Спосіб одержання ацетиленової сполуки





Формула / Реферат
1. Спосіб одержання ацетиленової сполуки формули (3)
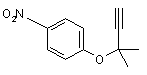 , (3)
, (3)
який відрізняється тим, що проводять взаємодію 4-нітрофторбензолу формули (1)
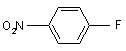 (1)
(1)
з алкоксидом 2-метил-3-бутин-2-олу формули (2)
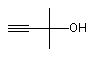 (2)
(2)
при температурі від -20 до 10 °С в розчиннику.
2. Спосіб одержання ацетиленової сполуки за п. 1, який відрізняється тим, що як розчинник використовують розчинник типу аміду.
3. Спосіб одержання ацетиленової сполуки за п. 1 або 2, який відрізняється тим, що кількість розчинника за масою в 2 рази більша кількості 4-нітрофторбензолу.
Текст
Даний винахід відноситься до способу отримання ацетиленової сполуки з 4-нітрофторбензолу і 2-метил-3бутин-2-олу. Дана сполука використовується як проміжна сполука для синтезу, наприклад, антифібриляторних агентів [див. JP-A-2001-151767] або гіпотензивних агентів [див. J. Med. Chem., 1983, Vol. 26, No. 11, 15821589]. Як відомий спосіб отримання ацетиленової сполуки формули (3) можна назвати спосіб, в якому 4нітрофенол (сполука (5)) взаємодіє з 2-метил-3-бутин-2-хлоридом (сполука (6)) в присутності основи [див., наприклад, J. Med. Chem., 1983, Vol. 26, No. 11, p. 1582 і JP-A-58-188880]. Відомий також спосіб, в якому 4-нітрофенол (сполука (5)) взаємодіє з 2-метил-3-бутин-2-хлоридом (сполука (6)) в присутності каталізатора йодиду міді, йодиду калію і карбонату калію [див., наприклад, Synthesis, 1995, Vol. 6, p.707]. Крім того, відомий спосіб, в якому 4-нітрофенол (сполука (5)) взаємодіє з похідним 2-метил-3-бутин-2-олу (сполука (7)) в присутності мідного каталізатора і DBU (1,8-діазабіцикло[5.4.0]ундец-7-ен) [див., наприклад, Tetrahedron Lett., 1994, Vol. 35, р.6405]. Крім того, відомий спосіб, в якому 4-нітрофенол (сполука (5)) взаємодіє з 2-метил-3-бутин-2-олом в присутності трифеніл фосфіну і DEAD (діетилазодикарбоксилату) [реакція Mitsunobu, див., наприклад, Synth. Commun.f 1989, vol. 19, р.1255]. Відомий спосіб з використанням 4-нітрофторбензолу як вихідної речовини, в якому 2-метил-3-бутин-2-ол використовують замість розчинника, і вихідна речовина взаємодіє з калієвим алкоксидом 2-метил-3-бутин-2олу [див., наприклад, J. Org. Chem., 1972, vol. 37, р.841]. (де X означає СІ, -ОСО2СН3 або -OCOCF3). При здійсненні способу, описаного в [J. Med. Chem., 1983, vol. 26, No. 11, p. 1582 і JP-A-58-188880], виникають деякі проблеми, такі як низький вихід, і використання 2-метил-3-бутин-2-хлориду (сполука (6)), що є відносно нестабільним. Хоча спосіб, описаний в [Synthesis, 1995, vol.6, p.707], дає збільшений до 89% вихід завдяки застосуванню мідного каталізатора, він має деякі недоліки, такі як видалення мідного каталізатора, що є важким металом, і застосування великої кількості йодиду калію, про що важко сказати, що це недорого, у випадку, коли застосовують йодид калію і т.п. Крім того, залишається проблема, пов'язана зі стабільністю 2-метил-3-бутин-2хлориду. Далі, існує проблема з точки зору вартості і можливостей методики, така як застосування 2-метил-3бутин-2-хлориду в кількості 2 моль на моль 4-нітрофторбензолу (сполука (5)). Спосіб, описаний в [Tetrahedron Lett., 1994, vol. 35, р.6405], подібний до вищезазначеного способу, але дає вихід 81% в більшості випадків, коли використовують 2-метил-3-бутин-2-хлорид; застосування трифторацетату, яке забезпечує найвищий вихід (88%), є явно невигідним з точки зору витрат, і DBU, що використовується, також дорогий. Тому даний спосіб непридатний як промисловий спосіб. Спосіб, описаний в [Synth. Commun., 1989, vol. 19, p. 1255], також непридатний як промисловий спосіб з точки зору низького виходу (45%), вартості дорогого DEAD і т.