Заміщені імідазоли, спосіб їх одержання, фармацевтична композиція на їх основі та спосіб лікування
Номер патенту: 63875
Опубліковано: 16.02.2004
Автори: Шелдрейк Пітер Вілльям, Бендер Пол Елліот, Боем Джефрі Чарльз, Гаріжіпаті Раві Шенкер, Галлагхер Тімоті Франсіс, Адамс Джеррі Лерой








Формула / Реферат
1. Замещенные имидазолы формулы (І):
 , (І)
, (І)
в которой
R1 представляет собой 4-пиридил, пиримидинил, хинолил, изохинолинил, хиназолин-4-ил, 1-имидазолил или 1-бензимидазолил, в которых гетероарильное кольцо может быть замещено одним или двумя заместителями, каждый из которых выбирается независимо друг от друга из C1-4-алкила, галогена, гидроксила, C1-4-алкоксигруппы, C1-4-алкилтиогруппы, C1-4-алкилсульфинила, CH2OR12, NR10R20 или N-гетероциклического кольца, которое содержит от 5 до 7 атомов и необязательно включает дополнительный гетероатом, выбранный из кислорода, серы или NR15;
R4 представляет собой фенил, нафт-1-ил, или нафт-2-ил или гетероарил, который может быть необязательно замещен одним или двумя заместителями, каждый из которых выбирается независимо друг от друга и который, в случае 4-фенильного, 4-нафт-1-ильного, 5-нафт-2-ильного или 6-нафт-2-ильного заместителей, является галогеном, цианогруппой, нитрогруппой, -C(Z)NR7R17, -C(Z)OR16, -(CR10R20)mCOR12, -SR5, -SOR5, -OR12, галогензамещенным C1-4-алкилом, C1-4-алкилом, -ZC(Z)R12, -NR10C(Z)R16 или -(CR10R20)mNR10R20, а в случае других заместителей является галогеном, цианогруппой, -C(Z)NR13R14, -C(Z)OR3, -(CR10R20)mCOR3, -S(О)mR3, -OR3, галогензамещенным C1-4-алкилом, C1-4-алкилом, -(CR10R20)mNR10C(Z)R3, -NR10S(О)mR8, -NR10S(О)m'NR7R17, -ZC(Z)R3 или -(CR10R20)mNR13R14;
R2 представляет собой C1-10-алкил N3, -(CR10R20)n'OR9, гетероциклил, гетероциклил-С1-10-алкил, C1-10-алкил, галогензамещенный C1-10-алкил, С2-10-алкенил, С2-10-алкинил, С3-7-циклоалкил, С3-7-циклоалкил-С1-10-алкил, С5-7-циклоалкенил, С5-7-циклоалкенил-С1-10-алкил, арил, арил-С1-10-алкил, гетероарил, гетероарил-С1-10-алкил, (CR10R20)nOR11, (CR10R20)nS(О)mR18, (CR10R20)nNHS(О)2R18, (CR10R20)nNR13R14, (CR10R20)nNО2, (CR10R20)nCN, (CR10R20)n'SО2R18, (CR10R20)nS(О)m'NR13R14, (CR10R20)nC(Z)R11, (CR10R20)nOC(Z)R11, (CR10R20)nC(Z)OR11, (CR10R20)nC(Z)NR13R14, (CR10R20)nC(Z)NR11OR9, (CR10R20)nNR10C(Z)R11, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nN(OR6)C(Z)R13R14, (CR10R20)nN(OR6)C(Z)R11, (CR10R20)nC(=NOR6)R11, (CR10R20)nNR10C(=NR19)NR13R14, (CR10R20)nOC(Z)NR13R14, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nNR10C(Z)OR10, 5-(R18)-1,2,4-оксадиазол-3-ил или 4-(R12)-5-(R18R19)-4,5-дигидро-1,2,4-оксадиазол-3-ил, при этом циклоалкильная, циклоалкилалкильная, арильная, арилалкильная, гетероарильная, гетероарилалкильная, гетероциклическая и гетероциклоалкильная группы могут быть необязательно замещены,
при условии, что R2 не может быть метилом, когда R1 - 4-пиридил,
n представляет собой целое число, имеющее значение от 1 до 10,
n’ обозначает 0 или представляет собой целое число, имеющее значение от 1 до 10,
m представляет собой 0 или целое число 1 или 2,
Z обозначает кислород или серу,
m’ обозначает 1 или 2,
R3 представляет собой гетероциклил, гетероциклил-С1-10-алкил или R8,
R5 представляет собой водород, C1-4-алкил, С2-4-алкенил, С2-4-алкинил или NR7R17, при этом радикал -SR5 не может представлять собой -SNR7R17, а радикал -SOR5 не может быть -SОН,
R6 представляет собой водород, фармацевтически приемлемый катион, C1-10-алкил, С3-7-циклоалкил, арил, арил-С1-4-алкил, гетероарил, гетероарилалкил, гетероциклил, ароил или C1-10-алканоил,
R7 и R17 выбирают независимо друг от друга из водорода или C1-4-алкила или R7 и R17, вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо, содержащее от 5 до 7 атомов, при этом упомянутое кольцо может необязательно включать дополнительный гетероатом, выбранный из кислорода, серы или NR15,
R8 представляет собой C1-10-алкил, галогензамещенный C1-10-алкил, С2-10-алкенил, С2-10-алкинил, С3-7-циклоалкил, С5-7-циклоалкенил, арил, арил-С1-10-алкил, гетероарил, гетероарил-С1-10-алкил, (CR10R20)nOR11, (CR10R20)nS(О)mR18, (CR10R20)nNHS(О)2R18, (CR10R20)nNR13R14, при этом арил, арилалкил, гетероарил и гетероарилалкил могут быть необязательно замещены,
R9 представляет собой водород, -C(Z)R11 или необязательно замещенный C1-10-алкил, S(О)2R18, необязательно замещенный арил или необязательно замещенный арил-С1-4-алкил,
R10 и R20 выбирают независимо друг от друга из водорода и C1-4-алкила,
R11 представляет собой водород, C1-10-алкил, С3-7-циклоалкил, гетероциклил, гетероциклил-C1-10-алкил, арил, арил-С1-10-алкил, гетероарил или гетероарил-С1-10-алкил,
R12 представляет собой водород или R16,
R13 и R14 выбирают независимо друг от друга из водорода или необязательно замещенного C1-4-алкила, необязательно замещенного арила или необязательно замещенного арил-С1-4-алкила, или вместе с атомом азота, к которому они присоединяются, образуют гетероциклическое кольцо, содержащее от 5 до 7 атомов, при этом упомянутое кольцо может необязательно включать дополнительный гетероатом, выбранный из кислорода, серы или NR9,
R15 представляет собой R10 или С(Z)-С1-4-алкил,
R16 представляет собой C1-4алкил, галогензамещенный C1-4-алкил или С3-7-циклоалкил,
R18 представляет собой С1-10-алкил, С3-7-циклоалкил, гетероциклил, арил, арилалкил, гетероциклил, гетероциклил-С1-10-алкил, гетероарил или гетероарилалкил,
R19 представляет собой водород, цианогруппу, C1-4-алкил, С3-7-циклоалкил или арил,
или их фармацевтически приемлемые соли.
2. Соединение по п.1, в котором R1 представляет собой необязательно замещенный 4-пиридил или 4-пиримидинил.
3. Соединение по п. 2, в котором заместитель необязательно представляет собой метил или аминогруппу.
4. Соединение по п. 2, в котором R4 представляет собой необязательно замещенный фенил.
5. Соединение по п. 4, в котором фенил замещен в одном или более положениях независимо друг от друга галогеном, -SR5, -S(О)R5, -OR12, галогензамещенным C1-4-алкилом или C1-4-алкилом.
6. Соединение по п.1, в котором R2 выбирается из C1-10-алкила, необязательно замещенного гетероциклила, необязательно замещенного гетероциклил-С1-10-алкила, (CR10R20)nNS(О)2R18, (CR10R20)nS(О)mR18, арил-С1-10-алкила, (CR10R20)nNR13R14, необязательно замещенного С3-7-циклоалкила или необязательно замещенного С3-7-циклоалкил-С1-10-алкила.
7. Соединение по п. 6, в котором циклоалкильная группа представляет собой С5- или С6-кольцо, которое может быть необязательно замещено в одном или нескольких положениях галогеном, гидроксилом, С1-10-алкоксигруппой, S(О)m-алкилом (где m означает 0, 1 или 2), NR7R17, C1-10-алкилом, галогензамещенным алкилом, гидроксилзамещенным С1-10-алкилом, -С(О)ОR11 или необязательно замещенным арилом.
8. Соединение по п. 6, в котором необязательно замещенный гетероциклический или гетероциклил-С1-10-алкильный радикал необязательно содержит от 1 до 4 заместителей, независимо друг от друга представляющих собой водород, C1-4-алкил, арил, арилалкил, С(О)ОR11, С(О)Н, С(О)С1-4-алкил, гидроксилзамещенный C1-10-алкил, C1-4-алкоксигруппу, S(О)m-алкил (где m обозначает 0, 1 или 2) или NR10R20, где R10 и R20 независимо друг от друга представляют собой водород или C1-4-алкил.
9. Соединение по п.1, в котором R2 представляет собой морфолинопропил, пиперидин, N-метилпиперидин или N-бензилпиперидин.
10. Соединение по п.1, представляющее собой:
1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-хлорпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-азидопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-аминопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-метилсульфонамидопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(N-фенилметил)аминопропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(N-фенилметил-N-метил)аминопропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(1-пирролидинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-диэтиламинопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(1-пиперидинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(метилтио)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[2-(4-морфолинил)этил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(4-морфолинил)пропил]-4-(3-метилтиофенил)-5-(4-пиридил)имидазол,
1-[3-(4-морфолинил)пропил]-4-(3-метилсульфинилфенил)-5-(4-пиридил)имидазол,
1-[3-(N-метил-N-бензил)аминопропил]-4-(3-метилтиофенил)-5-(4-пиридил)имидазол,
1-[3-(N-метил-N-бензил)аминопропил]-4-(3-метилсульфинилфенил)-5-(4-пиридил)имидазол,
1-[4-(метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[4-(метилсульфинил)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(метилсульфинил)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[2-(метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[2-(метилсульфинил)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[4-(4-морфолинил)бутил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-циклопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-изопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-циклопропилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-трет-бутил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(2,2-диэтоксиэтил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-формилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-гидроксииминилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-цианометил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(2-метилпирид-4-ил)имидазол,
4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-хлорпиридин-4-ил)имидазол,
4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-амино-4-пиридинил)имидазол,
1-(4-карбоксиметил)пропил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(4-карбоксипропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-карбоксиметил)этил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-карбокси)этил-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(1-бензилпиперидин-4-ил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]имидазол,
5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-бензилпиперидин-4-ил)имидазол,
5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(2-пропил)имидазол,
5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(циклопропилметил)имидазол,
5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-карбоксиэтил-4-пиперидинил)имидазол,
5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол,
(+/-)-4-(4-фторфенил)-1-[(3-метилсульфонил)пропил]-5-(4-пиридил)имидазол,
4-(4-фторфенил)-1-[(3-метилсульфинил)пропил]-5-(4-пиридил)имидазол,
1-(3-феноксипропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(фенилтио)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(4-хинолил)имидазол,
(+/-)-1-(3-фенилсульфинилпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-этоксипропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(3-фенилсульфонилпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-[3-(4-морфолинил)пропил]-4-(3-хлорфенил)-5-(4-пиридил)имидазол,
1-[3-(4-морфолинил)пропил]-4-(3,4-дихлорфенил)-5-(4-пиридил)имидазол,
4-[4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-пиримид-2-он-4-ил)имидазол,
4-(4-фторфенил)-5-[2-(метилтио)-4-пиримидинил]-1-[3-(4-морфолинил)пропил]имидазол,
4-(4-фторфенил)-5-[2-(метилсульфинил)-4-пиримидинил]-1-[3-(4-морфолинил)пропил]имидазол,
(Е)-1-(1-пропенил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
1-(2-пропенил)-4-(4-фторфенил)-5-(4-пиридил)имидазол,
5-[(2-N,N-диметиламино)пиримидин-4-ил]-4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]имидазол,
1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[4-(трифторметил)фенил]имидазол,
1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[3-(трифторметил)фенил]имидазол,
1-(циклопропилметил)-4-(3,4-дихлорфенил)-5-(4-пиридинил)имидазол,
1-(циклопропилметил)-4-(3-трифторметилфенил)-5-(4-пиридинил)имидазол,
1-(циклопропилметил)-4-(4-фторфенил)-5-(2-метилпирид-4-ил)имидазол,
1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-(3,5-бистрифторметилфенил)имидазол,
5-[4-(2-аминопиримидинил)]-4-(4-фторфенил)-1-(2-карбокси-2,2-диметилэтил)имидазол,
1-(1-формил-4-пиперидинил-4-(4-фторфенил)-5-(4-пиридинил)имидазол,
5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(1-метил-4-пиперидинил)имидазол,
1-(2,2-диметил-3-морфолин-4-ил)пропил-4-(4-фторфенил)-5-(2-амино-4-пиримидинил)имидазол,
4-(4-фторфенил)-5-(4-пиридил)-1-(2-ацетоксиэтил)имидазол,
или их фармацевтически приемлемые соли.
11. Фармацевтическая композиция, содержащая соединение по любому из пп.1-10 и фармацевтически приемлемый наполнитель или разбавитель.
12. Способ лечения цитокин-медиированного заболевания у нуждающегося в нем млекопитающего, в котором осуществляют введение упомянутому млекопитающему эффективного количества соединения формулы (I) по п.1.
13. Способ по п.12, в котором млекопитающее страдает цитокин-медиированным заболеванием из группы, включающей ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие состояния артритной природы, сепсис, септический шок, эндотоксический шок, сепсис, вызванный грам-отрицательными бактериями, синдром токсического шока, астму, респираторный дистресс-синдром взрослых, церебральную малярию, хроническое воспаление легких, силикоз, легочный саркоидоз, заболевание резорбции кости, остеопороз, реперфузионное повреждение, реакцию «трансплантат против хозяина», реакцию отторжения аллотрансплантата, болезнь Крона, язвенный колит или гипертермию.
14. Способ по п.12, в котором болезненное состояние медиируется ИЛ-1, ИЛ-6, ИЛ-8 или фактором некроза опухолевых клеток.
15. Способ по п.13, в котором цитокин-медиируемое болезненное состояние представляет собой астму или артрит.
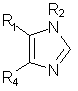
16. Способ получения соединения (I) по п.1, в котором проводят взаимодействие соединения формулы (II):
 II
II
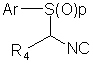
с соединением формулы (III):
 , III
, III
где р обозначает 0, 1 или 2, R1, R2 и R4 соответствуют определению, данному в п.1 или являются предшественниками групп R1, R2 и R4, а Ar представляет собой необязательно замещенную фенильную группу,
а затем, в случае необходимости, предшественник R1, R2 и R4 группы превращают в соответствующую R1, R2 и R4 группу.
17. Cпособ получения соединения по п.16, отличающийся тем, что в соединении формулы (II), когда ArS(О)p представляет собой тозил, R4 не является незамещенным фенилом.
18. Cпособ получения соединения по п.16, отличающийся тем, что в соединении формулы (II) р обозначает 0 или 2.
19. Способ ингибирования цитокинов у млекопитающего, нуждающегося в таком ингибировании, в котором осуществляют введение упомянутому млекопитающему эффективного количества 4-фенил-5-(4-пиридил)имидазола.
Текст
Настоящее изобретение относится к новой группеимидазольных соединений, к способу их получения и использованию для лечения заболеваний, медиированных цитокинами, а также к фармацевтическим композициям, применяемым для такой терапии. Интерлейкин-1(ИЛ-1) и фактор некроза опухолевых клеток(ФНО) представляют собой биологические вещества, продуцируемые различными клетками, такими, как моноциты или макрофаги. Было показано, что ИЛ-1 опосредует большое число видов биологической активности, которые выполняют существенную роль в иммунорегуляции, а также в случае других физиологических состояний, таких, как воспаление [см. Dinarello et al., Rev Infect Disease, 1984, 6, 51] Перечень разнообразной биологической активности, присущей ИЛ-1, включает, в том числе, активацию Т-хелперных клеток, индукцию развития лихорадочного состояния, стимуляцию образования простагландина или коллагеназы, хемотаксис нейтрофилов, индукцию белков острой фазы и супрессию уровня железа в плазме крови. Было продемонстрировано существование множества болезненных состояний, в ходе которых избыточное или нерегулируемое образование ИЛ-1 приводит к обострению и/или индуцированию заболевания. Перечень таких заболеваний включает ревматоидный артрит, остеоартрит, эндотоксемию и/или синдром токсического шока, ряд других состояний, возникающих при остром или хроническом воспалении, такие как воспалительные реакции, индуцированные эндотоксином, или воспалительные заболевания кишечника; туберкулез, атеросклероз, мышечная дегенерация, истощение, псориатический артрит, синдром Рейтера, подагра, травматический артрит, артрит, возникший в результате перенесенной краснухи, и острый синовит. Результаты последних исследований выявляют также очевидную связь активности ИЛ-1 с диабетом и панкреатическими b клетками. Динарелло [Dinarello et al., 1985, 5, 5, 287 – 297] сделал обзор данных по биологической активности, свойственной ИЛ-1. Следует отметить, что некоторые из таких эффектов были ранее описаны другими авторами в качестве непрямого воздействия ИЛ-1. Избыточное или нерегулируемое образование ФНО опосредует течение или обострение множества заболеваний, включающих ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие состояния артритной природы; сепсис, септический шок, эндотоксический шок, сепсис, вызванный грам-отрицательными бактериями, синдром токсического шока, респираторный дистресссиндром взрослых, церебральную малярию, хроническое воспаление легких, силикоз, легочный саркоидоз, резорбцию кости, реперфузионное повреждение, реакцию "трансплантат против хозяина", отторжение аллотрансплантата, лихорадку и миалгии, связанные с инфекцией, такой, как грипп, истощение как последствие инфекции или злокачественного заболевания, истощение как следствие синдрома приобретенного иммунодефицита(СПИД), СПИД, состояния, связанные со СПИДом(АRС), образование келлоидной ткани, образование зарубцевавшейся ткани, болезнь Крона, язвенный колит или гипертермия. СПИД развивается в результате инфекции Т-лимфоцитов вирусом иммунодефицита человека(ВИЧ). При этом было идентифицировано, по крайней мере, три вида штаммов ВИЧ, а именно: ВИЧ-1, ВИЧ-2 и ВИЧ-3. Вследствие ВИЧ инфекции, связанные с иммунитетом Т-клетки, повреждаются, а инфицированный организм проявляет признаки тяжелой условно-патогенной инфекции и/или необычных опухолей. Для внедрения ВИЧ в Т-лимфоцит необходима активация Т-лимфоцита. Другие вирусы, такие, как ВИЧ-1 и ВИЧ-2, инфицируют Т-лимфоциты после Т-клеточной активации, при этом такой процесс Т-клеточной активации вызывает или поддерживает экспрессию вирусного белка и/или репликацию. Как только активированный Т-лимфоцит инфицируется ВИЧ, упомянутый Т-лимфоцит должен поддерживаться в активированном состоянии с целью осуществления экспрессии ВИЧ гена и/или ВИЧ репликации. Монокины, представляющие собой специфические ФНО, участвуют в экспрессии белков ВИЧ, опосредованной активированными Т-клетками и/или репликацией вируса через включение в механизм поддержания активированного состояния Т-лимфоцитов. В этой связи создание помех для проявления монокинами активности посредством, например, ингибирования образования монокинов, в частности, ФНО, в ВИЧ инфицированном организме приводит к ограничению процесса поддержания Т-клеточной активации, что, в свою очередь, снижает распространение ВИЧ инфекции на неинфицированные до этого клетки, приводя в результате к снижению или полностью к подавлению прогрессирования иммунной дисфункции, вызванной ВИЧ инфекцией. Было показано, что моноциты, макрофаги и родственные клетки, такие, как клетки Куппфера и нейроглиальные клетки, участвуют в поддержании ВИЧ инфекции. Такие клетки, как Т-клетки, представляют собой мишени для репликации вирусов, при этом уровень вирусной репликации зависит от состояния активности клеток. [см. Rosenberg et al., The Immunofathogenesis of HIV Infection, Advances in Immunology, 1989, vol. 57]. Было показано, что монокины, такие, как ФНО, активируют ВИЧ репликацию в моноцитах и/или макрофагах [cм., Poli et al., Proc. Natl, Acad. Sci., 1990, 87 782 - 784], поэтому ингибирование образования или активности монокинов так же, как это было показано выше относительно Т-клеток, помогает ограничивать прогрессирование ВИЧ. ФНО также включается в механизм развития других вирусных инфекций, выполняя различные роли на основании приведенных выше свойств и функций, в частности, инфекций, вызванных вирусом цитомегалии(CMV ), вирусом гриппа и вирусом герпеса. Интерлейкин-8(ИЛ-8) представляет собой фактор хемотаксиса, впервые идентифицированный и охарактеризованный в 1987г. ИЛ-8 продуцируют несколько типов клеток, в том числе моноядерные клетки, фибробласты, эндотелиальные клетки и кератиноциты. Его образование эндотелиальными клетками индуцируется ИЛ-1, ФНО или липополисахаридом(ЛПС). Было показано, что человеческий ИЛ-8 действует на нейтрофилы мыши, морской свинки, крысы и кролика. ИЛ-8 было присвоено множество различных наименований, таких, как нетрофильный аттрактант/нейтрофил-активирующий белок-1(НАБ-1), полученный из моноцита нейтрофильный фактор хемотаксиса(ΜΠΗΦΧ), фактор активации нейтрофилов(НАФ) и фактор хемотаксиса Т-клеточного лимфоцита. ИЛ-8 стимулирует множество функций in vitro. Было показано, что он обладает хемоаттрактивными свойствами в отношении нейтрофилов, Т-лимфоцитов и базофилов. Кроме того, он индуцирует высвобождение гистамина из базофилов, как в случае здоровых, так и атопических организмов, а также высвобождение лизосомального фермента и респираторный импульс из нейтрофилов. Было также показано, что ИЛ-8 вызывает повышение экспрессии МАс-1(СD11b/СD18) на поверхности нейтрофилов, в отсутствие синтеза белка de novo, что может внести вклад в повышение адгезии нейтрофилов с эндотелиальными клетками сосудов. Большое число заболеваний характеризуется наличием массивной инфильтрации нейтрофилов. Условия, связанные с повышением образования ИЛ-8 (который ответственен за хемотаксис нейтрофилов в место воспаления), включает участие соединений, оказывающих супрессивноe воздействие на образование ИЛ-8. ИЛ-1 и ФНО воздействуют на большое число клеток и тканей, при этом упомянутые цитокины, а также другие цитокины, полученные из лейкоцитов, представляют собой существенно важные медиаторы в развитии множества болезненных состояний и условий. Ингибирование таких цитокинов позволяет достичь контроля, снижения выраженности патологических признаков и облегчения состояния в случае таких заболеваний. Таким образом, сохраняется потребность в лечении, а в этой связи, в соединениях, которые представляют собой препараты, оказывающие супрессивное воздействие на цитокин, т. е. потребность в соединениях, которые способны ингибировать цитокины, такие, как ИЛ-1, ИЛ-6, ИЛ-8 и ФНО. Настоящее изобретение относится к новым соединениям формулы(1) и к фармацевтическим композициям, содержащим соединение формулы(1), а также к фармацевтически приемлемому разбавителю или наполнителю. Настоящее изобретение также относится к способу ингибирования цитокинов и к лечению умлекопитающих при наличии в этом потребности, заболевания, опосредованного цитокином, при этом описываемый способ лечения включает введение упомянутомумлекопитающему эффективного соединения формулы(1). Более специфично, настоящее изобретение относится к способу ингибирования.