Арилалкіламіни, спосіб їх одержання та фармацевтична композиція
Номер патенту: 27224
Опубліковано: 15.08.2000
Автори: Гуляуік П'єр, Проєтто Вінченцо, Ван Брук Дідьє, ЕМОН-АЛЬТ Ксав'є






Формула / Реферат
1. Арилалкиламины общей формулы (І)
в которой Y представляет собой либо группу Cy-N, где Су является фенилом, незамещенным или замешенным гидроксилом, (С1-С4) алкоксигруппой, (С3-С6) циклоалкилом, пиримидилом, либо группу
в которой Аг является фенилом, незамещенным или однократно либо многократно замещенным одним из заместителей, выбираемых среди, атома галогена, гидроксила, (С1-С4) алкокси, трифтормети, (С1-С4) алкила причем заместители могут быть идентичными или различными, пиридилом или тиенилом,
- х равно 0 или 1,
- X является гидроксилом, (С1-С4) алкоксигруппой, гидроксиалкилом, в котором алкил является (С1-С4) алкилом, (С1-С4) ацилокси, карбокси, (С1-С4) карболкокси, цианогруппой, аминоалкиленом с (С1-С3) алкиленом, группой -NH-CO-Alk, где Alk представляет собой (С1-С4) алкил, группой Alk1-NH-CO-Alk2, где Alk2 представляет (С1-С3) алкилен, a Alk2 представляет собой (С1-С4) алкил или (С1-С4) ацил, или X вместе с атомом углерода, с которым он связан в гетероцикле, образует двойную связь, m равно 2 или 3,
- Аг представляет собой фенил, незамещенным или однократно либо многократно замещенным галогеном, предпочтительно хлором, бензотиенил, нафтил, индолил или, индолил, N-замещенный (С1-С3) алкилом,
- R является водородом, (С1-С3) алкилом,
- Т является -СО- или -CO-NH-
- Z является М или ОМ, когда Т представляет -СО-, или М, когда Т представляет -CO-NH-,
- М представляет собой (С1-С4) алкил, фенил, который может быть однократно либо многократно замещен галогеном, (С1-С4) алкилом или (С1-С4) алкоксигруппой, нафтил, который может быть замещен галогеном, фенилалкил, в котором алкил является (С1-С3) и который может быть замещен на ароматическом цикле галогеном, (С1-С4) алкилом или (С1-С4) алкоксигруппой, тиенил, фурил, пирролил, тиадиазолил, а также их соли органических или неорганических кислот.
2. Соединение по п. 1, в котором Аг является 3,4-дихлорфенильной группой или одной из ее солей неорганической или органической кислоты.
3. Соединение по любому из пп. 1, 2, в котором X является гидроксилом, ацетилоксигруппой или группой -NH-C(О)-AIk1 где Alk представляет собой (С1-С4) алкил, или одна из его солей органической или неорганической кислоты.
4. Соединение по любому из пп. 1-3, в котором R является метилом, или одна из его солей органической или неорганической кислоты.
5. Соединение по любому из пп. 1-4, в котором Т является группой -СО-, или одна из его солей органической или неорганической кислоты.
6. Соединение по любому из пп. 1-5, в котором Т является группой -СО-, a Z является тиенилом, или одна из его солей органической или неорганической кислоты.
7. Соединение по любому из пп. 1-6, в котором Z является фенилом, возможно двузамещенный галогеном, таким как хлор, или одна из его солей органической или неорганической кислоты.
8. Соединение по любому из пп. 1-7, отличающееся тем, что оно является N-метил-N-[4-(4-фенил-4-аце-тиламинопиперидинил-1)-2-(3,4-дихлорфенип)бутил]бензамидом в форме рацемата, или одной из его солей органической или неорганической кислоты.
9. Соединение по любому из пп. 1-7, отличающееся тем, что оно является (-)-N-метил-N-[4-(4-фенил-4-ацетиламинопиперидинил-1)-2-(3,4-дихлорфенил)-бутил]бензамидом или его солью органической или неорганической кислоты.
10. Соединение по любому из пп. 1-7 отличающееся тем, что оно является (+)-N-метил-N-[4-(4-фенил-4-ацетиламинопиперидинип-1)-2-(3,4-дихлорфенил)-бутил]бензамидом или его солью органической или неорганической кислоты.
11. Способ получения арилалкиламинов общей формулы (I)
в которой Y представляет собой либо группу Cy-N, где Су является фенилом, незамещенным или замещенным гидроксилом, (С1-С4) алкоксигруппой, (С3-С6) циклоалкилом, пиримидилом, либо группу
в которой
- Аг является фенилом, незамещенным или однократно либо многократно замещенным одним из заместителей, выбираемых среди атома галогена, гидроксила (С1-С4) алкокси, трифторметила (С1-С4) алкила причем заместители могут быть идентичными или различными, пиридилом или тиенилом,
- х равно 0 или 1,
- X является гидроксилом, (С1-С4) алкоксигруппой, гидроксиалкилом, в котором алкил является (С1-С4) алкилом, (С1-С4) ацилокси, карбокси, (С1-С4) карбалкокси, цианогруппой, аминоалкиленом с (С1-С3) алкиленом, группой -NH-CO-Alk, где Alk представляет собой (С1-С4) алкил, группой Alk1-NH-CO-Alk2, где Alk1 представляет (С1-С3) алкилен, a Alk2 представляет (С1-С4) алкил или (С1-С4) ацил, или X вместе с атомом углерода, с которым он связан в гетероцикле, образует двойную связь, m равно 2 или 3,
- Аг представляет собой фенил, незамещенным или однократно либо многократно замещенным галогеном, предпочтительно хлором, бензотиенил, нафтил, индолил или индолил, N-замещенный (С1-С3) алкилом,
- R является водородом, (С1-С3) алкилом,
- Т является -СО- или -CO-NH-,
- Z является М или ОМ, когда Т представляет -СО-, или М, когда Т представляет -СО-NH-,
- М представляет собой (С1-С4) алкил, фенил, который может быть однократно либо многократно замещен галогеном, (С1-С4) алкилом или (С1-С4) алкоксигруппой, нафтил, который может быть замещен галогеном, фенилалкил, в котором алкил является (С1-С3), и который может быть замещен на ароматическом цикле галогеном, (С1-С4) алкилом или (С1-С4) алкоксигруппой, тиенил, фурил, пирролил, тиадиазолил, а также их солей органических или неорганических кислот, отличающийся тем, что
а) производят обработку свободного амина формулы
где т, Аг и R такие, как определено выше, а Е обозначает О-защитную группу, такую, например, как тетрагидропиранил-2-окси или группу
в которой Y является таким, как определено выше, причем, когда Y представляет группу
где X является гидроксилом, этот гидроксил является защищенным, либо функциональным производным кислоты формулы
в которой Z является таким, как определено выше, когда требуется получить соединение формулы (I), где Т является -СО-, либо изоцианатом формулы
в которой Z является таким, как определено выше, когда требуется получить соединение, описываемое формулой (I), где Т является -C(O)-NH-, для получения соединения формулы
b) затем, если Е является тетрагидропиранилокси, удаляют тетрагидропиранильную группу обработкой кислотой,
с) производят обработку полученного таким образом N-замещенного алканоламина формулы
метансульфонилхлоридом,
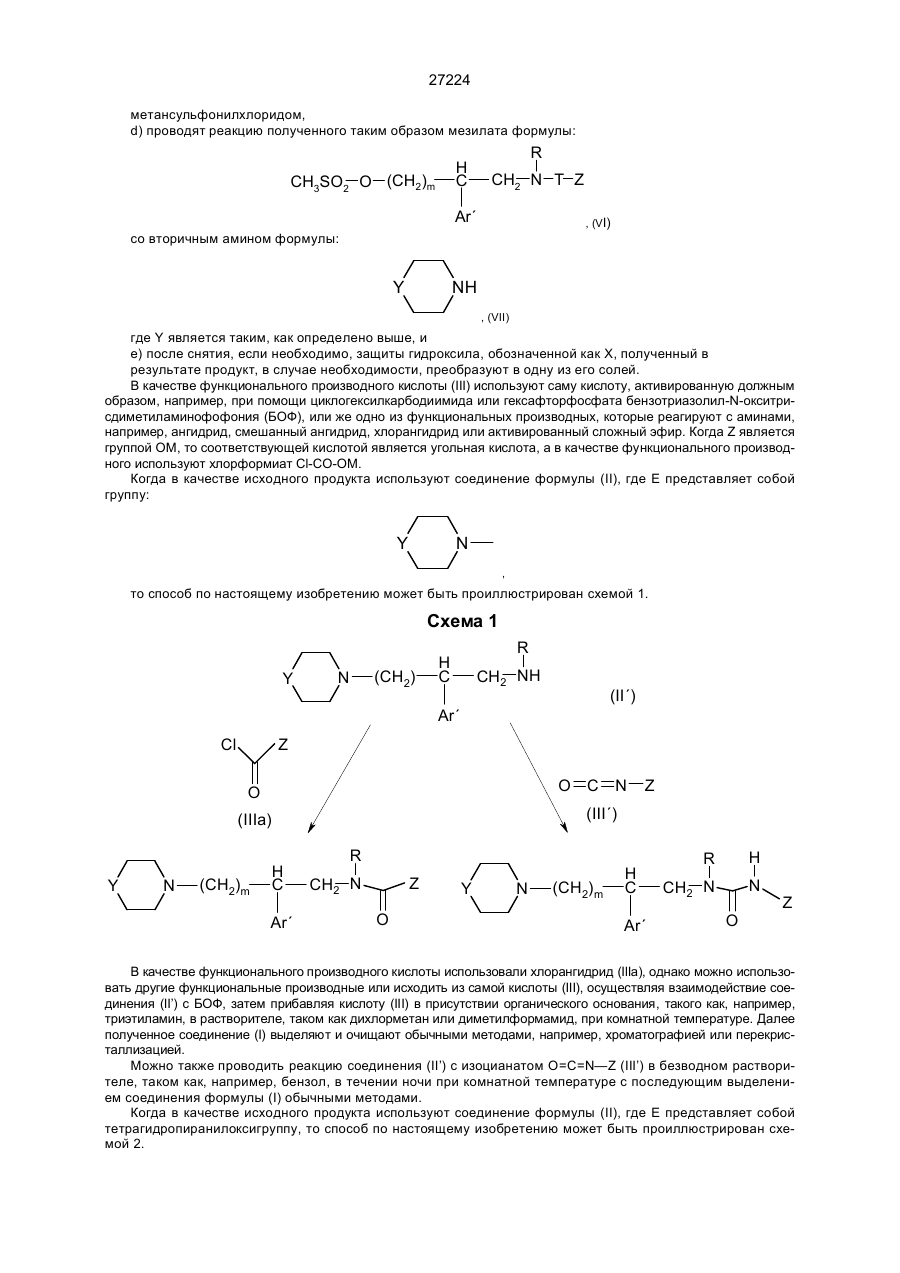
d) проводят реакцию полученного таким образом
мезилата формулы
со вторичным амином формулы
где Y является таким, как определено выше, и
е) после снятия, если необходимо, защиты гидроксила, обозначенной как X, полученный в результате продукт, в случае необходимости, преобразуют в одну из его солей.
