок очищали, використовуючи фле ш-хроматографію на силікагелі, використовуючи 20-25% EtOAc у гексані як елюент. Відповідні фракції збирали та концентрували, одержуючи 2',5'-біс-О-(третбутилдиметилсиліл)- і 3',5'-біс-О-(трет-бутилдиметилсиліл)захищені нуклеозиди у вигляді безбарвної піни (2,2г і 1,0г, відповідно). Стадія С: 7-[2,5-біс-О-(трет-бутилдиметилсиліл)-3-О-тозил-b-D-рибофуранозил]-4-[ди(4метоксифеніл)фенілметил]аміно-7Н-піроло[2,3-d]піримідин До охолодженого льодом розчину 2',5'-біс-О-(трет-бутилдиметил-силіл)захищеного нуклеозиду стадії В (2,0г, 2,5моль) у піридині (22мл) додавали п-толуолсульфонілхлорид (1,9г, 9,8моль). Реакційну суміш перемішували при кімнатній температурі протягом 4 діб. Потім її ретельно розтирали з водним піридином (50%, 10мл) і екстрагували ефіром (3x50мл), який містить невелику кількість СН 2Сl2 (10мл). Органічний шар промивали бікарбонатом натрію та водою (3x30мл). Органічний шар сушили над безводним Na2SO4 і концентрували. Піридин видаляли при спільному випаровуванні з толуолом (3x25мл). Олію, що залишилася, фільтрували через шар силікагелю, використовуючи гексан:етилацетат (70:30) як елюент; ви хід 1,4г. Стадія D: 4-[ди(4-метоксифеніл)фенілметил]аміно-7-[3-О-тозил-b-D-рибофуранозил-7Н-піроло[2,3d]піримідин Розчин сполуки стадії С (1,0г, 1,1моль) і ТГФ (10мл) перемішували з тетрабутиламонійфторидом (1М розчин у ТГФ, 2,5мл) протягом 0,5 години. Суміш охолоджували та розбавляли простим ефіром (50мл). Розчин промивали водою (3x50мл), сушили над безводним Na2SO4 і концентрували до олії. Залишок очищали, пропускаючи через шар силікагелю, використовуючи гексан:етилацетат (1:1) як елюент; вихід 780мг. Стадія Е: 4-аміно-7-(3-дезокси-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин і 4-аміно-7-(3дезокси-2-С-метил-b-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідин Розчин CH3MgI (3,0Μ розчин у простому ефірі, 3,0мл) у безводному толуолі (3,75мл) охолоджували у бані з льодом. До цього додавали розчин сполуки стадії D (500мг, 0,8моль) у безводному толуолі (3,7мл). Одержану суміш перемішували при кімнатній температурі протягом 3,5 годин. її охолоджували та обробляли водним розчином NH4Cl і екстрагували простим ефіром (50мл, що містить 10мл СН 2Сl2). Органічний шар виділяли і промивали сольовим розчином (2x30мл) і водою (2x25мл), сушили над безводним Na2SO4 та концентрували до олії, яку очищали, використовуючи флеш-хроматографію на силікагелі, використовуючи 4% МеОН у СН 2Сl2, одержуючи сполуку 2-С-a-метил (149мг) і сполуку 2-С-b-метил (34мг). Дані похідні окремо обробляли 80% оцтовою кислотою і реакційну суміш перемішували при кімнатній температурі протягом 2,5 годин. Оцтову кислоту видаляли при повторних спільних випаровуваннях з етанолом і толуолом. Залишок розділяли між хлороформом і водою. Водний шар промивали хлороформом і концентрували. Після випарювання залишок очищали на силікагелі, використовуючи 5-10% МеОН у СН 2Сl2 як елюент, одержуючи потрібні сполуки у вигляді твердої речовини білого кольору. 4-аміно-7-(3-дезокси-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин (90мг) 1 Н-ЯМР (ДМСО-d6): δ 0,74 (с, 3Н, СН3), 1,77 (дд, 1H, Н-3¢), 2,08 (т, 1Н, Н-3"), 3,59 (м, 1H, Н-5¢), 3,73 (м, 1H, Н-5"), 4,15 (м, 1H, Н-4¢), 5,02 (т, 1H, ОН-5¢), 5,33 (с, 1H, ОН-2¢), 6,00 (с, 1Н, Н-1¢), 6,54 (д, 1H, Н-7), 6,95 (шир.с, 2Н, ΝΗ2), 7,47 (д, 1Н, Н-8), 8,00 (с, 1Н, Н-2). ES-MS: 263,1 [М-Н]. 4-аміно-7-(3-дезокси-2-С-метил-b-D-арабінофуранозил)-7Н-піроло[2,3-d]піримідин (15мг) 1 H-ЯМР (ДМСО-d 6): δ 1,23 (с, 3Н, СН3), 2,08 (ддд, 2Н, Н-3¢ і 3"), 3,57 (м, 2Н, Н-5¢ і 5"), 4,06 (м, 1Н, Н-4), 5,10 (с, 1H, ОН-2¢), 5,24 (т, 1Н, ОН-5¢), 6,01 (с, 1Н, Н-1¢), 6,49 (д, 1Н, Н-7), 6,89 (шир.с, 2Н, ΝΗ2), 7,35 (д, 1Н, Н-8), 8,01 (с, Ш, Н-2). ES-MS: 265,2 [М+Н]. Приклад 28 4-аміно-7-(2,4-С-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 5-дезокси-1,2-О-ізопропіліден-D-ксилофураноза 1,2-О-ізопропіліден-О-ксилофуранозу (38,4г, 0,2моль), 4-диметиламіно-піридин (5г), триетиламін (55,7мл, 0,4моль) розчиняли у дихлорметані (300мл). Додавали п-толуолсульфонілхлорид (38,13г, 0,2моль) і реакційну суміш перемішували при кімнатній температурі протягом 2 годин. Реакційну суміш потім виливали у насичений водний розчин бікарбонату натрію (500мл) і два шари розділялися. Органічний шар промивали водним розчином лимонної кислоти (20%, 200мл), сушили (Na2SO4) і випарювали, одержуючи тверду речовину (70,0г). Тверду частину розчиняли у сухому ТГФ (300мл) і порціями протягом 30 хвилин додавали LiAlH4 (16,0г, 0,42моль). Суміш перемішували при кімнатній температурі протягом 15 хвилин. Протягом 30 хвилин по краплях додавали етилацетат (100мл) і суміш фільтрували через шар силікагелю. Фільтрат концентрували і одержану олію обробляли хроматографічно на силікагелі (ЕtOАс/гексан 1/4), одержуючи продукт у вигляді твердої речовини (32,5г). Стадія В: 3,5-біс-О-(2,4-дихлорфенілметил)-1-О-метил-4-метил-a-D-рибофураноза Оксид хрому (50г, 0,5моль), оцтовий ангідрид (50мл, 0,53моль) і піридин (100мл, 1,24моль) додавали до дихлорметану (1л) у бані з льодом і суміш перемішували протягом 15 хвилин. Додавали 5-дезокси-1,2-Оізопропіліден-О-ксилофуранозу (32г, 0,18моль) у дихлорметані (200мл) і суміш перемішували при тій же температурі протягом 30 хвилин. Реакційний розчин розбавляли етилацетатом (1л) і фільтрували через шар силікагелю. Фільтрат концентрували, одержуючи олію жовтого кольору. Дану олію розчиняли у 1,4-діоксані (1л) та формальдегіді (37%, 200мл). Розчин охолоджували до 0°С і додавали твердий КОН (50г). Суміш перемішували при кімнатній температурі протягом ночі і потім екстрагували етилацетатом (6x200мл). Після концентрування залишок обробляли хроматографічно на силікагелі (ЕtOАс), одержуючи продукт у вигляді олії (1,5г). Дану олію розчиняли у 1-метил-2-піролідиноні (20мл) і додавали 2,4-дихлорфенілметилхлорид (4г, 20,5моль) і