умлекопитающего, при наличии такой потребности, образования ИЛ-1, при этом данный способ включает введение упомянутомумлекопитающему эффективного количества соединения формулы(1). Более специфично, настоящее изобретение относится к способу ингибирования умлекопитающего, при наличии такой потребности, образования ИЛ-8, при этом данный способ включает введение упомянутомумлекопитающему эффективного количества соединения формулы(1). Более специфично, настоящее изобретение относится к способу ингибирования умлекопитающего, при наличии такой потребности, образования ФНО, при этом данный способ включает введение упомянутомумлекопитающему эффективного количества соединения формулы(1). В соответствии с вышеописанным, настоящее изобретение относится к соединению формулы(1): R1 представляет собой 4-пиридил, пиримидинил, хинолил, изохинолил, хиназолин-4-ил, 1-имидазолил или 1-бензимидазолил, в которых гетероарильное кольцо может быть замещено одним или двумя заместителями, каждый из которых отбирается независимо друг от друга из группы, включающей С1-4алкил, галоген, гидроксил, С1-4-алкокси, С1-4-алкилтио, С1-4-алкилсульфинил, СН2ОR12, NR10R20 или Nгетероциклическое кольцо, упомянутое кольцо которого может содержать от 5 до 7 членов и факультативно включать дополнительный гетероатом, отобранный из кислорода, серы или NR15 R4 представляет собой фенил, нафт-1-ил или нафт-2-ил или гетероарил, который может быть факультативно замещен одним или двумя заместителями, каждый из которых отбирается независимо друг от друга, и который, в случае 4-фенильного, 4-нафт-1-ильного, 5-нафт-2-ильного или 6-нафт-2ильного заместителя, является галоген-, циано-, нитро-, -С(Z)NR7R17, -C(Z)OR16, -(CR10R20)m COR12, -SR5, SOR5, OR12, галоген-замещенным-С1-4-алкил, -С1-4-aлкил, -ZC(Z)R12, -NR10C(Z)R16 или -(CR10R20)m NR10R20 paдикалом и который для других позиций замещения представляет собой галоген-, циано-, -С(Z)NR13R14, C(Z)OR3, -(СR10R20)m СОR3, -S(O)mR3, -OR3, галоген-замещенный С1-4-алкил -С1-4-алкил, -(СR10R20)m, NR10С(Z)R3, -NR10 S(О)m R8, -NR10S(O)m, NR7R17, -ZC(Z)R3 или -(CR10R20)m NR13R14 радикал; R2 представляет собой С1-1()-алкил-N3, -(СR10R20)n OR9, гетероциклил, гетероциклил-С1-10-алкил, С1-10, алкил, галоген-замещенный-С1-10-алкил, С2-10-алкенил, С2-10-алкинил, С3-7-циклоалкил, С3-7-циклоалкил-С1С5-7-циклоалкенил, С5-7-циклоалкенил-С1-10-алкил, арил, арил-С1-10-алкил, гетероарил, 10-алкил, гетероарил-С1-10-алкил, (CR10R20)nOR11, (CR10R20)n S(O)m R18, (СR10R20)n NHS(О)2R18, (CR10R20)n NR13R14, (СR10R20)n NO2, (CR10R20)n CN, (СR10R20)n'S(O)2R18, (CR10R20)n S(O)m', NR13R14, (CR10R20)nC(Z)R11, (CR10R20)nOC(Z)R11, (CR10R20)n C(Z)OR11, (CR10R20)n C(Z) NR13R14, (CR10R20)n С(Z)NR11OR9, (CR10R20)nNR10C(Z)R11, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nN(OR6)C(Z)NR13R14, (CR10R20)nN(OR6)C(Z)R11, (CR10R20)n C(=NOR6)R11, (CR10R20)nNR10C(=NR19)NR13R14, (CR10R20)nOC(Z) NR13R14, (CR10R20)n NR10C(Z)NR13R14, (CR10R20)nNR10C(Z)OR10, 5-(R18)-1,2,4-оксадиазол-3-ил или 4-(R12)-5-(R18R19)-4,5-дигидро-1,2,4-оксадиазол-3-ил; при этом арильная, арилалкильная, гетероарильная, гетероарилалкильная, гетероциклическая и гетероциклоалкильная группы могут быть факультативно замещены: n представляет собой целое число, имеющее значение от 1 до 10; n' обозначает О или представляет собой целое число, имеющее значение от 1 до 10; m представляет собой О или целое число 1 или 2; z представляет кислород или серу; m' обозначает 1 или 2; R3 представляет собой гетероциклил, гетероциклилС1-10-алкил или R8; R5 представляет собой водород, С1-4-алкил, С2-4-алкенил, С2-4-алкинил или NR7R17, при этом частицаSR5 не может представлять собой – SR7R17, а частица – SOR5, не может быть - SОН; R6 представляет собой водород, фармацевтически приемлемый катион, С1-10-алкил, С3-7-циклоалкил, арил, арилС1-4алкил, гетероарил, гетероарилалкил, гетероциклил, ароил или С1-10алканоил; R7 и R17 отбираются независимо друг от друга из водорода или С1-4алкила или R7 и R17 соединяются вместе через азот, к которому они прикрепляются, с образованием гетероциклического кольца, содержащего от 5 до 7 членов, при этом упомянутое кольцо может факультативно включать дополнительный гетероатом, отобранный из кислорода, серы или NR15; R8 представляет собой С1-10алкил, галоген-замещенный С1-10алкил: С2-10алкенил, С2-10алкинил, С37циклоалкил, С5-7циклоалкенил, арил, арилС1-10алкил, гетероарил, гетероарилС1-1()алкил, (СR10R20)n OR11, (CR10R20)nS(O)m R18, (CR10R20)nNHS(O)2R18, (CR10R20)nNR13R14, при этом арил, aрилалкил, гетероарил, гетероарилалкил могут быть факультативно замещены; R9 представляет водород, -С(Z)R11 или факультативно замещенный С1-10алкил, S(O)2R18, факультативно замещенный арил или факультативно замещенный арил-С1-4алкил; R10 и R20 отбираются независимо друг от друга из водорода или С1-4алкила; R11 представляет собой водород, С1-10алкил, С3-7циклоалкил, гетероциклил, гетероциклилС1-10алкил, арил, арилС1-10алкил, гетероарил или гетероарилС1-10алкил; R12 представляет собой водород или R16 R13 и R14 отбираются независимо друг от друга из водорода или факультативно замещенногоС14алкила, факультативно замещенного арила или факультативно замещенного арилС1-4алкила или вместе с атомом азота, к которому они присоединяются, образуют гетероциклическое кольцо, содержащее от 5 до 7 членов, при этом упомянутое кольцо может факультативно включать дополнительный гетероатом, отобранный из кислорода, серы или NR9; R15 представляет собой R1() или С(Z)-С1-4алкил; R16 представляет собой С1-4алкил, галоген-замещенный С1-4алкил или С3-7циклоалкил; R18 представляет собой С1-10алкил, С3-7циклоалкил, гетероциклил, арил, арилалкил, гетероциклил, гетероциклилС1-10алкил, гетероарил или гетероарилалкил; R19 представляет собой водород, циано-, С1-4алкил, С3-7циклоалкил или арил; или их фармацевтически приемлемые соли. Новые соединения формулы(1) могут быть также использованы для лечения не только людей, но и в ветеринарии для лечения другихмлекопитающих, при необходимости ингибирования активности или образования цитокина. В частности, к таким заболеваниям животных, опосредованным цитокином, в случае которых с целью терапии или профилактики могут использоваться указанные выше соединения, относятся такие болезненные состояния, которые указаны ранее в разделе "Способы лечения", но, в первую очередь, к ним относятся вирусные инфекции. Примеры таких вирусов включают, не ограничиваясь ими, лентивирусные инфекции, такие, как, например, инфекционный вирус анемии лошадей, козлиный вирус артрита, висна-вирус или меди-вирус, или ретровирусные инфекции, такие, как кошачий вирус иммунодефицита(FIV), бычий вирус иммунодефицита или вирус иммунодефицита собак, а также другие ретровирусные инфекции. В формуле(1) подходящие R1-заместители включают 4-пиридил, 4-пиримидинил, 4-хинолил, 6изохинолинил, 4-хиназолннил, 1-имидазолил и 1-бензимидозолил, при этом 4-пиридил, 4-пиримидинил и 4-хинолил предпочтительны. Более предпочтительными являются факультативно замещенный 4пиримидинил и факультативно замещенный 4-пиридил, а наиболее предпочтительным является факультативно замещенное 4-пиримидильное кольцо. Подходящими заместителями для R1 гетероарильных колец являются С1-4алкил, галоген, -ОН, С14алкокси-, С1-4алкилтио-, С1-4алкилсульфинил, CH2OR12, NR10R20 или N-гетероциклильное кольцо, при этом упомянутое кольцо содержит от 5 до 7 членов и факультативно включает дополнительный гетероатом, отобранный из кислорода, серы или ΝR15. Предпочтительным заместителем для всех R1 радикалов является С1-4алкил, в частности, метил, а также NR10R20, в котором предпочтительно, чтобы R10 и R20 были представлены водородом или метилом, более предпочтительно, чтобы R10 и R20 были представлены водородом. Наиболее предпочтительным заместителем является группировка NR10R20. Предпочтительным месторасположением заместителя R1 в кольце 4-пиридильного производного является 2-ое положение, такое, как в случае 2-метил-4-пиридила. Предпочтительное расположение кольца на 4пиримидиниле также представляет собой 2-ую позицию, в частности, как в 2-метил-пиримидине или в 2амино-пиримидине. Соответственно, R4 представляет собой фенил, нафт-1-ил или нафт-2-ил или гетероарил, которые факультативно замещены одним или двумя заместителями. В более предпочтительном случае R4 представляет собой фенильное или нафтильное кольцо. Подходящими заместителями для R4 в том случае, когда он является 4-фенилом, 4-нафт-1-илом, 5-нафт-2-илом или 6-нафт-2-илом являются один или два заместителя, каждый из которых независимо друг от друга отбирается из группы, включающей галоген, -SR5, -SOR5, -OR6, -СF3, или -(CR10R20)m NR10R20, тогда как для других позиций замещения в этих кольцах предпочтительными заместителями являются галоген, -S(О)m R3, -ОR3, -CF3, -(CR10R20)m NR13R14, NR10C(Z)R3 и -NR10S(O)m R3. Предпочтительные заместители в 4-ом положении в фениле и нафт-1-иле, а также в 5-ом положении в нафт-2-иле, включают галоген, в особенности фтор и хлор, а также –SR5 и – SOR5, где R5 представляет собой в предпочтительном варианте С1-2алкил, более предпочтительно метил; при этом фтор и хлор являются более предпочтительными, а наиболее предпочтительным является фтор. Предпочтительные заместители для 3-го положения в фенильном и нафт-1-ильном кольцах включают: галоген, в частности, фтор и хлор; -OR3, в особенности С1-4алкокси-; СF3, NR10R20, такой, как амино-; -NR10C(Z)R3, в особенности –NHCO3(С1-10алкил); -NR10S(O)m R8, в особенности NHSO2(С1-10алкил), и -SR3 и -SOR3, где R3 является предпочтительно С1-2aлкилом, а более предпочтительно - метилом. В том случае, когда фенильное кольцо имеет двойное замещение, предпочтительно, чтобы это были две независимые галогеновые группы, такие, как фтор и хлор, предпочтительно дихлор- и более предпочтительно в 3-ем и 4-ом положении. Предпочтительно, чтобы группировка R4, представляла собой незамещенную или замещенную фенильную группу. Более предпочтительно, когда R4. представляет собой фенил или фенил, замещенный в 4-ом положении фтором и/или замещенный в 3-ем положении фтор-, хлор -, С1-4алкокси-, метансульфонамидо- или ацетамидо-группой, или когда R4. представляет собой фенил, дважды замещенный в 3-ем и 4-ом положении хлором или фтором, более предпочтительно - хлором. Наиболее предпочтительно, чтобы R4 представлял собой 4-фторфенил. В формуле(1) Z относится к кислороду. Соответственно, R2 представляет собой С1-10алкил N3, -(СR10R20)nOR9, гетероциклил, гетероциклилС110алкил, С1-10алкил, галоген-замещенный С1-10алкил, С2-10алкенил, С2-10алкинил, С3-7циклоалкил, С37циклоалкилС1-10алкил, С5-7циклоалкенил, С5-7циклоалкенилС1-10алкил, арил, арилС1-10-алкил, гетероарил, гетероарилС1-10алкил, (CR10R20)nOR11, (CR10R20)nS(O)m R18, (CR10R20)nS(O)m R18, (CR10R20)nNR13R14, (CR10R20)nNO2, (CR10R20)nCN, (CR10R20)n'SO2R18, (CR10R20)n'S(O)m'NR13R14, (CR10R20)nC(Z)R11, (CR10R20)nOC(Z)R11, (CR10R20)nC(Z)OR11, (CR10R20)nC(Z)NR13R14, (CR10R20)nC(Z)NR11OR9, (CR10R20)nNR10C(Z)R11, (CR10R20)n'NR10C(Z)NR13R14, (CR10R20)n'N(OR6)C(Z)NR13R14, (CR10R20)nN(OR6)C(Z)R11, (CR10R20)nC(=NOR6)R11, (CR10R20)nNR10C(=NR19)NR13R14, (CR10R20)nOC(Z)NR13R14, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nNR10C(Z)OR10, 5-(R18)-1,2,4-оксадиазол-3-ил или 4-(R12)-5-(R18R19)-4,5-дигидро-1,2,4оксадиазол-3-ил; при этом арильная, арилалкильная, гетероарильная, гетероарилалкильная, гетероциклическая и гетероциклоалкильная группы могут быть факультативно замещены; при этом n представляет собой целое число со значением от 1 до 10, m обозначает 0 или целое число 1 или 2; n' обозначает 0 или целое число, принимающее значения от 1 до 10; а m' обозначает 1 или 2. Предпочтительно n является целым числом от 1 до 4. Предпочтительно R2 представляет собой факультативно замещенное гетероциклическое кольцо и факультативно замещенный гетерсциклилС1-10алкил, факультативно замещенный С1-10алкил, факультативно замещенный С3-7циклоалкил, факультативно замещенный С3-7циклоалкилС1-10алкил, (CR10R20)nC(Z)OR11 группу, (CR10R20)nNR13R14, (CR10R20)nNHS(O)2NR18, (CR10R20)nS(O)m R18, факультативно замещенный арилС110алкил, (CR10R20)nOR11, (CR10R20)nC(Z)R11 или (CR10R20)n(=NOR6)R11 группу. В более предпочтительном варианте R2 представляет собой факультативно замещенное гетероциклильное кольцо, а также факультативно замещенный гетероциклилС1-10алкил, факультативно замещенный арил, (CR10R20)nNR13R14 или (CR10R20)nC(Z)R11 группу. В том случае, когда R2 представляет собой гетероциклил, предпочтительно, чтобы кольцо представляло собой морфолино-, пирролидинило- или пиперидинило-группу. В том случае, когда кольцо факультативно замещено, заместители могут быть непосредственно присоединены к свободному азоту, как в случае пиперидинило-группы или пиррольного кольца, или на само кольцо. Предпочтительно, чтобы кольцо представляло собой пиперидин или пиррол, более предпочтительно пиперидин. Гетероциклическое кольцо может быть факультативно замещено от одного до четырех раз независимо друг от друга галогеном; С1-4-алкилом, арилом, таким, как фенил; арилалкилом, таким, как бензил- при этом арил- или арилалкил-группы могут быть сами факультативно замещены(как описано ниже в разделе определений); С(О)ОR11, таким, как С(О)С1-4-алкил или С(О)ОН группы; С(О)Н; С(О)С1-4алкилом; гидроксизамещенным С1-4алкилом; С1-4алкокси; S(O)m C1-4алкилом(где m обозначает 0, 1 или 2); NR10R20 (где R10 и R20 представляют собой независимые друг от друга водород или С1-4алкил). Предпочтительно, в том случае, если кольцо представляет собой пиперидин, чтобы кольцо соединялось симидазолом в 4-ом положении, а заместители присоединялись непосредственно к доступному азоту, то есть с образованием 1-формил-4-пиперидина, 1-бензил-пиперидина, 1-метил-4-пиперидина, 1-этоксикарбонил-4-пиперидина. Если кольцо замещено алкильной группой и кольцо присоединяется в 4-ом положении, то предпочтительно, чтобы оно было замещено во 2-ом или 6-ом положении или в обоих этих положениях, подобно, например, 2,2,6,6-тетрамeтил-4-пиперидину. Сходным образом, если кольцо представляет собой пиррол, это кольцо присоединяется кимидазолу в 3-ем положении, а заместители, так же непосредственно, присоединяются к свободному азоту. Когда R2 представляет собой факультативно замещенную гетероциклил С1-10алкильную группу, кольцо представляет собой, предпочтительно, морфолино-, пирролидинило- или пиперидинилогруппу. Предпочтительно, чтобы такая алкильная часть содержала от 1 до 4 атомов углерода, более предпочтительно 3 или 4, и наиболее предпочтительно 3, как это имеет место в случае пропильной группы. Предпочтительные гетероциклические алкильные группы включают, не ограничиваясь ими, морфолиноэтильную, морфолинопропильную, пирролидинилпропильную и пиперидинилпропильную части. Гетероциклическое кольцо также может быть факультативно замещено образом, аналогичным тому, что был приведен выше, относительно непосредственного прикрепления гетероциклила. В том случае, когда R2 представляет собой Факультативно замещенный С3-7алкил или факультативно замещенный С3-7циклоалкил, C1-10алкил, циклоалкильная группа представляет собой предпочтительно от С5 до С6 кольцо, при этом упомянутое кольцо может быть факультативно замещено в одном или более случаев независимо друг от друга галогеном, таким, как фтор, хлор, бром или иод; гидроксилом; С110алкоксигруппой, такой, как метокси- или этоксигруппа; S(O)m алкилом, где m имеет значение 0, 1 или 2, таким, как метилтио-, метилсульфинил- или метилсульфонилгруппа; амином, моно- или дизамещенным амином, таким, как NR7R17 группа; или R7R17 могут образовывать кольцо при участии атома азота, к которому они прикрепляются, с образованием 5-7-членного кольца, которое может факультативно включать дополнительный гетероатом, отобранный из О/N/S; С1-10алкилом, таким, как метил-, этил-, пропил-, изопропил- или t-бутил; галоген-зaмещенным алкилом, тaким, как CF3; гидрокси-замещенным С110алкилом; С(О)OR11, таким, как свободная кислота или ее производное в виде метилового эфира; факультативно замещенным арилом, таким, как фенил; факультативно замещенным арилалкилом, таким, как бензил или фенетил; кроме того, указанные арильные части могут быть также замещены в одном или в двух случаях галогеном; гидроксилом; С1-10алкокси-группой; S(O)m алкилом; аминогруппой, моно или дизамещенной амино-группой, такой, как в NR7R17 группе; алкилом или галоген-замещенным алкилом. В том случае, когда R2 представляет собой (СR10R20)nNR13R14, R13 и R14 соответствуют определениям, данным для формулы (1), в соответствии с которым R13 и R14 отбираются независимо друг от друга из водорода, факультативно замещенного С1-10алкила, факультативно замещенного арила или факультативно замещенного арил С1-10алкила, или вместе с атомом азота, к которому они прикрепляются, образуют гетероциклическое кольцо, содержащее от 5 до 7 членов, при этом упомянутое кольцо может факультативно включать дополнительный гетероатом, отобранный из кислорода, серы или NR9. Известно, что в ряде случаев может происходить образование такой же группировки, как и упомянутая выше С110алкильная часть, которая также представляет собой подходящую R2 переменную. Предпочтительно, чтобы R13 и R14 представляли собой независимо друг от друга водород, С1-10алкил, предпочтительно метил или бензил. Значение переменной n предпочтительно соответствует от 1 до 4, более предпочтительно равняется 3 или 4 и наиболее предпочтительно - 3, как в случае пропильной группы. Предпочтительные группы включают, не ограничиваясь ими, аминопропил, (N-метил-Nбензил)аминопропил, (N-фенилметил) амино-1-пропил или ди-этиламинопропил. В том случае, когда R2 обозначает (СR10R20)nС(Z)OR11-группу, R11 представляет собой, соответственно, водород, С1-10алкил, в особенности - метильную группу. Значение n соответствует, предпочтительно, от 1 до 4,более предпочтительно равно 2 или 3, как в случае этильной или пропильной группы. Предпочтительные группы включают, не ограничиваясь ими, карбоксиметил-1-бутил-, карбокси-1пропил- или 2-ацетоксиэтил-группу. В том случае, когда R2 представлен (CR10R20)nS(O)m R18-группой, m принимает значение 0, 1 или 2, а R18 представляет собой предпочтительно арил, в особенности фенил, или С1-10-алкил, в особенности метил. Значение n соответствует, предпочтительно, от 1 до 4. Более предпочтительно равняется 2 или 3, как в случае этильной или пропильной группы. В том случае, когда R2 представляет собой (CR10R20)nOR11, групу R11 представляет собой, соответственно, водород, арил, в особенности фенил, или С1-10алкильную группу, в особенности метильную или этильную. Значение n предпочтительно колеблется от 1 до 4, более предпочтительно равняется 2 или 3, как в случае этильной или пропильной группы. В том случае, когда R2 обозначает (CR10R20)nNHS(O)2R18 группу, R18 представляет собой, соответственно, алкил, в особенности метил. Значение n соответствует, предпочтительно, от 1 до 4, более предпочтительно равняется 2 или 3, как в случае этильной или пропильной группы. Когда R2 представляет собой факультативно замещенный арил, этот арил предпочтительно является фенилом. Арильное кольцо может быть факультативно замещено в одном или в двух случаях, предпочтительно одним или более заместителем, независимо отобранным из С1-10алкила, галогена, в особенноcти фтора или хлора, (CR10R20)tOR11, -(CR10R20)t NR10R20, в особенности аминогруппы или моноили диалкиламиногруппы, -(СR10R20)tS(O)m R18, где m представляет собой 0, 1 или 2; -SН-, -(CR10R20)n NR13R14, NR10C(Z)R3 (такой как -NHCO(С1-10алкил); -NR10S(O)m R8 (такой, как -NHSO2 (С1-10алкил); где t представляет собой 0 или целое число от 1 до 4. Предпочтительно, чтобы фенильная группа была замещена в 3-ем или 4-ом положении (CR10R20)t, S(O)m R18 радикалом, а R18 представляет собой, предпочтительно, С1-10-алкил, в особенности метил. В том случае, когда R2 представляет собой факультативно замещенную гетероарильную или гетероарилалкильную группу, кольцо может быть факультативно замещено в одном или большем количестве случаев, предпочтительно одним или двумя