12. Фармацевтическая композиция, обладающая антагонистической активностью к рецепторам нейрокинина А, содержащая активный ингредиент и фармацевтически приемлемый наполнитель, отличающаяся тем, что в качестве активного ингредиента она содержит арилалкиламин общей формулы (I)
в которой Y представляет собой либо группу Cy-N, где Су является фенилом, незамещенным или замещенным гидроксилом, (С1-С4)алкоксигруппой, (С3-С6) циклоалкилом, пиримидилом, либо группу
в которой
- Аг является фенилом, незамещенный или однократно либо многократно замещенный одним из заместителей, выбираемых среди атома галогена гидроксила, (С1-С4) алкокси, трифторметила, (С1-С4) алкила, причем заместители могут быть идентичными или различными, пиридилом или тиенилом,
- х равно 0 или 1,
- X является гидроксилом, (С1-С4) алкоксигруппой, гидроксиалкилом, в котором алкил является (С1-С4) алкилом, (С1-С4) ацилокси, карбокси, (С1-С4) карбалкокси, цианогруппой, аминоалкиленом с (С1-С3) алкиленом, группой -NH-CO-Alk, где Alk представляет собой (С1-С4) алкил, группой Alk1-NH-CO-Alk2, где Alk1 представляет (С1-С3) алкилен, a Alk2 представляет собой (С1-С4) алкил или (С1-С4) ацил, или X вместе с атомом углерода, с которым он связан в гетероцикле, образует двойную связь, m равно 2 или 3,
- Аг представляет собой фенил, незамещенные или однократно либо многократно замещенный галогеном, предпочтительно хлором, бензотиенил, нафтил, индолил или индолил, N-замещенный (С1-С3) алкилом,
- R является водородом, (С1-С3) алкилом,
- Т является -СО- или -CO-NH-,
- Z является М или ОМ, когда Т представляет -СО-, или М, когда Т представляет -CO-NH-, М представляет собой (С1-С4) алкил, фенил, который может быть однократно либо многократно замещен галогеном, (С1-С4) алкилом или (С1-С4) алкоксигруппой, нафтил который может быть замещен галогеном, фенилалкил, в котором алкил является (С1-С3), и который может быть замещен на ароматическом цикле галогеном, (С1-С4) алкилом или (С1-С4) алкоксигруппой, тиенил, фурил, пирролил, тиадиазолил, а также его соль с органической или неорганической кислотой, в количестве 0,25-250 мг на единичную дозу.
Текст
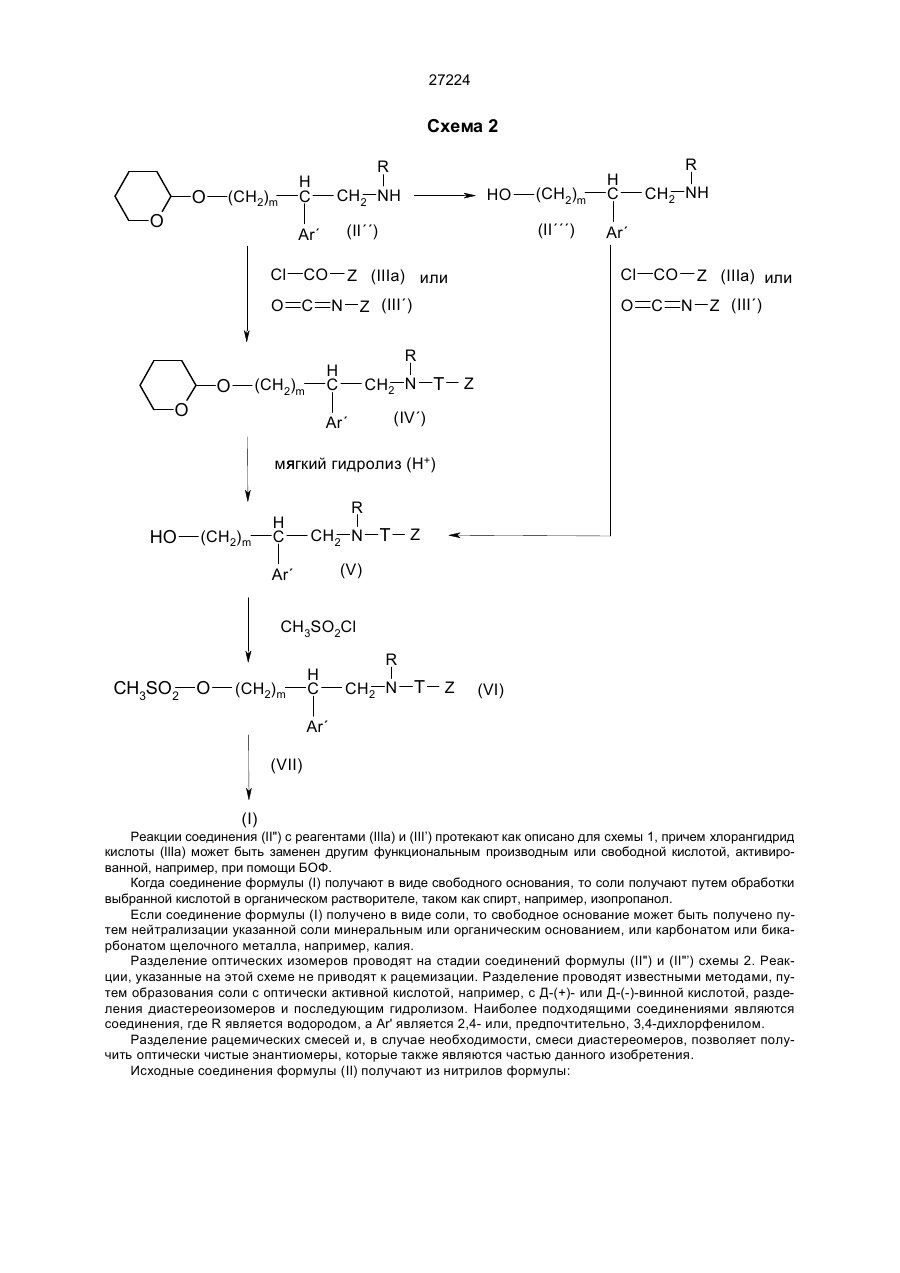
27224 Настоящее изобретение относится к новым ароматическим производным, замещенным аминогруппой и различными функциями сложных эфиров, аминов и амдов. Настоящее изобретение также относится к применению соединений данного изобретения в композициях для терапевтического использования, в частности при патологиях, которые затрагивают систему нейрокининов. Эндогенные лиганды в рецепторах нейрокининов были описаны ранее, например: Вещество Р (SP), нейрокинин А (НКА) (J.I. Bailey et al., Substance P, P.Surabancu ed., 16-17, Boole Press, Dubl, 1983) и нейрокинин В (НКВ) (S.P. Watson, Life Sciences, 1983, 25, 797-808). Рецепторы нейрокинина были известны для многочисленных препаратов и в настоящее время разделяются на три типа: HK1, HK2 и НК3. В то время как большинство изученных до настоящего времени препаратов имеют несколько типов рецепторов, такие как подвздошная кишка морской свинки (HK1, НК2 и НК3), некоторые имеют только один тип. Такие как сонная артерия собаки (HK1), легочная артерия кролика лишенная эндотелия (НК2) и воротная вена крысы (НК3) (D. Regoli et al.. Trends Pharmacol. Sci, 1988, 9, 290295 и Pharmacology, 1989, 38, 1-15). Более точная характеристика различных рецепторов становится возможной в результате недавнего синтеза селективных антагонистов. Так, например, [Sar9, Met-(O2)11]-BP, [NІе10]-НКА4-10 и [MePhe7]-HKB имеют селективность по отношению к рецепторам HK1, HK2 и НК3, соответственно (см. цитированные выше D. Regoli 1988 и 1989). Недавно было найдено, что некоторые аминосодержащие ароматические производные, замещенные различным способом, имеют интересные фармакологические свойства, как, например, свойство антагонистов рецептора нейрокинина А, и являются особенно полезными при лечении любой патологии зависящей от нейрокинина А. Рецепторы НК2 и нейрокинин А затрагиваются, например, при нейрогенных воспалениях дыхательных путей (Р. J. Basnes, Arch., Int. Pharmacodyn, 1990, 303. 67-82, G.F. Joos et al., Arch., Int. Pharmacodyn, 1990, 303,132-146). К настоящему времени были описаны только пептидные антагонисты рецепторов HK2. В публикации С.A. Maggi et al., Br. J. Parmacol., 1990, 100. 588-592 описаны пептиды, которые являются селективными антагонистами рецепторов HK2. В заявке на европейский патент 0347802 описаны производные пептидных антагонистов нейрокинина А, полезные в качестве иммуносуппресоров при лечении артрита, астмы, болей при воспалениях, желудочно-кишечной гипермоторики, болезни Ханингтона, психозов, гипертонии, мигрени, крапивницы и т.д. В заявке на европейский патент 0336230 также описаны производные пептидных антагонистов вещества Р и для нейрокинина А, полезные при лечении и профилактике астмы. Таким образом, одним из аспектов настоящего изобретения является ароматические арилалкиламины общей формулы (I): Y (CH2)m N H C R CH2 N T Z Ar´ , (I) в которой Y представляет собой либо группу Cy-N, где Су является фенилом, незамещенным или замещенным гидроксилом, (С1-С4)алкоксигруппой, (С3-С6)циклоалкилом, пиримидилом, либо группу Ar (CH2)x C X , в которой Аr является фенилом, незамещенным или однократно либо многократно замещенным одним из заместителей, выбираемых среди: атома галогена, гидроксила, (С1-С4)алкокси, трифторметила, (С1-С4)алкила, причем заместители могут быть идентичными или различными, пиридилом или тиенилом, х=0 или 1, Х является гидроксилом, (С1-С4)алкоксигруппой, гидроксиалкилом, в котором алкил является (С1-С4)алкилом, (С1-С4)ацилокси, карбокси, (С1-С4)карбалкокси, цианогруппой, аминоалкиленом с (С1-С3)алкиленом, группой -NH-CO-Alk, где Alk представляет собой (С1-C4)алкил, группой Alk1-NH-CO-Alk2, где Alk1 представляет (С1-С3)алкилен, a Alk2 представляет собой (С1-С4)алкил или (С1-С4)ацил, или Х вместе с атомом углерода, с которым он связан в гетероцикле, образует двойную связь, m равно 2 или 3, Аr' представляет собой фенил, незамещенным или однократно либо многократно замещенным галогеном, предпочтительно хлором, бензотиенил, нафтил, индолил или индолил, N-замещенным (С1-С3)алкилом, R является водородом, (С1-С3)алкилом, Т является -СО- или -CO-NH-, Z является М или ОМ, когда Т представляет –СО–, или М, когда Т представляет –CO–NH–, 27224 М представляет собой (С1-С4)алкил, фенил, который может быть однократно либо многократно замещен галогеном, (С1-С4)алкилом или (С1-С4)алкоксигруппой, нафтил, который может быть замещен галогеном, фенилалкил, в котором алкил является (С1-С3) и который может быть замещен на ароматическом цикле галогеном, (С1-С4)алкилом или (С1-С4)алкоксигрупой, тиенил, фурил, пирролил, тиадиазолил, а также их соли органических или неорганических кислот. Соли соединений формулы (І), в соответствии с настоящим изобретением, включают как соли минеральных или органических кислот, которые обеспечивают разделение или кристаллизацию соединений формулы (I), таких как пикриновая кислота, щавелевая кислота или оптически активные кислоты, например, миндальная или камфорсульфоновая кислота, так и фармацетически приемлемые соли, такие как гидрохлорид, гидробромид, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфат, малеат, фумарат, цитрат, 2-нафтилсульфонат, гликолят, глюконат, изотионат. Особенно предпочтительными соединениями является N-метил-N-[4-(4-фенил-4-ацетиламинопиперидинил-1)-2-(3,4-дихлорфенил)бутил]бензамид в виде рацемата или одного из энантиомеров (+) или (-), а также одна из его солей органической или неорганической кислоты. Другим аспектом настоящего изобретения является способ получения соединений формулы (I) отличающийся тем, что: а) производят обработку свободного амина формулы: E R H C (CH2)m CH2 NH Ar´ , (II) где m, Аr' и R такие, как определено выше, а Е обозначает O-защитную группу, такую, например, как тетрагидропиранил-2 окси или группу: N Y , в которой Y является таким, как определено выше, причем когда Y представляет группу Ar (CH2)x C X , где Х является гидроксилом, этот гидроксил является защищенным, – либо функциональным производным кислоты формулы: HO — CO — Z , (III) в которой Z является таким, как определено выше, когда требуется получить соединение формулы (I), где Т является -СО-, – либо изоцианатом формулы: O === C === N — Z , (III’) в которой Z является таким, как определено выше, когда требуется получить соединение, описываемое формулой (I), где Т является -C(0)-NH-, для получения соединения формулы: E (CH2)m H C R CH2 N T Z Ar´ , (IV) b) затем, если Е является тетрагидропиранилокси, удаляют тетрагидропиранильную группу обработкой кислотой, c) производят обработку полученного таким образом N-замещенного алканоламина формулы: HO (CH2)m H C Ar´ R CH2 N T Z , (V) 27224 метансульфонилхлоридом, d) проводят реакцию полученного таким образом мезилата формулы: R H C CH3SO2 O (CH2)m CH2 N T Z Ar´ , (VI) со вторичным амином формулы: NH Y , (VII) где Y является таким, как определено выше, и е) после снятия, если необходимо, защиты