NaH (60%, 0,8г). Суміш перемішували протягом ночі і розбавляли толуолом (100мл). Потім суміш промивали насиченим водним бікарбонатом натрію (3x50мл), сушили (Na2SO4) та випарювали. Залишок розчиняли у метанолі (50мл) і додавали НСl у діоксані (4М, 2мл). Розчин перемішували протягом ночі та випарювали. Залишок обробляли хроматографічно на силікагелі (ЕtOАс/гексан: 1/4), одержуючи потрібний продукт у вигляді олії (2,01г). Стадія С: 3,5-біс-O-(2,4-дихлорфенілметил)-2,4-ди-С-метил-1-О-метил-a-D-рибофураноза Продукт (2,0г, 4,0моль) стадії В та періодинан Dess-Martin (2,0г) у дихлорметані (30мл) перемішували протягом ночі при кімнатній температурі і потім концентрували при зниженому тиску. Залишок ретельно розтирали з простим ефіром (50мл) і фільтрували. Фільтрат промивали розчином Nа2S2О3×5Н2О (2,5г) у насиченому водному розчині бікарбонату натрію (50мл), сушили (MgSO4), фільтрували та випарювали. Залишок розчиняли у безводному Et2O (20мл) і по краплях додавали до розчину Me MgBr у Et 2O (3Μ, 10мл) при -78°С. Реакційну суміш залишали нагріватися до -30°С і перемішували при температурі від -30°С до -15°С протягом 5 годин, потім виливали у насичений водний розчин амонійхлориду (50мл). Два шари розділяли і органічний шар сушили (MgSO4), фільтрували та концентрували. Залишок обробляли хроматографічно на силікагелі (ЕtOАс/гексан: 1/9), одержуючи зазначену у заголовку сполуку у вигляді сиропу (1,40г). Стадія D: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2,4-ди-С-метил-b-D-рибофуранозил]-7Н-піроло[2,3d]піримідин До сполуки стадії С (0,70г, 1,3моль) додавали НВr (5,7Μ в оцтовій кислоті, 2мл). Одержаний розчин перемішували при кімнатній температурі протягом 1 години, випарювали у вакуумі та випарювали разом з безводним толуолом (3x10мл). 4-хлор-7Н-піроло[2,3-d]піримідин (0,5г, 3,3моль) і порошок КОН (85%, 150мг, 2,3моль) перемішували в 1-метил-2-піролідиноні (5мл) протягом 30 хвилин і суміш випарювали разом з толуолом (10мл). Одержаний розчин виливали у вказаний вище бромцукровий залишок і суміш перемішували протягом ночі. Суміш розбавляли толуолом (50мл), промивали водою (3x50мл) і концентрували при зниженому тиску. Залишок обробляли хроматографічно на силікагелі, елююючи сумішшю EtOАс/гексан (15/85), одержуючи тверду речовину (270мг). Стадія Е: 4-аміно-7-(2,4-С-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Сполуку стадії D (270мг) розчиняли у діоксані (2мл) і додавали рідкий аміак (20г) в автоклав з нержавіючої сталі. Суміш нагрівали при 100°С протягом 15 хвилин, потім охолоджували та випарювали. Залишок обробляли хроматографічно на силікагелі (EtOAc), одержуючи тверду речовину (200мг). Тверду частину (150мг) і Pd/C (10%, 150мг) у метанолі (20мл) струшували в атмосфері Н 2 (30 псі) протягом 3 годин, фільтрували і випарювали. Залишок обробляли хроматографічно на силікагелі (МеОН/СН2Сl2: 1/9), одержуючи потрібний продукт у вигляді твердої речовини (35мг). 1 H-ЯМР (ДМСО-d6): δ 0,65 (с, 3Н), 1,18 (с, 3Н), 3,43 (м, 2Н), 4,06 (д, 1H, J=6,3 Гц), 4,87 (с, 1Н), 5,26 (шир., 1H), 5,08 (д, 1H, J=6,3 Гц), 5,25 (т, 1Н, J=3,0 Гц), 6,17 (с, 1Н), 6,54 (д, 1Н, J=3,5 Гц), 6,97 (шир.с, 2Н), 7,54 (д, 1Н, J=3,4 Гц), 8,02 (с, 1Н). 13 С-ЯМР (ДМСО-d 6): δ 18,19, 21,32, 65,38, 73,00, 79,33, 84,80, 90,66, 99,09, 102,41, 121,90, 149,58, 151,48, 157,38. РХ-МС: знайдено: 295,1 (М+Н+), розраховано для C13Η18Ν4Ο4+Η+: 295,1. Приклад 29 4-аміно-7-(3-дезокси-3-фтор-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 3-дезокси-3-фтор-1-О-метил-5-О-толуоїл-a-D-рибофураноза 1,2-О-ізопропіліден-О-ксилофуранозу (9,0г, 50моль) і п-толуоїлхлорид (7,0мл, 50моль) у піридині (50мл) перемішували протягом 30 хвилин. Додавали воду (10мл) і суміш концентрували при зниженому тиску. Залишок розчиняли у толуолі (500мл) і розчин промивали водою (200мл) і насиченим водним розчином бікарбонату натрію (200мл). Два шари розділяли і органічний шар випарювали. Залишок розчиняли у метанолі (100мл) і додавали НСl у діоксані (4M, 10мл). Суміш перемішували при кімнатній температурі протягом ночі і потім випарювали при зниженому тиску. Одержану олію обробляли хроматографічно на силікагелі (ЕtOАс/гексан: 1/1), одержуючи олію (10,1г). Дану олію розчиняли у дихлорметані (100мл) і додавали діетиламіносульфуртрифторид (DAST) (5,7мл). Суміш перемішували протягом ночі і потім виливали у насичений водний розчин бікарбонату натрію (100мл). Суміш екстрагували толуолом (2x50мл) і об'єднані органічні шари концентрували. Залишок обробляли хроматографічно на силікагелі (ЕtOАс/гексан: 15/85), одержуючи зазначену у заголовку сполуку у вигляді олії (1,50г). Стадія В: 3-дезокси-3-фтор-2-С-метил-1-О-метил-5-О-толуоїл-a-D-рибофураноза Продукт стадії А (1,0г, 3,5моль) та періодинан Bess-Martin (2,5г) у дихлорметані (20мл) перемішували протягом ночі при кімнатній температурі і потім концентрували при зниженому тиску. Залишок ретельно розтирали з простим діетиловим ефіром (50мл) і фільтрували. Фільтрат промивали розчином Na2S2O3×5H2O (12,5г) у насиченому водному бікарбонаті натрію (100мл), сушили (MgSO4), фільтрували і випарювали. Залишок розчиняли у безводному ТГФ (50мл). При -78°С додавали ТіСl4 (3мл) і метилмагнійбромід у простому етиловому ефірі (3М, 10мл) та суміш перемішували при температурі від -50 до -30°С протягом 2 годин. Суміш виливали у насичений водний розчин бікарбонату натрію (100мл) і фільтрували через целіт. Фільтрат екстрагували толуолом (100мл) і випарювали. Залишок обробляли хроматографічно на силікагелі (ЕtOАс/гексан: 15/85), одержуючи зазначену у заголовку сполуку у вигляді олії (150мг). Стадія С: 4-аміно-7-(3-дезокси-3-фтор-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Продукт стадії В (150мг, 0,5моль) розчиняли у НВr (30%) в оцтовій кислоті (2мл). Через одну годину суміш випарювали при зниженому тиску і випарювали разом з толуолом (10мл). 