заместителями, отобранными независимо друг от друга в одном или большем числе случаев, из группы, включающей С1-4алкил, галоген, в особенности фтор или хлор, (CR10R20)t OR11 -(CR10R20)t NOR10 R20 особенности амино или моно- или диалкиламино (CR10R20)t S(O)m R18 где m обозначает 0, 1 или 2; -SH-,-(CR10R20)n -NR13R14, -NR10C(Z)R3 (такой, как NНСО(С1-10)алкил)); -NR10S(O)m R8 (такой, как –NHSO2 (С1-10алкил); t обозначает 0 или целое число от 1 до 4. Каждый специалист, обладающий средним уровнем знаний в даной области, может без труда определить, что в том случае, когда R2 представляет собой (CR10R20)nOC(Z)R11 или (CR10R20)nOC(Z) NR13R14 часть или любую другую сходным образом замещенную группу, n представляет собой в предпочтительном варианте по крайней мере 2 для того, чтобы сделать возможным проведение синтеза стабильных соединений. R2 представляет собой предпочтительно С1-4алкил(разветвленный или неразветвленный, в особенности метил, метил-тиопропил, метилсульфинилпропил, аминопропил, N-метил-бензил аминопропильную группу, диэтиламинопропил, цикло-пропилметил, морфолинилбутил, морфолинилпропил, морфолинил-этил, пиперидин или замещенный пиперидин. Более предпочтительно, R2 представляет собой метил, изопропил, бутил, t-бутил, н-пропил, метилтиопропил или метилсульфинилпропил, морфолинопропил, морфолинилбутил, фенил, замещенный галогеном, тиоалкилом или сульфинилалкилом, такими, как метилтио-, метилтиосульфинильная или метилтиосульфонильная часть; пиперидинил, 1-формил-4-пипeридин, 1-бензил-4- пипeридин, 1-бензил-4-пиперидин, 1-метил-4-пиперидин или 1-этоксикарбонил-4-пиперидин. Во всех приведенных здесь случаях при наличии алкенильной или алкинильной части в качестве замещающей группы, предпочтительна незамещенная связь, то есть виниленовая или ацетиленовая связь, которая не связана непосредственно с частями, содержащими азот, кислород или серу, например, с OR3 или, в некоторых случаях, с R2 частями. Применяемое в настоящем контексте выражение "факультативно замещенные", если ничего не оговорено особо, обозначает такие группы, как галоген, то есть фтористую, хлористую, бромистую или йодистую группы; гидроксил; гидрокси-замещенный С1-10алкил; С1-10алкоксигруппу, в частности, например, метокси- или этоксигруппу; S(O)m алкил, в котором обозначает 0, 1 или 2, такой, как метилтио, метилсульфинил или метилсульфонил; амино моно и ди-замещенная аминогруппа, такая, как NR7R17группа; или R7R17 могут соединяться вместе через азот, к которому они прикрепляются с образованием 57-членного кольца, которое может факультативно включать дополнительный гетероатом, отобранный из группы, включающей О/N/S; С1-10-алкил; циклоалкильную или циклопропилалкильную группу, такую, как метил, этил, пропил, изопропил, t-бутил и т. д. или циклопропилметильную группу; галоген-замещенный С1-10алкил, такой, как CF3; факультативно замещенный арил, такой, как фенил, или факультативно замещенный арилалкил, такой, как бензил или фенетил, при этом указанное арильные части могут быть также замещены в одном или двух случаях галогеном; гидроксил; гидрокси-замещенный алкил; С110алкоксигруппу; S(O)m алкил; амино, моно и ди-замещенную аминогруппу, такую, как в NR7R17 группе; алкил или CF3. В предпочтительных подсемействах соединений формулы (1) R1 представляет собой 4-пиридил, 2алкил-4-пиридил, 4-хинолил, 4-пиримидинил или 2-амино-4-пиримидинил; R2 представляет собой морфолинилпропил, аминопропил, пиперидинил, N-бензил-4-пиперидин или N-метил-4-пиперидин; а R4 представляет собой фенил или фенил, замещенный в одном или двух случаях фтором, хлором, С14алкоксигруппой, -S(O)m. алкилом, метансульфонамидогруппои или ацетамидогруппой. Предпочтительными подгруппами соединений формулы (1) являются те, в которых R2 отличается от метила, тогда как R1 представляет собой пиридил, а R4 представляет собой факультативно замещенный фенил. Подходящие фармацевтически приемлемые соли хорошо известны каждому специалисту, обладающему средним уровнем знаний в данной области, и включают основные соли неорганических и органических кислот, таких, как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, метансульфоновая кислота, этансульфоновая кислота, уксусная кислота, яблочная кислота, винная кислота, янтарная кислота, молочная кислота, щавелевая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, бензойная кислота, салициловая кислота, фенилуксусная кислота и миндальная кислота. Кроме того, фармацевтически приемлемые соли соединений формулы (1) могут быть также образованы с помощью фармацевтически приемлемых катионов, например, в том случае, когда группа заместителя включает карбокси-группу. Подходящие фармацевтически приемлемые катионы хорошо известны каждому специалисту, обладающему средним уровнем знаний в данной области, и включают щелочные и щелочноземельные катионы, аммоний и четвертичные катионы аммония. Следующие термины, используемые в данном контексте, обозначают следующее: "гало-группа" или "галогены" включают галогены: хлор, фтор, бром и йод. "С1-10алкил" или "алкил" - оба относится к линейным или разветвленноцепочечным радикалам, содержащим от 1 до 10 атомов углерода; и в том случае, если длина цепи каким-либо образом не ограничена, включают, не ограничиваясь ими, метильную, этильную, н-пропильную, изопропильную, нбутильную, сек-бутильную, изобутильную, трет-бутильную, н-пентильную и другие подобные им группы. Используемый в настоящем контексте термин "циклоалкил" означает циклические радикалы, содержащие предпочтительно, от 3 до 8 атомов углерода, которые включают, не ограничиваясь ими, циклопропил, циклопентил, циклогексил и им подобные группы. Используемый в настоящем контексте термин "циклоaлкенил" означает циклические радикалы, содержащие, предпочтительно, от 5 до 8 атомов углерода, которые содержат, по крайней мере, одну связь и включают, не ограничиваясь ими, циклопентил, циклогексенил и им подобные группы. Используемый в настоящем контексте термин "алкенил" во всех случаях означает линейную или разветвленноцепочечную радикальную цепь, содержащую 2-10 атомов углерода, и в случае, если какимлибо образом длина цепи не ограничена, включают, не органичиваясь ими, этенил, 1-пропенил, 2пропенил, 2-метил-1-пропенил, 1-бутенил и им подобные группы. "арил" обозначает фенил и нафтил. "гетероарил"(сам по себе или в любой комбинации, такой, как "гетероарилокси" или "гетероарилалкил") представляет собой 5-10-членную ароматическую кольцевую систему, в которой имеется одно или более колец, которые, в свою очередь, содержат один или более гетероатомов, отобранных из группы, состоящей из N, О или S, и включают, не ограничиваясь ими, пиррол, пиразол, фуран, тиофен, хинолин, изохинолин, хиназолинил, пиридин, пиримидин, оксазол, тиазол, тиадиазол, триазол,имидазол или бензимидазол. "гетероциклический"(сам по себе или в любой комбинации, такой, как "гетероциклилалкил") представляет собой насыщенную или частично ненасыщенную 4-10-членную кольцевую систему, в которой одно или более колец содержат один или большее число гетероатомов, отобранных из группы, состоящей из Ν, О или S; таких, как, не ограничиваясь ими, пирролидин, пиперидин, пиперазин, морфолин, тетрагидропиран илиимидазолидин. Используемые в настоящем контексте термины "аралкил" или "гетероарилалкил" обозначают С14алкил, определенный выше, присоединенный к арильной, гетероарильной или гетероциклической группировке, которая также определена здесь, в том случае, если каким-либо образом не указывается иное. "сульфинил" обозначает оксид S(О) соответствующего сульфида, термин "тио" относится к сульфиду, а термин "сульфонил" относится к полностью окисленной S(О)2 части. "ароил" обозначает С(О) Аr, при этом Ar обозначает фенильное, нафтильное или арилалкильное производное, такое, как было определено выше; указанная группа включает, не ограничиваясь ими, бензил и фенетил. "алканоил" обозначает С(О)С1-10aлкил, при этом алкил соответствует приведенному выше определению. Соединения настоящего изобретения могут содержать один или более асимметрических атомов углерода и могут существовать в виде рацемических или оптически активных форм. Все эти соединения включены а рамки настоящего изобретения. Приведенные примеры соединений формулы (1) включают: 1-(3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(3-хлорпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(3-азидопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(3-аминопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(3-метилсульфонамидопропил)-4-(4-фторфенил)-5-(4-пиридил)-имидазол; 1-(3-(N-фенилметил)аминопропил]-4-(4-фторфенил)-5-(4-пиридил)-имидазол; 1-(3-(N-фенилметил-N-метил)аминопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[3-(1-пирролидинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-{3-диэтиламинопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[3-(1-пиперидинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[3-(метилтио)пропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[2-(4-морфолинил)этил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[3-(4-морфолинил)пропил]-4-(3-метилтиофенил)-5-(4-пиридил)имидазол; 1-[3-(4-морфолинил)пропил]-4-(3-метилсульфинилфенил)-5-(4-пиридил)имидазол; 1-[3-(N-метил-N-бензил)аминопропил]-4-(3-метилтиофенил)-5-(4-пиридил)имидазол; 1-[3-(N-метил-N-бензил)аминопропил]-4-(3-метилсульфинил-фенил)-5-(4-пиридил)имидазол; 1-[4-(метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[4-(4-метилсульфинил)фенил|-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[3-(метилтио)фенил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[3-метилсульфинил)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[2-(метилтио)фeнил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[2-(метилсульфинил)фeнил]-4-(4-фторфенил)-5-((4-пиридил)имидазол; 1-[4-(4-морфолинил)бутил]-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-циклопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-изопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-циклопропилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-трет-бутил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(2,2-диэтоксиэтил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-формилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-гидроксииминилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-цианометил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-[3-(4-морфолинил)пропил-4-(4-фторфенил)-5-(2-метилпирид-4-ил)-имидазол; 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-хлорпири-дин-4-ил)имидазол; 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-амино-4-пиридинил)имидазол; 1-(4-карбоксиметил)пропил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(4-карбоксипропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(3-карбоксиметил)этил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(3-карбокси)этил-4-(4-фторфенил)-5-(4-пиридил)имидазол; 1-(1-бензилпиперидин-4-ил)-4-(4-фторфенил)-5-(4-пиридил)имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-[3-(4-морфолинил)пропил]имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-бензилпипери-дин-4-ил)имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(2- припил)имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-(циклоприпилметил)имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-карбоксиэтил-4-пиперидинил)имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол; 1-метил-4-фенил-5-(4-пиридил)имидазол; 1-метил-4-[3-(хлорфенил)]-5-[4-пиридил]имидазол; 1-метил-4-[3-метилтиофенил)]-5-[4-пиридил]имидазол; 1-метил-4-(3-метилсульфинилфенил)-5-(4-пиридил)имидазол; (+/-)-4-(4-фторфeнил)-1-[3-(метилсульфинил)пропил]-5-(4-пиридинио)имидазол; 4-(4-фторфенил)-1-[(3-метилсульфонил)пропилі]-5-(4-пиридинил)имидазол; 1-(3-феноксипропил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 1-[3-(фенилтио)пропил]-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(4-хинолил)имидазол; (+/-)-1-(3-фенилсульфинилпропил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 1-(3-этоксипропил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 1-(3-фенилсульфонилпропил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 1-[3-(4-морфолинил)пропил]-4-(3-хлорфенил)-5-(4-пиридил)имидазол; 1-[3-(4-морфолинил)пропил]-4-(3,4-дихлорфенил)-5-(4-пиридил)имидазол; 4-[4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(пиримид-2-он-4-ил)-имидазол; 4-(4-фторфенил)-5-[2-(метилтио)-4-пиримидинил]-1-[3-(4-морфолинил)пропил]имидазол; 4-(4-фторфенил)-5-[2-(метилсульфинил)-4-пиримидинил]-1-[3-(4-морфолинил)пропил]имидазол; (Е)-1-(1-пропенил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 1-(2-пропенил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 5-[(2-N,N-димeтиламино)пиримидин-4-ил]-4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]имидазол; 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[4-(трифторметил)фенил]имидазол; 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[3-(трифторметил)фенил]имидазол; 1-(циклопропилметил)-4-(3,4-дихлорфенил)-5-(4-пиридинил)имидазол; 1-(циклопропилметил)-4-(3-трифторметилфенил)-5-(4-пиридинил)имидазол; 1-(циклопропилметил)-4-(4-фторфенил)-5-(2-метилпирид-4-ил)имидазол; 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-(3,5-бистрифторметилфенил)имидазол; 5-[4-(2-аминопиримидинил]-4-(4-фторфенил)-1-(2-карбокси-2,2-диметилэтил)имидазол; 1-(1-формил-4-пиперидинил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол; 5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(1-метил-4-пиперидинил)имидазол; 1-(2,2-диметил-3-морфолин-4-ил)пропил-4-(4-фторфенил)-5-(2-амино-4-пиримидинил)имидазол; 4-(4-фторфенил)-5-(4-пиридил)-1-(2-ацетоксиэтил)имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-бензилпирролин-3-ил)имидазол; 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(2,2,6,6-тетраметилпиперидин-4-ил)имидазол. Предпочтительные соединения формулы (1) включают: 5-[4-(2-амино)пиримидинил]-4-(4-фторфенил)-1-(4-N-морфолино-1-пропил)имидазол; 5-[4-(2-аминопиримидинил)-4-(4-фторфенил)-1-(1-бензил-4-пиперидинил)имидазол; 5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(4-пиперидинил)-имидазол; 5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(1-метил-4-пи-перидинил)имидазол; В другом своем аспекте настоящее изобретение относится к соединению, 4-фенил-5-[4пиридил)имидазолу. Еще один аспект настоящего изобретения представляет собой фармацевтическую композицию, включающую наполнитель или разбавитель и эффективное количество 4-фенил-5-[4пиридил)имидазола. Еще одним аспектом настоящего изобретения является новый метод лечения болезненных состояний, опосредованных цитокином, умлекопитающего, имеющего потребность в таком лечении, с применением эффективного количества 4-фенил-5-[4-пиридин)имидазола. Для описываемых в настоящем изобретении целей диапазон дозировок, подробности составления композиций и способы их приготовления аналогичны таковым для соединений формулы (1). Соединения формулы (1) могут быть получены с применением процедур синтеза, некоторые из которых проиллюстрированы на Схемах 1 и У. Приведенные на этих Схемах варианты синтеза применимы для получения соединений формулы (1), включающих множество различных R1, R2. и R4. реагирующих групп, которые несут соответствующим образом защищенные факультативно заместители, при этом достигается совместимость с приведенными в настоящем описании реакциями. Последующая депротекция таких защищенных соединений дает соединения в том виде, в котором они здесь в основном раскрываются. При этом, как только создается имидазольное ядро, дальнейшие на его основе соединения формулы (1) могут быть получены с использованием хорошо известной стандартной техники осуществления внутренней конверсии функциональных групп. Например: получение -С(О)NR13R14 из –CO2CH3 при нагревании в присутствии или в отсутствие каталитического количества цианида металла, например, NaCN и HNR13R14 в СН3ОН; -С(О)R3 из -ОН с участием, например, СІС(О)R3 в пиридине; -NR10-C(S)NR13R14 из – NHR10 алкилизотиоцианатом или тиоциaновой кислотой: NR6C(О)ОR6, из – NHR6 с применением алкилхлорформиата; - NР10С(О)NR13R14 из NHR10 при обработке изоцианатом, а именно, HN=С=O или R10N=C=O –NR10-C(O)R8 из NHR10. при обpаботке в пиридине; -C(=(NR10)NR13R14 из –C(NR13R14)SR3 при взаимодействии с Н3NR3+ОАс- при нагревании в спирте; -С(NR13R14)SR3 из -C(S)NR13R14 при взаимодействии с R6-1 в инертном растворителе, например, в ацетоне; -С(S)NR13R14(где R13 и R14 отличны от водорода) в ходе реакции С(S)NН2 с NHR13R14-C(=NCN)-NR13R14 из -С(=NR13R14)-SR3 с NH2CN при нагревании с безводным спиртом, альтернативно, из –С(=NH)-NR13R14 при обработке BrCN и NаОЕt в EtОН; -NR10)-С(=NCN)SR8 из – NR10 при обработке (R8S)2С=NCN; -NR10SO2R3 из NHR10 при обработке СlSO2R3 в ходе нагревания в пиридине; -NR10C(S)R3 из –NR10C(O)R8 при обpаботке реактивом Льюисона [2,4-бис(4-метоксифенил)-1,3,2,4дитиадифосфэтан-2,4-дисульфид]; -NΡ10SО2СF3 из –NHR6, в реакции с трифлевым ангидридом и основанием, при этом R3, R6, R10, R13 и R14 соответствуют определениям, данным здесь для формулы (1). Схема 1 Представленные на Схеме 1 соединения формулы (1) приготовлены соответствующим образом в ходе реакции соединения формулы (II) с соединением формулы (III), где p обозначает 0, 1 или 2, R1, R2 и R4 соответствуют приведенным выше определениям или являются предшественниками групп R1, R2 и R4, а Аr представляет собой факультативно замещенную фенильную группу, при этом далее, при необходимости, происходит превращение предшественника R1, R2 и R4 в группу R1, R2 и R4. Аr представляет собой фенил, факультативно замещенный С1-4алкилом, С1-4алкоксигруппой или галогруппой. В предпочтительном варианте Аr представляет собой фенил или 4-метилфенил. Реакция осуществляется при температуре окружающей среды или при охлаждении(то есть в диапазоне от -50° до 10°С) в инертном растворителе, таком, как метиленхлорид, тетрагидрофуран, толуол или диметоксиметан, в присутствии соответствующего основания, такого, как 1,8-диазабицикло [5,4,0] ундек-7eн (ДБУ), или гуанидинового основания, такого, как 1,5,7-триазабицикло [4.4.0] дек-5-ен (ТБД). Соответственно, p представляет собой 0 или 2, в предпочтительном варианте p обозначает О, в этом случае интермедиаты формулы (II) характеризуются высокой стабильностью и способностью храниться в течение длительного времени. Предшественники групп R1, R2 и R4 могут отличаться от групп R1, R2 и R4 в которые они могут быть превращены с применением различных стандартных методик, применяемых для проведения внутренней конверсии функциональных групп. Например, соединение формулы (1), в котором R2 представляет собой галоген-замещенный С1-10алкил, может быть превращено в соответствующее С1-10 алкил N3, производное при реакции с подходящей солью азида, а затем, при необходимости, может быть восстановлено в соответствующее С1-10алкил NH2 соединение, которое, в свою очередь, может взаимодействовать с R18S(O)2X где X обозначает галоген(в частности, хлор) с выходом соответствующего С1-10алкил NHS(O)2R18 соединения. Альтернативно, соединение формулы (1), в котором R2 представляет собой С1-10алкил, может взаимодействовать с амином R13R14NH с получением в результате соответствующего С1-10алкил- NR13R14 соединения или может вступать в реакцию с R18SH солью щелочноземельного металла с образованием в результате реакции соответствующего С1-10алкил SR18 соединения. В другом аспекте настоящее изобретение относится к соединениям формулы (II), определенным здесь при условии, что, когда АrS (О)p представляет собой тозил, R4 не может быть незамещенным фенилом. Соединения формулы (II), приведенные на Схеме 1, могут быть получены с применением методов Ван-Лейзена с сотрудниками(Van Leusen et еl., YOС., 1153 (1977)). Например, соединение формулы (II) может быть получено при дегидратировании соединения формулы (IV), в котором Аr, R4. и p соответствуют приведенным выше определениям. Подходящие для этой цели дегидратирующие агенты включают оксихлорид фосфора, оксалилхлорид или тозилхлорид в присутствии соответствующего основания, такого, как триэтиламин. Соединения формулы (IV) могут быть получены в ходе реакции соединения формулы (V) R4СНО, где R4 соответствует приведенному выше определению, с ArS(O)pН и формамидом в условиях дегидратирования при температуре окружающей среды или при повышенной температуре, то есть в диапазоне температур от 30 до 150°С, приемлема также температура кипения с обратным холодильником, факультативно в присутствии кислотного катализатора. Альтернативно, вместо кислотного катализатора может применяться триметилсилилхлорид. Примеры кислотных катализаторов включают камфор-10сульфоновую кислоту, п-толуолсульфоновую кислоту, соляную кислоту и серную кислоту. Соединения формулы (II), в которых p обозначает 2, могут быть получены в ходе реакции, идущей в присутствии сильного основания, соединения формулы (VI) R4CH2NC, c соединением формулы (VII) АrSО2L1, где R4 и Аr соответствуют данным здесь определениям, a L1 обозначает уходящую группу, такую, как галоген, в частности, фтор. Подходящие сильные основания включают алкилы лития, такие, как литийбутил или литий-диизопропиламид. Соединения формулы (VI) могут быть получены в ходе реакции соединения формулы (VIII) R4CH2NH2 с алкилформиатом(в частности, с этилформиатом) с получением промежуточного амида, который может быть превращен в нужный изонитрил в ходе реакции с дегидратирующим агентом, таким, как оксалилхлорид, фосфороксихлорид или тозилхлорид, в присутствии подходящего основания, такого, как