Спосіб одержання піперазинілалкіл-3(2н)піридазинонів або їх фармацевтично прийнятих солей
Номер патенту: 19756
Опубліковано: 25.12.1997
Автори: Харольд Феллієр, Хайнц Блашке, Хаймо Штроісніг, Ріта Енценхофер






































Формула / Реферат
Способ получения пиперазинилалкил-3(2Н)-пиридазинонов формулы I
где R1 - водород, фенил, (C1-C6) - алкил, незамещенный или замещенный гидроксилом или группой NR4R5, в которой R4 и R5 могут быть одинаковыми или различными и представляют водород, метил или этил, или оксигруппой, или фенил, или водород;
R2 или R3 - водород или галоген, причем, по меньшей мере, один из Ra или Яз водород;
R6 - водород;
В - C1-C4-алкилен;
R7 и R8 могут быть одинаковыми или различными и представляют водород или C1-C6-алкил;
Ζ - незамещенный или замещенный однократно или многократно C1-C6-алкилом, C1-C6-алкокси, бензилоксигруппой, трифторметилом, галогеном, нитрогруппой, фенил или пиридил, или их фармацевтически приемлемых солей, отличающийся тем, что соединение формулы II
где R1, R2 и R3 имеют указанные значения, а Μ обозначает отщепляемую группу, подвергают взаимодействию с соединением формулы II
где R6, В, R7, R8 и Ζ имеют указанные значения, с выделением целевого продукта, где один из остатков R2 или R3 представляют галоген, или с дегалоидированием и выделением целевого продукта, где R2 и R3 - водород, или в соединении формулы I, где R1 означает изопропил, втор.бутил или трет.бу-тил, группу R1 отщепляют с помощью кислоты.
Текст
Изобретение относится к способу получения новых пиперазинилалкил-3(2Н)-пиридазинонов общей формулы I: где R1 - (C1-C6)-алкил, незамещенный или замещенный группой NR4R5, в которой R4 и R5 могут быть одинаковыми или различными и представляют водород, метил или этил; или окси-группой, или фенил, или водород; R2 или R3 обозначают водород или галоген, причем по меньшей мере один из R2 или R3 - водород, R6 - водород; В-(Сі-С4)-алкилен. R7 и Ra могут быть одинаковыми или различными и представляют водород или (C1-C4)~ алкил, Ζ - незамещенный или замещенный однократно или многократно (C1-C6)-алкилом, (C1-C6)алкокси, бензилокси, трифтор-метилом, галогеном, нитрогруппой фенил; пиридил или их фармацевтически приемлемых солей. Новые соединения формулы I и их фармацевтически приемлемые соли показывают в моделях в пробирке исключительное торможение периферических альфарецепторов (альфа1-адреноцепторов). Дополнительно многие из исследованных веществ имеют хорошее действие на центральных 5НТ-1Арецепторах. На основании этих фармакологических свойств новые соединения можно применять в медикаментах, одни или в смеси с другими активными веществами в форме обычных галогеновых препаратов, при высоком кровяном давлении и при заболеваниях сердца. Цель изобретения - синтез новых пиперазинилалкил-3(2Н)-пиридазинонов, превосходящих по своей активности структурные аналоги с использованием известного способа алкилирования. Поставленная цель достигается предлагаемым способом получения соединений формулы I', заключающимся во взаимодействии соединения формулы ІІ I где R1, R2 и R3 имеют вышеуказанные значения, а М обозначает отщепляемую группу, с соединением формулы II где R6, В, R7, R8 и Ζ определены выше, с выделением целевого продукта, где один из остатков R 2 или R3 представляет галоген, или с дегалоидированием и выделением целевого продукта, где R2 и R3 - водород, или в соединении формулы I, где R1 означает изопропил, втор.бутил илитрет.