п. Спосіб, описаний в [J. Org. Chem., 1972, vol. 37, p. 841], може бути названий способом вигідним за витратами і можливостями методики, оскільки він використовує як вихідні матеріали 2-метил-3-бутин-2-ол і 4нітрофторбензол, які відносно недорогі і стабільні, і не використовує каталізатори, такі як важкі метали. Однак даний спосіб має недоліки, такі як низький вихід (35%), тривалий час реакції (3 доби при кімнатній температурі) і т.п. Для того щоб вирішити вищезазначені проблеми, автори даного винаходу детально досліджували умови реакції між 4-нітрофторбензолом і алкоксидом 2-метил-3-бутин-2-олу. Внаслідок цього був знайдений спосіб, який є чудовим з точки зору можливостей методики і дає цільові сполуки з хорошим виходом, і в результаті був здійснений даний винахід. Даний винахід відноситься до способу отримання ацетиленової сполуки формули (3) який відрізняється взаємодією 4-нітрофторбензолу формули (1) з алкоксидом 2-метил-3-бутин-2-олу формули (2) при температурі від -20 до 10°С. Крім того, було знайдено, що спосіб згідно з даним винаходом може інгібувати утворення побічної сполуки (сполуки (4)), яка не може бути легко видалена на подальших стадіях. Найкращий спосіб здійснення винаходу Нижче пояснюється спосіб отримання ацетиленової сполуки формули (3). Ацетиленова сполука формули (3) може бути отримана з хорошим виходом шляхом взаємодії алкоксиду 2-метил-3-бутин-2-олу формули (2) з 4-нітрофторбензолом формули (1) в розчиннику при температурі від -20 до 10°С. У даному винаході використовують алкоксид 2-метил-3-бутин-2-олу формули (2) так, як звичайно використовують алкоксиди металів, і як метал в алкоксиді металу переважні лужні метали, такі як натрій, калій або літій, і т.п., і натрій є більш переважним з точки зору легкості поводження з ним і реакційної здатності. Використовувана кількість алкоксиду 2-метил-3-бутин-2-олу формули (2) складає від 0,5 до 20 молів на моль використовуваної кількості 4-нітрофторбензолу формули (1). Тим часом, оскільки вихід знижується при використовуванні кількості 1 моль або менше, кількість в 1 моль або більше є переважною, і кількість від 1 до 3 молів є більш переважною з точки зору витрат. Що стосується методики реакції, переважно додавати по краплях 4-нітрофторбензол формули (1) до розчину, що складається з розчинника і алкоксиду 2-метил-3-бутин-2-олу формули (2). Час, необхідний для додавання по краплях, складає переважно від 0,5 до 5 годин, хоча він не обмежений, до настання швидкого зростання температури в реакційній системі і поки утримується задана температура. Розчинники, що використовуються в даному винаході, включають розчинники типу аміду, такі як Ν,Νдиметилацетамід, Ν,Ν-диметилформамід, N-метилпіролідон, Ν,Ν'-диметилімідазилідинон або подібні, розчинники типу ароматичних вуглеводнів, таких як толуол, ксилол або подібні, розчинники типу аліфатичних вуглеводнів, такі як гексан, гептан або подібні, розчинники типу галогенвмісних вуглеводнів, таких як дихлорметан, хлороформ або подібні, і змішані або численні розчинники, згадані вище. Переважні розчинники включають розчинники типу аміду, більш переважно Ν,Ν-диметилацетамід і