триэтиламин. Альтернативно, соединение формулы (VII) может быть превращено в соединение формулы (VI) посредством реакции с хлороформом и гидроксидом натрия в водном дихлорметане с применением катализатора фазового перехода. Соединения формулы (III) могут быть получены при взаимодействии соединения формулы R1СНО с первичным амином R2ΝΗ2 Амино-соединения формулы (VIII) хорошо известны или могут быть получены из соответствующих спиртов, оксимов или амидов с использованием стандартных методов конверсии функциональных групп. В ходе дальнейшего процесса соединения формулы (1) могут быть получены посредством связывания подходящего производного соединения формулы (IX): где Т1 обозначает водород, а Т4 обозначает R4, или, альтернативно, Т1 обозначает R1, а Т4 является Н, при этом R1, R2 и R4 соответствуют данным ранее определениям; при условии: (1) когда Т1 обозначает водород, для облегчения присоединения гетероарильного кольца R1 к имидазольному ядру в положении 5, в условиях, благоприятствующих связыванию кольца, подходящим производным гетероарильного кольца является R1Н; (2) в том случае, когда Т4 представляет собой водород, для облегчения присоединения арильного кольца R4 с имидазольным ядром в положении 4 в условиях, благоприятствующих связыванию с кольцом, подходящим производным арильного кольца является R4Н. Такие арил/гетероарил реакции связывания хорошо известны каждому специалисту, обладающему средним уровнем знаний в данной области. В целом, происходит связывание металлорганического синтетического эквивалента аниона одного компонента с реакционно-способным производным второго компонента в присутствии подходящего катализатора. Анионный эквивалент может быть получен либо на основе имидазола формулы (IX), в этом случае арил/гетероарильное соединение дает реакционно способное производное. Соответственно, подходящие производные соединения формулы (IX) или арил/гетероарильные кольца включают металлоорганические производные, такие, как магнийорганические, цинк-органические, олово-органические и производные на основе борной кислоты, кроме того, подходящие производные включают бромистые, йодистые, трисульфонатные и трифторметансульфонатные производные. Приемлемые для таких случаев процедуры описаны в WO 91/19497, раскрытие которого приведено в настоящем описании в качестве ссылки. Подходящие магний-органические и цинк-органические производные соединения формулы (IX) могут вступать в реакцию с галогеновым, трисульфонатным или трифлатным производным гетероарильного или арильного кольца, в присутствии катализатора связывания, такого, как катализатор на основе палладия (О) или палладия (II) с применением процедуры Кумада с соавт. (Kumada et al., Tetrahedron Letters, 1981, 22, 5319). Подходящие для использования в таких процедурах катализаторы включают тетракис(трифенилфосфин)палладий и PdCl2[1,4-бис-(дифенилфосфино)-бутан], факультативно, в присутствии хлорида лития и основания, такого, как триэтиламин. Кроме того, в соответствии с процедурой Придгена (Pridqen, У. Orq, Chem., 1982, 47, 4319), для связывания арильного кольца может использоваться никель (II) катализатор, такой, как Ni(II)Cl2 (1,2-дифенилфосфино)этан. Подходящие для использования в такой реакции растворители включают гексаметилфосфорамид. В том случае, когда гетероарильное кольцо представляет собой 4-пиридил, подходящие производные включают 4-бром- и 4йод-пиридин, а также фторсульфонатные и трифлатные эфиры 4-гидроксипиридина. Аналогично, подходящие производные, в том случае, когда арильное кольцо представляет собой фенил, включают бромистые, фторсульфонатные, трифлатные, и, предпочтительно, йодистые производные. Подходящие магний-органические и цинк-органические производные могут быть получены при обработке соединения формулы (IX) или его бромистого производного алкиллитиевым соединением с получением соответствующего литиевого реагента, соответственно при депротонировании или трансметаллировании. Литиевый интермедиат может быть затем обработан избытком галогенида магния или галогенида цинка с получением соответствующего металлоорганического реагента. Триалкилтиновое производное соединения формулы (IX) может быть обработано бромидным, трифторсульфонатным, трифталатным или предпочтительно, йодистым производным соединения на основе арильного или гетероарильного кольца в инертном растворителе, таком, как тетрагидрофуран, содержащем предпочтительно 10% гексаметилфосфорамида, в присутствии подходящего связывающего катализатора, такого, как палладиевый (О) катализатор, например, тетракис(трифенилфосфин)палладия по описанному в литературе методу (Stille, J. Amer. Chem. Soc., 1987, 109, 5478, Патенты США №4719218 и 5002942) или с применением палладиевого (II) катализатора в присутствии хлорида лития, факультативно с добавлением основания, такого, как триэтиламин, в инертном растворителе, таком, как диметилформамид. Триалкилтиновые производные могут быть легко получены при металлировании соотвествующего соединения формулы (IX) литиевым реагентом, таким, как s-бутил-литий или n-бутиллитий, в эфирном растворителе, таком, как тетрагидрофуран, или при обработке бромистого производного соответствующего соединения формулы (IX) алкиллитием, с последующей обработкой в каждом случае триалкилтиновым галогенидом. Альтернативно, бромистое производное соединения (IX) может быть обработано соответствующим гетероарил- или арил-содержащим триалкилтиновым соединением в присутствии катализатора, такого, как тетракис-(трифенилфосфин)палладий, в условиях, близких к описываемым выше. Производные борной кислоты также находят применение і Отсюда, может проводиться реакция взаимодействия подходящего производного соединения (IX), такого, как бромистое, йодистое, трифлатное или фторсульфонатное производное, с гетероарил- или арилборной кислотой, в присутствии палладиевого катализатора, такого, как тетракис-(трифенилфосфин)-палладий или PdCl2[1,4-бис(дифенилфосфино)-бутан] в присутствии основания, такого, как бикарбонат натрия, в условиях нагревания при температуре кипячения с обратным холодильником, и в присутствии растворителя, такого, как диметоксиэтан (см. Fisher and Haviniga, Ree. Trav. Chem. Pays. Bas., 1965, 84, 439; Snieckus, V., Tetrahedron Lett., 1988, 29, 2135; и Тerashimia. M., Pharm. Bull., 1985, 11, 4755). При этом могут также использоваться неводные условия проведения реакции, в частности, растворитель, такой, как ДМФ, при температуре около 100°С, в присутствии Рd(II) катализатора (см. Thompson W.J, et al., J. Org. Chem., 1984, 49, 5237). Подходящие производные могут быть получены при обработке магниевого или литиевого производного триалкилборатным эфиром, таким, как триэтил-, три-изо-пропил- или трибутилборат, в соответствии со стандартными процедурами. При проведении таких реакций связывания следует уделять особое внимание функциональным группам, имеющимся в соединения формулы (IX). Так, в целом, аминоили серoсодержащие заместители должны быть в неокисленной форме или соответствующим образом защищены. Соединения формулы (1) представляют собой имидазолы, которые могут быть получены с помощью любой процедуры, ранее приведенной здесь для целей приготовления соединений формулы (1). В частности, a-гало-кетон или любые другие соответствующим образом активированные кетоны R4СОСН2Наl(для соединений формулы (IX), в которых Т1 представляет собой водород) или R1COCH2HAl(для соединений формулы (IX), в котором Т4 обозначает водород) может вступать в реакцию с амидином формулы R2NН-С=NH, где R2 соответствует определению, данному для соединения формулы (1), или обозначает его соль, в инертном растворителе, таком, как галогенированный углеводородный растворитель, например, хлороформ, при умеренно повышенной температуре и, если необходимо, в присутствии подходящего конденсирующего агента, такого, как основание. Получение подходящих a-галокетонов описано в WO 91/19497. Подходящие реакционноспособные эфиры включают эфиры сильных органических кислот, таких, как низшая алкансульфоновая или арилсульфоновая кислота, например, метан- или n-толуолсульфоновая кислота. Амидин применяется преимущественно в виде соли, пригодна, в частности, гидрохлористая соль, которая затем может быть превращена в свободный амидин, in situ, при использовании двухфазной системы, в которой реакционноспособный эфир находится в инертном растворителе, таком, как хлороформ, а соль - в водной фазе, к которой медленно, при энергичном перемешивании, добавляют водный раствор основания, в двумолярном количестве. Подходящие амидины могут быть получены с применением стандартных методов [см., например, Garigipatr R., Tetrahedron Letters, 1989, 31, 190]. Соединения формулы (1) могут быть также получены с применением способа, который включает реакцию взаимодействия соединения формулы (IX), в котором Τ1 обозначает водород, с N-ацил гетероарильной солью, в соответствии с методом, раскрытым в Патенте США 4803279, Патенте США 4719218 и в Патенте США 5002942, с получением интермедиата, в котором гетероарильное кольцо присоединено к имидазольному ядру и которое представляет собой его 1,4-дигидропроизводное, далее интермедиат должен быть помещен в условия окисления-деацилирования(Схема 11). Гетероарильная соль, в частности, пиридиниевая соль, может быть либо заранее приготовлена, либо, более предпочтительно, получена in situ при добавлении замещенного карбонилгалогенида(такого, как ацилгалогенид, ароилгалогенид, арилалкил-галоформиатный эфир или, предпочтительно, алкилгалоформиатный эфир, такой, как ацетилбромид, бензоилхлорид, бензил-хлорформиат или, предпочтительно, этилхлорформиат) к раствору соединения формулы (IX) в гетероарильном соединении R1Η или в инертном растворителе, таком, как метиленхлорид, к которому добавляют гетероарильное соединение. Условия, подходящие для деацилирования и окисления, описаны в Патентах США № 4803279, 4719218 и 5002942, ссылки на которые приведены в настоящем описании. Подходящие для окисления условия включают серу в инертном растворителе или смеси растворителей, такой, как декалин, и диглим, п-цимол, ксилол или мезитол, в условиях нагревания при температуре кипячения с обратным холодильником, или, предпочтительно, калий t-бутоксид в t-бутаноле вместе с сухим воздухом или кислородом. Схема ll В другом способе, приведенном на Схеме lll, ниже, соединения формулы (1) могут быть получены при обработке соединения формулы (X) термически или при помощи циклизующего агента, такого, как оксихлорид фосфора или пентахлорид фосфора [см. Engel and Steglich, Liebigs Ann. Chem., 1978, 1916; Strzybny et al., J. Org. Chem.,1963, 28, 3381]. Соединения формулы (1) могут быть получены, например, при ацилировании соответствующего a-кето-амина активированным производным формиата, таким, как соответствующий ангидрид, при соблюдении стандартных условий для проведения ацилирования, с получением в дальнейшем имина при действии R2NH2. Аминокетоновое производное может быть получено из исходного кетона при оксаминировании и восстановлении, а необходимый кетон может быть, в свою очередь, получен при декарбоксилировании бета-кетоэфира, полученным посредством конденсации арильного(гетероарильного) уксусного эфира с R1СОХ компонентом. Схема lll На приведенной ниже Схеме lV показаны два различных способа, в которых используют кетон(формулы IX) для получения соединения формулы (1). Гетероциклический кетон (XI) получают при добавлении анионной формы алкил-содержащего гетероцикла, такого, как 4-метилхинолин(приготовленного при обработке его алкил-литием, таким, как н-бутил-литий) к N-алкил-Оалкоксибензамиду, эфиру, или любому другому активированному производному, взятому на одной и той же стадии окисления. Альтернативно, анион может быть конденсирован с бензальдегидом с образованием спирта, который затем окисляется в кетон (XI). Схема ІV По другому способу, N-замещенные соединения формулы (1) могут быть получены при обработке аниона амида формулы (Хll): R1CH2NR2COH (XII), где R1 и R2 соответствуют приведенным ранее определениям: (а) нитрилом формулы (Хlll): R4CN (Xlll) где R4. соответствует приведенному ранее определению, или (б) избытком ацил-галогенида, ацилхлорида, формулы (ХlV): R4COHal (ХIV)„ где R4 соответствует данному ранее определению, а Hal представляет собой галоген или соответствующий ангидрид, с получением бис-ацилированного интермедиата, который затем обрабатывают источником аммония, таким, как ацетат аммония. Схема V Один вариант такого подхода был проиллюстрирован выше, на Схеме V. Первичный амин (R2NH2) обрабатывают гало-метил-содержащим гетероциклическим соединением формулы R1CH2X с получением вторичного амина, который превращают далее с помощью стандартной техники в амид. Альтернативно, амид может быть получен, в соответствии со Схемой V, при алкилировании формамида с помощью R1CH2Х. При депротонировании этого амида с применением сильного основания амида, такого, как литий ди-изо-пропиламид или натрий бис-(триметилсилил)амид, с последующим добавлением избытка ароилхлорида получают бис-ацилированное соединение, которое затем, при нагревании в уксусной кислоте, содержащей ацетат аммония, закрывают в имидазольное соединение формулы (1). Альтернативно, анион амида может вступать в реакцию с замещенным арил-нитрилом с получением непосредственно имидазола формулы (1). В технике известны подходящие защищающие группы для гидроксильных групп и для азота имидазола, которые описаны в литературе [в частности, в Protecting Groups in Organic Synthesis, Greene T. W., Wiley-Interschience, New York, 1981]. Примеры подходящих защитных групп для гидроксила включают силиловые эфиры, такие, как tбутилдиметил или t-бутил-дифенил, а также алкильные эфиры, такие, которые включают метил, связанный с алкильной цепью различной связью, (CR10R20)n. Примеры групп, подходящих для защиты азота имидазола включают тетрагидропиранил. Фармацевтические соли добавления кислоты соединений формулы (1) могут быть получены известным способом, например, при обработке исходного соединения соответствующим количеством кислоты в присутствии подходящего растворителя В приведенных примерах все значения температур представлены в градусах по стоградусной шкале(°С). Масс-спектры исследовали на масс-електрометре VG Zab с использованием бомбардировки быстрыми атомами, если особо не оговорено иное. 1Η ЯМР спектр(далее обозначаемый просто "ЯМР") записывают при 250МГц с применением спектрометра Брукер AM 250 или Am 400 (Bruker AM 250 или Am 400). Мультиплетность указывается следующим образом: с. = синглет, д. = дуплет, т. = триплет, к. = квартет, м. = мультиплет, тогда как ш. указывает на наличие широкого сигнала. Обозначение "Сат." относится к насыщенному раствору, экв. обозначает пропорцию молярных эквивалентов реагента относительно основного реагирующего вещества. Флeш-хроматографию проводят на Силикагеле 60, Мерк(230-400меш). Примеры синтеза Описание настоящего изобретения сопровождается далее ссылками на следующие примеры, которые являются лишь иллюстративными и ни в коей мере не должны рассматриваться как ограничивающие область настоящего изобретения. Все значения температур приведены в градусах по стоградусной шкале, все применяемые растворители соответствуют максимально возможной степени чистоты, а все реакции проводятся в безводных условиях в атмосфере аргона, если при этом не оговаривается иное. Пример 1 1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол а) 4-фторфенил-толилтиометилформамид Объединяют раствор п-фторбензальдегида(13,1миллилитра(далее -мл); 122миллимолей(далее ммоль), тиокрезола(16,64грамма(далее - г): 122ммоль), формамида(15,0мл; 445ммоль) и толуола(300мл), и нагревают полученную смесь в течение 18 часов при температуре кипения толуола с обратным холодильником с азеотропным удалением воды. Охлажденную реакционную смесь разбавляют EtOAc(500мл) и промывают насыщенным водным раствором Na2CO3(3 х 100мл), насыщенным водным раствором NаСl(100мл), высушивают(Nа2SО4) и концентрируют. Остаток растирают с петролейным эфиром, фильтруют и высушивают под вакуумом с получением 28,50г целевого соединения в виде белого твердого вещества(85%), точка плавления(далее - Тпл.) = 119-120°С. б) 4-фторфенил-толилтиометилизоцианид Соединение примера 1(а)(25г; 91ммоль) в СН2Сl2(300мл) охлаждают до температуры –30°С и при механическом перемешивании добавляют по каплям вначале РОСL(11мл; 110ммоль), а затем, тоже по каплям, Еt3N(45мл; 320ммоль), поддерживая температуру ниже –30°С. Смесь перемешивают в течение 30 минут при –30°С и в течение 2 часов при 5°С, разбавляют ее СН2Сl2(300мл) и промывают 5%-ным водным Na2СO3(3 x 100мл), высушивают(Na2CО3) и концентрируют до объема 500мл. Затем полученный раствор фильтруют через силикагелевый цилиндр размером 12 x 16см в большой воронке из спекшегося стекла, применяя СН2Сl2 и получая при этом 12,5г (53%) очищенного изонитрила в виде слегка коричневатого воскообразного твердого вещества. ИК (СН2Сl2) 2130см-1. в) Пиридин-4-карбоксальдегид-[4-морфолинилпроп-3-ил]имин Смешивают пиридин-4-карбоксальдегид(2,14г; 20ммоль), 4-(3-аминопропил) морфолин(2,88г, 20ммоль), толуол(50мл) и МgSО4(2г), и перемешивают полученную смесь в атмосфере аргона в течение 18 часов. Отфильтровывают ΜgSO4·, а полученный фильтрат концентрируют, и остаток повторно концентрируют из СН2СI2 с получением 4,52г(97%) целевого соединения в виде желтого масла, содержащего по данным 1Η ЯМР менее 5% альдегида. 1Н ЯМР(СD3СI): д. 8,69(д., J=4,5Гц, 2Н); 8,28(с.,1Н); 7,58(д., J=4,5Гц, 2Н); 3,84(м., 6Н); 2,44(м., 6Н); 1,91(м.,2Н). г) 1-[3-(4-морфолинил) пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол Соединение примера 1(б)(1,41г; 5,5ммоль), соединение примера 1(в)(1,17г; 5,0ммоль) и CH2Cl2(10мл) охлаждают до темпаратуры 5°С. Добавляют 1,5,7-триазабицикло [4,4,6] дек-5-ен, далее обозначаемый как ТБД(0,71г; 5,0ммоль), и выдерживают реакционную смесь при температуре 5°С в течение 16 часов; разбавляют ЕtОАс(80мл) и промывают насыщенным водным раствором Na2СО3(2 x 15мл). Фракцию EtOAc экстрагируют IN НСI(3 x 15мл), кислую фазу промывают EtOAc(2 x 25мл), наслаивают EtOAc(25мл) и подщелачивают вначале с помощью К2СО3 до рН 8,0, а затем с помощью 10%-ного NаОН до рН 10. Полученные фазы разделяют и экстрагируют водную фазу дополнительным количеством EtOAc(3 x 25мл). Экстракты высушивают(К2СО3), концентрируют, и остаток кристаллизуют в смеси ацетон/гексан с получением 0,94г(51%) целевого соединения, Тпл. = 149-150°С. Пример 2 1-(3-хлорпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид(3-хлорпропил)имин К солянокислому 3-хлорпропиламину(15,1г; 0,120 молей(далее - моль)) в Н2О(100мл) добавляют пиридин-4-карбоксальдегид(9,55мл; 0,100 моль), потом К2СО3(8,28г; 0,060 моль), затем CH2Cl2(100мл) и перемешивают полученную смесь в течение 40 минут. Разделяют полученные фазы„ и водную фазу экстрагируют дополнительным количеством CH2Cl2(2 x 50мл), высушивают(Na2SO4) и концентрируют с получением 17,1г(94%)(целевого соединения). 1 Н ЯМР(СD3СІ): д., 8,69(д., J=4,5Гц, 2Н); 8,32(с. 1Н); 7,58(д., J=4,5Гц, 2Н); 3,71(м., 2Н); 3,63(т., J=6Гц, 2Н); 2,24(т., J=6Гц, 2Н). Данные. 1Η ЯМР указывают на очевидное присутствие 9% альдегида. б) 1-(3-хлорпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол Соединение примера 1(б)(6,85г; 26,6ммоль), соединение примера 2(а)(6,32г; 34,6ммоль), CH2Cl2(70мл) и ТБД(4,07г; 28,4ммоль) вводят в реакцию взаимодействия в соответствии с процедурой примера 1(г) с получением 3,19г(38%)(целевого продукта). Тпл. = 139-140°С. Пример 3 1-(3-азидопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол К раствору соединения примера 2(б)(250 миллиграмм(далее -мг); 0,79ммоль) в ДМФ(5мл) добавляют NaN3(256мг; 3,95ммоль) и Nal(12мг; 0,08ммоль), нагревают полученную смесь до 90°С и выдерживают при этой температуре до тех пор, пока данные ТСХ анализа не покажут завершения реакции(19:1, CH2Cl2/MeOH). Охлажденную реакционную смесь прибавляют к 5%-ному водному раствору Nа2СО3(20мл) и экстрагируют EtОАс(3 x 25мл). Объединенные экстракты промывают Н2О(3 x 25мл) и проводят флэшхроматографию(колонка 2,2 x 10см) с использованием 0-1% МеОН в СН2СI2, получая 254мг(100%) целевого соединения в виде белого твердого вещества. Тпл. = 64-65°С. Пример 4 1-(3-аминопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол Описанное выше соединение примера 3(254мг; 0,79ммоль) растворяют в ТГΦ(2мл) и добавляют по каплям к находящемуся при температуре 0°С раствору 1N LiAlH4(1,2мл; 1,2ммоль), перемешивают при температуре 0°С в течение 15 минут, осторожно добавляют EtOAc(4мл) и прибавляют полученную смесь к охлажденному на льду 10%-ному раствору NaOH(15мл), после чего продукт экстрагируют ΕtOAc(4 x 25мл), высушивают(K2CO3) и концентрируют с получением воскообразного твердого вещества(175мг; 75%). Тпл. = 81-82°С. Пример 5 1-(3-метилсульфонамидопропил)-4-(4-фторйенил)-5-(4-пиридил)имидазол К описанному выше соединению примера 4(79мг; 0,26 мыоль) в СН2СI2(0,5мл) добавляют Et3N(72мкл; 0,52ммоль) и затем метансульфонилхлорид(25мкл; 0,31ммоль). Реакция проходит с выделением тепла до температуры кипения СН2СI2 с обратным холодильником, в течение короткого времени. Реакция завершается в течение 1мин на основании данных ТСХ(19:1, СН2СI2/МеОН), после чего смесь выливают в 10%-ный NaOH(5мл) и экстрагируют EtOAc(3 x 20мл). Экстракты промывают Н2О(10мл) и насыщенным водным NaCl(10мл), высушивают(NаSO4), концентрируют и проводят флэш-хроматографию(колонка с силикагелем 1 x 10см) с использованием 0-8% МеОН в СН2СI2 получая 63мг(65%)(целевого соединения). Тпл. = 18б-187°С. Пример 6 1-[3-(N-фенилметил)аминопропид]-4-(4-фторфенил)-5-(4-пиридил)имидазол Используя процедуру описанного выше примера 3, за исключением применения бензиламина в качестве нуклеофильного агента и очистки грубого продукта растиранием с горячим гексаном, получают целевое соединение в виде белого твердого вещества(выход 32%). Тпл. = 125-126°С. Пример 7 1-[3-(N-фенилметил-N-метил)аминопропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол Применяют процедуру примера 3, за исключением использования N-бензилметиламина в качестве нуклеофильного агента и очистки грубого продукта растиранием с горячим гексаном, получая целевое соединение в виде белого твердого вещества(выход 42%). Тпл. = 90-91°С. Пример 8 1-[3-(1-пирролидинил)пропил] -4-(4-фторфенил)-5-(4-пиридил)имидазол Используя процедуру описанного выше примера 3, за исключением применения пирролидина в качестве нуклеофильного агента и очистки грубого продукта растиранием с горячим гексаном, получают целевое соединение в виде белого твердого вещества(выход 35%).