бутил, гр уппу R 1 отщепляют с помощью кислоты. Пример 1.2-Метил-5-бром-4-(2-((4-(2-метоксифенил) пиперазинил-1)этил)амино) -3(2Н)-пиридазинон и 2метил-4-бром-5-{(2(4-{2-метоксифенил)пип ерази нил-1 )этил)-амино) -3(2Н)-пиридазинон - 3,0 г (0,0112 моль) 2-метил-4,5-дибром-3(2Н)-пиридазино-на, 2,64 г (0,0112 моль) 1-(2-аминоэтил)-4-(2-метоксифенил)пиперазина и 1,2 г (0,0112 моль) тонко измельченного в порошок бикарбоната калия нагревают в 100 мл диметилфор-мамида при хорошем перемешивании 20 ч до 60°С; затем отсасывают в горячем состоянии от неорганической части и сгущают на переструйном насосе. Остающееся коричневое масло растворяют в 0,5 и. HCI, экстрагируют 3 раза простым эфиром, подщелачивают водную фазу и помещают в воду и хлороформ. После сушки с сульфатом натрия и сгущения фазы хлороформа остаются 4,74 г коричневого масла, которое разделяют на силикагеле (0,20-0,045 мм) препаративной хроматографией на колонке с хлористым метиленом-метанолом 40:1,5. В качестве первой фракции появляются 0,67 г 2-метил-5-бром-4-((2-(4-(2метоксифенил)пи-перазинил-1) -этил)амино)-3(2Н)-пиридази-нона, 14,2% от теории; смешиванием с эквивалентным количеством фумаровой кислоты в абсолютном этаноле получают фумарат как бесцветное, кристаллическое вещество с точкой плавления 185-186°С; С 49,5%; Η 5,3%; Вг 15,3%; N 12,6%; О 17,3%; ультрафиолетовый спектр в 0,1 н. HCI; 208(4,63), 226(S 4,40), 286 (S 3,95), 302 (4,07). Дальнейшим элюированием получают в качестве 2-й фракции 2,07 г 2-метил-4-бром-5-((2-(4-(2метоксифенил)пиперазинил-1)-этил) амино-3(2Н)-пиридазинона, который растворяют в абсолютном этаноле и смешивают с фумаровой кислотой. Получают бесцветный кристаллический фумарат (2,3 эквивалента) с точкой плавления 125-129°С, 43,8% от теории: С 46,3%, Η 5,0%, Вr 11,9%, Ν 9,9%, О 26,9%; ультрафиолетовый спектр в 0,1 н. HCI: 212 (4,63), 226 (S 4,40), 282 (S 3,83), 302 (S 3,71). Пример 2. 2-Метил-5-хлор-4-{(2-(4-(2-метоксифенил) пиперазинил-1)этил)амино)-3(2Н)-пиридазинон и 2метил-4-хлор-5-((2-(4-2-метоксифенил)пиперазинил-1)этил)-амино)- 3(2Н)пиридазинон. 10,0 г (0,559 моль) 2-метил-4,5-дихлор-3(2Н>пиридазинона, 13,2 г (0,059 моль) 1-(2-аминоэтил)-4-(2 метоксифенил)-пиперазина и 5,6 г (0,559 моль) бикарбоната калия нагревают в 200 мл ацетонитрила при перемешивании 20 ч при флегме, отсасывают в горячем состоянии от неорганической части и охлаждают. Осаждаются 7,7 г 2-метил-5-хлор-4-((2-(4-метоксифенил)пиперазинил-1)- этил) амино-3(2Н)-пиридазинона, 36% от теории, как бесцветный кристаллический осадок, который после перекристаллизации из этанола дает 6,8 г (32,2%) чистого основания. Обработкой эфирным раствором HCI в этаноле его превращают в дигидрохлорид, точка плавления 210-220°С; С 45,9%; Η 5,7%;Сl (общий) 23,6%; Сl- 16,0%; бесцветное кристаллическое вещество; ультрафиолетовый спектр в 0,1 н. HCI; 210 (4,55), 230 (4,30), 300 (4,17). Охлаждением ацетонитрила - маточного раствора получают белый кристаллический осадок 4,5 г 2-метил-4хлор-5-((2-((4-2-метоксифенил)пиперазинил-1) этил)амино-3(2Н)-пиридазинона, 21,3% от теории, который растворением в изопропаноле и смешиванием с эфирным раствором соляной кислоты превращают в дигидрохлорид с точкой плавления 218-225°С и получают в чистом виде перекристаллизацией из изопропанола, точка плавления 223-227°С, бесцветные кристаллы, 14,3% от теории; С 48,0%; Η 5,7%; СІ (весь) 23,5%; Сl- 15,7%; Ν 15,0%; О 7,8%. Ультрафиолетовый спектр в этаноле: 210 (4,5), 230 (4,57), 286 (4,00). 304 (S 3,81). Пример 3.2-Трет.-бутил-4-хлор-5-((2-{3-(3-трифторметилфенол)пиперазинил-1)этил)-амино-3(2Н)пиридазинон и 2-трет.бутил-5-хлор-4((2-(3-(3-трифторметилфенил)пипера-зинил-1) этил)амино)-3(2Н)пиридазинон. 15,0 г(0,055 моль) 1-аминоэтил-4(3-триф-торметилфенил)-пиперазина и 15,2 г (0,069 моль) 2-трет.-бутил4,5-дихлор-3(2Н)- пири-дазинона нагревают с 6,9 г (0,069 моль) тонко измельченного в порошок бикарбоната калия в 100 мл ацетонитрила, при исключении влаги, 96 ч при флегме и при хорошем перемешивании до кипения: отфильтровывают твердое вещество, сгущают в вакууме, обрабатывают эфиром и 1 н. НСІ, экстрагируют кислую фазу е ще 2 раза простым эфиром, затем устанавливают щелочную среду посредством натронового щелока и экстрагируют снова 3 раза хлороформом, сушат органическую фазу сульфатом натрия и выпаривают растворитель: остаток весит 30,1 г и подвергается препаративной хроматографии на колонке с силикагелем (Matrex Silica Si60 0,020-0,045 мм) с растворителем хлористым метиленом-метанолом 40:1. Получают 18,8 г 2-трет.