Ν,Ν'диметилімідазилідинон, з точки зору виходу ацетиленової сполуки формули (3). Використовувана кількість розчинника переважно в 2 або більше разів більше по масі кількості 4нітрофторбензолу формули (1) і, більш переважно, наприклад, від 2 до 4 мас, разів або, наприклад від 2 до 3мас., разів з точки зору витрат. Хоча температура реакції варіюється в інтервалі від -20°С до 10°С, переважним є інтервал від -10°С до 0°С з точки зору подовження часу реакції завдяки зниженню температури реакції і інгібуванню утворення побічного продукту формули (4). І час реакції і температура реакції не можуть бути вказані в загальному випадку, оскільки вони залежать від використовуваної кількості алкоксиду або подібного. Ацетиленова сполука формули (3) може бути отримана у вигляді сирого продукту шляхом додання води з подальшою екстракцією органічним розчинником, таким як толуол, промивання і потім відгонки розчинника. Сирий продукт може бути використаний як такий для отримання бензопіранової проміжної сполуки, і він може бути очищений колонковою хроматографією або дистиляцією і т.п., якщо необхідно. Алкоксид 2-метил-3-бутин-2-олу формули (2), будучи вихідною речовиною в даному винаході, звичайно може бути отриманий переробкою 2-метил-3-бутин-2-олу формули (2) з гідридом металу, таким як гідрид натрію, гідрид калію або подібні, або з металом, таким як металевий натрій, металевий калій, металевий літій або подібні. Ацетиленова сполука формули (3), отримана згідно з даним винаходом, веде до похідних бензопірану, тобто до проміжної сполуки для синтезу дефібрилуючого агенту або гіпотензивного агента препаративним методом, показаним в наступній схемі реакції. Інакше кажучи, ацетиленова сполука формули (3) може бути перетворена в проміжну сполуку для синтезу вищезазначеного дефібрилюючого агента або гіпотензивного агента циклізацією його при нагріванні для отримання бензопіранової сполуки і відновленням і ацетилуванням отриманої сполуки. Побічний продукт формули (4), що утворився при отриманні ацетиленової сполуки формули (3), не може бути повністю видалений навіть кристалізацією вищезазначеної ацетиламінової форми. Тому для підвищення ефективності подальшого приготування важливо інгібувати утворення побічного продукту формули (4) при отриманні ацетиленової сполуки формули (3). Даний винахід описаний далі конкретно згідно з прикладами, які не обмежують винахід. Під час дослідів відносну площу ВЕРХ вимірювали при наступних умовах аналізу: Колонка: L-Column ODS φ4,6 x 250мм (виготовлена в Chemical Evaluation Research Institute, Japan); Елююючий розчин: від 0 до 45xв.CH3CN-0,01 Μ AcONH4 (45/55 об./об.) від 45 до 65хв. CH3CN-0,01 Μ AcONH4 (45/55 об./об. ->95/5 об./об.) від 65 до 85хв. CH3CN-0,01 Μ AcONH4 (95/5 об./об.); Детектування: УФ (245 нм); Витрата: 1мл/хв.; Температура колонки: 40°С. Приклад 1 У 300мл реакційну колбу, забезпечену термометром, мішалкою і краплинною лійкою, додавали 96,0г Ν,Νдиметилацетаміду (ДМА) і 11,6г (290ммоль) 60%-ного гідриду натрію (суспензія в мінеральному маслі) і додавали по краплях при перемішуванні і охолоджуванні льодом 25,2г (300ммоль) 2-метил-3-бутин-2-олу для отримання алкоксиду (час додавання по краплях: 2 години). Після перемішування протягом 30 хвилин додавали по краплях 33,8г (240ммоль) 4-нітрофторбензолу (при перемішуванні і охолоджуванні льодом; час додавання по краплях: 1,5 години) і після завершення додавання по краплях отриману суміш перемішували при тій же температурі