Тпл. = 105-107°С. Пример 9 1-(3-диметиламинопропил)-4-(4-5-(4-пиридил)имидазол Используя процедуру описанного выше примера 3, за исключением применения диэтиламина в качестве нуклеофильного агента и выделения продукта путем экстракции диэтиловым эфиром, получают целевое соединение в виде белого твердого вещества(выход 21%). Тпл. = 94-95°С. Пример 10 1-[3-(1-пиперидинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол Используя процедуру описанного выше примера 3, за исключением применения пиперидина в качестве нуклеофильного агента и очистки грубого продукта растиранием с горячим гексаном, получают целевое соединение в виде белого твердого вещества(выход 63%). Тпл. = 105-108°С. Пример 11 1-[3-(метилтио)пропил]-4-(4-фторфенил)-5-(4-пиридил)имидазол Используя процедуру описанного выше примера 3, за исключением применения тиометана натрия в качестве нуклеофильного агента, опуская этап обработки йодидом натрия, с последующей очисткой грубого продукта растиранием с горячим гексаном, получают целевое соединение в виде белого твердого вещества(выход 50%). Тпл. = 85-86°С. Пример 12 1-[2-(4-морфолинил)этил]-4-(4-фторфенил)-5-(4-пиридил)имидазол а) пиридин-4-карбоксальдегид-[2-(4-морфолинил)этил]имин С применением процедуры примера 1(в), за исключением использования 4-(2-аминоэтил)морфолина в качестве амина, получают целевое соединение в виде масла слегка желтоватого цвета(100%), содержащего менее 10% альдегида на основании данных 1H ЯМР. 1Н ЯМР(СD3СI): д. 8,68(д., J=6Гц, 2Н); 8,28(с., 1Н);7,58(д., J=6Гц, 2Н); 3,82(м., 2Н); 3,72(м., 4Н)·, 2,72. (м., 2Н), 2,55(м., 4Н). б)1-[2-(4-морфолинил)этил]-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения примера 2(а) в качестве имина, получают целевое соединение в виде белого твердого вещества(21%). Тпл = 114-115°С. Пример 13 1-[3-(4-морфолинил) пропил]-4-(3-метилтиофенил)-5-(4-пиридил)имидазол а) N-[3-метилтиофенил-(толилтио) метил]формамид С использованием процедуры примера 1(а), за исключением использования мметилтиобензальдегида в качестве альдегида, получают целевое соединение в виде белого твердого вещества(73%). Тпл. = 103-104°C б) 3-метилтиофенил-(толилтио)мeтилизоцианид С примененеим процедуры примера 1(б), за исключением использования соединения предыдущей стадии в качестве формамида, получают целевое соединение в виде масла слегка коричневого цвета(77%). ИК(СН2СI2) 2120см-1. в) 1-[3-(4-морфолинил)пропил]-4-(3-метилтиофенил)5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве изонитрила, получают целевое соединение в виде твердого вещества(31%). Тпл. = 105106°С. Пример 14 1-[3-(4-морфолинил)пропил]-4-(3-метилсульфинилфенил)-5-(4-пиридил)имидазол Соединение примера 13(в)(200мг; 0,49ммоль) растворяют в НОАс(4мл). Затем добавляют растворенный в Н2О K2S2O8(151мг; 0,56ммоль) и перемешивают раствор в течение 16 часов; затем вливают его в 10%-ный водный NaOH(50мл)(полученный раствор имеет рН более 10) и проводят экстракцию с использованием EtOAc(3 x 25мл). Экстракты высушивают(К2СО3), концентрируют и остаточное масло кристаллизуют из смеси ацетон/гексан с получением 87мг(42%) белого твердого вещества. Тпл. = 117-118°С. Пример 15 1-[3-(N-метил-N-бензил) аминопропил]-4-(3-метилтиофенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид-3-(N-метил-N-бензиламинопропил)имин С применением процедуры примера 1(в), за исключением использования 3-(N-метил-N-бензиламино) пропиламина в качестве амина(Vedа, Т.; Ishizaki, K.; Chem. Pharm. Bull., 1967, 15, 228-237), получают целевое соединение в виде масла светло-желтого цвета(100%), содержащего не менее 10% альдегида на основании данных 1Η ЯМР. 1Η ЯМР(CD3СI): д. 8,65(д., J=7Гц, 2Н); 8,21(с. 1Н); 7,54(д., J=4,5Гц, 2Н);7,52(м., 5Н); 3,69(т., J=11Гц, 2Н); 3,48(с., 2Н); 2,44(т. J = 11Гц, 2Н); 2,18(с., 3Н); 1,91(м., 2Н). б) 1-[3-(N-метил-N-бензил)аминопропил]-4-(3-метилотиофенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения примера 13(б) в качестве изонитрила, а также соединения, полученного на предыдущей стадии в качестве имина, получают целевое соединение в видe белого твердого вещества(36%) Тпл. = 87-88°С. Пример 16 1-[3-(N-метил-N-бензил)аминопропил]-4-(3-метилсульфинилфенил)-5-(4-пиридил)имидазол С применением процедуры примера 14, за исключением использования соединения примера 15(б) в качестве сульфида, получают целевое соединение в виде белого твердого вещества(97%).Тпл. = 84-85°С. Пример 17 1-[4-(Метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид(4-метилтиофенил)имин С применением процедуры примера 1(в), за исключенем использования 4-(метилтио)анилина в качестве амина, получают масло светло-желтого цвета(100%), в котором на основании данных 1Η ЯМР не обнаруживается определяемых количеств альдегида. 1Н ЯМР(СD3СI): д. 8,75(д., J=6Гц, 2Н); 8,47(с, 1Н); 7,74(д., J =6Гц, 2Н) і 7,30(д., J=8Гц, 2Н); 7,22(д., J=8Гц, 2Н); 2,52(с, 3Н). б) 1-[4-(метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(27%). Тпл. = 172-173°С. Пример 18 1-[4-(метилсульфинил)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 14, за исключением использования соединения примера 17(б) в качестве сульфида, получают целевое соединение в виде белого твердого вещества(67%). Тпл. = 202203°С. Пример 19 1-[3-(метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид(3-метилтиофенил)имин С применением процедуры примера 1(в), за исключением использования 3-(метилтио)анилина в качестве амина, получают(98%) масло светло-желтого цвета, содержащее приблизительно 2,5% альдегида на основании данных 1Η ЯМР. 1Η ЯМР(CD3CI): д. 8,76(д., J=6Гц, 2Н); 8,44(с. 1Н) 7,74(д., J=6Гц, 2Н); 7,30(д., J=8Гц, 2Н); 7,34-6,98(м., 4Н); 2,52(с., 3Н). б) 1-[3-(метилтио)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(42%). Тпл. = 155-156°С. Пример 20 1-[3-(метилсульфинил)фенил] -4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 14, за исключением использования соединения примера 19(б) в качестве сульфида, получают целевое соединение в виде белого твердого вещества(67%). Тпл. = 233234°C. Пример 21 1-[2-(метилтио)фенил]-4-(4-фторфенил-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид(2-метилтиофенил)имин С применением процедуры примера 1(в), за исключением использования 2-(метилтио)анилина в качестве амина, получают(98%) масло светло-желтого цвета, содержащее приблизительно 8% альдегида на основании данных 1Η ЯМР. 1H ЯМР(CD3CI): д 8,75(д J=6Гц 2Н); 8,41 (с., 1Н) 7,79(д., J = 6Гц, 2Н); 7,367,00(м., 4Н); 2,47(с., 3Н). б) 1-[2-(метилтио)фенилу]-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина и очистки с помощью флэш-хроматографии с использованием 0-1% МеОН в СH2СI2, полyчают целевое соединение в виде некристаллического белого пенообразного вещества(53%). Тпл. = 59-60°С. Пример 22 1-[2-(метилсульфинил)фенил]-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 14, за исключением использования соединения примера 21(б) в качестве сульфида и очистки с помощью флэш-хроматографии с использованием 0-4% МеОН в СН2СI2, получают целевое соединение в виде некристаллического белого пенообразного вещества(52%). Тпл. = 60-165°С.(Столь большой разброс в определении Тпл., по-видимому, является результатом присутствия смеси конформационных изомеров, что четко определяется на 1Η и 13С ЯМР-спектрах этого соединения). Пример 23 1-(3-хлорпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол (см. также выше, Пример 2, содержащий альтернативный способ получения) а) 4-фторфенил-тозилметилформамид К раствору гидрата натриевой соли толуолсульфиновой кислоты(120г) в воде(750мл) добавляют концентрированную серную кислоту(16мл). Затем добавляют дихлорметан(500мл) и разделяют органическую и водную фазы; водный слой экстрагируют дихлорметаном(2 x 200мл). Объединенные органические экстракты высушивают(Na2SO4) и выпаривают досуха с получением твердой сульфиновой кислоты(71,79г; 0,46моль). Этот продукт добавляют к п-фторбензальдегиду(57,04г; 0,46 моль) и формамиду(62,1г; 1,38моль) и перемешивают полученную смесь с камфор-10-сульфоновой кислотой(21,3г; 0,092моль) при температуре 60-65°С в атмосфере азота в течение 22 часов. После этого раствор бикарбоната натрия(33,6г; 0,40моль) в воде(400мл) прибавляют к охлажденному на льду твердому продукту, который разрушается, и продолжают перeмешивание в течение 30 минут. Грубый продукт собирают и промывают ацетоном(220мл), а затем эфиром(3 x 220мл), после чего высушивают с получением искомого продукта, 91,5г; 64,8%. б) 4-фторфенил-тозилметилизоцианид К суспензии соединения предыдущей стадии(3,22г; 10,5ммоль) в диметоксиэтане(21мл), перемешиваемой при температуре –10°С, добавляют по каплям в течение 5 минут оксихлорид фосфора(2,36мл; 25,3ммоль). Затем добавляют по каплям в течение 10 минут триэтиламин(7,35мл; 52,5ммоль) и вливают реакционную смесь в насыщенный раствор бикарбоната натрия(100мл), после чего маслянистый продукт экстрагируют дихлорметаном(2 x 30мл). Органические экстракты выпаривают с получением масла черного цвета(3,51г), которое элюируют на основном алюмине(Grade 111)(60г) с использованием дихлорметана. Объединенные фракции продукта выпаривают и добавляют эфир для кристаллизации искомого продукта, 1,735г; 57%. в) 1-(3-хлорпропил)-4-(4-фторфенил)-5-(4-пиридил)имидазол К раствору соединения предыдущей стадии(1,183г; 4,09ммоль) и соединения примера 2(а)(1,122г; 6,15ммоль) в диметоксиэтане(15мл) при комнатной температуре добавляют по каплям в течение 10 минут раствор ДБУ(0,67мл; 4,51ммоль) в диметоксиэтане(10мл). Реакционную смесь перемешивают при комнатной температуре в течение 1-1/2 часа и затем выпаривают с получением масла, которое элюируют на основном алюмине( Grade 111)(100г) с получением искомого продукта, 1,096г; 85%. Пример 24 1-[4-(4-морфолинил) бутил]-4-(4-фторфенил)-5-(4-пиридил)имидазол а) 4-(4-морфолино)бутил-1-фталимид Смешивают 4-бромбутил-1-фталимид(5,0г; 17,7ммоль) и морфолин(20мл) и перемешивают полученную смесь в течение 3 часов, разбавляют ее Et2(200мл) и фильтруют. Твердое вещество еще раз промывают Et2О и объединяют фильтраты, которые затем экстрагируют 3N НСI(3 x 25мл). Объединенные кислые фазы промывают Et2О(3 x 50мл), наслаивают Et OAc и подщелачивают сначала добавлением твердого К2СО3 до прекращения пенообразования, а затем добавлением 10%-ного водного NaOH до достижения рН выше 10. Далее проводят экстракцию с помощью Et OAc(3 x 100мл), высушивание(K2СО3), концентрирование и флэш-фильтрацию; при этом 1л силикагеля промывают сначала 0-4% МеОН с СН2СI2, а затем проводят элюцию продукта 4% МеОН и 1% Et3N в СН2СI2 с получением 5,52г(54%) целевого соединения в виде белого твердого вещества. б) 4-(4-морфолино)бутиламин Смешивают соединение примера 24(а)(1,0г; 3,47ммоль), моногидрат гидразина(190мкл; 3,82ммоль) и СН3ОН(20мл)., и перемешивают полученную смесь в течение ночи при 23°С. СН3ОН удаляют под вакуумом, а остаток концентрируют досуха из EtОН. Остаток объединяют с 2N НСI(20мл) и перемешивают в течение 2 часов, затем фильтруют, а твердое вещество промывают Н2О. Объединенные фильтраты концентрируют под вакуумом и дважды подвергают повторной концентрации из Εt ОН с получением белой пены, которую растворяют в смеси СН2СI2/СН3ОН(3:1), затем перемешивают с твердым К2СО3 в течение 5 минут и фильтруют. Фильтрат концентрируют с получением 0,535г(80%) масла коричневого цвета. 1Η ЯМР(СD3СI): 3,7-3,2(м., 6Η); 2,7-2,2(м., 6); 1,6-1,3(м., 6К). в) Пиридин-4-карбоксальдегид[4-(4-морфолинил) бутил]имин С применением процедуры примера 1(в), за исключением использования соединения примера 24(б) в качестве амина, получают целевое соединение в виде масла бледно-желтого цвета(100%), содержащего 30% альдегида на основании данных 1Н ЯМР. 1Н ЯМР(СD3СI): 8,60(д.,J=6Гц, 2Н); 8,19(с.,1H); 7,51(д.,J=6Гц, 2Н); 3,7 - 3,2(м.,6Н); 2,5 - 2,2(м.,6); 1,7 - 1,4(М.,4Н). г) 1-[4-(4-форфолинил)бутил]-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения примера 24(в) в качестве имина, получают целевое соединение(38%). Тпл. = 103 – 104°С Пример 25 1-циклопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид циклопропиламин С применением процедуры примера 1(в), за исключением использования 100%-гo избытка летучего циклопропиламина, получают целевое соединение в виде масла бледно-желтого цвета(100%). 1Н ЯМР(СD3СI): 8,65(д.,J=6Гц, 2Н); 8,40(с, 1Н); 7,51(д.,J=6Гц, 2Н); 3,07(м.,1Н); 1,01(м.,4Н). б) 1-циклопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол Охлаждают до температуры 0°С смесь соединения предыдущей стадии(20ммоль), соединения примера 1(б) (5,65г; 22ммоль) и СH2СI2(20мл), после чего добавляют ТБД(2,84г; 20ммоль). Перемешивают полученную смесь при 5°С в течение 2 часов и при 23°С в течение 48 часов и нагревают с обратным холодильником до температуры кипeния в течение 4 часов. Проводят флэш-фильтрацию грубой реакционной смеси через воронку из спекшегося стекла, заполненную силикагелем(1л силикагеля), элюируя 0 - 4%СН3ОН в CH2Cl2. Кристаллизуют из смеси гексан/ацетон с получением 839мг(15%) целевого соединения. Тпл. = 129,0 - 129,5°С. Пример 26 1-изопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид изопропилимин С применением процедуры примера 1(в), за исключением использования изопропиламина в качестве амина, получают целевое соединение в виде масла бледно-желтого цвета(100%). 1Н ЯМР(CD3Cl): 8,67(д.,J=4,4Гц, 2Н); 8,27(с., 1Н); 7,59(д., J=4,43Гц, 2Н); 3,59(м.,1Н); 1,27(д., J=6,3Гц, 6Н). б) 1-изопропил-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), заменяя имин на соединение предыдущей стадии, получают препарат, используя модифицированный вариант флэш-фильтрации грубой реакционной смеси через силикагель(0 - 4% СН3ОН в СН2СI2). Две кристаллизации из смеси гексан/ацетон дают целевое соединение в виде рыжевато-коричневых игольчатых кристаллов(30%) Тпл. = 179,0 - 179,5°С. Пример 27 1-циклопропилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид циклопропилметилимин С применением процедуры примера 1(в), за исключением использования циклопропилметиламина в качестве амина, получают целевое соединение в виде масла бледно-желтого цвета(100%). 1Н ЯМР(CD3Cl): 8,69(д., J=4,5Гц, 2Н); 8,27(с, 1Н); 7,61(д., J=4,5Гц, 2Н); 3,55(д., J=6,7Гц, 2Н); 1,15(м.,1Н); 0,57(м., 2Н); 0,27(м., 2Н). б) 1-циклопропилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), заменяя имин на соединение предыдущей стадии, получают препарат, используя модифицированный вариант флэш-фильтрации грубой реакционной смеси через силикагель(0 - 4% СH3ОН в СН2СI2). Кристаллизация из смеси гексан/ацетон дает целевое соединение в виде белых хлопьев(62%). Тпл. = 162,0 - 162,5°С. Пример 28 1-трет-бутил-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид трет-бутилимин С применением процедуры примера 1(в), за исключением использования трет-бутиламина в качестве амина, получают целевое соединение в виде масла бледно-желтого цвета(100%), 1Н ЯМР(CD3Cl): 8,67(д.,J=4,4Гц, 2Н); 8,22(с.,1Н); 7,61(д., J=4,4Гц, 2Н); 1,30(с., 9Н). б) 1-трет-бутил-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), заменяя имин на соединение предыдущей стадии, получают препарат, используя модифицированный вариант флэш-фильтрации грубой реакционной смеси через силикагель(0 - 4% СН3ОН в СН2СI2), приводящий к получению целевого соединения в виде порошка рыжевато-коричневого цвета(16%).Тпл = 199,0 - 200,0°С. Пример 29 1-(2,2-диэтоксиэтил)-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид 2,2-диэтоксиэтиламин С применением процедуры примера 1(в), за исключением использования 2,2-диэтоксиэтиламина в качестве амина, получают целевое соединение в виде масла бледно-желтого цвета(100%). 1Н ЯМР(СD3СI): 8,69(д., J=4,4Гц, 2Н); 8,28(с, 1Н); 7,60(д., J=4,4Гц, 2Н); 4,82(т., J=5,1Гц, 1Н); 3,82(д., J=5,1Гц, 1Н); 3,72(м., 2Н); 3,72(м., 2Н); 3,57(м., 2Н); 1,21(т., J=7,3Гц, 6Н). б) 1-(2,2-диэтоксиэтил)-4-(4-фторфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), заменяя имин на соединение предыдущей стадии, получают препарат, используя модифицированный вариант флэш-фильтрации грубой реакционной смеси через силикагель(0-4% СН3OН в CH2Cl2) с последующей флэш-хроматографией на силикагеле(с примнением 25100% EtOAc в гексане) и растиранием полученной камеди с гексаном, что приводит к получению целевого соединения в виде белого порошка(47%). Тпл. = 69,5-70,0°С. Пример 30 1-формилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол Смешивают продукт примера 29(б)(400мг; 1,13ммоль), H2O(10мл), ацетон(10мл) и концентрированную H2SО4(1мл) и нагревают полученную смесь при температуре кипения с обратным холодильником в течение 24 часов. Большая часть ацетона удаляется под вакуумом, а остаток объединяют с 5%-ным водным Na2CO3, и экстрагируют EtOAc; высушивают(Na2SО4), концентрируют и кристаллизуют из ацетона с получением целевого соединения в виде белого порошка(47%). Тпл. = 118,5-119°C Пример 31 1ггидроксииминилметил-4-(4-фторфенил)-5-(4-пиридил)имидазол Смешивают продукт примера 30(317мг; 1,13ммоль), гидроксиламинагидрохлорид(317мг), пиридин(317мкл) и EtOH(3,8мл) и нагревают при температуре кипения с обратным холодильником в течение 3 часов, после чего вливают полученную смесь в 5%-ный водный Nа2СО3 и экстрагируют ΕtОАс; высушивают(Na2SO4) и проводят флэш-фильтрацию с использованием 0-4% МеОН в СН2СI2, получая при этом 261мг(78%) целевого соединения в виде белого порошка. Тпл. = 184,0-185,0°С. Пример 32 1-цианометил-4-(4-рторфенил)-5-(4-пиридил)имидазол Смешивают продукт примера 31(250мг; 0,84ммоль) и СuSО4, после чего полученную смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов. Проводят флэш-фильтрaцию охлажденной реакционной смеси с использованием 0-4% МеОН в СН2СI2, получая в результате 129мг(55%) целевого соединения в виде белого порошка. Тпл. = 132,0-133,0°С. Пример 33 1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(2-метилпирид--4-ил)имидазол а) 4-формил-2-метилпиридин В соответствии с описанной в литературе методикой [Yamanaka, H., Abe, H., Sakamoto, T.; Hidetoshi, Hiranuma, H.; Kamata, A/ Chem. Pharm. Bull. 25(7), 1821-1826] получают 4-циано-2-метилпиридин на основе 2,6-лютидина. Далее, к охлажденноному до –78°С раствору 4-циано-2-метилпиридинг(0,367г; 3,11ммоль) и толуола(3,5мл) прибавляют по каплям с помощью шприц-насоса 1M DIBAL в гексaне(3,6мл; 3,6ммоль)(температура ниже -65°С). Затем реакционную смесь нагревают до 5°С и перемешивают в течение 5 минут; вновь охлаждают до температуры -78°С и добавляют СН3ОН(3,5мл)(температура ниже 40°С); нагревают до температуры 5°С и перемешивают в течение 5 минут, а затем добавляют 25%-ный водный раствор соли Рошеллa, перемешивают в течение 3 минут и подкисляют смесь до рH менее 1,0 с помощью 10%-ной водной H2SO4. Водный слой подщелачивают путем добавления твердого К2СO3 и экстрагируют EtOAc. Экстракты высушивают(Na2SO4), концентрируют и фильтруют через силикагсль(2% MeOH в СН2СI2) с получением 253мг(84%) альдегида. 1H ЯMP(СD3СI): д. 10,05(с. 1H); 8,74(д., J = 7Гц 1Н); 7,51(с. 1Н); 7,30(д., J = 7Гц, 1H): 2,68(с., 3Н). б) Пиридин-4-кaрбоксальдегид[3-(4-морфолинил)пропил]имин С применением процедуры примера 1(в) проводят реакцию взаимодействия продукта, полученного на предыдущей стадии, и 4-(3-аминопропил)морфолинa с получением целевого соединения в виде масла желтого цвета, не содержащего альдегида на основании 1Н ЯMР. 1Н ЯMР(СD3СI): д. 8,57(д., J =5,0Гц 1Н) 8,25(с, 1H); 7,46(с. 1Н); 7,36(д, J=5,0Гц 1H); 3,71(м.,6Н); 2,60(с. 1Н) 2,35(м., 6Н); 1,90(м., 2Н). 1-[3-(морфолинил)пропил]-4-(4-фторфенил)-5-(2-метилпирид-4-ил)имидазол С применением процедуры примера 1(г) проводят реакцию взаимодействия соединения, полученного на предыдущей стадии, с соединением примера 1(б), что дает в итоге целевое соединение в виде белого твердого вещества [51% из 33(a)] Тпл. = 116-117°С. Пример 34 4-(4-фтopфeнил)-1-[3-(4-морфолинил)пропил]-5-(2-хлорпиридин-4-ил)имидазол а) 2-хлорпиридин-4-карбоксальдегид [3-(4-морфолинил)пропил]имин 2-хлорпиридин-4-карбоксальдегид получают в соответствии с описанием, имеющимся в патентной литературе(WPl Ace. №88-258820/37), указанный патент включен полностью в качестве ссылки в настоящее описание. Этот альдегид взаимодействует с 4-(3-aминопропил)морфолином по процедуре примера 1(в) с образованием целевого соединения в виде желтого масла. 