-бутил-4-хлор-5- ((2-(3-(3-трифтор-метилфенил)пиперазинил-1) -этил)амино)-3(2Н)пиридазинона как первую фракцию, 74,6% от теории. Отсюда 3,80 г растворяют в 50 мл ацетона и при помощи эфирного раствора соляной кислоты переводят в 3,55 г легко водорастворимого, бесцветного кристаллического гидрохлорида (2,8 HCI-эквивалента) с точкой плавления 124-127°С; 54,0% от теории, С42,4%; Η 6,0%; СІ (общий) 23,0%; СІ" 16,7%; F9,2%; N 11,9%; 0 7,5%; ультрафиолетовый спектр в этаноле: 206 (4,39), 210 (4,4), 216 (4,37), 258 (4,08), 304 (4,12). В качестве второй фракции элюируют 8,3 г изомерного 2-трет.-бутил -5-хлор-4-{(2-(3-( 3 трифторметилфенил)пиперазинил-1)-этил)амино) -3(2Н)-пиридазинона; 32,9% от теории; 1,50 г этой фракции осаждают в 50 мл абсолютного этанола и с избытком эфирного раствора соляной кислоты и получают 1,20 г дигидрохлорида с точкой плавления 187-190°С как легко растворимое в воде, бесцветное кристаллическое вещество ; 22,6% от теории; С 47,4%; Η 5,5; СІ (весь) 20,0%; Сl- 13,2%; F 10,3%; N 13,2%; О 3,6%; ультрафиолетовый спектр в этаноле: 212 (S 4,39), 232 (4,52), 256 (4,19), 290 (3,97), 304 (S 3,86). Пример 4. 2-Метил-4-хлор-5-((3-(4-(2-метоксифенил) пиперазинил-1)пропил)ами-но)-3(2Н)-пиридазинон и 2-метил-5-хлор-4-{(3-(4-{2-метоксифенил)пиперазинил-1)про-пил)амино)-3(2Н)-пиридазинон. 10,0 г (0,040 моль) 1-аминопропил-4-{2-метоксифенил)-пиперазина и 7,9 г (0,044 моль)2-метил-4,5дихлор-3(2Н)-пиридазино-на нагревают вместе с 4,4 г (0,044 моль) бикарбоната калия в 100 мл свежеотогнанного диоксана 10 ч до 80°С и затем перемешивают 3 дня при комнатной температуре. После отфильтровывания неорганического материала сгущают в вакууме, остаток растворяют в водном растворе соляной кислоты и экстрагируют несколько раз простым эфиром; водную фазу доводят до щелочной среды при помощи натрового щелока, экстрагируют 3 раза путем встряхивания с хлороформом, сушат с сульфатом натрия и получают после сгущения в вакууме 15,7 г смеси изомеров. Проводят разделение препаративной хроматографией на колонке с силикагелем (Matrex Silica SI60 0.020-0,045 мм) с простым эфиром-метанолом 40:5 в качестве элюента. В качестве 1-й фракции элюируют 7,43 г 2-метил-4-хлор-5-((3-(4-(2метоксифенил)пипе-разинил-1)пропил)амино)-3(2Н)-пиридазино-на 47,5% от теории. Из них 5,0 г растворяют в абсолютном этаноле и смешивают с раствором соляной кислоты в этаноле и получают 5,6 г дигидрохлорида с точкой плавления 205-22О°С; С 48,7%; Η 6,5%; СІ (весь) 22,8%, Сl- 15,3%; N 15,0%; 0 7,0%; ультрафиолетовый спектр в 0,1 н. НСІ: 210 (4,49), 230 (4,54), 282 (3,93), 302 (3,85). При беспрерывном элюировании выделяют в качестве 2-й фракции 5,95 г 2-метил-5-хлор-4-((3-(4-(2-метоксифенил) пиперазинил1)пропил)амино)-3(2Н)-пиридазинона, 38,1% от теории. После растворения в абсолютном этаноле и смешивания с раствором соляной кислоты в этаноле 4,0 г этого продукта давали 3,9 г дигидрохлорида с точкой плавления 226-228°С; 37,1% от теории; С 49,0%; Η 6,5%; СІ (общий) 22,9%; Сl- 15,3%; N 14,8%; О 6,8%; ультрафиолетовый спектр в 0,1 н. НСІ: 204 (4,48), 230 (S, 4,54), 286 (S 3,95), 302 (3,85), 312 (S, 4,04). Пример 5. 2-Метил-4-хлор-5-((6-(4-(2-метоксифенил) пиперазинил-1)гексил)ами-но)-3(2Н)-пиридазинон и 2-метил-5-хлор-4-((6-{4-{2-метоксифенил)пиперазин ил-1 )гексил)-амино)-3(2Н)-пиридазинон. 