4-хлор-7Н-піроло[2,3-d]піримідин (0,5г, 3,3моль) і КОН у вигляді порошку (85%, 150мг, 2,3моль) перемішували у ДМФ (3мл) протягом 30 хвилин і суміш випарювали разом з толуолом (2мл). Одержаний розчин виливали у вказану вище суміш бромцукру і перемішували протягом ночі. Суміш розбавляли толуолом (50мл), промивали водою (3x50мл) і концентрували при зниженому тиску. Залишок обробляли хроматографічно на силікагелі (ЕtOАс/гексан: 15/85), одержуючи олію (60мг). Дану олію розчиняли у діоксані (2мл) і рідкий аміак (20г) додавали в автоклав з нержавіючої сталі. Суміш нагрівали при 85°С протягом 18 годин, потім охолоджували і випарювали. Залишок обробляли хроматографічно на силікагелі (метанол/дихлорметан: 1/9), одержуючи зазначену у заголовку сполуку у вигляді твердої речовини (29мг). 1 H-ЯМР (ДМСО-d6): δ 0,81 (с, 3Н), 3,75 (м, 2Н), 4,16 (м, 1H), 5,09 (дд, 1Н, J=53,2, 7,8 Гц), 5,26 (шир., 1Н), 5,77 (с, 1H), 6,15 (д, 1Н, J=2,9 Гц), 6,59 (д, 1Н, J=3,4 Гц), 7,02 (шир.с, 2Н), 7,39 (д, 1H, J=3,4 Гц), 8,06 (с, 1H). 13 С-ЯМР (ДМСО-d6): δ 19,40, 59,56, 77,24, 79,29, 90,15, 91,92, 99,88, 102,39, 121,17, 149,80, 151,77, 157,47. 19 F-Я МР (ДМСО-d 6): δ 14,66 (м). ES-MS: знайдено: 283,1 (М+Н+); розраховано для C12H15FN4 O3+H+: 283,1. Приклад 30 4-аміно-7-(2-С,2-О-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 4-хлор-7-[3,5-біс-О-(2,4-дихлорфенілметил)-2-С,2-О-диметил-b-D-рибофуранозил]-7Н-піроло[2,3d]піримідин До попередньо охолодженого (0°С) розчину сполуки прикладу 2, стадії D (618мг, 1,0моль) у ТГФ (8мл) додавали метилйодид (709мг, 5,0моль) і NaH (60% у мінеральному маслі) (44мг, 1,1моль). Одержану суміш перемішували протягом ночі при кімнатній температурі і потім виливали у суміш, що перемішується, насиченого водного амонійхлориду (50мл) і дихлорметану (50мл). Органічний шар промивали водою (50мл), сушили (MgSO 4) і випарювали у вакуумі. Одержаний сирий продукт очищали на силікагелі, використовуючи етилацетат/гексан як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (735мг) у вигляді безбарвної піни. Стадія В: 4-аміно-7-[3,5-біс-0-(2,4-дихлорфенілметил)-2-С,2-O-диметил-b-D-рибофуранозил]-7Нпіроло[2,3-d]піримідин До сполуки стадії А (735мг, 1,16моль) додавали метанольний аміак (насичений при 0°С) (20мл). Суміш нагрівали в автоклаві з нержавіючої сталі при 80°С протягом ночі, потім охолоджували і вміст випарювали у вакуумі. Сиру суміш очищали на силікагелі, використовуючи етилацетат/гексан як елюент. Фракції, які містять продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт (504мг) у вигляді безбарвної піни. Стадія С: 4-аміно-7-(2-С,2-О-диметил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Суміш продукту стадії С (64мг, 0,1моль), МеОН (5мл), Et3N (0,2мл) і 10% Pd/C (61мг) гідрували в апараті Пара при 50 псі при кімнатній температурі протягом ночі. Суміш фільтрували через целіт, випарювали у вакуумі та фільтрували через шар силікагелю, використовуючи 2% метанол у дихлорметані як елюент. Потрібний продукт збирали і випарювали у вакуумі. Дану сполуку знову розчиняли у метанолі (10мл) і додавали 10% Pd/C (61мг). Суміш гідрували в апараті Пара при 55 псі при кімнатній температурі протягом двох тижнів. Суміш фільтрували через целіт, випарювали у вакуумі та очищали на силікагелі, використовуючи 10% метанол у дихлорметані як елюент. Фракції, які містять продукт, об'єднували і випарювали у вакуумі, одержуючи потрібний продукт (110мг) у вигляді безбарвної піни. 13 С-ЯМР (ДМСО-d6): δ 0,68 (с, 3Н), 3,40 (с, 3Н), 3,52-3,99 (такий, що перекривається м, 4Н), 4,92 (д, 1H), 5,07 (т, 1Н), 6,26 (с, 1H), 6,55 (д, 1Н), 7,00 (шир.с, 2Н), 7,46 (д, 1Н), 8,05 (с, 1H). РХ-МС: знайдено: 293,1 (М-Н+); розраховано для C12H16N4O4-H+: 293,12. Приклад 31 4-метиламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Сполуку стадії Ε прикладу 2 (200мг, 0,67моль) додавали до метиламіну (5мл, конденсованого у невеликому автоклаві з нержавіючої сталі) і нагрівали при 85°С протягом 48 годин, потім охолоджували та випарювали у вакуумі. Сиру суміш очищали на силікагелі, використовуючи етанол як елюент і одержуючи зазначену у заголовку сполуку, яку виділяли у ви гляді аморфної твердої речовини після обробки MeCN. Аморфну тверду речовину розчиняли у воді та ліо філізували, одержуючи безбарвний порошок (144мг). 1 Н-ЯМР (ДМСО-d 6): δ 0,63 (с, 3Н, СН3), 3,32 (с, 3Н, N-CH3), 3,58-3,67 (м, 1Н, Н-5¢), 3,79-3,39 (м, 3Н, Н-5", Н4¢, Н-3¢), 5,03 (с, 1H, 2'-ОН), 5,04-5,11 (1H, 3'-ОН, 1Н, 5'-ОН), 6,14 (с, 1H, Н-11), 6,58 (д, 1H, J 5,6 =3,6 Гц, Н-5), 7,46 (д, 1Н, Н-6), 7,70 (шир.с, 1Η,ΝΗ), 8,14 (с, 1H,Н-2). РХ-МС: знайдено: 295,1 (М-Н+); розраховано для C13H18N4O4+H+: 294,3. Приклад 32 4-диметиламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Сполуку стадії Ε прикладу 2 (200мг, 0,67моль) додавали до диметиламіну (5мл, конденсованого у невеликому автоклаві з нержавіючої сталі) і нагрівали при 85°С протягом 48 годин, потім охолоджували та випарювали у вакуумі. Сиру суміш очищали на силікагелі, використовуючи етанол як елюент і одержуючи зазначену у заголовку сполуку, яку виділяли у ви гляді аморфної твердої речовини після обробки MeCN. Аморфну тверду речовину розчиняли у воді та ліо філізували, одержуючи безбарвний порошок (164мг). 1 H-ЯМР (ДМСO-d6): δ 0,64 (с, 3Н, СН3), 3,29 (с, 3Н, N-CH 3), 3,32 (с, 3Н, N-СН3), 3,60-3,66 (м, 1H, Н-5'), 3,773,97 (м, 3Н, Н-5", Н-4¢, Н-3'), 5,04 (с, 1H, 2'-ОН), 5,06-5,11 (1Н, 3'-ОН, 1Н, 5¢-ОН), 6,21 (с, 1Н, Н-1¢), 6,69 (д, 1H, J5,6=3,6 Гц, Н-5), 7,55 (д, 1Н,Н-6),8,13(с, 1Н,Н-2). РХ-МС: знайдено: 309,3 (М-Н+), розраховано для C14H20N4O4+H+: 308,33. Приклад 33 4-циклопропіламіно-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Сполуку стадії Ε прикладу 2 (200мг, 0,67моль) додавали до циклопропіламіну (5мл, конденсованого у невеликому автоклаві з нержавіючої сталі) і нагрівали при 85°С протягом 48 годин, потім охолоджували та випарювали у вакуумі. Сиру суміш очищали на силікагелі, використовуючи етанол як елюент і одержуючи зазначену у заголовку сполуку, яку виділяли у ви гляді аморфної твердої речовини після обробки MeCN. Аморфну тверду речовину розчиняли у воді та ліо філізували, одержуючи безбарвний порошок (148мг). 