гидроксила, обозначенной как X, полученный в результате продукт, в случае необходимости, преобразуют в одну из его солей. В качестве функционального производного кислоты (III) используют саму кислоту, активированную должным образом, например, при помощи циклогексилкарбодиимида или гексафторфосфата бензотриазолил-N-окситрисдиметиламинофофония (БОФ), или же одно из функциональных производных, которые реагируют с аминами, например, ангидрид, смешанный ангидрид, хлорангидрид или активированный сложный эфир. Когда Z является группой ОМ, то соответствующей кислотой является угольная кислота, а в качестве функционального производного используют хлорформиат Сl-СО-ОМ. Когда в качестве исходного продукта используют соединение формулы (II), где Е представляет собой группу: N Y , то способ по настоящему изобретению может быть проиллюстрирован схемой 1. Схема 1 Y N (CH2) R H C CH2 NH (II´) Ar´ Z Cl O C N O (III´) (IIIa) Y N (CH2)m H C Ar´ Z R Z CH2 N O Y N (CH2)m H C Ar´ R H CH2 N N O Z В качестве функционального производного кислоты использовали хлорангидрид (IlIa), однако можно использовать другие функциональные производные или исходить из самой кислоты (III), осуществляя взаимодействие соединения (II’) с БОФ, затем прибавляя кислоту (III) в присутствии органического основания, такого как, например, триэтиламин, в растворителе, таком как дихлорметан или диметилформамид, при комнатной температуре. Далее полученное соединение (I) выделяют и очищают обычными методами, например, хроматографией или перекристаллизацией. Можно также проводить реакцию соединения (II’) с изоцианатом O=C=N—Z (ІІI’) в безводном растворителе, таком как, например, бензол, в течении ночи при комнатной температуре с последующим выделением соединения формулы (I) обычными методами. Когда в качестве исходного продукта используют соединение формулы (II), где Е представляет собой тетрагидропиранилоксигруппу, то способ по настоящему изобретению может быть проиллюстрирован схемой 2. 27224 Схема 2 O R H C (CH2)m O (II´´) Cl CO O C O Ar´ CH2 NH Cl N Z (III´) CO O Z (IIIa) или C Z (IIIa) или N Z (III´) R H C (CH2)m O (CH2)m (II´´´) HO CH2 NH Ar´ R H C CH2 N T Z (IV´) Ar´ мЯгкий гидролиз (H+) R (CH2)m CH2 N T Ar´ HO H C (V) Z CH3SO2Cl CH3SO2 O (CH2)m H C R CH2 N T Z (VI) Ar´ (VII) (I) Реакции соединения (II") с реагентами (IlIa) и (III’) протекают как описано для схемы 1, причем хлорангидрид кислоты (IlIa) может быть заменен другим функциональным производным или свободной кислотой, активированной, например, при помощи БОФ. Когда соединение формулы (I) получают в виде свободного основания, то соли получают путем обработки выбранной кислотой в органическом растворителе, таком как спирт, например, изопропанол. Если соединение формулы (I) получено в виде соли, то свободное основание может быть получено путем нейтрализации указанной соли минеральным или органическим основанием, или карбонатом или бикарбонатом щелочного металла, например, калия. Разделение оптических изомеров проводят на стадии соединений формулы (II") и (II"’) схемы 2. Реакции, указанные на этой схеме не приводят к рацемизации. Разделение проводят известными методами, путем образования соли с оптически активной кислотой, например, с Д-(+)- или Д-(-)-винной кислотой, разделения диастереоизомеров и последующим гидролизом. Наиболее подходящими соединениями являются соединения, где R является водородом, а Аr' является 2,4- или, предпочтительно, 3,4-дихлорфенилом. Разделение рацемических смесей и, в случае необходимости, смеси диастереомеров, позволяет получить оптически чистые энантиомеры, которые также являются частью данного изобретения. Исходные соединения формулы (II) получают из нитрилов формулы: 27224 H C (CH2)m E CN Ar´ , (VIII) где заместители имеют значения указанные выше, путем восстановления. Для получения соединений (II), где R является водородом, исходные нитрилы гидрируют в спирте, таком как этанол, в присутствии катализатора, такого как никель Ренея, а свободный первичный амин может быть выделен обычными методами. Для получения соединений (II), где R является отличным от водорода, то первичный амин, полученный гидрированием, обрабатывают хлорформиатом Cl-CO-OR, где R имеет значения указанные выше, с целью получения карбаматов формулы: E (CH2)m H C H OR CH2 N Ar´ O , которые затем восстанавливают известными способами, например, гидридом металла или гидридом бора в растворителе, например, в эфире, толуоле или тетрагидрофуране, при температуре от комнатной до 60°С, полученный амин выделяют обычными способами. Можно также соединение формулы: E (CH2 )m H C H CH2 N T Z Ar´ , где заместители имеют значения, указанные выше, обрабатывать алкилгалогенидом в присутствии основания, такого как, например, гидрид натрия, в инертном растворителе, таком как тетрагидрофуран, при кипячении, с получением соединений формулы (IV), в которых R отличается от водорода. Нитрилы формулы (VIII) получают из нитрилов формулы: Ar´—CH2—CN, (XI) алкилированием соединением формулы E—(CH2)m—G, (XII) где G является атомом галогена, например, брома, или защищенной гидроксигруппой, а остальные заместители имеют значения, указанные выше. Соединения настоящего изобретения исследовались путем биохимических и фармакологических опытов. Антагонистические свойства по отношению к рецепторам НК2 были исследованы при помощи опытов проведенных на слизистой оболочке двенадцатиперстной кишки крысы в соответствии с L. Bergstom et al., Pharmacol. 1987, 32, 764-771. Также опыты проводились на легочной артерии кролика, лишенной эндотелия, которая имеет peцепторы НК2, активация которых приводит к мышечным сокращениям. Опыты на различных изолированных органах проводились в соответствии с D. Regoli et al., Trends Pharmacol. Sci., 1988, 9, 290-295 и Pharmacology. 1989, 38, 1-15. Опыты в отношении бронхоспазма у морской свинки, вызванного агонистом рецептора НК2, были проведены в соответствии с Н. Konzett et al., Exp. Path. Pharm., 1940, 195. 71-4. Соединения настоящего изобретения вытесняют [2-125I гистидил]-нейрокинин А из его рецепторов с Км порядка от 3 до 0.50 нМ. Те же самые соединения в опытах, проведенных на легочной артерии кролика, показали рА2 от 10.4 до 9. Те же самые соединения в опытах, проведенных в отношении бронхоспазмов у морской свинки, показали антагонистическую активность [NIe10]-нейрокинина А при внутривенном введении при дозе 200 мкг/кг. Учитывая антагонистические свойства к найрокинину А, которыми обладают соединения данного изобретения, они могут быть полезны при любых патологиях, зависящих от нейрокинина А, и, в частности, при нейрогенных воспалениях дыхательных путей, таких как, например, астма или бронхоконстрикция. Соединения по настоящему изобретению являются малотоксичными, их малая токсичность совместима с их применением в качестве медикаментов, для такого применения вводя млекопитающимся эффективное количество соединения формулы (I) или одной из его фармацевтически приемлемых солей. Еще одним аспектом данного изобретения являются фармацевтические композиции, содержащие в качестве активного ингредиента эффективное количество соединения формулы (I) или одной из его фарма 27224 цевтических приемлемых солей. В фармацевтических композициях по настоящему изобретению для орального, подъязычного, подкожного, внутримышечного, внутривенного,трансдермального локального или ректального введения активные ингредиенты вводятся в смеси с классическими фармацевтическими носителями. С целью получения желаемого эффекта доза активного вещества может варьироваться от 0,25 до 1000 мг в день предпочтительно от 2 до 250 мг. Каждая единичная доза может содержать от 0,25 до 250 мг активного компонента, предпочтительно от 1 до 250 мг, в комбинации с фармацевтически приемлемым носителем. Эта единичная доза может вводиться от 1 до 4 раз в день. Для таблеток смешивают активный ингредиент с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарт магния, тальк, гумиарабик или аналогичные. На таблетки можно наносить покрытие из сахарозы или других соответствующих веществ, или же обрабатывать таким образом, чтобы они имели пролонгированную активность или активность замедленного действия, или чтобы они непрерывно высвобождали предопределенное количество активного ингредиента. Препарат в желатиновых капсулах получают, смешивая активный ингредиент с разбавителем и переливая полученную смесь в мягкие или твердые желатиновые капсулы. Препарат в виде сиропа или эликсира может содержать активный компонент вместе с подсластителем, предпочтительно некалорийным, с метилпарабеном и пропилпарабеном в качестве антисептика, а также с вкусовыми добавками и подходящим красителем. Порошки и гранулы, диспергируемые в воде, могут содержать активный ингредиент смеси с диспергаторами или смачивающими агентами или агентами для перевода в суспензию, например, поливинилпирролидон, а также с подсластителями или корректорами вкуса. Для ректального введения используют свечи, приготовленные со связующим, плавящимся при ректальной температуре, например, масло-какао или полиэтиленгликоли. Для парентерального, интраназального или внутриглазного введения используют водные суспензии, солевые изотонические растворы или смачивающие агенты, например, пропиленгликоль или бутиленгликоль. Для ингаляций используют аэрозоль, содержащий, например, триолеат сорбината или олеиновую кислоту, а также трихлорфторметан, дихлордифторметан, дихлотетрафторэтан или другой биологически совместимый выталкивающий газ. Активный компонент может быть также прописан в виде микрокапсул, при необходимости, с одним или несколькими носителями или добавками. Указанные композиции могут содержать другие активные соединения, например, бронходилятаторы, противокашлевые или антигистаминные средства. ПРИМЕР ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ Желатинозная капсула, содержащая 100 мг химического соединения по примеру 72: – химическое соединение по примеру 72 (взято за основу)........................ 