5,8 г (0,020 моль) 1-аминогексил-4-(2-ме-токсифенил)~пиперазина и 4,45 г (0,025 моль) 2-метил-4,5дихлор-2-метил-3(2Н)-пиридазинона нагревают до кипения с 2,50 г (0,025 моль) тонко измельченного в порошок бикарбоната калия в 100 мл абсолютного этанола, при исключении влаги, 48 ч при флегме, при хорошем перемешивании; удаляют неорганический осадок фильтрованием, сгущают фильтрат в вакууме, подкисляют при помощи 1 н. НСІ, экстрагируют кислую водную фазу 3 раза простым эфиром, затем устанавливают щелочную среду при помощи натрового щелока и экстрагируют снова 3 раза хлороформом, сушат органическую фазу суль фатом натрия и сгущают растворитель в вакууме; остаток 10,0 г подвергают препаративной хроматографии на колонке с силикагелем (Waters Prep-Pak) с растворителем хлористым метиленом-метанолом-концентрированным аммиаком 40:1,5-0,1. Сначала элюируют 3,70 г 2-метил-4-хлор-5((6(4-(2-пи-перазинил-1)-гексил)амино)-3(2Н)- пирида-зинона как 1-ю фракцию; 42,6% от теории. Из них 2,00 г растворяют в 50 мл этанола р.А. и превращают с эфирным раствором соляной кислоты в 2,20 г водорастворимого бесцветного дигидрохлорида с точкой плавления 160-175°С; 38,0% от теории; С 50,3%; Η 6,8%; СІ (весь) 19,4%; СГ 13,1%; N 13,2%; О 10,3%; ультрафиолетовый спектр в этаноле: 212 (4,46), 216 (4,45); 234 (4,50), 286 (3,96, 304 (3,87, S). В качестве второй фракции элюируют 4,1 г изомерного 2-метил-5хлор-4-((6-(4-(2-метоксифенил) пиперизинил-1)-гексил)амино-3(2Н)-пиридазинона; 47,2% от теории. Из этой фракции растворяют 2,00 г в 50 мл р.А. этанола и превращают с эфирным раствором соляной кислоты в 1,50 г водорастворимого, бесцветного кристаллического дигидрохлорида с точкой плавления 153-165°С; 19,7% от теории; С 52,3%; Η 6,8; СІ (общий) 20,6%; Сl- 13,8%; N 13,9%; О 6,4%; ультрафиолетовый спектр в этаноле: 212 (4,47), 216 (4,44), 240 (4,29), 302 (414), 312 (3,89, S). Пример 6. 2-Метил-4-хлор-5-((4-(4-(2-метоксифенил) пиперазинил-1)бутил)ами-но)-3(2Н)-пиридазинон и 2метил-5-хлор-4-((4-(4-(2-метоксифенил) пиперазинил-1) бутил)амино)-3(2Н)-пиридазинон. 10,0 г (0,038 моль) 4-аминобутил-2-ме-токсифенил -пиперазина и 8,5 г (0,048 моль) 2-метил-4,5-дихлор3(2Н)-пиридазинона растворяют вместе с 4,75 г (0,048 моль) бикарбоната калия в 70 мл безводного диметилсульфоксида и выдерживают 15 ч при 80°С; разбавляют 200 мл воды и экстрагируют несколько раз хлороформом. Органическую фазу дополнительно промывают 3 раза водой, затем экстрагируют при помощи 1 н. Η Сі. Доводят водную фаз у до щелочной среды, экстрагируют п утем встряхи вания с хлороформом, сушат сульфатом натрия и получают после сгущения в вакууме 16,9 г смеси продукта. Дальнейшее разделение осуществляют методом препаративной хроматографии на колонке с силикагелем (Matrex Silica Si 60 0,0200,045 мм) с простым эфиром-метанолом 40:5 в качестве подвижной фазы. Как 1-ю фракцию выделяют 5,50 г (35,7% от теории) 2-метил-5-хлор-4-((4~(4-(2-метоксифенил) пиперазинил-1) бутил)амино)-3(2Н)пиридазинона, 30,1% от теории; растворяют в абсолютном этаноле и смешивают с раствором соляной кислоты в этаноле и получают дигидрохлорид с точкой плавления 205-207°С; С 50,1 %; Η 6,5%; СІ (весь) 21,5%; СІ* 14,5%; Ν 14,4%; О 7,0%; ультрафиолетовый спектр в этаноле: 206 (4,43), 210 (4,50), 244 (4,15), 296 (4,12), 312 (4,09). После дальнейшего элюирования появляются в качестве 2-й фракции 8,40 г 2-метил-4-хлор5-((4-(4-(2-метоксифенил) пиперазинил-1) амино)-3(2Н)-пиридазинона, 54,6% от теории, который после растворения в абсолютном этаноле и смешивания с раствором соляной кислоты в этаноле дает бесцветный кристаллический дигидрохлорид с точкой плавления 183-192°С; С 50,10%; Η 6,1%; Сl (общий) 21,8%; Сl14,9%; N 14,9%; О 7,0%. Ультрафиолетовый спектр в этаноле: 210 (4,41), 218 (4,42), 232 (4,46), 236 (4,45), 286 (3,96). Пример 7.2-Метил-4-хлор-5-((2-(4-{2,6-диметилфенил) пиперазинил-1)этил)амино)-3(2Н)-пиридазинон и 2метил-5-хлор-4-((2-(4-(2,6-диметилфенил) пиперазинил-1) этил)амино)-3(2Н) -пиридазикон. 