1 Н-ЯМР (ДМСО-d 6): δ 0,51-0,58 (м, 2Н), 0,64 (с, 3Н, СН3), 0,74-0,76 (м, 2Н), 3,62-3,67 (м, 1Н, Н-5¢), 3,79-3,82 (м, 3Н, Н-5"), 3,92-3,96 (м, Н-4¢, Н-3¢), 5,03 (с, 1Н, 2'-ОН), 5,05-5,10 (1Н, 3'-ОН, 1H, 5'-ОН), 6,15 (с, 1H, Н-1¢), 7,48 (д, 1H, J5,6=3,6 Гц, Н-5), 7,59 (д, 1Н, Н-6), 8,13 (с, 1H, Н-2). РХ-МС: знайдено: 321,1 (М-Н+), розраховано для C15H20N4+H+: 320,3. Приклад 34 4-аміно-7-(3-С-метил-b-D-ксилофуранозил)-7Н-піроло[2,3-d]піримідин Стадія А: 7-[2,5-біс-О-(трет-бутилдиметилсиліл)-b-D-рибофуранозил]-4-[(4метоксифеніл)дифенілметил]аміно-7Н-піроло[2,3-d]піримідин і 7-[3,5-біс-О-(трет-бутилдиметилсиліл)-b-Dрибофуранозил]-4-[(4-метоксифеніл)дифеніл метил]аміно-7Н-піроло[2,3-d]піримідин До розчину суміші сполук стадії А прикладів 26 і 27 (0,32г, 0,65моль) у безводному піридині (6мл) додавали монометокситритилхлорид (0,30г, 0,98моль) і реакційну суміш перемішували при кімнатній температурі протягом ночі. Суміш потім концентрували і залишок розділяли між СН 2Сl2 (70мл) і водою (20мл). Органічний шар промивали водою та сольовим розчином, сушили (Na2SO4) і концентрували. Залишок очищали на колонці з силікагелем, використовуючи 5-13% ЕtOАс у гексані як елюент. Відповідні фракції збирали і концентрували, одержуючи 2',5'-біс-О-(трет-бутилдиметилсиліл)- і 3',5'-біс-О-(трет-бутилдиметилсиліл)захищені нуклеозиди у вигляді безбарвних пін (343мг і 84мг, відповідно). Стадія В: 7-[2,5-біс-О-(трет-бутилдиметилсиліл)-b-D-еритропентофураноз-3-улозил]-4-[(4метоксифеніл)дифенілметил]аміно-7Н-піроло[2,3-d]піримідин До суспензії, яка ретельно перемішується, триокису хрому (91мг, 0,91моль) у СН 2Сl2 (4мл) при 0°С додавали піридин (147мкл, 1,82моль) і потім оцтовий ангідрид (86мкл, 0,91моль). Суміш перемішували при кімнатній температурі протягом 0,5 години. Потім додавали 2',5'-біс-О-(трет-бутилдиметил-силіл)захищений нуклеозид стадії А (343мг, 0,45моль) у СН 2Сl2 (2,5мл) і суміш перемішували при кімнатній температурі протягом 2 годин. Потім суміш виливали в охолоджений льодом EtOAc (10мл) і фільтрували через коротку колонку з силікагелем, використовуючи EtOAc як елюент. Фільтрат випарювали і залишок очищали на колонці з силікагелем, використовуючи гексан і гексан/ЕtOАс (7/1) як елюент і одержуючи зазначену у заголовку сполуку (180мг). Стадія С: 7-[2,5-біс-О-(трет-бутилдиметилсиліл)-3-С-метил-b-D-рибо-фуранозил]-4-[(4метоксифеніл)дифенілметил]аміно-7Н-піроло[2,3-d]піримідин і 7-[2,5-біс-О-(трет-бутилдиметилсиліл)-3-Сметил-b-D-ксилофуранозил]-4-[(4-метоксифеніл)дифеніл метил]аміно-7Н-піроло[2,3-d]піримідин До суміші Me MgBr (3,0Μ розчин у простому ефірі; 0,17мл, 0,5моль) у безводному гексані (1,5мл) при кімнатній температурі по краплях додавали розчин сполуки стадії В (78мг, 0,1моль) у безводному гексані (0,5мл). Після 2 годин перемішування при кімнатній температурі реакційну суміш виливали в охолоджену льодом воду (10 мл) та розбавляли EtOAc (20мл), потім фільтрували через целіт і потім ретельно промивали EtOAc. Шари розділяли і органічний шар промивали сольовим розчином, сушили (Na2SO 4) та концентрували. Залишок очищали на колонці з силікагелем, використовуючи 8-25% EtOAc у гексані як елюент і одержуючи 3С-метилксило- (60 г) і 3-С-метилрибоізомер (20мг). Стадія D: 4-аміно-7-(3-С-метил-b-D-ксилофуранозил)-7Н-піроло[2,3-d]піримідин До охолодженого льодом розчину 3-С-метилксилоізомеру стадії С (60мг, 0,08моль) у ТГФ (2мл) додавали TBAF (1Μ у ТГФ; 0,32мл, 0,32моль). Реакційну суміш перемішували при кімнатній температурі протягом 5 годин і потім розбавляли СН2СІ2 (50мл), промивали водою (3x15мл), сушили та випарювали. Залишок розчиняли у діоксані (0,3мл) і додавали 80% оцтову кислоту (3мл). Реакційну суміш перемішували при кімнатній температурі протягом 1 доби і потім випарювали. Залишок випарювали разом з діоксаном, поміщали у воду (50мл) та промивали СН 2Сl2 (2x10мл). Водний шар концентрували, і потім сушили виморожуванням. Залишок очищали на колонці з силікагелем, використовуючи СН 2Сl2/МеOН (20/1 і 10/1) як елюент і одержуючи зазначену у заголовку сполук у у вигляді білої пухнатої сполуки після сушіння виморожуванням (10мг). 1 H-ЯМР (CD3CN): δ 1,28 (с, 3Н, СН3), 3,56 (шир.с, 1H, ОН), 3,78 (м, 3Н, Н-4¢, Н-5', Н-5"), 4,10 (шир.с, 1H, ОН), 4,44 (д, 1H, J2¢1¢=3,9 Гц, Н-2¢), 5,58 (д, 1H, Н-1¢), 5,85 (шир.с, 2Н, NH2), 6,15 (шир.с, 1Н, ОН), 6,48 (д, Ш, J5¢6=3,7 Гц, Н-5), 7,23 (д, 1Н,Н-6), 8,11(с, 1H,Н-2). ES-MS:281 [MH]+. Приклад 35 4-аміно-7-(3-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин У рибоізомера (20мг) стадії С прикладу 32 видаляли захисні групи, використовуючи спосіб, описаний на стадії D прикладу 32, і одержуючи зазначену у заголовку сполук у (4мг). 1 Н-ЯМР (CD3CN): δ 1,43 (с, 3Н, СН3), 3,28 (шир.с, 1H, ОН), 3,58 (м, 2Н, Н-5', Н-5"), 3,99 (м, 1H, Н-4·), 4,10 (шир.с, 1H, ОН), 4,62 (д, 1H, J2¢1¢=8,1 Гц, Н-2¢), 5,69 (д, 1H, Н-1¢), 5,88 (шир.с, 3Н, ОН, NH2), 6,45 (шир.с, 1H, ОН), 6,51 (д, 1H, J 5,6=3,7 Гц, Н-5), 7,19 (д, 1H, Н-6), 8,12 (с, 1H, Н-2). ES-MS: 281[MH]+. Приклад 36 2,4-діаміно-7-(2-с-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Суміш продукту стадії В прикладу 4 (24мг) у водному аміаку (30%, 10мл) нагрівали в автоклаві з нержавіючої сталі при 100°С протягом ночі, потім охолоджували та випарювали. Залишок очищали на колонці з силікагелем, використовуючи СН 2Сl2/МеOН (10/1 і 5/1) як елюент і одержуючи зазначену у заголовку сполуку (15мг). 1 Н-ЯМР (ДМСО-(і6): δ 0,68 (с, 3Н, СН3), 3,48-3,58 (м, 1H, Н-5¢), 3,68-3,73 (м, 2Н, Н-5", Н-4¢), 3,84 (м, 1Н, Н3'), 4,72 (с, 1H, 2'-ОН), 4,97-5,03 (м, 2Н, 3'-ОН, 5¢-ОН), 5,45 (шир.