100 мг – моногидрат лактозы ................................... 170 мг – натриевая карбоксиметилцеллюлоза сетчатая(200 меш) ......................................... 3,4 мг – стеарат магния ............................................ 1,7 мг – очищенная вода*.........................................QSP** для белой прозрачной капсулы размера № 3 заполнение на ......................... 170 мг *Удаляется при сушке после влажного гранулирования **QSP: В достаточном количестве. Примеры Спектры ЯМР снимали при 200 МГц в дейтрированном диметилсульфоксиде. Пример 1. Гидрохлорид N-[2-(3,4-ихлофенил)-4-(4-гидрокси-4-фенилпипперидинил)бутил]-2,4-дихлорбензамида. 27224 HO Cl ; Ar´= Y N N = ; Cl m=2; R=H; T Z= Cl ; O Cl а) a-(2-теграгидропиранилоксиэтил)-3,4-дихлорбензоацетонитрил. 16,5 г гидрида натрия с содержанием 80 % в жидком масле переводят в суспензию в 200 мл сухого тетрагидрофурана. При температуре 20°С прибавляют по капле за 30 минут раствор, содержащий 100 г 3,4дихлорбензолацетонитрила в 600 мл тетрогидрофурана, затем перемешивают реакционную смесь при комнатной температуре в течение 2 часов. Смесь охлаждается до температуры -20°С и прибавляют раствор, содержащий 118 г I-бром-2-тетрагидропиранилоксиэтана в 100 мл тетрагидрофурана, дают смеси вернуться к комнатной температуре и спустя 2 часа прибавляют раствор, содержащий 50 г хлорида аммония в 3 литрах воды. Экстрагируют 3 литрами эфира, промывают насыщенным раствором хлорида натрия, декантирут, сушат на MgSO4 и концентрируют под вакуумом. Остаток хроматографируется на силикагеле, элюент: дихлорметан, затем дихлорметан-этилацетат 96-5 (об/об). Фракции чистого продукта концентрируются под вакуумом, в результате чего получают 118 г жидкого масла. в) b-(2-тетрагидропиранилоксиэтил)-3,4-дихлорбензолэтанамин. Растворяют 118 г полученного перед тем нитрида в 700 мл абсолютного этанола. Прибавляют 300 мл концентрированного аммиака, затем в атмосфере азота добавляют никель Ренея (10% от количества исходного нитрила). Затем гидрируют в атмосфере водорода при комнатной температуре и обычном давлении. За 4 часа поглощаются 16 литров водорода. Катализатор отделяют фильтрованием на целите, фильтрат концентрируют под вакуумом, остаток промывается насыщенным раствором хлорида натрия. После экстракции эфиром и сушки на MgSO4 получают 112 г жидкого масла. с) N-[2-(3,4-дихлорфенил-4-(2-тетрагидропиранилокси)бутил]-2,4-дихлорбензамид. Растворяют 70 г полученного перед этим амина в 800 мл дихлорметана. Раствор охлаждают до температуры 0°С, прибавляют 38,4 мл триэтиламина, затем 66 г хлорангидрида 2,4-дихлорбензойной кислоты. Потом реакционную смесь перемешивают при комнатной температуре в течение одного часа, затем промывают водой. Органическую фазу декантируют, сушат на MgSO4 и концентрируют под вакуумом, в результате чего получат 120 г жидкого масла. d) N-[2-(3,4-дихлорфенил)-4-гидроксибутил]-2,4-дихлорбензамид. Растворяют 120 г полученного перед этим продукта в 1 литре метанола в присутствии 12 г паратолуолсульфоновой кислоты. Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, затем концентрировали под вакуумом. Остаток извлекают в дихлорметан и промывают 10% раствором карбоната натрия. Органическую фазу декантируют и сушат на MgSO4, в результате чего получают 106 г жидкого масла. е) N-[4-метансульфонилокси-2-(3,4-дихлорфенил)бутил]-2,4-дихлорбензамид. Растворяют 106 г полученного перед этим спирта в 1 л дихлорметана, затем прибавляют к охлажденному до температуры 0°С раствору 44 мл триетиламина и 24,2 мл метансульфонилхлорида. Реакционную смесь перемешивали при температуре 0°С в течение 46 минут, промывали 3 раза ледяной водой, декантировали, высушили на MgSO4 и концентрировали под вакуумом. Остаток перекристаллизовали из изопропилового спирта. Выход 95 г. Т.пл.-93°С. f) Соединение 1. Смесь, состоящая из 1 г полученного перед этим продукта, 0,8 г 4-гидрокси-4-фенилпиперидина и 1 мл диметилформамида, нагревают при температуре 60°С в течение 2 часов. После охлаждения разбавляют эфиром, промывают разбавленным раствором гидроксида натрия, затем водой. После сушки на MgSO4 упаривают растворители и хроматографируют остаток на 40 г кремнезема, элюент: дихлорметан, затем дихлорметан/метанол 90/10 (об./об.). Концентрирование чистых фракций приводят к получению 0,9 г продукта, который превращают в гидрохлорид в дихлорметане, прибавляя раствор хлороводорода в эфире до рН=1. Осадок отделяют фильтрованием. Выход 0.95 г. 1 Н-ЯМР: 8,75 (т, IH), 7,7-7 (м, 11Н), 5,4 (с, 1Н), 3,6-2,6 (м, 9Н), 2,6-1,6 (м, 6Н). Пример 2 Гидрохлорид N-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенилпиперидинил)-бутил] ацетамида. 27224 HO Cl ; Ar´= Y N N = ; Cl T R=H; m=2; CH3 ; Z= O N-[2-(3,4-диxлopфeнил)-4-мeзилoкcибyтил]aцeтaмид получают, как описано в примере 1 на стадиях а), в), с), d) и е), но заменяя на стадии а) хлорангидрид 2,4-дихлорбензойной кислоты на ацетилхлорид. Соединение 2. Нагревают до температуры 60°С смесь, состоящую из 6,5 г полученного перед этим продукта, 6,8 г 4гидрокси-4-фенилпиперидина и 10 мл диметилформамида. Спустя один час реакционную смесь выливают в воду и экстрагируют этилацетатом. Органическую фазу декантируют, сушат на MgSO4, фильтруют и концентрируют под вакуумом. Остаток очищают методом хроматографии на силикагеле, элюент: метанол/дихлорметан-10/90 (об/об). Концентрирование фракций чистого продукта приводит к остатку, который превращается в соль при помощи раствора хлороводорода в эфире, в результате получают 6 г гидрохлорида. 1 Н-ЯМР: 7,95 (т, 1Н), 7,7-7,0 (м, 8Н), 3,6-2,6 (м, 9Н), 2,6-1,3 (м, 9Н). Пример 3. Гидрохлорид N-этил-N-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенилпиперидинил) бутил]-2-тиофенкарбоксамида. HO Cl ; Ar´= Y N Cl m=2; N = ; O R=CH2CH3; T Z= S ; а) Гидрохлорид N-этил-[4-гидрокси-4-фенилпиперидинил]-b-этил-3,4-дихлорбензолэтанамина. К суспензии, состоящей из 0,96 г алюмогидрида лития в 10 мл тетрагидрофурана, прибавляют раствор, содержащий 5,5 г полученного перед этим соединения 2 в 20 мл тетрагидрофурана, затем реакционную смесь кипятят в течение 2 часов. Потом охлаждают и гидролизуют при помощи 4 мл 4Н раствора гидроксида натрия, фильтруют от глинозема, промывают тетрагидрофураном и выпаривают. После образования соли при помощи раствора хлороводорода в эфире гидрохлорид кристаллизуют из смеси изопропанол/изопропиловый эфир; получают 4,7 г продукта. в) Соединение 3. При температуре 0°С к раствору, содержащему 2,45 г полученного перед этим продукта в дихлорметане, прибавляют 2,75 мл триэтиламина, затем 0,8 г 2-тиеноилхлорида. После гидролиза 0,1Н раствором гидроксида натрия и экстракции дихлорметаном, продукт очищают методом хроматографии на кремнеземе Н; элюент: метанол/дихлорметан-2,5/97,5 (об/об). Чистый продукт образует соль при действии раствора хлороводорода в эфире и получают 1,0 г гидрохлорида. 1 Н-ЯМР: 7,75-6,9 (м, 11Н), 5.36 (с, 1Н), 3.9-2,55 (м, 11Н), 2,55-1,5 (м, 6Н) 0,95 (т, ЗН). Пример 4 Гидрохлорид N-этил-Н-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенилпиперидинил)-бутил]-4-метоксибензамида. Соединение 4 получают по способу примера 3, исходя из соединения, полученного на стадии а), и заменяя 2-тиеноилхлорид на хлорангидрид 4-метоксибензойной кислоты. Т.пл.=165°С. Пример 5 Дигидрохлорид N-[4-{4-гидрокси-4-(2-пиридинилметил) пиперидинил}-2-(3,4- дихлорфенил)бутил]-2,4дихлорбензамида. 