9,2 г (0,039 моль) 1-аминоэтил)-4-{2.6-ди-метилфенил)-пиперазина и 8,8 г (0,049 моль) 2-метил-4,5дихлор-3(2Н)-пиридазинона нагревают до кипения вместе с 4,9 г (0,049 моль) тонко измельченного в порошок бикарбоната калия в 100 мл толуола, при исключении влаги, 20 ч при флегме и при хорошем перемешивании; отфильтровывают от неорганического материала, сгущают в вакууме, растворяют остаток в 1 н. HCI, экстрагируют 3 раза простым эфиром, затем доводят водную фазу до щелочной среды, экстрагируют снова 3 раза хлороформом, сушат органическую фазу с сульфатом натрия и выпаривают растворитель; остаток 15,7 г подвергают препаративной хроматографии на колонке с силикагелем (Waters Prep-Pak) с растворителем хлористым метиленом-метанолом 40:1. Получают 5,70 г 2-метил-4-хлор-5-((2-(2,6диметилфенил)пиперазинил-1)-этил)-амино)-3(2Н)-пиридазинона, 32,6% от теории, как 1-ю фракцию; из них 3,80 г растворяют в 50 мл абсолютного этанола, смешивают с раствором соляной кислоты в простом эфире и переводят в 3,00 г легко водорастворимого, бесцветного кристаллического дигидрохлорида с точкой плавления 235-242°С; 32,6% от теории; С 50,7%; Η 6,3%; СІ (весь) 23,2%; Сl- 15,4%; N 15,6%; 0 4,2%. Ультрафиолетовый спектр в этаноле: 220 (4,40), 232 (4,49), 290 (3,85), 311 (3,81). В качестве второй фракции колонки получают 7,40 г 2-метил-5-хлор-4-((2-(4-{2,6-диметилфенил)пиперазинил-1)этил)-амино)-3(2Н)-пиридазинона, 42,3% от теории; 4,0 г растворяют в 50 мл абсолютного этанола, осаждают избытком раствора соляной кислоты в простом эфире и получают 2,40 г легко водорастворимого бесцветного кристаллического гидрохлорида с точкой плавления 225-232°С, 24,9% от теории; С 55,5%; Η 6,8%; СІ (общий) 17,3%; Сl- 6,6%; N 17,2%; 0 3,2%. метил-4-((2-(4-(2-метоксифенил)пиперази-нил-1)этил)амино)-3(2Н)-пиридазинона (38,9% от теорий). Отсасывают, сушат, растворяют в ацетоне и переводят добавкой фумаровой кислоты при температуре флегмы в 1,50 г (24,5% от теории) соли фумаровой кислоты, точка плавления 144-148°С. С 54,0%; Η 6,1%, N 12,5%; О 27,4% ультрафиолет в 0,1 н. НС1: 208 (4,42), 226 (S, 4,22), 300 (4,47). Пример 11. 5-Хлор-4-((2-(4-(3-трифтор-метилфенил)пиперазинил-1)этил)амино)-3(2Н)-пиридазинон. 4,25 г (0,0093 мол) 2-трет.-бутил-5-хлор-4-((2-(4-(3-трифторметилфенил)-пиперази-нил-1)этил)амино)3(2Н)-пиридазинона перемешивают с 50 мл концентрированного водного раствора соляной кислоты при комнатной температуре в течение 72 часов, устанавливают основную среду и экстрагируют 3 раза хлороформом, сгущают органическую фаз у, сушат с сульфатом натрия и растирают остаток с ацетоном. Кристаллический осадок 5-хлор-4-((2-(4-(3-трифторметилфенил)пиперазинил-1)этил)-амино-3(2Н)пиридазинона весит 2,40 г (47,7% от теории). Переводят растворением в 100 мл горячего абсолютного спирта и добавкой эфирного раствора соляной кислоты в 2,20 г (46,7% от теории) дигидрохлорида с точкой плавления 220-223°С. С 40,6%, Η 4,2%, СІ (общий) 21,1%, СІ" 14,2%, F 11,5%, Ν 13,9%, 0 8,7%; ультрафиолет в 0,1 н. НСl: 208(4,42), 226 (S, 4,22), 300 (4,47). Аналогичным образом получают следующее вещство: 4-хлор-5-((2-(4-(2-метоксифенил)пиперазинил)этил)амино)-3(2Н)-пиридазинон ультрафиолет: растворитель: 0,1 н. НСl 210 (4,38), 228 (4,42), 280 (3,81), 304 (3,72) Пример 12. 2-Метил-6-хлор-4-((3-{4-{2-метоксифенил)пиперазинил-1)пропил)амино)-3(2Н)-пиридазинон. 6,0 г (0,0159 мол) тонко растертого 6-хлор-4-((3-(4-(2-метоксифенил)пиперазинил--1)пропил)амино)-3(2Н)пиридазинона суспендируют в 100 мл 2 н. NaOH и перемешивают при 60°С с 1,51 мл диметилсульфата (0,0159 мол) 2 часа, а затем охлаждают; экстрагируют несколько раз хлороформом, сушат органическую фазу и сгущают. Из маслянистого остатка кристаллизуются в течение ночи 3,50 г (56,2% от теории) нечистого 2-метил-6хлор-4-((3-(4- (!2-метоксифенил)пипера-зинил-1)пропил)амино)-3(2Н)-пиридазинона, который очищают препаративной хроматографией на силикагеле (Waters-Prep-Pak). Чистая фракция растворяется в изопропаноле, смешивается с эфирным раствором соляной кислоты и дает 0,85 г (11,2% от теории) белого, кристаллического дигидрохлорида, точка плавления 218-229°С. С 40,6%, Η 4,2%, СІ (общий) 21,1%, Сl- 14,2%, F 11,5%, N 13,9%, О 8,7%. Пример 13. 