с, 2Н, ΝΗ2), 6,00 (с, 1Н, Η-1¢), 6,28 (д, 1Н, J=3,7 Гц, Н-5), 6,44 (шир.с, 2Н, NH2), 6,92 (д, 1H, J=3,7 Гц, Н-6). ES-MS: 294,1 (М-Н+). Приклад 37 4-амшо-2-фтор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До розчину суміші HF/піридин (70%, 2мл), розбавленого піридином (1мл), при -30°С додавали сполуку прикладу 36 (60мг, 0,2моль) у 0,5мл піридину, а потім трет-бутилнітрит (36мкл, 0,3моль). Перемішування продовжували протягом 5 хвилин при -25°С. Потім розчин виливали у суміш води з льодом (5мл), нейтралізували 2н водним NaOH та випарювали досуха. Залишок очищали на колонці з силікагелем, використовуючи СН 2Сl2/МеОН (20/1 і 10/1) як елюент і одержуючи зазначену у заголовку сполуку. Приклад 38 4-аміно-5-фтор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин Стадія A: 4-ацетиламіно-7-(2,3,5-три-О-ацетил-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До розчину сполуки стадії F прикладу 2 (280мг, 1,00моль) у піридині додавали оцтовий ангідрид (613мг, 6,0моль). Одержаний розчин перемішували протягом ночі при кімнатній температурі, випарювали у вакуумі та одержану сиру суміш очищали на силікагелі, використовуючи етилацетат/гексан як елюент. Фракції, які містять потрібний продукт, об'єднували і випарювали у вакуумі, одержуючи потрібний продукт. Стадія В: 4-ацетиламіно-5-бром-7-(2,3,5-три-O-ацетил-2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3d]піримідин До попередньо охолодженого розчину (0°С) сполуки стадії А (460мг, 1,00моль) у ДМФ додавали Nбромсукцинімід (178мг, 1,0моль) у ДМФ. Одержаний розчин перемішували при 0°С протягом 30 хвилин, потім при кімнатній температурі протягом ще 30 хвилин. Реакцію гасили, додаючи метанол, і випарювали у вакуумі. Одержану сиру суміш очищали на силікагелі, використовуючи етилацетат/гексан як елюент. Фракції, які містять потрібний продукт, об'єднували та випарювали у вакуумі, одержуючи потрібний продукт. Стадія С: 4-аміно-5-фтор-7-(2-С-метил-b-D-рибофуранозил)-7Н-піроло[2,3-d]піримідин До попередньо охолодженого (-78°С) розчину сполуки стадії В (529мг, 1,00моль) у ТГФ додавали бутиллітій (2 Μ у гексані) (0,5мл, 1,00моль). Одержаний розчин перемішували при -78°С протягом 30 хвилин і потім гасили, використовуючи N-фторбензолсульфонімід (315мг, 1,00моль) у ТГФ. Одержаному розчину давали дуже повільно охолонути до кімнатної температури і потім виливали у суміш, що перемішується, насиченого водного розчину амонійхлориду і дихлорметану. Органічну фазу випарювали у вакуумі та обробляли гідроксидом амонію при 55°С у закритому контейнері протягом ночі. Одержану сиру суміш очи щали на силікагелі, використовуючи як елюент дихлорметан/метанол. Фракції, які містять потрібний продукт, об'єднували і випарювали у вакуумі, одержуючи потрібний продукт. Біологічні тести Далі описуються тести, які використовували для визначення інгібування HCV NS5B полімерази і HCV реплікації. Ефективність сполук даного винаходу як інгібіторів HCV NS5B РНК-залежної РНК полімерази (RdRp) визначали, використовуючи наступний аналіз. А. Аналіз інгібування HCV NS5B полімерази Даний аналіз використовували для визначення здатності нуклеозидних похідних даного винаходу інгібувати ферментативну активність РНК-залежної РНК полімерази (NS5B) вірусу гепатиту С (HCV) на гетеромірній матриці РНК. Процедура: Буфер для: (50мкл - всього на реакцію) 20мМ Tris, pH 7,5 50мкМ EDTA 5мМ DTT 2мМ MgCl2 80мМ КСl 0,4од/мкл PHKsin (RNAsin) (Promega, вихідний об'єм 40од/мкл) 0,75мкг t500 (500-нуісііеотидна РНК, одержана з використанням Т7 runoff-транскрипції з послідовністю з NS2/3 ділянки геному вірусу гепатиту С) 1,6мкг очищеної NS5B вірусу гепатиту С (одержана зі зрізуванням по С-кінцю 21 амінокислоти) 1мкМ A,C,U,GTP (н уклеозидна трифосфатна суміш) [альфа-32Р]-ОТР або [альфа-33Р]-GТР Сполуки тестували при різних концентраціях аж до 100мкМ кінцевої концентрації. Готували необхідний об'єм реакційного буфера, що включає фермент і матрицю t500. Нуклеозидні похідні даного винаходу поміщали піпеткою в ямки 96-ямкового планшету. Готували суміш н уклеозидних трифосфатів (NTP's), включаючи радіомічений GTP, і піпеткою вводили в ямки 96-ямкового планшету. Реакцію ініціювали, додаючи фермент-матричний реакційний розчин, і залишали для протікання реакції при кімнатній температурі на 1-2 години. Реакцію гасили, додаючи 20мкл 0,5Μ EDTA, pH 8,0. Здійснювали також сліпі реакції, в яких розчин, що гасить, додавали до NTPs до додавання реакційного буфера. 50мкл погашеного реакційного розчину наносили плямами на DE81 фільтрувальні диски (Whatman) і залишали для висихання на 30 хвилин. Фільтри промивали 0,3Μ амонійформіатом, pH 8 (150мл/промивку доти, доки число імпульсів за хвилину (срm) у 1мл промивки не стане менше 100, звичайно 6 промивок). Імпульси на фільтрах визначали у 5-мл сцинтиляційній рідині у сцинтиляційному лічильнику. Відсоток інгібування розраховували відповідно до наступного рівняння: % інгібування = [1-(срm у тестовій реакції - срm у сліпій) / (срm у контрольній реакції - срm у сліпій)]х100. Представницькі сполуки, протестовані в аналізі HCV NS5B полімерази, демонстрували величину ІК50 менше ніж 100мкмоль. В. Аналіз інгібування HCV PHK реплікації Сполуки даного винаходу оцінювали також відносно їх здатності впливати на реплікацію РНК вірусу гепатиту С у клітинах гепатоми (НuН-7), які культивуються, що містять субгеномний HCV реплікон. Подробиці аналізу розкриті далі. Цей репліконовий аналіз являє собою модифікацію аналізу, описаного V. Lohmann, F. Komer, J-O. Koch, U. Herian, L. Theilmann, and R. Bartenschlager, "Replication of a Sub-genomic Hepatitis С Virus RNAs in a Hepatoma Cell Line", Science 285: 110 (1999). Протокол: Аналіз являє собою in situ захист рибонуклеази, аналіз планшетів на основі Scintillation Proximity (SPA). Від 10000 до 40000 клітин висівали у 100-200мкл середовища, яке містить 0,8мг/мл G418 у 96-ямкових cytostar планшетах (Amersham). Сполуки