1H ЯМР(СD3СI): ы 8,45(д., J = 5,1Гц 1Н); 8,24(С. 1Н); 7,63(с. 1Н); 7,51(д., J = 5,1Гц 1Н), 3,72(м., 6H); 2,44(м., 6H); 1,91(м., 2Н). б) 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-хлор-4-пиридинил)имидазол С применением процедуры примера 1(г), за исключением использования соединения, полученного на предыдущей стадии, в качестве имина, получают целевое соединение в виде белого твердого вещества(93%). Тпл. = 97,0-97,5°С. Пример 35 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-амино-4-пиридинил)имидазол а) 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-гидразинил-4-пиридинил)имидазол Соединение примера 34(б) (872мг; 2,18ммоль) и 98%-ный гидрат гидразина(9мл) нагревают до температуры 115°С(температура бани) в течение 20 часов, затем охлаждают до 23°С, смешивают с H2O(20мл) и экстрагируют EtОАс(3 x 25мл). Объединенные экстракты промывают H2О(2 x 20мл) и высушивают(Na2SO4). В результате флэш-хроматографии с применением 0-8% СН3ОН в СH2СI2 получают 547мг(63%) целевого соединения в виде белого твердого вещества. б) 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(2-амино-4-пиридинил)имидазол Соединение, полученное на предыдущей стадии(100мг; 0,25ммоль), абсолютный ΕtОН(15мл) и Раней Ni(Raney Ni)(0,4мл) взбалтывают в атмосфере водорода(45фунт/кв.дюйм) в течение 4 часов. В результате флэш-хроматографии с использованием 0-8% СH3ОН в СH2СI2 получают 34мг(37%) целевого соединения в виде белого твердого вещества. Тпл. = 186-187°С. Пример 36 1-(4-карбоксиметил)пропил-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид(4-карбоксиметилбутил)имин Проводят реакцию пиридин-4-кгрбоксальдегида с метил-4-aминобутирaтом с применением процедуры примера 1(в), получая при этом целевое соединение в виде желтого масла. 1H ЯМP(СD3СI): ы 8,69(д., J=5,8Гц, 2Н); 8,27(с., 1Н); 7,56(д., J=5,8Гц, 2Н); 3,70(м., 2Н); 2,31(т., J=8,0Гц, 2Н); 2,08(м.,2Н). б) 2-(4-карбоксиметил)пропил-4-(4-фторфeнил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г), за исключением использования соединения, полученного на предыдущей стадии, в качестве имина, получают целевое соединение в виде белого твердого вещества(35%). Тпл. = 69,0-70,0°С. Пример 37 1-(4-Карбоксипропил)-4-(4-оторфенил)-5-(4-пиридил)имидазол Смешивают соединение примера 36(100мг, 0,29ммоль), CH3OH(3мл) и ТГФ(1,5мл) и обрабатывают полученный раствор раствором LiОН(62мг; 1,5ммоль) в H2O(1,5мл); полученную смесь перемешивают в течение 4 часов. Затем проводят удаление летучих компонентов под вакуумом, перерастворяют остаток в Н2O и хроматографируют его на НР-20 вначале с использованием Н2O до достижения нейтральной реакции элюатов, а затем 25%-ным водным МеОН, что приводит к получению целевого соединения в виде соли лития; 65мг(68%). ЕS(+) ΜS m/е=326(МН+). Пример 38 1-(3-карбоксиметил)этил-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид(3-карбоксиметил)этилимин С применением процедуры примера 1(в) проводят реакцию взаимодействия пиридин-4карбоксальдегида с метиловым эфиром β-аланина, получая в результате целевое соединение в виде масла желтого цвета. 1H ЯМР(СD3СI): ы 8,68(д., J=4,5Гц, 2Н); 8,33(с. 1Н); 7,57(д., J=4,5Гц, 2Н); 3,93(t., J=6,7Гц, 2Н); 3,68(с., 3Н); 2,76(т., J=6,7Гц, 2Н). б) 1-(3-карбоксиметил)этил-4-(4-Фторфенил)-5-(4-пиридил)имидазолSB-219302 С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(40% на основе амина). Тпл. = 119,0-120,0°С. Пример 39 1-(3-карбокси)этил-4-(4-фторфенил)-5-(4-пиридил)имидазол С использованием процедуры примера 37 подвергают гидролизу соединение примера 38(б) с получением целевого соединения в виде соли лития:(71%). ЕS(+)МS m/е = 312(МН+). Пример 40 1-(1-бензилпиперидин-4-ил)-4-(4-фторфенил)-5-(4-пиридил)имидазол а) Пиридин-4-карбоксальдегид(1-бензилпиперидин-4-ил)имин; Проводят реакцию взаимодействия пиридин-4-карбоксальдегида с 4-амино-N-бензилпиперидином с использованием процедуры примера 1(в), получая целевое соединение в виде желтого масла. б)1-(1-бензилпиперидин-4-ил)-4-(4-фторфенил)-5-(4-пиридил)имидэзол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии примера в качестве имина, получают целевое соединение в виде белого твердого вещества(9% на основании амина). ЕS(+)МS m/e=413(MН+). Пример 41 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]имидазол а) 2-аминопиримидин-4-карбоксальдегид диметилацеталь Смешивают диметилацеталь диметилформамида(55мл; 0,41моль) и диметилацеталь пировиноградного альдегида(50мл; 0,41моль) и нагревают полученную смесь до 100°С в течение 18 часов. Под вакуумом удаляют метанол с получением масла. Добавляют раствор NaOH(18г; 0,45 моль) в H2О(50мл) к раствору солянокислого гуанидина(43г; 0,45моль) и затем приливают полученный раствор к описанному выше маслу. Полученную смесь перемешивают при температуре 23°С в течение 48 часов. Фильтрованием этой смеси получают целевое соединение в количестве 25г(50%). б) 2-аминопиримидин-4-карбоксальдегид Смешивают соединение, полученное на предыдущей стадии(1,69г; 10ммоль) и 3н НСІ(7,3мл; 22ммоль) и нагревают полученную смесь до 48°С в течение 14 часов; охлаждают, наслаивают EtOAc(50мл) и затем нейтрализуют добавлением малых порций NaHCO3(2,1г; 25ммоль). Экстрагируют водную і фазу с применением EtOAc(5 x 50мл), после чего экстракты высушивают(Na2SO4) и концентрируют с получением 0.793г, (64%) целевого соединения. в) 2-аминопиримидин-4-карбоксальдегид [3-(4-морфолинил)пропил]имин С применением процедуры примерa 1(в) проводят реакцию с участием соединения, приготовленного на предыдущей стадии, и 4-(3-аминопропил)морфолина с получением целевого соединения в виде желтого пасла. г) 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-3-(4-морфолинил)пропил]имидазол SВ 216385 С применением процедуры примера 1(г), за исключением использования соединения, приготовленного на предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества. 1Η ЯМР(СD3Cl): ы 8,15(д., J=5,4Гц, 1Н); 7,62(с. 1Н) 7,46(дд., 2Н); 7,00(т., J=8,6Гц, 2Н); 6,50(д., J=5,4Гц 1Н); 5,09(шд.,с., 2Н); 4,34(т., J=7,0Гц, 2Н); 3,69(м., 4Н); 2,35(шд., c., 4Н); 2,24(т., J=4,6Гц, 2Н); 1,84(м., 2Н). Пример 42 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-бензилпиперидин-4-ил)имидазол а) 2-аминопиримидин-4-карбоксальдегид 1-(1-бензилпиперидин-4-ил)имин С применением процедуры примера 1(в) проводят реакцию взаимодействия 2-аминопиримидин-4карбоксальдегида с 4-амино-бензил-пиперидином с получением целевого соединения в виде желтого масла. б) 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-бензилпиперидин-4-ил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(31% на основании амина). Тпл. = 227-229°С(разлож.). Пример 43 5-2-аминопиримидин-4-ил)-4-(4-Фторфенил)-1-(2-пропил)имидaзол а) 2-аминопиримидин-4-карбоксальдегид 1-(2-пропил)имин С применением процедуры примера 1(в) проводят реакцию взаимодействия 2-аминопиримидин-4карбаксальдегида с 2-пропиламином с получением целевого соединения в виде желтого масла. б) 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(2-пропил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(32% на основании 2-аминопиримидинальдегида). Тпл. = 201-202°С. Пример 44 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(циклопропилметил)имидазол а) 2-аминопиримидин-4-карбоксальдегид(циклопропилметил)имин С применением процедуры примера 1(в) проводят реакцию взаимодействия 2-аминопиримидин-4карбоксальдегида с 2-циклопропилметиловым амином с получением целевого соединения в виде желтого масла. б) 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(циклопропилметил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(38% на основании 2-аминопиримидинальдегида). Тпл. = 187-188°С. Пример 45 5-(2-аминопиримидин-4-ил)-4-(4-Фторфенил)-1-(1-карбоксиэтил-4-пиперидинил)имидазол а) 2-аминопиримидин-4-карбоксальдегид(1-карбокси-этил-4-пиперидинил)имин С применением процедуры примера 1(в) проводят реакцию взаимодействия 2-аминопиримидин-4карбоксальдегида с 1-карбоксиэтил-4-аминопиперидином с получением целевого соединения в виде желтого масла. б) 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-карбоксиэтил-4-пиперидинил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(26% на основании 2-аминопиримидинальдегида). Тпл. = 216-218°С(разлож). Пример 46 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол а) 2-аминопиримидин-4-карбоксальдегид(1-е-бутокси-карбонил-4-аминопиперидин)имин С применением процедуры примера 1(в) проводят реакцию взаимодействия 2-аминопиримидин-4карбоксальдегида, приготовленного в примере 41, с 1-t-бутоксикарбонил-4-иминопиперидином(Mach R. H, et all., J. Med. Chem, 36, р.3707-3719, 1993) с получением целевого соединения в виде желтого масла, б) 5-[4-(2-амино)пиримидинил]-4-(4-фторфенил)-1-(1-t-бутоксикарбонилпиперидин-4-ил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(27% на основании 2-аминопиримидинальдегида). в) 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол SВ 220025 Соединение, приготовленное на предыдущей стадии, смешивают с 4н HСI в диоксане(5мл), перемешивают в течение 10 минут, разбавляют EtОАс и декантируют жидкую фазу. Твердое вещество дважды промывают с помощью Εt2О(25мл), и декантируют жидкую фазу. Затем проводят растирание с EtOH(адс) и с Et2O далее высушивают под вакуумом при температуре 50°С в течение 16 часов, получая в итоге целевое соединение в виде тригидрохлорида(41%). Тпл. = 265-275°С(разлож.). Пример 47 1-метил-4-фенил-5-(4-пиридил)имидазол С применением процедуры примера 48(б), за исключением использования бензонитрила, получают целевоe соединение в виде белого твердого вещества. Тпл. = 161-162°С. Пример 48 1-метил-4-[3-(хлорфенил)]-5-(4-пиридинил)имидaзол а) N-(4-пиридинилметил)-N-метилформамид К перемешиваемому раствору 4-пиколилхлорид-НСI(15г; 91,4ммоль) и N-метилформамида(53,4мл; 914ммоль) в 300мл ТГФ, через который продувают аргон, добавляют при комнатной температуре порциями в течение 20 минут суспензию 80%-ного NaH(5,48г; 183ммоль). Реакцию oстанавливают через 18 часов добавлением льда; смесь распределяют между метиленхлоридом и водой, промывают водой и солевым раствором, высушивают над MgSO4 и выпаривают досуха с получением темного масла. С применением флэш-хроматографии на силикагеле получают 10,5г(76%) целевых соединений в виде масла бледно-желтого цвета. ТСХ; силикагель(9:1 СНСI/МеОЕ) Rf = 0,54. б) 1 -метил-4-[3-(хлорфенил)]-5-(4-пиридинил)имидазол При перемешивании под аргоном при температуре –78°С к раствору диизопропиламина лития(далее ЛДА),(полученного из 11,2мл диизопропиламида в 150мл тетрaгидрофурана(далее ТГФ) добавлением 31,9мл 2,5М n-BuLi в гексанах), добавляют порциями продукт, полученный в результате предыдущей реакции(10г; 66,5ммоль) в 100мл ТГФ. Продолжают перемешивание полученного красновато-коричневого раствора при температуре –78°С в течение 40 минут; с этого момента добавляют по каплям в течение 20 минут раствор 3-хлорбензонитрила(18,3г; 133ммоль) в 100мл ТГФ. Температуру реакционной смеси повышают до комнaтной, перемешивают смесь в течение одного часа и нагревают с обратным холодильником до температуры кипения в течение 12 часов. Затем реакционную смесь охлаждают и обрабатывают тем же образом, что и в предыдущей реакции. Далее проводят флэш-хроматографию на силикагеле с получением 2,15г масла, которое кристаллизуют при растворении с нагреванием в 10мл этилацетата. После кристаллизации собирают твердое вещество, промывают и высушивают(0,4мм ртутного столба) с получением 1,43г(8%) целевого соединения в виде твердого вещества слегка рыжеватого цвета. Тпл. = 119-121°С. Пример 49 1-метил-4-(3метилтиофенил)-5-(4-пиридил)имидазол С применением процедуры примера 48(б), за исключением использования соединения примера 13(б) в качестве арилнитрила, получают целевое соединение в виде белого твердого вещества. ЕS(+) MS m/е = 281(МН+). Пример 50 1-метил-4-(3-метилсульфинилфенил)-5-(4-пиридил)имидазол С применением процедуры примера 14, за исключением использования соединения примера 49 в качестве сульфида, получают целевое соединение в виде белого твердого вещества. ΕS(+) MS m/е = 297(МH+). Пример 51 (+/-)-4-(4-фторфеиил)-1-[3-(мeтилсульфонил)пропил]-5-(4-пиридинил)имидазол С применением процедуры примера 14, за исключением использования соединения примера 11 в качестве сульфида и остановки реакции добавлением насыщенного водного NН4ОH, получают целевое соединение в виде белого твердого вещества(0,87г; 80%). Тпл. = 122-123°C Пример 52 4-(4-фторфенил)-1-[3-(метилсульфонил)пропил]-5-(4-пиpидинил)имидазол Соединение примера 51(0,5087г, 1,48ммоль) растворяют в метаноле(8мл) и охлаждают до температуры 0°С. Добавляют трифторуксусную кислоту(0,12мл), затем по каплям прибавляют метахлорпероксибензойную кислоту(0,23г; 2,22ммоль), растворенную с СН2СI2(10мл). После перемешивания в течение 1 часа выпаривают растворители. Остаток распределяют между H2O и EtOAc и подщелачивают водную фазу добавлением 2N NаОH. Отделяют органическую фазу, высушивают(MgSО4) и концентрируют, а остаток чистят с применением флэш-хроматографии(силикагель, 5% МеОН/СН2СІ2) с получением целевого соединения(0,37г; 69%). Тпл. = 146-147°С. Пример 53 1-(3-феноксипропил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол К раствору соединения из примера 2(б)(0,22г; 70ммоль) в ацетонитриле(10мл) добавляют К2СО3(0,19г; 1,40ммоль) и фенол(0,10г; 1,05ммоль). После перемешивания при температуре 70°С в течение 24 часов реакционную смесь разбавляют H2О. Органическую фазу отделяют, концентрируют, а остаток чистят с помощью флэш-хроматографии(силикагель, 5% MeOH/CH2Cl2), после чего проводят перекристаллизацию в гексане с получением целевого соединения(0,22г; 8%) в виде белого твердого вещества. Тпл. = 95-96°С. Пример 54 1-[3-(фенилтио)пропил]-4-(4-фторфенил)-5-(4-пиридинил)имидазол Применяют процедуру примерa 3, за исключением использования тиофeнола в качестве нуклeофильного агента, добавляя 2,2экв. К2СО3, и исключая обработку N al · Реакционную смесь охлаждают, разбавляют 10%-ным NaΟΗ и экстрагируют продукт эфиром. После этого проводят Флэшхроматографию с последующей перекристаллизацией из гексака с получением целевого соединения(0,13г; 53%) в виде белых игольчатых кристаллов. Тпл. = 98-99°С. Пример 55 1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(4-хинолил)имидазол а) Хиноил-4-карбоксальдегид [3-(4-морфолинил)пропил]имин С применением процедуры примера 1(в) проводят реакцию взаимодействия хиноил-4карбоксальдегида с 4-(3-аминопропил)морфолином с получением целевого соединения в виде желтого масла. б) 1-[3-(4-морфолинил)пропил]-4-(4-фторфенил)-5-(4-хинолил)имидaзол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(48% на основании амина). Тпл. = 139,5-140,0°С. Пример 56 (+/-) -1-(3-фенилсульфинилпропил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол Применяя процедуру примера 14, зa исключением использования соединения примера 54 в качестве сульфида, и останавливая реакцию добавлением насыщенного водного NH4OH, получают целевое соединение в виде белого твердого вещества. Тпл. = 146,5-148°С. Пример 57 1-(3-этоксипропил)-4-(4-фторфенил)-5-(4-пиридинил)имидaзол К раствору примера 2(б)(0,40г; 1,26ммоль) в этаноле(25мл) добавляют этокенд натрия(0,8мл; 21 вес.% в этаноле). После нагревания при температуре кипения с обратным холодильником в течение 16 часов смесь охлаждают, разбавляют Н2O и экстрагируют EtOAc. Далее проводят концентрирование растворителя и очистку с применением флэш-хроматографии(силикагель, 5% МеОН/СН2СI2) с получением в результате целевого соединения(0,05г; 12%). Тпл. = 85-86°С. Пример 58 1-(3-фенилсульфонилпропил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол С применением процедуры примера 52, за исключением использования соединения примера 56 в качестве сульфоксида и повторной кристаллизации из гексана с последующей хроматографией, получают целевое соединение в виде белого твердого вещества. Тпл. = 109-110°С. Пример 59 1-[3-(4-морфолинил)пропил]-4-(3-хлорфенил)-5-(4-пиридил)имидазол а) 3-хлорфенил-толилтиометилизоцианид С применением процедуры примера 1(а,б), за исключением использования трихлорбензальдегида в качестве альдегидного компонента, получают целевое соединение. б) 1-[3-(4-морфолинил)пропил]-4-(3-хлорфенил)-5-(4-пиридинил)имидазол С применением процедуры примера 1(г) с заменой на изоцианид, приготовленный на предыдущей стадии, получают целевое соединение. MS-DCI NH3=383[M+H]. Пример 60 1-[3-(4-морфолинил)пропил]-4-(3,4-дихлорфенил)-5-(4-пиридил)имидазол С применением процедуры примера 1(г) с заменой на изоцианид, приготовленный в примере 67(а), получают целевое соединение. Тпл. = 106°С. Пример 61 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(пиримид-2-он-4-ил)имидазол а) 2-метилтиопиримидин-4-карбоксальдегид [3-(4-морфолинил)пропил]имин С применением процедуры примера 1(в), за исключением использования 2-метилтиопиримидин-4карбоксальдегида(Bredereck H. Et. Al., Chem. Ber., 1964, 3407) получают целевое соединение в виде желтого масла. б) 4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]-5-(пиримид-2-он-4-ил)имидазол Добавляют концентрированный водный гидроксид аммония(2мл) к 4-(4-фторфенил)-5-[2(метилсульфинил)-4-пиримидинил]-1-[3-(4-морфолинил)пропил]) имидазолу(0,14г; 0,37ммоль) [полученному в примере 63 ] и нагревают полученную смесь до 150°С в течение 18 часов. Затем охлаждают смесь до комнатной температуры и декантируют гидроксид аммония. Остаток чистят флэшхроматографией с применением для элюции последовательно 4% и 10% метанола в дихлорэтане, а затем смесей 90/10/1 и 70/30/3 хлороформ/метанол/концентрированный гидроксид аммония. Растирают с эфиром получая и целевое соединение в виде твердого вещества грязновато-белого цвета(0,035г; 24%). ESMS(m/z) 384(М++H). Пример 62 4-(4-фторфенил)-5-[2-(метилтио)-4-пиримидинил]-1-[3-(4-морфолинил)пропил]имидазол С применением процедуры примера 1(г), за исключением использования 2-метилтиопиримидин-4Кaрбоксальдегид-[3-(4-мoрфoлинил)пропил]имина [полученного в примере 61(а)], получают целевое соединение в виде желтого масла. 1Η ЯMР(СD3СI): ы 8,31(д., J=7Гц 1Н); 7,64(с. 1Н); 7,46(дд., 2Е); 7,05(т., J=8Гц, 2Н); 6,81(д., J=5Гц 1Н): 4,42(т., J=7,5Гц, 2Н); 3,71(т., j=5Гц, 4Н); 2,58(с., 3Н); 2,27(т., J=6Гц, 2Н); 1,85(м., 2Η). Пример 63 4-(4-фторфенил)-5-[2-(метилсульфинил)-4-пиримидинил]-і-[3-(4-морфолинил) пропил]имидазол Раствор К2S2O8(0,20г; 0,73ммоль) в воде(5мл) добавляют к 4-(4-фторфенил)-5-[2-(метилтио)-4пиримидинил]-1-[3-(4-морфолинил)пропил]имидазолу(0,20г; 0,48ммоль) в ледяной уксусной кислоте(10мл). После перемешивания при комнатной температуре в течение 72 чесов реaкционную смесь выливают в воду нейтрализуют с применением водного концентрированного раствора гидроксидa аммония и четырежды экстрагируют дихлорметаном. Органические фазы объединяют и выпаривают· Остаток чистят флэш-хроматографией с применением для элюции последовательно 1%, 2%, 4% и 10% метанола в дихлорметaне с получением в итоге целевого соединения в виде прозрачного масла(0,15г; 73%). 