6-Хлор-2-этил-4-((3-{4-(2-метоксифенил)пиперазинил-1)пропил)амино)-3(2Н)-пиридазинон. 1,80 г (0,00474 мол) тонко растертого 6-хлор-4-((3-{4--(2-метоксифенил)пиперазинил--1)пропил)амино)3(2Н)- пиридазинона суспендируют в 80 мл 2 н. NaOH, добавляют 1,8 мл (0,014 мол) этилйода и перемешивают при комнатной температуре 90 минут; после этого снова добавляют 1,8 мл этилйода и перемешивают еще 2 часа. Выпаривают растворитель, поглощают в воде и экстрагируют хлороформом. Из высушенной органической фазы остается при сгущении коричневое масло, которое при растворении в этаноле и смешивании с этаноловым раствором соляной кислоты дает 0,70 г (30,6% от теории) чистого дигидрохлорида 6-хлор-2-этил-4- ((3-(4-{2-метоксифенил)пиперазинил-1)пропил)амино-3(2Н)- пиридазинона. 202-207°С; как белое кристаллическое вещество: С 49,4%, Η 6,4%, СІ (общий) 21,8%, Сl- 14,6%, N14.3%, О 7,7%. Пример А. Определение сродства соединений формулы к альфа-адреноцепторам. Сродство соединений общей формулы альфа-адреноцепторам определяли по известному методу. По этому методу измеряют конкурентное вытеснение насыщенного тритием працозина (2-{4-{2-фуроил)-1-пиперазинил-4-амино-6,7-диметокси-хиназолина) на сердечных мембранах тест-веществами и определяют как ІС50 (торможение концентрации 50%) ту концентрацию, которая вызывает 50%-ное торможение специфической связи насыщенного тритием працозина на альфа-адреноцепторах в сердечных мембранах крыс. Из ІС50- величин определяли независимые от концентрации константы ингибиторов Ki-альфа.
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for preparation of piperazinyle-alkyl-3(2n)pyridazinones or pharmaceutically acceptable salts thereof
Назва патенту російськоюСпособ получения пиперазинилалкол-3(2н)пиридазинонов или их фармацевтически приемлемых солей
МПК / Мітки
МПК: C07D 403/14, C07D 403/12, A61K 31/501, A61P 9/00
Мітки: піперазинілалкіл-3(2н)піридазинонів, спосіб, одержання, прийнятих, фармацевтично, солей
Код посилання
<a href="https://ua.patents.su/38-19756-sposib-oderzhannya-piperazinilalkil-32npiridazinoniv-abo-kh-farmacevtichno-prijjnyatikh-solejj.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання піперазинілалкіл-3(2н)піридазинонів або їх фармацевтично прийнятих солей</a>
Спосіб одержання похідних (1н-імідазол-1-ілметіл)замішаного бензімідазола або їх фармацевтично прийнятих солей кислоти, або солей металів, або стереоізомерів

Номер патенту: 2706
Опубліковано: 26.12.1994
Автори: Жерар Шарль Санз, Альфонс Герман Маргарета Реймакерс, Едді Жан Едгард Фрейн
МПК: A61P 17/00, C07D 409/14, C07D 403/06, A61K 31/425, A61K 31/443, A61P 5/00, A61P 19/06, C07D 403/14, A61K 31/4427, C07D 405/14, C07D 521/00, C07D 417/14, A61P 35/00, C07D 401/14, A61K 31/44, A61K 31/4433, A61K 31/47, A61P 43/00, A61K 31/415
Мітки: 1н-імідазол-1-ілметіл)замішаного, кислоти, прийнятих, фармацевтично, спосіб, солей, похідних, бензімідазола, металів, одержання, стереоізомерів
Формула / Реферат:
Способ получения производных (1Н-имидазол-1-илметил)-замещенного бензимидазола общей формулы где R2 — водород, С1— С6-алкил, С3— С7-цикло-алкил, фенил, необязательно замещенный двумя заместителями, выбранными из гало-, С1— С4-алкила, С1— С4-алкилоксикарбонила, карбоксила или С1— С4-алкилокси, тиенилфуранил, галофуранил, имидазолил или пиридинил, R1 — водород, С3— С7 - циклоалкил, фенил, С4 - С6-алкил, необязательно замещенный...