додавали до клітин у різних концентраціях, аж до 100мкМ у 1% ДМСО у моменти часу від 0 до 18 годин, а потім культивували протягом 24-96 годин. Клітини фіксували (20 хвилин, 10% формалін), робили їх проникними (20 хвилин, 0,25% Triton X-100/PBS) та гібридизували (протягом ночі, 50°С) з одноланцюговим 33Р РНК зондом, комплементарним (+) ланцюгу NS5B (або інших генів), який міститься у РНК вірусного геному. Клітини промивали, обробляли РНКазою, промивали, нагрівали до 65°С і підраховували у Top-Count. Інгібування реплікації визначали як зменшення числа імпульсів за хвилину (срm). Людські клітини гепатоми НuН-7, які були відібрані як такі, що містять субгеномний реплікон, містили цитоплазмічну РНК, яка складається з HCV 5і нетрансльованої ділянки (NTR), маркера, що неоміцинселектується, EMC V IRES (вн утрішній сайт входження рибосоми) (internal ribosome entry site), i HCV неструктурних білків NS3-NS5B, з наступним 3' NTR. Представницькі сполуки, протестовані у реплікаційному аналізі, демонстрували ЕС50 менше ніж 100мкмоль. Нуклеозидні похідні даного винаходу оцінювали також на клітинну токсичність і антивірусну специфічність у скринінгу можливого інгібування (counterscreens), що описується далі. С. Counterscreens Здатність нуклеозидних похідних даного винаходу інгібувати людські ДНК полімерази визначали, використовуючи наступні аналізи. а. Інгібування людських ДНК альфа- і бета-полімераз: Умови реакції Реакційний об'єм 540мкл Компоненти реакційного буфера: 20мМ Tris-HCl, pH 7,5 200мкг/мл альбуміну телячої сироватки 100мМКСІ 2мМ b-меркаптоетанолу 10мM MgCl2 1,6мкM dA,dG,dC,dTTP a-33P-dATФ Фермент і матриця 0,05мг/мл матриці ДНК сперми риб з гепами 0,01од/мкл ДНК a- або b-полімераз Одержання матриці ДНК сперми риб з гепами Додавали 5мкл 1Μ MgCl2 до 500мкл активованої ДНК сперми риб (USB 70076); Нагрівали до 37°С і додавали 30мкл 65од/мкл екзонуклеази III (GibcoBRL 18013-011); Інкубували 5 хвилин при 37°С; Закінчували реакцію, нагріваючи до 65°С протягом 10 хвилин; Вводили 50-100мкл аліквоти у хроматографічні колонки Bio-spin 6 (Bio-Rad 732-6002), врівноважені 20мМ Tris-HCl, pH 7,5; Елюювали центрифугуванням при 1000xg протягом 4 хвилин; Об'єднували елюат і вимірювали поглинання на 260 нм для визначення концентрації. ДНК матрицю розбавляли до відповідного об'єму 20мМ Tris-HCl, pH 7,5, і фермент розбавляли до відповідного об'єму 20мм Tris-HCl, що містить 2мМ b-меркаптоетанолу, і 100 мм КСІ. Матрицю і фермент поміщали піпеткою в ампули для мікроцентрифугування або 96-ямковий планшет. Готували також сліпі реакції без ферменту і контрольні реакції без тестової сполуки, використовуючи буфер для розбавлення ферменту і розчинник для тестової сполуки, відповідно. Реакцію ініціювали реакційним буфером з перерахованими вище компонентами. Реакційну суміш інкубували протягом 1 години при 37°С. Реакцію гасили, додаючи 20мкл 0,5Μ EDTA. 50мкл погашеної реакційної суміші наносили плямою на диски фільтрів Whatman DE81 і сушили на повітрі. Диски фільтрів повторно промивали 150мл 0,3Μ амонійформіату, pH 8, доти, доки число імпульсів у 1мл промивки не складало менше 100срm. Диски промивали двічі 150 мл абсолютного етанолу та один раз 150мл безводного простого ефіру, сушили і імпульси підраховували у 5мл сцинтиляційної рідини. Відсоток інгібування розраховували відповідно до наступного рівняння: % інгібування = [1-(срm у тестовій реакції - срm у сліпій) / (срm у контрольній реакції - срm у сліпій)]х100. b. Інгібування людської ДНК гамма-полімерази Ефективність інгібування людської ДНК гамма-полімерази визначали у реакціях, які включають 0,5нг/мкл ферменту; 10мкМ dATФ, dGTP, dCTP і ТТР; 2мкКюрі/реакцію [a-33P]-dATФ, і 0,4мкг/мкл активованої ДНК сперми риб (постачання US Biochemical) у буфері, що містить 20мМ Tris, pH 8, 2мМ b-меркаптоетанолу, 50мМ КСІ, 10мМ MgCl2 і 0,1 мкг/мкл BSA. Реакції залишали протікати протягом 1 години при 37°С і гасили, додаючи 0,5Μ EDTA до кінцевої концентрації 142мМ. Утворення продукту визначали кількісно, використовуючи зв'язування з аніонообмінним фільтром і підрахунок сцинтиляцій. Сполуки тестували аж до концентрації 50мкм. Відсоток інгібування розраховували, використовуючи наступне рівняння: % інгібування = [1-(срm у тестовій реакції - срm у сліпій) / (срm у контрольній реакції - срm у сліпій)]х100. Здатність нуклеозидних похідних даного винаходу інгібувати ВІЛ (HIV) інфективність і поширення ВІЛ визначали у наступних аналізах: c. Аналіз ВІЛ інфективності Аналіз здійснювали з типом клітин HeLa Magi, що експресували як CXCR4, так і CCR5, вибрані для низького фону експресії b-галактозидази (b-gal). Клітини інфікували протягом 48 годин і b-gal продукування з інтегрованого ВІЛ-1 LTR промотору підраховували, використовуючи хемілюмінесцентний субстрат (Galactolight Plus, Tropix, Bedford, MA). Інгібітори титрували (у дублікаті) дворазовими серійними розведеннями, починаючи з 100мкМ; відсоток інгібування для кожної з концентрацій розраховували стосовно контрольної інфекції. d. Інгібування поширення ВІЛ Здатність сполук даного винаходу інгібувати поширення вірусу імунодефіциту людини (ВІЛ) визначали за способом, описаним у патенті США №5413999 (9 травня 1995р.) і J.P. Vacca, e t al., Proc. Natl. Acad. Sei., 91: 4096-4100 (1994), що включені в опис як посилання. Нуклеозидні похідні даного винаходу також піддавали скринінгу відносно цитотоксичності проти клітин, які культивуються, гепатоми (НuН-7), що містять субгеномний HCV реплікон, використовуючи аналіз на основі MTS клітин, як описано в аналізі далі. НuН-7 клітинна лінія описана в Н. Nakabayashi, et al., Cancer Res., 42: 3858 (1982). e. Аналіз цитотоксичності Клітинні культури готували у відповідному середовищі у концентраціях приблизно 1,5x105 клітин/мл для суспензійних культур для 3 діб інкубування і 5,0x10 4 клітин/мл для адгезійних культур для 3 діб інкубування. 