1Η ЯМР(СD3СI): ы 8,57(д., J=7Гц, 1H); 7,77(с. 1Н); 7,47(дд., 2Н); 7,18(д., J=5Гц 1Н); 7,09(τ., J=9Гц, 2К); 4,56(м., 2Н); 3,72(т., J=5Гц, 4Н); 3,00(с., 3Н); 2,40(шд.,с., 4H); 2,33(т., J=8Гц, 2Н); 1,94(м., 2Н). Пример 64 (E)-1-(1-пропенил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол а) Пиридин-4-карбоксальдегид(2-пропенил)имин С применением процедуры примера 1(в) проводят реакцию взаимодействия пиридин-4карбоксальдегида с 2-пропиниламином с получением целевого соединения в виде желтого масла. б) (Е)-1-(1-пропенил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают смесь целевого соединения и 1-(2-пропенил)-4-(4-фторфенил)-5-(4пиридинил)имидззола. После хроматографирования смеси с применением для элюции 0-50% EtOAc в гексанах получают целевое соединение(43%). Тпл. = 173,5-174,0°С. Пример 65 1-(2-пропенил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол Дальнейшее хроматографирование смеси из примера 64(б) приводит к получению целевого соединения(54%). Тпл. = 116-117°С Пример 66 5-[(2-N,N-диметиламино)пиримидин-4-ил]-4-(4-фторфенил) 1-[3-(4-морфолинил)пропил]имидазол С применением процедуры примера 61(б), за исключением использования водного диметиламина, получают целевое соединение в виде желтой стеклообразной массы. ESMS(m/z): 411(М++Н). Пример 67 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[4-(трифторметил)-фенил]имидазол а) 4-трифторметилфенил-толилтиометилизоцианид С применением процедуры примера 1(а, б), за исключением использования 4трифторметилбензальдегида в качестве альдегидного компонента, получают целевое соединение. б) 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[4-(трифторметил)фенил]имидазол С применением процедуры примера 1(г) проводят реакцию взаимодействия имина, приготовленного в примере 1(в), с изоцианидом, приготовленным на предыдущей стадии, получая в результате целевое соединение. Тпл. = 133°С. Пример 68 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[3-(трифторметил)-фенил]имидазол а) 3-трифторметилфенил-толилтиометилизоцианид С применением процедуры примера 1(а, б), за исключением использования 3трифторметилбензальдегида в качестве альдегидного компонента, получают целевое соединение. б) 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-[3-(трифторметил)фенил]имидазол С применением процедуры примера 1(г) проводят реакцию взаимодействия имина, приготовленного в примере 1(в), с изоцианидом, приготовленным на предыдущей стадии, получая в результате целевое соединение. ΕSMS = 417(M+H). Пример 69 1-(циклопропилметил)-4-(3,4-дихлорфенил)-5-(4-пиридинил)имидазол а) 3,4-дихлорфенил-толилтиометилизоцианид С применением процедуры примера 1(а, б). за исключением использования 3,4-дихлорбензальдегида в качестве альдегидного компонента, получают целевое соединение. б) 1-(циклопропилмeтил)-4-(3,4-дихлорфенил)-5-(4-пиридинил)-имидазол Применяя процедуру примера 1(г), проводят реакцию взаимодействия имина, приготовленного в примере 27(г), с изоцианидом, приготовленным на. предыдущей стадии, в результате чего получают целевое соединение. Тпл. = 145 5°С. Пример 70 1-(циклопропилмeтил)-4-(3-трифторметилфенил)-5-(4-пиридинил)-имидазол Применяя процедуру примера 1(г), проводят реакцию взаимодействия имина, приготовленного в примере 27(г), с изоцианидом, приготовленным в примере 68(а), получая в результате целевое соединение. Тпл. = 105,5°С. Пример 71 1-(циклопропилпетил)-4-(4-фторфенил)-5-(2-метилпирид-4-ил)имидазол а) 2-метилпиридин-4-карбоксальдегид(циклопропилмeтил)имин С применением процедуры примера 1(в) проводят реакцию взаимодействия 4-формил-2метилпиридина, приготовленного в примере 33(а), с циклопропилметиламином с получением в результате целевого соединения в виде желтого масла. б) 1-(циклопропилметил)-4-4-фторфенил)-5-(2-метилпирид-4-ил)-имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(62% на основании 2-аминопиримидинальдегида). Тпл. = 141,0 - 141,5°С. Пример 72 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-(3,5-бистри-фторметилфенил)имидазол а) 3,5-бистрифторметилфенил-толилтиометилизоцианид С применением процедуры примера 1(а, б), за исключением использования 3,5бистрифторметилбензальдегида в качестве альдегидного компонента, получают целевое соединение. б) 1-[3-(4-морфолинил)пропил]-5-(4-пиридинил)-4-(3,5-бистрифторметилфенил)имидазол Применяя процедуру примера 1(г) с заменой на имин, приготовленный в примере 1(в), и изоцианид, приготовленный на предыдущей стадии, получают целевое соединение. Тпл. = 136,5 - 137,5°C. Пример 73 5-[4-(2-аминопиримидинил)]2-4-(4-фторфенил)-1-(2-карбокси-2,2-диметилэтил)имидазол а) 2-аминопиримидин-4-карбоксальдегид (этил-3-амино-2,2-диметилпропионат С применением процедуры примера 1(г) проводят реакцию взаимодействия 2-аминопиримидин-4карбоксальдегида с этил-3-амино-2,2-диметилпропионатом с получением целевого соединения в виде желтого масла. б) 5-[4-(2-аминопиримидинил)-4-(4-фторфенил)-1-(2-карбоксиэтил)-2,2-диметилпропил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(11% на основании амина). в) 5-[4-(2-аминопиримидинил)]-4-(4-фтороенил)-1-(2-карбокси-2,2-диметилэтил)имидазол, литиевая соль С применением процедуры примера 37 проводят гидролиз соединения 73(б) с получением целевого соединения в виде литиевой соли; (67%). ЕS(+)MS m/е = 356. Пример 74 1-(1-формил-4пиперидинил)-4-(4-фторфенил)-5-(4-пиридинил)имидазол 1-(1-бензилпиперидин-4-ил)-4-(4-фторфенил)-5-(4-пиридил)-имидазол(100мг; приготовлен в примере 40) растворяют в смеси 10%-ная муравьиная кислота/метанол в атмосоeре аргона и добавляют палладий черный(100мг), перемешанный со смесью 10%-ная муравьиная кислоте/метанол. Реакционную смесь перемешивают в атмосфере аргона при комнаткой температуре в течение 16 часов. Затем реакционную смесь выпаривают, а остаток перемешивают со смесью H2О/этилацетат, доводя рН до 10. Слои разделяют, и экстрагируют водную базу этилацетатом. Объединенные органические фракции выпаривают, а остаток подвергают флэш-хроматографии (силикагель/метиленхлорид/метанол) с получением целевого соединения в виде твердого вещества грязновато-белого цвета. ЕS(+)MS m/е = 351(MΗ+). Пример 75 5-(2-амино-4-пиримидицил)-4-(4-фторфенил)-1-(1-метил-4-пиперидинил)имидазол а) 4-амино-1-метилпиперидин Смешивают 1-метилпиперидин-4-он(4,22г; 37ммоль) и ледянок раствор 1н НСI в Et2O(37мл; 37ммоль). Проводят растирание с последующим выпариванием Еt2О при температуре 23°С в потоке аргона с получением гидрохлорида. После этого добавляют MeОН(114мл), безводный NН4ОАс(28,7; 373ммоль) и молекулярное сито 3А. Полученную смесь перемешивают в течение 10 минут, после чего добавляют NаСNRH3(2,33г; 37ммоль) и перемешивают реакционную смесь в течение еще одного часа. Затем подкисляют смесь до рН меньше 1, применяя концентрированную НСI, и промывают Еt2O. Полученную смесь подщелачивают с помощью 50%-ного водного NaOH и экстрагируют EеОАс; высушивают(К2СО3) и отгоняют(Ткип. = 55 - 60°С, 15мм) с получением 3,88г(88%) целевого соединения. б) 2-аминопиримидин-4-карбоксальдегид(1-метилпиперидин-4-ил)-имин С применением процедуры примера 1(в) проводят реакцию взаимодействия 2-аминопиримидин-4карбоксальдегида и соединения предыдущей стадии с получением целевого соединения з виде желтого масла. с) 5-(2-аминопиримидин-4-ил)-4-(4-фторфенил)-1-(1-бензилпиперидил-4-ил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение после очистки с применением хроматографии на силикагеле с использованием 0 - 10% MеОH и 0 - 1% Εt3N в СН2СI2 с последующим фракционным осаждением из MеОH с помощью Et2O; твердое вещество желтого цвета(20% по амину). Тпл. = 235 237°С(разлож.). Пример 76 1-(2,2-диметил-3-морфолин-4-ил)пропил-4-(4-фторфенил)-5-(2-амино-4-пиримидинил)имидазол а) N-1-амино-2,2-диметлпропил)морфолин 2,2-димeтил-3-N-морфолинилпропионовый альдегид (Cheney L. L., J. Amer. Chem. Soc., 1951, 73, p.685-686)(855мг, 5,0ммоль) растворяют в Et2O(2мл) и добавляют 1N HCI в Et2Ο(5мл; 5ммоль). Пeремeшивaют полученную смесь в течение 5 минут и выпаривают Et2О в струе aргона. Твердый продукт растворяют в безводном МеОН(15мл), затем добавляют безводный NH4OAc(3,85г; 50ммоль) и молекулярные, сита 3А. Перемешивaют полученную смесь в течение 5 минут, после чего добавляют NаСNВН3(0,314г; 4,0ммоль). Перемешивают реакционную смесь в течение. 45 минут и добавляют концентрированную НСI до тех пор, пока pН реакционной смеси не станет ниже 1. Удаляют МeOН под вакуумом, а остаток растворяют в Н2О(15мл) и экстрагируют Et2О(25мл). На водную фазу наслаивают новую порцию Εt2O и подщелачивают с помощью добавления 50%-ногo водного NaОH до достижения рН более 10. Затем экстрагируют с помощью Et2О(3 x 40мл), высушивают(К2СО3) и концентрируют с получением целевого соединения(86%). б) 2-аминопиримидин-4-карбоксальдегид[3-(4-морфолинил)-2,2-диметилпропил]имин С применением процедуры примерa 1(в) проводят реакцию взаимодействия 2-аминопиримидин-4карбоксальдегида и продукта предыдущей стадии с получением целевого соединения в виде желтого масла. в) 1-(2,2-диметил-3-морфолин-4-ил)пропил-4-(4-фторфенил)-5-(2-амино-4-пиримидинил)имидазол С применением процедуры примера 1(г), за исключением использования соединения предыдущей стадии в качестве имина, получают целевое соединение в виде белого твердого вещества(16% на основании амина). Тпл. = 242-245°С(разлож.). Пример 77 4-(4-фторфенил)-5-(4-пиридил)-1-(2-ацетоксиэтил)имидазол 500мг 4-(4-фторфенил)-5-(4-пиридил)имидазола высушивают при температуре 50°С под вакуумом в течение ночи, после чего помещают его в колбу, содержащую 20мл высушенного(сито) диметилформамида(в дальнейшем - ДМФ), и обрабатывают NaH(при 0°С); затем перемешивают при комнатной температуре и добавляют по каплям 2-ацетоксиэтилбромид. По прошествии трех дней реакционную смесь вливают в ледяную воду и экстрагируют метиленхлоридом; водную фазу промывают водой, высушивают над сульфатом натрия и отгоняют под вакуумом. Далее проводят флэшхроматографию остатка на силикагеле с использованием смеси СН2СI2-ацетон(85:15) и элюирование CH3ΟΗ в концентрации, повышающейся от 0 до 10%. Получают две основных фракции продукта, чистые фракции объединяют, получая смесь более медленно и более быстро элюируемых изомеров. Эти изомеры отгоняют и перекристаллизовывают из смеси EtОАс-гексан с получением минорного изомера(движущегося медленнее) и быстрого основного изомера(целевое соединение). ЯМР (250МГц, СDСI3) показывает СН2СН2 в виде синглета при ы 4,1 ppm, очень чистый, Н-орто к F, триплет т. при 6,9 ррm.. По расчету(в %): С - 66,60; Η - 4,86 N - 12,92. Обнаружено: С - 67,10; 67,03; Η - 5,07; 4,94; N - 13,08; 13,09. ИК (nujol mull) показывает 1740см-1(острый, эфир). Методы обработки Соединение формулы(1) или их фармацувтически приемлемые соли могут быть использованы в производстве медикамента для профилактики или терапевтического применения в случае любого болезненного состояния у человека или другого млекопитающего, которое усугубляется избыточным или нерегулируемым образованием цитокина в таких клетках млекопитающих, как моноциты и/или макрофаги, не ограничиваясь только названными клетками. Соединения формулы(1) обладают способностью ингибировать предвоспалительные цитокины, такие, как ИЛ-1, ИЛ-6, ИЛ-8 и ФНО и в этой связи используются в терапии. ИЛ-1, ИЛ-6, ИЛ-8 и ФНО воздействуют на множество клеток и тканей, поэтому эти цитокины, так же, как и другие цитокины, производные от лейкоцитов, представляют собой существенно важные медиаторы воспаления, сопровождающие большое число болезненных состояний и условий. Ингибирование таких предвоспалительных цитокинов приводит к достижению контроля, снижения выраженности и облегчения течения многих из этих воспалительных состояний. В соответствии с этим, настоящее изобретение относится к способу лечения цитокин-опосредованного заболевания, который включает введение с целью подавления цитокина эффективного количества соединения формулы(1) или его фармацевтически приемлемой соли. В частности, соединения формулы(1) или их фармацевтически приемлемые соли используются в профилактике или лечении любого заболевания у человека или другого млекопитающего, которое либо вызывается либо ухудшается в случае избыточной или нерегулируемой продукции ИЛ-1, ИЛ-8 или ФНО такими клетками млекопитающих, которые представляют собой, не ограничиваясь ими, моноциты и/или макрофаги. В соответствии с этим, другим аспектом настоящего изобретения является способ ингибирования образования ИЛ-1 у млекопитающего, при наличии потребности в таком ингибировании, который включает введение указанному млекопитающему эффективного количества соединения формулы(1) или его фармацевтически приемлемой соли. Избыточное или нерегулируемое образование ИЛ-1 опосредует возникновение или обострение множества заболеваний, включающих ревматоидный артрит, остеоартрит, эндотоксемию и/или синдром токсического шока, другие острые или хронические воспалительные состояния, такие, как воспалительная реакция, индуцированная эндотоксином, или воспаление кишечника, туберкулез, атеросклероз, мышечная дегенерация, множественный склероз, истощение, резорбцию кости, псориатический артрит, синдром Рейтера, подагру, травматический артрит, артрит, возникший в результате перенесенной краснухи, и острый синовит. Результаты последних исследований выявляют также очевидную связь активности ИЛ-1 с диабетом и панкреатическими β-клетками. В другом своем аспекте настоящее изобретение отнгосится к способу ингибирования у млекопитающего, при наличии такой потребности, образования ФНО, при этом данный способ включает введение упомянутому млекопитающему эффективного количества соединения формулы(1) или его фармацевтически приемлемой соли. Избыточное или нерегулируемое образование ФНО опосредует возникновение или обострение множества заболеваний, включающих ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие состояния артритной природы, сепсис, септический шок, эндотоксический шок, сепсис, вызванный грам-отрицательными бактериями, синдром токсического шока, респираторный дистресс-синдром взрослых, церебральную малярию, хроническое воспаление легких, силикоз, легочныйсаркоидоз, резорбцию кости, такую, как остеопороз, реперфузионное повреждение, реакцию "трансплантат против хозяина", отторжение аллотрансплантата, лихорадку и миалгии, связанные с инфекцией, такой, как грипп, истощение вследствие инфекции или злокачественного заболевания, истощение, вторичное относительно синдрома приобретенного иммунодефицита(СПИД), СПИД, состояния, связанные со СПИДом(ARC), образование келлоидной ткани, образование зарубцевавшейся ткани, болезнь Крона, язвенный колит и гипертермия. Соединения формулы(1) используются также для лечения вирусных инфекций, в тех случаях, когда вирусы проявляют чувствительность к позитивной регуляции под действием ФИО или обнаруживают образование ФНО in vivo. Рассматриваемые в настоящем описании вирусы, которые поддаются предлагаемому способу лечения, включают те из них, которые чувствительны к ингибированию, такому, как снижение репликации, прямое или непрямое, посредством ФНО-ингибирующих соединений формулы(1). Такие вирусы включают, не ограничиваясь ими, ВИЧ-1, ВИЧ-2 и ВИЧ-3, а также вирус цитомегалии(CMV ), вирус гриппа, аденовирус и вирусы группы герпеса, которые включают, не ограничиваясь ими, вирус Зостер и простой вирус. В соответствии с этим в другом своем аспекте настоящее изобретение относится к способу лечения млекопитающего, пораженного вирусом иммунодефицита человека(ВИЧ), который включает введение млекопитающему, при необходимости такого лечения, эффективного количества ФНО-ингибирующего соединения формулы(1) или его фармацевтически приемлемой соли. Соединения формулы(1) могут также находить применение при лечении других, отличных от человека млекопитающих, в сочетании с используемыми в ветеринарии способами, при необходимости ингибировать образование ФНО. Перечень заболеваний животных, опосредованных ФНО, в случае которых показано применение описываемых соединений, с целью терапии или профилактики, включает отмеченные выше болезненные состояния, и в особенности вирусные инфекции. Примеры таких вирусов включают, не ограничиваясь ими, лентивирусные инфекции, такие, как вирус инфекционной анемии лошадей, козлиный вирус артрита, висна вирус, а также медивирусные или ретровирусные инфекции, которые включают, не ограничиваясь ими, вирус иммунодефицита кошек(ВИК), бычий вирус иммунодефицита или вирус иммунодефицита собак, а также другие ретровирусные инфекции. Соединения формулы(1) могут также находить применение при местном использовании для лечения или профилактики тех местных болезненных состояний, течение или обострение которых связано с избыточным образованием цитокина, таких, как, соответственно, ИЛ-1 или ФНО, которые включают, в частности, воспаление связок, экзему, псориаз, а также другие воспалительные состояния кожи, такие, как солнечный ожог; воспалительные заболевания глаз, включающие конъюнктивит; гипертермию, боль и другие состояния, связанные с развитием воспаления. Было также показано, что соединения формулы(1) ингибируют образование ИЛ-8(интерлейкин-8, НАБ). Соответственно, в другом своем аспекте настоящее изобретение относится к способу ингибирования образования ИЛ-8 у млекопитающего, при необходимости такого ингибирования, который включает введение упомянутому млекопитающему эффективного количества соединения формулы(1) или его фармацевтически приемлемой соли. Имеется множество болезненных состояний, в обострении и/или возникновении которых принимает участие избыточное или нерегулируемое образование ИЛ-8. Эти болезни характеризуются массивной инфильтрацией нейтрофилов, к числу которых относится, например, псориаз, воспаление кишечника, астма, реперфузионное повреждение сердца и почек, респираторный дистресс-синдром взрослых, тромбоз и гломерулонефрит. Все эти заболевания связаны с увеличенной продукцией ИЛ-8, который отвечает за хемотаксис нейтрофилов к месту воспаления. В отличие от других воспалительных цитокинов(ИЛ-1, ФНО и ИЛ-6), ИЛ-8 обладает уникальным свойством способствовать транспорту нейтрофилов и их активации. В этой связи, ингибирование образования ИЛ-8 должно вести к прямому снижению инфильтрации нейтрофилов. Соединения формулы(1) вводятся в количестве, достаточном для ингибирования цитокина, в частности, ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, продукция которых снижается при такой регуляции до нормального уровня, или в некоторых случаях, до субнормальных уровней с тем, чтобы смягчить течение или предупредить развитие болезненного состояния. Аномальные уровни ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, в контексте настоящего изобретения включают: (1) уровень свободного(не связанного с клеткой) ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, который выше или равен 1 пикограмму на мл; (2) связанный с клеткой ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, любой из них; или (3) мРНК к ИЛ-1, ИЛ-6, ИЛ-8 или ФНО в количестве, превышающем их уровень в тех клетках и тканях, в которых соответственно ИЛ-1, ИЛ-6, ИЛ-8 или ФНО продуцируются. Открытие того факта, что соединения формулы(1) представляют собой ингибиторы цитокинов, а именно: ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, основывается на воздействии соединений формулы(1) на образование ИЛ-1, ИЛ-8 и ФНО в исследованиях in vitro, которые приведены в настоящем описании. Приведенный в настоящем описании термин "ингибирующий образование ИЛ-1 ИЛ-6, ИЛ-8 или ФНО) относится к: а) снижению избыточного in vivo уровня цитокина(ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) в организме человека до нормального или cубнормального уровней посредством ингибирования in vivo высвобождения цитокина всеми клетками, включая, но не ограничиваясь ими, моноциты или макрофаги; б) регуляции по типу отрицательной обратной связи на уровне генома избыточного in vivo уровня цитокина(ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) в организме человека до нормального или субнормального уровней; в) регуляцию по типу отрицательной обратной связи, посредством ингибирования прямого синтеза цитокина(ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) на посттранляционном уровне; или г) регуляции по типу отрицательной обратной связи на уровне трансляции избыточного in vivo уровня цитокина(ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) в организме человека до нормального или субнормального уровней. Примененный в настоящем описании термин "болезнь или болезненное состояние, опосредованные ФНО" относился к любому или всем болезненным состояниям, в которых функционирует ФНО, либо посредством образования самого ФНО, либо посредством воздействия через ФНО на высвобождение монокина, такого, как ИЛ-1, ИЛ-6 или ИЛ-8, не ограничиваясь ими. Болезненное состояние, в котором, например, ИЛ-1 представляет собой основной компонент, а образование или действие связано с ФНО, будет, в этой связи, рассматриваться как болезненное состояние, опосредованное ФНО. Примененный в настоящем описании термин "цитокин" относится к любому секретируемому полипептиду, который воздействует на функции клетки, или представляет собой молекулу, модулирующую взаимодействие между клетками в случае иммунной, воспалительной или гемопоэтической реакции. Цитокин включает, не ограничиваясь ими, монокины и лимфокины, независимо от того, какие клетки продуцируют их. Например, считается, что монокин продуцирует и секретирует моноядерная клетка, такая, в частности, как макрофаг или моноцит. Однако, многие другие клетки также могут продуцировать монокины, такие, как естественные киллерные клетки, фибробласты, базофилы, нейтрофилы, эндотелиальные клетки, астроциты мозга, стромальные клетки костного мозга, эпидермальные кератиноциты и В-лимфоциты. Считается, что лимфокины продуцируют лимфоцитарные клетки. Примеры цитокинов включают, не ограничиваясь ими, интерлейкин-1(ИЛ-1), интерлейкин-6(ИЛ-6), интерлейкин-8(ИЛ-8), альфа фактор некроза опухолевых клеток(ФНО-a) и бета фактор некроза опухолевых клеток(ФНО-b). Примененный в настоящем описании термин "цитокин-подавляющее" или "цитокин-супрессивное количество" относится к эффективному количеству соединений формулы(1), которое вызывает снижение in vivo уровня цитокина до нормального или субнормального уровня, в случае его введения пациенту с целью профилактики или лечения болезненного состояния, которое обостряется или вызывается избыточным или нерегулируемым образованием цитокина. В контексте настоящего описания под цитокином в следующей фразе "ингибирование цитокина с целью использования для лечения ВИЧ-инфицированного организма человека" понимают цитокин, который участвует в (а) инициации и/или поддержании активации Т-клеток и/или экспрессии ВИЧ гена, опосредованной активированными Т-клетками и/или репликации и/или (б) любого цитокинопосредованного осложнения болезни, такого, как истощение или мышечная дегенерация. Поскольку ΦΗΟ-b(известный также как лимфотоксин) имеет тесную структурную гомологию с ФНОa(известным так же, как кахектин) и поскольку каждый из них индуцирует сходные биологические реакции и связывается с одними и теми же клеточными рецепторами, и ФНО-a, и ΦΗΟ-b ингибируются соединениями настоящего изобретения и обозначаются в целом как "ФНО", если в описании не оговорено иное. Для целей использования соединения формулы(1) или его фармацевтически приемлемой соли в терапии в соответствии со стандартной фармацевтической практикой его вводят в состав фармацевтической композиции. Поэтому настоящее изобретение относится также к фармацевтической композиции, включающей эффективное нетоксичное количество соединения формулы(1) и фармацевтически приемлемый носитель или разбавитель. Соединения формулы(1), их фармацевтически приемлемые соли и включающие их фармацевтические композиции могут быть легко введены любым способом, традиционно используемым для введения лекарственных препаратов, например, перорально, местно, парентерально или путем ингаляции. Соединения формулы (1) могут быть введены в виде традиционных дозированных форм, приготовленных при сочетании соединения формулы (1) со стандартным фармацевтическим наполнителем, в соответствии с известными процедурами. Соединения формулы (1) могут быть также введены в стандартных дозировках в сочетании с известным вторым терапевтически активным соединением. Такие процедуры могут включать смешивание, гранулирование и прессование или растворение ингредиентов в зависимости от того способа, который подходит для приготовления нужного препарата. Следует при этом иметь в виду, что форма и характер фармацевтически приемлемого наполнителя или разбавителя определяются количеством активного ингредиента, с которым он должен быть объединен, способом введения и другими хорошо известными переменными. Носитель(и) должен быть "приемлемым" в том смысле, что должен быть совместим с другими ингредиентами композиции и не должен приносить вреда реципиенту. Используемый фармацевтический носитель может представлять собой либо твердое вещество, либо жидкость. Примеры твердых носителей включают лактозу, терра альбу, сахарозу, тальк, желатин, агар, пектин, аравийскую камедь, стеарат магния, стеариновую кислоту и др. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, вода и т.д. Аналогично, носитель или разбавитель может включать известный в технике материал, задерживающий разложение, такой, как глицерил моностеарат или глицерил дистеарат, сами по себе или в сочетании с воском. Может быть использовано множество фармацевтических форм. Так, если применяют твердый наполнитель, препарат может иметь форму таблеток, может быть помещен в твердую желатиновую капсулу, в виде порошка или пеллета, или в форме пастилки или лепешки. Количество твердого напoлнителя может широко варьировать, но предпочтительно лежит в пределах от 25мг до примерно 1г. В случае использования жидкого носителя препарат может представлять собой сироп, эмульсию, мягкую желатиновую капсулу, стерильную инъецируемую жидкость, в таком виде, как ампула или неводная жидкая суспензия. Соединения формулы(1) могут вводиться местным способом, который представляет собой не системный способ введения. Он включает нанесение соединения формулы (1) наружно на эпидермис или в щечный карман или закапывание такого соединения в ухо, глаз, нос, так, что соединение не может в значительном количестве поступать в ток крови. В отличие от него, системное введение относится к пероральному, внутривенному, внутрибрюшинному и внутримышечному способам. Композиции, пригодные для местного введения, включают жидкие или полужидкие препараты, способные проникать через кожу к месту воспаления, такие, как линименты, лосьоны, кремы, мази или пасты, а также капли, пригодные для введения в ухо, глаз или нос. Активный ингредиент может составлять для целей местного введения от 0,001% до 10%(вес/вес), в частности, от 1% до 2% по весу относительно всей композиции. Однако он может составлять до 10%(вес/вес), но предпочтительно составляет менее 5%(вес/вес), более предпочтительно от 0,1% до 1%(вес/вес) от всей композиции. Лосьоны, в соответствии с натоящим изобретением, включают такие из них, которые пригодны для внесения на кожу или в глаза. глазной лосьон может при этом включать стерильный водный раствор, факультативно содержащий бактерицидное вещество, и может быть приготовлен с помощью методов, аналогичных тем, которые применяют для изготовления капель. Лосьоны или линименты для нанесения на кожу могут также включать агент, способствующий высушиванию и охлаждению кожи, такой, как спирт или ацетон, и/или увлажнитель, Такой, как глицерин или масло, в частности, касторовое масло или арахисовое масло. Кремы, мази или пасты, в соответствии с настоящим изобретением, представляют собой полутвердые композиции, содержащий активный ингредиент, для наружного нанесения. Они могут быть получены при смешивании активного ингредиента в мелкоизмeльченнои или порошковой форме, самого по себе или в растворе, или в суспензии в водной или неводной жидкости, при использовании подходящего прибора, с жирным или нежирным основанием. Основание может включать углеводороды, такие, как твердый, мягкий или жидкий парафин, глицерин, пчелиный воск, металлическое мыло; слизь; масло натурального происхождения, такое, как миндальное, кукурузное, арахисовоe, касторовое или оливковое масло; ланолин или его производные или жирная кислота, такая, как стеариновая кислота или олеиновая кислота вместе со спиртом, таким, как пропиленгликоль или макрогель.. Композиция может включать любой поверхностно активный агент, такой, как анионное, катионное или неионное поверхностно активное вещество, в частности, сорбита - новый эфир или его полиоксиэтиленовое производное, в композицию могут быть также включены суспендирующие агенты, такие, как натуральные смолы, производные целлюлозы или неорганические материалы, такие, как кремнезем, и другие ингредиенты, в частности, ланолин. Капли, в соответствии с настоящим изобретением, могут включать стерильные водные или масляные растворы или суспензии и могут быть приготовлены при растворении активного ингредиента в подходящем водном растворе бактерицидного и/или фунгицидного агента и/или любого подходящего стабилизатора, включая предпочтительно поверхностно-активный агент. Полученный раствор может быть затем осветлен при фильтровании, перенесен в подходящий контейнер, который запечатывается и стерилизуется при автоклавировании или с применением любого другого способа поддержания температуры 98-100°С в течение получаса. Альтернативно, раствор может быть простерилизован при фильтровании и перенесен в контейнер с помощью асептической техники. Примеры бактерицидных и фунгицидных агентов, подходящих для включения в состав капель, включают ртутьнитрафенил или ртутьацетатфенил(0,002%), хлорид бензалкония(0,01%) и ацетат хлоргексидина(0,01%). Подходящие растворители для приготовления масляного раствора включают глицерин, разбавленный спирт и пропиленгликоль. Соединения формулы (1) могут быть введены парентерально, т. е. с помощью внутривенного, внутримышечного, подкожного, внутриназального, внутрирeктального, внутривагинального или внутрибрюшинного способов введения. Обычно в качестве предпочтительных форм парентерального введения используют подкожный и внутримышечный способ. Для целей такого введения подходящие дозированные формы могут быть получены с помощью традиционной техники. Соединения формулы (1) могут быть также введены с помощью ингаляции, т.е. с помощью внутриназальной и оральной техники ингаляционного введения. Соответствующие дозированные формы для такого введения, в частности, в виде аэрозольной композиции или дозированного ингалятора, могут быть получены с применением традиционной техники. Для всех методов использования описанных в настоящем изобретении соединений формулы(1) режим дневного перорального дозирования варьирует предпочтительно от 0,1 до примерно 80мг/кг общего веса тела, предпочтительно от 0,2 до 30мг/кг, более предпочтительно от 0,5 до 15мг. Дневной режим дозирования при парентеральном введении составляет предпочтительно от 0,1мг до примерно 80мг/кг общего веса тела, предпочтительно от 0,2 до примерно 30мг/кг и более предпочтительно, от примерно 0,5мг до 15мг/кг. Дневной режим дозирования при местном введении составляет предпочтительно от 0,1мг до 150мг, при условии введения от одного до четырех, а предпочтительно от двух до трех раз в день. Дневной режим дозирования при ингаляции составляет предпочтительно от 0,01мг/кг до примерно 1мг/кг в день. При этом каждому специалисту со средним уровнем знаний в данной области должно быть ясно, что оптимальное количество и форма представления индивидуальной дозы соединения формулы(1) или его фармацевтически приемлемой соли могут быть определены природой и условиями того заболевания, которое предстоит лечить, формой, способом и местом введения, а также особенностями состояния больного, подвергающегося рассматриваемой терапии, при этом такие оптимальные показатели могут быть определены с применением традиционной техники. Кроме того, каждый специалист со средним уровнем знаний в данной области может оценить оптимальный способ лечения, т.е. количество доз соединения формулы(1) или его фармацевтически приемлемой соли, которое следует вводить в день в течение определенного количества дней, причем упомянутый способ может быть определен таким специалистом со средним уровнем знаний в данной области с использованием традиционного метода тестирования. Далее настоящее изобретение описывается со ссылкой на биологические примеры, которые являются только лишь иллюстративными и которые ни в коем случае не следует рассматривать как ограничивающие область настоящего изобретения. Биологические примеры Цитокин-ингибирующее действие соединений настоящего изобретения было определено in vivo в следующих исследованиях: Интерлейкин-1(ИЛ-1) Моноциты из периферической крови человека были выделены и очищены либо из свежих препаратов крови добровольных доноров, либо из лейкоцитных пленок в банке крови в соответствии с процедурой Колотта с сотр. [Colotta et al, J Immumol, 1984, 132, 936]. Эти моноциты(1 x 106) помещают на 24-гнездное плато в концентрации 1 - 2 миллиона/мл в расчете на одну ячейку. Клеткам позволяют слипаться в течение 2 часов, после чего неслипшиеся клетки удаляют при осторожном промывании. За 1 час до внесения липополисахаридов(50нг/мл) к клеткам добавляют исследуемые соединения, и инкубируют культуры при температуре 37°С еще в течение 24 часов. По окончании этого периода обтирают культуральные супернатанты и очищают их от клеток и других инородных тел. После этого клеточные супернатанты сразу же исследуют на биологическую активность ИЛ-1 либо по методу Симона с соавт. [Simon et al, J Immunol Nethods, 1985, 84, 85] (на основе способности ИЛ-1 стимулировать интерлeйкин-2продуцирующую клеточную линию(ЕL-4)) (секретировать ИЛ-2 во взаимодействии с А23187 ионофором) или по методу Ли с соавт. [Lee t al., J. Immuno Therapy, 1990, 6(1), 1-12] (исследование по методу ЭЛИЗА). Было показано, на основе данных Примеров 1 - 24, что соединения формулы(1) являются ингибиторами in vitro ИЛ-1, продуцируемого моноцитами человека. Фактор некроза опухолевых клеток(ФНО): Моноциты из периферической крови человека были выделены и очищены либо из лейкоцитных пленок из банка крови, либо из тромбаферезных остатком, в соответствии с процедурой Колотта с соавт. [Colotta et al/, J Immunol, 1984, 132(2), 936]. Моноциты помещают с плотностью 1 x 106 клеток/мл среды/ячейку в 24-ячеечную чашку Петри. Клеткам дают слипнуться в течение 1 часа, после чего супернатант отсасывают, добавляют свежую среду [1мл, ЯРМ1-1640, Витакер Байомедикал Продактс, Витакер, СА [Whitaker, Biomedical Products, Whitaker СА], в которую добавлены 1% околоплодной сыворотки теленка плюс пенициллин и стрептомицин(10 единиц/мл). Клетки инкубируют в течение 45 минут в присутствии или в отсутствие тестируемого соединения в диапазоне дозировок 1нМ 10мм(соединения солюбилизируют в диметилсульфоксид/этаноле, так, чтобы конечная концентрация растворителя в культуральной среде составляла 0,5% диметилсульфоксидa/0,5% этанола). Затем добавляют бактериальный липополисахарид [Е. Coli 055; В5 [ЛПС] от Сигма Кемикалс Ко.(Sigma Chemicals Со)] (100нг/мл в 10мл фосфатно-буферного раствора), и инкубируют клетки в течение 16 - 18 часов при температуре 37°С в инкубаторе, содержащем 5% СО2. К концу инкубационного периода отделяют клеточный супернатант от клеток и центрифугируют со скоростью 3000об/мин для удаления клеточных остатков. После этого супернатант анализируют на наличие ФНО активности с использованиeм либо радиоиммунного метода, либо метода ЭЛИЗА, как описано в W0 92/10190, а также в работе Беккера с соавт. [Becker et al, J Immunol, 1991, 147, 4307]. Соединения формулы(1), кaк следует из Примеров 1 - 24, являются ингибиторами in vitro ФHО, продуцируемого моноцитами человека. По-видимому, ингибирующая активность в отношении ИЛ-1 и ФНО не коррелирует со способностью соединений формулы(1) участвовать в ингибировании метаболизма арахидоновой кислоты. Более того, способность ингибировать образование простагландина и/или синтеза лейкотриена нестероидными противовоспалительными лекарственными препаратами с мощной циклооксигеназной и липоксигеназной ингибирующей активностью не означает, что такое соединение также обязательно будет ингибировать образование ФНО или ИЛ-1 в нетоксичных дозах. Интерлейкин-8(ИЛ-8) Первичные эндотелиальные клетки пупочного канатика человека(HUVEC) [Селл Системз, Кирланд, WA(Cell Systems, Kirland, WA] поддерживают на культуральной среде, содержащей 15% околоплодной сыворотки теленка и 1% СS – HBGF, состоящего из фактора роста фибробластов(FGF) и гепарина. Затем клетки разбавляют в 20 раз перед помещением их(250мкл) на 96-гнездные плато, покрытые желатином. Перед использованием проводят замещение культуральной среды свежей средой(200мкл). Затем к каждой ячейке добавляют либо буфер, либо исследуемое соединение(25мкл, в концентрации от 1 до 10мкМ) и инкубируют плато в течение 6 часов в увлажненном инкубаторе при температуре 37°С в атмосфере 5% СО2 По окончании инкубационного периода супернатант отбирают, определяя в нем концентрацию ИЛ-8 с использованием набора ЭЛИЗА для ИЛ-8, полученного от R&D Системз(Миннеаполис, MN) [R&D Systems(Minntapolis, MN)]. Все приведенные данные представляют собой полученные на основе стандартной кривой средние значения(нг/мл) множества исследованных образцов, где это приемлемо, значения ИК50 выведены с использованием методов нелинейного регрессивного анализа. Тест на специфическое связывание цитокина с белком Был разработан метод конкурентного связывания с использованием радиоактивных соединений для проведения первичного скрининга при исследовании взаимосвязи структура-активность с получением хорошо воспроизводимых результатов. Данный тест имеет ряд преимуществ перед традиционно применяемыми традиционными методами анализа в связи с тем, что предполагает использование свежевыделенных из организма человека моноцитов в качестве источника цитокинов, а также метод ЗЛИЗА для их количественной характеристики. Кроме того, что упомянутый метод более прост в применении, результаты описываемого теста на связывание отличаются высокой корреляцией с данными биотеста. Был разработай специфичный, воспроизводимый тeст на связывание ингибитора цитокина(СSAlDTM) с использованием для этого цитозольной фракции из ТНР.1 клеток и радиоактивного меченого соединения. Так, например, в качестве подходящего представителя радиоактивно меченого соединения из такого СSAlDTM класса ингибиторов цитокина может использоваться 4-(фторфенил)-2-(4гидроксифенил-3,5-t2)-5-(4-пиридил)имидазол. Вкратце, ТНР.1 цитозоль готовят рутинным способом из клеточного лизата, полученного при препаровке азотом с последующим низкоскоростным 10К х g и высокоскоростным 100К х g- центрифугированием, полученный при этом супернатант рассматривают как цитозольную фракцию. ТНР.1 цитозоль инкубируют с соответствующим образом разбавленным лигандом при комнатной температуре в течение заранее определенного периода времени для достижения равновесного связывания. После этого образец добавляют к G-10 колонке и проводят элюцию 20мМ TRN, 50мм b-меркаптоэтанола, NaN3. Собирают фракцию, включающую безнасадочный объем и оценивают уровень радиоактивности посредством счета в жидком сцинтилляторе. Было показано, что она отражает количество связанного радиолиганда, поскольку радиоактивный сигнал аннулируется в присутствии избытка холодного лиганда в инкубационной смеси или в случае отсутствия цитозольной фракции.
ДивитисяДодаткова інформація
Назва патенту англійськоюSubstituted imidazoles, a process for the preparation thereof, a pharmaceutical composition based thereon and a method for treatment
Назва патенту російськоюЗамещенные имидазолы, способ их получения, фармацевтическая композиция на их основе и способ лечения
Автори російськоюAdams, Jerry, Leroy
МПК / Мітки
МПК: A61K 31/505, C07D 239/42, A61P 11/00, A61P 21/00, C07D 405/14, A61Q 1/00, A61K 31/4439, C07D 401/12, A61K 31/44, A61P 43/00, A61Q 19/00, A61P 19/00, C07C 323/45, A61P 25/28, A61P 3/10, A61P 17/06, A61K 31/5377, A61K 8/20, A61P 31/12, A61K 31/4545, C07D 213/53, A61P 37/00, A61P 31/18, A61P 1/00, C07C 319/00, A61K 31/4427, A61P 9/10, A61K 8/00, A61P 29/00, C07D 239/38, C07B 61/00, A61P 31/04, C07D 215/12, C07D 401/04, C07D 413/14, A61P 1/04, C07D 213/61, C07D 401/14, C07D 403/14, C07C 317/40, A61Q 1/12, A61K 31/445, A61P 3/00, C07D 403/04, A61P 19/02, C07C 323/41, A61K 31/535, A61K 8/23, C07C 315/00, A61K 31/506, A61K 8/19
Мітки: спосіб, фармацевтична, лікування, заміщені, одержання, імідазоли, основі, композиція
Код посилання
<a href="https://ua.patents.su/33-63875-zamishheni-imidazoli-sposib-kh-oderzhannya-farmacevtichna-kompoziciya-na-kh-osnovi-ta-sposib-likuvannya.html" target="_blank" rel="follow" title="База патентів України">Заміщені імідазоли, спосіб їх одержання, фармацевтична композиція на їх основі та спосіб лікування</a>
Похідні аміно(тіо)ефірів, спосіб їх одержання (варіанти), фармацевтична композиція на їх основі і спосіб її одержання

Номер патенту: 45952
Опубліковано: 15.05.2002
Автори: Грайнер Хартмут, Цайе Жан-Жак, Бруне Мішель, Бартосцук Герд, Нобле Марк, Бьоттхер Хеннінг, Бертелон Жан-Жак, Деван Ральф
МПК: C07D 405/12, C07C 255/54, C07D 333/20, C07D 311/58, C07D 213/38, C07C 217/16, C07D 311/64, C07C 217/20
Мітки: одержання, основі, фармацевтична, варіанти, похідні, композиція, аміно(тіо)ефірів, спосіб
Формула / Реферат:
1. Производные амино(тио)эфиров формулы I,где X представляет собой кислород, серу, или, если R0 и R1 вместе не являются алкиленовой цепью с 1-3 атомами, то СН2;Z представляет собой -(СН2)n1-(СНА)n2-(СН2)n3, причем n1 = 0, 1, 2 или 3; n2 = 0 или 1; n 3 = 0, 1, 2 или 3, при условии, что n1 + n2 + n3 < 4;R0 представляет собой водород или А;R1 представляет собой водород, А, ОА, фенокси, Ph, ОН, F, Cl, Br, CN,...
Похідні еритроміцину, спосіб їх одержання та фармацевтична композиція

Номер патенту: 58504
Опубліковано: 15.08.2003
Автори: Шанто Жан-Франсуа, Агурідас Константен
МПК: A61K 31/7048, C07H 17/08, A61K 31/00, A61P 31/00, A61K 31/7042, A61K 31/70, A61P 31/04
Мітки: одержання, композиція, похідні, еритроміцину, фармацевтична, спосіб
Формула / Реферат:
1. Производные эритромицина формулы (I): , (I)в которых:Χ представляет собой радикал CH2 или SO2, или атом кислорода,Υ представляет собой радикал (CH2)m-(CH=CH)n(CH2)o, где m+n+о8, n = 0 или 1,Аr представляет собой гетероарил, моно- или бициклический, содержащий от 1 до 4 атомов азота, возможно, замещенный,W представляет собой атом водорода или остаток карбаматной функции –CO–NHR", где...
Заміщені гідроксилом азетидинони, придатні як гіпохолестеринемічний засіб, фармацевтична композиція на їх основі та спосіб лікування або профілактики

Номер патенту: 41948
Опубліковано: 15.10.2001
Автори: Кладер Джон В., Розенблум Стюарт Б, Мекіттрік Брайєн А., Дугар Сандіп, Бьорнетт Дуане А.
МПК: C07D 205/00, A61K 31/395, A61P 9/10, A61K 31/397
Мітки: основі, композиція, азетидинони, лікування, профілактики, спосіб, гідроксилом, гіпохолестеринемічний, придатні, засіб, фармацевтична, заміщені
Формула / Реферат:
1. Замещенные гидроксилом азетидиноны общей формулыили их фармацевтически приемлемые соли, где:Аr1 и Аr2 независимо друг от друга выбраны из группы, включающей арили замещенный остатком R4 арил,Аr3 - арил или замещенный остатком R5 арил,X, Y и Z независимо друг от друга выбраны из группы, включающей-СН2-, -СН(низший алкил)-, -С(ди-низший алкил)-,R и R2 независимо друг от друга выбраны из...
Похідні еритроміцину, спосіб їх одержання, фармацевтична композиція на їх основі та аміни як проміжні сполуки
Номер патенту: 51618
Опубліковано: 16.12.2002
Автори: Гуен Д'Амбрієр Солянж, Дені Алексіс, Шанто Жан-Франсуа, Ле Мартре Оділь, Агурідас Константін
МПК: C07H 17/08, A61K 31/7048, A61K 31/04
Мітки: одержання, фармацевтична, проміжні, основі, спосіб, похідні, композиція, аміни, сполуки, еритроміцину
Формула / Реферат:
1. Производные эритромицина формулы (I):, (І)в которой R представляет радикал , в котором n представляет число 3, 4 или 5 и Ar представляет гетероциклический радикал, несущий, в случае необходимости, один или несколько заместителей, представляющих собой алкил, алкокси, трифторметокси или галоген, и выбираемый в группе...
N-заміщені азагетероциклічні карбонові кислоти та їх ефіри, спосіб їх одержання та фармацевтична композиція на їх основі (варіанти), спосіб її одержання та спосіб лікування нейрогенного запалення

Номер патенту: 47396
Опубліковано: 15.07.2002
Автори: Грьонвальд Фредерік Крістіан, Андерсен Хенрік Суне, Йоргенсен Тіне Крогх, Андерсен Кнуд Ерік, Сонневальд Урсула, Петерсен Ханс, Ольсен Уффе Банг
МПК: A61K 31/55, A61P 11/00, A61K 31/445, C07D 211/78, C07D 409/06, A61P 1/00, C07D 403/06, A61K 31/4433, A61K 31/00, C07D 211/60, C07D 417/06, A61P 25/04, A61K 31/4427, A61P 17/00, A61P 43/00, C07D 413/06, A61K 31/54, A61P 29/00, A61K 31/535, C07D 401/06
Мітки: фармацевтична, запалення, лікування, карбонові, основі, одержання, нейрогенного, спосіб, кислоти, ефіри, азагетероциклічні, композиція, n-заміщені, варіанти
Формула / Реферат:
1. N-замещенные азагетероциклические карбоновые кислоты и их эфиры формулы (I):, (I)в которой R1 и R2 независимо представляют собой атом водорода, атом галогена, трифторметил, С1 - С6 - алкильная или С1 - С6 - алкоксильная группа, Y - это группы > N-CH2-, > СН- CH2- или > С=СН-, в которых только подчеркнутый атом участвует в циклической системе, Х - это группы -О-, -S-, -CR7R8-, -СН2-СН2-, СН=СН-СН2-, -СН2-СН=СН-,...
Попередній патент: Похідні 1-(2-оксоацетил)-піперідин-2-карбонової кислоти, спосіб одержання, фармацевтична композиція та спосіб лікування
Наступний патент: Спосіб лікування шизофренії та захворювань шизофренічної форми
Випадковий патент: Скиртоутворювач