Спосіб одержання похідних бензоксазоламіна або бензотиазоламіна, або їх фармацевтично прийнятих солей, або їх стереоізомерів

Номер патенту: 2698
Опубліковано: 26.12.1994
Автори: Реймонд Антуан Стокброекс, Марсель Геребернус Марія Льюікс, Франс Едуард Жанссенс
МПК: C07D 413/12, C07D 513/04, A61K 31/435, A61P 25/28, C07D 417/12, C07D 207/08, A61K 31/445, C07D 498/04, A61K 31/4468, C07D 413/14, C07D 519/00, C07D 451/04, A61K 31/46, C07D 471/04, C07D 211/56, C07D 407/04, C07D 319/00, C07D 417/14
Мітки: фармацевтично, солей, бензотиазоламіна, одержання, прийнятих, стереоізомерів, похідних, бензоксазоламіна, спосіб
Формула / Реферат:
Способ получения производных бензоксазоламина или бензотиазоламина общей формулыгде — А1 = А2—А3=А4 — двухвалентный радикал, имеющий формулы:где один или два атома водорода в радикале каждый независимо друг от друга может быть замещен галогеном, С1— С6- алкилом, гидроксилом или С1— С6-алкоксигруппой, Z — 0 или S; n—0 или 1; R — заместитель, выбираемый из группы, состоящей из водорода, С1— С6-алкила, гидроксила и...
Спосіб одержання похідних 4-аміно-2-(піперазін1-іл)- або 4-аміно-2-(гомопіперазін-1-іл)хіназоліна або їх фармацевтично прийнятих солей з кислотами

Номер патенту: 8324
Опубліковано: 29.03.1996
Автор: Саймон Фрейзер Кемпбелл
МПК: C07D 239/95, C07D 405/14, C07D 319/00, A61P 9/00, C07D 405/12, A61K 31/505, A61P 9/12
Мітки: одержання, солей, спосіб, прийнятих, 4-аміно-2-(піперазін1-іл, кислотами, 4-аміно-2-(гомопіперазін-1-іл)хіназоліна, фармацевтично, похідних
Формула / Реферат:
Способ получения производных 1-амино-2-(пиперазин-1-ил)- или 1-амино-2-(гомопипера-зин-1-ил)хиназолина общей формулыгде (R)n-6,7-ди-(низший алкокси) или 6,7,8-три-(низший алкокси), m равно 1 или 2, Х - означает группу -CHR1, в которой R1 - атом водорода или метил; R2 и R3 одинаковы или различны и обозначают каждый атом водорода или атом хлора, или их фармацевтически приемлемых солей с кислотами, отличающийся тем, что хиназолин...
Спосіб отримання похідних імідазола або його фармацевтично прийнятих солей

Номер патенту: 2697
Опубліковано: 26.12.1994
Автори: Дейвід Джон Керіні, Джон Джонес Вітотес Данкіе
Мітки: фармацевтично, спосіб, похідних, імідазола, отримання, солей, прийнятих
Текст:
...5 Гц). пластинках для ТСХ. Масс-спектр 325. ЯМР (200 мГц, Е. 1~(4~Аминобензил)~5-гидроксиме3 э 8,00-6,80 (м, 8Н), тил-2-(2*-метоксиэтил~-4-«зшоримидазол. 5,15 ( с , 2Н>, 4,45 ( с , 2Н), 3,60 ( т , Соединение СИНТеЗИРУЮТ П Примеру О 2Н, 5 Г ц ) , 3,15 ( с , ЗН), 2,75 54, Е из 5-тидроксимет'ил—2~(2-меток~ ( т , 2Н, 5 Г ц ) 0 П р и м е р ы сиэгил)— 1 — (4—нитроб ензил) — 4—хлорими— 57—71„ Соединения, синтезированные по дазола ( 2 , 2 г, 6,75...