99мкл клітинної культури переносили в ямки 96-ямкового планшету і додавали 1мкл 100-кратної кінцевої концентрації тестової сполуки у ДМСО. Планшет інкубували при 37°С і 5% СО2 протягом визначеного проміжку часу. Після закінчення періоду інкубування у кожну комірку додавали 20мкл реагенту для аналізу клітинної проліферації (CellTiter 96 Aqueous One Solution Cell Proliferation Assay reagent) (MTS) (Promega) і планшет інкубували при 37°С і 5% СО2 протягом додаткового проміжку часу аж до 3 годин. Вміст планшету перемішували для одержання однорідної суміші та вимірювали поглинання при довжині хвилі 490нм, використовуючи пристрій, що зчитує, для планшету. Стандартну криву для клітин суспензійних культур одержували для відомої кількості клітин безпосередньо перед додаванням реагенту MTS. Метаболічно активні клітини змінюють MTS до формазану. Формазан поглинає при довжині хвилі 490нм. Поглинання при довжині хвилі 490нм у присутності сполуки порівнювали з поглинанням в ямках, в які не додавали ніякої сполуки. Посилання: Cory, A.H. et al., "Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture", Cancer Commun. 3:207(1991). Для визначення активності сполук даного винаходу проти інших РНК-залежних РНК вірусів використовували наступні аналізи. a. Визначення in vitro противірусної активності сполук відносно риновірусу (аналіз інгібування цитопатичного ефекту) Умови проведення аналізу описані Sidwell і Huffman, "Use of disposable microtissue culture plates for antiviral and interferon induction studies", Appl. Microbiol. 22: 797-801 (1971). Віруси Риновірус типу 2 (RV-2), штам HGP, використовували з клітинми KB і середовищем (0,1% NaHCO3, без антибіотиків) як зазначено у посиланні Sidwell і Huffman. Даний вірус, одержаний з АТСС, був одержаний з мазка з горла дорослого чоловіка з помірно гострим запаленням верхніх ди хальних шляхів. Риновірус типу 9 (RV-9), штам 211, і риновірус типу 14 (RV-14), штам Tow, також були одержані з Американської Колекції Типових Культур (АТСС) у Роквілі, MD. RV-9 був одержаний зі змиву горла людини і RV-14 був одержаний з мазка з горла молодої людини з захворюванням верхніх дихальних шля хів. Обидва ці віруси використовували з клітинами HeLa Ohio-1 (Dr. Fred Hayden, Univ. of VA), що являють собою людські клітини епітелоїдної карциноми. Як ростове середовище використовували MEM (мінімальне необхідне середовище Ігла) з додаванням 5% фетальної телячої сироватки і 0,1% NaHCO3. Середовище для противірусного тесту для всіх трьох типів вірусів було MEM з 5% FBS, 0,1% NaHCO3, 50мкг гентаміцину/мл і 10мМ MgCl2. Концентрація 2000мкг/мл була найвищою з використаних концентрацій для аналізу сполук даного винаходу. Вірус додавали в аналітичні планшети приблизно через 5 хвилин після сполуки, що тестується. Здійснювали також відповідні контрольні вимірювання. Аналізовані планшети інкубували у вологому повітрі з вмістом СО2 5% при 37°С. Цитотоксичність визначали у контрольних клітинах, досліджуючи морфологічні зміни за допомогою мікроскопу. Регресійний аналіз результатів СРЕ для вірусів і контрольні результати токсичності давали значення ED50 (50% ефективна доза) і СС50 (50% цитотоксична концентрація). Показник селективності (SI) розраховували за формулою: SI=CC50/ED50. b. Визначення in vitro противірусної активності сполук відносно лихоманки Денге. Банзі та жовтої лихоманки (аналіз інгібування СРЕ) Подробиці аналізу представлені у зазначеному вище посиланні Sidwell і Huffman. Віруси Вірус лихоманки Денге типу 2, штам з Нової Гвінеї, був одержаний з Центру боротьби із захворюваннями (Center for Disease Control). Дві лінії ниркових клітин африканських зелених мавп були використані для культур вірусу (Vero) і для здійснення противірусних тестів (МА-104). Як вірус жовтої лихоманки, штам 17D, одержаний з мозку інфікованої миші, так і вірус лихоманки Банзі, штам Η 336, виділений з сироватки хворого лихоманкою хлопчика у Південній Африці, були одержані з АТСС. Для обох цих вірусів і для аналізів були використані Vero клітини. Клітини і середовища МА-104 клітини (BioWhittaker, Inc., Walkersville, MD) i Vero клітини (АТСС) були використані у середовищі 199, доповненому 5% FBS і 0,1% NaHCO3 і без додавання антибіотиків. Як аналітичне середовище для вірусів ли хоманки Денге, Банзі та жовтої лихоманки використовували MEM, 2% FBS, 0,18% NaHCO3 і 50мкг гентаміцину/мл. Тести на противірусну активність сполук даного винаходу здійснювали відповідно до посилань Sidwell і Huffman і аналогічно до вказаного вище тестування риновірусів. Адекватні значення для цитопатичного ефекту (СРЕ) досягалися через 5-6 діб для кожного з даних вірусів. c. Визначення in vitro противірусної активності сполук відносно вірусу За хідного Нілу (West Nile Virus) (аналіз інгібування СРЕ) Деталі аналізу представлені у вказаному вище посиланні Sidwell і Huffman. Вірус Західного Нілу, ізолят з Нью-Йорка, одержаний з курячого мозку, був одержаний з Центру боротьби із захворюваннями (Center for Disease Control). Vero клітини вирощували і використовували, як зазначено вище. Як тестове середовище використовували MEM, 1% FBS, 0,1% NaHCO3 і 50мкг гентаміцину/мл. Тестування противірусної активності сполук даного винаходу здійснювали відповідно до способів Sidwell і Huffman, що аналогічні тим, які використовували в аналізах активності проти риновірусів. Адекватні значення для цитопатичних ефектів (СРЕ) досягалися через 5-6 діб. d. Визначення in vitro противірусної активності сполук відносно риновірусів, вірусу жовтої лихоманки, вірусів Денге, Банзі та вірусу Західного Нілу (аналіз захоплення нейтрального червоного) Після здійснення зазначеного вище аналізу інгібування СРЕ використовували додатковий метод
ДивитисяДодаткова інформація
Назва патенту англійськоюNucleoside derivatives as inhibitors of rna-dependent rna viral polymerase
Автори англійськоюOlsen David B.