протягом 18 годин. До реакційної суміші додавали 480мл води і 480мл толуолу, і отриману суміш струшували. Після відстоювання суміш розділяли на дві фази і толуольну фазу відбирали. Водну фазу знову екстрагували 240мл толуолу і отриману толуольну фазу об'єднували з раніше отриманою толуольною фазою, промивали 240мл води і потім відганяли розчинник, отримуючи сирий продукт (63,0г) ацетиленової сполуки формули (3), що є цільовим продуктом. Сирий продукт очищали колонковою хроматографією на силікагелі, отримуючи 44,0г (вихід 90%) цільового продукту у вигляді жовтого маслянистого продукту. 1 Н-ЯМР (CDCl3) ч/млн: 8,18 (2Н, д, J=9,2 Гц), 7,30 (2Н, д, J=9,2 Гц), 2,68 (1H, с), 1,73 (6Н, с). Приклад 2 У 2 л реакційну колбу, забезпечену термометром, мішалкою і краплинною лійкою, додавали 283г Ν,Νдиметилацетаміду (ДМА), отриману суміш охолоджували до (-13)-(-12°С) і до неї додавали 34,3г (856ммоль) 60%-ного гідриду натрію (суспензія в мінеральному маслі). Потім додавали по краплях 74,5г (886ммоль) 2метил-3-бутин-2-олу для отримання алкоксиду (внутрішня температура: (-10)-(-8°С), час додавання по краплях: 3,5 години). Після перемішування протягом 1,5 годин додавали по краплях 100г (709ммоль) 4-нітрофторбензолу (внутрішня температура: (-10)-(-5°С), час додавання по краплях: 1,5 години) і після завершення додавання по краплях отриману суміш перемішували при тій же температурі протягом 38 годин. За температури нижче 10°С до реакційної суміші додавали 1420мл води, після перемішування протягом 1 години до неї додавали 1420мл етилацетату і струшували; отриманій суміші давали відстоятися, розділяли на дві фази і етилацетатну фазу відбирали. Водну фазу екстрагували 709мл етилацетату і отриману етилацетатну фазу об'єднували з раніше отриманою етилацетатною фазою, промивали 709мл води і потім відганяли розчинник, отримуючи сирий продукт (177г) ацетиленової сполуки формули (3), що є цільовим продуктом. Відносні проценти площі при ВЕРХ ацетиленової сполуки формули (3) і побічного продукту формули (4) після реакції протягом 38 годин і в сирому продукті наведені в таблиці 1. Таблиця 1 Реакція 38 годин Сирий продукт (3) (3) 95,0% 93,8% (4) 0,5% 0,5% Посилальний приклад 1 У 1л реакційну колбу, забезпечену термометром, мішалкою, конденсатором Дімрота і краплинною лійкою, додавали 162г о-дихлорбензолу, нагрітого до 170°С, і протягом 3год. 40хв. туди додавали по краплях всю кількість сирого продукту ацетиленової сполуки формули (3), отриманої в прикладі 2, розчиненої в 186г одихлорбензолу (внутрішня температура: 168-176°С). Після завершення додавання по краплях отриману суміш перемішували при тій же температурі протягом 1 години і відганяли розчинник, отримуючи 169г сирого продукту 2,2-диметил-6-нітро-2Н-1-бензопірану, що є цільовим продуктом. Сирий продукт розчиняли при нагріванні в змішаному розчиннику з 317г метанолу і 56г води і поступово охолоджували до 2°С, і потім піддавали кристалізації протягом 2год. 30хв. при температурі від 0 до 5°С. Отримані кристали відбирали фільтруванням і промивали, потім сушили при зниженому тиску при 50°С, отримуючи 137г (вихід: 94%) 2, 2диметил-6-нітро-2Н-1-бензопірану. Оскільки кристали були забруднені мінеральним маслом гідриду натрію і тому подібним, вони були піддані визначенню з внутрішнім стандартом і мали чистоту 89,4%. Отже, вихід в сукупності двох стадій становив 84%. Значення фізичних властивостей зразка, отриманого очищенням колонковою хроматографією з силікагелем, були наступними: Т.