Спосіб одержання сульфінільних похідних гетероциклічних сполук або їх фармацевтично прийнятих солей
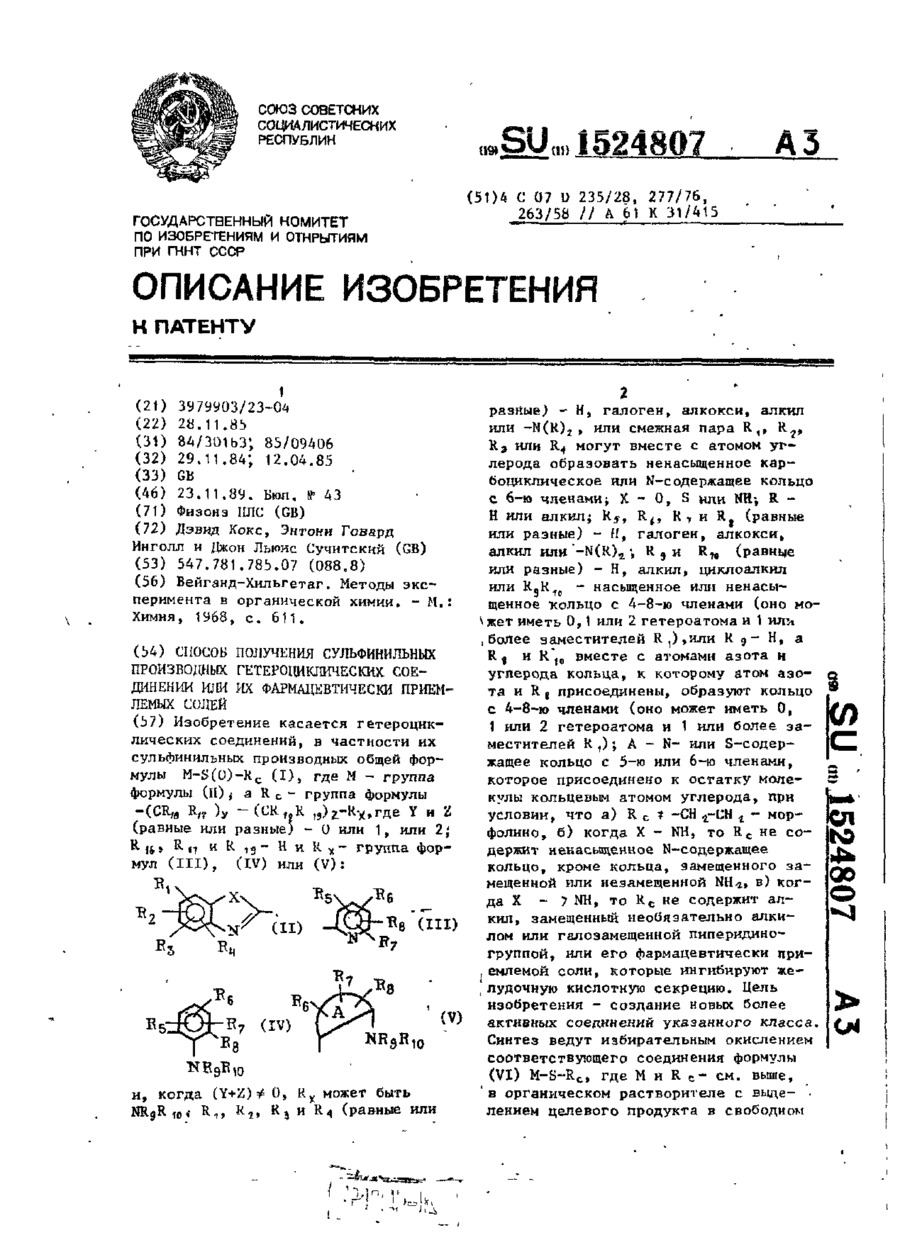
Номер патенту: 1944
Опубліковано: 20.12.1994
Автори: Девід Кокс, Джон Льюіс Сучитский, Ентоні Говард Інголл
Мітки: сполук, похідних, солей, прийнятих, одержання, спосіб, фармацевтично, гетероциклічних, сульфінільних
Формула / Реферат:
Формула изобретенияСпособ получения сульфинильных производных гетероциклических соединений общей формулыRC - группа формулы (CR16, R17)y -( CR18R19)z- Rх, где Y и Z - 0 или 1, или 2 каждый, одинаковые или разные;R16, R17, R18 и R19 – каждый водород;Rх – кольцо формули, когда (Y+Z)≠0, Rх может быть NR9R10;R1-R4 - одинаковые или разные, каждый - водород, галоген, алкокси, алкил или...
Попередній патент: Установка для заглиблення паль у грунт
Наступний патент: Спосіб одержання пектину
Випадковий патент: Пристрій для подрібнення сипучого матеріалу