Назва патенту російськоюПроизводные нуклеозида как ингибиторы рнк-зависимой рнк вирусной полимеразы
Автори російськоюОлсен Девид Б.
МПК / Мітки
МПК: A61K 31/7068, C07H 19/04, C07D 487/04, A61K 31/519, C07H 19/19, A61K 31/708, C07H 19/16, C07H 19/10, A61K 31/7064, A61P 1/16, C07H 19/067, A61P 43/00, C07H 19/09, A61P 31/14, C07H 19/14, C07H 19/20, A61K 45/00, C07H 19/06, A61K 38/21, C07H 19/167, C07H 19/173
Мітки: вірусної, інгібітори, нуклеозиду, полімерази, рнк-залежної, похідні, рнк
Код посилання
<a href="https://ua.patents.su/31-73843-pokhidni-nukleozidu-yak-ingibitori-rnk-zalezhno-rnk-virusno-polimerazi.html" target="_blank" rel="follow" title="База патентів України">Похідні нуклеозиду як інгібітори рнк-залежної рнк вірусної полімерази</a>
Похідні пуринового l-нуклеозиду та фармацевтична композиція на їх основі

Номер патенту: 63984
Опубліковано: 16.02.2004
Автори: Аверетт Деврон, Ванг Гуанжі, Тем Роберт
МПК: C07H 19/23, A61K 45/06, C07H 19/04, A61P 19/06, A61P 29/00, A61P 43/00, A61K 31/7052, C07H 19/14, A61K 31/7064, A61K 31/7056, A61K 31/7115, A61P 3/10, A61K 31/7076, A61K 31/708, A61K 31/7072, A61P 17/06, A61K 31/7042, A61P 11/06, A61P 35/00, C07H 19/16, A61P 19/02, A61P 1/04, A61K 31/522, A61P 37/06, A61P 31/00, A61K 31/706, A61P 37/02, A61K 31/70, A61P 17/00, C07H 19/24, A61P 33/00, C07H 19/02, A61K 31/52
Мітки: фармацевтична, l-нуклеозиду, композиція, пуринового, основі, похідні
Формула / Реферат:
1. Похідні пуринового L-нуклеозиду, що мають структуру згідно з формулою I:, І де R1, R2', R3' і R4 - H;R2, R3 і R5 - OH;Z1 - N;Z2 вибраний із групи, що містить N і CH;Z3 - із групи, що містить -NR-, -C(R)2-, -S-, де R, однакові чи різні, вибрані з групи, що містить H, Br, NH2, алкіл і алкеніл;Z4 - із групи, що містить -C=O, -NR-, -C(R)2-, де R, однакові чи різні, вибрані з групи, що містить H...
Складні ефіри нуклеозиду та його похідні, фармацевтична композиція на їх основі

Номер патенту: 41296
Опубліковано: 17.09.2001
Автори: СТУККЕ Кьєлль Тургейр, Берретсен Бернт, МЮХРЕН Фінн, ДАЛЕН Аре
МПК: A61K 31/7072, A61K 31/7068, C07D 239/54, C07H 19/06, A61K 31/70, C07H 19/16, A61K 31/7052, C07D 473/18, A61P 31/12, A61K 31/7042, A61K 31/505, C07H 19/02, C07H 19/19, C07D 473/34, C07D 473/00, A61K 31/7064, A61K 31/52, A61K 31/522, C07H 19/09, A61K 31/7076, A61K 31/708
Мітки: фармацевтична, ефіри, основі, композиція, нуклеозиду, складні, похідні
Формула / Реферат:
1. Сложные эфиры нуклеозида и его производные общей формулы (I) Nu-О-Fa, (I)в которой О представляет собой кислород,Nu представляет собой нуклеозид или его аналог иFa представляет собой ацильную группу мононенасыщенной С18 или C20 жирной кислоты, причем жирная кислота этерифицирована гидроксильной группой, находящейся в 5-положении сахарного фрагмента нуклеозида или его аналога, или гидроксильной группой,...
Похідні піридину як інгібітори pde iv

Номер патенту: 66842
Опубліковано: 15.06.2004
Автори: Діас-Мартінес Адольфо, Фреіне Едді Жан Едгар, Матесанс-Баллестерос Марія Енкарнасіон, Дієлс Гастон Станіслас Марселла
МПК: A61K 31/4439, A61K 31/4427, A61P 17/00, C07D 405/14, A61P 37/08, C07D 401/06, A61K 31/443, A61P 29/00, A61P 11/06, A61P 43/00
Мітки: піридину, інгібітори, похідні
Формула / Реферат:
1. Похідне піридину формули:, (I)деL являє собою водень; С1-6алкіл; С1-6алкіл, заміщений одним або двома замісниками, вибираними з групи, що містить гідрокси, С1-4алкілокси, С1-4алкілоксикарбоніл, моно- та ді(С1-4алкіл)аміно, арил та Het2; С3-6алкеніл; С3-6алкеніл, заміщений арилом; С1-6алкілкарбоніл; С1-6алкілоксикарбоніл; піперидил; піперидил,...
Дициклічні гетероароматичні сполуки як інгібітори білкової тирозинкінази

Номер патенту: 66827
Опубліковано: 15.06.2004
Автори: Сміт Катрін Джейн, Кокерілл Джордж Стюарт, Лекі Керін Елізабет, антріп Стівен Беррі, Картер Малколм Клайв
МПК: C07D 405/14, C07D 239/94, A61K 31/517, C07D 405/04, A61K 31/4709, A61P 43/00, A61P 29/00, A61P 35/00, A61P 17/06, A61K 31/519, C07D 417/04, C07D 417/14, C07D 471/04
Мітки: білкової, дициклічні, гетероароматичні, сполуки, інгібітори, тирозинкінази
Формула / Реферат:
1. Дициклічні гетероароматичні сполуки формули (І): (І)або їх солі чи сольвати як інгібітори білкової тирозинкінази,деY - CR1, a V - N,або Y - CR1, a V - CR2,R1 - група CH3SO2CH2CH2NHCH2-Ar, причому Аr вибрано з групи, яка складається з фурану та тіазолу, кожний з яких заміщено, як варіант, одним чи двома галогенами,...
Галоїдовані похідні амідиноамінокислот як інгібітори no-синтетази

Номер патенту: 67767
Опубліковано: 15.07.2004
Автори: Халлінан Е. Енн, Тот Міхалі В., Пітцел Барнетт С., Бергманіс Арійя А., Авасті Алік К., Хейген Тімоті Дж., Цимбалов Софья, Хансен Доналд У., мол., Спенглер Дейл П., Уеббер Р. Кіт
МПК: C07D 333/38, A61K 31/381, A61K 31/198, A61P 43/00, C07C 257/00, C07D 333/24, C07C 323/58
Мітки: no-синтетази, похідні, інгібітори, амідиноамінокислот, галоїдовані
Формула / Реферат:
1. Галоїдована похідна амідиноамінокислот формули (І): (I)або її фармацевтично прийнятна сіль, де:R1 вибирають з групи, що включає водень, С1-С10 алкіл, С2-С10 алкеніл і С2-С10 алкініл;R2 вибирають з групи, що включає водень, С1-С10 алкіл, С2-С10 алкеніл, С2-С10 алкініл і R- або S-альфа-амінокислоту;R3 і R4 незалежно один від одного...
Попередній патент: Спосіб пуску прямоструминного котла енергоблока на ковзному тиску
Наступний патент: Спосіб формування заряду водонаповненої вибухової речовини
Випадковий патент: Спосіб виготовлення труб з прецизійних сплавів