27224 N HO Cl ; Ar´= Y N N = ; Cl T R=H; m=2; Cl ; Z= O Cl а) N-[2-(3,4-дихлорфенил)-4-(4,4-этилендиоксипиперидинил)бутил] -2,4-дихлорбензамид. Нагревают при температуре 100°С в течение 15 минут смесь, состоящую из 12,1 г мезилата, полученного в примере 1 на стадии е) и 8,6 г 4,4-этилендиоксипиперидина. Смесь охлаждают, экстрагируют дихлорметаном, промывают водой. Органическую фазу декантируют, сушат на MgSO4, фильтруют и концентрируют под вакуумом. Остаток хроматографируют на силикагеле, элюент: метанол/дихлорметан-2/98 (об/об). Концентрирование чистых фракций приводит к получению 12,15 г ожидаемого продукта. в) N-[2-(3,4-дихлорфенил)-4-(4-оксопиперидинил)бутил]-2,4-дихлорбензамид. Растворяют полученный перед этим продукт в 100 мл ацетона, затем прибавляют 100 мл 6Н хлороводородной кислоты. Спустя 2 часа прибавляют 1 литр воды и 1 литр эфира и извлекают водную фазу. Последняя затем доводится до значения рН=10, прибавлением гидроксида натрия, потом экстрагируют одним литром эфира. После сушки и выпаривания органической фазы получают 9,7 г чистого продукта. с) Соединение 5. При температуре 25°С в атмосфере азота к раствору содержащему 1 г полученного перед этим продукта в 5 мл тетрагидрофурана, прибавляют 2,15М раствор 2-пиколиллития в тетрагидрофуране до устойчивого красного окрашивания (смотри Synthesis, страница 43, 1974). После гидролиза и экстракции в эфире остаток хроматографируют на силикагеле; элюент: метанол/дихлорметан-15/85 (об/об). После выпаривания чистых фракций и образования соли из остатка путем действия раствора хлороводорода в эфире получают 400 мг пенообразного вещества белого цвета. 1 Н-ЯМР: 8,8-7,15 (м, 11Н), 5.3 (широкий с, 1Н), 4-2.55 (м, 11Н), 2,35-1,4 (м, 6Н). Пример 6 Гидрохлорид N-[4-(4-бензил-4-гидроксипиперидинил)-2-(3,4-дихлорфенил)бутил]-N’-нафт-1-илмочевина. HO Cl ; Ar´= Y N N = ; Cl m=2; R=H; T H N Z= ; O а) N-[4-(2-тетрагидропиранилокси)-2-(3,4-дихлорфенил)бутил]-N’-нафт-1-илмочевина. К раствору, содержащему 7,6 г нафт-1-илизоцианата в 50 мл толуола, прибавляют 12 г b-(2-тетрагидропиранилоксиметил)-3,4-дихлорбензолэтанамина в 50 мл толуола. Перемешивают реакционную смесь в течении 10 минут, затем прибавляют 50 мл метанола и концентрируют под вакуумом. в) N-[4-гидрокси-2-(3,4-дихлорфенил)бутил]-N'-нафт-1-илмочевина. К раствору, полученного перед этим продукта, прибавляют 2 г п-толуолсульфоновой кислоты и нагревают смесь до кипения в течение 10 мин. Раствор промывают бикарбонатом натрия и упаривают досуха. После очистки остатка методом хроматографии на силикагеле, элюент: этилацетат, получают 13,1 г бесцветного жидкого масла. c) N-[2-(3,4-дихлорфенил)-4-мезилоксибутил]-N'-нафт-1-илмочевина. К раствору, содержащему 13,1 г полученного перед этим продукта в 100 мл дихлорметана, прибавляют при температуре 0°С 6,37 мл триэтиламина, затем 2,77 мл мезилхлорида. Потом раствор промывают 3 раза 100 мл ледяной воды, затем органическая фаза сушится и выпаривается. Потом остаток перекристаллизовывают из изопропанола, затем фильтруют и промывают изопропиловым эфиром. Выход=8 г Т.пл.=120°С d) Соединение 6. 27224 Нагревают при температуре 100°С в течении 20 минут смесь, состоящую из 4 г полученного перед этим продукта, 4 г 4-гидрокси-4-бензилпиперидина и 4 мл диметилформамида. Все это затем выливают в воду и экстрагируют дихлорметаном. Потом остаток очищают методом хроматографии на силикагеле, элюент: смесь метанол/дихлорметан-4/96 (об/об). После образования соли при действии раствора хлороводорода в эфире получают 1,0 г чистого гидрохлорида. 1 Н-ЯРМ:8,7 (с,1Н) 8,2-6,8 (м,11Н), 3,6-2,6 (м, 11Н), 2,3-1,3(м, 6Н). Соединения, осписанные ниже в таблицах 1, 2 и 3 были синтезированы тем же способом, что и в примерах 1-5. Все соединения являются гидрохлоридами. Таблица 1 HO Ar (CH2)2 N (CH2)x H C H CH2 N Cl O *HCl Cl Cl Cl Пример № Ar x 1 Спектр H—ЯМР 8,54 (т, 2H), 7,7-7 (м, 11H), 4,8 (широкий с, 1H), 3,8-2,55 (м, 11H), 2,4-1,25 (м, 6H) 7 1 8,6 (т, 1H), 7,8-7,2 (м, 10H), 4,9 (с, 1H), 3,7-2,6 (м, 11H), 2,2-1,4 (м, 6H) Cl 8 0 9 8,55 (т, 1H), 7,8-7,2 (м, 10H), 5,70 (с, 1H), 3,6-2,6 (м, 9H), 2,6-1,6 (м, 6H) 1 8,7 (д, 2H), 8,45 (т, 1H), 7,85 (д, 2H), 7,6-7 (м, 6H), 5,3 (широкий с, 1H), 4,02,3 (м, 11H), 2,3-0,6 (м, 6H) CF3 10 N 27224 Таблица 2 (CH2)2 N Y H H C CH2 N Cl O *HCl Cl Cl Cl Пример № N Y 1 Спектр H—ЯМР 8,4-8,8 (м, 3H), 7,2-7,8 (м, 6H), 6,8 (т, 1H), 4,7 (д, 2H), 2,6-3,8 (м, 11H), 2,2 (м, 2H) 11 Таблица 3 HO R1´ H S CH2 N (CH2)2 C N (CH2)x O CH3 *HCl R1 Cl Cl Пример № R 12 H Y 1 7,8-6,8 (м, 11H), 4,75 (с, 1H), 4,0-2,5 (м, 14H), 2,3-1,3 (м, 6H) 13 4—OCH3 H 0 6,9-7,4 (м, 5H), 3,9-4,9 (м, 1H), 0,6-2,6 (м, 11H) 1 R 1´ 1 x Спектр H—ЯМР Пример 14 Гидрохлорид N-метил-N-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенил)бутил]-бензамида HO Cl ; Ar´= Y N N = ; Cl m=2; R=CH3; T ; Z= O а) N-[2-(3,4-дихлофенилфенил)-4-(2-теграгидропиранилокси)бутил]этилкарбамат. К охлажденному до -20°С раствору, содержащему 80 г амина, полученного в примере 1 на стадии в), и 39 мл триэтиламина в 800 мл дихлорметана, прибавляют по капле 26,4 мл этилхлорформиата. Спустя 30 минут промывают два раза водой, затем буферным раствором с рН=2. Декантируют органическую фазу и 11 27224 сушат на MgSO4, затем концентрируют под вакуумом, в результате чего получают 98 г продукта в виде жидкого масла. в) N-метил-(2-тетрагидропиранилокси)-b-этил-3,4-дихлорбензолэтанамин. В трехгорлую колбу объемом 2 литра, продуваемую азотом, вводят 20 г алюмогидрида лития в виде суспензии в 200 мл тетрагидрофурана. При температуре 20°С прибавляют раствор, содержащий 98 г полученного перед этим карбамата в 600 мл тетрагидрофурана. Осторожно нагревают до кипения и кипятят в течение 18 часов. Охлаждают до 0°С и гидролизуют при помощи 35 мл воды, а затем смесью, состоящей из 17 мл концентрированного раствора гидроксида натрия и 150 мл воды. Отделяют твердые вещества фильтрованием, затем концентрируют под вакуумом, в результате чего получают 80,5 г продукта в виде жидкого масла. c) Гидрохлорид N-метил-b-гидроксиэтил-3,4-дихлорбензолэтанамина. К 50 г полученного перед этим защищенного аминоспирта, растворенного в 500 мл этанола, прибавляют 20 мл концентрированной хлороводородной кислоты. Спустя 2 часа 30 минут концентрируют под вакуумом, растворяют остаток в 200 мл ацетонитрила, затем медленно добавляют 350 мл эфира. Перемешивают в течение одного часа, отфильтровывают кристаллы, промывают эфиром. Выход=32,8 г. Т.пл.=152°С. d) N-метил-N-[2-(3,4-дихлорфенил)-4-гидроксибутил]-трет-бутилкарбамат. К 32,6 г гидрохлорида полученного перед этим продукта, растворенного в 300 мл диоксана и 30 мл воды, прибавляют 20 мл триэтиламина. Потом добавляют 27 г Вос2О (ди-трет-бутил-дикарбоната), затем перемешивают при комнатной температуре в течение 16 минут. Нагревают при температуре 60°С в течение 30 минут. После концентрирования досуха экстрагируют эфиром, промывают водой, затем буферным раствором с рН=2, потом опять водой. Сушат на MgSO 4, упаривают под вакуумом, в результате чего получают 40 г жидкого масла. e) N-метил-N-[2-(3,4-дихлорфенил)-4-метансульфонилоксибутил]-трет-бутилкарбамат. К 40 г полученного перед этим спирта, растворенного в 400 мл дихлорметана, прибавляют 17 мл триэтиламина. Охлаждают до температуры 0°С и добавляют по капле 9,3 мл мезилхлорида. Спустя 15 минут промывают 2 раза водой, сушат на MgSO4, концентрируют досуха, в результате чего получают 49 г жидкого масла. f) Гидрохлорид N-метил-N-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенилпиперидинил)-бутил]-трет-бутилкарбамата. Нагревают смесь, состоящую из 20 г полученного перед этим продукта и 18 г 4-гидрокси-4фенилпиперидина в 40 мл диметилформамида, при температуре 70°С в течение 1 часа 30 минут. Затем приливают раствор к 300 мл ледяной воды, отфильтровывают осадок и промывают водой. Твердое вещество потом экстрагируют эфиром, сушат на MgSO4 и упаривают. Сырой продукт очищают методом хроматографии на силикагеле, элюент: градиент метанола/дихлорметан (до 5%). Получают 22 г чистого продукта. g) Дигидрохлорид N-метил-(4-гидрокси-4-фенилпиперидинил)-b-этил-3,4-дихлорбензолметанамин. К раствору, содержащему 22 г полученного перед этим производного в 100 мл метанола, прибавляют 100 мл концентрированной хлороводородной кислоты в 20 мл воды. Спустя один час реакционную смесь упаривают под вакуумом. Получают пенообразное вещество, которое превращают в порошок в эфире, затем сушат. Выход=20,7 г. h) Соединение 14 К раствору, содержащему 2 г полученного перед этим продукта и 2 мл триэтиламина в 20 мл дихлорметана, при температуре -78°С в атмосфере азота прибавляют 0,51 мл бензилхлорида и оставляют с перемешиванием в течение 10 минут. После гидролиза 0,1Н раствором гидроксида натрия и экстракции дихлорметаном продукт очищают методом хроматографии, элюент: смесь метанол/дихлорметан-10/90 (об/об). Получают таким образом 1,37 г чистого продукта, который превращают в гидрохлорид, прибавляя раствор хлороводорода в эфире до рН=1. Получают 1,40 г гидрохлорида в виде пенообразного вещества. 1 Н-ЯМР: 7.7-6,6 (м, 13Н), 5.35 (с, 1Н), 3,8-2,5(м, 12Н), 2.5-1,5 (м, 6Н). Пример 15 Гидрохлорид N-метил-N-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенилпиперидинил) бутил]бензилмочевины. HO Cl ; Ar´= N Y N = Cl m=2; R=CH3; T H N Z= O 12 ; C H2 ; 27224 При температуре 0°С в атмосфере азота к раствору, содержащему 2 г продукта, полученного в соответствии с примером 14 на стадии g), к 1,2 мл триэтиламина в 20 мл дихлорметана, прибавляют 0,60 мл бензилизоцианата и оставляют смесь при перемешивании в течение 1 часа. После промывания 0,1Н раствором гидроксида натрия продукт очищают методом хроматографии на силикагале, элюент: метанол/дихлорметан-6/94 (об/об). Затем получают соль продукта действием раствора хлороводорода в эфире и получают 1,8 г гидрохлорида. 