пл.: 74,6-74,7°С 1 H-ЯМР (CDCl3) ч/млн: 8,02 (1H, дд, J=8,9, 2,8 Гц), 7,89 (1H, д, J= 2,8 Гц), 6,81 (1Н, д, J=8,9 Гц), 6,36 (1Н, д, J=9,9 Гц), 5,75 (1H, д, J=9,9 Гц), 1,48 (6Н, с). Приклади 3-5 (Вплив температури реакції і кількості розчинника) Реакцію проводили в тих же умовах, як в прикладі 2, за винятком температури реакції і кількості розчинника, які були змінені. Відносні проценти площі при ВЕРХ ацетиленової сполуки формули (3) і побічного продукту формули (4) після завершення реакції і в сирому продукті наведені в таблиці 2. Під час дослідів час реакції змінювали в залежності від умов реакції. Крім того, кількість розчинника наведена в масових частинах на частину використаної кількості 4нітрофторбензолу формули (1) Для порівняння приведені також результати прикладу 2. Приклад 6 (Приклад, в якому як розчинник використаний Ν,Ν¢-диметилімідазолідинон (ДМІ)) У 300мл реакційну колбу, забезпечену термометром, мішалкою і краплинною лійкою, додавали 96,0г Ν,Ν'даметилімідазолідинон (ДМІ) і 11,6г (290ммоль) 60%-ного гідриду натрію (суспензія в мінеральному маслі) і додавали по краплях при перемішуванні і охолоджуванні льодом 25,2г (300ммоль) 2-метил-3-бутин-2-олу для отримання алкоксиду (внутрішня температура: 10°С або менше, час додавання по краплях: 1,5 години, перемішування протягом 30хв. після додавання по краплях). Потім додавали по краплях 33,8г (240 ммоль) 4-нітрофторбензолу, і отримана суміш реагувала при перемішуванні протягом 18 годин при охолоджуванні льодом. До реакційної суміші додавали 480мл толуолу і 480мл води і отриману суміш струшували. Після відстоювання суміш розділяли на дві фази і толуольну фазу відбирали. Водну фазу знову екстрагували 240мл толуолу і отриману толуольну фазу об'єднували з раніше отриманою толуольною фазою. Об'єднану толуольну фазу сушили над безводним сульфатом натрію і фільтрували, потім відганяли розчинник, отримуючи сирий продукт ацетиленової сполуки формули (3), що є цільовим продуктом. Сирий продукт піддавали колонковій хроматографії на силікагелі (500г силікагелю, елююючий розчин: етилацетат-гексан=1/20), отримуючи 43,5г (вихід 89%) очищеного продукту ацетиленової сполуки формули (3) у вигляді жовтого маслянистого продукту. Аналіз ВЕРХ показав, що відносний процент площі цільового продукту становив 91,2% і відносний процент площі 4-нітрофторбензолу становив 7,9%. Оскільки 4-нітрофторбензол показав відношення чутливості в 4 рази, коректована ВЕРХ чистота становила 97,9%. Порівняльний приклад 1 У 1л реакційну колбу, обладнану термометром, мішалкою і краплинною лійкою, додавали 96г Ν,Νдиметилацетаміду (ДМА), отриману суміш охолоджували до 3°С і додавали до неї 11,6г (289ммоль) 60% гідриду натрію (суспензія в мінеральному маслі). Потім по краплях додавали 25,2г (299ммоль) 2-метил-3бутин-2-олу для отримання алкоксиду (внутрішня температура: від 3 до 10°С, час додавання по краплях: 1 година). Потім по краплях додавали 33,8г (239ммоль) 4-нітрофторбензолу (внутрішня температура: від 2 до 5°С, час додавання по краплях: 1,5 години) і після завершення додавання по краплях отриману суміш перемішували при 10-15°С протягом 20 годин. Після цього, подібно до методики прикладу 2, був отриманий сирий продукт ацетиленової сполуки формули (3) Відносні проценти площі ВЕРХ ацетиленової сполуки формули (3) і побічного продукту формули (4) після реакції протягом 18 годин і в сирому продукті наведені в таблиці 3. Таблиця 3 Реакція 18 годин Сирий продукт (3) (3) 72,2% 83,1% (4) 1,2% 1,5% Порівняльний приклад 2 (Реакція між 4-нітрофенолом і 2-метил-3-бутин-2-хлоридом) У 3л реакційну колбу, забезпечену термометром і мішалкою, додавали 148г (1,06моль) нітрофенолу і 1000мл води, додавали і розчиняли в цьому водний розчин гідроксиду натрію [NaOH 64,7г (1,62моль)/100мл Н2О] (температура підіймалася до 35°С завдяки екзотермічній реакції). Потім послідовно додавали 1100мл дихлорметану, 166г (1,62моль) 2-метил-3-бутин-2-хлориду і 92,0г гідроксиду триметилбензиламонію (40% метанольний розчин), і отриману суміш перемішували при кімнатній температурі протягом 5 діб. Перемішаному розчину давали відстоятися, ділили на 2 фази і відбирали дихлорметанову фазу. Водну фазу екстрагували 500мл хлороформу і отриману хлороформну фазу об'єднували з раніше отриманою дихлорметановою фазою. Об'єднану фазу промивали послідовно 1000г 10% водного розчину гідроксиду натрію, 700мл води і 500мл води і потім сушили над безводним сульфатом натрію. Отриманий розчин піддавали фільтруванню і потім відганяли розчинник, отримуючи 71,3г (вихід 33%) ацетиленової сполуки формули (3), що є цільовим продуктом, у вигляді темно-коричневого маслянистого продукту. Даний винахід може забезпечити процес отримання ацетиленової сполуки формули (3), що використовується як проміжна сполука фармацевтичних субстанцій, з 4-нітрофторбензолу, який є доступним і недорогим, промисловим економічно вигідним шляхом. Застосовність в промисловості Ацетиленова сполука формули (3), отримана згідно з способом за даним винаходом, використовується, наприклад, як проміжна сполука для синтезу дефібрилюючого агента або гіпотензивного агента, і тому є корисним для фармацевтичної промисловості і т.п.
ДивитисяДодаткова інформація
Назва патенту англійськоюProcess for production of an acetylenic compound
Автори англійськоюYamada Osamu, Matsumoto Hiroo, Shimizu Takanori
Назва патенту російськоюСпособ получения ацетиленового соединения
Автори російськоюЯмада Осаму, Мацумото Хироо, Симизу Таканори
МПК / Мітки
МПК: C07C 201/00, C07C 205/00
Мітки: одержання, ацетиленової, сполуки, спосіб
Код посилання
<a href="https://ua.patents.su/5-78876-sposib-oderzhannya-acetilenovo-spoluki.html" target="_blank" rel="follow" title="База патентів України">Спосіб одержання ацетиленової сполуки</a>
Спосіб одержання похідної сполуки хіноліну та проміжна сполука

Номер патенту: 65627
Опубліковано: 15.04.2004
Автори: Охара Йошіо, Сузукі Мікіо, Янагава Йошінобу, Такада Ясутака
МПК: C07D 215/14, C07D 215/12
Мітки: спосіб, хіноліну, одержання, сполуки, похідної, сполука, проміжна
Формула / Реферат:
1. Проміжна сполука нітрилу формули (1). (1)2. Спосіб одержання похідного хіноліну (3), що проводять за допомогою сполуки нітрилу, одержуваної в результаті реакції сполуки альдегіду, представленої формулою (2), з діетилціанометилфосфонатом (2) (1) (3).
Триазолопіримідинові сполуки, спосіб їх одержання (варіанти) та проміжні сполуки (варіанти)

Номер патенту: 73182
Опубліковано: 15.06.2005
Автори: Пальмгрен Андреас, Ларссон Ульф, Мусіль Тібор, Магнуссон Маттіас
МПК: C07D 317/44, C07D 239/47, C07D 405/12, C07B 61/00, C07D 487/04, C07D 239/56
Мітки: триазолопіримідинові, одержання, сполуки, варіанти, спосіб, проміжні
Формула / Реферат:
1. Сполука формули (І): (I).2. Спосіб одержання сполуки формули (І), що включає взаємодію сполуки формули (II): (II)з сіллю сполуки формули (III): (III).3. Спосіб одержання сполуки формули...