1 Н-ЯМР: 7,7-6,9 (м, 13Н), 6,75 (т, 1Н), 5,4 (широкий с, 1Н), 4,1 (м, 2 Н), 3,7-2,5 (м, 2Н), 2,5-1,4 (м, 6Н). Пример 16 Гидрохлорид N-метил-N-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенилпиперидинил) бутил]этилкарбамата. HO Cl ; Ar´= N Y N = ; Cl m=2; T R=CH3; O Z= CH2CH3 ; O При температуре -78°С в атмосфере азота к раствору, содержащему 2 г продукта, полученного перед этим в соответствии с примером 14 на стадии g), к 2 мл триэтиламина в 20 мл дихлорметана, прибавляют 0,44 мл этилхлорформиата. Спустя 5 минут гидролизуют 0,1Н растором гидроксида натрия и экстрагируют дихлорметаном. Затем продукт очищают методом хроматографии на силикагеле, элюент: метанол/дихлорметан-8/92 (об/об). Чистые фракции концентрируют под вакуумом в результате прибавления раствора хлороводорода в эфире получают 1,1 г гидрохлорида в виде пенообразного вещества белого цвета. 1 Н-ЯМР: 7,7-7,1 (м, 8Н), 5,45 (с, 1Н), 4,1-2,6 (м, 14Н), 2,6-1,6 (м. 6Н), 1,1 (т, ЗН). Соединения, описанные в таблицах 4 и 5, были синтезированы в соответствии с примерами 14-16. Эти соединения являются гидрохлоридами. Таблица 4 HO H (CH2)m C N CH3 CH2 O N M *HCl R1 Cl Cl Пример № M 17 –CH3 7,8-7,1 (м, 8H), 3,7-2,65 (м, 12H), 2,65-1,6 (м, 9H) 18 –CH2–CH2–CH3 7,7-7,0 (м, 8H), 3,7-2,55 (м, 12H), 2,55-0,5 (м, 13H) R 1 1 m Спектр H—ЯМР или Т.пл., °С CH3 19 7,8-7,1 (м, 8H), 5,5 (широкий с, 1H), 3,9-2,65 (м, 13H), 2,650,5 (м, 12H) CH CH3 H 2 7,7-6,8 (м, 13H), 5,35 (с, 1H), 3,7-2,5 (м, 14H), 2,5-1,5 (м, 6H) 20 21 4–Cl 22 H 7,8-6,9 (м, 12H), 5,6 (с, 1H), 3,9-2,6 (м, 12H), 2,6-1,6 (м, 6H) 3 148 13 27224 Пример № M R 1 1 m 2 23 Спектр H—ЯМР или Т.пл., °С 198-200 S 24 4–CH3 7,9-7,0 (м, 10H), 5,40 (м, 1H), 3,9-2,6 (м, 12H), 2,6-1,6 (м, 9H) S 25 O 26 7,8-6,4 (м, 11H), 5,3 (с, 1H), 3,9-2,6 (м, 12H), 2,4-1,6 (м, 6H) H N 27 7,6-7,2 (м, 8H), 6,8 (с, 1H), 6,4 (с, 1H), 6,05 (с, 1H), 5,4 (с, 1H), 3,7 (м, 2H), 3,4-2,6 (м, 7H), 3,0 (с, 3H), 2,4 (м, 2H), 2,1 (м, 2H), 1,7 (м, 2H) N N S 28 2 203 H 29 198-200 30 180 (с разложением) F Таблица 5 HO N H (CH2)2 C CH3 O CH2 N OM Cl Cl Пример № 1 М Спектр Н-ЯМР 14 *HCl 27224 Пример № 1 М Спектр Н-ЯМР 31 7,6-6,9 (м, 13Н), 5,35 (широкий с, 1Н), 5,1-4,6 (м, 2Н), 3,7-2,5 (м, 12Н), 2,5-1,5 (м, 6Н) 32 7,7-7,0 (м, 11Н), 6,85 (д, J=8 Гц, 1Н), 6,75 (д, J=8 Гц, 1Н), 5,35 (с, 1Н), 3,8-2,6 (м, 12Н), 2,4 (м, 2Н), 2,1 (м, 2Н), 1,7 (м, 2Н) Пример 33 Гидрохлорид N-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-фенилпиперидинил)бутил]-N-метил-2-тиофенкарбоксамида. HO Cl ; Ar´= Y N Cl m=2; N = ; O R=CH3; T Z= S ; a) N-[2-(3,4-диxлopфeнил)-4-(2-тeтpaгидpoпиpaнилoкcи)бyтил]-2- тиoфeнкapбoкcaмид. При комнатной температуре перемешивают смесь, состоящую из 4,77 г амина, полученного в соответствии с примером 1 на стадии в), и 1,7 г триэтиламина и 50 мл дихлорметана. Потом прибавляют по каплям при комнатной температуре 2,19 г 2-тиеноилхлорида, растворенного в 20 мл дихлорметана, и оставляют при комнатной температуре на ночь. Растворитель упаривают под вакуумом, остаток промывают водой, экстрагируют эфиром, промывают 5% раствором бикарбоната натрия, затем насыщенным раствором хлорида натрия, органическую фазу декантируют, сушат на Na2SO4, фильтруют и упаривают под вакуумом. Остаток хроматографируют на силикагеле, элюент: метанол/дихлорметан-98/2 (об/об). Чистые фракции собирают и концентрируют под вакуумом. Остаток промывают 5% раствором гидроксида натрия, экстрагируют эфиром и сушат. Получат 4,6 г бесцветного жидкого масла. в) N-[2-(3,4-дихлорфенил)-4-(2-тетрагидропиранилокси)бутил]-N-метил-2- тиофенкарбоксамид. При комнатной температуре перемешивают смесь, состоящую из 3,4 г полученного перед этим амида и 0,45 г гидрида натрия с содержанием 55% в 10 мл тетрагидрофурана. Реакционная смесь становится прозрачной жидкостью оранжевого цвета. Затем прибавляют 1,23 г иодометана в 10 мл тетрагидрофурана и потом перемешивают в течение 1 часа при комнатной температуре и кипятят в течение 1 часа. Упаривают тетрагидрофуран, остаток разбавляют водой, экстрагируют в эфире, промывают второй раз раствором хлорида натрия и концентрируют под вакуумом. Выход-3,4 г. c) N-[2-(3,4-дихлорфенил)-4-гидроксибутил]-N-метил-2-тиофенкарбоксамид. Растворяют 3,5 г полученного перед этим продукта в 30 мл метанола в присутствии 0,35 г ионообменной смолы (Амберлист Н-15 Олдридж, сухая кислотная сульфосмола) и кипятят смесь в течение 1 часа 30 минут. Смесь фильтруют на целите, фильтрат упаривают под вакуумом, остаток промывают гексаном, затем экстрагируют смесью эфир/гексан. Получают 2,6 г кристаллов белого цвета. Т.пл.-107-109°С. d) N-[2-(3,4-дихлорфенил)-4-метансульфонилоксибутил]-N-метил-2-тиофен-карбоксамид. При комнатой температуре перемешивают 2 г полученного перед этим спирта и 0,65 г триэтиламина в 30 мл дихлорметана. Затем прибавляют по капле раствор, содержащий 0,69 г мезилхлорида в 10 мл дихлорметана. По окончании прибавления, кипятят в течение 30 минут. Упаривают дихлорметан под вакуумом, промывают остаток водой, экстрагируют этилацетатом, промывают 5 % раствором бикарбоната натрия, затем насыщенным раствором хлорида натрия, сушат на Na2SO4, упаривают растворитель. Выход-1,2 г. e) Соединение 33. Смесь, состоящая из 1 г полученного перед этим продукта, 1 г 4-гидрокси-4-фенилпиперидина в 2 мл диметилформамида, нагревают при 60°С в течение 2 часов. После охлаждения разбавляют эфиром, промывают водой, затем разбавленным раствором гидроксида натрия. Сушат на MgSО4, затем выпаривают растворитель. Остаток хроматографируют на силикагеле элюент: градиент дихлорметан – дихлорметан с добавкой 2,6% метанола. Получают 0,7 г продукта, который превращается в гидрохлорид, после растворе 15 27224 ния в дихлорметане прибавляют раствор хлороводорода в эфире до рН=1, затем концентрируют под вакуумом. Гидрохлорид растирают с эфиром. Выход-0,74 г. 1 Н-ЯМР: 7.8-6.8 (м, IIН), 5,3 (с, ІН), 3,8-2,5 (м, 12Н), 2,5-1.4 (м, 6Н). Соединения, описанные в таблице 6, были получены в соответствии с примером 33. Все эти соединения являются гидрохлоридами. Пример 34 Гидрохлорид N-метил-[2-(3,4-дихлорфенил)-4-(4-гидрокси-4-(4-гидроксимфенил) пиперидинил)бутил]-2тиофенкарбоксамида. HO Cl ; Ar´= Y N = HO Cl m=2; N ; O R=CH3; T Z= S ; а) Получение амина: 4-гидрокси-4-(4-гидроксимфенил)пиперидин. Стадия 1 4-бензилоксибромбензол. Перемешивают 32,6 г 4-бромфенола, 34,2 г бензилбромамида и 42 г карбоната калия в 160 мл диметилформамида при температуре 40°С в течение двух часов. Раствор упаривают под вакуумом, остаток промывают водой и экстрагируют эфиром, промывают водой, декантируют, сушат на MgSO4 и упаривают под вакуумом. Остаток перекристаллизовывают из изопропанола. Выход - 30 г. Т.пл.=61°С. Стадия 2 1-бензил-4-(4- бензилоксифенил)-4-гидроксипиперидин. Растворяют 14 г полученного перед этим продукта в 100 мл тетрагидрофурана и прибавляют к 1,2 г магния, находящегося в 20 мл тетрагидрофурана, при температуре 60°С. По окончании прибавления поддерживают температуру 60°С в течении 2 часов и охлаждают затем до -10°С. Потом прибавляют по каплям раствор 10 г 4-бензилпиперидина и дают вернуться к комнатной температуре. Смесь промывают насыщенным раствором хлорида аммония, экстрагируют эфиром, промывают водой, декантируют, сушат на MgSO 4 и упаривают под вакуумом. Остаток хроматографируют на силикагеле, элюент: дихлорметан/метанол -97,6/2,5 (об/об). Упаривание чистых фракций дает 9 г ожидаемого продукта. Т.пл.-104-107°С. Стадия 3. 4-гидрокси-4-(4-гидроксифенил)пиперидин. При комнатной температуре, атмосферном давлении 6 г полученного перед этим продукта, растворенного в 200 мл этанола, гидрируют в присутствии 10% палладия на угле. Когда теоретический объем водорода поглощается, отфильтровывают катализатор, концентрируют фильтрат под вакуумом, извлекают остаток эфиром и отфильтровывают кристаллы. Выход-1,1 г Т.пл.-232-235°С. в) Соединение 34. Растворяют 2,1 г продукта, полученного ранее в соответствии с примером 33 на стадии d), 1 г амина, полученного выше на стадии 3, и 1,1 г триэтиламина в 5 мл диметилформамида и нагревают при температуре 80°С в течение 2 часов. Концентрируют смесь под вакуумом, остаток разбавляют водой и подкисляют до значения рН=3 раствором хлороводородной кислоты, экстрагируют этилацетатом, декантируют, сушат на MgSO4 и упаривают под вакуумом. Остаток разбавляют ацетоном и осаждается из эфира. Выход - 0,7 г. 1 Н-ЯМР: 9.3 (с,. 1Н), 6.6-8 (м, 11 Н), 5.2 (с, 1Н), 2,6 - 4 (м, 12Н), 1.6-2,4 (м. 6 Н). Таблица 6 16 27224 N Y CH3 H (CH2)2 C CH2 N O S *HCl Cl Cl Пример № Y N 1 Спектр - Н-ЯМР HO 35 Cl 7,8-6,8 (м, 10Н), 5,5 (м, 1Н), 4,0-2,6 (м, 12Н), 2,61.5 (м, 6Н) N HO N 36 8-7 (м, 10Н), 5,8 (с, 1Н), 4-2,7 (м, 12Н), 2,7-1,7 (м, 6Н) CF3 HO 37 N Cl 7,85 (с, 1Н), 7,75-7,2 (м, 7Н), 7,05 (с, 1Н), 5,8 (с, 1Н), 3,8-2,6 (м, 12Н), 2,6-1,6 (м, 6Н) CF3 38 N N 6,8-7,8 (м, 6Н), 2,6-4 (м, 17Н), 0,8-2,2 (м, 18Н) 39 N N 8,2 (с, 2Н), 6,7-7,8 (м, 1Н), 2-4 (м, 18Н) N N 40 6,7-7,8 (м, 10Н), 1,8-4 (м, 10Н) OMe N 41 7-8 (м, 10Н), 6,35 (с, 1Н), 2-4,2 (м, 16Н) CF3 Пример 42 Гидрохлорид N-[4-(4-бензил-4-ацетилоксипиперидинил)-2-(3,4-дихлорфенил)бутил]-2,4-дихлорбензамида. 