Піримідини як інгібітори сорбітдегідрогенази, фармацевтична композиція, що їх містить, проміжні сполуки та спосіб одержання проміжної сполуки

Номер патенту: 71951
Опубліковано: 17.01.2005
Автори: Зембровскі Уільям Джеймс, Міларі Банавара Лакшман, Чу-Моєр Маргарет Юхуа, Маррі Джеррі Ентоні
МПК: A61K 31/5377, C07D 417/14, C07D 451/02, A61K 31/53, C07D 491/048, C07D 491/04, A61P 43/00, C07D 405/12, C07D 491/20, C07D 521/00, C07D 487/04, C07D 498/04, A61P 9/10, C07D 413/12, A61P 3/10, A61K 31/506, C07D 491/10, C07D 417/12, C07D 513/10, C07D 451/06, C07D 401/12, A61K 31/517, C07D 401/14, C07D 471/04, C07D 487/08, C07D 239/42, C07D 403/12, C07D 403/14, C07D 409/14, C07D 401/04, C07D 405/14, C07D 409/12, C07D 471/08, C07D 403/04
Мітки: проміжної, сполуки, інгібітори, спосіб, піримідини, містить, композиція, одержання, сорбітдегідрогенази, фармацевтична, проміжні
Формула / Реферат:
1. Похідне піримідину формули I: , Iйого пролікарська форма або фармацевтично прийнятна сіль згаданої сполуки або згаданої пролікарської форми, де: R1 є (R)-1-гідроксіетилом; R2 є воднем; R3 є; іR9 є піримідилом або триазинілом; згаданий піримідил або...
Похідні ехінокандину, спосіб їх одержання, їх застосування, проміжні сполуки та фармацевтична композиція

Номер патенту: 72200
Опубліковано: 15.02.2005
Автори: Мелон Мангер Домінік, Куртен Олів'є, Фово Патрік, Маркус Астрид, Мішель Жан-Марк, Шио Лоран
МПК: A61P 31/10, A61K 38/04, C07K 7/56
Мітки: ехінокандину, композиція, одержання, фармацевтична, похідні, проміжні, сполуки, спосіб, застосування
Формула / Реферат:
1. Похідні ехінокандину формули (І):(І),їх можливі ізомерні форми або їх суміші, в яких:або R1 і R2, однакові або відмінні один від одного, означають атом водню, гідроксил; лінійний, розгалужений або циклічний алкільний, алкенільний або алкінільний радикал, що містить до 8 атомів вуглецю, що можливо переривається атомом кисню, можливо заміщений атомом...
Спосіб одержання циталопраму та проміжні сполуки для його одержання

Номер патенту: 63034
Опубліковано: 15.01.2004
Автори: Сван Хенрік, Петерсен Ханс, Рок Майкл Харольд
МПК: C07D 307/87, C07B 61/00, A61K 31/343, A61P 29/00, A61P 25/24, C07D 307/81
Мітки: одержання, сполуки, спосіб, проміжні, циталопраму
Формула / Реферат:
1. Спосіб одержання циталопраму, під час якого здійснюють взаємодію сполуки формули IV:, IVу якій R являє собою галоген або групу СF3-(СF2)n-SO2-O-, де n являє собою ціле число в інтервалі 0-8, включно, із джерелом ціаніду в присутності паладієвого каталізатора і каталітичної кількості Сu+ або Zn2+, або з Zn(CN)2 у присутності паладієвого каталізатора, і виділяють відповідну 5-ціаносполуку, тобто циталопрам:, (I)у...
Попередній патент: Спосіб попереднього біологічного очищення стічних вод
Наступний патент: Матриця для виготовлення пластмасових труб відцентровим методом
Випадковий патент: Пристрій для креслення кривих