17 27224 AcO Cl ; Ar´= Y N N = ; Cl Cl ; T Z= R=H; m=2; O Cl К 0,4 г гидрохлорида N-[4-(4-бензил-4-гидроксипиперидинил)-2-(3,4-дихлорфенил) бутил]-2,4-дихлорбензамида (соединение 7, полученное в соответствии с примером 1), растворенного в 10 мл дихлорметана в присутствии двух эквивалентов триэтиламина прибавляют 0,12 г ацетилхлорида. После перемешивания в течение 1 часа, при комнатной температуре промывают водой, декантируют органическую фазу, сушат на MgSO4 и упаривают подвакуумом. Остаток хроматографируют на силикагеле, элюент: дихлорметан, затем дихлорметан/метанол-96/9 (об/об). Фракции чистого продукта упаривают под вакуумом, подкисляют до рН=1 и упаривают под вакуумом. Гидрохлорид выпадает из эфира. Выход 0,26 г. 1 Н-ЯМР: 8.5 (т, 1Н), 7,7-6,98 (м, 11Н), 3,6-2,6 (м, 11Н), 2,4-1,8 (м, 9Н). Соединения, описанные в таблице 7, были получены в соответствии с примерами 1-42. Таблица 7 X CH3 H (CH2)2 C N CH2 O S N *HCl R1 Cl Cl Пример № R 43 H 44 4–Cl 1 1 X O Спектр H—ЯМР CH3 7,7-6,9 (м, 10H), 3,8-2,5 (м, 12H), 2,5-2,1 (м, 6H), 2,0 (с, 3H) O 45 7,9-6,9 (м, 11H), 4,0-2,65 (м, 12H), 2,68-1,8 (м, 9H) –CN 7-7,9 (м, 1H), 2-4,1 (м, 18H) CH3 46 7-7,8 (м, 11H), 1,8-4 (м, 21H) O H OCH2CH3 47 7-7,9 (м, 11H), 2-4,3 (м, 20H), 1,15 (т, 3H) O Таблица 8 18 27224 HO N (CH2)2 O R H C Z1 CH2 N Z2 *HCl Ar´ Пример № Ar´ R 48 Z 1 Z 2 1 Спектр H—ЯМР или Т.пл., °С CH3 186 H H 49 H 148-152 50 CH3 144-146 51 H 8,4 (д, J=8 Гц, 1H), 8,0-7,7 (м, 4H), 7,7-7,2 (м, 8H), 6,6 (м, 2H), 5,4 (с, 1H), 4,1-2,8 (м, 9H), 3,8 (с, 3H), 3,6 (с, 3H), 2,5-2,2 (м, 4H), 1,8 (м, 2H) OCH3 OCH3 52 140-145 (с разложением) 53 118 S CH3 54 H H 7,7-7,6 (м, 15H), 5,4 (м, 1H), 4,0-2,6 (м, 15H), 2,4-1,6 (м, 6H) N CH3 Таблица 9 Y N H (CH2)2 C CH3 CH2 N Cl Cl 19 O *HCl 27224 Пример № Y N 1 Спектр - Н-ЯМР или Т.пл., °С O H3C 55 7,8-6,9 (м, 12Н), 3,9-2,7 (м, 12Н), 2,7-1,8 (м, 9Н) O Cl N HO 56 F 200 N H3C 57 7,8-6,8 (м, 13Н), 4,6 (м, 1Н), 3,8-1,8 (м, 20Н) N CH3CH2 O N 58 7,8-6,9 (м, 13Н), 3,8-2,6 (м, 14Н), 2,5-1,9 (м, 6Н), 1,1 (т, J=6 Гц, 3H) O 59 H3C 203 O N O CH3CH2 60 198 O N O O 61 140 N H2N 62 163 N 20 27224 Пример № Y O 63 N 1 Спектр - Н-ЯМР или Т.пл., °С H N H3C 144 N S N 64 188-190 HO N 65 N 134 OMe 66 N MeO N 114-116 OH 67 N 128-130 N Таблица 10 X N Ar (CH2)x H (CH2)2 C R CH2 N O Z *HCl Ar´ X Пример № N 68 R N Ar (CH2)x Z CH3 7,7-6,8 (м, 12Н), 4,7 (с, 1Н), 3,62,4 (м, 16Н), 2,2-1,4 (м, 6Н) N 69 1 Спектр - Н-ЯМР или Т.пл., °С 7,7-6,8 (м, 22Н), 3,8-2,5 (м, 14Н), 2,6-2,0 (м, 6Н) CH 21 27224 X Пример № Ar (CH2)x N R Z N 70 1 Спектр - Н-ЯМР или Т.пл., °С 232 H3COCO Пример 71 Гидрохлорид N-метил-N-[4-(4-фенил-4-ацетиламинопиперидинил)-2-(3,4-дихлорфенил)бутил]бензамамида. А/ Получение амина : Гидрохлорид 4-ацетамидо-4-фенилпиперидина. а) Гидрохлорид 4-ацетамидо-4-фенил-1-бензилпиперидина. Прибавляют по каплям 260 мл 95% серной кислоты к 69 г 4-гидрокси-4-фенил-1-бензилпиперидину, в виде суспензии в 300 мл ацетонитрила, поддерживая температуру между 25 и 30°С. Потом реакционную смесь перемешивают при комнатной температуре в течение 4 часов, затем постепенно выливают на лед и нейтрализуют 30% раствором гидроксида натрия. Осадок отделяют путем фильтрования, промывают водой, затем сушат ацетоном. Выход-58 г. Т.пл. =180,6-182°С. в) Гидрохлорид 4-ацетамидо-4-фенилпиперидина. К 58 г полученного перед этим продукта, растворенного в 600 мл метанола, прибавляют эфир, насыщенный хлороводородом до рН=1. Затем гидрируют при атмосферном давлении и комнатной температуре в присутствии 10% палладия на угле. Когда поглотится теоретический объем водорода, отделяют катализатор путем фильтрования, упаривают фильтрат под вакуумом и перекристаллизовывают остаток из этанола. Выход-45 г. Т.пл. =286,5-288°С. В) Получение соединения 71. а) N-метил-N-[4-метансульфонилокси-2-(3,4-дихлорфенил)бутил]бензамамид Это соединение получают в соответствии с примером 1 стадия е). Т.пл.=100-102°С в) Соединение 71. При температуре 90°С в течение 2 часов нагревают 1,4 г 4-ацетамидо-4-фенилпиперидина и 1,4 г полученного выше мезилата в 3 мл ДМФА. Прибавляют лед и экстрагируют дихлорметаном. Органическую фазу декантируют и последовательно промывают водой, затем насыщенным раствором NaCL и сушат на MgSO4. Упаривают под вакуумом и остаток хроматографируют на силикагеле, элюент: дихлорметан/метанол 97/3 (об/об). Упаривание фракций чистого продукта дает остаток, который растворяют в метаноле. Гидрохлорид получают путем прибавления хлороводорода в эфире. Выход-0,8 г. 1 Н-ЯМР: ЗН при 2 (с, СН3-С(О)-), 18Н при 2,10-3,9 (м, N-СН3, все СН3, СН-С6Н5), 13 Н между 7,00 и 7,80 (м, Н ароматические), 1Н при 8,20 (с, NH-C(O)-). Пример 72 Гидрохлорид (-)-N-метил-N-[4-(4-фенил-4-ацетиламинопиперидинил)-2-(3,4-дихлорфенил)бутил]бензамамида. Стадия 1. a-(2-тетрагидропиранилоксиэтил)-3,4-дихлорбезолацетонитрил. Это соединие получают в соответствии с примером 1, стадия а). Стадия 2. b-(2-тетрагидропиранилоксиэтил)-3,4-дихлорбезолэтанамин. Это соединие получают в соответствии с примером 1, стадия в). Стадия 3. b-гидроксиэтил-3,4-дихлорбезолэтанамин. Растворяют 81 г продукта, полученного перед этим в соответствии со стадией 2, в 38 мл метанола. Прибавляют 90 мл раствора эфира, насыщенного хлороводородом, поддерживая температуру между 20 и 25°С. Перемешивают в течение 30 минут при комнатной температуре, затем концентрируют досуха. Растворяют остаток в 250 мл воды, промывают 2 раза эфиром, подщелачивают раствором NaOH, экстрагируют дихлорметаном. После сушки на MgSO4 концентрируют досуха, разбавляют 800 мл изопропилового эфира, удаляют нерастворимую часть фильтрованием на целите, концентрируют под вакуумом, вводят за 22 27224 травку (кристаллы аминоспирта) для кристаллизации, при перемешивании в течение ночи. Фильтруют, промывают изопропиловым спиртом, затем пентаном. Получают 30,2 г ожидаемого продукта. Т.пл.=90-91°С. Стадия 4. (+)-b-гидроксиэтил-3,4-дихлорбезолэтанамин. К кипящему раствору, содержащему 29 г (-)-Д-винной кислоты, в 800 мл метанола, прибавляют раствор, содержащий 44,7 г продукта, полученного в соответствии с предыдущей стадией 3, в 300 мл метанола. Дают вернуться к комнатной температуре и перемешивают в течение 4 часов. Фильтруют, промывают этанолом, затем эфиром. Получают 34,1 г тартрата. Перекристаллизовывают из 1,75 л метанола, в результате чего получают 26,6 г тартрата. aд25=-4.2 (c=1, H2O). Тартрат растворяют в 120 мл воды. Подщелачивают раствором NaOH, экстрагируют 2 раза дихлорметаном, сушат на MgSO4, упаривают досуха. Разбавляют небольшим количеством изопропилового эфира, прибавляют пентан, фильтруют, в результате чего получают 16,4 г продукта. Т.пл.=79-80°С. aд25 =+9.4 (с=1, МеОН). Стадия 5. N-(4-гидрокси-2-(3,4-дихлорфенил)бутил)этилкарбамат. Растворяют 15 г продукта, полученного в соответствии с предыдущей стадией 4, в 200 мл дихлорметана. Прибавляют 9,9 мл триэтиламина. Охлаждают до температуры 0°С и прибавляют по капле при этой температуре раствор, содержащий 6,3 мл этилхлорформиата в 30 мл дихлорметана. Спустя 15 минут промывают водой, затем разбавленной НСl, потом насыщенным водным раствором NаНСО3. После сушки на MgSO4 упаривают досуха, в результате получают 20 г продукта в виде жидкого масла. Стадия 6. (+)-М-метил-(3-гидроксиэтил-3,4-дихлорбезолэтанамин. К 5,1 г алюмогидрида лития, суспензия в 60 мл безводного ТГФ, прибавляют раствор, содержащий 20 г продукта, полученного в соответствии с предыдущей стадией 5, в 150 мл безводного ТГФ. Кипятят в течение 1 часа. Гидролизуют 20 мл воды, отфильтровывают твердый остаток, упаривают досуха. Полученное жидкое масло растворяют в 100 мл ацетона. Прибавляют хлороводород в эфире до рН=1, затем эфир до образования мути. Перемешивают в течение 1 часа, отфильтровывают кристаллы, промывают небольшим количеством ацетона, затем эфира, в результате чего получают 11 г ожидаемого продукта. Т.пл.=129°С aд25 =+8.4 (с=1, МеОН). Стадия 7. (-)-N-метил-N-[4-гидрокси-2-(3,4-дихлорфенил)бутил]бензамамид. К 8,1 г продукта, полученного в соответствии с предыдущей стадией 6, в виде суспензии в 120 мл дихлорметана, прибавляют 3,4 мл триэтиламина. Охлаждают до температуры 0°С и прибавляют по капле раствор, содержащий 3,4 мл бензоилхлорида в 35 мл дихлорметана. Спустя 15 минут промывают водой, затем разбавленной НСl, потом водным раствором NaHCO3. Сушат на MgSO4, концентрируют досуха. Получают твердое вещество, которое переносят в эфир и фильтруют. Выход= 9.0 г. Т.пл.=129°С. aд25 =-19 (с=1, МеОН). Стадия 8. (-)-N-метил-N-[4-метансульфонилокси-2-(3,4-дихлорфенил)бутил]бензамамид. К 10,5 г продукта, полученного в соответствии с предыдущей стадией 7, растворенного в 120 мл дихлорметана, прибавляют 4,8 мл триэтиламина. Охлаждают до температуры 0°С и прибавляют по капле 2,7 мл метансульфонилхлорида. Спустя 15 минут промывают 2 раза водой, затем насыщенным водным раствором NaCl. Сушат на MgSО4, концентрируют досуха, в результате чего получают пенообразное вещество. aд25 =-2,3 (с=1, СНСІ3). Стадия 9. Соединение 72. Растворяют 22,7 г гидрохлорида 4-фенил-4-ацетиламинопиперидина в 20 мл воды. Прибавляют 10 мл концентрированного раствора гидроксида натрия. Экстрагируют 2 раза дихлорметаном, сушат на MgSO4. Полученный раствор прибавляют к продукту, полученному на стадии 8. Концентрируют досуха, прибавляют 30 мл ДМФА и нагревают при температуре 70°С в течение 1 часа 30 минут. Очень медленно приливают раствор к 30 мл смеси воды и льда. Отфильтровывают осадок, промывают его за несколько раз водой и центрифугируют (обезвоживают) его. Очищают методом хроматографии на кремнеземе, элюент: чистый дихлорметан, затем дихлорметан с добавкой метанола до 10%. Гидрохлорид: основание растворяют в ацетоне. Прибавляют эфир, насыщенный хлороводородом до рН=1. Приливают раствор к изопропиловому эфиру, фильтруют, сушат. Выход 11г. aд25 =-29,5 (с=1, МеОН). 1 Н-ЯМР: ЗН при 1,95 (с, СН3-С(О)-), 18 Н между 2,00 и 3,76 (м, N-СН3, все СН2, СН-С6Н5), 13Y при 6,80 и 7,70 (м, Н ароматические), 1 Н при 8,10 (с, NH-C(O)-). 23 27224 Пример 73. Гидрохлорид (+)-Н-метил-[4-(4-фенил-4-ацетиламинопиперидинил)-2-(3,4-дихлорфенил)бутил]бензамамида. Энантиомер (+) получают в соответствии с той же самой методикой, что и для (-) энантиомер, описанной выше в примере 41, заменяя на стадии 4 (-)-Д-винную кислоту на (+)-L-винную кислоту. aд25 =+30.6 (с=1. МеОН). 1 Н-ЯМР: 3 Н при 1,85 (с, СН3-С(О)-), 18 Н между 2,00 и 3,75 (м. N-СН3, все СН3, СН-С6Н5), 13 Н между 6,80 и 7,70 (м, Н ароматические), 1 Н при 8,10 (с, NH-C(O)-). Пример 74 Гидрохлорид (-)-N-метил-N-[4-(4-гидрокси-4-фенилпиперидинил)-2-(2,4-дихлорфенил)бутил]-2-тиофенкарбоксамида. Это соединение получают при проведении синтеза в соответствии с примером 72, приведенным выше. aд25 =-51,0 (с=1, МеОН). Пример 75 Гидрохлорид (+)-N-метил-N-[4-(4-гидрокси-4-фенилпиперидинил)-2-(2,4-дихлорфенил)бутил]-2-тиофенкарбоксамида. Это соединение получают при проведении синтеза в соответствии с примерами 72 и 73, приведенными выше. aд25 =+51,7 (с=1, МеОН). Спирты, синтезированные в соответствии с примером 1, стадия d), приведенными выше, или в соответствии с примером 42, являются ключевыми промежуточными соединениями для получения соединений формулы (I).В приведенной ниже таблице А описаны различные спирты, полезные для получения соединений формулы (I). Таблица A Промежуточные соединения HO (CH2)2 H C R CH2 N O M Cl Cl Пример № M 1 R (a) Спектр H—ЯМР 6,8-7,8 (м, 8H), 4,5 (шс, 1H), 2,6-4 (м, 8H), 1,3-2,1 (м, 2H) H3C (b) 6,8-7,6 (м, 6H), 3-4,2 (м, 5H), 2,4 (с, 3H), 1,4-2,2 (м, 8H) H3C CH3 S (c) 6,8-7,8 (м, 6H), 4,4 (т, 1H), 2,6-4 (м, 8H), 1,4-1,9 (шс, 2H) (d) 1,3 (м, 2H), 2,6-5 (м, 9H), 8,2-6,2 (м, 10H), 5-2,6 (м, 9H), 1,3 (м, 2H) 24 27224 Тираж 50 екз. ДП «Український інститут промислової власності» (Укрпатент) Україна, 01133, м. Київ-133, бул. Л. Українки, 26 (044) 295 – 81 – 42 (044) 295 – 61 – 97 25
ДивитисяДодаткова інформація
Назва патенту англійськоюArylalkylamines, process for preparing them and pharmaceutical composition based on thereof
Автори англійськоюEmon-Alt Ksave, Gulyauik Per, Proietto Vinchentso, Van Bruk Dide
Назва патенту російськоюАрилалкиламины, способ их получения и фармацевтическая композиция
Автори російськоюЭМОН-АЛЬТ Ксавье, Гуляуик Пъер, Проетто Винченцо, ван Брук Дидье
МПК / Мітки
МПК: C07D 295/12, A61K 31/341, C07D 401/12, C07D 209/14, C07D 211/64, C07D 213/74, C07D 211/62, C07D 409/12, C07D 405/12, C07D 211/26, A61K 31/451, C07D 401/06, C07D 409/06, C07C 233/69, A61K 31/495, C07D 211/22, C07D 213/36, C07D 295/125, A61K 31/4402, C07D 211/34, A61P 11/08, C07D 333/38, A61K 31/505, A61K 31/4433, A61K 31/38, C07D 211/44, C07D 307/68, A61K 31/445, A61K 31/34, C07D 211/48, C07D 239/26, A61P 43/00, C07D 409/04, C07D 295/13, C07D 239/42, C07D 211/52, A61K 31/4427, A61K 31/443, A61K 31/381, C07D 333/20, C07D 333/58, C07D 417/12, C07D 211/32, A61K 31/4406, C07D 403/12
Мітки: одержання, композиція, спосіб, арилалкіламіни, фармацевтична
Код посилання
<a href="https://ua.patents.su/25-27224-arilalkilamini-sposib-kh-oderzhannya-ta-farmacevtichna-kompoziciya.html" target="_blank" rel="follow" title="База патентів України">Арилалкіламіни, спосіб їх одержання та фармацевтична композиція</a>
Похідні n-алкіленпіперидинів як антагоністи рецептора нейрокініну, спосіб їх одержання, проміжні сполуки та фармацевтична композиція

Номер патенту: 26894
Опубліковано: 29.12.1999
Автори: Мартінез Серж, Емонд-Альт Ксавьє, Пруаєтто Венсанзо, ван Броєкк Дідьє
МПК: A61K 31/4468, A61K 31/4433, C07D 401/06, A61K 31/445, C07D 401/12, A61P 43/00, C07D 211/58, A61K 31/454, C07D 211/46, A61K 31/4427, A61K 31/4465, C07D 409/12, C07D 403/12, A61K 31/44, A61K 31/4545, C07D 211/54
Мітки: n-алкіленпіперидинів, композиція, проміжні, фармацевтична, похідні, одержання, сполуки, нейрокініну, антагоністи, рецептора, спосіб
Текст:
...где Alk является (С,-Сэ)алкильной группой, R представляет собой атом водорода 40 или С^-С^алкил, Z представляет собой фенил, незамещенный или моно- или дизамещенный галогеном или (С^-Сзіалкоксигруппой, наф 26894 7 тил, замещенный галогеном, фенилметильной группой, замещенной на фениле (С,С4}алкоксигруппой, * обозначает хиральный центр, в виде рацемата или оптически чистых изомеров, или их соли с минеральными или органическими кислотами,...
3-циклоалкілпропанамід, що проявляє протизапальну і імунодепресивну активність, спосіб його одержання, ціанамід і фармацевтична композиція

Номер патенту: 26393
Опубліковано: 30.08.1999
Автори: Кей Девід Пол, Куо Елізабет Анн, Хеджекок Чарльз Джон Роберт, Хамблетон Філіп Томас, Туллі Вільфред Роджер
МПК: C07C 255/27, A61K 31/16, C07C 271/26
Мітки: одержання, 3-циклоалкілпропанамід, ціанамід, протизапальну, активність, проявляє, композиція, спосіб, фармацевтична, імунодепресивну
Формула / Реферат:
1. 3-Циклоалкилпропанамид общей формулы l:где R1 - C1-C6-циклоалкил;R2 - атом водорода, C1-C6-алкил;R3, R4, R5, R6 и R7 - одинаковые или различные - атом водорода, атом галогена, прямолинейный или разветвленный алкил или алкоксигруппа с 1 - 6 атомами углерода, алкилтиогруппа с 1 - 6 атомами углерода, радикал -(CH2)n, -CF3, -O-(CH2)m- -CF3, -S-(CH2)m-CF3, где m - целое число между 0 и 3,...
Похідні триазолопіримідину, які мають антагоністичну активність до рецепторів ангіотензину іі, та фармацевтична композиція, що їх містить

Номер патенту: 26627
Опубліковано: 11.10.1999
Автори: Ніколя Ерік, Брю-Маньє Ніколь, Тьолон Жан-Марі
МПК: A61P 9/04, A61P 43/00, A61P 9/00, A61P 9/12, C07D 487/04, A61K 31/519, A61P 9/10, A61K 31/505
Мітки: антагоністичну, активність, рецепторів, містить, іі, ангіотензину, фармацевтична, мають, похідні, композиція, триазолопіримідину
Формула / Реферат:
1. Производные триазолопиримидина общей формулы lгде R1 является низшим алкилом с 1 - 6 атомами углерода или C3-C7-циклоалкилом;R2 является атомом водорода, низшим алкилом с 1 - 6 атомами углерода, группой NH-NH2, группой (CH2)mOR4, (CH2)mSR4;R4 является атомом водорода, низшим алкилом с 1 - 6 атомами углерода и m равно целому числу от 0 до 2;группа является радикалом, выбранным среди следующих...
Сіль, яка утворена ранітидином та комплексом вісмуту з карбоновою кислотою, спосіб її одержання, фармацевтична композиція на її основі

Номер патенту: 26669
Опубліковано: 12.11.1999
Автор: Клітроу Джон Уотсон
МПК: C07D 307/52
Мітки: кислотою, яка, композиція, основі, сіль, спосіб, фармацевтична, ранітидином, карбоновою, вісмуту, комплексом, одержання, утворена
Формула / Реферат:
1. Соль, образованная ранитидином и комплексом висмута с карбоновой кислотой, или сольват такой соли, где карбоновая кислота выбрана из лимонной или винной кислот, обладающая терапевтической активностью.2. Соль по п.1, где она представляет собой N-[2-[[[5-[(диметиламино)метил]-2-фуранил]метил]тио]этил]-N'-метил-2-нитро-1,1-этендиамин-2-гидрокси-1,2,3-пропантрикарбоксилат висмута (3+) и ее сольваты.3. Соль по п.1, где она...
Похідні n-заміщеного 4-феніл-4-піперидинкарбоксаміду та їх фармацевтично прийнятні солі, що проявляють анестезувальну та анальгезувальну дію, спосіб їх одержання та фармацевтична композиція

Номер патенту: 26403
Опубліковано: 30.08.1999
Автори: Аск Анна-Лена, Сандберг Руне
МПК: A61P 29/00, A61P 25/04, A61K 31/451, C07D 211/64, C07D 211/52, A61K 31/445
Мітки: спосіб, солі, анальгезувальну, прийнятні, анестезувальну, дію, 4-феніл-4-піперидинкарбоксаміду, похідні, одержання, проявляють, фармацевтична, n-заміщеного, композиція, фармацевтично
Формула / Реферат:
1. Производные N-замещенного 4-фенил-4-пиперидинкарбоксамида общей формулыгде R1 представляет собой алкильную группу с 2 - 6 атомами углерода или алкоксиалкильную группу R4O(CH2)m, в которой R4 представляет собой алкильную группу с 1 - 4 атомами углерода и m равно 2 - 4;R2 и R3 являются одинаковыми или различными и каждый представляет собой алкильную группу с числом атомов углерода до 6, или R2 и R3 образуют вместе цепь...
Попередній патент: Спосіб безперервного виготовлення панелей типу “сендвіч” та пристрій для його здійснення
Наступний патент: Спосіб каталітичного очищення автомобільних відпрацьованих газів під час холодного пуску двигуна
Випадковий патент: Спосіб довгострокового збереження живців плодово-ягідних культур